7 minute read
Kardiale Genetik – hereditäre Kardiomyopathien und Ionenkanalerkrankungen

Dagmar Iris Keller Lang, Christiane Gruner und Ardan Saguner
Gemeinsamkeiten hereditärer Arrhythmien und Kardiomyopathien
Advertisement
7 Ausgeprägte klinische und genetische Heterogenität 7 Können zum Sudden Cardiac Death (SCD) führen 7 Verhinderung des SCD: am effektivsten durch ICDImplantation (subkutaner ICD in Einzelfällen möglich) 7 Vererbung meist autosomaldominant
Rolle der Genetik
7 Bestätigung der klinischen Diagnose resp. des Phänotyps beim
IndexPatienten 7 Deshalb: Genetische Testung beim Indexpatienten nur bei vorhandenem Phänotyp 7 Identifikation von Genotyppositiven Familienmitglieder (auch ohne Phänotyp) 7 RisikoStratifizierung für den SCD aufgrund einer Genmutation nur in seltenen Fällen
Long QT Syndrom (LQTS)
Das LQTS ist charakterisiert durch eine verzögerte Repolarisation im Myokard, welche sich im 12AbleitungsEKG als QTVerlängerung und TWellen Abnormität manifestiert.
Diagnose LQTS
IndexPatient: QTc ≥480ms in mindestens zwei 12Kanal EKGs oder LQTS Risikoscore >3 oder bei eindeutig pathogener Mutation unabhängig vom QTc (Schwartz P.J. Am. Heart J. 1985; 2: 399–411)
Familienangehörige: QTc Männer ≥470 ms/ Frauen ≥480ms oder falls QTc in Ruhe kleiner: Ergometrie: 4min. der Erholungsphase QTc ≥445 ms
Klinik
Klinische Manifestationen sind Synkopen, Krampfanfälle und der plötzliche Herztod meist aufgrund einer Torsade de pointes Tachykardie. Die Prävalenz beträgt ca. 1:2500.
Gene und Proteine
LQTS Major Genes KCNQ1 (LQTS1) KCNH2 (LQTS2) SCN5A (LQTS3) LQTS Minor Genes AKAP9 CACNA1C CALM1 CALM2 CAV3 KCNE1 KCNE2 KCNJ5 SCN4B SNTA1 AnkyrinB Syndrome ANK2 AndersenTawil Syndrome KCNJ2 Timothy Syndrome CACNA1C Iks potassium channel alpha subunit (KvLQT1) Ikr potassium channel alpha subunit (hERG) Cardiac sodium channel (Nav1.5)
Yotiao Voltage-gated L-type calcium channel (Cav1.2) Calmodulin Calmodulin Caveolin-3 Kv7.1 potassium channel beta subunit (MinK9) KV11.1 potassium channel beta subunit (MiRP1) Potassium inwardly-rectifying channel (Kir3.4) Sodium channel beta 4 subunit Syntrophin-alpha 1
Ankyrin B
Ik1 potassium channel (Kir2.1)
Voltage-gated L-type calcium channel (Cav1.2)
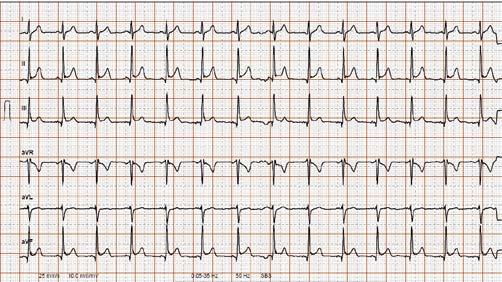
Mutationen in den 3 Hauptgenen KCNQ1, KCNH2 und SCN5A sind für die grosse Mehrheit (90%) der LQTS verantwortlich. Diese können gelegentlich auf dem EKG unterschieden werden (Referenzableitung II oder V2/V3; Abb. 1): LQTS1: breite TWelle LQTS2: niedrige TWellenAmplitude, double notch TWelle LQTS3: später Beginn der TWelle, Morphologie frequenzabhängig
Abbildung 1: Verschiedene Typen des Long-QT-Syndroms
Auslöser/Trigger für ventrikuläre Rhythmusstörungen
7 LQTS1: Sport, v.a. Schwimmen 7 LQTS2: Laute Geräusche in Ruhe (z.B. Wecker) 7 LQTS3: Bradykardien, emotionaler Stress
Genetische Testung bei LQTS
Immer nach obligatorischer genetischer Beratung in einer Sprechstunde für Rhythmologie/Kardiogenetik. Genetische Testung in einem Institut für Medizinische Molekulargenetik. Eine Kostengutsprache ist obligatorisch.
Das BrS ist charakterisiert durch eine typische zeltförmige STStreckenhebung (covedtype) mit präterminal negativer TWelle in den rechtspräkordialen Ableitungen V1V2 (PseudoRechtsschenkelblock) im 12KanalEKG (BrS Typ 1 EKG; Abb 2). Typisch sind folgende Symptome 1. Rhythmogene Synkopen 2. Nächtliche Agonie 3. Plötzlicher Herztod Die Prävalenz beträgt ca. 1:1000–10000 (je nach Region, in Südostasien gehäuft)
EKG Phänotypen
Typ 1 JWellen Amplitude >2mm TWelle negativ STKonfiguration zeltförmig STSegment terminal deszendierend Typ 2 erhöht positiv/biphasisch sattelförmig erhöht >0.5mm

Abbildung 2: Brugada Syndrom EKG Das früher definierte Typ 3 Brugada EKG wurde in den aktuellen Konsensuspapieren abgeschafft. Ein BrS Typ 2 EKG ist nicht diagnostisch für ein Brugada Syndrom; ein Typ 1EKG muss demaskiert werden durch protokollierte i.v. Gabe eines Klasse 1A oder 1C Antiarrhythmikums (z.B. Ajmalin oder Flecainid; nur bei monitorisierten Patienten in ReanimationsBereitschaft!).
Gene SCN5A GPD1-L SCN1B SCN3B SCN2B RANGRF SLMAP KCNE3 KCNJ8 HCN4 KCNE5 KCND3 CACNA1C CACNB2B CACNA2D1 TRPM4 Proteine Nav1.5 glycerol-3-P-DH-1 NavB1 NavB3 NavB2 RAN-G-release factor sarcolemma associated protein MiRP2 Kv6.1 Kir6.1 hyperpolarization cyclic nucleotide-gated 4 K voltage-gated subfamily E member 1 like Kv4.3 Kir4.3 Cav1.2 voltage-dependent B-2 voltage-dependent A2/D1 transient receptor potential M4
Mutationen im Gen SCN5A sind für ca. 30% des BrS verantwortlich
Genetische Testung bei BrS
Immer nach obligatorischer genetischer Beratung in einer Sprechstunde für Rhythmologie/Kardiogenetik. Genetische Testung in einem Institut für Medizinische Molekulargenetik.
Catecholaminerge Polymorphe Ventrikuläre Tachykardie (CPVT)
Die CPVT manifestiert sich v.a. bei Kindern und jüngeren Patienten durch emotionale und körperliche Anstrengung (Sport) mit polymorphen ventrikulären Rhythmusstörungen, welche zu Synkopen und plötzlichem Herztod führen können. Schwimmen ist ein Trigger für Rhythmusstörungen. Das 12KanalRuhe EKG ist bei der CPVT normal. Diagnostisch ist die Ergometrie/Stresstest mit ventrikulären Rhythmusstörung (ventrikuläre Ektopie, polymorphe VT, bidirektionale VT, selten Kammerflimmern) während und in der frühen Erholungsphase nach der Belastung.
Gene Protein RyR2 cardiac ryanodine receptor CASQ2 calsequestrin-2 protein TRDN tradin CALM1 calmodulin
Genetische Testung bei CPVT
Immer nach obligatorischer genetischer Beratung in einer Sprechstunde für Rhythmologie/Kardiogenetik. Genetische Testung in einem Institut für Medizinische Molekulargenetik.
Hypertrophe Kardiomyopathie (HCM)
Die HCM ist die häufigste hereditäre Kardiomyopathie und ist definiert als: 7 Unerklärte linksventrikuläre (LV) Hypertrophie in Abwesenheit einer anderen Ursache (arterielle Hypertonie, Aortenstenose,
Speicherkrankheiten, Amyloidose u.a.m.) 7 Maximale LV Wanddicke >15 mm 7 Kardiale Symptome wie Tachyarrhythmien, Dyspnoe und/oder
Angina in 50% der Fälle; die übrigen 50% werden im Rahmen des Familienscreenings oder Abklärung von EKGVeränderungen diagnostiziert 7 EKGAbnormitäten (Abb. 3) Der klinische Phänotyp variiert stark von asymptomatisch bis hin zu Herzinsuffizienz und zum plötzlichen Herztod als Erstmanifestation. Der morphologische Phänotyp divergiert zwischen minimer bis extensiver Hypertrophie mit anatomischen Subtypen, wenig oder massiver myozytärer Desorganisation/Fibrose, fehlender oder massiver LVOT Obstruktion. Die Prävalenz beträgt ca. 1:500.
Komplikationen der hypertrophen Kardiomyopathie
7 Obstruktion des linksventrikulären Ausflusstrakts (50–75% der
Fälle) • Therapie: Medikamente (negativ inotrop und negativ chronotrop; Vermeidung von Nachlastsenkung, zu starke Vorlastsenkung), invasive Optionen bei NYHA III trotz ausgebauter
Medikation ins Auge fassen (Septalalkoholablation, chirurgische Myektomie) in hierfür erfahrenen Zentren.
7 Vorhofflimmern (25% der Fälle) • Orale Antikoagulation unabhängig von CHA2DS2VAsc Score wegen hohem Thrombembolierisiko, Rhythmuskontrolle ist wichtig! (keine ICAntiarrhythmika, kein Sotalol) 7 Plötzlicher Herztod (Risiko ca. 0,6%/Jahr; Normalbevölkerung ca. 0,3%/Jahr) • Regelmässige Risikostratifzierung (Faktoren s. unten) 7 Systolische Herzinsuffizienz (ca. 5–10% der Fälle) • Bereits bei einer LVEF <50% liegt eine Herzinsuffizienz vor • Bei LVEF <50% wird eine primärprophylaktische
ICDImplantation empfohlen unabhängig von den untenstehenden Risikofaktoren. • Medikation gemäss den Richtlinien der Herzinsuffizienz 7 Diastolische und mikrovaskuläre Dysfunktion (50% der Fälle) • Therapie: Betablocker oder Kalziumantagonisten vom
Verapamiltyp

Abbildung 3: 12-Kanal EKG eines Patienten mit Hypertropher Kardiomyopathie
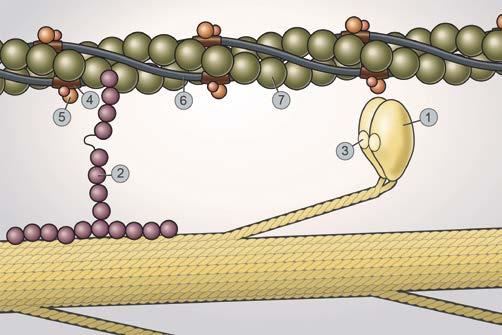
Abbildung 4: Mutationen in den Genen für verschiedener kontraktile Proteine bei hypertropher Kardiomyopathie: 1 β-Myosin heavy chain; 2 Myosin-binding protein-C; 3 Myosin light chain 2 and 3; 4 Troponin T; 5 Troponin I; 6 Tropomyosin; 7 Actin
Risikostratifizierung für den SCD
Hauptrisikofaktoren SEKUNDÄRPROPHYLAXE Überlebter SCD, VF oder anhaltende VT PRIMÄRPROPHYLAXE Positive Familienanamnese für SCD (erstgradige Familien-Angehörige <50 Jahre) Maximale linksventrikuläre Wanddicke >30 mm Nicht-erklärbare Synkopen in den letzten 6–12 Monaten Nicht-anhaltende Kammertachykardien Abnormales BD-Verhalten während Belastungstest (Blutdruckabfall, oder Anstieg <20mmHg) Risikomodifizierende Faktoren LVOT-Obstruktion Mindestens moderate myokardiale Fibrose (late gadolinium enhancement) im Herz-MRI Linksventrikuläres apikales Aneurysma LVEF <50% >2 krankheitsverursachende Gen-Mutationen
Neben obgenannten Faktoren, gibt es neu auch einen Risikokalkulator der ESCGuidelines, welcher für die Beurteilung des Risikos für plötzlichen Herztod herangezogen werden kann (Link: www.doc2do.com/hcm/webHCM.html). Wichtig ist, dass die Beurteilung des Risikos für plötzlichen Herztod vor allem in Grenzfällen durch einen in HCM erfahrenen Kardiologen erfolgt.
Genetik bei HCM
Die HCM wird autosomaldominant vererbt mit variabler Expressivität und altersabhängiger Penetranz. Heute sind mind. 27 Gene involviert, wobei bei max. 30–40% aller Fälle eine Mutation identifiziert wird wobei die meisten (70%) Mutationen in den 2 Hauptgenen MYH7 und MYBPC3 gefunden werden (Abb. 4). Bisher ist es so, dass die genetische Untersuchung vor allem durchgeführt werden sollte bei fraglicher Diagnose (wenn morphologisch auch Speicherkrankheiten in Frage kommen), da die HCMPanels immer auch die Gene der Phänokopien enthalten (z.B. Morbus Fabry, TransthyretinAmyloidose, PRKAG2Kardiomyopathie, Morbus Danon). Ansonsten ist die Genetik momentan v.a. für das Familienscreening hilfreich, wenn eine krankheitsverursachende Mutation bekannt ist! Eine genetische Testung sollte nur nach obligatorischer genetischer Beratung in spezialisierten Zentren erfolgen.
Familienscreening
Unabhängig von der Genetik ist es von grösster Wichtigkeit, dass die erstgradigen Familienangehörigen in regelmässigen Abständen (alle 3–5 Jahre!) mittels EKG (Abb. 3) und Echokardiographie untersucht werden, da sich die Krankheit in jedem Lebensalter manifestieren kann. Kinder und Jugendliche in der Adoleszenz sollten alle 12–18 Monate untersucht werden, da sich die Krankheit gehäuft in der Phase des Längenwachstums ausbildet.
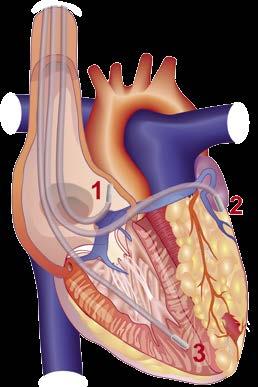
Die ARVC wird meist autosomaldominant vererbt und ist charakterisiert durch einen Ersatz des rechtsventrikulären (RV) und selten auch linksventrikulären Myokards durch Fibrose und Fett (Abb. 5). Klinisch präsentiert sich die ARVC heterogen mit einem breiten SymptomSpektrum von ventrikuärer Arrhythmien, die zu Palpitationen, Schwindel, Synkopen und dem SCD führen, bis hin zur biventrikulären Herzinsuffizienz. Die Prävalenz beträgt ca. 1:2000–1:5000. Die seltene (1:1 Mio.) NaxosErkrankung ist eine autosomalrezessive Form der ARVC charakterisiert durch zusätzliche palmoplantare Keratosen und wolligem Kopfhaar. Mutationen werden bei IndexPatienten mit ARVC in ca. 50–60% der Fälle identifiziert, und betreffen am häufigsten das desmosomale Gen PKP2.

Abbildung 5: Morphologisches Bild der arrhythmogenen rechtsventrikulären Cardiomyopathie mit Fettumbau und Fibrose vor allem des rechten Ventrikels.
EKG
Negative TWellen in V1–V3 (Abb. 6 rechts) sowie verzögerter «Szacken upstroke» in V1–V3 (+/− Epsilonwelle; Abb. 6 links).

Abbildung 6: ARVC EKG (Repolarisationsstörungen, rechts, und Depolarisationsstörungen, links)
Morphologie
In der Bildgebung rechtsventrikuläre Dilatation und Aneurysmata (v.a. subtrikuspidal) mit im Verlauf eingeschränkter Funktion, die z.B. im MRI visualisiert werden kann.
Wichtigste Gene und Proteine
Gene PKP2 DSG2 DSC2 DSP JUP TGFB-3 RyR2 Transmembrane Proteine Plakophilin-2 Desmoglein-2 Desmocollin-2 Desmoplakin Plakoglobin Transforming growth factor beta-3 Ryanodine receptor protein 43
Genetische Testung bei ARVC
Immer nach genetischer Beratung in einer spezialisierten ARVC Sprechstunde. Eine Kostengutsprache ist obligatorisch.