

www.humani-corporis.com
















Organização do sistema linfo-hemático
Órgãos linfáticos primários:
- medula óssea; - timo.
Órgãos linfáticos secundários:
- baço;
- linfonodos (e vasos linfáticos);
- tonsilas (anel linfático faríngeo);
- nódulos linfáticos (tecido linfático associado a mucosas).

Medula óssea
Características morfofuncionais gerais:
- órgão amorfo, difuso, acomodado no interior dos ossos;
- rica vascularização (irrigação por ramos de aa. nutrícias, metafisiais, epifisiais e/ou periosteais e drenagem venosa para plexos periosteais);
- exibe lipossubstituição progressiva com a idade.


Medula óssea
Características morfofuncionais gerais:
- compõe-se de tecido linfo-hematopoético sustentado por fibras reticulares, com proporções variáveis de tecido adiposo (conforme idade); - produz as células que compõe os tecidos sanguíneo e linfático (linfócitos T terminam seu desenvolvimento no timo).



Aspecto microscópico da medula óssea em exame citológico de amostra obtida por punção aspirativa (mielograma; à direita, acima) e histológico de fragmento incisional (à direita, abaixo).
Medula óssea
Características morfofuncionais gerais Citologia


A medula óssea é composta por uma variedade de células maduras e imaturas que integram diferentes linhagens de desenvolvimento (1: precursor mieloide eritroide; 2: precursor mieloide granuloide; 3: megacariócito; 4: neutrófilo imaturo).
Medula óssea
Linfo-hematopoese normal

Doenças neoplásicas
Linfoma de Hodgkin:
- neoplasia maligna derivada de células da linhagem B maduras;
- tumor com características morfopatológicas únicas;
- representa 10% dos linfomas;
- em geral em torno dos 30 anos.


O patologista britânico Thomas Hodgkin, na década de 1830, descreveu a doença que carregaria seu nome.
Doenças neoplásicas
Linfoma de Hodgkin:
- linfonodomegalia cervical é a manifestação clínica mais comum; - sintomas sistêmicos inespecíficos (febre, sudorese noturna, perda ponderal) podem ser observados na doença mais avançada; - em geral bom prognóstico (sobrevida em 5 anos maior que 70%).

Nodulações cervicais podem ser secundárias ao linfoma de Hodgkin.
Doenças neoplásicas
Linfoma de Hodgkin:
- as células neoplásicas teriam origem a partir de linfócitos infectados mutantes (EBV tem sido implicado);
- citocinas e fatores de crescimento produzidos pelas células neoplásicas atraem vigorosamente macrofagócitos, leucócitos e fibroblastos, que compõem mais de 90% da massa tumoral;
- as células tumorais induzem as células recrutadas a produzir fatores que favorecem sua subsistência e proliferação.


Clássica célula de Reed-Sternberg, (aspecto de "olhos de coruja"), cercada por leucócitos.
Doenças neoplásicas
Linfoma de Hodgkin:
- acomete, em geral isoladamente, cadeias linfonodais axiais: cervicais (75% dos casos), mediastinais, para-aórticas ou axilares; - a disseminação costuma respeitar a organização anatômica das cadeias linfonodais (de uma cadeia para a subsequente);
- comprometimento esplênico em 20% dos casos, ao qual tipicamente se sucede o comprometimento hepático e medular;
- doença extralinfonodal primária é rara.


Extenso comprometimento mediastinal no linfoma de Hodgkin (à esquerda) e metástases hepáticas (à direita).
Doenças neoplásicas
Linfoma de Hodgkin:
- à microscopia, as volumosas células de Reed-Sternberg (e suas variantes mononucleadas, células de Hodgkin) permeiam maciço infiltrado inflamatório (cenário patognomônico);
- diversos subtipos histopatológicos podem ser distinguidos (esclerosante nodular é o mais comum).


Aspecto microscópico (panorâmico, à esquerda; em maior aumento, à direita) do linfoma de Hodgkin esclerosante nodular.
Doenças neoplásicas
Linfoma difuso de grandes células B:
- neoplasia maligna derivada de células da linhagem B maduras;
- linfoma "não Hodgkin" mais comum;
- mais frequente em torno dos 60 anos;
- doença de comportamento agressivo (sobrevida variável; pode haver cura em até 50% dos casos).
Doenças neoplásicas
Linfoma difuso de grandes células B: - etiopatogênese permanece em investigação; - EBV parece associado, mas seu potencial papel é incógnito; - mutações no gene BCL6 (controle da diferenciação celular e apoptose) têm sido implicadas.
Doenças neoplásicas
Linfoma difuso de grandes células B: - comprometimento pode ser linfonodal (mais frequente) ou extralinfonodal (é o linfoma extralinfonodal mais comum); - pode haver comprometimento de cadeias linfonodais em topografias diversas (não respeita a hierarquia anatômica);
- sítios extralinfonodais mais comuns são o tubo digestório, tonsilas e baço, seguidos pelo fígado, rins, tireoide, glândulas salivares e ossos.



Aspecto macroscópico do linfoma difuso de grandes células B comprometendo o intestino grosso (à esquerda), o baço (centro) e o rim (à direita).
Doenças neoplásicas
Linfoma difuso de grandes células B: - à microscopia, proliferação de células linfoides volumosas (até 5x o tamanho de um linfócito), com imunofenótipo compatível com a linhagem B (CD19, CD20, CD22).

Aspecto microscópico do linfoma difuso de grandes células B (seta maior: célula neoplásica; seta menor: macrofagócito; cabeça de seta: linfócito)
Doenças neoplásicas
Linfoma difuso de grandes células B:


À imuno-histoquímica, marcação positiva para CD20 (típico de linfócitos B, à esquerda) e negativa para CD3 (típico de linfócitos T, à direita) são característicos do linfoma difuso de grandes células B.
Doenças neoplásicas
Linfoma de Burkitt:
- neoplasia maligna derivada de células da linhagem B maduras; - maioria crianças e jovens; - altamente agressivo, constitui o tumor de crescimento mais rápido conhecido.
Doenças neoplásicas
Linfoma de Burkitt:
- etiopatogênese associada a translocações cromossômicas envolvendo o proto-oncogene MYC e, provavelmente, infecção pelo EBV; - geralmente sítios extralinfonodais, principalmente ossos da face e vísceras abdominais; - à microscopia, células linfocitoides densamente agrupadas permeadas por macrofagócitos de citoplasma amplo (padrão em "céu estrelado"), repletos de corpos apoptóticos; mitoses são frequentes.

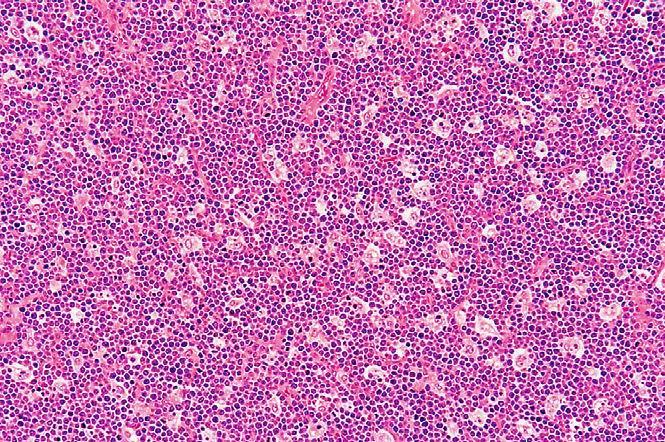

Doenças neoplásicas
Leucemia linfoblástica/Linfoma linfoblástico: - neoplasia maligna de células imaturas da linhagem linfoide (linfoblastos), em geral (85% dos casos) pré-linfócitos B; - se envolvimento prioritário da medula óssea, leucemia (maioria dos casos); se de tecidos linfáticos extra-medulares, linfoma; - câncer mais comum da infância; - sinônimos: leucemia linfoide/linfocítica aguda (LLA).
Doenças neoplásicas
Leucemia linfoblástica/Linfoma linfoblástico:
- em geral antes dos 5 anos, como leucemia B; na adolescência (menos frequente), como linfoma T do timo (picos fisiológicos);
- etiopatogênese pouco elucidada (diversas alterações gênicas que comprometem o desenvolvimento normal da linhagem linfoide já foram identificadas);
- associada (10% dos casos) à presença do cromossomo "Filadélfia".
Doenças neoplásicas
Leucemia linfoblástica/Linfoma linfoblástico:
- sintomas (na leucemia) associam-se à falência da hematopoese normal: fadiga (anemia), hemorragias (trombocitopenia), febre (neutropenia e infecções);
- pode haver dor óssea (infiltração periosteal), linfonodomegalias, esplenomegalia e hepatomegalia;
- curso clínico agudo (dias a poucas semanas);
- bom prognóstico na infância (˃ 2 anos), mas desfavorável no adulto.



Petéquias, gengivorragia e hematúria são possíveis manifestações da leucemia linfoblástica.
Doenças neoplásicas
Leucemia linfoblástica/Linfoma linfoblástico:
- à microscopia, proliferação de células com aspecto imaturo (blastoide) e imunofenótipo compatível com a linhagem linfoide (CD45+, TdT+); - distinção entre linhagens B e T por imunomarcadores.


Medula óssea repleta de células linfoblastoides (à esquerda), também presentes em esfregaço de sangue periférico (à direita).
Doenças neoplásicas
Leucemia linfocítica (crônica)/Linfoma linfocítico de pequenas células:
- neoplasia maligna de células maduras da linhagem B;
- se envolvimento prioritário da medula óssea, leucemia; se de tecidos linfáticos extra-medulares, linfoma;
- leucemia mais comum do adulto;
- maioria em torno dos 60 anos;
- sinônimos: leucemia linfoide/linfocítica crônica (LLC).
Doenças neoplásicas
Leucemia linfocítica (crônica)/Linfoma linfocítico de pequenas células: - etipatogênese permanece em investigação (mutações de genes implicados na proliferação de linfócitos B foram identificadas); - à microscopia, adensamentos de células linfocitoides vagamente organizadas em pseudofolículos (centros de proliferação).


Densas proliferações linfocitoides em medula óssea (à esquerda) e linfonodo (à direita), exibindo aspecto heterogêneo, com áreas empalidecidas (pseudofolículos) coalescidas de permeio.
Doenças neoplásicas
Leucemia linfocítica (crônica)/Linfoma linfocítico de pequenas células: - no sangue periférico, linfocitose com presença de "manchas de Gumprecht" (linfócitos esmagados, por sua fragilidade característica na doença).

Sangue periférico com linfocitose e manchas de Gumprecht (setas).
Doenças neoplásicas
Leucemia linfocítica (crônica)/Linfoma linfocítico de pequenas células: - a identificação do imunofenótipo (imuno-histoquímica ou citometria de fluxo) corrobora com o diagnóstico.


Imuno-histoquímica com característica expressão concomitante (aberrante) de CD5 (à esquerda) e CD20 (à direita).
Doenças neoplásicas
Leucemia linfocítica (crônica)/Linfoma linfocítico de pequenas células: - em geral assintomática ao diagnóstico (investigação motivada pela linfocitose, em média em torno de 50.000 células/ml), mas pode haver sintomas inespecíficos (fadiga, perda ponderal, inapetência); - prognóstico variável; sobrevida em média ultrapassa 5 anos.
Doenças neoplásicas
Leucemia mieloblástica:
- neoplasia maligna de células imaturas da linhagem mieloide (mieloblastos);
- múltiplos subtipos, conforme a sublinhagem comprometida e defeitos genéticos subjacentes;
- maioria após os 60 anos;
- tabagismo dobra do risco para a doença;
- sinônimos: leucemia mieloide/mielógena/mielocítica aguda (LMA).
Doenças neoplásicas
Leucemia mieloblástica:
- etiopatogênese pouco elucidada, associada a mutações em genes que regulam a diferenciação da linhagem mielocítica; - à microscopia, proliferação de células com aspecto imaturo (blastoide) e imunofenótipo compatível com células da linhagem mieloide (CD45+, TdT-).


Medula óssea repleta de células mieloblastoides (à esquerda), também presentes em esfregaço de sangue periférico (à direita).
Doenças neoplásicas
Leucemia mieloblástica:
- os sintomas relacionam-se à falência da hematopoese normal: fadiga (anemia), hemorragias (trombocitopenia), febre (neutropenia e infecções);
- curso clínico agudo (semanas);
- 60% dos casos tratados evoluem com remissão, porém, recorrências são frequentes.
Doenças neoplásicas
Leucemia mieloide (crônica):
- neoplasia maligna de células relativamente imaturas da medula, da linhagem mieloide (patamar mielocítico/metamielocítico);
- 15-20% das leucemias do adulto;
- maioria após os 40 anos;
- sinônimos: leucemia mielógena/mielocítica crônica (LMC).
Doenças neoplásicas
Leucemia mieloide (crônica):
- associada (90% dos casos) à presença do gene BCR-ABL, no cromossomo "Filadélfia" (produto de uma translocação entre os cromossomos 9 e 22); - o gene BCR-ABL associa-se a maior atividade mitogênica, insensibilidade a citocinas inibidoras do crescimento celular e resistência à apoptose.

Crom. 9
Crom. 22
Translocação
Crom. 9 alterado

Cariograma com presença do cromossomo Filadélfia.
Crom. 22 alterado (Filadélfia)
Doenças neoplásicas
Leucemia mieloide (crônica): - marcada hipercelularidade medular às custas de precursores granulocíticos é característica, com presença de micromegacariócitos; - no sangue periférico, importante leucocitose (com desvio mieloide à esquerda, mas não mais de 10% de blastos), que pode superar 100.000 células/ml.


Aspecto da medula óssea (à esquerda) e esfregaço de sangue periférico (à direita) na leucemia mieloide crônica.
Doenças neoplásicas
Leucemia mieloide (crônica):
- fase crônica marcada por hipercelularidade medular, mas sem aumento de células imaturas (blastos);
- fase blástica (após alguns anos) marcada por significativo aumento de células imaturas (blastos) na medula (assemelhandose à leucemia mieloblástica).
Doenças neoplásicas
Leucemia mieloide (crônica):
- em geral assintomática ao diagnóstico (investigação motivada pela leucocitose);
- evolução lenta (anos), com manifestações inespecíficas (fadiga, inapetência, perda ponderal);
- esplenomegalia importante é comum, associada ou não à hepatomegalia (hematopoese extramedular compensatória à substituição medular);
- pacientes jovens tendem a responder bem ao transplante (ao menos na fase crônica).
Doenças neoplásicas
Mieloma múltiplo:
- neoplasia maligna de plasmócitos (linhagem B);
- representa 15% das neoplasias hematopoéticas;
- maioria após os 60 anos;
- compromete a medula óssea em múltiplos sítios (5% das neoplasias plasmocitárias são solitárias: mielomas solitários ou plasmocitomas).
Doenças neoplásicas
Mieloma múltiplo:
- etiopatogênese parcialmente compreendida; - alterações envolvendo os genes MYC e TP53 podem ser identificadas (associam-se a pior prognóstico); - a proliferação das células neoplásicas é especialmente dependente de IL-6 (produzida pelo próprio tumor) e altos níveis séricos se associam a pior prognóstico.
Doenças neoplásicas
Mieloma múltiplo:
- sítios mais comuns: crânio, vértebras, costelas (ossos axiais), pelve, fêmur, clavícula, escápula; - neoplasia osteolítica (ativadora de osteoclastos, inibidora de osteoblastos), progride da cavidade medular aos tecidos periosteais; - a destruição óssea (hipercalcemia) promove dor e favorece fraturas patológicas.
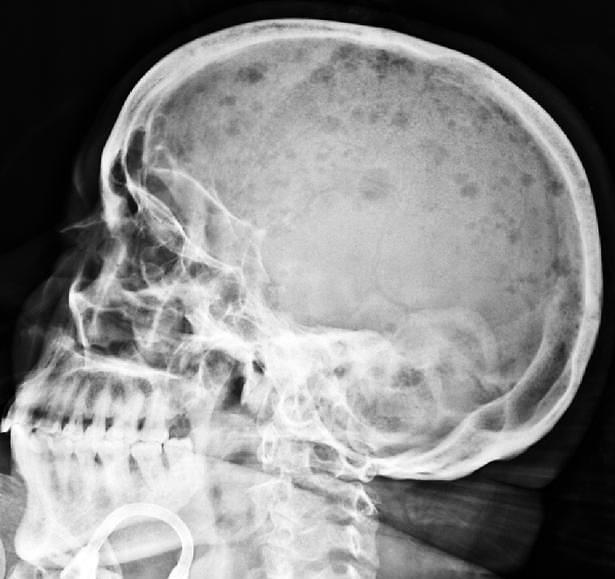


Lesões osteolíticas à radiografia (setas), ao exame macroscópico do crânio e à dissecção da coluna vertebral, no mieloma múltiplo.
Doenças neoplásicas
Mieloma múltiplo:
- hipercelularidade medular às custas de células plasmocitoides; - presença de elevadas concentrações séricas de imunoglobulina monoclonal (mais comumente IgG) é característica;
- uma queda na produção de imunoglobulinas normais leva à debilidade humoral e favorece infecções recorrentes;
- prognóstico em geral reservado (sobrevida menor que 5 anos).


Aspirado medular exibindo múltiplas células plasmocitoides (neoplásicas); à direita, eletroforese de proteínas plasmáticas sinalizando hipergamaglobulinemia monoclonal.
Referências bibliográficas
Gerais:
1. Kumar V, Abbas A, Aster J. Robbins & Cotran: Pathologic basis of disease. 9th ed. Elsevier; 2016.
2. O'Dowd G, Bell S, Wright S. Wheather's Pathology: A text, atlas and review of histopathology. 9th ed. Elsevier; 2020.
3. Klatt E. Robbins & Cotran: Atlas of pathology. 3rd ed. Elsevier; 2015.
4. Buja L, Krueger G. Netter's Illustrated human pathology. 2nd ed. Elsevier; 2011.
5. Goldblum J, et al. Rosai & Ackerman's Surgical pathology. 11th ed. Elsevier; 2017.
Específicas:
1. Jaffe ES, Arber DA, Campo E, et al. Hematopathology. 2nd ed. Elsevier; 2017.
2. Fletcher CDM. Diagnostic histopathology of tumors. 4th ed. Elsevier; 2013.
Referências bibliográficas
Complementares:
1. Sociedade Brasileira de Anatomia. Terminologia Anatômica. Manole; 2001.
2. Federative International Committee on Anatomical Terminology. Terminologia Histologica. LWW; 2008.
3. Ovalle WK, Nahirney PC. Netter: Bases da histologia. Elsevier; 2008.
4. Ross MH, Pawlina W. Histologia texto e atlas. 6ª ed. Guanabara Koogan; 2012.
5. Mills S. Histology for pathologists. 4th ed. LWW; 2012.
6. Alberts B, et al. Biologia molecular da célula. 6ª ed. Artmed; 2017.
7. Standring S. Gray's Anatomia: a base anatômica da prática clínica. 40ª ed. Elsevier; 2011.
8. Schunke M, Schulte E, Schumacher U. Prometheus: Atlas de anatomia. Guanabara Koogan; 2007.
9. Netter FH. Atlas de anatomia humana. 7ª ed. Elsevier; 2018.
10.Jansen JT, Netter FH. Netter's Clinical anatomy. 4nd ed. Elsevier; 2019.
11.Moore KL, Persaud PVN. Embriologia clínica. 10ª ed. Elsevier; 2016.
12.Boron W, Boulpaep E. Medical Physiology. 3rd ed. Elsevier; 2017.
13.Coico R, Sunshine G. Imunologia. 6ª ed. Guanabara Koogan; 2010.
14.Porto C, Porto A. Semiologia médica. 8ª ed. Guanabara Koogan; 2019.
15.Longo DL, Fauci AS, Kasper DL, et al. Medicina interna de Harrison. 18ª ed. Artmed; 2013.


