Ein Expert:innenkonsens – gerichtet an Allgemeinmediziner:innen
Wachstumshormonmangel: Auch psychosoziale Aspekte bedenken.
Diagnose-Algorithmus für Allgemeinmediziner:innen bei Verdacht auf ATTR-Amyloidose.
IMPRESSUM
Herausgeber und Medieninhaber: RegionalMedien Austria Gesundheit – RMA Gesundheit GmbH, Am Belvedere 10 / Top 5, 1100 Wien, Tel. 01/74321708114, office@gesund.at. Geschäftsführung: Mag.a Birgit Frassl, Marlis Rumler. Redaktionsleitung: Mag.a Karin Martin. Redaktion: Mag.a Karin Martin, Anna Schuster, BSc, Mag.a Ines Pamminger, BA, Margit Koudelka. Lektorat: Mag.a Katharina Maier. Produktion & Grafik: Helena Valasaki, BA. Cover-Foto: unsplash.com/Vlad Hilitanu Verkaufsleitung: Mag.a Birgit Frassl, birgit.frassl@regionalmedien.at. Kundenbetreuung: Mag.a Dagmar Halper, dagmar.halper@regionalmedien.at, Ornela-Teodora Chilici, BA, ornela-teodora.chilici@regionalmedien.at. Druckerei: Bösmüller Print Management GesmbH & Co. KG. Verlags- und Herstellungsort: Wien.
Grundlegende Richtung: Unabhängige österreichische Fachzeitschrift für niedergelassene Ärztinnen und Ärzte.
Die HAUSÄRZT:IN – Praxis-Magazin für Primärversorgung –ist ein interdisziplinäres Informations- und Fortbildungsmedium.
Aus Gründen der besseren Lesbarkeit verzichten wir in den Artikeln teilweise auf die gendergerechte bzw. gänzlich orthografisch/grammatikalisch korrekte Schreibweise. Sofern nicht anders vermerkt, gelten alle Bezeichnungen für sämtliche Geschlechter.
Namentlich gekennzeichnete Beiträge geben nicht unbedingt die Meinung der Redaktion oder des Verlages wieder, sondern fallen in den Verantwortungsbereich der Autor:innen. Der Inhalt von entgeltlichen Einschaltungen und Beilagen sowie die Angaben über Dosierungen und Applikationsformen liegen außerhalb der Verantwortung der Redaktion oder des Verlages und sind vom/von der jeweiligen Anwender:in im Einzelfall auf ihre Richtigkeit zu überprüfen. Nachdruck nur mit schriftlicher Genehmigung des Verlages. Alle Rechte, insbesondere das Recht der Vervielfältigung und Verbreitung sowie der Übersetzung, vorbehalten. Kein Teil des Werkes darf in irgendeiner Form (Fotokopie, Mikrofilm oder ein anderes Verfahren) ohne schriftliche Genehmigung des Verlages reproduziert oder unter Verwendung elektronischer Systeme gespeichert, verarbeitet, verviel-fältigt, verwertet oder verbreitet werden.
Mit „Bezahlte Anzeige“ gekennzeichnete
Beiträge/Seiten sind gemäß §26 Mediengesetz bezahlte Auftragswerke. Offenlegung: gesund.at/impressum
Haus Ärzt:in DIALOGTAG
Hausärzt:in trifft Kliniker:in
Moderne Schmerzmedizin –Möglichkeiten & Grenzen
PräsenzFortbildung Sa., 3. Juni 2023
IN LINZ
Themen (mit Fallbeispielen aus der Allgemeinpraxis):
Chronischer Schmerz – eine Herausforderung: von der Diagnose bis zur Therapie
Geriatrie & Palliativmedizin – Schmerztherapie ist Teamarbeit
Pädiatrie – Kleiner Mensch, großer Schmerz
Podiumsdiskussion:
Ambulante Schmerztherapie heute – Was tun gegen Lücken in der Versorgung?
Programm und Anmeldung: meinmed.at/dialogtag-linz
7 DFP-Punkte in Planung
Teilnahmegebühr:
OBGAM-, ÖGAM-, JAMÖ-Mitglieder 65€, Nichtmitglieder 85€ Rückfragen an info@meinmed.at
Mit freundlicher Unterstützung von:
Veranstalter:innen:
Änderungen vorbehalten.
Wenn die Diagnose im Kindesalter ausbleibt
Herausforderungen bei Erwachsenen mit Lennox-Gastaut-Syndrom
Das Lennox-Gastaut-Syndrom (LGS) zählt zu den am schwierigsten zu identifizierenden – und zu behandelnden – epileptischen Störungen. Diese seltene, im Verlauf schwere, Erkrankung beginnt meist im Kindesalter. Hierbei treten multiple epileptische Anfallsformen überwiegend in tonischer Form oder mit atypischen Absencen auf. Oft passieren sie im Schlaf und werden daher übersehen.1 Hinzu kommt die Problematik, dass die Erscheinungsformen und Verläufe insbesondere im Erwachsenenalter differieren können. Diesen speziellen diagnostischen Herausforderungen widmeten sich drei Experten im Rahmen eines Pressebriefings im November 2022.*
Diagnosekriterien
Das Lennox-Gastaut-Syndrom kann im Wesentlichen an klinischen Merkma-
len festgemacht werden. „Wir ordnen das LGS den generalisierten Epilepsien mit Entwicklungsverzögerungen, Intelligenzminderung und zusätzlichen Verhaltensstörungen und einem charakteristischen Anfalls- bzw. auch EEGMuster zu“, erklärt Priv.-Doz. Dr. Felix von Podewils, MHBA, Leiter des interdisziplinären Epilepsiezentrums der Klinik und Poliklinik für Neurologie an der Universitätsmedizin Greifswald. In der Regel manifestiere sich diese Epilepsieform vor dem achten Lebensjahr, mit einem Peak zwischen dem dritten und fünften Lebensjahr. „ Fünf Jahre nach Epilepsiebeginn haben 75-95 Prozent der Patientinnen und Patienten kognitive Störungen und/oder Verhaltensstörungen“, führt der Experte weiter aus. Dies spreche dafür, dass neben einer Grundveranlagung, die wahrscheinlich syndromabhängig sei, auch die epileptischen Anfälle Auswirkungen auf die
kognitive Leistungsfähigkeit hätten. Durch eine adäquate Behandlung könne man also einen Teil dieser kognitiven Entwicklungsstörungen wieder abfangen – so die Hypothese. „ Deshalb ist es so wichtig, die Diagnose eines LennoxGastaut-Syndroms so früh wie möglich zu stellen“, schlussfolgert der Facharzt für Neurologie. Dass eine adäquate Therapie Erfolg in puncto sozialer Integration und kognitiver Leistung haben könne, hätten bereits die Daten einer Studie mit einem Beobachtungszeitraum von drei Jahren gezeigt:2 Die Alltagsadaption habe sich durch die Medikation deutlich verbessern lassen.
Doz. Podewils zufolge gilt die LennoxEnzephalopathie als Syndrom des Kindesalters – die Diagnose im Erwachsenenalter ist bislang unüblich. Als Take-home-Message hebt er hervor: „I m Fokus der Diagnose eines LennoxGastaut-Syndroms sollten die charakteristischen Anfallsformen stehen “ Sowohl EEG-Veränderungen als auch kognitive Leistungen könnten sich nämlich sehr variabel gestalten.
LEITKRITERIEN
Häufig: atonische und (teils schlafgebundene) tonische Anfälle, atypische Absencen
Epileptische Spasmen
Myoklonische Anfälle, Sturzanfälle
Nonkonvulsiver Status epilepticus (50-75 %)
Bewusstseinstrübung, z. T. unterbrochen von kurzen tonischen Anfällen
Fokale klonische Anfälle, z. T. mit Übergang in bilateral tonisch-klonische Anfälle
EEG:
Diffuse Slow-Spike-Wave-Komplexe (< 3/s) im Wachen
Mit zunehmendem Lebensalter nicht mehr nachweisbar
(Non-REM)-schlafgebundene schnelle Rhythmen (10-20/s als Korrelat der tonischen Anfälle)
Vorliegen einer Intelligenzminderung ist kein Diagnosekriterium
Quelle: Vortrag von Prof. Dr. Adam Strzelczyk/Kerling und Rauch, Epileptische Anfälle und Epilepsien im Erwachsenenalter, 2021.
SCREENING-INSTRUMENT
Das „Refractory Epilepsy Screening Tool for LGS“ (REST-LGS) wurde von einer Gruppe von Expert:innen entwickelt, um sowohl die Identifizierung als auch die Behandlung der Erkrankung zu verbessern. REST-LGS soll dabei helfen, zwischen dem Lennox-GastautSyndrom und anderen refraktären Epilepsien zu unterscheiden, indem auch wichtige – auf LGS hindeutende – Nebenkriterien erfasst werden.
Publikation:
Piña-Garza JE et al., Epilepsy Behav. 2019 Jan;90:148-153.
Variables EEG
Als EEG-Charakteristika dieser seltenen Epilepsieform können irreguläre 1,5 bis 2,5 Herz Slow-Spike-Waves (SSW), generalisierte Polyspike-Paroxysmen und generalisierte Low-Voltage-Fast-Activity genannt werden. Laut Dr. Volker Sepeur, Ärztlicher Leiter des Epilepsiezentrums am Christlichen Klinikum Unna, können die typischen SSW-Komplexe jedoch mit dem Alter abnehmen: „ Bei bis zu 20 % der Patienten lassen sich LGS-typische Muster nach zehn bis 20 Jahren nicht mehr nachweisen.“
Zudem hänge die epileptische Aktivität im EEG von der Aufmerksamkeit der zu untersuchenden Person ab und könne beispielsweise durch Stress unterdrückt werden. Zu sehen sei sie zumeist bei Müdigkeit oder im Schlaf.
Jedoch: „ F ür LGS-Patienten ist ein Langzeit-Video-EEG häufig nicht tolerabel“, berichtet der Vortragende aus Erfahrung. In seinem Fazit stellt Dr. Sepeur folglich den Stellenwert der Elektroenzephalographie bei der Diagnose dieser therapierefraktären Epilepsieform in Frage.
Späte Krankheitserkennung ist möglich
Prof. Dr. Adam Strzelczyk, Leitender Oberarzt am Epilepsiezentrum der Universitätsklinik Frankfurt, schloss die Vortragsreihe mit einem Fallbeispiel ab: „ Die Patientin war seit ihrem sechsten Lebensjahr bei Kollegen in Behandlung – mit der Diagnose einer fokalen Epilepsie. Im Rahmen der Transition wurde diese nochmals überprüft, auf Grund häufiger Sturzanfälle wurde ein LGS in Betracht gezogen “ Die Betroffene erlitt 15-25 Anfallstage pro Monat, darunter multiple Stürze. Letztere sind mit einem hohen Verletzungsrisiko verbunden. Nach Überprüfung der Diagnose wurden die Medikation und die sozialmedizinische Situation verbessert. Letztendlich konnte die Betroffene von modernen Therapeutika profitieren und die Anfallsfrequenz wurde deutlich reduziert.
Mag.a Ines Pamminger, BA
Epidyolex® wird als Zusatztherapie von Krampfanfällen im Zusammenhang mit dem Lennox-Gastaut-Syndrom (LGS) oder dem DravetSyndrom (DS) in Verbindung mit Clobazam bei Patienten ab 2 Jahren angewendet.1
1 00 mg/ml Lösung zum Einnehmen Cannabidiol
100 mg/ml Lösung zum Einnehmen Cannabidiol
Epidyolex® wird als Zusatztherapie von Krampfanfällen im Zusammenhang mit Tuberöser Sklerose (TSC) bei Patienten ab 2 Jahren angewendet.1
Epidyolex®, das erste und bisher einzige zugelassene CBD-Medikament bei LGS, DS und TSC in Europa.1
Epidyolex® ist seit 1.4.2021 in der dunkelgelben Box.
1. Epidyolex® Fachinformation. Stand 01/22 Datum der Erstellung: September 2022. AT-EPX-2200197
* Online-Pressebriefing von Jazz Pharmaceuticals, vom 22.11.22: „Herausforderungen der Diagnostik bei Erwachsenen mit einem nicht diagnostizierten Lennox-Gastaut-Syndrom“.
2 Patel AD et al., Epilepsia. 2021 Sep;62(9):2228-2239.
Erfahren Sie mehr auf www.epidyolex.at
Bezeichnung des Arzneimittels: Epidyolex 100 mg/ml Lösung zum Einnehmen. Qualitative und quantitative Zusammensetzung: Jeder ml der Lösung zum Einnehmen enthält 100 mg Cannabidiol. Sonstiger Bestandteil mit bekannter Wirkung: Jeder ml Lösung enthält: 79 mg Ethanol, 736 mg raffiniertes Sesamöl, 0,0003 mg Benzylalkohol. Liste der sonstigen Bestandteile: Raffiniertes Sesamöl, Ethanol, Sucralose (E955), Erdbeer-Aroma (enthält Benzylalkohol). Anwendungsgebiete: Epidyolex wird als Zusatztherapie von Krampfanfällen im Zusammenhang mit dem Lennox-Gastaut-Syndrom (LGS) oder dem Dravet-Syndrom (DS) in Verbindung mit Clobazam bei Patienten ab 2 Jahren angewendet. Epidyolex wird als Zusatztherapie von Krampfanfällen im Zusammenhang mit Tuberöser Sklerose (TSC) bei Patienten ab 2 Jahren angewendet. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Patienten mit erhöhten Transaminasewerten, die das Dreifache der oberen Normgrenze (ULN) übersteigen, und deren Bilirubinwerte das Zweifache der ULN übersteigen. Pharmakotherapeutische Gruppe: Antiepileptika, andere Antiepileptika, ATC-Code: N03AX24. Inhaber der Zulassung: GW Pharma (International) B.V., Amersfoort A1, Databankweg 26, 3821AL Amersfoort, Niederlande. Rezeptpflicht/ Apothekenpflicht: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. Weitere Informationen zu den Abschnitten Warnhinweise und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen, Nebenwirkungen und Gewöhnungseffekte sowie zu Fertilität, Schwangerschaft und Stillzeit entnehmen Sie bitte der veröffentlichten Fachinformation. Stand der Information: 01/2022 Darreichungsform: Eine 100-ml-Flasche; jeder ml der Lösung zum Einnehmen enthält 100 mg Cannabidiol. Die Flasche ist in einem Karton mit zwei 5-ml- und zwei 1-ml- Applikationsspritzen für Zubereitungen zum Einnehmen und zwei Flaschenadaptern verpackt. Die 5-ml-Spritzen sind in Schritten von 0,1 ml und die 1-ml-Spritzen in Schritten von 0,05 ml unterteilt. Für mehr Information kontaktieren Sie bitte: medinfo@gwpharm.com
Jazz Pharmaceuticals Austria GmbH, Gertrude-Fröhlich-Sandner-Straße 2-4, Turm 9/9, 1100 Wien Kontakt für Österreich: Dr. Maria Heinrich, Tel.: + 43 664 1372 758, E-Mail: maria.heinrich@jazzpharma.com
Das Kleinbleiben verhindern
Kinder mit Hyposomatotropismus erkennen und ihnen eine Behandlung ermöglichen
Ein Kleinwuchs kann bei Kindern diverse Gründe haben. Im Falle eines Wachstumshormonmangels („G rowth Hormone Deficiency“ – GHD) besteht die Möglichkeit einer GH-Substitution, mit welcher sich zumeist ein Aufholwachstum und in der Folge eine normale Erwachsenengröße erreichen lassen. Entscheidend hierfür sind allerdings Faktoren wie ein rechtzeitiger Behandlungsbeginn und die Therapieadhärenz.
Wann hellhörig werden?
Für das Längenwachstum spielt Somatotropin nicht von Geburt an die zentrale Rolle – die wechselseitige Abhängigkeit der beiden nimmt im Laufe der Entwicklung zu. So lässt sich von Geburtsgröße und -gewicht im Kontext des Hyposomatotropismus prinzipiell nicht auf die Erwachsenengröße schließen. Bei Säuglingen und Neugeborenen können Hypoglykämien Anzeichen für einen Wachstumshormonmangel sein, da Somatotropin den Blutzuckerspiegel reguliert. Im Kleinkindalter stellt dann das Fallen der Wachstumskurve unter den Normbereich oder unter den Bereich der genetisch logischen Körpergröße das wichtigste und typische Symptom von GHD dar. Häufig wirken Kinder mit isoliertem Wachstumshormonmangel besonders klug, da man auf
den ersten Blick von einem jüngeren Alter ausgeht.
Ist ein Kind in der Perzentilenkurve deutlich kleiner als 97 Prozent seiner Altersgenossen, sollten betreuende Ärztinnen und Ärzte jedenfalls hellhörig werden und eine weiterführende Abklärung in die Wege leiten. Zunächst gilt es, differentialdiagnostisch vorzugehen und andere mögliche Gründe für die Wachstumsstörung auszuschließen, beispielsweise eine Schilddrüsenproblematik, eine Nierenerkrankung oder bei Mädchen ein Turner-Syndrom. In puncto GHD-Abklärung kommt etwa der GHRH(„G rowth Hormone-Releasing Hormone“)-Arginin-Test zum Einsatz, er gibt Aufschluss über die Fähigkeit des Kindes, Somatotropin auszuschütten. Überwiegend liegt ein angeborener idiopathischer (und isolierter) Hyposomatotropismus vor. Seltener sind Ursachen wie anlagebedingte Fehlbildungen der Hypophyse, genetische Veränderungen, die einen GH-Mangel bewirken, oder erworbene Formen – beispielsweise aufgrund von Verletzungen oder Tumoren der Hypophyse.
Psychosoziale Aspekte bedenken
Besteht ein gesicherter Wachstumshormonmangel, ist ab dem vierten Lebensjahr die Gabe von rekombinantem
menschlichem Wachstumshormon („recombinant human Growth Hormone“ – rhGH) indiziert. Die Behandlung wird bis zum Verschluss der Wachstumsfugen fortgeführt, üblicherweise also bis zum Einsetzen der Pubertät. In der Regel erfolgt die Diagnose im Kleinkind- bis Vorschulalter – was wichtig ist, denn so bleibt genügend Zeit bis zum Pubertätsbeginn und der Wachstumsrückstand kann meist völlig aufgeholt werden. Die erforderliche Langzeittherapie kann jedoch herausfordernd sein, denn das tägliche Spritzen des Hormons ist vor allem bei kleinen Kindern für die Eltern bzw. betreuenden Personen oft schwierig. Mittlerweile wurde auch ein langwirksames Wachstumshormon zugelassen, bei dem eine Verabreichung einmal wöchentlich ausreicht.
Empfehlenswert ist jedenfalls eine umfassende Beratung und Betreuung in einem spezialisierten Zentrum für pädiatrische Endokrinologie – nicht zuletzt vor dem Hintergrund, dass eine persistierende Mikrosomie bei Betroffenen mit erheblichen psychosozialen Belastungen verbunden sein kann, etwa das Selbstbewusstsein, die Berufs- und Partnerwahl betreffend. Eine gute Versorgung kann vielen Kindern den Weg zu einem möglichst normalen „G roßwerden“ ebnen.
Anna Schuster, BSc
Das 1 x wöchentliche Wachstumshormon im praktischen Fertigpen*
* Zur Behandlung von Kindern und Jugendlichen ab einem Alter von 3 Jahren mit Wachstumsstörung durch unzureichende Ausschüttung von Wachstumshormon
Morbus Fabry: Betrifft Männer und Frauen!
Die Frühsymptome der seltenen lysosomalen Speichererkrankung können uncharakteristisch sein. Bei Leitsymptomen wie akralem Schmerz, Hitzeempfindlichkeit durch vermindertes Schwitzen, gastrointestinalen Beschwerden sowie einer familiären Häufung von Herz- und Nierenerkrankungen sollte Morbus Fabry differenzialdiagnostisch berücksichtigt werden.1
Morbus Fabry zählt mit einer Inzidenz von etwa 1 : 40.000 zu den Seltenen Erkrankungen. 1 Pathogenetisch liegt der lysosomalen Speichererkrankung eine Mutation im α-Galaktosidase-A-Gen (GLA-Gen) zugrunde, das auf dem XChromosom lokalisiert ist. Betroffen können sowohl Männer als auch Frauen sein (Details: siehe Kasten 1).
Ablagerungen von Speichermaterial
Die verringerte oder fehlende Funktion des Enzyms α-Galaktosidase A führt zu einer fortschreitenden Akkumulation des charakteristischen Speichermaterials Globotriaosylceramid (Gb3; in der deacetylierten Form: Lyso-Gb3) in verschiedenen Zelltypen. Es kommt zu progressiven funktionalen Defiziten, Insuffizienzen und Fibrosierungen in den betroffenen Organen. Das klinische Erscheinungsbild ist entsprechend variabel, typische Manifestationen betreffen jedoch Niere, Herz, Blutgefäße sowie zentrales und peripheres Nervensystem.1
Heterogene Symptomatik
Die Frühsymptome eines Morbus Fabry können uncharakteristisch sein, was die Diagnose erschwert und verzögert. Richtungsweisend können folgende Symptome sein, vor allem wenn diese in Kombination auftreten:
• unklare, brennende Schmerzen in Händen und Füßen,
• gastrointestinale Beschwerden,
• Hautveränderungen in Form von Angiokeratomen,
• Tinnitus oder zunehmender Hörverlust,
• linksventrikuläre Hypertrophie ohne Hypertonie,
KASTEN 1
Beide Geschlechter können erkranken! Morbus Fabry: X-chromosomaler
• Mikroalbuminurie/Proteinurie,
• charakteristische Trübung der Hornhaut durch winzige feine Ablagerungen („Cornea verticillata“), die im Rahmen einer augenärztlichen Spaltlampenuntersuchung sichtbar werden.
Erbgang
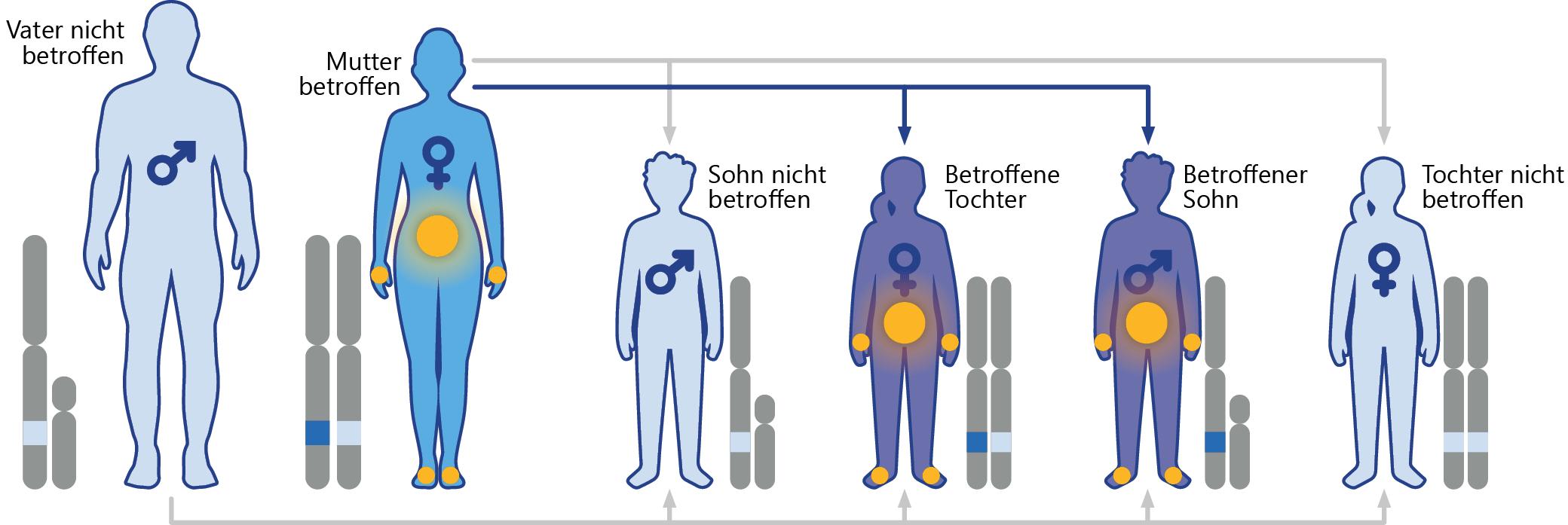
Abb.1.: X-chromosomaler Erbgang bei Morbus Fabry (Beispiel: Mutter betroffen) 4
Für die seltene multisystemische lysosomale Speichererkrankung Morbus Fabry ist eine X-chromosomal vererbte Mutation im Alpha-Galaktosidase-A-Gen (GLA-Gen) ursächlich.4 Folgende Erbgänge sind möglich:
Die Mutter ist Trägerin einer Fabry-Mutation Frauen mit einer GLA-Mutation können diese über das X-Chromosom sowohl an ihre Söhne als auch an ihre Töchter weitergeben, wobei die Wahrscheinlichkeit, dass heterozygote Frauen ein krankes Chromosom vererben, bei 50 % liegt (siehe Abb. 1).4
Der Vater ist Träger einer Fabry-Mutation
Männer mit einer GLA-Mutation im X-Chromosom geben dieses immer an ihre Töchter weiter, denn ein Mädchen erhält das einzige X-Chromosom ihres Vaters, dazu eines der beiden X-Chromosomen der Mutter. Söhne erhalten von ihren Vätern immer das nicht betroffene Y-Chromosom und können daher nicht erkranken, sofern die Mutter ohne Fabry-Erkrankung ist.4
Beide Elternteile sind Träger einer Fabry-Mutation
Tragen sowohl die Mutter als auch der Vater eine Fabry-Mutation im X-Chromosom, haben 50 % der Söhne auch eine Fabry-Mutation und alle Töchter eine Fabry-Mutation in einem oder beiden X-Chromosomen.4 Wegen der zufälligen Inaktivierung eines der beiden X-Chromosomen in jeder Zelle während der frühen Embryogenese können auch Frauen eine typische Morbus-FabrySymptomatik aufweisen.1
Auf Grund des X-chromosomalen Erbgangs sind Männer jedoch meist früher und schwerer von Fabry-spezifischen Symptomen betroffen als Frauen. Das klinische Erscheinungsbild bei Frauen ist deutlich variabler: Klinisch unauffällige Verlaufsformen sind ebenso möglich wie ähnlich schwere Krankheitsausprägungen wie bei Männern.4
Die genannten Beschwerden können bereits im Kindes- und Jugendalter auftreten, insbesondere neuropathische und gastrointestinale Beschwerden sogar schon bei Kleinkindern.1,2
Auch Hinweise auf eine positive Familienanamnese (z. B. familiäre Häufung von Herz- oder Nierenerkrankungen oder von Schlaganfällen in jüngeren Lebensjahren bei nahen Angehörigen) erfordern eine weitere diagnostische Abklärung.1,2
Trockenblutkarte bei Erstverdacht
Bei Verdacht auf Morbus Fabry können Hausärztinnen und -ärzte kostenlos eine Trockenblutkarte anfordern (Kontakt: www.archimedlife.com ). Damit wird die Aktivität der α-Galaktosidase A gemessen. Ist diese pathologisch erniedrigt, steht die Diagnose bei Männern fest. Bei Frauen ist jedoch eine Analyse des GLAGens notwendig, da die Enzymaktivität bei Frauen auch im Normalbereich liegen kann. 3 Auch bei Männern mit erniedrigter Enzymaktivität schließt sich standardmäßig ebenfalls eine genetische Untersuchung an, um die Mutation im GLA-Gen zu bestimmen. Die Mutation kann Einfluss auf die zur Verfügung stehenden Therapieoptionen haben. Eine Einbeziehung des Krankheitsmarkers Lyso-Gb3 kann die Diagnostik unterstützen. 3
Genetik am Fabry-Zentrum
Bei bestehendem oder bestätigtem Verdacht sollte die weitere Diagnostik zusammen mit einem Fabry-Zentrum durchgeführt werden. Inzwischen sind mehr als 1.000 Fabry-Mutationen bekannt, wobei viele Patienten sogenannte „private“ –also familientypische – Mutationen aufweisen. 1,3,4 Ein Überblick über die gängigen diagnostischen Kriterien für Morbus Fabry wird in Tabelle 1 gegeben.5,6 Falls die Diagnose Morbus Fabry gestellt wurde, sollte unter Berücksichtigung des X-chromosomalen Erbganges (siehe Kasten 1) ein Familienscreening durchgeführt werden. 3,4 Eine frühzeitige Diagnose kann entscheidend für die weitere Prognose sein: Denn die verfügbaren spezifischen Therapieoptionen bei Morbus Fabry können umso effektiver wirken, je früher mit der Behandlung dieser chronisch progredienten Erkrankung begonnen wird (Kasten 2). 7
KASTEN 2
Morbus Fabry: Behandlungsmöglichkeiten
Für die kausale Therapie des Morbus Fabry stehen zwei etablierte Therapieansätze zur Verfügung: die Enzymersatztherapie als Infusion, die alle 14 Tage verabreicht wird, und für Patienten mit geeigneten Mutationen auch eine pharmakologische Chaperon-Therapie in Form einer Kapsel zur oralen Einnahme jeden zweiten Tag.1
1 Lenders M, Brand E, Drugs 2021; 81:635–645.
2 Laney DA et al., Genet Med 2015; 17(5):323–330.
3 Ortiz A et al., Mol Genet Metab 2018; 123: 416–27.
C: Familienmitglied mit bestätigter Fabry-Diagnose und derselben GLA-Mutation
Tabelle 1: Adaptiert nach: Biegstraaten M, 2015 5; und Smid BE, 2014 6
Web-Tipps für Morbus Fabry
https://morbus-fabry.eu: Selbsthilfegruppe Morbus Fabry auch für Patienten und Angehörige www.orpha.net: Portal für Seltene Krankheiten und Orphan Drugs www.erknet.org:
Informationen zu Seltenen Erkrankungen wie Morbus Fabry aus nephrologischer Sicht (European Rare Kidney Disease Reference Network)
Die Progression verlangsamen
Ein Klettverschlussgeräusch könnte ein Indiz für eine idiopathische Lungenfibrose sein
Bislang gilt die idiopathische pulmonale Fibrose (IPF) als eine seltene unheilbare Erkrankung. Die gemeldeten Inzidenzen reichen von 0,2 bis 94 pro 100.000 Einwohner pro Jahr.1 Sowohl die therapeutischen Möglichkeiten, als auch jene eine frühzeitige Diagnose zu stellen sind begrenzt. Sie stellen aber einen wesentlichen Faktor dar, um die Lebenszeit von betroffenen Personen zu verlängern. Die mediane Überlebenszeit ohne Behandlung beträgt zwei bis fünf Jahre ab dem Zeitpunkt der Diagnose.1 Durchschnittlich vergehen jedoch vom Beginn der Beschwerden bis zur Diagnose 1,5 Jahre. Zuletzt hat die Awareness für die Erkrankung – auch dank der mittlerweile verfügbaren Behandlungsoptionen –zugenommen. Aktuell wird ein neuer Therapieansatz zur Erhaltung der Lungenfunktion erforscht.2
Lungenauskultation könnte erste Hinweise geben
Die idiopathische Lungenfibrose ist durch eine progredient verlaufende chronische
Fibrose der Lunge charakterisiert. „Diese geht mit einer großen Symptomlast und einer sehr schlechten Prognose einher“, weiß Dr. Mathis Hochrainer von der Abteilung für Innere Medizin und Pneumologie an der Klinik Floridsdorf in Wien. „Auch wenn wir die Ursache der IPF nicht kennen, sind Risikofaktoren bekannt: Betroffen sind vor allem Männer über 60 Jahre mit Raucheranamnese“, so der Experte. Häufig werden die Patienten falsch diagnostiziert – ein Umstand, der unter anderem auf die unspezifische Symptomatik zurückzuführen ist. Menschen mit IPF stellen sich meist mit einem trockenen Husten und einer Belastungsdyspnoe vor. Das typische Fibroseknistern ist bei der Auskultation der Lunge vor allem in den unteren hinteren Lungenabschnitten hörbar.1 Dieses ähnelt dem Geräusch eines sich öffnenden Klettverschlusses. Die pulmonale Auskultation kann im niedergelassenen Bereich für die Frühdiagnose von größter Bedeutung sein. Ebenso können Trommelschlägelfinger und Uhrglasnägel als Folgen einer chronischen Hypoxämie beobachtet werden.
Eine Untersuchung der Lungenfunktion ergibt in der Regel eine restriktive Ventilationsstörung und eine verminderte Diffusionskapazität.
Ausschlussdiagnose
Zu einer Vernarbung der Lungen können neben der IPF auch sekundäre Lungenfibrosen führen, welche auf Autoimmunerkrankungen, medikamentös-toxische Reaktionen, häusliche oder berufliche Umwelteinflüsse, Strahlenschäden sowie Infektionen zurückzuführen sind. Bei Verdacht auf eine idiopathische Lungenfibrose gilt es daher, zunächst bekannte Ursachen einer interstitiellen Lungenerkrankung (ILD) auszuschließen. Wenn diese nicht in Frage kommen, erfolgt im nächsten Schritt eine multidisziplinäre Falldiskussion im Rahmen eines interdisziplinären ILD-Boards mit Spezialisten aus Pneumologie, Radiologie und Pathologie. Diese stellt den Goldstandard in der Diagnostik der IPF dar. Auf eine Biopsie kann verzichtet werden, wenn ein typisches UIP-Muster („usual interstitial pneumo-
nia“) in der hochauflösenden Computertomographie (HRCT) der Lunge vorliegt. Ein typisches UIP-Muster ist durch basal betonte retikuläre Veränderungen, Traktionsbronchiektasen und honigwabenartige Zysten (Honeycombing) charakterisiert. Handelt es sich hingegen um ein wahrscheinliches UIP-Muster, wird anhand der klinischen Vortestwahrscheinlichkeit individuell entschieden, ob bzw. welche Form der Biopsie notwendig ist.
EXPERTE:
psychosoziale Kompetenz von Hausärztinnen und Hausärzten notwendig. Sie fungieren einerseits in vielen Fällen als erste Ansprechpersonen, andererseits übernehmen sie eine koordinierende Rolle – auch bei der Behandlung von Komorbiditäten.
en Therapieansatz bieten. Sie weisen neben einer antientzündlichen auch eine antifibrotische Wirkung auf. In einer Phase-2-Studie wurde bereits gezeigt, dass der PDE4B-Inhibitor BI 1015550 die Lungenfunktion von Patientinnen und Patienten mit IPF stabilisieren kann.2 Mag.a Ines Pamminger, BA
Bisher stehen die beiden Antifibrotika Pirfenidon und Nintedanib für die Behandlung zur Verfügung. Sie können den fibrotischen Prozess zwar verlangsamen, aber nicht stoppen. Aus diesem Grund sollte die medikamentöse Therapie möglichst schnell nach Diagnosestellung eingeleitet werden, betont Dr. Hochrainer. „Wichtige Bestandteile der Behandlung von Patientinnen und Patienten mit IPF sind außerdem die symptomatische Therapie von Husten und Dyspnoe, die etwaige Verordnung von Langzeitsauerstoff und die palliative Therapie“, ergänzt der Facharzt. Psychologische Hilfe kann die Krankheitsverarbeitung verbessern. Um Patientinnen und Patienten mit dieser stark einschränkenden Erkrankung zu behandeln, ist außerdem eine ausgeprägte
Zusätzlich zu den bisher verfügbaren antifibrotischen Medikamenten könnten in Zukunft Substanzen, die das Enzym Phosphodiesterase Typ 4 hemmen, einen möglichen neu-
Symptomatik
DAS WICHTIGSTE IN KÜRZE
Referenzen:
1 Idiopathic pulmonary fibrosis, Stand: Juli 2020, orpha.net (abgerufen am 24.2.23).
2 Richeldi L et al., N Engl J Med. 2022 Jun 9;386(23):2178-2187. doi: 10.1056/NEJMoa2201737.
Belastungsdyspnoe, trockener Husten und Abgeschlagenheit zählen zu den typischen Symptomen. Klinisch findet sich in der Auskultation der Lunge in vielen Fällen eine Sklerophonie. Die chronische Hypoxämie kann Uhrglasnägel und Trommelschlägelfinger verursachen.
(Späte) Diagnose
Es handelt sich um eine Ausschlussdiagnose. Oftmals verzögert sich die Diagnosestellung aufgrund der unspezifischen Symptomatik und der potenziell unauffälligen Thoraxröntgen-Befunde in der Frühphase der Erkrankung. Durchschnittlich vergehen ab Beschwerdebeginn 1,5 Jahre, bis eine IPF erkannt wird.
Therapie
Das Behandlungsziel ist es, den Verlust an Lungenfunktion zu bremsen und somit das Überleben zu verlängern. Die Therapie mit Antifibrotika sollte daher unmittelbar nach Diagnosestellung initiiert werden. Für die optimale Betreuung von Menschen mit IPF ist die Zusammenarbeit zwischen dem niedergelassenen Bereich und ILD-Spezialambulanzen in Krankenhäusern essenziell.
Wissenswertes für Patient:innen
Betroffene finden hilfreiche Informationen auf der Seite der Interessenvertretung: lungenfibroseforum.at
Beim Kurzdarmsyndrom ist Teamwork gefragt
Das Therapieregime wird in Abhängigkeit von der intestinalen Adaptation angepasst – es erfordert diätologische, medikamentöse und chirurgische Maßnahmen
Beim Kurzdarmsyndrom handelt es sich um eine seltene Entität – man geht von einer Prävalenz von 34 Betroffenen pro einer Million Einwohner aus. Bei dieser Form des Darmversagens ist der Dünndarm so verkürzt, dass seine absorptive Funktion reduziert ist – zum Beispiel infolge mesenterialer Durchblutungsstörungen, wiederholter Resektionen eines komplizierten Morbus Crohn oder einer Strahlenenteritis. Folgen wie eine eingeschränkte Nährstoffresorption, vermehrte intestinale Flüssigkeitsverluste sowie osmotische oder sekretorische Diarrhoe machen aus dem Kurzdarmsyndrom ein komplexes Krankheitsbild, welches ein interdisziplinäres Therapiemanagement erfordert. Im Wesentlichen zielt die Behandlung auf eine Gewichtsstabilisierung, eine bedarfsgerechte Energie- und Nährstoffversorgung sowie auf eine weitgehende Symptomfreiheit ab.
Anatomie des
Restdarms entscheidend
Eine Darmresektion kann aufgrund angeborener Anomalien oder erworbener Ursachen notwendig werden – in Frage kommen vaskuläre und entzündliche Erkrankungen sowie Traumen oder Tumoren. Um mögliche klinische Konsequenzen besser abschätzen zu können, ist eine genaue Kenntnis der Art des operativen Eingriffs erforderlich: Der intraoperative Situs und die Länge der verbliebenen Darmabschnitte sollten möglichst gemessen und dokumentiert werden. „Das metabolisch-nutritive Defizit bei Patientinnen und Patienten ist höchst individuell“, erklärt Dr. Felix Harpain von der Abteilung für Allgemeinchirurgie am AKH Wien. In Hinblick auf den Schweregrad der Symptomatik hat sich der Erhalt von Kolon und Ileozäkalklappe als günstig erwiesen, wohingegen die Resektion des
Iliums als ungünstig einzustufen ist. „Die verbleibende Dünndarmlänge ist ganz entscheidend für das Outcome“, bestätigt Dr. Harpain. Diese sei Prädiktor für die Notwendigkeit einer parenteralen Ernährungs- und Flüssigkeitstherapie.
Phasengerechte Ernährung
Nach einer Darmresektion werden drei Stadien der Adaptation durchlaufen. Die erste Phase ist durch Hypersekretion und einen intestinalen Flüssigkeits- sowie Elektrolytverlust charakterisiert. Im Zuge von Adaptionsvorgängen sind eine Vertiefung der Krypten und eine Verlängerung der Villi zu beobachten, wodurch sich die Resorptionsfläche des verbliebenen Darms vergrößert und sich konsekutiv auch die Resorptionsleistung verbessert. In Hinblick auf den Krankheitsverlauf ist eine phasengerechte Ernährung unabdingbar. „Unmittelbar nach der Operation benöti-
Zugelassen für Patienten ab dem Alter von 1 Jahr mit Kurzdarmsyndrom (KDS) nach einer Phase der intestinalen Adaption.1
Freiheit ist möglich:
• Reduktion des parenteralen Ernährungsvolumens1
•Reduktion der Infusionsdauer 1
• Zunahme der enteralen Kalorienzufuhr 1
1 Revestive® Fachinformation Stand Mai 2022
C-APROM/AT/REV/0035, Juni 2022
Fachkurzinformation siehe Seite 1 18
www.takeda.at
Führt zu einer Zunahme der Darmzottenhöhe und Darmkryptentiefe
gen Betroffene in der Regel eine totale parenterale Ernährung“, sagt Elisabeth Hütterer, Diätologin an der Medizinischen Universität Wien. „Ziel ist jedoch, diese ehebaldigst wieder stufenweise auszuschleichen.“
In der Adaptionsphase wird begleitend zur parenteralen Ernährung eine Sonden- oder Trinknahrung verabreicht. Zu beachten ist, dass große Trinkmengen zu einer vermehrten Dünndarmund Magensekretion und damit auch zu einer gehäuften Ausscheidung führen. Die Diätologin erachtet es daher für sinnvoll, kleine Schlucke isotonischer Lösungen zu trinken. „Die Evaluation der Flüssigkeitszufuhr und -ausscheidung über 24 Stunden bildet die Grundlage für die Beurteilung der Flüssigkeitshomöostase“, betont Diätologin Hütterer und empfiehlt, dahingehend Protokoll zu führen. Entscheidend seien eine ausreichende Urinmenge von mehr als einem Liter pro Tag und ein stabiles Körpergewicht.
Bis zu zwei Jahre nach dem Eingriff ist das Maximum der Anpassungsfähigkeit des Restdarms erreicht. Je nach individueller Verträglichkeit kann der Kostaufbau nach dem Prinzip der Leichten Vollkost gesteigert und bedarfsweise durch eine enterale oder parenterale Ernährung sowie durch eine adaptierte Trinknahrung ergänzt werden. Mikronährstoffe und spezifische Verluste müssen jedoch selbst bei vollständiger oraler Autonomie lebenslang substituiert werden.
Medikamentöse und chirurgische Behandlungsoptionen
Um die Notwendigkeit einer Substitution zu erkennen und eine solche zu steuern, sollten die Serumwerte betreffend Elektrolyte, Nierenretentionsparameter, Albumin und Gesamteiweiß sowie Vitamine und Spurenelemente auch im Verlauf zwei bis viermal jährlich bestimmt werden. „Mängel treten vor allem bei den
ANLAUFSTELLEN FÜR BETROFFENE
Die Plattform ced-kompass.at bietet – in Kooperation mit der ÖMCCV (Österreichische Morbus Crohn und Colitis ulcerosa Vereinigung) – umfangreiche Services wie z. B. Infomaterial, Podcasts, Videos und auch eine telefonische Helpline an.
Die Patientenorganisation „Die Chronischen Experten“ (chronisch.at) hat sich speziell auf KDS-Betroffene fokussiert. Bei Bedarf gibt sie auch Ratschläge durch spezialisierte Fachärzt:innen bzw. Diätolog:innen.
Als Ansprechpartnerin von der Klinischen Abteilung für Gastroenterologie und Hepatologie am AKH Wien können Ärzt:innen Priv.-Doz.in Dr.in Stefanie Dabsch kontaktieren: stefanie.dabsch@meduniwien.ac.at
Das Kurzdarmsyndrom für Patient:innen erklärt:
Link: meinmed.at/gesundheit/kds-behandlung/2700
EXPERT:INNEN-TEAM:
Dr. Felix Harpain Abteilung für Allgemeinchirurgie, AKH Wien
Priv.-Doz.in Dr.in Stefanie Dabsch Klinische Abteilung für Gastroenterologie und Hepatologie, MedUni Wien
Elisabeth Hütterer Diätologin, MedUni Wien
fettlöslichen Vitaminen A, E, D und K auf“, weiß Priv.-Doz.in Dr.in Stefanie Dabsch von der Klinischen Abteilung für Gastroenterologie und Hepatologie an der Medizinischen Universität Wien. Das Augenmerk gelte es außerdem auf die ausreichende Versorgung mit Folsäure, Eisen, Selen und Zink zu richten. Eine Kontrolle der Knochendichte sollte im Abstand von zwölf Monaten vorgenommen werden.
Eine zusätzliche medikamentöse Therapie mit Loperamid oder Tinctura opii kann die intestinale Motilität hemmen, wodurch sich die Verweildauer des Speisebreis und die Zeit des Kontakts von Darmmukosa, Verdauungsenzymen und Chymus verlängern. Insbesondere während der postoperativen Periode und zu Beginn der oralen Ernährung tritt bei vielen Patientinnen und Patienten eine gesteigerte Magensäuresekretion auf, die eine Diarrhö verstärken und die Nährstoffresorption durch Inaktivierung der Pankreaslipase sowie Dekonjugation der Gallensalze verschlechtern kann. Protonenpumpeninhibitoren wirken dem entgegen. „Pankreasenzyme können mit dem Ziel einer besseren Fettabsorption substituiert werden“, so die Fachärztin für Innere Medizin. Bei Patienten nach Ileozäkalklappenresektion kann es zu einer aszendierenden bakteriellen Überwucherung des Dünndarms kommen, die eine antibiotische Behandlung indiziert. Durch die Therapie mit Teduglutid, einem Analogon des Peptidhormons Glucagon-like Peptid 2 (GLP-2), kann der Bedarf der Patienten an parenteraler Ernährung verringert werden. Das Hormon GLP-2 wird in enteroendokrinen L-Zellen des Ileums und proximalen Kolons gemeinsam mit GLP-1 aus einem Vorläuferprotein gebildet. „Teduglutid führt zu einem Zottenwachstum und damit zu einer größeren Darmoberfläche, es erhöht den enteralen Blutfluss und verbessert die Aufnahme von Nährstoffen“, erklärt Doz.in Dabsch. Die Lebensqualität der Betroffenen könne dadurch verbessert werden.
Auf chirurgischer Ebene ist es in manchen Fällen möglich, die Resorptionskapazität durch Wiederherstellung der Kontinuität distaler ausgeschalteter Darmanteile zu optimieren. In diesem Kontext ebenfalls von Bedeutung sind der „Verschluss von Fisteln, das Aufheben von blinden Schlingen und eine Infektsanierung im Abdomen“, nennt Dr. Harpain operative Optionen. „Die bei komplikationsträchtigen Krankheitsverläufen mögliche Dünndarmtransplantation wird derzeit nur bei einer Minderheit der Patientinnen und Patienten und in wenigen Zentren durchgeführt – in Österreich gar nicht“, so der Chirurg. Mag.a Sylvia Neubauer
Die Diagnose von Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) erfolgt in vielen Fällen verzögert oder wird gänzlich übersehen.
ACHTEN SIE AUF DIE HINWEISE:
HFpEF* bei Patient*innen, die typischerweise über 60 Jahre alt sind2
HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients typically over 60 1-3
HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients typically over 60 1-3
HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients typically over 60 1-3
INTOLERANZ
HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients typically over 60 1-3
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients typically over 60 1-3
gegenüber Herzinsuffizienzbehandlung wie z.B.:
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
ACE-Hemmer oder Beta Blocker3
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
I NTOLERANCE to standard heart failure therapies, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers 7-9
DISKREPANZ
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
zwischen Niedervoltage und erhöhter linksventrikulärer Wanddicke3
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
DISCORDANCE between QRS voltage on electrocardiography (ECG) and left ventricular (LV) wall thickness seen on echocardiography 10,11
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
DIAGNOSE
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
eines Karpaltunnelsyndroms oder einer Lumbalstenose1,3
Diagnosis of CARPAL TUNNEL SYNDROME or LUMBAR SPINAL STENOSIS8,14,16-22
AUTONOMIC NERVOUS SYSTEM dysfunction, including gastrointestinal complaints or unexplained weight loss 2,22,27,28 HEART FAILURE WITH PRESERVED EJECTION FRACTION in patients
AUTONOMIC NERVOUS SYSTEM dysfunction, including gastrointestinal complaints or unexplained weight loss 2,22,27,28
NERVENSYSTEM
AUTONOMIC NERVOUS SYSTEM dysfunction, including gastrointestinal complaints or unexplained weight loss 2,22,27,28
AUTONOMIC NERVOUS SYSTEM dysfunction, including gastrointestinal complaints or unexplained weight loss 2,22,27,28
AUTONOMIC NERVOUS SYSTEM dysfunction, including gastrointestinal complaints or unexplained weight loss 2,22,27,28
Dysfunktion des autonomen Nervensystems einschließlich gastrointestinaler Beschwerden und unerklärbarem Gewichtsverlust2
– hier erfahren Sie mehr über
Fehldiagnosen bei ATTR-Amyloidose vermeiden
Ein Expert:innenkonsens – gerichtet an Allgemeinmediziner:innen
Amyloidose, eine Gruppe von Seltenen Erkrankungen, ist auf die Ablagerung von fehlgefalteten Proteinen zurückzuführen. Diese Amyloidablagerungen zerstören die physiologische Gewebestruktur und können lebensbedrohliche Organdysfunktionen verursachen. Die Therapiemöglichkeiten orientieren sich am Subtyp der Erkrankung – dementsprechend essenziell ist die exakte und zeitnahe Diagnose bzw. Zuweisung durch Allgemeinmedizinerinnen und -mediziner an spezialisierte Zentren. Hilfreiche Empfehlungen hierfür gibt der Expertenkonsens von Gertz et al., 20201
Formen und Subtypen
Grundsätzlich werden zwei Hauptformen der systemischen Amyloidose unterschieden – AL-Amyloidose und ATTR-Amyloidose. Bei einer AL-Amyloidose produzieren krankhafte Plasmazellen im Knochenmark strukturell veränderte Leichtketten. AL-Amyloidose ist eine akut
lebensbedrohliche Erkrankung und erfordert eine rasche onkologische Abklärung. Die Transthyretin-Amyloidose (ATTRAmyloidose) wiederum kann sich in einer Polyneuropathie (ATTR-PN) oder einer Kardiomyopathie (ATTR-CM) manifestieren – Mischformen treten jedoch häufig auf. In beiden Fällen kommt es zur Ablagerung von Transthyretin(TTR)-Fibrillen, also fehlgefaltetem Transthyretin, im Gewebe verschiedener Organe. ATTR-Amyloidose kann einerseits durch eine Mutation des TTR-Gens verursacht werden (hereditäre TransthyretinAmyloidose = mATTR), andererseits durch idiopathische Veränderungen von Transthyretin – diese Form wird als Wildtyp bezeichnet (wtATTR). Besteht der Verdacht auf eine ATTR-Amyloidose, werden jedenfalls Gentests empfohlen, welche die Diagnose unterstützen. Ist das Ergebnis positiv, können sich Familienmitglieder für eine genetische Beratung entscheiden und sich selbst testen lassen.
Das Bewusstsein schärfen
Lange Zeit wurde die ATTR-Amyloidose als nicht behandelbar angesehen. Mittlerweile stehen jedoch krankheitsmodifizierende Therapien zur Verfügung. So zielt die Arbeit von Gertz et al. darauf ab, das allgemeine Bewusstsein für die ATTR-Amyloidose zu schärfen und den Weg zur Diagnose zu verkürzen. Die Krankheitserkennung stellt nicht zuletzt aufgrund der klinischen Heterogenität eine Herausforderung dar. Aus der Anhäufung von TTR-Fibrillen resultiert eine multisystemische Dysfunktion, welche mit klinischen Manifestationen im Herz, im muskuloskelettalen System, sowie im peripheren und im vegetativen Nervensystem einhergeht. Zwar sind bei der ATTR-Amyloidose meist das Herz und das periphere Nervensystem betroffen,
INFO
Laut Positionspapier der DGK sollte an eine ATTR-CM bei deutlicher LV-Wandverdickung ohne hypertensive Herzerkrankung bzw. wenn einer der folgenden Punkte vorliegt, gedacht werden:
Alter von > 60 Jahren, Herzinsuffizienzsymptomatik und normal große Ventrikel
Niedervoltage bzw. Nachweis eines AV- oder Schenkel-Blockes im Ruhe-EKG
Nachweis eines Perikardergusses, einer interatrialen Verdickung, einer echoreichen Myokardtextur, einer RV-Wandverdickung bzw. einer Klappenverdickung oder eines „apical sparing“
Pathognomonische Makroglossie mit Einkerbungen im Bereich der Zungenwand
Periorbitale Purpura (typischerweise schon nach Bagatellverletzungen)
• gastrointestinale Symptome, z. B. frühe Sättigung, Übelkeit, Erbrechen und/oder veränderte Stuhlgewohnheiten
• chirurgische Korrektur eines Karpaltunnelsyndroms in der Anamnese und/oder (lumbale) Spinalkanalstenose
Echo und EKG kardiale MRT Knochenszintigraphie mit 99mTc-PYP/99mTc-DPD
Überweisung zur Kardiologie
Symptome peripherer Neuropathie
progressive sensible Polyneuropathie, autonome Dysfunktion (z. B. chronische Diarrhoe, Obstipation oder alternierend Diarrhoe und Obstipation, erektile Dysfunktion und orthostatische Hypotonie), Schmerzen in Händen oder Füßen und Gangstörung
andere systemische Zeichen:
• Gewichtsverlust unklarer Genese
• kardiale Symptome
• okuläre Manifestationen
• renale Anomalien
• chirurgische Korrektur eines Karpaltunnelsyndroms in der Anamnese und/oder (lumbale) Spinalkanalstenose
genetische Testung auf TTR-Mutation
Überweisung zur Neurologie und genetischen Beratung
Quelle: Adaptiert nach Gertz M et al., BMC Family Practice (2020) 21:198.
Abbildung: Diagnose-Algorithmus für Allgemeinmediziner:innen bei Verdacht auf ATTR-Amyloidose. Abkürzungen: Echo: Echokardiographie, EKG: Elektrokardiographie, MRT: Magnetresonanztomographie, 99mTc-PYP: 99mTechnetium-markiertes Pyrophosphat, 99mTc-DPD: 99mTechnetium-markierte 3,3-Diphosphono-1,2-Propanodicarbonsäure.
häufig leiden die Patientinnen und Patienten aber auch an gastrointestinalen und anderen systemischen Manifestationen. Daher wird die ATTR-Amyloidose oft als eine gängigere Erkrankung fehldiagnostiziert. Nach wie vor gilt der histologische Nachweis als Goldstandard für die Bestätigung einer Transthyretin-Amyloidose, jedoch gibt es mittlerweile viele Verfahren, welche dazu beitragen, eine frühzeitige und genaue Diagnose zu stellen. Insbesondere bei der Beurteilung von Patientinnen und Patienten mit ungeklärter peripherer Neuropathie sollte eine genetische Abklärung in Erwägung gezogen werden – so der Konsens.
Noch viel Potential
Durch wirksame krankheitsmodifizierende Mittel – zum Beispiel Vyndaqel® (Tafamidis) – für die Therapie von mATTR- oder ATTRwt-Kardiomyopathie und mATTRbedingter peripherer Neuropathie* hat sich die Bandbreite der Behandlungsoptionen inzwischen erweitert. Auch die Prävalenz der ATTR-Amyloidose ist höher als bisher
angenommen. So ergab eine Studie zu Obduktionen2, dass ATTRwt bei bis zu 25 % der über 80-jährigen Personen vorliegen kann. Eine separate Autopsie-Studie3 stellte fest, dass ATTRwt bei 5 % der Patientinnen und Patienten mit HFpEF als Ursache für die Herzinsuffizienz mit erhaltener Ejektionsfraktion in Frage kommt.
Bei der exakten Diagnosestellung der Transthyretin-Amyloidose gibt es somit noch großes Verbesserungspotential. Zu den häufigsten Fehldiagnosen zählen laut Expertenkonsens im Spektrum der Kardiologie neben einer nicht näher definierten HFpEF etwa die hypertensive Herzerkrankung, die hypertrophische Kardiomyopathie oder die unkomplizierte degenerative Aortenstenose. Im gastrointestinalen Bereich besteht die Gefahr einer Verwechslung u. a. mit dem Reizdarmsyndrom und bei neurologischer Beteiligung z. B. mit diabetischer Neuropathie oder amyotropher Lateralsklerose. In einer Befragung4 von Patientinnen und Patienten mit ATTR-Amyloidose wurde mATTR nur in
35 % der Fälle innerhalb von sechs Monaten erkannt, bei ATTRwt waren es 46 %. Viele der Betroffenen mussten mindestens fünf Ärztinnen oder Ärzte aufsuchen, um die exakte Diagnose zu erhalten.
Diagnose-Algorithmus
Aus diesen Gründen haben Expertinnen und Experten einen diagnostischen Algorithmus (siehe Abbildung) entwickelt, welcher auf Red Flags von ATTR-Amyloidose bei kardialer und neurologischer Beteiligung basiert. Dieser erleichtert der Hausärztin bzw. dem Hausarzt die Identifizierung klinisch verdächtiger Patientinnen und Patienten und folglich die zeitnahe Überweisung an ein multidisziplinäres spezialisiertes medizinisches Zentrum.
* Vyndaqel® 20 mg Zulassung bei symptomatischer Polyneuropathie im Stadium 1 bei Erwachsenen.
Quellen:
1 Gertz M et al., BMC Family Practice (2020) 21:198.
2 Tanskanen M et al., Ann Med. 2008;40(3):232-9.
3 Mohammed SF et al., JACC Heart Fail. 2014 Apr;2(2):113-22.
4 Lousada I et al., Orphanet J Rare Dis. 2015 Nov 2;10(Suppl 1):P22. Unterstützt von Pfizer Corporation Austria.
IL-5-Hemmung bei eosinophilen Erkrankungen
Nucala® (Mepolizumab): Zielgerichtete Therapien für EGPA- und HES-Betroffene
Eosinophile Granulomatose mit Polyangiitis (EGPA) und das HypereosinophilieSyndrom (HES) sind potentiell lebensbedrohliche und seltene Erkrankungen, die durch Gewebeentzündungen ausgelöst werden.1,2,3 Diese Entzündungen bringen eine Reihe von Symptomen mit sich, teilweise mit schwerem Verlauf. Mepolizumab ist die erste zugelassene zielgerichtete Behandlung für EGPA1,3,4 und die erste anti-IL-5-Biologika-Behandlung für Patientinnen und Patienten mit HES2,4,5 und CRSwNP6,4,5 in Europa. Das Arznei-
mittel ist als Zusatztherapie für Personen ab sechs Jahren mit schubförmig remittierender oder refraktärer EGPA und für Erwachsene mit unzureichend kontrolliertem HES ohne erkennbare nicht-hämatologische sekundäre Ursache zugelassen.4,5 EGPA ist durch eine Vaskulitis, eine weit verbreitete Entzündung der Wände kleiner Blutgefäße, gekennzeichnet. Die Krankheit kann mehrere Organsysteme betreffen und mit Symptomen wie Müdigkeit, Muskel- und Gelenkschmerzen und Gewichtsverlust einhergehen.1,3
Beim Hypereosinophilie-Syndrom können die Komplikationen von Fieber und Unwohlsein bis hin zu Atemwegs- und Herzproblemen reichen.1,2 Die Symptome können sich zunehmend verschlimmern und lebensbedrohlich sein.
Referenzen:
1 Pavord ID et al., Allergy. 2022 Mar;77(3):778-797.
2 Roufosse F et al., Allergy Clin Immunol. 2020 Dec;146(6):1397-1405.
Eine im Jahr 2000 eingeführte EU-Verordnung über Arzneimittel für seltene Erkrankungen wird derzeit überarbeitet. Die neuen Regelungen müssen so gestaltet sein, dass die Entwicklungsarbeit, etwa durch Anreize, weiterhin gefördert und nicht durch zu restriktive Vorgaben eingeschränkt wird – heißt es in einer Aussendung der Pharmig. Ebenso sollte
die Arbeit der Expertisezentren in das nationale Sozialversicherungssystem eingebettet werden, damit die dort erbrachten Leistungen auch entsprechend vergütet werden. Die Unternehmen, die sich in der Therapieentwicklung engagieren, sind doppelt gefordert: einerseits wegen des sehr hohen Risikos, dass Forschungsprojekte scheitern, andererseits wegen des limitierten Umsatzes aufgrund der geringen Anzahl an Betroffenen. „Um weiterhin und mit möglichst hohem Tempo wissenschaftliche Erkenntnisse in Therapien
zu verwandeln, sind daher umsichtige politische Entscheidungen, nachhaltige Investitionen und klare Förderrichtlinien notwendig. Denn nur, wenn es die Rahmenbedingungen zulassen, kann die Forschungsgeschwindigkeit erhöht werden, damit in Zukunft noch mehr Betroffene behandelt werden können“, so Mag. Alexander Herzog, Generalsekretär der Pharmig, anlässlich des Welttags der seltenen Erkrankungen.
Quelle: PHARMIG – Verband der pharmazeutischen Industrie Österreichs
Therapie seltener Tumorerkrankungen
OGNMB: Immenses Potenzial der Nuklearmedizin in der Präzisionsmedizin
Anlässlich des „Rare Disease Day 2023“ wies die Österreichische Gesellschaft für Nuklearmedizin und molekulare Bildgebung (OGNMB) auf das enorme Potenzial in der Präzisionsmedizin hin. Zu den seltenen Erkrankungen zählen unter anderem neuroendokrine Tumoren (NET). In Österreich sind etwa 4.500 Menschen von solchen Tumoren betroffen. „Da diese zu 70 Prozent asymptomatisch verlaufen und meistens sehr langsam wachsen, werden sie meist sehr spät entdeckt, sodass sich bereits Metastasen gebildet haben“, berichtete Ao. Univ.-Prof. Dr. Rainer W. Lipp, Onkologe und Nuklearmediziner an der Klinischen Abteilung für Onkologie an der
Med Uni Graz. Ein NET ist eine bösartige Neubildung aus neuroendokrinen Zellen, die überall im Körper vorkommen können. Daher gibt es unterschiedliche Arten von NET, zu denen im klassischen Sinne gastrointestinale und pankreatische sowie NET der Lunge gehören. Metastasen gelten trotz hervorragender Forschung noch immer als größte Gefahr einer Krebserkrankung. Mit einer Radionuklidtherapie können jedoch Tumoren wie Metastasen gezielt behandelt werden. Patientinnen und Patienten wird ein radioaktiver Stoff verabreicht, der sich im Tumor anreichert: Radioaktive Strahlung zerstört damit die Tumorzellen von innen heraus.
Diese Methode der Radionuklidtherapie bezeichnet man als Theranostik. Diese zählt zu den großen Errungenschaften der Nuklearmedizin. „Patientinnen und Patienten mit einem NET können mit dieser Therapie selbst in einem fortgeschrittenen Tumorstadium oftmals viele Jahre – mitunter Jahrzehnte – bei guter Lebensqualität leben. Die Betroffenen haben gelernt, mit Krebs als chronische Krankheit umzugehen“, so Univ.-Prof. Dr. Michael Gabriel, Vorstand des Instituts für Nuklearmedizin und Endokrinologie am Kepler Universitätsklinikum und Vorstandsmitglied der OGNMB.
Quelle: Österreichische Gesellschaft für Nuklearmedizin und molekulare Bildgebung
BESSERUNG, DIE BLEIBT.1,2
DAUERHAFT
EFFEKTIV #,1 12
Fachkurzinformation siehe Seite 18
DUPIXENT® zur Langzeitbehandlung der Atopischen Dermatitis*,1
Für alle Patienten ab 6 Jahren*
ÜBERZEUGEND PRAKTISCH +,1 MIT ÜBERLEGENER SICHERHEIT °,13 15
Allein aus Gründen der besseren Lesbarkeit wurde auf die gleichzeitige Verwendung geschlechtsspezifischer Sprachformen verzichtet. Sämtliche Personenbezeichnungen gelten aber selbstverständlich für alle Geschlechter. * DUPIXENT® wird angewendet zur Behandlung von mittelschwerer bis schwerer Atopischer Dermatitis (AD) bei Erwachsenen und Jugendlichen ab 12 Jahren und bei Kindern ab 6 Jahren mit schwerer AD, die für eine systemische Therapie in Betracht kommen. # Zeigte sich zum Beispiel in der CHRONOS-Studie im Behandlungsarm Dupilumab 300 mg Q2W + TCS durch ein anhaltendes, multidimensionales Ansprechen von Woche 16–52 der Hautläsionen (durchschnittliche %-Verbesserung im EASI-Score im Vergleich zu Baseline in Woche 16: 76,7 %; in Woche 52: 78,3 %), des Juckreizes (durchschnittliche %-Verbesserung im Peak-Pruritus-NRS-Score im Vergleich zu Baseline in Woche 16: 56,2 %; in Woche 52: 56,2 %) sowie der Lebensqualität (durchschnittliche Verbesserung im DLQI-Score im Vergleich zu Baseline in Woche 16: 9,7 Punkte; in Woche 52: 10,9 Punkte). + Begründet sich auf die Tatsache, dass vor der Therapie mit Dupilumab keine Voruntersuchungen und Laborkontrollen nötig und keine klinisch relevanten Wechselwirkungen zu beachten sind. Darüber hinaus kann Dupilumab unabhängig von Komorbiditäten und Risikofaktoren eingesetzt werden. ° Zeigte sich an der niedrigeren Rate behandlungsbedingter unerwünschter Ereignisse (TEAEs) unter Dupilumab als unter Upadacitinib bzw. Abrocitinib. In der Heads-Up-Studie lag die Rate an TEAEs mit begründeter Möglichkeit eines Arzneimittelbezugs im Dupilumab-Arm bei 35,5 % und im Upadacitinib-30-mg-Arm bei 44 %. In der JADE-DARE-Studie lag die Rate an TEAEs im Dupilumab-Arm bei 65 % verglichen mit 74 % im Abrocitinib-200-mg-Arm. Darüber hinaus liegen Langzeitsicherheitsdaten zu Dupilumab in der Behandlung der mittelschweren bis schweren AD über 4 Jahre vor. 1 DUPIXENT® Fachinformation, Stand Jänner 2023; 2 Blauvelt A et al. Lancet 2017; 389:2287–2303; 3 Silverberg JI et al. J Am Acad Dermatol 2020; 82:1328–1336; 4 Canonica GW et al. EAC 2020; Poster; 5 Cork MJ et al. J Dermatol Treat 2020; 31:606–614; 6 Paller AS et al. Am J Clin Dermatol 2020; 21:119–131; 7 Cork MJ et al. JDDG FV02/07; ISSN 1610-0379; 2021; 8 Paller AS et al. J Am Acad Dermatol 2020; 83:1282–1293; 9 Siegfried E et al. PA & NP Fall Clinical Dermatology Virtual Conference 2020; Poster; 10 Thaçi D et al. J Dermatolog Sci 2019; 94: 266–275; 11 Barbarot S et al. J Dermatolog Treat. 2022; 33(1):266–277; 12 Barbarot S et al. EADV 2020 (virtuell); Poster: P0237; 13 Blauvelt A et al. JAMA Dermatol 2021; 157:1047–1055; 14 Reich K et al. Lancet 2022; 400:273–282; 15 Beck LA et al. Am J Clin Dermatol 2022; 23:393–408.