JAHRGANG 34
HEFT 2
März 2025

JAHRGANG 34
HEFT 2
März 2025

Eosinophile Erkrankungen verstehen und adäquat therapieren
Immunglobulin Hizentra® in der Fertigspritze erleichtert die Behandlung primärer und sekundärer Immundefekte
Zystische Fibrose: Biomarker quantifizieren die Wiederherstellung der CFTR-Funktion durch CFTR-Modulatoren
FSME: Encepur® für eine schnelle Grundimmunisierung und langjährigen Schutz
Vitiligo: Verlängerte Therapie mit Ruxolitinib-Creme bewirkt dauerhafte Repigmentierung
Mit der Triple-Kombination BUD/GLY/FORM die Mortalität der COPD reduzieren
Sacituzumab govitecan: Das Antibody-Drug-Conjugate überzeugt beim HR+/HER2– metastasierten Brustkrebs
Palforzia® – die erste orale Immuntherapie für Kleinkinder mit Erdnussallergie
Exagamglogene autotemcel – eine innovative Gentherapie bei Sichelzellkrankheit und Beta-Thalassämie
Mirikizumab wirkt schnell und anhaltend in der First-Line-Therapie der Colitis ulcerosa
Feuchte altersabhängige Makuladegeneration: PULSAR-Studie bestätigt die 3 Jahre anhaltende Wirksamkeit und Sicherheit von Aflibercept 8 mg

Blue trägt seinen vierjährigen Bruder Sunday sieben Kilometer dorthin, wo er die einzige Mahlzeit des Tages erhält – in die Schule. Blue ist unterernährt. Sunday ist bereits lebensbedrohlich unterernährt und ihm fehlt jegliche Kraft zum Laufen. Beide leiden sehr unter der aktuellen Hungerkrise.
Helfen Sie mit, noch mehr Kinder zu ernähren.
Immer mehr Menschen hungern: 733 Millionen Menschen sind laut Welternährungsorganisation (FAO) davon betroffen. Einer von elf Menschen weltweit, einer von fünf in Afrika.
Deshalb geben wir als größter Partner des Welternährungsprogramms der Vereinten Nationen so viel wie möglich: Alle 60 Sekunden erhält ein hungriges Kind eine Mahlzeit.
Helfen Sie mit, damit es reicht. worldvision.de/es-ist-genug
Unglaublich! Jetzt ist es schon ein halbes Jahrzehnt her, als sich eine Lawine ungeahnten Ausmaßes auf uns alle zuzubewegen begann. Nur wenige Wochen lagen zwischen dem ersten gemeldeten CoronaFall in Deutschland (Ende Januar 2020 beim Autozulieferer Webasto) und dem ersten landesweiten Corona-Lockdown am 22. März. Was geblieben ist, ist vor allem Kritik am Umgang mit der Seuche. Dabei ärgert es mich schon, wenn Neunmalkluge jetzt alles besser wissen als seinerzeit diejenigen, die damals Entscheidungen treffen mussten, deren Bedeutung und Tragweite im Voraus keiner absehen konnte.
Die vom Robert Koch-Institut (RKI) wöchentlich herausgegebenen Zahlen legten im April 2020 eine Mortalität bei Corona-Infektion von nicht weniger als 5 % und mehr nahe, und wir alle erinnern uns wohl noch mit Schaudern an die gespenstigen Szenarien aus der italienischen Stadt Bergamo, als Kolonnen von Fahrzeugen CoronaOpfer aus der Stadt brachten. Interessanterweise hat es gar nicht so lange gedauert, bis sich abzeichnete, dass das „neuartige Virus SARS-CoV-2“ eben eine Virusinfektion ist. „Neuartig“ war das Virus nicht zuletzt für das menschliche Immunsystem. Schnell wurde klar, dass eine Corona-Infektion für Menschen in der ersten Lebenshälfte ziemlich sicher kein lebensbedrohliches Ereignis sein dürfte, sehr wohl aber für ältere Menschen. Bis Ende 2020 zählte das RKI über 50.000 Corona-Tote, 99 % davon waren über 50 Jahre alt und 70 % über 80 Jahre. Wer vor diesem Hintergrund heute die damalige Schließung von Schulen und Kindergärten anprangert, nimmt billigend in Kauf, dass eine ungehinderte Ausbreitung unter jungen Menschen für diese zwar wenig dramatische Folgen gehabt hätte, wohl aber für die ältere Hälfte der Bevölkerung. Seit zu Beginn des Jahres 2021 in Rekordzeit entwickelte Impfstoffe
Was war, was ist, was bleibt?
auf den Markt kamen, begann mit zunehmender quantitativer Verfügbarkeit ihre Schutzfunktion gerade für ältere Menschen immer besser zu greifen. Masken hätten diesbezüglich wohl noch mehr leisten können, wäre die Kommunikation ihrer Potenziale besser gelungen [1]: Medizinische („chirurgische“) Masken schützen andere, FFP2Masken auch den Träger selbst – vorausgesetzt, Ein- und Ausatemluft können nicht an der Maske vorbei, d.h., Mund und Nase sind beide effektiv bedeckt. Obwohl die Bedeutung von Aerosolen für die Übertragung ebenfalls schon bald gut verstanden wurde [2], blieben die logischen Konsequenzen für den Umgang in der Pandemie lange aus.
Bis Ende 2020 wurden dem RKI 1,78 Millionen Corona-Fälle gemeldet, bis Ende 2021 waren es 7,2 Millionen und bis Ende 2022 37,4 Millionen (bei gleichzeitig rapide steigender Dunkelziffer). Spätestens Ende 2022 war das Immunsystem der meisten Menschen in Deutschland wohl mit dem Virus in Berührung gekommen und etwa 4 von 5 Menschen hatten ihr Immunsystem durch eine oder mehrere Impfungen einschlägig vorbereitet. Endete 2020 noch für etwa jeden 40. Infizierten Covid-19 tödlich (2,3 %) und in 2021 für jeden 80. Infizierten (1,23 %), so sank die Letalität in 2022 auf etwa 1:500 (0,2 %) und näherte sich damit der der Influenza an.
Eine essenzielle Lehre aus der Pandemie liegt sicherlich in der Erkenntnis der Wichtigkeit von gut dokumentierten Beobachtungsstudien, die nahezu zeitgleich mit dem Geschehen charakteristische As-

Prof. Dr. med. Karl-Ludwig Resch
pekte zu Symptomen wie zu möglichen Potenzialen der notwendigerweise ex juvantibus erfolgenden therapeutischen Ansätze zuließ. Frühe Mitteilungen zur Bedeutung von Kortikoiden in Bezug auf die Überlebenswahrscheinlichkeit haben wohl Millionen Menschen das Leben gerettet. Erste (belastbare) Erkenntnisse aus hochwertigen, vergleichenden Interventionsstudien (randomisiert-kontrollierten Studien) wurden erst veröffentlicht, als die Pandemie bereits weitgehend „durch war“…
Inzwischen zwar aus den Schlagzeilen verschwunden, aber immer noch ein medizinisches wie volkswirtschaftlich relevantes Problem sind persistierende Folgen einer Infektion mit SARS-CoV-2, das sog. Long Covid. Grundsätzlich waren derartige langandauernde Komplikationen von anderen Viruserkrankungen schon lange bekannt und beschrieben. Bereits seit 1989 finden sich im Schlagwortkatalog der
Datenbank Medline (MeSH Headings) unter dem Begriff „Chronic Fatigue Syndrome“ die Begriffe „Postviral Fatigue Syndrome“ und „Myalgic Encephalomyelitis“ als Synonyme [3]. Erst die um Dimensionen höhere Infektionsinzidenz ließ die typische Problematik sichtbar werden: „Symptoms are not caused by ongoing exertion; are not relieved by rest; and result in a substantial reduction of previous levels of occupational, educational, social, or personal activities. Minor alterations of immune, neuroendocrine, and autonomic function may be associated with this syndrome.“ Überzeugende Therapieansätze fehlen bis heute. Die stationäre Rehabilitation kann von ihrem Ansatz her (Wiederherstellen von Fähigkeiten durch gezieltes Trainieren) wie von den Ressourcen (fachübergreifende Abteilungen für die Behandlung von Long Covid) nur den berühmten Tropfen auf den heißen Stein leisten. Die wohnortnahen Versorgungsstrukturen (inkl. wohnortnahe ambulante Reha) sind angesichts der Komplexität in jeder Hinsicht überfordert. Für innovative, integrative Ansätze fehlen offensichtlich weiter (fächerübergreifende) Ideen sowie formale Zugänge. So werden wir wohl noch längerer Zeit mit den Folgen der Pandemie leben müssen, auch wenn Corona für die Medien kein Thema mehr ist. Karl-Ludwig Resch, Nürnberg
Quellen
1 Resch KL. Corona und die Maskenpflicht: ein Trauerspiel ohne Ende. J Pharmakol Ther 2021;30:1-2
2 Resch KL. Aktueller Stand der Erkenntnis zu den Übertragungsmechanismen von SARS-CoV-2 und der Wirksamkeit präventiver Maßnahmen in geschlossenen Räumen mit besonderem Fokus auf Aerosole: Systematischer Review. J Pharmakol Ther 2020;29:76-82
3 https://www.ncbi.nlm.nih.gov/mesh? term=D015673
Eosinophile Erkrankungen verstehen und adäquat therapieren 36 Brigitte Söllner
Immunglobulin Hizentra® in der Fertigspritze erleichtert die Behandlung primärer und sekundärer Immundefekte
41
Zystische Fibrose: Biomarker quantifizieren die Wiederherstellung der CFTR-Funktion durch CFTR-Modulatoren 42
FSME: Encepur® für eine schnelle Grundimmunisierung und langjährigen Schutz
Vitiligo: Verlängerte Therapie mit Ruxolitinib-Creme bewirkt dauerhafte Repigmentierung
48
Mit der Triple-Kombination BUD/GLY/FORM die Mortalität der COPD reduzieren 50
Das Antibody-Drug-Conjugate überzeugt beim HR+/HER2– metastasierten Brustkrebs 52
Palforzia® – die erste orale Immuntherapie für Kleinkinder mit Erdnussallergie 55
Exagamglogene autotemcel – eine innovative Gentherapie bei Sichelzellkrankheit und Beta-Thalassämie 56
Mirikizumab wirkt schnell und anhaltend in der First-Line-Therapie der Colitis ulcerosa 60
Feuchte altersabhängige Makuladegeneration: PULSAR-Studie bestätigt die 3 Jahre anhaltende Wirksamkeit und Sicherheit von Aflibercept 8 mg 62
45, 46, 47, 61
Wie wird man 100 Jahre alt? Und was hat eine Sommer-Auszeit am Goldenen Energieberg in Lech damit zu tun? Ein Wort: Longevity.
Der Sommer ist eine ganz besondere Zeit am Arlberg.
Besonders belebend. Besonders natürlich. Besonders kraftvoll.
Die grüne Bergwelt lockt, sich in ihr zu verlieren. Stundenlang in der Natur zu baden. Sich ganz dem guten, bewussten Leben hinzugeben.

Dass das dem Körper und der Seele etwas gibt, was von enormer Bedeutung ist, weiß Daniela Pfefferkorn schon lang. Longevity – dem Begriff, der das gesunde Leben und Altern beschreibt –schenkt sie in ihrem Hotel Goldener Berg in Oberlech deshalb nun jenen Platz, der ihm gebührt. „Wir in der westlichen Welt Lebenden müssen einiges dafür tun, dass wir gesund ins hohe Alter kommen“. Neben dem Körper braucht hierzu auch die Seele Zuneigung, und schließlich kommt es auch auf gesunde Gedanken an. Der Sommerurlaub auf ihrem feinen Energieberg bringt dafür die allerbesten Voraussetzungen mit. (Long) mountain living at its best!
Ganzheitliche Methoden & Wellness der Superlative
Frische Luft, gesundes Essen, wohltuender Schlaf – sie sind für Daniela Pfefferkorn die absoluten Basics einer ganzheitlichen Auszeit. Mountain Selfcare im Goldenen Berg geht deshalb noch weit darüber hinaus – ganz im Sinne des LongevityGedankens:
Achtsame Energiemediziner und Coaches nehmen sich der Seele an, kümmern sich um den Geist und die (guten) Gedanken.
Großartige Therapeuten verhelfen zu achtsamer Bewegung in der unfassbar schönen Bergwelt des Arlbergs.
Umfassende Detox-Angebote helfen dabei, sich von Stress und schädlichen Toxinen freizumachen.
Letztlich – und damit kann sich jeder einfach selbst etwas Gutes tun – schenkt der großzügige Wellness und Holistic Selfcare Bereich mit Infinitypool, Panorama-Sauna, Yoga- und Meditationsräumen viel Platz zum Entfalten.

„Um zum Glück und dem ausgeglichenen Dasein zu finden, muss die Energie frei fließen können. Müssen Körper, Geist und Seele im Einklang sein. Muss man auch einfach einmal loslassen können.“ Ein Leichtes während des Sommers im Goldenen Berg.


Kontakt: Hotel Goldener Berg Oberlech 117
A-6764 Lech am Arlberg Tel. +43 (0) 5583/22050 happy@goldenerberg.at www.goldenerberg.at
Eosinophile Granulozyten (EOS) spielen eine zentrale Rolle bei entzündlichen Prozessen und sind entscheidend für die Pathogenese verschiedener Erkrankungen wie dem schweren eosinophilen Asthma (SEA) und der eosinophilen Granulomatose mit Polyangiitis (EGPA) [1]. Als Bestandteile des Immunsystems sind EOS auch an der Abwehr von parasitären, bakteriellen und viralen Infektionen beteiligt. Sie sammeln sich in verschiedenen Zielorganen des Körpers an und werden dort aktiviert [2, 3]. Ist die Regulation der EOS-Aktivität gestört, kann es zur vermehrten Ausschüttung proinflammatorischer Zytokine kommen, was wiederum zu Krankheiten in verschiedenen Organen führen kann – z.B. zu Asthma, chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP), eosinophilen gastrointestinalen Störungen, EGPA oder dem hypereosinophilen Syndrom (HES) [1, 2, 3]. In der Pathophysiologie dieser Erkrankungen spielen EOS eine zentrale Rolle, denn sie triggern die zugrundeliegende Inflammation und die Schädigung des Gewebes. Als Teil der Typ-2-Immunantwort werden die Signale der EOS über die Interleukine(IL)-4 und -13 sowie insbesondere über das Zytokin IL-5 vermittelt. IL-5 ist maßgeblich an der Vermehrung, Reifung, Aktivierung und Anreicherung von EOS beteiligt [1].
Asthma (SEA)
Bei mehr als 80 % der Patienten mit schwerem Asthma liegt ein eosinophiler Phänotyp vor [4]. Hierbei reagiert der Körper auf Reize wie beispielsweise Infekte, indem er vermehrt eosinophile Granulo-
Brigitte Söllner, Erlangen
zyten produziert. Diese sammeln sich im Lungengewebe und rufen dort eine Entzündung hervor [5, 6]. Diese Entzündungsreaktion führt zu Gewebeschäden, einer erhöhten Schleimproduktion sowie zu einer Verengung der Atemwege und damit zu den typischen Asthmasymptomen wie Atemnot, Husten und Engegefühl in der Brust [5–7]. Die Patienten haben häufig Exazerbationen, signifikante Einschränkungen der Lungenfunktion und eine verringerte Lebensqualität [8, 9, 10]. Eine erhöhte Anzahl von EOS im Blut gilt daher als ein zuverlässiger Prädiktor und Biomarker für das Risiko einer Exazerbation und korreliert mit einer schlechteren Asthmakontrolle [11, 12]. Außerdem ist eine erhöhte EOS-Konzentration im Sputum mit einer stärkeren Atemwegsobstruktion und einem Abfall der Einsekundenkapazität (FEV1) assoziiert [13, 14, 15].
Eosinophile Granulomatose mit Polyangiitis (EGPA)
EGPA, früher als Churg-StraussSyndrom bezeichnet, ist eine seltene, komplexe Autoimmunerkrankung mit langem Leidensweg und teilweise lebensbedrohlichen Situationen für die Patienten. Sie kann verschiedene Organe, einschließlich der Lunge, Herz, Haut,
Magen-Darm-Trakt und Nerven, schädigen [16, 17]. EGPA tritt fast immer (≥95 %) zusammen mit Asthma auf und verursacht eine Entzündung kleiner bis mittelgroßer Blutgefäße, begleitet von einer Eosinophilie im Blut und Gewebe [18, 19]. Zu den häufigsten Symptomen zählen extreme Müdigkeit, Gewichtsverlust, Muskel- und Gelenkschmerzen, Hautausschläge, Nervenschmerzen sowie Nasenund Atemwegsbeschwerden. Ohne Behandlung kann die Krankheit tödlich verlaufen [20].
Zwar ist noch nicht vollständig verstanden, wie die EGPA entsteht, man geht jedoch davon aus, dass EOS für die mit EGPA verbundenen Organschäden verantwortlich sein können, indem sie das Gewebe infiltrieren und durch zytotoxische Effekte wie die Freisetzung des eosinophilen kationischen Proteins (ECP) und des Major Basic Protein (MBP) direkt schädigen [18, 21]. Außerdem rekrutieren sie auch andere Immunzellen wie Basophile und ILC2, die die Entzündungsreaktion verstärken und die Gewebeschädigung fördern [22, 23].
Die EGPA entwickelt sich typischerweise in 3 Phasen, wobei die einzelnen Phasen nicht bei allen Patienten und nicht in einer bestimmten Reihenfolge auftreten oder sich teilweise überschneiden. EOS sind an allen 3 Phasen betei-
Benralizumab (Fasenra®) ist ein anti-eosinophiler, humanisierter, afucosylierter, monoklonaler Antikörper (IgG1, kappa). Dieser bindet mit hoher Affinität und Spezifität an die Alpha-Untereinheit des menschlichen Interleukin-5-Rezeptors (IL-5Rα). Der IL-5-Rezeptor wird spezifisch auf der Oberfläche von Eosinophilen und Basophilen exprimiert. Das Fehlen von Fucose im Fc-Bereich von Benralizumab führt zu einer hohen Affinität von Benralizumab zu FcγRIII-Rezeptoren auf Immuneffektorzellen, wie z.B. natürlichen Killerzellen (NKZellen). Dies führt durch eine verstärkte antikörperabhängige zellvermittelte Zytotoxizität zur Apoptose von Eosinophilen und Basophilen, wodurch die eosinophile Entzündung reduziert wird [29].
Benralizumab ist zugelassen als Add-on-Erhaltungstherapie bei erwachsenen Patienten mit schwerem eosinophilem Asthma (SEA), das trotz hochdosierter inhalativer Kortikosteroide plus langwirksamer Beta-Agonisten unzureichend kontrolliert ist, sowie als Add-on-Therapie bei erwachsenen Patienten mit rezidivierender oder refraktärer eosinophiler Granulomatose mit Polyangiitis (EGPA) [29].
Benralizumab wird mittels Fertigspritze oder Pen subkutan appliziert. Bei Asthma beträgt die empfohlene Dosis von Benralizumab 30 mg als subkutane Injektion, die ersten 3 Dosen in einem Abstand von 4 Wochen und anschließend alle 8 Wochen. Bei EGPA beträgt die empfohlene Dosis von Benralizumab 30 mg als subkutane Injektion alle 4 Wochen [29].
Die Behandlung mit Benralizumab führt bei SEA innerhalb von 24 Stunden nach der ersten Dosis zu einer nahezu vollständigen Depletion der Eosinophilen im Blut, die über die gesamte Behandlungsdauer erhalten bleibt. Bei Patienten mit EGPA war die Depletion der Eosinophilen im Blut konsistent mit dem in den Asthma-Studien beobachteten Effekt [29].
ligt, dabei korreliert der Grad der Bluteosinophilie mit der Krankheitsaktivität [22]:
1. In der Prodromalphase stehen Asthma und Allergiesymptome im Vordergrund, oft verbunden mit chronischer Rhinosinusitis und Nasenpolypen (CRSwNP). Typisch sind erhöhte EOS-Zahlen im Sputum und erhöhte Spiegel von aus EOS ausgeschütteten Mediatoren [16, 21, 24].
2. In der zweiten, eosinophilen infiltrativen Phase erhöht sich die Eosinophilenzahl im Blut und Gewebe, was Entzündungen und Gewebeschäden verursacht [21, 24].
3. Die dritte, vaskulitischen Phase ist durch eine nekrotisierende Vaskulitis gekennzeichnet mit Entzündungen der kleinen Arterien und Venen, die zu weiteren Organschäden bis hin zu lebensbedrohenden Situationen führen kann [24].
Im Durchschnitt vergehen 3 – 9 Jahre von Beginn des Asthmas bis zur vaskulitischen Phase [16].
Nachteile der Therapie mit hochdosierten Kortikosteroiden
Die Behandlung der EGPA hat das Ziel, eine langfristige Remission zu erreichen. Allerdings gelingt das bei etwa 40 % der Patienten mit den bisherigen Behandlungsoptionen nicht [25]. Laut Leitlinie bilden bislang orale Kortikosteroide (OCS) die Basis der Behandlung [26]. Diese sind aber in der Langzeitanwendung mit erheblichen Nebenwirkungen und Schäden für die Patienten verbunden, sodass die Leitlinien dazu raten, die OCSDosis soweit möglich zu reduzieren [26]. Außerdem kommt es beim Absetzen von OCS häufig zu einem Rezidiv, das mögli -
cherweise auf eine entstehende gewebespezifische Kortikosteroidresistenz zurückzuführen ist – ein Teufelskreis entsteht [27, 28].
Mit Benralizumab die eosinophile Entzündungsreaktion gezielt stoppen
Da eosinophile Graulozyten bei der EGPA die treibende Kraft für die Inflammation sind, rücken Therapieoptionen, die direkt oder indirekt auf die EOS abzielen und somit nicht nur die Symptome, sondern auch die Ursache der eosinophilen Entzündung bekämpfen, immer mehr in den Fokus. Ein Wirkstoff, der diese Anforderungen erfüllt, ist Benralizumab (Fasenra ®). Dieser monoklonale Antikörper blockiert gezielt den Interleukin-(IL-)5-Rezeptor auf der Oberfläche von Eosinophilen
(95 %
50 %
%
%
4 mg/Tag
Abbildung 1: Ergebnisse der MANDARA-Studie für den sekundären Endpunkt OCS-Reduktion. Ein nominell größerer Anteil der Patienten unter Benralizumab erreichte eine vollständige OCS-Reduktion im Vergleich zur Mepolizumab-Gruppe [33].
Test auf Überlegenheit von Benralizumab gegenüber Mepolizumab, alle p
und Basophilen und stoppt so die eosinophile Entzündungskaskade.
Da bei den Eosinophilen gleichzeitig durch die Rekrutierung von Immuneffektorzellen die Apoptose eingeleitet wird, werden die EOS in Blut und Gewebe bereits innerhalb von 24 Stunden nahezu vollständig entfernt [29]. Dieser Effekt bleibt über die gesamte Behandlungsdauer erhalten und ermöglicht damit den Patienten das Erreichen einer Remission sowie eine Reduktion oder sogar das Absetzen von OCS [1].
Überzeugende Studiendaten für die Behandlung von SEA und EGPA
Benralizumab wird aufgrund seiner hohen Wirksamkeit bei der Reduzierung der EOS schon seit
über 7 Jahren zur Behandlung des schweren eosinophilen Asthmas (SEA) eingesetzt, das trotz hochdosierter inhalativer Kortikosteroide plus langwirksamer BetaAgonisten unzureichend kontrolliert ist. Bereits in den Studien PONENTE und SHAMAL bei Patienten mit unkontrolliertem SEA wurde gezeigt, dass die Steroidgabe durch die Behandlung mit Benralizumab reduziert werden konnte [30, 31]. In PONENTE konnten 63 % der Patienten die OCS vollständig absetzen und 92 % ihre OCS-Dosis auf ≤5 mg pro Tag senken. Bei Patienten mit Nebenniereninsuffizienz war bei 82 % ein vollständiges Absetzen der OCS bzw. eine Reduktion auf unter 5 mg pro Tag möglich. Auch mit der reduzierten OCS-Dosis blieben 75 % der Patienten exazerbationsfrei [30].
SHAMAL ist die erste randomisierte, kontrollierte Studie, die die Möglichkeit einer Reduktion der Hintergrundmedikation mit ICS/ LABA unter der Therapie mit Benralizumab zeigen konnte. Unter Benralizumab reduzierten 92 % der Patienten erfolgreich die Erhaltungsdosis HD-ICS/LABA bei gleichbleibender Asthmakontrolle während der 32-wöchigen Reduktionsphase [31].
Langzeitstudien zum Sicherheitsprofil bei SEA zeigten außerdem, dass der Wirkstoff bei diesen Patienten über Jahre hinweg angewendet werden konnte – ohne zusätzliche Sicherheitssignale [32].
Seit dem 24.10.2024 ist der in der Therapie des unkontrollierten SEA bewährte Anti-IL-5-R-Antikörper auch für die Behandlung der rezidivierenden oder refraktären EGPA zugelassen [29]. Die Indikationser-
Eosinophilenzahlen im Blut
Abbildung 2: Ergebnisse der MANDARA-Studie für den sekundären Endpunkt, die Reduktion der Eosinophilen-Zahl im Blut. Unter der Be handlung mit Benralizumab erfolgte die EOS-Depletion schneller und vollständiger als unter Mepolizumab [33].

weiterung basiert auf den Ergebnissen der Nichtunterlegenheitsstudie MANDARA, der ersten und einzigen Head-to-head-Studie zweier Biologika bei EGPA, die die Wirksamkeit und Verträglichkeit von Benralizumab und Mepolizumab bei Patienten mit rezidivierender oder refraktärer EGPA verglich [33]. Die teilnehmenden 140 Patienten erhielten über 52 Wochen alle 4 Wochen entweder 30 mg Benralizumab als einzelne s.c. Injektion oder 300 mg Mepolizumab als 3 separate s.c. Injektionen. Alle Studienteilnehmer bekamen eine stabile orale Kortikosteroid(OCS)-Dosis von ≥7,5 bis ≤50 mg/ Tag. Primärer Endpunkt war der Anteil der Patienten, die sowohl in Woche 36 als auch in Woche 48 eine Remission erreichten, definiert als Birmingham Vasculitis Activity Score (BVAS) = 0 (d.h. keine aktive Vaskulitis) plus Prednisolon/Prednison-Dosis ≤4 mg/ Tag [33].
Beide Biologika erzielten vergleichbare Remissionsraten: 58 % unter
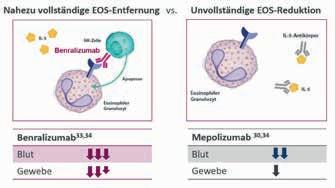
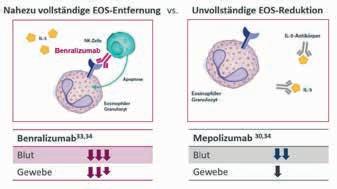
Abbildung 3: Die schnellere und vollständigere EOS-Reduktion unter Benralizumab im Vergleich zu Mepolizumab könnte auf die unterschiedlichen Wirkmechanismen zurückzuführen sein. Mepolizumab bindet an IL-5, verhindert dessen Bindung an die EOS und vermindert so deren Produktion und Überleben. Benralizumab hingegen bindet direkt an den IL-5-Rezeptor auf den EOS und leitet gleichzeitig die Apoptose von Eosinophilen und Basophilen ein [29].
Benralizumab und 57 % unter Mepolizumab. Der Anteil der Patienten, die ihre OCS-Dosis in den Wochen 48 – 52 vollständig absetzen konnten, war jedoch mit Benralizumab höher (41 % vs. 26 %; 95-%-KI: 0,67–30,71; nominaler p = 0,04)
(Abb. 1). Benralizumab erwies sich als gut verträgliche Therapieoption und zeigte zudem auch eine schnelle und nachhaltige Reduktion der EOS im Blut über 52 Wochen hinweg (Abb. 2 und 3) [33].
Eosinophile Granulozyten, aktiviert durch IL-5, sind der Drehund Angelpunkt bei inflammatorischen Prozessen, so auch bei schwerem, nicht kontrollierbarem eosinophilem Asthma (SEA) und der oft rezidivierenden oder refraktären eosinophilen Granulomatose mit Polyangiitis (EGPA). Für die davon betroffenen Patienten, die häufig unter den Nebenwirkungen der meist hochdosierten Glukokortikoidtherapie leiden, bietet der Anti-IL-5-R-Antikörper Benralizumab (Fasenra®) eine willkommene, langfristig wirksame und sichere Alternative, um Langzeitschäden an Organen zu verhindern und ihre Lebensqualität zu verbessern. Denn Benralizumab kann durch die Blockade des IL-5-Rezeptors die eosinophile Entzündungskaskade in Blut und Gewebe stoppen, sodass die Patienten eine Remission erreichen und die Glukokortikoide reduzieren oder sogar ganz absetzen können.
Brigitte Söllner, Erlangen
Literatur
1 Wechsler ME et al. Mayo Clin Proc 2021; 96:2694-2707
2 Ramirez GA et al. Biomed Res Int 2018; 2018:9095275
3 Jackson DJ et al. Eur Respir Rev 2022; 31:210150
4 Heaney LG et al. Chest 2021;160:814830
5 McBrien CN et al. Front Med (Lausanne) 2017;4:93
6 Brusselle GG et al. N Engl J Med 2022; 386:157-171
7 Porsbjerg C et al. Lancet 2023;401:858873
8 Chung KF et al. Eur Respir J 2014;43: 343-373
9 Wenzel S. Am J Respir Crit Care Med 2005;172:149-160
10 Fernandes AGO et al. J Bras Pneumol 2014;40:364-372
11 Price D et al. J Asthma Allergy 2016;9:112
12 Price DB et al. Lancet Respir Med 2015; 3:849-858
13 Broekema M et al. Respir Med 2010; 104:1254-1262
14 Talini D et al. BMJ Open 2015;5:e005748
15 Woodruff PG et al. J Allergy Clin Immunol 2001;108:753-758
16 Chakraborty RK et al. Churg-Strauss Syndrome. In: StatPearls. Treasure Island (FL): StatPearls Publishing; March 23, 2023
17 Trivioli G et al. Rheumatology 2020;59 (Suppl 3):iii84-iii94
18 Furuta S et al. Allergol Int 2019;68:430436
19 Cottin V et al. Eur Respir J 2016;48: 1429-1441
20 American Partnership for Eosinophilic Disorders. Eosinophilic Granulomatosis with Polyangiitis (EGPA). https://apfed. org/about-ead/eosinophilic-granulomatosis-withpolyangiitis/
21 Khoury P et al. Nat Rev Rheumatol 2014; 10:474-483
22 Fagni F et al. Front Med 2021;8:627776
23 Fijolek J et al. Front Med 2023;10: 1145257
24 Gioffredi A et al. Front Immunol 2014; 5:549
25 Jakes RW et al. ERJ Open Res 2024; 10:00912-02023
26 Holle JU et al. S3-Leitlinie Diagnostik und Therapie der ANCA-assoziierten Vaskulitiden. Version: 1.1. Stand: August 2024. https://register.awmf.org/de/leitlinien/ detail/060-012
27 Jackson DJ et al. Eur Respir Rev 2022; 31:210150
28 Volmer T et al. Eur Respir J 2018;52: 1800703
29 Fachinformation Fasenra®; Stand: Oktober 2024
30 Menzies-Gow A et al. Lancet Respir Med 2022;10:47-58
31 Jackson DJ et al. Lancet 2024;403:271281
32 Korn S et al. J Allergy Clin Immunol Pract 2021;9:4381-4392.e4
33 Wechsler ME et al. N Engl J Med 2024; 390:911-921
Anschrift der Verfasserin: Brigitte Söllner Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen brigitte.soellner@online.de
Hizentra® ist die deutschlandweit am häufigsten verschriebene subkutane Immunglobulintherapie (SCIg) und hat sich seit über einem Jahrzehnt als bewährte Behandlungsoption bei verschiedenen primären und sekundären Immundefekten etabliert. Menschen mit diesen Erkrankungen leiden unter einer verminderten Antikörperproduktion, die häufig zu schweren und wiederkehrenden Infektionen führt [1].
Hizentra®
Durch die regelmäßige Gabe von Immunglobulinen wird der Antikörpermangel ausgeglichen und so das Infektionsrisiko bei geeigneten Patienten gesenkt [2].
Bewährte Therapie jetzt noch flexibler und komfortabler
Mit Hizentra® stehen in Deutschland nun erstmals Immunglobulin-Fertigspritzen zur Substitu-
Hizentra® wird aus dem Plasma menschlicher Spender hergestellt. 1 ml enthält 200 mg normales Immunglobulin vom Menschen (mindestens 98 % sind Immunglobuline Typ G) [3].
Es ist zugelassen zur
• Substitutionstherapie bei Erwachsenen, Kindern und Jugendlichen (0 – 18 Jahre) mit:
– primären Immundefektsyndromen (primary immunodeficiency syndromes, PID) mit geminderter Antikörperproduktion
– sekundären Immundefekten (secondary immunodeficiencies, SID) bei Patienten mit schweren oder wiederkehrenden Infektionen, ineffektiver antimikrobieller Behandlung und entweder nachgewiesenem ungenügendem Anstieg von Impfantikörpern (proven specific antibody failure, PSAF*) oder einem IgG-Serumspiegel <4 g/l
• Immunmodulationstherapie bei Erwachsenen, Kindern und Jugendlichen (0 – 18 Jahre) mit chronischer inflammatorischer demyelinisierender Polyneuropathie (CIDP) als Erhaltungstherapie nach Stabilisierung mit einer intravenösen Immunglobulin-Ersatztherapie (IVIg) [3].
* PSAF = Ausbleiben eines mindestens 2-fachen Anstiegs der IgG-Antikörperkonzentration auf Pneumokokken-Polysaccharid- und Polypeptid-Antigen-Impfstoff.
tionstherapie bei Erwachsenen und Kindern mit primärer oder sekundärer Immundefizienz zur Verfügung. Diese Innovation vereint die bewährte Wirksamkeit von Hizentra® mit einer neuen Darreichungsform. Die Fertigspritzen sind in den Größen 2 g/10 ml und 4 g/20 ml erhältlich [3].
Die Einführung der Hizentra®Fertigspritze bringt bedeutende Vorteile für die Patienten: Sie ist sofort einsatzbereit, da das Aufziehen der Immunglobulinlösung aus Durchstechflaschen entfällt. Dank ihrer einfachen Handhabung ermöglicht sie es den Patienten, die Therapie optimal in ihren Alltag zu integrieren [4].
Nach Einleitung der Therapie und der Schulung durch qualifiziertes medizinisches Fachpersonal können die Patienten die Fertigspritze sowohl mit einer Infusionspumpe als auch mittels „Manual Push“ – also per manueller Infusion –eigenständig anwenden. Die Immunglobuline können subkutan in die Unterhaut, etwa am Bauch, Oberschenkel oder Oberarm, injiziert werden [3]. Durch die anwenderfreundlicheTherapie mit den bruchsicheren Fertigspritzen lassen sich bei adäquater Dosierung von Hizentra® unphysiologisch niedrige IgG-Spiegel in den Normbereich anheben, stabile IgG-Spiegel aufrechterhalten und dadurch Infektionen bekämpfen.
Langjährige Erfahrung und fundierte Studiendaten
Hizentra® ist seit 2011 zur Substitutionstherapie von verschiedenen primären und sekundären Immundefekten bei Erwachsenen und Kindern im Alter von 0 – 18 Jahren zugelassen. Die Wirksamkeit und Sicherheit der Therapie sind durch fundierte Studiendaten und langjährige klinische Erfahrung belegt [5–8].
Brigitte Söllner, Erlangen
DLiteratur
1 McCusker C et al. Allergy Asthma Clin Immunol 2018;14(Suppl 2):61
2 Arbeitsgemeinschaft Pädiatrische Immunologie (API) e. V., S3-Leitlinie „Therapie primärer Antikörpermangelerkrankungen”, AWMF-Registriernummer: 189-001, Stand 04/2019. https://register.awmf.org/ de/leitlinien/detail/189-001
3 Fachinformation Hizentra®; Stand: 08/ 2024
4 Harris Survey Selected Results, data on file at CSL Behring
5 Jolles S et al. Clin Immunol 2011;141:90102
6 Jolles S et al. Clin Immunol. 2014; 150:161-169
7 Kanegane H et al. J Clin Immunol 2014;3 4:204-211
8 Hagan JB et al. J Clin Immunol 2010; 30:734-745
ie Zystische Fibrose (CF, Mukoviszidose) wird durch Mutationen im Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)-Gen verursacht, die zu einem defekten oder fehlenden CFTR-Protein führen. Dieses reguliert den Fluss von Chlorid-Ionen und infolgedessen auch von Wasser in und aus den Epithelzellen. Bei gestörter oder fehlender CFTR-Funktion werden Körpersekrete dickflüssig und zäh, sodass lebenswichtige Organe in ihrer Funktion beeinträchtigt werden. So kann in den Atemwegen die Ansammlung eines dickflüssigen, zähen Schleims chronische Lungeninfektionen und eine fortschreitende Schädigung der Lunge verursachen, die schließlich zum Tod führt [1]. Das mittlere Sterbealter an Mukoviszidose erkrankter Menschen liegt in Deutschland derzeit bei 39 Jahren [2].
Mit CFTR-Modulatoren wie Kaftrio®, einer Dreifachkombination aus Ivacaftor, Tezacaftor und Elexacaftor(IVA/TEZ/ELX), steht seit einigen Jahren ein kausaler Therapieansatz zur Verfügung. CF-Patienten profitieren davon z.B. durch eine Verbesserung der Lungenfunktion sowie die Verringerung bzw. sogar das Ausbleiben von pulmonalen Exazerbationen.
Letztendlich verbessert sich die CFTR-Funktion – mit weiterem Therapiepotenzial hin zur Normalisierung.
Schweißtest als Maß für die Funktionsstörung des CFTR-Proteins
Eine direkte Messung der CFTRFunktion ist über die nasale Potenzialdifferenzmessung (NPD) [3] oder die intestinale Strommessung (ICM) [4] möglich. Beide Biomarker erlauben eine sensitive Messung der CFTR-Funktion, anhand derer im Behandlungsverlauf auch die Wiederherstellung der normalen Aktivität durch CFTR-Modulatoren quantifiziert werden kann [5].
Als Goldstandard für die Diagnose der CF ist seit Jahrzehnten jedoch der Schweißtest etabliert. Er kann bei Patienten (fast) jeden Alters durchgeführt werden [6] und wird in großen klinischen Studien als Biomarker für die CFTR-Funktion eingesetzt [7].
Die diagnostische Schwelle für CF ist ein Schweißchloridwert (SwCl) von ≥60 mmol/l [8], wobei Werte zwischen 30 und 59 mmol/l anzeigen, dass eine CF vorliegen könnte und weitere Tests zur Be-
stätigung der Diagnose erforderlich sind [8]. Ein SwCl-Wert von <30 mmol/l wird bei gesunden Menschen gemessen, aber auch bei Menschen, die eine Kopie einer CFTR-Genmutation tragen, aber keine CF-typischen Krankheitsmanifestationen zeigen [9]. Höhere SwCl-Werte werden auf Populationsebene dagegen mit einer schwereren Erkrankung in Verbindung gebracht, wohingegen ein Abfall des SwCl-Wertes und damit eine Verbesserung der CFTRFunktion mit besseren klinischen Ergebnissen korreliert, wie z.B. einer besseren und stabileren Lungenfunktion, weniger pulmonalen Exazerbationen und einem längeren Überleben [10]. Da SwClWerte unter 30 mmol/l als normal gelten und typisch für CF-Träger sind, die nicht erkrankt sind, wird als zentrales Ziel der Behandlung mit Kaftrio® die Senkung des SwCl-Wertes auf unter 30 mmol/l angestrebt.
Abnahme des SwCl-Wertes unter der Therapie mit IVA/TEZ/ELX
Der Schweißtest ist als Biomarker in klinischen Studien etabliert und wurde auch in den Studien mit Ivacaftor/Tezacaftor/Elexacaftor angewendet. Unter der IVA/TEZ/ ELX-Therapie kam es zu einer Abnahme des SwCl-Wertes und damit zu einer Verbesserung der CFTRFunktion, wobei die Ergebnisse der SwCl-Messung anhand wei-
terer Biomarker bestätigt wurden [11, 12]. IVA/TEZ/ELX erhöhte die CFTR-Funktion im Nasen- und Darmepithel von Patienten mit mindestens einer F508del-Mutation auf 40 – 50 % der normalen CFTR-Aktivität [11, 13]. Wirksame CFTR-Modulatoren können demnach zur Wiederherstellung der CFTR-Funktion in den Epithelien beitragen.
CFTR-Funktion korreliert mit klinischem Verlauf
Was bedeutet es für die Betroffenen, wenn sich die CFTR-Funktion verbessert? Auf Populationsebene ist der SwCl-Wert prädiktiv für die CF-Prognose, wie Registerdaten aus den USA zeigen [14]. Im Zeitverlauf kommt es mit Zunahme der Effizienz einzelner CFTR-Modulatoren zu einer Verbesserung der CFTR-Funktion, gemessen mittels SwCl, bis hin zu einer Normalisierung bei einzelnen Betroffenen. In einer klinischen Phase-III-Studie mit Patienten, die 2 F508del-Mutationen aufwiesen, erreichten 79 % unter der Behandlung mit IVA/ TEZ/ELX einen SwCl-Wert von <60 mmol/l und damit den diagnostischen Schwellenwert der CF. 23 % erreichten sogar SwCl-Werte von <30 mmol/l, d.h. den Bereich, der für Menschen ohne CF typisch ist, einschließlich gesunden Genträgern [15]. Die Verbesserung spiegelt sich auch in den klinischen Endpunkten wider: Je höher die
CFTR-Funktion, gemessen mittels niedrigerer SwCl-Werte, war, desto deutlicher fiel die Verbesserung bei der Lungenfunktion, beim Anstieg des BMI und der Verringerung der jährlichen Rate pulmonaler Exazerbationen aus [16].
Elisabeth Wilhelmi, München
Literatur
1 Elborn JS. Lancet 2016;388:2519-2531
2 Mukoviszidose e.V. Deutsches Mukoviszidose-Register, Berichtsband 2023
3 Cystic Fibrosis Foundation. Sweat test. www.cff.org/intro-cf/sweattest
4 Sontag MK. Am J Respir Crit Care Med 2016;194:1311-1313
5 Ratjen F et al. Nat Rev Dis Primers 2015; 1:15010
6 Cholon DM et al. J Cyst Fibros 2018; 17:S52-S60
7 Gräber SY et al. Am J Respir Crit Care Med 2018;197:1433-1442
8 S2k-Leitlinie Diagnose der Mukoviszidose, AWMF Registernummer 026-023
9 Sermet-Gaudelus E et al. J Cystic Fibros 2022;21:922-936
10 McKone EF et al. J Cyst Fibros 2015; 14:580-586
11 Middleton PG et al. N Engl J Med 2019; 381:1809-1819
12 Gräber SY et al. Am J Respir Crit Care Med 2022;206:311-320
13 Gräber SY et al. Am J Respir Crit Care Med 2022;205:540-549
14 McKone EF et al. J Cyst Fibros 2015; 14:580-586
15 Sutharsan S et al. Lancet Respir Med 2022;10:267-277
16 Mayer-Hamblett N et al. Greater reductions in Sweat Chloride with CFTR modulator use are associated with improved clinical outcomes. Poster 694 presented at NACFC 2022
Laut Robert Koch Institut wurden im Jahr 2024 insgesamt 686 FSME-Erkrankungen gemeldet, was einer Zunahme von 44 % gegenüber dem Vorjahreswert von 478 FSME-Erkrankungen entspricht. Damit wurde nach dem bislang fallstärksten Jahr 2020 mit 718 FSME-Erkrankungen ein zweiter Höchstwert seit Beginn der Datenerfassung erreicht. Ein Grund für die steigenden Fallzahlen kann die mittlerweile ganzjährige Aktivität von Zecken sein, die Hauptüberträger der Krankheit sind (Abb. 1). Bei 59 % der 2024 übermittelten Fälle wurde ein klinisches Bild mit Manifestationen einer Meningitis, Enzephalitis oder Myelitis angegeben – ein höherer Anteil als im Vorjahr 2023 mit 53 % [1]. Die Impfung – auch für Kinder – ist daher wichtiger denn je.
STIKO empfiehlt
FSME-Impfung ab 12 Monaten
Da es für die FSME keine kausale Therapie gibt, empfiehlt die Ständige Impfkommission (STIKO) die FSME-Impfung als zuverlässigsten Schutz vor einer Erkrankung für Erwachsene und Kinder ab 12 Monaten, die in Risikogebieten leben, arbeiten oder hinreisen und dabei ein Risiko für Zeckenstiche haben [2].
Für die Impfung können die FSME-Impfstoffe Encepur® für
Kinder von 1 – 11 Jahren und Encepur® für Erwachsene und Jugendliche ab 12 Jahren eingesetzt werden. Encepur® zeichnet sich durch eine lange Haltbarkeit von 36 Monaten aus und ist in plastikfreien Großpackungen z.B. mit 20
Fertigspritzen für eine nachhaltige und wirtschaftliche Verordnung verfügbar. Der Impfstoff bietet einen wirksamen Schutz und ist gut verträglich. Das belegen mehr als 30 Jahre Erfahrung in der Anwendung [3, 4].
Differenzialdiagnostisch immer auch an FSME denken
Weil auch in Nicht-Risikogebieten Fälle von FSME auftreten und die unspezifischen Symptome in der ersten Krankheitsphase eine eindeutige Diagnose erschweren, sollten Ärzte überall in Deutschland differenzialdiagnostisch an FSME denken. Nach dem ersten Erkrankungsgipfel mit grippeähnlichen Beschwerden kann im weiteren Krankheitsverlauf hohes Fieber (über 40°C) auftreten. Bei 5 – 30 % der Erkrankten kommt es zu Entzündungen der Hirnhäute, des Gehirns und des Rückenmarks. Spätfolgen einer FSME-Infektion können langanhaltende Kopfschmerzen, Gedächtnis- und Konzentrationsschwäche sowie Lähmungen sein. Bei schweren Verläufen besteht insbesondere bei Erwachsenen, aber auch bei Kindern die Gefahr von persistierenden und folgenschweren neurologischen Ausfällen. In seltenen Fällen verläuft die FSME sogar tödlich [2].

Abbildung 1: FSME und verwandte Virusenzephalitiden werden durch das TickBorne Encephalitis-(TBE-)Virus verursacht. Dieses wird meist durch Zecken auf den Menschen übertragen, in Deutschland überwiegend durch die Spezies Ixodes ricinus (Quelle: iStock Ladislav Kubeš).
Encepur®-Schnellschema
ermöglicht den Abschluss der Grundimmunisierung in 21 Tagen
Die Encepur®-Impfung gemäß konventionellem Impfschema ist für Menschen geeignet, die einem kontinuierlichen Infektionsrisiko ausgesetzt sind. Die Grundimmunisierung mit 3 Impfdosen innerhalb weniger Monate sollte bevorzugt in der kälteren Jahreszeit erfolgen, um einen Impfschutz in der Hauptrisikozeit (Frühjahr/ Sommer) zu gewährleisten. Eine erste Auffrischung ist bei der konventionellen Impfung nach 3 Jahren zu empfehlen.
Wenn kein FSME-Impfschutz besteht, eine Reise in FSMERisikogebiete aber kurzfristig ansteht, kann auch das Encepur®Schnellschema verwendet werden. Dieses ermöglicht eine vollständige Grundimmunisierung bereits innerhalb von 21 Tagen nach der 1. Impfung. Eine erste Auffrischimpfung erfolgt in diesem Fall nach 12 – 18 Monaten.
Je nach Impfschema werden 3 Wochen nach der 3. Impfung folgende Serokonversionsraten erzielt: Konventionelles Schema 100 % (Erwachsene und Kinder), beschleunigtes konventionelles Schema 100 % (Erwachsene und Kinder), Schnellschema 97 % (Erwachsene) und 99 % (Kinder) [3, 4]. Daten bei Erwachsenen und Jugendlichen über 12 Jahren zeigen, dass unabhängig vom Schema der Grundimmunisierung 10 Jahre nach der Auffrischimpfung über 97 % der Geimpften noch FSMEAntikörpertiter im Neutralisationstest ≥10 haben [3].
Mehr Aufklärung in der Arztpraxis tut not
98 % der 2024 an das RKI übermittelten FSME-Erkrankten waren gar nicht oder unzureichend geimpft [1]. Die Steigerung der Impfquoten könnte einen erheblichen Teil der Erkrankungen verhindern. Um die Impfbereitschaft zu erhöhen und mögliche Impfbarrieren, z. B. ein geringes Risikobewusstsein und die Angst vor Impfnebenwirkungen zu reduzieren, ist eine Aufklärung in den ärztlichen Praxen wesentlich. Für die Information von Patienten zu FSME und die Impfberatung stellt Bavarian Nordic kostenfreies Informationsmaterial zur Verfügung. Ärzte können dieses per E-Mail anfordern unter service@bavarian-nordic.com Brigitte Söllner, Erlangen
Abirasolon® – ein praktischer Kombinationsblister für die Therapie des mCRPC
Literatur
1 RKI. Epidemiologisches Bulletin 9/2025. FSME-Risikogebiete in Deutschland, Zur FSME-Situation in Deutschland im Jahr 2024. www.rki.de/DE/Aktuelles/Publikationen/Epidemiologisches-Bulletin/2025/09_25.pdf?__blob=publication File&v=4
2 https://www.rki.de/DE/Themen/Infektionskrankheiten/Impfen/Informationsmaterialien/Faktenblaetter-zum-Impfen/ FSME.html?nn=16907268
3 Fachinformation Encepur® Erwachsene. Stand: Mai 2023
4 Fachinformation Encepur® Kinder. Stand: Mai 2023
Zur Behandlung des metastasierenden kastrationsresistenten Prostatakarzinoms (mCRPC) ist die Wirkstoffkombination Abirateron/ Prednisolon eine Therapieoption, die sowohl das Gesamtüberleben als auch das progressionsfreie Überleben der Betroffenen verlängern kann. Bei 11 % der mCRPC-Patienten liegen BRCA1/2Mutationen vor, die mit einem schlechteren klinischen Outcome assoziiert sind. Falls klinisch eine Chemotherapie nicht indiziert ist, profitieren diese Patienten besonders von einer Kombinationstherapie aus Abirateron/Prednisolon und einem PARP-Inhibitor. Nach der aktuellen S3-Leitlinie kann Abirateron/Prednisolon beim mCRPC aber auch ohne Nachweis einer BRCA1/2-Mutation mit dem PARP-Inhibitor Olaparib kombiniert werden. Für diese Therapie bietet Abirasolon® den Vorteil, in einem übersichtlichen Blister sowohl die Abirateron-Filmtabletten 500 mg als auch die notwendigen Prednisolon-Tabletten 5 mg zu enthalten. Diese Verpackung ermöglicht eine einfachere und damit zuverlässigere Medikamenteneinnahme und unterstützt so die Adhärenz. Insbesondere ältere Patienten profitieren von möglichst einfach gestalteten Blistern. Neben der Kombipackung für einen Monat (4 Blister; 1 Blister pro Woche) ist das Präparat auch als 2-MonatsPackung (8 Blister; 1 Blister pro Woche) erhältlich. Damit muss nur noch alle 2 Monate ein Rezept ausgestellt werden.
S. M.
S3Leitlinie Chronische Lymphatische Leukämie umfassend aktualisiert
Das Leitlinienprogramm Onkologie hat die S3-Leitlinie zur Chronischen Lymphatischen Leukämie (CLL) umfassend überarbeitet. Einige Kapitel wurden umstrukturiert. Zudem wurden unter anderem die Inhalte zur Therapie aktualisiert und ein Kapitel zur Behandlung der Richter-Transformation neu aufgenommen. Die S3Leitlinie entstand unter Federführung der Deutschen Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) sowie unter Mitwirkung von 31 weiteren Fachgesellschaften und Organisationen. Finanziert wurde die Leitlinie von der Deutschen Krebshilfe im Rahmen des Leitlinienprogramms Onkologie.
Evidenzbasierte
Behandlungsmöglichkeiten
Die CLL ist die häufigste Form einer bösartigen Neubildung des lymphatischen Systems und macht etwa 36 % aller Leukämien aus. Nach Zahlen des Robert Koch-Instituts erkrankten in Deutschland im Jahr 2022 etwa 5.200 Menschen neu an einer CLL.
Das klinische Erscheinungsbild und die Prognose können stark variieren. Einige Patienten haben jahrelang keine behandlungsbedürftigen Symptome und eine normale Lebenserwartung. Bei anderen treten schnell Krankheitsanzeichen wie infektiöse Komplikationen auf und sie sterben trotz der Behandlung innerhalb weniger Jahre. Diese Patienten sollten frühzeitig
durch verbesserte diagnostische Verfahren identifiziert werden, um die Therapie entsprechend anzupassen und damit die Heilungsrate und das Gesamtüberleben zu verbessern. Für Betroffene mit einer günstigeren Prognose sollen dagegen die Akut- und Langzeittoxizitäten der Behandlung sowie das Auftreten von Sekundärtumoren minimiert werden. Dafür zeigt die S3-Leitlinie evidenzbasierte Behandlungsmöglichkeiten und Entscheidungskriterien auf.
Risikofaktoren zur Bestimmung der Therapieverfahren
Vor dem Start einer neuen Therapielinie sollen der TP53-Deletions- und Mutationsstatus und zusätzlich der IGHV-Mutationsstatus erhoben werden. In der Vergangenheit wurde lediglich die Bestimmung des TP53-Mutationsstatus empfohlen. „Die Ergebnisse beider Untersuchungen liefern uns wichtige Informationen zum Risiko der Patienten und damit zur Therapieplanung“, so Professorin Barbara Eichhorst, Mit-Koordinatorin der Leitlinie. „Bei entsprechendem Risikoprofil können wir mit einer intensiveren Überwachung und möglicherweise aggressiveren Therapien reagieren. Damit bieten wir Erkrankten mit einem hohen Risiko einer Progression bessere Überlebenschancen und können gleichzeitig Personen mit einer besseren Prognose mit schonenderen Therapien behandeln.“
Zielgerichtete Substanzen statt Chemo(immun)therapie
Eine Chemotherapie – oder die Chemoimmuntherapie, bei der Zytostatika in Kombination mit Anti-
körpern verabreicht werden – wirkt gegen die Krebszellen, schädigt als Nebenwirkung aber auch gesundes Gewebe. Seit dem Erscheinen der ersten Fassung der CLL-Leitlinie (2018) hat die Verfügbarkeit von Signalweginhibitoren – in diesem Fall BTK- oder Bcl-2-Inhibitoren – neue Therapieabläufe bei der CLL ermöglicht. Eichhorst: „Wir empfehlen in der Überarbeitung der Leitlinie für die Erstlinien- und auch die Rezidivtherapie nur noch chemotherapiefreie, zielgerichtete Therapien. Studien zeigen, dass diese den Chemoimmuntherapien bei der Behandlung der CLL überlegen sind. Selbst die Prognose der Hochrisiko-Patienten hat sich deutlich verbessert. Das ist ein großer Erfolg.“ Eine Chemoimmuntherapie ist nach der Leitlinie nun nur noch in Ausnahmefällen eine Option.
Neues Kapitel: Therapie der Richter-Transformation
Komplett neu aufgenommen wurden Behandlungsempfehlungen zur Richter-Transformation (RT). Dies ist die Entwicklung der CLL zu einem aggressiven Lymphom – meist einem diffus großzelligen B-Zell Lymphom (DLBCL). Oftmals zeigen die Patienten dabei eine schnelle klinische Verschlechterung. Das Kapitel der Leitlinie zur RT umfasst Empfehlungen zu Risikofaktoren, Diagnostik und Therapie der RT.
DGHO
Die aktualisierte S3-Leitlinie ist abrufbar unter: www.leitlinienprogramm-onkologie.de/leitlinien/chronische-lymphatischeleukaemie-cll
Schubförmige Multiple Sklerose:
5JahresDaten bestätigen hohe Wirksamkeit von Ublituximab
Ublituximab (Briumvi®) ist der erste glykoengineerte Anti-CD20Antikörper zur Behandlung erwachsener Patienten mit aktiver schubförmiger Multipler Sklerose (RMS) und seit Februar 2024 in Deutschland verfügbar. Auf dem 97. Kongress der Deutschen Gesellschaft für Neurologie (DGN) wurden neue 5-Jahres-Daten der Open-Label-Extensionsphase der Zulassungsstudien ULTIMATE-I und ULTIMATE-II vorgestellt. Sie zeigen, dass im 5. Behandlungsjahr eine niedrige jährliche Schubrate von 0,021 erreicht werden kann. Dies entspricht durchschnittlich einem Schub alle 50 Patientenjahre bei RMS.
Auch nach 5 Jahren waren 97 % der Patienten noch progressionsfrei
In den beiden doppelt-verblindeten Zulassungsstudien, in denen Ublituximab versus Teriflunomid untersucht wurde, konnte unter der Therapie mit Ublituximab die CD19+-B-Zellzahl bereits nach der ersten Ublituximab-Infusion um 97 % gesenkt werden, wobei die Depletion bis zum Studienende anhielt.
An die zweijährige Studienphase der ULTIMATE-Studien wurde eine dreijährige Open-LabelExtension (OLE) angeschlossen, in der die Studienteilnehmer die Behandlung mit Ublituximab fortsetzen (UBL-UBL) oder von Teriflunomid zu Ublituximab wechseln (TER-UBL) konnten. Die Patienten in der UBL-UBL-
Gruppe wiesen über 5 Jahre eine stetig sinkende jährliche Schubrate mit einem Wert in Jahr 5 von 0,021 vs. 0,045 in der TER-UBLGruppe auf (p = 0,0727). Lediglich 8 % der kontinuierlich mit Ublituximab Behandelten zeigten dabei eine bestätigte Behinderungsprogression, die 24 Wochen andauerte, verglichen mit 14,3 % in der TER-UBL-Gruppe (p = 0,0032). Bis zum fünften Jahr blieben 92 % der Behandelten in der UBL-UBLGruppe progressionsfrei. Auch im Hinblick auf eine Behinderungsverbesserung, die 24 Wochen anhielt, schnitt die UBL-UBL-Gruppe mit 17 % besser ab im Vergleich zu 12,2 % in der TER-UBL-Gruppe (p = 0,0249).
In der OLE wurden keine neuen Sicherheitssignale beobachtet. Die Immunglobulinspiegel blieben stabil, die mittleren IgM- und IgGSpiegel über der unteren Normgrenze.
Weniger Fatigue und mehr Lebensqualität
In den ULTIMATE-Zulassungsstudien verbesserten sich unter der Ublituximab-Behandlung auch die Fatigue und Lebensqualität signifikant gegenüber Teriflunomid. Als weitere Vorteile des Anti-CD20-Antikörpers erwiesen sich das lange Applikationsintervall und die kurze Infusionsdauer. Diese liegt ab der zweiten Infusion bei etwa einer Stunde, ohne die Notwendigkeit einer systematischen Nachbeobachtung nach der dritten Infusion, wenn bei vorangehenden Infusionen keine Infusionsreaktionen und/oder Überempfindlichkeit beobachtet wurden. Damit ist Ublituximab auch im ambulanten Setting wirtschaftlich einsetzbar.
Glycoengineering in der RMS-Therapie
Ublituximab ist der erste AntiCD20-Antikörper in der MSTherapie, der durch Glycoengineering gezielt optimiert wurde. Bei diesem Verfahren werden störende Zuckerreste vom Antikörper entfernt, die die Effektorfunktionen des Antikörpers beeinträchtigen können. Aus immunologischer Sicht ist das Glycoengineering ein bedeutsamer Fortschritt, da es zu einer verstärkten antikörperabhängigen zellulären Zytotoxizität (ADCC) führt und eine präzise Interaktion mit den Fc-Rezeptoren ermöglicht. Dies kann die Effektivität und gezielte Wirkung dieses Anti-CD20-Antikörpers verbessern.
Leitlinien stützen frühen Einsatz hochwirksamer Arzneimittel wie Ublituximab
Die neuen Daten zeigen, dass die jährliche Schubrate niedriger war, je früher Ublituximab initiiert wurde, und unterstützen damit den Ansatz „hit hard and early“, dem sich inzwischen auch die aktuellen Leitlinien stärker geöffnet haben. Ublituximab wird darin der höchsten Wirksamkeitskategorie zugeordnet. Daher sollte bereits zu Therapiebeginn immer sorgfältig evaluiert werden, ob ein hochwirksames Arzneimittel wie Ublituximab eingesetzt werden kann.
B. S.
Quelle: Cree B et al. Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); Poster #P324.
Vitiligo ist eine chronische Autoimmunerkrankung der Haut, die mit einem Pigmentverlust einhergeht und durch weiße Flecken der Haut sichtbar wird. Bei der Erkrankung schädigen körpereigene Immunzellen die Pigmentzellen der Haut. Vitiligo zählt zu den häufigsten Pigmentierungsstörungen mit einer geschätzten Prävalenz zwischen 0,5 und 2 %. Das entspricht bis zu 150 Millionen Betroffenen weltweit. Sie tritt in verschiedenen Ausprägungen auf, mit 85 % ist die nichtsegmentale Vitiligo (NSV) die häufigste Form.
Das Therapieziel bei Vitiligo ist das Erreichen und der Erhalt einer gleichmäßigen Repigmentierung. Zur Behandlung steht mit Opzelura® eine innovative Cremeformulierung des selektiven JAK1/ JAK2-Inhibitors Ruxolitinib zur Verfügung, der erste und einzige in der EU zugelassene topische JAKInhibitor, der für die Behandlung der nichtsegmentaler Vitiligo mit Beteiligung des Gesichts bei Erwachsenen und Jugendlichen ab 12 Jahren indiziert ist [1]. Auf dem Kongress der European Academy of Dermatology and Venereology (EADV) vorgestellte neue Studiendaten zeigen, wie sich das Erwartungsmanagement und – damit verbunden – die Therapieadhärenz bei der Behandlung mit Opzelura® verbessern lassen.
Fortführung der Behandlung lohnt sich
Im Rahmen der zulassungsrelevanten Phase-III-Studien TRuE-V1/ V2 und ihrer Verlängerungsphase TRuE-V-LTE wurde die faziale Repigmentierung bei Patienten mit nichtsegmentaler Vitiligo in Behandlungsphasen mit Ruxoliti-
nib-Creme 1,5 %, 2 täglich im Vergleich zu einem wirkstofffreien Vehikel über einen Beobachtungszeitraum von insgesamt 104 Wochen erfasst [2–5]. Nach 52 Wochen war bei 30,3 % der Patienten eine vollständige oder fast vollständige (≥90 %) Verbesserung der Pigmentierung im Gesicht erreicht, entsprechend einem Facial Vitiligo Area Scoring Index von 90 (F-VASI90) [2]. Diese Patienten wurden erneut randomisiert einer Behandlungsgruppe (Ruxolitinib-Creme 1,5 %, 2tägl.) oder Vehikelgruppe zugeordnet [3]. In der Behandlungsgruppe konnten 61,8 % der Patienten ihr F-VASI90-Ansprechen beibehalten, in der Vehikelgruppe waren es dagegen nur 21,4 %. Dies zeigt, dass eine fortgesetzte Therapie mit Opzelura® Rückfällen effektiv vorbeugen kann [3].
Aber auch die Gesamtdauer der Anwendung von RuxolitinibCreme hat einen Einfluss auf die Stabilität des Ansprechens [6]. In der Verlängerungsstudie TRuE-VLTE wurden insgesamt 57 Patienten mit F-VASI90 der Vehikelgruppe zugeordnet: 45 Patienten waren von Tag 1 der TRuE-V-Studie in der Behandlungsgruppe und hatten Ruxolitinib-Creme insgesamt 52 Wochen angewendet. Von diesen 45 Patienten hatten 25 vor/in Woche 24 ein F-VASI90-Ansprechen gezeigt, 20 erreichten diesen Grad der Repigmentierung vor/in
Woche 52. 12 Patienten waren zu Studienbeginn in der Vehikelgruppe und hatten Ruxolitinib-Creme nur in der unverblindeten Verlängerungsphase für 28 Wochen angewandt. Patienten mit langer Anwendungszeit und frühem Ansprechen konnten ihr F-VASI90Ansprechen im Median 12 Monate aufrechterhalten, bei den Patienten mit der kürzeren Behandlungsdauer lag der Median bei 3 Monaten. Außerdem zeigten Patienten mit einem schnellen Ansprechen einen besseren Erhalt der Pigmentierung im Vergleich zu Patienten, die später ein F-VASI90-Ansprechen hatten [6].
Einflussfaktoren auf die Repigmentierung unter Behandlung mit Opzelura®
Zusätzlich zur Anwendungsdauer wurden weitere demographische und krankheitsbezogene Faktoren auf einen möglichen Zusammenhang mit der Wahrscheinlichkeit für einen Rückfall untersucht [7]. Von den 57 Patienten, die in Woche 52 ein F-VASI90-Ansprechen hatten und der Vehikelgruppe zugeordnet worden waren, hatten in Woche 104 12 Patienten ihr F-VASI90und 20 Patienten ein F-VASI75Ansprechen aufrechterhalten, umgekehrt zeigten 31 Patienten einen Rückgang der Pigmentierung (< F-VASI90) und 14 Patienten
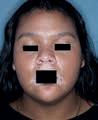
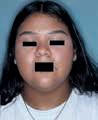
Baseline Woche 24 Woche 52
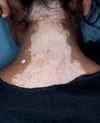
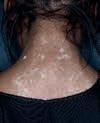
Baseline Woche 24 Woche 52
Abbildung 1: Eine längere Behandlungszeit ging in den TruE-V-Zulassungsstudien mit einer allmählichen und kontinuierlich zunehmenden Repigmentierung einher (© Incyte).
Ruxolitinib-Creme 15 mg/g
Ruxolitinib-Creme (Opzelura®), eine innovative Cremeformulierung des selektiven JAK1/JAK2-Inhibitors Ruxolitinib, ist der erste und einzige in der EU zugelassene topische JAK-Inhibitor, der für die Behandlung von nichtsegmentaler Vitiligo mit Beteiligung des Gesichts bei Erwachsenen und Jugendlichen ab 12 Jahren indiziert ist [1]. Opzelura® wird zweimal täglich in einer dünnen Schicht auf die depigmentierten Hautareale aufgetragen, maximal dürfen 10 % der Körperoberfläche gleichzeitig behandelt werden. Eine zufriedenstellende Repigmentierung kann eine Behandlung mit Opzelura® über mehr als 24 Wochen erfordern. Eine Überwachung der Laborwerte ist unter Therapie mit Opzelura® nicht erforderlich [1].
hatten einen Rückfall (definiert als < F-VASI75). Eine post hoc durchgeführte Stratifizierung der Patienten in Subgruppen deutet darauf hin, dass einige demographische Faktoren, wie Alter oder Geschlecht, und Charakteristika der individuellen Krankengeschichte, darunter das Ausmaß der Vitiligoläsionen
im Gesicht (F-BSA), die Erkrankungsdauer oder vorangegangene Therapieversuche, Einfluss auf den langfristigen Erhalt der Repigmentierung nach Absetzen der Behandlung haben könnten [7].
Eine weitere Analyse verglich die Ansprechrate von Patienten, die von Beginn der TRuE-V1/V2-
Studien an Ruxolitinib-Creme 1,5 %, 2 täglich, angewandt hatten (RUX-RUX-Gruppe), mit der von Patienten, die nach initialer 24-wöchiger Vehikelbehandlung in der unverblindeten Verlängerungsphase auf Ruxolitinib-Creme 1,5 %, 2 täglich, umgestellt wurden (VEH-RUX-Gruppe) [8]. Alle Patienten zeigten eine kontinuierliche Verbesserung der Pigmentierung im Gesicht und am Körper durch Ruxolitinib-Creme. Jedoch war das Ansprechen von Patienten der VEH-RUX-Gruppe in Woche 104 geringer als bei Patienten der RUX-RUX-Gruppe nach 80 Wochen, was der gleichen Behandlungsdauer mit dem Prüfpräparat entspricht.
Während des gesamten Beobachtungszeitraums von 2 Jahren wurde Ruxolitinib-Creme generell gut vertragen. Die häufigste behandlungsbedingte Nebenwirkung war Akne an der Applikationsstelle bei 5 % der Patienten [8].
Elisabeth Wilhelmi, München
Literatur
1 Fachinformation Opzelura®, aktueller Stand
2 Rosmarin D et al. N Engl J Med 2022; 387:1445-1455
3 Harris JE. Presented at: AAD 2023, Abstract #46159
4 Rosmarin D. Presented at: AAD 2023, Abstract #46163
5 Wolkersdorfer A. Presented at: EADV 2023, #Abstract 6479
6 Rosmarin D. Presented at: EADV 2024, Presentation #D2T01.3E
7 Rosmarin D et al. Presented at: EADV 2024, Poster P2984
8 López Estebaranz JL et al. Presented at EADV 2024 Poster P2983
In Deutschland leben schätzungsweise 6,8 Millionen Menschen mit chronisch obstruktiver Lungenerkrankung (COPD), von denen nur ca. 50 % diagnostiziert sind [1, 2]. Zur Unterdiagnose kommt eine Unterversorgung hinzu: Fast die Hälfte der Patienten erhält 12 Monate nach der Diagnose noch keine inhalative Therapie, nach 3 Jahren ist es immer noch etwa ein Drittel [3]. Im letzten Jahr vor ihrem Tod wird mehr als die Hälfte der Patienten nicht leitliniengerecht behandelt – daher stirbt auch heute noch alle 15 Minuten ein Mensch in Deutschland an COPD [2]. Die Ursache sind nicht fehlende Medikamente, sondern deren mangelnder Einsatz. Eine Senkung der Mortalität wäre also durch die konsequente Umsetzung der Therapieempfehlungen erreichbar. Wie der neu erschienene GOLD Report 2025 betont, sollte dabei kardiovaskulären Komorbiditäten mehr Aufmerksamkeit geschenkt werden [4]. Denn kardiovaskuläre Komorbiditäten und COPD-Exazerbationen stehen in einer Wechselbeziehung, die das Risiko für schwere kardiovaskuläre Ereignisse mit Todesfolge erhöht [5].
Exazerbationen durch TherapieEskalation vermeiden
Exazerbationen gehen mit einer akuten Verschlechterung der Symptome einher, beeinträchtigen den Gesundheitszustand der Patienten nachhaltig und verschlechtern deren Prognose [4]. Eine große retrospektive Kohortenstudie aus Deutschland verdeutlicht die Gefahr: Über einen Zeitraum von im Median 36 Monaten erlitten 38,6 % der COPD-Patienten ein schweres kardiovaskuläres Ereig-
nis oder verstarben nach einer Exazerbation [6]. Mit jeder Exazerbation steigt außerdem das Risiko für weitere Exazerbationen, wie die Studie AvoidEx zeigt: Bei 82 % der COPD-Patienten mit mehr als 2 mittelschweren Exazerbationen oder einer schweren Exazerbation in der Vorgeschichte kam es innerhalb von 3 Jahren zu einer weiteren Exazerbation [7]. Exazerbationen werden jedoch oft unterschätzt, obwohl sie die Mortalität erhöhen. Sie signalisieren, dass Patienten nicht ausreichend auf ihre Therapie ansprechen und eine Eskalation von einer dualen Kombination auf eine Triple-Therapie erforderlich ist.
Mit BUD/GLY/FORM
Exazerbationsrisiko und Mortalität senken
Einen Beitrag zur Senkung der Gesamtmortalität und der kardiovaskulären Mortalität kann die Triple-Therapie Trixeo Aerosphere® mit Budesonid/Glycopyrronium/ Formoterol (BUD/GLY/FORM) leisten. In der KRONOS-Studie reduzierte BUD/GLY/FORM die Rate mittelschwerer oder schwerer Exazerbationen im Vergleich zur LAMA/LABA-Kombination GLY/FORM über einen Zeitraum von 24 Wochen signifikant um 52 % bei einer Number Needed to Treat (NNT) von 3, d.h., nur 3 Patienten müssen mit BUD/GLY/
FORM behandelt werden, um eine mittelschwere oder schwere Exazerbation zu verhindern. Das bedeutet, dass nahezu jeder Patient von der Therapie profitieren kann [8, 9]. Dass die Reduktion des Exazerbationsrisikos mit BUD/GLY/ FORM auch mit einem Überlebensvorteil einhergeht, zeigte eine Post-hoc-Analyse der ETHOSStudie: Über 52 Wochen wurde die Gesamtmortalität versus LAMA/ LABA (GLY/FORM) signifikant um 49 % reduziert (absolute Todesrate 1,4 % vs. 2,6 %; HR: 0,51; 95%-KI: 0,33 – 0,80), was hauptsächlich auf den Rückgang der Sterblichkeit durch kardiovaskuläre Ereignisse zurückzuführen war [9]. Diese Daten aus der ETHOSStudie trugen wesentlich dazu bei, dass das Senken der Mortalität seit November 2022 fest als Therapieziel in den GOLD-Reports verankert ist [3].
Exazerbationen sind Haupttreiber für kardiovaskuläre Ereignisse und umgekehrt [10]. Wie eine weitere Post-hoc-Analyse der ETHOSStudie zeigte, hatten mehr als 70 % der COPD-Patienten mindestens einen kardiovaskulären Risikofaktor und somit ein hohes pulmokardiales Risiko [11]. Dieses kann mit BUD/GLY/FORM gesenkt werden, da die Triple-Therapie den Hauptursachen von Exazerbationen entgegenwirkt [12]. Das enthaltene inhalative Kortikosteroid Budesonid wirkt antiinflammatorisch in der Lunge und kann so die
Trixeo Aerosphere® ist eine Dreifach-Fixkombination aus Budesonid (BUD, 160 µg), Glycopyrronium (GLY, 7,2 µg) und Formoterol (FORM, 5 µg) als Dosieraerosol. BUD/GLY/FORM ist als Erhaltungstherapie bei erwachsenen Patienten mit moderater bis schwerer chronisch obstruktiver Lungenerkrankung (COPD) zugelassen, die mit einer Kombination aus einem inhalativen Kortikosteroid (ICS) und einem langwirksamen Beta-2-Rezeptoragonisten (LABA) oder einer Kombination aus einem LABA und einem langwirksamen Muskarin-Rezeptorantagonisten (LAMA) nicht ausreichend eingestellt sind [12]. Dies ist z. B. der Fall, wenn die Betroffenen weiterhin an Exazerbationen leiden.
Die Triple-Therapie vereint die Wirkmechanismen des ICS Budesonid und der beiden langwirksamen Bronchodilatatoren Glycopyrronium und Formoterol:
• Budesonid wirkt in den großen und kleinen Atemwegen antiinflammatorisch.
• Formoterol entfaltet über die Stimulation von Beta-2-Rezeptoren in der glatten Muskulatur seine bronchodilatatorische Wirkung.
• Glycopyrronium wirkt bronchodilatatorisch, indem es die Muskarin-Rezeptoren in der glatten Muskulatur blockiert.
Die Fixkombination bewirkt eine Symptomkontrolle über 24 Stunden bei 2x täglicher Gabe.
Hierbei kommt die Aerosphere™ Delivery Technology zum Einsatz, die mit 37,7 % eine überdurchschnittlich hohe Deposition von Wirkstoffen in der Lunge ermöglicht. Sie nutzt als Trägerstoff poröse Mikropartikel aus Phospholipiden, die auch einen überwiegenden Teil des natürlichen Lungensurfactants darstellen. Mit einem MMAD (mass median aerodynamic diameter) von ca. 3 μm haben sie eine optimale aerodynamische Größe, um an ihren Zielort zu gelangen.
Ausbreitung der Entzündung auf andere Organe verhindern [12]. Die Bronchodilatatoren GLY/ FORM reduzieren den Atemwegswiderstand und die Lungenüberblähung, was den Herzmuskel entlasten kann [13]. In Kombination kann BUD/GLY/FORM das Ventilations-Perfusions-Verhältnis verbessern und die Hypoxämie verringern [14, 15].
Lungendeposition ist entscheidend für die Wirksamkeit
Voraussetzung, dass BUD/GLY/ FORM wesentlich zur Errei-
chung der GOLD-Therapieziele Symptomlinderung, Exazerbationsvermeidung und vor allem Senkung der Mortalität beiträgt, ist, dass die Wirkstoffe da ankommen, wo sie ihre Wirkung entfalten sollen. Diesbezüglich schneidet BUD/GLY/ FORM im Vergleich mit anderen Triple-Kombinationen besser ab: In-silico-Daten liefern Hinweise dafür, dass BUD/GLY/FORM eine deutlich bessere Lungendeposition aufweist als andere ICS/ LAMA/LABA-Kombinationen wie Beclometasondipropionat (BDP)/GLY/FORM oder Fluticasonfuroat/Umeclidiniumbromid/
Vilanterol (FF/UMEC/VI). Im Vergleich zu BDP/GLY/FORM war die Gesamtdeposition aller 3 Komponenten von BUD/GLY/ FORM in der Lunge um ca. 40 % höher. In den kleinen Atemwegen erreichte BUD/GLY/FORM eine um ca. 20 % höhere Lungendeposition als BDP/GLY/FORM. Auch gegenüber FF/UMEC/VI war BUD/GLY/FORM überlegen. Die Gesamtdeposition aller Komponenten war bei BUD/GLY/FORM in etwa doppelt so hoch wie bei FF/UMEC/VI [16].
Fabian Sandner, Nürnberg
Literatur
1 Pritzkuleit R et al. Pneumologie 2010;64: 535-540
2 Wissenschaftliches Institut der AOK (Wido). Gesundheitsatlas Deutschland, 2023
3 Buhl R et al. Int J Chron Obstruct Pulmon Dis 2022;17:2355-23567
4 Global Initiative for Chronic Obstructive Lung Disease. GOLD Report 2025
5 Solidoro P et al. Front Med 2022;9: 816843
6 Vogelmeier CF et al. Increased risk of severe cardiovascular events following exacerbations of COPD: a multi-database cohort study. ERS Congress 2023
7 Vogelmeier CF et al. Int J Chron Obstruct Pulmon Dis 2021;16:2407-2417
8 Ferguson GT et al. Lancet Respir Med 2018;6:747-758
9 Martinez FJ et al. Am J Respir Crit Care Med 2021;203:553-564
10 Stolz D et al. Lancet. 2022;400:921-972
11 Singh D et al. Effect of triple-inhaled therapy with budesonide/glycopyrrolate/ formoterol fumarate on cardiopulmonary events in chronic obstructive pulmonary disease: a post-hoc analysis of ETHOS. ATS 2024; Poster 913
12 Fachinformation Trixeo Aerosphere®; Stand: August 2023
13 Kent BD et al. Int J Chron Obstruct Pulmon Dis 2011;199
14 Hwang HJ et al. Int J Chron Obstruct Pulmon Dis 2019;14:2195-2203
15 Voskrebenzev A et al. Radiol Cardiothorac Imaging 2022;4:e210147
16 Marshall J et al. Poster P919, präsentiert auf der American Thoracic Society (ATS) International Conference, San Diego 2024
Das Hormonrezeptor-positive, HER2-negative Mammakarzinom, das durch die Expression von Östrogen- oder Progesteron-Rezeptoren (HR+) auf mindestens 1 % der Tumorzellen [1] und die fehlende bzw. geringe Expression des humanen epidermalen Wachstumsfaktor-Rezeptors 2 (HER2-) gekennzeichnet ist, ist die mit Abstand häufigste Form des Mammakarzinoms und betrifft rund 70 % der Patientinnen mit malignen Brusttumoren [2, 3]. Damit ist HR+/HER2– Brustkrebs etwa 7-mal häufiger als die HR–/ HER2– sowie die HR+/HER2+ Subtypen des Mammakarzinoms und etwa 17-mal häufiger als der HR–/HER2+ Subtyp [2].
Hoher medizinischer Bedarf in der endokrin-resistenten Situation
Während die relativen 5-JahresÜberlebensraten für die Patientinnen mit metastasiertem HR+/ HER2– Mammakarzinom (HR+/ HER2– mBC), deren Tumoren noch lokalisiert sind bzw. nur regionale Lymphknoten betreffen, mit 100 % bzw. 90,3 % sehr gut sind, verschlechtert sich die Prog-
nose der Erkrankten bei Vorliegen von Fernmetastasen deutlich: Hier liegt die 5-Jahres-Überlebensrate nur noch bei 34 % [2]. Basis der Behandlung in der metastasierten Situation ist die endokrine Therapie in Kombination mit einem CDK4/6-Inhibitor in der Erstlinie [4]. Dank dieser modernen Therapiestrategien konnte das mediane Überleben der betroffenen Patientinnen im Erstliniensetting auf über 5 Jahre verbessert werden [5]. Besonders herausfordernd ist allerdings die Therapiesituation, in der es zu einem Progress unter endokriner Therapie kommt. Dann benötigen die Patientinnen alternative Therapieoptionen, die das Überleben verlängern, Symptome vermindern, Komplikationen
Trop-2 als Target in der Onkologie
Das Oberflächenprotein Trop-2 wird auf zahlreichen soliden Tumoren überexprimiert und gilt damit als wichtiges Target, um Krebszellen zielgerichtet zu eliminieren [10, 14]. Das Protein ist bei verschiedenen Tumorarten stark ausgeprägt und häufig mit einer ungünstigen Prognose verbunden [16]. Das gilt auch für Mammakarzinome – insbesondere den triple-negativen Subtyp [10]. So belegen Studiendaten, dass die überwiegende Mehrheit der Patientinnen eine Trop2-Expression aufweist und nur wenige keine bzw. lediglich eine sehr schwache Expression (IHC1+) [12]. Das Protein stellt daher auch in dieser Indikation ein zuverlässiges Target bei einer zielgerichteten Tumortherapie dar [15]. Trop-2 als Target wird in derzeit laufenden Studien auch in anderen Entitäten solider Tumore wie Lungen- oder Urothelkarzinom untersucht.
vorbeugen und die Lebensqualität bestmöglich erhalten [6]. Weitere Behandlungsoptionen beschränken sich hier bislang weitgehend auf Monochemotherapieregime, die allerdings mit sinkenden Ansprechraten, geringerer Krankheitskontrolle sowie erhöhter Toxizität verbunden sind [5]. Das mediane Gesamtüberleben (mOS) unter Chemotherapie nach endokriner Therapie liegt bei nur rund einem Jahr [6]. In dieser Behandlungssituation bieten AntibodyDrug-Conjugates (ADC) wie Sacituzumab govitecan (Trodelvy®) zusätzliche Optionen.
Ab der Zweitlinie ADC Sacituzumab govitecan
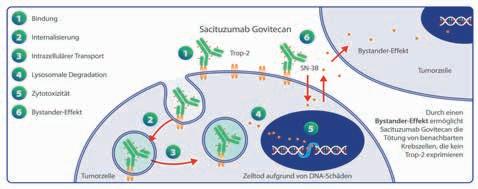
Seit Juli 2023 steht mit dem gegen das Trophoblasten Oberflächenantigen 2 (Trop-2) gerichteten Antibody-Drug-Conjugate (ADC) Sacituzumab govitecan (Sg) eine Behandlungsoption zur Verfügung, die den hohen medizinischen Bedarf beim HR+/HER2– mBC adressieren kann. Gemäß Zulassung kann Sg bei HR+/HER2– mBC nach 2 systemischen Therapien in der metastasierten Situation eingesetzt werden, sodass eine Chemotherapie in diesem Setting nicht zwingend erforderlich ist [7]. Die Arbeitsgemeinschaft Gynäkologische On-
Sacituzumab govitecan – ein Antibody-Drug-Conjugate der 3. Generation
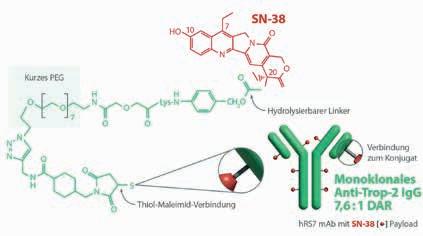
Sacituzumab govitecan (Trodelvy®) gehört zur Arzneimittelkategorie der ADCs. Diese bestehen aus einem monoklonalen Antikörper, der über einen Linker mit einem zytotoxischen Wirkstoff verbunden ist. Sacituzumab ist der gegen das Oberflächenantigen Trop-2 gerichtete Antikörper, govitecan die aus dem Linker und SN-38 bestehende chemische Einheit [9]. SN-38 ist der aktive Metabolit von Irinotecan, einem Topoisomerase-I-Inhibitor [10, 11].
kologie e.V. (AGO), Kommission
Mamma, empfiehlt den zulassungskonform frühzeitigen Einsatz von Sg mit ihrem höchsten Empfehlungsgrad „++“ [4]. Denn in Studien hat der ADC signifikante Vorteile gegenüber der klassischen Chemotherapie (CTx) gezeigt [5, 8].
Signifikante Verbesserung des progressionsfreien und des Gesamtüberlebens
In der Phase-III-Studie TROPiCS-02 wurde untersucht, ob die Therapie mit Sacituzumab govitecan (Sg) im Vergleich zu einer Monochemotherapie nach Wahl des Arztes (Eribulin, Vinorelbin, Gemcitabin oder Capecitabin) bei Patientinnen mit einem endokrinresistenten, chemotherapeutisch
vorbehandelten, inoperablen lokal fortgeschrittenen oder metastasierten Hormonrezeptor-positiven, HER2-negativen Brustkrebs (HR+/ HER2– mBC) zu einer Verbesserung des progressionsfreien Überlebens (Progression Free Survival, PFS) sowie des Gesamtüberlebens (Overall Survival, OS) führt [5, 8]. Eingeschlossen wurden 543 Patientinnen mit nicht resezierbarem oder metastasiertem HR+/ HER2– BC, die bereits mindestens 2 – 4 Chemotherapie-Regime im fortgeschrittenen Stadium und mindestens eine endokrine Vortherapie, ein Taxan und einen CDK4/6Inhibitor unabhängig vom Setting erhalten hatten. Sie wurden 1:1 auf die Behandlung mit Sg (n = 272) oder eine Monochemotherapie (n = 271) randomisiert. Die i.v. Verabreichung von Sg 10 mg/kg KG
erfolgte an Tag 1 und 8 im Rahmen eines 21-Tage-Zyklus. Die Monochemotherapie wurde gemäß der Fachinformation verabreicht. Die Patienten wurden so lange behandelt, bis eine Krankheitsprogression oder eine inakzeptable Toxizität auftrat. Primärer Endpunkt war das PFS, wichtigster sekundärer Endpunkt das OS [5]. Nach einem medianen Followup von 10,2 Monaten zeigte die 1. Interimsanalyse für die mit Sg behandelten HR+/HER2– mBCPatientinnen im Vergleich zur Monochemotherapie nach ärztlicher Wahl signifikante Verbesserungen beim medianen progressionsfreien Überleben mit 5,5 vs. 4,0 Monaten (HR: 0,66; 95%-KI: 0,53 – 0,83; p = 0,0003) wie auch beim medianen Gesamtüberleben mit 14,4 vs. 11,2 Monaten (HR:0,79; 95%-KI:
Verglichen mit anderen ADCs weist Sacituzumab govitecan besonders viele zytotoxische Moleküle pro Antikörper auf: Das Verhältnis beträgt 7,6:1, was die Pharmakokinetik nicht beeinträchtigt. Nach Bindung an Trop-2 wird der gesamte Komplex internalisiert und zu den Lysosomen transportiert. Durch hydrolytische Spaltung wird SN-38 freigesetzt, gelangt in den Zellkern, verhindert dort die Reparatur von DNA-Schäden und löst damit den programmierten Zelltod aus [11]. Eine weitere Rolle spielt der sog. „Bystander-Effekt“: Durch Hydrolyse liegt SN-38 auch extrazellulär vor. Es reichert sich in der Mikroumgebung des Tumors an und induziert nach Permeation der Zellmembran auch in benachbarten Tumorzellen die Apoptose – selbst wenn diese kein Trop-2 exprimieren [9, 11]. Durch diesen Effekt scheint Sacituzumab govitecan für Tumoren mit heterogenen Oberflächenantigenmustern eine effektive Therapiestrategie darzustellen.
Sacituzumab govitecan ist seit Juli 2023 als Monotherapie zur Behandlung von erwachsenen Patientinnen mit nicht resezierbarem oder metastasiertem Hormonrezeptor (HR)-positivem, HER2-negativem Mammakarzinom zugelassen, die eine Endokrin-basierte Therapie und mindestens 2 zusätzliche systemische Therapien bei fortgeschrittener Erkrankung erhalten haben. Bereits seit 2021 hat das ADC die EU-Zulassung zur Behandlung von erwachsenen Patienten mit metastasiertem triple-negativem-Brustkrebs (mTNBC) nach ≥2 systemischen Therapien (darunter ≥1 gegen die fortgeschrittene Erkrankung). Auch in dieser Indikation belegen sowohl Studien- als auch RealWorld-Daten signifikante Vorteile beim mPFS und mOS gegenüber einer Standard-CTx [12, 13].
0,65 – 0,96; p = 0,02). Demnach reduzierte Sg das Sterberisiko um 21 % (, p = 0,02) [5].
Die häufigsten Nebenwirkungen von Sg waren Neutropenie (67,6 %), Übelkeit (62,6 %), Diarrhoe (62,5 %), Fatigue (61,5 %) und Alopezie (45,6 %) [7]. Für Sg zeigte sich zudem eine längere Zeitspanne bis zur Verschlechterung der Lebensqualität der Patientinnen gegenüber einer CTx [5].
Brigitte Söllner, Erlangen
1 Allison KH et al. J Clin Oncol 2020;38: 1346-1366
2 National Cancer Institute. Cancer Stat Facts: Female Breast Cancer Subtypes. https://seer.cancer.gov/statfacts/html/ breast-subtypes.html
3 Andrahennadi S et al. Curr Oncol 2021; 28:1803-1822
4 AGO e. V. Kommission Mamma, Guidelines Breast Version 2023.1D. https:// www.ago-online.de/fileadmin/ago-online/ downloads/_leitlinien/kommission_mamma/2023/Einzeldateien/AGO_2023D_ 26_Therapiealgorithmen.pdf
5 Rugo H et al. J Clin Oncol 2022;40:33653376
6 Planchat E et al. Breast 2011;20:574-578
7 Fachinformation Trodelvy®; Stand: Juli 2023
8 Rugo HS et al. Lancet 2023;402:14231433
9 Nagayama A et al. Oncology 2021;35: 249-254
10 Bardia A et al. NEJM 2021;384:15291541
11 Rugo HS et al. Future Oncol 2020;16: 705-715
12 Bardia A et al. J Clin Oncol 2024;42: 1738-1744
13 Schäffler H et al. Geburtshilfe Frauenheilkd 2024; 84:855-865
14 Goldenberg DM et al. Oncotarget 2018; 9:28989-29006
15 Lenárt S et al. Cancers 2020;12:3328
Die Erdnussallergie ist eine der häufigsten Nahrungsmittelallergien im Kindesalter und die Ursache vieler Notaufnahmebesuche wegen einer Anaphylaxie. Um das Risiko einer schweren allergischen Reaktion bei versehentlichem Verzehr von Erdnüssen zu verringern, ist eine frühzeitige Behandlung entscheidend. Ein mittlerweile in der Praxis etablierter Ansatz besteht darin, durch eine frühe und konsequente Exposition gegenüber dem Allergen die Entwicklung und Aufrechterhaltung einer Erdnusstoleranz bei den Kindern zu fördern. Es hat sich gezeigt, dass eine orale Immuntherapie mit Erdnussallergenpulver (Palforzia® = Erdnussprotein als entfettetes Pulver von Arachis hypogaea L. semen) bei kontrollierter Exposition gegenüber steigenden Allergendosen die Immunantwort verändert und schrittweise die Schwelle erhöht, bei der die Patienten auf Erdnüsse reagieren [1].
Desensibilisierung jetzt bereits ab dem 2. Lebensjahr möglich
Palforzia® ist die erste orale Immuntherapie der Erdnussallergie, die von der EMA für Patienten im Alter von 4 – 17 Jahren zugelassen wurde. In mehreren randomisierten Studien mit Kindern und jungen Erwachsenen im Schulalter ließ sich bei der Mehrheit (50 – 70 %) der behandelten Teilnehmer eine Desensibilisierung erzielen, wobei die erhöhte Reaktionsschwelle auch nach Beendigung der Therapie für eine unterschiedliche Zeitspanne erhalten blieb. Auffällig war, dass von der Desensibilisierung vor allem die jüngsten Studienteilnehmer profitierten. Daher wurden in der Folge auch Studien mit Kleinkindern im Alter von 1 – 3 Jahren
durchgeführt. Sie zeigten zum einen, dass die orale Erdnussimmuntherapie bei kleinen Kindern sicher angewendet werden kann [2], und zum anderen, dass die Remissionsrate im 2. Lebensjahr am höchsten war [3], das ideale Alter, in dem mit der Desensibilisierung begonnen werden kann, demnach die Säuglingsphase und frühe Kindheit sein müsste.
Die randomisierte, doppelblinde, placebokontrollierte Phase-IIIStudie POSEIDON untersuchte daher die Wirksamkeit der bereits bei älteren Kindern und Jugendlichen bewährten niedrig dosierten (300 mg) oralen Erdnussimmuntherapie mit Palforzia® bei Säuglingen und Kleinkindern im Alter von 1 – 3 Jahren, die im Rahmen einer doppelblinden, placebokontrollierten Nahrungsmittelprovokation (DBPCFC) ab ≤300 mg Erdnussprotein dosislimitierende Symptome zeigten. Sie erhielten 1 Jahr lang randomisiert Erdnussallergenpulver oder Placebo im Verhältnis 2:1. Nach Studienende wurden alle Teilnehmer einer abschließenden DBPCFC unterzogen. Primärer Endpunkt war die Desensibilisierung (d.h. die Verträglichkeit einer Einzeldosis Erdnussprotein von ≥600 mg bei nur leichten Allergiesymptomen).
Die Studie erreichte ihren primären Endpunkt: 73,5 % der Kinder, die eine orale Erdnussimmuntherapie erhalten hatten, vertrugen eine Einzeldosis von 600 mg Erdnussprotein bei der Abschluss-DBPCFC
gegenüber 6,3 % in der PlaceboGruppe. Dabei tolerierte die Mehrheit (61,2 %) der mit Erdnussallergenpulver behandelten Teilnehmer die höchste ermittelte Dosis (explorativer Endpunkt einer Einzeldosis von 2000 mg). Aufgrund der kumulativen Natur des DBPCFC konsumierten Teilnehmer, die die 2000-mg-Dosis erreichten, insgesamt über 4 g Erdnussprotein, das entspricht über 12 Erdnusskernen, verglichen mit einer mittleren maximal tolerierten Dosis von 30 mg (ungefähr ¹/10 eines Erdnusskerns) vor der Behandlung [4].
Aufgrund dieser überzeugenden Studienergebnisse hat die Europäische Kommission im Januar 2025 die Erweiterung der Indikation von Palforzia® für die Behandlung von Kleinkindern im Alter von 1 – 3 Jahren mit einer bestätigten Diagnose einer Erdnussallergie genehmigt. Damit ist es möglich, die Desensibilisierung bereits ab dem 2. Lebensjahr zu beginnen und bei Kleinkindern das Risiko schwerer allergischer Reaktionen bei versehentlichem Kontakt mit Erdnussallergenen zu verringern.
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Palforzia®; Stand: Dezember 2024
2 Vickery BP et al. J Allergy Clin Immunol 2017;139:173-181
3 Jones SM et al. Lancet 2022;399:359-371
4 Du Toit G et al. NEJM Evid 2023;2:EVIDoa2300145. doi: 10.1056/EVIDoa2300 145
Seit dem 15. Januar 2025 ist mit Exagamglogene autotemcel (kurz: Exa-cel, Handelsname Casgevy®) in der EU die erste Gentherapie für die Behandlung geeigneter Patienten ab 12 Jahren mit schwerer Sichelzellkrankheit (SCD) mit rezidivierenden vasookklusiven Krisen (VOC) oder transfusionsabhängiger Beta-Thalassämie (TDT) zur Verfügung, die für eine Transplantation von hämatopoetischen Stammzellen (HSZ) geeignet sind und für die
Sichelzellkrankheit
kein humaner Leukozyten-Antigen (HLA)-kompatibler, verwandter HSZ-Spender verfügbar ist [1]. Exa-cel beruht auf der CRISPR/ Cas9-Geneditierung und eröffnet, wie die Ergebnisse der Zulassungsstudien zeigen, für die von TDT und SCD betroffenen Patienten die Möglichkeit einer an den Grundmechanismen der beiden Krankheiten angreifenden, potenziell dauerhaften funktionellen Heilung.
Maßgeschneiderte Therapie mit CRISPR/Cas9
Die Ursachen von TDT und SCD sind seit Jahrzehnten bekannt und die SCD gilt als die erste auf molekularer Ebene beschriebene Krankheit. Bislang wurde jedoch noch keine kausale Therapie dafür gefunden. Mit der Entwicklung der CRISPR-Geneditierung besteht nun die Möglichkeit, die Ursache der Erkrankungen anzugehen.
Die Sichelzellkrankheit (Sickle Cell Disease, SCD) ist eine autosomal-rezessiv vererbte, progrediente und lebensverkürzende Erkrankung der roten Blutkörperchen. Ursache ist eine Punktmutation auf dem Gen, das für die Hämoglobin-Beta-Kette (HBB) codiert. Folge ist eine veränderte Struktur des Hämoglobins, die zu einer Veränderung der Erythrozyten-Form führt: Die roten Blutkörperchen sind nicht mehr rund und elastisch, sondern sichelförmig und starr. Diese Sichelzellen schädigen das Endothel und können kleine Blutgefäße verstopfen. Die daraus resultierenden Gefäßverschlüsse führen zu einer verminderten Durchblutung von Geweben und Organen und letztlich zum Organversagen. Da die Sichelzellen sehr schnell abgebaut werden, kann sich zusätzlich eine hämolytische Anämie entwickeln [2]. Typische Symptome reichen vom Hand-Fuß-Syndrom (eine durch Gefäßverschlüsse verursachte schmerzhafte Schwellung der Finger- und Zehenknochen) und einer Milzsequestrationskrise im Kleinkindalter über vasookklusive Schmerzkrisen des Stammskeletts und der langen Röhrenknochen im Erwachsenenalter bis hin zu einer Einschränkung der Nierenfunktion, pulmonaler Hypertonie, einem paralytischen Ileus, ZNS-Infarkten und schließlich einem lebensbedrohlichen Organversagen [2]. Die Lebenserwartung beträgt nur rund 55 Jahre [3].
Die einzige kurative Therapie der SCD ist bislang die Stammzelltransplantation, die aber nur für einen kleinen Teil der Betroffenen infrage kommt. In den meisten Fällen konzentriert sich die Therapie auf die Schmerzlinderung und die Begrenzung von Organschäden, was oft eine lebenslange Gabe von Medikamenten, manchmal auch monatliche Bluttransfusionen und häufige Klinikaufenthalte erfordert. SCD ist in den tropischen Anteilen Afrikas, im Mittleren Osten, in weiten Teilen Indiens, in der Ost-Türkei und geographisch begrenzt in Griechenland und Süditalien endemisch. Die meisten in Deutschland lebenden Patienten stammen aus Afrika, aus dem Mittleren Osten und aus der Türkei [4]. Die Inzidenz liegt weltweit bei 300.000 – 400.000 betroffenen Neugeborenen pro Jahr. In Deutschland leiden etwa 2.500 Menschen an SCD [4].
Auch die transfusionsabhängige Beta-Thalassämie (TDT) ist eine Erbkrankheit, die durch eine Mutation im HBB-Gen ausgelöst wird. Sie führt zu einer zu geringen oder fehlenden Produktion von Betaglobin, einem wichtigen Baustein des Hämoglobins. Ein erheblicher Teil der Betroffenen ist wegen der schweren Anämie alle 2 Wochen auf Bluttransfusionen angewiesen und benötigt eine lebenslange symptomatische Eisenchelat-Therapie zur Vorbeugung einer Schwermetallvergiftung [5]. Die einzige Chance auf eine Heilung der TDT besteht bislang in einer Stammzelltransplantation, die aber nur für einen kleinen Teil der an TDT erkrankten Menschen infrage kommt. Die Lebenserwartung ist mit rund 55 Jahren stark reduziert [6]. Weltweit leiden etwa 288.000 Menschen an TDT, in Deutschland sind geschätzt 250 Menschen davon betroffen [7].
Exa-cel ist eine auf der CRISPR/ Cas9-Geneditierung (vgl. Insert auf S. 58) basierende Ex-vivoTherapie, bei der autologe hämatopoetische Stammzellen aus dem Blut der Patienten gewonnen und mithilfe der CRISPR/Cas9-Technologie genetisch so modifiziert werden, dass diese wieder fetales Hämoglobin (HbF) bilden.
HbF ist eine natürliche Form des Hämoglobins, das bei allen Menschen im Mutterleib und nach der Geburt vorhanden ist. Es funktioniert selbst dann ordnungsgemäß, wenn SCD- oder TDT-Mutationen vorliegen. Etwa 3 – 6 Monate nach der Geburt geht der Körper aber von der Produktion des fetalen Hämoglobins auf das adulte Hämoglobin über, weshalb die Symptome von SCD und TDT häufig erstmals im Alter von etwa 3 Monaten auftreten. Die Erhöhung der HbF-Produktion bei Patienten mit SCD bzw. TDT zielt darauf ab, eine signifikante Reduktion oder gar Beseitigung der krankheitsbedingten Symptome zu erreichen [8, 9].
Dazu wird in den Stammzellen des Patienten ein bestimmtes regulatorisches Enhancer-Segment des BCL11A-Gens durch einen präzisen Doppelstrangbruch editiert. BCL11A ist ein Transkriptionsfaktor, der die Expression von Gamma-Globin, einem Bestandteil von
HbF, hemmt. Durch die Editierung von BCL11A wird das HbF reaktiviert und nach der Re-Infusion der Zellen kommt es zu einer erhöhten Produktion von HbF in den roten Blutzellen, das die bei SCD und TDT mutierte Beta-Variante des Hämoglobins ersetzt.
Weniger vasookklusive Krisen und Transfusionen
Die Zulassung von Exa-cel durch die Europäische Kommission basiert auf den Daten der Studien CLIMB-111, -121 und -131 [12, 13]. Deutschland war wegweisend bei dieser klinischen Forschung, und die entscheidenden Studien wurden in 3 spezialisierten Studienzentren in Deutschland durchgeführt. Die bisherigen Studiendaten stützen den potenziell dauerhaften klinischen Vorteil von Exa-cel. Die längste Nachbeobachtungszeit, sowohl für SCD- als auch für TDT-Patienten, beträgt mittlerweile mehr als 5 Jahre, mit einem Median von 33,2 bzw. 38,1 Monaten.
In CLIMB-121 sowie in der laufenden offenen Langzeitstudie CLIMB-131 waren 39/42 (93 %)
der eingeschlossenen SCD-Patienten mit auswertbaren Daten (mindestens 16 Monate Nachbeobachtung) für mindestens 12 aufeinanderfolgende Monate (VF12) frei von vasookklusiven Krisen (VOC). Die durchschnittliche Dauer der VOC-Freiheit betrug 30,9 Monate, mit einem Maximum von 59,6 Monaten. Jene 3 Patienten mit auswertbaren Daten, die VF12 nicht erreichten, erlangten einen bedeutenden klinischen Nutzen, u.a. durch eine Verringerung der Hospitalisierungsrate für VOC um 91 %, 71 % und 100 %.
In CLIMB-111 und CLIMB-131 erreichten 53/54 (98 %) der Patienten mit auswertbaren Daten (mit einer Nachbeobachtung von mindestens 16 Monaten) eine Transfusionsunabhängigkeit von mindestens 12 aufeinanderfolgenden Monaten mit einem gewichteten durchschnittlichen Hämoglobinwert von mindestens 9 g/dl (TI12). Die mittlere Dauer der Transfusionsunabhängigkeit betrug 34,5 Monate, mit einem Maximum von 64,1 Monaten. Ein Studienteilnehmer, der TI12 noch nicht erreicht hat, zu dem aber auswertbare Daten vorliegen, ist seit 8,2 Monaten transfusionsfrei.
CRISPR steht für Clustered Regulatory Interspaced Short Palindromic Repeats, also kurze Abschnitte sich wiederholender DNA, die bestimmte Bakterien als Teil ihrer Virenabwehr nutzen.
Cas9 ist eine an den CRISPR-Abschnitt gekoppelte Endonuklease bzw. ein Enzym, das von einer Guide-RNA entweder genau zu der Stelle in der DNA geführt wird, die mit der „Genschere“ durchtrennt werden soll, um eine Änderung im Genom vorzunehmen, oder zu einer anderen Region transportiert wird, welche die betroffenen Gene reguliert, um dort den Gendefekt über Entfernen oder Einfügen eines DNA-Stückes zu beheben. In beiden Fällen lassen sich die Gene entweder ex vivo oder in vivo verändern [10].
Mit CRISPR/Cas9 können 3 Formen genetischer Editierung durchgeführt werden [11]:
• Durchtrennen: Wird nur ein einzelner Schnitt ausgeführt, können mittels einer nicht homologen Endenverknüpfung Basenpaare hinzugefügt oder entfernt werden. Dadurch wird die ursprüngliche DNA-Sequenz so nachhaltig verändert, dass das Gen inaktiviert wird.
• Deletion: Werden 2 Guide-RNAs verwendet, die auf 2 unterschiedliche Stellen abzielen, lässt sich durch die anschließende nicht homologe Verknüpfung der freien Enden die ursprünglich dazwischen gelegene Sequenz entfernen.
• Insertion: Fügt man dem CRISPR/Cas9-System zusätzlich noch eine DNA-Sequenz als Vorlage hinzu, ermöglicht man der Zelle, ein Gen entweder zu korrigieren oder über den Prozess der Homologie gesteuerten Reparatur sogar ein neues Gen zu ergänzen.
Verbesserte Lebensqualität und stabile HbF-Werte
Sowohl SCD- als auch TDT-Patienten berichteten über anhaltende und klinisch bedeutsame Verbesserungen ihrer Lebensqualität, einschließlich des körperlichen, emotionalen und sozialen/familiären Wohlbefindens sowie des
allgemeinen Gesundheitszustands. Die Patienten zeigen in den Studien weiterhin stabile Werte für das fetale Hämoglobin (HbF) und die Allel-Editierung über alle Altersgruppen und Genotypen hinweg. Das Sicherheitsprofil von Exa-cel ist bislang im Allgemeinen konsistent mit der myeloablativen Konditionierung mit Busulfan und
der autologen hämatopoetischen Stammzelltransplantation.
Für Patienten mit Sichelzellkrankheit oder Beta-Thalassämie sind die Therapiemöglichkeiten bisher sehr begrenzt – die einzige kurative Behandlung besteht in einer Stammzelltransplantation, die jedoch nur für wenige Betroffene infrage kommt und mit immunologisch bedingten Komplikationen wie Transplantat-Abstoßung und Spender-gegen-EmpfängerErkrankung einhergehen kann. Vor diesem Hintergrund ist die von Vertex Pharmaceuticals entwickelte Gentherapie Exa-cel (Casgevy®), die auf einer Ex-vivo-Geneditierung der blutbildenden Stammzellen basiert, ein bemerkenswerter vielversprechender Ansatz, um die Krankheitsursache – den Gendefekt – zu beheben.
Die Therapie mit Exa-cel reduziert die Symptome sowie die Abhängigkeit von Bluttransfusionen und erhöht damit nicht nur die Lebensqualität der Patienten, sondern könnte auch die langfristigen Behandlungskosten senken. Eine lebenslange Transfusionstherapie kostet durchschnittlich 5,4 Millionen Dollar, für den US-Markt hat Vertex Pharmaceuticals einen Preis von 2,2 Millionen Dollar pro Exacel-Behandlung festgelegt [14].
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Casgevy®; Stand: März 2024
2 Gesellschaft für Pädiatrische Onkologie und Hämatologie. AWMF-Leitlinie 025/016 „Sichelzellkrankheit“, Version 2.0 vom 2. Juli 2020
3 MedlinePlus. Sichelzellkrankheit. https:// medlineplus.gov/genetics/condition/sickle-cell-disease/
4 Kunz JB et al: Sickle cell disease in Germany: Results from a national registry. Pediatr Blood Cancer 2020;67:e28130
5 https://www.onkopedia.com/de/onkopedia/guidelines/beta-thalassaemie/@@guideline/html/index.html
6 Ladis V, et al. Survival in a large cohort of Greek patients with transfusion-dependent beta thalassaemia and mortality ratios compared to the general population. Eur J Haematol 2011;86:332-338
7 IQWIG. https://www.iqwig.de/download/ g23-07_exagamglogen-autotemcel_einschaetzung-der-patientenzahlen_v1-0.pdf
8 Jinek M et al. RNA-programmed genome editing in human cells. eLife 2013; 2:e00471
9 Frangoul H et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med 2021;384: 252-260
10 Jinek M et al. A programmable dualRNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337:816821
11 Doudna J et al. The new frontier of genome engineering with CRISPR/Cas9. Science 2014;346:1258096
12 Locatelli F et al. Exagamglogene autotemcel for transfusion-dependent β-thalassemia. N Engl J Med 2024;390: 16631676
13 Frangoul H et al. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med 2024;390:1649-1662
14 US Securities and Exchange Commission. Casgevy. 2023. https://www.sec.gov/ ix?doc=/Archives/edgar/data/000087532 0/000087532023000054/vrtx-20231208.
Zulassungserweiterung von Cetuximab ermöglicht zweiwöchentliche Gabe
Der EGFR-Antikörper Cetuximab (Erbitux®) wurde von der Europäischen Arzneimittelagentur nun auch für die zweiwöchentliche Verabreichung beim metastasierten kolorektalen Karzinom (mCRC) sowie beim rezidivierenden und/ oder metastasierenden Plattenepithelkarzinom im Kopf- und Halsbereich (R/M SCCHN) zugelassen. Mit der Zulassungserweiterung ist Cetuximab die einzige Anti-EGFR-Therapie, die bei mCRC und R/M SCCHN sowohl wöchentlich (Q1W) als auch zweiwöchentlich (Q2W) eingesetzt werden kann. Für die Behandlung des lokal fortgeschrittenen Plattenepithelkarzinoms des Kopf- und Halsbereichs mit Cetuximab in Kombination mit einer Strahlentherapie ist weiterhin ausschließlich die wöchentliche Verabreichung zugelassen.
Gleichbleibende Wirksamkeit und Verträglichkeit
Studiendaten zur zweiwöchentlichen Verabreichung von Cetuximab in der Erstlinienbehandlung
Cetuximab (Erbitux®) ist ein monoklonaler Antikörper vom Typ IgG1, der gezielt den epidermalen Wachstumsfaktorrezeptor (EGFR) blockiert. Dadurch werden die Aktivierung des Rezeptors und der nachgeschaltete Signaltransduktionsweg gehemmt, sodass sowohl die Invasion der Tumorzellen in gesundes Gewebe als auch die Metastasierung vermindert werden. Außerdem werden die Fähigkeit der Tumorzellen, die durch Chemo- und Strahlentherapie verursachten Schäden zu reparieren, sowie die Ausbildung neuer Blutgefäße in den Tumoren unterbunden, was zu einer generellen Hemmung des Tumorwachstums führt.
des mCRC in Kombination mit verschiedenen ChemotherapieBackbones wie FOLFOX und FOLFIRI zeigten eine vergleichbare Wirksamkeit und Verträglichkeit wie bei der bislang praktizierten wöchentlichen Gabe. So war in der randomisierten Phase-II-Studie CECOG-CORE, die die Erstli-
nientherapie mit Cetuximab bei ein- und zweiwöchentlicher Gabe in Kombination mit FOLFOX verglich, die Gesamtansprechrate mit 62 % unter der zweiwöchentlichen und 53 % unter der wöchentlichen Verabreichung vergleichbar. Auch bei der Verabreichung von Cetuximab in Kombination mit Irinotecan in späteren Therapielinien des mCRC wurden unter den Dosierungsschemata Q1W und Q2W vergleichbare Ergebnisse erzielt. In der Erhaltungstherapie des R/M SCCHN mit Cetuximab erwiesen sich beide Verabreichungsschemata ebenfalls als vergleichbar wirksam und verträglich. In den Studien DIRECT und TPExtreme ergaben sich keine signifikanten Unterschiede im Gesamtüberleben zwischen den Behandlungsarmen mit wöchentlicher und zweiwöchentlicher Gabe.
Fazit für die Praxis
Die Zulassungserweiterung von Erbitux® schafft mehr Flexibilität in der täglichen Versorgung und kann zu einer größeren Therapieadhärenz sowie letztlich auch zu einer Verbesserung der Lebensqualität der Patienten beitragen.
B. S.
Eine erhöhte Stuhlfrequenz, Bauchschmerzen, Durchfälle oder die oft besonders belastende Bowel Urgency (BU, imperativer Stuhldrang) – die Symptome bei Colitis ulcerosa (CU) können vielfältig sein und die Lebensqualität enorm beeinträchtigen [1]. Um den Betroffenen eine möglichst rasche Rückkehr in einen normalen Alltag zu ermöglichen, ist es entscheidend, schnell eine starke und anhaltend wirksame Therapie bei guter Verträglichkeit zu finden.
Leitliniengerecht und im Rahmen der Zulassung kann der Interleukin(IL)-23p19-Inhibitor Mirikizumab (Omvoh®) [2] als First-Line-Biologikum bei mittelschwerer bis schwerer aktiver CU eingesetzt werden* [1, 3] – mit langfristigem Erfolg, wie die neuen 3-Jahres-Daten belegen, die auf der „United European Gastroenterology Week“ in Wien vorgestellt wurden. Demnach kann Mirikizumab als stark wirksame Therapie über 152 Wochen eine anhaltende Symptomkontrolle bei guter Verträglichkeit ermöglichen – und das steroidfrei [4].
Steroidfreie klinische Remission über 3 Jahre
Die langanhaltende klinische und endoskopische Remission ohne die zusätzliche Einnahme von Kortikosteroiden ist eines der zentralen Ziele in den Leitlinien zur Behandlung der CU. [5] Unter der Therapie mit Mirikizumab befanden sich nach 3
* Omvoh® ist angezeigt für die Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer aktiver Colitis ulcerosa (CU), die auf eine konventionelle Therapie oder eine Biologika-Behandlung unzureichend angesprochen haben, nicht mehr darauf ansprechen oder eine Unverträglichkeit zeigen [2].
Jahren 79 % der Biologika-naiven Patienten in steroidfreier klinischer Remission**[6]. 83 % dieser Population erzielten darüber hinaus eine endoskopische Verbesserung (ES = 0 oder 1, ohne Schleimhautvulnerabilität) in Woche 152 [4]. Ein Blick auf die Gesamtpopulation zeigt die kontinuierliche Wirksamkeit von Mirikizumab: 81 % der Patienten konnten die klinische Remission von Woche 52 bis Woche 152 aufrechterhalten. 98 % dieser Patienten benötigten keine zusätzlichen Steroide, blieben also steroidfrei in Remission.
Frühe und langanhaltende Kontrolle belastender Symptome
Bereits in den LUCENT-Zulassungsstudien konnte Mirikizumab mit einer frühen und anhaltenden Symptomkontrolle überzeugen –mit signifikanten Verbesserungen der Symptome Stuhlfrequenz, rektale Blutungen und BU gegenüber Placebo bereits in Woche 2 [7]. Die aktuellen 3-Jahres-Daten zeigen, dass die Patienten von einer anhaltenden Kontrolle der Symptome profitieren können, auch mit Blick auf die für sie oft belastende BU: Die Verbesserungen blieben über den gesamten Zeitraum stabil [8]. Weiterhin wiesen 72 % der Biologika-naiven Patienten nach 3 Jahren eine Remission der BU auf (Bewertung des StuhldrangSchweregrads UNRS mit 0 oder 1).
Disease Clearance –neue, ambitionierte Ziele in der CU-Therapie
Dass dank moderner Therapieoptionen wie Mirikizumab ambitioniertere Therapieziele möglich sind, unterstrich eine weitere auf der UEGW präsentierte Auswertung zur Disease Clearance. Diese beschreibt das gleichzeitige Vorliegen einer symptomatischen Remission und kombinierten histologisch-endoskopischen Schleimhautremission (HEMR) und ist ein ambitioniertes patientenrelevantes Behandlungsziel bei CU [9]. So ist beispielsweise das Vorliegen einer Disease Clearance bei CU mit einem geringeren Risiko für Krankenhausaufenthalte und Operationen assoziiert [10].
In einer Post-hoc-Auswertung der Zulassungsstudien sowie der ersten Interimsanalyse von LUCENT-3 (nach 2 Jahren) wurden die Ergebnisse von Mirikizumab mit Blick auf eine Disease Clearance betrachtet [7]:
• Ein höherer Anteil der mit Mirikizumab behandelten Patienten erreichte in Woche 12 (16 % vs. 7,1 %; p < 0,0001) und in Woche 52 (36,4 % vs. 19,6 %; p < 0,001) eine Disease Clearance im Vergleich zu Placebo.
• Nach 2 Jahren MirikizumabBehandlung erreichten 41 % der Patienten (NRI-Auswertung) eine Disease Clearance.
• Die Analyse ergab zudem, dass eine Disease Clearance mit der Verbesserung verschiedener Patient-Reported Outcomes (PROs) assoziiert ist.
Diese Ergebnisse und die aktuellen 3-Jahres-Daten unterstreichen die Bedeutung von Mirikizumab bei der Behandlung der CU. Mit dem IL-23p19-Inhibitor können die Patienten von einer frühen und anhaltenden Symptomkontrolle profitieren und das bei Steroidfreiheit und guter Verträglichkeit – auch im Langzeitverlauf wurden keine neuen Sicherheitssignale entdeckt [11].
Fabian Sandner, Nürnberg
Literatur
1 Knowles SR et al. Inflamm Bowel Dis 2018; 4:742-751
2 Fachinformation Omvoh®, aktueller Stand
3 Deutsche Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten e.V. (DGVS). Aktualisierte S3-Leitlinie Colitis ulcerosa (Version 6.2); Januar 2024 – AWMF-Registriernummer: 021009. URL: https://register.awmf.org/assets/guidelines/021-009l_S3_Colitis-ulcerosa_2024-06.pdf
4 Sands BE et al. Inflamm Bowel Dis 2024; izae253 (DOI: 10.1093/ibd/izae253)
5 Kucharzik T et al. Aktualisierte S3-Leitlinie Colitis ulcerosa (Version 6.2). https:// register.awmf.org/assets/guidelines/021009l_S3_Colitis-ulcerosa_2024-06.pdf
6 Data on File, Eli Lilly & Company
7 Danese S et al. J Crohns colitis 2024; jjae088 (Online ahead of print)
8 Travis S et al. Inflamm Bowel Dis 2024;30:939-949
9 Colombel JF et al. Poster: The impact of mirikizumab induction, maintenance and long-term treatment on disease clearance in patients with moderately to severely active ulcerative colitis: Post Hoc Analysis of LUCENT-Trials. UEGW 2024; MP615
10 D’Amico F et al. United European Gastroenterol J 2022;10:777-784
11 D’Haens G et al. N Engl J Med 2023; 388:2444-2455 (plus supplementary appendix)
Chronische Insomnie: Langzeitdaten bestätigen die Wirksamkeit von Daridorexant
Daridorexant (Quviviq™) ist bislang die einzige Therapieoption zur Behandlung der chronischen Insomnie bei Erwachsenen, die ohne maximale Behandlungsdauer zugelassen ist und für eine zeitlich uneingeschränkte Verordnung zu Lasten der gesetzlichen Krankenkassen zur Verfügung steht. Der duale Orexin-Rezeptor-Antagonist (DORA) greift direkt in der Pathophysiologie der Insomnie ein, denn er blockiert die OrexinRezeptoren und verhindert so die Weiterleitung von Wachheitssignalen durch Orexin. Dies kann das Einschlafen erleichtern und das Durchschlafen ermöglichen, ohne das Verhältnis der natürlichen Schlafphasen zu verändern.
Kürzere Einschlafzeiten, besseres Durchschlafen
Zwei Jahre nach der Markteinführung von Daridorexant in Deutschland bestätigen Daten aus der Phase-III-Verlängerungsstudie mit insgesamt 550 Studienteilnehmern die Ergebnisse der Zulassungsstudien. Die Post-hoc-Analyse zeigte, dass Daridorexant 50 mg über einen Zeitraum von 40 Wochen signifikant die Einschlafzeit verkürzt und das Durchschlafen sowie die subjektiv empfundene Gesamtschlafdauer und die Tagesaktivität verbessert. Eine sekundäre Datenanalyse der Phase-III-Studie belegt außerdem die gute Wirksamkeit bei älteren Patienten >65 Jahre. Diese konnte
Daridorexant
Daridorexant (Quviviq™) blockiert die Rezeptoren der Neuropeptide Orexin A und Orexin B und unterbindet damit die Weiterleitung von Signalen an die Neuronen im Wachsystem, die das Wachsein fördern.
über einen langen Zeitraum aufrechterhalten werden, und das bei gleichzeitig guter Verträglichkeit und Sicherheit ohne Anzeichen von körperlicher Abhängigkeit oder Toleranzentwicklung. Das günstige Sicherheitsprofil ermöglicht es auch, Patienten mit einer komorbiden chronischen Insomnie, z.B. beim obstruktiven Schlafapnoesyndrom oder Restless-Legs-Syndrom, längerfristig mit Daridorexant zu behandeln. Denn anders als bei herkömmlichen Hypnotika ist die Behandlungsdauer für Daridorexant nicht begrenzt.
Daridorexant wird angewendet zur Behandlung von Erwachsenen mit Insomnie, deren Symptome seit mindestens 3 Monaten anhalten und eine beträchtliche Auswirkung auf die Tagesaktivität haben. Die empfohlene Tagesdosis für Erwachsene beträgt 50 mg und wird abends innerhalb von 30 Minuten vor dem Schlafengehen eingenommen. Da die Halbwertszeit von Daridorexant etwa 8 Stunden beträgt, sollte das Präparat nicht zu spät bzw. nicht nachts eingenommen werden. Der Effekt von Daridorexant setzt bei einigen Patienten sofort ein, bei anderen kann es bis zur vollen Wirkung 3 Wochen dauern. Deshalb ist ein ausreichend langes Zeitfenster zur Bewertung des Therapieerfolgs zu Beginn wichtig.
B. S.
Auf der 22. Angiogenesis Tagung in Miami haben Bayer und Regeneron die Ergebnisse der offenen Verlängerung der klinischen Studie PULSAR bei Patienten mit neovaskulärer (feuchter) altersabhängiger Makuladegeneration präsentiert. Die Daten bestätigen die langanhaltende Wirkung von Aflibercept 8 mg (Eylea® 8 mg, 114,3 mg/ml Injektionslösung) sowie die Langzeitsicherheit nach 3 Jahren. Aflibercept 8 mg ist die einzige Anti-VEGF-Therapie, die für Behandlungsintervalle von bis zu 5 Monaten für die beiden Anwendungsgebiete feuchte altersabhängige Makuladegeneration und diabetisches Makulaödem in der EU, Großbritannien und anderen Märkten zugelassen ist.
Überzeugende Daten der PULSAR-Verlängerungsstudie ...
Ziel der offenen Verlängerungsstudie war es, die langfristige Wirksamkeit und Sicherheit von Aflibercept 8 mg bei Patienten mit neovaskulärer (feuchter) altersabhängiger Makuladegeneration (nAMD) von Woche 96 bis Woche 156 zu untersuchen. Die 208 Patienten, die zuvor mit Aflibercept 2 mg behandelt wurden, konnten
Altersabhängige Makuladegeneration
Die altersabhängige Makuladegeneration (AMD) ist eine chronische Erkrankung der Netzhautmitte, der Macula lutea (gelber Fleck), die in verschiedenen Formen auftritt:
• Bei der trockenen AMD sterben die Zellen in der Makula, dem Teil der Netzhaut, der für das scharfe zentrale Sehen und das Erkennen feiner Details verantwortlich ist, infolge der Ablagerung von Stoffwechselendprodukten (Lipofuszinen) sowie Durchblutungsstörungen in der Aderhaut nach und nach ab. Die Degeneration der Netzhautmitte geht ohne Flüssigkeitsansammlungen einher und schreitet langsam voran. Die Sehschärfe kann daher über viele Jahre stabil bleiben, bevor die Patienten hochgradig sehbehindert werden und erblinden.
• Anders ist das bei der feuchten (neovaskulären) nAMD, bei der Blutgefäße in die Netzhaut einwachsen und Blut sowie Flüssigkeit im Bereich der Makula austreten. Folgen sind eine Netzhautverdickung oder ein Netzhautödem sowie sub-/intraretinale Blutungen, die zum Verlust der Sehschärfe führen. Unbehandelt schreitet die nAMD rasch fort, sodass der Patient bereits nach wenigen Monaten erblindet. Dieser Prozess lässt sich durch eine rechtzeitige Injektionstherapie aufhalten. Bei dieser Anti-VEGFTherapie wird eine Substanz (z.B. Aflibercept) in den Glaskörper des Auges gespritzt, die das Gefäßwachstum durch Hemmung des Gefäßwachstumsfaktors VEGF (Vascular Endothel Growth Factor) unterdrückt sowie den Flüssigkeitsaustritt und die Schwellung verringert, sodass der Sehverlust verhindert wird.
Weltweit leben 170 Millionen Menschen mit AMD. Etwa 10 % der Menschen mit AMD entwickeln die fortgeschrittene Form nAMD.
auf Aflibercept 8 mg umgestellt werden und wurden sofort einem 12-wöchigen Behandlungsintervall zugewiesen (es waren keine monatlichen Initialdosen erforderlich). Die 417 Patienten, die in Woche
96 bereits mit Aflibercept 8 mg behandelt wurden, setzten ihr zuletzt zugewiesenes Intervall fort. 90,2 % der Patienten, die in die offene Verlängerungsstudie aufgenommen wurden, beendeten die Studie.
Das diabetische Makulaödem (DMÖ) ist eine häufige Komplikation in den Augen von Menschen, die an Diabetes leiden. Es tritt auf, wenn hohe Blutzuckerwerte zu Schädigungen der Blutgefäße im Auge führen, aus denen dann Flüssigkeit in die Makula austritt. Dies kann zum Verlust des Sehvermögens und in einigen Fällen zur Erblindung führen. Weltweit leben derzeit 146 Millionen Menschen mit einer diabetischen Retinopathie, die sich zu einem diabetischen Makulaödem entwickeln kann, und etwa 27 Millionen Menschen sind von DMÖ betroffen.
Aflibercept (Eylea®)ist ein Fusionsprotein, das die relevantesten extrazellulären Anteile der VEGF-Rezeptoren VEGFR-1 und VEGFR-2 mit einem IgG1 Fc verbindet. Durch diese molekulare Struktur fungiert Aflibercept als Köderrezeptor, der VEGF-A-Isoformen sowie VEGF-B und den Plazenta-Wachstumsfaktor (PlGF) abfangen kann. VEGF-A und PlGF sind an der Stimulation des abnormalen Wachstums von Blutgefäßen bei Patienten mit AMD, bestimmten Arten von Makulaödem und kurzsichtiger choroidaler Neovaskularisation beteiligt. Durch Blockade dieser Faktoren reduziert Aflibercept die krankhafte Gefäßneubildung und kontrolliert die erhöhte Gefäßpermeabilität.
In der Ophthalmologie wird Aflibercept angewendet bei Erwachsenen zur Behandlung
• der neovaskulären (feuchten) altersabhängigen Makuladegeneration (nAMD),
• einer Visusbeeinträchtigung aufgrund eines Makulaödems infolge eines retinalen Venenverschlusses,
• einer Visusbeeinträchtigung aufgrund eines diabetischen Makulaödems (DMÖ),
• einer Visusbeeinträchtigung aufgrund einer myopen choroidalen Neovaskularisation.
Die anhand festgelegter, klinisch relevanter Kriterien zur Änderung des Krankheitsverlaufs (DRM) bewertete Krankheitsaktivität wurde bis Woche 108 alle 4 Wochen und danach alle 12 Wochen erhoben. Bei Patienten ohne Krankheitsaktivität wurde das Behandlungsintervall in 2-Wochen-Schritten bis zu einem maximalen Intervall von 24 Wochen (6 Monaten) verlängert. Bei Patienten mit Krankheitsaktivität wurde das Behandlungsintervall in 2-Wochen-Schritten ver-
kürzt, mit einem Mindestintervall von 8 Wochen (2 Monaten).
Unter der Therapie mit Aflibercept 8 mg erreichten die Patienten nachhaltige Visusgewinne und eine Flüssigkeitskontrolle über 3 Jahre. Robuste Verringerungen der mittleren zentralen Netzhautdicke (central retinal thickness, CRT) zu Studienbeginn blieben mit Afliber-
cept 8 mg über 3 Jahre erhalten. Die Wirksamkeit von Aflibercept 8 mg, die anhand der mittleren Veränderung der bestkorrigierten Sehschärfe (BCVA) bewertet wurde, blieb im 3. Jahr (Woche 156) im Vergleich zum Beginn der Verlängerungsstudie (Ausgangswert in Woche 96) erhalten.
Die große Mehrheit der Patienten, die seit Studienbeginn mit Aflibercept 8 mg behandelt wurden, hatte nach 3 Jahren ein letztes zugewiesenes Behandlungsintervall von ≥3 Monaten, 40 % der Patienten, die von Beginn an mit Aflibercept 8 mg behandelt wurden, hatten nach 3 Jahren ein letztes zugewiesenes Behandlungsintervall von ≥5 Monaten und 24 % von 6 Monaten.
Das Sicherheitsprofil von Aflibercept 8 mg blieb auch über 3 Jahre hinweg günstig und entspricht dem gut etablierten Sicherheitsprofil von Aflibercept 2 mg. Die Daten zur Langzeitsicherheit ergaben keine neuen Signale für eine der Behandlungsgruppen (Patienten, die zu Studienbeginn einer Behandlung alle 12 oder 16 Wochen zugeordnet wurden, und Patienten, die von Aflibercept 2 mg auf Aflibercept 8 mg umgestellt wurden). Die Häufigkeit von behandlungsbedingten unerwünschten Ereignissen des Auges war in allen Behandlungsgruppen ähnlich. Es traten keine Fälle von okklusiver Vaskulitis auf. Die Häufigkeit von intraokularen Entzündungen war über die 3 Jahre hinweg niedrig: 2,4 % bei den Patienten, die auf Aflibercept 8 mg umgestellt wurden, und 1,9 % bei den Patienten, die von Beginn an auf Aflibercept 8 mg randomisiert wurden).
Angesichts einer alternden Bevölkerung und der steigenden Prävalenz der feuchten altersabhängigen Makuladegeneration sowie des diabetischen Makulaödems besteht ein dringender Bedarf an langwirksamen Therapien. Wie die Langzeitdaten zur Behandlung mit Aflibercept 8 mg belegen, kann ein relevanter Anteil der Patienten von langen Behandlungsintervallen mit nur einer Injektion alle 6 Monate profitieren. Basierend auf den positiven Daten hat Aflibercept 8 mg das Potenzial, die neue Standardtherapie für die Behandlung von Netzhauterkrankungen zu werden, was für die Patienten eine geringere Krankheitslast bei vergleichbarer Wirksamkeit und Sicherheit im Vergleich zur derzeitigen Standardtherapie Aflibercept 2 mg bedeutet.
Brigitte Söllner, Erlangen
Neu diagnostiziertes Multiples Myelom: Isatuximab in Kombination mit VRd als neue Therapieoption
Seit dem 22. Januar 2025 ist Isatuximab (Sarclisa®) in Kombination mit Bortezomib, Lenalidomid und Dexamethason (IsaVRd) zur Behandlung des neu diagnostizierten Multiplen Myeloms (NDMM) bei Erwachsenen zugelassen, die für eine autologe Stammzelltransplantation nicht geeignet sind. Die Vierfachkombination wird bereits in den neuesten NCCN-Leitlinien und in den im Oktober 2024 aktualisierten Onkopedia-Leitlinien zum Multiplen Myelom empfohlen.
Dass die Zulassung von IsaVRd einen neuen Standard in der Erstlinie setzt und sich dadurch neue Perspektiven für die Patienten ergeben – darin waren sich Dr. Lisa Leypoldt, Hamburg, und Dr. Jens Kisro, Lübeck, bei einer Pressekonferenz von Sanofi einig.
on, gefolgt von Rd. Primärer Endpunkt war das progressionsfreie Überleben (PFS). „Mit einem medianen Alter von 72 Jahren bildete das Studien-Kollektiv die klinische Praxis gut ab“, betonte Kisro.
IsaVRd: Prognostiziertes medianes PFS von etwa 7,5 Jahren
Quelle: Wong TY. Three-year outcomes of aflibercept 8 mg in nAMD: safety and efficacy results from the PULSAR extension study. Presented at the 22nd Annual Angiogenesis Meeting in Miami.
IMROZ-Studie mit praxisnaher Patientenklientel
Grundlage der Zulassung sind die Daten der Phase-III-Studie IMROZ. In der Studie wurde IsaVRd mit Bortezomib, Lenalidomid und Dexamethason (VRd) bei neu diagnostizierten Patienten mit Multiplem Myelom verglichen, die nicht für eine autologe Stammzelltransplantation geeignet waren. Die eingeschlossenen 446 Patienten erhielten randomisiert entweder IsaVRd als Induktion, gefolgt von Isatuximab in Kombination mit Lenalidomid und Dexamethason (IsaRd), oder VRd in der Indukti-
Nach einer medianen Beobachtungsdauer von 59,7 Monaten war das mediane PFS (mPFS) unter IsaVRd noch nicht erreicht und betrug unter VRd 54,3 Monate. Unter IsaVRd zeigte sich eine Risikoreduktion für Krankheitsprogression oder Tod von 40 % (HR: 0,596; 98,5%-KI: 0,41 – 0,88; p = 0,001). Die PFS-Rate nach 5 Jahren betrug für Patienten im IsaVRd-Arm 63,4 % vs. 45,2 % bei den Patienten im VRd-Arm. Ausgehend vom derzeitigen Trend wird unter IsaVRd ein medianes PFS von 7,5 Jahren bzw. 90 Monaten prognostiziert. „Diese Ergebnisse sind praxisverändernd. Sollte sich das prognostizierte mediane PFS von 90 Monaten bestätigen, wäre dies das längste PFS, das wir bislang in diesem Patientenkollektiv gemessen haben. IsaVRd ist damit ein neuer Standard für Patienten mit neu diagnostiziertem Multiplen Myelom, die nicht transplantiert werden“, resümierte Kisro.
Tiefes und langanhaltendes Ansprechen bei guter Verträglichkeit
74,7 % der Patienten, die mit IsaVRd behandelt wurden, erzielten eine Komplettremission (CR), über die Hälfte 55,5 % erreichten sowohl eine MRD-Negativität als auch eine CR. Auch hinsichtlich der anhaltenden MRD-Negativität
von mindestens 12 Monaten war IsaVRd gegenüber VRd überlegen. IsaVRd wurde insgesamt gut vertragen, die darunter beobachteten Nebenwirkungen standen im Einklang mit dem bekannten Sicherheitsprofil von Isatuximab und VRd. Behandlungsassoziierte unerwünschte Ereignisse des Grades ≥3 traten bei 91,6 % der mit IsaVRd und bei 84 % der mit VRd behandelten Patienten auf. Ereignisse vom Grad 5 wurden bei 11,0 % der mit IsaVRd und bei 5,5 % der mit VRd behandelten Patienten beobachtet. „Diese numerischen Unterschiede liegen

Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin
Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
weitestgehend an der längeren Behandlungsdauer im IsaVRd-Arm“, erläuterte Kisro (53,2 Monate unter IsaVRd vs. 31,3 Monate im VRd-Arm). Adjustiert ergebe sich eine Rate von 0,03 Ereignissen pro Patientenjahr unter IsaVRd versus 0,02 unter VRd.
Vierfachkombination IsaVRd ermöglicht neue Perspektiven beim NDMM
„Jede weitere Therapielinie erreichen 15 – 35 % weniger Patienten“, berichtete Leypoldt, die einen Überblick zum Multiplen Mye-
lom gab. „Mit der Zulassung von IsaVRd schlagen wir hier ein neues Kapitel in der Erstlinienbehandlung von Patienten mit Multiplem Myelom auf, die nicht transplantiert werden“. Da die Erkrankung mit jedem Rezidiv aggressiver verläuft und somit schwerer zu behandeln ist, ist eine solche effektive Erstlinientherapie umso bedeutsamer“, betonte die Expertin. „Zudem weist IsaVRd im IMROZProtokoll eine hohe Wirtschaftlichkeit auf, die ebenso von großer Bedeutung ist“, bemerkte Dr. Kisro abschließend.
Elisabeth Wilhelmi, München
Titelbild: Die Frühsommermeningoenzephalitis SME) wird durch das Tick-Borne Encephalitis-(TBE-)Virus verursacht. Dieses wird meist durch Zecken auf den Menschen übertragen, in Deutschland überwiegend durch die Spezies Ixodes ricinus (Quelle: iStock Ladislav Kubeš).
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz:
HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R.
Roland Welker Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de
Stabil therapiert 3,4
Zuverlässig geschützt1,3
Individuell appliziert1,2
Alle Vorteile der Fertigspritze finden Sie hier auf einen Blick:
Sekundäre/erworbene Immundefekte
Hizentra® 200 mg/ml Lösung zur subkut. Injektion. Wirkstoff: Normales humanes Immunglobulin. Zusammensetzung: 1 ml Hizentra® enth. 200 mg normales humanes Immunglobulin (Reinheitsgrad mind. 98 % IgG). Verteilung der IgG-Subklassen: IgG1 ca. 69 %, IgG2 ca. 26 %, IgG3 ca. 3 %, IgG4 ca. 2 %. IgA max. 50 μg/ml. Sonst. Bestandteile: L-Prolin, Polysorbat 80, Wasser für Injektionszwecke, Chlorwasserstoffsäure, Natriumhydroxid. Anwendungsgebiete: Substitutionstherapie bei prim. Immundefektsyndromen mit verminderter Antikörperprodukt. sowie bei sekund. Immundefekten mit schweren oder wiederkehrenden Infekt., ineffektiver antimikrobieller Behandlung und entweder ungenügendem Anstieg von Impfantikörpern gegen Pneumokokken-Polysaccharide oder IgG-Serumspiegel < 4 g/l. Immunmodulationstherapie bei chron. inflammator. demyelinisierender Polyneuropathie (CIDP) als Erhaltungstherapie nach Stabilisierung mit IVIG. Gegenanzeigen: Überempfindlichk. gg. den Wirkstoff oder einen der sonst. Bestandteile, Hyperprolinämie Typ I od. II. Nebenwirkungen (Häufigkeit pro Infusion): Sehr häufig: Reakt. an der Infusionsstelle. Gelegentl.: Kopfschmerzen, Durchfall, Bauchschmerzen, Hautausschlag, Schmerzen im Bewegungsapparat, Arthralgie, Müdigk. (einschl. Unwohlsein), Fieber, niedriger Blutdruck, Rückenschmerzen. Selten: Überempfindlichk., Schwindel, Migräne, Tremor, (einschl. psychomotor. Hyperaktivität), Hypertonie, Hautrötung, Übelk., Erbrechen, Pruritus, Urtikaria, Muskelspasmen und -schwäche, Schmerzen im Brustraum, grippeähnl. Symptome, Schmerzen, Schüttelfrost (einschl. Unterkühlung), erhöhtes Kreatinin. Sehr selten: asept. Meningitis, Tachykardie. In Einzelfällen: Anaphylakt. Schock. Häufigk. nicht bekannt: Anaphylakt. Reakt., Brennen, embol. und thrombot. Ereignisse, Ulkus an der Infusionsstelle. Verschreibungspflichtig. Pharmazeutischer Unternehmer: CSL Behring GmbH, Emil-von-Behring-Str. 76, 35041 Marburg. Stand: August 2024.