Druckerei: Bösmüller Print Management GesmbH & Co. KG. Verlags- und Herstellungs ort: Wien. Grundlegende Richtung: Unabhängige österreichische Fachzeitschrift für niedergelassene Ärztinnen und Ärzte. Die HAUSÄRZT:IN – Praxis-Magazin für Primärversorgung – ist ein interdisziplinäres Informations- und Fortbildungsmedium. Aus Gründen der besseren Lesbarkeit verzichten wir in den Artikeln teilweise auf die gendergerechte Schreibweise. Sofern nicht anders vermerkt, gelten alle Bezeichnungen für sämtliche Geschlechter. Namentlich gekennzeichnete Beiträge geben nicht unbedingt die Meinung der Redaktion oder des Verlages wieder, sondern fallen in den Verantwortungsbereich der Autor:innen. Der Inhalt von entgeltlichen Einschaltungen und Beilagen sowie die Angaben über Dosierungen und Applikationsformen liegen außerhalb der Verantwortung der Redaktion oder des Verlages und sind vom/von der jeweiligen Anwender:in im Einzelfall auf ihre Richtigkeit zu überprüfen. Nachdruck nur mit schriftlicher Genehmigung des Verlages. Alle Rechte, insbesondere das Recht der Vervielfältigung und Verbreitung sowie der Übersetzung, vorbehalten. Kein Teil des Werkes darf in irgendeiner Form (Fotokopie, Mikrofilm oder ein anderes Verfahren) ohne schriftliche Genehmigung des Verlages reproduziert oder unter Verwendung elektronischer Systeme gespeichert, verarbeitet, vervielfältigt, verwertet oder verbreitet werden. Mit „Bezahlte Anzeige“ gekennzeichnete Beiträge/Seiten sind gemäß §26 Mediengesetz bezahlte Auftragswerke. Offenlegung: gesund.at/impressum

Das Seltene im Blick RARE DISEASE DIALOG 11/2022 Akromegalie: Früherkennung und Management im Fokus Wachstum Disproportionales Diagnostisches Licht ins Dunkel bringen Netzhautdystrophien Hereditäre IMPRESSUM: Herausgeber und Medieninhaber: RMA Gesundheit GmbH, Am Belvedere 10 / Top 5, 1100 Wien, Tel. 01/74321708114, office@gesund.at. Geschäftsführung: Mag.a Birgit Frassl, Marlis Rumler. Redaktionsleitung: Mag.a Karin Martin. Redaktion: Mag.a Karin Martin, Anna Schuster, BSc, Mag.a Ines Pamminger, BA, Margit Koudelka. Lektorat: Mag.a Katharina Maier. Produktion & Grafik: Helena Valasaki, BA. Cover-Foto: shutterstock.com/AnnaVel Verkaufsleitung: Mag.a Birgit Frassl, birgit.frassl@ regionalmedien.at. Kundenbetreuung: Mag.a Dagmar Halper, dagmar.halper@regionalmedien.at, Ornela-Teodora Chilici, BA, ornela-teodora.chilici@regionalmedien.at.
Rare Disease
03 Teamwork gefragt
Kurzdarmsyndrom: Therapieregime erfordert diätologische, medika mentöse und chirurgische Maßnahmen
06 Der Weg bis zur Diagnose ist oft lange Idiopathische pulmonale Fibrose: Update Diagnostik und Therapie
08 „Das Ziel der antiepileptischen Medikation rückt näher“ Pädiatrische Epileptologie bei seltenen Erkrankungsformen
12 Disproportionales Wachstum
1. November ist WeltAkromegalie-Tag
15 Diagnostisches Licht ins Dunkel bringen
Lege artis muss bei hereditären Netzhaut dystrophien ein Gentest erfolgen
18 „Schön langsam wächst die Awareness“
Transthyretin-Amyloidose mit Kardiomyopathie frühzeitig erkennen und therapieren

Hausärzt:in Inhaltsverzeichnis
2 November 2022
08
Seltene Epilepsiesyndrome bei Kindern.
© shutterstock.com/WindNight
Beim Kurzdarmsyndrom ist Teamwork gefragt
Das Therapieregime wird in Abhängigkeit von der intestinalen Adaptation ange passt – es erfordert diätologische, medikamentöse und chirurgische Maßnahmen
eine totale parenterale Ernährung“, sagt Elisabeth Hütterer, Diätologin an der Medizinischen Universität Wien. „Ziel ist jedoch, diese ehebaldigst wieder stufen weise auszuschleichen.“
Beim Kurzdarmsyndrom handelt es sich um eine seltene Entität – man geht von einer Prävalenz von 34 Betroffenen pro einer Million Einwohner aus. Bei dieser Form des Darmversagens ist der Dünn darm so verkürzt, dass seine absorptive Funktion reduziert ist – zum Beispiel infolge mesenterialer Durchblutungsstö rungen, wiederholter Resektionen eines komplizierten Morbus Crohn oder einer Strahlenenteritis. Folgen wie eine einge schränkte Nährstoffresorption, vermehr te intestinale Flüssigkeitsverluste sowie osmotische oder sekretorische Diarrhoe machen aus dem Kurzdarmsyndrom ein komplexes Krankheitsbild, welches ein interdisziplinäres Therapiemanagement erfordert. Im Wesentlichen zielt die Be handlung auf eine Gewichtsstabilisie rung, eine bedarfsgerechte Energie- und Nährstoffversorgung sowie auf eine weit gehende Symptomfreiheit ab.
Anatomie des Restdarms entscheidend
Eine Darmresektion kann aufgrund an geborener Anomalien oder erworbener Ursachen notwendig werden – in Frage kommen vaskuläre und entzündliche Er krankungen sowie Traumen oder Tumo ren. Um mögliche klinische Konsequen zen besser abschätzen zu können, ist eine genaue Kenntnis der Art des operativen Eingriffs erforderlich: Der intraoperati
ve Situs und die Länge der verbliebenen Darmabschnitte sollten möglichst gemes sen und dokumentiert werden. „Das me tabolisch-nutritive Defizit bei Patientin nen und Patienten ist höchst individuell“, erklärt Dr. Felix Harpain von der Abtei lung für Allgemeinchirurgie am AKH Wien. In Hinblick auf den Schweregrad der Symptomatik hat sich der Erhalt von Kolon und Ileozäkalklappe als günstig erwiesen, wohingegen die Resektion des Iliums als ungünstig einzustufen ist. „Die verbleibende Dünndarmlänge ist ganz entscheidend für das Outcome“, bestätigt Dr. Harpain. Diese sei Prädiktor für die Notwendigkeit einer parenteralen Er nährungs- und Flüssigkeitstherapie.

Phasengerechte Ernährung
Nach einer Darmresektion werden drei Stadien der Adaptation durchlaufen. Die erste Phase ist durch Hypersekretion und einen intestinalen Flüssigkeits- sowie Elektrolytverlust charakterisiert. Im Zuge von Adaptionsvorgängen sind eine Vertie fung der Krypten und eine Verlängerung der Villi zu beobachten, wodurch sich die Resorptionsfläche des verbliebenen Darms vergrößert und sich konsekutiv auch die Resorptionsleistung verbessert. In Hinblick auf den Krankheitsverlauf ist eine phasengerechte Ernährung un abdingbar. „Unmittelbar nach der Ope ration benötigen Betroffene in der Regel
In der Adaptionsphase wird begleitend zur parenteralen Ernährung eine Son den- oder Trinknahrung verabreicht. Zu beachten ist, dass große Trinkmengen zu einer vermehrten Dünndarm- und Magensekretion und damit auch zu ei ner gehäuften Ausscheidung führen. Die Diätologin erachtet es daher für sinnvoll, kleine Schlucke isotonischer Lösungen zu trinken. „Die Evaluation der Flüssig keitszufuhr und -ausscheidung über 24 Stunden bildet die Grundlage für die Be urteilung der Flüssigkeitshomöostase“, betont Diätologin Hütterer und emp fiehlt, dahingehend Protokoll zu führen. Entscheidend seien eine ausreichende Urinmenge von mehr als einem Liter pro Tag und ein stabiles Körpergewicht. Bis zu zwei Jahre nach dem Eingriff ist das Maximum der Anpassungsfähigkeit des Restdarms erreicht. Je nach individu eller Verträglichkeit kann der Kostaufbau nach dem Prinzip der Leichten Vollkost gesteigert und bedarfsweise durch eine enterale oder parenterale Ernährung so wie durch eine adaptierte Trinknahrung ergänzt werden. Mikronährstoffe und spezifische Verluste müssen jedoch selbst bei vollständiger oraler Autonomie le benslang substituiert werden.
Medikamentöse und chirurgische Behandlungsoptionen
Um die Notwendigkeit einer Substitution zu erkennen und eine solche zu steuern, sollten die Serumwerte betreffend Elektrolyte, Nierenretentionsparameter, Al bumin und Gesamteiweiß sowie Vitami ne und Spurenelemente auch im Verlauf zwei bis viermal jährlich bestimmt werden. „Mängel treten vor allem bei den fettlösli chen Vitaminen A, E, D und K auf“, weiß Priv.-Doz.in Dr.in Stefanie Dabsch von der Klinischen Abteilung für Gastroenterolo
3 Hausärzt:in Rare Disease November 2022 >
© shutterstock.com/KAZLOVA
IRYNA
gie und Hepatologie an der Medizinischen Universität Wien. Das Augenmerk gelte es außerdem auf die ausreichende Versorgung mit Folsäure, Eisen, Selen und Zink zu richten. Eine Kontrolle der Knochendichte sollte im Abstand von zwölf Monaten vorge nommen werden.
Eine zusätzliche medikamentöse Therapie mit Loperamid oder Tinctura opii kann die intestinale Motilität hemmen, wodurch sich die Verweildauer des Speisebreis und die Zeit des Kontakts von Darmmukosa, Verdauungsenzymen und Chymus verlän gern. Insbesondere während der postoperativen Periode und zu Beginn der oralen Ernährung tritt bei vielen Patientinnen und Patienten eine gesteigerte Magensäuresekretion auf, die eine Di arrhö verstärken und die Nährstoffresorption durch Inaktivie rung der Pankreaslipase sowie Dekonjugation der Gallensalze verschlechtern kann. Protonenpumpeninhibitoren wirken dem entgegen. „Pankreasenzyme können mit dem Ziel einer besse ren Fettabsorption substituiert werden“, so die Fachärztin für Innere Medizin. Bei Patienten nach Ileozäkalklappenresektion kann es zu einer aszendierenden bakteriellen Überwucherung des Dünndarms kommen, die eine antibiotische Behandlung indiziert. Durch die Therapie mit Teduglutid, einem Analogon des Peptidhormons Glucagon-like Peptid 2 (GLP-2), kann der Bedarf der Patienten an parenteraler Ernährung verringert wer den. Das Hormon GLP-2 wird in enteroendokrinen L-Zellen des Ileums und proximalen Kolons gemeinsam mit GLP-1 aus einem Vorläuferprotein gebildet. „Teduglutid führt zu einem Zottenwachstum und damit zu einer größeren Darmoberfläche,

EXPERT:INNEN-TEAM:
MedUni Wien/Matern
©
Priv.-Doz.in Dr.in Stefanie Dabsch Klinische Abteilung für Gastroenterologie und Hepatologie, MedUni Wien



Hütterer Diätologin, MedUni Wien
es erhöht den enteralen Blutfluss und verbessert die Aufnahme von Nährstoffen“, erklärt Doz.in Dabsch. Die Lebensqualität der Betroffenen könne dadurch verbessert werden. Auf chirurgischer Ebene ist es in manchen Fällen möglich, die Resorptionskapazität durch Wiederherstellung der Kontinuität distaler ausgeschalteter Darmanteile zu optimieren. In diesem Kontext ebenfalls von Bedeutung sind der „Verschluss von Fisteln, das Aufheben von blinden Schlingen und eine Infektsanierung im Abdomen“, nennt Dr. Harpain operative Opti onen. „Die bei komplikationsträchtigen Krankheitsverläufen mögliche Dünndarmtransplantation wird derzeit nur bei einer Minderheit der Patientinnen und Patienten und in wenigen Zentren durchgeführt – in Österreich gar nicht“, so der Chirurg.
Mag.a Sylvia Neubauer
Dank www.symptomsuche.at finden Sie Seltene Erkrankungen online! Nach Eingabe der Symptome werden mögliche Erkrankungen angezeigt und Sie können die Ursachen der Beschwerden früher eingrenzen. So ist es möglich, Seltene Erkrankungen rascher zu diagnostizieren und zu therapieren –und Sie ersparen Ihren Patient:innen unnötige Irrwege.
Für einzelne Krankheits bildbeschreibungen gibt es DFP-Fortbildungen mittels Online-Test auf www.meindfp.at.
Hausärzt:in Rare Disease
Elisabeth
WELLDONE Rare
Dr. Felix Harpain Abteilung für Allgemein chirurgie, AKH Wien ribbon
© zVg © Bernhard Noll
disease
Seltene Erkrankungen häufiger erkennen
Zugelassen für Patienten ab dem Alter von 1 Jahr mit Kurzdarmsyndrom (KDS) nach einer Phase der intestinalen Adaption.1


























































Freiheit ist möglich:
• Reduktion des parenteralen Ernährungsvolumens1 •Reduktion der Infusionsdauer 1 • Zunahme der enteralen Kalorienzufuhr 1
Führt zu einer Zunahme der Darmzottenhöhe und Darmkryptentiefe
1 Revestive® Fachinformation Mai 2020 C-APROM/AT//1674; 08/2020 Fachkurzinformation siehe Seite 2 www.takeda.at
Der Weg bis zur Diagnose ist oft lange
Idiopathische pulmonale Fibrose: Update zur aktuellen Diagnostik und Therapie





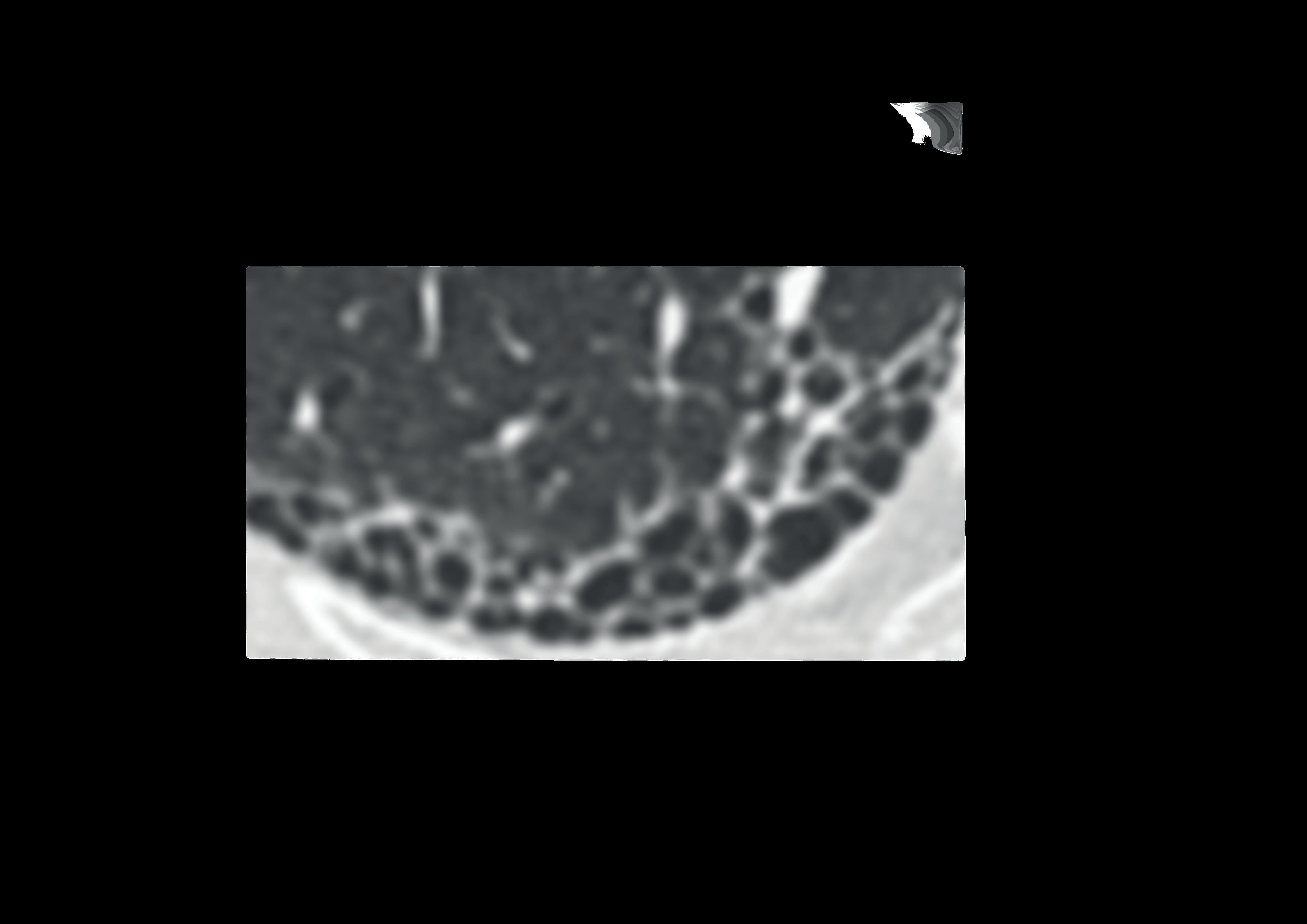
hochauflösende Computertomographie (HRCT) der Lunge durchgeführt (siehe Abbildung 1). Hier zeigt sich das radio logische Muster einer gewöhnlichen in terstitiellen Pneumonie („ u sual intersti tial pneumonia – UIP “)
Fallbeispiel: Ein 70-jähriger Mann sucht wegen langsam zunehmender Dyspnoe – so wie monatelangen trockenen Hustens – seine Hausärztin auf. Er ist Ex-Raucher (50 Pa ckungsjahre) und hat bis auf einen arteriellen Bluthoch druck keine bekannten Vorer krankungen. In der klinischen Untersuchung fallen Trom melschlägelfinger mit Uhrglasnägeln auf. In der Auskultation der Lun ge ist basal beidseits feines Knisterras seln hörbar, das dem Geräusch eines sich öffnenden Klettverschlusses ähnelt. Die periphere Sauerstoffsättigung liegt bei 91 %. Zur weiteren Abklärung wer den ein Lungenröntgen sowie eine Spi rometrie veranlasst. Während das Rönt gen einen unauffälligen Befund ergibt, zeigt die Spirometrie eine restriktive Ventilationsstörung. Aufgrund dieser Befundkonstellation verdichtet sich der Verdacht auf eine interstitielle Lun generkrankung (ILD) und es wird eine
Der Fall wird nun in einer multidisziplinären Fallkonferenz (ILD-Board) besprochen. Nach Ausschluss einer dem radiologischen UIP-Muster zugrunde liegenden Systemerkrankung wird die Diagnose einer idiopathischen pulmo nalen Fibrose (IPF) gestellt und eine antifibrotische The rapie eingeleitet.
Risikogruppen
Die IPF ist per definitionem eine idiopathische Erkran kung. Sie ist durch eine chro nische Fibrose der Lunge charakterisiert, die immer progredient verläuft, mit einer hohen Symptomlast und einer sehr schlechten Prognose einhergeht. Auch wenn wir die Ursache der IPF nicht kennen, sind Risikofaktoren be kannt: Betroffen sind vor allem Männer über 60 Jahre mit Raucheranamnese.
Diagnostische Maßnahmen bei Verdacht auf IPF

Die Symptomatik der IPF ist unspezi fisch. In den meisten Fällen stehen pro grediente Dyspnoe, welche initial bei Belastung, später auch in Ruhe auftritt, trockener Husten und Abgeschlagen
heit im Vordergrund. Einen Hinweis auf das Vorliegen einer fibrosierenden Lungenerkrankung kann die klinische Untersuchung liefern: In der Auskulta tion der Lunge manifestiert sich in vie len Fällen ein charakteristisches Knis tern, die Sklerophonie. Als Ausdruck einer chronischen Hypoxämie kann es darüber hinaus zur Ausbildung von Uhrglasnägeln und Trommelschlägel fingern kommen. Die Lungenfunktion zeigt in der Regel eine restriktive Ven tilationsstörung sowie eine verminder te Diffusionskapazität.

Liegt der Verdacht auf eine IPF vor, müssen zunächst bekannte Ursachen in terstitieller Lungenerkrankungen (z. B. häusliche, berufliche oder Umweltein flüsse, rheumatologische Erkrankun gen oder Medikamententoxizität) aus geschlossen werden, da es sich bei der IPF um eine – per definitionem – idio pathische Erkrankung handelt.
DAS WICHTIGSTE IN KÜRZE
Der Weg bis zur Diagnose der IPF ist oft lange. Im Schnitt vergehen vom Beginn der Beschwerden bis zur endgültigen Diagnosestellung ca. 1,5 Jahre. Eine unspe zifische Symptomatik (Belastungsdyspnoe, trockener Husten, Abgeschlagenheit) und in der Frühphase potenziell unauffällige Thoraxröntgen-Befunde tragen dazu bei, dass die Erkrankung häufig lange nicht diagnostiziert wird.
Bei der optimalen Betreuung von Patien tinnen und Patienten mit IPF kommt der Zusammenarbeit von niedergelassenem Bereich und ILD-Spezialambulanzen in Krankenhäusern eine besonders große Bedeutung zu.
Die multidisziplinäre Falldiskussion im Rah men eines ILD-Boards mit Spezialisten aus Pneumologie, Radiologie und Pathologie ist Goldstandard in der Diagnostik interstitieller Lungenerkrankungen.
Abbildung 1
Abbildung 1: Die HRCT zeigt ein typisches UIP-Muster. Dieses ist durch retikuläre Veränderungen mit einer basalen, subpleuralen Prädominanz, das Auftreten von honigwabenartigen Zysten (Honeycombing) und durch das Fehlen von Hinweisen, die eine alternative Diagnose nahelägen, charakterisiert.
3. Relativ neue antifibrotische Medikamente können den jährlichen Verlust an Lungen funktion bremsen und dadurch das Überle ben mit IPF verlängern. Mit der Einleitung der antifibrotischen Therapie sollte in der Regel möglichst schnell nach Diagnosestel lung begonnen werden.
Hausärzt:in Rare Disease
GASTAUTOR: Dr. Mathis Hochrainer Abt. f. Innere Medizin und Pneumologie, Klinik Floridsdorf, Wien
© privat
Picture Retikuläres Muster 1 2
© shutterstock.com/Ground
M.Hochrainer
Goldstandard in der Diagnostik der IPF ist das interdisziplinäre ILD-Board, in welchem der jeweilige Fall von Ex pertinnen und Experten aus Pneumolo gie, Radiologie, Pathologie und häufig auch Rheumatologie diskutiert wird. Im Zentrum der Diagnostik steht hier die HRCT der Lunge. Anhand genau definierter Kriterien wird das radiolo gische Bild in eine von vier Kategorien eingeteilt: UIP-Muster, wahrscheinli ches UIP-Muster, unbestimmt für ein UIP-Muster oder Muster, das eine al ternative Diagnose nahelegt. Die Di agnose der IPF erfordert neben dem Ausschluss bekannter Ursachen inter stitieller Lungenerkrankungen entwe der das Vorliegen eines UIP-Musters in der HRCT oder eine spezifische Kom bination von Radiologie und Histolo gie. Im ILD-Board wird diskutiert, ob bereits anhand von Klinik, Anamnese, Serologie und Radiologie die Diagnose einer IPF mit ausreichender Sicherheit gestellt werden kann oder ob weitere histo- bzw. zytologische Untersuchun gen erforderlich sind. Ist eine Biopsie notwendig, stehen je nach Befundkon stellation bronchoskopische (broncho alveoläre Lavage (BAL), Kryobiopsie) und thoraxchirurgische Verfahren („v i deo-assisted thoracic surgery “ , VATS) zur Verfügung (Abbildung 2). Hierbei hat die bronchoskopische Kryobiopsie in den letzten Jahren an Bedeutung gewonnen und wird mittlerweile in in ternationalen Leitlinien der VATS vor gezogen.
Krankheitsverlauf und Therapieoptionen
Die IPF ist per se eine unheilbare Er krankung. Ihre Verlaufsformen sind he terogen. Langsam progrediente Verläufe über mehrere Jahre sind genauso mög lich wie sehr rasche Verlaufsformen und akute Exazerbationen, die mit einem mittleren Überleben von 3-4 Monaten einhergehen. Da die IPF vor allem im höheren Lebensalter auftritt und in den meisten Fällen spät nach Symptombe ginn diagnostiziert wird, ist die Lungen transplantation in der klinischen Praxis nur selten eine Option. Seit 2011 bzw. 2015 sind in der EU zwei antifibrotische Medikamente zugelassen: Pirfenidon
und Nintedanib. Mit Hilfe dieser kann der jährliche Verlust an Lungenfunktion nicht verhindert, aber verlangsamt wer den, was statistisch mit einem längeren Überleben einhergeht. Die Therapie mit einem der beiden antifibrotischen Me dikamente sollte möglichst schnell nach Diagnosestellung eingeleitet werden.
Der Einsatz weiterer antifibrotischer Therapieoptionen ist Inhalt aktueller kli nischer Studien.
Neben antifibrotischen Therapien sind die symptomatische Behandlung von Husten und Dyspnoe, die etwaige Ver ordnung von Langzeitsauerstoff und die palliative Therapie wichtige Bestandtei le der Behandlung von Patientinnen und Patienten mit IPF.
Verdacht auf IPF
Anamnese, Klinik, Serologie, HRCT Non-IPF-ILD? ILD-Board HRCT: UIP-Muster HRCT: wahrscheinliches UIP-Muster HRCT: ubestimmtes Muster Alter > 60, männlich, Ex-Raucher? BAL & TB KryoBx Hinweise für Non-IPF-ILD BAL ILD-Board
nein nein
IPF
nein ja ja
Alternative Diagnose Unklarer Befund Chirurgische Bx
Abbildung 2: Diagnosealgorithmus bei IPF. Nach Ausschluss bekannter Ursachen einer ILD kann –wie im eingangs angeführten Fallbeispiel – bei Vorliegen eines typischen UIP-Musters in der HRCT auf eine Biopsie verzichtet werden (roter Pfeil). Liegt ein wahrscheinliches UIP-Muster vor, soll anhand der klinischen Vortestwahrscheinlichkeit individuell entschieden werden, ob bzw. welche Form der Biopsie notwendig ist. ILD = interstitielle Lungenerkrankung, BAL = bronchoalveoläre Lavage, TB KryoBx = transbronchiale Kryobiopsie, Bx = Biopsie. (Modifiziert nach Behr et al. Pneumologie 2020).
Hausärzt:in Rare Disease
<
„Das Ziel der antiepileptischen Medikation rückt näher“
Herausforderungen der pädiatrischen Epileptologie bei seltenen Erkrankungsformen
Die seltenen Epilepsieerkrankungen im Kindesalter – Lennox-Gastaut-Syndrom (LGS), Dravet-Syndrom (DS) und Tuberöse Sklerose (TSC) – weisen eine therapierefraktä re Natur auf. Betroffene, Ange hörige, aber auch behandeln de Ärztinnen und Ärzte sind daher mit diversen Heraus forderungen konfrontiert. Im Gespräch mit der Hausärzt:in berichtet Priv.-Doz.in OÄ Dr.in Gudrun Gröppel vom Kepler Universitätsklinikum unter anderem über die derzeitigen Behandlungsmöglichkeiten dieser Epilepsieformen und die Einsatzmöglichkeiten von CBD.
EXPERTIN:
Priv.-Doz.in OÄ Dr.in Gudrun Gröppel Klinik für Neurologie 1 & Universitätsklinik für Kinder- und Jugend heilkunde am Kepler Uniklinikum, Linz

welche maßgeschneidert für die zugrun deliegende Ursache der Erkrankung ist. Epilepsien direkt am Entste hungspunkt der Anfälle be handeln zu können – und nicht nur das „Symptom“ –, ist eine spannende Perspektive. Somit rückt das Ziel endlich näher, eine antiEPILEPTISCHE Me dikation zu haben – und nicht nur eine AntiANFALLSmedi kation. Zusätzlich ist zu erwar ten, dass auf diesem Weg auch die Komorbiditäten und vor allem die epileptische Enze phalopathie, welche für die Le bensqualität oft eine deutliche Einschränkung darstellt, hintangehalten werden könnten.
leisten, ist die Basis für eine adäquate weitere Behandlung. Was können sie tun? Wachsam sein, den Familien zuhö ren, Videos der Anfälle einfordern und sich nicht scheuen, Kontakt mit einem tertiären Zentrum aufzunehmen.
Was ist bei der Anwendung von und bei Therapiebeginn mit CBD-Fertigarznei mitteln zur Behandlung von Epilepsie bei Kindern zu beachten?
HAUSÄRZT:IN: Welche Fortschritte konnten in den letzten Jahren in puncto Diagnose und Behandlungs möglichkeiten in der pädiatrischen Epileptologie erzielt werden? Welche positiven Entwicklungen sind in naher Zukunft in Sicht? Priv.-Doz.in GRÖPPEL: Der größte Fortschritt ist sicherlich im Bereich der genetischen Diagnostik zu verzeichnen, welche in der Folge eine gezielte Thera pie ermöglichen wird. Wir bewegen uns nun auf dem Weg zur Präzisionsmedizin,
Eine frühe Diagnose und somit eine rasche Behandlung sind entscheidend. Wie können Fachärzt:innen für Kinder- und Jugendheilkunde und Allgemeinmediziner:innen dazu bei tragen?
Die niedergelassenen Kolleginnen und Kollegen sind oft die erste Anlaufstelle und somit eine wichtige Verbindung zwi schen sogenannten tertiären Zentren wie der Univ.-Kinderklinik und den Familien. Der Beitrag, welchen die Kolleg:innen
CBD hat sich als äußerst wirkungsvoll in der Behandlung von epileptischen Anfällen bei Dravet-Syndrom, LennoxGastaut-Syndrom und Tuberöser Skle rose herausgestellt. Vor dem Start der Behandlung ist es wichtig, die Familien über mögliche Nebenwirkungen auf zuklären und über eine genaue Anam nese der anderen Medikation inklusive des Medikamentenspiegels zu verfügen. Dies dient dazu, mögliche Wechselwir kungen abschätzen und schon früh auf Schwankungen im Medikamentenspie gel reagieren zu können. Zusätzlich gilt es, die Diagnose zu sichern. Es ist aber anzumerken, dass es sich in diesen Fäl len um schwer therapierbare Patienten handelt und somit eine Vorstellung in ei nem Expertisezentrum vor Etablierung einer Therapie mit CBD-Fertigarznei mitteln wichtig ist.

8 Hausärzt:in Rare Disease November 2022
© shutterstock.com/WindNight
© KUK
CBD steht für die Behandlung der seltenen Epilepsieerkrankungen im Kindesalter ab zwei Jahren zur Verfü gung. Welche Therapiemöglichkeiten gibt es für jüngere Kinder bzw. Säuglinge?
Für die Behandlung von seltenen Epilepsieerkrankungen stehen glücklicherweise immer mehr Therapien zur Ver fügung. Allen ist aber gemeinsam, dass man den genauen Krankheitsmechanismus kennen sollte, um diese auch ein setzen zu können. Da jene Epilepsieformen selten sind, gibt es auch oft wenig Erfahrung mit den einzelnen Krankheiten. Aus diesem Grund ist die Behandlung dieser Kinder in Ex pertisezentren wie der Univ.-Klinik für Kinder- und Jugend heilkunde wesentlich.
Wie können Angehörige von betroffenen Kindern unterstützt werden?
Die psychosoziale Versorgung von Familien mit chro nisch kranken Kindern ist wichtig und eine Investition in die Zukunft. Es geht nicht nur darum, dass die Pati enten eine adäquate Physio-, Ergo- oder logopädische Therapie bekommen, sondern auch darum, den Angehö rigen selbst beizustehen, um eventuell die Eltern in der Erwerbstätigkeit zu halten, bzw. psychologische Unter stützung bieten zu können. Leider zeigt uns die Realität, dass dies im Moment kaum oder unzureichend angeboten werden kann. Der Andrang in unsere Spezialambulan zen ist groß und wir können den einzelnen Familien nicht annähernd so viel Zeit widmen, wie wir gerne würden und wie es notwendig wäre. Die Gründung von sozialpädiatrischen Zentren ist eine absolut notwendige Investi tion, um diesen Bereich abdecken zu können. Leider hängt auch diese Zukunftsperspektive von den Förderungsmög lichkeiten der öffentlichen Hand ab.
Das Interview führte Mag.a Ines Pamminger, BA.
SELTENE EPILEPSIESYNDROME IM PÄDIATRISCHEN SETTING
Lennox-Gastaut-Syndrom (LGS)
Das LGS geht mit verschiedenen Anfallstypen in hoher Anfallsfrequenz einher – eine eingehende Anamnese ist daher erforderlich. Bei den meisten Patienten besteht zusätzlich eine Entwicklungsverzögerung. Mit einem prolongierten Video-EEG-Monitoring werden tonische Anfäl le im Schlaf festgestellt. Die Ursachen sind verschiedenartig.
Dravet-Syndrom (DS)
Eine rechtzeitige Identifikation von fieberprovozierten Anfällen in hoher Frequenz als Dravet-Syndrom ist wesentlich, diese sollten nicht als unkomplizierte Fieberkrämpfe eingestuft werden. Eine genetische Abklärung ist bei hochfrequent auftretenden fieberprovozierten Anfällen zu bedenken. Komorbiditäten sind zu eruieren. Medikamente, welche hemmend auf Natriumkanäle wirken, sollten vermieden werden, da diese einen Status epilepticus auslösen können.
Tuberöse Sklerose (TSC)
Tuberöse Sklerose ist eine Multiorganerkrankung, welche fast immer mit einer schweren Epilepsie einhergeht. Alle Organe sowie die Haut (Wood-Licht) sind zu untersuchen. Zusätzlich sind jährliche Kontrolluntersuchungen notwendig, um etwaige Organkomplikationen recht zeitig zu bemerken und behandeln zu können.
Unklare Schmerzen als Schlüssel zu einer Seltenen Erkrankung
Morbus Fabry: Das Leitsymptom Schmerz gehört wohl zum Berufsalltag in jeder hausärztlichen Praxis. Nicht immer sind die Ursachen sofort erkennbar, insbesondere wenn eine Seltene Erkrankung vorliegt.
Dazu zählt die lysosomale Speicherkrankheit Morbus Fabry. Mit einer Inzidenz von etwa 1 : 40.0001 sollte es in Österreich daher etwa 200 Betroffene geben – genauere Zahlen liegen nicht vor. Morbus Fabry geht auf eine X-chromosomal vererbte Mutation im AlphaGalaktosidase-A-Gen (GLA-Gen) zurück. Für den Morbus Fabry gilt, dass nicht nur Män ner, sondern auch heterozygote Frauen er kranken können.2 Durch die Mutation geht die Funktion des Enzyms Alpha-Galaktosida se A (α-GAL A) in den Lysosomen ganz oder teilweise verloren. Wegen der daraus resultie renden fortschreitenden intrazellulären Akku mulation des charakteristischen Speicherma
terials Globotriaosylceramid (Gb3) kommt es zu Inflammation, Hypertrophie und letztlich Fibrosierung mit progredienten Zell- und Or gandysfunktionen sowie irreversiblen Spät schäden.1
Oft Jahrzehnte bis zur Diagnose Schmerzen können eines der ersten Symp tome des Morbus Fabry sein und schon im Kindes- und Jugendalter auftreten.3 Meist werden brennende, stechende oder auch einschießende Schmerzen in Händen und/ oder Füßen beschrieben. Morbus-Fabry-Pati enten leiden daher häufig an unspezifischen, nicht zuordenbaren Schmerzen. Oft dauert
MORBUS FABRY
es Jahre oder gar Jahrzehnte, bis die Erkran kung richtig diagnostiziert und behandelt wird. Richtungsweisend sind daher weitere Fabry-typische Symptome3 (siehe auch Skiz ze)4: Dazu zählen neben neuropathischen Schmerzen in Händen und Füßen und Hitzeintoleranz in Verbindung mit mangelnder Fähigkeit zu schwitzen auch gastrointesti nale Beschwerden. Auffällig können zudem die rötlich-violetten Hautflecken, sog. Angio keratome, sein. Im Rahmen einer Spaltlam penuntersuchung beim Augenarzt können häufig charakteristische Trübungen der Hornhaut durch winzige feine Ablagerungen, die Cornea verticillata, beobachtet werden.4
Eine Krankheit - Viele Manifestationen
PSYCHE
Depression, Fatigue
AUGEN
Cornea verticillata
HERZ
Hypertrophe Kardiomyopathie, Myokardinfarkt, Arrhythmien, Herzinsuffizienz

ZENTRALNERVENSYSTEM
Transitorische ischämische Attacke, Apoplex, Marklagerläsionen
Hörverlust, Tinnitus, Schwindel
NIERE PERIPHERES NERVENSYSTEM
Mikroalbuminurie, Proteinurie, Niereninsuffizienz
Neuropathische Schmerzen als Brennschmerz in Händen und Füßen, episodische Schmerzkrisen, Parästhesien, Temperaturintoleranz
Dyspnoe bei Belastung, chronisch obstruktive Lungenerkrankung
Nausea, Emesis, Diarrhö, Obstipati on, Bauchkrämpfe, Bauchschmerzen
OHREN LUNGE GI-TRAKT HAUT
Angiokeratome, Hypohidrose
BEZAHLTE ANZEIGE 10 Hausärzt:in Rare Disease November 2022
Abklärung in Spezialambulanz
Bei unspezifischen Schmerzen in Verbin dung mit einem oder mehreren typischen Symptomen der chronisch progredienten Multiorganerkrankung Morbus Fabry sollte der Patient umgehend an eine Spezialam bulanz zur weiteren Abklärung überwiesen werden. Bei Verdacht auf Morbus Fabry wird mittels Trockenblutkarte die Aktivität des Enzyms α-GAL A in Leukozyten be stimmt und gegebenenfalls eine moleku largenetische Untersuchung durchgeführt. Entscheidende Hinweise bei unerklärlichen Schmerzen kann auch eine genauere Fa milienanamnese geben: Sind bei Eltern, Großeltern oder Geschwistern Herz- oder
BETROFFENENBERICHTE*
Morbus Fabry: Behandlungsmöglichkeiten
Für die kausale Therapie des Morbus Fabry stehen zwei etablierte Therapieansätze zur Verfügung: die Enzymersatztherapie als Infu sion, die alle 14 Tage verabreicht wird, und für Patienten mit geeigneten Mutationen auch eine pharmakologische Chaperon-Therapie in Form einer Kapsel zur oralen Einnahme jeden zweiten Tag.1
Nierenerkrankungen oder ein Schlaganfall in jüngeren Lebensjahren bekannt, sollte Morbus Fabry in die differenzialdiagnosti schen Überlegungen einfließen. Die nach
folgenden Fallbeispiele veranschaulichen die heterogene Symptomatik, die Famili enzusammenhänge und den langen Weg zur Diagnose.
Nähere Informationen für Patienten und An gehörige stehen zum Beispiel auch auf der Website der Selbsthilfegruppe Morbus Fabry (https://morbus-fabry.eu) zur Verfügung.
1 Lenders M, Brand E. Fabry Disease: The Current Treatment Landscape. Drugs 2021; 81:635–645.
2 Germain DP, Fabry Disease, Orphanet J Rare Dis 2010; 5–30.
3 Politei J et al., Clin Genet 2016; 89: 5–9.
4 Ortiz A et al., Mol Genet Metab 2018; 123: 416– 27.
Die 16-jährige Laura leidet an Morbus Fabry. Ein Jahr zuvor wurde zufällig vom Augenarzt eine Cornea verticillata entdeckt, der zur genau eren Abklärung eines Sehfehlers auch eine Spaltlampenuntersuchung durchführte. Laura weist sonst keine typischen Fabry-Symptome auf, wenngleich sie im Gegensatz zu ihren Freundinnen nicht gerne Sport treibt und als „Stubenhockerin“ gilt, da sie rasch erschöpft ist und daher früh ins Bett geht.
Nach Lauras Diagnose an einer Universitätsklinik wurde der Familie empfohlen, sich ebenfalls auf Morbus Fabry untersuchen zu lassen. Tatsächlich wurde bei der Mutter, bei einer Tante und bei der Oma von Laura dieselbe Fabry-Mutation wie bei Laura nachgewiesen. Die Sym ptome der Familienmitglieder von Laura waren jedoch so heterogen, dass in all den Jahren nicht an einen Zusammenhang oder gar eine Erbkrankheit gedacht wurde.
Mutter Susanne (46 Jahre) lei det seit der Kindheit an unklaren und unspezifischen gastrointestinalen Beschwerden und an Taub heitsgefühlen in den Fingern und ist inzwischen wegen einer mit telschweren chronischen Niereninsuffizienz in Behandlung. Bei Tante Anita, die gerade ihren 50er gefeiert hat, dominiert die kardiale Komponente. Ihre hypertrophe Kardiomyopathie verur sacht jedoch noch keine funktionellen Einschrän kungen. Bei Oma Erika (73 Jahre alt) sind deutliche ZNS- Manifestationen erkennbar: Sie klagt seit Jahren über starke Kopfschmerzen, wirkt erschöpft und leidet an schweren Depressionen. Vor fünf Jahren hatte sie einen leichten zerebralen Insult. Alle Familienmitglie der sowie Laura erhalten inzwischen eine Fabry-spezifische Therapie.*
Kurt wirkt fit und gesund. Ein drahtiger, sportlicher, braungebrannter Anfang-60er mit Energie und positiver Lebenseinstellung. Doch Kurt ist eigenen Angaben zufolge nur „fast gesund“. Denn Kurt leidet an Morbus Fabry, was er allerdings erst seit etwa 15 Jahren weiß. Er weist eine Mutation des GLA-Gens auf, die ihm die Behandlung mit einer pharmakologischen Chaperon-Therapie ermöglicht. Heute lebt Kurt „fast normal“, wie er betont.
Die Jahrzehnte bis zur Diagnose waren jedoch von vielen Rückschlä gen und Herausforderungen geprägt. Schon als junger Erwachsener wunderte er sich, warum er, der begeisterte Läufer, trotz intensiven Trainings immer wieder unerklärliche physische Einbrüche während des Laufens erlebte. Hände und Füße brannten und juckten immer wieder grundlos, zum Teil bestehen diese Symptome nach wie vor, trotz Therapie. Zahlreiche Un tersuchungen blieben zunächst ohne Ergebnisse. Die Unge wissheit, was mit ihm tatsächlich los sei, hat Kurt während seines jahrelangen Leidensweges auch psychisch belastet.
Nach einem EKG, einem Ultraschall und einer MRT an einer kardiologischen Uni versitäts klinik bestand der hochgradige Verdacht auf Morbus Fabry. Die Diagnose wurde schließlich durch weitere Untersu chungen und einen Gentest bestätigt. Er nahm die Diagnose zum Anlass, einige Verwandte mütterlicher seits nach vielen Jahren wieder zu kontaktieren. Inzwischen hat sich die Diagnose bei einigen Angehörigen bestätigt, alle können behandelt werden.*
* Namen und biographi sche Daten der beschriebenen Patien ten wurden verändert, die medizini schen Befunde nur auszugsweise zusammengefasst.
BEZAHLTE ANZEIGE 11 Hausärzt:in Rare Disease November 2022
Laura und ihre Tanten Kurt, der begeisterte Läufer
INFO
© shutterstock.com/JoyCrew NP-GA-AT-00010922
Disproportionales Wachstum
Die Schuhe sind zu klein, der Ring passt nicht mehr, der Hemdkragen ist zu eng und die Gesichtszüge wir ken gröber ... Auf den ersten Blick mögen jene Symptome harmlos erscheinen, allerdings können sie Ausdruck einer ernstzunehmenden Erkran kung sein. Die Akromegalie ist durch einen Überschuss an Wachstumshormon (GH, Somatotropin) gekennzeich net, der wiederum zu einer gesteigerten Produktion von IGF-1 („I nsulin-like growth factor 1“) in der Leber führt –verantwortlich ist dafür fast immer ein GH-sezernierendes Hypophysenade nom. In der Folge entstehen zahlreiche Beschwerden und Komorbiditäten. Die seltene Erkrankung schreitet langsam voran und wird häufig erst Jahre nach dem retrospektiv ermittelten Krank
heitsbeginn diagnostiziert. Zwar können die Behandlungsoptionen eine Heilung oder gute Kontrolle erzielen, aber die Progredienz assozi ierter Erkrankungen ist frühestmöglich zu drosseln, um Folgeschäden zu verhindern.

Wegweisende Klinik
Anders als beim Gigantismus, welcher in der Kindheit durch eine GH-Hypersekretion ent steht, tritt im Erwachsenen alter kein Längenwachstum auf, dafür aber die namensge bende Vergrößerung der Akren. Auffäl lig sind bei Akromegalie-Patientinnen und -Patienten beispielsweise größere Hände und Füße, ein Wachstum der Mandibula und der Nase sowie Ge lenkveränderungen. Zudem kann es zu einer Vergrößerung der inneren Orga
ne kommen. Assoc. Prof.in Priv.-Doz.in Dr.in Greisa Vila, Oberärztin an der Kli nischen Abteilung für Endokrinologie und Stoffwechsel der MedUni Wien, schildert typische durch den Hormonex zess verursachte Probleme: „ Besonders im jungen Alter stehen etwa Hypogona dismus und seine Folgen wie Hyperhidrose, Libidoverlust oder extreme Müdigkeit im Vordergrund. Bei Frauen kommt es oftmals zu Menstruationsstörungen. Orthopädische Beschwerden sind sehr häufig und viele Betroffene klagen über Kopfschmerzen. Große Adenome kön nen zudem Gesichtsfeldausfälle her vorrufen.“ Im Durchschnitt werde die Erkrankung zwischen dem 40. und dem 50. Lebensjahr diagnostiziert. Umso äl ter die Betroffenen, desto häufiger sei en Komorbiditäten wie Hypertonie und Diabetes mellitus. Weitere Begleiterkrankungen (siehe Infobox, S. 14) stellen zum Beispiel

12 Hausärzt:in Rare Disease November 2022 >
EXPERTIN: Assoc. Prof.in Priv.-Doz.in Dr.in Greisa Vila Klinische Abteilung für Endokrinologie und Stoffwechsel, MedUni Wien
© MedUni Wien, Matern
1. November ist Welt-Akromegalie-Tag: frühe Diagnose und Management im Fokus
© shutterstock.com/Wysocka Malgorzata
das Karpaltunnelsyndrom, Schild drüsenknoten oder kardiovaskuläre Komplikationen wie Kardiomyopa thien, Herzinsuffizienz, Arrhythmien oder Herzklappenerkrankungen dar. Prof.in Vila zufolge geht die Akromegalie auch mit einer hohen Anfälligkeit für Tumorerkrankungen einher, außerdem mit einem erhöhten Risiko, Frakturen zu erleiden, da sich die Knochenstruk tur verändert. „ D ie Häufigkeit und Schwere von Komorbiditäten hängt einerseits von der Ausprägung des Wachstumshormonüberschusses ab, an dererseits davon, wie lange die Erkran kung unerkannt und somit unbehandelt geblieben ist“, gibt die Endokrinologin zu bedenken.
Diagnose sichern
Aufgrund der weitreichenden Pathologie betrifft die Akromegalie eine Bandbreite von Medizinerinnen und Medizinern, die bereits bei der Diagnosestellung gefragt sind – etwa Internisten, Endokrinologen/ Schilddrüsenspezialisten, Augenärzte und Orthopäden. „Den Verdacht auf Akro megalie können auch Zahnärzte äußern, wenn eine Malokklusion durch vergrö ßerte Zahnlücken besteht“, erläutert die Expertin und weist darauf hin, dass typische Symptome nicht bagatellisiert werden sollten. „Erstaunlich oft wird zum Beispiel das Wachstum der Füße als eine Alterserscheinung verkannt. Natürlich stimmt es, dass die Füße im Alter größer werden, aber nur minimal “ Prof.in Vila betont zudem die zentrale Rolle von Hausärztinnen und Hausärz ten. „Idealerweise initiiert der Allge meinmediziner nach einer ausführlichen Anamnese bei Verdacht auf Akromega lie die Labordiagnostik “ Sie umfasst die Bestimmung der Nüchternspiegel von Wachstumshormon und IGF-1 im Serum sowie die Überprüfung der GH-Sup pression im oralen Glukosetoleranztest. „Wenn die Biochemie eine Akromegalie bestätigt, sollte eine Hypophysen-MRT veranlasst werden“, so Prof.in Vila.
Therapiemöglichkeiten ausschöpfen
In erster Linie wird die Akromegalie mittels transsphenoidaler Entfernung
des Hypophysenadenoms behandelt. Der Erfolg hängt maßgeblich von der Größe des Tumors ab – bei Mikroade nomen, also jenen Adenomen mit einer Größe von < 1 cm, liegt die Heilungsrate bei über 80 Prozent. Im Falle größerer Tumoren und unvollständiger Resektion oder wenn eine Operation nicht mög lich ist, bedarf es einer medikamentösen Therapie. Hierfür stehen zunächst So matostatinanaloga wie Octreotid oder Lanreotid zur Verfügung, bei nur gering erhöhten IGF-1-Konzentrationen außer dem der Dopaminagonist Cabergolin. Wachstumshormon-Rezeptorantagonis ten wie Pegvisomant stellen die SecondLine-Therapie dar, mit zusätzlicher posi tiver Wirkung auf den Glukosehaushalt. Prof.in Vila führt aus: „Wenn sich der IGF-1Spiegel durch Somatostatinanaloga nicht normalisiert, ergänzen wir die Behand lung um Pegvisomant oder wir stellen den Patienten bei Nebenwirkungen oder Komplikationen auf eine Monotherapie mit den Wachstumshormon-Rezeptorantagonisten um “ Die medikamentöse
Behandlung ist in den meisten Fällen le benslang fortzuführen. U. a. aus diesem Grund kann letztendlich auch eine radio chirurgische Intervention sinnvoll sein.
Interdisziplinäre Nachsorge
IM ÜBERBLICK
Symptome (Auswahl)
charakteristisches akrales Wachstum: Hände, Füße, Stirnhöhlen, vergröberte Gesichtszüge mit größerer Nase und prägnanten Wangenknochen, mandibuläre Vergrößerung mit Auseinanderweichen der Zähne, Malokklusion
Makroglossie, tiefere Stimme
Gelenkschmerzen, Kopfschmerzen, Müdigkeit, Hyperhidrose, Menstruationsstörungen, Libido- und Potenzstörungen etc.
bei großen Hypophysenadenomen bitemporale Hemianopsie durch Komprimierung des Chiasma opticum
Assoziierte Komorbiditäten (Auswahl)
arterielle Hypertonie
Diabetes mellitus
sekundärer Hypogonadismus
kardiovaskuläre Komplikationen
Karpaltunnelsyndrom
Arthropathie
Schlafapnoe
Struma
Malignome
Kolon-, Gallenblasenpolypen
„Ist der Patient nach der Operation ge heilt, sollte er einmal pro Jahr endokrino logisch kontrolliert werden. Betroffene, die medikamentös eingestellt sind und wieder normale IGF-1-Werte aufweisen, benötigen alle sechs bis zwölf Monate eine Kontrolle im Zentrum, beim Allge meinmediziner alle drei Monate“, erklärt Prof.in Vila. Die Hausärztin, der Hausarzt überprüft nicht nur die Laborwerte –z. B. Blutzuckerwerte, Leberwerte –, son dern unterstützt die Patienten auch beim Management der Nachsorge. Neben der Behandlung von etwaigen bereits mani festen Komorbiditäten benötigen Akro megalie-Patienten der Expertin zufolge regelmäßig eine MRT der Hypophyse und wesentlich früher diverse Vorsor geuntersuchungen, beispielsweise eine Koloskopie, als es für die Normalbevöl kerung empfohlen ist. Auch ein Röntgen der Wirbelsäule sei indiziert, um ver steckte Frakturen auszuschließen. „Im Zentrum der MedUni Wien gehen wir bei der jährlichen Kontrolle eine Check liste mit dem Patienten durch und halten fest, welche Untersuchungen durchge führt wurden und welche ausständig sind. Die Nachsorge bei den Fachärztinnen und -ärzten organisiert der Patient, na türlich mithilfe des Hausarztes. Der En dokrinologe behält den Überblick und bekommt alle Befunde.“
Abschließend thematisiert Prof.in Vila die Auswirkungen der Akromegalie auf die Lebensqualität der Patientinnen und Patienten: „Ein Hypogonadismus kann zum Beispiel Beziehungsprobleme ver ursachen. Beruflich entstehen mitunter zeitliche Konflikte aufgrund der großen Anzahl von ärztlichen Untersuchungen, feinmotorische Tätigkeiten sind zum Teil nicht mehr ausführbar, wenn die Finger ‚bamstig‘ sind.“ So könne in manchen Fällen eine psychologische Betreuung empfehlenswert sein. „Aber wenn die Er krankung adäquat behandelt wird, führt der Großteil der Betroffenen meiner Er fahrung nach ein normales Leben.“
Anna Schuster, BSc
14 Hausärzt:in Rare Disease November 2022
Diagnostisches Licht ins Dunkel bringen

Lege artis muss bei hereditären Netzhautdystrophien ein Gentest erfolgen
Stellenwert der genetischen Abklärung
Morphologie und Funktion weiterhin wichtig
Mit einer Prävalenz von 1 : 2.000 bis 1 : 3.000 stellen erbliche Netzhauterkran kungen bei Kindern, Jugendlichen und jungen Erwachsenen eine häufige Ursa che schwerer Sehbehinderungen dar, die bis hin zur Erblindung führen können.1,2,3 Es handelt sich allerdings um eine sehr heterogene Gruppe von Erkrankungen. Historisch betrachtet gibt es rund 100 klinische Entitäten, wozu etwa Retinitis pigmentosa (RP) oder Morbus Stargardt zählen. „Die moderne Augenheilkunde geht allerdings dazu über, die kausalen Gene zu benennen, statt sich mit den phänotypischen Beschreibungen zufrie den zu geben“, erklärt Assoz. Prof. Priv.-Doz. Dr. Markus Ritter, Leiter der Ambulanz für erbliche Netzhauterkran kungen – Elektrophysiologie, MedUni Wien/AKH Wien. „L ege artis müssen alle Patien tinnen und Patienten genetisch abgeklärt werden. Das Ergeb nis wird dann in Synopsis mit dem klinischen Phänotypen interpretiert.“
Derzeit sind mehr als 270 ver schiedene Gene bekannt, wel che hereditäre Netzhautdys trophien verursachen können, wobei die dort vorhandenen Mutationen auch zu jeweils unterschiedlichen Phänotypen führen können.2 „ Augenärztinnen und Augenärzte haben leider eine zu geringe Awareness in Bezug auf diese Art von Erkrankungen. Bis zur korrekten Diag nose vergehen oft viele Jahre“, bedauert Prof. Ritter.
In einer rezenten Befragung von Betrof fenen gaben nur zwei Drittel an, sich ei ner genetischen Testung unterzogen zu haben. Lediglich 47 % hatten eine hu mangenetische Beratung erhalten.4 Hier appelliert Prof. Ritter an die Allgemein medizinerinnen und -mediziner: „ Jede Hausärztin und jeder Hausarzt sollte bei Personen mit bekannter Diagnose einer hereditären Netzhauterkrankung hinterfragen, ob diese schon einmal genetisch abgeklärt wurden, und dar an erinnern, dass die Patientin bzw. der Patient Anspruch darauf hat “ Erst das Auffinden des kausalen Gens erlaube es, z. B. durch bereits publizierte Fallserien abzuschätzen, wie schnell sich das Sehen verschlechtern werde, ob es assoziierte Erkrankungen gebe und ob der Verer bungsmodus rezessiv, dominant, mito chondrial, X-chromosomal etc. sei. Jedoch müssen Ärztinnen und Ärzte den Betroffenen bewusst machen, dass nicht jede genetische Ursache mit den der zeitigen Analysemethoden identifiziert werden kann. Bei der Analyse des gesamten Exoms handelt es sich um eine Breitspektrumanalyse, bei der jegliches in Proteine umgesetzte gene tische Material erfasst wird. Untersucht man hingegen das gesamte Genom, lassen sich auch tiefe intronische Verände rungen identifizieren. Mit der Whole-Exome-Analysis liegen die Chancen, das ursächliche Gen aufzufinden, bei 70 %, mit der meist nur in Studien eingesetzten Whole-GenomeAnalysis bei etwa 80 %. „Das heißt, es bleibt in Standarduntersuchungen im mer noch gut ein Drittel der Betroffenen übrig, bei dem man zwar mit relativer Si cherheit sagen kann, dass es sich um eine genetisch bedingte Netzhauterkrankung handelt, aber deren Ursache lässt sich noch nicht sicher festmachen“, gibt Prof. Ritter zu bedenken.
„B ei einer schweren Sehbeeinträch tigung im Kindes- und Jugendalter, die bisher nicht eindeutig zugeord net werden konnte, empfehle ich also dringend, dass man auch an hereditäre Netzhautdystrophien denkt “ , fasst der Experte zusammen. Natürlich könn ten z. B. neurologische oder entzünd liche Veränderungen ebenfalls zu den Sehproblemen führen. Trotzdem sei es ratsam, an ein Zentrum oder eine Spe zialklinik zu überweisen, die auch das Spektrum der Netzhauterkrankungen abklären könnten, weil sie über die passenden Spezialgeräte verfügten. „Vor allem eine unklare reduzierte Sehschärfe, Nachtsichtstörungen und Sehstörungen im Rahmen von Syndro men sind Warnsignale“, macht Prof. Ritter aufmerksam.
Die Identifikation des ursächlichen Gens allein reicht für die Diagnostik nicht aus. Die Untersuchung von Mor phologie und Funktion der Retina be halte ihren Stellenwert, so der Experte und er fügt hinzu: „ A ls Augenärztin oder Augenarzt muss man immer fra gen, ob die Mutationen wirklich zu je ner Morphologie und Funktion passen, die wir messen bzw. darstellen kön nen – man muss den klinischen und den genetischen Befund in Einklang bringen können.“ Denn es gebe auch genetische Varianten unklarer Signi fikanz, bei denen noch nicht erwiesen sei, dass sie die Krankheit tatsächlich verursachten.

15 Hausärzt:in Rare Disease November 2022
© shutterstock.com/AnnaVel
EXPERTE:
Assoz. Prof. Priv.-Doz. Dr. Markus Ritter Leiter der Ambulanz für erbliche Netzhauterkrankungen – Elekt rophysiologie, MedUni Wien/AKH Wien
© privat
>
„Jede Hausärztin und jeder Hausarzt sollte bei Personen mit bekannter Diagnose einer hereditären Netzhauterkrankung hinterfragen, ob diese schon einmal genetisch abgeklärt wurden.“
Notwendige Untersuchungen umfassen etwa:
• bezüglich der Morphologie: u. a. optische Kohärenz-Tomographie (OCT), Fundus-Autofluoreszenz (FAF), • bezüglich der Funktion: u. a. Elektrore tinogramm (ERG).
Abklärung vor wichtigen Lebensentscheidungen
Zwei Lebensphasen stellen besondere Knackpunkte dar: Schuleintritt und Be rufswahl. „Ich lege Familien nahe, wo möglich betroffene Kinder vor Eintritt in die Schule umfassend untersuchen zu lassen, da die Kindheit schwierig wer den kann, wenn das Kind z. B. im Sport tollpatschig ist oder mit dem Lesen Probleme hat und man nicht weiß, was dahintersteckt. Das Schulamt vermittelt eigene Sozialarbeiterinnen und Sozial arbeiter, damit das Kind die beste Aus bildung erhält“, weiß Prof. Ritter. Der zweite Punkt betreffe die Berufswahl: Hier eigneten sich etwa Berufe mit Bildschirmarbeit gut, da es Softwarepro gramme gebe, die das Lesen erleichter ten, wohingegen feinmechanische Be rufe oder solche, bei denen das Lenken von Fahrzeugen im Vordergrund stehe, ungeeignet seien. „ Natürlich kann man die Eltern nicht dazu zwingen, ihr Kind untersuchen zu lassen, aber man muss es mit ihnen besprechen“, so der Ex perte. Zusätzlich empfiehlt er, die ge netische Beratung in Kooperation mit einem Institut für medizinische Gene tik durchführen zu lassen. In Bezug auf Datenschutz, Befundübermittlung und -aufbewahrung gibt es hierzu sehr stren ge Regularien.
Gentherapie als Hoffnung
Patientinnen und Patienten mit bialleli schen Mutationen im RPE65-Gen kön nen seit Ende 2018 von der Gentherapie mit Voretigen Neparvovec profitieren. Adeno-assoziierte Virusvektoren beinhalten eine intakte Kopie des Gens. „Während der Operation spritzt die Ärz tin oder der Arzt die Suspension mit den Virusvektoren unter die Netzhaut, so dass sich die Viren in die Pigmentepithel zellen einbauen können. Dadurch kann die Produktion eines gesunden RPE65-
MD
CRX*, RPGRIP1 ABCA4, PROM1, PRPH2
LCA
CRB1, CWC27, IFT140, IMPDH1, LRAT, MERTK, RDH12, RPE65, SPATA7, TULP1
C8orf37, CDHR1, RPGR, SEMA4A NR2E3, NRL
BEST1, FSCN2, GUCA1B, 1MP62, RP1L1
Häufige IRD-Phänotypen
MD: Makuladystrophie
ZSD: Zapfen-StäbchenDystrophie
ESCS CSNB RP
ADGRA3*, AGBL5, AHI1, ARHGEF18, ARL2BP, ARL3*, ARL6, BBS1, BBS2, BBS9, CRorf71, CA4, CERKL, CLN3, CLRN1, CNGA1, CNGB1, DHDDS, DHX38*, EMC1*, EYS, FAM161A, HGSNAT, HK1, IDH3A, IDH3B, IFT172, KIAA1549*, KIZ, KLHL7, MAK, MVK, NEK2, NEUROD1*, OFD1, PANK2, PDE6A, PDE6G, POMGNT1, PRCD, PRPF3, PRPF4, PRPF6, PRPFR8, PRF31, RBP3, RDG11, REEP6, RGR, RGR, ROM1, RP1, RP2, RP9, SAMD11, SLC7A14, SNRNP200, TOPORS, TTC8, USH2A, ZNF408, ZNF513
GNAT1, PDE6B, RHO, RLBP1, SAG
* Kandidatengene für nichtsyndromische RP Quelle: Nachbau: Verbakel SK et al., Progress in Retinal and Eye Research 2018; 66: 157-186.
codierten Eiweißmoleküls, welches eine wichtige Rolle im Sehzyklus spielt, ange kurbelt werden“, beschreibt Prof. Ritter die Vorgehensweise. Voraussetzung für die Gentherapie ist, dass die oder der Betroffene noch über genügend funkti onsfähige Netzhautzellen verfügt, die das Virus aufnehmen und eine verbesserte Sehkraft erzielen können.
Weitere Behandlungsoptionen
„Aber es gibt auch andere Therapien in der Pipeline, die schon in recht fort geschrittenen Stadien sind, was die Er probung an Patientinnen und Patienten betrifft“, betont der Facharzt. Als Bei spiele für Erkrankungen, die erforscht werden, nennt er Formen der RP, welche durch Mutationen im RPGR-Gen ver ursacht werden, sowie Choroideremie und Achromatopsie. Weiters gibt es Behandlungsansätze, die z. B. den Verlauf der Erkrankung ver langsamen sollen, etwa bei Morbus Star gardt. Ebenfalls zu nennen sind Antisen se-Oligonukleotide. „ Diese kann man als eine Art Genpflaster betrachten, mit dem sich gewisse Mutationen aus schalten lassen“, erläutert Prof. Ritter. Teilweise gebe es auch genunspezifische Ansätze. „ E s wird viel zu diesen Erkran kungen geforscht, aber es ist keine ein fache Thematik“, meint der Spezialist.
Fazit
„Abschließend sollte man wissen, dass hereditäre Netzhautdystrophien rein
LCA: Leber'sche kongenitale Amaurose
CSNB: Kongenitale stationäre Nachtblindheit
RP: Retinitis pigmentosa
okuläre Erkrankungen sein, aber auch als Teil eines Syndroms auftreten kön nen“, unterstreicht Prof. Ritter. „ Au ßerdem darf man als Behandelnde oder Behandelnder nicht vergessen, dass die Betroffenen Mitglied einer Familie sind – auch wenn sie vielleicht die Einzigen sind, die derzeit Symptome aufweisen. Zuletzt soll man sich nicht mit einer rei nen klinischen Diagnose zufriedenge ben, sprich: einer Diagnose ohne geneti sche Abklärung “ Dies entspreche nicht mehr dem State of the Art, resümiert der Experte.
Mag.a Marie-Thérèse Fleischer, BSc
Quellen:
1 Birtel J et al., Klin Monatsbl Augenheilkd 2021; 238: 249-260.
2 García Bohórquez B et al., Front Cell Dev Biol 2021; 9: 645600.
3 Deutsche Ophthalmologische Gesellschaft (DOG) et al., S1-Leitlinie: Erbliche Netzhaut-, Aderhaut- und Sehbahnerkrankungen, Stand: 09/2021.
4 Kellner U et al., Die Ophthalmologie 2022; 119: 820-826.
ANAMNESTISCH RELEVANTE FRAGEN3
Inwiefern ist das Sehvermögen beein trächtigt? Z. B. Farbsehschwäche, Nachtblindheit, Gesichtsfeldausfälle
Wann haben die Beschwerden begonnen bzw. haben sie sich im Lauf der Zeit verändert?
Gibt es in der Familie bekannte Sehstörungen?
Liegen weitere Beeinträchtigungen vor? Z. B. Hör-, Riech-, neurologische Störungen, Stoffwechselerkrankungen etc.
16 Hausärzt:in Rare Disease November 2022
ZSD
1. Chung et al. Am J Ophthalmol. 2019; 199: 58-70. 2. Chacon-Camacho et al. WJCC. 2015; 3.2: 112.































Erbliche Netzhauterkrankungen erkennen und genetisch abklären
© 2022 Novartis Pharma GmbH, Jakov-Lind-Str. 5/3.05, A-1020 Wien, HG Wien FN41622i
07/2022, AT2207216777 WeitereInformationenaufwww.novartis.at Häufige Anzeichen und Symptome1,2 Nachtblindheit Verlangsamte Dunkeladaption Nystagmus (unkontrollierte/repetitive Augenbewegungen) Lichtempfindlichkeit Sehschärfeverlust N R DOVHR HSDK CKZO
„Schön langsam wächst die Awareness“
Aufs Herz gebracht: Transthyretin-Amyloidose mit Kardiomyopathie frühzeitig erkennen und therapieren
Die kardiale Manifestation der ATTRAmyloidose ist in der Vergangenheit aufgrund der Heterogenität der Symp tome oft übersehen bzw. erst spät dia gnostiziert worden – mit entsprechend negativen Konsequenzen für die Prog nose der betroffenen Patientinnen und Patienten. Das hat sich erfreulicherwei se in den letzten Jahren verändert. Für die Früherkennung und Begleitung der Patienten ist die Zusammenarbeit zwi schen Fachexperten unterschiedlicher Disziplinen essenziell.
HAUSÄRZT:IN: Warum hat die kardiale ATTR-Amyloidose jüngst an Aufmerksamkeit gewonnen?
OA Dr. EBNER: Weil die Diagnostik einfacher wurde und weil uns heute ver schiedene Therapiemöglich keiten zur Verfügung stehen. Grundsätzlich unterscheiden wir ja zwei Formen der Amy loidose, beide sind Eiweiß speichererkrankungen: die AL-Amyloidosen, bei denen krankhafte Plasmazellen im Knochenmark strukturell ver änderte Leichtketten produ zieren – sie werden von den Kolleginnen und Kollegen der Onkologie behandelt –, und die beiden Manifestationen der ATTR-Amyloidose, bei der es zur Ablagerung von fehlgefalte tem Transthyretin kommt, welches in der Leber gebildet wird: die primär neu rologische Form mit Polyneuropathie (ATTR-PN) sowie die kardiale mit Kar diomyopathie (ATTR-CM) – entweder als mutierte (mATTR) oder als WildtypForm (wtATTR). Erstere kann schon in jüngeren Jahren auftreten und vererbt werden. Die Wildtyp-Form hingegen tritt erst im höheren Alter auf, etwa von 70 Jahren aufwärts. Früher hat man sie auch als senile Amyloidose bezeichnet. Wichtig ist zu betonen: Mischformen sind häufig. Die fehlgefalteten Proteine wer den auch bei der primär kardialen Ma nifestation nicht allein im Herz abgelegt,

EXPERTE:
sondern im ganzen Körper – die Amylo idose ist, wenn man so will, eine ubiqui täre Erkrankung. Dass viele Betroffene ein Herzproblem bekommen, hängt oft damit zusammen, dass dieses sie auf der Endstufe der Erkrankung symptoma tisch macht.
Auf welche Red Flags können Hausund niedergelassene Fachärzt:innen achten?
Erkennungszeichen gibt es meist schon lange vor den ersten kardialen Sympto men. Die Verlaufsformen können sehr unterschiedlich sein. Polyneuropathien sind z. B. ein Red Flag, werden aber nicht immer gleich mit einer Amyloido se assoziiert. Das ist verständlich. Denn Polyneuropathien sind für Neurologen keine seltene Erkrankung.
Nur ein sehr kleiner Anteil davon tritt im Vorfeld bei Amyloidosen auf. So haben etwa auch unsere kardiologi schen Patienten immer wie der Gefühlsstörungen in den Beinen.
Ebenfalls ein Red Flag ist das Karpaltunnelsyndrom: Die Gewebe saugen sich bei einer Amyloidose mit den Eiweiß körpern an, ebenso das Bänd chen im Handgelenk. So kann ich bei einem 80-jährigen Patienten mit Herzschwäche/Atemnot, der einen deutlich verdickten Herz muskel im Ultraschall hat, außerdem ein Karpaltunnelsyndrom ein- oder beidseitig, schon darauf tippen, dass eine kardiale Amyloidose vorliegt. Es gibt also Pre-Marker, sie werden aber immer erst im Kontext des Gesamtbil des stimmig.
Manche Betroffene haben – eher un charakteristisch – auch Kopfschmerzen oder Magen-Darm- oder Augen-Pro bleme, wenn es in diesen Körperbe reichen zu Ablagerungen im Gewebe kommt. Theoretisch könnte auch ein Augenarzt die Erstdiagnose stellen. Wichtig ist, dass Haus- und niedergelas
sene Fachärzte eine gewisse Awareness für die Seltene Erkrankung entwickeln.
Worauf ist bei der Diagnose besonders zu achten?
Bei Verdacht auf Amyloidose ist es wichtig, zuerst die hämatoloonkologi sche Form auszuschließen. Denn die AL-Amyloidose ist eine sehr gefährli che, rasch fortschreitende Erkrankung. Früher musste man dazu eine Endomyo kardbiopsie durchführen. Heute reicht meist die Kombination verschiedener nichtinvasiver diagnostischer Maßnah men. Ist der Herzmuskel im Ultraschall verdickt, so folgen in der Regel Labor untersuchungen und parallel eine Kno chenszintigrafie. Die Erhöhung des kar dialen Biomarkers NT-proBNP ist ein wichtiges Anzeichen für eine ATTR-CM. Die Graduierung der Ergebnisse der Ske lettszintigrafie erfolgt nach dem PeruginiScore (Grad 0 bis 3). Bei Patienten mit einer ATTR-Amyloidose sind die kar dialen Einlagerungen signifikant höher als bei jenen mit einer AL-Amyloidose. Bei größer/gleich Grad 2 können wir von einer ATTR-CM ausgehen. Bei Grad 1 setzen wir zur Absicherung der Diagnose zusätzlich auf eine Endomyokardbiopsie. Zusätzlich wird eine kardiale Magnetre sonanztomografie durchgeführt. Passen die Ergebnisse der drei Untersuchungen zusammen, können wir heute direkt mit der Therapiestrategie starten.
Gibt es einen kurativen Ansatz? Bei der hereditären Form ja, die Leber transplantation. Man ersetzt das Or gan, in dem das verursachende Protein gebildet wird. Allerdings gibt es viele verschiedene genetische Formen – mit günstiger und weniger günstiger Prog nose. Die TTR-Mutationen können vor ab durch einen Gentest nachgewiesen werden. Kommt der Patient erst in einer späteren Phase seiner Erkrankung, hat er schlechte Parameter – und ist er be reits malnutriert, dann sind auch durch die Lebertransplantation keine großen Erfolge zu erwarten.
18 Hausärzt:in Rare Disease November 2022
Dr. Christian W. Ebner Oberarzt an der 2. Internen Abteilung am Ordensklinikum - Krankenhaus der Elisabethinen in Linz
© KH der Elisabethinen, Linz
Bei der genetischen Form sollten auch Verwandte untersucht werden … Unbedingt! Die hereditäre ATTR-Amy loidose wird autosomal-dominant ver erbt. Fällt der Gentest negativ aus, kann man die Verwandten beruhigen. Wird das Gen frühzeitig nachgewiesen, kann man rasch mit einer Therapie beginnen, die es in der Zwischenzeit gibt.
Welche medikamentöse Therapie kommt in Frage?
Von den Genetic Silencers stehen der zeit zwei Präparate zur Verfügung: Pa tisiran und Inotersen. Beide inhibieren die Produktion von ATTR-CM in der Leber und werden entweder intravenös oder subkutan verabreicht. Die entspre chenden Studien dazu hatten vorrangig neurologische Endpunkte. Bei beiden Präparaten wurden aber auch Unter gruppen mit ATTR-Kardiomyopathie angeschaut. Und es zeigte sich, dass die zwei Präparate auch hier wirken. Derzeit sind sie für genetische Varianten zugelas sen. Wahrscheinlich würden sie auch ge gen den Wildtyp helfen. Das ist natürlich immer auch eine Kostenfrage …
Ein weiteres wichtiges Präparat, das in den letzten Jahren Erfolge gezeigt hat, ist Tafamidis. Was kann es? Es ist ein sogenannter TTR-Stabilisator. Das Medikament kann also die Krank
heit nicht heilen, aber diese zu dem Zeitpunkt einfrieren, zu dem die Thera pie begonnen wird. In Studien hat man gesehen, dass es bis zu zwölf Monate dauert, bis sich Erfolge der Therapie ge genüber jenen Patienten zeigen, die das Medikament nicht bekommen. Dann sind sie allerdings signifikant.
Je früher die Erkrankung entdeckt wird, umso größer ist also die Chance, dass der Patient das Medikament bewilligt bekommt und noch möglichst lange bei möglichst guter Lebensqualität leben kann.
Worauf ist bei Medikamenten fürs Herz zu achten?
Die üblichen Herzinsuffizienz-Medika mente wie Betablocker und ACE-Hem mer greifen bei unseren Patienten meist nicht bzw. werden sie schlecht vertragen. Auch das kann ein Erkennungszeichen sein. Der Einsatz von Diuretika steht deshalb im Vordergrund.
Worauf kommt es bei der Zusammenarbeit mit niedergelassenen Kolleg:innen an?
Der Zeitfaktor ist sehr wichtig für die Prognose. Die Erstdiagnose kann im Grunde auch der niedergelassene Arzt stellen. Um die innovativen Medika mente ansuchen können allerdings nur von der Kasse auserkorene Zentren. Die
niedergelassenen Kollegen weisen uns Verdachtsfälle idealerweise zu. Erfreu lich ist: Wir bekommen immer wieder Patienten zugewiesen, die bereits vordi agnostiziert wurden. Nicht zuletzt wissen viele niedergelassene Kollegen mittler weile um den Wert der Knochenszinti grafie als Diagnostikum. Schön langsam wächst also die Awareness.
Die Patienten müssen in der Folge alle drei bis sechs Monate zur Kontrolle ins Zentrum kommen. Wichtig ist für uns, dass sie dazwischen von ihrem Haus arzt/ihrem Internisten betreut werden.
Die Betroffenen sind keine einfachen Patienten: Dekompensationen sind häufig. Sie haben viele Komorbiditäten wie Rhythmusstörungen oder Herz klappenfehler. Wir sind also auf die gute Zusammenarbeit mit den Haus ärzten angewiesen.
Was erwarten/erhoffen Sie sich von der Zukunft?
Derzeit haben wir Möglichkeiten der Behandlung: bei der hereditären Form die Lebertransplantation und die Me dikation, beim Wildtyp hauptsächlich Tafamidis. Auch zu Medikamentenkom binationen und zu innovativen Ansätzen wie In-vivo-Gen- oder Antikörperthera pien wird geforscht. Es gibt also Licht am Ende des Tunnels!
Das Interview führte Mag.a Karin Martin.
Kampagne „Portraits of Progress“...
... startet europaweit
Von der Entdeckung der Hämophilie in den 1940er Jahren1 bis heute wurden unglaubliche Fortschritte im Verständ nis und in der Behandlung der Krank heit erzielt. Medizinische Forschung und Innovation sind jedoch nur ein Teil der Geschichte: Die täglichen Auswir kungen auf Patient:innen, ihre Angehö rigen und Betreuer:innen sind ebenso wichtig, um das Gesamtbild wirklich zu verstehen.
Mit dem Start der europaweiten Kam pagne „ Portraits of Progress“ möchte CSL Behring Aufmerksamkeit auf die Hämophilie-Gemeinschaft richten und zeigen, wie weit das Wissen zur Erkran
kung und die Behandlung der Krankheit in den letzten Jahren gekommen sind. Die Kampagne – in deren Mittelpunkt Porträtaufnahmen von Patient:innen, Pflegepersonal und medizinischen Fach kräften des renommierten Fotografen Rankin stehen – ist ab sofort auf der europäischen Website verfügbar und er zählt die Geschichten von Patient:innen in Fotos und Videos.
Dies sind die „ Portraits of Progress“ –sehen Sie selbst, wie weit wir bis heute gekommen sind: www.portraitsofprogress.eu

1 Wolfgang S. The history of haemophilia - a short review. Thromb Research 2014; 134(1): S4-S9.
Quelle: CSL-Behring, September 2022 EUR-OTH-0173 / AUT-GEN-0057
19 November 2022 Hausärzt:in Rare Disease
© Rankin/portraitsofprogress.eu
