FÜR PHARMAKOLOGIE UND THERAPIE
JOURNAL OF PHARMACOLOGY AND THERAPY
COVID-19: Frühzeitig einsetzende antivirale Therapie reduziert postakute Folgeerkrankungen, schwere Verläufe und Gesamtletalität

Hidradenitis suppurativa: Anhaltende Symptomverbesserungen unter der Therapie mit Secukinumab
Epilepsie: Neue Erkenntnisse zum Einsatz von Perampanel
Neue Leitlinie zur Diagnostik und Therapie myasthener Syndrome
Urothelkarzinom: Neue Daten untermauern den Stellenwert der ErstlinienErhaltungstherapie mit Avelumab
Multiple Sklerose: Patienten profitieren von der subkutanen Gabe von Natalizumab
Upstaza™ – eine neue intraputaminale Gentherapie bei AADC-Mangel
Upadacitinib bei atopischer Dermatitis: Stark wirksam und allgemein gut verträglich
Voclosporin – der erste in Europa zugelassene Calcineurininhibitor zur Behandlung der Lupus-Nephritis
32
2 April 2023 ISSN 1432-4334
JAHRGANG
HEFT
VERL AG PERFUSION


Es handelt sich hier um die Darstellung einer fiktiven Patientin. HS: Hidradenitis suppurativa, auch als Acne inversa bezeichnet. Referenzen: 1. Garg A et al. J Am Acad Dermatol 2020; 82(2): 366–376. 2. Kimball AB, Jemec GBE, eds. Hidradenitis Suppurativa: a Disease Primer. Springer International Publishing AG; 2017. 7 bis 10 Jahre bleibt die Erkrankung bei den meisten Patienten unerkannt.1 Handeln Sie jetzt und verhindern Sie das Fortschreiten der Symptome! 2
Sie
Verborgenen
Novartis Pharma GmbH – Roonstr. 25 – 90429 Nürnberg
Erkennen
HS?
Krankheit im Eine
Wenn sich etwas ändern soll, sind Förderprogramme oft eine gute Sache. Das gilt als „Motivationshilfe“ etwa für den Umstieg auf erneuerbare Energien, die Förderung des ländlichen Raums, mehr Bildung oder körperliche Aktivität und vieles mehr. Allen diesen Beispielen gemeinsam ist, dass wir grundsätzlich die Wahl haben, uns für oder gegen etwas zu entscheiden.
Ganz anders verhält es sich mit Kriterien, die sich grundsätzlich nicht beeinflussen lassen wie Geschlecht, Abstammung, Rasse, Sprache, Heimat und Herkunft (wörtlich entnommen: Artikel 3, Absatz 3, Deutsches Grundgesetz). Aktuell von Protagonistinnen und Protagonisten einer „feministischen Politik“ allenthalben in den Raum gestellte Behauptungen, „eine weiblich dominierte Welt sähe anders aus“ (= besser), ist mitnichten evidenzbasiert, sondern baut auf das Axiom, wer von seinen Eltern ein XY-Chromosom in die Wiege gelegt bekommen hat, ist deshalb ein Mensch mit gesellschaftlich weniger positiven Eigenschaften.
Auch wenn das höchstwahrscheinlich nicht bewusst so gemeint ist, scheint es mir doch wichtig, auf diesen gedanklichen Irrweg hinzuweisen. Und auch darauf, dass selbst „verordnete Gleichheit“ im täglichen Leben belanglos ist, wenn sie nicht gelebt wird. In den USA reifte irgendwann die Erkenntnis, dass es Unrecht ist, dass Herkunft und Hautfarbe entscheiden, wer frei und wer Sklave ist. Wäre den Nordstaatlern damals die Denke der „feministischen Politik“ zu eigen gewesen, wäre nicht die Abschaffung der Sklaverei die Konsequenz gewesen, sondern die Umkehr der Verhältnisse. Wieso thematisiert niemand, dass mit der Forderung nach Quoten junge Merkmalsträger der aktuell in der Überzahl be-

Feminismus: Chauvinismus 2.0!
findlichen Merkmalsvariante systematisch diskriminiert werden? Würden wir diese Quotenideologie konsequent in allen Bereichen zur Anwendung bringen, dann müssten nicht nur DAX-Unternehmen verpflichtet werden, erst dann wieder Menschen mit XY-Chromosom in den Vorstand zu berufen, wenn „genügend“ Menschen mit XX-Chromosom berufen wurden, sondern erst dann wieder Kindergärtnerinnen oder Krankenschwestern, wenn eine vergleichbare XY-Quote erreicht ist. In beiden genannten und allen anderen vergleichbaren Fällen würden junge Menschen aufgrund ihres eben nicht frei gewählten Geschlechts diskriminiert.
Vor diesem Hintergrund zeigt das Beispiel der Abschaffung der Sklaverei in den USA auch exemplarisch, dass eine staatlich verordnete Gleichberechtigung so lange kein ein besseres Miteinander garantiert, solange die Botschaft nicht in den Köpfen angekommen ist und im beruflichen, gesellschaftlichen und privaten Leben gelebt wird. Das gilt natürlich nicht nur für das Ende der Sklaverei, sondern auch für die Betreuung im Kindergarten oder die Pflege. Und deshalb bringt es mich auch so auf, dass die Politik dabei ist, weitreichende neue Ungerechtigkeiten zu schaffen und eine gesellschaftsspaltende Klientelpolitik damit letztlich zu zementieren. Ich wünsche mir weder eine männlich noch eine weiblich dominierte Welt. Auch keine der Weißen oder Schwarzen, der Jungen oder Alten. Ich wünsche
mir, dass Diskriminierung jedweder Merkmale, insbesondere solcher, für die die Merkmalsträger nichts können, die sie weder wählen noch beeinflussen können, eben gerade nicht im Fokus steht, sondern geächtet wird. Empathie, die übrigens eigentlich die Basis unserer Kultur des „christlichen Abendlandes“ ist, gilt es zum hehren Ziel aller Bemühungen zu machen. Das Gesetz wäre so einfach, es ist seit Langem hinlänglich bekannt, die einfache wie geniale Botschaft lautet: „Liebe Deinen Nächsten wie Dich selbst“ (Quelle: Bibel) bzw. „Handle nur nach derjenigen Maxime, durch die du zugleich wollen kannst, dass sie ein allgemeines Gesetz werde“ (Immanuel Kant). Für mich liegt der Schlüssel in einer Reform der Bildung und Erziehung junger Menschen. Es muss sichergestellt werden, dass Kinder nicht nur das und so viel mitbekom-
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH EDITORIAL
Prof. Dr. med. Karl-Ludwig Resch
37
men, wie in ihrer Familie zufällig prävalent ist (was ja großenteils auch nicht Konsequenz einer informierten Willensentscheidung ist ...).
Wie wäre es mit einem Menschentag statt einem Frauentag, einem Tag der Bürgerrechte, Tagen der irgendwelchen Partikularinteressen?
Vielleicht fragen Sie sich, was ein solches politisches Statement an dieser Stelle zu suchen hat. Sehr viel! Wir alle, die wir qua Approbation bzw. Facharztanerkennung die „Lizenz zum Helfen und Heilen“ bekommen haben, sind Merkmalsträger dieser Qualifikation. Das Geschlecht des jeweiligen Merkmalsträgers sollte für die kompetente Entscheidung keine Rolle spielen, ebenso wenig wie Hautfarbe und Herkunft (oder auch Glauben und andere persönliche Prioritätensetzungen fürs eigene Leben u.v.m.). Es wäre skandalös, würden wir eine chauvinistische oder feministische oder sonst diskriminierende Patientenversorgung betreiben. Wir sind für alle da. Das schwören wir seit zweieinhalb Tausend Jahren im Hippokratischen Eid. Ich fürchte, das reicht nicht mehr. Mischen wir uns ein, melden wir uns zu Wort, machen wir Druck, dass die Welt menschlicher wird, nicht weniger männlich, weiblich, schwarz oder weiß!
Karl-Ludwig Resch, Nürnberg
ORIGINALARBEIT
COVID-19: Frühzeitig einsetzende antivirale Therapie reduziert postakute Folgeerkrankungen, schwere Verläufe und Gesamtletalität 40 Barbara John
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
NEUE UND BEWÄHRTE ARZNEIMITTEL
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 38 INHALT RUBRIKEN Wissenswertes 54, 56 Kongresse 65 Multiple Sklerose: Patienten profitieren von der subkutanen Gabe von Natalizumab 55 Upstaza™ – eine neue intraputaminale Gentherapie bei AADC-Mangel 58 Upadacitinib bei atopischer Dermatitis: Stark wirksam und allgemein gut verträglich 60 Voclosporin – der erste in Europa zugelassene Calcineurininhibitor zur Behandlung der Lupus-Nephritis 62
Hidradenitis suppurativa: Anhaltende Symptomverbesserungen unter der Therapie mit Secukinumab 46 Epilepsie: Neue Erkenntnisse zum Einsatz von Perampanel 48 Neue Leitlinie zur Diagnostik und Therapie myasthener Syndrome 51 Urothelkarzinom: Neue Daten untermauern den Stellenwert der Erstlinien-Erhaltungstherapie mit Avelumab 52

SEIN?
häufigsten Symptome sind:1-3
Krise
Kongestion Erfahren Sie mehr auf www.aadc-mangel.de und www.aadc-testen.de Referenzen: 1. Manegold C, Hoffmann GF, Degen I, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32(3):371-380. 2. Wassenberg T, Molero-Luis M, Jeltsch K, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12(1):12. doi: 10.1186/s13023-016-0522-z. 3. Brun L, Ngu LH, Keng WT, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology. 2010;75(1):64-71. ©2022 PTC Therapeutics. PTC2105KK504
MUSKELHYPOTONIE BEI KINDERN? KÖNNTE ES EIN AADC-MANGEL
Die
Muskelhypotonie Entwicklungsverzögerungen Bewegungsstörungen Okulogyre
Nasale
ZUSAMMENFASSUNG
Postakute Folgeerkrankungen („Long COVID“) betreffen derzeit geschätzte 65 Millionen Menschen, die sich mit dem SARS-CoV-2-Virus infiziert haben. Populationsbasierte Studien und Meta-Analysen untersuchten die Wirksamkeit von Nirmatrelvir/r in der klinischen Routinepraxis. Den Ergebnissen zufolge reduziert dessen frühzeitiger Einsatz unabhängig vom Impfstatus das Risiko für schwere Verläufe, Hospitalisierungen, postakute Folgeerkrankungen und Todesfälle. Die Ergebnisse sind konsistent mit den Daten der Zulassungsstudie EPIC-HR.
Schlüsselwörter: SARS-CoV-2, Coronavirus, COVID-19, postakute Folgeerkrankungen, PostCOVID-19-Syndrom, Long COVID, Nirmatrelvir/Ritonavir
SUMMARY
Post-acute sequelae affect millions of people who contract an infection with the SARS-CoV-2 respiratory coronavirus. A number of large population-based studies and meta-analyses examined the efficacy of nirmatrelvir/r in routine clinical practice. Early use of this specific antiviral agent reduces the risk of severe disease, hospitalization, post-acute sequelae, and death, regardless of the vaccination status. These data are consistent with the results of EPIC-HR, the pivotal trial of nirmatrelvir/r.
Keywords: SARS-CoV-2, coronavirus, COVID-19, post-acute sequelae, post COVID 19 syndrome, long COVID, nirmatrelvir/ritonavir
COVID-19: Frühzeitig einsetzende antivirale
Therapie reduziert postakute Folgeerkrankungen, schwere Verläufe und Gesamtletalität
Barbara John Abteilung für Gastroenterologie, Onkologie und Palliativmedizin, Krankenhaus Leonberg, Klinikverbund Südwest
Postakut auftretende und chronische Folgeerkrankungen einer Infektion mit dem Coronavirus SARS-CoV-21 betreffen Millionen Menschen, die sich mit dem Virus anstecken. Aktuellen Schätzungen zufolge leiden 10 % der bislang mindestens 651 Millionen Menschen, bei denen eine COVID-19-Erkrankung2 dokumentiert wurde, unter lang anhaltenden Symptomen [1]. Der Prävention solcher Infektionsfolgen kommt daher eine herausragende Bedeutung für die Reduktion der mit COVID-19 assoziierten Krankheitslast zu.
Long COVID
Der Begriff Long COVID bezeichnet alle gesundheitlichen Beeinträchtigungen, Erkrankungen und Verschlechterungen vorbestehender Beschwerden im Zusammenhang mit einer SARS-CoV-2-Infektion, die nach Ende der akuten Krankheitsphase (definitionsge-
mäß mindestens 4 Wochen nach Auftreten der ersten Symptome) in Erscheinung treten oder bestehen bleiben [2]. Er umfasst damit alle mit einer SARS-CoV-2-Infektion assoziierten gesundheitlichen Beeinträchtigungen jenseits der akuten Infektion.
Schätzungen des European Centre for Disease Prevention and Control (ECDC) zufolge entwickelt im Mittel jede zweite an COVID-19 erkrankte Person mindestens eine Form von Post-COVID-Symptomatik, die per definitionem über mindestens 12 Wochen nach der akuten Infektion fortbesteht und nicht anderweitig erklärt werden kann [3]. Das ECDC weist jedoch darauf hin, dass in populationsbasierten Studien ohne nicht infizierte Vergleichspopulation die Häufigkeit von Symptomen, die ursächlich auf eine SARS-CoV2-Infektion zurückzuführen sind, überschätzt werden kann. So ermittelte eine mit Kontrollpopulation durchgeführte prospektive Kohortenstudie aus den Niederlanden für die Allgemeinbevölkerung einen Anteil von 12,7 % mit PostCOVID-Symptomatik [4].
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 40 ORIGINALARBEIT
1 SARS-CoV-2 = Severe Acute Respiratory Syndrome Coronavirus Type 2
2 COVID-19 = Coronavirus disease 2019
Antivirale Medikamente
Zur ursächlichen Therapie der akuten Coronavirus-Erkrankung (COVID-19) sind in Deutschland derzeit zwei kleinmolekulare Substanzen zugelassen, welche die Replikation von SARS-CoV-2 hemmen: der Proteaseinhibitor Nirmatrelvir/r3 sowie das Nukleosidanalogon Remdesivir. Nirmatrelvir inhibiert die 3CLProtease von SARS-CoV-2 und verhindert damit die Prozessierung des viralen Polyproteinvorläufers in seine funktionsfähigen Untereinheiten. Nirmatrelvir/r ist zugelassen für die Behandlung von COVID-19 bei Erwachsenen, die zwar keine Sauerstoffsubstitution benötigen, jedoch ein erhöhtes Risiko haben, einen schweren COVID-19-Verlauf zu entwickeln [5] (Tab. 1).
Eine Reihe großer populationsbasierter Studien und Meta-Analysen untersuchte die Wirksamkeit von Nirmatrelvir/r in der klinischen Routinepraxis. Damit steht zur Beurteilung der Wirksamkeit und Sicherheit dieser Therapieoption über die Zulassungsstudien hinaus eine breitere Datenbasis zur Verfügung. Die folgende Übersicht stellt repräsentative Studien vor.
US-amerikanische Kohortenstudie zur Therapie mit Nirmatrelvir/r während der akuten Infektion
Eine anhand der medizinischen Datenbank des US-amerikanischen US Department of Veterans Affairs (VA) durchgeführte große retrospektive Kohortenstudie [6] analysierte die längerfristigen Therapieergebnisse bei Patienten,
3 Nirmatrelvir wird mit dem gegen SARSCoV-2 inerten Ritonavir (/r) als pharmakokinetischem Booster angewendet.
Risikofaktoren für einen schweren COVID-19-Verlauf
• Alter >60 Jahre
• Body Mass Index >25 kg/m2
• Raucher oder Exraucher
• Maligne Erkrankung
• Kardiovaskuläre Erkrankung
• Nierenerkrankung
• Lungenerkrankung
• Diabetes mellitus
• Hypertonus
• Immunologische Dysfunktion
Tabelle 1: In der VA-Studie vordefinierte Risikofaktoren für die Progression der SARS-CoV-2-Infektion zur schwerwiegenden COVID-19-Erkrankung [6].
die im Zeitraum von März bis Juni 2022 im Rahmen der klinischen Routinetherapie während der akuten Phase einer SARS-CoV2-Infektion eine Behandlung mit Nirmatrelvir/r erhalten hatten.
In die Kohorte eingeschlossen wurden insgesamt 281.793 initial ambulant betreute Patienten mit SARS-CoV-2-Infektion und mindestens einem der vordefinierten Risikofaktoren (Tab. 1) für die Progression zu einer schwerwiegenden COVID-19-Erkrankung. Die Patienten der Nirmatrelvir/rGruppe (n = 35717) begannen binnen 5 Tagen nach dem Datum des ersten positiven PCR-basierten Nachweises von SARS-CoV-2 eine fünftägige ambulante Therapie mit Nirmatrelvir/r in der zugelassenen Dosierung (300/100 mg alle 12 Stunden).
Die Autoren verglichen die Verlaufsdaten dieser mit Nirmatrelvir/r therapierten Gruppe mit denen der kontemporären antiviral unbehandelten Kontrollgruppe von 246.076
Nirmatrelvir/r versus keine antivirale Therapie [* Reduktion statistisch signifikant anhand des 95%-Konfidenzintervalls]
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 41 ORIGINALARBEIT
Hazard Ratio Absolute Risikoreduktion bis Tag 180 [%] Postakutes Ereignis Tod 0,53 –0,65* Hospitalisierung 0,76 –1,72* Tod oder Hospitalisierung 0,74 –2,15* Postakute Erkrankung/Symptome 0,74 –4,51* Herzrhythmusstörung 0,73 –1,05* Myokardischämie 0,71 –0,51* Lungenembolie 0,61 –0,27* Tiefe Venenthrombose 0,30 –0,20* Fatigue 0,79 –0,94* Lebererkrankung 0,91 –0,14 Akute Nierenfunktionsstörung 0,67 –0,38* Myalgie 0,65 –0,72* Diabetes mellitus 0,98 –0,03* Neurokognitive Beeinträchtigung 0,74 –0,45* Dysautonomie 0,86 –0,16* Kurzatmigkeit 0,89 –0,51* Husten 0,96 –0,15
Tabelle 2: Postakute Ereignisse, Erkrankungen und Symptome im Gefolge einer SARS-CoV2-Infektion, mit Auftreten binnen 30 bis 180 Tagen bei Personen mit Nirmatrelvir/r-Behandlung versus Personen ohne antivirale Therapie [6].
Patienten aus derselben Kohorte. Diese hatten keine Therapie mit antiviralen Substanzen oder Antikörpern gegen SARS-CoV-2 in den ersten 30 Tagen der Infektion erhalten. Die Vergleichbarkeit der beiden Gruppen hinsichtlich ihrer Risikofaktoren wurde mit der Methode des Inverse Probability Weighting gewährleistet.
Analyse der postakuten Folgeerkrankungen und Todesfälle
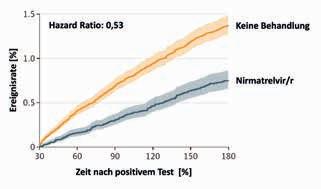
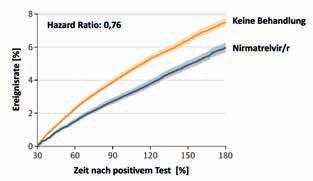
Das primäre Zielkriterium der Auswertung war das Auftreten von mindestens 1 von insgesamt 13 verschiedenen postakuten Folgeerkrankungen (PAFE) der SARSCoV-2-Infektion im Zeitraum von 30 bis 180 Tagen nach der Infektion (Tab. 2). Bei Analyse der Verlaufsdaten zeigte sich, dass die früh einsetzende Behandlung mit Nirmatrelvir/r das Risiko für eine PAFE signifikant um 26 % reduziert hatte (Hazard Ratio [HR]: 0,74; 95%-Konfidenzintervall [KI]: 0,69 – 0,81). Das Risiko einer postakuten Hospitalisierung war um 24 % vermindert (relatives Risiko [RR]: 0,76; Abb. 1A), das eines letalen Verlaufs um 47 % (RR: 0,53; Abb. 1B).
Für 11 der 13 vor der Analyse definierten postakuten Folgeerkrankungen bzw. Folgesymptome der SARS-CoV-2-Infektion wurde eine signifikante Risikoreduktion beobachtet. Diese war in den Subpopulationen der ungeimpften, geimpften und mehrfach geimpften Patienten gleichermaßen nachweisbar, ebenso bei Patienten mit primärer SARS-CoV-2-Infektion oder Reinfektion.
Das Ausmaß der Risikoreduktion war in moderatem Umfang abhängig von der Art der Grunder-
A. Postakute Hospitalisierung
B. Tod
Abbildung 1: Die Rate postakuter Hospitalisierungen war nach Behandlung mit Nirmatrelvir/r um etwa 30 % niedriger als in der Kontrollgruppe ohne antivirale Therapie (A). Das Risiko für einen postakut letalen Verlauf war um annähernd die Hälfte geringer (B). Halbtransparente Bereiche: 95%-Konfidenzintervall).
krankung und demographischen Merkmalen (Bereich der HR: 0,66 – 0,80). Sie war am stärksten in der Altersgruppe über 70 Jahre (HR = 0,66) und bei Patienten mit chronischen Nierenerkrankungen (HR = 0,65).
Ergebnisse einer großen Kohortenstudie aus Kanada
Schwartz und Kollegen [7] untersuchten eine Kohorte von
177.545 Patienten mit SARSCoV-2-Infektion, davon 8.876 mit Nirmatrelvir/r behandelte Patienten und 168.669 nicht damit therapierte Kontrollpersonen. Die beiden Gruppen waren in Bezug auf demographische und klinische Merkmale nach IPTW-Gewichtung (Inverse Probability of Treatment Weighting) gut ausgeglichen. In der mit Nirmatrelvir/r behandelten Gruppe waren die Raten von stationären Aufnahmen und/ oder Todesfällen innerhalb von 30
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 42 ORIGINALARBEIT
Tagen nach Diagnose der Infektion im Vergleich zu der nicht therapierten Gruppe um signifikant 43 % reduziert (2,1 % versus 3,7 %; Odds Ratio [OR]: 0,56; 95%-KI: 0,47 – 0,67). Auch die Gesamtmortalität war um etwa die Hälfte vermindert (1,6 % versus 3,3 %; OR: 0,49; 95%-KI: 0,39 – 0,62).
Die Zahl der Behandlungen mit Nirmatrelvir/r, die den beobachteten Verläufen zufolge erforderlich war, um eine schwere COVID19-Erkrankung (d.h. mit stationärer Aufnahme oder letalem Verlauf) zu verhindern, betrug 62 (95%-KI: 43 – 80). Die entsprechenden Ergebnisse waren in den Subkohorten mit verschiedenem Alter (≥70 bzw. <70 Jahre), Impfstatus (0 vs. 1 – 2 bzw. ≥3 Dosen) und Komorbiditäten (n = 3 bzw. <3) jeweils konsistent [7].
Kohortenstudien aus den USA beobachten Effekt auf die Hospitalisierungsrate
In einer großen Kohortenstudie [8] des Kaiser Permanente Healthcare System (USA) verglichen Lewnard und Kollegen die Erkrankungsverläufe in gematchten Kohorten von SARS-CoV-2-infizierten Personen mit versus ohne Nirmatrelvir/r-Therapie (n = 7.274 bzw. 126.152). Eine bis zu 5 Tage nach Auftreten der ersten Symptome begonnene 5-tägige Therapie mit Nirmatrelvir/r reduzierte die Wahrscheinlichkeit einer Hospitalisierung und/oder des Versterbens binnen 30 Tagen nach dem ersten positiven SARS-CoV-2Test um 79,6 %.
Eine Analyse der Subpopulation, die im Vorfeld ihrer Infektion ≥2 Dosen eines SARS-CoV-2-Impfstoffs erhalten hatte (93,3% der Gesamtkohorte), ergab ähnliche Werte der Risikoreduktion.
Zhou und Mitarbeiter [9] stellten die Interimsanalyse einer laufenden populationsbasierten Kohortenstudie auf Basis von Daten des Optum-Gesundheitsnetzwerks in den USA vor. Die ausgewerteten Daten wurden im Zeitraum von Dezember 2021 bis Juni 2022 erhoben. Hauptkriterien für die Aufnahme in die Kohorte waren ein Alter ≥12 Jahre, ein positiver SARS-CoV-2-Test, eine COVID-19-Diagnose sowie ein hohes Risiko für einen schweren Erkrankungsverlauf auf Basis demographischer und/oder klinischer Merkmale. Imbalancen der Patientenmerkmale zwischen der Gruppe, die Nirmatrelvir/r erhielt (n = 2.808), und der Kontrollpopulation (n = 10.849) wurden mittels Propensity Score Matching abgeglichen.
Primäres Zielkriterium der Analyse war die Rate von infektionsbedingten stationärer Aufnahmen. Diese betrug binnen 30 Tagen nach COVID-19-Diagnose in der Nirmatrelvir/r-Gruppe 1,2 % gegenüber 6,9 % bei den nicht Behandelten, was einer Reduktion des Hospitalisierungsrisikos um 84 % entspricht (HR: 0,16; 95%KI: 0,11 – 0,22).
Ähnliches ergab die Auswertung des Zeitraums bis 15 Tage nach der COVID-19-Diagnose: In der Nirmatrelvir/r-Gruppe wurden 0,8 % stationäre Aufnahmen verzeichnet gegenüber 6,5 % in der Kontrollgruppe, entsprechend einer Risikoreduktion um 89 % (HR: 0,11; 95%-KI: 0,07 – 0,17).
Die relative Risikoreduktion war über alle Altersgruppen hinweg vergleichbar ausgeprägt und bei geimpften wie ungeimpften Patienten ähnlich. Die Effektstärke entsprach den Beobachtungen in der Zulassungsstudie EPIC-HR zu Nirmatrelvir/r (vgl. Insert) [10].
Nirmatrelvir/r bei Patienten mit hohem Risiko für Hospitalisierung und letalen Verlauf einer SARSCov-2-Infektion: Meta-Analyse von 10 Beobachtungsstudien
Mittlerweile wurde eine Reihe methodisch hochwertiger Studien zum Einsatz von Nirmatrelvir/r bei SARS-Cov-2-infizierten Patienten mit hohem Risiko für eine Hospitalisierung und/oder einen letalen Verlauf durchgeführt. Um deren Ergebnisse in der Gesamtschau zu erschließen, erstellten
Ergebnisse der zulassungsrelevanten Studie EPIC-HR
• In der randomisierten Studie [10] mit 2.246 Patienten reduzierte Nirmatrelvir/r (150 mg/100 mg) das Risiko für den Endpunkt „schwerer COVID-19-Verlauf“ versus Placebo um 87,8 %, wenn die Therapie binnen 5 Tagen nach Auftreten erster Symptome begonnen wurde (jeweils p < 0,001).
• Nirmatrelvir/r war in allen untersuchten Subpopulationen signifikant überlegen. Alle dokumentierten Todesfälle traten in der Placebogruppe auf.
• Am deutlichsten war die Risikoreduktion bei Patienten der Altersgruppe ab 65 Jahren (Reduktion um den Faktor 19,2 bzw. 6,6 bei jüngeren Patienten).
• Unter Nirmatrelvir/r war die Viruslast am 5. Behandlungstag um den Faktor 7,4 bzw. 4,9 geringer als in der Placebogruppe (p < 0,001).
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 43 ORIGINALARBEIT
Cheema et al. [11] die bislang umfangreichste gemeinsame Meta-Analyse der beiden Zulassungsstudien (EPIC-HR [10] und EPIC-SR [12]) mit 10 beobachtenden Kohortenstudien zur Behandlung mit Nirmatrelvir/r. Insgesamt wurden die Daten von über 360.000 Patienten ausgewertet.
Die primären Zielkriterien waren die Hospitalisierungsrate und die Gesamtmortalität. Für ihre Vergleichsberechnungen verwendeten die Autoren die validierte GRADE-Methodik des Cochrane Network für Meta-Analysen [13]. Den Ergebnissen zufolge verminderte Nirmatrelvir/r in der Gesamtpopulation signifikant sowohl die Gesamtmortalität (relative Reduktion um 76 %) als auch die Hospitalisierungsrate (relative Reduktion um 59 %) im Zusammenhang mit SARS-CoV-2 Infektionen. Die Befunde der eingeschlossenen Beobachtungstudien waren konsistent mit denen der beiden Zulassungsstudien zu Nirmatrelvir/r (EPIC-HR und EPIC-SR). Übereinstimmung bestand auch in den Patientengruppen mit und ohne vorbestehende Immunität und in allen untersuchten Altersgruppen. Es gab keine Hinweise auf einen Publikationsbias [11].
Ergebnisse für die wichtigsten Subpopulationen
Die Reduktion der Hospitalisierungsrate durch die Therapie mit Nirmatrelvir/r war erwartungsgemäß bei Personen mit gesicherter Infektion, aber ohne nachweisbare Antikörper gegen SARS-CoV-2 stärker ausgeprägt als bei Patienten, die bereits eine humorale Immunität gegen das Virus aufgebaut hatten (RR: 0,27 gegenüber 0,45);
der Unterschied war jedoch nicht signifikant. Das Alter der Patienten (≥65 versus <65 Jahre) hatte keinen wesentlichen Einfluss auf die Reduktion der Hospitalisierungsrate durch Nirmatrelvir (RR: 0,26 bzw. 0,29). Das Risiko für einen letalen Verlauf hingegen war bei Patienten ab einem Alter von 65 Jahren um die Hälfte reduziert (RR: 0,49). In eine der ausgewerteten Studien waren gezielt Patienten ab 40 Jahren aufgenommen worden – auch hier war die Reduktion des Risikos eines letalen Verlaufs (RR: 0,21) statistisch signifikant (p ≤ 0,05). Hinsichtlich therapieassoziierter unerwünschter Ereignisse (UE) und schwerwiegender UE bestanden keine signifikanten Unterschiede zwischen mit Nirmatrelvir/r behandelten Patienten und der Vergleichspopulation [11].
Empfehlungen des RKI
Große populationsbasierte Studien und Meta-Analysen von Be-
Antivirale Therapie bei Long COVID?
Der Einsatz von Nirmatrelvir/r bei Patienten mit bereits bestehendem Long COVID-Syndrom wird ab Anfang 2023 im Rahmen prospektiver randomisierter Studien untersucht – ausgehend von der Hypothese, dass eine anhaltende virale Antigenämie zu der protrahierten Symptomatik beiträgt [19, 20]. Publizierte Fallberichte legen nahe, dass Patienten mit länger persistierenden Beschwerden von der antiviralen Therapie profitieren können [21, 22].
obachtungsstudien stützen den frühen Einsatz von Nirmatrelvir/r bei Patienten mit SARS-CoV2-Infektion und Risikofaktoren für einen schweren Verlauf der COVID-19-Erkrankung. Den Ergebnissen zufolge ist die Gabe von Nirmatrelvir/r assoziiert mit signifikant reduzierten Risiken für eine
Risikofaktoren für einen schweren Verlauf, die laut STAKOB eine antivirale Frühtherapie begründen
• Alter ab 50 – 60 Jahre
• Immunsuppression (durch Grunderkrankung oder medikamentös bedingt, etwa bei rheumatologischen Erkrankungen, oder nach Stammzell- oder Organtransplantation)
• Aktive Tumorerkrankung
• Chronische Nierenerkrankungen, auch Dialysepflichtigkeit
• Kardiovaskuläre Erkrankungen
• Diabetes mellitus
• Chronische Lungen- und Lebererkrankungen
• Sichelzellanämie oder Thalassämie
• Trisomie 21
• Adipositas mit Body Mass Index >35
Tabelle 3: Risikofaktoren für einen schweren Verlauf von COVID-19, die unabhängig vom Impfstatus eine Indikation zur antiviralen Therapie mit Nirmatrelvir/r (oder Molnupiravir*) in der Frühphase einer Antigen-positiven SARS-CoV-2 Infektion begründen – nach Angaben der Fachgruppe STAKOB (Ständiger Arbeitskreis der Kompetenz- und Behandlungszentren für Krankheiten durch hochpathogene Erreger) des Robert-Koch Instituts [18].
* Nach Angabe von STAKOB unterlegen im indirekten Vergleich.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 44 ORIGINALARBEIT
postakute Folgeerkrankung sowie für schwerwiegende und letale Verläufe. Übereinstimmend mit den Daten der hier vorgestellten Studien und weiterer populationsbasierter Analysen [14, 15] sowie basierend auf der vorangegangenen prospektiven Zulassungsstudie [10] empfiehlt die Fachgruppe COVRIIN des Robert Koch-Instituts den Einsatz von Nirmatrelvir/r als ein Medikament der ersten Wahl zur antiviralen Frühtherapie bei Patienten mit Risikofaktoren für einen schweren Verlauf (Tab. 3) und Symptombeginn bzw. vermutetem Infektionszeitpunkt von bis zu 5 Tagen [16, 17].
Literatur
1 Davis HE, McCorkell L, Vogel JM et al. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023;21:133-146
2 Koczulla AR, Ankermann T, Behrends U et al. AWMF S1-Leitlinie Long/Post-COVID. Robert-Koch-Institut, Stand 17. August 2022
3 European Centre for Disease Prevention and Control (ECDC). Prevalence of post COVID-19 condition symptoms: A systematic review and meta-analysis of cohort study data stratified by recruitment setting. Oktober 2022; https://www.ecdc.europa.eu/sites/default/files/documents/Prevalence-post-COVID-19-condition-symptoms.pdf
4 Anaya JM, Rojas M, Salinas ML et al. Post-COVID syndrome. A case series and comprehensive review. Autoimmun Rev 2021;20:102947
5 Fachinformation Paxlovid® (Nirmatrelvir/ Ritonavir); Stand: Januar 2023
6 Xie Y, Choi T, Al-Aly Z. Association of treatment with nirmatrelvir and the risk of post-COVID-19 condition. JAMA Intern Med 2023; e230743
7 Schwartz KL, Wang J, Tadreous M, et al. Real-world effectiveness of nirmatrelvir/ ritonavir use for COVID-19: a population-based cohort study in Ontario, Canada. Can Med Ass J (CMAJ) 2023; 195: E220-E226.
8 Lewnard JA, McLaughlin JM, Malden D et al. Effectiveness of nirmatrelvir-ritonavir in preventing hospital admissions and deaths in people with COVID-19: a cohort study in a large US health-care system. Lancet Infect Dis 2023;S14733099(23)00118-4
9 Zhou X, Scott PK, Liang C et al. Realworld effectiveness of nirmatrelvir/ritonavir in preventing hospitalization among patients with COVID-19 at high risk for severe disease in the United States: a nationwide population-based cohort study. https://doi.org/10.1101/2022.09.13. 22279908 (preprint)
10 Hammond J, Leister-Tebbe H, Gardner A et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N Engl J Med 2022;386:1397-1408
11 Cheema HA, Jafar U, Sohail A et al. Nirmatrelvir-ritonavir for the treatment of COVID-19 patients: a systematic review and meta-analysis. J Med Virol 2023; 95:e28471
12 Evaluation of protease inhibition for COVID-19 in standard-risk patients (EPICSR). https://clinicaltrials.gov/ct2/show/ NCT05011513
13 The GRADE working group 2022. https://www.gradeworkinggroup.org; Zugriff 6.2.2023
14 Aggarwal NR, Molina KC, Beaty LE et al. Real-world use of nirmatrelvir/ritonavir in outpatients with COVID-19 during the era of omicron variants including BA.4 and BA.5 in Colorado, USA: a retrospective cohort study. Lancet Infect Dis 2023;S1473-3099(23)00011-7
15 Dryden-Peterson S, Kim A, Kim AY et al. Nirmatrelvir plus ritonavir for early COVID-19 in a large U.S. health system: a population-based cohort study. Ann Intern Med. 2023;176: 77-84
16 Fachgruppe COVRIIN des Robert-KochInstituts. COVID-19: Medikamentöse und nicht-medikamentöse Therapieempfehlungen nach Erkrankungsphase. Orientierungshilfe für Ärztinnen und Ärzte (pdf; Stand 6.12.2022; Zugriff 03.03.2023)
17 Fachgruppe COVRIIN des Robert-KochInstituts. Hinweise zu Therapie und Versorgung bei COVID-19; https://www.rki. de/DE/Content/Kommissionen/COVRIIN/Therapie_Versorgung/FG_COVRIIN_ Therapie_Versorgung_node.html; Zugriff 10.3.2023
18 Ständiger Arbeitskreis der Kompetenzund Behandlungszentren für Krankheiten durch hochpathogene Erreger am RobertKoch-Institut (STAKOB). Hinweise zu Erkennung, Diagnostik und Therapie von Patienten mit COVID-19. Stand: 8.2.2023
19 SARS CoV-2 viral persistence study (PASC) – study of long COVID-19 (PASC); clinicaltrials.gov/ct2/show/NCT 05595369 (Zugriff 6.2.2023)
20 A decentralized phase 2 efficacy and safety study of nirmatrelvir/ritonavir in adult participants with long COVID (PAXLC). https://clinicaltrials.gov/ct2/ show/NCT05668091 (Zugriff 6.2.2023)
21 Peluso MJ, Anglin K, Durstenfeld MS et al. Effect of oral nirmatrelvir on long COVID symptoms: 4 cases and rationale for systematic studies. Pathog Immun 2022;7:95-103
22 Visvabharathy L, Orban ZS, Koralnik IJ. Case report: treatment of long COVID with a SARS-CoV-2 antiviral and IL-6 blockade in a patient with rheumatoid arthritis and SARS-CoV-2 antigen persistence. Front Med (Lausanne) 2022; 9:1003103
Anschrift der Verfasserin:
Dr. med. Barbara John
Geschäftsführende Chefärztin
Abteilung für Gastroenterologie, Onkologie und Palliativmedizin
Krankenhaus Leonberg, Klinikverbund
Südwest
Rutesheimer Straße 50
71229 Leonberg
b.john@klinikverbund-suedwest.de
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 45 ORIGINALARBEIT
Die Hidradenitis suppurativa (HS, auch Acne inversa genannt) ist eine systemische, chronisch-entzündliche Hauterkrankung, die zu schmerzhaften, potenziell irreversiblen entstellenden Läsionen führen und damit nicht nur die Lebensqualität, sondern auch die psychische Gesundheit der Betroffenen stark beeinträchtigen kann. Kennzeichnend sind wiederkehrende entzündliche Knoten unter der Haut, die sich zu schmerzhaften kutanen bis subkutanen Abszessen mit Eiterbildung und schließlich zu entzündeten Hauttunneln bzw. Fisteln entwickeln, die häufig übelriechende Sekrete abgeben (Abb. 1). Prädilektionsstellen sind die Axilla, die Perinealregion, die Genitalregion und der Kopf [1]. Die Hidradenitis suppurativa tritt in der Regel nach der Pubertät auf und begleitet die Patienten meist über mehrere Jahrzehnte.

In Deutschland sind aktuell etwa 25.000 bis 50.000 Personen von der Erkrankung betroffen, wobei eine hohe Dunkelziffer vermutet wird [2]. Zwischen der Erstmanifestation und der Diagnosestellung vergehen in manchen Fällen bis zu 10 Jahre [3]. Die derzeit verfügbaren medikamentösen Therapieoptionen sind begrenzt und können ergänzend eine weit ausgedehnte chirurgische Exzision mit anschließender Transplantatdeckung der betroffenen Bereiche erfordern. Alternativ kann eine ablative Lasertherapie versucht werden [4]. Um den Betroffenen ein von der Erkrankung möglichst unbeeinträchtigtes Leben zu ermöglichen, bedarf es daher neuer, lang anhaltend wirksamer und verträglicher Therapien.
Vielversprechend ist der Einsatz von Biologika, die durch Hemmung der entzündungsfördernden
Hidradenitis suppurativa: Anhaltende Symptomverbesserungen unter der Therapie mit Secukinumab
Zytokine Linderung bringen [1]. Wie aktuelle, im Lancet publizierte Studiendaten zeigen, lassen sich durch die Therapie mit dem Interleukin(IL)-17A-Inhibitor Secukinumab (Cosentyx®) die Abszesse und Entzündungsherde bei Patienten mit mittelschwerer bis schwerer Hidradenitis suppurativa signifikant verringern und die Schmerzen anhaltend lindern [5].
Hohe Ansprechraten und anhaltende Wirksamkeit
Die kurz- (16 Wochen) und langfristige (bis zu 52 Wochen) Wirk-
samkeit von Secukinumab bei erwachsenen Patienten mit mittelschwerer bis schwerer Hidradenitis suppurativa wurde in den beiden randomisierten, placebokontrollierten Parallelgruppenstudien SUNSHINE [6] und SUNRISE [7] (NCT03713632) untersucht. Mit mehr als 1.000 Studienteilnehmern in 40 Ländern stellen sie das größte Phase-IIIProgramm mit HS-Patienten dar. An der SUNSHINE-Studie nahmen 541, an der SUNRISE-Studie 543 Patienten teil.
In jeder Studie wurden die Teilnehmer nach dem Zufallsprinzip einem der 3 Studienarme zugeteilt:
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 46 AKTUELLE THERAPIEKONZEPTE
FÜR DIE PRAXIS
Abbildung 1: Kennzeichen der Hidradenitis suppurativa sind Abszesse mit Eiterbildung, die sich zu entzündeten Fisteln entwickeln können.
• Secukinumab 300 mg zweiwöchentlich (q2w) nach einer wöchentlichen Initialdosis in den Wochen 0, 1, 2, 3 und 4
• Secukinumab 300 mg vierwöchentlich (q4w) nach einer wöchentlichen Initialdosis in den Wochen 0, 1, 2, 3 und 4
• Placebo-Dosis zweiwöchentlich nach einer wöchentlichen Initialdosis in den Wochen 0, 1, 2, 3 und 4
Primärer Endpunkt war das klinische Ansprechen gemäß HS Clinical Response (HiSCR) nach 16 Behandlungswochen. HiSCR war definiert als Rückgang der Abszesse und Entzündungsherde um mindestens 50 % ohne Zunahme der Anzahl der Abszesse und/oder Fisteln im Vergleich zum Ausgangswert. Zu den wichtigsten sekundären Endpunkten zählten die prozentuale Veränderung der Anzahl der Abszesse und entzündlichen Knoten in Woche 16 gegenüber dem Ausgangswert sowie der Anteil der Patienten, bei denen über 16 Wochen ein Krankheitsschub auftrat. Ein weiterer wichtiger sekundärer Endpunkt war der Anteil der Patienten, der in Woche 16 einen NRS30-Score (30%igen Reduktion der Schmerzen) aufwies, bestimmt anhand einer numerischen 11-Punkte-Bewertungsskala (berichtet als gepoolte Daten aus beiden Studien).
In Woche 16 der SUNSHINEStudie erreichten 45,0 % der Teilnehmenden unter Secukinumab 300 mg q2w eine HiSCR (vs. Placebo 33,7 %; p = 0,0070), in der SUNRISE-Studie erzielten 42,3 % der entsprechenden Behandlungsgruppe eine HiSCR (vs. Placebo 31,2 %; p = 0,0149).
Secukinumab 300 mg q4w war in der SUNRISE-Studie in Bezug auf das Erreichen der HiSCR im Vergleich zu Placebo überlegen
Stadieneinteilung
Entsprechend der Schwere des Krankheitsbildes können nach Hurley 3 Stadien unterschieden werden [1]:
Stadium I
Auftreten von Riesenkomedonen. Einzelne oder multiple Läsionen ohne Vernarbung und Fistelbildung. Derbe, tastbare Knoten in der Haut. Verwechslung mit Acne conglobata oder Furunkeln möglich. Die Therapie erfolgt je nach Schweregrad mit einer topischen und/ oder systemischen Antibiotikagabe.
Stadium II
Ein oder mehrere Abszesse mit abgekapselten Eiteransammlungen und Fistelgängen sowie Narbenbildung. Auf manuellen Druck kann sich Talg, Eiter oder übelriechendes Sekret entleeren. Zur Behandlung werden systemischen Antibiotika über einen längeren Zeitraum verabreicht. Inzision und Drainage der Entzündungsherde können die Symptomatik lindern, Fistelgänge sollten exzidiert, tiefere Läsionen ggf. plastisch gedeckt werden.
Stadium III: Diffuser, flächiger Befall mit multiplen Abszessen, die zu großen Strängen zusammenfließen können. Zahlreiche, tief reichende und miteinander kommunizierende Fistelgänge sowie narbige Areale mit ausgebrannten Entzündungsherden machen die Gabe von antiinflammatorisch wirkenden Biologika nötig. Oft ist eine ausgedehnte chirurgische Exzision mit anschließender Transplantatdeckung der betroffenen Bereiche erforderlich. Alternativ kann eine ablative Lasertherapie versucht werden.
Secukinumab
Secukinumab (Cosentyx®) ist ein vollhumaner, monoklonaler Antikörper, der direkt gegen IL-17A gerichtet ist. Das Zytokin IL-17A ist an Entzündungsprozessen und der Entstehung von Plaque-Psoriasis (PsO), Psoriasis-Arthritis (PsA) und der axialen Spondyloarthritis (axSpA) beteiligt. Secukinumab wird seit mehr als 14 Jahren klinisch untersucht. Der IL-17A-Inhibitor verfügt über eine umfangreiche klinische Evidenz für die 3 Indikationen PsO, PsA und axSpA sowie Daten aus der klinischen Praxis. Außerdem liegen Daten zu 2 Unterformen der juvenilen idiopathischen Arthritis (JIA), der Enthesitis-assoziierten Arthritis (EAA) und der juvenilen PsoriasisArthritis (jPsA) vor. Cosentyx wird mittels Fertigpen verabreicht [8].
(46,1 % vs. 31,2 %; p = 0,0022]), zeigte in der SUNSHINE-Studie hingegen keine statistische Signifikanz gegenüber Placebo (41,8 % vs. 33,7 %; p = 0,0418).
Wie eine explorativen Analyse beider Studien ergab, verbesserte sich die Ansprechrate auf die Behandlung mit Secukinumab über den primären Endpunkt in Woche
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 47 AKTUELLE THERAPIEKONZEPTE
FÜR DIE PRAXIS
16 hinaus: Mehr als 55 % der Patienten erreichten in Woche 52 ein klinisches Ansprechen auf Secukinumab, gemessen an der HSiSCR. Zudem waren in Woche 52 mehr als 60 % der Patienten frei von Schüben. Über die Hälfte berichtete darüber hinaus von einer mindestens 30%igen Reduktion ihrer Schmerzen (NRS30) im Vergleich zu dem Zeitpunkt vor Therapiebeginn. In beiden Studien wurden keine neuen Sicherheitssignale beobachtet [5].
Die Ergebnisse wurden vom Hersteller Novartis bei der Europäischen Arzneimittel-Agentur (EMA) zur Zulassung eingereicht. Die Entscheidung wird für das Jahr 2023 erwartet.
Brigitte Söllner, Erlangen
Epilepsie: Neue Erkenntnisse zum Einsatz von Perampanel
Zur Behandlung verschiedener Formen der Epilepsie stehen heute mehr als 20 anfallssupprimierende Medikamente zur Verfügung, sodass sich häufig die Frage stellt, welche Substanz wo im Therapie-Algorithmus zu verorten ist. Lange Zeit war es gängige Praxis, später zugelassene Wirkstoffe auch später im Therapieverlauf einzusetzen. Dies wird jedoch dem Potenzial moderner anfallssupprimierender Medikamente nicht gerecht. Dies trifft auch auf den Wirkstoff Perampanel (Fycompa®) zu, für den es mittlerweile eine breite Datenlage und ausreichend Evidenz dafür gibt, dass ein früher Einsatz dieser Substanz zu einem besseren Ansprechen und höheren Retentionsraten führt [1, 2, 3].
Frühzeitige Gabe bringt signifikante Vorteile
Literatur
1 MedLine Plus. Hidradenitis suppurativa. https://medlineplus.gov/hidradenitissuppurativa.html
2 Kirsten N et al. Arch Dermatol Res 2021;313:95-99
3 Kokolakis G et al. Dermatol 2020;236: 421-430
4 Cramer P et al. Hautarzt 2021;72;692-699
5 Kimball AB et al. Lancet 2023; doi: 10.1016/S0140-6736(23)00022-3
6 ClinicalTrials.gov. This is a study of efficacy and safety of two secukinumab dose regimens in subjects with moderate to severe hidradenitis suppurativa (HS) (SUNSHINE). NCT03713619. https://clinicaltrials.gov/ct2/show/NCT03713619
7 ClinicalTrials.gov. Study of efficacy and safety of two secukinumab dose regimens in subjects with moderate to severe hidradenitis suppurativa (HS) (SUNRISE). NCT03713632. https://clinicaltrials.gov/ ct2/show/NCT03713632
8 Fachinformation Cosentyx®; Stand: Januar 2023
Dass die Patienten von einem frühzeitigen Einsatz von Perampanel
deutlich profitieren können, zeigen unter anderem die Ergebnisse der 12-monatigen, prospektiven Beobachtungsstudie PERADON, in die 113 Patienten mit fokalen Anfällen mit oder ohne sekundäre Generalisierung eingeschlossen wurden [1]. Die Studienteilnehmer erhielten Perampanel entweder als erste oder als zweite Zusatztherapie. Der frühere Einsatz führte zu höheren Ansprech- (76,2 % vs. 63,4 %) und Anfallsfreiheitsraten (38,1 % vs. 19,7 %) (Abb. 1).
Auch in der retrospektiven Beobachtungsstudie COM-PER bei Patienten mit fokalen Anfällen zeigte sich der Vorteil der frühen Perampanel-Gabe [2]. Während von den 21 Patienten, die den Wirkstoff als erste Zusatztherapie erhielten, 71,4 % anfallsfrei wurden, erreichten dies in der späten Zusatztherapie (Vortherapie mit im Mittel 5 verschiedenen Wirkstoffen) nur 13,3 % von 60 Patienten. Gleichzeitig belegte die Studie, dass selbst Patienten mit frustraner
Abbildung 1: In der PERADON-Studie führte Perampanel, wenn es als erste Zusatztherapie gegeben wurde, zu einer höheren Ansprech- und Anfallsfreiheitsrate als bei der späteren Gabe [1].
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 48 AKTUELLE THERAPIEKONZEPTE FÜR
DIE PRAXIS
Abb. 1: PERADON: Wirksamkeit von Perampanel als erste vs. zweite Zusatztherapie (modifiziert nach Jaramillo et al. Epilepsy Behav. 2020; 102: 106655)
0 10 20 30 40 50 60 70 80 90 100 Anfallsreduktion ≥ 50% Anfallsfreiheit Diagrammtitel 1.Zusatztherapie 12 Mon. (n = 42) 2.Zusatztherapie 12 Mon. (n = 71) 76,2 % 63,4 % 38,1 % 19,7 % p = 0,158 p = 0,033 Patienten (%)
Wirksamkeit von Perampanel als erste vs. zweite Zusatztherapie
Therapieanamnese nach Einsatz einer Vielzahl von anfallssupprimierenden Medikamenten noch von Perampanel profitieren können. Damit ist der Wirkstoff nicht nur eine gute Option in der frühen Zusatztherapie, sondern hat auch einen hohen Stellenwert in schwierigen Konstellationen.
Inwiefern sich diese Erkenntnisse bereits in der Behandlungspraxis widerspiegeln, wird aktuell – unter verschiedenen anderen Aspekten – in der prospektiven, nicht interventionellen Studie PERPRISE (NCT04202159) in Deutschland untersucht [4]. Eingeschlossen wurden Patienten mit einer aktiven Epilepsie mit Grand-mal-Anfällen, die Perampanel als einzige Zusatztherapie erhalten. Ziel ist es, zu erfahren, welchen Stellenwert Perampanel für die behandelnden Ärzte hat, wie und mit welchen Kombinationspartnern es im Therapieverlauf eingesetzt wird – und mit welchem Erfolg.
Den Anforderungen junger Patienten gerecht werden
Kinder und Jugendliche stellen besondere Anforderungen an die Therapie. So sollten die Einnahmeschemata einfach sein (möglichst mit Einmalgabe), um bei den jungen Patienten eine gute Compliance zu erzielen. Außerdem muss bei dieser Altersgruppe auf Verhaltensänderungen und eine Beeinträchtigung der Kognition geachtet werden, die unter der anfallssupprimierenden Medikation auftreten können.
Die Langzeit-Wirksamkeit und -Verträglichkeit von Perampanel in dieser Altersgruppe wurden z.B. in der retrospektiven Langzeit-PhaseIV-Studie PROVE mit einer Subgruppe von 151 Kindern unter 12
Patienten (%)
Perampanel
Perampanel (Fycompa®) ist ein hoch selektiver, nicht kompetitiver AMPA-Rezeptor-Antagonist (α-Amino-3-hydroxy-5-methyl-4isoxazolpropionsäure), der seine Wirksamkeit in der Reduktion von epileptischen Anfällen in Studien der Phasen II und III demonstriert hat. AMPA-Rezeptoren, die weithin in fast allen exzitatorischen Neuronen vorhanden sind, übertragen Signale, die vom Neurotransmitter Glutamat im Gehirn vermittelt werden. Es wird davon ausgegangen, dass sie eine Rolle bei Erkrankungen des zentralen Nervensystems spielen, die sich durch übermäßige exzitatorische Signalübertragung auszeichnen, wie z.B. Epilepsie.
Perampanel ist in Deutschland als Zusatztherapie für Patienten ab 4 Jahren mit fokalen Anfällen mit oder ohne sekundäre Generalisierung zugelassen. Außerdem ist es indiziert bei Patienten ab 7 Jahren zur Zusatzbehandlung primär generalisierter tonisch-klonischer Anfälle bei idiopathisch generalisierter Epilepsie [13].
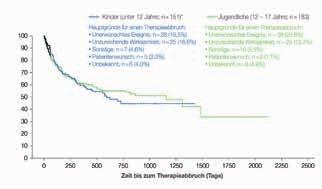
Jahren und 183 Jugendlichen im Alter von 12 – 17 Jahren untersucht [5]. Nach etwa 4 Jahren verblieb noch etwa jeder zweite Studienteilnehmer auf Perampanel. 18,5 % bzw. 20,8 % brachen die Therapie aufgrund unerwünschter Ereignis
se, 16,6 % bzw. 13,7 % aufgrund mangelnder Anfallskontrolle ab (Abb. 2). Insgesamt zeigte Perampanel also auch im Langzeitverlauf eine sehr gute Retentionsrate und Anfallsreduktion bei Kindern und Jugendlichen.
Die Verträglichkeit von Perampanel hinsichtlich Verhalten und Kognition untersuchte eine retrospektive Beobachtungsstudie bei 37 jugendlichen Patienten mit fokalen Anfällen mit oder ohne sekundäre Generalisierung anhand zweier Scores: dem EpiTrack® Junior und dem CBCL (Child Behavior Checklist) [6]. Dabei zeigten sich keine signifikanten Veränderungen zur Baseline-Untersuchung.
Dass es durch die Zusatztherapie mit Perampanel auch zu keiner Be-
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 49 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
-
Abbildung 2: Ergebnisse der PROVE-Studie: Retentionsrate und Gründe für einen Therapieabbruch bei Kindern und Jugendlichen [5].
* Responderrate** Anfallshäufigkeit unverändert Anfallshäufigkeit erhöht
Abbildung 3: Subgruppenanalyse aus PERMIT und PROVE: Responder- und Anfallsfreiheitsraten bei Älteren (≥ 65 Jahre). * Keine Anfälle seit der letzten Kontrolle, ** 50 % verringerte Anfallshäufigkeit gegenüber Baseline [12].
einträchtigung der Lebensqualität kommt, belegen die Ergebnisse der Post-hoc-Analyse einer unverblindeten Phase-III-Studie mit 115 pädiatrischen Patienten im Alter von 4 – 11 Jahren, die unter fokalen Anfällen mit oder ohne sekundäre Generalisierung bzw. primär generalisierten Anfällen litten [7]. Die Lebensqualität blieb unter Perampanel insbesondere dann konstant, wenn eine gute Anfallskontrolle erreicht werden konnte.
Bei älteren Patienten steht die Verträglichkeit im Fokus
Bei den Senioren, einer durch den demografischen Wandel stetig wachsenden Gruppe, gilt es eine Reihe von Besonderheiten [8] zu beachten, die spezielle Anforderungen an die Therapie stellen. So zeigen sich bei Pharmakokinetik und Pharmakodynamik altersabhängige Effekte: Ältere Menschen weisen stoffwechselbedingt eine erhöhte Arznei-Sensitivität hinsichtlich der Wirkung, aber auch Unverträglichkeiten auf. Außerdem sind sie durch Anfälle stärker gefährdet, haben eine erhöhte Verletzungsgefahr infolge von Stür-
zen, insbesondere unter einer Antikoagulation.
Wie eine Studie belegt, hat die anfallssupprimierende Medikation einen Einfluss auf die Lebenserwartung bei Älteren [9]. Dabei könnten in diesem Kollektiv häufige Begleiterkrankungen und eine Polytherapie eine verträgliche Anfallskontrolle zusätzlich verkomplizieren, was in klinischen Studien möglicherweise nicht ausreichend berücksichtig wird. Daher sind Daten zur Wirksamkeit und Verträglichkeit von Antiepileptika für diese Patientengruppe besonders wichtig.
In einer aktuellen Beobachtungsstudie aus Italien zu Perampanel mit 92 älteren Epilepsie-Patienten haben sich unter einer niedrigen Dosierung von 6 mg (Median) eine mit anderen Altersgruppen vergleichbare Wirksamkeit und Verträglichkeit gezeigt [10]. Aus der großen internationalen RealWorld-Studie PERMIT [3] und der amerikanischen Phase-IV Studie PROVE [11] gingen Daten von 394 Patienten ab 65 Jahren (Median 72,5 Jahre) in eine altersspezifische Analyse der Therapie mit Perampanel ein [12]. Die Patienten erhielten im Mittel 5,1 mg Perampanel, am häufigsten kom
biniert mit Levetiracetam oder Lamotrigin. Die Retentionsrate, der kombinierte Endpunkt für Wirksamkeit und Verträglichkeit, lag nach 12 Monaten bei 60,9 %. Die häufigsten Gründe für einen Therapieabbruch waren unerwünschte Ereignisse (22,4 %) und mangelnde Wirksamkeit (6,1 %). Die Ansprechraten lagen bei 69,1 % (fokale Anfälle) bzw. 70,6 % (generalisierte Anfälle) (Abb. 3). Aus diesen Daten lässt sich schließen, dass Perampanel bei älteren Patienten unter Praxisbedingungen insbesondere in niedrigen Dosierungen eine gute Wirksamkeit und Verträglichkeit zeigt.
Fabian Sandner, Nürnberg
Literatur
1 Jaramillo JA et al. Epilepsy Behav 2020; 102:106655
2 Canas N et al. Seizure 2021;86:109-115
3 Villanueva V et al. J Neurol 2022; 269:1957-1977
4 https://www.clinicaltrials.gov/ct2/show/ NCT04202159?term=NCT04202159&dr aw=2&rank=1
4 Segal E et al. J Child Neurol 2022; 8830738211047665
6 Operto FF at al. Epilepsy Behav 2020; 103:106879
7 Trigg A et al. Epilepsy Behav 2021;118: 107938
8 Trinka E et al. Acta Neurol Scand 2003: 108 (Suppl. 180):33-36
9 Werhan KJ et al. Epilepsia 2015;56:450459
10 Leppik IE et al. Epilepsy Res 2015; 110:216-220
11 Wechsler RT et al. Epilepsia Open 2022; 7:293-305
12 Inaji et al. AES 2022, Nashville, TN, USA. 2–6 December 2022
13 Fachinformation Fycompa®; Stand: August 2022
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 50 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
-
Fokale Anfälle Generalisierte Anfälle 100 90 80 70 60 50 40 30 20 10 0 12,1 � 11,8 � 8,2 � 0 � 69,1 � 70,6 � 39,0 � 52,6 � 2/17 12/17 10/19 19/231 28/231 161/233 96/246 0/17
Patienten (%)
Anfallsfreiheitsrate
Die Deutsche Gesellschaft für Neurologie (DGN) hat die neue S2k-Leitlinie „Diagnostik und Therapie myasthener Syndrome“ publiziert (www.dgn. org/leitlinien). Basierend auf den pathophysiologischen Erkenntnissen der letzten Jahre und den damit verbundenen Arzneimittelzulassungen wurden neue Therapieziele sowie -konzepte für die generalisierte Myasthenia gravis (gMG) erarbeitet. Die gMG ist eine seltene, chronische, neuromuskuläre Autoimmunerkrankung, die durch eine belastungsabhängige im Tagesverlauf zunehmende Muskelschwäche gekennzeichnet ist. Bei ca. 80 % der gMG-Patienten lassen sich Autoantikörper gegen den Acetylcholin-Rezeptor (AChR) nachweisen, die zu einer Störung der Erregungsübertragung an der postsynaptischen Membran der neuromuskulären Endplatte führen.
Paradigmenwechsel durch ein an der Krankheitsaktivität orientiertes Therapieprozedere
Als übergeordnetes Therapieziel definiert die neue S2k-Leitlinie die bestmögliche Krankheitskontrolle unter Wiederherstellung bzw. Er-
Ravulizumab
DIE PRAXIS
Neue Leitlinie zur Diagnostik und Therapie myasthener Syndrome
halt der Lebensqualität. Um das zu erreichen, wird ein Therapieprozedere empfohlen, das sich neben dem Antikörperstatus und der Thymuspathologie auch zunehmend an der Krankheitsaktivität orientieren soll. So erfolgt nun anhand des Schweregrads der klinischen Symptomatik, ihrer Dauer und Rückbildungstendenz sowie der klinischen Residuen und des Vorhandenseins bzw. der Zahl krisenhafter Verschlechterungen/Krisen eine Einstufung in eine milde/moderate versus (hoch-)aktive Erkrankung. Dabei wird die (hoch-)aktive generalisierte MG (inklusive „therapierefraktärer“ MG) in der neuen Leitlinie wie folgt definiert:
• Symptomatik (≥ MGFA IIa) und/oder mindestens 2 rezidivierende schwere Exazerbationen/myasthene Krisen mit Notwendigkeit der therapeutischen Intervention (i.v. Immun-
Ravulizumab (Ultomiris®) ist ein humanisierter monoklonaler IgGAntikörper und der erste und einzige langwirksame terminale C5Komplementinhibitor, der zu einer sofortigen, vollständigen und langanhaltenden Komplementhemmung führt. Der Wirkmechanismus beruht auf einer hochselektiven Hemmung des C5-Proteins in der terminalen Komplementkaskade. Unkontrolliert aktiviert kann das Komplementsystem, Teil der körpereigenen Immunabwehr, zu einer Zerstörung gesunder körpereigener Zellen führen. In der EU ist Ravulizumab seit September 2022 als Zusatztherapie zu einer Standardtherapie bei erwachsenen Patienten mit AchRAntikörper-positiver generalisierter Myasthenia gravis (gMG) zugelassen. Nach Gabe einer Initialdosis wird Ravulizumab erwachsenen gMG-Patienten alle 8 Wochen i.v. verabreicht.
globuline, Plasmapherese, Immunadsorption innerhalb eines Jahres nach Diagnosestellung trotz adäquater verlaufsmodifizierender und symptomatischer Therapie)
• Oder anhaltende alltagsrelevante Symptomatik (≥ MGFA IIa) und schwere Exazerbation/ myasthene Krise innerhalb des letzten Jahres trotz adäquater verlaufsmodifizierender und symptomatischer Therapie
• Oder anhaltende alltagsrelevante auch milde/moderate Symptomatik (≥ MGFA IIa) über mehr als 2 Jahre trotz adäquater verlaufsmodifizierender und symptomatischer Therapie
Bei der verlaufsmodifizierenden Therapie der milden/moderaten AChR-Antikörper-positiven gMG empfehlen die Leitlinien weiterhin Glukokortikoide und/oder Azathioprin. Bei der Behandlung der (hoch-)aktiven (inkl. therapierefraktären) AChR-Antikörper-positiven gMG soll nun die Klasse der C5-Komplementinhibitoren – mit Eculizumab* und Ravulizumab** – neben FcRn-Modulatoren (Efgartigimod**) mit oder ohne Thymektomie eingesetzt werden.
Fabian Sandner, Nürnberg
* Der C5-Komplementinhibitor Eculizumab (Soliris®) steht seit 2017 für die Behandlung der therapierefraktären gMG bei AChR-Antikörper-positiven Patienten in Deutschland zur Verfügung; er wird alle 2 Wochen verabreicht.
** Ravulizumab und Efgartigimod sind als Add-on-Therapie für die AChR-Antikörperpositive gMG zugelassen.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 51 AKTUELLE THERAPIEKONZEPTE
FÜR
Weltweit ist Blasenkrebs die zehnthäufigste Krebserkrankung. Im Jahr 2020 wurden weltweit mehr als eine halbe Million Neuerkrankungen diagnostiziert und rund 200.000 Todesfälle entfielen auf diese Erkrankung [1]. Das Urothelkarzinom macht etwa 90 % aller Blasenkrebsfälle aus [2]. Seine Behandlung wird schwieriger, je weiter die Erkrankung fortschreitet, da sich der Tumor durch die Schichten der Blasenwand hindurch ausbreitet [3]. Nur etwa ein Drittel der Patienten erhält nach Erstlinien-Chemotherapie eine Zweitlinientherapie [4]. In der Zweitlinie ist derzeit neben der erneuten Chemotherapie mit platinhaltigen Zytostatika
Urothelkarzinom: Neue Daten untermauern den Stellenwert der ErstlinienErhaltungstherapie mit Avelumab
oder Vinflunin auch die Gabe von Immuncheckpoint-Inhibitoren praxisrelevant [5]. Bei Patienten mit fortgeschrittenem Urothelkarzinom beträgt die 5-Jahres-Überlebensrate lediglich 7,7 % [6] – der Bedarf an innovativen Therapien ist entsprechend hoch.
Ergebnisse der Studie JAVELIN Bladder 100 markieren einen Wendepunkt
Vor 2 Jahren wurde Avelumab (Bavenico®) in der EU zur Therapie des lokal fortgeschrittenen bzw. metastasierten Urothelkarzinoms bei Patienten ohne Krankheitsprogression nach platinbasierter
Chemotherapie zugelassen. Damit besteht nun die Möglichkeit für ein völlig neues Behandlungsschema, bei dem die Patienten eine Erstlinien-Erhaltungstherapie mit Avelumab erhalten [7]. Mit diesem Ansatz wurde in der zulassungsrelevanten Phase-III-Studie JAVELIN Bladder 100 im Vergleich zur alleinigen Best Supportive Care (BSC) ein signifikant verlängertes medianes Gesamtüberleben (mOS) erzielt. Zum Datenschnitt 19.01.2020 lag es im AvelumabArm bei 22,1 Monaten, gegenüber 14,6 Monaten im Kontroll-Arm und war damit unter Avelumab um 7,5 Monate verlängert (HR für Tod: 0,70; 95%-KI: 0,56 – 0,86; p = 0,0008). Die mOS-Rate
[9].
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 52 AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
nach
Subgruppen Cisplatin + Gemcitabin (n = 389) Carboplatin + Gemcitabin (n = 269) Therapie-Arme Avelumab + BSC (n = 183) BSC (n = 206) Avelumab + BSC (n = 183) BSC (n = 122) Medianes OS* (95%-KI) 25,1 Monate (19,3 – 30,9) 17,5 Monate (13,7 – 24,2) 20,8 Monate (17,9 – 28,7) 13,0 Monate (9,4 – 16,1) HR: 0,79; 95%-KI: 0,611 – 1,020 HR: 0,69;95%-KI: 0,516 – 0,925) Medianes PFS* (95%-KI) 5,7Monate (4,6 – 7,5) 2,0 Monate (1,9 – 3,6) 3,7 Monate (3,6 – 5,6) 2,0 Monate (1,9 – 3,0) HR: 0,56; 95%-KI: 0,446 – 0,713 HR: 0,48; 95%-KI: 0,362 – 0,640 Behandlungsbedingte unerwünschte Ereignisse ≥Grad 3 30 (16,5 %) 0 32 (22,5 %) 0
Studie JAVAELIN Bladder 100 nach ≥38 Monaten Nachbeobachtungszeit.
Gemessen
Erstlinien-Erhaltungstherapie
Tabelle 1: Subgruppenanalyse der
*
vom Start der
einem Jahr ab Randomisierung betrug 71,3 % für Avelumab vs. 58,4 % in der Kontroll-Gruppe [8]. Vom Überlebensvorteil unter Avelumab profitierten sowohl Patienten, die eine Cisplatin- als auch solche, die eine Carboplatin-basierte Erstlinien-Chemotherapie erhalten hatten. Er war außerdem unabhängig von Alter, ECOG-Status, Baseline-Metastasierung oder Nierenfunktion und über alle präspezifizierten Patientensubgruppen hinweg mit einem guten Erhalt der Lebensqualität verbunden [8]. Unter Therapie traten bei 47,4 % der Patienten schwerwiegende unerwünschte Ereignisse (Grad ≥3) auf. Die häufigsten (>1 %) davon waren Harnwegsinfekte (4,4 %), Anämie (3,8 %), Fatigue und Hämaturie (jeweils 1,7 %), Rückenschmerzen und Erbrechen (jeweils 1,2 %) [8].
Überzeugende Langzeitdaten
Auf dem diesjährigen Amerikanischen GU Krebskongress wurden nun Langzeitdaten der Studie JAVELIN Bladder 100 vorgestellt, die nach einer medianen Nachbeobachtungszeit von ≥38 Monaten (Datenschnitt 04.06.2021) erhoben wurden [9]. Sie zeigen, dass die mit Avelumab + BSC behandelten Patienten in beiden Subgruppen (Erstlinien-Chemotherapie: Cisplatin + Gemcitabin oder Carboplatin + Gemcitabin) ein längeres medianes Gesamtüberleben und medianes progressionsfreies Überleben erreichten als die Patienten, die nur BSC erhielten (Tab. 1). In der Gesamtpopulation betrug das mediane Gesamtüberleben nach Start der Erstlinien-Chemotherapie zum Zeitpunkt des Datenschnitts
29,7 Monate (95%-KI: 25,2 – 34,0) im Avelumab + BSC-Arm gegen-
FACTSHEET
Avelumab (Bavencio®)

Avelumab
Wirkstoffklasse
Avelumab ist ein humaner monoklonaler Antikörper, der gegen den programmierten ZelltodLiganden 1 (PD-L1) gerichtet ist.1 Er zählt zu den sogenannten Checkpoint-Inibitoren.1
Wirkprinzip
PD-L1 wird unter anderem auf Tumorzellen exprimiert, der entsprechende Rezeptor PD-1 auf T-Zellen 2 Durch die Bindung von PD-L1 an PD-1 werden die betroffenen T-Zellen in ihrer Aktivität unterdrückt.2 Damit kann sich der Krebs einer anti-tumoralen Immunantwort entziehen.
Avelumab (Bavenico®) ist ein humaner monoklonaler Antikörper, der gegen den programmierten Zelltod- Liganden 1 (PD-L1) gerichtet ist. Er zählt zu den sogenannten Checkpoint-Inhibitoren. PDL1 wird unter anderem auf Tumorzellen exprimiert, der entsprechende Rezeptor PD-1 auf T-Zellen. Durch die Bindung von PD-L1 an PD-1 werden die betroffenen T-Zellen in ihrer Aktivität unterdrückt, sodass sich der Krebs einer antitumoralen Immunantwort entziehen kann.
Avelumab bindet spezifisch an PD-L1 und blockiert so die Bindung zwischen PD-L1 und PD-1 1 Dadurch bleiben tumorspezifische T-Zellen aktiviert und können eine entsprechende Immunantwort einleiten (Abb. 1) 1
Avelumab bindet spezifisch an PD-L1 und blockiert so die Bindung zwischen PD-L1 und PD-1. Dadurch bleiben tumorspezifische TZellen aktiviert und können eine entsprechende Immunantwort einleiten [7].
In präklinischen Studien zeigte sich außerdem, dass Avelumab mittels antikörperabhängiger zellulärer Zytotoxizität (Antibody Dependent Cell Cytotoxicity, ADCC) zu einer direkten Tumorzelllyse führt, die durch Natürliche Killerzellen (NK-Zellen) vermittelt wird.1
In präklinischen Studien zeigte sich außerdem, dass Avelumab mittels antikörperabhängiger zellulärer Zytotoxizität zu einer direkten Tumorzelllyse führt, die durch natürliche Killerzellen vermittelt wird [7].
über 20,5 Monaten (95%-KI: 19,0 – 23,5) im BSC-Arm. Der Unterschied war signifikant (HR: 0,77; 95%-KI: 0,635 – 0,921) [9]. Diese Daten bestätigen nicht nur die bisherigen Ergebnisse für die Erstlinien-Erhaltungstherapie mit Avelumab beim lokal fortgeschrittenen bzw. metastasierten Urothelkarzinom, sie untermauern auch die Bedeutung von Avelumab als neuem Behandlungsstandard in dieser Indikation.
1
AVENANCE-Studie unterstreicht Wirksamkeit und Verträglichkeit von Avelumab in der Praxis
Ebenfalls auf dem Amerikanischen GU Krebskongress wurde die laufende, nicht interventionelle Studie AVENANCE vorgestellt [10]. Diese Erhebung aus dem Praxisalltag in Frankreich umfasst 593 Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom, die keine Progression
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 53 AKTUELLE THERAPIEKONZEPTE FÜR
DIE PRAXIS
Abbildung 1: Wirkmechanismus Avelumab (modifiziert nach 1,2)
nach einer Platin-basierten Erstlinien-Chemotherapie aufwiesen und eine vorangegangene, laufende oder geplante Erstlinien-Erhaltungstherapie mit Avelumab erhalten bzw. erhalten haben. Primärer Endpunkt ist das Gesamtüberleben gemessen vom Beginn der Erstlinien-Erhaltungstherapie mit Avelumab. Sekundäre Endpunkte sind u.a. progressionsfreies Überleben, Dauer der Behandlung und Sicherheit [10].
Zum Zeitpunkt des Datenschnitts betrug die mediane Nachbeobachtungszeit 15,2 Monate Das mediane OS lag ab Beginn der Behandlung mit Avelumab bei 20,7 Monaten bei einer 12-MonatsGesamtüberlebensrate von 65,4 %.
Kaia COPD: Digitales Therapeutikum mit messbaren Erfolgen
Ende Dezember 2022 hat das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) die als Medizinprodukt zugelassene App „Kaia COPD: Meine aktive COPD-Therapie“ im DiGAVerzeichnis gelistet. Ab sofort können COPD-Patienten die App auf Rezept erhalten. Kaia COPD besteht aus individuell zusammengestellten Bewegungseinheiten, einer ausführlichen Schulung zum Umgang mit der Krankheit sowie Atem- und Entspannungsübungen. Kernelemente der pneumologischen Rehabilitation sind dadurch jederzeit digital verfügbar und können aktiv in den Alltag integriert werden.
Die App basiert auf Behandlungsempfehlungen zur Unterstützung
Das mediane progressionsfreie Überleben betrug 5,7 Monate [1]. Die Daten der AVENANCE-Studie bestätigen die klinische Wirksamkeit und das akzeptable Sicherheitsprofil von Avelumab in einer heterogenen Realworld-Kohorte und unterstreichen den Stellenwert der Erstlinien-Erhaltungstherapie mit Avelumab in der klinischen Praxis.
Brigitte Söllner, Erlangen
3 American Cancer Society. What is bladder cancer? https://www.cancer.org/cancer/bladdercancer/about/what-is-bladdercancer.html
4 Niegisch G et al. J Cancer 2018;9:13371348
5 Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S3-Leitlinie Früherkennung, Diagnose, Therapie und Nachsorge des Harnblasenkarzinoms, Langversion 2.0, 2020, AWMF-Registrierungsnummer 032/038OL, https://www.leitlinienprogramm-onkologie.de/leitlinien/harnblasenkarzinom/.
6 SEER. Cancer stat facts: bladder cancer. https://seer.cancer.gov/statfacts/html/ urinb.html
7 Fachinformation Bavencio®; Stand: Dezember 2022
Literatur
1 Sung H et al. CA CANCER J CLIN 2021;71:209-249
2 Cancer.net. Bladder cancer: introduction. https://www.cancer.net/cancer-types/bladdercancer/introduction
8 Powles TB et al. N Engl J Med 2020; 383:1218-1230
9 Sridhar SS et al. ASCO GU 2023, Poster/ Abstract 508
10 Barthélémy P et al. ASCO GU 2023, Poster/Abstract 471
der nicht medikamentösen Therapie der COPD, wie sie z.B. in der nationalen Versorgungsleitlinie COPD formuliert sind, und wurde gemeinsam mit führenden Experten aus dem Bereich der pneumologischen Rehabilitation entwickelt. Die Verordnung von Kaia COPD ist für gesetzlich versicherte Patienten mit einer COPD-Diagnose ganz einfach per Rezept (Muster 16) möglich. Die Kosten werden von allen gesetzlichen Krankenkassen übernommen und belasten derzeit das Arzneimittel- und Heilmittel-Budget der verordnenden Ärzte nicht.
Mehr körperliche Aktivität, weniger Symptomlast
Wie die Ergebnisse der AMOPURStudie zeigen, ließ sich die in der pneumologischen Rehabilitati-
on erreichte körperliche Aktivität nach 6 Monaten bei den Patienten, die die App nutzten, im Vergleich zur Kontrollgruppe ohne App signifikant besser erhalten. Auch die empfundene Symptomlast, gemessen anhand des CAT-Scores, war bei den App-Nutzern signifikant geringer als in der Kontrollgruppe. Insgesamt hatten die Patienten bei regelmäßiger Nutzung der App ihre Erkrankung besser im Griff und waren aktiver als die Patienten ohne App.
Unabhängig von der Prüfung des BfArM hat „Kaia COPD: Meine aktive COPD-Therapie“ nach Bewertung durch Patienten, Ärzte und Therapeuten das „PneumoDigital-Siegel“ der Deutschen Atemwegsliga e. V. erhalten.
S. M.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 54
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Um den an Multipler Sklerose (MS) Erkrankten mehr Flexibilität und zeitliche Freiräume zu verschaffen, hat Biogen sein bewährtes MS-Medikament Natalizumab (Tysabri®), das im Jahr 2006 als erster monoklonaler Antikörper zur Behandlung der hochaktiven schubförmig remittierenden MS (RRMS) zugelassen wurde [1], neben der i.v. Verabreichungsform auch als subkutane Applikation auf den Markt gebracht.
Tysabri® s.c. liegt in Form von 2 Fertigspritzen vor, die für eine vollständige Dosis beide verabreicht werden müssen [2]. Wie die bisherige i.v. Applikationsform wird auch Natalizumab s.c. alle 4 Wochen injiziert. Ein Wechsel zwischen den Verabreichungsformen ist jederzeit möglich.
Patienten bevorzugen die subkutane Applikation
Um festzustellen, ob die MS-Patienten die s.c. Applikation der i.v. Infusion vorziehen, wurde 2022 die Real-World-Studie SISTER [3, 4] initiiert, die bis Ende März 2024 abgeschlossen sein soll. Im Rahmen dieser 12-monatigen Studie wird geprüft, wie sich die subkutane Anwendungsform im Praxisalltag bewährt. Eingeschlossen werden sollen insgesamt 500 Teilnehmer, darunter mit Natalizumab i.v. vorbehandelte Patienten, die
Multiple Sklerose: Patienten profitieren von der subkutanen Gabe von Natalizumab
sich entschieden haben, auf Natalizumab s.c. zu wechseln, sowie Natalizumab-naive Patienten, die eine Therapie mit Natalizumab begonnen haben und sich für die s.c. oder i.v. Anwendung entscheiden können. Primärer Endpunkt ist die Präferenz der Patienten hinsichtlich der Art der Anwendung von Natalizumab nach 6 und 12 Monaten. Auf der ECTRIMS 2022 wurden erste Zwischenergebnisse der SISTER-Studie präsentiert [5]. Bis Juli 2022 wurden 206 Patienten in die Studie eingeschlossen: 158 Patienten wechselten von Natalizumab i.v. auf Natalizumab s.c. (mediane Dauer der NatalizumabTherapie vor dem Wechsel: 3,1 Jahre), 22 Patienten begannen die Therapie mit der i.v. und 26 Patienten mit der s.c. Anwendungsform. Als Hauptgründe für den Beginn mit bzw. den Wechsel auf die s.c. Applikation wurden die kürzere Dauer der Verabreichung und die komfortablere Anwendung im Vergleich zur i.v. Infusion angegeben. Den für die i.v. Infusion von Natalizumab erforderlichen Zeitaufwand bezifferten die Studienteilnehmer mit 2,8 Stunden, verglichen mit nur 1,3 Stunden bei der subkutanen Applikation. Dies entspricht einer Zeitersparnis von 1,5 Stunden [5]*. So können sowohl Ärzte als auch Patienten von einem verringerten Zeitaufwand und mehr Flexibilität profitieren. Außerdem wird in der Praxis kein Infusionsplatz benötigt. Die Ver-
abreichung der Injektionen kann überdies durch eine MS-Nurse erfolgen.
93,3 % der behandelnden Neurologen beurteilten die s.c. und i.v. Anwendung als vergleichbar für den Therapiebeginn mit Natalizumab bei Betroffenen, die bisher nicht mit dem monoklonalen Antikörper behandelt wurden. Bei der Befragung zwischen der 1. und 4. Natalizumab-Gabe (Baseline der SISTER-Studie) bevorzugten 89,6 % der Patienten die s.c. Gabe gegenüber der i.v. Infusion, nach 6 Monaten betrug der Anteil 96,4 %. Außerdem gaben 98,7 % (Baseline) bzw. 98,1 % (nach 6 Monaten) der Patienten, die die s.c. Gabe erhielten, an, dass sie mit der Art der Anwendung zufrieden sind [5]. Auch die Verträglichkeit wurde untersucht: 8,4 % der Patienten berichteten von unerwünschten Ereignissen im Zusammenhang mit der Natalizumab-Injektion (Schmerzen an der Injektionsstelle, Reaktionen an der Injektionsstelle, Rötungen, Juckreiz, Entzündung). Die Mehrzahl dieser Ereignisse war mild [5].
* Bei der s.c. Verabreichung von Tysabri® ist nach den ersten 6 Applikationen eine Nachbeobachtungszeit von 1 Stunde nötig, bei allen weiteren Applikationen liegt die Nachbeobachtung im Ermessen des Arztes. Bei der i.v. Verabreichung ist nach den ersten 12 Applikationen eine Nachbeobachtungszeit von 1 Stunde nötig, bei allen weiteren Applikationen liegt die Nachbeobachtung im Ermessen des Arztes.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 55 NEUE UND BEWÄHRTE ARZNEIMITTEL
Vergleichbarer therapeutischen Nutzen
Aufgrund der vergleichbaren pharmakodynamischen und -kinetischen Eigenschaften der beiden Anwendungsformen ist der therapeutische Nutzen von Natalizumab i.v. auf Natalizumab s.c. übertragbar [2]. Im Rahmen der Studie TOP wurde über einen Zeitraum von 10 Jahren eine gesamte jährliche Schubrate von 0,15 beobachtet, dies entspricht einer Reduktion um 92,5 % im Vergleich zum Jahr vor Therapiebeginn [6].
Klinische Verbesserungen können sich auch in der Lebensqualität niederschlagen: So wurden in verschiedenen Studien positive Aus-
wirkungen einer Behandlung mit Natalizumab u.a. auf die Kognition, die Fatigue und die Arbeitsfähigkeit beobachtet [7–12]. Wie u. a. Daten aus der Real-World-Kohorte MS PATHS andeuten, hatte Natalizumab in der klinischen Praxis auch einen positiven Einfluss auf verschiedene Aspekte der physischen, kognitiven und emotionalen Lebensqualität [13]. Weltweit wurden bislang bereits mehr als 248.000 Patienten über mehr als eine Million Patientenjahre mit dem Antikörper behandelt [5].
Brigitte Söllner, Erlangen
2 Fachinformation Tysabri™ 150 mg Injektionslösung in einer Fertigspritze; Stand: Mai 2022
3 Paul-Ehrlich-Institut (PEI). Nicht-interventionelle Studie (Anwendungsbeobachtung) NIS-Nr.: 611. https://www.pei.de/ SharedDocs/awb/nis-0601-0700/0611. html?nn=173138
4 ClinicalTrials.gov; NCT05304520
5 Gold R et al. ECTRIMS 2022; P365
6 Butzkueven H et al. J Neurol Neurosurg Psychiat 2020;91:660-668
7 Svenningsson A et al. PLoS One 2013; 8:e58643
8 Stephenson JJ et al. Health Qual Life Outcomes 2012;10:155
9 Rudick RA et al. Ann Neurol 2007; 62:335-346
10 Olofsson S et al. BioDrugs 2011;25:299306
11 Kreimendahl F et al. Value Health 2014; 17:A400
Digitale Gesundheitsanwendungen (DiGA) können Ärzte im Versorgungsalltag entlasten und Patienten aktiv in die Behandlung einbeziehen. Gerade für Menschen mit psychischen Gesundheitsproblemen, wie z.B. einer Depression, können internetbasierte Interventionen wie das Online-Therapieprogramm deprexis® eine wichtige Komponente einer multimodalen, leitliniengerechten Therapie darstellen. Zudem ermöglicht es diese innovative Therapieform, die aktuell bestehenden z.T. monatelangen Wartezeiten auf einen Psychotherapieplatz zu überbrücken und den Patienten die dringend notwendige therapeutische Unterstützung zu geben.
Dennoch wird das Potenzial der DiGA bisher noch nicht ausgeschöpft: Wie eine Erhebung der Techniker Krankenkasse er-
Literatur
1 Fachinformation Tysabri™ 300 mg i.v.; Stand: Mai 2022
gab, erhalten in Deutschland nur 0,0023 % der depressiven Patienten eine DiGA.
Nachgewiesener medizinischer Nutzen
Digitale Therapieprogramme haben den Vorteil, ohne Wartezeiten und Stigma-Empfinden zeitlich und örtlich flexibel genutzt werden zu können und obendrein noch kostengünstig zu sein. Das therapeutische Potenzial digitaler Anwendungen zur Behandlung von Depressionen wird auch von der kürzlich aktualisierten S3-Leitlinie/Nationale VersorgungsLeitlinie „Unipolare Depression“ hervorgehoben: Zum ersten Mal empfiehlt die Leitlinie den Einsatz internetund mobilbasierter Interventionen als eine Alternative zur konventionellen Psychotherapie und damit als eine wesentliche Säule neben der Psychopharmakotherapie.
12 Wilken J et al. Int J MS Care 2013;15: 120-128
13 Hersh CM et al. Mult Scler J Exp Transl Clin 2021;7:20552173211004634
Zu den am besten untersuchten DiGA überhaupt gehört das Online-Therapieprogramm deprexis® zur Therapieunterstützung bei Depression. Eine Metaanalyse aus 12 randomisierten Studien mit insgesamt 2900 Teilnehmenden bestätigte signifikante und klinisch relevante Verbesserungen der depressiven Symptome – unabhängig davon, ob die Patienten das interaktive Therapieprogramm eigenständig oder mit ärztlicher Begleitung (Blended Care) nutzten*. Aufgrund der nachgewiesenen Evidenz wurde deprexis® dauerhaft in das DiGA-Verzeichnis (https://diga.bfarm.de/de) aufgenommen. Es ist für alle Schweregrade der Depression zugelassen und erfüllt sowohl die Anforderungen deutscher Datenschutzgesetze als auch die von der DGPPN definierten Qualitätskriterien für wirksame und sichere Online-Interventionen.
* Twomey C et al. PLoS One 2020;15: e0228100
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 56 NEUE UND BEWÄHRTE ARZNEIMITTEL / WISSENSWEERTES
DiGA deprexis® bewährt sich bei Depression
Einfache, intuitive Anwendung
deprexis® basiert überwiegend auf den anerkannten Methoden der kognitiven Verhaltenstherapie. Es passt sich individualisiert den Bedürfnissen und Reaktionen der Patienten an und leitet sie in einem virtuellen Dialog dynamisch durch die 10 verschiedenen Module. Neben Informationen zur Depression werden alltagsrelevante Übungen zur Depressionsbewältigung vermittelt. Das passwortgeschützte Online-Therapieprogramm ist überall und jederzeit auf allen Endgeräten (PC, Laptop, Tablet und Smartphone) nutzbar und steht in 9 Sprachen zur Verfügung.
App auf Rezept
Niedergelassene Ärzte und Psychotherapeuten können deprexis® nach Diagnose einer Depression zu Lasten der Gesetzlichen Krankenkassen rezeptieren (PZN 17265872). Nachdem der Patient das Rezept bei seiner Krankenkasse eingereicht hat, erhält er einen Freischaltcode. Mit diesem Code kann er die App aktivieren und sofort mit der Anwendung beginnen. Auch im Rahmen des Entlassmanagements in der Klinik kann deprexis® verschrieben werden.
B. S.
Durchbruch bei der Lupus-
Behandlung?
Mit aufgerüsteten
Immunzellen gegen die chronische Entzündung
CAR-T-Zellen zählen seit einigen Jahren zu den Hoffnungsträgern in der Krebsmedizin. Am Universitätsklinikum Erlangen sind sehr erfolgreich erstmals auch Patienten
mit schwerem systemischem Lupus erythematodes (SLE) mit den Immunzellen behandelt worden. Neue, wirksame Behandlungsoptionen für die mitunter schwer zu therapierende entzündlich-rheumatische Autoimmunerkrankung werden dringend gebraucht, so die Deutsche Gesellschaft für Rheumatologie e.V. (DGRh). Wenn sich die ersten Erfolge auch bei größeren Patientenkollektiven bestätigten, komme dies einer Revolution der SLE-Therapie gleich.
Maßgeschneiderte Immunzellen
Individuell, aufwändig und – noch – sehr teuer: CAR-T-Zellen müssen für jeden Patienten „maßgeschneidert“ werden. Zunächst werden dafür körpereigene T-Zellen aus dem Blut des Patienten isoliert und im Labor dann gentechnologisch so verändert, dass sie die namensgebenden „Chimären Antigen-Rezeptoren“ (CAR) auf ihrer Oberfläche ausbilden. „Solche Rezeptoren können nahezu beliebig viele Zielstrukturen, z.B. Eiweiße, auf anderen Zellen erkennen und dann eine Immunreaktion auslösen“, erläutert Professor Georg Schett, einer der federführenden Autoren der Studie. Für die Therapie der B-Zell-Leukämien wurde das für B-Zellen charakteristische Oberflächeneiweiß CD19 als Ziel ausgewählt. Weil auch der SLE mit einer gesteigerten B-Zell-Aktivität einhergeht, lag es nahe, dieselbe genetische Modifikation der CAR-T-Zellen auch hierfür einzusetzen. Die so modifizierten CAR-T-Zellen erhält der Patient über eine Infusion. Zuvor erfolgt, bei SLE ebenso wie bei Leukämie, eine Chemotherapie. Sie hemmt die Aktivität des körpereigenen Immunsystems, um die spätere Arbeit der CAR-T-Zellen zu erleichtern.
Weltweit erste CAR-T-Zelltherapie bei 5 SLE-Patienten
Nach vielversprechenden Vorversuchen an Mäusen bewährten sich die Design-Zellen auch bei den 5 Erlanger Patienten. Vier Frauen und ein Mann erhielten als weltweit erste von SLE Betroffene eine CAR-T-Zelltherapie. Die zuvor hohe Krankheitsaktivität, die bereits die Nieren in Mitleidenschaft gezogen hatte, ging durch die Therapie drastisch zurück. „Sowohl die krankheitstypischen Antikörper als auch Symptome wie Müdigkeit und Abgeschlagenheit nahmen stark ab, zugleich besserte sich die Nierenfunktion deutlich“, berichtet Studienleiter Schett. Besonders beeindruckend: Noch Monate nach der einmaligen CAR-T-Zell-Infusion konnten die Patienten auf ihre zuvor eingenommenen Medikamente verzichten, der SLE kehrte dennoch nicht wieder zurück.
„Die neue Therapie scheint wie ein Reset-Knopf zu wirken, der dem entgleisten Immunsystem einen Neustart ermöglicht“, kommentiert Professor Christof Specker, Präsident der DGRh. Mit großen Erwartungen wird nun verfolgt, wie die Therapie sich in größeren Patientenkollektiven bewährt – und wie es den zuerst Behandelten weiter ergeht. Ob man von einer dauerhaften Heilung eines SLE durch eine solche Therapie sprechen kann, muss trotz der vielversprechenden Ergebnisse noch abgewartet werden. Die Nachbeobachtungszeit der ersten so behandelten Patienten beträgt bislang erst 13 – 23 Monate. DGRh
Quelle: Mackensen A et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med 2022;28:2124-2132. https://doi.org/10.1038/s41591-022-02017-5
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 57 WISSENSWERTES
Der Mangel an AromatischerL-Aminosäure-Decarboxylase (AADC) ist eine seltene, häufig tödlich verlaufende neurometabolische autosomal-rezessiv vererbte Erkrankung, die in der Regel schon in den ersten Lebensmonaten zu erheblichen körperlichen und kognitiven Beeinträchtigungen und Verhaltensauffälligkeiten führt [1, 2]. Verursacht wird sie durch verschiedene Mutationen im Dopa-Decarboxylase (DDC)-Gen [3]. Dieses Gen codiert für das Enzym AADC, das eine Schlüsselrolle in der Neurotransmittersynthese spielt. In der Folge kommt es zu einem kombinierten Mangel an Dopamin, Serotonin, Adrenalin und Noradrenalin [4]. Das Fehlen dieser Neurotransmitter hemmt die postsynaptische neuronale Signalübertragung im Zentralnervensystem, die für die motorische Entwicklung, das Verhalten, und die autonome Funktion erforderlich ist [5]. Ohne Dopamin können die Kinder keine Meilensteine in der motorischen Entwicklung erreichen – sie können den Kopf nicht heben, nicht sitzen, stehen, gehen oder gar sprechen [6]. Oft ist das Leben der kleinen Patienten durch belastende Symptome, lebensbedrohliche Komplikationen und unzählige medizinische Maßnahmen geprägt. Zudem benötigen sie eine 24-Stunden-Betreuung für alle Aspekte des täglichen Lebens – eine enorme Belastung, nicht zuletzt für die Eltern.
Bislang gab es keine wirksame Behandlung für diese Erkrankung. Aber nun steht für die betroffenen Kinder erstmalig eine wirklich lebensverändernde Therapieoption zur Verfügung: Seit Juli 2022 ist die Gentherapie Upstaza™ als erste krankheitsmodifizierende Behandlung von Patienten im Alter ab 18 Monaten mit einer klinisch,
Upstaza™ – eine neue intraputaminale Gentherapie bei AADC-Mangel
molekularbiologisch und genetisch bestätigten Diagnose eines AADCMangels mit einem schweren Phänotyp zugelassen [7].
Upstaza™ korrigiert den zugrunde liegenden genetischen Defekt
Upstaza™ (Eladocagene exuparvovec) ist eine einmalige Genersatztherapie, die für die Behandlung von Patienten ab 18 Monaten mit einer klinisch, molekularbiologisch und genetisch bestätigten
Diagnose eines AADC-Mangels mit einem schweren Phänotyp indiziert ist [7]. Upstaza™ enthält die für das humane AADC-Enzym kodierende cDNA (complementary DNA) unter Kontrolle eines Zytomegalievirus-Promoters, eingebettet in den viralen Vektor rAAV2 (rekombinanter adenoassoziierter Serotyp-2-Vektor). Dabei handelt es sich um ein kleines, nicht replizierendes und nicht pathogenes Parvovirus [5].
Upstaza™ wird im Rahmen eines stereotaktischen neurochirurgischen Verfahrens einmalig bila-
Frühe Symptome, auf die geachtet werden sollte
Die wichtigsten Symptome des seltenen AADC-Mangels, auf die Eltern bei ihrem Baby achten sollten und die den Kinderarzt hellhörig werden lassen sollten sind die sehr ausgeprägte Muskelhypotonie, die Augenmuskelstörungen, d.h. die okulogyren Krisen. Auffällig sind zudem eine ständig verstopfte Nase sowie eine nach der Geburt auftretende Hypoglykämie. Das sind vier Aspekte, die zusammengenommen – nicht einzeln – durchaus ein klares Bild ergeben können.
AADC-Mangel diagnostizieren
Biochemisch kann durch die einfache Bestimmung des 3-O-Methyldopas (3-OMD) im Trockenblut eine erste Abklärung erfolgen (Patienten <18 Jahre). Dafür können Trockenblutkarten z.B. im Stoffwechsellabor der Universität Heidelberg angefordert werden.
Zur Bestätigung der Diagnose AADC-Mangel werden 3 Tests angewandt:
• Messung der AADC-Enzymaktiviät im Blutplasma
• Liquoruntersuchung
• Gentest
2 der 3 diagnostischen Haupttests sollten positiv sein, um die Diagnose AADC-Mangel zu bestätigen.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 58 NEUE UND BEWÄHRTE ARZNEIMITTEL
gischer Erkrankungen.10,13
Upstaza™ zielt also auf den zugrunde liegenden genetischen Defekt ab, indem ein funktionierendes DDC-Gen direkt in das Putamen eingebracht wird. Dadurch kommt es zur Exprimierung des AADC-Enzyms, was wiederum zur Produktion von Dopamin und folglich zur Entwicklung der motorischen Funktionen bei Patienten mit AADC-Mangel führt.1
Signifikante Verbesserung der motorischen Fähigkeiten
Abb. 2: Zielstruktur der Infusionstherapie ist das Putamen. Die Behandlung erfolgt unter MRT-Kontrolle (mod. nach2). hAADC = humanes AADC-Enzym;
[Abb. PTC Therapeutics]
Abbildung 1: Zielstruktur der Infusionstherapie ist das Putamen. Der stereotaktische neurochirurgische Eingriff mit bilateraler Platzierung zweier Kanülen in 2 Bereichen pro Putamen erfolgt unter MRT-Kontrolle. hAADC = humanes AADC-Enzym, rAAV2 = rekombinanter adenoassoziierter Serotyp-2-Vektor (© PTC Therapeutics).
Peabody Developmental Motor Scale (PDMS-2)
PDMS-2 ist die zweite Auflage einer Skala zur Bewertung der motorischen Entwicklung eines Kindes. Dabei werden mittels Tests in verschiedenen untergeordneten Bereichen sowohl die grob- als auch die feinmotorischen Fähigkeiten bewertet. Zu den Unterbereichen gehören:
• Visuelle motorische Integration (Hand-Augen-Koordination)
• Stillhalten (z.B. selbstständig aufrecht sitzen)
• Objektmanipulation (je nach Alter z.B. Fangen, Werfen oder Treten eines Balls)
• Fortbewegung (Krabbeln, Gehen, Laufen, Hüpfen)
• Reflexe (bei Kindern ab 12 Monaten)
• Greifen
Auf der PDMS-2 können 0 – 2 Punkte erreicht werden, wobei 2 Punkte dem vollständigen Beherrschen des jeweiligen Meilensteins entsprechen, 1 Punkt dem weitgehenden Beherrschen und 0 Punkte dem Fehlen der Fertigkeit.
teral in das Putamen infundiert (Abb. 1) [7]. Pro putaminaler Infusionsstelle werden 0,08 ml (0,45 × 1011 Vg) Upstaza™ infundiert, bei insgesamt 4 Infusionen mit einem Gesamtvolumen von 0,320 ml (1,8 × 1011 Vg) [7]. Das Putamen ist verantwortlich für die motorische Kontrolle, das Lernen, Verhalten und Emotionen. Im Gegensatz zu anderen potenziellen Targets ist das Putamen scharf begrenzt und aufgrund seiner Größe leichter
zugänglich. Somit ist es ein besonders gut geeignetes Ziel bei Patienten mit AADC-Mangel [8]. Durch das Einbringen des funktionierenden DDC-Gens direkt in das Putamen wird der zugrunde liegende genetische Defekt korrigiert. In der Folge wird das AADCEnzym exprimiert, was wiederum zur Produktion von Dopamin und schließlich zur Entwicklung der motorischen Funktionen bei den Patienten führt.
Die Wirksamkeit und das Sicherheitsprofil von Upstaza™ wurden in 2 klinischen Studien sowie im Rahmen einer CompassionateUse-Studie nachgewiesen [7, 9]. Primärer Endpunkt war das Erreichen von motorischen Meilensteinen 24 Monate nach der Behandlung mit Upstaza™. Die Bewertung erfolgte mittels Peabody Development Motor Scale, Version 2 (PDMS-2). Damit ein Meilenstein als erreicht bewertet wurde, musste das Kind dafür die maximale Punktzahl von 2 erhalten (siehe Insert).
Erste Effekte auf die motorischen Fähigkeiten zeigten sich bei einigen der mit der Gentherapie behandelten Patienten, die vorher keinerlei motorische Meilensteine erreicht hatten, bereits 3 Monate nach der Behandlung. Diese Verbesserungen setzten sich bis zu einem Beobachtungszeitraum von über 5 Jahren nach der Behandlung fort [9]. Auch die kognitiven Fähigkeiten verbesserten sich bei allen behandelten Patienten. Zudem reduzierten sich Symptome wie z.B. okulogyre Krisen (unwillkürliches Augenverdrehen). Ein wichtiger Aspekt ist auch, dass sich nach der Gentherapie die Lebensqualität sowohl der Kinder als auch der Eltern deutlich verbesserte [7, 9].
UpstazaTM wurde allgemein gut vertragen [9]. Die häufigsten Nebenwirkungen, die infolge der Behandlung auftraten, waren Dyskinesien (bei 92 % der Patienten, n = 26) sowie Pyrexie (bei 96 % der Patienten, n = 26) [9]. Alle Dyskinesie-Ereignisse, die im Zusammenhang mit Upstaza™ aufgetreten waren, legten sich innerhalb von 10 Monaten nach der
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 59 NEUE UND BEWÄHRTE ARZNEIMITTEL
ung onkolo-
MRT = Magnetresonanztomographie.
Behandlung und konnten mit einer routinemäßigen medizinischen Versorgung behandelt werden [9].
Erfahrungen mit der Gentherapie in der Praxis
In der Neurochirurgischen Klinik am Universitätsklinikum Heidelberg wurde das erste Kind mit AADC-Mangel in Deutschland mit UpstazaTM behandelt. Wie der behandelnde Arzt Professor Thomas Opladen, ärztlicher Leiter der pädiatrischen Stoffwechselambulanz, berichtete [10], fiel die Patientin bereits in den ersten 3 Lebensmonaten durch fehlende Kopfkontrolle sowie ein erhöhtes Vanillinlaktat im Urin auf. Das MRT des Schädels und das EEG waren normal. Weitere Laboruntersuchungen und eine Genanalyse führten schließlich zur Diagnose AADC-Mangel. Mit 4 Monaten zeigte das Kind zwar ein interaktives Lächeln, aber weder spontane Bewegungen, aktive Lautäußerungen noch Kopfkontrolle. Das heißt, es hatte keine Meilensteine der Entwicklung erreicht. Darüber hinaus bestanden eine signifikante Muskelhypotonie, eine Ptosis und eine nasale Kongestion. Besonders belastend für Kind und Eltern waren die wiederholten ausgeprägten okulogyren Krisen sowie Schlafstörungen und Stimmungsschwankungen. Die Ernährung erfolgte ausschließlich parenteral, dennoch war die Gewichtszunahme sehr zögerlich. Unter der Standardbehandlung über Jahre konnte keine relevante Besserung der Symptome erzielt werden.
Im Alter von 3 Jahren und 4 Monaten wurde das Mädchen dann im September 2021 im Rahmen eines individuellen Heilversuchs in Heidelberg mit Upstaza™ behandelt.
Es hat die stereotaktische Operation und die intraputaminale Infusion gut vertragen. Vorübergehend traten in den ersten Wochen nach der Therapie Dyskinesien auf. Bei allen Nachuntersuchungsterminen (nach 4 und 9 Wochen, 3, 7 und 12 Monaten) zeigt das Kind eine zunehmende Entwicklung insbesondere der Motorik und des Muskeltonus. Ein Jahr nach der Gentherapie kann das Mädchen seinen Kopf halten, selbstständig sitzen und mit Hilfe stehen. Es schläft viel besser und hat weniger Stimmungsschwankungen. Und vor allem die belastenden okulogyren Krisen haben sich in Dauer und Intensität deutlich gebessert. Ptosis und nasale Kongestion sind jedoch unverändert und es ist noch keine expressive Sprache vorhanden. Mittlerweile isst das Kind auch selbstständig und nimmt weiter an Gewicht zu. Heute besucht das Mädchen einen integrativen Kindergarten. Auch für die Eltern hat durch die Gentherapie „ein neues Leben“ begonnen.
Brigitte Söllner, Erlangen
Die atopische Dermatitis betrifft verschiedene Körperregionen. Exponierte Bereiche wie etwa Kopf und Hals oder auch die Hände sind Umweltstressoren wie Kälte, trockener Luft, Allergenen oder Reizstoffen besonders stark ausgesetzt und sprechen mitunter weniger gut auf eine Therapie an. Vor allem die im Bereich des Gesichts auftretenden stigmatisierenden Hautläsionen und Rötungen werden von den Betroffenen als sehr belastend empfunden. Daher kommt einem guten Therapieansprechen in diesem Bereich des Körpers eine besonders große Bedeutung zu.
Für den selektiven JAK-Inhibitor Upadacitinib (Rinvoq®), der sich bereits in der Behandlung der rheumatoiden Arthritis, der aktiven Psoriasis-Arthritis und der axialen Spondyloarthritis bewährt hat, liegen jetzt auch vielversprechende Studiendaten für die Therapie der atopischen Dermatitis vor.
Überlegenheit gegenüber Dupilumab
Literatur
1 Manegold C et al. J Inherit Metab Dis 2009;32:371-380
2 Brun L et al. Neurology 2010;75:64-71
3 Chien YH et al. Mol Genet Metab 2016; 118:259-263
4 Wassenberg T et al. Orphanet J Rare Dis 2017;12:12
5 Hwu WL et al. JIMD Rep 2018;40:1-6
6 Fachinformation Upstaza™; Stand: September 2022
7 Hwu PWL et al. EMBO Mol Med 2021;13:e14712
8 Tai CH et al. Mol Ther 2022;30:509-518
9 Präsentation auf der Launch-Pressekonferenz von PTC Therapeutics, 29.11.2022
Die Studie Heads Up [1], eine randomisierte, doppelblinde, wirkstoffkontrollierte Double-DummyStudie der Phase IIIb, verglich die Wirksamkeit von Upadacitinib und Dupilumab bei Erwachsenen mit mittelschwerer bis schwerer atopischer Dermatitis. Die Patienten erhielten randomisiert über 24 Wochen entweder Upadacitinib (30 mg oral einmal täglich) oder Dupilumab (600 mg als Induktionsdosis bei Baseline, danach 300 mg als subkutane Injektion alle 2 Wochen). Für die Applikationsart des jeweils anderen Behandlungsarms bekamen die Studienteilnehmer ein entsprechendes Placebo. Primärer Endpunkt war der Anteil
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 60 NEUE UND BEWÄHRTE ARZNEIMITTEL
Upadacitinib bei atopischer Dermatitis: Stark wirksam und allgemein gut verträglich
der Patienten, die zu Woche 16 einen EASI 75 erreichten.
Nach 16 Wochen zeigte die Therapie mit Upadacitinib eine stärkere Wirksamkeit als die mit Dupilumab: Mit Upadacitinib 30 mg erreichten im Vergleich zu Dupilumab mehr Patienten sogar den EASI 90 bei diesen schwer zu behandelnden und zu Stigmatisierung führenden Läsionen (p ≤ 0,001).
Diese überlegene Wirksamkeit zeigte sich sowohl für den Kopfund Halsbereich (53 % vs. 38 %) als auch für den Oberkörper (61 % vs. 44 %) sowie die oberen und unteren Gliedmaßen (58 % vs. 43 % und 62 % vs. 45 %). Zusätzlich wurde bei den Patienten im Kopfund Halsbereich mit Upadacitinib häufiger eine Abnahme der Rötungen beobachtet [1].
Die in der Heads-Up-Studie erfassten Sicherheitsdaten stimmten mit denen überein, die in den globalen zulassungsrelevanten Phase-IIbund Phase-III-Studien beobachtet wurden. Dieses Ergebnis wird jetzt von aktuellen Langzeit-Sicherheitsdaten untermauert.
Langzeit-Sicherheitsdaten
über bis zu 2,8 Jahre
Eine aktuelle umfangreiche Sicherheitsanalyse der 3 klinischen Studien AD UP [2], MEASUREUp 1 & 2 [3] belegt die allgemein
gute Verträglichkeit von Upadacitinib über einen Behandlungszeitraum von bis zu 2,8 Jahren bei Patienten mit atopischer Dermatitis. Dabei wurden die Daten von mehr als 2.690 Patient berücksichtigt, die Upadacitinib 15 mg (2.035,8 Patientenjahre) oder Upadacitinib 30 mg (2.118,0 Patientenjahre) erhalten hatten. Im Vergleich zu früheren Auswertungen wurden keine neuen Sicherheitssignale identifiziert [4].
Indikationsübergreifende Sicherheitsanalyse bei mehr als 6.900 Patienten
Insgesamt liegen zu Upadacitinib nun Langzeit-Sicherheitsdaten von mehr als 6.900 Patienten mit einer Therapiedauer bis zu 5,5 Jahren bei rheumatoider Arthritis (RA, n = 3.209), Psoriasis-Arthritis (PsA, n = 907), axialer Spondyloarthritis (AS, n = 182) sowie bei atopischer Dermatitis (AD n=2.693) vor [4]. Das entspricht einer Exposition von 15.425 Patientenjahren über alle 4 Indikationen hinweg. Die integrierte Sicherheitsanalyse zeigt keine erhöhten Raten für MACE, Malignität (exkl. NMSC) und VTE unter Upadacitinib bei RA und PsA im Vergleich zu Adalimumab und Methotrexat. Die beobachteten Eventraten von Malignitäten (exkl. NMSC) unter
Upadacitinib bei rheumatischen Indikationen wurden vergleichbar häufig berichtet wie unter Vergleichstherapien (Adalimumab und Methotrexat) und sind vergleichbar mit den Raten in der Allgemeinbevölkerung [4].
Literatur
1 Thyssen J et al. Efficacy and safety of upadacitinib vs dupilumab treatment for moderate to severe atopic dermatitis in four body regions – analysis from the Heads Up Study. Virtueller EADV Kongress 2021; Präsentation FC0103
2 Reich K et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021;397:2169-2181
3 Guttman-Yassky E et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 2021;397:2151-2168
4 Burmester GR et al. Safety profile of upadacitinib over 15 000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open 2023;9:e002735
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 61 NEUE UND BEWÄHRTE ARZNEIMITTEL
Fabian Sandner, Nürnberg
Der systemische Lupus erythematodes (SLE) ist eine chronisch-entzündliche Autoimmunerkrankung, die in Schüben verläuft und durch ein heterogenes klinisches Erscheinungsbild gekennzeichnet ist. Eine der schwerwiegendsten Organmanifestationen des SLE ist eine Beteiligung der Nieren, die Lupus-Nephritis (LN) [1]. Sie entwickelt sich bei bis zu 40 % der SLE-Patienten [2] und geht mit einer Proteinurie, Hämaturie und eingeschränkter Nierenfunktion einher. Bei nahezu 20 % der LN-Patienten kommt es innerhalb von 10 Jahren nach Diagnosestellung zum terminalen Nierenversagen [3].
Leitlinien empfehlen die frühzeitige Reduktion der Proteinurie
Die wichtigsten Therapieziele bei der Behandlung der LupusNephritis sind der Erhalt der Nierenfunktion, die Senkung des Mortalitätsrisikos bei gleichzeitiger Minimierung behandlungsbedingter unerwünschter Ereignisse sowie eine Verbesserung der Lebensqualität. Eine frühzeitige Verringerung der Proteinurie gilt dabei als bester einzelner Prädiktor für verbesserte Langzeitergebnisse bei Lupus-Nephritis [4]. Die EULAR/ ERA-EDTA-Leitlinie-2019 [2] und die KDIGO-Leitlinie-2021 [5] empfehlen daher eine frühzeitige Reduktion der Proteinurie in den ersten 12 Monaten nach Behandlungsbeginn (Abb. 1). Allerdings können bis zu 60 % der LN-Patienten dieses Behandlungsziel mit den derzeitigen therapeutischen Optionen nicht erreichen [4].
Daher besteht ein offensichtlicher Bedarf an wirksameren Therapien mit raschem Ansprechen und
Voclosporin – der erste in Europa zugelassene Calcineurininhibitor zur Behandlung
der Lupus-Nephritis
der Möglichkeit einer Steroidreduzierung. Eine neue Perspektive bietet in dieser Hinsicht der neuartige Calcineurininhibitor (CNI) Voclosporin (Lupkynis®) [6].
Voclosporin – eine effektive Therapieoption mit raschem Wirkeintritt
Der neuartige Calcineurininhibitor Voclosporin (Lupkynis®) wurde im September 2022 in Kombina-
tion mit Mycophenolat-Mofetil (MMF) als erste orale Therapie für Erwachsene mit aktiver LupusNephritis der Klassen III, IV oder V (einschließlich gemischter Klassen III/V und IV/V) zugelassen [6]. Basis der Zulassung waren die positiven Ergebnisse der PhaseIII-Studie AURORA 1 [4], die die Wirksamkeit und Sicherheit von Voclosporin bei Patienten mit Lupus-Nephritis der Klassen III, IV oder V (einschließlich gemischter Klassen III/V und IV/V) unter-
Abbildung 1: Empfehlung zur frühzeitigen Reduktion der Proteinurie in den ersten 12 Monaten nach Behandlungsbeginn nach der EULAR/ERA-EDTA-Leitlinie-2019 sowie der KDIGO-Leitlinie-2021 [2, 4].
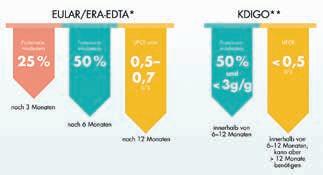
* Bei Patienten mit einer Proteinurie im nephrotischen Bereich zu Beginn der Behandlung kann es weitere 6 – 12 Monate dauern, bis ein vollständiges klinisches Ansprechen erreicht wird; in solchen Fällen ist ein sofortiger Wechsel der Therapie nicht notwendig, wenn sich die Proteinurie verbessert.
** Bei Kindern <18 Jahren ist das vollständige Ansprechen definiert als Proteinurie <0,5 g/1,73 m2/d oder <300 mg/m2/d basierend auf einer 24-Stunden-Urinprobe.
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 62 NEUE UND BEWÄHRTE ARZNEIMITTEL
suchte [4]. Eingeschlossen in die randomisierte, placebokontrollierte und doppelblinde Studie wurden 357 Patienten, die 1:1 randomisiert ein Jahr lang entweder zweimal täglich 23,7 mg Voclosporin (n = 179) oder Placebo (n = 178) erhielten, jeweils in Kombination mit MMF und rasch reduzierten niedrigdosierten Steroiden. Primärer Endpunkt war das komplette renale Ansprechen in Woche 52, was die folgenden Parameter beinhaltete:
• Urin-Protein-Kreatinin-Ratio (UPCR) ≤0,5 mg/mg
• eGFR ≥60 ml/min/1,73 m2 oder keine bestätigte Abnahme der eGFR von >20 % gegenüber dem Ausgangswert
• Keine Gabe von Reservemedikamenten
• Nicht mehr als 10 mg/d Prednisonäquivalent an ≥3 aufeinanderfolgenden Tagen oder an ≥7 Tagen insgesamt in den Wochen 44
52
In der Voclosporin-Gruppe zeigte sich nach 52 Wochen eine signifikante Verbesserung des kompletten renalen Ansprechens im Vergleich zur Kontroll-Gruppe (41 % vs. 23 %; OR: 2,65; 95%-KI: 1,64 – 4,27; p < 0,0001; Abb. 2).

Außerdem kam es unter Voclosporin zu einer raschen Reduktion der Proteinurie: Patienten aus der Voclosporin-Gruppe erreichten doppelt so schnell wie die Kontrollen eine 50%ige Reduktion der UrinProtein-Kreatinin-Ratio (UPCR)
(29 vs. 63 Tage, p < 0,001) sowie eine UPCR ≤0,5 mg/mg (169 vs. 372 Tage; p < 0,001). Hervorzuheben ist auch die rasche Reduktion der oralen Steroide: Bei mehr als 80 % der Patienten aus beiden Behandlungsgruppen konnte in der Behandlungswoche 16 die Steroiddosis auf ≤2,5 mg/d reduziert werden.
Abbildung 2: In der Studie AURORA 1 erreichten signifikant mehr Patienten in der Voclosporin-Gruppe als in der Kontroll-Gruppe ein komplettes renales Ansprechen in Woche 52 [4].
Voclosporin
Voclosporin (Lupkynis®) ist ein immunsupprimierender Calcineurininhibitor, der Calcineurin dosisabhängig bis zu einer Maximaldosis von 1,0 mg/kg hemmt. Die Aktivierung der Lymphozyten beinhaltet einen Anstieg der intrazellulären Kalziumkonzentrationen. Calcineurin ist eine Kalzium-Calmodulin-abhängige Phosphatase, deren Aktivität bei der Induktion der Produktion von T-Zell-Lymphokinen und der Proliferation erforderlich ist. Die immunsupprimierende Aktivität von Voclosporin führt zur Hemmung der Lymphozytenproliferation, T-Zell-Zytokinproduktion und Expression von T-Zell-aktivierenden Oberflächenantigenen.
Im Vergleich zu Ciclosporin zeigt Voclosporin eine stärkere Calcineurin-Hemmung und ein stabiles pharmakokinetisches und pharmakodynamisches Profil, sodass kein therapeutisches Drug Monitoring erforderlich ist [8]. Voclosporin hat außerdem keinen Einfluss auf die Konzentration von Mycophenolsäure, dem aktiven Bestandteil von MMF. Darüber hinaus zeigt Voclosporin eine günstige Auswirkung auf die Lipidwerte und hat keinen signifikanten Einfluss auf den Glukosespiegel [6].
Lupkynis® wird in Form von Weichkapsel verabreicht. Jede Weichkapsel enthält 7,9 mg Voclosporin. Die empfohlene Dosis beträgt zweimal täglich 23,7 mg Voclosporin (3 Weichkapseln à 7,9 mg). Lupkynis® muss in Kombination mit Mycophenolat-Mofetil (MMF) angewendet werden [6].
Das Sicherheitsprofil der beiden Behandlungsgruppen war vergleichbar. Die beobachteten uner-
wünschten Ereignisse entsprachen den Erwartungen für die Population und Therapie [4].
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 63 NEUE UND BEWÄHRTE ARZNEIMITTEL
–
Überzeugende Langzeit-Sicherheit
AURORA 2, die Fortsetzungsstudie von AURORA 1, untersuchte die Langzeit-Sicherheit und -Verträglichkeit sowie den langfristigen Behandlungsnutzen von Voclosporin bei Patienten mit Lupus-Nephritis, die nach Abschluss der AURORA 1-Studie weitere 24 Monate behandelt wurden [7]. Die Patienten setzten die gleiche verblindete Therapie wie am Ende von AURORA 1 fort: Entweder 23,7 mg Voclosporin (3 Kapseln à 7,9 mg) (n = 116) oder Placebo (n = 100) zweimal täglich, jeweils in Kombination mit 2 g MMF (1 g zweimal täglich) und niedrigdosierten Steroiden.
Neben der Sicherheit und Verträglichkeit wurden auch die geschätzte glomeruläre Filtrationsrate (eGFR), das Urin-ProteinKreatinin-Verhältnis (UPCR) und das Serum-Kreatinin (SCr) beurteilt [7].
Über den gesamten Behandlungszeitraum von 3 Jahren (AURORA 1 und 2) wurde Voclosporin gut vertragen, es wurden keine unerwarteten Sicherheitssignale festgestellt und die Gesamtzahl der unerwünschten Ereignisse (AEs) ging im Laufe der Zeit zurück. Die Rate der gemeldeten schwerwiegenden AEs war in beiden Behandlungsarmen ähnlich: Voclosporin-Gruppe 18,1 % vs. Kontroll-Gruppe 23,0 % [7].
Die geschätzte glomeruläre Filtrationsrate und das Serum-Kreatinin blieben während des gesamten Studienzeitraums stabil. Zugleich wurden die signifikanten und bedeutsamen Reduktionen der Proteinurie, die in AURORA 1 erreicht wurden, in AURORA 2 beibehalten (Abb. 3) – ein überzeugender Beleg für den langfristigen Behandlungsnutzen von Voclosporin bei Patienten mit Lupus-Nephritis .
Brigitte Söllner, Erlangen
Literatur
1 Anders HJ et al. Lupus nephritis. Nat Rev Dis Primers 2020;6:7
2 Fanouriakis A et al. 2019 Update of the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis 2020;79:713-723
3 Anders HJ et al. A pathophysiologybased approach to the diagnosis and treatment of lupus nephritis. Kidney Int 2016;90:493-501
4 Rovin BH et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2021;397: 2070-2080

5 Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021;100:S1–S276
6 Fachinformation Lupkynis®; Stand: September 2022
7 Saxena A et al. POS0186 Voclosporin for lupus nephritis: results of the two-year AURORA 2 continuation study. Ann Rheum Dis 2022;81:325
8 Li Y et al. Pharmacokinetic disposition difference between cyclosporine and voclosporin drives their distinct efficacy and safety profiles in clinical studies. Clin Pharmacol 2020;12:83-96
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 64 NEUE UND BEWÄHRTE ARZNEIMITTEL
Abbildung 3: LS-Mittelwert UPCR in AURORA 1, AURORA 2 sowie dem Nachsorgetermin 4 Wochen nach Absetzen des Studienmedikaments [7].
Metastasiertes hormonsensitives
Prostatakarzinom:
Aktuelle Daten zur Therapie mit Apalutamid
Zum Androgenrezeptor-Antagonisten Apalutamid (Erleada®), der u.a. in Kombination mit einer Androgendeprivationstherapie (ADT) in der Therapie des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC) zum Einsatz kommt, wurden auf dem Genitourinary Cancers Symposium der American Society of Clinical Oncology (ASCO-GU) in San Francisco die Ergebnisse einer USamerikanische Real-World-Studie und einer Post-hoc-Analyse der SPARTAN-Studie vorgestellt.
Real-World-Studie zur mHSPC-Therapie
Die Kastrationsresistenz ist ein wichtiger klinischer Indikator für das Fortschreiten der Krankheit und die Verschlechterung des Gesamtüberlebens bei Patienten mit mHSPC. Eine retrospektive Längsschnitt-Kohortenstudie aus den USA* analysierte daher anhand der Daten des Flatiron Metastatic Prostate Cancer Core Registry Electronic DataMart (Auswertungszeitraum: 01/2013–07/2022) den Progress zur Kastrationsresistenz sowie das Gesamtüberleben (OS) bei 1251 Patienten mit metastasiertem Prostatakarzinom ohne Anzeichen einer Kast-
* Lowentritt B et al. Real-world time-tocastration resistance among patients with metastatic castration-sensitive prostate cancer initiating apalutamide, enzalutamide, or abiraterone acetate from an oncology database. J Clin Oncol 2023;41(Suppl 6): Abstract 65 & Poster Session
rationsresistenz, die einer Therapie mit Apalutamid (n = 155), Enzalutamid (n = 385) oder Abirateron (n = 711) zugeführt wurden. Alle Patienten wurden bis zum Ende der klinischen Aktivität (Eintreten der Kastrationsresistenz oder Tod) oder bis zum Ende der Datenverfügbarkeit nachverfolgt. Anhand von Kaplan-Meier-Analysen wurde für jede Kohorte unabhängig das OS und der Anteil derjenigen Patienten ermittelt, die bis zu 24 Monate nach Therapiestart NHT eine Kastrationsresistenz entwickelten.
Nach 2 Jahren Therapie war der Anteil der mHSPC-Patienten mit einem Progress zur Kastrationsresistenz Apalutamid mit 26,2 % geringer als unter den beiden anderen untersuchten neuen Hormontherapien Enzalutamid (36,3%) und Abirateron (32,8 %). Dabei entsprach der unter Alltagsbedingungen beobachtete Prozentsatz bei den mit Apalutamid behandelten Patienten in etwa dem Anteil der Patienten mit Progress zur Kastrationsresistenz in der Phase-III-Studie TITAN, auf der die Zulassung von Apalutamid für die Therapie des mHSPC basierte.
Ähnliches galt für das Gesamtüberleben: Hier lag der Anteil der Studienteilnehmer, die in der USReal-World-Studie nach 24 Monaten noch lebten, unter Apalutamid mit 88,1 % deutlich über dem Ergebnis für Enzalutamid (71,0 %) und Abirateron (77 %).
Post-hoc-Analyse
gibt Hinweise zur Sequenztherapie
Die SPARTAN-Studie untersuchte die Wirksamkeit und Sicherheit von Apalutamid (plus Androgendeprivationstherapie) im Vergleich zu Placebo. Es zeigte sich eine si-
gnifikante Überlegenheit von Apalutamid: Der Median des metastasenfreien Überlebens betrug 40,5 Monate im Vergleich zu 16,2 Monaten in der Placebogruppe (HR für Metastasierung oder Tod: 0,28; 95%-KI: 0,23 – 0,35; p < 0,001) und auch die Zeit bis zur Entdeckung der ersten Metastase, das progressionsfreie Überleben und die Zeit bis zur Progression der Symptome waren unter Apalutamid statistisch signifikant länger als unter Placebo (p < 0,001).
Eine aktuelle Post-hoc-Analyse der SPARTAN-Studie** liefert ergänzend weitere Hinweise zur Wirksamkeit einer Sequenztherapie beim fortgeschrittenen Prostatakarzinom, die mit Apalutamid/ ADT beginnt. Daten zur Sequenz gewinnen zunehmend an Bedeutung, weil für die Behandlung der fortgeschrittenen Stadien des Prostatakarzinoms eine Vielzahl von Wirkstoffen zur Verfügung steht, die in der Regel nacheinander zum Einsatz kommen.
Für Apalutamid/ADT war bereits sowohl in der TITAN-Studie als auch in der SPARTAN-Studie, auf der die Zulassung für die Therapie des nicht metastasierten kastrationsresistenten Prostatakarzinoms (M0CRPC) mit hohem Metastasierungsrisiko basiert, eine signifikante Überlegenheit gegenüber Placebo/ADT beim progressionsfreien Überleben in der zweiten Therapielinie (PFS2, explorativer Endpunkt) gezeigt worden. Demnach konnten hier erste Hinweise generiert werden, dass nach Apalutamid/ADT die Wirksamkeit von
** Oudard S et al. Efficacy of subsequent treatments in patients who progressed to mCRPC following treatment with apalutamide for nonmetastatic castration-resistant prostate cancer (nmCRPC): A post-hoc analysis of the SPARTAN phase III trial. J Clin Oncol 2023;41(Suppl 6): Abstract 157 & Poster Session
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 65 KONGRESSE
Folgetherapien im Stadium des metastasierten kastrationsresistenten Prostatakarzinoms (mCRPC) erhalten blieb.
Die aktuelle Post-hoc-Analyse der SPARTAN-Studie weist nun erstmals darauf hin, dass nach Progress unter Apalutamid/ADT das Gesamtüberleben unter der ersten Folgetherapie im mCRPC (sOS) bei verschiedenen Wirkstoffen bzw. Wirkstoffklassen vergleichbar war. Nach Einsatz von Apalutamid/ ADT beim Hochrisiko-M0CRPC und Progress zum mCRPC betrug das mediane Gesamtüberleben unter einer mCRPC-Erstlinientherapie mit Enzalutamid (n = 20) 17,0 Monate, mit Docetaxel (n = 29)
18,2 Monate, mit Abirateron plus Prednison/Prednisolon (n = 241)
20,2 Monate und mit anderen Therapien (n = 21) 21,6 Monate. Auch das progressionsfreie Überleben unter diesen Folgetherapien (sPFS) war vergleichbar. In dieser Post-hoc-Analyse der SPARTANStudie konnte demnach bei Patienten mit schlechter Prognose und Progress unter Apalutamid/ADT kein wesentlicher Unterschied in der Wirksamkeit von neuen Hormontherapien und Chemotherapie in der mCRPC-Erstlinientherapie beobachtet werden.
Fabian Sandner, Nürnberg
Hautkrebs – die unterschätzte Berufskrankheit
„Die Fälle von hellem Hautkrebs steigen aufgrund des Klimawandels und der demographischen Entwicklung jährlich um etwa 10 %“, berichtete Professor Swen Malte John, Osnabrück, auf einem von der Bayer Vital GmbH veranstalte-
ten virtuellen Meet-the-Expert. Ein besonders hohes Risiko, an hellem Hautkrebs zu erkranken, haben Menschen mit beruflich bedingter hoher UV-Belastung. Dazu gehören unter anderem Beschäftigte im Baugewebe, der Landwirtschaft und im öffentlichen Dienst (z.B. in Kindergärten, bei der Müllabfuhr und in Badeanstalten).
Ein Fall für die Gesetzliche Unfallversicherung
Beruflich bedingter Hautkrebs ist seit 2015 bei langjährig sonnenexponierten Außenbeschäftigten als Berufskrankheit (BK 5103) anerkannt und seither zur zweithäufigsten anerkannten Berufskrankheit in Deutschland aufgestiegen. Von den 6.000 anerkannten Fällen pro Jahr haben etwa 800 Betroffene erhebliche Rentenansprüche. John unterstrich: „Mit der Anerkennung als beruflich bedingte Hauterkrankung können dann auch Therapieverfahren zulasten der Gesetzlichen Unfallversicherung (DGUV) erbracht werden, die im Bereich der Versorgung durch die Gesetzlichen Krankenkassen (GKV) so nicht möglich wären – einschließlich der ablativen Lasertherapie nebst leitliniengerechter Nachsorge.“
Leitliniengerechte Nachsorge nach Lasertherapie
Die im März 2022 erschienene S2k-Leitlinie „Lasertherapie der Haut“ empfiehlt für die Nachbehandlung einer ablativen Lasertherapie den Einsatz eines Dexpanthenol-haltigen Externums wie z.B. Bepanthen® Wund- und Heilsalbe. Hintergrund der Empfehlung sind die positiven Ergebnisse einer klinischen Studie, die das Dexpanthe-
nol-haltige Externum mit Vaseline verglich. Studien am Hautmodell bestätigten, dass Dexpanthenol die Abheilung nach einer Behandlung mit einem ablativen CO2- oder Er:YAG-Laser (Erbium:YttriumAluminium-Granat-Laser) fördert und beschleunigt (vgl. Abb. 1).
Beruflich wie privat Handekzeme vermeiden
Eine weitere häufige Berufskrankheit sind Handekzeme (BK 5101). Besonders oft betroffen sind Beschäftigte in Berufen mit häufiger Feuchtarbeit wie Pflegepersonal, Friseure und Beschäftigte in der Gastronomie.
„Natürlich sollte im Vorfeld alles daran gesetzt werden, Handekzeme im beruflichen Bereich durch frühzeitige gezielte Prävention wie Handschuhe und Hautschutzpräparate zu vermeiden“ erklärte John und ergänzte: „Hände werden aber nicht nur beruflich oft stark strapaziert, auch die Feuchtarbeit im Haushalt ist eine Stresssituation für die Hände.
Um die Heilung von oberflächlichen Hautschädigungen zu unterstützen, kann den Betroffenen u.a. Bepanthen® Wund- und Heilsalbe empfohlen werden.“ Bei Juckreiz und zu Ekzemen neigender Haut bietet sich Bepanthen® Sensiderm Creme an, denn sie unterstützt die Regeneration der Hautschutzbarriere und enthält u.a. Lipide in hautähnlicher, schichtartiger Struktur (Lipid-Lamellen-Technologie) sowie Feuchthaltefaktoren.
Elisabeth Wilhelmi, München
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 66 KONGRESSE
Weltweit ein Problem: Die nicht alkoholische Fettlebererkrankung und ihre Folgen
Maßgeblich getriggert durch Risikofaktoren wie Übergewicht, metabolisches Syndrom und Typ2-Diabetes stellen die nicht alkoholische Fettlebererkrankung (NAFLD) und ihre potenziell lebensbedrohlichen Folgeerkrankungen nicht nur Hepatologen vor immer größere Probleme. Eine der zentralen medizinischen Herausforderungen besteht darin, eine Krankheitsprogression hin zu Leberzirrhose und hepatozellulärem Karzinom zu verhindern oder zumindest frühzeitig zu erkennen, wie bei einem von der Falk Foundation e.V. veranstalteten Symposium im Rahmen der 39. Jahrestagung der German Association for the Study of the Liver (GASL) in Bochum deutlich wurde.
Leberzellkarzinom auch ohne vorherige Leberzirrhose
Nach heutigem Verständnis gilt die NAFLD als die hepatische Manifestation des metabolischen Syndroms – dem überlappenden Auftreten von Erkrankungen wie Adipositas, Bluthochdruck, Störung des Blutzuckerstoffwechsels oder eben auch der Fettleber. In Ländern der westlichen Welt sind nach PD Dr. Jan Best, Bochum, etwa 25 – 30 % der erwachsenen Bevölkerung von einer NAFLD betroffen. Die Diagnose kann dann gestellt werden, wenn sich mittels Biopsie oder geeigneter Bildgebung ein Leberfettanteil (Steatose) von mindestens 5 % in Abwesenheit anderer Leberpathologien (z.B. hoher Alkoholkonsum) nachweisen lässt.

Mit dem Fortschreiten der Erkrankung kommt es bei etwa jedem vierten der NAFLD-Patienten zu
einer nicht alkoholischen Leberentzündung (NASH), die schließlich eine Leberzirrhose oder ein hepatozelluläres Karzinom (HCC) nach sich ziehen kann. Die Entwicklung einer Leberzirrhose ist dabei kein obligater Zwischenschritt der im Einzelnen noch nicht zureichend verstandenen HCCKarzinogenese. Im Fokus des wissenschaftlichen Interesses stehen in den letzten Jahren diesbezüglich vor allem diverse Genpolymorphismen (z.B. PNPLA3 rs738409 Polymorphismus) oder auch der Einfluss der gastrointestinalen Mikrobiota.
Unstrittig ist hingegen dies: Ob in den USA, dem Vereinigten Königreich oder in Deutschland – überall beobachtet man einen deutlichen Anstieg von NAFLD/NASH-asso-
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 67 KONGRESSE
Abbildung 1: Dexpanthenol (Bepanthen® Wund- und Heilsalbe) beschleunigt die Abheilung nach einer Behandlung mit einem ablativen CO2oder Er:YAG-Laser.
Rasante Zunahme des NAFLD-HCC
ziierten Leberzellkarzinomen. So berichtete Professorin Helen Reeves, Newcastle upon Tyne, dass unter den dem Tumorzentrum der Newcastle University zugewiesenen HCC-Patienten der Anteil der NAFLD-assoziierten Fälle zwischen 2000 und 2010 kontinuierlich von 0 auf 35 % gestiegen ist und damit bereits mit Abstand an der Spitze der HCC-Ätiologien lag.
2018 berichtete die American Cancer Society zwischen den Jahren 2010 und 2015 über eine Zunahme NASH-bezogener HCCs in Höhe von 68 %, womit die nicht alkoholische Leberentzündung auch in den USA inzwischen nicht nur die am rasantesten angestiegene Ursache von Leberzellkarzinomen geworden ist, sondern auch die absolut gesehen häufigste.
Vergleichsweise schlechte Prognose
Sei es aufgrund eines besonders aggressiven Tumorwachstums von NAFLD-assoziierten Leberzellkarzinomen oder aufgrund offenkundiger Defizite in Sachen Screening und medizinischer Überwachung von NAFLD-Patienten – die (Überlebens)-Prognose von Betroffenen mit NAFLD-basiertem HCC ist vergleichsweise schlecht. Wie die Experten berichteten, erhalten die Betroffenen etwa im Vergleich zu Patienten mit HCV-assoziierten Leberzellkarzinomen nur selten eine spezifische HCC-Behandlung. Zudem erfüllen nach Maßgabe der Milan-Kriterien zum Zeitpunkt der Erstdiagnose
eines HCC vergleichsweise wenige Patienten die Voraussetzung für eine Lebertransplantation. Nach aktuellen Daten eines US-amerikanischen Transplantationsregisters sind an NAFLD erkrankte OPKandidaten alt, übergewichtig und häufiger von Diabetes betroffen. So verwundert es auch nicht, dass nach den von Best vorgestellten Daten einer deutschen Multicenterstudie die Gesamtüberlebensrate bei NAFLD/NASH-HCCPatienten niedriger lag als bei Nicht-NASH-HCC-Patienten.
NAFLD: Häufig lange übersehen
Wird die Erstdiagnose eines NAFLD-HCC gestellt, so ist Best zufolge bei einem großen Teil der Patienten die Diagnose der zugrunde liegenden NAFLD noch gar nicht gestellt worden – ein Beleg für die (zu) geringe Beachtung dieses häufigen hepatologischen Krankheitsbildes. Dabei würde die Frühdiagnose von NAFLD und NASH die Voraussetzungen dafür schaffen, durch geeignete Präventivmaßnahmen die weitere Krankheitsprogression hin zu Fibrose und HCC zu stoppen bzw. eine Fibrose rückgängig zu machen. Mangels zugelassener Medikamente bieten sich dafür derzeit vor allem eine Lebensstiländerung und das optimale Management von oft vorhandenen Komorbiditäten wie Hypercholesterinämie oder Typ-2-Diabetes an. Bei der medikamentösen Therapie hält der Gastroenterologe neben Metformin, Pioglitazon oder GLP-1-Rezeptoragonisten auch Obetichol-
säure – ein synthetisches Derivat der physiologischen Gallensäure Chenodesoxycholsäure – für einen hoffnungsvollen Kandidaten. Tatsächlich gibt es bei NAFLD-Patienten Hinweise auf ein verändertes Gallensäuremuster und einen veränderten Gallensäuremetabolismus, woraus sich in mehrfacher Hinsicht therapeutische Perspektiven eröffnen könnten.
Therapeutisches Vorgehen nach Maßgabe der BCLC-Algorithmen
Auch bei Patienten mit NAFLDHCC orientiert sich das therapeutische Vorgehen laut Reeves grundsätzlich an den aktuellen BCLC (Barcelona Clinic Liver Cancer prognosis and treatment strategy)Algorithmen. Dezidierte Aussagen zu den Überlebenschancen dieser Patienten werden dadurch erschwert, dass diese Subgruppe im Rahmen klinischer Studien zumeist entweder gar nicht eigens ausgewiesen oder nur spärlich vertreten ist. Während TyrosinkinaseInhibitoren ähnlich effektiv sind wie beim Nicht-NAFLD-HCC, gibt es Hinweise auf eine verminderte Wirksamkeit von ImmunCheckpoint-Inhibitoren. Wirksamkeit und Sicherheit vorhandener und kommender Medikamente bei Leberzellkarzinom sollten im Rahmen klinischer Studien künftig gezielt für Subgruppen wie die immer häufiger vertretene NAFLD/ NASH untersucht werden.
Elisabeth Wilhelmi, München
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 68 KONGRESSE
REPUBLIK MOLDAU: Raisa Pavlova flieht vor den Kämpfen in der Ukraine, unsere Mitarbeiterin Svetlana Bujac bietet ihr Hilfe an. © Peter Bräunig

KRIEGEN SETZEN WIR HOFFNUNG ENTGEGEN
Mit Ihrer Spende rettet ÄRZTE OHNE GRENZEN Leben: Mit 52 Euro können wir zum Beispiel 40 Menschen auf der Flucht drei Monate lang mit den wichtigsten Medikamenten versorgen.
Private Spender*innen ermöglichen unsere weltweite Hilfe –jede Spende macht uns stark
Spendenkonto: Bank für Sozialwirtschaft
IBAN: DE72 3702 0500 0009 7097 00
BIC: BFSWDE33XXX www.aerzte-ohne-grenzen.de/spenden
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
Titelbild: Quelle: istockfoto.com
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung: Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung:
DRUCK_INFORM_W.R.
Roland Welker
Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20
90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de
JOURNAL PHARMAKOL. U. THER. 2/2023 · 32. JAHRGANG © VERLAG PERFUSION GMBH 69 VERL AG PERFUSION Journal
2 JAHRE1
SUBKUTAN
2Fertigsprit zen = 1Dosis
MEHR FLEXIBILITÄT
a
Mehrheit der Patient*innen präferiert die subkutane Gabe1
> 16.000 mit TYSABRI™ s.c. behandelte Patient*innen in Europa seit März 2021b,1
PZN:
Für TYSABRI™ Patient*innen entwickelt 2,3

1 Mio. Tests durchgeführtc,4
Tysabri® 300 mg Konzentrat zur Herstellung einer Infusionslösung / Tysabri® 150 mg Injektionslösung in einer Fertigspritze. Wirkstoff: Natalizumab. Zusammensetzung: 300 mg Konzentrat: Ein Milliliter Konzentrat enthält 20 mg Natalizumab (300 mg Natalizumab pro Durchstechflasche); 150 mg Injektionslösung: Ein Milliliter Lösung enthält 150 mg Natalizumab. Sonstige Bestandteile: Natriumdihydrogenphosphat 1 H2O, Dinatriumhydrogenphosphat 7 H2O, Natriumchlorid, Polysorbat 80 (E 433), Wasser für Injektionszwecke. Anwendungsgebiete: Krankheitsmodifizierende Monotherapie bei Erwachsenen mit hochaktiver, schubförmig remittierend verlaufender Multipler Sklerose (RRMS) bei folgenden Patientengruppen: Patienten mit hochaktiver Erkrankung trotz Behandlung mit einem vollständigen und angemessenen Zyklus mit mindestens einer krankheitsmodifizierenden Therapie (DMT) oder Patienten mit rasch fortschreitender RRMS. Gegenanzeigen: Überempfindlichkeit gegen Natalizumab oder einen der sonstigen Bestandteile; Progressive Multifokale Leukenzephalopathie (PML); Patienten mit einem erhöhten Risiko für opportunistische Infektionen, wie immungeschwächte Patienten (einschließlich solcher Patienten, die aktuell eine immunsuppressive Behandlung erhalten oder durch frühere Therapien immungeschwächt sind); Kombination mit anderen krankheitsmodifizierenden Therapien (DMTs); bekannte aktive Malignome (Ausnahme: Basaliom). Nebenwirkungen: Sehr häufig: Nasopharyngitis, Harnwegsinfektion, Infusionsbedingte Reaktionen, Übelkeit, Fatigue, Schwindelgefühl, Kopfschmerzen, Arthralgie; Häufig: Herpes-Infektion, Überempfindlichkeit, Anämie, Leberenzym erhöht, Arzneimittelspezifischer Antikörper nachweisbar, Dyspnoe, Erbrechen, Fieber, Schüttelfrost, Reaktion an der Infusionsstelle, Reaktion an der Injektionsstelle, Pruritus, Ausschlag, Urtikaria, Flush; Gelegentlich: Progressive Multifokale Leukenzephalopathie (PML), Anaphylaktische Reaktion, Immunrekonstitutionssyndrom, Thrombozytopenie, Immunthrombozytopenische Purpura (ITP), Eosinophilie, Gesichtsödem; Selten: Herpes des Auges, hämolytische Anämie, Kernhaltige Erythrozyten, Hyperbilirubinämie, Angioödem; Häufigkeit nicht bekannt: Herpes-Meningoenzephalitis, JCV Körnerzellen- Neuronopathie, Nekrotisierende Herpesretinopathie, Leberverletzung; Fälle von: Wirkungen auf Labortests. Warnhinweis: 150 mg Injektionslösung: 2 Spritzen mit 150 mg verwenden. Vollständige Dosis = 300 mg. Verschreibungspflichtig. Weitere Angaben: Siehe Fachinformation. Biogen Netherlands B.V., Prins Mauritslaan 13, 1171 LP Badhoevedorp, Niederlande. Stand: August 2021.
Biogen GmbH Riedenburger Straße 7 • 81677 München • www.biogen.de
16825458 | a Durch kürzere Dauer der Verabreichung im Vergleich zur intravenösen Infusion1,5,6 | b Stand September 2022 | c Stand November 2021 1. Gold R et al. ECTRIMS 2022, P365 | 2. Lee P et al. J Clin Virol. 2013; 57(2): 141 – 146 | 3. Ho PR et al. Lancet Neurol. 2017; 16(11): 925 – 933 | 4. Unilabs, data on file | 5. TYSABRI™ s.c. Fachinformation, Stand Mai 2022 | 6. TYSABRI™ i.v. Fachinformation, Stand Mai 2022
DER
™ EFFEKT
TYSABRI
Wirksamkeit in Aktion
Biogen-102336
