SELTENE ERKRANKUNGEN

Eosinophile Granulomatose mit Polyangiitis (EGPA) Vanessa Rennspieß hat EGPA und wünscht sich bessere Aufklärung und kürzere Diagnosewege, um die Belastung Betroffener zu mindern.
Seite 05
Morbus Fabry und Morbus Pompe
Im Interview spricht Dr. med. Christina Lampe über diese seltenen lysosomalen Speichererkrankungen.
Seite 10
"Trotz aller Herausforderungen empfinde ich mein Leben als wunderbar!"
Sara Franke ist von der seltenen Knochenstoffwechselstörung XLH betroffen und berichtet im Interview über ihr Leben mit dieser seltenen Erkrankung. Seite 14
EINE UNABHÄNGIGE KAMPAGNE VON MEDIAPLANET Lesen Sie mehr auf www.seltenekrankheiten.de
VERANTWORTLICH FÜR DEN INHALT IN DIESER AUSGABE
Miriam Hähnel
Die vier Millionen Menschen in Deutschland, die von einer seltenen Erkrankung betroffen sind, müssen gehört werden.
Schenken Sie ihnen Aufmerksamkeit: Auch über den Tag der Seltenen Erkrankungen hinaus!
IN DIESER AUSGABE


06
Leben mit NDM
Manuela Albert hat eine Nichtdystrophe Myotonie und erzählt von ihrem schwierigen Weg zur richtigen Diagnose.
08

“Ich bin wie alle!“
Felix hat das seltene Alagille-Syndrom. Er und seine Mutter berichten, was die Krankheit für ihn und ihre Familie bedeutet.

12
“Die besten Informationen bekommen Patienten von anderen Betroffenen.“
Natascha Sippel-Schönborn, Morbus Fabry-Betroffene und Geschäftsführerin der Morbus Fabry Selbsthilfegruppe e. V., im Interview.
Director Business Development Health: Miriam Hähnel, Geschäftsführung: Richard Båge (CEO), Henriette Schröder (Managing Director), Philipp Colaço (Director Business Development), Lea Hartmann (Head of Design), Cover: Privat Mediaplanet-Kontakt: de.redaktion@ mediaplanet.com
Alle Artikel, die mit “In Zusammenarbeit mit“ gekennzeichnet sind, sind keine neutrale Redaktion der Mediaplanet Verlag Deutschland GmbH. Aus Gründen der besseren Lesbarkeit wird auf die gleichzeitige Verwendung der Sprachformen männlich, weiblich und divers (m /w/d) verzichtet. Alle Personenbezeichnungen gelten gleichermaßen für alle Geschlechter.
Forschung braucht Vernetzung, Förderung und eine strukturelle Finanzierung
Liebe Leserinnen und Leser, der letzte Tag im Februar ist der Tag der Seltenen Erkrankungen. Betroffene Menschen, Angehörige und Unterstützer weltweit schaffen Aufmerksamkeit für ein Thema, das in der Forschung, der Politik, im Gesundheitswesen und der öffentlichen Wahrnehmung viel zu oft in den Schatten gedrängt wird: Seltene Erkrankungen. Die Errungenschaften der letzten Jahre wie die Etablierung der Zentrenstruktur, neue Therapieansätze, einige Medikationen sowie eine gewachsene Medienaufmerksamkeit können nicht darüber hinwegtäuschen, dass das Leid der vier Millionen Menschen, die in Deutschland von Seltenen Erkrankungen betroffen sind, noch immer groß ist.
Die ACHSE, Dachverband und Netzwerk von und für Menschen mit Seltenen Erkrankungen, bündelt die Anliegen und Bedarfe der Betroffenen seit nunmehr 20 Jahren und verschafft ihnen Gehör. Da ist der Kampf um Heil- und Hilfsmittel, um Arzneimittel und pflegerische Unterstützung. Finanzielle Nöte, auch weil Kind oder Partner zu Hause gepflegt werden. Viele Betroffene fühlen sich allein gelassen, es gibt kaum strukturelle Entlastung. Pandemie, Krieg und Krisen hinterlassen Spuren, zusätzliche Einschnitte sind allseits spürbar. Dazu der Mangel an Fachkräften im Bereich Pflege und Medizin. Nun sind es die Weiterentwicklung des Gesundheitswesens sowie geplante Reformen, die geradezu existenzielle Ängste bei den Betroffenen hervorrufen. Eltern schwerkranker Kinder fürchten, dass die Chancen auf Therapieentwicklungen weiter sinken. Viele fragen sich, ob sie ihre Medikamente in Zukunft noch kaufen können oder diese gar erhalten.
Fragen Sie Betroffene, was diese sich am meisten wünschen, so ist es „Heilung“ oder zumindest eine Therapie, die ihr Leid, das des Kindes, des Partners oder der Partnerin lindert. Es sind Fortschritt und Innovation, die neue Diagnoseverfahren, Therapie- und Behandlungsmöglichkeiten schaffen. Für Kinder und Erwachsene mit Seltenen Erkrankungen bedeuten sie eine Chance auf ein längeres, besseres Leben und Hoffnung auf Heilung. Digitalisierung, Genomsequenzierung, Neugeborenen-Screening sind nur drei Bereiche, die neue Türen öffnen.
Wir danken folgenden Partnern für die Zusammenarbeit:
Amicus Therapeutics GmbH
www.amicusrx.de
Chiesi GmbH
www.chiesirarediseases.de
Dr. Falk Pharma GmbH
www.drfalkpharma.de
Doch Forschung braucht Vernetzung, Förderung und eine strukturelle Finanzierung. Die Zentren für Seltene Erkrankungen, die nun zum Teil seit mehr als zehn Jahren ausgezeichnete Arbeit leisten, benötigen endlich eine nachhaltige Finanzierung, auch um ihren Forschungsauftrag durchführen zu können. Im Eckpunktepapier für die geplante Krankenhausreform kommen die Menschen mit Seltenen Erkrankungen nicht vor. Das muss geändert werden.
Immer am letzten Tag im Februar richten wir die Scheinwerfer auf die Seltenen Erkrankungen. Die Anliegen und Bedarfe der Betroffenen gelten 365 Tage im Jahr.
Ihre Eva Luise Köhler

Es sind Fortschritte und Innovationen, die neue Diagnoseverfahren, Therapie- und Behandlungsmöglichkeiten schaffen.
Eva Luise Köhler, Schirmherrin der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V.
Menschen mit Seltenen Erkrankungen brauchen die ACHSE. Unterstützen Sie die ACHSE mit Ihrer Spende!
Spendenkonto: Bank für Sozialwirtschaft
IBAN: DE89 3702 0500 0008 0505 00
Weitere Informationen: achse-online.de/jetzt_spenden
DEUTSCHEFACHPFLEGE www.deutschefachpflege.de
GlaxoSmithKline GmbH & Co. KG de.gsk.com
Hormosan Pharma GmbH www.hormosan.com
Kyowa Kirin GmbH www.kyowakirin.com
Mirum Pharmaceuticals Germany GmbH www.mirumpharma.com
Novartis Pharma GmbH www.novartis.de
Sächsisches Staatsministerium für Wissenschaft, Kultur und Tourismus www.smwk.sachsen.de
Lesen Sie mehr auf seltenekrankheiten.de 2 Please recycle facebook.com/MediaplanetStories @Mediaplanet_germany
Text Eva Luise Köhler
FOTO: ANDREA KATHEDER
ORPHAN DRUGS –
viele Patienten warten auf Therapien!
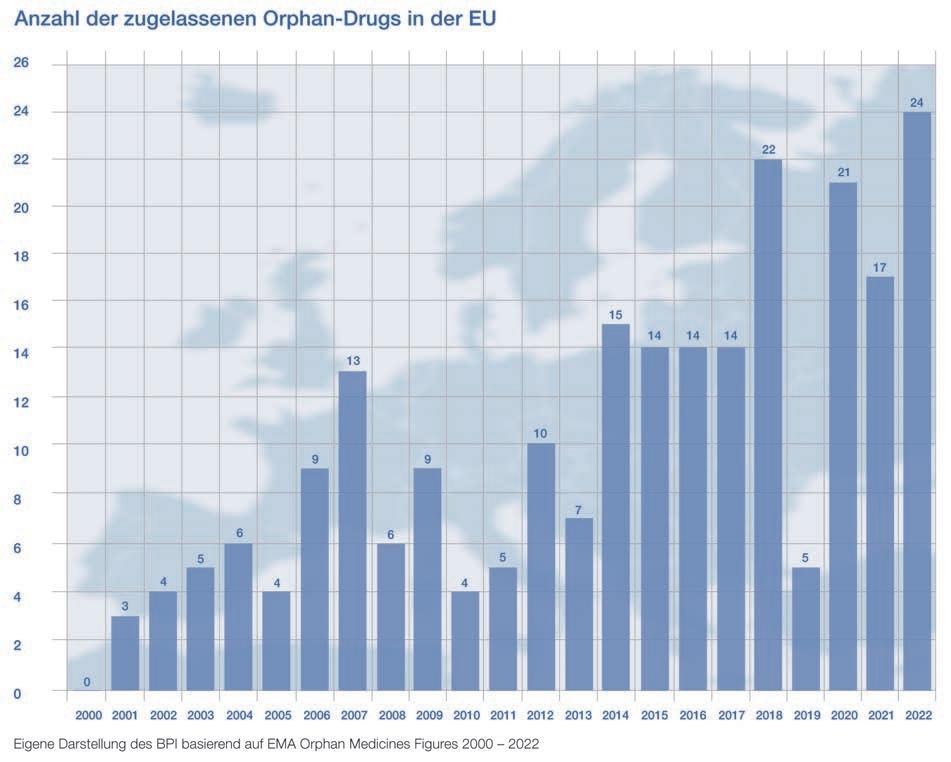
Derzeit gibt es ca. 200 Arzneimittel zur Behandlung eines Seltenen Leidens (Orphan Drug). Etwa 8.000 verschiedene Seltene Erkrankungen sind bekannt. Der therapeutische Bedarf ist also weiterhin groß! Orphan Drugs können erheblich zu einer Verbesserung der Lebensqualität von Patientinnen und Patienten beitragen. Sie wirken sich positiv auf den nachfolgenden Pflege- und Behandlungsaufwand der Betroffenen aus.
Text Dr. Matthias Wilken
Neue Therapien stehen Patientinnen und Patienten in Deutschland so schnell und umfassend zur Verfügung wie in keinem anderen europäischen Staat. Eine gute Nachricht –denn ein rascher Zugang ist im Sinne der Patientinnen und Patienten. Pharmazeutische Unternehmen tun alles dafür, dieser Verpflichtung auch in Zukunft nachzukommen. Denn Menschen mit einer Seltenen Erkrankung haben das gleiche Recht auf eine gute Gesundheitsversorgung wie Menschen mit häufigen Erkrankungen.
Das Besondere bei der Forschung im Bereich der Seltenen Erkrankungen ist, dass die Erkenntnisse weit über ihr Indikationsgebiet hinaus strahlen. Das gewonnene Wissen aus dem Krankheitsgeschehen einer Seltenen Erkrankung lässt sich meist auch auf andere Krankheitsbilder übertragen. Je mehr Faktoren eines Krankheitsverlaufs bekannt sind, umso eher können pharmazeutische Unternehmen präzise Therapien entwickeln. Forschung an Seltenen Erkrankungen ist also auch „das Labor“ für häufige Erkrankungen –es profitieren mittelbar auch Menschen mit anderen Erkrankungen.

Stabile politische Rahmenbedingungen sind für die Arzneimittelentwicklung das A und O.
Dr. Matthias Wilken Geschäftsführer BPI (Bundesverband der Pharmazeutischen Industrie e.V.) , Market Access, Märkte und Versorgung
Wichtig ist, dass der Arzneimittelmarkt preispolitisch so aufgestellt bleibt, dass es Unternehmen möglich ist, Therapien für nur wenige Patientinnen und Patienten zu entwickeln. Stabile politische Rahmenbedingungen sind dafür das A und O.
Leider weisen aktuelle Entwicklungen in eine andere Richtung: Auf europäischer Ebene werden Einschnitte im Anreizsystem zur Entwicklung von Orphan Drugs diskutiert. Und auch auf nationaler Ebene wird der sozialrechtliche Sonderstatus immer wieder in Frage gestellt.
Da im Vergleich zu häufigen Erkrankungen wie Diabetes oder Hypertonie (Bluthochdruck) nur sehr wenige Patientinnen und Patienten diese speziellen Arzneimittel brauchen, entwickelt die pharmazeutische Industrie Orphan Drugs unter ganz besonderen Bedingungen. Forschende stehen bei der Vorbereitung und Durchführung von Studien vor großen Herausforderungen. Das fängt schon bei der Datengenerierung an: Meist ist das Wissen zu der jeweiligen Erkrankung sehr begrenzt. Die Patientenpopulationen sind klein, heterogen und geografisch oft weit verteilt. Die Rekrutierung von Probanden ist daher sehr schwierig und kostenintensiv – das Investitionsrisiko für pharmazeutische Unternehmen wiederum sehr hoch. Entscheidet sich ein Unternehmen, Therapieansätze für eine Seltene Erkrankung zu erforschen, sucht es nach der Stecknadel im Heuhaufen. Auch die Entwicklung eines Orphan Drugs ist sehr aufwändig: Sie kann bis zu 15 Jahre dauern und mehrere hundert Millionen bis Milliarden Euro kosten. Hersteller müssen also in der Lage sein, diese Kosten zu refinanzieren. Da der Absatzmarkt durch kleine Patientenpopulationen aber stark begrenzt ist, ergeben sich auch höhere Preise.
Als Bundesverband der Pharmazeutischen Industrie (BPI) engagieren wir uns seit Jahren für verbesserte Diagnose- und Therapieoptionen. Die Versorgung ist ein Prozess: Entscheidend ist eine frühzeitige Diagnose. Das setzt voraus, dass Ärztinnen und Ärzte bei der Diagnosestellung auch an Seltene Erkrankungen denken. Oft vergehen etliche Jahre, bis Patientinnen und Patienten die zutreffende Diagnose erhalten. Aufgrund der Seltenheit und Komplexität der Krankheitsbilder ist es für viele Menschen ein langer Weg, bis sie von einer adäquaten Therapie profitieren können. Patientenlotsen in Zentren für Seltene Erkrankungen können bei unklaren Diagnosen helfen, diesen Prozess zu beschleunigen.
Therapien machen dann den Unterschied – doch Patientinnen und Patienten, die von ihnen profitieren, müssen erst einmal „gefunden“ werden. Um diesen Prozess integrativ zu denken, machen wir uns als BPI im Rahmen des Nationalen Aktionsbündnisses für Menschen mit Seltenen Erkrankungen (NAMSE) stark. Im Zusammenschluss mit Bundesministerien und über 20 Bündnispartnern setzen wir uns dafür ein, dass alle Patientinnen und Patienten eine Chance auf eine zeitnahe Diagnose und Therapie ihrer Seltenen Erkrankung erhalten.
Bei Seltenen Erkrankungen handelt es sich oft um genetisch bedingte Erkrankungen, oft sind Kinder betroffen. Für viele schwerkranke Patientinnen und Patienten, bei denen andere Therapien meist ausgeschöpft sind, können mitunter Arzneimittel für neuartige Therapien (ATMP) zum Einsatz kommen.
WEITERE INFORMATIONEN ZUM THEMA FINDEN SIE IM BPI-THEMENDIENST „ATMP“
UND IM BPI-THEMENDIENST „SELTENE ERKRANKUNGEN“
Lesen Sie mehr auf seltenekrankheiten.de 3
FOTO: BPI/KRUPPA


Weil Pflege so viel mehr ist.
"Auch digital lassen sich besondere Momente mit Angehörigen teilen."
Text Hanna Sinnecker
Herr Klein, die außerklinische Versorgung steht bei der DEUTSCHENFACHPFLEGE besonders im Fokus. Was sind die Herausforderungen in der Intensivpflege von Menschen mit Seltenen Erkrankungen? Im Grundsatz sind die Anforderungen nicht anders als bei allem, was wir ohnehin tun. Die Menschen, die sich uns anvertrauen, sollen sich in ihren individuellen Bedürfnissen verstanden und gut betreut fühlen können. Dieser zwischenmenschliche Aspekt steht bei uns im Mittelpunkt. Das ist eine anspruchsvolle Aufgabe. Denn in der außerklinischen Intensivpflege befinden sich die Menschen und ihre Angehörigen häufig in einer Ausnahmesituation, die sie psychisch mitnimmt und oft überfordert. Wir tun sehr viel, um unsere Pflegekräfte dafür zu sensibilisieren. Diese zwischenmenschlichen Aspekte, Einfühlungsvermögen und Geduld sind fundamental. Deshalb müssen unsere Pflegekräfte besonders geschult sein und auch die Besonderheiten der Seltenen Erkrankungen kennen.

Die Verbesserung der Pflege ist ein Prozess, der nie abgeschlossen sein wird.
Daniel Klein CEO DEUTSCHE FACHPFLEGE
Seltene Erkrankungen sind oft sehr komplex und erfordern ein hohes Maß an Wissen um die Erkrankung. Wie stellen Sie sicher, dass Menschen mit Seltenen Erkrankungen bestmöglich versorgt werden können?
Das A und O in der außerklinischen Intensivpflege ist die Schulung unserer Pflegekräfte. Die bieten wir auch über unsere hausinterne Pflegeleicht Akademie an. Das geht bei uns weit über die gesetzlichen Mindestanforderungen hinaus. Wir unternehmen aber auch große Anstrengungen, um die Pflegekräfte durch digitale
Innovationen und Künstliche Intelligenz zu unterstützen. So arbeiten wir daran, dass wir künftig die Vitalwerte durch digitale Lösungen rund um die Uhr überwachen können. Dadurch werden unsere Pflegekräfte früh auf mögliche Abweichungen von den Normalwerten aufmerksam gemacht und können rasch intervenieren. Das zweite große Gebiet ist das Fachteam medizinische Behandlungspflege, das wir in den vergangenen Jahren aufgebaut haben. Eine große Herausforderung bei der Pflege von Menschen mit Seltenen Erkrankungen ist, dass neben der Hauptdiagnose häufig unterschiedlichste Nebendiagnosen festgestellt werden. Dadurch gleicht keine Betreuung der anderen. Im FmB, wie wir das abkürzen, bringen wir für jeden betreuten Menschen individuell die notwendigen Fachkompetenzen zusammen: Atmungstherapeuten, Fachkrankenpfleger und andere Spezialisten. Auch mit den Fachärzten kooperieren wir dabei eng. Im Jahr 2020 haben wir diese Innovation eingeführt – und die Ergebnisse geben uns recht. Schließlich ist ein zentraler Bestandteil für uns das Qualitätsmanagement, in das wir große Ressourcen investieren. Die Verbesserung der Pflege ist ein Prozess, der nie abgeschlossen sein wird. Neben dem Qualitätsmanagement pflegen wir deshalb einen engen Kontakt zu Forschung und Wissenschaft, um fortwährend neueste wissenschaftliche Erkenntnisse oder das Wissen anderer Fachgebiete in unsere Pflegekonzepte einfließen zu lassen.
Menschen mit Seltenen Erkrankungen haben oft viele Krankenhausaufenthalte hinter sich und fühlen sich daher im häuslichen Umfeld wohler. Wie können Krankenhauseinweisungen minimiert werden? Manche Vertreter im Gesundheitswesen halten die stationäre Betreuung für überlegen. Das ist meiner Meinung nach falsch. Unsere Erhebungen sprechen eine andere Sprache: Die Menschen, die auf außerklinische Intensivpflege angewiesen sind, wollen vor allem Selbstbestimmung über ihr Leben, das steht für sie an oberster Stelle. An zweiter Stelle kommt Nähe zu den Angehörigen. Wir überfordern unser Gesundheitssystem, wenn wir glauben, wir könnten in Deutschland flächendeckend genügend Heime zur Betreuung dieser Personengruppe aufbauen. Deshalb sollte die außerklinische Intensivpflege einen angemessenen Stellenwert im Gesundheitswesen bekommen.
Ich bin der Meinung, dass jeder selbst die Möglichkeit haben sollte, zu entscheiden, ob er oder ob sie lieber in einem Heim, in der eigenen Häuslichkeit oder in einer wohnortnahen Wohngemeinschaft betreut werden möchte. Ich wünsche mir ein System, in dem ideologiefrei die verschiedenen Betreuungsformen nebeneinander existieren. Denn ich bin der festen Überzeugung: Das Selbstbestimmungsrecht ist unantastbar. Wir können nicht wegdiskutieren, dass Lebensqualität in unmittelbarem Zusammenhang mit dem häuslichen Milieu steht.
Das FmB und eine akkurate Analyse der Diagnosen sind neben einer qualitativ hochwertigen Pflege aus meiner Sicht die wesentlichen Elemente, um den Zustand der Menschen, die sich uns anvertrauen, zu stabilisieren und, wenn möglich, zu verbessern. So können wir Krankenhausaufenthalte reduzieren, die für die betroffenen Menschen, aber auch für die Krankenhäuser belastend sind.
Die Digitalisierung spielt auch bei der Intensivversorgung von Menschen mit Seltenen Erkrankungen eine immer größere Rolle. Können Sie uns dazu mehr erzählen?
Lassen Sie mich einen Vergleich aus dem Sport nehmen: Bisher wurden Laktatwerte mit Blutproben am Ohrläppchen nach dem Training gemessen. Bis dahin sind die Spitzenbelastungen jedoch schon abgeklungen. Mit digitalen Lösungen können Sie Blutwerte während der Belastung messen. Die sind viel genauer. Genauso können Sie auch mit klugen digitalen Lösungen und Künstlicher Intelligenz die Vitalwerte von Menschen, die Intensivpflege benötigen, fortwährend messen und Veränderungen unmittelbar und schneller wahrnehmen. Das ist wichtig, um Komplikationen vorzubeugen, damit wirkliche Risiken gar nicht erst entstehen. Das ist das Ziel: Wir wollen durch Künstliche Intelligenz Risiken und Komplikationen offensiv und gezielt entgegenwirken. Wie können Angehörige von Betroffenen in den Pflegeprozess eingebunden und vor allem entlastet werden?
Die Angehörigen sind in der außerklinischen Intensivpflege enorm wichtig. Wenn Menschen, die Intensivpflege benötigen, in einem häuslichen Umfeld leben, in dem sie sich wohlfühlen, verbessert sich ihre gesamte psychische und mentale Grundeinstellung. Und das kann sich nur positiv auf ihre gesundheitliche Entwicklung auswirken.
Ganz wichtig ist aber auch, dass sich die Angehörigen nicht überfordert fühlen. Sie brauchen eine Unterstützung, die ihnen nicht nur Aufgaben abnimmt. Sie muss auch fachlich so hochwertig sein, dass sich die Angehörigen entlastet und gut aufgenommen fühlen. Dieser Aspekt ist ganz wichtig, wenn wir eine Rückzugspflege erfolgreich durchführen wollen. Und wir wollen die Menschen ja darin unterstützen, wieder ein Mehr an Autonomie zu gewinnen. Manchmal überfordert aber auch eine häusliche Unterbringung die Angehörigen. Deshalb haben wir in 15 der 16 Bundesländer Intensivpflege-Wohngemeinschaften gegründet, in denen die Menschen selbstbestimmt in der Nähe ihrer Angehörigen leben können. Das bietet einen unglaublichen Gewinn an Lebensqualität.
Lesen Sie mehr auf seltenekrankheiten.de 4
Weitere Informationen: www.deutschefachpflege.de
Artikel ist in Zusammenarbeit mit der DEUTSCHENFACHPFLEGE entstanden.
Dieser
FOTOS: DEUTSCHE FACHPFLEGE HOLDING GMBH
„Unsicherheit und Angst sind meine ständigen Begleiter.“
Rheumatische Erkrankungen - da denkt man zunächst an eine Volkskrankheit, von der viele Menschen betroffen sind. Doch es gibt auch eine Reihe seltener rheumatischer Erkrankungen, zu denen die sogenannten Vaskulitiden gehören, die durch eine Entzündung der Blutgefäße gekennzeichnet sind. Vanessa Rennspieß ist von der eosinophilen Granulomatose mit Polyangiitis (kurz EGPA) betroffen und spricht mit uns über ihr Leben mit der Krankheit und ihre Wünsche für Betroffene.
Text Alexandra Lassas
Frau Rennspieß, sie haben EGPA und ihre Beschwerden haben bereits vor 26 Jahren begonnen. Mit welchen Symptomen hat sich die Erkrankung gezeigt?
Die Symptome meiner EGPA-Erkrankung sind schwer eindeutig zuzuordnen, da ich bereits an einer systemischen Sklerose litt und der Weichteilrheumatismus meine Beschwerden verstärkte. Die Beschwerden schlichen sich nach und nach in mein Leben ein, begleitet von häufigen, plötzlich auftretenden Nasennebenhöhlenentzündungen. Selbst sportliche Aktivitäten wie regelmäßiges Walken wurden durch die Verschlechterung der Atmung in Verbindung mit dem Heuschnupfen zur Qual und es entwickelte sich Asthma. Die Abstände zwischen den Phasen, in denen es mir gut ging, wurden immer kürzer.
Auch für erfahrene Ärzte ist es nicht leicht, die Symptome in Zusammenhang zu bringen und die richtige Diagnose zu stellen. Können Sie uns von Ihrem Weg zur Diagnose erzählen?
Nach einem Aufenthalt in einer Rehaklinik wurde mir die Diagnose Raynaud-Syndrom mitgeteilt. Zu diesem Zeitpunkt hatte ich bereits offene Stellen an den Händen, Schmerzen bei einfachsten Bewegungen und beim Atmen. Die offenen Stellen schränken mich in meiner Freizeit und im Haushalt sehr stark ein.
Normale Dinge wie Staubsaugen, Kochen oder Putzen gehen zum Teil nur mit Handschuhen. Auch bei meiner Arbeit stoße ich an meine Grenzen. Kurz nach der Geburt meiner Tochter traten häufig blaue, taube Finger auf. In den Jahren darauf verschlimmerten sich alle Symptome. Zahlreiche Arztbesuche und Überweisungen brachten keine eindeutige Diagnose, bis ich schließlich einen Rheumatologen aufsuchte, der mich gründlich untersuchte und die systemische Sklerose diagnostizierte. Parallel dazu wurde ich in der Uniklinik wegen meiner offenen Hautstellen behandelt, erhielt aber von der Dermatologie viele Diagnosen, mit denen ich wenig anfangen konnte. Im Alltag musste ich lernen, mit den Symptomen zu leben. Ständig neue Diagnosen machten mich unsicher und verzweifelt.

Dieser
FOTO: PRIVAT

Erst 2017 wurde die Diagnose EGPA gestellt, als meine Blutwerte, insbesondere die Eosinophilen, auffällig waren. Eine Nasenblutentnahme in Kombination mit einer Chemotherapie brachte schließlich die genaue Diagnose.
Wie sind Sie mit der permanenten Ungewissheit umgegangen?
Das Gefühl ist erdrückend. Die intensive Suche nach den Ursachen macht unruhig und führt dazu, dass jede Kleinigkeit im Alltag überdacht wird. Ich habe z. B. oft meine Ernährung umgestellt und Routineverhalten geändert, um herauszufinden, was meine Symptome verschlimmert. Auch die Sorge, die Krankheit vielleicht an meine Tochter weiterzugeben, war ein Gedanke, der mir oft den Schlaf raubte. Kaum ein Monat verging, ohne dass ich einen Arzt aufsuchte oder zumindest einen Arztbesuch plante. Jeder Arztbesuch brachte mehr Informationen, aber oft auch mehr Unsicherheit. Unsicherheit und Angst sind meine ständigen Begleiter.
Generell wünsche ich mir, dass seltene Erkrankungen frühzeitig erkannt werden, um die Unsicherheit und Belastung der Patienten zu minimieren.
Wie geht es Ihnen jetzt?
Ich gehe regelmäßig zum Kardiologen, weil ich unter Herzrhythmusstörungen leide. Außerdem sind immer wieder Darmspiegelungen notwendig, um Probleme mit meinem Verdauungstrakt zu überwachen.
Auch Lungenfunktionstests und Besuche in der Uniklinik gehören zu meinen regelmäßigen Terminen. Da meine Erkrankungen sehr selten und unterschiedlich ausgeprägt sind, ist es schwierig, das richtige Medikament zu finden. Viele negative Nebenwirkungen sind teilweise sehr schwer auszuhalten. Zum Glück hilft mir ein Biologikum, meine Lunge in Remission zu halten.
Wegen schlechter Thrombozyten- und Leukozytenwerte muss ich regelmäßig Blut abnehmen lassen, um Veränderungen zu beobachten. Vor diesen Terminen bin ich immer nervös, weil ich nie genau weiß, wie sie verlaufen und welche Ergebnisse sie bringen werden. Die Ergebnisse wirken sich direkt auf meine Stimmung aus. Der schwierige Heilungsprozess und die langen Termine stellen insgesamt eine enorme körperliche und emotionale Belastung dar.
Forschung im Bereich Seltener Erkrankungen ist lebenswichtig für Betroffene: Sie beteiligen sich z. B. selbst an Studien rund um Ihre Krankheit. Was wünschen Sie sich als Betroffene bezüglich der Patientenversorgung, abgesehen von anhaltender Forschung?
Generell wünsche ich mir, dass Seltene Erkrankungen frühzeitig erkannt werden, um die Unsicherheit und Belastung der Patienten zu minimieren. Dazu wäre es wichtig, die Ärzte in diesen Fragen besser zu schulen. Die Erfahrungen und Ergebnisse bei Arztbesuchen sind oft unterschiedlich, so dass man sich oftmals nicht ernst genommen oder ausreichend untersucht fühlt. Trifft man jedoch auf einen geschulten Arzt, der sich mit Seltenen Erkrankungen auskennt, kann dies zu einem Durchbruch in der eigenen Diagnose führen. Eine weitere wichtige Komponente wäre die Unterstützung bei der Beantragung von Maßnahmen, die den Alltag erleichtern. Oft sind mehrere Anträge oder ein paar Tipps nötig, um mit Frührente, Zusatzurlaub und Schwerbehindertenausweis leichter durch den Alltag zu kommen.
Der Formwandler unter den rheumatischen Erkrankungen
Unser Immunsystem ist ein komplexes Netzwerk. Seine Aufgabe ist es, Viren, Bakterien oder Parasiten zu bekämpfen. Diese Abwehrschlacht tobt meist unmerklich, und nur wenn es besonders heiß hergeht, spüren wir sie in Form von Fieber oder einer laufenden Nase. Doch wie jedes komplexe System, ist auch dieses fehleranfällig. Dann kann es zu Autoimmunerkrankungen kommen, bei denen unsere Abwehr gegen körpereigene Zellen vorgeht, wie im Falle des hypereosinophilen Syndroms (HES) oder der eosinophilen Granulomatose mit Polyangiitis (EGPA).
Herr Dr. Aries, Sie und Ihr Team vom Immunologikum Hamburg sind auf Immunerkrankungen sowie entzündliche rheumatischen Erkrankungen spezialisiert. Was ist die besondere Herausforderung dabei? Diese sehr komplexen Krankheitsbilder sind teils schwer zu diagnostizieren und zu behandeln. Betrachtet man z. B. das HES oder die EGPA, treten bei diesen die Eosinophile – eine Unterart der weißen Blutkörperchen – in erhöhter Konzentration auf und lösen an unterschiedlichen Organen Entzündungsprozesse aus.
Je nach betroffenem Organ treten Symptome auf, die auch auf andere Erkrankungen deuten. Im Falle der EGPA tritt in frühen Erkrankungsphasen z.B. häufig ein Asthma auf. Der Betroffene erhält daraufhin oft eine Asthmabehandlung, die den Krankheitsmechanismus der EGPA aber nicht berücksichtigt, so dass dieser weiter fortschreiten und weitere Organe angreifen kann. Häufig kommt es auch zu Problemen der Nebenhöhlen, wie z.B. Nasenpolypen, oder des Nervensystems.
Wie finden Betroffene den Weg zu Ihnen?
In der Hausarztpraxis ist es extrem schwer, solche seltenen Erkrankungen zu erkennen. Meist erfolgt zunächst eine Überweisung an einen Facharzt. Fällt aber im Behandlungsverlauf auf, dass z. B. ein Asthma nicht auf die übliche Behandlung anspricht oder ggf. weitere Entzündungssymptome an anderen Organen auftreten, liegt der Verdacht nahe, dass die Befunde einen gemeinsamen Nenner haben. An dieser Stelle werden die Patient*innen an uns überwiesen. Wir erheben dann Biomarker per Differentialblutbild, nehmen ggf. Gewebeproben und untersuchen die Patient*innen sprichwörtlich von der Locke bis zur Socke, um ein ganzheitliches Bild zu bekommen und die richtige Diagnose stellen zu können.
Welche Behandlungsmöglichkeiten gibt es heute für solche Autoimmunerkrankungen?
Bei der Therapie von Erkrankungen wie HES und EGPA, geht es nicht nur darum die Symptome zu beseitigen, sondern vor allem um die Entzündungshemmung.

Dazu ist im ersten Schritt meist der Einsatz von Kortison notwendig, eventuell von weiteren Immunsuppressiva. Das Kortison setzen wir aber wenn möglich ebenso schnell wieder ab, weil es mittel- und langfristig sehr viele Nebenwirkungen mit sich bringt. Dann kommen zielgerichtetere Therapien zum Einsatz, die etwas Zeit brauchen, bis sie ihre Wirkung entfalten, dafür langfristig eingenommen werden können. Die therapeutischen Möglichkeiten sind aktuell schon relativ gut, doch es wird es noch weitere Forschung benötigen, die einer engen Zusammenarbeit mit den Betroffenen und ihrer Behandlungsteams bedarf.
Weitere Informationen: www.immunologikum.de
Lesen Sie mehr auf seltenekrankheiten.de 5
Text Hanna Sinnecker
Artikel ist in Zusammenarbeit mit der GlaxoSmithKline GmbH & Co. KG entstanden.
NP-DE-EOS-ADVR-240001; 01/2024
Dr. med. Peer M. Aries Internist, Rheumatologe und Immunologe
Vanessa Rennspieß EGPA-Patientin
FOTO: ASJA CASPARI

“Meine Diagnose: Eine seltene Krankheit mit lehrbuchfremdem Verlauf“
Die drei Buchstaben NDM stehen für eine Gruppe seltener erblicher Erkrankungen, die sogenannten Nicht-dystrophen Myotonien. Manuela Albert ist davon betroffen. Allerdings zeigt sich die NDM bei der 43-jährigen Mutter von vier Kindern lehrbuchfremd, was die an und für sich einfache Diagnosestellung erschwerte. Das ist Manuelas Geschichte.
Text Doreen Brumme
Manuela, wann zeigte sich Ihre NDM erstmals?
Ich erinnere mich, dass ich auf dem Schulhof der Grundschule häufig stürzte. Häufiger, als meine Mitschüler. Es war, als würde mir jemand aus dem Nichts heraus mitten im Lauf ein Bein stellen. Von 100 auf 0. Nur, dass da niemand war. Noch heute habe ich Narben an Händen und Knien von den andauernden Schürfwunden damals.
Gab es bereits NDM in Ihrer Familie?
Meine Mutter hat typische klinische Anzeichen einer leichten Myotonie. Sie kann Muskeln, die zum Bewegen angespannt werden, teilweise nicht mehr entspannen. Auch meine Oma und mein Urgroßvater mütterlicherseits hatten solche Symptome.
Haben Sie Ihre Symptome fortan stets begleitet?
Nein. Mit Beginn der Pubertät waren sie verschwunden.
Wie kam es zur Diagnose NDM?
Vor zwei Jahren wollte ich eines Nachts aufs Klo und kam gar nicht erst aus dem Bett. Das ist Stress, dachte ich. Dann fiel ich quasi aus der Dusche und stieß mir den Kopf am Türrahmen. Wenige Minuten später stürzte ich noch einmal. Daraufhin fuhr ich ins Krankenhaus. Der Verdacht auf einen Schlaganfall bestätigte sich zum Glück nicht. Ebenso wenig der auf Multiple Sklerose. Ich berichtete dem Arzt in der Notaufnahme jedoch von meinen Unfällen und der familiären Belastung: Der schickte mich geistesgegenwärtig in die Neurologie.
Die Ärzte dort machten zwei schnelle Tests mit mir, die bei einem NDM-Verlauf nach Lehrbuch oft schon zur Diagnose führen: Faust ballen und öffnen sowie Augen zukneifen. Beide Tests waren bei mir unauffällig. Auch beim EMG, wo Nadelelektroden in meine Muskeln gestochen wurden, fiel nichts auf. (Mit einer Elektromyografie misst man die natürliche elektrische Aktivität eines Muskels. – Anm. d. Red.).
Seit ich meine Diagnose erhalten habe und in Behandlung bin, habe ich wieder Handlungsspielräume. Ich kann sogar wieder in Vollzeit arbeiten.
Manuela Albert NDM-Betroffene

FOTO: PRIVAT
Die mich behandelnde Ärztin erklärte meine Probleme als psychosomatisch. Ich war verzweifelt und recherchierte auf eigene Faust im Internet.
Dort stieß ich auf den Verein „Mensch und Myotonie e. V.“, eine Patientenorganisation. Ich nahm Kontakt auf und bekam den Namen eines NDM-Experten, den ich anschrieb. Er antwortete mir unerwartet schnell. Meine Symptome und medizinische Familiengeschichte sprächen durchaus für eine Myotonie, schrieb er. Vier Monate später war ich in seiner Klinik und wurde dort einmal komplett auf links gedreht.
Das gründliche EMG ergab diesmal pseudomyotone Entladungen. Da meine Symptome so lehrbuchfremd waren, riet man mir zu einem Gentest. Der bestätigte mir eine Myotonia Congenita Thomsen.
Lesen Sie mehr auf seltenekrankheiten.de 6
FOTO: SHUTTERSTOCK_ 789680077
Manuela Albert NDM-Patientin
Wie sieht Ihr Alltag mit NDM heute aus?
Sie kennen vielleicht den schmerzhaften Wadenkrampf nach einem langen Tag oder nach körperlicher Anstrengung, der einen plötzlich nachts im Bett wachhält...? Den hatte ich dreißig Mal am Tag in unterschiedlichen Muskeln, gleichwohl im Lehrbuch steht, dass die Thomsen-Myotonie schmerzfrei verläuft.
Ich nehme seit der Diagnose zweimal täglich das Medikament, das in Deutschland für die Behandlung von NDM zugelassen ist. Das mindert meine Muskelkrämpfe und Schmerzen für einige Stunden. Und diese Zeit nutze ich. Ich arbeite in Vollzeit als Sekretärin in einem Baubetrieb. Meinem Arbeitgeber und meinen Kollegen habe ich von meiner Diagnose erzählt und ernte Verständnis und Unterstützung.
Daheim zeigt sich die NDM oft beim Zubereiten der Mahlzeiten für meine Großfamilie. Sobald ich merke, dass ich die Arme gleich nicht mehr bewegen kann, um in Topf und Pfanne umzurühren, rufe ich laut nach Hilfe. Das Medikament verschafft mir Handlungsspielraum. Seit Februar bin ich zudem im E-Rolli unterwegs und genieße die wiedergewonnene Bewegungsfreiheit.
Ich leide außerdem am Erschöpfungssyndrom Fatigue. Einmal zuhause die Treppe hoch und runter – und ich bin fertig! Die Erschöpfung betrifft aber auch meinen Kopf: Immer wieder spüre ich nach körperlicher Anstrengung so einen Nebel. Dann höre ich zwar, wie jemand mich was fragt, brauche aber mitunter mehrere Minuten, um zu antworten. Draußen auf der Straße ist diese verzögerte Reaktion lebensgefährlich.
Was wünschen Sie sich und anderen NDMBetroffenen?
Weitere Informationen finden Sie auf der Website der Patientenorganisation Mensch und Myotonie e. V. unter: www.menschundmyotonie.de
Kontakt
Mensch & Myotonie e. V.
Postfach 16 03 30, 44333 Dortmund
1. Vorsitzender: Volker Kowalski
E-Mail: vokiko@online.de
Tel.: 0231-803290 ( ab 12 Uhr )



Im Internet stieß ich auf den Verein „Mensch und Myotonie e. V.“, eine Patientenorganisation. Ich nahm Kontakt auf und bekam den Namen eines NDM-Experten, der mir vier Monate später eine Myotonia Congenita Thomsen bestätigte.
• Ich wünsche uns, dass die Ärzte uns genau zuhören, auch wenn die Zeit knapp und der Stress groß sind. Meine NDM ist so untypisch, und doch ist sie real. Das schnelle Abtun als „psychosomatisch“ erleben leider viele NDM-Betroffene.
• Ich wünsche uns ein gründliches Abklären unserer Symptome seitens der Ärzte.
• Ich wünsche uns bessere Aufklärung. Auch im Internet finden sich scheinbar seriöse Quellen, die die NDM teils falsch, teils lückenhaft beschreiben.
officialmyotonia.orga
www.instagram.com/officialmyotonia.orga/ Mensch & Myotonie e. V.
www.facebook.com/Myotonien @myotonia.org
www.tiktok.com/@myotonia.org
Auch auf der Website der Deutschen Gesellschaft für Muskelkranke e. V. finden Sie weitere Informationen: www.dgm.org
Kontakt
Bundesgeschäftsstelle der DGM
E-Mail: info@dgm.org Tel.: 07665 94470
Ständig unter Strom, und doch blockiert

Ich bin sehr muskulös, habe aber keine Kraft. Mein Nachbar hält mich für einen Macho, weil meine Frau die Getränkekisten trägt….
Die Musik ist mein Leben: die erste Geige im Orchester spielen – ein Traum, der mit einer wirksamen Therapie Realität werden könnte.
Als ich die Hand meines neuen Chefs nicht loslassen konnte, wäre ich am liebsten im Boden versunken. Ihm nicht die Hand zu geben war keine Option!
Kälte verstärkt meine Symptome. Wintersport –ohne wirksame Therapie ist das undenkbar!
Meine Eltern hielten mich für bockig, weil ich vor der Treppe stehen blieb und nicht hochgehen konnte.
Die Unfähigkeit, einen Muskel nach Anspannung schnell wieder zu entspannen, beeinträchtigt unser Leben in vielerlei Hinsicht. Alltägliche Dinge wie Händeschütteln, Treppensteigen, nach dem Bus Rennen, sogar Aufstehen und einfach Loslaufen stellen enorme Herausforderungen dar und bedeuten emotionalen Stress für uns. Äußerlich wirken wir gesund, teilweise sogar athletisch, was oft Unverständnis bei Außenstehenden hervorruft und uns zusätzlich belastet.
Wir lassen Sie nicht allein!
ANZEIGE Lesen Sie mehr auf seltenekrankheiten.de 7
DE-NDM-2401-00002
„Ich bin wie alle!“
Felix* bekam mit sechs eine neue Leber, denn er leidet am seltenen Alagille-Syndrom, das Probleme an Leber, Herz, Nieren und Knochen macht. Was die Erbkrankheit für ihn und seine Familie bedeutet, berichten Felix und seine Mutter hier.
Text Doreen Brumme
Frau Steinbach*, Felix leidet am AlagilleSyndrom. Wann fiel Ihnen auf, dass er Beschwerden hat – und wie sahen diese aus? Felix hat vier ältere Geschwister, ich hatte also bereits Erfahrung mit Neugeborenen. Mir fiel sofort auf, dass er sich mit dem Stillen schwertat. Mit der Hebamme zusammen versuchte ich alles, um ihn zum Trinken zu bewegen – vergebens. Zudem war Felix ungewöhnlich lange nach der Geburt noch sehr gelb – als Krankenschwester wusste ich, dass das mit der Leber zusammenhing. Bei der ersten Untersuchung bei unserer Kinderärztin wurde deshalb gleich ein Bluttest gemacht und der ergab dann die Diagnose.
Wie wurde Felix behandelt?
Felix‘ Leber wurde fortan engmaschig kontrolliert. Da bei der Erbkrankheit oft auch Herz, Nieren und Knochen in Mitleidenschaft geraten, machte man eine Herzkatheter-Untersuchung und stellte Gefäßveränderungen fest. Die Leberwerte von Felix waren jahrelang auf einem schlechten Niveau, das gerade noch geduldet werden konnte. Mit vier Jahren hatte Felix bereits etliche Knochenbrüche hinter sich. Das hieß: Arztbesuche bestimmten seine Kindheit. Immer wieder stand eine neue Leber im Raum. Mit sechs Jahren brauchte Felix eine neue Leber. Kurz nach Neujahr kam der Anruf... Wir hatten nicht mal eine Kliniktasche gepackt. Den Moment, als Felix in den OP geschoben wurde, werde ich nie vergessen. Ich hatte selbst an Transplantationen mitgewirkt. Ich wusste, was ihm, was uns bevorstand. Ich kannte die Risiken. Ich höre noch heute, wie der operierende Arzt mir versicherte, es sei eine gute Leber, die er für Felix hätte. Das half, die 14 Stunden Warten auszuhalten.
Felix, du bist inzwischen 14 Jahre alt. Wirkt sich deine Erkrankung auf deinen Alltag aus – und fühlst du dich anders als gesunde Gleichaltrige?
Ich bin ganz normal und werde so auch wahrgenommen. Als ich neulich beim Schulausflug meinen Behindertenausweis zeigte, um den günstigeren Eintritt zu bekommen, fragten viele, warum ich den habe. Ab und zu sprechen mich manche auch auf meine grau verfärbten Zähne an. Dann sage ich, dass das von den Medikamenten kommt. Ansonsten bin ich wie alle. Ich kann machen, was meine Freunde auch tun: Ich fühle mich nicht im Abseits. Ich bin sehr sportlich, mache seit
Jahren Akrobatik im Zirkus. Ich weiß ziemlich gut, was ich mir zumuten kann und was nicht. Die vermeintlich „normalen“ Dinge wie Rauchen, Saufen und Kiffen lasse ich gerne aus.
Was bedeutet Felix‘ Diagnose für die Familie?
Frau Steinbach: Felix war von Anfang an unser Päppelkind. Jahrelang ging es bei ihm vor allem darum, dass er genug isst, zunimmt und wächst. Das hat den Takt der Familie bestimmt. Er war lange zu klein... Felix: Jetzt bin ich einer der Großen!

Frau Steinbach: Felix hatte in der Grundschule eine Begleitung an der Seite, die seinen Ranzen trug, ihm schwere Türen öffnete. Doch je weiter die OP zurück lag, desto normaler wurde sein Leben und unser Familienleben. Die Geschwister hatten ja auch ihre Themen mit Schule, Abschluss ... Felix: Wir sind alle frühreif.
Frau Steinbach: Das stimmt. Alle sind an der Situation gewachsen. Mitunter sorge ich mich, dass die Krankheit Felix und seinen Geschwistern die Leichtigkeit aus dem Leben nimmt. Doch dann sage ich mir: Was ist, ist kein Zufall. So, wie’s ist, ist’s gut. Unser, also vor allem mein nächstes Thema wird sein, Felix in sein eigenes Leben zu entlassen.
Vielleicht wäre es irgendwann mal ganz schön, jemanden zu sprechen, der Ähnliches erlebt hat.
Wo bekommen Sie Unterstützung?
Frau Steinbach: Ich bin mit meinen fünf Kindern allein. Doch ich habe eine große Familie, Freunde und Nachbarn, auf die immer Verlass ist. Wir sind in unserer freikirchlichen Gemeinde sehr gut eingebunden und finden dort immer Hilfe. Mir ist klar, dass das nicht selbstverständlich ist. Ich bin dankbar für die Gewissheit, mit allem in Gottes Hand zu sein und bin mir bewusst, dass das ein Geschenk ist.
*Namen von der Redaktion geändert
Andere, insbesondere Alleinerziehende, haben solch ein Hilfenetz wie ich vielleicht nicht. Allein kommt man beim Betreuen eines chronisch kranken Kindes rasch an seine Grenzen. Es kostet Kraft, zu erkennen, dass man Hilfe braucht. Und es kostet noch mehr Kraft, Hilfe zu holen. Da können Selbsthilfegruppen eine Unterstützung sein. Felix: Vielleicht wäre es irgendwann mal ganz schön, jemanden zu sprechen, der Ähnliches erlebt hat. Aber momentan geht’s mir gut. Ich habe Freunde, die alles von mir wissen. Mit denen kann ich reden, wenn mir danach ist.
Der Verein Leberkrankes Kind e. V. Seit 1987 gibt es den Verein Leberkrankes Kind e. V. Gegründet wurde der Verein von Eltern leberkranker Kinder, die das Bedürfnis hatten, sich mit anderen betroffenen Familien auszutauschen. Heute hat unser Verein rund 300 Mitglieder. Wir informieren über Krankheitsbilder und über Unterstützungsmöglichkeiten für Familien schwer kranker Kinder. Der Verein fördert verschiedene Projekte an Kliniken, unterstützt Ferienfreizeiten oder ermöglich transplantierten Kindern die Teilnahme an den World Transplant Games.
Wir als Verein möchten informieren, Mut machen und unsere Erfahrungen teilen. Jede und jeder ist herzlich als Mitglied in unserem Verein willkommen.
Je mehr wir sind, desto mehr können wir bewirken –gemeinsam für unsere Kinder!
Weitere Informationen unter: www.leberkrankes-kind.de
„Eine frühe Diagnose des Alagille-Syndroms ist entscheidend“
Das Alagille-Syndrom ist eine seltene angeborene System-Erkrankung, die hauptsächlich die Leber und oft das Herz betrifft. Wir sprachen mit PD Dr. Eberhard Lurz, Facharzt Kinder- und Jugendmedizin, Zusatz-Weiterbildung Kinder-Gastroenterologie, am LMU Zentrum für Entwicklung und komplex chronisch kranke Kinder im Dr. von Haunerschen Kinderspital, 2. Vorsitzender der GPGE e. V., über die Symptome und die derzeitigen Behandlungsmöglichkeiten.
Text Katharina Lassmann
Herr Dr. Lurz, was passiert beim Alagille-Syndrom im Körper Betroffener und wie äußert sich die Erkrankung?
Das Alagille-Syndrom manifestiert sich meist unmittelbar nach der Geburt oder in der Säuglingsphase und ist durch eine deutliche Lebererkrankung mit Gelbsucht und Juckreiz gekennzeichnet, oft betrifft sie aber auch Herz oder Nieren. Verantwortlich hierfür ist ein Defekt in einem der beiden Strukturgene JAG1 bzw. NOTCH2, der zu einer fehlerhaften Entwicklung spezifischer Zellen führt.
Dadurch kommt es zu einer unzureichenden Entwicklung der Gallengänge in der Leber und Symptome wie Gelbsucht, Juckreiz und Leberprobleme können entstehen. Die Galle kann nicht regelrecht aus der Leber ausgeschieden werden, sodass es langsam zu einer Vernarbung der Leber kommt. Gemäß einer aktuellen internationalen (GALA) Registerstudie überleben weniger als 50% der Kinder mit ihrer eigenen Leber und eine Lebertransplantation wird im Verlauf notwendig. Manche Patienten haben einen sehr milden Krankheitsverlauf ohne relevante Symptome und die Diagnose wird erst zu einem späteren Zeitpunkt gestellt, z. B. wegen auffälligen Gesichtsmerkmalen wie einem spitzen Kinn mit breiter
Stirn, kleinen Fettknötchen (Xanthomen) an den Augen oder der Haut, oder eigenem Nachwuchs mit AlagilleSyndrom. Eine endgültige Diagnose erfolgt in der Regel mittels genetischer Tests.
Was sind die größten Herausforderungen für Patienten und ihre Angehörigen?
Neugeborene zeigen oft in den ersten beiden Wochen eine gelbliche Verfärbung der Haut oder Skleren, welche man gut beobachten und spätestens nach dem 14. Lebenstag mit einer Blutuntersuchung und BilirubinBestimmung abklären lassen sollte. Manche Neugeborenen haben auch sehr hellen, kalkfarbenen oder entfärbten Stuhlgang, bei dem man die Blutuntersuchung sofort durchführen sollte. Durch diese konsequente Untersuchung kann die Diagnose eines Alagille-Syndroms oder anderer Gelbsuchterkrankungen der Leber möglichst früh gestellt werden. Eine späte Diagnose birgt größere Belastungen für das Kind und die Familie, wie starken Juckreiz und Schlafprobleme. Hemmung des Wachstums und eine Entwicklungsstagnation können auftreten. Durch einen möglichen Mangel der fettlöslichen Vitamine besteht auch das Risiko für z. B. eine Vitamin K Mangel-bedingte Hirnblutung.
Welche Behandlungsoptionen gibt es derzeit?

Die Therapie beginnt mit der Verabreichung von fettlöslichen Vitaminen, um einen Mangel an den Vitaminen A, D, E und K zu verhindern. Zusätzlich versucht man durch die Gabe einer künstlich hergestellten Gallensäure, die Löslichkeit der Gallenflüssigkeit und damit Abfluss dieser aus der Leber zu optimieren. Die körperliche Entwicklung des Kindes wird engmaschig kontrolliert und die Ernährung ggf. angepasst und auf eine ausreichende Kalorienzufuhr geachtet. Teilweise kratzen sich Kinder mit Alagille-Syndrom täglich blutig und können nachts nicht schlafen. Ein neues Medikament ist seit letztem Jahr verfügbar und für die Behandlung dieses Juckreizes zugelassen. Dieses Medikament blockiert die Aufnahme der Gallensäuren im Dünndarm, sodass diese im Blut gesenkt werden und sich der Juckreiz mindert. Eventuell wird sogar die Leber entlastet und das Überleben mit der eigenen Leber verbessert. Eine frühe Diagnose und damit früher Beginn aller verfügbaren Therapieoptionen ist somit sehr wichtig für betroffene Kinder, um schwere Komplikationen zu vermeiden und ihnen eine altersentsprechende Entwicklung mit maximaler Lebensqualität zu ermöglichen.
Lesen Sie mehr auf seltenekrankheiten.de 8
FOTO: LMU KLINIKUM MÜNCHEN
FOTO: SHUTTERSTOCK_465884015
Polycythaemia Vera –
Betroffene spielen eine wichtige Rolle in der Therapie
Myeloproliferative Neoplasien (MPN) sind eine Gruppe von seltenen Erkrankungen des Knochenmarkes. Charakteristisch für diese Krankheitsbilder ist eine gesteigerte Produktion von Blutzellen, was sich in einer Vielzahl von Symptomen äußern kann. Wir sprachen mit Frau Prof. Dr. med. Haifa Kathrin Al-Ali über die Symptome und Behandlungsmöglichkeiten der Polycythaemia Vera (PV), die zu den MPN zählt.
Text Alexandra Lassas
Frau Prof. Al-Ali, wie sehen die Symptome einer PV aus und auf welche Symptomkonstellationen sollten Mediziner achten?
Symptome lassen sich in allgemeine Beschwerden und durch die Komplikationen verursachte Probleme unterteilen. Allgemeine Symptome sind schwer zu erkennen und von den Patienten kaum mit der Erkrankung in Verbindung zu bringen, wie z. B. Kopfschmerzen, Müdigkeit und Konzentrationsstörungen. Die Symptome sind unspezifisch, aber ihre Auswirkungen sind enorm und beeinträchtigen die Lebensqualität erheblich. Zusätzlich treten spezifische Beschwerden wie Sehstörungen und Juckreiz auf, der bei 14% der Patienten auftritt, obwohl auf der Haut keine sichtbaren Anzeichen vorhanden sind. Viele Patienten durchlaufen einen langen Leidensweg, bis die Krankheit korrekt diagnostiziert wird, und manche kämpfen jahrzehntelang mit den Symptomen. Aufgrund der erhöhten Dichte der roten Blutkörperchen im Körper sehen die Betroffenen äußerlich gesund aus, fühlen sich aber genau das Gegenteil. Dies hat Auswirkungen auf die psychische Verfassung, da viele nicht ernst genommen werden.
Welche Untersuchungsmöglichkeiten hat der Arzt, um eine PV zu diagnostizieren und wie ginge es dann weiter?
Der Arzt kann eine PV anhand des Blutbildes schnell und eindeutig diagnostizieren. Erhöhte Werte von Hämoglobin und Hämatokrit sind dabei ein deutlicher Hinweis. Eine PCR-Analyse des Blutes kann zusätzlich die

Wie merkt der Patient, dass sich die Symptome verändern/verschlechtern und es z. B. nicht um weitere Veränderungen des Alters geht?
JAK2-Mutation nachweisen, die die Diagnose PV bekräftigt und eine Untersuchung des Knochenmarks rundet das diagnostische Vorgehen ab. Es ist auch möglich, dass eine PV ohne auffällige Blutwerte vorliegt. Insbesondere bei jungen Menschen können plötzliche und ungewöhnliche Thrombosen auf eine vorhandene JAK2-Mutation hinweisen. Grundsätzlich hat die Erkrankung eine gute Prognose, wenn sie frühzeitig diagnostiziert wird. In Absprache mit dem Patienten sollte dann eine geeignete Therapie gefunden werden.
Warum sollten Betroffene nach Diagnose oder unter Therapie regelmäßig zu Kontrolluntersuchungen gehen?
Regelmäßige Kontrolluntersuchungen sind für Betroffene von großer Bedeutung. Ein nicht gut kontrollierter Hämatokritwert erhöht z. B. das Risiko von Thrombosen. Eine regelmäßige Überwachung des Blutbildes ist daher unverzichtbar. Auch die Lebensqualität und eine gute Kontrolle der Beschwerden können nur durch gute Verlaufskontrollen gewährt werden. Eine Vergrößerung der Milz kann u. a. ein Anzeichen für das Fortschreiten der Krankheit sein und möglicherweise eine Anpassung der Behandlung erfordern. Zusätzlich müssen die auftretenden Nebenwirkungen der verwendeten Medikamente beobachtet werden: Z. B. ist bei der PV insbesondere während der Behandlung auf unerwünschte Hautreaktionen wie Geschwüre an den Beinen (Beinulzerationen) und hellen Hautkrebs zu achten. Ein regelmäßiger Hautcheck ist daher sehr wichtig.
Leben mit MPN –Umfassende Hilfe für Betroffene
Seltene Krankheiten stehen oft im Schatten. Dabei ist es von entscheidender Bedeutung, das Bewusstsein für seltene Erkrankungen zu schärfen und Solidarität mit Betroffenen zu zeigen. Der Rare Disease Day bietet auch dieses Jahr eine einzigartige Gelegenheit, auf seltene Erkrankungen wie die Polycythaemia vera aufmerksam zu machen. Das forschende Pharmaunternehmen Novartis denkt Medizin neu, um besonders auch Menschen mit seltenen Erkrankungen mit innovativen Therapien zu mehr Lebensqualität zu verhelfen und ihnen mit umfangreichen Unterstützungs- und Informationsangeboten zur Seite zu stehen.
Speziell für Menschen, die an einer Myeloproliferativen Neoplasie (MPN) wie der Myelofibrose, der Polycythaemia Vera oder der Chronischen Myeloischen Leukämie leiden, hat Novartis für Patient:innen und deren Angehörige umfangreiche Informationsinitiativen ins Leben gerufen, die Betroffenen und deren Angehörigen wissenschaftlich fundiertes Wissen zur Erkrankung und zum Umgang damit zur Verfügung stellen.
Symptome erkennen – und richtig in Zusammenhang bringen
Da die verschiedenen Symptome der MPN sehr vielschichtig sind und mit Fortschreiten der Erkrankung stärker werden, sind fundierte Informationen zu den möglichen Beschwerden für Patient:innen und deren Angehörige sehr wichtig. Das macht das Beispiel der Polycythaemia Vera deutlich: denn Beschwerden wie chronische Müdigkeit, Schmerzen im linken Oberbauch, verstärktes nächtliches Schwitzen, Juckreiz besonders nach Kontakt mit Wasser und Appetitlosigkeit lassen oft nicht direkt an eine schwere Erkrankung denken. Gerade Frauen denken oftmals eher an die Wechseljahre und nicht an eine seltene Bluterkrankung. Auch Seh- und Konzentrationsstörungen, Ohrensausen, trockene Haut werden eher auf das Alter zurückgeführt und nicht in Kombination betrachtet. Die Folge: der Arztbesuch bleibt aus, die PV bleibt unentdeckt und somit auch unbehandelt, schwere Komplikationen können auftreten.
Zunehmende Beschwerden ernst nehmen
Aber auch wenn die Diagnose bereits gestellt wurde, sollten Betroffene die Symptome im Blick behalten. Gerade wenn die Symptomlast zunimmt oder Nebenwirkungen auftreten, sollten Betroffene das Gespräch mit dem Behandlungsteam suchen. Manche Begleiterkrankungen oder Komplikationen können für Betroffene im schlimmsten Fall lebensbedrohlich werden, weshalb ein schnelles Gegensteuern entscheidend ist. Ist der Betroffene gut informiert, kann er bei der Wahl und Durchführung der passenden Therapie intensiv mit einbezogen werden. Die Patient:innen sollten immer ein offenes Ohr finden, wenn Handlungsbedarf besteht. Das gilt auch für die Angehörigen der Betroffenen, denn sie können eine große Stütze sein: Auch wenn es darum geht, körperliche und seelische Beschwerden oder eine Verschlechterung des Zustandes frühzeitig zu erkennen. Sie spielen also eine tragende Rolle, wenn es darum geht, Betroffene zu unterstützen und ihre Lebensqualität zu verbessern.
Für Patienten stehen international anerkannte ragebögen zur Verfügung, die sie regelmäßig während des Kontakts mit ihrem behandelnden Arzt ausfüllen sollten. Durch den Vergleich der Werte über einen längeren Zeitraum können Verschlechterungen oder Veränderungen erkannt werden. Symptome wie Müdigkeit und Juckreiz lassen sich so über einen längeren Zeitraum besser beurteilen. Zudem wird dadurch das Ausmaß der Beschwerden deutlich und es kann eine klare Abgrenzung zu altersbedingten schleichenden Veränderungen erfolgen. Wie sollten sich Betroffene verhalten, wenn sie Veränderungen oder neue Beschwerden feststellen? Es ist ratsam, sofort den behandelnden Arzt aufzusuchen. Durch die Auswertung des Fragebogens erhält der Patient einen umfassenden Überblick über die Symptome, und der Arzt kann entsprechende therapeutische Maßnahmen ergreifen oder die Behandlung anpassen. Der Austausch mit anderen Betroffenen spielt ebenfalls eine wichtige Rolle. Das MPN-Netzwerk bietet die Möglichkeit, das Verständnis für die Krankheit zu verbessern und Kontakt zu anderen Betroffenen aufzunehmen.
Wie können behandelnde Ärzte erkennen, wann eine Anpassung der Therapie notwendig ist?
Der Arzt kann durch die Auswertung des Blutbildes eingreifen und die Therapie entsprechend anpassen. Anhand der Fragebögen können alternative Therapiemöglichkeiten zur Verbesserung der Lebensqualität des Patienten gesucht werden. PV ist eine äußerst vielfältige Erkrankung, und die Probleme und Beschwerden jedes einzelnen Patienten sind unterschiedlich. Als Arzt ist es wichtig, alle Parameter im Blick zu behalten, sie individuell auf den Patienten abzustimmen und gemeinsam an der Therapie zu arbeiten. Bei der Anpassung der Behandlung sollten auch die emotionalen Aspekte berücksichtigt werden. Die Verbesserung der Lebensqualität sollte gemeinsam angestrebt werden.

Die einzelnen Initiativen www.leben-mit-myelofibrose.de, www.leben-mit-pv.de und www.leben-mit-cml.de möchten Betroffene deshalb über alle Facetten der Erkrankung informieren. Hier finden sich auch Patienten-Erfahrungsberichte und Expertenbeiträge zu verschiedenen krankheitsrelevanten Schwerpunkten. Zudem finden Patient:innen ausführliche Checklisten, die ihnen die Gespräche mit dem Behandlungsteam erleichtern können: denn die Patient:innen selbst spielen eine wesentliche Rolle bei der Wahl und Durchführung der geeigneten Therapie. Dazu kann auch eine Anpassung der bestehenden Therapie gehören, wenn die bestehende Behandlung nicht den gewünschten Erfolg erzielt. Dabei kann auch der MPN-Tracker unter www.mpntracker.com helfen, der Patient:innen in Form eines Therapietagebuches bei der Dokumentation zur Entwicklung ihrer Erkrankung unterstützt.
Zusammen stärker
Auch der Austausch mit anderen Betroffenen, Selbsthilfeorganisationen und Fachärzt:innen stärkt Patient:innen und ihre Angehörigen im Umgang mit der Erkrankung. Seit 2016 können MPN-Betroffene einen bundesweit etablierten Treffpunkt nutzen: die MPN-Patient:innentage. Diese finden mehrmals im Jahr an immer anderen Standorten statt, damit möglichst viele Betroffene teilnehmen können. Seit 2020 ist für einige der Termine auch eine Online-Teilnahme möglich. Die Teilnahme an den MPN Veranstaltungen ist kostenlos. Auf www.mpn-patiententage.de findet man die Anmeldung für den nächsten Patient:innentag sowie weitere Informationen und einen kleinen Rückblick auf vergangene Veranstaltungen.
Scannen Sie den QR-Code und lesen Sie mehr zu uns auf unserer Webseite unter https://www.leben-mit-pv.de/sp1

Lesen Sie mehr auf seltenekrankheiten.de 9
FOTO: NOVARTIS PHARMA GMBH
ANZEIGE
UNIVERSITÄTS- MEDIZIN HALLE
FOTO:
Morbus Fabry und Morbus Pompe -
„Die Lebensqualität der Betroffenen steht immer im Vordergrund“
Morbus Fabry und Morbus Pompe zählen zu den lysosomalen Speichererkrankungen, einer Gruppe von seltenen angeborenen Stoffwechselerkrankungen. Werden die Erkrankungen nicht behandelt, schreiten sie unaufhaltsam fort und beeinträchtigen das Leben Betroffener stark. Wir sprachen mit Dr. Christina Lampe über die Wichtigkeit einer frühen Diagnose und die derzeitigen Therapieoptionen.
Text Hanna Sinnecker
Frau Dr. Lampe, Morbus Fabry und Morbus Pompe sind seltene Stoffwechselerkrankungen. Was passiert dabei im Körper Betroffener?
Die Erkrankungen äußern sich sehr unterschiedlich: Morbus Pompe ist eine Muskelerkrankung, Morbus Fabry betrifft das Herz, die Niere und das Nervensystem. Sie gehören aber beide zu den lysosomalen Speichererkrankungen. Das Grundprinzip ist so: Durch eine Genveränderung wird ein Enzym nicht korrekt gebildet. Enzyme benötigt man, damit in den Zellen der Organe Abfallstoffe (die nun mal bei der Zellerneuerung anfallen) zerkleinert und ausgeschieden werden können. Fehlt das entsprechende Enzym oder wird es nur unzureichend gebildet, verbleiben die Abfallprodukte in den Zellen und stören die Funktion der Organe. Je mehr Abfallprodukt abgelagert wird, desto schwerer die Erkrankung oder anders gesagt, je weniger Enzym im Körper vorhanden ist, desto schwerer ist der Patient betroffen.
Die Genveränderungen sind meist ererbt. Man erbt immer ein Gen vom Vater und eins von der Mutter. Bei den meisten seltenen Erkrankungen benötigt man 2 kranke Gene, um krank zu sein, so bei Morbus Pompe. Hat man nur ein krankes Gen, ist man Träger der Erkrankung, ist aber gesund. Bei Morbus Fabry ist das anders: Die Genveränderung liegt auf dem weiblichen Geschlechtschromosom, dem X-Chromosom. Männer tragen ein X- und ein Y-Chromosom, Frauen zwei X-Chromosomen. Daher können Frauen manchmal etwas weniger betroffen sein, da das gesunde XChromosom ausgleichen kann.
Wie sehen die Symptome eines Morbus Fabry aus und wann zeigen sie sich typischerweise?
Die Symptome können von Patient zu Patient unterschiedlich sein, nicht jeder Betroffene zeigt alle Symptome. In der Kindheit stehen die brennenden Schmerzen in Händen und Füßen, eine verminderte Fähigkeit zu schwitzen, Bauchschmerzen und Durchfall sowie unerklärbare Fieberschübe im Vordergrund, im Jugendalter kommen ein Tinnitus, ein Hörsturz, Schwindel, eine verminderte Leistungsfähigkeit, Müdigkeit und kleine rote Pünktchen im Bereich des Nabels, der Leisten oder im Gesäßbereich hinzu (Angiokeratome). Auch eine Augenveränderung, die sogenannte Cornea verticillata, kann auftreten. Im Erwachsenenalter können die brennenden Schmerzen weniger werden. Neben den genannten Symptomen kann es nun zu Nierenfunktionsstörungen, Auffälligkeiten am Herzen wie eine Linksherzvergrößerung, Herzinfarkt oder Schlaganfall vor dem 55. Lebensjahr kommen. Die Patienten berichten z. B. über Kälte und/oder Hitzeunverträglichkeit, Atemnot bei Belastung, Abnahme der Belastungsfähigkeit, Abgeschlagenheit/Fatigue.
Welche Symptome sind für Morbus Pompe typisch? Grundsätzlich muss man 2 Formen des Morbus Pompe unterscheiden: die infantile (kindliche) Form, auch IOPD genannt und die jugendliche oder erwachsene (spät einsetzende, late onset) Form, die LOPD. Eine
IOPD fällt schon im Säuglingsalter auf. Typisch sind eine Trinkschwäche mit frühzeitiger Erschöpfung und starkem Schwitzen, eine Kopfhalte- und stammbetonte Muskelschwäche, eine Entwicklungsverzögerung und Gedeihstörung sowie eine Ateminsuffizienz. Die betroffenen Kinder haben eine massive Herzvergrößerung und Herzinsuffizienz. Das sind Kinder, die keine Körperspannung haben, sich kaum bewegen und schlapp wirken. Unbehandelt sterben die betroffenen Kinder meist im ersten Lebensjahr.
Bei der juvenilen und erwachsenen Form fehlt die Herzbeteiligung. Betroffene Kinder fallen häufig hin, machen unsichere Bewegungen beim Klettern und Spielen, haben Schwierigkeiten beim schnellen Laufen und Treppensteigen. Auch scheint der Gang watschelnd, das Aufstehen vom Boden bereitet ihnen Probleme, es sind „Sport-Bewegungsmuffel“. Hinzu können feinmotorische Probleme wie Schwierigkeiten beim Schuhe binden, Ausmalen oder Rucksackpacken kommen. Sie haben eine Entwicklungsstörung. Zudem können sie Schwierigkeiten beim Atmen oder eine Atemnot im Liegen haben, über morgendlichen Kopfschmerz oder Müdigkeit bzw. Antriebslosigkeit klagen. Die Nackenmuskulatur ist schwach ausgebildet, viele Kinder haben eine Skoliose. Bei betroffenen Erwachsenen fällt ein wiegender Gang auf, sie haben Probleme beim Treppensteigen, beim Aufstehen aus dem Liegen oder aus der Hocke und Schwierigkeiten beim Anheben von Lasten. Hinzu kommen ein morgendlicher Kopfschmerz, Tagesschläfrigkeit, Atemnot bei Belastung und beim Liegen und eine allgemeine Schwäche.
Wie lange dauert es durchschnittlich, bis eine Diagnose gestellt wird?
Bei Morbus Fabry geht man von etwa 16 Jahren bei Frauen und 14 Jahren bei Männern zwischen Symptombeginn und Diagnose aus. Bei Morbus Pompe hängt es von der Verlaufsform ab. Bei der schweren infantilen Form (IOPD) sind es etwa 6 Monate, bei der später einsetzenden Form, der late Onset Form (LOPD) etwa 12 Jahre. In manchen Ländern gibt es für Fabry und Pompe ein Neugeborenenscreening, in Deutschland ist das leider noch nicht der Fall. Die Herausforderung für Ärzte ist daher, die Symptome richtig in Zusammenhang zu bringen, um den Erkrankungen auf die Spur zu kommen.
Warum ist eine möglichst frühe Diagnose so wichtig und welche Rolle spielt die Familienanamnese bei erblich bedingten Erkrankungen wie Morbus Fabry und Morbus Pompe?
Da es sich um chronisch fortschreitende Erkrankungen handelt, bedeutet eine möglichst frühe Diagnose und Behandlung, dass potenziell weniger Gewebe oder gar Organe unwiderruflich geschädigt werden. Man kann also therapeutisch eingreifen, bevor schwere Organschäden entstehen. Zwar gibt es noch keine Therapien, die die Erkrankungen heilen, aber man kann das Fortschreiten der Erkrankungen verlangsamen oder manchmal auch für eine Zeit stoppen. Das wirkt sich natürlich positiv auf die Lebensqualität der Betroffenen aus.

Die Familienanamnese ist deshalb so wichtig, da die beiden Erkrankungen ererbt sind, das heißt, die Genveränderung liegt in der Familie.
Dr. med. Christina Lampe Oberärztin am Zentrum für seltene Erkrankungen Gießen (ZSEGI), Abteilung Kinderneurologie, Sozialpädiatrie und Epileptologie, Zentrum Kinderheilkunde und Jugendmedizin (Univ.-Klinikum Giessen / Marburg; Standort Giessen)
Die Familienanamnese ist deshalb so wichtig, da die beiden Erkrankungen ererbt sind, das heißt, die Genveränderung liegt in der Familie. Mit einer Stammbaumanalyse kann man somit weitere Betroffene herausfinden, bevor sie Krankheitssymptome haben, diese noch nicht bemerkt haben oder auch Symptome haben, deren Ursache bislang noch nicht gefunden wurde. Sowohl für Morbus Fabry als auch für Morbus Pompe stehen glücklicherweise gut wirksame Behandlungsoptionen zur Verfügung. Können Sie uns erklären, wie diese wirken und wie sie sich auf die Lebensqualität Betroffener auswirken können? Es gibt derzeit 2 Enzymersatztherapien und eine Kombinationstherapie (Enzymersatztherapie in Kombination mit einer oral einzunehmenden Kapsel) bei Morbus Pompe und 3 Enzymersatztherapien sowie eine Chaperontherapie bei Morbus Fabry. Bei der Enzymersatztherapie wird das fehlende Enzym künstlich hergestellt und als Infusion über die Vene verabreicht. Damit wird das Enzym, welches vom Köper nicht ausreichend oder gar nicht gebildet wird, ersetzt. So können zwar bereits entstandene Schäden nicht rückgängig gemacht werden, aber es können weitere Schäden verhindert werden. So ist eine Verlangsamung des Fortschreitens der Erkrankung, manchmal sogar ein Aufhalten möglich. Bei Morbus Fabry gibt es auch eine orale Therapie, eine sogenannte Chaperontherapie. Sie kann aber nur bei bestimmten Genveränderungen angewendet werden, nämlich solchen, die zu einer gestörten Faltung des Enzyms führen. Hier kann durch die Chaperontherapie die Struktur des Enzyms wieder hergestellt werden, damit es wirken kann. Diese Kapsel wird alle 2 Tage eingenommen. Bei allen Therapien handelt es sich um lebenslange Therapien.
Neuere Therapieansätze sind beispielsweise Gentherapien. Hierbei wird das Erbmaterial so verändert, dass es selbständig das entsprechende Enzym produzieren kann. Aber diese Therapieansätze sind noch in klinischer Erprobung. Neben der Verhinderung weiterer Schäden durch ein Fortschreiten der Erkrankung und weiteren Symptomen steht natürlich die Lebensqualität der Betroffenen im Vordergrund. Viele Patienten berichten, dass sie sich besser fühlen, mehr Energie und Ausdauer haben, aktiver am sozialen Leben teilnehmen können. Und das ist ja das Ziel.
Lesen Sie mehr auf seltenekrankheiten.de 10
FOTO: UK GIESSEN

G„Es braucht viel mehr Aufklärung zu meiner Erkrankung!“
Gabi D. dachte den größten Teil ihres Lebens, dass sie vollkommen gesund sei: Bis die Diagnose Morbus Pompe sie vollkommen unvermittelt traf. Wie es ihr heute geht und was ihr im Umgang mit ihrer Erkrankung hilft, erzählte sie uns im Interview.
Text Hanna Sinnecker
abi, Sie haben Morbus Pompe. Wie und wann ist man Ihrer Erkrankung auf die Spur gekommen?
Ich habe 55 Jahre lang ein ganz normales Leben geführt und dachte, ich wäre kerngesund. Ich war immer sehr aktiv und sportlich, habe als Kind und Jugendliche Geräteturnen und später regelmäßig Step-Aerobic, Zumba und Spinning gemacht. Seit 2017 wurde ich immer wieder auf meine schwankende Art zu gehen angesprochen. Ich schob das auf das Alter. Bis dann Knieschmerzen auftraten, die ich beim Orthopäden untersuchen ließ: Es konnte aber nichts festgestellt werden, ich bekam eine Bandage und das war‘s. Zwei Jahre später habe ich ein Urlaubsvideo von mir gesehen und bin sehr erschrocken: Mein Gang war wirklich stark verändert, ich ging unsicher und wackelig. Ich machte erneut einen Termin in der Orthopädie, wo die Ärztin weitere Tests durchführte und eine Muskeldystrophie vermutete, sie überwies mich in die Neurologie. Dort hatte ich das große Glück, dass der junge und engagierte Neurologe eine genetisch bedingte Muskelerkrankung vermutete. Im Januar 2020 bestätigte dann ein Bluttest, ein EMG und ein Gentest die Diagnose Morbus Pompe. Das war ein Schock für mich, da es in meiner Familie bisher keine ähnlichen Fälle gab.
Ich dachte direkt: Was ist mit meinen Kindern? Glücklicherweise sind sie gesund, sie können aber Träger des defekten Gens sein.
Die Erkrankung wird Sie Ihr ganzes Leben begleiten: Wie bestreiten Sie Ihren Alltag und wo ist der Morbus Pompe ggf. ein Hindernis für Sie? Ich arbeite im Home Office als technische Zeichnerin, bin dadurch flexibel und habe zudem einen höhenverstellbaren Schreibtisch, der mir bei plötzlich auftretenden Muskel- oder Rückenschmerzen sehr hilft. Sportlich musste ich sehr viel kürzer treten. Stattdessen mache ich nun Rehasport, Physiotherapie und gehe spazieren. Alle zwei Wochen benötige ich eine ca. 4,5-stündige Infusion für die Enzymersatztherapie.
Ich wünsche mir, dass die Erkrankung irgendwann heilbar oder leichter behandelbar sein wird.
Gabi D. Morbus Pompe-Patientin
Die ersten sechs Monate musste ich dafür ins Uniklinikum fahren, was sehr aufwendig war und viel Zeit gekostet hat. Mittlerweile konnte ich aber auf eine Heiminfusion umstellen, was viel komfortabler ist.
Sie sind im UK Giessen bei Frau Dr. Lampe in Behandlung: Wie geht es Ihnen unter Therapie? Durch die Therapie sind meine Beschwerden soweit gut kontrollierbar, und auch die moderate sportliche Betätigung hilft mir sehr. Ich habe einen stabileren Gang und bin insgesamt beweglicher. Zudem hilft mir der Austausch mit anderen Betroffenen sehr. Bei meinen ersten Recherchen zur Krankheit bin ich auf die Selbsthilfegruppe Pompe Deutschland e. V. gestoßen und wurde dort Mitglied. Gerade am Anfang hat man viele Fragen. Andere Betroffene können helfen, Tipps geben, verstehen die Probleme, die man hat. Das ist unglaublich viel wert!
Was wünschen Sie sich in Zukunft an Verbesserungen für Betroffene, sei es bzgl. der Versorgung oder auch der Forschung?
Haus- und Fachärzte benötigen mehr Informationen zu seltenen Krankheitsbildern, damit Diagnosen schneller gestellt und Betroffene professionell und einfühlsam versorgt werden können. Auch ein flächendeckenderes Netz an spezialisierten Zentren wäre wichtig, denn oft sind leider lange Anfahrtswege für die Therapie und die Kontrolltermine die Regel. Und natürlich wünsche ich mir, dass die Erkrankung irgendwann heilbar oder leichter behandelbar sein wird. Es befinden sich ja bereits erste gentherapeutische Therapieansätze und neue Medikamente in der Entwicklung, das macht Hoffnung!
Morbus Fabry und Morbus Pompe: Den Erkrankungen auf die Spur kommen
Morbus Fabry und Morbus Pompe sind seltene genetische Erkrankungen, die sich durch eine Vielzahl an Symptomen bemerkbar machen können. Es handelt sich um Multisystemerkrankungen: es können also Schäden an verschiedenen Organen auftreten. Da die Erkrankungen unbehandelt weiter fortschreiten, sich die Beschwerden weiter verschlechtern und die Lebensqualität Betroffener zunehmend einschränken, ist eine frühe Diagnose von entscheidender Bedeutung.
Mögliche Symptome bei Morbus Fabry
NERVENSYSTEM
• Starke Schmerzen, die Minuten bis Stunden andauern
• Hörverlust, Tinnitus
• Hitze- oder Kälteunverträglichkeit oder Belastungsintoleranz
• Transitorisch-ischämische Attacke (TIA) und Schlaganfall
• Brennen der Hände und Füße, auch als Akroparästhesie bezeichnet
• Schwindel
HERZ • Unregelmäßiger Herzschlag (schnell/langsam)
• Herzanfall oder -versagen
• Vergrößertes Herz
NIEREN
• Eiweiß im Urin
• Verminderte Nierenfunktion
• Nierenversagen
MAGEN-DARM
• Übelkeit und Erbrechen
• Durchfall und/oder Verstopfung
• Bauchschmerzen
• Blähungen
HAUT
• Vermindertes Schwitzen
• Kleine dunkelrote Punkte, die als Angiokeratome bezeichnet werden, vor allem zwischen Bauchnabel und Knien
AUGEN
• Wirbelförmiges Muster auf der Hornhaut
• Fabry-Katarakt (eine bestimmte Form der Linsentrübung)
PSYCHOSOZIALE ASPEKTE
• Depression • Angstzustände
• Panikattacken
• Isolation
Morbus Fabry
Morbus Fabry wird auch als das Chamäleon unter den seltenen Erkrankungen bezeichnet, da er diverse Symptome verursachen kann, die zunächst auf die falsche Fährte locken können. Betroffene haben oft eine jahrelange Odyssee von Arzt zu Arzt hinter sich, in der häufig Fehldiagnosen gestellt werden, verbunden mit einer Fehlbehandlung, die für die Patienten oft wirkungslos bleibt. Zu den körperlichen Symptomen kommen daher nicht selten psychische Beschwerden hinzu.
Wenn also verschiedene unspezifische Symptome auftreten (s. Grafik links), sollten diese in Kombination betrachtet werden, um der Erkrankung möglichst früh auf die Spur zu kommen.
Sind zudem bereits ähnliche Fälle in der Familie aufgetreten, sollte man umso hellhöriger werden. Bei männlichen Patienten kann bereits ein einfacher Bluttest zur Bestimmung der Enzymaktivität zur Diagnose führen, die durch eine genetische Testung abgesichert werden kann. Bei weiblichen Patienten kann die genetische Testung die Diagnose sichern. Je früher die Erkrankung erkannt wird, umso schneller kann die passende Therapie in die Wege geleitet werden. Nur so können Beschwerden eingedämmt, mögliche Folgeschäden verhindert und die Lebensqualität Betroffener verbessert werden.
Morbus Pompe
Wie der Morbus Fabry ist auch der Morbus Pompe eine Erkrankung mit vielen Gesichtern, die sich durch eine Vielzahl an Symptomen bemerkbar machen kann (s. Grafik rechts). Da die Beschwerden auch auf andere neuromuskuläre Erkrankungen hindeuten können, dauert es oft bis die richtige Diagnose gestellt ist. Die Erkrankung schreitet weiter voran und kann die Lebensqualität stark beeinträchtigen.
Oft kommen seelische Beschwerden durch den anhaltenden Leidensdruck und die langen Diagnosewege hinzu. Die einzelnen Symptome sollten also unbedingt in Gänze betrachtet werden.
Morbus Pompe ist eine genetisch bedingte Erkrankung. Besteht der Verdacht auf Morbus Pompe, ist der erste Schritt ein einfacher Bluttest. Verhärtet sich der Verdacht, entscheidet der Arzt, ob eine genetische Testung anzuraten ist, die den Morbus Pompe final bestätigen kann.
Der Zeitpunkt der Diagnose ist entscheidend für den Behandlungserfolg, da bereits entstandene Schäden oft irreversibel sind. Um weitere Organschäden zu verhindern sowie die Lebensqualität Betroffener langfristig zu verbessern, sollte bei einer entsprechenden Symptomkombination ein Arzt aufgesucht werden.
Mögliche Symptome bei adultem Morbus Pompe
SKELETT UND MUSKELN
• Muskelschwäche, vor allem der rumpfnahen Muskulatur
• Rückenschmerzen
• Körperliche Aktivität ist nicht mehr oder eingeschränkt möglich
• Schwierigkeiten beim Treppensteigen
• Gangstörungen (Watschelgang)
• Gelenksteifheit
• Flügelartiges Abstehen der Schulterblätter
• Eingeschränkte Beweglichkeit der Wirbelsäule
• Abnorme Krümmung der Wirbelsäule
• Motorische Einschränkungen
LUNGE
• Atemschwäche
• Atembeschwerden
• Häufige Atemwegsinfekte
• Schlafapnoe
• Tagesschläfrigkeit
• Morgendliche Kopfschmerzen
VERDAUUNGSTRAKT
• Schwierigkeiten beim Kauen und Schlucken
• Unzureichende Gewichtszunahme
• Chronische Verstopfung
• Harn- und Stuhlinkontinenz
Weitere Informationen unter: www.fabryfamilytree.de und www.amicusrx.de

ANZEIGE Lesen Sie mehr auf seltenekrankheiten.de 11
NP-NN-DE-00010124(v1.0)-02/24
FOTO: PRIVAT
„Die besten Informationen bekommen Morbus Fabry-Patienten von anderen Betroffenen.“
Morbus Fabry ist eine erblich bedingte Stoffwechselstörung, die das Leben Betroffener stark beeinträchtigen kann.
Natascha Sippel-Schönborn ist selbst betroffen von der Erkrankung und Geschäftsführerin der Morbus Fabry Selbsthilfegruppe (MFSH) e. V. Warum anhaltende Forschung im Sinne der Patienten so wichtig ist und welche Rolle die Selbsthilfe hier einnimmt, erzählt sie uns im Interview.
Text Hanna Sinnecker
Frau Sippel-Schönborn, Sie sind selbst betroffen von Morbus Fabry. Wie gehen Sie persönlich mit Ihrer Erkrankung um?
Nachdem ich mit Verdacht auf Herzinfarkt ins Uniklinikum Mainz eingeliefert wurde, war ein Arzt direkt hellhörig, da ich keinen Gefäßverschluss hatte, der für einen Infarkt typisch wäre. Als er hörte, dass mein verstorbener Vater mehrere Herzinfarkte, Nierenprobleme und Schlaganfälle hatte, riet er mir zu weiteren Untersuchungen und einer genetischen Analyse. Es war großes Glück, dass der Arzt schon einmal von Morbus Fabry gehört hatte, die Puzzleteile richtig kombinierte und so die Diagnose gestellt werden konnte. Meinen ersten Behandlungstermin im Zentrum für seltene Erkrankungen Mainz hatte ich dann sechs Monate später: die Wartezeit war schrecklich und ich hatte große Angst, dass zwischenzeitlich wieder etwas passiert. Ich leide zum Glück nicht unter Schmerzen, aber habe Einlagerungen im Herzen, eine Herzwandverdickung und Herz-Rhythmus-Störungen. Die medikamentöse Behandlung hält das Fortschreiten meiner Erkrankung auf und bringt mir die Sicherheit, nicht von Krankheitsschüben überrollt zu werden. Ich habe außerdem einen Herzschrittmacher. Mir geht es dadurch recht gut und ich lebe nicht mehr in ständiger Angst.
Sie sind Geschäftsführerin der MFSH e. V. Warum ist die Vernetzung unter Patienten so wichtig?
Nach meiner Diagnose musste ich feststellen, dass sehr wenige Ärzte Morbus Fabry kennen. Ich war vergeblich auf der Suche nach Informationen für mich und meine vier Kinder, die alle von Morbus Fabry betroffen sind. Leider hatte ich das damals jährliche Treffen der Selbsthilfegruppe gerade verpasst. Als die damalige Vorsitzende der Selbsthilfegruppe zurücktrat, rückten Herr Dr. Wilden als Vorsitzender und Herr Landgraf als stellvertretender Vorsitzender nach. Sie fragten mich, ob ich den nächsten Fabry-Frauenworkshop besuchen und dabei die MFSH vorstellen könne: mein erster Schritt in die Selbsthilfearbeit. Ich bekam von den Teilnehmenden sehr positives Feedback und besuchte weitere Workshops, in denen ich kleine Vorträge hielt. Mir wurde bald klar, dass ich eine Teilzeitarbeit nicht mehr mit der Arbeit in der Selbsthilfe unter einen Hut bekomme.

Leben mit Morbus Fabry –
Die Patient:innen im Fokus
Die ungedeckten Bedürfnisse von Menschen mit seltenen Erkrankungen besser zu verstehen und zu erfüllen: Das geht nur im engen Austausch mit den Betroffenen selbst. Das forschende Pharmaunternehmen Chiesi, das stets an der Entwicklung neuer Behandlungsoptionen und der Bereitstellung von Unterstützungsangeboten für Menschen mit seltenen Erkrankungen arbeitet, hat sich genau das zum Ziel gesetzt. Derzeit liegt der Schwerpunkt des Unternehmens unter anderem auf lysosomalen Speicherkrankheiten, wie Morbus Fabry.
Also habe ich erst einen Minijob in der Selbsthilfegruppe übernommen und arbeite inzwischen halbtags als Geschäftsführerin der MFSH e. V. Meine Ambition war von Anfang an, mehr Informationen verfügbar zu machen und engmaschigere Austauschmöglichkeiten für Betroffene zu schaffen. Im Alltag sind Betroffene meist auf sich gestellt. Die besten Tipps bekommen sie von anderen Patienten, die ihre Beschwerden nachvollziehen können.
Meine Ambition war von Anfang an, mehr Informationen verfügbar zu machen und engmaschigere Austauschmöglichkeiten für Betroffene zu schaffen.
Dieser Austausch ist extrem wichtig, insbesondere für Kinder: wie geht man z. B. im Sportunterricht damit um, wenn man Schmerzen hat, ohne als Drückeberger abgestempelt zu werden? Wie erkläre ich anderen meine Erkrankung? Unsere Jugendtreffen geben Raum für diesen Austausch. Erwachsene Betroffene haben wieder andere Hürden: Viele können z. B. nicht in Vollzeit arbeiten und brauchen Unterstützung bei sozialrechtlichen Fragen. Bei unseren Online-Treffen, die zweimal monatlich stattfinden, bekommen sie schnell Antworten auf ihre Fragen. Sie vernetzen auch die Forschung mit Betroffenen. Wie können Betroffene sich hier aktiv beteiligen und welche Rolle übernimmt dabei die MFSH e. V.? Patienten sind sehr interessiert daran, was sich in der Forschung tut, da sie wissen, dass sie davon profitieren, wenn Therapien weiter verbessert werden. Wir sehen es als unsere Aufgabe, mit medizinischen Zentren und der Pharmaindustrie in Kontakt zu treten, um Patienten z. B. zu Studien zu informieren, an denen sie teilnehmen können. Den Betroffenen ist sehr bewusst, welche wichtige Rolle sie in der Forschung spielen, gerade weil die Patientenzahl klein ist.
Unterstützung für Morbus Fabry-Patient:innen
Das Leben mit seltenen Erkrankungen kann herausfordernd sein. Deshalb tritt Chiesi in den Dialog mit Betroffenen, um herauszufinden, welche Hindernisse Morbus Fabry birgt, und welche durch gemeinsame Projekte Stück-für-Stück bewältigt werden können. So ist beispielsweise nicht nur ein Bereich für Fachpersonal auf der neuen Website des Unternehmens zu finden, sondern auch ein Patient:innenbereich, der in Zusammenarbeit mit Betroffenen entstanden ist.
Unter morbus-fabry.chiesirarediseases.de wird sowohl
Patient:innen als auch ihren Angehörigen Hilfestellung dabei angeboten, die Erkrankung zu verstehen und mit ihr umzugehen. Denn sowohl für den Patient oder die Patientin selbst als auch für die Angehörigen kann es zunächst schwer sein, mit einer solche Diagnose zurecht zu kommen. Auf der Website finden sie nicht nur grundlegende Informationen zu Symptomen sowie Diagnose- und Behandlungsmöglichkeiten, sondern vor allem Berichte von Betroffenen und ihre konkreten Erfahrungen mit der Erkrankung, die besonders neu diagnostizierten Patient:innen eine große Hilfe sein können. Zudem sind unter anderem Anlaufstellen für die spezialärztliche Versorgung in der Nähe zu finden, und auch zu sozialrechtlichen Themen wie der Beantragung eines Schwerbehindertenausweises oder einer Erwerbsminderungsrente finden Betroffene und ihre Angehörigen hier wertvolle Tipps. Patient:innen sind Expert:innen ihrer Erkrankung und wissen selbst am besten, welche Unterstützung sie benötigen. Chiesi möchte ihnen deshalb mehr Gehör verschaffen.

Natascha Sippel-Schönborn Geschäftsführerin der Morbus Fabry Selbsthilfegruppe (MFSH) e. V. und selbst Morbus Fabry-Patientin
Die große Hoffnung der Patienten ist, dass es irgendwann eine Möglichkeit gibt, die Erkrankung ursächlich zu bekämpfen. Zudem werden wir zunehmend gefragt, was wir Patienten brauchen, um den Alltag mit der Erkrankung zu bestreiten und zu welchen Themen wir Informationen benötigen. Diese Themen bringen wir dann bei Ärzten und Pharmaunternehmen zur Sprache, die dann z. B. Workshops und Vorträge für Betroffene organisieren. Auch beim Erstellen von Infomaterial werden wir intensiv mit einbezogen, da uns hierfür oftmals die Ressourcen fehlen. Wir stellen dann gern die gesammelten Infos der Pharmaunternehmen zur Verfügung und freuen uns, dass unser Patienteninput wertgeschätzt wird. Trotzdem bleiben wir dabei immer eine unabhängige Anlaufstelle.
Eines Ihrer Ziele ist es, über die Erkrankung aufzuklären. Sie haben eine tolle Lichterkettenaktion gestartet: können Sie uns dazu etwas mehr erzählen?
Bei Morbus Fabry geht man von einer hohen Dunkelziffer Betroffener aus. D. h. es gibt vermutlich viele nicht- oder falschdiagnostizierte Patienten. Daher ist es sehr wichtig, die Erkrankung bekannter zu machen, und Aktionen zum Rare Disease Day tragen dazu bei.
Wir haben von einer Patientin inspiriert begonnen, Ampullen der Fabry-Medikamente zu sammeln und daraus Lichterketten in den Farben des Rare Disease Days zu basteln, die in Apotheken zusammen mit Infoflyern ausliegen. So konnten wir zudem noch Spendengelder z. B. für unsere Jugendtreffen sammeln. Wir haben hunderte Ampullen verarbeitet! Das schafft nicht nur Aufmerksamkeit, sondern bringt auch Patienten in dieser gemeinsamen Mission zusammen.
Weitere Informationen unter: www.fabry-shg.org
Aktive Forschung im Sinne der Betroffenen
Und diese Arbeit im Sinne der Patient:innen bleibt nicht stehen: Derzeit werden von Chiesi u.a. verschiedene Studienprojekte gefördert, mit dem Ziel, unter anderem die Diagnostik der Erkrankung zu verbessern. Dabei bleibt immer eines im Fokus: Die Bedürfnisse der Betroffenen und ihrer Angehörigen und die weitere Verbesserung ihrer Lebensqualität. Nur, wenn ihre Stimmen gehört werden, können Betroffene aktiv mit einbezogen werden: bei der Forschung und Entwicklung, bei der so wichtigen Aufklärungsarbeit rund um ihre Erkrankung und bei allen Fragen zu einer passgenauen Versorgung und Unterstützung von Patient:innen und den ihnen Nahestehenden.

Weitere Informationen finden Sie unter morbus-fabry.chiesirarediseases.de
Lesen Sie mehr auf seltenekrankheiten.de 12 ANZEIGE
FOTO: PRIVAT
„Für mich war es immer wichtig zu wissen, dass ich mit meinen Beschwerden nicht allein bin.“
Vermutlich jeder kennt das Gefühl: Man isst zu schnell und hat das Gefühl, dass einem der Bissen förmlich im Hals stecken bleibt. Treten Schluckbeschwerden häufiger auf und werden von Schmerzen begleitet, kann auch eine ernsthafte Erkrankung dahinterstecken: Die eosinophile Ösophagitis (kurz EoE). Carla Oberth ist erst 23 Jahre alt, lebt seit über zehn Jahren mit einer EoE und erzählte uns ihre Geschichte.
Text Hanna Sinnecker
Frau Oberth, sie haben eine EoE, die Sie bereits seit der Kindheit begleitet. Wie ist man Ihrer Erkrankung auf die Schliche gekommen?
Ich hatte bei meiner Geburt einen Speiseröhrenverschluss (eine sogenannte Ösophagusatresie), der operativ geweitet werden musste. Es blieb eine Engstelle in der Speiseröhre, die jährlich überprüft wurde. Bei einer dieser Routineuntersuchungen wurde festgestellt, dass meine Speiseröhre Längsrillen aufweist, was typisch für die EoE ist. Zudem hatte ich immer das Gefühl, einen Kloß im Hals zu haben: ein weiteres deutliches Zeichen, das auf eine EoE deutet. Es wurden dann Gewebeproben entnommen und untersucht. Das Ergebis war, dass meine Eosinophilen (eine bestimmte Art weißer Blutkörperchen) deutlich erhöht sind, was das entscheidende klinische Merkmal für eine EoE ist. So bekam ich im Alter von elf Jahren meine Diagnose.
Gerade im Kindes- und Jugendalter ist es nicht leicht, anders zu sein: Wie sind Sie in so jungen Jahren mit Ihrer Erkrankung umgegangen und zurecht gekommen?
Natürlich war es nicht angenehm. Aber da ich seit meiner Geburt auf Hilfsmittel angewiesen war und auf meine Ernährung achten musste, war das für mich Normalzustand. Durch die EoE, die dann dazukam, war ich natürlich noch ein Stück weiter eingeschränkt, ich musste eine strikte Diät einhalten und auf viele Nahrungsmittel verzichten. Aber ich habe immer versucht, positiv zu bleiben, da das zunächst gut funktionierte, um die Symptome zu kontrollieren.
Als ich etwa 19 Jahre alt war, wurden die Symptome stärker, die Diät wurde weiter verschärft. Ich verzichtete auf alle weizen- und zuckerhaltigen Produkte, habe quasi vegan gelebt. Zudem wurde ich dann auch medikamentös behandelt, aber mein Arzt meinte schon bald, dass ich für eine dauerhafte medikamentöse Therapie zu jung sei.
Ich habe immer versucht, positiv zu bleiben.
Carla Oberth, EoE-Patientin
Heute geht es mir soweit gut, ich halte aber weiter eine strikte Auslassungsdiät ein. Ich achte darauf, wenn möglich zu Hause zu essen, spontane Abendessen z. B. im Restaurant versuche ich zu vermeiden. Das schränkt mich durchaus ein.
Welche Rolle spielt für Sie der Austausch mit anderen Betroffenen, z.B. über den KEKS e. V.? Für mich war es immer sehr wichtig zu wissen, dass ich mit meinen Beschwerden nicht allein bin. Der Austausch mit anderen Betroffenen bietet mir die Möglichkeit, mich auszutauschen zu Fragen im Umgang mit meiner Erkrankung, zu Behandlungsoptionen und Anlaufstellen. Ich bin deswegen sowohl beim KEKS e. V. als auch bei SoMA e. V. aktiv, helfe bei Vorträgen und Workshops. Für mich ist das essenziell!

KEKS e. V. - Die Patienten- und SelbsthilfeOrganisation für Kinder und Erwachsene mit kranker Speiseröhre Der KEKS e. V. unterstützt im Schwerpunkt Menschen, die mit einer nicht durchgängigen Speiseröhre (Ösophagusatresie) oder Verengung (Stenose) geboren wurden. Hier finden auch Betroffene mit eosinophiler Ösophagitis und Verätzungen der Speiseröhre Unterstützung. Was 1984 als Gemeinschaft von Eltern mit von einer Ösophagusatresie betroffenen Kindern begann, hat sich heute zu einer professionellen, bundesweit tätigen und international vernetzten Patientenorganisation entwickelt.
Weitere Informationen unter: www.keks.org
Die Symptome der Eosinophilen Ösophagitis (EoE)
Die eosinophile Ösophagitis (kurz EoE) bezeichnet eine seltene immunvermittelte Erkrankung, bei der die Speiseröhre (Ösophagus) chronisch entzündet ist. Auch wenn sie zu den seltenen Erkrankungen zählt, ist die EoE die zweithäufigste Erkrankung der Speiseröhre.
ACHTUNG – VERWECHSLUNGSGEFAHR!
Aufgrund der unspezifischen Symptome kommt es nach wie vor häufig zu Fehldiagnosen. Daher ist es wichtig, die Symptome zu kennen und im Patientengespräch die richtigen Fragen zu stellen, um den Betroffenen durch eine korrekte Diagnose und Therapie zu mehr Lebensqualität zu verhelfen.
Oftmals wird die EoE zunächst mit der gastroösophagealen Refluxkrankheit (GERD) verwechselt. Es können aber auch andere Erkrankungen wie eine eosinophile Gastroenteritis, parasitäre Infektionen oder auch Morbus Crohn als Fehldiagnosen gestellt werden. Daher ist es sehr wichtig, die rechts aufgeführten Symptome zu kennen und bei Alarmzeichen hellhörig zu werden.
Die Diagnose sollte in jedem Fall durch eine*n Gastroenterolog*in erfolgen, der/die nach einem ausführlichen Patientengespräch eine endoskopische Untersuchung durchführt, bei der mehrere Gewebeproben aus der Speiseröhre entnommen und unter dem Mikroskop analysiert werden. Die Diagnose kann dann zusammen mit dem endoskopischen Bild und durch den Nachweis eosinophiler Granulozyten (einer speziellen Art weißer Blutkörperchen) zweifelsfrei gestellt und eine Therapie eingeleitet werden.

Hauptsymptome der EoE bei Erwachsenen Schluckbeschwerden, vor allem beim Verzehr fester Speisen und faseriger/trockener Lebensmittel
• Unangenehmes oder schmerzhaftes Gefühl, als würde ein Bissen im Hals stecken bleiben
• Brennen hinter dem Brustbein
• Bolusimpaktion: Nahrungsbissen bleibt im Hals stecken
ACHTUNG! Dies kann einen medizinischen Notfall hervorrufen!
Betroffene leiden oft zusätzlich an allergischen Erkrankungen wie Heuschnupfen, Asthma oder atopischen Ekzemen.
Hauptsymptome der EoE bei Kindern
• Übelkeit
• Erbrechen
• Bauchschmerzen
• Nahrungsverweigerung Wachstumsstörungen
Lesen Sie mehr auf seltenekrankheiten.de 13
Weitere Informationen finden Sie unter www.schluckbeschwerden.de
BILD: SHUTTERSTOCK_2238871153
FOTO: PRIVAT ANZEIGE
Leben mit Phosphatdiabetes“Gestalten statt hinnehmen“
Die x-chromosomale Hypophosphatämie (kurz XLH) ist eine seltene Störung des Knochenstoffwechsels.
Die Mutation, die XLH hervorruft, kann vererbt werden oder als spontane Mutation auftreten, was die Diagnose erschweren kann. XLH führt zu einem Phosphatmangel, weshalb die Erkrankung auch die Bezeichnung Phosphatdiabetes trägt. Auch wenn die Erkrankung entdeckt und behandelt wird, leiden Betroffene an ihren Folgen: dazu zählen u. A. starke Schmerzen, enorme Bewegungseinschränkungen und schlecht verheilende Knochenbrüche. Wir sprachen mit Sara Franke über ihr Leben mit der Erkrankung.
Text Alexandra Lassas
Frau Franke, Sie haben die Erkrankung XLH. Wann und wie hat sich die Erkrankung bei Ihnen geäußert und gibt es in Ihrer Familie weitere Betroffene?
Bei mir wurde XLH bereits im Kleinkindalter sichtbar. Mit dem Beginn des Laufens zeigten sich deutliche Verformungen meiner Beine. Meine Mutter bemerkte das und zog sofort Parallelen zu ihrer eigenen Kindheit, in der sie ähnliche Verformungen hatte. Die Krankheit, die auch meine Mutter und meine beiden Geschwister betrifft, wurde bei meiner Mutter damals fälschlicherweise als Knochenfehlbildung diagnostiziert. Damals versuchte man durch verschiedene OPs, ihre Beine zu korrigieren, was zu starken Verformungen und irreversiblen Schäden bei ihr führte. Glücklicherweise wurde durch die Hartnäckigkeit meiner Eltern bei meinen Schwestern und mir die korrekte Diagnose gestellt, als ich drei Jahre alt war.
Was waren und sind für Sie die größten Herausforderungen aufgrund Ihrer Erkrankung?
In meiner Kindheit kämpfte ich mit Zahnabszessen und den Einschränkungen durch O-Beine, besonders beim Laufen und Springen. Aber unsere Familie lebte nach dem Motto: Jeder gestaltet sein Leben so gut wie möglich. In meiner Jugend führten Operationen zur Korrektur der Beine und weitere Eingriffe zu vielen Fehlzeiten in der Schule. Es war sehr schwer, als "behindert" abgestempelt und von Mitschülern und Lehrern in meinen Fähigkeiten unterschätzt zu werden. Doch dank der Unterstützung meiner Familie und Freunde konnte ich mein Selbstbewusstsein stärken.
Mein gesamtes Skelett ist deformiert, was zu zahlreichen Begleiterkrankungen führte. Ärzte behandeln diese meist nur symptomatisch: ein zeitaufwendiger und kraftraubender Prozess. Die wirksame Behandlung begann erst, als ich gute Beziehungen zu den Ärzten aufbaute. Es gibt nicht viele Patienten mit dieser Krankheit, und es dauert oft zu lang, bis man als erwachsener Patient einen Spezialisten findet. Bis dahin versuchte ich, meine Schmerzen mit Schmerzmitteln zu lindern. Als lebensfroher Mensch stieß ich oft auf

„Meine Hautprobleme begannen 2004“, berichtet eine Patientin. Sie litt zu dieser Zeit unter wiederkehrenden Hautausschlägen und Schmerzen. Erst Jahre später wurde bei ihr eine Mycosis fungoides diagnostiziert, eine Krebserkrankung, die in Europa weniger als einen von 110.000 Menschen betrifft.1
„Ich befand mich fast zehn Jahre lang in einer Grauzone“, erinnert sie sich. Ihre anfänglichen Symptome wurden zunächst als Ekzem erkannt. Erst ein Zufallsbefund führte zur richtigen Diagnose. Diese Geschichte ist kein Einzelfall: Der Weg bis zum Befund dauert bei diesem Krankheitsbild durchschnittlich zwei bis sieben Jahre.2
Kyowa Kirin ist ein global tätiges biopharmazeutisches Unternehmen, das die Versorgung von Menschen mit seltenen Erkrankungen verbessern möchte. Es wurde
Quellen:
Schwierigkeiten, die Intensität meiner Schmerzen den Ärzten zu vermitteln und die Dringlichkeit meiner Situation zu betonen. Aber nicht nur erkrankte Kinder, sondern auch erwachsene Patienten benötigen weiterhin eine adäquate Betreuung, um Beschwerden zu lindern!
XLH ist zwar noch nicht heilbar, aber behandelbar: wie geht es Ihnen unter Therapie?
Mir geht es ganz gut. Natürlich begleiten mich ständig Schmerzen, die wohl auch bleiben werden. Das Bücken und Knien wird mir immer verwehrt bleiben. Trotz dieser Herausforderungen empfinde ich mein Leben als wunderbar. Seit einem Jahr spritze ich mir ein Medikament, das auch für Erwachsene zugelassen ist und mir Tage mit erträglichen Schmerzen schenkt.
Sie sind 2022 Mutter geworden: Hatten Sie Sorge, dass ihr Kind auch von XLH betroffen sein könnte und wie verlief Ihre Schwangerschaft?
Mein Mann und ich haben ausführlich darüber gesprochen und uns entschlossen, unsere Lebensfreude und unsere Träume nicht von einer Erkrankung beeinträchtigen zu lassen. Als das neue Medikament zugelassen wurde und die Erfolge bei Kindern vielversprechend waren, wurde unser Wunsch nach einer Familie nur stärker. Meine Frauenärztin war hervorragend über meine Krankheit informiert und hat mich jederzeit einfühlsam unterstützt. Nun sind wir stolze Eltern eines kerngesunden Sohnes.
Wie geht es Ihnen heute und was möchten Sie anderen Betroffenen mit auf den Weg geben?
In jedem von uns schlummert etwas, für das es sich zu kämpfen lohnt. Jeder, der betroffen ist, sollte die Kraft und Geduld aufbringen, Ärzte aufzusuchen, bei denen er sich wohl fühlt. Unterstützung ist von entscheidender Bedeutung. Zudem sollte man sich bemühen, sich von der Erkrankung nicht übermäßig einschränken zu lassen. Es ist von grundlegender Bedeutung, fest an sich selbst zu glauben. Wir verdienen es, wertgeschätzt zu werden. Wir sind so viel mehr als die Erkrankung!
Dieser

Symptome der XLH im Überblick
Mögliche Symptome bei Kindern:
• Fehlstellungen der Beine (O- oder X-Beine)
• Deformierungen der Knochen und Achsenfehlstellungen
• Weiche Knochen (Rachitis)
• Verspäteter Laufbeginn, Gangbildveränderungen, „Watschelgang“
• Verzögertes/Vermindertes Wachstum (Kleinwuchs)
• Dysproportionen
• Knochen- und Gelenkschmerzen
• Muskelschmerzen und -schwäche
• Schädeldeformationen: Craniosynostose (verfrühter Verschluss der Schädelnähte), Chiari Malformation (Fehlbildung des Übergangs zwischen Hinterhaupt und Wirbelsäule)
• Spätes Sekundärgebiss und Zahnprobleme (z. B. Abszesse und Fisteln)
Zusätzliche mögliche Symptome bei Erwachsenen:
• Veränderungen des Gehörs: Schwerhörigkeit bis hin zum Hörverlust, Tinitus, Schwindel
• Frakturen und Pseudofrakturen durch unzureichende Knochenmineralisation Osteomalazie (Knochenerweichung)
• Spinalkanalstenosen (Verengungen des Wirbelkanals)
• Früh einsetzende Arthrose oder Knochen- und Gelenkentzündungen
• Bewegungseinschränkungen und Steifigkeit
• Mineralisierung (Verkalkung) von Sehnen und Bändern
• Dauerhafte Verkürzung von Sehnen, Muskeln und Bändern Reduzierte Belastbarkeit, Erschöpfung
Phosphatdiabetes e. V. Die Patientenorganisation Phosphatdiabetes e. V. ist eine Gemeinschaft von Betroffenen und Angehörigen, die Informationen bereitstellt, Hilfestellung anbietet und durch persönlichen Erfahrungsaustausch im Umgang mit der Erkrankung unterstützt. Die Belange aller Altersstufen – von Kindern, Jugendlichen und Erwachsenen – finden Beachtung.
Weitere Informationen unter: www.phosphatdiabetes.de
Einsatz für Menschen mit seltenen Erkrankungen
1949 in Japan gegründet und entwickelt seit dieser Zeit innovative Therapien in den Bereichen Nephrologie, Neurologie, Onkologie und Immunologie. Die Forschung, Entwicklung und Wirkstoffproduktion stützen sich auf Verfahren der Spitzenbiotechnologie aus eigenem Hause.
Kyowa Kirin ist ein global tätiges Unternehemen, das die Versorgung von Menschen mit seltenen Erkrankungen verbessern möchte.
So gilt das Unternehmen als Pionier in der Behandlung des nur selten auftretenden Phosphatdiabetes. Ein weiterer Schwerpunkt ist die Behandlung seltener Krebserkrankungen wie der Mycosis fungoides und des Sézary-Syndroms – beides Unterformen des kutanen T-Zell-Lymphoms (CTCL).
Kyowa Kirin möchte sämtlichen Menschen, mit denen es sich im Austausch befindet, ein Lächeln schenken – nicht nur durch die Bereitstellung neuer Wirkstoffe, sondern auch durch gelebte Partnerschaften. Das Unternehmen sucht weltweit den Austausch mit Betroffenen und Beteiligten, um gemeinsam bessere Antworten auf Patientenbedürfnisse zu finden, getrieben von dem Ansporn „Making people smile“.
Weitere Informationen: www.kyowakirin.com

Lesen Sie mehr auf seltenekrankheiten.de 14
Text
Katharina Lassmann
ist in Zusammenarbeit
entstanden.
Artikel
mit der Kyowa Kirin GmbH
https://tinyurl.com/4vr9ar9v 2 – CL Foundation https://tinyurl.com/mvk67utw
1 – Orphanet
FOTO: PRIVAT
Sara Franke XLH-Patientin
FOTO: GPOINTSTUDIO
Dieser Beitrag ist in Zusammenarbeit mit dem Sächsischen Staatsministerium für Wissenschaft, Kultur und Tourismus entstanden.
„Die interdisziplinäre Expertise ist der entscheidende Vorteil eines Zentrums für Seltene Erkrankungen“
Zentren für Seltene Erkrankungen (ZSEs) sind eine wichtige Anlaufstelle für Menschen mit Seltenen Erkrankungen. Deutschlandweit gibt es mittlerweile über 30 ZSEs. Eines davon ist das UniversitätsCentrum für Seltene Erkrankungen (USE) in Dresden. Wir sprachen mit Prof. Dr. med. Reinhard Berner, Sprecher des USEs Dresden, über die wichtige Rolle der ZSEs.
Text Hanna Sinnecker
He rr Prof. Berner, Seltenen Erkrankungen auf die Spur zu kommen ist nicht leicht. Wie sehen die Diagnosewege derzeit aus?
Die Zugangswege sind ganz unterschiedlich. Ein sehr großer Teil der Seltenen Erkrankungen hat eine genetische Ursache, sodass Betroffene oft schon im Säuglingsoder Kleinkindalter klinische Auffälligkeiten zeigen. Wenn deren Ursache nicht zu klären ist, wird heute in der Pädiatrie relativ schnell auch auf Seltene Erkrankungen hin untersucht, die zur Symptomatik passen könnten. Bei Kindern ist der Zugangsweg also meist relativ direkt. Bei erwachsenen Betroffenen sieht das oft anders aus, da die Symptomatik unklar ist, sie häufig von Facharzt zu Facharzt überwiesen werden und man oft erst spät an eine Seltene Erkrankung denkt. Sie warten oft sehr lange auf ihre Diagnose oder erhalten sie schlimmstenfalls nie. Sie verfolgen am USE einen interdisziplinären Ansatz bei der Diagnostik und Therapie: Wie funktioniert die Zusammenarbeit im Netzwerk genau und welche Rolle spielt dabei moderne Technik?
Wenn wir einen Patienten übermittelt bekommen, wird der Fall in einer interdisziplinären Fallkonferenz besprochen. Dabei sind je nach Symptomatik Mediziner unterschiedlicher Fachbereiche eingebunden, die so einen ganzheitlichen Blick, aber von verschiedenen Seiten auf den Patienten, seine Beschwerden und seine Krankheitsgeschichte werfen. Diese strukturierte interdisziplinäre Expertise ist der entscheidende Vorteil eines ZSEs. Verhärtet sich der Verdacht auf eine Seltene Erkrankung und die Fallkonferenz kommt zu der Entscheidung, dass eine genetische Untersuchung sinnvoll ist, dann kann diese im Regelfall direkt in die Wege geleitet werden.

Prof. Dr. med. Reinhard Berner Sprecher des UniversitätsCentrums für Seltene Erkrankungen am Universitätsklinikum Carl Gus tav Carus Dresden
FOTO: UNIVERSITÄTSKLINIKUM DRESDEN
Die Möglichkeiten der genetischen Diagnostik sind ein enormer medizinischer Fortschritt, der besonders auch Menschen mit Seltenen Erkrankungen zugutekommt.
Diese Möglichkeiten der genetischen Diagnostik sind ein enormer medizinischer Fortschritt, der besonders auch Menschen mit Seltenen Erkrankungen zugutekommt. Mittlerweile können wir an den Zentren für Seltene Erkrankungen annähernd die Hälfte der Patienten diagnostizieren, aber unser Anspruch ist es natürlich, diese Zahl auch unter Einsatz von Bioinformatik, neuen Labor-, Analyse- und Auswertemethoden und KI in Zukunft weiter zu erhöhen.
Welche Schwerpunkte haben Sie in der Arbeit mit Patienten und ihren Angehörigen, und wo sehen Sie Potenziale, sie noch stärker einzubinden und zu unterstützen?
Am USE haben wir hier schon verschiedene Projekte
erfolgreich umgesetzt, allen voran das Projekt Translate NAMSE. Der Fokus war hier ganz klar, Diagnosewege zu verkürzen und Betroffenen eine Behandlung zu ermöglichen, wenn es für die Erkrankung zielgerichtete Therapien gibt.
Aber auch wenn die diagnostizierte Erkrankung noch nicht zielführend therapiert werden kann, haben viele der am Projekt beteiligten Familien berichtet, dass es bereits eine große Erleichterung für sie war, dass der Erkrankung ein Name gegeben werden konnte. Und vielen Betroffenen konnten dadurch auch neue Wege der Begleitung und Therapieunterstützung ermöglicht werden. Dabei ist es wichtig herauszufinden – und dafür brauchen wir gerade die Patienten und ihre Angehörigen auch dringend und müssen sie einbinden – wo sie denn in der Betreuung, die ja häufig auch interdisziplinär und multidimensional sein muss, Unterstützungsbedarfe sehen. Ein weiterer wichtiger Punkt des Projektes war es, den Übergang vom Kindes- und Jugendalter zum Erwachsenenalter zu begleiten. Hier sind wir dabei, strukturierte Programme zu entwickeln, um die Betroffenen bei diesem Übergang zu unterstützen und ihre Weiterversorgung zu sichern. Dieser Prozess ist aber sehr aufwendig und derzeit in der normalen Regelversorgung nicht abbildbar. Umso wichtiger sind solche Projekte, da sie zeigen, dass diese sog. Transitionsprogramme von unschätzbarem Wert für Betroffene und ihre Angehörigen und auf lange Sicht eben doch kosteneffizient sind. Insofern müssen sie mittelfristig in die Regelversorgung überführt werden.
Das USE ist auch in der Forschung breit aufgestellt. In welchen Bereichen liegt Ihre besondere Expertise? Wie praktisch alle ZSEs fokussieren wir uns auf spezielle Krankheitsgebiete, auch wenn wir in unserer koordinierenden Funktion natürlich Ansprechpartner für alle Patienten sind. Am USE in Dresden konzentrieren wir uns zum einen auf Erkrankungen des Immunsystems. Gerade bei Kindern gibt es angeborene Immundefekterkrankungen, bei denen ein Teil des Immunsystems fehlt oder nicht vollständig funktionsfähig ist. Aber auch Fehlregulierungen des Immunsystems wie z. B. autoinflammatorische Erkrankungen oder Autoimmunerkrankungen gehören zu unserer Expertise; ein weiterer Schwerpunkt sind seltene angeborene Entwicklungsstörungen.
Gibt es Bereiche, in denen Sie besondere Forschungsfortschritte verzeichnen konnten?
In den eben genannten Bereichen forschen wir ganz aktiv daran, die Gene zu identifizieren, deren Fehlfunktion für die entsprechenden immunologischen Erkrankungen verantwortlich sind. Wir möchten herausfinden, an welcher Stelle der Signalweg zur Regulation des Immunsystems unterbrochen wird, um den Krankheitsmechanismus zu verstehen. Denn wenn man die Mechanismen versteht, kann man u. U. gezielt bestimmte Moleküle einsetzen, die diese fehlregulierten Signalwege „umprogrammieren“. Das hilft bei der Entwicklung ganz neuer Therapiekonzepte und kann Hinweise liefern, ob bereits zugelassene Medikamente, die vielleicht für ganz andere Zwecke entwickelt worden sind, hier zum Einsatz kommen können, weil deren Wirkmechanismus an der entsprechenden Stelle ansetzt.
Die Zukunft passiert nicht einfach, hier wird sie gestaltet!
•
Das Wissenschaftsland Sachsen besitzt eine enorme Vielfalt, Attraktivität und Exzellenz und belegt in vielen Bereichen Spitzenpositionen.
Ein ganz wichtiger und oft vergessener Punkt dabei: Mit den Erkenntnissen, die wir anhand der Aufklärung seltener Krankheitsbilder gewinnen, können vielfach auch häufiger auftretende Erkrankungen und ihre Wirkmechanismen besser verstanden und Therapie- oder Präventionskonzepte entwickelt werden.
Von der Forschung an seltenen Erkrankungen profitieren also am Ende sehr viel mehr Menschen als nur die Betroffenen selbst!

Forscher und Ärzte arbeiten beständig daran, Diagnosen schneller und genauer stellen zu können und Behandlungsmöglichkeiten weiterzuentwickeln.
Ein Gastbeitrag von Sachsens Wissenschaftsminister Sebastian Gemkow
Vernetzung und Kooperation von Expertinnen und Experten – das ist der Schlüssel!
Die Fortschritte in der Biotechnologie und anderen Forschungsgebieten in der Medizin in den vergangenen Jahren tragen inzwischen Früchte und helfen Patienten mit Seltenen Erkrankungen heute viel besser als noch vor wenigen Jahren. Sachsen trägt seinen Teil dazu bei: Mit gleich zwei universitären Zentren für Seltene Erkrankungen. Neben dem UniversitätsCentrum für Seltene Erkrankungen am Uniklinikum „Carl Gustav Carus“ Dresden (USE) arbeiten Forscher und Ärzte auch am Universitären Zentrum für Seltene Erkrankungen Leipzig (UZSE Leipzig) beständig daran, Diagnosen schneller und genauer stellen zu können und Behandlungsmöglichkeiten weiterzuentwickeln. Der Schlüssel zum Erfolg ist aber nicht der technologische Fortschritt allein. Vielmehr ist es die Vernetzung und Kooperation mit Expertinnen und Experten verschiedenster Disziplinen und Professionen unter Einbindung von Patientennetzwerken und wiederum spezialisierten Zentren für unterschiedliche Erkrankungen. Der Vorteil: Mit der Vernetzung werden Erkenntnisse zusammengeführt und in Verbindung mit modernster Ausstattung in der Medizin-Technik neue Forschungsansätze generiert. Neues Wissen vervollständigt das Bild, das sich Ärztinnen und Ärzte von Seltenen Erkrankungen machen. Solange, bis es eines Tages komplett ist und damit Verlauf, Symptome, biologische, chemische und physikalische Abläufe der jeweiligen Erkrankung verstanden sind. Dieses Wissen kommt den Patientinnen und Patienten in der Versorgung unmittelbar zugute. Es verbessert die Lebensqualität mit der Erkrankung oder kann sogar heilen. Der Freistaat Sachsen unterstützt deshalb kontinuierlich mit Investitionen in die Universitätsmedizin.





























•
•




















Lesen Sie mehr auf seltenekrankheiten.de 15
BEN BIERIG ANZEIGE
FOTO:
Robotik & Mensch-Maschine-Interaktion
Biotechnologie & Genetik
Pharmazie & Krebsforschung
Energie & Wassersto
Künstliche Intelligenz & Quantencomputing
•
•
•
•
Mikroelektronik
Materialforschung & Leichtbau
das Wissenschaftsland Sachsen entdecken!
Jetzt
Alle Infos auf: SPIN2030.com





ANZEIGE
