Kreislauf- und Stoffwechselerkrankungen in Klinik und Praxis
Jahrgang 36, Heft 3
August 2023
VERL AG PERFUSION
Offizielles Organ der Deutschen Gesellschaft für Arterioskleroseforschung
Current Contents/ Clinical Medicine
ORIGINALARBEIT
Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
FOREN
Forum Lipidstoffwechselstörungen:
Kardiovaskulärer Risikofaktor Lp(a) wird in Deutschland zu selten bestimmt
Forum Schlaganfall:
Virtual-Reality-Simulationstraining für die Schlaganfallbehandlung
Forum cardiologicum:
• Tragbare Defibrillatorweste kann kardiovaskuläre
Hochrisikopatienten vor dem plötzlichen Herztod schützen
• Herzinsuffizienz: CardioMEMS™-Sensor verbessert
Lebensqualität und senkt Risiko für Hospitalisierung
Forum nephroticum:
Empagliflozin erhält Zulassung zur Behandlung der chronischen Niereninsuffizienz bei Erwachsenen
REDAKTIONELLER TEIL
Mitteilungen, Kongressberichte
3 2023
ISSN 0935-0020
Besuchen Sie uns auf dem DGRh-KONGRESS / STAND 32 30.08. – 02.09.23 in Leipzig
Lupkynis®: Der erste zugelassene CNI für Lupus Nephritisa,b,1
Ermöglichen Sie Ihren Patient:innen mit Lupus Nephritis von einer Lupkynis®-Therapie mit MMF und niedrig dosierten Steroiden zu profitieren:a,c,1,2
Überlegenes komplettes renales Ansprechenc,1,2
• 41 % vs. 23 % nach 52 Wochen; p < 0,0001d
Doppelt so schnelle Reduktion der Proteinuriec,1,2
• 50 %ige UPCR-Reduktion: 29 vs. 63 Tage; p < 0,001e
• UPCR ≤ 0,5 mg/mg: 169 vs. 372 Tage; p < 0,001f
Rasche Steroidreduktion auf ≤ 2,5 mg/Tag in Woche 16 in beiden Behandlungsgruppen; Beibehaltung dieser niedrigen Dosis bis Woche 52c,g,1,2
Vergleichbares Sicherheitsprofil zwischen beiden Behandlungsgruppenc,2
Behandlung von erwachsenen Patient:innen mit aktiver Lupus Nephritis (LN) der Klassen III, IV oder V (einschließlich gemischter Klassen III/V und IV/V).1; b Lupkynis® ist der erste in der EU zugelassene CNI für Lupus Nephritis.1; c Lupkynis® (Voclosporin) vs. Placebo, jeweils kombiniert mit MMF und niedrig dosierten Steroiden1,2; d OR: 2,65 [95%-KI: 1,64;4,27], p < 0,00011,2; e HR: 2,05 [95%-KI: 1,62;2,60], p < 0,0011,2; HR: 2,02 [95%-KI: 1,51;2,70], p < 0,0011,2; g Reduktion der oralen Steroiddosis auf ≤ 2,5 mg/Tag bei > 80 % der Patient:innen aus beiden Behandlungsgruppen in Woche 16, Beibehaltung dieser niedrigen Dosis bis Woche 52 bei 75 % der Patient:innen in der Lupkynis®-Gruppe und 73 % der Patient:innen in der KontrollGruppe2
CNI: Calcineurininhibitor; HR: Hazard Ratio; KI: Konfidenzintervall; MMF: Mycophenolat-Mofetil; OR: Odds Ratio; UPCR: Protein/Kreatinin-Verhältnis im Urin.
Referenzen: 1. Fachinformation Lupkynis®, Stand Februar 2023. 2. Rovin BH et al. Lancet 2021; 397:2070–2080. Lupkynis® 7,9 mg Weichkapseln
Wirkstoff: Voclosporin. Zusammensetzung: Wirkstoff: Jede Weichkapsel enthält 7,9 mg Voclosporin; sonstige Bestandteile: Ethanol, Tocofersolan, Polysorbat 40, Mittelkettige Triglyceride, Gelatine, Sorbitol, Glycerol, gereinigtes Wasser, Titandioxid (E 171), Eisen(III)-oxid (E 172), Eisen(III)-hydroxid-oxid x H 20 (E 172), (3- sn -Phosphatidyl)cholin (Soja). Anwendungsgebiete: Lupkynis wird angewendet in Kombination mit Mycophenolat-Mofetil zur Behandlung von erwachsenen Patienten mit aktiver Lupus-Nephritis (LN) der Klassen III, IV oder V (einschl. gemischter Klassen III/V und IV/V). Gegenanzeigen: Überempfindlichkeit gegen Voclosporin oder einen der sonst. Bestandteile; gleichzeitige Anwendung von Voclosporin mit starken CYP3A4-Inhibitoren (z. B. Ketoconazol, Itraconazol, Clarithromycin).
Nebenwirkungen: Sehr häufig: Infektion der oberen Atemwege (auch virale und bakterielle Infektion der oberen Atemwege), Anämie, Kopfschmerzen, Hypertonie (auch Blutdruck erhöht, Blutdruck diastolisch erhöht), Husten, Diarrhoe, Abdominalschmerz (auch Schmerzen Oberbauch, abdominale Beschwerden), glomeruläre Filtrationsrate vermindert (auch Nierenfunktionsbeeinträchtigung, Kreatinin im Blut erhöht). Häufig: Grippe, Herpes zoster, Gastroenteritis, Harnwegsinfektion, Hyperkaliämie, Appetit vermindert, Krampfanfall, Tremor, Übelkeit, Zahnfleischhyperplasie (auch Gingivitis, Zahnfleischbluten/-hypertrophie/-schwellung), Dyspepsie, Alopezie, Hypertrichose (auch Hirsutismus), akute Nierenerkrankung und akute Nierenschädigung (auch Nierenfunktionsbeeinträchtigung).
Warnhinweise: Enthält Alkohol (Ethanol), Sorbitol und kann Spuren von (3-sn -Phosphatidyl)cholin (Soja) enthalten. Arzneimittel für Kinder unzugänglich aufbewahren. Pharmazeutischer Unternehmer: Otsuka Pharmaceutical Netherlands B.V., Herikerbergweg 292, 1101 CT, Amsterdam, Niederlande. Örtliche Vertretung in D: Otsuka Pharma GmbH, Europa-Allee 52, 60327 Frankfurt am Main. Stand: September 2022
Weitere Einzelh. u. Hinweise siehe Fach- u. Gebrauchsinformation. Verschreibungspflichtig! www.otsuka.de

DE-LUP-2300143
a Lupkynis® wird angewendet in Kombination mit Mycophenolat-Mofetil zur
EDITORIAL
What food is healthy?
Much is being written about the importance of diet on our health. Today, we know only too well what food is unhealthy. Yet, far less is known about what food might keep us fit and well. Therefore, it seems reasonable to ask whether there is a diet that is demonstrably healthy. A recent investigation attempted to answer this question.
This study by Mente et al. (Eur Heart J 2023;44:2560-2579) was aimed at developing a healthy diet score that is associated with health outcomes and is globally applicable. It used data from the Prospective Urban Rural Epidemiology (PURE) study and tried to replicate it in 5 independent studies on a total of 245,000 people from 80 countries.
A healthy diet score was developed on the basis of the data from 147,642 people from the general population, from 21 countries in the PURE study. The consistency of the associations of the score with events was examined in 5 large independent studies from 70 countries.
The healthy diet score was developed based on 6 foods each of which has been associated with a significantly lower risk of mortality (i.e. fruit, vegetables, nuts, legumes, fish, and dairy [mainly whole-fat]; range of scores, 0–6). The main outcome measures were all-cause mortality and major cardiovascular events (cardiovascular disease, CVD).
During a median follow-up of 9.3 years in PURE, compared with a diet score of ≤1 point, a diet score of ≥5 points was associated with a lower risk of:
• mortality (HR: 0.70; 95% CI: 0.63–0.77)
•
•
•
In 3 independent studies with vascular patients, similar results were found, with a higher diet score being associated with lower mortality (HR: 0.73; 0.66–0.81), CVD (HR: 0.79; 0.72–0.87), myocardial infarction (HR: 0.85; 0.71–0.99), and a non-statistically significant lower risk of stroke (HR: 0.87; 0.73–1.03). Additionally, in 2 case-control studies, a higher diet score was associated with lower first myocardial infarction (OR: 0.72; 0.65–0.80] and stroke (OR: 0.57; 0.50–0.65). A higher diet score was associated with a significantly lower risk of death or CVD in regions with lower than with higher gross national incomes (p for heterogeneity <0.0001). The PURE score showed slightly stronger associations with death or CVD than several other common diet scores (p < 0.001 for each comparison).
The authors rightly stress that their analyses have a number of limitations:
First, diet was self-reported and variations in reporting might lead to
Fruits and vegetables 4 – 5 servings daily
Legumes 3 – 4 servings weekly
Nuts 7 servings weekly
Fish 2 – 3 servings weekly
Dairy 14 servings weekly
Whole grain Moderate amounts (e.g. 1 serving daily) can be part of a healthy diet
Unprocessed meat Moderate amounts (e.g. 1 serving daily) can be part of a healthy diet
Table 1
random errors that could dilute real associations between diet scores and clinical outcomes. Therefore, the beneficial effects of a healthier diet may be larger than estimated.
Second, the researchers did not examine the role of individual types of fruits and vegetables as components in the diet score.
Third, in observational studies, the possibility of residual confounding from unquantified or imprecise measurement of covariates cannot be ruled out.
Fourth, the use of the median intake of each food component as a cut-off in the scoring scheme for each diet may not reflect the full range of consumption or provide a meaningful indicator of consumption associated with the disease.
Fifth, the level of intake to meet the cut-off threshold for each food group in the diet score may differ between countries.
Sixth, misclassification of exposures cannot be ruled out as repeat measures of diet were not available in all studies.
Seventh, one unique aspect of the study is the focus on only protective foods, i.e. a dietary pattern score that highlights what is missing from the food supply, especially in poorer world regions, but this does not negate the importance of limiting the consumption of harmful foods such as highly processed foods.
So, what should we, according to these findings, be looking for and how much of it should we consume?
Table 1 might answer these questions.
Edzard Ernst Emeritus Professor, University of Exeter

1 medium apple, banana, pear; 1 cup leafy vegs; 1/2 cup other vegs
1/2 cup beans or lentils
1 oz., tree nuts or peanuts
3 oz. cooked (pack of cards size)
1 cup milk or yogurt; 1 ½ oz. cheese
1 slice (40 g) bread; ½ medium (40 g) flatbread; ½ cup (75–120 g) cooked rice, barley, buckwheat, semolina, polenta, bulgur, or quinoa
3 oz. cooked red meat or poultry
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 3/2023 69
Prof. Dr. med. E. Ernst, Exeter, U.K.
CVD (HR: 0.82; 0.75–0.91)
myocardial infarction (HR: 0.86; 0.75–0.99)
stroke (HR 0.81; 0.71–0.93)
Offizielles Organ der
Deutschen Gesellschaft
Arterioskleroseforschung Current Contents/Clinical Medicine 80 Forum Lipidstoffwechselstörungen 84 Forum Schlaganfall 88, 91 Forum cardiologicum 93 Forum nephroticum 82, 87, Mitteilungen 94, 99 95 Kongressberichte 80 Forum lipid metabolism disorders 84, 87 Forum stroke 88, 91 Forum cardiologicum 93 Forum nephroticum 82, 87, Informations 94, 99 95 Congress reports Heft 3 August 2023 INHALT CONTENTS EDITORIAL 69 What food is healthy? E. Ernst ORIGINAL PAPER 72 Mavacamten – the first targeted therapy for hypertrophic obstructive cardiomyopathy B. Söllner EDITORIAL 69 Welche Nahrung ist gesund? E. Ernst ORIGINALARBEIT 72 Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie B. Söllner
für
®
Diabetes in Balance halten

Mit der richtigen Blutzuckereinstellung können Sie Ihre Patient*innen effektiv vor Folgeerkrankungen schützen.1–5
Wirkstoff: Insulin glargin. Zusammens.: 300 Einheiten Insulin glargin/ml (entsprechend 10,91 mg) SoloStar-Pen: Ein Pen enthält 1,5 ml Injektionslösung, entsprechend 450 Einheiten. DoubleStar-Pen: Ein Pen enthält 3 ml Injektionslösung, entsprechend 900 Einheiten. Sonst. Bestandt.: Zinkchlorid, Metacresol (Ph.Eur.), Glycerol, Salzsäure, Natriumhydroxid, Wasser für Injektionszwecke. Anw.-Geb.: Diabetes mellitus bei Erwachsenen, Jugendlichen u. Kindern ab 6 Jahren. Gegenanz.: Überempfindlichk. gegen d. Wirkstoff/sonstige Bestandt. Warnhinw. u. Vorsichtsm.: Arzneimittel für Kinder unzugänglich aufbewahren. Nur in diesem Pen anwenden, sonst kann schwere Überdosierung auftreten. Nur klare und farblose Lösungen verwenden. Nebenwirk.: Immunsyst.: Selten allerg. Reaktionen. Stoffwechsel/Ernährungsstör.: Sehr häufig Hypoglykämie. Nervensyst.: Sehr selten Geschmacksstör. Augen: Selten Sehstörungen, Retinopathie. Haut/Unterhautzellgeweb.: Häufig Lipohypertrophie, gelegentl. Lipoatrophie, nicht bekannt kutane Amyloidose. Skelettmusk./Bindegew./Knochen: Sehr selten Myalgie. Allg./Verabr.ort: Häufig Reakt. a. d. Einstichstelle, selten Ödeme. Verschreibungspflichtig. Sanofi-Aventis Deutschland GmbH D 65926 Frankfurt am Main, Deutschland. Stand: Juli 2020
Suliqua® 100 Einheiten/ml + 33 Mikrogramm/ml Injektionslösung im Fertigpen.
Wirkstoffe: Insulin glargin und Lixisenatid. Zusammens.: Insulin glargin 100 E/ml und Lixisenatid 33 µg/ml. Jeder Fertigpen enthält 300 Einheiten Insulin glargin und 100 Mikrogramm Lixisenatid in 3 ml Lösung. Sonst. Bestandt. m. bekannt. Wirkung: Metacresol 2,7 mg/ml. Sonst. Bestandt.: Glycerol 85 %, Methionin, Metacresol, Zinkchlorid, Salzsäure, Natriumhydroxid, Wasser für Injektionszw. Anw.geb.: Verbesserung der Blutzuckerkontrolle als Ergänzung zu Diät u. Bewegung in Kombination mit Metformin ± Natrium-Glucose-Cotransporter-2-(SGLT-2-)Inhibitoren bei erw. Pat. mit unzureichend kontrolliertem Diabetes mellitus Typ 2. Gegenanz.: Überempfindlichk. gegen die Wirkstoffe/sonstig. Bestandt. Warnhinw. u. Vorsichtsm.: Arzneimittel für Kinder unzugänglich aufbewahren. Nur klare und farblose Lösung verwenden. Nur in diesem Pen verwenden. Nebenwirk.: Infekt. u. parasitäre Erkr.: Gelegentlich: Nasopharyngitis, Infekt. der oberen Atemwege. Immunsyst.: Gelegentlich: Urtikaria. Stoffwechsel/ Ernährungsstör.: Sehr häufig: Hypoglykämie. Nervensyst.: Häufig: Schwindel. Gelegentlich: Kopfschm. Gastrointestinaltrakt Häufig: Übelkeit, Diarrhö, Erbrechen. Gelegentlich: Dyspepsie, Abd.schmerz. Selten: Verzögerte Magenentleerung. Leber- und Gallenerkr. Gelegentlich: Cholelithiasis, Cholezystitis. Haut/Unterhautzellgew.: Nicht bekannt: Kutane Amyloidose, Lipodystrophie. Allg./Verabr.ort: Häufig: Reaktionen a. d. Inj.stelle. Gelegentlich: Ermüdung. Verschreibungspflichtig Pharmazeutischer
Unternehmer: Sanofi Winthrop Industrie, 82 avenue Raspail, F-94250 Gentilly, Frankreich. Örtlicher Vertreter d. Zulassungsinhabers: Sanofi-Aventis Deutschland GmbH, D-65926 Frankfurt am Main. Stand: Mai 2023 Apidra® 100 Einheiten/ml Injektionslösung in einer Patrone · Apidra® 100 Einheiten/ml Injektionslösung in einer Durchstechflasche · Apidra® SoloStar® 100 Einheiten/ml Injektionslösung in einem Fertigpen
Wirkstoff: Insulinglulisin. Zusammens.: 1 ml enthält 100 Einheiten Insulinglulisin (entsprechend 3,49 mg). Sonstige Bestandteile: Metacresol, NaCl, Trometamol, Polysorbat 20, Salzsäure 36%, NaOH, Wasser für Injektionszwecke. Anw.-geb.: Zur Behandlung von Erwachsenen, Jugendlichen u. Kindern. ab 6 J. mit Diabetes mellitus, sofern die Behandlung mit Insulin erforderlich ist. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile, Hypoglykämie. Warnhinw. u. Vorsichtsmaßn.: Arzneimittel für Kinder unzugänglich aufbewahren. Nur klare u. farblose Lösung verwenden. Apidra® SoloStar: Nur Nadeln verwenden, die für SoloStar geeignet sind. Nebenwirkungen: Stoffwechsel, Ernähr.: Sehr häufig Hypoglykämie. Unbekannt Hyperglykämie (kann zu diabetischer Ketoazidose führen). Haut, Unterhautzellgew.: Häufig Reaktionen an der Injektionsstelle, lokale Überempfindlichkeitsreaktionen. Selten Lipodystrophie. Nicht bekannt kutane Amyloidose. Allgemein: Gelegentlich systemische Überempfindlichkeitsreaktionen. Verschreibungspflichtig. Sanofi-Aventis Deutschland GmbH, D-65926 Frankfurt am Main. Stand: Juli 2020 Insulin aspart Sanofi® 100 Einheiten/ml Injektionslösung in einer Durchstechflasche • Insulin aspart Sanofi® 100 Einheiten/ml Injektionslösung in einer Patrone • Insulin aspart Sanofi® 100 Einheiten/ml Injektionslösung im Fertigpen Wirkstoff: Insulin aspart. Zusammens.: 1 ml enthält 100 Einheiten (3,5 mg) Insulin aspart. Sonst. Bestandt.: Phenol, Metacresol (Ph.Eur.), Zinkchlorid, Polysorbat 20, Natriumchlorid, Salzsäure 36 % und Natriumhydroxid zur Einstellung des pH, Wasser für Injektionszwecke. Anw.-Geb: Zur Behandlung von Diabetes mellitus bei Erwachsenen, Jugendlichen und Kindern ab dem Alter von 1 Jahr. Gegenanz.: Überempfindlichk. gegen d. Wirkstoff/sonstige Bestandt. Nebenwirk.: Immunsyst.: Gelegentlich: Urtikaria, Exanthem, Hautausschlag; sehr selten: Anaphylaktische Reaktionen. Stoffwechsel/ Ernährungsstör.: Sehr häufig: Hypoglykämie. Nervensyst.: Selten: Periphere Neuropathie (schmerzhafte Neuropathie). Augen: Gelegentlich: Refraktionsanomalien, diabetische Retinopathie. Haut/Unterhautgeweb.: Gelegentlich: Lipodystrophie, nicht bekannt: kutane Amyloidose. Allg./Verabr.ort: Gelegentlich: Reakt. a. d. Injektionsstelle, Ödeme. Verschreibungspflichtig. Pharmazeutischer Unternehmer: Sanofi Winthrop Industrie, 82 avenue Raspail, 94250 Gentilly, Frankreich. Örtlicher Vertreter d. Zulassungsinhabers: Sanofi-Aventis Deutschland GmbH, D-65926 Frankfurt am Main. Stand: Dezember 2022
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.
Insulin lispro Sanofi® 100 Einheiten/ml Injektionslösung in einer Patrone · Insulin lispro Sanofi® 100 Einheiten/ml Injektionslösung in einer Durchstechflasche · Insulin lispro Sanofi® 100 Einheiten/ml Injektionslösung im Fertigpen
Wirkstoff: Insulin lispro. Zusammens.: 1 ml enthält 100 Einheiten (3,5 mg) Insulin lispro. Sonst. Bestandt.: m-Cresol, Glycerol, Dinatriumhydrogenphosphat 7 H2O, Zinkoxid, Wasser für Injektionszwecke, Salzsäure 36 % und Natriumhydroxid zur Einstellung des pH. Anw.-geb.: Zur Behandlung von Erwachsenen und Kindern mit Diabetes mellitus, die Insulin für die Aufrechterhaltung eines normalen Glucosehaushaltes benötigen. Ebenfalls angezeigt bei Ersteinstellung des Diabetes mellitus. Gegenanz.: Hypoglykämie, Überempfindlichkeit gegen den Wirkstoff oder einen sonstigen Bestandteil. Warnh. u. Vorsichtsmaßn.: Arzneimittel für Kinder unzugänglich aufbewahren. Nebenw.: Stoffwechsel/Ernährungsstör.: Häufigste Nebenwirkung jeder Insulinbehandlung ist Hypoglykämie. Schwere Hypoglykämien können zu Bewusstlosigkeit und im Extremfall zum Tod führen. Immunsyst.: Häufig: lokale allerg. Reaktionen; Selten: systemische Allergie. Haut (Unterhautzellgeweb.): Gelegentlich: Lipodystrophie; Nicht bekannt: kutane Amyloidose. Allg./Verabr.ort: Nicht bekannt: Ödeme. Verschreibungspflichtig. Pharmazeutischer Unternehmer: Sanofi Winthrop Industrie, 82 avenue Raspail, 94250 Gentilly, Frankreich. Örtlicher Vertreter d. Zulassungsinhabers: Sanofi-Aventis Deutschland GmbH, D-65926 Frankfurt am Main. Stand: Dezember 2022
© Sanofi 2023. Alle Rechte vorbehalten. Sanofi-Aventis Deutschland GmbH | Lützowstraße
MAT-DE-2301007-2.0-07/2023
| 10785
|
Insulin glargin (100 E/ml) und Lixisenatid 52 52 010 | www.sanofi.de
107
Berlin
Telefon 0800
1. Stratton IM et al. BMJ 2000 ; 321: 405–12; 2. Aiello LP et al. Diabetes Care 2014; 37(1): 17–23; 3. ADVANCE Collaborative Group. N Engl J Med 2008; 358: 2560–72;
4. Martin CL et al. Diabetes Care 2014; 37(1): 31–8; 5. Paul SK et al. Cardiovasc Diabetol 2015; 14: 100.
Toujeo® 300 Einheiten/ml SoloStar®, Injektionslösung in einem Fertigpen · Toujeo® 300 Einheiten/ml DoubleStar™, Injektionslösung in einem Fertigpen
Insuline aus dem Sanofi-Portfolio
PERFUSION 2023; 33: 72 – 78
Die hypertrophe Kardiomyopathie (HCM) ist die häufigste erbliche Herzmuskelerkrankung [1]. Charakteristisches Merkmal der chronischen, progredient verlaufenden Erkrankung ist eine Hypertrophie des linken Ventrikels mit einer Wanddicke von ≥15 mm, die nicht durch systemische oder andere kardiale Erkrankungen wie arterielle Hypertonie oder Klappenerkrankungen zu erklären ist [2, 3]. Schwerwiegende Folgen können Angina pectoris, Vorhofflimmern, Schlaganfall, Herzinsuffizienz und ein erhöhtes Mortalitätsrisiko sein. Die bisherigen Therapiemöglichkeiten sind limitiert, denn sie zielen nur auf die Linderung der Symptome ab. Um die Prognose dieser Patienten zu verbessern, bedarf es daher neuer Therapien, die mit kausalem Ansatz zielgerichtet in das Krankheitsgeschehen eingreifen.
hypertrophen obstruktiven Kardiomyopathie
Häufigste Ursache der HCM sind mutierte Sarkomerproteine
Die mit bis zu 60 % der Fälle weitaus häufigste Ursache der HCM sind Genmutationen, die für die kontraktilen Sarkomerproteine codieren
ÜBERSICHTSARBEIT
Mavacamten (CAMZYOS®) ist die bislang erste und einzige in medikamentöse Therapie zur Behandlung Erwachsener mit obstruktiver Kardiomyopathie (HOCM) der Klasse II-III der New Die HOCM ist eine chronisch progrediente Erkrankung, die zu führt und den Ausdehnungs- und Füllungsprozess des Herzens selektiver, allosterischer und reversibler kardialer Myosin-Inhibitor zugrundeliegenden Pathomechanismen der HOCM ab.2
 Brigitte Söllner, Erlangen
Brigitte Söllner, Erlangen
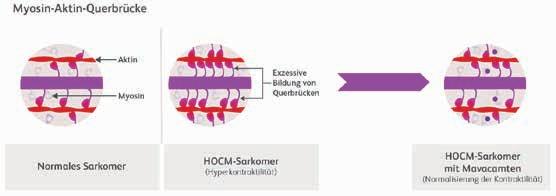
[2]*. Infolge dieser Mutationen werden vermehrt aktive Myosinköpfe gebildet, die zu einem Überschuss an Aktin-Myosin-Querbrücken führen (Abb. 1) [4]. Als Folge verändern sich die Herzstruktur und Herzfunktion. Es kommt zur Hyperkontraktilität mit nachfolgender Hypertrophie der Ventrikelwände, zu
einer verminderten Relaxation und erhöhten Steifigkeit des kardialen Gewebes sowie zu einem erhöhten Energiebedarf des Herzmuskels [5, 6]. Histopathologische Kennzeichen sind eine gestörte myokardiale Ordnung mit vergrößerten, ungeordneten Kardiomyozyten und vermehrter Fibrose [7].
Mechanistische Kennzeichen der HOCM sind die übermäßige Querbrücken und die Dysregulation des entspannten Zustands. Hyperkontraktilität, beeinträchtigter Relaxation, übermäßigem myokardialem Wandstress führen (Abb. 1).1 Mavacamten moduliert Myosinköpfchen, die einen energiebereitstellenden Zustand die Wahrscheinlichkeit für die Bildung von kraftentwickelnden während der Systole und der Enddiastole reduziert (bzw. bei bewirkt ferner eine Verschiebung des Gesamtmyosins hin zu rekrutierbaren, superrelaxierten Zustand.1 Bei HOCM-Patienten kardialen Myosins durch Mavacamten zu einer Normalisierung Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung Füllungsdrücke.1
Abbildung 1: Kennzeichnend für die HCM sind die übermäßige Bildung von MyosinAktin-Querbrücken und die Dysregulation des entspannten Zustands. Folgen sind eine kardiale Hyperkontraktilität, eine beeinträchtigte Relaxation, ein übermäßiger Energieverbrauch und myokardialer Wandstress [5, 6].
* Daneben existieren auch weitere genetische und nicht genetische Formen, die für etwa 5 – 10 % der HCM-Fälle verantwortlich sind. Dazu gehören u.a. metabolische und neuromuskuläre Erkrankungen wie Morbus Fabry, Friedreich-Ataxie oder Amyloidosen. Bei 20 – 25 % der Fälle sind die Ursachen der HCM unbekannt [2].
72 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
1
Abb. 1: Wirkmechanismus von Mavacamten.1, 2
* Die zentrale Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), Europäischen Union in Norwegen, Liechtenstein und Island zugelassen.
Nicht-obstruktive HCM (HNCM) Obstruktive HCM
Der LV-Muskel ist auf ≥15 mm verdickt, der Blutfluss aus dem linken Ventrikel wird wenig oder gar nicht behindert.
Der Peak-Gradient im linken Ausflusstrakt (LVOT) beträgt in Ruhe und unter Belastung <30 mmHg.
Tabelle
Die Wahrscheinlichkeit für die Vererbung des Gendefekts liegt bei etwa 50 % [2]. Betroffen sind vor allem Menschen im Alter von 40 – 60 Jahren, die HCM kann aber in jedem Alter auftreten. Die klinische Ausprägung der Erkrankung kann unterschiedlich verlaufen: Generell wird zwischen einer nicht obstruktiven (HNCM) und einer obstruktiven Form (HOCM) unterschieden (Tab. 1). Bei etwa 70 % der HCM-Patienten liegt eine obstruktive Form vor, die durch eine dynamische Obstruktion des linksventrikulären Ausflusstraktes (LVOT) von mindestens 30 mmHg in Ruhe oder nach Provokation definiert ist [8]. Sie entsteht, wenn aufgrund des hypertrophierten Ventrikelseptums das linksventrikuläre Volumen abnimmt, sich der LVOT verengt und infolgedessen der Blutfluss eingeschränkt wird [2]. Die dynamische Verengung kann mit einer systolischen anterioren Bewegung bereits in Ruhe oder unter Belastung einhergehen, bei der sich das vordere Mitralsegel gegen das Septum wölbt. Auch eine Abnahme der Vor- und Nachlast sowie eine Zunahme der Kontraktilität können die Obstruktion weiter verstärken, sodass in der Hälfte der Fälle ein Peak-LVOTGradient ≥30 mmHg erst unter körperlicher Belastung auftritt [7, 8].
(HOCM)
Die Hypertrophie des Ventrikelseptums verursacht eine Obstruktion im LVOT, die den Blutfluss aus dem linken Ventrikel verringert.
Der Peak-LVOT-Gradient in Ruhe oder unter Provokationsmanöver beträgt ≥30 mmHg.
Hoher therapeutischer Bedarf für eine kausale Therapie
Häufige Symptome einer HCM sind Angina pectoris, Dyspnoe, Palpitationen, Synkopen, Müdigkeit und körperliche Belastungsintoleranz. Über alle Altersgruppen hinweg ist das Morbiditäts- und Mortalitätsrisiko im Vergleich zur Allgemeinbevölkerung erhöht, vor allem bei Patienten mit einer obstruktiven HCM besteht ein hohes Risiko für Vorhofflimmern, Schlaganfall, Herzinsuffizienz und – wenn auch selten – für einen plötzlichen Herztod [7]. Die bisherigen Therapiemöglichkeiten sind limitiert, im Vordergrund steht die Linderung der Symptome. Die Therapiekaskade der aktuell geltenden Leitlinien sieht zunächst eine medikamentöse Therapie mit kardioselektiven, nicht vasodilatierenden Betablockern wie Metoprolol oder Bisoprolol vor. Sind diese kontraindiziert oder nicht wirksam, können Kalziumantagonisten wie Verapamil oder Diltiazem eingesetzt werden. Persistiert die Symptomatik, kommen interventionelle Verfahren zur Septumreduktion wie z.B. die chirurgische Myektomie oder die Alkoholseptumablation zum Einsatz. Dennoch ist die Prognose dieser Patienten schlecht [2], sodass ein hoher Bedarf an neuen zielgerichteten Therapien besteht.
Mavacamten – ein kardialer Myosin-Inhibitor, der gezielt in den Pathomechanismus der HOCM eingreift
Im Juni 2023 hat die Europäische Kommission Mavacamten (Camzyos®, 2,5 mg-, 5 mg-, 10 mg- und 15 mg-Kapseln) zur Behandlung der symptomatischen hypertrophen obstruktiven Kardiomyopathie der NYHA-Klasse II–III bei erwachsenen Patienten zugelassen. Damit ist Mavacamten der erste allosterische, reversible und selektive kardiale Myosin-Inhibitor, der ursächlich auf die zugrunde liegenden Pathomechanismen der HOCM abzielt und in allen Mitgliedsstaaten der Europäischen Union zugelassen ist [9].
Wie oben beschrieben, werden bei der HOCM infolge einer Genmutation übermäßig viele MyosinAktin-Querbrücken im Sarkomer gebildet, die die Muskelrelaxation beeinträchtigen. Mavacamten moduliert die Anzahl der Myosinköpfchen, die einen energiebereitstellenden Zustand erreichen können (Abb. 2). So wird die Wahrscheinlichkeit für die Bildung von kraftentwickelnden Querbrückenverbindungen während der Systole und der Enddiastole reduziert (bzw. bei HOCM normalisiert). Außerdem bewirkt Mavacamten eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber rekrutierbaren, superrelaxierten Zustand [9].
Bei HOCM-Patienten führt die Inhibition des kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungs-
73 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
1: Formen der hypertrophen Kardiomypathie (HCM) [2, 8].
Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition des durch Mavacamten zu einer Normalisierung der Kontraktilität, einer dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen
Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition des Mavacamten zu einer Normalisierung der Kontraktilität, einer LVOT-Obstruktion und einer Verbesserung der kardialen
Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition des durch Mavacamten zu einer Normalisierung der Kontraktilität, einer dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen
bewirkt ferner eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, rekrutierbaren, superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition
kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungsdrücke.1
der LVEF und der Mavacamten-
Wirkmechanismus von Mavacamten.1, 2
Wirkmechanismus von Mavacamten.1, 2
Abb. 1: Wirkmechanismus von Mavacamten.1, 2
Abbildung 2: Wirkmechanismus von Mavacamten (Camzyos®): Der selektive kardiale Myosin-Inhibitor reduziert die Anzahl der Myosinköpfchen im Sarkomer, die einen energiebereitstellenden Zustand erreichen können [9, 10].
Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich zur Norwegen, Liechtenstein und Island zugelassen.
Mavacamten.1, 2 den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich zur Liechtenstein und Island zugelassen.
Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. Mavacamten ist zusätzlich zur Norwegen, Liechtenstein und Island zugelassen.
drücke. Dies bestätigen die beiden zulassungsrelevanten Phase-IIIStudien EXPLORER-HCM [10, 11] und VALOR-HCM [12], in denen der Myosin-Inhibitor klinisch bedeutsame und anhaltende Verbesserungen kardialer Parameter der chronischen Herzmuskelerkrankung erzielen konnte.
* Die zentrale Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich Europäischen Union in Norwegen, Liechtenstein und Island zugelassen.
oder bei Provokation zum Zeitpunkt der Diagnose). Zusätzliches Einschlusskriterium war ein LVOTGradient von ≥30 mmHg nach Valsalva-Manöver während des Screenings [11].
kungen auf den LVOT-Spitzengradienten nach Belastung, die pVO2, die NYHA-Klasse sowie den Kansas City Cardiomyopathy Questionnaire-Clinical Summary Score (KCCQ-CSS**) und den Hypertrophic Cardiomyopathy Symptom Questionnaire Score (HCMSQ***) in Woche 30.
EXPLORER-HCM:
Mavacamten in allen Wirksamkeits-Endpunkten überlegen
Die zulassungsrelevante Studie
EXPLORER-HCM [10, 11] war eine randomisierte, placebokontrollierte, doppelblinde Phase-IIIStudie, in die insgesamt 251 Erwachsene mit symptomatischer (NYHA Klasse II oder III) obstruktiver HCM eingeschlossen wurden. Alle Teilnehmer hatten bei Studienbeginn eine messbare linksventrikuläre Ejektionsfraktion (LVEF) ≥55 % und einen Druckgradienten im linksventrikulärer Auswurftrakt (LVOT) von ≥50 mmHg (in Ruhe
Zu Studienbeginn gehörten etwa 73 % der randomisierten Patienten der NYHA-Klasse II und 27 % der NYHA-Klasse III an. Die mittlere LVEF betrug 74 %, und der mittlere LVOT-Gradient nach Valsalva-Manöver lag bei 73 mmHg. Der durchschnittliche Kansas City Cardiomyopathy Questionnaire 23-item
Clinical Summary Score (KCCQ23 CSS**) betrug 71. 92 % der Patienten erhielten eine Hintergrundtherapie mit Betablockern oder Kalziumantagonisten.
Die Studienteilnehmer wurden 1 : 1 auf Mavacamten 1 × täglich oral oder Placebo randomisiert. Startdosis waren 5 mg Mavacamten mit anschließender Dosistitration. Die Dosisanpassung erfolgte auf der Grundlage von echokardiographischen Messungen des LVOT-Gradienten nach Valsalva-Manöver,
Die Studie erreichte alle primären und sekundären Endpunkte mit statistischer Signifikanz.
Verbesserung der Leistungsfähigkeit und Symptomatik
Den primären Endpunkt erreichten in Woche 30 signifikant mehr Patienten unter Mavacamten im Vergleich zu Placebo: 37 % versus 17 % (Differenz +19,4 %; 95%-KI: 8,67–30,13; p = 0,0005; Abb. 3) [10, 11].
** Der KCCQ-23 CSS wird aus dem Total Symptoms Score (TSS) und dem Physical Limitations (PL) Score des KCCQ-23 abgeleitet. Der CSS reicht von 0 bis 100, wobei höhere Werte einen besseren Gesundheitszustand ausdrücken.
*** Der HCMSQ SoB Domain Score misst die Häufigkeit und den Schweregrad der Kurzatmigkeit. Der Bereichsscore reicht von 0 bis 18, wobei niedrigere Werte weniger Kurzatmigkeit symbolisieren.
74 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
1
1
1
1
Mavacamten (n = 123)
Placebo (n = 128)
Mavacamten (n = 123)
Placebo (n = 128)
37 %
17
%
HCM [12] ging der Frage nach, ob die Behandlung mit Mavacamten bei Erwachsenen mit symptomatischer HOCM, die für eine Septumreduktionstherapie (SRT) infrage kommen, die Notwendigkeit eines solchen invasiven Verfahrens mit chirurgischer Myektomie oder Alkoholseptumablation vermeiden kann.
% Primärer kombinierter Endpunkt
n = 45 n = 22
kombinierter Endpunkt
Abbildung 3: Ergebnisse der EXPLORER-HCM-Studie: Im Erreichen des primären Studienendpunkts bis Woche 30 war Mavacamten Placebo signifikant überlegen [10, 11]. # Patienten mit einem nicht bewertbaren primären Endpunkt wurden als Non-Responder betrachtet; die Ansprechraten wurden mit dem N-Wert als Nenner berechnet.
Verbesserung der LVOT-Gradienten und des Gesundheitszustands
Auch in den sekundären Endpunkten war Mavacamten in Woche 30 Placebo signifikant überlegen (Tab. 1) [10, 11]:
• Der durchschnittliche LVOTSpitzengradient nach Belastung verringerte sich in der Verumgruppe bis Woche 30 um –47 mmHg versus –10 mmHg in der Placebogruppe (95%-KI: –43 bis –28; p < 0,0001).
• Die Peak-VO2) erhöhte sich unter Mavacamten um 1,4 ml/kg/ min versus –0,05 ml/kg/min in der Placebogruppe (95%-KI: 0,6–2; p = 0,0006).
• 65 % der Patienten der Interventionsgruppe verbesserten sich um ≥1 NYHA-Klasse versus 31 % in
der Placebogruppe (95%-KI: 22–45; p < 0,0001).
• Auch auf die Lebensqualität wirkte sich die Behandlung mit Mavacamten positiv aus und verbesserte den KCCQ-CSS signifikant um 14 versus 4 Punkte (95%-KI: 5–13; p < 0,0001).
• Der Schweregrad der HCMSymptome gemäß HCMSQ SoB verbesserte sich um –2,8 Punkte in der Interventionsgruppe versus –0,9 Punkte in der Placebogruppe (95%-KI: –2,4 bis –1,2; p < 0,0001).
VALOR-HCM: Medikamentöse Therapie oder Intervention?
Die doppelblinde, placebokontrollierte Phase-III-Studie VALOR-
An der Studie nahmen Patienten mit symptomatischer obstruktiver HCM und NYHA-Klasse III–IV oder Klasse II mit Belastungssynkope oder Beinahe-Synkope teil, die die Leitlinienkriterien für eine SRT erfüllten und für einen invasiven Eingriff überwiesen worden waren.
Die Studiendauer war auf bis zu 138 Wochen ausgelegt, einschließlich einer 2-wöchigen ScreeningPhase.
Die 112 Studienteilnehmer wurden im Verhältnis 1 : 1 randomisiert und erhielten – zusätzlich zu Betablockern, Kalziumantagonisten und Disopyramid als Monotherapie oder in Kombination – Mavacamten (2,5 – 15 mg) oder Placebo über 16 Wochen. Eine Dosisanpassung erfolgte nach Bedarf in Woche 4, 8 und 12.
Der primäre kombinierte Endpunkt setzte sich zusammen aus der Anzahl von Patienten, die sich vor oder in Woche 16 für eine geplante SRT entschieden, und der Anzahl an Patienten, die in Woche 16 in der MavacamtenGruppe im Vergleich zur Placebogruppe weiterhin für die SRT infrage kamen (LVOT-Gradient ≥50 mmHg und NYHA-Klasse III–IV oder Klasse II mit Belastungssymptomen einer Synkope oder Beinahe-Synkope).
75 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
#
17
Δ 19,4 % (95 % -KI 8,67; 30,13) p = 0,0005
Entweder Zunahme Peak VO2 um ≥ 1,5 ml/kg/min mit Verbesserung ≥ 1 NYHA-Klasse oder Zunahme Peak VO2 ≥ 3,0 ml/kg pro min ohne Verschlechterung der NYHA-Klasse #
n = 45 n = 22 37 %
n
= 45 n = 22
Primärer
Entweder Zunahme Peak VO2 um ≥ 1,5 ml/kg/min mit Verbesserung ≥ 1 NYHA-Klasse oder Zunahme Peak VO2 ≥ 3,0 ml/kg pro min ohne Verschlechterung der NYHA-Klasse Δ 19,4 % (95 % -KI 8,67; 30,13) p =
37 %
0,0005 n
= 45 n = 22
37 %
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
Tabelle 1: Analyse der kombinierten primären und sekundären Endpunkte der EXPLORER-HCM-Studie [9, 10].
Notwendigkeit einer Septumreduktionstherapie signifikant verringert
Die VALOR-HCM-Studie erreichte alle primären und sekundären Endpunkte mit statistischer Signifikanz [13]. Nur 17,9 % der mit dem Myosin-Inhibitor behandelten Patienten im Vergleich zu 76,8 % der Patienten in der Placebogruppe
entschieden sich nach Woche 16 für eine SRT oder waren nach Leitlinien für eine SRT geeignet (95%-KI: 44,0–73,9; p < 0,0001; Abb. 4). Auch bei den sekundären Endpunkten zeigte sich der Nutzen der zielgerichteten Therapie mit Mavacamten (Tab. 2):
• Der LVOT-Spitzengradient unter Belastung (Valsalva-Manöver) konnte gegenüber Placebo
um –37,2 mmHg gesenkt werden (p < 0,0001).
• 62,5 % der Patienten verbesserten sich unter Mavacamten um mindestens eine NYHA-Klasse im Vergleich zu 21,4 % in der Placebogruppe.
• Das NTpro-BNP veränderte sich in Woche 16 gegenüber dem Ausgangswert um 0,35 ng/l versus 1,13 ng/l (p < 0,0001).
76 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Mavacamten n = 123 Placebo n = 128 Patienten, die in Woche 30 den primären Endpunkt erreichten 45 (37 %) 22 (17 %) Behandlungsunterschied (95%-KI) 19,4 (8,67–30,13) p-Wert 0,0005 Veränderung des LVOT-Spitzengradienten nach Belastung von Baseline bis Woche 30 [mmHg] n = 123 n = 128 Mittelwert (SD) −47 (40) −10 (30) Behandlungsunterschied (95%-KI) −35 (−43 bis −28) p-Wert <0,0001 Veränderung der pVO2 von Baseline bis Woche 30 [ml/kg/min] n = 123 n = 128 Mittelwert (SD) 1,4 (3) −0,05 (3) Behandlungsunterschied (95%-KI) 1,4 (0,6–2) p-Wert <0,0006 Patienten mit Verbesserung der NYHAKlasse um ≥1 in Woche 30 n = 123 n = 128 n (%) 80 (65 %) 40 (31 %) Behandlungsunterschied (95%-KI) 34 (22–45) p-Wert <0,0001 Veränderung des KCCQ-23 CSS von Baseline bis Woche 30 n = 92 n = 88 Mittelwert (SD) 14 (14) 4 (14) Behandlungsunterschied (95%-KI) 9 (5–13) p-Wert <0,0001 Baseline n = 99 n = 97 Mittelwert (SD) 71 (16) 71 (19) Veränderung des HCMSQ-Scores, SoBDomäne, von Baseline bis Woche 30 n = 85 n = 86 Mittelwert (SD) −2,8 (2,7) −0,9 (2,4) Behandlungsunterschied (95%-KI) −1,8 (−2,4 bis −1,2) p-Wert <0,0001 Baseline n = 108 n = 109 Mittelwert (SD) 4,9 (2,5) 4,5 (3,2)
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
p-Wert < 0,0001
Tabelle 2: Analyse der kombinierten primären und sekundären Endpunkte der VALOR-Studie [12, 13].
• Das kardiale Troponin I veränderte sich in Woche 16 gegenüber dem Ausgangswert um 0,50 ng/l versus 1,03 ng/l (p < 0,0001).
• Die Daten zur Lebensqualität und zur Sicherheit fielen ebenfalls positiv aus: Der KCCQ-23 CSS verbesserte sich zu Woche 16 gegenüber Placebo um 9,5 Punkte (p < 0,0001).
Weitere Reduktion der Notwendigkeit einer Septumreduktionstherapie nach 32 Wochen
Nach 32 Wochen erfüllten nur noch 6 von 56 Patienten (10,7 %) in der
77 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Mavacamten n = 56 Placebo n = 56 Patienten, die in Woche 16 den primären kombinierten Endpunkt erreichten 10 (17,9 %) 43 (76,8 %) Behandlungsunterschied (95%-KI) 58,9 (44,0–73,9) p-Wert <0,0001 Entscheidung des Patienten, mit der SRT fortzufahren 2 (3,6) 2 (3,6) Für eine SRT gemäß Leitlinien-Kriterien geeignet 8 (14,3) 39 (69,6) SRT-Status nicht auswertbar (wird als Erreichen des primären Endpunkts gewertet) 0 (0,0) 2 (3,6) Veränderung des LVOT-Spitzengradienten nach Belastung von Baseline bis Woche 16 [mmHg] n = 55 n = 53 Mittelwert (SD) −39,1 (36,5) −1,8 (28,8) Behandlungsunterschied (95%-KI) −37,2 (−48,1 bis −26,2) p-Wert <0,0001 Patienten mit Verbesserung der NYHA-Klasse um ≥1 in Woche 16 n = 55 n = 53 n (%) 35 (62,5 %) 12 (21,4 %) Behandlungsunterschied (95%-KI) 41,1 (24,5 % bis 57,7 %) p-Wert <0,0001 Veränderung des KCCQ-23 CSS von Baseline bis Woche 16 n = 55 n = 53 Mittelwert (SD) 10,4 (16,1) 1,8 (12,0) Behandlungsunterschied (95%-KI) 9,5 (4,9–14,0) p-Wert <0,0001 Baseline n = 56 n = 56 Mittelwert (SD) 69,5 (16,3) 65,6 (19,9) Veränderung des NT-proBNP von Baseline bis Woche 16 n = 55 n = 53 ng/l geometrisches Mittelwertverhältnis 0,35 1,13 Geometrisches Mittelwertverhältnis Mavacamten/Placebo (95 %-KI) 0,33
p-Wert
Veränderung des kardialen Troponin I von Baseline bis Woche 16 n = 55 n = 53 ng/l geometrisches Mittelwertverhältnis 0,50 1,03 Geometrisches Mittelwertverhältnis Mavacamten/Placebo (95%-KI)
(0,27–0,42)
<0,0001
0,53 (0,41–0,70)
ursprünglichen Mavacamten-Gruppe und 7 von 52 Patienten (13,5 %) in der Placebo-Crossover-Gruppe die Leitlinienkriterien für eine SRT oder entschieden sich für die Intervention (Abb. 4) [14]. Somit konnte Mavacamten den primären kombinierten Endpunkt von Woche 16 bis Woche 32 weiter reduzieren. Diese Ergebnisse ermöglichen es Patienten, invasive Eingriffe durch den Einsatz der neuen Therapieoption zu vermeiden.
während der Behandlung zu einer Verringerung der LVEF <50 %. Diese war bei allen Betroffen reversibel und diese schlossen die Studie unter Behandlung ab [9].
Literatur
1 Semsarian C, Ingles J, Maron MS et al. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015;65:1249-1254
Gepoolte Sicherheitsdaten aus beiden Studien belegen die gute Verträglichkeit von Mavacamten
Die am häufigsten gemeldeten unerwünschten Nebenwirkungen bei den 179 Patienten, die in den beiden zulassungsrelevanten Phase-IIIStudien mit Mavacamten behandelt wurden, waren Schwindel (17 %), Dyspnoe (12 %), systolische Dysfunktion (5 %) und Synkopen (5 %) [9]. In diesen klinischen Studien kam es bei 5 % (9/179) der Patienten in der Mavacamten-Gruppe
2 Elliott PM, Anastasakis A, Borger MA et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733-2779
3 Ommen SR, Mital S, Burke MA et al. 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/ American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020;142:e558-e631
4 Trivedi DV, Adhikari AS, Sarkar SS et al. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev 2018;10:27-48
5 Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch 2019; 471:701-717
6 Sequeira V, Bertero E, Maack C. Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett 2019;593:16161626
7 Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017;121:749770
8 Batzner A, Schäfers H-J, Borisov KV et al. Hypertrophic obstructive cardiomyopathy. Dtsch Arztebl Int 2019;116:4753
9 Fachinformation Camzyos® 2,5 mg, 5 mg, 10 mg, 15 mg, aktueller Stand
10 Olivotto I, Oreziak A, Barriales-Villa R et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebocontrolled, phase 3 trial. Lancet 2020; 396:759-769
11 Ho CY, Olivotto I, Jacoby D et al. Study design and rationale of EXPLORER-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy. Circ Heart Fail 2020;13:e006853
12 Desai MY, Wolski K, Owens A et al. Study design and rationale of VALORHCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am Heart J 2021;239:80-89
13 Desai MY, Owens A, Geske JB et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol 2022;80:95-108
14 Desai MY, Owens A, Geske JB et al. Dose-blinded myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy: outcomes through 32 weeks. Circulation 2023;147:850-863
Anschrift der Verfasserin: Brigitte
78 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
B. Söllner: Mavacamten – die erste zielgerichtete Therapie der hypertrophen obstruktiven Kardiomyopathie
Abbildung 4: Ergebnisse der VALOR-HCM-Studie bis Woche 16 bzw. 32 [13, 14].
Lektorin
10 91058 Erlangen E-Mail:brigitte.soellner@online.de 17,9% 10,7% 76,8% 13,5% 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Woche 16 Placebo zu Mavacamten Crossover-Gruppe Woche 32 Anteil Studienteilnehmer (%) Mavacamten
Söllner Medizinjournalistin und Wissenschaftliche
Lärchenweg
Placebo Studienteilnehmer,
die eine SRT hatten oder nach Leitlinie dafür geeignet waren
VALOR-HCM: Notwendigkeit einer Septumreduktionstherapie (SRT) mit Mavacamten signifikant verringert
Bühne frei für die zielgerichtete MyosinInhibition




bei HOCM.¹
CAMZYOS® verbessert die Lebensqualität * und Leistungsfähigkeit # von Patient : innen mit hypertropher obstruktiver Kardiomyopathie.²,³


www.hypertrophekardiomyopathie.de
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Fachinformation.
CAMZYOS 2,5 mg, 5 mg, 10 mg, 15 mg Hartkapseln.
Wirkstoff: Mavacamten; Zusammensetzung: 2,5 mg bzw. 5 mg bzw. 10 mg bzw. 15 mg
Mavacamten. Sonst. Bestandteile: Siliciumdioxid-Hydrat, Mannitol, Hypromellose, Croscarmellose-Natrium, Magnesiumstearat, Gelatine, Titandioxid, Eisenoxide, Schellack, Propylenglycol, Ammoniak-Lösung, Kaliumhydroxid. Anwendungsgebiete: CAMZYOS wird angewendet bei erwachsenen Patienten zur Behandlung der symptomatischen (NYHA Klasse II – III) hypertrophen obstruktiven Kardiomyopathie (HOCM). Gegenanzeigen: Überempfindlichkeit gg. den Wirkstoff o.e.d. sonst. Bestandteile. Schwangerschaft und bei gebärfähigen Frauen, die keine
zuverlässige Empfängnisverhütung anwenden; gleichzeitige Behandlung mit starken CYP3A4Inhibitoren (Patienten mit CYP2C19-Metabolisierer Phänotyp „langsam“ und nicht bestimmtem CYP2C19-Phänotyp); gleichzeitige Behandlung mit Kombination starker CYP2C19-Inhibitor und starker CYP3A4-Inhibitor. Nebenwirkungen: Sehr häufig: Schwindel; Dyspnoe. Häufig: Synkope; Systolische Dysfunktion. Weitere Hinweise: siehe Fachinformation. Für jede verschriebene Dosis ist eine einzelne Kapsel zu verwenden. Verschreibungspflichtig. Pharmazeutischer Unternehmer: Bristol-Myers Squibb Pharma EEIG, Plaza 254 - Blanchardstown

In der EXPLORER-HCM Studie mit n = 251 Patient:innen erreichten den kombinierten primären Endpunkt 37 % der Patient:innen im CAMZYOS®-Arm vs. 17 % im Placebo-Arm.² * gemessen mittels Kansas City Cardiomyopathy Questionnaire, KCCQ Overall Score mit einem Unterschied zwischen den Gruppen von +9,1 (95 % KI 5,5 bis 12,8; p<0,0001) zugunsten von CAMZYOS® in Woche 30.³ # gemessen im kombinierten primären Endpunkt bestehend aus Erfassung des Peak-VO₂ und der NYHA-Klasse mit einem Unterschied von 19,4 % (95 % KI 8,7 bis 30,1; p=0,0005) zugunsten von CAMZYOS®.² HOCM: hypertrophe obstruktive Kardiomyopathie.

Referenzen: 1 CAMZYOS® (Mavacamten) Fachinformation, aktueller Stand. 2 Olivotto I, Oreziak A, Barriales-Villa R, et al. Lancet. 2020;396(10253):759 – 769. 3 Spertus JA, Fine JT, Elliott P, et al. Lancet. 2021;397(10293):2467 – 2475.









Park 2 - Dublin 15, D15 T867, Irland. Stand: V01.
Corporate
3500-DE-2300009 06.2023
© 2023 MyoKardia, Inc., a Bristol-Myers Squibb Company Alle Rechte vorbehalten.
Für Schlaganfall-Patienten zählt jede Minute [1]. Routinierte Arbeitsabläufe tragen dazu bei, unnötig Zeit zu verschwenden. Das praktische Erlernen von Kompetenzen im Bereich Schlaganfall war bis dato nur theoretisch oder in einem Rollenspiel (simuliertem Training) im Krankenhaus möglich. Das band Räumlichkeiten und Ressourcen. Simulierte Trainings sind für angehende Fachärzte jedoch von entscheidender Bedeutung, um ihre Fähigkeiten zu trainieren und sie auf realistische Anwendungsfälle vorzubereiten [2]. Sie bieten den Assistenzärzten ein sicheres Umfeld, um Erfahrungen zu sammeln, routinierte Arbeitsabläufe zu erlernen und Feedback aus verschiedenen Perspektiven zu erhalten. Dass sich ein Simulationstraining positiv auf die Bewertung und Behandlung akuter Schlaganfälle auswirkt, zeigten Bohmann et al. in der STREAM-Studie [3]. Dabei konnten über einen Zeitraum von fast 2 Jahren an mehreren Kliniken mit simulationserfahrenen Teams signifikant kürzere Door-to-Groin-Zeiten erzielt werden: Die Door-to-GroinZeit nahm im Durchschnitt um 21 Minuten ab. Zudem verbesserten sich das Sicherheits- und Teamarbeitsklima sowie die Arbeitszufrie-
denheit der Teilnehmer [3]. Es ist jedoch anzumerken, dass es sich bei dieser Studie nicht um ein Virtual-Reality-Simulationstraining handelte.
die Abläufe können gezielt erprobt werden. Ziel ist es, die Arbeitsabläufe beim Schlaganfall zu verinnerlichen, um im Ernstfall routiniert handeln zu können.
VR-Technologie eröffnet neue Möglichkeiten der Fortbildung
Pfizer investiert seit Jahren in die Fortbildung junger Ärzte im Bereich Schlaganfall und konzipiert diese im Zuge des aktuellen digitalen Wandels neu. Dabei wird verstärkt darauf geachtet, neue Formen der Zusammenarbeit einzubinden und Fortbildungsinhalte für die Generation Z einfach, ortsunabhängig, spielerisch, in Echtzeit, realitätsnah und vor allem digital darzustellen [4]. Eine spannende Entwicklung in diesem Bereich ist das VirtualReality-Simulationstraining zum Thema Schlaganfall. Es ermöglicht es den Assistenzärzten, dank der immersiven Wirkung der VR-Technologie Situationen sehr realistisch wahrzunehmen und Szenarien zu erleben, die in der Realität nur schwer oder kostenintensiv simuliert werden können [5].
Durch realitätsnahe Simulationen [6] lässt sich die Stresssituation eines Schlaganfalls nachstellen und
Realistische VR-Simulation eines schweren Hemisphärensyndroms nach Schlaganfall zur Förderung der Zusammenarbeit im Team
In diesem Virtual-Reality-Simulationstraining wird ein Schlaganfall im Zeitfenster mit einem schweren Hemisphärensyndrom rechts dargestellt. Für einen immersiven Einstieg ins Training schildert ein Notarzt den fiktiven Fall eines 78-jährigen Patienten mit Sprachstörungen und Lähmungserscheinungen.
Am Rollenspiel nimmt ein interdisziplinäres Team aus 7 Personen teil, denen die folgenden Rollen zugeteilt werden: der Instruktor, der Patient, der Pfleger, der Neurologe, der Radiologe sowie der Beobachter für die Kommunikation und der Beobachter für die Arbeitsabläufe (Abb. 1). In der Simulation tragen der Patient, der Pfleger, der Neurologe und der Radiologe eine VR-Brille, während die Beobachterrollen die Handlungen über eine
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 84 FORUM SCHLAGANFALL
Virtual-Reality-Simulationstraining für die Schlaganfallbehandlung
Servet Yilmaz, Jan Sobesky, Waltraud Pfeilschifter, Annica Stähly, Joris Klause, Daniel Kalanovic
Simulationstraining
Bildschirmübertragung an einem Monitor verfolgen. Die Assistenzärzte in der VR befinden sich räumlich zuerst im Schockraum einer Notaufnahme, bevor sie weiter in den CT-Untersuchungsraum gehen. Über Controller an den Händen werden entsprechende Handlungen gesteuert. Dabei können die Teilnehmer nicht nur die Hände ihres eigenen Avatars sehen, sondern auch die der anderen Team-Mitglieder, mit denen sie die gesamte Zeit kommunizieren und interagieren. Die Assistenzärzte können sich frei im Trainingsbereich bewegen. Alle Schritte von der Notaufnahme, über Anamnese und Bildgebung bis hin zur Therapieentscheidung werden geübt.
Während der Anamnese werden bereits die nächsten Schritte geplant: Der Radiologe, der Teil des Teams ist, wird im Rollenspiel telefonisch kontaktiert, die Werte werden durchgegeben und das CT
wird vorbereitet. Das Ganze geschieht unter großem Zeitdruck – 10 Minuten bleiben dem Team im Schockraum der Notaufnahme, bevor es in die Radiologie geht, wo sich aufgrund des CT-Befundes der Verdacht eines Gefäßverschlusses erhärtet und die ersten Maßnahmen zur Thrombektomie, dem mechanischen Entfernen des Blutgerinnsels, eingeleitet werden.
Trainingsablauf in drei Phasen
Das Training besteht aus drei Phasen und dauert insgesamt 60 Minuten:
• Phase 1 beinhaltet ein kurzes technisches Tutorial und einen emotionalen Einstieg in das Szenario durch die Übergabe des Patienten durch den Rettungsdienst.
• In Phase 2 findet das medizinische Training wie oben beschrieben statt. Es umfasst die
Bereiche Notaufnahme, Anamnese, Bildgebung und Therapieentscheidung. Abbildung 2 zeigt die Situation bei der Anamnese. • In Phase 3 des Trainings findet eine umfassende Auswertung statt. Neben der wichtigen Doorto-Needle Time werden die Kompetenzen und Leistungen aus verschiedenen Perspektiven in einem 360°-Feedback bewertet. Dazu gehören die Erkenntnisse der Beobachter, die die Kommunikation und Arbeitsabläufe während des Trainings evaluiert haben. Das Training des Notfalls mithilfe von Virtual-Reality-Simulationen wurde bei Pilotveranstaltungen von allen Teilnehmern positiv bewertet.
Bildungsziele und Kompetenzen
Prozessabläufe
Das Virtual-Reality-Training bietet eine eigenständige Simulationsübung, bei der sowohl das klinische Wissen getestet als auch technische Fähigkeiten erweitert werden können. Durch die realitätsnahe Umgebung werden Assistenzärzte dazu ermutigt, ihre Aufgaben und Denkansätze direkt umzusetzen. Der innovative Ansatz ermöglicht es den Teilnehmern, zeitkritische Abläufe in der Schlaganfallversorgung praktisch zu trainieren. Es geht darum, die Tätigkeitsabfolgen sinnvoll auszuführen, unnötige Zeitverzögerungen zu erkennen, diese beim nächsten Durchlauf zu vermeiden sowie eine richtige Therapieentscheidung zu treffen.
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 85 FORUM SCHLAGANFALL
Instruktor
Patient Pfleger
Beobachter Arbeitsabläufe
Beobachter Kommunikation
Radiologe
Neurologe
Abbildung 1: Simulationstraining mit 7 Rollen, wobei 4 Rollen eine VR-Brille tragen.
Abbildung 2: Die Phase 2 des Simulationstrainings der Schlaganfall-Akutversorgung ermöglicht es dem Neurologen, den Patienten zu anamnestizieren.
Das Simulationstraining legt besonderen Fokus auf das Prinzip von „Trial-and-Error“ [7], da die Virtual-Reality-Umgebung ein sicheres Lernumfeld bietet, ohne einen Patienten real zu gefährden. Den Teilnehmern ist es – anders als in der Realität – erlaubt, Fehler zu machen und daraus zu lernen. Dabei steht jederzeit der Patient im Fokus und es wird gemeinsam nach

Lösungen gesucht, um die Therapie zu optimieren.
Kommunikation
Ein weiterer wichtiger Aspekt der Simulation ist die Förderung der Kommunikationsfähigkeiten jedes einzelnen Teilnehmers. Im interdisziplinären Team ist eine effek-
tive und klare Kommunikation essenziell, um gemeinsame Ziele zu erreichen und eine optimale Versorgung des Patienten zu gewährleisten. Durch das Simulationstraining lernen die Assistenzärzte, sich verständlich auszudrücken, aktiv zuzuhören und aufmerksam auf die Bedürfnisse anderer einzugehen. Die Simulation bietet außerdem die Möglichkeit, den Umgang mit Fairness und Rücksichtnahme zu üben. Die Teilnehmer lernen, auf die Bedürfnisse ihrer Teammitglieder einzugehen und ihre eigenen Interessen zugunsten des Teamerfolgs zurückzustellen. Dadurch werden nicht nur die sozialen Kompetenzen der Assistenzärzte gestärkt, sondern auch ein respektvolles Umgangsverhalten gefördert. Der Gamification-Ansatz [8] ermöglicht es den Teams, ihre Aufmerksamkeit gezielt auf die spielerischen Elemente zu lenken. Dadurch werden nicht nur die Lerninhalte besser verinnerlicht, sondern auch der Spaßfaktor und die Motivation für das Teamspiel erhöht.
Neue Perspektiven gewinnen
Die Virtual-Reality-Simulation bietet den Assistenzärzten die Möglichkeit sich in verschiedene Rollen hineinzuversetzen. Das fördert die sozial-emotionale Entwicklung [9]. Die Assistenzärzte vollziehen einen Perspektivwechsel. Sie können sich in die Lage des Patienten oder der Pflegekraft versetzen, was eine Grundvoraussetzung für das soziale Miteinander ist.
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 86 FORUM SCHLAGANFALL
Gerade im stressigen Klinikalltag sind solche Rollenspiele von großer Bedeutung. Die Teilnehmer haben die Gelegenheit, in andere Personen zu schlüpfen, das Verhalten anderer zu verstehen und angemessen darauf zu reagieren. Die Rolle des Instruktors spielt hierbei eine wichtige Rolle, da er einen sicheren Raum für die Teilnehmer schafft. Er leitet die Simulation, hält die Gruppendynamik aufrecht und fungiert als persönlicher Betreuer, der bei Fragen und Unsicherheiten zur Seite steht.
Mit
Jeder Dritte leidet nach einem Schlaganfall unter Depressionen. Bisher konnte man nicht sicher voraussagen, welche Patienten eine Post-Stroke-Depression entwickeln werden. Aktuelle sonografische Untersuchungen im Rahmen der PROMoSD-Studie* zeigen nun, dass es dafür einen Risikofaktor geben könnte, der sich einfach per Ultraschall ermitteln lässt.
Raphe-Kerne des Hirnstamms als potenzieller Biomarker für Depression
Bei etwa jedem 4. SchlaganfallPatienten sind die mesenzephalen Raphe-Kerne des Gehirns strukturell verändert – und in dieser Region des Hirnstamms finden sich
Literatur
1 Meretoja A et al. Save a Minute, Save a Day. Stroke 2014;4:1053-1058
2 Deinzer R et al. Endocrine and psychological stress response in simulated doctor-patient interactions in medical education. Psychoneuroendocrinology 2019;105:172-177
3 Bohmann FO et al. Simulation-based training improves process times in acute stroke care (STREAM). Eur J Neurol 2021;29:138-148
4 Hentschel C et al. Systematische Innovation. In: Kamiske GF, Hrsg. TRIZ –Innovation mit System. München: Hanser; 2010:7-14
5 Dörner R et al. Einführung in Virtual und Augmented Reality. In: Dörner R et al., Hrsg. Virtual and Augmented Reality (VR/AR). Berlin: Springer; 2019:141
6 Langer E. Medieninnovationen AR und VR, Erfolgsfaktoren für die Entwick-
auch bei fast 70 % aller untersuchten Menschen mit einer unipolaren Depression Anomalien. Feststellen lassen sich diese anatomischen Auffälligkeiten mittels transkraniellem Ultraschall (TCS) – einer neuen schnell verfügbaren, kostengünstig und ohne Nebenwirkungen durchführbaren Neuroimaging-Methode. Laut Professor Christos Krogias, Leiter der DEGUM-Sektion Neurologie, legen die Beobachtungen nahe, dass es eine physiogenetische Veranlagung für Depressionen in diesem Areal des Hirnstamms zu geben scheint und dass zwischen der Hirnstammveränderung und der Entwicklung einer Post-StrokeDepression ein Zusammenhang besteht.
* Richter D et al. Prognostic markers of post-stroke depression (PROMoSD): study protocol of a prospective singlecenter observational study on raphe hypoechogenicity as a predictor of poststroke depression. Neurol Res Pract 2022; 4, 59
lung von Experiences. Berlin: Springer Vieweg; 2020:61-65
7 Pförtsch WA et al. Das neue MarketingMindset, Management, Methoden und Prozesse für ein Marketing von Mensch zu Mensch, Wiesbaden: Springer Gabler; 2019:9-45
8 Alter A. Irresistible, The Rise of Addictive Technology and the Business of Keeping Us Hooked. New York: Penguin; 2017:291-314
9 Blank-Mathieu M. Spielformen in Bezug auf Bildungsprozesse 2007. http:// www.schola-europaea.eu/ELC/popups/ 08.pdf
Für die Verfasser:
Servet Yilmaz
Customer Education Manager
Medical Affairs Germany
Pfizer Pharma GmbH
Linkstraße 10 10785 Berlin
E-Mail: servet.yilmaz@pfizer.com
Nach Schlaganfall Gefäße und Hirnstamm screenen
Diese Erkenntnis könnte dazu beitragen, eine Post-Stroke-Depression künftig besser vorherzusagen. „Bestätigen sich die bisherigen Ergebnisse, wäre eine standardisierte Ultraschalluntersuchung des Hirnstamms absolut empfehlenswert, um Risikopatienten frühzeitig zu identifizieren“, erklärt Dr. Daniel Richter, Erstautor der PROMoSDStudie.
Die Betroffenen könnten gezielter präventiv betreut werden, engmaschige Kontrolluntersuchungen und ggf. eine medikamentöse oder psychotherapeutische Behandlung könnten die Depression abmildern oder gar verhindern. Das würde die Lebensqualität und -erwartung der Patienten deutlich verbessern.
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 87 FORUM SCHLAGANFALL
Ultraschall frühzeitig PostStroke-Depressionen erkennen
DEGUM
Krankheiten des Herz-KreislaufSystems sind laut Statistischem Bundesamt für 35 % der Todesfälle in Deutschland verantwortlich – das entspricht 338.000 Todesfällen pro Jahr [1]. Davon entfallen 20 %, also 65.800 Todesfälle, auf den plötzlichen Herztod (Sudden Cardiac Death, SCD) [2]. Pro Tag erleiden demnach 180 Menschen in Deutschland einen SCD. Häufigster Auslöser sind schnelle Herzrhythmusstörungen wie anhaltende Kammertachykardien und Kammerflimmern. Das höchste SCD-Risiko haben Patientinnen und Patienten mit Herzinsuffizienz und hochgradig eingeschränkter Pumpfunktion in den ersten Monaten nach der Diagnosestellung. Eine tragbare Defibrillatorweste (Wearable Cardioverter Defibrillator, WCD, LifeVest®) kann entscheidend dazu beitragen, Risikopatienten vor dem plötzlichen Herztod zu bewahren, wie die Daten von mehr als 20.000 Teilnehmern aus retrospektiven und prospektiven Registern [3–6] sowie die Ergebnisse einer großen randomisierten kontrollierten Studie [7] umfassend belegen. Aber wie sind diese Risikopatienten am besten zu identifizieren?
auch für Patienten mit einer LVEF zwischen 40 und 49 % ist das Risiko, einen plötzlichen Herztod zu erleiden, je nach Alter, Allgemeinzustand und Begleitumständen noch hoch [8]. Daher ist die LVEF der wichtigste Risikomarker für den plötzlichen Herztod – hat ein Patient eine schlechte Pumpfunktion, hat er eine schlechte Prognose. Die Risikostratifizierung lässt sich durch weitere Untersuchungen verbessern, z.B. durch die Berücksichtigung der Parameter des autonomen Tonus, die Identifikation der Narbe durch Bildgebung sowie ein frühzeitiges Rhythmus-Monitoring.
Eingeschränkte Herzleistung ist Risikofaktor Nr. 1
Trotz optimaler medikamentöser Therapie (OMT) verstirbt einer von 20 Herzinfarktpatienten mit einer linksventrikulären Ejektionsfraktion (LVEF) unter 35 % in den ersten 90 Tagen und in rund 50 % der Fälle ist die Ursache ein SCD [4]. Aber
Vulnerable Phase der Medikamenteneinstellung sicher überbrücken
Prinzipiell können Risikopatienten mit einem implantierbaren Cardioverter-Defibrillator (ICD) vor dem plötzlichen Herztod geschützt werden. Allerdings empfiehlt die ESC-Guideline zur Behandlung der akuten und chronischen Herzinsuffizienz [9], vor der Versorgung mit
einem ICD eine Wartezeit einzuhalten: nach Myokardinfarkt ohne Revaskularisierung 40 Tage bzw. 3 Monate mit Revaskularisierung, bei nicht ischämischer Kardiomyopathie mindestens 3 Monate unter optimaler medikamentöser Therapie. Hierdurch soll der medikamentösen Therapie mit den neuen verfügbaren hochpotenten Herzinsuffizienz-Medikamenten, den sog. fantastischen Vier (RAS-Blocker, MRAs, Betablocker und SGLT2-Hemmer), Zeit gegeben werden, ihre volle Wirkung am Herz zu entfalten. Die medizinische Optimierung und Stabilisierung einer Herzinsuffizienz kann 3 Monate oder länger dauern. Studien zeigen, dass wirksame Dosen von Betablockern bei Herzinsuffizienz in der Regel nach 8 – 12 Wochen erreicht werden und frühestens nach 3 Monaten zu einem Mortalitätsvorteil führen [10–12]. Und auch die Kombination von Sacubitril/Valsartan verbessert zwar die systolische Funktion bei Herzinsuffizienz mit reduzierter Auswurffraktion (HFrEF), aber es kann bis zu 6 Monate bis zur vollen Wirkung dauern [13]. Das Rapid
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 88 FORUM CARDIOLOGICUM
Tragbare Defibrillatorweste kann kardiovaskuläre Hochrisikopatienten vor dem plötzlichen Herztod schützen
LifeVest®
Die Bestandteile der LifeVest
Die LifeVest besteht aus einem Stoffteil, einem Elektrodengürtel und einem Monitor.
Die LifeVest®-Defibrillatorweste überwacht das Herz des Patienten kontinuierlich. Wird ein lebensgefährlicher Herzrhythmus erkannt, gibt das Gerät einen elektrischen Behandlungsschock ab, um den normalen Herzrhythmus wiederherzustellen. Das gesamte Ereignis dauert in der Regel etwa eine Minute.
STOFFTEIL
Wird unter der normalen Kleidung direkt auf der Haut getragen
Enthält den Elektrodengürtel
ELEKTRODENGÜRTEL
Erkennt, wenn die Herzfrequenz in einen gefährlichen Rhythmus gewechselt hat
Gibt einen Behandlungsschock ab
MONITOR
Wird an der Taille oder mit einem Schultergurt getragen
Zeichnet die Herzfrequenz kontinuierlich auf
In den ESC-HF-Guidelines [9] hat die Defibrillatorweste für ischämische und nicht ischämische Patienten eine IIb-Empfehlung mit Evidenzgrad B. Bei Post-Myokardinfarkt-Patienten ist der WCD mit einer IIb-Empfehlung mit Evidenzgrad B in den aktuellen VT/VF-Guidelines [17] aufgeführt.
Ein wesentliches Merkmal der LifeVest sind laute Ansagen und Vibrationsalarme, die Ihnen zeigen, dass das Gerät ordnungsgemäß funktioniert. Allerdings ist zu beachten, dass laute Umgebungen oder Vibrationen es erschweren können, einen Alarm zu hören. Wenn Sie sich also in einer solchen Umgebung oder Situation befinden, sollten Sie Ihre LifeVest besonders beachten, um auf mögliche Alarme reagieren zu können. Zum Beispiel können beim Motorradfahren oder beim Rasenmähen Vibrationen erzeugt werden, die es erschweren, einen Alarm wahrzunehmen. Dies könnte dann zu einer unerwünschten Behandlung führen.
Sequencing der 4 Medikamentenklassen ermöglicht es, die Patienten innerhalb von 3 – 6 Monaten medikamentös stabil einzustellen. In nahezu 50 % der Fälle führt dies dazu, dass sich die Pumpleistung des Herzens verbessert und das Langzeitrisiko eines plötzlichen Herztodes sinkt, sodass kein ICD implantiert werden muss [14].
Gerade in der Zeit direkt nach einem kardialen Ereignis und damit in der empfohlenen Wartezeit bis zur ICD-Implantation besteht jedoch das höchste Risiko für einen plötzlichen Herztod [15, 16]. Da-
her sollten Risikopatienten in dieser vulnerablen Phase der medikamentösen Therapieeinstellung mit einer tragbaren Defibrillatorweste wie der LifeVest® geschützt werden [9, 17]. Umgekehrt macht der WCD die moderne medikamentöse Therapie der Herzinsuffizienz erst möglich.
Überzeugende wissenschaftliche Datenlage
Umfangreiche Daten aus retrospektiven und prospektiven Registern mit mehr als 20.000 Patienten
belegen die klinische Wirksamkeit der LifeVest® bei der Terminierung ventrikulärer Tachyarrhythmien bei gleichzeitig sehr niedrigem Risiko inadäquater Therapien [5, 6, 18, 19].
Eine Metaanalyse mit annährend 20.000 Patienten zeigte erfolgreiche VT/VF-Terminierungen in 96 % der Fälle [18]. Nach den Ergebnissen des WEARIT II-Registers benötigen nur 42 % der Patienten mit ischämischer, nicht ischämischer oder kongenitaler Kardiomyopathie und eingeschränkter linksventrikulärer Funktion nach einer mittleren Tragedauer des WCD von 90 Tagen (22,5 Stunden/Tag) noch einen ICD [20]. In einer deutschen Registerstudie mit 6.000 Patienten betrug die Tragezeit 23,1 Stunden pro Tag (Median) – die Compliance war also sehr hoch [5].
Dass die LifeVest® bei konsequenter Anwendung einen klaren Überlebensvorteil schafft, verdeutlichen auch die Ergebnisse der randomisierten, kontrollierten Studie „Vest Prevention of Early Sudden death Trial“ (VEST), die 2.302 Patienten mit stark eingeschränkter Pumpfunktion nach Myokardinfarkt einschloss [4]. In der Intention-totreat-Analyse konnte zwar keine signifikante Senkung des primären Endpunkts arrhythmische Mortalität gezeigt werden (relatives Risiko, 0,67; p = 0,18). Die Gesamtmortalität hingegen – der wichtigste sekundäre Endpunkt – wurde signifikant reduziert (relatives Risiko, 0,64; p = 0,04). Dieses Ergebnis wurde in einer im Jahr 2020 veröffentlichten Per-Protocol-Analyse der VEST-Studie nochmals näher beleuchtet. In diese Analyse wurden

Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 89 FORUM CARDIOLOGICUM
13
alle Patientendaten vom Zeitpunkt der Randomisierung bis zum letzten protokollgemäßen Tragetag des WCD eingeschlossen, nicht aber diejenigen Tage nach einem vorzeitigen Abbruch, z.B. wegen ICDImplantation. Die Per-ProtocolAnalyse zeigte, dass bei Patienten unter konsequenter WCD-Therapie nicht nur die Gesamtmortalität um 75 % (p < 0,001), sondern auch der primäre Endpunkt eines arrhythmischen Todes um 62 % (p = 0,02) signifikant reduziert wurde. Die tägliche Tragezeit in der Per-Protocol-Analyse (Median) lag bei ≥22 Stunden [21].
Die 2021 publizierte PROLONGII-Studie [7] schloss 353 Patienten mit neu diagnostizierter Herzinsuffizienz (mit ischämischer [ICM] oder nicht ischämischer Kardiomyopathie [NICM]; LVEF 25 ± 8 %) ein, die zwischen 2012 und 2017 mit einem WCD versorgt worden waren. Die mittlere Tragezeit pro Patient lag bei 104 ± 76 Tagen. Bei guter Compliance (22 h pro Tag) erhielten 4 % der ICM- und 4 % der NICM- Patienten aufgrund lebensbedrohlicher ventrikulärer Arrhythmien einen Behandlungsschock durch die LifeVest®. Nach Abgabe des WCD hatten 53 % der Patienten keine ICD-Indikation mehr (Verbesserung der LVEF auf ≥35 %). Anschließend wurden die Patienten 2,8 ± 1,5 Jahre (Maximum: 6,8 Jahre) nachbeobachtet. Dabei zeigte sich, dass eine Defibrillation durch den WCD keinen Einfluss auf die Mortalität im weiteren Followup hatte. Darüber hinaus zeigte die Studie, dass sich bei 53 % der LifeVest®-Patienten durch die opti-
Kostenerstattung
Die LifeVest® Defibrillatorweste ist seit 2005 im Hilfsmittelverzeichnis des GKV-Spitzenverbandes gelistet und wird als Hilfsmittel verordnet. Die Kosten werden in der Regel von den Krankenkassen erstattet. Im Juli 2019 wurde das Hilfsmittelverzeichnis aktualisiert und ergänzend alle primärprophylaktischen Indikationen mit einer hochgradig eingeschränkten Pumpfunktion (LVEF ≤35 %) aufgenommen. Hierzu zählen v.a. Patienten mit einem temporär erhöhten Risiko für einen plötzlichen Herztod nach akutem Myokardinfarkt (<40 Tage zurückliegend), einer erwarteten LVEF-Verbesserung bei akuter Myokarditis oder bei Erstdiagnose einer dilatativen Kardiomyopathie. Weitere Einsatzbereiche sind die peripartale Kardiomyopathie, die fortgeschrittene koronare Herzkrankheit während der Wartezeit auf eine geplante Herzoperation sowie nach einer PTCA oder Bypass-Operation (ACVB). Weiterhin wird eine prolongierte Risikostratifizierung mit der Defibrillatorweste aufgeführt.
male medikamentöse Therapie die LVEF auf >35 % verbessern konnte. Somit war bei diesen Patienten keine ICD-Indikation mehr gegeben [7].
Sektorenübergreifendes Konzept für den Einsatz der LifeVest® in der Praxis
In die klinische Versorgung eines Herzpatienten sind verschiedene spezialisierte Teams und Funktionsbereiche involviert, wie Herzkatheterlabor, Elektrophysiologie, Intensivstation, Echolabor oder Telemetriestation. Im St. VinzenzHospital Köln wurden diese mithilfe eines funktionsbereichübergreifenden „WCD-Kompetenzteams“ verbunden, sodass der Weg jedes Herzpatienten von der Aufnahme über die Diagnose und Schulung bis hin zum strukturierten Entlassmanagement genau festgelegt ist [22]. So können Risikopatienten rasch identifiziert und entsprechend
mit einem WCD versorgt werden. Auch die ambulante Anbindung der Patienten ist klar geregelt: Ein strukturiertes Entlassmanagement gewährleistet eine optimale Weiterversorgung. Dazu gehört auch ein strukturiertes Follow-up durch spezialisierte Pflegekräfte. Diese Heart Failure Nurses rufen die Patienten an und unterstützen bei der medikamentösen Aufdosierung sowie beim WCD-basierten Telemonitoring. Über das ZOLL Patient Management (ZPM) Network kann via Telemonitoring auf aktuelle Gesundheitsdaten des Patienten während der WCD-Tragezeit zugegriffen und u.a. Herzfrequenz, körperliche Aktivität und Körperposition erfasst werden. Die Heart Failure Nurses und die Schrittmacherambulanz des Krankenhauses fungieren als koordinierendes Zentrum und als Ansprechpartner für die niedergelassenen Ärzte in der ambulanten Weiterbetreuung – somit können die Patienten sektorenübergreifend versorgt
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 90 FORUM CARDIOLOGICUM
werden und der stationäre und ambulante Bereich arbeiten Hand in Hand.
Brigitte Söllner, Erlangen
Herzinsuffizienz:
CardioMEMS™-Sensor verbessert

Lebensqualität und senkt Risiko für Hospitalisierung
Literatur
1 Statistisches Bundesamt, https://www. destatis.de/DE/Presse/Pressemitteilungen/2021/11/PD21_505_23211.htm
2 Empana JP et al. J Am Coll Cardiol 2022;79:1818-1827
3 Halkin A et al. J Am Coll Cardiol 2005; 45:1397-1405
4 Olgin JE et al. N Engl J Med 2018; 379:1205-1215
5 Wäßnig NK et al. Circulation 2016; 134:635-643
6 Epstein AE et al. J Am Coll Cardiol 2013;62:2000-2007
7 Mueller-Leisse J et al. ESC Heart Fail 2021;8:5142-5148
8 Chatterjee NA et al. JAMA Cardiol 2018;3:591
9 McDonagh TA et al. Eur Heart J 2021; 42:3599-3726
10 Hjalmarson A et al. Lancet 1999;353: 2001-2007
11 Packer M et al. N Engl J Med 1996; 334:1349-1355
12 Lechat P et al. Lancet 1999;353:9-13
13 Guerra F et al. Eur J Clin Pharmacol 2021;77:1835-1842
14 Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (Hrsg.). Ventrikuläre Arrhythmien und Prävention des plötzlichen Herztodes, ESC Pocket Guidelines, Version 2015
15 Adabag AS et al. JAMA 2008;300: 2022-2029
16 Solomon SD et al. N Engl J Med 2005;352:2581-2588
17 Zeppenfeld K et al. Eur Heart J 2022;43:3997-4126
18 Nguyen E et al. J Innov Card Rhythm Manag 2018;9:3151–3162
19 Deneke T et al. Kardiologe 2017;11:2743
20 Kutyifa V et al. Circulation 2015;132: 1613-1619
21 Olgin JE et al. J Cardiovasc Electrophysiol 2020;31:1009-1018
Traditionell konzentrieren sich Ärzte auf physiologische Parameter (Patientengewicht, Blutdruck usw.) und die kardialen Marker BNP und NT-proBNP, um eine Verschlechterung des Zustands ihrer Herzinsuffizienz-(HF-)Patienten zu erkennen. Allerdings treten diese Marker erst spät im Dekompensationsverlauf auf, sodass nur wenig Zeit bleibt, adäquat auf sie zu reagieren, bevor ein Krankenhausaufenthalt erforderlich ist. Eine innovative Möglichkeit, HF-Patienten engmaschig zu überwachen, bietet das CardioMEMS™ HF-System von Abbott: Per Fernzugriff kontrolliert ein dauerhaft in der distalen Pulmonalarterie implantierter Sensor (Abb. 1) kontinuierlich den Pulmonalarteriendruck des Patienten und signalisiert dem behandelnden Arzt Druckveränderungen, die ein Indikator für Wasseransammlungen in der Lunge und damit als frühe Anzeichen einer sich verschlechternden Herzinsuffizienz zu werten sind. Diese frühen Veränderungen lassen sich häufig durch einfache Anpassungen der Versorgung, wie z.B. die Titration der Medikamente, beheben, sodass einer Progression vorgebeugt wird, ohne dass ein Termin mit dem Patienten erforderlich ist.
Abbildung 1: Der CardioMEMS™Sensor hat mit einer Länge von 15 mm in etwa die Größe einer Büroklammer. Nach der minimalinvasiven Platzierung in der Zielpulmonalarterie misst er kontinuierlich den Pulmonalarteriendruck und überträgt die Messwerte drahtlos an den betreuenden Arzt. Auf diese Weise kann dieser den Zustand des Patienten von praktisch überall aus überwachen (© Abbott).
Überzeugende Studienergebnisse
Auf der diesjährigen ESC-Tagung in Prag wurden die Daten der niederländischen Studie MONITOR-HF vorgestellt, die als dritte randomisierte, kontrollierte Studie weltweit einen signifikanten gesundheitlichen Nutzen der Fernüberwachung mit dem CardioMEMS™ HF-System für HF-Patienten belegt [1, 2]. Eingeschlossen wurden 348 Patienten, die an einer Herzinsuffizienz der NYHA-Klasse III litten, mit einer Kombination von bis zu
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 91 FORUM CARDIOLOGICUM
4 Herzmedikamenten leitlinienkonform behandelt und innerhalb des letzten Jahres wegen einer Herzinsuffizienz hospitalisiert worden waren oder eine Notfallbehandlung mit intravenösen Diuretika benötigt hatten. Ihre mittlere Ejektionsfraktion betrug 30 % (23 – 40 %).
Die Patienten erhielten randomisiert entweder eine hämodynamische Überwachung mit dem CardioMEMS™-Sensor (n = 176) oder eine leitliniengerechte medikamentöse Standardbehandlung der Herzinsuffizienz (n = 172). Alle Studienteilnehmer wurden nach 3 und 6 Monaten und danach halbjährlich (bis zu 48 Monate lang) untersucht, die Patienten, denen der CardioMEMS™-Sensor implantiert wurde, wurden mindestens ein Jahr lang überwacht.
Die MONITOR-HF-Studie erreichte ihren primären Endpunkt durch den Nachweis einer klinisch bedeutsamen, signifikanten Verbesserung der Lebensqualität bei den Teilnehmern mit CardioMEMS™Sensor-Überwachung gegenüber der Kontrollgruppe ohne Drucksensor: Während im CardioMEMS™Studienarm ein durchschnittlicher Anstieg von 7 Punkten im Kansas City Cardiomyopathy Questionnaire (KCCQ) zu verzeichnen war, verschlechterte sich der Durchschnittswert in der Kontrollgruppe (Unterschied = 7,13 Punkte; 95%KI: 1,51 – 12,75; p = 0,013) [1, 2]. Der KCCQ-Fragebogen gilt als klinische Standardbewertung bei Herzinsuffizienz und wird zur Erfassung und Messung der Betroffenen-berichteten Versorgungsqualität empfohlen [3]. Dabei werden
die Patienten gefragt, wie schwierig es für sie ist, alltägliche Aktivitäten zu bewältigen, wie beispielsweise kurze Strecken zu Fuß, Hausarbeiten oder sich mit der Familie und Bekannten zu treffen. Ein weiteres, überzeugendes Ergebnis war, dass sich die HF-bedingten Krankenhausaufenthalte bei den HF-Patienten unter der leitliniengerechten medikamentösen Therapie um 44 % verringerten [1, 2]. Außerdem war bei den Patienten, denen der CardioMEMS™-Sensor implantiert und die mindestens 1 Jahr lang betreut wurden, das Risiko für Komplikationen mit 2,3 % gering und in Übereinstimmung mit früheren Untersuchungen [1, 2].
Fazit
Die MONITOR-HF-Studie wurde von der Erasmus Universität in Rotterdam konzipiert und durchgeführt. Wie der leitende Prüfarzt Dr. Jasper Brugts bei seiner Präsentation auf dem ESC abschließend betonte, „ist das niederländische Gesundheitssystem für seinen strukturierten Ansatz bei der Behandlung von Herzinsuffizienz in ambulanten Kliniken mit einem allgemeinen Zugang zu leitliniengerechten medizinischen Therapien bekannt. Dieser Ansatz legt die Messlatte höher und macht die Ergebnisse der MONITOR-HF-Studie noch aussagekräftiger, indem Patienten, die bereits einen hohen Pflegestandard erhielten, bei Verwendung des CardioMEMS™ HF-Systems über signifikante Verbesserungen ihrer Lebensqua-
lität berichteten.” Die Relevanz der Studie auch für die deutsche Versorgungslandschaft bestätigte Professor Stefan Störk, Leiter des Departments Klinische Forschung und Epidemiologie am Deutschen Zentrum für Herzinsuffizienz des Universitätsklinikums Würzburg: „Die MONITOR-HF Studie hat eindrucksvoll gezeigt, dass ein telemedizinisch unterstütztes Monitoring, verbunden mit einer sehr guten Nachsorge, die immer noch viel zu hohe Hospitalisierungsrate bei Herzinsuffizienz substanziell reduzieren kann. Davon profitieren unsere Patienten ganz unmittelbar, und auch für unser Gesundheitssystem erwarten wir positive Effekte.“
Literatur
1 Brugts JJ et al. Remote haemodynamic monitoring of pulmonary artery pressures in patients with chronic heart failure. Presented at the European Society of Cardiology Heart Failure Association annual meeting, Prague, Czech Republic, May 20, 2023
2 Brugts JJ et al. Remote haemodynamic monitoring of pulmonary artery pressures in patients with chronic heart failure (MONITOR-HF): a randomised clinical trial. Lancet 2023;401:21132123
3 Spertus JA et al. Interpreting the Kansas City Cardiomyopathy Questionnaire in clinical trials and clinical care: JACC State-of-the-Art Review. J Am Coll Cardiol 2020;76:2379-2390
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 92 FORUM CARDIOLOGICUM
Brigitte Söllner, Erlangen
Im Juli 2023 hat die Europäische Kommission Empagliflozin (Jardiance®) zur Behandlung von Erwachsenen mit chronischer Niereninsuffizienz (Chronic Kidney Disease, CKD) zugelassen, die allein in Deutschland etwa 8 – 10 Millionen Menschen betrifft [1]. Mit der Zulassungserweiterung kann der hochselektive Natriumglucose-Cotransporter 2 (SGLT2)Inhibitor ab sofort in den Indikationen Typ-2-Diabetes, chronische Herzinsuffizienz (NYHA-Klasse II–IV) oder chronische Niereninsuffizienz sowie einer Kombination dieser Erkrankungen eingesetzt werden [2]. Für die Patienten ist das ein wichtiger Meilenstein, da diese Erkrankungen sehr häufig sind und sich oft gegenseitig bedingen. Das Herz-Kreislauf-System, Nieren und Stoffwechsel sind physiologisch eng miteinander verbunden und haben viele gemeinsame Risikofaktoren entlang des Krankheitskontinuums. Eine Funktionsstörung in einem System kann das Auftreten anderer Störungen beschleunigen. Umgekehrt können Verbesserungen in einem System zu positiven Effekten in den anderen Systemen führen [3, 4].
Die Behandlung mit Empagliflozin kann nun in allen zugelassenen Indikationen bis zu einer glome-
Empagliflozin erhält Zulassung
zur Behandlung der chronischen
Niereninsuffizienz bei Erwachsenen
rulären Filtrationsrate von 20 ml/ min/1,73m2 begonnen werden [2].
Signifikante Reduktion der Progression der Niereninsuffizienz
Die Zulassung basiert auf der Studie EMPA-KIDNEY®, der bisher größten und umfassendsten Studie eines SGLT2-Inhibitors bei chronischer Niereninsuffizienz [5]. An der Studie nahmen 6609 Patienten mit einer Vielzahl zugrundeliegender Ursachen der CKD teil, darunter diabetische und hypertensive Nephropathie sowie Glomerulonephritiden und IgA-Nephropathie –also auch Erkrankungen, die bisher in klinischen Studien mit SGLT2Inhibitoren zur chronischen Niereninsuffizienz unterrepräsentiert waren.
Einschlusskriterien waren eine geschätzte glomeruläre Filtrationsrate (eGFR) von mindestens 20, aber <45 ml/min/1,73 m2 oder eine eGFR von mindestens 45, aber <90 m/min/1,73 m2 mit einem UrinAlbumin-Kreatinin-Verhältnis von mindestens 200. Die Studienteilnehmer erhielten zusätzlich zum aktuellen Behandlungsstandard randomisiert entweder Empagliflozin 10 mg oder Placebo.
Der primäre Endpunkt war definiert als die Zeit bis zum Tod durch kardiovaskuläre Ursachen oder bis zum Fortschreiten der Niereninsuffizienz; diese war definiert als Niereninsuffizienz im Endstadium (Notwendigkeit einer Nierenersatztherapie wie Dialyse oder Nierentransplantation), eine anhaltende Abnahme der eGFR auf <10 ml/min/1,73 m2, Tod renaler Ursache oder ein anhaltender Rückgang der eGFR um ≥40 % nach Randomisierung.
Diesen Endpunkt erreichten im Verlauf von 2 Jahren 13,1 % der Patienten, die mit Empagliflozin behandelt wurden, und 16,9 % der Patienten in der Placebo-Gruppe. Dies entspricht einer signifikanten Risikoreduktion des primären Endpunkts von relativen 28 % unter Empagliflozin gegenüber Placebo (HR: 0,72; 95%-KI: 0,64 – 0,82; p < 0,000001). Der Unterschied beim kombinierten primären Endpunkt war mehrheitlich auf eine signifikante Reduktion der Progression der Niereninsuffizienz zurückzuführen. Die Behandlungsergebnisse waren unabhängig von der zugrundeliegenden Ursache der chronischen Niereninsuffizienz konsistent.
Auch im sekundären Endpunkt, den Hospitalisierungen jedweder Ursache, war der SGLT2-Inhibitor
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 93 FORUM NEPHROTICUM
überlegen: Gegenüber Placebo zeigte sich eine signifikante relative Risikoreduktion von 14 % (HR: 0,86; 95%-KI: 0,78 – 0,95; p = 0,0025) [5]. Vor dem Hintergrund, dass bei Patienten mit CKD das Risiko einer Hospitalisierung im Vergleich zu Menschen ohne CKD doppelt so hoch ist [6], kommt diesem Ergebnis eine besondere Bedeutung zu, zumal in der EU Hospitalisierungen für bis zu 70 % der gesamten Gesundheitsausgaben für Menschen mit CKD verantwortlich sind [7, 8]. Die allgemeinen Daten zur Sicherheit standen im Einklang mit vorherigen Studienergebnissen und bestätigten das gut etablierte Sicherheitsprofil von Empagliflozin [5].
Fazit
Eine chronische Niereninsuffizienz steht in engem Zusammenhang mit anderen kardiorenalen und metabolischen Erkrankungen wie Typ2-Diabetes und Herzinsuffizienz. Ein integrierter Ansatz ist daher für die Verbesserung von Behandlungsstrategien für diese miteinander verbundenen Erkrankungen ausschlaggebend. Empagliflozin hat das Potenzial, das Gleichgewicht zwischen den miteinander verbundenen kardiorenal-metabolischen Systemen wiederherzustellen und das Risiko schwerer Komplikationen zu verringern.
Fabian Sandner, Nürnberg
MITTEILUNGEN
Literatur
1 Girndt M et al. Dtsch Arztebl Int 2016;113:85-91
2 Fachinformation Jardiance®, aktueller Stand
3 García-Donaire A et al. Int J Nephrol 2011:975782
4 Leon M et al. World J Diabetes 2015;6:1246-58
5 Herrington W et al. N Engl J Med 2023;388:117-127
6 USRDS. 2021 Annual Report. https:// usrds-adr.niddk.nih.gov/2021/chronickid-ney-disease/3-morbidity-and-mortality-in-patients-with-ckd.
7 Turchetti G et al. Eur J Health Econ 2017;18:847-858
8 Gandjour A et al. PLOS-ONE 2020;15: e0231375
Arzneimittel-Nutzenbewertung: Vutrisiran erhält
Der Gemeinsame Bundesausschuss (G-BA) hat dem RNA-InterferenzTherapeutikum Vutrisiran (Amvuttra®) einen „Hinweis auf einen geringen Zusatznutzen“ gegenüber Patisiran (Onpattro®), das bereits mit einem beträchtlichen Zusatznutzen bewertet wurde, zugesprochen.
Vutrisiran wird zur Behandlung der hereditären Transthyretin-Amyloidose (hATTR-Amyloidose) bei
erwachsenen Patienten mit Polyneuropathie der Stadien 1 oder 2 angewendet. Der Wirkstoff wurde im September 2022 als Orphan Drug in der Europäischen Union zugelassen. Trotz des Status als Orphan Drug, entschied sich Alnylam dazu, für Vutrisiran eine reguläre Nutzenbewertung durch das IQWiG (Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen) unter Vorlage vergleichender Daten gegenüber der zweckmäßigen Vergleichstherapie durchführen zu lassen.
Die Nutzenbewertung von Vutrisiran basiert auf den Ergebnissen der Zulassungsstudie HELIOS-A. Deren Resultate belegen unter an-
derem die statistisch signifikanten Vorteile von Vutrisiran gegenüber Patisiran in der Kategorie Nebenwirkungen. So traten unter Vutrisiran insgesamt weniger schwerwiegende und schwere unerwünschte Ereignisse auf. Zudem gingen aus der G-BA-Bewertung weitere Vorteile von Vutrisiran gegenüber Patisiran im Hinblick auf verschiedene spezifische unerwünschte Ereignisse hervor.
Auch für Alnylams Arzneimittel Patisiran wurde im März 2019 bereits ein beträchtlicher Zusatznutzen beschlossen.
Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH 94 FORUM NEPHROTICUM
„Hinweis auf einen geringen Zusatznutzen“ gegenüber Patisiran
S.
F.
KONGRESSE
Weniger Herzinfarkte: Kombinationstherapie mit Alirocumab zur LDL-C-Senkung lohnt sich
Die Kardioprotektion durch Senkung des LDL-Cholesterins (LDLC) stand im Fokus zahlreicher Vorträge auf der 89. Jahrestagung der Deutschen Gesellschaft für Kardiologie (DGK). Auf einem Sanofi-Symposium wurde deutlich: Noch immer erreichen nur wenige kardiovaskuläre Hochrisikopatienten die von den europäischen Fachgesellschaften ESC und EAS empfohlenen LDL-C-Zielbereiche. Einer der Gründe hierfür: Kombinationstherapien, z.B. mit Alirocumab (Praluent®), einem Inhibitor der Proproteinkonvertase-Subtilisin/Kexin-Typ-9 (PCSK9), werden sehr zurückhaltend eingesetzt, obwohl sich mit ihnen auch sehr niedrige LDL-C-Werte erreichen lassen. Neue Daten der Post-hoc-Analyse von ODYSSEY OUTCOMES zeigen, dass sich bereits eine zeitlich begrenzte Anwendung der Kombinationstherapie für einen langfristigen klinischen Effekt lohnt.
Lebensstil-Optimierung ist die Basis, aber meist nicht ausreichend
„Die Optimierung des Lebensstils bildet die Basis, um das kardiovaskuläre Risiko zu senken“, betonte Professor Ingo Hilgendorf, Freiburg/Bad Krozingen. Dazu zählen vor allem regelmäßige Bewegung, mediterrane Ernährung und der
Verzicht auf Tabakprodukte. „Für einen Rauchstopp ist es nie zu spät – die E-Zigarette ist keine Alternative“, betonte Hilgendorf. Neben Diabetes, Hypertonie und Rauchen ist LDL-C der wichtigste kardiovaskuläre Risikofaktor, der eine kausale Rolle bei der Entwicklung atherosklerotischer kardiovaskulärer Erkrankungen (ASCVD) spielt. Jedoch kann mit Lebensstilmaßnahmen alleine dieser Risikofaktor im Allgemeinen nicht ausreichend adressiert werden – eine patientenindividuelle cholesterinsenkende Therapie muss die Lifestyle-Modifikation ergänzen.
Es gibt kein One-size-fits-all
Wie tief das LDL-C gesenkt werden sollte, hängt vom kardiovaskulären Gesamtrisiko ab. „Es gibt kein One-size-fits-all“, betonte Hilgendorf. Mit einer LDL-C-senkenden Therapie sollte frühzeitig begonnen werden, denn die atherogenen Wirkungen akkumulieren über die Jahre hinweg. „Für die LDL-C-Senkung haben wir eine große Toolbox“, so Hilgendorf. Allerdings: Die üblichen oralen LDLC-senkenden Medikamente stoßen bei hohen Ausgangswerten und bei Menschen mit sehr hohem kardiovaskulärem Risiko an ihre Grenzen. Ein sehr hohes Risiko besteht z.B. nach einem Myokardinfarkt. In dieser Situation werden LDL-C-Werte <55 mg/dl (<1,4 mmol/l) plus eine Reduktion vom Ausgangswert um mindestens 50 % empfohlen. Bei Menschen, die innerhalb von 2 Jahren einen erneuten Myokardinfarkt erlitten haben, soll das LDLC sogar in Bereiche <40 mg/dl (<1,0 mmol/l) gesenkt werden.
Im Versorgungsalltag wird zu selten kombiniert
Diese niedrigen LDL-C-Zielwerte werden jedoch nur von einem geringen Anteil der Hochrisikopatienten erreicht. Dafür sprechen die Ergebnisse der in 14 europäischen Ländern durchgeführten Beobachtungsstudie SANTORINI, die bei 9.602 Erwachsenen mit sehr hohem und hohem kardiovaskulärem Risiko untersuchte, inwieweit die 2019 eingeführten Leitlinien umgesetzt wurden und das Lipidmanagement bei kardiovaskulären Hochrisikopatienten verbessert haben. Die Ergebnisse sind ernüchternd: In Deutschland wurden die in den ESC/EAS-Leitlinien empfohlenen LDL-C-Zielwerte nur in 20,1 % der Fälle erreicht. „Einer der Gründe hierfür: Die zur Verfügung stehenden Therapieoptionen werden nicht ausgeschöpft“, erklärte Professor Ulrich Laufs, Leipzig. Insbesondere werden zu selten Kombinationstherapien eingesetzt.
Starke LDL-C-Senkung mit Alirocumab schützt bei sehr hohem Risiko
Der PCSK9-Inhibitor Alirocumab (Praluent®) bewirkt eine starke LDL-C-Senkung bei zusätzlicher Gabe zu einer optimierten Lipidtherapie, die sich in der großen Endpunktstudie ODYSSEY OUTCOMES in eine bessere Prognose umsetzte. Aktuelle Daten einer Post-hoc-Analyse von ODYSSEY OUTCOMES zeigten, dass sich auch eine befristete LDL-C-Senkung in sehr niedrige Bereiche (LDL-C <15 mg/dl bzw. <0,29 mmol/l) in einen klinischen
95 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Benefit übersetzt und über mehrere Jahre hinweg erhalten bleibt. Vor diesem Hintergrund erscheint es sinnvoll, bei hohem kardiovaskulärem Risiko bereits früh im LDLC-senkenden Therapiealgorithmus eine PCSK9-hemmende Strategie in die Kombinationstherapie zu integrieren.
Elisabeth Wilhelmi, München
Transthyretin-Amyloidose mit Kardiomyopathie Strukturiertes
Vorgehen beim Verdacht auf ATTRCM empfohlen
Wichtige Hinweise auf die immer noch unterdiagnostizierte Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) zeigen sich bereits bei gängigen kardiologischen Untersuchungsmethoden. Dennoch benötigt die Erkrankung mehr Aufmerksamkeit in der täglichen Praxis – darin waren sich die Experten bei einem Pfizer-Symposium im Rahmen der diesjährigen DGK-Jahrestagung einig. Unter dem Vorsitz von Professor Norbert Frey, Heidelberg, und Professor Christoph Stellbrink, Bielefeld, diskutierten sie den aktuellen Stand bei der Diagnose und Therapie der Herzinsuffizienz. Besonderes Augenmerk galt dabei der interdisziplinären Diagnosesicherung der ATTR-CM. „Schauen Sie mal auf das EKG“, forderte Stellbrink und riet außerdem: „Erstellen Sie sich einen Diagnosepfad. Dieser schließt Echokardiografie, KardioMRT, Szintigrafie und ggf. auch mal Genetik und Biopsie ein.“
Professor Wilhelm Haverkamp, Berlin, fasste die Verdachtsmomente im Elektrokardiogramm (EKG) zusammen und betonte, dass sich die ATTR-CM von einer normalen Hypertrophie unterscheidet – sowohl pathologisch als auch im EKG-Befund. Wie der Experte betonte, sind die Reduktion der RAmplitude (periphere Niedervoltage) und Pseudoinfarkt-Muster häufige Anzeichen. Zudem präsentiert sich die Erkrankung auch durch Leitungsstörungen und Schenkelblockierungen. „Das EKG trägt ganz wesentlich dazu bei, Amyloidosen zu diagnostizieren“, hob der Kardiologe den Stellenwert dieser gängigen Methode hervor. Gleichzeitig erinnerte er daran, dass das EKG nur ein diagnostisches Puzzleteil von vielen ist und weitere Verfahren, wie z.B. die Echokardiografie, zur Abklärung notwendig sind. Wie Untersuchungen zeigen, weist die Kombination aus EKG und Echokardiografie neben einer diagnostischen Genauigkeit von 98 % eine sehr hohe Sensitivität (100 %) und Spezifität (95 %) bei der Differenzierung zwischen ATTR-CM und einer Herzinsuffizienz auf. Für den Experten bietet sich mit der Unterstützung durch Künstliche Intelligenz (KI) zur EKG-Auswertung ein spannender Blick in die zukünftige Diagnostik der ATTR-CM.
„Red Flags“ gezielt erfragen
Wie es nach dem EKG im Diagnosealgorithmus weitergeht, erläuterte Dr. med. Regina Gaub, Potsdam. Sie empfiehlt bei Verdacht auf ATTR-CM den Einsatz bildgebender Verfahren wie PW-Doppler,
Gewebe-Doppler und Messung des 2D global longitudinal Strains. „Mit letztgenannter technischer Methode können Sie ganz klar verifizieren, dass der Strain in den basalen Segmenten verkürzt ist, hingegen aber in den apikalen Segmenten wunderbar funktioniert“, erklärte die Expertin. Für ein einheitliches, strukturiertes Vorgehen ist es empfehlenswert, die erforderlichen methodischen Abläufe zu dokumentieren. Diese Arbeitsanweisungen (SOPs) führen alle Kollegen Schritt für Schritt durch die Diagnose und erinnern auch daran, während der Echokardiografie weitere „Red Flags“ abzufragen, beispielsweise orthopädische Manifestationen wie das beidseitige Karpaltunnelsyndrom – eine typische frühe Symptomatik bei der ATTR-CM. Ein wichtiger Bestandteil der Untersuchungen ist auch die zeitnahe Analyse einer Urin- und Serumprobe zum Ausschluss bzw. Nachweis einer Leichtketten (AL)-Amyloidose. Neben der guten Kommunikation und Dokumentation innerhalb der Praxis ist die interdisziplinäre Kommunikation, beispielsweise zur Nuklearmedizin, wichtig. Hier ist es ratsam, die erforderliche Methodik (Skelettszintigrafie) und den notwendigen Tracer klar zu kommunizieren. Als beweisend für die ATTR-CM gilt die kardiale TracerAnreicherung Perugini-Grad 2 – 3 in der Skelettszintigrafie zusammen mit dem Ausschluss einer ALAmyloidose.
Symptomatische Therapie der Herzinsuffizienz
Professor Harald Rittger, Fürth, stellte die leitlinienbasierte, sym-
96 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
ptomatische Therapie der Herzinsuffizienz vor und betonte die Bedeutung der Therapie mit ACE-I/ ARNI, Betablockern, MRA und SGLT2i, auch „Fantastic Four“ genannt. Dabei sollte stets zeitgleich eine maximale Dosierung aller 4 Medikamentenklassen angestrebt werden. „Und wenn das nicht möglich ist, dann ist es besser, mit niedriger Dosis alle Targets zu tangieren, anstatt mit einem Medikament die volle Dosis aufzutitrieren.“ In den Augen des Experten ist das ein Paradigmenwechsel, da praktizierende Ärzte einer größeren Anzahl von Patienten gerecht werden könnten. Auch ältere Patienten könnten von dieser Therapie profitieren. „Es gibt nach vielen Jahrzehnten, in denen nicht viel passiert ist, nun viele neue therapeutische Möglichkeiten, die wir ausnutzen sollten und müssen, um unseren Patientinnen und Patienten gerecht zu werden“, so Rittger.
Tafamidis 61 mg – die kausale Therapie der ATTR-CM
Wie PD Dr. med. Bettina Heidecker, Berlin, ausführte, ist Tafamidis 61 mg (Vyndaqel®) das derzeit einzige zugelassene Medikament zur Therapie der ATTR-Amyloidose mit Kardiomyopathie. Tafamidis hat eine Klasse-I-Empfehlung bei hereditärer und Wildtyp-ATTRCM bei NYHA-Klasse I oder II. Bei NYHA-Klasse III verwies Heidecker auf die amerikanischen Leitlinien, die Tafamidis auch für diese Patienten empfehlen. In der Phase-III-Studie ATTRACT wurden die 441 Teilnehmer im Verhältnis 2:1:2 auf TafamidisMeglumin 80 mg* (verabreicht als
4 20 mg) versus Tafamidis-Meglumin 20 mg versus Placebo randomisiert. Die beiden Tafamidis-Meglumin-Studienarme wurden gepoolt analysiert. Die ATTR-ACT-Studie zeigte, dass Tafamidis das Mortalitätsrisiko in Monat 30 um 30 % und das Risiko für kardiovaskulär bedingte Hospitalisierungen um 32 % senken konnte. „Zudem war die Lebensqualität der Patienten, die mit Tafamidis behandelt wurden, im Vergleich zur Placebogruppe deutlich besser“, berichtete Heidecker und ergänzte: „Tafamidis hat das Potenzial, die symptomatische Therapie der ATTR-CM nun zu einer kausalen Therapie zu machen.“ Die höhere Awareness für die Erkrankung spiegelt sich sich schon jetzt in einem Anstieg der publizierten Studien wider.
In der abschließenden Diskussion hob Heidecker hervor, dass die Verordnung von Tafamidis 61 mg –insbesondere bei der altersbedingten ATTRwt-CM – ihrer Meinung nach nicht zwangsläufig an eine Altersgrenze gebunden sei. Vielmehr müsse individuell in Anbetracht der Prognose entschieden werden, auch vor dem Hintergrund von Komorbiditäten und dem Erhalt von Lebensqualität.
Elisabeth Wilhelmi, München
Quelle: Symposium „Herzinsuffizienz von A wie Amyloidose bis Z wie Zucker“ anlässlich der 89. Jahrestagung der Deutschen Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. am 13. April 2023 in Mannheim. Veranstalter: Pfizer.
Mit ultraschnellem Mahlzeiteninsulin mehr Flexibilität und Kontrolle beim Diabetesmanagement
Das ultraschnelle Mahlzeiteninsulin Lyumjev®, eine weiterentwickelte Formulierung von Insulin lispro, hat vor 3 Jahren Einzug in die Therapie des Diabetes mellitus gehalten. Bei einer Veranstaltung von Lilly gab Professor Oliver Schnell, München, ein Update zum Einsatz des Mahlzeiteninsulins. Er erläuterte, dass Lyumjev® im Vergleich zum herkömmlichen Insulin lispro eine verbesserte postprandiale Glukosekontrolle ermöglicht und Patienten praktische Vorteile wie eine höhere Flexibilität bei den Mahlzeiten bietet, da kein SpritzEss-Abstand eingehalten werden muss.
Noch genaueres Imitieren der physiologischen Insulinwirkung
* Die für die Behandlung der ATTR-CM zugelassene Formulierung Tafamidis 61 mg ist dosisäquivalent zu 80 mg Tafamidis-Meglumin (4 20 mg).
Lyumjev® zeichnet sich aufgrund seiner Formulierung im Vergleich zum herkömmlichen Insulin lispro durch einen schnelleren Wirkeintritt und eine verkürzte Wirkdauer aus. Somit imitiert es die physiologische Insulinwirkung noch genauer. In den Studien PRONTO-T1D und -T2D zeigten sich unter Lyumjev® im direkten Vergleich weniger postprandiale Glukosespitzen und ein insgesamt flacherer Glukoseverlauf (Abb. 1). Auch die Zeit im Zielbereich kann durch den Einsatz sowohl bei Typ-1- also auch Typ2-Diabetes verlängert werden. Bei Patienten mit Typ-2-Diabetes wurde eine Verbesserung des HbA1cWerts beobachtet. Bei Typ-1-Dia-
97 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
Abbildung
betikern sank mit Lyumjev® zudem in der späten postprandialen Phase (>4 Stunden nach der Mahlzeit) das Risiko von Hypoglykämien. „Durch diese Eigenschaften ermöglicht es Lyumjev® Menschen mit Diabetes, eine bessere Glukosekontrolle zu erreichen. Auf diese Weise kann auch das Risiko von Folgeerkrankungen sinken, die mit starken Glukoseschwankungen assoziiert werden,“ erläuterte Schnell.
Praktischer Nutzen im Alltag
Dank des schnellen Wirkeintritts von Lyumjev® ergeben sich auch praktische Vorteile für die Patienten: Lyumjev® bietet Menschen mit Diabetes mehr Flexibilität bei den Mahlzeiten, da kein Spritz-EssAbstand eingehalten werden muss. Das Insulin kann direkt vor oder falls erforderlich noch bis zu 20 Minuten nach der Mahlzeit verabreicht
werden. „Das ist es, was sich die meisten Patienten wünschen – jederzeit flexibel und ohne Wartezeiten essen zu können“, so die Erfahrung von Schnell. Darüber hinaus ist die Therapie auch gut steuerbar, da wegen der kurzen Wirkdauer von Lyumjev® Korrekturboli gegeben werden können, ohne dass das Risiko für Hypoglykämien infolge überlappender Insulindosen steigt. „Die Patienten erhalten damit mehr Kontrolle über ihr Diabetesmanagement im Alltag, was sich positiv auf Lebensqualität und Motivation auswirken kann“, erklärte Schnell.
Positive Effekte bei einer Umstellung auf Lyumjev®
In einer von Schnell vorgestellten Fallgeschichte konnte ein 62-Jähriger mit Typ-2-Diabetes, der Angst vor Hypoglykämien hatte, von der Umstellung auf Lyumjev® profi-
tieren. Infolge seiner Angst verabreichte er sich einen Korrekturbolus oft erst ab einem Wert von über 250 mg/dl. Auch vergaß er manchmal die Bolusgabe vor den Mahlzeiten. Der Patient wurde aufgrund der hohen postprandialen Glukosewerte auf Lyumjev® umgestellt. Die Werte im ambulanten Glukoseprofil verbesserten sich nach der Umstellung sichtbar. Mehr Zeit im Zielbereich sowie weniger postprandiale Glukosespitzen und Hypoglykämien führten zu einer größeren Zufriedenheit mit der Insulintherapie.
„Insgesamt hat sich Lyumjev® als verträgliches und wirksames Mahlzeiteninsulin erwiesen“, so das Fazit von Schnell. „Es lässt sich durch seine Flexibilität und gute Steuerbarkeit den individuellen Bedürfnissen der Patientinnen und Patienten anpassen und kann so das Diabetesmanagement insgesamt verbessern.“
Elisabeth Wilhelmi, München
98 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH
Kongresse
1: Lyumjev® ist Humalog® in der Kontrolle der postprandialen Glukose signifikant überlegen. * p > 0,05 vs. Humalog® (Quellen: Klaff L et al. Diabetes Obes Metab 2020;22:1799-1807 und Blevins T et al. Diabetes Care 2020;43:2991-2998; © Lilly Deutschland GmbH).
MITTEILUNGEN
Sacubitril/Valsartan jetzt auch zur Behandlung der HFrEF bei Kindern zugelassen
Die chronische Herzinsuffizienz mit linksventrikulärer Dysfunktion (HFrEF) ist eine häufige Ursache für Morbidität und Mortalität im Kindesalter – bis zu einem Drittel der kardiologischen Hospitalisierungen bei pädiatrischen Patienten sind auf eine Herzinsuffizienz zurückzuführen. Diese Hospitalisierungen können mit einem 20-fachen Anstieg des Sterberisikos im Vergleich zu hospitalisierten Kindern ohne Herzinsuffizienz verbunden sein.
Viele Kinder, bei denen eine Herzinsuffizienz diagnostiziert wird, benötigen eine Herztransplantation, bevor sie 5 Jahre alt sind, und fast ein Drittel der Kinder mit Herzinsuffizienz stirbt innerhalb eines Jahres.
Die Erkrankung wird bisher weitestgehend mit Therapien behandelt, die nicht für Kinder zugelassen sind und auf Daten aus Studien mit Erwachsenen beruhen. Daher ist es ein wichtiger Meilenstein,
* Dies gilt bei der Verordnung einer üblichen Anfangsdosis (24 mg/26 mg oder 49 mg/51 mg). Bei pädiatrischen Patienten mit einem Körpergewicht von 40 kg und <50 kg, die keinen ACE-Hemmer oder Angiotensin-II-Rezeptorblocker (ARB) eingenommen oder niedrige Dosierungen dieser Arzneimittel angewendet haben, mit Nierenfunktionsstörung (eGFR <60 ml/min/1,73 m2) oder mittelschwerer Leberfunktionsstörung wird eine halbe Anfangsdosis empfohlen. Diese ist als Granulat zu verabreichen (voraussichtlich verfügbar ab Q4/2023).
dass die Europäische Kommission Sacubitril/Valsartan (Entresto®), das seit 2015 für Erwachsene mit HFrEF verfügbar ist, nun auch für die Behandlung von Patienten mit symptomatischer, chronischer Herzinsuffizienz mit linksventrikulärer Dysfunktion im Alter von 1 bis <18 Jahren in der Europäischen Union zugelassen hat. Der Angiotensin-Rezeptor-Neprilysin-Inhibitor (ARNI) kann bei Kindern und Jugendlichen mit einem Körpergewicht >40 kg in Tablettenform verabreicht werden*. Eine spezielle gewichtsadaptiert zu dosierende Darreichungsform (ein Granulat) für Kinder mit einem Körpergewicht <40 kg wird voraussichtlich im 4. Quartal 2023 zur Verfügung stehen.
Sacubitril/Valsartan
Bedeutender klinischer Nutzen im Vergleich zu bestehenden Therapien
Die Zulassung basiert auf den Ergebnissen der 52-wöchigen PhaseIII-Studie PANORAMA-HF, der größten bislang durchgeführten Studie zur pädiatrischen Herzinsuffizienz, sowie auf der Extrapolation der Daten von Erwachsenen mit Herzinsuffizienz aus der Phase-IIIStudie PARADIGM-HF (vgl. Insert auf S. 100) auf pädiatrische Patienten.
PANORAMA-HF war eine zweiteilige Studie. Ihr erster Teil bestand aus einer offenen Studie zur Dosisfindung. Der zweite Teil war eine randomisierte, doppelblinde, 52-wöchige Studie zum Vergleich von Sacubitril/Valsartan mit dem
Sacubitril/Valsartan (Entresto®) war bisher ausschließlich zugelassen zur Behandlung von Erwachsenen mit HFrEF. Der Angiotensin-Rezeptor-NeprilysinInhibitor (ARNI) entlastet den insuffizienten Herzmuskel auf zweierlei Weise:
• Der Neprilysin-Inhibitor Sacubitril unterstützt die natürliche Wirkung des natriuretischen Peptid-Systems (NPS). Dieses ist für die Produktion von natriuretischen Peptiden (z.B. atrial natriuretic peptide, ANP, brain natriuretic peptide, BNP und C-type natriuretic peptide, CNP) zuständig, die kardioprotektiv wirken, indem sie die Natriurese und Diurese fördern, den Blutdruck senken, volumen- und druckentlastend wirken und das Herz vor der Entwicklung einer bei Herzinsuffizienz auftretenden Fibrose schützen. Das Enzym Neprilysin baut diese Peptide bereits nach kurzer Zeit wieder ab, weshalb sie nur eine begrenzte Wirksamkeit haben. Durch die Blockade von Neprilysin kann Sacubitril diesem Effekt entgegenwirken.
• Der Angiotensin-II-Rezeptorantagonist Valsartan blockiert selektiv den AT1-Rezeptor, der für die blutdrucksteigernden Effekte des Gebwebhormons Angiotensin II verantwortlich ist, das am Renin-Angiotensin-Aldosteron-Systems (RAAS) beteiligt ist. Auf diese Weise verringert Valsartan die schädlichen Effekte des dauerhaft aktiven RAAS auf das Herz. Denn das RAAS reagiert als Folge mangelhafter Durchblutung des Herzens mit einer Reihe von Gegenmaßnahmen: Vasokonstriktion (und dadurch Erhöhung des Blutdrucks) sowie vermehrte Ausschüttung von Aldosteron (und dadurch erhöhte Wasser- und Natriumretention in der Niere).
99 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
Ergebnisse der PARADIGM-HF-Studie
Die Ergebnisse der PARADIGM-HF-Studie mit 8.442 Patienten zeigten im Vergleich zu Enalapril, dass Sacubitril/Valsartan
• das Risiko für kardiovaskulären Tod um 20 % (p < 0,00004) senkte,
• das Risiko für Herzinsuffizienz-bedingte stationäre Einweisungen um 21 % (p < 0,00004) reduzierte und
• das Gesamtsterblichkeitsrisiko um 16 % (p < 0,0005) verringerte.
Insgesamt wurde durch die Behandlung mit Sacubitril/Valsartan das Risiko für den kombinierten primären Endpunkt aus kardiovaskulärem Tod oder Herzinsuffizienz-bedingter stationärer Einweisung um 20 % reduziert (p < 0,0000004).
Im Vergleich zu Enalapril brachen weniger Patienten die Behandlung mit Sacubitril/Valsartan aufgrund von unerwünschten Ereignissen ab. In der Sacubitril/Valsartan-Gruppe wurden häufiger Hypotonien und nicht schwerwiegende Angioödeme, aber seltener Nierenfunktionsstörungen, Hyperkaliämien und Husten im Vergleich zur Enalapril-Gruppe beobachtet.
(Quelle: McMurray J et al. N Engl J Med 2014;371:993-1004)
aktiven Vergleichspräparat Enalapril bei Patienten im Alter von 1 Monat bis <18 Jahren mit Herzinsuffizienz (NYHA/Ross-Klasse II–IV) aufgrund einer systemischen linksventrikulären systolischen Dysfunktion (LVEF <45 % oder fraktionelle Verkürzung ≤22,5 %). Primärer Endpunkt war ein GlobalRank Endpunkt.
Der kardiale Biomarker NT-proBNP (n-terminales pro-B-Typ natriuretisches Peptid), der üblicherweise zur Beurteilung des Schweregrads und der Prognose von Herzinsuffizienz verwendet wird, diente in der Studie als Bridging Biomarker für die Extrapolation von Erwachsenen auf die pädiatrische Population. Die Studie wurde in 30 Ländern und 105 Studienzentren in Nordamerika, Europa, Asien und Lateinamerika durchgeführt.
Die Ergebnisse von PANORAMA HF und der Extrapolation der Daten aus PARADIGM-HF zeigen, dass Sacubitril/Valsartan sowohl bei erwachsenen als auch bei pädiatrischen Patienten im Alter von
1 bis <18 Jahren mit HFrEF zu einer ähnlichen klinisch bedeutsamen Verringerung des NT-proBNP im Vergleich zum Ausgangswert führte. Diese Erkenntnis ermöglicht eine Extrapolation der nachgewiesenen Wirksamkeit bei Erwachsenen auf pädiatrische Patienten. Im Vergleich zum ACE-Hemmer Enalapril, der derzeitigen Standardtherapie bei pädiatrischer Herzinsuffizienz (die jedoch nicht speziell für pädiatrische Herzinsuffizienz zugelassen ist), erzielte Sacubitril/ Valsartan bei dem primären „Global Rank“-Endpunkt eine numerisch deutlichere sowie bei den sekundären Endpunkten NYHA/ Ross-Klasse und Patient Global Impression of Severity (PGIS)Score-Veränderung vergleichbare Verbesserungen gegenüber dem Ausgangswert.
Die Sicherheit und Verträglichkeit von Sacubitril/Valsartan bei pädiatrischen Patienten entsprach derjenigen, die bei Erwachsenen beobachtet wurde
B. S.
Herzinsuffizienz nach Dekompensation: Neues Positionspapier der ESC empfiehlt Vericiguat bei Worsening Heart Failure
Das Potenzial von Vericiguat (Verquvo®) zur Risikoreduktion von kardiovaskulärem Tod oder herzinsuffizienzbedingter Hospitalisierung bei Erwachsenen mit Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF) nach Dekompensation wurde in einem wichtigen neuen Positionspapier anerkannt. Die Heart Failure Association (HFA) der European Society of Cardiology (ESC) empfiehlt den Therapiebeginn mit Vericiguat als Erweiterung der „vier Säulen der HFrEF-Therapie“ bei symptomatischen Patienten mit einer linksventrikulären Ejektionsfraktion (LVEF) <45 % nach einer Verschlechterung der Herzinsuffizienz (worsening heart failure, WHF).
Ein WHF-Ereignis ist eine Eskalation der Herzinsuffizienz bei Patienten mit bereits bestehender Herzinsuffizienz, die eine Intensivierung der Therapie erfordert. Auch bei Patienten, die eine leitliniengerechte Vierfachtherapie erhalten, bleibt das Risiko für WHF-Ereignisse und Tod bestehen, was die Notwendigkeit weiterer Maßnahmen – wie eine Behandlung mit Vericiguat – verdeutlicht. Patienten mit Herzinsuffizienz werden durchschnittlich einmal pro Jahr ins Krankenhaus eingeliefert und mit jeder weiteren Dekompensation verschlechtert sich die Herzfunktion unwiederbringlich. Trotz der Fortschritte in der Behandlung sterben etwa 30 % der Patienten mit diagnostizierter Herzinsuffizienz innerhalb eines Jahres.
100 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
Vericiguat
Vericiguat (Verquvo®) ist ein einmal täglich oral zu verabreichender Stimulator der löslichen Guanylatzyklase (sGC), einem wichtigen Enzym eines für die Herzinsuffizienz entscheidenden Signalweges (NO-sGC-cGMP). Wenn NO an sGC bindet, katalysiert das Enzym die Synthese von intrazellulärem zyklischem Guanosinmonophosphat (cGMP), einem zweiten Botenstoff, der eine zentrale Rolle bei der Regulierung der Funktion des Herzens und der Blutgefäße spielt. Herzinsuffizienz ist mit einer verminderten NO-Synthese und Aktivität der sGC verbunden, was zu einer Beeinträchtigung der Herzund Gefäßfunktion führen kann. Durch die direkte Stimulierung der sGC, unabhängig von und synergistisch mit NO, erhöht Vericiguat die intrazellulären cGMP-Spiegel, was der Beeinträchtigung von Herz und Gefäßen entgegenwirkt. In der EU ist Vericiguat zugelassen zur Behandlung der symptomatischen, chronischen Herzinsuffizienz bei erwachsenen Patienten mit reduzierter Ejektionsfraktion, die nach einem kürzlich aufgetretenen Dekompensationsereignis, das eine i.v.-Therapie erforderte, stabilisiert wurden.
Das neue Positionspapier der HFA ESC speziell zur WHF ist erstmals von der Fachgesellschaft herausgegeben worden. Die fortschreitende Herzinsuffizienz stellt bekanntermaßen eine große Belastung für die Gesundheitssysteme dar, damit ist die Verhinderung von WHF-Ereignissen ein Hauptziel der Herzinsuffizienzbehandlung. Nach einem WHF-Ereignis müssen 56 % der Patienten innerhalb von 30 Tagen erneut hospitalisiert werden.
E. W.
IMPRESSUM
OFFIZIELLES ORGAN DER DEUTSCHEN GESELLSCHAFT FÜR ARTERIOSKLEROSEFORSCHUNG
Herausgeber:
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School,Salmon Pool Lane, Exeter EX2 4SG, UK
Prof. Dr. med. Wolfgang Koenig Deutsches Herzzentrum München
Technische Universität München
Lazarettstraße 36 80636 München
Wissenschaftlicher Beirat:
Prof. Dr. med. T. von Arnim (Kardiologie), München
Prof. Dr. med. G. V. R. Born (Arterioskleroseforschung), London
Prof. Dr. med. C. Diehm (Angiologie), Karlsbad
Priv.-Doz. Dr. med. Dr. phil. C. Drosde (Kardiologie), Freiburg
Dr. med. J. Dyerberg MD, Ph. D. (Klin. Chemie), Aalborg Sygehus, Dänemark Univ.-Prof. Dr. med. H. W. Eichstädt, (Kardiologie), Berlin
Doz. Dr. rer. nat. F.-D. Ernst (Hämorheologie), Dresden
Dr. med. J. Gehring (Kardiologie, Rehabilitation), München
Prof. Dr. med. J. D. Gruß (Gefäßchirurgie), Kassel
Prof. Dr. J. Harenberg (Hämostaseologie), Mannheim
Prof. Dr. med. L. Heilmann (Gynäkologie), Rüsselsheim
Prof. Dr. med. H. M. Hoffmeister (Kardiologie), Solingen
Prof. Dr. med. H. U. Janka (Diabetologie), München
Dr. med. J. Janzen MPhil (Pathologie), Bern, Schweiz
Prof. Dr. med. L. Kollár M.D., PhD (Gefäßchirurgie), Universität Pécs, Ungarn Prof. Dr. med. M. Marshall (Phlebologie), Rottach Egern
Prof Dr. med. J. Matsubara (Chirurgie), Ishikawa, Japan
Prof. Dr. med. G. Mchedlishvilli (Mikrozirkulation), Tbilisi, Georgien
Prof. Dr. med. V. Mitrovic (Kardiologie, Klinische Pharmakologie), Bad Nauheim
Prof. Dr. med. H. Mörl (Angiologie), Mannheim
Prof. Dr. med. F. J. Neumann (Kardiologie), Bad Krozingen
Prof. Dr. med. K. L. Resch (Medizin-Statistik), Bad Elster
Prof. Dr. med. G. Rettig (Kardiologie), Homburg
PD Dr. med. Rainer Röttgen (Radiologie), Berlin
Prof. Dr. med. G. Schmid-Schönbein (Biomechanik), La Jolla, USA
Prof. Dr. med. H. Schmid-Schönbein (Physiologie), Aachen
Prof. Dr. med. A. Schrey (Pharmakologie), Düsseldorf
Prof. Dr. med. H. Sinzinger (Nuklearmedizin), Wien, Österreich
Prof. Dr. med. T. Störk (Kardiologie, Angiologie), Göppingen
Prof. Dr. med. I. Szirmai M.D. (Neurologie), Universität Budapest, Ungarn
Prof. Dr. med. G. Trübestein (Angiologie), Bonn
Prof. Dr. med. B. Tsinamdzvrishvili (Kardiologie, Hypertonie), Tbilisi, Georgien
Prof. Dr. med. W. Vanscheidt (Dermatologie), Freiburg
Prof. Dr. med. H. Weidemann (Kardiologie, Sozialmedizin), Bad Krozingen
Schriftleitung:
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School, Salmon Pool Lane, Exeter EX2 4SG, UK
E-Mail: Edzard.Ernst@pms.ac.uk
Tel: +44 (0) 1392 726029
Fax: +44 (0) 1392 421009
Die Zeitschrift erscheint 6-mal im Jahr; Jahresabonnement 27,–; Einzelheft 5,50, inklusive MwSt., zuzüglich Versandspesen. Der Abonnementpreis ist im voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout:
HGS5 GmbH – Rolf Wolle (verantwortlich) Schwabacherstr. 117, 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna (verantwortlich) Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden in der PERFUSION personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz:
Rolf Wolle, Schwabacherstr. 117, 90763 Fürth
Druck und Verarbeitung:
DRUCK_INFORM_W.R.
Roland Welker
Austraße 7, 96114 Hirschaid
PERFUSION is listed in Current Contents/Clinical Medicine (CC/CM) and listed in The Genuine Article.
Verlag PERFUSION
101 Perfusion 3/2023 36. Jahrgang © Verlag PERFUSION GmbH Mitteilungen
GmbH Storchenweg 20 90617 Puschendorf Telefon: 09101/990 11 10 Fax: 09101/990 11 19 www.Verlag-Perfusion.de E-Mail: perfusion@t-online.de
PERFUSION VERL AG PERFUSION Journal
LDL-C
Effektive und langanhaltende LDL-C-Senkung a, 1
Zwei Injektionen pro Jahr (nach Initialdosis) b, 2
LEQVIO wird bei Erwachsenen mit primärer Hypercholesterinämie (heterozygot familiär und nicht-familiär) oder gemischter Dyslipidämie zusätzlich zu diätetischer Therapie angewendet.2, §
AM-RL: Arzneimittelrichtlinie, LDL-C: Low Density Lipoprotein Cholesterin a Daten einer gepoolten Analyse der drei zulassungsrelevanten Studien ORION-9, -10 und -11 zeigten eine zeitlich gemittelte, placebokorrigierte LDL-C-Senkung um 50,5 % (p < 0,0001) zwischen Monat 3 und 18 (Tag 90–540) im Vergleich zum Ausgangswert zusätzlich zu einer maximal tolerierten Statin-Therapie und ggf. weiteren lipidsenkenden Medikamenten. LEQVIO wurde an Tag 1 und Tag 90, gefolgt von zusätzlichen Injektionen in 6-Monats-Intervallen an Tag 270 und Tag 450, verabreicht.1 b Einzelne subkutane Injektion zu Behandlungsbeginn, nach 3 Monaten und danach alle 6 Monate.2 § Verordnungskriterien entsprechend AM-RL Anlage III Nr. 35c.
1. Wright RS, et al. J Am Coll Cardiol. 2021;77(9):1182–1193. https://doi.org/10.1016/j.jacc.2020.12.058.
LEQVIO® 284 mg Injektionslösung in einer Fertigspritze
2.

Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Wirkstoff: Inclisiran. Zusammensetzung: Arzneimittel wirksame Bestandteile: Jede Fertigspritze enthält Inclisiran-Natrium entsprechend 284 mg Inclisiran in 1,5 ml Lösung. Jeder Milliliter enthält Inclisiran-Natrium entsprechend 189 mg Inclisiran. Sonstige Bestandteile: Wasser für Injektionszwecke, Natriumhydroxid, konzentrierte Phosphorsäure. Anwendungsgebiete: Leqvio wird bei Erwachsenen mit primärer Hypercholesterinämie (heterozygot familiär und nicht-familiär) oder gemischter Dyslipidämie zusätzlich zu diätetischer Therapie angewendet: in Kombination mit einem Statin oder einem Statin mit anderen lipidsenkenden Therapien bei Patienten, die mit der maximal tolerierbaren Statin-Dosis die LDL-C-Ziele nicht erreichen, oder allein oder in Kombination mit anderen lipidsenkenden Therapien bei Patienten mit Statin-Intoleranz oder für welche ein Statin kontraindiziert ist. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Häufig: Reaktionen an der Injektionsstelle. Verschreibungspflichtig. Weitere Hinweise: Siehe Fachinformation. Stand: März 2022 (MS 02/22.3). Novartis Pharma GmbH, Roonstr. 25, 90429 Nürnberg. Tel.: (0911) 273-0, Fax: (0911) 273-12 653. www.novartis.de
Licensed from Alnylam Pharmaceuticals, Inc.
LEQVIO® aktuelle Fachinformation.







