Lesen Sie mehr



Lesen Sie mehr
NICHT VERPASSEN:
Alagille-Syndrom
Wenn das eigene Kind eine seltene Lebererkrankung hat Seite 04
Morbus Fabry
Eine Erbschaft mit Folgen Seite 12
Das Bardet-Biedl-Syndrom
Im Fokus der Forschung steht, die enorme Last Betroffener zu mindern Seite 14
"Ich versuche, meine Erkrankung ganzheitlich zu sehen und sie in mein Leben zu integrieren –ohne mich dabei von ihr einschüchtern zu lassen."
Stefanie Peheim über ihr Leben mit Primärer Myelofibrose.
VERANTWORTLICH FÜR DEN INHALT IN DIESER AUSGABE FEBRUAR 2023
Miriam Hähnel
Das Thema Seltene Erkrankungen gehört in die Öffentlichkeit: auch über den Rare Disease Day hinaus. Denn hinter jeder und jedem Betroffenen steht ein persönliches Schicksal.

IN DIESER AUSGABE

Wissen bündeln, Situation für Betroffene verbessern Kai Pilgermann ist betroffen von GIST und engagiert sich als Patientenvertreter der Deutschen Sarkom-Stiftung.

Leben ohne Sicht heißt nicht: aussichtslos!
Linda Meschke hat Retinitis Pigmentosa, eine seltene Netzhauterkrankung, und engagiert sich in der Patientenselbsthilfe der PRO RETINA e. V.
Director Business Development Health: Miriam Hähnel Geschäftsführung: Richard Båge (CEO), Philipp Colaço (Managing Director), Alexandra Lassas (Content and Production Manager), Henriette Schröder (Sales Director), Lea Hartmann (Grafik), Cover: Stefanie Peheim von Melanie Peterseil
Mediaplanet-Kontakt: de.redaktion@mediaplanet.com Alle Artikel, die mit "in Zusammenarbeit mit" gekennzeichnet sind, sind keine neutrale Redaktion der Mediaplanet Verlag Deutschland GmbH.
Aus Gründen der besseren Lesbarkeit wird auf die gleichzeitige Verwendung der Sprachformen männlich, weiblich und divers (m/w/d) verzichtet. Sämtliche Personenbezeichnungen gelten gleichermaßen für alle Geschlechter.


Eva Luise Köhler Schirmherrin der Allianz Chronischer Seltener Erkrankungen e. V.
Weitere Informationen finden Sie unter: www.achse-online.de
Liebe Leserinnen und Leser, etwa 8000 Seltene Erkrankungen wurden bisher entdeckt, stetig kommen neue hinzu. 4 Millionen Menschen in Deutschland sind betroffen, darunter besonders viele Kinder. Zum Tag der Seltenen Erkrankungen, der am letzten Tag im Februar begangen wird und mittlerweile fest in vielen Kalendern verankert ist, ist die Aufmerksamkeit für dieses Thema besonders groß. Redaktionen erkundigen sich nach den Entwicklungen.
Gerne berichte ich über die Erfolge, auf die wir mittlerweile blicken können: Da sind die 35 Zentren für Seltene Erkrankungen bundesweit, die zudem europäisch vernetzt und qualitätsgeprüft ihre wertvolle Arbeit bei der Diagnosefindung und Behandlung leisten. Das Nationale Aktionsbündnis für Menschen mit Seltenen Erkrankungen NAMSE ist nach zehn Jahren immer noch die Vernetzungsplattform aller relevanten Akteure, stellt sicher, dass die Maßnahmen aus dem Nationalen Aktionsplan weiter vorangetrieben und neue eruiert werden. Zahlreiche Innovationsfondprojekte ermöglichen die Erprobung von Versorgungskonzepten, die dann in die Regelversorgung übergehen sollen, wie mit TRANSLATE-NAMSE schon geschehen. Und: Die Seltenen Erkrankungen sind ein Thema – in Politik, Öffentlichkeit, Gesundheitswesen etc. Dies ist auf nationaler Ebene vor allem den mittlerweile 130 Selbsthilfeorganisationen zu verdanken, die sich unter dem Dach der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V. zusammengeschlossen haben und die gemeinsam mit ihrem Netzwerk die wichtigen Anliegen aller betroffenen Menschen und deren Angehöriger vorantreiben.
richtigen Diagnose durch unser Gesundheitssystem irren, um zu erfahren, dass es weder Medikation, noch eine Therapie oder gar Heilung gibt. Angehörige, die mit der Pflege allein gelassen sind, die um Heil- und Hilfsmittel kämpfen, mit Kassen, die ihre Erkrankung nicht kennen und Anträge immer wieder ablehnen. Sie alle haben keine Zeit zu verlieren.
Schenken Sie den Seltenen Erkrankungen Ihre Aufmerksamkeit und den Menschen echtes Interesse. Drücken Sie Ihre Solidarität mit den Betroffenen aus und signalisieren Sie ihnen Ihre Unterstützung.
Text
Eva Luise Köhler
Wir danken folgenden Partnern für die Zusammenarbeit.
Albireo Pharma, Inc. www.albireopharma.com
Amicus Therapeutics GmbH www.amicusrx.de
BioCryst Pharma Deutschland GmbH www.biocryst.de
Diese Anliegen wollen wir am Tag der Seltenen Erkrankungen ganz besonders in den Mittelpunkt der Aufmerksamkeit rücken. Denn den meisten Betroffenen läuft die Zeit davon. Es sind die Eltern, die um ihr kleines Kind bangen, dessen Krankheit keiner kennt, weil sie nicht erforscht ist. Betroffene, die immer noch jahrelang vergeblich auf der Suche nach der
Deciphera Pharmaceuticals Germany GmbH www.deciphera.com
Dr. Falk Pharma GmbH www.drfalkpharma.de
GlaxoSmithKline GmbH & Co. KG de.gsk.com
Hormosan Pharma GmbH www.hormosan.com
Für die nahe Zukunft wünsche ich mir, dass wir die Kräfte in der Politik, im Gesundheitswesen, in Wissenschaft und Forschung noch viel stärker bündeln. Dass Strukturen geschaffen oder vorhandene so vernetzt, genutzt und gefördert werden, dass die vielen betroffenen Menschen mit Seltenen Erkrankungen eine adäquate Versorgung erhalten und eine Chance auf Heilung. Dafür setze ich mich ein, als Schirmherrin der ACHSE und mit der Eva Luise und Horst Köhler Stiftung. Was können Sie tun? Schenken Sie den Seltenen Erkrankungen Ihre Aufmerksamkeit und den Menschen echtes Interesse. Drücken Sie Ihre Solidarität mit den Betroffenen aus, signalisieren Sie ihnen Ihre Unterstützung, informieren und verbreiten Sie das Wissen, dass es Seltene Erkrankungen gibt – zum Tag der Seltenen Erkrankungen und darüber hinaus.
Mehr zum Thema erfahren Sie in dieser Sonderbeilage, ich wünsche Ihnen eine erhellende Lektüre.
Ihre Eva Luise Köhler (Schirmherrin ACHSE e. V.)
Janssen-Cilag GmbH www.janssen.com/germany
Novartis Pharma GmbH www.novartis.de
Rhythm Pharmaceuticals, Inc. www.rhythmtx.com
Vertex Pharmaceuticals (Germany) GmbH www.vrtx.de
Die Zahl derer, denen die seltene Speiseröhrenerkrankung eosinophile Ösophagitis (EoE) Schluckbeschwerden macht, steigt nachweislich und vor allem in Industrieländern. Als Ursache werden Allergene in der Nahrung und der Luft vermutet. Im Interview berichtet Prof. Dr. Ahmed Madisch, Facharzt für Gastroenterologie und EoE-Spezialist, wie sich die belastende Krankheit gut in Schach halten lässt.
Text Doreen Brumme
 Prof. Dr. Ahmed Madisch Centrum Gastroenterologie Bethanien, Agaplesion Krankenhaus Bethanien
Prof. Dr. Ahmed Madisch Centrum Gastroenterologie Bethanien, Agaplesion Krankenhaus Bethanien
Prof. Dr. Madisch, was passiert bei der EoE im Körper Betroffener?
Bei EoE-Betroffenen ist die Barrierewirkung der Schleimhaut der Speiseröhre gestört. Das macht die Schleimhaut durchlässig für Allergene, mit denen sie über Speisen, Getränke und die Luft in Kontakt kommt. Die Allergene dringen in die Schleimhaut ein und verursachen lokale Entzündungen, die mit der Zeit die Gewebestruktur verändern können, sodass die natürliche Schluckbewegung beeinträchtigt und auch schmerzhaft ist – insbesondere, wenn man Gröberes wie Fleisch oder Trockenes wie Brot isst. Schlimmstenfalls bleiben Speisebrocken in der Speiseröhre stecken und müssen in einer Notfallendoskopie entfernt werden.
Wie wird die EoE diagnostiziert und was erschwert die Diagnose mitunter?
Ein Verdacht auf EoE lässt sich beim Gastroenterologen mit Gewebeproben der Speiseröhre schnell und sicher bestätigen. Allerdings kommt dieser Verdacht nicht sofort auf. Denn Betroffene passen ihre Ernährungsweise oft lange an, indem sie auf bestimmtes Essen ganz verzichten, stets sehr gut kauen und mit viel Flüssigkeit „spülen“.
Und selbst wenn sie mit ihren Beschwerden zum Arzt gehen, beschreiben sie diese mitunter
ungenau, sodass selbst der Arzt, dem die seltene Erkrankung EoE ein Begriff ist, nicht sofort an diese denkt. Verwechslungen mit der Refluxkrankheit sind nicht selten.
Gibt es den „typischen EoE-Patienten“?
Ja. Am häufigsten bekommen Männer zwischen 30 und 40 Jahren die Diagnose EoE, Frauen sind eher seltener betroffen. Typisch sind begleitende Allergien und Erkrankungen wie Neurodermitis, Heuschnupfen und Asthma.
Einmal im Jahr sollte ein Gastroenterologe den Verlauf checken.
„Es war mir schon länger klar, dass irgendetwas nicht stimmt. Aber es hat lange gedauert, mir einzugestehen, dass meine Schluckbeschwerden zunehmen und ich zum Arzt muss“, erzählt Jasmin. Die inzwischen 39-jährige Krankenschwester litt vier Jahre an chronischen Schluckstörungen, bevor sie einen Arzt aufsuchte.
„Angefangen hat alles in einem chinesischen Restaurant“, berichtet Jasmin. „Ich habe ein Reisgericht gegessen und urplötzlich ist ein Bissen nicht mehr weitergerutscht und regelrecht im Hals stecken geblieben“. Es dauerte eine Weile, bis der schmerzhafte Vorfall vorüber war und der Reis die Speiseröhre passierte. Ein Abend, auf den sie sich gefreut hatte, wurde zum Alptraum.
Krampfartige Schmerzen und Angst zu ersticken
Die Schluckbeschwerden blieben und wurden häufiger. Die junge Frau versuchte alles, um das Problem zu kompensieren: Langsames Essen, sorgfältiges Kauen, bestimmte Lebensmittel wie Reis nur noch mit viel Soße und reichlichem Trinken zu den Mahlzeiten. Wenn sie gemeinsam mit Freunden oder Bekannten aß, wurde sie oft gefragt, warum sie so oft das Gesicht verziehe. „Das waren Momente, in denen die Schluckbeschwerden stark waren und ich krampfartige Schmerzen hatte“, erklärt Jasmin.
Doch ihre Strategien während des Essens halfen nicht. Immer häufiger blieb die Nahrung im Hals stecken und Jasmin bekam panische Angst, weil sie keine Luft mehr bekam. Vor allem Reis, Nudeln, Brot und andere Backwaren konnte Jasmin kaum mehr zu sich nehmen.
Eine Untersuchung schafft Klarheit
Jasmin begann im Internet nach Antworten zu suchen. Das schürte ihre Ängste und führte schließlich dazu, dass Jasmin einen Arzt aufsuchte. Eine Magenspiegelung wurde vorgenommen, in der die Speiseröhre bis auf einige weißliche Ablagerungen zunächst weitgehend unauffällig aussah. In der Untersuchung der entnommenen Gewebeproben zeigte sich jedoch eine ausgeprägte Entzündung und es wurde die Diagnose einer „eosinophilen Ösophagitis“, kurz EoE, gestellt. Der Arzt verordnete Jasmin ein Medikament, das sie zwölf Wochen lang einnehmen sollte. Die Schluckbeschwerden bildeten sich rasch zurück und schon bald konnte die Mutter von zwei kleinen Kindern wieder ganz normal essen.
Welche Therapien gibt es für Betroffene und wie bewerten Sie diese?
Wir behandeln die EoE mit einem lokal wirkenden Kortison in Tablettenform. Schmelztabletten mit Brauseeigenschaften werden morgens und abends in den Mund gelegt, wo sie sich auflösen. Bei über 90 Prozent der damit Behandelten normalisiert sich das Entzündungsgeschehen, sodass sie beschwerdefrei leben können.
Wer auf das Kortison nicht anspricht oder es nicht verträgt, kann die EoE auch mit einer Eliminationsdiät gut behandeln. Diese ist aber mit teilweise erheblichen Einschränkungen im täglichen Leben verbunden und nur mit sehr viel Disziplin durchzuhalten. In Kürze kommt zudem eine Antikörpertherapie auf den Markt, die als Reservetherapie angewendet werden kann. Die Antikörper werden einmal pro Woche per Spritze über die Bauchdecke verabreicht.
Worauf kommt es an, wenn man die EoE erfolgreich in Schach halten möchte?
Die EoE ist eine sich langsam einschleichende chronische Erkrankung. Deshalb bleibt nur beschwerdefrei, wer nach der ersten Akuttherapie dauerhaft gegenhält. Einmal im Jahr sollte ein Gastroenterologe den Verlauf checken.

Die Beschwerden kommen zurück
Nach dem Absetzen des Medikaments war jedoch schnell alles wieder beim Alten. „Mein Arzt wollte mir das Medikament allerdings nicht weiter verordnen, weil in der Kontrolluntersuchung zuvor die Magenspiegelung keinen krankhaften Befund mehr gezeigt hatte. Er motivierte mich vielmehr, eine Auslassdiät zu machen“, so Jasmin. Das aber war ihr durch ihren unregelmäßigen Tagesrhythmus und die Doppelbelastung als Mutter und Krankenschwester nicht möglich. Wenn die Schluckbeschwerden besonders stark waren, suchte sie das WC auf, um den steckengebliebenen Nahrungsbissen zu erbrechen.
Unterstützung durch andere Betroffene
Unterstützung fand sie in einer WhatsApp-Gruppe zur EoE und in anderen sozialen Medien. In den Gruppen wurden Online-Informationsabende mit EoE-Expert*innen organisiert. „Das war sehr hilfreich“, sagt Jasmin. In den Gruppen wurde sie von anderen Betroffenen ermuntert, sich mit den Schluckstörungen nicht abzufinden. Daher erkundigte sich Jasmin bei ihrem Arbeitgeber nach einem niedergelassenen Gastroenterologen, um eine Zweitmeinung einzuholen.
Der Gastroenterologe bestätigte die Diagnose EoE und verordnete ihr das Medikament, das ihr so gut geholfen hatte, zur langfristigen Erhaltungstherapie. Jasmin: „Wenige Tage später war der Spuk wieder vorbei. Ich nehme das Medikament seither regelmäßig und kann wieder ganz normal essen. Das ist für mich und auch für meine Familie ein wichtiges Plus an Lebensqualität!“, berichtet Jasmin abschließend. Anderen Betroffenen rät die junge Frau, keinesfalls aufzugeben, die Beschwerden ernst zu nehmen, da sie langfristig zu massiven Veränderungen an der Speiseröhre führen können, sich umfassend über das Krankheitsbild zu informieren und im Gespräch mit Ärztinnen und Ärzten selbstbewusst auf eine effektive Therapie zu bestehen.
Emily (19) leidet am Alagille-Syndrom Im Interview erzählt uns ihre Mutter, Deyna Dost (44), Emilys Geschichte, die selbst für eine seltene Erkrankung außergewöhnlich selten ist.
Text Doreen Brumme
studiolh
Deyna, Ihre Tochter leidet am seltenen AlagilleSyndrom. Wann fiel Ihnen das erste Mal auf, dass mit Emily etwas nicht stimmt?
Als ich im siebten Monat mit Emily schwanger war, bekam ich plötzlich vorzeitige Wehen. Und in mir machte sich das Gefühl breit, dass irgendetwas nicht in Ordnung ist. Als Emily in der 37. Schwangerschaftswoche per Kaiserschnitt geholt wurde, wirkte sie ganz normal. Weil sie zu schwach war zum Stillen und auch durch Flaschennahrung nicht genug zunahm, wurde sie durch eine Sonde ernährt. Die Ärztinnen und Ärzte entdeckten zudem ein unklares Herzgeräusch, beruhigten mich jedoch damit, dass das bei Neugeborenen nicht ungewöhnlich sei. Als Emily eine Gelbsucht entwickelte, hieß es wieder: alles normal. Die Neugeborenengelbsucht verging, kam allerdings nach zwei Wochen wieder. Das beunruhigte mich. Unser Kinderarzt schickte uns für einen Lebercheck zu Spezialisten, da auch der Stuhl des Babys auffallend hell war.
Wie kam es zur Diagnose?
Wir hatten Riesenglück: In der Uniklinik trafen wir auf eine Ärztin, die zum Alagille-Syndrom forschte. Nachdem sie Emilys Untersuchungsergebnisse gelesen und sich mein Baby angeschaut hatte, äußerte sie ihren Verdacht. Emily hatte viele der für die seltene Erkrankung typischen Symptome: eine hohe Stirn, weit auseinanderstehende Augen, einen Herzfehler. Der Gentest bestätigte den Verdacht. Wobei Emily ihr Alagille-Syndrom nicht geerbt, sondern spontan entwickelt hatte – eine Laune der Natur, die alles veränderte.
Was macht Emilys Geschichte außergewöhnlich?
Emilys Leberwerte waren nicht die besten, lagen aber im Normbereich. Man schickte uns mit dem Rat nach Hause, Emily nicht zu fett zu ernähren. Ansonsten gab es keine Einschränkungen. Sie wirkte gesund, war nur etwas blass. Mit sechs Monaten fing mein kleines Mädchen plötzlich an, sich am ganzen Körper zu kratzen. Zu sehen war nichts, doch der Juckreiz muss unerträglich gewesen sein. Sie kratzte sich rund um die Uhr. An Schlaf war nicht zu denken. Unsere Ärztinnen und Ärtze hier konnten nichts für Emily tun. Schließlich landeten wir in der Uniklinik in Hamburg und standen schnell vor der Entscheidung zwischen Leben und Tod: Emily brauchte eine neue Leber. Die Transplantation fand statt, als sie 22 Monate alt war. Sie hatte gerade laufen gelernt. Leider verlief die OP schlecht, sodass Emily im Februar 2005 innerhalb einer Woche eine neue Leber transplantiert werden musste. Doch auch diese führte zu vielen Komplikationen. Ich verbrachte Monate mit Emily im Krankenhaus in Hamburg, habe mein Leben hintenangestellt, auch meine Ehe zerbrach. Im August 2005 erhielt Emily zum dritten Mal eine neue Leber. Die OP verlief zum Glück wie im Bilderbuch. Mit dieser Leber lebt Emily bis heute. Sie hat damit keinerlei Einschränkungen zu befürchten, kann ganz normal leben, beruflich alles machen, Sport treiben, alt werden.

Wie wirkt sich die Erkrankung auf Emilys Alltag aus?
Emily musste nach dem langen Krankenhausaufenthalt neu laufen lernen. Wegen der Medikamente, die sie seit der Transplantation ununterbrochen nimmt, damit ihr Körper die fremde Leber nicht abstößt, ist ihr Immunsystem geschwächt. Sie war bis zu ihrem sechsten Geburtstag immer wieder im Krankenhaus, weil sie jeden Infekt mitnahm. Darunter litt ihr soziales Leben. Mit zwölf Jahren verlor Emily plötzlich alle Haare am Körper und diese wuchsen drei Jahre lang nicht nach. Niemand konnte uns erklären, warum, geschweige denn etwas dagegen tun. Eine Katastrophe für eine Pubertierende. Emily konnte sich nicht mehr im Spiegel anschauen, mied Menschen irgendwann ganz und stürzte in ein Loch, in dem sie noch immer steckt. Sie wurde schwer depressiv. Die Depression beeinträchtigt sie so sehr, dass sie bis heute nicht in der Lage ist, ihr Leben auf eigene Füße zu stellen. Emily würde gerne im medizinischen Bereich arbeiten, um anderen Menschen in ähnlicher Situation, wie sie sie erlebt, zu helfen. Doch für depressive Menschen wie Emily, die große Probleme mit festen Strukturen im Alltag haben, gibt es leider kaum Chancen in unserem auf Leistung getrimmten System.
Was möchten Sie anderen Eltern mit auf den Weg geben, deren Kind die Diagnose Alagille-Syndrom erhalten hat?
1
Scheuen Sie sich nicht, eine zweite medizinische Meinung einzuholen!
Wechseln Sie den Arzt, wenn Sie nicht ernst genommen werden oder dieser kein Spezialist für Ihren Fall ist
Gehen Sie in den Austausch mit anderen betroffenen Eltern und teilen Sie Ihr Schicksal. Gemeinsam erträgt es sich leichter.
Suchen Sie sich psychologische Unterstützung für Ihr krankes Kind und für sich selbst. Sie müssen das nicht allein durchstehen, es gibt professionelle Hilfe!
Fordern Sie die Vereinbarkeit von Pflege eines kranken Kindes und Beruf ein.
Die Leber ist das größte innere Organ des menschlichen Körpers. Sie besteht aus mehr als 300 Milliarden Zellen, die zusammen jede Menge Aufgaben lösen müssen.
Die Leber:
• Prüft alle Nahrungsbestandteile und wandelt sie in brauchbare Substanzen um
• Filtert Schadstoffe und Gifte
• Stellt Proteine her, mit deren Hilfe Kinder wachsen
• Produziert Vitamin K, das die Blutgerinnung ermöglicht
Speichert Zucker, Kupfer, Eisen und Vitamine und gibt diese Stoffe bei Bedarf ab
• Kontrolliert den Flüssigkeits- und Hormonspiegel
Im Gegensatz zu den anderen inneren Organen ist die Leber in der Lage, sich zu regenerieren. Sie übernimmt selbst mit einem kleinen Anteil an gesunden Zellen ihre Aufgaben über lange Zeit. Die Leber wächst sogar wieder nach, wenn ein Teil operativ entfernt wurde. Voraussetzung ist natürlich, dass die verbliebenen Leberzellen gesund sind. Bei so vielen Funktionen der Leber ist es nicht verwunderlich, dass es mehr als 100 verschiedene Lebererkrankungen bei Kindern gibt.
Die großen Gruppen der Lebererkrankungen sind:
• entzündliche Lebererkrankungen
• Stoffwechselerkrankungen
• angeborene Fehlanlagen der Gallenwege und andere Gallenwegserkrankungen
Unbehandelt ist den meisten dieser Erkrankungen langfristig die Entwicklung einer Leberzirrhose gemeinsam, eines narbigen Umbaus der Leber mit Verlust an funktionierenden Leberzellen.
So unterschiedlich die Ursache einer Lebererkrankung sein kann, so unterschiedlich sind auch die möglichen Behandlungen. In jedem Fall ist die Betreuung durch spezialisierte Ärzte oder Kliniken erforderlich, denn durch die Vielfältigkeit der Lebererkrankungen und ihre insgesamt geringe Häufigkeit können nur hier die nötigen Erfahrungen gesammelt und die geeignete Therapie gefunden werden.
Seit 1987 gibt es den Verein Leberkrankes Kind e. V. Gegründet wurde er von Eltern leberkranker Kinder, die das Bedürfnis hatten, sich mit anderen betroffenen Familien auszutauschen – vor der Zeit von Internet und Social Media. Wenn ein Kind schwer erkrankt, steht die gesamte Familie vor großen Herausforderungen im Alltag. Hier unterstützt der Verein durch Einzelfallhilfen, Informationen, Beratung und sein großes Netzwerk.
Einmal im Jahr veranstaltet der Verein einen Familientag an einem der Kinder-Leberzentren und gibt eine Mitgliederzeitschrift heraus. Zudem lädt er seine Mitglieder zu Regionalgruppen-Treffen ein und vernetzt so betroffene Familien in der Nähe ihres Wohnortes. Heute hat der Verein rund 300 Mitglieder. Der Mitgliedsbeitrag von 65 Euro pro Familie und Jahr kommt direkt den Kinderkliniken zugute, die auf Lebererkrankungen bei Kindern spezialisiert sind. Auch Fördermitgliedschaften für Privatpersonen und Unternehmen sind möglich (ab 40 Euro pro Jahr).
Spendenkonto
Commerzbank Rastatt
IBAN: DE43 660 400 180 250 108 800
BIC: COBADEFFXXX
Weitere Informationen unter: www.leberkrankes-kind.de
Quelle: Verein Leberkrankes Kind e. V.
Dieser Artikel ist in Zusammenarbeit mit Albireo Pharma entstanden.
Gallenstau: Mögliche Ursache kann eine seltene Erkrankung sein
Wenn Neugeborene durch eine gelbe Hautfärbung auffallen, steckt manchmal eine seltene Erkrankung wie das Alagille-Syndrom dahinter. Eine frühzeitige Diagnose ist wichtig, um den Leidensdruck zu verringern. Gentests können hierbei Licht ins Dunkel bringen.
Text Julia Brandt
“Eine gesunde Hautfarbe haben“ – dieser geflügelte Begriff hat tatsächlich einen wahren Hintergrund. Denn oft gibt eine ungewöhnliche Färbung der Haut Hinweise auf eine mögliche Erkrankung.
So ist es zum Beispiel bei Neugeborenen, die durch eine gelblich gefärbte Haut auffallen. Eine solche Neugeborenengelbsucht ist in der Regel harmlos. Dauert sie länger als zwei Wochen an, werden Ärzte jedoch hellhörig. Möglicherweise leiden die betroffenen Babys unter einem angeborenen Gallenstau. Hierbei fließt die Gallenflüssigkeit nicht richtig, sondern staut sich in der Leber oder in den Gallengängen. Dies kann die Leberzellen schädigen – und viele weitere Organe und Gewebe beeinträchtigen.
Häufige Ursachen einer solchen Gallenstauung sind Infektionen oder Stoffwechselerkrankungen. Sie kann bei Neugeborenen aber auch als Folge des sogenannten Alagille-Syndroms (kurz: ALGS) auftreten.
Die Ursache dieser Krankheit liegt in den Genen: Bei den Betroffenen ist eine bestimmte Erbinformation so verändert, dass die Gallengänge in der Leber nicht richtig gebildet werden.


ALGS ist gekennzeichnet durch unterschiedlichste Symptome
Das Alagille-Syndrom ist eine angeborene Erkrankung, die sehr selten auftritt. Schätzungen zufolge kommt in Deutschland nur etwa eines von 50.000 Neugeborenen damit zur Welt. Alagille-Patienten, bei denen die Leber betroffen ist, zeigen Symptome wie Gelbsucht, Wachstumsverzögerungen sowie Cholesterinablagerungen in der Haut. Viele leiden zudem unter starkem Juckreiz –was die Lebensqualität stark beeinträchtigt. Die betroffenen Kinder und Säuglinge sind durch den ständigen Juckreiz leicht reizbar, unruhig und schlafen kaum. Eine Belastung für Eltern und Kinder.
Auch körperliche Merkmale deuten auf das AlagilleSyndrom hin: Viele Patienten fallen durch Kleinwuchs sowie charakteristische Gesichtszüge auf: eine große Stirn sowie weit auseinander- und tiefliegende Augen. Das typische Aussehen, ist für die Ärzte ein Hinweis, dass eine Lebererkrankung möglicherweise durch das Alagille-Syndrom verursacht wird. In den meisten Fällen verschafft ein Gentest Klarheit, ob diese Erbkrankheit vorliegt. Da es sich bei dem ALGS um eine seltene Erkrankung handelt, vergehen in manchen Fällen Monate oder Jahre, bis die korrekte Diagnose gestellt wird.
Behandlungsoptionen des Alagille Syndroms
Die Symptome des Alagille-Syndroms sind individuell verschieden ausgeprägt. Bei einigen Patienten schränken sie den Alltag kaum ein, andere verspüren hingegen einen starken Leidensdruck. So unterschiedlich wie der Verlauf der Erkrankung ist auch ihre Behandlung. Viele Patienten bekommen Medikamente, die Symptome wie Juckreiz lindern. Vitaminpräparate oder eine spezielle Ernährung können außerdem dazu beitragen, ALGSFolgen abzumildern. Zusätzlich gibt es spezielle Medikamente, die den Gallefluss verbessern – das schützt die Leber vor schädigenden Stoffen.
Ist die Leber stark angegriffen oder lassen sich Symptome wie Juckreiz nicht durch die Behandlung lindern, kommt möglicherweise eine Lebertransplantation in Betracht. Hierbei bekommen die kleinen Patienten entweder die Leber eines Fremden oder einen Teil der Leber eines Elternteils. Im Anschluss an die Operation müssen sie lebenslang Medikamente einnehmen, die ihr Immunsystem unterdrücken, sonst würde ihr Körper das fremde Organ abstoßen. Die Chancen und Risiken eines solchen Eingriffs werden daher immer im Einzelfall sorgfältig abgewogen.
FREIGABENUMMER: DE-BV-23-00002 |
Kai Pilgermann, Patientenvertreter der Deutschen Sarkom-Stiftung, ist selbst von einem Gastrointestinalen Stromatumor (GIST) betroffen. Er war sehr jung, als er die Diagnose bekam. Im Interview berichtet Kai Pilgermann über seinen eigenen Weg mit der Erkrankung und die Stiftung, die Patienten, Ärzte, Forscher, Angehörige und Vertreter des Gesundheitswesens zusammenführt, um die Situation für Betroffene zu verbessern.
Text Miriam RauhHerr Pilgermann, wann bekamen Sie die Diagnose „Gastrointestinaler Stromatumor“?
Ich war damals erst 27 Jahre alt. Das ist ungewöhnlich, Betroffene haben meist ein deutlich höheres Lebensalter. Es war ein Zufallsbefund, der Tumor wurde im Rahmen einer Blinddarm-OP entdeckt. Die Operateure haben allerdings nicht mehr geschafft, eine Gewebeprobe zu entnehmen oder ihn gleich zu entfernen, weswegen ich für eine weitere Operation in die Klinik musste. Im Nachgang wurde bei der pathologischen Untersuchung des entnommenen Gewebes festgestellt, dass es sich tatsächlich um GIST handelt. Warum dauert es bei GIST in vielen Fällen so lange bis zur Diagnose?
Die meisten Gastrointestinalen Stromatumoren wachsen außerhalb eines Organs oder am Dünndarm und machen im Bauchraum nach außen hin in der Regel zunächst wenig Beschwerden. Wenn Beschwerden auftreten, sind die Tumore häufig schon relativ groß. Hat man den Tumor aber entdeckt, geht die Diagnose an sich recht schnell.
Wenn Sie zurückblicken – gab es bei Ihnen Anzeichen für die Erkrankung? Was berichten andere Betroffene?
Im Normalfall gehen Betroffene mit Beschwerden des Verdauungstrakts zum Arzt, aber diese sind diffus und weisen nicht sofort auf GIST hin. Beschwerden beim Stuhlgang beispielsweise, Völlegefühl, eine gewisse Müdigkeit – das alles kann auch andere Ursachen haben. Sehr eindeutige Symptome gibt es nicht.
Wie ging es nach der Diagnose für Sie weiter? Wie wurde GIST bei Ihnen therapiert?
Meine Onkologin kannte sich gut mit dem Thema aus und wusste bereits von dem neuen Medikament, das damals erst seit zwei Jahren eingeführt war. Sie hat mich auch direkt damit behandelt. Das war großes Glück. Insgesamt rate ich Betroffenen, sich wenn mög-

lich in einem auf Sarkome spezialisierten Zentrum behandeln zu lassen. Als meine Diagnose gestellt wurde, gab es solche Zentren noch nicht – heute gibt es sie an mehreren Orten in Deutschland.
Die Erkrankung hat Sie zur Deutschen SarkomStiftung gebracht. Was sind Ihre Aufgaben als Patientenvertreter?
Es gab eine Vorläuferorganisation, das Lebenshaus, eine reine Patientenorganisation, in der ich mich bereits engagierte. Wir haben mit dem Lebenshaus schon einiges erreicht, wurden auch von Experten unterstützt, diese waren aber nie Teil der Organisation. Das wollten wir ändern und haben beschlossen, gemeinsam mit Experten die Deutsche Sarkom-Stiftung aufzubauen, um sie fest zu integrieren. Die Deutsche Sarkom-Stiftung ist ein Zusammenschluss aus Ärzten, Zentren und Patienten, um die Diagnose-Situation und die Behandlungsqualität für GIST und Sarkome in Deutschland zu verbessern. Für Betroffene bieten wir auch Webinare an, derzeit online, um neueste Erkenntnisse zu GIST zu präsentieren und einen Rahmen zum Austausch mit Ärzten zu schaffen.
Was empfehlen Sie anderen Betroffenen im Umgang mit der Erkrankung?
Grundsätzlich ist es sehr wichtig, sich um die Erkrankung zu kümmern. Es ist gut, sich zu informieren und Hintergrundwissen anzueignen. Manchmal kann es helfen, nicht alleine zu Terminen zu gehen, sondern einen Angehörigen, einen guten Freund oder eine Freundin mitzunehmen. Und es schadet nicht, im Zweifelsfall eine Zweitmeinung einzuholen. Wenn man nicht in der Nähe eines spezialisierten Sarkom-Zentrums wohnt, kann man vielleicht den Schwerpunkt der Behandlung bei einem niedergelassenen Onkologen oder einer Onkologin durchführen lassen, sich für besondere Fragestellungen aber an ein Sarkom-Zentrum wenden. Bei der Deutschen Sarkom-Stiftung erhalten Betroffene viele wertvolle Tipps, finden neueste Studienergebnisse und viele Informationen. Sie können dort die Zentren und auch niedergelassene Onkologen finden, die sich gut mit der Erkrankung auskennen.
Grundsätzlich ist es sehr wichtig, sich um die Erkrankung zu kümmern. Es ist gut, sich zu informieren und Hintergrundwissen anzueignen.
Die Deutsche Sarkom-Stiftung ist eine gemeinsame Organisation von Patienten und Experten. Die Stiftung setzt sich dafür ein, die Situation für Sarkom-Patienten in Deutschland zu verbessern. Dafür engagiert sie sich in verschiedenen Bereichen: Information, Forschung, Fortbildung, Versorgungsstrukturen inkl. Etablierung von spezialisierten Sarkom-Zentren, Diagnose- und Behandlungsqualität wie auch Patienteninformation und Interessenvertretung.
Weitere Informationen unter: www.sarkome.de

Gastrointestinale Stromatumoren (GIST) sind sehr seltene Weichteilsarkome, die im Magen-Darm-Trakt entstehen. In Deutschland erkranken pro Jahr ein bis zwei von 100.000 Menschen, die meisten sind bei Diagnosestellung 60 Jahre alt oder älter.
PD Dr. med. Reichardt leitet das Sarkomzentrum Berlin-Buch und erklärt, was die Herausforderungen bei der Diagnose sind und wie Betroffene heute behandelt werden können.
Text Miriam Rauh
PD Dr. med. Peter Reichardt
Chefarzt der Klinik für Onkologie und Palliativmedizin am Helios Klinikum Berlin-Buch und Leiter des Sarkomzentrums Berlin-Buch
Herr Dr. Reichardt, was sind die Herausforderungen bei der Diagnose von GIST und im Verlauf der Erkrankung?
Die Beschwerden sind in der Regel eher unspezifisch. Aus diesem Grund wird ein Gastrointestinaler Stromatumor oft zufällig entdeckt, bspw. im Rahmen einer Magenspiegelung, Ultraschalluntersuchung oder Computertomographie. Wichtig ist, dass neben der pathologischen Diagnose auch eine Mutationsanalyse gemacht wird, da die genaue Kenntnis der zugrundeliegenden Mutationen für die Therapieplanung entscheidend ist; zudem hat sie Einfluss auf die Prognose. Die Feindiagnostik sollte in einem erfahrenen Referenzzentrum durchgeführt werden, um Inkorrektheiten auszuschließen.
Wie ist die Prognose?
Man muss hier zwischen lokalisierter Erkrankung und fortgeschrittener Erkrankung unterscheiden. Die Prognose des
fortgeschrittenen, metastasierten GIST hat sich in den letzten Jahren durch zunehmende therapeutische Optionen kontinuierlich verbessert; seit ca. einem Jahr steht mit Ripretinib eine Viertlinientherapie zur Verfügung. Mittlerweile können wir bei einer metastasierten Erkrankung eine mittlere Lebenserwartung von sechs oder sieben Jahren erwarten.
Bei einer lokalisierten Erkrankung, die operativ behandelt wurde, können wir recht genau vorhersagen, wie groß das Risiko eines Patienten für Metastasen bzw. ein Rezidiv ist. Hiervon abhängig ist die Indikation einer vorbeugenden, adjuvanten Therapie. Als Richtwert gilt ein Rezidivrisiko in der Größenordnung über 50 Prozent, sofern der Tumor eine Imatinibsensitive Mutation aufweist.
Die Prognose des fortgeschrittenen, metastasierten GIST hat sich in den letzten Jahren durch zunehmende therapeutische Optionen kontinuierlich verbessert.
Welche Therapieoptionen gibt es derzeit, um GIST zu behandeln, und wie ist deren Stellenwert?
Imatinib stellt nach wie vor den Standard in der Erstlinientherapie und in der adju-
vanten Therapie dar. Bei einer ImatinibIntoleranz oder einem Krankheitsprogress unter Imatinib ist die Zweitlinientherapie Sunitinib vorgesehen. Wenn auch diese nicht mehr wirkt, kommen Regorafenib und schließlich Ripretinib in der Drittund Viertlinie zum Einsatz. Für die sehr seltene D842V-Mutation steht mit Avapritinib seit einiger Zeit erstmals eine wirksame Therapie zur Verfügung.
Bei der Therapie spielen für Betroffene in den verschiedenen Phasen der Erkrankung neben Wirksamkeit auch Verträglichkeit und Lebensqualität eine Rolle. Wie sieht es bei den Behandlungsoptionen gerade in späteren Stadien aus?
Die für die Therapie des fortgeschrittenen GIST etablierten Medikamente sind unterschiedlich gut verträglich, was angesichts der häufig langfristigen Einnahme von besonderer Bedeutung ist. Imatinib, Standard in der Erstlinientherapie, ist in der Regel gut verträglich. Sunitinib ist etwas schlechter verträglich als Imatinib, was sich in Durchfällen, Abgeschlagenheit, Müdigkeit oder Hautreizung an Händen und Füßen bemerkbar machen kann, auch Blutdruck und Schilddrüsenfunktion sollten überwacht werden. Regorafenib ist vom Nebenwirkungsspektrum dem Sunitinib ähnlich, mit einer häufig ausgeprägteren Tendenz zu Nebenwirkungen; eine individuelle Einstellung ist bei diesen Medikamenten besonders wichtig.
Das Medikament der Viertlinientherapie, Ripretinib, ist wiederum in aller Regel besser verträglich. Dies erhöht auch die Lebensqualität der Patienten.

Unter Myeloproliferativen Neoplasien (MPN) versteht man eine Gruppe von seltenen Erkrankungen des Knochenmarks, pro Jahr erkranken in Deutschland ein bis zwei Menschen pro 100.000 Einwohner. Charakteristisch für diese Krankheitsbilder ist eine gesteigerte Produktion von Blutzellen, was sich in einer Vielzahl von Symptomen äußern kann, die das Leben Betroffener zum Teil stark beeinträchtigen.
Zu den MPN zählt auch die Primäre Myelofibrose (PMF), von der Stefanie Peheim betroffen ist. Sie erzählt uns von ihrem Weg zur Diagnose und ihrem Leben mit dieser seltenen chronischen Erkrankung.
Text Miriam RauhFrau Peheim, Sie sind betroffen von der Primären Myelofibrose. Können Sie uns erzählen, wann erstmals Beschwerden aufgetreten sind und wie diese aussahen?
Etwa ein bis zwei Jahre vor der Diagnose bemerkte ich erste Beschwerden wie Taubheit in den Fingern, auch Müdigkeit am Tag, besonders gegen Mittag. Ich habe sehr viel Schlaf gebraucht, den brauche ich nach wie vor. MPN sind von Mensch zu Mensch in der Ausprägung sehr verschieden. Wann wurde die richtige Diagnose gestellt?
Das war im Jahr 2020, ich war 26 Jahre alt. Ich hatte Blut gespendet, im Anschluss erhielt ich eine Auswertung meiner Blutwerte. Weil mein Thrombozytenwert erhöht war, wurde mir geraten, ihn noch mal beim Hausarzt kontrollieren zu lassen. Mein Hausarzt empfahl mich dann weiter an eine Spezialistin, dort erhielt ich einen Monat später die Diagnose.
Gab es direkt eine passende/individuelle Behandlungsoption für Sie?
Mir wurde gut erklärt, was es mit der Erkrankung auf sich hat, wie sie sich auf mein Leben auswirkt und welche Möglichkeiten es gibt. Da ich keine großen Beschwerden hatte und auch die Werte nicht dramatisch waren, habe ich in Rücksprache mit meiner Ärztin anfangs keine Medikamente genommen, sondern ging nur regelmäßig zur Kontrolle. Erst mal abzuwarten, war für mich der richtige Weg. Im Herbst 2022 haben sich die Werte etwas verschlechtert und ich habe begonnen, Medikamente zu nehmen. Die Einstellungsphase dauert mindestens drei bis vier Monate, bei manchen durchaus auch länger. In dieser Phase muss man sowohl die Blutwerte als auch mögliche Nebenwirkungen monitoren. Erst nach dieser Phase kann man beurteilen, wie der Körper die Medikamente annimmt und welche Therapie die individuell passende ist. In dieser Phase befinde ich mich.

Zudem wurde mir direkt zu Beginn meiner Therapie von einer Studie berichtet, an der ich seitdem teilnehme. Vor allem bei seltenen Krankheiten wie der PMF ist das sehr wichtig, damit an den Medikamenten geforscht werden kann und Betroffene direkt in das Forschungsgeschehen mit einbezogen werden können.
Ich muss gut auf meinen Körper aufpassen und dafür sorgen, dass er bekommt, was er braucht.
Was sind für Sie persönlich die größten Belastungen und Herausforderungen, die mit der Erkrankung einhergehen?
Ich habe eine Weile gebraucht, bis ich realisierte, dass ich eine Erkrankung habe, anfangs habe ich es verdrängt. Erst im letzten Jahr wurde mir richtig bewusst, dass ich auf meinen Körper aufpassen und gut dafür sorgen muss, dass er bekommt, was er braucht.
Die Müdigkeit ist sehr präsent – auch wenig Energie kenne ich sonst gar nicht von mir, ich habe immer viel unternommen. Jetzt muss ich konsequent auf meinen Körper hören und sehen, wo meine Grenzen sind. Grenzen zu stecken und genau hinzusehen, was mir guttut, das ist derzeit die größte Herausforderung für mich. Auch habe ich häufig mit schweren Beinen zu kämpfen: Bei meiner Tätigkeit als Konditorin merke ich das oft schon nach zwei bis drei Stunden, da ich ja viel im Stehen arbeite. Durch die medikamentöse Behandlung hat sich das aber bereits gebessert.
Wie wirkt sich die Erkrankung auf Ihr Berufsleben aus?
Die Diagnose hat mich in meiner Berufswahl einmal mehr bestätigt. Ich wollte etwas tun, das mir zu 100 Prozent Freude macht, und bin seit knapp zwei Jahren als Konditorin selbstständig. Meine Arbeitsstätte ist in der Nähe, ich bekomme auch sehr viel Unterstützung durch meine Familie. Anders würde es nicht funktionieren.
Wie gehen Sie mit der Last Ihrer Erkrankung um, und was hilft Ihnen im Umgang mit der PMF? Abgesehen davon, dass es mir hilft, meine Zeit und Energie gut einzuteilen, schätze ich den Austausch mit anderen Betroffenen sehr. Bis zur Diagnose war ich nie wirklich krank, ich musste auch nie Medikamente nehmen. Durch den Austausch bekomme ich einen besseren Einblick in den Alltag mit der Erkrankung. Wie geht es anderen Betroffenen damit, was machen sie? Man unterstützt sich gegenseitig sehr.
Eine solche Erkrankung betrifft auch indirekt die Angehörigen. Wie geht Ihr Umfeld mit Ihrer Erkrankung um?
Insgesamt sehr gut, ich bekomme viel Unterstützung. Bei mir ist die Krankheit aber auch derzeit kein großes Thema, ich habe wenig Beschwerden.
Welche Rolle spielt für Sie die Vernetzung in der Selbsthilfegruppe?
Diese Möglichkeit empfinde ich als sehr wertvoll. Die Krankheit ist noch relativ wenig erforscht, es gibt kein Patentrezept für den Umgang, vieles muss individuell betrachtet und angepasst werden. Manchmal haben andere Betroffene ergänzend zu Ärzten wertvolle Tipps, einfach aus der Alltagserfahrung heraus – z. B. wann die beste Tageszeit für die Einnahme der Medikamente ist. Meine Ärztin hat mich auf eine Selbsthilfegruppe in Österreich aufmerksam gemacht, dort bin ich Mitglied. Durch eigene Recherche habe ich auch Gruppen auf Facebook gefunden, in denen ich aktiv bin. Was haben Sie aus Ihrer Erfahrung mit der Krankheit gelernt, was würden Sie an andere Betroffene weitergeben?
Es ist wichtig, sich nicht einschüchtern zu lassen. Man sollte nicht ängstlich an das Thema herangehen, sondern sich an die Situation anpassen und sie ins Leben integrieren. Ich versuche, die Krankheit ganzheitlich zu sehen. Nicht nur Medikamente können helfen, eine Erkrankung hat auch eine psychologische Komponente. Diesen ganzheitlichen Ansatz würde ich sehr empfehlen.

FOTO: ELISABETH PEHEIM
Das MPN-Netzwerk e. V. ist eine Selbsthilfeinitiative für Menschen mit Myeloproliferativen Neoplasien (MPN) und ihre Angehörigen. Wir stellen fundierte, allgemein verständliche Informationen zu MPN-Erkrankungen zur Verfügung und bieten Patient:innen und deren Angehörigen die Möglichkeit, sich miteinander auszutauschen und zu vernetzen. Zudem arbeiten wir eng mit einschlägigen Expert:innen für die MPN-Erkrankungen zusammen, um die Forschung weiter voranzutreiben.
Weitere Informationen finden Sie unter www.mpn-netzwerk.de
„Patienten tragen heute entscheidend zu unserem Gesundheitswesen bei.“
Die Myeloproliferativen Neoplasien (MPN) sind eine Gruppe von seltenen Erkrankungen des Knochenmarkes, zu denen auch die Polycythaemia Vera (PV) gehört. Wir sprachen mit Werner Zinkand über die Last der Erkrankung und die wichtige Rolle der Patientenselbsthilfe.
Text Hanna Sinnecker Werner Zinkand Vorsitzender der internationalen MPN-Advocates
Werner Zinkand Vorsitzender der internationalen MPN-Advocates
Herr Zinkand, Sie sind betroffen von der seltenen Erkrankung Polycythaemia Vera. Wie hat sich die Erkrankung bemerkbar gemacht und wann haben Sie Ihre Diagnose erhalten?
Im Jahr 2000 war ich zum Gesundheitscheck bei meiner Hausärztin, da war ich 47 Jahre alt. Meine Ärztin hatte zu hohe Thrombozyten festgestellt und mir ASS (einen Blutverdünner) verschrieben, das habe ich einige Jahre genommen. Aber es kamen mit der Zeit Sehstörungen dazu: ich habe verschwommen oder Doppelbilder gesehen, nach einer Minute war das meist wieder vorbei. Die Ärztin konnte das nicht einordnen. Schrecklich war auch ein extremer, stechender Juckreiz, besonders nach Wasserkontakt nach dem Duschen. Juckreiz ist ein deutliches Symptom der PV, der die Patienten verrückt machen kann. Aber mein Dermatologe kam nicht auf PV. Die richtige Diagnose kam durch Zufall: 2011, zehn Jahre nach den ersten Beschwerden, bin ich auf die Schulter gestürzt, es wurde ein MRT gemacht. Dem Radiologe fiel mein Knochenmark auf, es war marmoriert. Im Knochenmark bilden sich die Blutzellen. Meine Hausärztin hat mich dann zum Hämatologen überwiesen, der die Diagnose Polycythaemia Vera gestellt hat.
Weitere Informationen finden Sie unter www.mpnadvocates.net
Was sind für Sie als Betroffener die größten Herausforderungen und wie wirkt sich die Erkrankung auf Ihr Leben aus?
Wegen des Juckreizes kann man nachts nicht schlafen
Patienten müssen gehört werden.
und ist tagsüber kaputt. Die Diagnose selbst ist ein Schock: man hat über Nacht, für den Rest des Lebens, eine unbekannte chronische Krankheit. Wenn man in der Hämatologie und Onkologie behandelt wird, bekommt man Angst, wird mit schlimmen Schicksalen konfrontiert und fragt sich: wie geht es jetzt weiter? Habe ich jetzt Krebs? Mit der Antwort tun sich die Ärzte schwer, denn ja, es handelt sich um eine chronische Blutkrebserkrankung. Chronisch heißt aber, dass sie in den meisten Fällen langsam voranschreitet. Die Zellen des Blutes vermehren sich unkontrolliert, unbehandelt haben wir ein hohes Thrombose- oder Embolierisiko. Mittlerweile gibt es Medikamente, mit denen die Beschwerden gelindert werden können. Ein klassisches Medikament, eine leichte Chemotherapie, ist seit Jahrzehnten auf dem Markt. 2012 kam ein sogenannter Inhibitor dazu, der später eingesetzt wird, wenn man die Erstlinientherapie nicht verträgt oder sie nicht mehr reicht. Aktuell werden mehr Medikamente zugelassen, alle wirken verschieden. Ein erfahrener Hämatologe kann unsere Beschwerden meist gut kontrollieren – das ist eine wichtige Information für Betroffene.
Das forschende Pharmaunternehmen Novartis denkt Medizin neu, um besonders auch Menschen mit seltenen Erkrankungen mit innovativen Therapien zu mehr Lebensqualität zu verhelfen und ihnen mit umfangreichen Unterstützungs- und Informationsangeboten zur Seite zu stehen.
Speziell für Menschen, die an einer Myeloproliferativen Neoplasie (MPN) wie der Myelofibrose, der Polycythaemia Vera oder der Chronischen Lymphatischen Leukämie leiden, hat Novartis für Patient:innen und deren Angehörige umfangreiche Informationsinitiativen ins Leben gerufen, die Betroffenen und deren Angehörigen wissenschaftlich fundiertes Wissen zur Erkrankung und zum Umgang damit zur Verfügung stellen.
Symptome erkennen – und richtig in Zusammenhang bringen Da die verschiedenen Symptome der MPN sehr vielschichtig sind und mit Fortschreiten der Erkrankung stärker werden, sind fundierte Informationen zu den möglichen Beschwerden für Patient:innen und deren Angehörige sehr wichtig. Das macht das Beispiel der Polycythaemia Vera deutlich: denn Beschwerden wie chronische Müdigkeit, Schmerzen im linken Oberbauch, verstärktes nächtliches Schwitzen, Juckreiz besonders nach Kontakt mit Wasser und Appetitlosigkeit lassen oft nicht direkt an eine schwere Erkrankung denken. Gerade Frauen denken oftmals eher an die Wechseljahre und nicht an eine seltene Bluterkrankung. Auch Seh- und Konzentrationsstörungen, Ohrensausen, trockene Haut werden eher auf das Alter zurückgeführt und nicht in Kombination betrachtet. Die Folge: der Arztbesuch bleibt aus, die PV bleibt unentdeckt und somit auch unbehandelt, schwere Komplikationen können auftreten.
Zunehmende Beschwerden ernst nehmen
Aber auch wenn die Diagnose bereits gestellt wurde, sollten Betroffene die Symptome im Blick behalten. Gerade wenn die Symptomlast zunimmt oder Nebenwirkungen auftreten, sollten Betroffene das Gespräch mit dem Behandlungsteam suchen. Manche Begleiterkrankungen oder Komplikationen können für Betroffene im schlimmsten Fall lebensbedrohlich werden, weshalb ein schnelles Gegensteuern entscheidend ist. Ist der Betroffene gut informiert, kann er bei der Wahl und Durchführung der passenden Therapie intensiv mit einbezogen werden. Die Patient:innen sollten immer ein offenes Ohr finden, wenn Handlungsbedarf besteht. Das gilt auch für die Angehörigen der Betroffenen, denn sie können eine große Stütze sein: Auch wenn es darum geht, körperliche und seelische Beschwerden oder eine Verschlechterung des Zustandes frühzeitig zu erkennen. Sie spielen also eine tragende Rolle, wenn es darum geht, Betroffene zu unterstützen und ihre Lebensqualität zu verbessern.
Sie sind sehr engagiert in der nationalen und internationalen MPN-Patientenselbsthilfe. Welche Rolle spielt diese aus Ihrer Sicht, wenn es um die Verbesserung der Lebensqualität Betroffener geht?
Wissen ist die beste Medizin. Die Selbsthilfe hilft Betroffenen, sich mit ihrer Erkrankung vertraut zu machen. Das kann ein Stück weit den Schrecken nehmen. Man fühlt sich zu Beginn sehr allein, besonders mit einer seltenen Erkrankung wie der PV. Ich hatte bald einen anderen Betroffenen kennengelernt, der eine kleine Selbsthilfegruppe gegründet hat, wir waren anfangs zu dritt. Der Erfahrungsaustausch war sehr wichtig für mich, deshalb engagierte ich mich neun Jahre lang im deutschen MPN-Netzwerk. Seit zwei Jahren bin ich Vorstand der internationalen MPN-Advocates, das ermöglicht mir eine größere Perspektive. Gemeinsam kann man viele positive Entwicklungen vorantreiben! Erfahrene Patient:innen sind heute gefragt, mehr denn je. Für Betroffene, die sich engagieren wollen, gibt es Schulungen. Wir arbeiten in nationalen und internationalen Gremien mit, unsere Erfahrungen helfen auch Pharmafirmen bei der Entwicklung neuer Medikamente. Heute tragen Patienten entscheidend zu unserem Gesundheitswesen bei.
Was ist bezüglich der Versorgung Betroffener wichtig, damit diese ihren Alltag bestmöglich meistern können?
Man muss zurückfinden ins Leben und lernen, die Krankheit als Teil des Lebens anzunehmen. Aber sie sollte in den Hintergrund treten. Neben der Medizin spielt auch die psychologische Betreuung eine große Rolle. Sie kann helfen, die Krankheit zu akzeptieren, ohne dass man die Hoheit über das eigene Leben verliert. Außerdem müssen wir Patienten gehört werden. Ärzte achten oft auf andere Aspekte als wir. Eine Umfrage ergab, dass Ärzte zuerst auf das Blutbild schauen, Patienten ist aber die Lebensqualität wichtiger. Und die korreliert nicht unbedingt mit guten Blutwerten.

Die einzelnen Initiativen www.leben-mit-myelofibrose.de, www.leben-mit-pv.de und www.leben-mit-cml.de möchten Betroffene deshalb über alle Facetten der Erkrankung informieren. Hier finden sich auch Patienten-Erfahrungsberichte und Expertenbeiträge zu verschiedenen krankheitsrelevanten Schwerpunkten. Zudem finden Patient:innen ausführliche Checklisten, die ihnen die Gespräche mit dem Behandlungsteam erleichtern können: denn die Patient:innen selbst spielen eine wesentliche Rolle bei der Wahl und Durchführung der geeigneten Therapie. Dazu kann auch eine Anpassung der bestehenden Therapie gehören, wenn die bestehende Behandlung nicht den gewünschten Erfolg erzielt. Dabei kann auch der MPN-Tracker unter www.mpntracker.com helfen, der Patient:innen in Form eines Therapietagebuches bei der Dokumentation zur Entwicklung ihrer Erkrankung unterstützt.
Zusammen stärker
Auch der Austausch mit anderen Betroffenen, Selbsthilfeorganisationen und Fachärzt:innen stärkt Patient:innen und ihre Angehörigen im Umgang mit der Erkrankung. Seit 2016 können MPN-Betroffene einen bundesweit etablierten Treffpunkt nutzen: die MPN-Patient:innentage. Diese finden mehrmals im Jahr an immer anderen Standorten statt, damit möglichst viele Betroffene teilnehmen können. Seit 2020 ist für einige der Termine auch eine Online-Teilnahme möglich. Die Teilnahme an den MPN Veranstaltungen ist kostenlos. Auf www.mpn-patiententage.de findet man die Anmeldung für den nächsten Patient:innentag sowie weitere Informationen und einen kleinen Rückblick auf vergangene Veranstaltungen.
Scannen Sie den QR-Code und lesen Sie mehr zu uns auf unserer Webseite unter https://www.leben-mit-pv.de/sp1


Schwellungsattacken
Das Hereditäre Angioödem (kurz HAE) ist eine seltene vererbbare Erkrankung, die sich durch wiederkehrende Schwellungen bemerkbar macht. Diese Schwellungen verursachen starke Schmerzen und können lebensbedrohlich werden, zum Beispiel wenn sie im Halsbereich auftreten und Betroffenen buchstäblich die Luft nehmen. Je früher die Erkrankung diagnostiziert wird, umso schneller kann eine Behandlung in die Wege geleitet werden. Denn mit der richtigen Therapie können Betroffene ein nahezu normales Leben führen. Was passiert bei HAE im Körper? HAE-Betroffene weisen eine Mutation auf dem Chromosom 11 auf, die einen Defekt des sogenannten SERPING1-Gens verursacht. Dieses Gen ist dafür zuständig, das Protein C1-INH zu produzieren, das bei HAE-Patienten nicht in ausreichender Menge oder gar nicht produziert wird. Das führt zu einer Störung des Enzyms Plasma-Kallikrein, was wiederum zu einer zu großen Menge des Gewebshormons Bradykinin führt. Bradykinin reguliert u. a. den Blutdruck und erhöht die Durchlässigkeit der Blutgefäße. Die Folge von zu viel Bradykinin: Die Blutgefäße werden durchlässiger für das Blutplasma. Es tritt aus den Gefäßen aus, lagert sich im Gewebe ein und führt zu den attackenartigen Schwellungen.
Wo können Schwellungen auftreten?
Die Schwellungen können nahezu überall auftreten. Besonders häufig sind Schwellungen der Haut, vor allem im Gesicht (Augen, Lippen), an den Händen, Armen, Füßen und Beinen. Auch an den Schleimhäuten im Magen-Darm-Trakt können
diese Schwellungen auftreten, wo sie Bauchschmerzen, kolikartige Krämpfe, Erbrechen und Durchfall auslösen können. Auch Kehlkopf, Genitalien, Harnblase, Muskulatur, Gelenke, Gehirn und Nieren können betroffen sein. Die Schwellungen treten meistens attackenartig oder in Schüben auf. Meist entwickeln sie sich über einen Zeitraum von 12 bis 36 Stunden und klingen unbehandelt innerhalb von 2 bis 5 Tagen wieder ab.
Schwellungsattacken kontrollieren –Lebensqualität steigern
Ist die Erkrankung diagnostiziert, lassen sich die Schwellungsattacken durch die verfügbaren Behandlungsoptionen gut kontrollieren. An spezialisierten Zentren für seltene Erkrankungen oder HAEZentren können der behandelnde Arzt und der Patient die Behandlungsmöglichkeiten besprechen, die immer darauf abzielen, die Erkrankung zu kontrollieren und im Idealfall Schwellungsattacken ganz zu vermeiden – immer mit dem Ziel, Betroffenen ein Leben zu ermöglichen, das so normal wie möglich verläuft.
Mittlerweile stehen mehrere Medikamente zur Verfügung, die prophylaktisch eingesetzt werden, um Schwellungsattacken gar nicht erst entstehen zu lassen.
Diese Medikamente unterscheiden sich lediglich in der Art der Anwendung und den Abständen der Verabreichung.
Da es trotz Prophylaxe dennoch sein kann, dass plötzlich eine Attacke auftritt, sollten Menschen mit HAE zusätzlich immer eine ausreichende Menge Akutmedikamente für mindestens zwei Attacken dabeihaben.
Wann haben Sie Ihre Diagnose erhalten?
Meine erste Attacke hatte ich mit 14 Jahren. Da meine Mutter ebenfalls betroffen ist, war schnell klar, dass ich auch HAE habe. Dieses „Glück“ hat ja aber nicht jeder. Ich weiß, dass viele Betroffene von Arzt zu Arzt laufen und es teilweise Jahre dauert, bis sie eine Diagnose erhalten.
Welche Herausforderungen gibt es für Menschen mit HAE?
Die Attacken machen das Leben weniger planbar und können theoretisch auch lebensbedrohlich werden. Persönlich habe ich mich aber nie wirklich eingeschränkt gefühlt. Durch meine familiäre Vorbelastung bin ich früh von Experten betreut worden, die sich gut mit HAE auskannten. Ich hatte immer meine Akutmedikation dabei und konnte ein relativ normales Leben führen. Aber als ich in eine andere Stadt gezogen bin, habe ich auch anderes erlebt. Da musste ich den Ärzten erklären, was HAE ist und auch, dass manche Therapievorschläge nicht helfen, beispielsweise Kortison.
Wenn die Diagnose einmal steht, ist die Herausforderung eher eine organisatorische.
Wenn die Diagnose einmal steht, ist die Herausforderung eher eine organisatorische. Ich nehme inzwischen regelmäßig ein Medikament zur Prophylaxe, habe aber vorsichtshalber auch immer meine Akutmedikation dabei. Aber davon abgesehen mache ich alles, was Nichtbetroffene auch können: Ich habe studiert, ich arbeite, mache Sport, gehe feiern, fahre in den Urlaub …
Warum sind Initiativen für Betroffene und ihre Angehörigen wichtig?
Patienteninitiativen mit Informationen rund um die Erkrankung und Tipps für ein Leben mit HAE machen Mut. Das ist vor allem für Menschen wichtig, die vielleicht noch gar nicht wissen, was sie haben, oder für solche, die gerade frisch diagnostiziert sind und sich fragen, wie es jetzt weitergehen soll. Ich konnte mich ja immer mit meiner Mutter austauschen, aber was machen andere, die sich ratlos und allein fühlen? Wäre ich damals bei meiner Diagnose in einer anderen Situation gewesen, hätte ich nach genau so etwas gesucht.

Franziska von Werder (27) hat mit 14 Jahren die Diagnose HAE erhalten. Sie lebt in Wiesbaden.
Menschen mit der seltenen chronischen Erkrankung Hereditäres Angioödem (HAE) leiden unter plötzlich auftretenden Schwellungsattacken, die den gesamten Körper betreffen können. Insbesondere im Kopf-Halsbereich kann es zu schweren, lebensbedrohlichen Attacken kommen. Doch Informationen zu dieser seltenen Erkrankung sind häufig schwer zu finden. Nun bietet die Initiative „HAEllo zum Leben“ umfangreiche Informationen zur Erkrankung, zu ihrem Management sowie Services und Hilfestellung.
Das Hereditäre Angioödem (engl. hereditary angioedema, kurz: HAE) ist eine chronische genetische Erkrankung, die schon in frühen Jahren auftreten kann. So wie beispielsweise bei Franziska, 27, aus Wiesbaden.
Inzwischen hat sie ihr HAE gut im Griff – ihre Geschichte macht Mut und ist unter www.haellozumleben.de zu sehen.
Denn mit der Diagnose HAE stellen sich plötzlich viele Fragen: Welche Auswirkungen hat HAE auf mein Leben? Wie lässt sich HAE kontrollieren? Was kann ich selbst tun, um mein Leben mit HAE zu verbessern? Welche Therapiemöglichkeiten habe ich? Die Initiative „HAEllo zum Leben“ von BioCryst Pharma bietet mit einer Website sowie den Social-Media-Kanälen Facebook und Instagram Informationen zur Erkrankung und ihrem Management, wie etwa den Behandlungsempfehlungen der aktuellen Leitlinie, Aktionswochen oder digitalen Experten-Sprechstunden sowie PatientenInsights und Tipps zum Umgang mit HAE.

Drei Fragen an Waldemar Heiduk, VP & General Manager DACH bei BioCryst Pharma Deutschland
Die Diagnose HAE ist oft schwierig. Warum? Die Symptome sind unspezifisch und ähneln stark anderen Erkrankungen. Oft werden sie als Lebensmittelunverträglichkeit, Allergie oder Blinddarmentzündung fehlgedeutet. Da HAE so selten ist, kann es schwierig sein, eine Ärztin oder einen Arzt zu finden, der oder die Symptome richtig deutet.


Warum ist eine Initiative wie „HAEllo zum Leben“ wichtig?
Solche Initiativen mit Tipps für ein Leben mit HAE machen Mut. Das ist vor allem für Menschen wichtig, die vielleicht noch gar nicht wissen, was sie haben, oder für solche, die gerade frisch diagnostiziert sind und sich fragen, wie es jetzt weitergehen soll.
Was raten Sie Betroffenen?
Wichtig ist, sich bei unklarer Diagnose rechtzeitig an ein Zentrum für seltene Erkrankungen oder ein HAE-Zentrum überweisen zu lassen. Eine Liste mit HAE-Behandlungszentren gibt es zum Beispiel bei der deutschen HAE-Patientenvereinigung unter: www.hae-online.de/behandlungszentren.
„Es ist ein Kraftakt, wieder am normalen Leben teilzunehmen.“
Rheumatische Erkrankungen - Dabei denken viele zunächst an eine Volkskrankheit, die eine Vielzahl an Menschen betrifft. Dabei gibt es auch eine beträchtliche Anzahl an seltenen rheumatischen Erkrankungen, zu denen auch die sogenannten Vaskulitiden gehören, die durch eine Entzündung der Blutgefäße charakterisiert sind. Vanessa Rennspieß ist betroffen von der Eosinophilen Granulomatose mit Polyangiitis (kurz EGPA) und sprach mit uns über Ihr Leben mit dieser seltenen Erkrankung.
Text Alexandra LassasFrau Rennspieß: Welche Erkrankungen haben Sie und wie hat sich diese geäußert?
Ich leide an der Gefäßentzündung EGPA, die sich vor 26 Jahren durch eine progressive systemische Sklerose, eine Verhärtung des Gefäß- und Bindegewebes der Haut geäußert hat. Die Beschwerden kamen schleichend in mein Leben. Ich hatte schon länger etwas Heuschnupfen, leichte Allergien und vermehrt taube, blaue Finger. Durch einen Aufenthalt in einer Rehaklinik erhielt ich die Diagnose Raynaud-Syndrom und hatte zu diesem Zeitpunkt schon offene Stellen an meinen Händen, vermehrt Schmerzen bei den einfachsten Bewegungen und beim Luftholen. Auch hormonelle Veränderungen durch die Geburt meiner Tochter verschlimmerten meine Symptome.
Wie lange hat es gedauert, bis nach den ersten Beschwerden die Diagnose gestellt wurde und was waren in dieser Zeit die größten Herausforderungen für Sie?
Die Symptome wurden von Jahr zu Jahr stärker und ich musste mein Leben komplett einschränken. 2014 war auch meine Lunge betroffen, Kalkablagerungen in meinem Körper führten zu einer Knie-OP und alles wurde als Folge der systemischen Sklerose gesehen. 2017 ver-
schlimmerten sich meine Luftprobleme, die in vielen Hustenanfällen endeten. Im März darauf begann ich eine Chemotherapie, danach musste ich in die Uniklinik und diese habe ich dann mehrere Monate nicht verlassen. Eine schwere Panzytopenie und eine Entzündung der Gallenblase folgten. Mit den verschiedenen Symptomen startete ein jahrlanger Arztmarathon. Das erfordert gute Koordination und gutes Zeitmanagement.
Dazu kommen die permanente Ungewissheit und die anhaltenden Symptome. Spazieren gehen, mein geliebtes Nordic Walking und überhaut Bewegung und Luft holen wurden zur Tortur. Zudem wurde die gemeinsame Zeit mit der Familie knapp. Das hat auch für mein Umfeld alles verändert. Mein Mann und meine Tochter haben stark darunter gelitten und sich um mich gesorgt.
Gibt es etwas, was Sie sich an Verbesserungen für Betroffene wünschen würden?
Vor 25 Jahren war die Forschung noch in den Kinderschuhen und man konnte nicht darauf schließen, dass ich unter EGPA leide. Mittlerweile gibt es mehrere Medikamente und Therapien, um die Symptome zu bekämpfen. Die Koordination der Ärzte und das Zeitmanagement raubt viel Kraft und Nerven, da brauch es einfach eine bessere Struktur in unserem Gesundheitssystem.
Dieser Artikel ist in Zusammenarbeit mit der GlaxoSmithKline GmbH & Co. KG entstanden.
Wie geht es Ihnen jetzt unter Therapie?
Ich bekomme einmal im Monat eine Spritze, ein Biologikum für EGPA. Das hält meine Lunge in Remission. Weiterhin gehe ich regelmäßig zur Kontrolle ins Uniklinikum. Aber ich bin nach wie vor krankgeschrieben. Das Leben ist nicht mehr das Gleiche. Auch jetzt noch brauche ich viel Kraft, um wieder normal am Leben teilzunehmen. Gemeinsame Unternehmungen mit meiner Familie und dem Arbeitsalltag mit Tatendrang gegenüberstehen: das ist mein Ziel.
Ist ihr Umfeld eine Stütze? Oder haben Sie Hilfe? Aktuell habe ich die Krankheit einigermaßen im Griff. Alle Dinge, die ich allein machen kann, versuche ich zu organisieren und damit mein Umfeld nicht zu belasten. Auch meine Therapie hat mir geholfen, mit der Schwere meiner Krankheit umzugehen und nach vorne zu schauen. Weiterhin helfen mir mein Job und meine Tiere, die Gedanken auf etwas anderes zu lenken. Durch die Uniklinik und die Ärzte vor Ort fühle ich mich gut betreut und biete gerne meine Hilfe an. Ich stelle mich den forschenden Studenten zur Verfügung, die mein Blut untersuchen und Informationen zu der Krankheit sammeln, um so einen Beitrag zur Erforschung der Krankheit zu leisten.
Weitere Unterstützung finden Betroffene und deren Angehörige bei dem bundesweit tätigen
Verein Vaskulitis e. V.
Hauptstraße 6, 54526 Landscheid/Eifel
Tel.: 06575-9014995
Fax: 06575-903794
Mail: info@vaskulitisverein-rlp.de
Mehr Informationen finden Sie auf unserer Webseite www.vaskulitisverein-rlp.de
Gerade in der kalten Jahreszeit leiden Asthma-Patienten häufig unter einer Verschlechterung ihrer Erkrankung oder treten Asthma-Symptome wie Atemnot mit oder ohne Reizhusten sowie ein Engegefühl in der Brust das erste Mal auf. Manchmal steckt hinter dem Asthma die Autoimmunerkrankung EGPA, die im Laufe der Zeit noch weitere Organe des Körpers angreift und bei besonders schweren Fällen tödlich verlaufen kann. Pro Jahr treten in Deutschland nur etwa 1.000 bis 1.500 neue Fälle von EGPA auf, was sie zu einer seltenen Erkrankung macht.
Frau Dr. Lampert, wie oft haben Sie in Ihrer Praxis bereits Patient:innen erlebt, hinter deren Asthma sich eine EGPA verbarg, und wie sind Sie ihr auf die Schliche gekommen?

Dr. Sabine
Lampert Fachärztin für Innere Medizin und Pneumologie (Lungenfachärztin) und Leiterin der Lungenpraxis „Lunge im Zentrum“
Tatsächlich habe ich das schon mehrfach erlebt. Asthma bronchiale ist eine Erkrankung mit ganz unterschiedlichen Ursachen. Bei einigen Patient:innen entwickelt es sich langsam aus einem Heuschnupfen, bei anderen beginnt es plötzlich z. B. nach einem Infekt. Bei allen wollen wir mit der Therapie das Asthma unter Kontrolle bringen, d. h. der/die Erkrankte nimmt seine Medikamente und spürt sonst nichts vom Asthma. Wenn das nicht gelingt, muss man überlegen, warum nicht und dabei auch an seltene Erkrankungen denken. Aber es gibt auch andere Szenarien, die einen als Arzt/ Ärztin aufhorchen und an eine EGPA denken lassen sollten. So erzählte mir ein Patient, der zwar seitens seines Asthmas beschwerdefrei war, von Herzproblemen und einer Nervenentzündung im Bein! Bei einer anderen Asthmatikerin fielen mir bestimmte Blutwerte im bei uns standardmäßig durchgeführten großen Blutbild auf. Es zeigte erhöhte Eosinophile, eine bestimmte Art der weißen Blutkörperchen, die zu bestimmen grundsätzlich wichtig für die Asthmatherapie ist und deren starke Erhöhung auf eine EGPA hinweisen kann.
Wieso wird die EGPA häufig erst so spät diagnostiziert? Was ist die besondere Schwierigkeit?
Ich glaube, das grundsätzliche Problem ist, dass die EGPA so unterschiedliche Beschwerden machen kann, die völlig unzusammenhängend erscheinen. Die Schwierigkeit für mich persönlich ist, unter den vielen Asthmatiker:innen, die ich jeden Tag sehe, den/diejenige mit EGPA herauszufinden. Man muss in dieser täglichen Routine hellhörig sein und genau hinsehen. Nicht nur das Asthma sehen und behandeln, sondern den Menschen mit dem Asthma. Das ist zwar eine Plattitüde, aber nichtsdestotrotz wahr.
Welche Behandlungsmöglichkeiten gibt es heute für die Betroffenen?
Das Asthma wird bei Vorliegen einer EGPA genauso behandelt, wie andere Asthmaformen auch. Bei einem unkontrollierbaren, schweren Verlauf stehen uns moderne Biologika zur Verfügung. Für die EGPA an sich ist häufig der Einsatz von Kortison notwendig, eventuell von weiteren Immunsuppressiva, und auch moderne zielgerichtete Therapien können eingesetzt werden. Dafür sind Rheumatologen die Experten und führen die Therapie. Die gute Zusammenarbeit mit ihnen ist enorm wichtig und ich bin sehr froh, dass dies mit meinen rheumatologischen Kollegen der Fall ist.


Frau Roth, wie zeigte sich Ihr Morbus Fabry und wann erhielten Sie Ihre Diagnose?
Ich hatte keinerlei beunruhigende Symptome. Gegen meinen Bluthochdruck nahm ich seit Jahren Medikamente. Bei der regelmäßigen Kontrolle beim Nephrologen fiel immer mal wieder zu viel Eiweiß im Urin auf, ich schluckte dann Antibiotika. Mit Mitte 40 spürte ich, dass ich körperlich nicht mehr ganz so fit war wie früher, ich wurde schneller müde, konnte mich mitunter nur schwer konzentrieren. Das schob ich aber auf die Wechseljahre. Im Jahr 2017 bekam meine Schwester plötzlich Herzprobleme, deren Ursache lange keiner erklären konnte – bis ein Gentest ihr schließlich einen Morbus Fabry bescheinigte.
Meine Schwester fand schnell heraus, dass dieser über das X-Chromosom des Vaters an alle Töchter vererbt wird, und sprach mich daraufhin an. Ein Gentest brachte mir meine Diagnose 2018. Bei den anschließenden Untersuchungen zeigten sich bei mir typische Symptome: Meine Herzwand war verdickt und der Herzmuskel vergrößert. Ein aktuelles Kopf-MRT (Magnet-ResonanzTomographie) ergab zudem leichte Ablagerungen im Gehirn, und neben dem Herz ist auch meine Niere inzwischen leicht betroffen.
Der Austausch mit anderen Betroffenen in der Morbus Fabry Selbsthilfegruppe ist eine große Hilfe für mich.
Was fühlten Sie in dem Moment der Diagnose?
Nach dem ersten Schock sagte ich mir: „Judith. Du hast 55 Jahre ohne große gesundheitliche Probleme gelebt. Das ist jetzt so. Da musst du künftig eben drauf achten.“ Ich war Arzthelferin bei einem Augenarzt – der „professionelle“ Hintergrund half mir, die Diagnose zu schlucken. Viel schwerer dagegen fiel es mir, meinen Kindern davon zu berichten und ihnen zu sagen, dass ich ihnen den Morbus Fabry vererbt haben könnte: Das Risiko lag bei 50:50.
Bestätigte sich Ihre Befürchtung?
Leider ja. Sowohl mein Sohn als auch meine Tochter, heute beide über 30, haben einen Morbus Fabry. Unsere „familiäre Mutation“ der Erkrankung ist zwar nicht ganz so gravierend, aber bei meinem Sohn zeigten sich bereits erste Anzeichen an den Nieren. Es ist krankheitstypisch, dass Männer meist früher und stärker davon betroffen sind. Bei meiner Tochter waren die Testbefunde glücklicherweise bislang negativ.
Der Morbus Fabry ist eine erblich bedingte Stoffwechselstörung. Betroffenen fehlt ein Enzym zum Aufspalten bestimmter Fette. Die lagern sich infolgedessen in verschiedenen Organen ab und schädigen sie zunehmend. Judith Roth bekam die Diagnose Morbus Fabry mit 55. Hier berichtet sie über ihren Alltag mit der lysosomalen Speicherkrankheit – einer Erbschaft mit Folgen.
Noch ist ein Morbus Fabry zwar unheilbar – doch er ist gut behandelbar. Lassen Sie sich therapieren, und wie geht es Ihnen unter der Therapie?
Ich bin, wie auch mein Sohn, seit drei Jahren in Behandlung, zuerst bei Spezialisten in Mainz, inzwischen in Heidelberg. Ich bekomme eine Enzymersatztherapie, das heißt, dass mir alle 14 Tage ein synthetisches Enzym in die Blutbahn gegeben wird, anfangs in der Klinik, mittlerweile zu Hause. Meine Tochter startet demnächst mit ihrer Therapie.
Die Infusionen vertrage ich gut. Manchmal bin ich danach etwas erschöpft, aber das hat sicher auch noch andere alltägliche Ursachen. Vergangenes Jahr hatte ich plötzlich Herzrhythmusstörungen, was für Menschen wie mich – mit „Baustelle am Herzen“ – nicht untypisch ist. Mit einer Kardioversion konnte der zu schnelle Herzrhythmus wieder normalisiert werden (Sinusrhythmus). Wirklich beeinträchtigt fühle ich mich von meinem Morbus Fabry nicht – noch ist er kein Störfaktor. Zum Glück ist er bislang auch schmerzlos.
Ein Morbus Fabry ist chronisch, er bleibt Ihr Leben lang. Wie läuft der Alltag damit?
Ich habe mich arrangiert. Es dauerte zwar, bis ich mir vor zwei Jahren eingestand, dass mir mein Job in der Augenarztpraxis zu stressig geworden war. Doch heute arbeite ich im Gemeindebüro einer evangelischen Kirche in Wiesbaden – und der Wechsel tat mir gut.
Die regelmäßige Heimtherapie ist ein Termin im Kalender wie jeder andere auch. Mit der Erschöpfung, die mich begleitet, habe ich umzugehen gelernt. Spüre ich sie, gebe ich meinem Körper, was er braucht: Ruhe. Ich lege mich hin und sage auch mal die eine oder andere geplante Unternehmung ab, gerade in für alle sowieso stressigen Zeiten wie vor Weihnachten: Da sinkt meine Belastbarkeit spürbar und ich bin auch psychisch schon mal etwas angeschlagen. Hilfreich ist für mich dann oft der Austausch mit anderen Betroffenen in der MorbusFabry-Selbsthilfegruppe.
In Deutschland sind derzeit etwa 1.200 Morbus FabryPatienten diagnostiziert, wobei eine hohe Dunkelziffer vermutet wird. Es ist eine Erbkrankheit, die zu Beginn sehr unspezifische Auswirkungen hat: Schmerzen in den Gelenken, Flecken auf der Haut oder extreme Müdigkeit. Auch Brennschmerzen in den Händen und Füßen, die bereits Betroffene im Kindesalter bemerken, können ein Hinweis auf die Erkrankung sein. So wird die Krankheit häufig erst festgestellt, wenn sie schon große Schäden angerichtet hat: starke Nierenschädigung, Schlaganfall in jungen Jahren oder extreme Vergrößerung des Herzmuskels. Unbehandelt kann sich die Lebenszeit Betroffener um bis zu 25 Jahre verkürzen. Seit 20 Jahren gibt es für Patienten mit Morbus Fabry wirkungsvolle Therapien, die die Erkrankung stoppen oder verlangsamen. Je früher sie erkannt wird, umso geringer sind die bleibenden Schäden. Doch gibt es nur wenige gute Behandlungszentren für diese seltene Erkrankung.
Es ist wichtig, dass wir als Gruppe von betroffenen Patienten sichtbarer werden, uns gegenseitig mit Informationen über Kliniken und neue Therapieansätze versorgen – auch im persönlichen Austausch. Mit mittlerweile 160 Mitgliedern versucht die Morbus Fabry Selbsthilfegruppe (MFSH) unter anderem, in der Politik und in der Forschung auf dieses Krankheitsbild aufmerksam zu machen. Zusammen sind wir stark: Je mehr Menschen uns als Mitglieder unterstützen, umso mehr Gehör bekommen wir!
Weitere Informationen unter: www.fabry-shg.org

Morbus Fabry ist eine seltene monogenetische Stoffwechselstörung , die zu den lysosomalen Speichererkrankungen gehört. Was viele Betroffene eint, ist der oftmals lange Leidensweg bis zur Diagnose. Wir sprachen mit Prof. Dr. Christine Kurschat , Leiterin der Morbus Fabry Spezialambulanz am UK Köln, über die seltene Erkrankung.
Text Miriam Rauh

Prof. Dr. Christine Kurschat Internistin, Nephrologin, Transplantationsmedizinerin, Hypertensiologin
DHL und Leiterin der Spezialambulanz Morbus Fabry am UK Köln
Morbus Fabry wird auch als das „Chamäleon unter den seltenen Erkrankungen“ bezeichnet. Warum?
Morbus Fabry kann sehr viele verschiedene Symptome in unterschiedlichen Bereichen hervorrufen, zum Beispiel an der Niere, in den Blutgefäßen oder am Herzen, auch frühe Schlaganfälle können ein Zeichen sein. Fabry-Patienten weisen aber nie alle Symptome auf und auch innerhalb der gleichen Familie sind die Symptome der einzelnen Betroffenen oft ganz unterschiedlich.
Was passiert bei der Erkrankung im Körper?
Bei Morbus Fabry kann ein bestimmter Stoff nicht abgebaut werden, weil das Enzym Alpha-Galaktosidase fehlt, bzw. nicht richtig funktioniert. Das führt dazu, dass die Fettstoffe, Glykosphingolipide, die das Enzym normalerweise spaltet, sich im Gewebe und in zahlreichen Organen anreichern, insbesondere das Globotriaosylceramid, GL-3 oder Gb3.
Diese Anreicherung lässt sich bereits in der Plazenta nachweisen. Die klinischen Auswir-

kungen zeigen sich erst später, allerdings kann Morbus Fabry schon im Kindes- und Jugendalter zu Beschwerden führen, wie brennenden Schmerzen an Händen und Füßen, die in Wellen auftreten und durch bestimmte Umstände wie körperliche Anstrengung oder fiebrige Infekte ausgelöst werden. Oft wird dies als Wachstumsschmerz abgetan, aber man sollte bei solchen Symptomen immer auch daran denken, dass eine seltene Erkrankung dahinterstecken kann.
Wie lange dauert es durchschnittlich bis zur Diagnose?
Bis zur Diagnose können durchaus zehn bis fünfzehn Jahre vergehen, manchmal mehr. Das liegt daran, dass die Symptome, die sich anfangs zeigen, meist sehr unspezifisch sind.
Es gibt ca. 8000 verschiedene seltene Erkrankungen; man denkt zunächst an die häufigen, bis man sich unter den seltenen auf Ursachenforschung macht. Wenn etwas nicht ins Bild passt, beispielsweise eine seltsame Hautveränderung, merkwürdige Einlagerungen in der Hornhaut oder ein dickeres Herz, ohne dass ein Bluthochdruck vorliegt, könnte dies auf Morbus Fabry hinweisen.
Morbus Fabry ist eine genetische Erkrankung, die über mehrere Generationen einer Familie vererbt werden kann. Das heißt: Wenn eine Person in einer Familie die Diagnose Morbus Fabry hat, können andere Familienangehörige ebenfalls betroffen sein. Eine ausführliche Analyse des Familienstammbaums ist daher sehr wichtig für Betroffene und deren Angehörige.
Ich bin betroffen – Was nun?
Ist die Diagnose Morbus Fabry gestellt, dann ist es für Betroffene wichtig zu wissen, was die eigene Diagnose für Familienangehörige bedeuten kann und wer aufgrund des Vererbungsmusters ein erhöhtes Risiko für Morbus Fabry hat. Hier kommt die neue Website www.fabryfamilytree.de ins Spiel, die Betroffenen umfassende Informationen und Hilfestellungen an die Hand geben möchte.
Dazu gehören grundlegende Informationen, wie die Erkrankung vererbt wird und wer in der Familie ein erhöhtes Risiko hat. Über ein Online Stammbaum-Tool kann man zusammen mit seinem behandelnden Arzt seinen individuellen FabryStammbaum erstellen und für die persönliche Nutzung herunterladen, um Angehörige mit erhöhtem Fabry-Risiko gezielt informieren zu können. Die Daten werden streng vertraulich behandelt. Die Website gibt professionelle Hilfestellung, wie man Angehörige mit erhöhtem Risiko dann darauf ansprechen und sie aufklären kann. Dazu gehört auch eine Briefvorlage, die man nutzen kann, wenn eine direkte Ansprache sich schwierig gestalten sollte.
PSYCHOSOZIALE ASPEKTE
• Depression
• Angstzustände
• Panikattacken
• Isolation AUGEN
• Wirbelförmiges Muster auf der Hornhaut
• Fabry-Katarakt (eine bestimmte Form der Linsentrübung)
NIEREN
• Eiweiß im Urin
• Verminderte Nierenfunktion
• Nierenversagen
HAUT
• Vermindertes Schwitzen
NERVENSYSTEM
• Starke Schmerzen, die Minuten bis Stunden andauern
• Hörverlust, Tinnitus
• Hitze- oder Kälteunverträglichkeit oder Belastungsintoleranz
• Transitorisch-ischämische Attacke (TIA) und Schlaganfall
• Brennen der Hände und Füße, auch als Akroparästhesie bezeichnet
• Schwindel
HERZ
• Unregelmäßiger Herzschlag (schnell oder langsam)
• Herzanfall oder Herzversagen
• Vergrößertes Herz
MAGEN-DARM
• Übelkeit und Erbrechen
• Durchfall und/oder Verstopfung
• Bauchschmerzen
• Blähungen
• Kleine dunkelrote Punkte, die als Angiokeratome bezeichnet werden, vor allem zwischen Bauchnabel und Knien
Welche Rolle spielt die Familienanamnese?
Da es sich um eine erbliche Erkrankung handelt, ist sie sehr wichtig. Im Fabry-Zentrum machen wir bei der Erstvorstellung immer eine ausführliche Familienanamnese und zeichnen auch den Stammbaum auf. Wenn ein Betroffener Morbus Fabry hat, ist die Wahrscheinlichkeit hoch, dass es innerhalb der gleichen Familie weitere Betroffene gibt, die eine Therapie benötigen.
Vor zwanzig Jahren wurde die erste kausale Therapie für Fabry-Betroffene zugelassen. Was hat sich seitdem getan?
Es ist gut, dass wir die Therapie haben, die Daten sind sehr überzeugend. Wir können Krankheitsverläufe verlangsamen und Lebenszeit verlängern. Allerdings kann man Morbus Fabry bislang nicht komplett zum Stillstand bringen. Für den Therapieerfolg spielt eine Rolle, ob die Erkrankung früh entdeckt wurde oder ob schon Organe geschädigt sind. Es müssen nicht alle Betroffenen therapiert werden.

Informationen für Familienangehörige mit erhöhtem Fabry Risiko
Auf der Website gibt es aber auch für Angehörige von Morbus Fabry-Patienten detaillierte Informationen, die dabei helfen sollen, die Erkrankung zu verstehen und warum sie selbst ein erhöhtes Risiko haben. Dabei ist eines sehr wichtig: ein erhöhtes Risiko bedeutet nicht zwangsläufig, dass man tatsächlich auch betroffen ist.
Daher sollten Angehörige, die laut Stammbaum ein erhöhtes Risiko haben, unbedingt einen Arzt ansprechen und weitere Untersuchungen durchführen lassen. Das kann der eigene Hausarzt oder aber der Fabry-Spezialist des betroffenen Angehörigen sein. Der Arzt entscheidet dann, ob ggf. auch eine genetische Testung sinnvoll ist.
Informationen für das Fachpersonal
Aber auch medizinisches Fachpersonal findet auf der Website Materialien und Hilfestellungen, wenn es darum geht, Fabry-Patienten oder deren Angehörige zu beraten und aufzuklären. Dazu gehört ebenfalls die Nutzung des Online StammbaumTools in Zusammenarbeit mit dem Patienten, sowie weitere Broschüren, die beim Familienscreening unterstützen sollen.
Informieren Sie sich unter www.fabryfamilytree.de
Neue Behandlungsansätze für das Bardet-Biedl-Syndrom: Im Fokus steht, die große Last der Erkrankung für Betroffene zu mindern
Das Bardet-Biedl-Syndrom ist eine seltene genetisch bedingte Erkrankung und gehört zu den sogenannten Ziliopathien. Sie wirkt sich als Multisystemerkrankung auf den gesamten Körper Betroffener aus. Wir sprachen mit Dr. Metin Cetiner, der sich unter anderem auf die Behandlung dieser sehr belastenden Erkrankung spezialisiert hat.

Dr. med.
Metin Cetiner Oberarzt und Facharzt für Kinderund Jugendmedizin, Kindernephrologe, Transplantationsmediziner, Pädiatrische Sonographie (Universitätsklinikum Essen)
Herr Dr. Cetiner, das Bardet-Biedl-Syndrom (BBS) ist eine seltene genetisch bedingte Erkrankung. Was macht die Diagnose für Mediziner so schwer?
Zunächst natürlich die Seltenheit. In unserem Register haben wir ca. 170 Betroffene in Deutschland, wobei die tatsächliche Zahl vermutlich etwa fünf- bis zehnmal so hoch ist. Zudem gibt es sechs Leitsymptome, die in verschiedenen Bereichen des Körpers und auch nicht gleichzeitig auftreten. Die Herausforderung ist daher, überhaupt erst einmal den richtigen Verdacht auf BBS zu haben.
Wie sehen diese Leitsymptome aus, und wie kann die Diagnose gestellt werden?

Patienten mit dem Bardet-Biedl-Syndrom leiden aufgrund einer genetischen Mutation unter einem unkontrollierbaren Hungergefühl, das bereits im Kindesalter zu sehr starkem Übergewicht führt.
tät (typischer Tunnelblick) und verschlechtern sich recht schnell, sodass Betroffene im Übergang zum Erwachsenenalter meist nur noch einen Visus von fünf bis zehn Prozent haben und somit per definitionem blind sind.
Die Diagnose an sich kann recht unkompliziert durch einen Gentest gestellt werden. Durch die Bandbreite der Symptome gibt es aber große Unterschiede, wann die Diagnose erfolgt. Wird das BBS nicht im Kindes- oder Jugendalter diagnostiziert, dann ist die Gefahr sehr hoch, dass die Betroffenen erst sehr spät oder nie diagnostiziert werden.
Wie sehen die Behandlungsmöglichkeiten derzeit aus, und können die verfügbaren Therapien die Lebensqualität Betroffener verbessern?
Betroffene leiden darunter oft sehr und fühlen sich isoliert.
Hier wurde kürzlich ein neues Medikament zugelassen, das die erste und einzige kausale Therapie gegen den spezifischen Gendefekt darstellt, der das Sättigungszentrum außer Kraft setzt. Dieses Medikament kompensiert den Gendefekt durch einen MC4R-Rezeptor-Agonisten, der das Sättigungszentrum wieder aktiviert, wodurch es laut aktueller Studienlage bei vielen Patientinnen und Patienten zu einem deutlich reduzierten Hungergefühl und infolgedessen zu einer starken Gewichtsreduktion kommt. Auf dieses Medikament setzen viele Betroffene sehr viel Hoffnung.
Weitere
Informationen zum NEOCYSTForschungsprogramm unter: www.neocyst.de
Weitere
Informationen zum
Patientenseminar des Arbeitskreises Bardet-BiedlSyndrom finden Sie im Veranstaltungskalender der PRO RETINA e. V. unter: www.pro-retina.de
Das erste Symptom ist die sog. Polydaktylie (Mehrfingrigkeit): Manche Betroffene haben bei Geburt einen ganzen Finger oder Zeh mehr, manche haben verkürzte oder zusammengewachsene Finger oder Zehen. Hinzu kommen Nierenauffälligkeiten wie zu große bzw. zu kleine Nieren mit veränderter Binnenstruktur und teilweise Zysten. Etwa ab dem 6. Lebensmonat zeigt sich zudem eine starke, fortschreitende Übergewichtigkeit, denn durch eine Genmutation funktioniert das Sättigungszentrum im Gehirn nicht, Betroffene kennen also das Gefühl „Ich bin satt“ nicht und entwickeln ein unkontrolliertes gesteigertes Essverhalten (sog. Hyperphagie). Insgesamt zeigt sich bei den Kindern eine Entwicklungsverzögerung speziell beim Laufen- und Sprechenlernen. Zudem kommen betroffene Kinder mit Veränderungen nicht gut zurecht, sind stark routineliebend und haben eine niedrige Frustrationsgrenze. Hinzu kommt der Hypogenitalismus, also eine Unterentwicklung der Geschlechtsorgane. Männliche Betroffene haben einen Mikropenis und die Hoden können in der Leiste verortet sein. Weibliche Betroffene können Veränderungen an der Vagina, den Schamlippen oder der Gebärmutter aufweisen, was aber häufiger übersehen wird, da ein Ultraschall vonnöten wäre, um diese Veränderungen zu entdecken. Das Symptom, das am spätesten auftritt, aber am deutlichsten auf ein BBS hinweist, ist die Netzhautdegeneration: Betroffene verlieren zunehmend ihre Sehfähigkeit. Das zeigt sich schon im Vorschulalter durch Nachtblindheit (Angst und Orientierungsschwäche im Dunkeln) und Lichtempfindlichkeit. Deutlich zeigen sich die Beschwerden dann in der Puber-
Bisher gibt es keine Therapie gegen das gesamte Symptomspektrum des BBS, aber es wird intensiv an einer möglichen Gentherapie geforscht. Bezüglich der Netzhautdegeneration kann man nur begleitende Maßnahmen in die Wege leiten, aufhalten kann man den Sehverlust bisher leider nicht. Auch die starke Übergewichtigkeit bedeutet eine enorme Last für die Betroffenen und ihre Familien. Eltern versuchen, das unter Kontrolle zu halten, sperren das Essen weg und schließen teils sogar den Kühlschrank ab, das Thema (zu viel) Essen ist allgegenwärtig. Betroffene Kinder ziehen sich zurück und leiden sehr stark unter ihrem Anderssein. Denn wir sprechen von schwerstem Übergewicht, das die Kinder und jungen Erwachsenen besonders in Kombination mit den anderen Symptomen vom normalen Leben ausschließt.
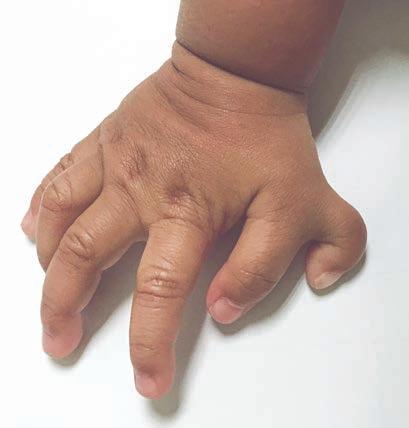
Die größte Unterstützung können sich Betroffene und Angehörige gegenseitig geben.
Betroffene und deren Familien/Angehörige erleben durch die Erkrankung eine starke Belastungssituation, die die Lebensqualität stark einschränken kann. Wo erfahren Betroffene Unterstützung?
Die größte Unterstützung können sich Betroffene und deren Angehörige gegenseitig geben. In der Patientenvereinigung PRO RETINA e. V. gibt es einen Arbeitskreis zum Bardet-Biedl-Syndrom, in dem Betroffene und Eltern betroffener Kinder erfahren: Wir sind nicht allein! Vom 12. bis 14. Mai 2023 wird es in Bonn ein Patientenseminar der PRO RETINA geben, das eine tolle Möglichkeit der Vernetzung darstellt.
Sie sind Ansprechpartner für das NEOCYSTForschungsprogramm, das sich u.a. auf die Erforschung des BBS fokussiert. Welche Vorteile hat ein Patient, der sich an diesem Programm beteiligt?
Wir vernetzen die behandelnden Ärzte und die Grundlagenforscher mit der BBS-Community, um die Erkrankung besser zu verstehen und im Idealfall neue Behandlungsansätze entwickeln zu können, die den Betroffenen zugutekommen. Dieser transparente Austausch schafft eine WinWin-Win-Situation und erhöht die Motivation auf allen Seiten! Zudem ist das BBS in vielerlei Hinsicht eine Modellerkrankung, denn die Forschung an dieser Erkrankung hat uns schon viele Erkenntnisse beschert, die auch auf andere Erkrankungen anwendbar sind. Teilnehmende Betroffene werden also aktiver Teil der Forschergemeinschaft!
Adipös, seheingeschränkt, entwicklungsverzögert –Wie das Bardet-Biedl-Syndrom das Leben Betroffener zu einer besonderen Herausforderung macht
Das Bardet-Biedl-Syndrom (kurz BBS) ist eine seltene genetisch bedingte Erkrankung, die das Leben Betroffener extrem beeinträchtigt. Besonders der Verlust der Sehfähigkeit, die zum Teil stark eingeschränkte Nierenfunktion, vielfältige Entwicklungsverzögerungen und das starke, genetisch bedingte Übergewicht sind extrem belastend und schränken den Alltag in vielen Bereichen ein. Hinzu kommt die Stigmatisierung von außen, denn die Symptome können sie zur Zielscheibe ihrer Mitmenschen machen. Wir sprachen mit Maximilian Kerber (BBS-Patient) und Andrea Kierek (Mutter von zwei betroffenen Kindern) über die extreme Last der Erkrankung und die große Hoffnung auf zielgerichtete Therapien.
Interviewpartner: Maximilian Kerber (Betroffener und Leiter des Arbeitskreises Bardet-Biedl-Syndrom der PRO RETINA e. V.) und Andrea Kierek (Mutter von zwei betroffenen Kindern)
Text Hanna Sinnecker
Herr Kerber, sie sind betroffen vom BardetBiedl-Syndrom (BBS). Wie und wann hat sich die Erkrankung bei Ihnen bemerkbar gemacht?
Meine Mutter hat schon früh gemerkt, dass etwas nicht stimmt, und ist mit mir von Arzt zu Arzt gelaufen. Ich war immer ein wenig tollpatschig, da meine Grob- und Feinmotorik nicht richtig funktioniert. Zudem hatte ich bei meiner Geburt einen sechsten Zeh, der operativ entfernt wurde. Im Grundschulalter kamen der Sehverlust und das starke Übergewicht hinzu. Meine Eltern haben daher immer auf genügend Bewegung und eine ausgewogene Ernährung geachtet, damit das Übergewicht nicht überhandnimmt.
Als ich sechs oder sieben war, hat erstmals ein Humangenetiker den Verdacht BBS geäußert. Damals, Anfang der 2000er, wurden meine Werte in die USA geschickt, wo dann auch die Diagnose gestellt wurde. Heute können bereits mehr als 20 verschiedene BBS-Gene ganz einfach erkannt werden. Da hat sich in den letzten Jahren vieles weiterentwickelt. Ab diesem Zeitpunkt wurde ich in einer Kinderklinik betreut. Später kam dann noch eine Entwicklungsverzögerung hinzu, meine Pubertät musste daher hormonell eingeleitet werden.
Herr Kerber, wie sieht Ihr Alltag mit der Erkrankung aus und was sind die größten Herausforderungen?
Das Thema der ausreichenden Bewegung, in Kombination mit dem Sehverlust und meiner gestörten Motorik, ist nicht immer einfach zu bewältigen, da man zusätzlich eingeschränkt und gehemmt ist. Die Gewichtskontrolle ist daher ein wichtiges Thema. Auch die Frage, welches Schulsystem für mich das richtige ist, war nicht leicht zu lösen. Ich habe das normale Regelschulsystem durchlaufen und mein Schulalltag war in bestimmten Bereichen auf mich angepasst, wo es notwendig war: Ich wurde z. B. beim Sportunterricht nicht nach Leistung beurteilt, oder ich habe Aufgabenstellungen aufgrund meiner Seheinschränkung größer ausgedruckt bekommen. Aber das Unverständnis seitens meiner Mitschüler hat mich durchgehend in der weiterführenden Schule begleitet. Später habe ich trotz der Erkrankung studiert und bin in den Beruf eingestiegen, was für viele Betroffene nicht oder nur zum Teil möglich ist. Was mich ständig begleitet, sind die massiven Einschränkungen durch den Sehverlust. Hier wird einem tagtäglich bewusst, dass man zu einem gewissen Teil eingeschränkt ist. Auch begleiten mich oft die Gedanken, wie es mit meiner noch gesunden Niere weitergehen wird, da dies auch ein häufiges Symptom der Erkrankung ist und auch erst zu einem
späteren Zeitpunkt auftreten kann. Wie stark die Erkrankung im Zusammenspiel der Symptome mich im Alltag einschränkt, ist mir aber tatsächlich erst in den letzten Jahren bewusst geworden. Für Außenstehende war z. B. nicht nachvollziehbar, dass ich aufgrund der Seheinschränkung gewisse Dinge nicht wahrnehme oder Menschen ungewollt anremple. Da stößt man auf Unverständnis und verärgerte Mitmenschen. Seit zwei Jahren habe ich einen Blindenstock, der auch als eine Art „Erkennungszeichen“ fungiert: Seitdem treffe ich auf viel größeres Verständnis und mehr Hilfsbereitschaft.
Frau Kierek, Sie sind Mutter von zwei betroffenen Kindern. Was macht eine solche Diagnose mit den Eltern?
Meine Kinder waren 11 und 14 bei der Diagnose, bis dahin hatten auch wir viele Ärzte und Kliniken gesehen. Zu erfahren, dass meine Kinder blind werden: Das war ein Schock. Da fragt man sich, wie die Kinder und man selbst das bewältigen soll. Auf der anderen Seite war es mit der Diagnose leichter, ihre Symptome zu erklären. Ich dachte schon immer, dass etwas Seltenes dahinterstecken könnte. Meine Tochter war wegen organischer Beschwerden, u.a. einer Nierentransplantation, oft im Krankenhaus. Dadurch war ihre Entwicklungsverzögerung leichter nachvollziehbar. Bei meinem Sohn war es schwieriger: Er hat autistische Züge und eine extreme Sprachbeeinträchtigung, das war komplizierter zu erklären. Und natürlich war auch das Übergewicht ein Problem, da es den Alltag sehr prägt und rund ums Essen viel Konfliktpotenzial bietet. In der Beziehung war die Diagnose schon eine gewisse Erleichterung, da wir nun wussten, was hinter den Beschwerden steckt, und etwas gelassener damit umgehen konnten.
Oftmals sind die direkten Angehörigen diejenigen, die sensibler für den Gesundheitszustand Betroffener sind. Ist das auch bei Ihnen der Fall, und wie gehen Sie damit um?
Kierek: Wir versuchen, unseren Kindern nicht all unsere Sorgen und Befürchtungen mitzuteilen, aber sprechen natürlich mit ihnen über die besonderen Herausforderungen, die sie haben. Wir versuchen, sie zu motivieren, dranzubleiben, auch wenn sie schon viele Dinge ausprobiert haben. Generell benötigen unsere Kinder aber sehr viel Betreuung und werden diese auch ihr Leben lang benötigen. Sie sind inzwischen 19 und 22 Jahre alt und werden nie so selbstständig sein wie Herr Kerber, der verheiratet ist, seiner Arbeit nachgeht und sein Leben selbstständig lebt. Von daher muss ich oft Entscheidungen für meine Kinder treffen.
Hatten Ihre Kinder auch mit Unverständnis im Umfeld zu kämpfen?
Mein Sohn ist generell eher zurückhaltend und hat sich mit seiner besonderen Rolle irgendwie arrangiert, er würde solche Punkte nie von sich aus ansprechen. Meine Tochter ist sehr kontaktfreudig und hatte stärker damit zu kämpfen, in der Schule wurde sie oft sehr gemobbt. Sie hat zwar immer irgendwie ihren Weg gefunden und auch viele einfühlsame und verständnisvolle Menschen getroffen. Aber da kommt man als Mutter schon an seine Grenzen, wenn die eigenen Kinder derartigen psychischen Belastungen ausgesetzt sind. Man versucht dann, die positiven Erlebnisse zu verstärken und das abzufedern.
Sie sind beide aktiv im Arbeitskreis Bardet-BiedlSyndrom in der PRO RETINA. Welche Rolle spielt für Sie beide die Vernetzung mit anderen Betroffenen, und was wünschen Sie sich hinsichtlich der Versorgung von Betroffenen?
Kierek: Für mich war das erste BBS-Patientenseminar der PRO RETINA ein beeindruckendes Erlebnis. Bei der PRO RETINA hatte ich zum ersten Mal das Gefühl: Die wissen, wovon ich spreche und wie es uns geht. Jetzt, wo ich aktiv im Arbeitskreis tätig bin, ist es toll zu sehen, was an Forschung geschieht, wie gefragt die Patientinnen und Patienten diesbezüglich sind und was im Miteinander erreichbar ist.
Kerber: Wir haben mit unserer Patientengruppe eine ganz tolle Gemeinschaft von Betroffenen, ihren Angehörigen und Forschern, die uns sehr unterstützen. Man ist nicht mehr allein und kann zudem die Forschung aktiv mitgestalten, wie zum Beispiel auch bei der ersten Therapie gegen die genetisch bedingte Adipositas in Zusammenhang mit dem BBS.
Da waren wir von Anfang an eng eingebunden, damit das patientennah geschieht und wir unsere Eindrücke und Aspekte mit einbringen können.
Deswegen ist die Finanzierung der Forschungsprojekte für mich ein ganz wichtiger Punkt. Es ist vieles auf den richtigen Weg gebracht, aber ich wünsche mir, dass es einfacher wird, Fördermittel zur Erforschung und Behandlung Seltener Erkrankungen zu bekommen.
Der größte Wunsch von ganz vielen Betroffenen ist aber sicher die Entwicklung von Therapien. Die neue Therapie gegen die Adipositas ist für uns ein erster Schritt in die richtige Richtung zur Behandlung des Bardet-BiedlSyndroms. Wenn es dann noch gelingt, eine Therapie gegen den fortschreitenden Sehverlust zu entwickeln, wäre das ein riesiger Erfolg für die Betroffenen!
Im Arbeitskreis Bardet-Biedl-Syndrom (BBS) haben sich Betroffene mit dieser Erkrankung und deren Angehörige zusammengeschlossen. Vielleicht haben Sie Fragen oder möchten gern Ihre Erfahrungen mit anderen Betroffenen austauschen. Der Erfahrungsaustausch in der PRO RETINA kann Eltern und Betroffenen helfen, diese Erkrankung anzunehmen, zu akzeptieren und zu meistern.
Für weitere Informationen zum Bardet-Biedl-Syndrom scannen Sie den QR-Code, oder melden Sie sich per E-Mail unter: bbs@pro-retina.de
Der Rare Diseases Run 2023: RUN FOR RARE!
Der Rare Diseases Run ist ein virtueller inklusiver Charity-Lauf, an dem jeder teilnehmen kann! Ein großer Teil der Teilnahmegebühr geht automatisch an verschiedene Organisationen, die sich mit seltenen Erkrankungen befassen, darunter auch die Bardet-Biedl-Patientengruppe.
Weitere Informationen zum Wettbewerb sowie Tickets finden Sie unter: www.laufenmachtgluecklich.de
FOTO:Die konsequente Durchführung der Behandlung soll Betroffenen ein normales und beschwerdefreies Leben ermöglichen
Mukoviszidose, auch zystische Fibrose (CF) genannt, ist eine autosomal-rezessiv vererbte Erkrankung, die unbehandelt tödlich verläuft. Warum eine frühe Diagnose und eine kontinuierliche Behandlung so wichtig sind, erklärt Prof. Dr. med. Marcus A. Mall im Interview.
Text Alexandra LassasHerr Prof. Mall, die Mukoviszidose ist eine seltene Multiorganerkrankung und eine der wenigen seltenen Erkrankungen, die im Neugeborenen-Screening abgebildet ist. Warum ist es so wichtig, die Erkrankung möglichst früh zu diagnostizieren?
Das Organ, das bei den meisten Patienten die stärksten Beschwerden verursacht, ist die Lunge. Dort entsteht aufgrund des Gendefektes, der der Mukoviszidose zugrunde liegt, ein besonders zäher Schleim, welcher die Atemwege verstopft. Das ist ein idealer Nährboden für Bakterien und führt bereits bei kleinen Kindern zu einer chronischen Infektion und Entzündung der Atemwege. Diese chronische Entzündung zerstört fortschreitend die Lunge. Unbehandelt erreichen betroffene Kinder kaum das Schulalter. Durch das Neugeborenen-Screening gibt es die Möglichkeit, die Erkrankung frühzeitig zu diagnostizieren und eine Therapie in die Wege zu leiten. Ein früher Therapiebeginn hat das Potenzial, die Entstehung von irreversiblen Organschäden, vor allem an der Lunge, zu verzögern oder gar zu vermeiden.
Mit welchen Beschwerden haben Betroffene zu kämpfen?
Neben der Lunge sind eine Reihe von weiteren Organsystemen betroffen: dazu gehören die Bauchspeicheldrüse, der Darm und die Leber. Etwa 85% der Betroffenen haben aufgrund angeborener Probleme mit der Bauchspeicheldrüse eine Verdauungsstörung, die sich durch Bauchschmerzen und chronische Durchfälle äußert. Hierdurch kommt es bereits bei kleinen Kindern zu einer Gedeihstörung, d.h. die Kinder nehmen nicht ausreichend an Gewicht zu. Weiterhin leiden sie durch die Schädigung der Lunge unter chronischem Husten und häufigen Infekten der Atemwege, bis hin zu Lungenentzündungen. Durch die Beeinträchtigung der Bauchspeicheldrüse kann zudem im weiteren Verlauf der Erkrankung ein Diabetes hinzukommen. Außerdem kann es zu Leberproblemen im Sinne einer Leberzirrhose kommen.
Wie sehen die derzeitigen Therapieoptionen aus?
Über lange Zeit konnten wir ausschließlich die Symptome der Erkrankung behandeln, das aber durchaus mit gutem Erfolg: denn so konnten wir die Lebenserwartung für Betroffene bereits auf über 40 Jahre steigern. Symptomorientiert bedeutet z. B. für die Verdauungsstörungen, dass die fehlenden Verdauungs-Enzyme der Bauchspeicheldrüse ersetzt werden, um damit die Durchfälle und Gedeihstörung zu behandeln. Für die Lunge bedeutet das eine lebenslange, schleimlösende Therapie: diese besteht zum einen aus einer schleimlösenden Inhalationstherapie unter Einsatz verschiedener schleimlösender Medikamente, und zum anderen aus Physiotherapie, um den Schleim aus der Lunge abzutransportieren. Die Atemwegsinfektionen werden mittels Inhalationen oder einer systematische Antibiotikagabe behandelt. Alle Inhalationen müssen mehrmals am Tag durchgeführt werden und beschäftigen die Betroffenen oft mehrere Stunden am Tag. Seit einigen Jahren gibt es einen kausalen Therapieansatz, der ein wahrer Durchbruch für Betroffene war, weil das eigentliche Problem an der Wurzel angepackt wird. Diese sogenannten CFTR-Modulatoren greifen an dem durch den Gendefekt fehlgefaltetem Protein an und setzen somit am Basisdefekt der Erkrankung an.
Das Ziel is, dass Betroffene möglichst lange ein normales, gesundes Leben führen können, ohne dass die Erkrankung das Steuer übernimmt.
Die digitale Plattform mit Informationen und Services rund um CF.

Seit einigen Jahren können wir so bis zu 90% der Betroffenen behandeln. Betroffene müssen dafür zweimal täglich Tabletten einnehmen, was gegenüber der rein symptomorientierten Behandlung einfach umzusetzen und viel weniger zeitintensiv ist.
Zudem hat die systemische Verabreichung in Form einer Tablette den Vorteil, dass jedes betroffene Organ erreicht wird. Das ist ein echter Fortschritt, der zu einer enormen Verbesserung der Lebensqualität und voraussichtlich auch der Lebenserwartung führt. Dadurch kann eine potenziell tödliche Erkrankung zu einer behandelbaren, chronischen Erkrankung werden.
Da es bisher noch keine Heilung für die Erkrankung gibt, müssen Betroffene ein Leben lang behandelt werden. Wie können Betroffene motiviert bleiben, an der Therapie dranzubleiben?
Der langfristige Behandlungserfolg hängt wesentlich von einer lebenslangen und regelmäßig durchgeführten Therapie ab. Durch den Fortschritt der angesprochenen Kausaltherapie werden die Beschwerden deutlich weniger, was aber nicht zum Vergessen oder Auslassen der Einnahme führen darf. Daher ist vor allem auch bei Kindern und Jugendlichen auf eine regelmäßige Therapie zu achten. Die Betroffenen und ihre Familien müssen daher auch in Zukunft engmaschig betreut werden. Die Herangehensweise sollte sein, dass man mit einem frühen Therapiebeginn vor Auftreten der Beschwerden präventiv tätig wird, anstatt wie früher den Problemen hinterherzulaufen.
Der Schlüssel ist zudem, sowohl die Kinder als auch ihre Familien in Schulungsprogrammen zu erklären, was im Körper von Betroffenen passiert, weshalb sie die Therapie durchführen, und was passieren kann, wenn sie hier nachlässig werden. Denn das Ziel ist ja, dass sie lange ein möglichst normales, gesundes Leben führen können, ohne dass die Erkrankung das Steuer übernimmt.

Prof. Dr. Marcus A. Mall Professor und Direktor der Klinik für Pädiatrie m. S. Pneumologie, Immunologie und Intensivmedizin, Ärztlicher Centrumsleiter des CharitéCentrum 17 für Frauen-, Kinder und Jugendmedizin mit Perinatalzentrum und Humangenetik, Charité - Universitätsmedizin Berlin

Linda Meschke (36) leidet an der erblichen Netzhauterkrankung Retinitis Pigmentosa Sie sprach mit uns über ihren Weg bis zur Diagnose, über derzeitige Behandlungsmöglichkeiten und ihren Alltag mit dieser seltenen Augenerkrankung.
Text Doreen BrummeLinda, wann traten Ihre Augenprobleme auf und wie kam es zur Diagnose Retinitis Pigmentosa?
Ich konnte schon als Kind nicht gut sehen, trug in der Schule eine Brille. Regelmäßige Besuche beim Augenarzt waren angesagt, irgendwann entdeckte dieser Verknöcherungen auf meiner Netzhaut und meinte, dass ich damit im Dunkeln ja gar nichts sehen müsste, was ich bejahte – für mich war das ein Normalzustand. Zudem hatte ich von Anfang an auf beiden Augen einen grauen Star, also eine Eintrübung meiner Augenlinsen, was mir mit 18 auch diagnostiziert wurde. Mit 27 wurde ich deshalb in der Uniklinik Dresden operiert, da ich im Alltag schlecht zurechtkam. In den Entlassungspapieren las ich zum ersten Mal die Diagnose: Retinitis Pigmentosa. Die gab ich bei Google ein und ließ mir erklären, was es damit auf sich hat. Nach der Recherche wusste ich zwei Dinge: Ich werde erblinden. Und es gibt nichts, was man dagegen tun kann. Erst zwei, drei Jahre später ließ ich einen Gentest machen, der die Diagnose bestätigte.
Ihre Netzhautzellen sterben nach und nach ab und mindern Ihre Sehfähigkeit zunehmend. Wie verändert das Ihren Alltag?
Die Veränderung von sehend zu blind verläuft in kleinen Schüben. Ich sehe die Welt inzwischen mit einem Tunnelblick. Das heißt, bei guter Beleuchtung erkenne ich Dinge in der Ferne noch sehr gut. Wobei die Betonung auf der guten Beleuchtung liegt, die selten herrscht. Auch die Nahsicht ist noch gut: Ich kann lesen und meinen Bürojob machen. Doch mein Sichtfeld ist mit 10 bis 15 Grad mittlerweile deutlich kleiner als das eines Augengesunden (180 Grad). Ich bin im Alltag deshalb oft auf Hilfe angewiesen, insbesondere dort, wo ich mich nicht auskenne oder wo viel los ist. Kaufe ich zum Beispiel ein, erschrecke ich, wenn plötzlich jemand von links oder rechts in meinen „Sichttunnel“ tritt, denn ich habe ihn nicht kommen sehen. Meine buchstäblich schwindende Aussicht lässt mich langsam das Vertrauen in mich selbst verlieren.

Die Retinitis Pigmentosa ist genetisch bedingt. Gab es in Ihrer Familie bereits vor Ihnen bestätigte Fälle oder Familienangehörige, die entsprechende Symptome gezeigt haben?
Mein Vater zeigt seit Langem zunehmende Symptome, hat das aber nie abklären lassen, sondern verdrängt.
Warum engagieren Sie sich in der Patientenselbsthilfe der PRO RETINA?
Die Gewissheit, zu erblinden, stellte mein Leben auf den Kopf. Zumal der individuelle Verlauf ungewiss ist. Bis zu der Erkenntnis, dass das Leben auch mit schlechter oder ohne Sicht nicht aussichtslos ist, war es für mich ein langer Weg mit so manchem tiefen Loch, in das ich fiel. Davor würde ich gerne andere Betroffene bewahren.
PRO RETINA e. V.
Der Selbsthilfeverein PRO RETINA Deutschland e. V. ist bundesweit die größte und älteste Patientenvereinigung von und für Menschen mit Netzhauterkrankungen und deren Angehörige. PRO RETINA unterstützt Betroffene und ihre Angehörigen nach dem Leitsatz „Forschung fördern, Krankheit bewältigen, selbstbestimmt leben“, fungiert als Bindeglied zwischen Patient und Arzt und unterstützt die Forschungsförderung, damit neue Therapien entwickelt werden. Für seine Arbeit ist der gemeinnützige Verein auf die Unterstützung von Spendern und Sponsoren angewiesen. Weitere Informationen unter: www.pro-retina.de
ANZEIGE
Für die meisten der bisher bekannten rund 8.000 seltenen Krankheitsbilder gibt es aktuell noch keine Therapieoption. Das Pharmaunternehmen Janssen hat den Anspruch, durch kontinuierliche Forschung einen Beitrag zu leisten, um Menschen mit seltenen Krankheiten eine Perspektive bieten zu können.

Mit freundlicher Unterstützung der Janssen-Cilag GmbH

„Wir forschen in den Bereichen, in denen der medizinische Bedarf hoch ist – unabhängig davon, wie häufig eine Krankheit ist“, erklärt Dr. med. Ursula Kleine-Voßbeck, medizinische Direktorin im Bereich Lungenhochdruck bei Janssen Deutschland. „Unser Ziel ist, da anzusetzen, wo wir einen entscheidenden Unterschied machen können.“ Für einige seltene Krankheitsbilder konnte Janssen bereits erfolgreich Therapien entwickeln, unter anderem im Bereich der Hämatologie, z. B. für Menschen mit Amyloidose und Morbus Waldenström sowie für Lungenhochdruck.
Lunge unter Druck
Eine spezielle Form des Lungenhochdrucks ist die pulmonal arterielle Hypertonie (kurz PAH). Bei dieser Krankheit stehen die Blutgefäße, die vom Herz zur Lunge führen, unter einem zu hohen Druck. In der Folge muss die rechte Herzhälfte immer stärker gegen diesen erhöhten Druck arbeiten. Auf Dauer kann das Herz diese Leistung nicht erbringen. Bleibt die Erkrankung unbehandelt, kann es zum Herzversagen kommen. Besonders tückisch ist, dass die Leitsymptome der PAH zu Beginn sehr unspezifisch sind (z. B. Atemnot, Druck auf der Brust, Erschöpfung) und Verwechslungsgefahr mit häufigeren Krankheiten wie Asthma oder COPD besteht.
Auch wenn eine PAH grundsätzlich jeden treffen kann, gibt es Risikogruppen: Ein erhöhtes PAH-Risiko haben beispielsweise Menschen mit einem angeborenen Herzfehler. Schätzungsweise entwickeln bis zu zehn Prozent der Betroffenen eine PAH – selbst Jahrzehnte nach erfolgreicher Korrektur des Herzfehlers. Außerdem sind chronische Bindegewebserkrankungen wie die systemische Sklerose mit einem erhöhten Erkrankungsrisiko verbunden. Für diese Risikogruppen ist daher ein regelmäßiger Check in spezialisierten Zentren zu empfehlen. Je frühzeitiger im Verlauf die PAH erkannt wird, desto besser. Die Krankheit ist aktuell nicht heilbar, aber es gibt mittlerweile gute Behandlungsmöglichkeiten.
Retinitis Pigmentosa: Gentest bringt Licht ins Dunkel Janssen forscht zudem an Therapieoptionen für 16 weitere seltene Krankheitsbilder, unter ihnen die seltene X-chromosomale Retinitis Pigmentosa. Bei dieser erblich bedingten Netzhautdegeneration werden die Photorezeptoren allmählich zerstört. Das erste Anzeichen ist eine stärker werdende Nachtblindheit, die Betroffene oft bereits vor dem 10. Lebensjahr bemerken können. Aufgrund des progressiven Verlaufs grenzt sich das Sichtfeld immer stärker ein, was bis zur Erblindung führen kann. Der Großteil der Betroffenen ist männlich. Frauen haben meist keine oder nur leichte Symptome, können aber Trägerin des mutierten Gens sein und die Retinitis Pigmentosa an ihre Kinder weitergeben. Ein Gentest ist für die Diagnose entscheidend: Denn nahezu 100 verschiedene Mutationen kommen als Auslöser der Retinitis Pigmentosa in Betracht. Erst wenn der Gentest die Diagnose sichert bzw. eingrenzt, kann über mögliche Behandlungsoptionen gesprochen werden. Selbst wenn es für die vorliegende Genmutation heute noch keine Therapiemöglichkeit gibt, ist die Testung sinnvoll. Betroffene können sich in ein Register eintragen und für spätere Behandlungsoptionen vormerken lassen. Außerdem bietet sich eventuell die Chance, an klinischen Studien für aufkommende Therapieoptionen teilzunehmen.
Unter www.janssenwithme.de/erkrankungen erhalten Sie umfangreiche Fakten und Hintergründe sowohl zur PAH als auch zur Retinitis Pigmentosa. Zudem finden Sie auf dem YouTube-Kanal von Janssen Deutschland in der Playlist „Pulmonale Hypertonie“ zahlreiche Videos, die über Lungenhochdruck sowie den Umgang mit der Erkrankung informieren.
Nicht-dystrophe Myotonien sind seltene, genetisch bedingte neuromuskuläre Erkrankungen. Das charakteristische Merkmal: Betroffene sind aufgrund der Krankheit nicht fähig, die der körperlichen Bewegung dienenden Muskeln (Skelettmuskulatur) nach der Kontraktion sofort wieder zu entspannen. Das kann die Lebensqualität Betroffener stark beeinträchtigen und sogar lebensgefährlich werden.
Text Miriam RauhCaro, du bist betroffen von einer nicht-dystrophen Myotonie, kurz NDM. Wann hast du gemerkt, dass etwas gesundheitlich nicht stimmt, und welche Beschwerden hattest du?
Bei mir war früh zu sehen, dass etwas nicht stimmt. Ich fing erst mit zwei Jahren an zu laufen, humpelte, wenn ich nach längerem Sitzen wieder aufstand, und lief insgesamt oft steif. Die Myotonie ist zwar eine Erkrankung, die die Muskulatur des ganzen Körpers betrifft, aber bei mir waren die Symptome in den Beinen am offensichtlichsten. Meine Eltern gingen von Kinderarzt zu Kinderarzt, immer hieß es, ich sei einfach zu faul oder das sei normal und würde sich mit dem Wachstum ändern. Es folgte die Fehldiagnose, es sei etwas mit meiner Hüfte. Ich erhielt mehrere Jahre Physiotherapie. Erst als ich neun war, äußerte ein Arzt Zweifel an der Hüft-These, als er meine Röntgenbilder sah. Es folgten weitere Untersuchungen und Tests und schließlich kam die Diagnose: NDM.
Durch die genetische Mutation kann ich meine Muskeln problemlos anspannen, aber nicht sofort entspannen. Ich bin auch sehr wetterempfindlich, bei Wärme geht es mir viel besser als bei Kälte. Die Krankheit hat eine psychische Komponente – wenn es mir gut geht, meine ich, ich könnte einen Marathon rennen, wenn es mir schlecht geht, geht fast nichts.
Wenn es mir gut geht, meine ich, ich könnte einen Marathon rennen, wenn es mir schlecht geht, geht fast nichts.

Was waren/sind die größten Herausforderungen im Zusammenhang mit der Erkrankung für dich?
Während ich noch im Wachstum war, wurde mir gesagt, dass die Krankheit entweder besser oder schlechter werden kann. Ich kenne mein Leben nicht ohne die Erkrankung, ich weiß nicht, wie es anders ist. Natürlich ist sie immer wieder anstrengend für mich, psychisch und physisch, auch weil sich die Krankheit bei mir verschlechtert hat. Als ich Kind war, spürte ich die NDM nur in den Beinen, im Wachstum wurden auch meine Hände langsam starr, dann meine Arme, mein Rücken und auch die Zunge. Bevor ich die Medikamente nahm, musste ich mich immer aufwärmen, wenn ich z. B. telefonieren wollte. An diese Verschlechterungen und Veränderungen musste ich mich gewöhnen, aber insgesamt habe ich es schnell und gut gemeistert. Zum Glück hatte ich auch die Unterstützung meiner Familie. Wesentlich schwieriger war und ist für mich der Umgang mit gesunden Menschen. Es ist nicht leicht, Außenstehenden zu vermitteln, wie sich diese Myotonie äußert. Ich gehe noch zur Schule, derzeit in die 11. Klasse. In diesem Jahr wurde vereinbart, dass ich in Sport keine Noten bekomme, mich aber trotzdem beteilige. Ich habe die Schule gewechselt, weil ich in der vorherigen stark gemobbt wurde.
Ich habe meinen neuen Klassenkameraden nichts von meiner Krankheit erzählt, weil ich die Erfahrung gemacht habe, dass mein Umfeld nicht versteht, was Myotonie ist. Mir würde es sehr weiterhelfen und mich auch entspannen, dass andere respektieren, wenn ich sage, dass ich etwas nicht machen kann, auch wenn ich fünf Minuten später losrenne, als wäre nichts. Das ist für Außenstehende schwer nachvollziehbar, aber so ist die Myotonie. Wenn ich aufgewärmt bin, kann ich einiges, was sonst nicht geht.
Wie wird deine Erkrankung behandelt und wie wirkt sich das auf deinen Alltag bzw. deine Lebensqualität aus?
Zurzeit bekomme ich Medikamente und werde durch ein Krankenhaus in Rom betreut. Hier in Deutschland hatte ich leider nicht sehr viel Glück mit Ärzten und es war schwer, die Krankenkasse zu überzeugen, dass ich die Medikamente brauche. Meine Mutter ist Italienerin, in Italien ging es sehr viel schneller. Ich nehme das Medikament, seitdem ich zwölf bin, mittlerweile dreimal am Tag 200 Milligramm, und sie haben meine Lebensqualität stark verbessert. Es gibt bessere und schlechtere Tage, aber ich habe keine Schmerzen mehr.
Ist es für dich wichtig, dich mit anderen Betroffenen über ihre Erfahrungen mit der Erkrankung auszutauschen?
Ich bin Mitglied im Verein „Mensch & Myotonie“. Es war eine lebensverändernde Erfahrung für mich, anderen Menschen zu begegnen, die das gleiche Schicksal haben wie ich. Wenn man sein Leben lang behandelt wurde, als würde man sich die Symptome ausdenken, fühlt es sich wunderbar an, Menschen zu treffen, die dasselbe durchmachen. Auch wenn die Erkrankung bei jedem anders ist, erzählen alle über ihre Erfahrungen recht ähnliche Geschichten und können gute Tipps geben.
Wenn man sein Leben lang behandelt wurde, als würde man sich die Symptome ausdenken, fühlt es sich wunderbar an, Menschen zu treffen, die dasselbe durchmachen.
Informationen zur Patientenorganisation „Mensch & Myotonie gem. e. V.“
Eine Mitgliedschaft in der ehrenamtlich von einer Myotonie -Betroffenen geführten Patientenorganisation „Mensch & Myotonie gem. e. V.“ ist komplett kostenlos. Jeder zusätzliche Beitritt stärkt uns, unsere Interessen in der Öffentlichkeit und bei Institutionen wahrzunehmen. Zusätzlich zu den „NDM“ engagieren wir uns auch für Betroffene von „Periodischen Paralysen“ sowie von „Neuromyotonien“.
Machen Sie mit – in Ihrem und unserem Interesse!
Weitere Informationen finden Sie unter: www.menschundmyotonie.de
Kontakt
Mensch & Myotonie e. V. Postfach 16 03 30 44333 Dortmund
1. Vorsitzender: Volker Kowalski
E-Mail: vokiko@online.de Tel.: 0231-803290 (ab 12 Uhr)



officialmyotonia.orga www.instagram.com/officialmyotonia.orga/
myotonia.org www.tiktok.com/@myotonia.org
Ich bin sehr muskulös, habe aber keine Kraft. Mein Nachbar hält mich für einen Macho, weil meine Frau die Getränkekisten trägt….

Die Musik ist mein Leben: die erste Geige im Orchester spielen – ein Traum, der mit einer wirksamen Therapie Realität werden könnte.
Als ich die Hand meines neuen Chefs nicht loslassen konnte, wäre ich am liebsten im Boden versunken. Ihm nicht die Hand zu geben war keine Option!
Kälte verstärkt meine Symptome. Wintersport –ohne wirksame Therapie ist das undenkbar!
Meine Eltern hielten mich für bockig, weil ich vor der Treppe stehen blieb und nicht hochgehen konnte.



Starke Erdbeben haben in der Türkei und Syrien ein unvorstellbares Ausmaß der Zerstörung hinterlassen. Viele Menschen sind tot und Tausende verletzt. Aktion Deutschland Hilft leistet Nothilfe. Mit Nahrungsmitteln, Trinkwasser und medizinischer Hilfe. Helfen Sie jetzt – mit Ihrer Spende!
Spendenkonto: DE62 3702 0500 0000 1020 30
Jetzt spenden: www.Aktion-Deutschland-Hilft.de