JAHRGANG 34
HEFT 4
Oktober 2025

JAHRGANG 34
HEFT 4
Oktober 2025
JOURNAL OF PHARMACOLOGY AND THERAPY
Kombinationen aus Thymiankraut und Primelwurzel bei akuter Bronchitis: Wirkeintritt im Vergleich mit Placebo
Chronische spontane Urtikaria: Aktuelle Daten bestätigen die Wirksamkeit von Remibrutinib
Plaque-Psoriasis: Gelenkbeteiligung und Sonderlokalisationen sprechen für einen frühzeitigen Biologika-Einsatz
Neovaskuläre altersabhängige Makuladegeneration: Faricimab reduziert retinale Flüssigkeit schnell und anhaltend
Impfung der Mutter mit Abrysvo® schützt Säuglinge vor RSV-Infektion
siRNA Givosiran bei akuter hepatischer Porphyrie: Real-World-Daten belegen klinischen Nutzen auch bei chronischem Verlauf
Lecanemab – ein Meilenstein in der Therapie der frühen Alzheimer-Krankheit
Givinostat – eine neue Option zur Behandlung der DuchenneMuskeldystrophie

Therapie auf www.trimbow.de
Trimbow® 88/5/9 im Pulverinhalator NEXThaler®

Trimbow® 87/5/9

Trimbow® 172/5/9

* Trimbow ® 88/5/9 Mikrogramm Pulver zur Inhalation ist zugelassen zur Erhaltungstherapie bei erwachsenen Patienten mit moderater bis schwerer COPD, die mit einer Kombination aus einem inhalativen Kortikosteroid und einem langwirksamen Beta-2-Agonisten oder einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem langwirksamen Muskarin-Rezeptor-Antagonisten nicht ausreichend eingestellt sind.
** Asthma: Trimbow 87/5/9 Mikrogramm Druckgasinhalation, Lösung ist zugelassen zur Erhaltungstherapie bei erwachsenen Patienten mit Asthma, die mit einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem mitteldosierten inhalativen Kortikosteroid nicht ausreichend eingestellt sind und bei denen im vergangenen Jahr mind. eine Asthma- Exazerbation aufgetreten ist. COPD: Trimbow ® 87/5/9 Mikrogramm Druckgasinhalation, Lösung ist zugelassen zur Erhaltungstherapie bei erwachsenen Patienten mit moderater bis schwerer COPD, die mit einer Kombination aus einem inhalativen Kortikosteroid und einem langwirksamen Beta-2-Agonisten oder einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem langwirksamen Muskarin-Rezeptor-Antagonisten nicht ausreichend eingestellt sind.
*** Trimbow ® 172/5/9 µ g Druckgasinhalation, Lösung ist zugelassen zur Erhaltungstherapie bei erwachsenen Patienten mit Asthma, die mit einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem hochdosierten inhalativen Kortikosteroid nicht ausreichend eingestellt sind und bei denen im vergangenen Jahr mind. eine Asthma-Exazerbation aufgetreten ist. Trimbow 87 Mikrogramm/5 Mikrogramm/9 Mikrogramm Druckgasinhalation, Lösung. Trimbow 172 Mikrogramm/5 Mikrogramm/9 Mikrogramm Druckgasinhalation, Lösung. Trimbow 88 Mikrogramm/5 Mikrogramm/9 Mikrogramm Pulver zur Inhalation. Zus.: Trimbow 87/5/9: Jede abgegebene Dosis (die das Mundstück verlässt) enthält 87 Mikrogramm Beclometasondipropionat (Ph.Eur.), 5 Mikrogramm Formoterolfumarat-Dihydrat (Ph.Eur.) u. 9 Mikrogramm Glycopyrronium (als 11 Mikrogramm Glycopyrroniumbromid (Ph.Eur.)). Jede abgemessene Dosis (die das Ventil verlässt) enthält 100 Mikrogramm Beclometasondipropionat (Ph.Eur.), 6 Mikrogramm Formoterolfumarat-Dihydrat (Ph.Eur.) u. 10 Mikrogramm Glycopyrronium (als 12,5 Mikrogramm Glycopyrroniumbromid (Ph.Eur.)). Trimbow 172/5/9: Jede abgegebene Dosis (die das Mundstück verlässt) enthält 172 Mikrogramm Beclometasondipropionat (Ph.Eur.), 5 Mikrogramm Formoterolfumarat-Dihydrat (Ph.Eur.) und 9 Mikrogramm Glycopyrronium (als 11 Mikrogramm Glycopyrroniumbromid (Ph.Eur.)). Jede abgemessene Dosis (die das Ventil verlässt) enthält 200 Mikrogramm Beclometasondipropionat (Ph.Eur.), 6 Mikrogramm FormoterolfumaratDihydrat (Ph.Eur.) und 10 Mikrogramm Glycopyrronium (als 12,5 Mikrogramm Glycopyrroniumbromid (Ph.Eur.)). Trimbow 88/5/9 Pulver zur Inhalation: Jede abgegebene Dosis (die das Mundstück verlässt) enthält 88 Mikrogramm Beclometasondipropionat (Ph.Eur.), 5 Mikrogramm Formoterolfumarat-Dihydrat (Ph.Eur.) u. 9 Mikrogramm Glycopyrronium (als 11 Mikrogramm Glycopyrroniumbromid (Ph. Eur.)). Jede abgemessene Dosis enthält 100 Mikrogramm Beclometasondipropionat (Ph.Eur.), 6 Mikrogramm Formoterolfumarat-Dihydrat (Ph.Eur.) u. 10 Mikrogramm Glycopyrronium (als 12,5 Mikrogramm Glycopyrroniumbromid (Ph.Eur.)). Sonst. Bestandteile: Trimbow 87/5/9 und Trimbow 172/5/9: Ethanol, Salzsäure, Norfluran (Treibmittel). Trimbow 88/5/9 Pulver zur Inhalation: Lactose-Monohydrat, Magnesiumstearat (Ph.Eur.). Anw.: Trimbow 87/5/9: COPD: Zur Erhaltungstherapie b. erw. Patienten mit moderater bis schwerer chronisch obstruktiver Lungenerkrankung (COPD), die mit einer Kombination aus einem inhalativen Kortikosteroid u. einem langwirksamen Beta-2 Agonisten od. einer Kombination aus einem langwirksamen Beta-2-Agonisten u. einem langwirksamen Muskarin-Antagonisten nicht ausreichend eingestellt sind. Asthma: Zur Erhaltungstherapie b. erw. Patienten mit Asthma, die mit einer Kombination aus einem langwirksamen Beta 2-Agonisten u. einem mitteldosierten inhalativen Kortikosteroid nicht ausreichend eingestellt sind u. bei denen im vergangenen Jahr mind. eine Asthma-Exazerbation aufgetreten ist. Trimbow 172/5/9: Zur Erhaltungstherapie b. erw. Patienten mit Asthma, die mit einer Kombination aus einem langwirksamen Beta-2-Agonisten u. einem hochdosierten inhalativen Kortikosteroid nicht ausreichend eingestellt sind u. bei denen im vergangenen Jahr mind. eine Asthma-Exazerbation aufgetreten ist. Trimbow 88/5/9 Pulver zur Inhalation: COPD: Zur Erhaltungstherapie bei erwachsenen Patienten mit moderater bis schwerer chronisch obstruktiver Lungenerkrankung (COPD), die mit einer Kombination aus einem inhalativen Kortikosteroid und einem langwirksamen Beta-2-Agonisten oder einer Kombination aus einem langwirksamen Beta-2-Agonisten und einem langwirksamen Muskarin-Antagonisten nicht ausreichend eingestellt sind. Gegenanz.: Überempfindlichkeit gegen die Wirkstoffe od. einen d. sonst. Bestandteile. Nebenw.: Trimbow 87/5/9 Druckgasinhalation, Trimbow 172/5/9 Druckgasinhalation u. Trimbow 88/5/9 Pulver zur Inhalation: Risiko eines paradoxen Bronchospasmus. Allergische Reakt. wie Hautallergien, Quaddeln, Hautjucken, Hautausschlag, Hautrötungen, Schwellung d. Haut od. Schleimhäute, insbes. im Augen-, Gesichts-, Lippen- u. Rachenbereich. Akutes Auftreten eines Engwinkelglaukoms mit Anzeichen wie Augenschmerzen od. -beschwerden, vorübergehend verschwommene Sicht, Sehen v. Lichtkreisen od. farbigen Bildern in Verb. m. geröteten Augen. Pneumonie mit Symptomen wie Fieber od. Schüttelfrost, vermehrter Bildung v. Schleim, Farbänderung des Schleims, stärkerem Husten od. verstärkten Atembeschwerden. Halsschmerzen, juckende, laufende od. verstopfte Nase u. Niesen, Pilzinfektionen (Mund, Hals, Ösophagus, vaginal, im Brustraum), Heiserkeit, Kopfschmerzen, Harnwegsinfektion. Grippe, Entzündung d. Nasennebenhöhlen, Ruhelosigkeit, Zittern, Schwindel, gestörter od. verminderter Geschmackssinn, Taubheitsgefühl, Ohrentzündung, unregelmäßiger Herzschlag, Veränderungen im EKG, ungewöhnlich schneller Herzschlag u. Herzrhythmusstörungen, Herzklopfen, Gesichtsrötung, erhöhte Durchblutung in bestimmten Körpergeweben, Asthmaanfall, Husten mit od. ohne Auswurf, Rachenreizung, Nasenbluten, Mundtrockenheit, Durchfall, Schluckbeschwerden, Übelkeit, Magenverstimmung, Magenbeschwerden nach einer Mahlzeit, brennendes Gefühl auf den Lippen, Zahnkaries, Hautausschlag, Quaddeln, Juckreiz, Entzündung d. Mundschleimhaut mit od. ohne Geschwüre, vermehrtes Schwitzen, Muskelkrämpfe u. -schmerzen, Schmerzen in Armen, Beinen, Muskeln, Knochen od. Gelenken des Brustraums, Müdigkeit, Anstieg des Blutdrucks, Abnahme einiger Blutwerte, z. B. Granulozyten, Kalium od. Cortisol, Anstieg einiger Blutwerte: Blutzucker, C-reaktives Protein, Anz. d. Blutplättchen, Insulin, freie Fettsäuren od. Ketone. Verminderter Appetit, Schlafstörungen, starke Brustschmerzen, Gefühl eines ausgebliebenen od. zusätzlichen Herzschlags, ungewöhnlich langsamer Herzschlag, Verschlechterung v. Asthma, Austreten v. Blut aus einem Gefäß in das umgebende Gewebe, Abfall des Blutdrucks, Schwäche, Schmerzen im hinteren Bereich v. Mund u. Rachen, Entzündung u. Rötung des Rachens, trockener Hals, Schmerzen u. Schwierigkeiten beim Wasserlassen u. häufiges Wasserlassen, Nierenentzündung. Niedrige Anz. an Blutplättchen, Atemnot od. Kurzatmigkeit, Anschwellen v. Händen u. Füßen, Wachstumsverzögerung bei Kindern u. Jugendl., verschwommenes Sehen. Bei langfristiger Anwendung hochdosierter inhalativer Kortikosteroide: Nebennierensuppression, Abnahme d. Knochenmineraldichte, Katarakt, bei Kindern häufiger: Depression, Angstgefühl, Nervosität, Erregtheit od. Reizbarkeit. Verschreibungspflichtig. Chiesi GmbH, Hamburg. Stand: März 2022
Vor gut 50 Jahren beklagte ein britischer Epidemiologe namens Archibald („Archie“) Cochrane, dass wissenschaftliche Erkenntnisse in der klinischen Forschung für den klinischen Alltag nicht oder nur mit großer zeitlicher Verzögerung zur Verfügung stehen [1]. Er benannte auch den Grund: „Es ist sicherlich ein großer Kritikpunkt an unserem Berufsstand, dass wir keine kritische Zusammenfassung aller relevanten randomisierten kontrollierten Studien nach Fachgebiet oder Unterfachgebiet erstellt und diese regelmäßig angepasst haben.“
20 Jahre später war die Zeit reif, dieser Kritik mit einer internationalen Initiative entgegenzuwirken, der Cochrane Collaboration (heute meist einfach als „Cochrane“ bezeichnet). Wissenschaftler auf der ganzen Welt waren eingeladen, sich der Initiative anzuschließen und zu konkreten, selbst wählbaren klinischen Fragestellungen eine umfassende Übersichtsarbeit zu erstellen. Neu war zu diesem Zeitpunkt, dass Suchkriterien und Suchmethoden – insbesondere die abgefragten Datenbanken, die konkret verwendete Such-Syntax und die Regeln der Bearbeitung – genau beschrieben werden mussten. So war es theoretisch für jeden Interessierten möglich, einen Cochrane-Review für sich selbstständig zu aktualisieren, indem die Suchanfragen für den Zeitraum, der im CochraneReview nicht mehr erfasst wurde, zur Anwendung gebracht und die zusätzlichen Originalarbeiten für eine aktualisierte Zusammenfassung mit herangezogen werden konnten. Neu war auch, dass ein Team, das ein Thema auf Antrag zugesprochen bekam, einwilligen musste, das Thema als „life long commitment“ in Abständen zu aktualisieren.
Schon für Archie Cochrane hatte die Studienmethodik der randomisiert-kontrollierten Studie (randomized controlled trial, RCT) für Fragestellungen zur Wirksamkeit einer Intervention eine herausragende Bedeutung. Die strikt zufällige Zuteilung der Teilnehmer zu zwei oder
mehreren Gruppen ist bis heute die einzige zuverlässige Methode, um (bei größeren Fallzahlen in den einzelnen Gruppen) „Strukturgleichheit“ herzustellen, also eine einigermaßen gleiche Verteilung aller Merkmale, also auch der gar nicht bekannten, in den verschiedenen Gruppen zu gewährleisten – die Grundvoraussetzung dafür, dass Unterschiede am Ende auf die verschiedenen Interventionen zurückgeführt werden können und nicht etwa durch „unkontrollierte Paralleleffekte“ gestört bzw. verzerrt werden.
Einer der Gründerväter der Cochrane Collaboration neben Iain Chalmers, der 1992 als erster Direktor der neu gegründeten UK Cochrane Centre berufen wurde, der Epidemiologe und Internist David Sacket, beschrieb zusammen mit weiteren zentralen Protagonisten in einem bis heute aktuellen und von der Cochrane Collaboration noch immer auf ihren Seiten zitierten Artikel im British Medical Journal Anliegen und Ziel der auf die Zusammenschau der wissenschaftlichen Erkenntnisse zu einer Fragestellung aufbauenden evidenzbasierten Medizin (EbM): „Evidenzbasierte Medizin ist die gewissenhafte, explizite und umsichtige Nutzung der aktuell besten Evidenz bei Entscheidungen über die Behandlung einzelner Patienten. Die Praxis der evidenzbasierten Medizin bedeutet die Integration individueller klinischer Expertise mit den besten verfügbaren externen klinischen Erkenntnissen aus systematischer Forschung.“ Seit dieser Zeit hat sich die EbMMethodik natürlich weiter entwickelt. Es wurde immer offensichtlicher, dass schlecht kontrollierte Störfaktoren Studienergebnisse beeinflussen können. Deshalb wur-

Seit in jüngster Zeit AnwendungsProf. Dr. med. Karl-Ludwig Resch
den die für den Review herangezogenen Einzelstudien immer rigideren „Qualitätskontrollen“ unterzogen [3]. Das wäre ja grundsätzlich zu begrüßen, wenn nicht parallel das „Lager der Methodiker“ immer deutlicher die Oberhand gegenüber dem „Lager der Kliniker“ gewonnen hätte. Auf gut deutsch: Die klinische Relevanz einer Fragestellung (externe Validität) wurde in den letzten 30 Jahren gegenüber der Qualität des methodologischen Ansatzes (interne Validität) immer mehr marginalisiert. Immer mehr Reviews kommen aus „Review-Fabriken“ („paper mills“ [4]) und sind folgerichtig methodisch exzellent, aus fachlicher Perspektive aber nicht selten peinlich oberflächlich bis falsch, da dieselben Methodiker sich ein Thema nach dem anderen vornehmen, ohne wirkliches Verständnis des klinischen Problems bzw. der „klinischen Dimension“. Keine Spur von der von Sackett beschriebenen „Integration“ (s.o.).
möglichkeiten im Bereich der sog. künstlichen Intelligenz (KI) immer mehr verfügbar werden, werden diese genau deshalb potenziell zur existenziellen Bedrohung für Cochrane in seiner heutigen Form. Mit KI meine ich natürlich nicht banale Tools wie den inzwischen z.B. von Google standardmäßig angeboten „AI overview“ über den eigentlichen Suchergebnissen. Ich meine vielmehr spezielle Dienste wie z.B. Elicit, das „automated search, screening, data extraction, and report generation“ als Dienstleistung anbietet. Wer zahlt, bekommt einen oberflächlich wirklich professionell wirkenden systematischen Review. Hochprofessionell, was die angebotene Dienstleistung anbelangt, vollständig unqualifiziert, was die „medizinische Beurteilung“ (das „clinical reasoning“) anbelangt. Handwerklich eher profunder als das, was typischerweise aus den Review-Fabriken kommt, mit vergleichbar nicht vorhandener klinischer Fachkompetenz, dafür aber unvergleichlich schneller. Keine gute Perspektive für die Cochrane Collaboration mit ihren aktuellen Prioritäten, wenn es nicht gelingt, wieder zuverlässig ausreichend klinische Fachkompetenz (und damit auch die persönliche berufliche Empirik) bei der Erstellung von Übersichtsarbeiten einzubinden.
Karl-Ludwig Resch, Nürnberg
Kombinationen aus Thymiankraut und Primelwurzel bei akuter Bronchitis: Wirkeintritt im Vergleich mit Placebo 100 Rainer Brünjes, Horst Lorenz
Quellen
1 Cochrane AL (1972) Effectiveness and efficiency: Random reflections on health services. Nuffield Trust. https:// www.nuffieldtrust.org.uk/sites/default/files/2017-01/effectiveness-andefficiency-web-final.pdf)
2 Sackett DL et al. Evidence based medicine: what it is and what it isn’t. BMJ 1996;312:71-72
3 Sterne JAC et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898
4 Aronson JK. When I use a word … Research integrity sleuths. BMJ 2025; 390:r1800
AKTUELLE THERAPIEKONZEPTE FÜR DIE PRAXIS
Chronische spontane Urtikaria: Aktuelle Daten bestätigen die Wirksamkeit von Remibrutinib 108
Plaque-Psoriasis: Gelenkbeteiligung und Sonderlokalisationen sprechen für einen frühzeitigen Biologika-Einsatz
Neovaskuläre altersabhängige Makuladegeneration: Faricimab reduziert retinale Flüssigkeit schnell und anhaltend
Impfung der Mutter mit Abrysvo® schützt Säuglinge vor RSV-Infektion
110
114
116
siRNA Givosiran bei akuter hepatischer Porphyrie: Real-World-Daten belegen klinischen Nutzen auch bei chronischem Verlauf 118
Lecanemab – ein Meilenstein in der Therapie der frühen Alzheimer-Krankheit 120
Givinostat – eine neue Option zur Behandlung der Duchenne-Muskeldystrophie 123
Wissenswertes 106, 113, 124 Kongresse 126























Erwägen Sie für Ihre geeigneten erwachsenen PAH-Patienten dem Behandlungsplan ®WINREVAIR hinzuzufügen, den ersten und einzigen zugelassenen Aktivin-Signalweg-Inhibitor bei 1,2PAH.

* Für den Claim „Zeit, dass sich was dreht.“ besteht keine Verbindung, keine geschäftliche Beziehung und keine Assoziation zu Urhebern, deren Werken, Marken oder sonstigen Schutzrechten. | ** WINREVAIR® ist, in Kombination mit anderen Therapien gegen pulmonale arterielle Hypertonie (PAH), für die Behandlung von PAH bei erwachsenen Patienten mit der WHO-Funktionsklasse (FK) II bis III zur Verbesserung der körperlichen Leistungsfähigkeit angezeigt.2
Referenzen: 1. European Medicines Agency (EMA), Positive CHMP opinion on first-in-class medicine to treat pulmonary arterial hypertension; www.ema.europa.eu/en/news/positive-chmpopinion-first-class-medicine-treat-pulmonary-arterial-hypertension (eingesehen am 30.07.2025) | 2. Fachinformation WINREVAIR®, Stand März 2025
www.msd.de
Winrevair® 45 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Winrevair® 60 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung

Wirkstoff: Sotatercept Zus.: Arzneil. wirks. Bestandt.: 1 Durchstechfl. enth. 45 bzw. 60 mg Sotatercept. Nach Rekonstitution enth. jeder ml 50 mg Sotatercept. Sonst. Bestandt.: Citronensäure-Monohydrat (E 330), Natriumcitrat (E 331), Polysorbat 80 (E 433), Saccharose, Wasser für Injekt.-zwecke. Anw.: In Komb. m. anderen Ther. gegen pulmonale arterielle Hypertonie (PAH) für die Behandl. von PAH bei erw. Pat. m. d. WHO-Funktionsklasse (FK) II bis III zur Verbesserung der körperlichen Leistungsfähigkeit. Gegenanz.: Überempf.-keit gg. d. Wirkstoff od. e. d. sonst. Bestandt. Pat. m. e. konstanten Thrombozytenzahl < 50 × 109/l vor Beginn d. Behandl. Vorsicht bei: Pat. m. Erythrozytose, bei d. ein erhöh. Risiko für thromboembolische Ereignisse besteht; Pat., d. zusätzlich eine Prostacyclin-Inf. erhalten; Pat., d. Antithrombotika erhalten; Pat. m. niedriger Thrombozytenzahl; Pat. ≥ 65 Jahre Nicht empf.: Anw. während Schwangerschaft u. bei Pat. im gebärfähigen Alter, d. nicht verhüten. Nebenw.: Sehr häufig: Thrombozytopenie; Hämoglobin erhöht. Schwindelgefühl; Kopfschmerz. Epistaxis. Diarrhoe. Teleangiektasie; Ausschlag. Häufig: Zahnfleischbluten. Erythem. Jucken an der Injekt.-stelle. Erhöhter Blutdruck. Warnhinw.: Stillen während Behandl. und für 4 Mon. nach der letzten Behandl.-dosis. unterbrechen. Verschreibungspflichtig. Bitte lesen Sie vor Verordnung von Winrevair® die Fachinformation! Pharmazeutischer Unternehmer: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, Niederlande; Lokaler Ansprechpartner: MSD Sharp & Dohme GmbH, Levelingstr. 4a, 81673 München MSD MedInfo: Tel.: +49 (0) 89 20 300 4500, E-Mail: medinfo@msd.de Stand: 03/2025 (RCN: 000027561-DE)
ZUSAMMENFASSUNG
Hintergrund: Die vorliegende statistische Re-Analyse von zwei klinischen Studien untersuchte die Anwendung von zwei pflanzlichen Hustentherapeutika (Bronchicum® Tropfen, BCUM-T05®, und Bronchicum® Elixir, BCUM-E05®) bei Patienten mit akuter Bronchitis. Das Ziel war es, mittels statistischer Analysen einen Vergleich der Wirksamkeit von Bronchicum® Elixir (BCUM-E05®) gegenüber Placebo zu ermöglichen.
Patienten und Methodik: Zunächst wurden die Baselinedaten ausgewählter klinischer Parameter beider Gruppen aus Studie 1 (150 Patienten) und Studie 2 (189 Patienten) auf statistisch signifikante Unterschiede geprüft. Für den Fall einer eindeutigen Homogenität zwischen allen Gruppen sollten dann die Daten der Placebogruppe aus Studie 1 und der Bronchicum® ElixirGruppe (BCUM-E05®) weiteren statistischen Analysen unterzogen werden. Dabei sollten die Daten aus den Patiententagebüchern verwendet werden (jeweils die ersten 5 Tage der Therapie). Ergebnisse: Die Baselinewerte aller 4 Gruppen aus den beiden Studien zeigten ein hohes Maß an Übereinstimmung. In beiden Studien nahm der mittlere BronchitisSeverity-Score (BSS) im betrachteten Zeitraum der ersten 5 Tage ab. Bei BCUM-T05® war vom zweiten Behandlungstag zur Nacht bis Tag 5 ein signifikanter Unterschied gegenüber Placebo nachweisbar. Bei BCUM-E05® war ab dem dritten Behandlungstag morgens bis Tag 5 ein signifikanter Unterschied gegenüber Placebo nachweisbar.
Schlussfolgerung: Durch den statistischen Vergleich wird die Annahme gestützt, dass auch BCUM-E05® in seiner Wirksam-
Rainer Brünjes1, Horst Lorenz2
1 Cassella-med GmbH & Co. KG, Köln
2 Büro für Biometrie und Statistik, Neuberg
Die akute Bronchitis ist eine häufige Erkrankung, die bei vielen Patienten dazu führt, ärztliche Hilfe zu suchen [1]. Eine medikamentöse Therapie der Hustensymptomatik ist mit chemischen oder pflanzlichen Wirkstoffen möglich [2]. In einer Übersichtsarbeit wurde eine starke Evidenz für die Wirksamkeit von Efeu/Primel/ Thymian-Präparaten auf Frequenz und Schwere von Hustensymptomen gesehen. Auch wurde ein signifikanter Unterschied gegenüber Placebo festgestellt [3].
Die beiden Phytotherapeutika Bronchicum® Tropfen (BCUMT05®) und Bronchicum® Elixir (BCUM-E05®) werden angewendet zur Behandlung der Symptome einer akuten Bronchitis und von Erkältungskrankheiten der Atemwege mit zähflüssigem Schleim. In einer klinischen, placebokontrollierten, doppelblinden, randomisierten, multizentrischen Studie wurde bei Patienten mit einer akuten Bronchitis BCUM-T05® mit Placebo verglichen (Studie 1). Es zeigte sich eine signifikante Über-
legenheit von BCUM-T05® gegenüber Placebo in der Verringerung des Bronchitis-Severity-Scores (BSS) ab der zweiten Untersuchung nach 3 – 5 Tagen [4].
In einer klinischen, einfachblinden, verumkontrollierten, randomisierten Studie wurden bei Patienten mit akuter Bronchitis BCUM-T05® und BCUM-E05® miteinander verglichen (Studie 2). Beide Arzneimittel führten zu einer deutlichen Verringerung des BSS und waren bei einem Test auf einseitige Äquivalenz statistisch vergleichbar [5].
In beiden Studien wurden neben den Arztvisiten die Einzelparameter des BSS (Husten, Sputum, Rasselgeräusch, Brustschmerz während des Hustens, Dyspnoe) jeweils morgens und zur Nacht über Patiententagebücher erfasst. Für die Studie BCUM-T05® vs. Placebo wurden diese Tagebuchdaten bereits nachträglich analysiert. Dabei war die Wirkung von BCUM-T05® beim BSS und beim Leitsymptom Husten bereits am zweiten Tag nach Beginn der Be-
handlung derjenigen von Placebo statistisch überlegen [6].
Bei der Beurteilung der Wirksamkeit von Hustentherapeutika ist ein starker Placebo-Effekt zu beachten, sodass für einen Placebovergleich ein umfangreiches Patientenkollektiv notwendig ist [7].
Für BCUM-E05® gibt es bisher nur den Nachweis einer einseitigen Äquivalenz zwischen den beiden Verum-Substanzen BCUM-T05® und BCUM-E05® (positiver Nachweis einer Nichtunterlegenheit).
Das Ziel der vorliegenden Auswertung war daher, für BCUM-E05® den Verlauf des BSS in den ersten Tagen hinreichend genau zu beschreiben und auf eine mögliche Überlegenheit zu Placebo hin zu prüfen.
Im Rahmen einer statistischen Post-hoc-Auswertung wurden die Daten der Patiententagebücher der beiden klinischen Studien (BCUMT05® versus Placebo und BCUMT05® versus BCUM-E05®) noch einmal ausgewertet, um den Bezug zu Placebo in beiden Studien insgesamt beurteilen und vergleichen zu können. Beide Studien waren randomisiert, mit vergleichbarem Design und mit identischem Datenlayout (Tagebücher) für die Behandlung von Patienten mit akuter Bronchitis. Damit konnten unter der Annahme von homogenen Ausgangsverteilungen in Studie 1 (BCUM-T05® versus Placebo) und Studie 2 (BCUM-T05® versus BCUM-E05®) für einige Variablen (z.B. Husten und BSS) die Ausgangsverteilungen als gemeinsamer Startpunkt betrachtet und zusätzliche (additive) Behandlungseffekte geschätzt werden.
100 ml BCUM-T05® enthalten 43,4 g Fluidextrakt aus Thymiankraut (1 : 2 – 2,5), Auszugsmittel Ammoniaklösung 10 % (m/m), Glycerol 85 %, Ethanol 90 % (V/V), Wasser (1 : 20 : 70 : 109) und 21,7 g Tinktur aus Primelwurzel (1 : 5), Auszugsmittel Ethanol 50 % (V/V).
100 ml BCUM-E05® enthalten 6,635 g Fluidextrakt aus Thymiankraut (1 : 2 – 2,5), Auszugsmittel Ammoniaklösung 10 % (m/m), Glycerol 85 %, Ethanol 90 % (V/V), Wasser (1 : 20 : 70 : 109) und 3,318 g Fluidextrakt aus Primelwurzel (1 : 2 – 2,5), Auszugsmittel Ethanol 70 % (m/m).
Untersuchte Parameter
Folgende Einzelparameter wurden in den Studien anhand einer 5-Punkte-Skala (0 = nicht vorhanden, 1 = leicht, 2 = mäßig, 3 = stark, 4 = sehr stark) zur Beurteilung der Wirksamkeit durch die Patienten eingeschätzt: Husten, Brustschmerz, Dyspnoe, Rasselgeräusche und Sputum. Die Werte wurden jeweils morgens und zur Nacht in die Patiententagebücher eingetragen. Aus den 5 Variablen wurde der Summenscore BSS berechnet. Während bei den beiden Ausgangspublikationen Auswertungen zu 3 Messpunkten (Baseline, nach 3 – 5 Tagen, nach 7 – 9 Tagen) dargestellt werden, wird in der vorliegenden Auswertung durch Auswertung der Tagebücher eine tageweise Betrachtung möglich. Dabei wurde der Zeitraum der ersten 5 Tage der Behandlung (Tag 1 = Behandlungsbeginn) betrachtet.
keit gegenüber Placebo überlegen ist. Die Re-Analyse der beiden klinischen Studien zeigte für beide Placebo-Vergleiche für BCUMT05® als auch BCUM-E05® einen signifikant verringerten BSS nach 2 bzw. 3 Tagen Behandlung.
Schlüsselwörter: akute Bronchitis, Bronchitis-Severity-Score, pflanzliche Hustentherapeutika, BCUMT05®, BCUM-E05®, Placebo SUMMARY
Background: The present statistical re-analysis of two clinical trials investigated the use of two herbal cough therapeutics (Bronchicum® Tropfen, BCUM-T05®, and Bronchicum® Elixir, BCUM-E05®) in patients with acute bronchitis. The aim was to use statistical analyses to compare the efficacy of Bronchicum® Elixir (BCUM-E05®) with placebo. Patients and methods: First, the baseline data of selected clinical parameters of both groups from Study 1 (150 patients) and Study 2 (189 patients) were examined for statistically significant differences. In the event of clear homogeneity between all groups, the data from the placebo group from Study 1 and the Bronchicum® Elixir group (BCUM-E05®) should then be subjected to further statistical analyses. Data from the patient diaries (each from the first 5 days of therapy) should be used. Results: The baseline values of all four groups from the two studies showed a high degree of agreement. In both studies, the mean Bronchitis Severity Score (BSS) decreased in the first 5 days of consideration. In BCUM-T05®, a significant difference compared to placebo was evident from the
second day of treatment at night to day 5. In BCUM-E05®, a significant difference compared to placebo was evident from the third day of treatment in the morning to day 5. Conclusions: The statistical comparison supports the assumption that BCUM-E05® is also superior to placebo in its effectiveness. The re-analysis of the two clinical studies revealed for both placebo comparisons of BCUM-T05® and BCUM-E05® significantly lowered BSS after a treatment of 2 and 3 days, respectively.
Key words: acute bronchitis, Bronchitis Severity Score, herbal cough therapeutics, BCUM-T05®, BCUM-E05®, placebo
Die Analysen wurden mithilfe der R-Software (https://www. r-project.org/, Version 3.6.3) und zugehörigen Programmpaketen (u.a. deskriptive Auswertungen, Missing-Value-Techniken, Konfidenzintervall-Bestimmungen) durchgeführt. Als Plot-Programme wurden die Möglichkeiten des RProgramm-Systems angewendet. Der Schwerpunkt der explorativen Arbeiten lag auf der Bestimmung von Punktschätzern und zugehörigen 95%-Konfidenzintervallen (KI) auf der Basis von Item-Mittelwerten zur Beschreibung und statistischen Bewertung der Differenzen zwischen den vorgegebenen Therapiegruppen und Veranschaulichung der Ergebnisse bezüglich einer möglichen Placebo-Überlegenheit der beiden Vera.
Es wurden die Daten von 150 Patienten aus Studie 1 (BCUM-T05® vs. Placebo) und 189 Patienten aus
Studie 2 (BCUM-E05® vs. BCUMT05®) statistisch ausgewertet und verglichen.
Ergebnisse der Studie 1 (BCUM-T05®/Placebo)
Im Mittel wurde zu Behandlungsbeginn ein BSS von 12 Punkten dokumentiert, was im Durchschnitt etwa 2,5 Scorepunkte pro Item bedeutete (d.h. „mäßig starke bis starke“ Intensität pro Item). Hinsichtlich der Anfangswerte (Tag 1, Beurteilung morgens) war kein signifikanter Gruppenunterschied nachweisbar (t-Test: p > 0,5; 95%KI:[–1,06, 1,84]). Vom zweiten Behandlungstag (Beurteilung zur Nacht) bis Tag 5 war ein signifikanter Unterschied zwischen beiden Behandlungsgruppen zugunsten der Verum-Gruppe nachweisbar.
Tabelle 1 und 2 enthalten statistische Kennziffern und Angaben zur Signifikanz der Werte. Abbildung 1 veranschaulicht den zeitlichen Verlauf des BSS in beiden Gruppen. Die Zahl der fehlenden Wer-
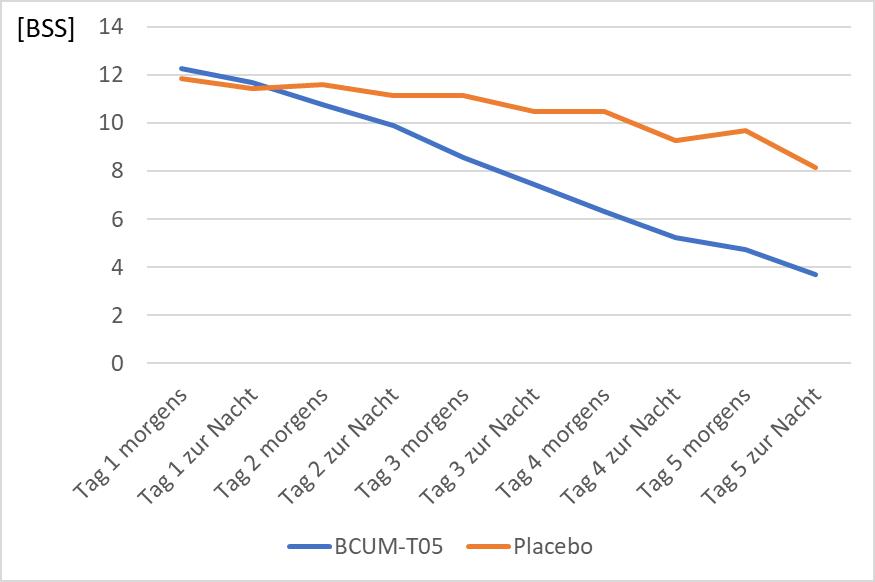
Abbildung 1: Zeitlicher Verlauf des BSS für BCUM-T05® und Placebo.
(n = 75)
(n = 75)
Tabelle 1: BSS während der Behandlung mit BCUM-T05® oder Placebo. n = auswertbare Patienten.
Behandlung1
95%-KI p-Wert morgens [–1,06, 1,84] p = 0,596 [–2,09, 0,38] p = 0,174 [–3,81,–1,39] p < 0,0001 [–5,36,–3,02] p < 0,0001 [–6,06,–3,83] p < 0,0001
95%-KI p-Wert zur Nacht [-0,95, 1,52] p = 0,646 [–2,30,–0,12] p = 0,0294
[–4,13,–2,00] p < 0,0001 [–4,97,–3,04] p < 0,0001 [–5,40,–3,51] p < 0,0001
Tabelle 2: Score-Differenzen beim BSS während der Behandlung (BCUM-T05® vs. Placebo). 1KI = Konfidenzintervall der Differenz (Verum–Placebo), signifikante p-Werte der t-Tests in Fettdruck (Tag 1 = Baseline).
Behandlung
Placebo (Studie 1) n = 72 n = 72
BCUM-T05® (Studie 1) n = 72 n = 72
Husten (Tag 1) BSS (Tag 1) 3,18 11,86 [2,99, 3,37] [10,84, 12,88]
Husten (Tag 1)
BCUM-T05® (Studie 2) n = 82 n = 82 Husten (Tag 1) BSS (Tag 1) 3,01
BCUM-E05® (Studie 2) n = 81 n = 81
[2,84, 3,18] [10,98, 12,95]
Husten (Tag 1) BSS (Tag 1) 3,01 11,83 [2,81, 3,21] [10,73, 12,93]
Tabelle 3: Baseline-Homogenität der Studien 1 und 2. 1KI = Konfidenzintervall (Tag 1) der Scorevariablen.
te lag auf einem niedrigen Niveau und beeinflusste die Ergebnisse nur unwesentlich.
Vergleich der Studie 2 (BCUM-E05®/BCUM-T05®) mit Studie 1
Die Datenstruktur von Studie 2 war identisch mit der von Studie 1. Die gleichen 5 Einzelparameter wurden erfasst: Husten, Brustschmerz, Dyspnoe, Rasselgeräusche, Sputum. Identische Scores (5-PunkteSkala) lagen vor, allerdings mit BCUM-E05® und BCUM-T05® als Prüfsubstanzen.
Durch eine Untersuchung der Homogenität des vorliegenden Datenmaterials zum Ausgangspunkt (Tag 1) musste geklärt werden, ob es sinnvoll war, die Daten der BCUM-E05®-Gruppe aus Studie 2 mit den Placebodaten aus Studie 1 paarweise zuzuordnen und dann bezüglich einer möglichen Placebo-Überlegenheit auszuwerten. Dazu wurden die Baseline-Mittelwerte der Variablen Husten und BSS mit den zugehörigen 95%-KI bestimmt und numerisch verglichen (Tab. 3).
Im Ergebnis waren die 95%-KI der berechneten Variablen-Mittelwerte überlappend für alle betrachteten
Vergleiche und zeigten damit keine bedeutsamen Unterschiede. Nachfolgend wurden für die Variablen Husten bzw. BSS die Gruppenvergleiche BCUM-E05® vs. Placebo ermittelt und diskutiert.
Husten
Im Mittel lagen „starke“ Hustenbeschwerden (Grad 3) bei Behandlungsbeginn vor, die im Mittel um weniger als eine halbe Scorestufe zwischen den Behandlungsgruppen abwichen (95%-KI der Gruppendifferenz: [–0,44, 0,11]. Beide Behandlungsgruppen waren damit
Behandlung
Tabelle 4: Einzelparameter „Husten“ während der Behandlung mit BCUM-E05® oder Placebo. n = auswertbare Patienten.
Behandlung1
95%-KI p-Wert morgens
95%-KI p-Wert zur Nacht
[–0,44, 0,11] p = 0,229 [–0,42, 0,08] p = 0,185 [–0,80,–0,27] p = 0,0001 [–1,10,–0,50] p < 0,0001 [–,31,–0,68] p<0,0001
[–,31, 0,14] p = 0,472 [–0,37, 0,11] p = 0,276 [–0,70, -0,20] p = 0,0005 [–0,75,–0,21] p = 0,0006 [–1,05,–0,47] p < 0,0001
Tabelle 5: Score-Differenzen beim Parameter „Husten“ (BCUM-E05® vs. Placebo). 1KI = Konfidenzintervall der Differenz (Verum–Placebo), signifikante p-Werte der t-Tests in Fettdruck (Tag 1 = Baseline).
an Tag 1 hinsichtlich der Verteilung des Husten-Schweregrades vergleichbar.
Nach dem zweiten Behandlungstag war bis zum Ende an Tag 5 eine signifikante, stetige Besserung der Beschwerden in der BCUM-E05®Gruppe um etwa einen Schweregrad erkennbar, wobei die Werte zur Nacht nach Tag 3 insgesamt etwas pessimistischer als am Morgen des jeweiligen Tages beurteilt wurden.
Tabelle 4 und 5 enthalten statistische Kennziffern sowie Angaben zur Signifikanz der Werte. Abbildung 2 veranschaulicht den zeitlichen Verlauf der Husten-Symptomatik.
Die Zahl der fehlenden Werte lag auf einem niedrigen Niveau und beeinflusste die Ergebnisse daher nur wenig. Es wurden 10 Patienten mit fehlenden Baseline-Werten bei der Auswertung nicht berücksichtigt.
Bronchitis-Severity-Score
Aus den 5 Einzelsymptomen der Studien wurde der zugehörige Summenscore BSS für die BCUME05®-Gruppe und die Placebogruppe bestimmt und in gleicher Weise wie vorher zwischen beiden Behandlungsgruppen ausgewertet. Im Mittel wurde bei Behandlungsbeginn ein Summenscore von etwa
12 Punkten dokumentiert, was im Durchschnitt ungefähr 2,5 Scorepunkte pro Item bedeutete (d.h. „mäßig starke bis starke“ Intensität pro Item). Hinsichtlich der Anfangswerte (Tag 1, Beurteilung morgens) war kein signifikanter Gruppenunterschied nachweisbar (t-Test: p > 0,5, 95%-KI: [–1,53, 1,46]).
Ab dem dritten Behandlungstag (Vormittags-Beurteilung) war bis
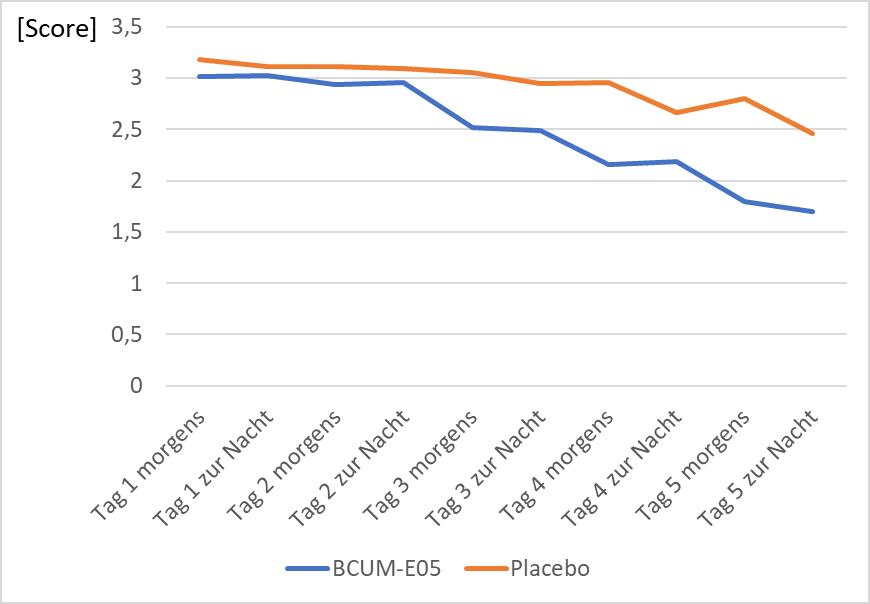
Abbildung 2: Zeitlicher Verlauf des Einzelparameters „Husten“ für BCUM-E05® und Placebo.
Tabelle 6: BSS während der Behandlung mit BCUM-E05® oder Placebo. n = auswertbare Patienten
Behandlung1
95%-KI p-Wert morgens
95%-KI p-Wert zur Nacht
[–1,53, 1,46] p = 0,964
[–0,68, 2,11] p = 0,311
[–1,81, 0,86] p = 0,482 [–3,27,–0,61] p = 0,0046 [–4,44,–1,73] p < 0,0001 [–5,55,–2,77] p < 0,0001
[–1,21, 1,32] p = 0,934
[–2,58,–0,14] p = 0,0294
[–3,37,–1,04] p = 0,0003
[–4,13,–1,69] p < 0,0001
Tabelle 7: Score-Differenzen beim BSS während der Behandlung (BCUM-E05® vs. Placebo). 1KI = Konfidenzintervall der Differenz (Verum–Placebo), signifikante p-Werte der t-Tests in Fettdruck (Tag 1 = Baseline).
Tag 5 ein signifikanter Unterschied zwischen beiden Behandlungsgruppen zugunsten von BCUME05® nachweisbar.
Tabelle 6 und 7 enthalten statistische Kennziffern sowie Angaben zur Signifikanz der Werte. Abbildung 3 veranschaulicht den zeitlichen Verlauf des BSS in beiden Gruppen. Die Zahl der fehlenden Werte lag nach der Korrektur der
Baselinewerte auf einem niedrigen Niveau und beeinflusste die Ergebnisse nur unwesentlich.
Die Daten aus den Patiententagebüchern von zwei kontrollierten, klinischen Studien zur Anwen-

Abbildung 3: Zeitlicher Verlauf des BSS für BCUM-E05® und Placebo.
dung von pflanzlichen Hustentherapeutika bei Patienten mit akuter Bronchitis wurden gemeinsam statistisch ausgewertet. Um Aussagen über den Wirkeintritt machen zu können, wurde der Behandlungszeitraum der ersten 5 Tage betrachtet. Je Behandlungstag wurden 2 Werte erfasst (morgens und zur Nacht).
Zunächst wurde der Behandlungserfolg von BCUM-T05® und Placebo anhand der Daten von Studie 1 untersucht. Der Summenscore BSS zeigte einen signifikanten Unterschied zugunsten von BCUMT05® ab Tag 2 (zur Nacht). Bis zum Tag 5 (zur Nacht) verringerte sich der BSS im Mittel um etwa 61 % des Anfangswertes, während er unter Placebo nur etwa 31 % niedriger lag.
Um auch für Studie 2 den Gruppenunterschied zwischen BCUME05® und Placebo abschätzen zu können, wurden 10 Patienten mit fehlenden Baselinewerten (tägliche Bewertung der Symptomatik) bei der Auswertung (Tag 1 – 5) nicht berücksichtigt. Das Ergeb-
nis waren annähernd konstante Patientenzahlen zu allen Auswertetagen, sodass der zeitliche Gruppenvergleich (BCUM-E05® (Daten Studie 2) mit Placebo (Daten der Studie 1) mit höherer Test-Power durchgeführt werden konnte.
Signifikante Veränderungen ergaben sich für den Einzelparameter Husten. Die Besserung der Symptome von Tag 1 bis Tag 5 (zur Nacht) betrug 0,5 – 1,1 Scorepunkte. Der BSS verringerte sich im Mittel unter der BCUM-E05®Behandlung in diesem Zeitintervall um 53 % gegenüber 31 % unter Placebo. Ab dem dritten Behandlungstag (Vormittags-Beurteilung) war bis Tag 5 ein signifikanter Unterschied zwischen beiden Behandlungsgruppen zugunsten von BCUM-E05® nachweisbar.
Bei BCUM-E05® fällt sowohl beim Einzelparameter Husten als auch beim Summenscore BSS der
Rückgang der Werte zwischen Abend- und Morgenbewertung stärker aus als zwischen Morgen- und Abendbewertung. Das gleiche Phänomen tritt auch bei BCUM-T05® auf, allerdings deutlich schwächer. Anhand der berechneten Konfidenzintervalle ist dieser Effekt jedoch insgesamt als nicht bedeutsam einzustufen. Da die Einnahme des Arzneimittels tagsüber mehrmals (4-mal) erfolgte, könnte sich trotzdem eine zusätzliche schleimlösende Wirkung in ihrer Auswirkung tagsüber zeigen.
Zusammenfassend zeigte die statistische Re-Analyse der Daten von zwei klinischen Studien für die beiden pflanzlichen Arzneimittel BCUM-T05® und BCUME05® eine gegenüber Placebo bessere Wirksamkeit. Beim BSS wurde Signifikanz gegenüber Placebo bei BCUM-T05® an Tag 2 zur Nacht und bei BCUM-E05® an Tag 3 morgens erreicht.
Literatur
1 Ebell MH, Lundgren J, Youngpairoj S. Ann Fam Med 2013;11:5-13
2 S2k-Leitlinie Fachärztliche Diagnostik und Therapie von erwachsenen Patienten mit Husten. AWMF-Registernummer: 020-003, Langversion 4.1 – Januar 2025
3 Akuter und chronischer Husten, S3-Leitlinie, AWMF-Register-Nr. 053-013, DEGAM-Leitlinie Nr. 11, Stand 2021
4 Grünwald J, Graubaum HJ, Busch R. Efficacy and tolerability of a fixed combination of thyme and primrose root in patients with acute bronchitis. Arzneimittelforschung 2005;55:669-676
5 Grünwald J, Graubaum HJ, Busch R. Evaluation of the non-inferiority of a fixed combination of thyme fluid and primrose root extract in comparison to a fixed combination of thyme fluid extract and primrose root tincture in patients with acute bronchitis. Arzneimittelforschung 2006;56:574-581
6 Heindl S, Hucke HP, Brünjes R. Wie schnell wirken Phytotherapeutika bei akuter Bronchitis? Zeitschrift für Phytotherapie 2017;38(Suppl. 1): S1-S44
7 Eccles R. The powerful placebo effect in cough: relevance to treatment and clinical trials. Lung 2020;198:13-21
Für die Verfasser: Dr. Rainer Brünjes Cassella-med GmbH & Co. KG Gereonsmühlengasse 1, 50670 Köln
Rauchfrei auf Rezept:
Vareniclin kann jetzt auf Kosten der gesetzlichen Krankenversicherung verordnet werden
Der Gemeinsame Bundesausschuss (G-BA) hat am 15. Mai 2025 eine neue Regelung zur Erstattung der Tabakentwöhnung beschlossen. Demnach haben bestimmte gesetzlich Krankenversicherte erstmals Anspruch auf eine Verordnung des Arzneimittels Vareniclin (Champix®) zur Raucherentwöhnung auf Kassenrezept. Nach rechtlicher Prüfung durch das Bundesministerium für Gesundheit (BMG) und Veröffentlichung im Bundesanzeiger ist der Beschluss des G-BA
am 20. August 2025 in Kraft getreten und bietet Ärzten ab sofort neue Möglichkeiten, Raucher und Raucherinnen mit einer schweren Tabakabhängigkeit auf dem Weg in ein rauchfreies Leben professionell zu unterstützen. Vareniclin ist seit Oktober 2006 in der EU zur Raucherentwöhnung bei Erwachsenen zugelassen und wird nach einer Vertriebspause seit April 2025 wieder von Pfizer in Deutschland vermarktet.
Voraussetzungen für Kostenübernahme
Grundlage für die Kostenübernahme der medikamentösen Raucher-
entwöhnung mit Vareniclin ist eine ärztlich festgestellte Diagnose „Psychische und Verhaltensstörungen durch Tabak; Abhängigkeitssyndrom“ (ICD-10: F17.2). Für diese müssen mindestens drei der folgenden Kriterien erfüllt sein:
• Starkes Verlangen oder Zwang, Tabak zu konsumieren
• Verminderte Kontrollfähigkeit über Beginn, Beendigung oder Menge des Konsums
• Entzugssymptome bei Reduktion oder Beendigung
• Toleranzentwicklung (steigende Mengen nötig)
• Vernachlässigung anderer Interessen zugunsten des Konsums
• Fortgesetzter Konsum trotz schädlicher Folgen Neben einer diagnostizierten schweren Tabakabhängigkeit ist außerdem die Teilnahme an einem evidenzbasierten Entwöhnungsprogramm notwendige Voraussetzung für die Erstattungsfähigkeit von Vareniclin. Eine schwere Tabakabhängigkeit ist definiert durch einen Punktwert von ≥6 im Fagerström-Test* zur Berechnung der Nikotinabhängigkeit oder durch einen fehlgeschlagenen Tabakverzicht trotz Risikokonstellationen wie COPD, Asthma oder koronare Herzkrankheit.
Als evidenzbasierte Programme zur Tabakentwöhnung gelten dem G-BA zufolge im Wesentlichen die Kriterien der bestehenden Präventionsprogramme (§20 Abs. 4 Nummer 1 SGB V). Auf Basis des aktuellen medizinischen Erkenntnisstandes müssen diese Programme den Nutzern z.B. Hintergrundwissen zum Rauchverhalten und zur Tabakentwöhnung vermitteln. Die strukturierten Entwöhnungsprogramme können entweder als kostenpflichtige Präventionskurse oder als verordnungsfähige digitale Gesundheitsanwendungen (DiGA) durchgeführt werden. Präventionskurse sind oft gruppenbasiert und können sowohl online als auch in Präsenz stattfinden. Die Kosten liegen in der Regel zwischen 75 und 150 Euro, wobei eine teilweise Erstattung durch die gesetzliche Krankenversicherung möglich ist.
DiGA wie SmokeFree oder NichtraucherHelden bieten eine individuelle, App-basierte Unterstützung und sind vollständig erstattungsfä-
* Der Fagerström-Test zur Berechnung der Nikotinabhängigkeit findet sich auf www. pfizerpro.de.
Vareniclin (Champix®) ist zur Raucherentwöhnung bei Erwachsenen angezeigt und wird 2 × täglich oral in der empfohlenen Dosierung von 1 mg eingenommen, sobald eine einwöchige Titrationsphase abgeschlossen wurde. Vareniclin blockiert Nikotin, um die positiven Verstärkungseffekte des Rauchens zu verringern. Es reduziert außerdem die belohnenden und verstärkenden Effekte des Rauchens, indem es die Bindung von Nikotin an die α4β2 neuronalen nikotinergen Acetylcholinrezeptoren verhindert (antagonistische Wirkung). Bei der Anwendung von Vareniclin wird weiterhin Dopamin freigesetzt, allerdings in geringerem Maße als bei Nikotin. Diese partielle Agonistenaktivität am α4β2-Nikotinrezeptor kann dazu beitragen, die Symptome des Verlangens und des Entzuges zu lindern.
Die Starterpackung (0,5/1 mg) sowie die Folgepackung (1 mg) sind mit jeweils 14 und 28 Tagesdosen verfügbar. Darüber hinaus kann auch eine wirkstoffreduzierte Folgepackung (0,5 mg) mit 28 Tagesdosen verordnet werden.
hig, wenn sie ärztlich oder psychotherapeutisch verordnet werden.
Klinische Evidenz spricht für den Einsatz von Vareniclin
Ein Cochrane-Review** von 41 klinischen Studien mit über 17.000 Teilnehmern zeigte mit hoher Evidenz für Vareniclin eine signifikant höhere Erfolgsquote im Vergleich zu Placebo (Risk Ratio: 2,3) bei dem Ziel, für mindestens 6 Monate mit dem Rauchen aufzuhören. Auch eine Meta-Analyse*** von 34 randomisierten Studien mit insgesamt mehr als 26.000 Rauchern belegt die klinische Evidenz von Vareniclin bei der Tabakentwöhnung von Erwachsenen. Sie ergab, dass die Einnahme von Vareniclin im Vergleich zu Bupropion, Nikotinersatztherapie, Beratung oder Placebo die Wahrscheinlichkeit
** Livingstone-Banks J et al. Cochrane Database of Systematic Reviews 2023, Issue 6. Art. No. CD006103
*** Guo K et al. Drug Alcohol Depend 2022;241:109672
erhöhte, mit dem Rauchen aufzuhören bzw. dauerhaft rauchfrei zu sein.
Zur medikamentösen Raucherentwöhnung sind neben Vareniclin auch Nicotin, Bupropion, und Cytisin zugelassen. Während die ärztliche Verordnung von Vareniclin und Nicotin mit dem aktuellen G-BA-Beschluss für bestimmte Patienten nun mittels eines Kassenrezeptes möglich und erstattungsfähig ist, werden Bupropion und Cytisin vom G-BA weiterhin als Lifestyle-Arzneimittel eingestuft, bei deren Anwendung eine Erhöhung der Lebensqualität im Vordergrund steht (§ 34 Absatz 1 Satz 7 SGB V). Die Kosten für die Präparate Bupropion und Cytisin müssen demzufolge von den Versicherten weiterhin selbst getragen werden.
B. S.
Die chronische spontane Urtikaria (csU) ist eine chronisch-entzündliche primär Mastzell-vermittelte Autoimmunerkrankung, die mit plötzlich auftretenden Angioödemen und/oder Quaddeln einhergeht [1]. Diese zum Teil sehr belastenden und unvorhersehbaren Beschwerden können die davon Betroffenen in ihrer Lebensqualität stark einschränken [2], weshalb die Patienten häufig auch psychische Begleiterkrankungen wie Depressionen oder Angstzustände entwickeln [3].
Aufgrund des hohen Leidensdrucks von csU-Patienten sind eine rechtzeitige Diagnose und adäquate Therapieoptionen entscheidend. Hilfreich zu wissen ist dabei, welche Auslöser und Triggerfaktoren für die Symptome verantwortlich sind und welcher Autoimmunmechanismus der csU zugrunde liegt: Typ I, die autoallergische Form, bei der die Entzündung durch IgEAutoantikörper gegen körpereigene Proteine getriggert wird, oder Typ IIb, die autoimmune Form, bei der die Mastzell-Aktivierung via IgG-Autoantikörper erfolgt, die gegen IgE oder seine Rezeptoren gerichtet sind [4].
Mittel der Wahl zur Behandlung der csU sind bisher H1-Antihistaminika, da sie Histaminrezeptoren blockieren und die entzündungsfördernde Wirkung von Histamin verhindern, die Juckreiz und
Schwellungen verursacht [5]. Bei über 50 % der Patienten lässt sich die csU jedoch allein durch H1Antihistaminika nicht kontrollieren [5]. In diesen Fällen können Medikamente eingesetzt werden, die auf die Hemmung der autoimmunen Prozesse abzielen, wie z.B. der Anti-IgE-Antikörper Omalizumab. Dabei ist zu beachten, dass die Ausgangswerte des IgE-Spiegels mit dem Behandlungserfolg korrelieren können: Ein niedriger Gesamt-IgE-Spiegel im Serum zu Beginn der Behandlung könnte möglicherweise ein schlechtes oder fehlendes Ansprechen auf reine IgE-Inhibitoren vorhersagen [6].
BTK-Inhibitor führt unabhängig vom IgESpiegel zu einer anhaltenden Symptomverbesserung
Einen Fortschritt in der Behandlung der csU verspricht der hochselektive Bruton-Tyrosinkinase (BTK)-Inhibitor Remibrutinib, da er unabhängig vom Ausgangs-IgESpiegel und dem zugrunde liegenden Endotyp die Urtikaria-Symptome früh und anhaltend lindern kann. Dies belegen die beiden randomisierten placebokontrollierten Phase-III-Studien REMIX-1 [7] und REMIX-II [8], in denen der BTK-Inhibitor bei Patienten mit
mittelschwerer bis schwerer csU, die nicht durch H1-Antihistaminika der zweiten Generation kontrolliert werden konnte, frühzeitig eine signifikante Verbesserung der Symptomkontrolle (p < 0,001) erzielte. Die Überlegenheit von Remibrutinib gegenüber Placebo zeigte sich bereits in Woche 1 (signifikant in Woche 12 und 24) und das Ansprechen unter Remibrutinib hielt bis in Woche 52 an [9].
Remibrutinib war gut verträglich und wies im Vergleich zu Placebo bis zu 52 Wochen lang ein günstiges Sicherheitsprofil auf, einschließlich ausgeglichener Leberfunktionstests. Die am häufigsten (≥5 %) beobachteten Nebenwirkungen in den Phase-III-REMIXStudien waren Infektionen der Atemwege (einschließlich COVID-19 und Nasopharyngitis) und Kopfschmerzen, alle vergleichbar mit Placebo [9].
Gepoolte Analysen untermauern die Phase-III-Ergebnisse
Im Rahmen des Kongresses der European Academy of Allergy and Clinical Immunology 2025 (EAACI) wurden zwei neue Subgruppenanalysen der Phase-III-Studien REMIX-1 und -2 präsentiert. In der ersten gepoolten Analyse wurden 606 Patienten im Remibrutinib-Arm mit 306 Patienten aus
Remibrutinib bindet kovalent und mit hoher Selektivität an die Bruton-Tyrosinkinase (BTK) und inhibiert somit sehr effektiv die Aktivierung der Basophilen und Mastzellen, unabhängig von dem der Erkrankung zugrunde liegenden Autoimmunmechanismus [12]. Die Aktivierung der BTK-Signalkaskade führt bei chronischer spontaner Urtikaria (csU) zur Ausschüttung von Histamin, das Quaddeln und Schwellungen verursacht. Durch die Blockade von BTK wird dieser Vorgang verhindert [13].
dem Placebo-Arm verglichen [10]. Dabei wurde die durchschnittliche Veränderung gegenüber dem Ausgangswert im wöchentlich erfassten Urtikaria-Aktivitäts-Score (UAS7) in den Wochen 1, 2, 12, 24 und 52 in Abhängigkeit vom Gesamt-IgE-Ausgangswert bewertet (verwendete Cut-offs: niedrig ≤43 IU/ml; normal/hoch >43 IU/ ml sowie alternativ ≤100 IU/ml; >100 IU/ml). Insgesamt hatten 25,9 % der Patienten unter Remibrutinib und 29,4 % unter Placebo einen niedrigen IgE-Ausgangswert (<43 IU/ml) [10].
Die Behandlung mit Remibrutinib führte zu einer frühen und anhaltenden Symptomverbesserung unabhängig vom Ausgangs-IgESpiegel. Bereits in Woche 1 betrug die mittlere UAS7-Reduktion bei niedrigem IgE –13,8 vs. –2,4 (Placebo) und bei normalem/hohem IgE –11,1 vs. –4,0 (Placebo). In Woche 12 verstärkte sich dieser Effekt auf –24,8 vs. –11,4 bzw. –20,0 vs. –13,7 [10].
Diese Ergebnisse zeigen, dass Remibrutinib zu einer anhaltenden Reduktion der Urtikaria-Sympto-
me unabhängig vom bestehenden IgE-Level führt. Eine weitere Subgruppenanalyse untersuchte, inwieweit sich unter der Behandlung mit Remibrutinib bestehende Angioödeme reduzieren ließen [11]. In die Analyse gingen alle Patienten ein, die einen Angioödem-Aktivitäts-Score 7 (AAS7) von >0 hatten. Zu Studienbeginn berichteten 374 Patienten in der Remibrutinib-Gruppe und 169 in der Placebo-Gruppe über Angioödeme. Der durchschnittliche AAS7-Ausgangswert betrug 43,3 respektive 39. Unter Remibrutinib verbesserte sich die AngioödemAktivität schnell und anhaltend: Bereits in Woche 1 erreichte ein höherer Anteil der Patienten unter Remibrutinib einen AAS7 = 0 (keine Angioödeme) im Vergleich zu Placebo. Dieser Effekt hielt bei 82 % der Patienten bis Woche 52 an [11].
Wirksamkeit des BTK-Inhibitors Remibrutinib auch in den untersuchten Subgruppen und zeigen ein konsistentes Sicherheitsprofil [10, 11]. Basierend auf diesen Ergebnissen hat Novartis bei den Gesundheitsbehörden in den USA, der EU und China die Zulassung von Remibrutinib beantragt.
Brigitte Söllner, Erlangen
Die auf dem EAACI 2025 präsentierten Daten bestätigen erneut die
Literatur
1 AWMF. https://register.awmf.org/assets/ guidelines/013-028l_S3_KlassifikationDiagnostik-Therapie-Urtikaria_2022-04. pdf
2 Fricke J et al. Allergy 2020;75:423-432
3 Peters EMJ. J Dtsch Dermatol Ges 2016; 14:233-252
4 Curch MK et al. Immunol Rev 2018; 282:232-247
5 Maurer M, Weller K, Bindslev-Jensen C, et al. Ungedeckter klinischer Bedarf bei chronischer spontaner Urtikaria. Ein Bericht der GA²LEN-Taskforce. Allergie 2011;66:317-330
6 Chuang KW et al. J Allergy Clin Immunol Pract 2023;11:2382-2389.e3
7 Novartis Pharmaceuticals. https://clinicaltrials.gov/study/NCT05030311
8 Novartis Pharmaceuticals. https://clinicaltrials.gov/study/NCT05032157
9 Metz M et al. EAACI 2024. Abstract
10 Reed J, et al. EAACI 2025, Abstract 001293
11 Hide M, et al. EAACI 2025, Abstract 000816
12 Angst D et al. J Med Chem 2020;63: 5102-5118
13 Maurer M et al. J Allergy Clin Immunol 2022;150:1498-1506.e2
Aus heutiger Sicht ist die Plaque-Psoriasis keine reine Hauterkrankung, sondern eine chronisch-entzündliche Systemerkrankung. Die systemischen Entzündungsreaktionen können unter anderem die Ursache dafür sein, dass die Plaque-Psoriasis mit zahlreichen Komorbiditäten assoziiert ist, wie z.B. kardiovaskulären und metabolischen Erkrankungen sowie der Psoriasis-Arthritis (PsA) [1, 2, 3]. Zur Eindämmung der systemischen Entzündung und ihren Folgeerscheinungen ist eine frühzeitige Therapie notwendig [4]. Wie diese im Versorgungsalltag umgesetzt wird und welche Faktoren bei einer frühzeitigen Behandlung der mittelschweren bis schweren Plaque-Psoriasis eine Rolle spielen, zeigt eine aktuelle, repräsentative Befragung von 100 niedergelassenen und 100 klinisch tätigen Dermatologen in Deutschland, die Novartis beim Marktforschungsinstitut Ipsos in Auftrag gegeben hat [5].
Was geben Dermatologen als Hauptgründe für eine frühzeitige Intervention an?
Eine frühzeitige Intervention bei mittelschwerer bis schwerer Plaque-Psoriasis wird von 65 % der befragten Ärzte als vorteilhaft hinsichtlich der Wirksamkeit angesehen, während 64 % eine Reduktion von Komorbiditäten betonen. Damit bestätigt die Befragung, dass die Plaque-Psoriasis auch im Versorgungsalltag größtenteils als systemische Erkrankung erkannt wird.
Dabei legen die befragten in der Klinik tätigen Dermatologen mit 50 % einen besonderen Wert auf die Vorteile der frühzeitigen Beeinflussung des Krankheitsverlaufs
zur Verhinderung der Ausbreitung, wohingegen 70 % der befragten niedergelassenen Dermatologen die Wirksamkeit und Symptomverbesserung als primäre Vorteile ansehen (Abb. 1) [5].
Komorbiditäten verhindern –Lebensqualität verbessern
von den Patienten berichtete verminderte Lebensqualität (93 %) eine wesentliche Rolle. Zwischen Klinikern und niedergelassenen
Umfrage unter deutschen Dermatolog*innen1
Wann setzen Dermatologen auf eine frühzeitige Intervention mit Biologika?
Die befragten Dermatolog*innen (n = 200) sehen durch eine frühzeitige Therapie Vorteile bei der Verbesserung von Symptomen – besonders deshalb, weil…
Sowohl die Schwere der Erkrankung als auch die Gelenkbeteiligung haben mit jeweils 99 % den größten Einfluss auf die Entscheidung für eine frühe Biologika-Intervention. Darüber hinaus spielen auch die Beteiligung von Nägeln und Kopfhaut (97 %) sowie die
Dermatologen bestehen demnach nur geringe Unterschiede. Auffällig ist jedoch, dass niedergelassene Dermatologen in der Befragung die von den Patienten berichtete Beeinträchtigung der Lebensqualität mit 70 % stärker in den Blick nehmen als die Kliniker mit 52 % [5].
…Komorbiditäten verhindert werden können.2
Welche Lokalisationen werden als besonders schwer zu behandeln wahrgenommen?
…die Symptom-Auslöser nachhaltig positiv verändert werden können.3 …die Lebensqualität verbessert werden kann.2
Besonders schwer zu behandeln sind nach Meinung der befragten
Vorteile einer frühzeitigen Therapiea
Wirksamkeit/gute Heilung/ Verbesserung der Symptome
Weniger Komorbiditäten
Frühzeitige Beeinflussung des Krankheitsverlaufes/ Verhinderung von Ausbreitung Verbesserte Lebensqualität
anur die vier häufigsten Antworten sind dargestellt
Gesamt niedergelassene Dermatolog*innen Klinik-Dermatolog*innen
Abbildung 1: Die befragten Dermatologen sprachen sich übereinstimmend für eine frühzeitige Therapie mit Biologika aus, da sie für den Patienten deutliche Vorteile bringen kann [5].
„Plaqueablagerungen an anderen Organen vermeiden, Hautbild schnell verbessern.“ (Klinik Derma) „Langfristige Krankheitskontrolle, weniger Risiko für eine Gelenkbeteiligung, Aufrechterhaltung
Ärzte die Nägel (95 %), der Genitalbereich (93 %) und die Kopfhaut (89 %) (Abb. 2). Bestimmte Hautareale wie die an Torso, Beinen und Armen werden im Vergleich dazu als weniger problematisch eingestuft. Dabei ergaben sich zwischen den Fachgruppen kaum Unterschiede hinsichtlich der Einschätzung der Behandlungsschwierigkeit [5]. Bei der Frage, ob sie Biologika verstärkt bei der Behandlung von Sonderlokalisationen wie Kopfhaut und Nägeln einsetzen, zeigte sich, dass deren Einsatz bei den befragten Klinikern weit verbreitet ist. Sonderlokalisationen scheinen aber eine so große Herausforderung zu sein, dass auch fast alle der befragten niedergelassenen Dermatologen auf Biologika zurückgreifen – und das sogar häufiger als Kliniker (95 % vs. 83 %) [5].
Nach welchen Kriterien suchen Dermatologen das passende Biologikum aus?
Für 70 % der befragten Dermatologen ist die Datenlage entscheidend für die Auswahl des Biologikums, das sie zur Behandlung von Sonderlokalisationen bei der PlaquePsoriasis einsetzen, gefolgt von der Wirksamkeit (52 %). Die niedergelassenen Dermatologen legen häufiger Wert auf die Zulassung (46 % gegenüber 30 % der Kliniker), die Sicherheit und Verträglichkeit (33 % vs. 18 %) sowie den Ausschluss von Kontraindikationen (23 % vs. 6 %).
Im Gegensatz dazu berücksichtigen die befragten Klinik-Dermatologen stärker ihre eigene Erfahrung (38 % vs. 23 %) sowie Begleiterkrankungen (30 % vs. 15 %) bei ihrer Entscheidung. Auch die Meinung von Kollegen spielt eine Rolle bei der Auswahl [5].
eingestuft. Zwischen den Fachgruppen gibt es kaum Unterschiede in der Einschätzung der Behandlungsschwierigkeit.1
Bei der Frage, ob Biologika verstärkt bei der Behandlung von Sonderlokalisationen wie Kopfhaut und Nägeln eingesetzt werden, zeigt sich, dass deren Einsatz bei den befragten Kliniker*innen weit verbreitet ist. Sonderlokalisationen scheinen aber eine so große Herausforderung zu sein, dass auch in der Niederlassung fast jede*r der Befragten auf Biologika zurückgreift – und das sogar häufiger als Kliniker*innen (95 % vs. 83 %) 1 So stellt sich insgesamt eine sehr hohe Anwendung von Biologika bei Sonderlokalisationen in der Praxis heraus
Nägel
Genitalbereich
Kopfhaut
Palmoplantar
Gesicht

Nur die vier häufigsten Antworten sind dargestellt
Gesamt niedergelassene Dermatolog*innen
Klinik-Dermatolog*innen
Abb 1: Besonders schwer zu behandelnde Lokalisationen bei Plaque-Psoriasis aus Sicht der befragten Ärzt*innen
Abbildung 2: Besonders schwer zu behandelnde Lokalisationen bei Plaque-Psoriasis aus Sicht der befragten Ärzte [5].
Quelle: Ipsos Medic*Bus, Basis: n=200 Dermatolog*innen (n=100 ndgl. Dermatolog*innen, n=100 Klinik Dermatolog*innen)
Secukinumab
Nach welchen Kriterien suchen Dermatolog*innen das passende Biologikum aus? Insgesamt geben 70 % der befragten Dermatolog*innen an, dass die Datenlage entscheidend für die Auswahl des richtigen Biologikums für Sonderlokalisationen bei Plaque-Psoriasis ist, gefolgt von der Wirksamkeit (52 %). Die interviewten niedergelassenen Dermatolog*innen legen häufiger Wert auf die Zulassung (46 % im Vergleich zu 30 % bei Klinik-
Secukinumab (Cosentyx®) ist ein vollhumaner, monoklonaler Antikörper, der direkt gegen Interleukin (IL)-17A gerichtet ist [11]. Das Zytokin IL-17A ist an Entzündungsprozessen und der Entstehung von Plaque-Psoriasis [12], Psoriasis-Arthritis (PsA) [13,14], axialer Spondyloarthritis (axSpA) [15,16] und Hidradenitis suppurativa (HS) [17] beteiligt.
Secukinumab ist seit 10 Jahren für mittlerweile 8 Indikationen zugelassen [11] und verfügt über umfangreiche klinische Evidenz in allen Indikationen: Dazu zählen die Plaque-Psoriasis [12], die pädiatrische Plaque-Psoriasis [18], HS [17], PsA [13,14], die nichtröntgenologische axSpA (nr-axSpA) [16], ankylosierende Spondylitis (AS, r-axSpA) [15] sowie die Enthesitis-assoziierte Arthritis (EAA) und die juvenile Psoriasis-Arthritis (JPsA), zwei Unterformen der juvenilen idiopathischen Arthritis (JIA) [19].
Secukinumab überzeugt durch evidente Datenlage
Mit dem größten Studienprogramm – über 200 Studien [6] –sowie weltweit mehr als 1,6 Millionen behandelten Patienten [7] liefert Secukinumab unter allen zugelassenen Biologika eine evidente Datenlage als entscheidendes Kriterium für die Wahl des passenden Biologikums.
Gerade Patienten mit schwierig zu therapierenden Sonderlokalisationen können von einer adäquaten Therapie profitieren, wie die Studien TRANSFIGURE bei Nagel-Psoriasis [8] und SCALP bei Kopfhaut-Psoriasis zeigten [9]. Das untermauert auch die Praxiserfahrung der befragten Dermatologen. Zudem lässt sich aus den Ergebnissen der randomisierten, placebokontrollierten Phase-IIIStudie FUTURE 5 schließen, dass
auch eine röntgenologische Progression der PsA mit Secukinumab verzögert werden kann [10].
Als Systemerkrankung stellt die Plaque-Psoriasis besondere Herausforderungen an die Therapie. Die Wahl eines geeigneten Biologikums sollte sich daher an der verfügbaren Evidenz orientieren, wobei Secukinumab durch eine besonders breit differenzierte Datenlage hervorsticht. Wie die Befragung der 200 Dermatologen deutlich macht, bieten sich durch eine frühzeitige Therapie Vorteile bei der Verbesserung von Symptomen, weil sich die Symptom-Aus-
löser nachhaltig positiv verändern und Komorbiditäten verhindert werden können und sich auch die Lebensqualität des Patienten verbessern lässt [5].
Fabian Sandner, Nürnberg
7 Novartis Pharma GmbH. https://www.novartis.com/sites/novartis_com/files/202410-interim-financial-report-en.pdf
8 Reich K et al. Br J Dermatol 2021; 184:425-436
9 Bagel J et al. J Am Acad Dermatol 2017; 77:667-674
10 Mease PJ et al. RMD Open 2021;7: e001600
11 Fachinformation Cosentyx®, aktueller Stand
attexis® – die erste dauerhaft gelistete DiGA für Erwachsene mit ADHS
Nach der Zulassung der DiGA hiToco® für Eltern von Kindern mit einer ADHS erweitert Medice sein digitales Produktportfolio mit attexis® nun um eine verordnungsfähige digitale Therapieoption für Erwachsene mit ADHS. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) hat attexis® dauerhaft die Zulassung als digitale Gesundheitsanwendung (DiGA) erteilt. attexis® ist extrabudgetär verordnungsfähig und seit 6. August 2025 übernehmen alle gesetzlichen Krankenkassen sowie viele private Versicherungen die Kosten für diese DiGA. attexis® ist flexibel einsetzbar zur Wartezeitüberbrückung zwischen den Therapiesitzungen, zur Therapiebegleitung oder Nachsorge und kann sowohl parallel zu einer
Literatur
1 Mrowietz U et al. Exp Dermatol 2014; 23:705-709
2 Augustin M et al. Acta Derm Venereol 2010;90:147-151
3 Mease PJ et al. J Am Acad Dermatol 2013;69:729-735
4 WHO. Global Report on Psoriasis. 2016. https://iris.who.int/handle/10665/204417
5 Ipsos Medic*Bus, n=200 Dermatolog*innen (n=100 ndgl. Dermatolog*innen, n=100 Klinik Dermatolog*innen) CATI Interviews, Erhebungszeitraum: 09.12.24 – 14.02.25
6 National Library of Medicine. https://clinicaltrials.gov/search?intr=Secukinumab
Pharmakotherapie als auch unabhängig davon verwendet werden. Die DiGA bietet ADHS-Patienten ab 18 Jahren eine individualisierte, digitale Therapie zur alltagsbegleitenden Unterstützung – mit Fokus auf effektive Alltagsstrukturierung, Symptombewältigung und Stärkung des Selbstmanagements im Einklang mit der S3-Leitlinie. Das Programm umfasst 6 interaktive Gespräche in Form von digitalen therapeutischen Dialogen. Zu Beginn beantworten die Nutzer eine Einstiegsfrage, auf deren Basis sie anschließend passende Inhalte erhalten, abgestimmt auf ihre individuellen Bedürfnisse und Herausforderungen. Im Verlauf der Gespräche können die Inhalte durch die Auswahl entsprechender Antworten weiter personalisiert werden. Wöchentliche Reflexionen und Fortschrittsübersichten machen Entwicklungsschritte sichtbar und fördern die Motivation. Mit Planungsfunktionen, Checklisten und Routinen unterstützt
12 Langley RG et al. N Engl J Med 2014; 371:326-338
13 Mease PJ et al. N Engl J Med 2015; 373:1329-1339
14 McInnes IB et al. Lancet 2015;386:11371146
15 Baeten D et al. N Engl J Med 2015; 373:2534-2548
16 Deodhar A et al. Arthritis Rheumatol 2021;73:110-120
17 Kimball AB et al. Lancet 2023;401:747761
18 Magnolo N et al. J Am Acad Dermatol 2022;86:122-130
19 Brunner HI et al. Ann Rheum Dis 2023; 82:154-160
attexis® die Tagesstruktur und den Transfer therapeutischer Inhalte in den Alltag. Praktische Materialien wie Übungen und Audio-Dateien erleichtern die Integration und fördern nachhaltige Veränderungen.
Die Wirksamkeit von attexis® wurde in einer randomisiert kontrollierten Studie nachgewiesen (ClinicalTrials.gov ID: NCT06221930): Die Nutzung von attexis® ergänzend zum „Treatment as usual“ (TAU) führte zu einer signifikanten Reduktion der ADHS-Kernsymptomatik mit großer Effektstärke im Vergleich zu TAU allein. Auch die sekundären Endpunkte „soziales & berufliches Funktionsniveau“, „gesundheitsbezogene Lebensqualität“, „depressive Symptome“ und „Selbstwertgefühl“ verbesserten sich signifikant im Vergleich zum TAU.
B. S.
Die Poliomyelitis („Kinderlähmung“) ist eine schwere neurologische Krankheit, die bei ungeimpften Menschen zu dauerhaften Lähmungen führen kann. Sie ist durch Schmierinfektion übertragbar und betrifft vor allem Kinder unter 5 Jahren. Doch auch Erwachsene können daran erkranken. Wie das Robert Koch-Institut (RKI) berichtete, sind in vielen deutschen Städten im Abwasser Polio-Viren entdeckt worden. Die nachgewiesenen Erreger (cVDPV2) stammen von Impfviren, die sich mit der Zeit so verändert haben, dass sie wieder pathogen sind, vor allem bei Menschen mit unzureichendem Impfschutz. Nach Einschätzung des RKI erscheint es „zunehmend wahrscheinlicher, dass derzeit in Deutschland zumindest lokal begrenzt eine Übertragung von cVDPV2 stattfindet“. Begründet wird dies mit der langen Dauer des Geschehens – über erste auffällige Wasserproben wurde bereits Ende 2024 in einem Großteil der nun betroffenen Städte berichtet – und mit dem Nachweis von cVDPV2 an verschiedenen Standorten. Auch wenn nach Experten-Einschätzung kein Risiko für eine Endemie besteht, ist es laut Prof. Dr. Uta Meyding-Lamadé, Mitglied der DGN-Kommission Neuroinfektiologie und stv. Vorsitzende der Nationalen Poliokommission des RKI, nicht mehr ausgeschlossen, dass es bei vulnerablen Men-
schen – dazu zählen Kinder und immundefiziente Erwachsene ohne Impfschutz – zu Infektionen kommen kann. Eine Infektion bedeutet zwar nicht automatisch eine Erkrankung, dennoch sind neurologische Folgen nicht ausgeschlossen. Daher rät die Deutsche Gesellschaft für Neurologie (DGN) Risikopersonen, ihren Impfstatus zu überprüfen.
Ein besonderes Augenmerk gilt dabei den Kindern – es gibt immer wieder Fälle, in denen aus verschiedensten Gründen Impftermine nicht wahrgenommen werden und die Kinder nicht ausreichend geschützt sind. Die zweite gefährdete Gruppe sind Erwachsene, bei denen die Immunkompetenz eingeschränkt ist, z.B. durch angeborene oder erworbene Störungen des Immunsystems, durch Erkrankungen wie beispielsweise Leukämien, oder durch die Gabe von Immunsuppressiva. „Ungeimpfte dieser Risikogruppe sollten eine Vakzinierung in Erwägung ziehen und vulnerable geimpfte Personen sollten prüfen, wann die nächste Auffrischungsimpfung fällig ist“, so Meyding-Lamadé.
Übertragungsrisiko minimieren
Des Weiteren verweist das RKI darauf, dass es sich in den meisten Fällen um eine Schmierinfektion handelt, deren Ausbreitung durch strenge Handhygiene vermieden werden kann. Die Viren werden mit dem Stuhl ausgeschieden und oft mit der Klinke in die Hand gegeben. Regelmäßiges Händewaschen und Handdesinfektionen minimieren das Übertragungsrisiko.
Dreiphasiger Verlauf und limitierte Behandlungsmöglichkeiten
Die Polio-Erkrankung verläuft in 3 klinischen Phasen. Zunächst kommt es zu Kopfschmerzen und Fieber, labordiagnostisch ist in diesem Stadium eine Liquorpleozytose, d.h. eine vermehrte Anzahl von Zellen in der Rückenmarksflüssigkeit, auffällig. Danach schließt sich das paralytische Stadium an. Wieder kommt es zu Fieber und durch die entzündliche Schädigung des Rückenmarks entwickeln sich die für die Kinderlähmung typischen asymmetrischen, proximal akzentuierten Lähmungen. Diese bilden sich dann einige Wochen später, im Reparaturstadium, meist nur unvollständig zurück.
„Gut ein Drittel der Betroffenen trägt schwere, dauerhafte Lähmungen davon“, erklärt DGN-Generalsekretär Prof. Dr. Peter Berlit. Hinzu kommen Spätkomplikationen wie das Post-Polio-Syndrom, das durch Fatigue und diffuse Schmerzen gekennzeichnet ist. Außerdem besteht ein hohes Risiko für die postpoliomyelitische spinale Muskelatrophie, bei der ein fortschreitender Muskelschwund erneut zu Lähmungen führt.
„Die Therapiemöglichkeiten einer akuten Poliomyelitis sind sehr limitiert. In Frage kommt lediglich die Gabe von Immunglobulinen, doch die Wirksamkeit ist bisher noch nicht ausreichend belegt. Das macht deutlich, wie wichtig die Prophylaxe durch die Impfung ist“, schlussfolgert Berlit.
DGN
Die neovaskuläre altersabhängige Makuladegeneration (nAMD), die durch eine intraretinale oder subretinale Flüssigkeitsansammlung gekennzeichnet ist, zählt zusammen mit dem diabetischen Makulaödem (DMÖ) zu den häufigsten Ursachen für den Verlust der Sehkraft. Mit dem bispezifischen Antikörper Faricimab (Vabysmo®), der direkt in den Glaskörper injiziert wird, ist seit 2022 eine Therapieoption verfügbar, die für erwachsene Patienten eine zeitgleiche Zulassung in beiden Indikationen erhalten hat.
Die Zulassung für die Therapie der nAMD basiert auf den beiden jeweils für 2 Jahre angelegten identisch konzipierten, randomisierten, doppelblinden, aktiv komparatorkontrollierten Phase-III-Studien TENAYA und LUCERNE, in denen Faricimab mit Aflibercept verglichen wurde [1, 2]. Die therapienaiven Studienteilnehmer erhielten randomisiert entweder Faricimab 6 mg intravitreal bis maximal alle 16 Wochen nach 4 initialen monatlichen Dosen oder Aflibercept 2 mg intravitreal alle 8 Wochen nach 3 initialen monatlichen Dosen. Primärer Endpunkt in beiden Studien war der Nachweis der Nicht-Unterlegenheit von Faricimab gegenüber Aflibercept in Bezug auf die Veränderung der bestkorrigierten Sehschärfe (Best-corrected Visual Acuity, BCVA) nach einem Jahr. Beide Studien erreichten den primären Endpunkt: Bei den Patienten, die mit Faricimab behandelt wurden, verbesserte sich der Visus nach 12 Monaten um durchschnittlich 5,8 Buchstaben in der TENAYA-Studie und um 6,6 Buchstaben in der LUCERNE-Studie. Unter Aflibercept waren es +5,6 bzw. +6,6 Buchstaben, was die Nichtunterlegenheit von Faricimab belegt [1]. Die Verbesserungen unter
Faricimab reduziert retinale Flüssigkeit
der Faricimab-Therapie blieben im zweiten Jahr bei durchschnittlich nur 3 Injektionen erhalten und
etwa 78 % der Patienten erreichten ein Behandlungsintervall von 3 oder 4 Monaten [2].
Antikörper mit bispezifischem Wirkansatz
Faricimab (Vabysmo®) ist der erste bispezifische Antikörper, der für die intravitreale Anwendung entwickelt wurde. Der IgG1-Antikörper richtet sich nicht nur – wie andere Therapieoptionen – gegen den vaskulären endothelialen Wachstumsfaktor (VEGF-A), sondern hemmt zusätzlich auch Angiopoietin-2 (Ang-2) [4].
VEGF-A stimuliert die pathologische Neubildung von Blutgefäßen im Bereich der Netzhaut. Diese sind jedoch weniger stabil und durchlässiger als die bereits vorhandenen Gefäße, sodass Flüssigkeit aus den Gefäßen austritt und sich in der Netzhaut ansammelt. Angiopoietin-2 sensibilisiert die Blutgefäße für die Aktivität des VEGF-A und verstärkt damit dessen negative Wirkung auf die Stabilität und Durchlässigkeit der Blutgefäße. Dadurch werden entzündliche Prozesse angeregt, die das Gewebe weiter schädigen und zum Sehverlust beitragen.
Indem Faricimab sowohl VEGF-A als auch Ang-2 blockiert, hemmt es beide Signalwege für die pathologische Angiogenese und die Destabilisierung der Gefäße. Diese bispezifische Wirkung könnte die Sehkraft von Menschen mit Netzhauterkrankungen länger erhalten als eine alleinige Anti-VEGF-Monotherapie und gleichzeitig die Häufigkeit der erforderlichen Augeninjektionen reduzieren [5, 6].
Die empfohlene Dosis von Faricimab beträgt 6 mg (0,05 ml Lösung), angewendet als intravitreale Injektion alle 4 Wochen für die ersten 3 Dosen. 16 und/oder 20 Wochen nach Einleitung der Behandlung sollte eine Beurteilung der Krankheitsaktivität basierend auf den anatomischen und/oder visuellen Befunden erfolgen, um die Behandlung individuell an den Patienten anpassen zu können. Bei Patienten ohne Krankheitsaktivität ist eine Verabreichung von Faricimab alle 16 Wochen (4 Monate) zu erwägen. Bei Patienten mit Krankheitsaktivität ist eine Behandlung alle 8 Wochen (2 Monate) oder alle 12 Wochen (3 Monate) zu erwägen [7].
Aktuelle Daten aus dem Praxisalltag bestätigen diese Ergebnisse und zeigen, dass jeder 2. Patient mit nAMD in der Routine-Versorgung bereits nach der ersten Injektion von der Behandlung mit Faricimab profitieren kann [3].
Flüssigkeitsreduktion nach nur einer Injektion
Die Anwesenheit retinaler Flüssigkeit, sei es intraretinal (IRF) oder subretinal (SRF), ist ein entscheidender Biomarker für die Krankheitsaktivität bei nAMD. Dass Faricimab auch in der Routineversorgung die Krankheitsaktivität entscheidend verbessern kann, indem es die retinale Flüssigkeit schnell und anhaltend reduziert, belegen die Ergebnisse der USReal-World-Studie TRUCKEE [3]. In dieser Studie wurden insgesamt 521 Augen von nAMD-Patienten mit Faricimab behandelt. Fast die Hälfte der Augen (258 von 521) wies initial eine klinisch relevante Flüssigkeitslast von über 10 nL auf. Bei fast 70 % dieser Augen kam es schon nach der ersten Faricimab-Injektion zu einer Flüssigkeitsreduktion. Bei der Hälfte aller untersuchten Augen (49,9 %) verringerte sich nach der ersten Faricimab-Injektion die retinale Flüssigkeit im Mittel um 60,7 nL [3]. Die Analyse nach Flüssigkeitskompartimenten zeigte ebenfalls konsistente Ergebnisse. Bei Augen mit sowohl intraretinaler als auch subretinaler Flüssigkeit zu Baseline wurde bei über 80 % die Flüssigkeit bereits nach der ersten Injektion reduziert – ein Effekt, der über 4 aufeinanderfolgende Behandlungen anhielt [3].
Hohe Wirksamkeit bei therapienaiven und vorbehandelten Patienten
Die Wirksamkeit von Faricimab zeigte sich sowohl bei zuvor nicht behandelten Patienten als auch bei jenen, die bereits eine Behandlung erhalten hatten: Bei therapienaiven Patienten führte die erste Faricimab-Behandlung bei 81,6 % aller Augen zu einer Flüssigkeitsreduktion, bei den Augen mit hoher Flüssigkeitslast in dieser Gruppe waren es sogar 91,2 %. Die durchschnittliche Flüssigkeitsreduktion bei unbehandelten Augen betrug –219,2 nL.
Bei den bereits mit Aflibercept 2 mg vorbehandelten Patienten reduzierte Faricimab bei der ersten Behandlung die Flüssigkeit bei der Hälfte der Augen (50 %). Im Durchschnitt betrug diese Reduktion –57,0 nL. Besonders deutlich war der Effekt bei den Augen, die eine messbare Flüssigkeitsmenge von über 10 nL aufwiesen: Hier führte Faricimab bei etwa zwei Drittel (67,6 %) dieser vorbehandelten Augen zu einer Flüssigkeitsreduktion [3].
Anhaltende Wirksamkeit ermöglicht verlängerte Behandlungsintervalle
Die flüssigkeitsreduzierenden Effekte von Faricimab waren anhaltend und nachhaltig. Der Anteil der Augen, in denen die Flüssigkeit verringert werden konnte, blieb von der 2. bis zur 5. Injektion stabil bei über 50 %. Aufgrund der hohen und konstanten Wirksamkeit konnten die Behandlungsintervalle kontinuierlich verlängert werden: von
53,4 Tagen nach der 2. Injektion auf 61,5 Tage nach der 5. Injektion [3] – ein wichtiger Schritt zur Verringerung der Behandlungslast für die Patienten.
Präzise Therapiedaten dank KI-gestützter Flüssigkeitsanalyse
Zur Quantifizierung der intraretinalen (IRF) und subretinalen Flüssigkeit (SRF) wurde der Notal OCT Analyzer (NOA) verwendet. Dieser auf künstlicher Intelligenz basierende Algorithmus ermöglicht eine automatische und exakte Quantifizierung von IRF und SRF in Nanolitern (nL) und liefert detaillierte Einblicke in das anatomische Ansprechen auf die Behandlung mit Faricimab, auch bei Patienten mit komplexen Krankheitsverläufen [2, 8].
Brigitte Söllner, Erlangen
Literatur
1 Heier JS et al. Lancet 2022;399:729-740
2 Khanani AM et al. Ophthalmology 2024; 131:914-926
3 Aziz et al. Eye (Lond) 2025;39(6):10991106
4 Khan M et al. Cells 2020;9:1869
5 Heier JS et al. Retina-J Ret Vit Dis 2021; 41:1-19
6 Sahni J et al. Ophthalmology 2019;126: 1155-1170
7 Fachinfornmation Vabysmo®; Stand: Mai 2025
8 Wong et al. Ophthalmology 2024;131: 708-723
Für Neugeborene ist ein ausreichender Schutz vor dem Respiratorischen Synzytial-Virus (RSV) von besonderer Bedeutung, denn Säuglinge sind wegen ihrer kleinen Atemwege und des noch nicht ausgereiften Immunsystems besonders anfällig für eine Infektion. Die RSV-Infektion ist vor allem in den ersten Lebensmonaten mit einer hohen Morbidität assoziiert [1].
Im ersten Lebensjahr infizieren sich 50 – 70 % aller Säuglinge und bis zum zweiten Geburtstag fast alle Kinder mit RSV [2]. Weltweit ist das Virus für zwei Drittel aller akuten Atemwegserkrankungen bei Säuglingen und Kindern verantwortlich [3]. Während der RSV-Saison ist fast die Hälfte der Hospitalisierungen auf akute RSVAtemwegsinfekte bei <1-Jährigen zurückzuführen [4]. Auch in der Saison 2024/25 waren die Fallzahlen akuter Atemwegserkrankungen in Deutschland auf einem hohen Niveau, betroffen dabei waren vor allem Kinder von 0 – 4 Jahren [5].
Maternale Antikörper bieten dem Neugeborenen 6 Monate Schutz
Zur Prävention von schweren Verläufen stehen verschiedene Möglichkeiten zur Verfügung, darunter der seit August 2023 in der Europäischen Union zugelassene RSV-Impfstoff Abrysvo®, der bei Schwangeren appliziert werden kann, um ihr Neugeborenes für die ersten 6 Lebensmonate zu schützen [6]. In der Zulassungsstudie MATISSE zeigte Abrysvo® bei Säuglingen eine hohe Wirksamkeit gegen RSV-bedingte Erkrankungen der unteren Atemwege [7]. Gegen schwere RSV-assoziierte, behandlungsbedürftige Erkrankungen der unteren Atemwege lag
die Wirksamkeit bei Säuglingen bei 82,4 % für die ersten 90 Tage und bei 70,0 % für die ersten 180 Tage nach der Geburt [7]. In vielen Ländern – darunter Polen, Großbritannien und Frankreich – wird die maternale Impfung mit dem bivalenten Präfusions-F-Impfstoff Abrysvo® daher bereits empfohlen, um Neugeborene zu schützen. Durch die Impfung bilden werdende Mütter vermehrt Antikörper, die sie über die Plazenta auf ihr Ungeborenes übertragen und die das Baby vom ersten Atemzug an über die ersten Lebensmonate hinweg schützen können [8]. Seit März 2024 gehört die maternale RSV-Impfung auch zum nationalen Impfprogramm in Argentinien. Im ersten Jahr der Implementierung wurde die Immunisierung bereits von 62,5 % der Schwangeren in Anspruch genommen [9].
Auch Real-World-Daten belegen die hohe Wirksamkeit
Real-World-Daten aus der multizentrischen Studie BERNI zeigen eine Effektivität in den ersten 6 Lebensmonaten von 71,3 % für Abrysvo® beim Schutz vor Hospitalisierungen aufgrund RSVassoziierter Erkrankungen der unteren Atemwege (lower respiratory tract illness, LRTI). Gegen schwere RSV-assoziierte LRTI, die eine Hospitalisierung erforderten, wies Abrysvo® für die ersten
6 Lebensmonate eine Effektivität von 76,9 % auf [10]. Weitere Studien aus Argentinien kamen zu vergleichbaren Ergebnissen [9, 11]. Auch Daten des US-amerikanischen Centers for Disease Control and Prevention (CDC) zeigen in der RSV-Saison 2024/25 für die maternale Impfung mit Abrysvo® eine Effektivität von 79 %* bzw. 70 %** gegen RSV-assoziierte Krankenhausaufenthalte bei Säuglingen [12].
Effektive Strategie mit zwei Präventionsmöglichkeiten
Neben der maternalen Immunisierung gibt es auch die Möglichkeit, Säuglinge nach der Geburt durch die Gabe monoklonaler Antikörper vor RSV zu schützen. Eine gemeinsame Präventionsstrategie, bei der beide Optionen gleichberechtigt empfohlen sind und den Eltern zur Wahl stehen, wird unter anderem in den USA verfolgt. Das Advisory Committee on Immunization Practices (ACIP) empfiehlt seit Anfang August 2023 den monoklonaler Antikörper und seit Ende September desselben Jahres die maternale Immunisierung mit Abrysvo®. Erste Real-World-Da-
* VISION: Auswertung elektronischer Gesundheitsakten aus 160 Notaufnahmen und 131 Krankenhäusern in 6 US-Bundesstaaten
** NVSV: Aktive Überwachung akuter Atemwegserkrankungen bei Kindern in 7 akademischen pädiatrischen Gesundheitssystemen in 7 US-Bundesstaaten
ten weisen darauf hin, dass sich der Ansatz mit dem Angebot verschiedener Präventionsmöglichkeiten bewährt [12, 13, 14]. So konnten RSV-Hospitalisierungen bei Säuglingen im Alter von 0 – 7 Monaten in den USA in der Saison 2024/25 im Vergleich zu 2017 – 2020 um 31 %*** bzw. 38 %** gesenkt werden [13].
Maternale Immunisierung –eine oft präferierte RSVPräventionsmethode
Umfragen aus den USA [15] und Deutschland [16] weisen auf eine breite Akzeptanz für die bestehenden Präventionsoptionen gegen RSV hin. So zeigte eine Umfrage unter Eltern aus Oregon und Washington eine vergleichbare Akzeptanz für den Schutz des Babys durch eine Impfung der werdenden Mutter (70 %) wie mit dem monoklonaler Antikörper nach der Geburt (68 %). Während 49 % für beide Optionen offen waren, präferierten 37 % die maternale Impfung und 12 % die passive Immunisierung des Babys [15]. Auch eine Umfrage aus Deutschland zeigt eine Präferenz der maternalen Immunisierung bei den Müttern: 46,5 % der Befragten bevorzugten die Impfung in der Schwangerschaft gegenüber 37,5 %, die den monoklonaler Antikörper präferierten [16]. Die deutsche Umfrage ergab zudem, dass 88,7 % der befragten Mütter RSV als ernstzunehmende Bedrohung für die Gesundheit ihres Neugeborenen betrachten. 59 % der Befragten würden sich bei künftigen Schwan-
*** RSV-Associated Hospitalization Surveillance Network (RSV-NET): aktive Surveillance laborbestätigter RSV-Hospitalisierungen an 300 Krankenhäusern in 13 Staaten
RSV-Impfung mit Abrysvo® für viele Menschen ab 60 Jahren nun deutschlandweit über Sprechstundenbedarf verordenbar
Nach Abschluss der letzten regionalen Impfvereinbarung kann die RSV-Impfung im Rahmen der STIKO-Empfehlung seit Juli deutschlandweit über den Sprechstundenbedarf (SSB) bezogen werden. Die Kostenübernahme basiert auf der Empfehlung der Ständigen Impfkommission (STIKO) für die einmalige RSV-Impfung für alle Personen ab 75 Jahren sowie Personen im Alter von 60 – 74 Jahren, die mit bestimmten Risikofaktoren für einen schweren RSV-Krankheitsverlauf und/oder in Einrichtungen der Pflege leben.
Zulassungserweiterung für Personen ab 18 Jahren Seit Ende März 2025 ist Abrysvo® auch für Personen ab 18 Jahren zugelassen. Dies ermöglicht eine individuelle Impfentscheidung über den RSV-Schutz bei Erwachsenen jeden Alters. Mehrere Fachgesellschaften, wie z.B. die Deutsche Gesellschaft für Pneumologie und Beatmungsmedizin e.V., sprechen sich bereits für eine RSVImpfung ab 18 Jahren aus, wenn schwere pulmonale oder kardiovaskuläre Vorerkrankungen oder deutliche Einschränkungen der Immunabwehr vorliegen.
Die RSV-Impfung mit dem bivalenten Präfusions-F-Impfstoff Abrysvo® kann ganzjährig erfolgen und gleichzeitig mit der saisonalen Grippeimpfung sowie einem COVID-19-mRNA-Impfstoff verabreicht werden. Die STIKO empfiehlt, die Impfung möglichst vor der RSV-Saison durchzuführen, damit der Schutz bereits bei Saisonbeginn besteht.
gerschaften gegen RSV impfen lassen, aktuell geben allerdings nur 17 % an, gegen RSV geimpft zu sein. Hauptgründe für die niedrige Impfrate waren, dass den Frauen die Impfung nicht ärztlicherseits empfohlen wurde (41 %), sowie eine allgemein fehlende Kenntnis (25,9 %) über die maternale RSVImmunisierung [16].
Aktuelle Empfehlungs- und Erstattungssituation in
Während die maternale RSV-Impfung in vielen Ländern bereits breit angewendet und erstattet wird, ist dies in Deutschland regulär noch nicht der Fall, da eine Empfeh-
lung der Ständigen Impfkommission (STIKO) aktuell aussteht. Die Deutsche Gesellschaft für Gynäkologie und Geburtshilfe (DGGG) sprach sich bereits im November 2023 zusammen mit mehreren perinatologischen Fachgesellschaften sowie dem Berufsverband der Frauenärzte für die RSV-Impfung von Schwangeren aus [17]. Die deutsche Gesellschaft für Perinatale Medizin forderte gemeinsam mit der DGGG die STIKO im September 2024 erneut dazu auf, sich zeitnah mit der maternalen RSVImpfung zu befassen [18].
Aktuell erstatten bereits über 30 Krankenkassen ihren schwangeren Versicherten die RSV-Impfung als freiwillige Satzungsleistung. Bei Privatversicherten ist die Er-
stattung an die individuelle Tarifleistung geknüpft. Darüber hinaus besteht die Möglichkeit der Beantragung einer Kostenübernahme im Rahmen der Einzelfallentscheidung.
Elisabeth Wilhelmi, München
Literatur
1 Pickles RJ et al. J Pathol 2015;235:266276
2 Robert Koch-Institut Epid Bull 2024;1:311
3 Piedimonte G et al. Pediatr Rev 2014; 35:519-30. Erratum in Pediatr Rev 2015; 36:85
4 Vasconcelos MK et al. BMJ Open Respir Res 2021;8:e000887
5 Robert Koch-Institut. ARE-Dashboard: Aktuelle Situation akuter respiratorischer Erkrankungen in Deutschland. Datenstand: 29.02.2025
6 Fachinformation Abrysvo®; Stand: April 2025
7 Simoes EAF et al. Obstet Gynecol 2025; 145:157-167
8 Kampmann B et al. N Engl J Med 2023; 388:1451-1464
9 Gentile A et al. Pediatr Infect Dis J 2025; doi: 10.1097/INF.0000000000004878
10 Pérez MG et al. Lancet Infect Dis 2025 doi: 10.1016/S1473-3099(25)00156-2
11 Razzini JL et al. VeriXiv 2025;2:34
12 CDC. ACIP 2025. https://www.cdc.gov/ acip/downloads/slides-2025-06-25-26/03MacNeil-MatPeds-RSV-508.pdf
13 Patton ME et al. MMWR 2025;74:273281
14 Litman EA et al. JAMA Netw Open 2025; 8:e2460729
15 Kuntz JL et al. Pediatr Infect Dis J 2025;44(2S):S162-S166
16 Civey-Konsumentenbefragung 05/2025 (data on file, accessible on request)
17 Deutsche Gesellschaft für Gynäkologie und Geburtshilfe (DGGG). Stellungnahme zur RSV-Impfung für Schwangere, November 2023. https://www.dggg.de/fileadmin/data/Stellungnahmen/GBCOG/2023/Stellungnahme_fur_RSVImpfung_fuer_Schwangere_final.pdf
18 Deutsche Gesellschaft für Perinatale Medizin. Stellungnahme zur spezifischen Prophylaxe von Infektionen mit Respiratorischen Synzytial Viren (RSV). www. dgpmonline.org/fileadmin/media/publikation/neuigkeiten/DGPM_Stellungnahme_ RSV_Prophylaxe.pdf
Den seltenen und erblich bedingten akuten hepatischen Porphyrien (AHP) liegt eine gestörte Häm-Biosynthese in der Leber zugrunde. Die Linderung der typischen Beschwerden mit heftigen Bauchschmerzen und Übelkeit bis hin zu Krämpfen war vor der Zulassung der small interfering (si)RNA Givosiran (Givlaari®) [1] im Jahr 2020 limitiert: Die Therapieoptionen waren auf akute Attacken beschränkt ohne jedoch chronische Symptome zu adressieren. Auch deshalb liefern aktuelle Real-World-Daten zur Therapie der akuten intermittierenden Porphyrie (AIP), der mit einem Anteil von 80 % häufigsten Unterform der AHP, wertvolle Erkenntnisse. Bei 75 % der 28 Patienten, die am Porphyrie-Zentrum des Klinikums Chemnitz und der Charité Berlin über einen Zeitraum von durchschnittlich 30 Monaten hinweg
mit Givosiran behandelt wurden, wurde sowohl bei akuter als auch chronischer Verlaufsform die Symptomatik gelindert. Außerdem berichteten die Studienteilnehmer von positiven Effekten auf ihre Lebensqualität und die psychische Gesundheit [2].
Auch Patienten mit chronischer AHP und seltenen Attacken profitieren
Die aktuelle Real-World-Studie weist eine wichtige Besonderheit auf: Sie schloss auch Patienten mit chronischen Symptomen und seltenen Attacken ein, die nicht den Einschlusskriterien der Zulassungsstudien entsprachen [3]. „Wir haben bereits vor Durchführung der Studie vermutet, dass auch Patienten mit chronischen Symptomen und seltenen Attacken einen
AHP: Potenziell lebensbedrohlich und bislang unterversorgt
Ursachen der AHP sind erbliche Gendefekte, die sich auf die Funktion der Enzyme der Hämsynthese auswirken. Infolgedessen kommt es zu einer Akkumulation von Aminolävulinsäure (ALA) und Porphobilinogen (PBG), die das neurologische und gastroenterale Beschwerdebild mit Symptomen wie heftigen Bauchschmerzen, Übelkeit, Erbrechen und Krampfanfällen hervorruft. Möglich sind zudem fortschreitende Lähmungen, die auch die Atemmuskulatur betreffen können und daher prinzipiell lebensbedrohlich sein können. Die Patienten berichten außerdem von Angstzuständen, Müdigkeit oder schmerzenden Gliedmaßen [4–9].
RNA-Interferenz
Als RNA-Interferenz (RNAi) wird ein natürlicher Mechanismus in den Zellen von Lebewesen bezeichnet, welcher der zielgerichteten Abschaltung von Genen dient. Die RNAi ist einer der derzeit vielversprechendsten und sich am schnellsten entwickelnden Bereiche in der Biologie und der Arzneimittelentwicklung. Die Entdeckung der RNAi wurde als „ein bedeutender wissenschaftlicher Durchbruch, der etwa einmal pro Dekade vorkommt“ gefeiert und 2006 mit dem Nobelpreis für Physiologie oder Medizin ausgezeichnet. Die neue Arzneimittelklasse der RNAi-Therapeutika nutzt den natürlichen Mechanismus in den Zellen. Bei der RNAi verbindet sich siRNA (small interfering RNA) mit Ribonukleinsäure-Molekülen und unterbindet so deren normale Funktion. siRNA bilden die therapeutische RNAi-Plattform von Alnylam Pharmaceuticals und wirken auf einer den heutigen Medikamenten vorgeschalteten Ebene. Dort bewirken sie eine Stummschaltung der jeweiligen Messenger-RNA (mRNA), die die Bauanleitung für die krankheitsverursachenden Proteine überträgt, und verhindern so deren Produktion. Dieser bahnbrechende Ansatz hat das Potenzial, die Behandlung von Patienten mit genetischen und anderen Erkrankungen grundlegend zu verändern.
Das auf dem Prinzip der RNA-Interferenz basierende Arzneimittel Givosiran (Givlaari®) führt zur Stummschaltung der ALAS-1 mRNA und damit zur Abnahme der Konzentrationen an ALA und PBG [1].
klinischen Nutzen aus der Therapie mit Givosiran ziehen könnten“, berichtete Professor Ulrich Stölzel, Porphyrie-Zentrum am Klinikum Chemnitz. „Nun haben wir den wissenschaftlichen Nachweis hierfür und konnten damit dazu beitragen, eine relevante Datenlücke zu verringern.“
Die Studie konnte zudem zeigen, dass sich neben den biochemischen Parametern die subjektive Bewertung der Lebensqualität durch die Patienten selbst verbesserte. „Das große therapeutische Potenzial der siRNA Givosiran liegt auch darin, dass die allgemeine Gesundheitsbelastung der Patienten mit chronischen Symptomen – zum Beispiel gemessen an der selbstberichteten Lebensqualität – unserer Studie zufolge stark reduziert wurde“, erläuterte Stölzel.
Alle Patienten berichteten von einer verbesserten Lebensqualität, einschließlich der Parameter psychische Gesundheit und Schmerzen – darunter auch Studienteilnehmer mit chronischen Symptomen, die nur sporadische Attacken hatten [2].
Zwei Studienteilnehmer beendeten die Therapie vorzeitig aufgrund einer therapieassoziierten Fatigue. Das Verträglichkeitsprofil war im Allgemeinen ausgewogen und es traten keine lebensbedrohlichen Nebenwirkungen auf [2].
„Vor der Zulassung von Givosiran basierte unser Vorgehen hauptsächlich darauf, Triggerfaktoren auszuschalten und Stress zu reduzieren. Mit der zielgerichteten Therapie hat sich die Versorgungssituation für Patienten mit AHP entscheidend verbessert“, fasste Stölzel zusammen [2, 10].
Elisabeth Wilhelmi, München
klinische Verbesserungen
Die in die Real-World-Studie eingeschlossenen Patienten erhielten 1 monatlich 2,5 mg/kg Givosiran subkutan injiziert, was bei 75 % der Patienten (n = 21) zu einer Verbesserung von chronischen und akuten Erkrankungssymptomen führte [2].
Nach 6 Therapiemonaten lagen die Aminolävulinsäure (ALA)-Werte aller Behandelten unter dem oberen Grenzwert der Normalwerte (<2 ULN/Upper Limit of Normal). Die Porphobilinogen (PBG)-Werte betrugen bei 60 % der Patienten <2 ULN (p < 0,001). Die annualisierte Rate der Attacken (AAR) sank von 2,9 vor Therapiebeginn auf 0,45 nach 6 Therapiemonaten (p < 0,01) [2].
Literatur
1 Fachinformation Givlaari®; Stand: November 2024
2 Kubisch I et al. J Clin Med 2024;13:6779
3 Kuter DJ et al. J Hepatol 2023;79:11501158
4 Bissell DM et al. Clin Transl Hepatol 2015;3:17-26
5 Ramanujam VM et al. Curr Protoc Hum Genet 2016;86:17.20.1-17.20.26
6 Gerischer LM et al. Brain Behav 2021; 11:e2389
7 Stölzel U et al. Internist 2021;62:937-951
8 Stölzel U et al. Gastroenterol 2019;157: 365-381.e4
9 https://www.livingwithporphyria.eu/de/ ueber-ahp; Stand: 18.06.2025
10 Dickey AK et al. Givosiran: a targeted treatment for acute intermittent porphyria. Hematology Am Soc Hematol Educ Program (2024) 2024;1:426-433
Bei der Alzheimer-Krankheit handelt es sich um eine primär degenerative zerebrale Erkrankung, die meist schleichend beginnt und sich langsam, aber stetig über einen Zeitraum von mehreren Jahren bis Jahrzehnten entwickelt. Deshalb versteht man sie heute als ein Kontinuum mit Progression von einer frühen asymptomatischen Phase über eine prodromale Phase der leichten kognitiven Störung (mild cognitive impairment, MCI) und schließlich bis zur leichten, mittelschweren und schweren Demenz [1, 2]. Leitsymptom der frühen Alzheimer-Krankheit ist die kognitive Beeinträchtigung, insbesondere der Gedächtnisleistung. Weitere mögliche Symptome sind Störungen der Aufmerksamkeit sowie der zeitlichen und/oder örtlichen Orientierung. Auch Sprachvermögen und ausführende Funktionen wie Planung oder Problemlösung können beeinträchtigt sein. Zu den nicht-kognitiven Symptomen zählen auch schon in frühen Krankheitsstadien neuropsychiatrische Auffälligkeiten wie Depression, Unruhe, Angst, Erregbarkeit, Teilnahmslosigkeit oder Schlafstörungen sowie Verlangsamung in Aktivität und Motivation [3].
Früherkennung hat einen hohen Stellenwert
Die neuropathologischen Veränderungen der kontinuierlich fort-
schreitenden Alzheimer-Krankheit sind bereits< 15 – 20 Jahre vor der Manifestation erster Symptome nachweisbar. Epidemiologische Daten weisen zudem darauf hin, dass die Mehrheit der Menschen mit Alzheimer-Pathologie (noch) nicht an Demenz leidet, sondern sich in einem frühen, präklinischen Stadium der Erkrankung befindet [4]. Vor diesem Hintergrund kommt der Früherkennung eine große Bedeutung zu, da sie eine frühe Behandlung und sekundärpräventive Maßnahmen in einem möglichst optimalen Zeitfenster ermöglicht. Dies gilt umso mehr im Zuge der Entwicklung neuer, krankheitsmodifizierender Behandlungsoptionen für frühe Stadien der Alzheimer-Krankheit. Ziel ist es, durch frühzeitige und indi-
vidualisierte Therapiemaßnahmen die Lebensqualität der Betroffenen möglichst lange zu erhalten sowie eine Progredienz der Symptomatik zu verlangsamen und bestenfalls zu verhindern [1, 3, 4].
Zielgerichtete Therapie mit Lecanemab reduziert pathologische Amyloid-Aggregate und -Plaques
In der EU waren bislang lediglich Arzneimittel zur symptomatischen Behandlung der Alzheimer-Krankheit im Stadium der Demenz zugelassen. Diese Medikamente zielen auf eine Besserung oder zumindest Stabilisierung der kognitiven Defizite und der Alltagskompetenz sowie eine Verminderung der mit
Die Alzheimer-Demenz ist mit einem Anteil von ca. 60 – 80 % die häufigste Form bzw. Ursache der Demenz und entspricht der letzten Phase der Alzheimer-Krankheit mit fortschreitendem Verlust der Gedächtnisleistung sowie der Alltagskompetenz und Selbstständigkeit [1]. Die geschätzte jährliche Inzidenz der AlzheimerDemenz in Deutschland liegt etwa zwischen 150.000 und 240.000 Fällen [3, 4]. Infolge der demografischen Entwicklung ist in den kommenden Jahren mit einer beträchtlichen Zunahme zu rechnen [4]. In mehr als 95 % der Fälle von Demenz aufgrund der Alzheimer-Krankheit handelt es sich um eine Form mit spätem Beginn, d.h. im Alter von ≥65 Jahren [3].
Der Demenz voraus geht eine prodromale Phase mit leichten kognitiven Beeinträchtigungen (mild cognitive impairment, MCI), aber mit im Wesentlichen erhaltener Alltagskompetenz. Die MCI ist ein wichtiges Früh- bzw. Risikosyndrom von Demenzerkrankungen [1, 3].
Neben laborchemischen Blutanalysen und bildgebenden Verfahren wie (bevorzugt) MRT oder CT zur differenzialdiagnostischen Abklärung und zum Ausschluss nicht-neurodegenerativer Ursachen werden heute zur Frühdiagnostik der Alzheimer-Erkrankung mittels Liquoranalyse und Positronen-EmissionsTomographie (PET) verschiedene Biomarker bestimmt, die auf charakteristische neuropathologische Veränderungen in Form der Proteine β-Amyloid und Tau hinweisen [1, 5]:
Biomarker-Gruppe
A (= Amyloid-Marker)
T (= Tau-Marker)
N (= Marker für Neurodegeneration)
Demenz einhergehenden Verhaltensstörungen ab [1, 4]. Eine darüber hinausgehende zielgerichtete krankheitsmodifizierende Therapie ermöglicht nun erstmals der humanisierte monoklonale Antikörper Lecanemab (Leqembi®), der im April 2025 von der Europäischen Kommission zur Behandlung von erwachsenen Patienten mit klinisch diagnostizierter leichter kognitiver Störung (MCI) und leichter Demenz aufgrund der AlzheimerKrankheit zugelassen wurde [6].
Voraussetzung für die LecanemabTherapie ist eine frühzeitige Diagnose noch vor dem Auftreten eines demenziellen Syndroms*. Außerdem muss eine Amyloid-Pathologie bestätigt werden und die Patienten müssen Apolipoprotein E ε4 (ApoE ε4)-Nichtträger oder heterozygote ApoE ε4-Träger sein** [7].
* Die beiden neuropathologischen Marker Veränderungen der Konzentration von β-Amyloid (Aβ) und intrazelluläre neurofibrilläre Tau-Protein-Konglomerate sind bereits Jahre vor dem Auftreten kognitiver Beeinträchtigungen oder erster klinischer Demenz-Symptomen im Gehirn bzw. im Liquor nachweisbar. Infolgedessen weist die Mehrzahl der Menschen mit Alzheimer-Pathologie (noch) keine Demenz auf, sondern befindet sich in einem frühen Stadium der Erkrankung [4, 10].
Messverfahren
Aβ42 oder Aβ42/Aβ40-Ratio im Liquor, Amyloid-PET
Phosphoryliertes Tau im Liquor, Tau-PET
Strukturelle MRT, FDG-PET, Gesamt-Tau im Liquor
Leqembi® ist die erste in Deutschland verfügbare Therapie der frühen Alzheimer-Krankheit, die auf zugrunde liegende Krankheitsprozesse abzielt [8]: Im Verlauf der Alzheimer-Pathogenese bilden sich im Gehirn durch Autoaggregation von Amyloid-Peptiden amyloide Plaques sowie hyperphosphorylierte Tau-Fibrillen. Mit fortschreitender Akkumulation setzen entzündliche Prozesse ein, die zum Untergang von Neuronen führen, was sich klinisch im Abbau kognitiver Fähigkeiten und einem zunehmenden Verlust der Selbsthilfefähigkeit manifestiert. Eine wichtige Erkenntnis ist, dass die Ansammlung zerebraler Amyloid-Ablagerungen der TauPathologie vorausgeht und eine Voraussetzung für die irreversible Progression der Neurodegenera-
** Apolipoprotein E ist ein Protein, das am Lipidstoffwechsel des Menschen beteiligt ist. Es wird mit der Alzheimer-Krankheit in Verbindung gebracht. Bei Menschen mit nur einem (heterozygot) oder keinem Allel (Nicht-Träger) des ApoE ε4-Gens ist die Wahrscheinlichkeit einer Amyloid-assoziierten Bildgebungsanomalien (ARIA) geringer als bei Menschen mit 2 ApoE ε4-Allelen (homozygot) [9]. ARIA sind bekannte Nebenwirkungen von Lecanemab, die Schwellungen und mögliche Blutungen im Gehirn umfassen [1, 9].
tion bei der Alzheimer-Krankheit ist.
Der monoklonale Antikörper Lecanemab richtet sich gezielt gegen beta-Amyloid (Aβ) im Gehirn. Er bindet sowohl an lösliche, toxische Aβ-Protofibrillen*** als auch an unlösliche Aβ-Plaques, reduziert deren Anreicherung und trägt so zur Minderung der neuronalen Schäden bei [7, 9].
Effektiv in allen Wirksamkeitsendpunkten
Basis der Zulassung von Lecanemab war die internationale randomisierte Studie Clarity AD, an der 1.795 Erwachsene (Einschlusskriterium Alter: 50 – 90 Jahre) mit früher symptomatischer Alzheimer-Krankheit teilnahmen
*** Protofibrillen gelten als eine der maßgeblich toxischen Formen von Aβ und tragen durch die Zunahme von unlöslichen AβPlaques, aber auch durch die direkte Schädigung von Zellmembranen und neuronalen Synapsen entscheidend zur Degeneration von Nervenzellen und damit zur kognitiven Verschlechterung bei der Alzheimer-Krankheit bei [11]. Es wird angenommen, dass die Reduktion von Protofibrillen das Fortschreiten der Alzheimer-Krankheit verlangsamen kann [1].


Verschlechterung
Adjustierte mittlere Veränderung gegenüber Baseline ( ± SE)
Placebo (n)
Lecanemab (n)
Adjustierter mittlerer Behandlungseffekt nach 18 Monaten: -0,54 95% CI: -0,78, -0,29 p=0.00001
Visiten (Monate)

31% Verlangsamung durch Lecanemab nach 18 Monaten
-PET
Abbildung 1: Lecanemab (Leqembi®) verlangsamte bei ApoE ε4-Nichtträgern und Heterozygoten nach 18 Monaten das Fortschreiten der Alzheimer-Krankheit im CDR-SB um 31 % verglichen mit Placebo [12].
Lecanemab verlangsamte das Fortschreiten der Krankheit im CDR -SB um 31% nach 18 Monaten verglichen mit Placebo bei ApoE ε4 Nichtträgern und Heterozygoten1
(Clinical Dementia Rating – Sum of Boxes), der die Kognition und Alltagsfunktion der Patienten anhand verschiedener Domänen abbildet [13].
– Die adjustierte mittlere Veränderung gegenüber Baseline nach 18 Monaten betrug 1,22 für Lecanemab und 1,75 für Placebo (Differenz: -0,54; 95% CI: -0,78 bis -0,29)1
– Insgesamt stimmen die Ergebnisse mit der Auswertung nach MMRM überein, nach der Lecanemab die klinische Verschlechterung im CDR-SB nach 18 Monaten gegenüber Placebo um 33% reduzierte (adjustierte mittlere Differenz gegenüber Baseline nach 18 Monaten : -0,58; 95% CI: -0,81, -0,35; p<0.00001) 2
Hinweis: Wirksamkeitsanalyse mittels multipler Imputationsmethode auf Basis der Werte der Kontrollgruppe unter Einbeziehung von 757 mit Lecanemab behandelten und 764 mit Placebo behandelten Patienten.
Lecanemab reduziert die fibrilläre Amyloid -Last zu jedem Beobachtungszeitpunkt ab 3 Monaten bei ApoE ε4 Nichtträgern und Heterozygoten 1
AD, Alzheimer’s disease, Alzheimer-Krankheit; CDR-SB, Clinical Dementia Rating-Sum of Boxes; CI, confidence interval, Konfidenzintervall; MMRM, mixed model repeated measures; SE, standard error, Standardfehler
1. Frölich L, ) UK Summary of Product Characteristics.


Weniger Amyloid Adjustierte mittlere Veränderung gegenüber
( ± SE) Placebo Lecanemab
<0,0001 -59,4% CL Differenz zugunsten Lecanemab nach 18 Monaten

Nach 18 Monaten hatte sich in der EU-indizierten Population (ApoE ε4-Nichtträger oder heterozygote ApoE ε4-Träger) unter der Behandlung mit Lecanemab im Vergleich zu Placebo die Verschlechterung in der CDR-SB, d.h. das Fortschreiten der Erkrankung, um 31 % reduziert (Abb. 1). Der mittlere CDR-SB-Wert zur Baseline lag in beiden Gruppen bei etwa 3,2. Die adjustierte mittlere Veränderung gegenüber Baseline nach 18 Monaten betrug 1,22 für Lecanemab und 1,75 für Placebo (Differenz: –0,54; 95%-KI: –0,78 bis –0,29; p = 0,00001) [12].

Die Behandlung mit Lecanemab verlangsamte die Progression der Erkrankung ähnlich stark auch in weiteren, bestimmte Teilaspekte beleuchtenden Scores [12]:
• Im ADAS-Cog14 (Alzheimer’s Disease Assessment Scale Cognitive Subscale 14), der speziell die kognitive Leistungsfähigkeit der Patienten abbildet, kam es zu einer Reduktion der Progression um 26 %.
Lecanemab 2,*

Abbildung 2: Lecanemab (Leqembi®) reduziert die fibrilläre Amyloid-Last zu jedem Beobachtungszeitpunkt ab 3 Monaten bei ApoE ε4-Nichtträgern und Heterozygoten [12].

AD, Alzheimer’s disease, Alzheimer-Krankheit; ApoE ε4, Apolipoprotein E ε4; CL Centiloid; PET, Positronen-Emissions-Tomographie *Bitte beachten Sie, dass die hier abgebildete Patientenpopulation auch homozygote ApoE ε4-Träger umfasst und damit von der Zulassung für Lecanemab in der EU abweicht. Homozygote ApoE ε4-Träger sind durch die Zulassung für Lecanemab in
der Behandlung ausgeschlossen.
[9]. Für die EU-Zulassung erfolgte eine Post-hoc-Analyse, bei der lediglich ApoE ε4-Nichtträger und heterozygote ApoE ε4-Träger (n = 1.521) berücksichtigt wurden [12]. Die Wirksamkeitsendpunkte in dieser Subpopulation waren konsistent mit den Ergebnissen der Gesamtkohorte der Clarity ADStudie.
Die Teilnehmer an der für die EUZulassung relevanten Studie (85 % der ursprünglichen CLARITY-AD Population) erhielten randomisiert entweder alle 2 Wochen Lecanemab 10 mg/kg, verabreicht als i.v. Infusion (n = 757), oder Placebo (n = 764). Die Studiendauer betrug 18 Monate. Primärer Endpunkt war der CDR-SB-Score
1. Frölich L, et al. Präsentation International Conference on Alzheimer’s and Parkinson’s Diseases (AD/PD). Wien, Österreich. 2025; 2. Bateman R. Präsentation Clinical Trials on Alzheimer’s Disease (CTAD) Congress San Francisco, USA. 2022.
• Im ADCS-MCI-ADL-Score (Alzheimer’s Disease Cooperative Study-Activities of Daily Living), der die Selbständigkeit der Patienten im Alltag bewertet, einschließlich der Fähigkeit, sich selbst anzuziehen, selbständig zu essen und an Gemeinschaftsaktivitäten teilzunehmen, kam es zu einer Verlangsamung der Verschlechterung um 33 %. Lecanemab reduzierte signifikant die Amyloid-Last im Gehirn, während diese in der Placebo-Gruppe leicht anstieg (Differenz: –59,4 Centiloid in Amyloid-PET nach 18 Monaten; p < 0,0001; Abb. 2) [12].
Die häufigsten Nebenwirkungen in der indizierten Population waren infusionsbedingte Reaktionen (26 % vs. 7 % unter Placebo), Amyloidassoziierte Bildgebungsanomalien mit Hamosiderinablagerung (ARIA-H), was Mikroblutungen und superfizielle Siderose umfasst (13 % vs. 7 %), Kopfschmerzen (11 % vs. 7 %) und Amyloid-assoziierte Bildgebungsanomalien mit Ödem (ARIA-E), die sich im MRT als Hirnödem oder Flüssigkeitsansammlungen im Bereich der Sulci darstellen (9 % vs. 1 %). Symptomatische ARIA-E traten bei 1,6 % der Studienteilnehmer auf (schwere symptomatische bei 0,3 %), symptomatische ARIA-H bei 0,8 %.
Insgesamt waren ARIA in der indizierten Teilpopulation nur etwa halb so häufig wie in der Gesamtpopulation [7, 9, 12].
Brigitte Söllner, Erlangen
Literatur
1 DGN e.V. & DGPPN e.V. (Hrsg.) S3Leitlinie Demenzen, Version 5.0
2 Jack CR Jr et al. Alzheimer’s Association 2024;20:5143-5169
3 Bleß H-H et al. Weißbuch Versorgung der frühen Alzheimer-Krankheit. Springer, München 2021
4 Riepe MW et al. Monitor Versorgungsforschung 03/2023:55-61
5 Jack CR Jr et al. Alzheimer’s Dement 2018;14:535-562
6 European Medicines Agency; EMA/ 63941/2025. https://www.ema.europa.eu/ en/documents/medicine-qa/questionsanswers-approval-marketingauthorisation-leqembi-lecanemab_en.pdf
7 Fachinformation Leqembi®; Juni 2025
8 www.g-ba.de/downloads/40-268-11631/ 2025-06-24_AMRL-III_SNV_Antidementiva-Lecanemab_TrG.pdf
9 van Dyck CH et al. New Engl J Med 2023;388:9-21
10 Pawlowski M et al. Internist 2022;63: 1000-1008
11 Ono K et al. Int J Mol Sci 2020;21:952
12 Froelich L et al. DGPPN 2024; Poster 17
13 Morris JC. Neurology 1993;43:24122414
Die Duchenne-Muskeldystrophie (DMD) ist eine seltene, X-chromosomal-rezessiv vererbte, fortschreitende neuromuskuläre Erkrankung, die durch Mutationen im DMD-Gen verursacht wird. Diese verhindern die Produktion von funktionsfähigem Dystrophin, eines Eiweißes, das mit zahlreichen weiteren Proteinen in der Muskelzelle vernetzt ist. Dieser Dystrophin-assoziierte Proteinkomplex (DAPC) sorgt für die mechanische Stabilität der Zellmembran während der Muskelkontraktion. Infolge des Dystrophinmangels bricht der DAPC zusammen, wodurch die Muskelfasern anfälliger für Schäden werden. Außerdem steigt der Spiegel der Histon-Deacetylase (HDAC) in den Muskelzellen, sodass die Aktivierung wichtiger Gene blockiert wird, die für die Erhaltung und Reparatur der Muskeln erforderlich sind. Infolgedessen werden die Muskelfasern ständig geschädigt, chronische Entzündungen und eine schlechte Regeneration führen schließlich zum Absterben der Muskelzellen und zum Umbau der Muskulatur in Fett- und Bindegewebe [1, 2, 3, 4].
DMD betrifft in erster Linie Jungen, wobei die Symptome typischerweise im Alter zwischen 2 und 5 Jahren auftreten. Mit fortschreitender Erkrankung verschlimmert sich die Muskelschwäche, was zu Schwierigkeiten beim Gehen und schließlich zum Verlust der Gehfä-
higkeit führt. Im weiteren Verlauf werden auch die Herz- und Atemmuskulatur in Mitleidenschaft gezogen, was die Hauptursache für einen vorzeitigen Tod ist [5]. DMD ist eine der schwersten und häufigsten Formen der Muskeldystrophie im Kindesalter und tritt bei der Geburt weltweit bei etwa 1 von 5.050 Jungen auf [6].
Bisherige Therapieansätze können Progression nicht stoppen
DMD ist nicht heilbar und die Lebenserwartung der Patienten ist deutlich reduziert – der Tod tritt meist zwischen dem 30. und 50. Lebensjahr ein. Bislang gibt es nur wenige zugelassene Behandlungen, die die Pathologie der DMD bei einem breiten Patientenspektrum adressieren und den Verlauf dieser schwerwiegenden Erkrankung signifikant beeinflussen können. Zur Basistherapie in jeder Krankheitsphase gehören supportive und symptomatische Maßnahmen, die medikamentöse (z.B. systemische Glukokortikoide, Ataluren), nichtmedikamentöse (z.B. Physiotherapie, Lagerungsschienen) und chirurgische Verfahren umfassen. Glukokortikoide stellen die medikamentöse Standardtherapie der DMD dar und sollten für jeden Patienten ab 5 Jahren erwogen werden, wobei die steroidtypischen Nebenwirkungen zu beachten sind.
Innovativer Histon-DeacetylaseInhibitor verzögert die Progression signifikant
Einen vielversprechenden neuen Therapieansatz stellen die HistonDeacetylase(HDAC)-Hemmer dar. Sie setzen an der unkontrollierten pathologischen Überaktivität der HDAC an, die zu Muskelschäden bzw. zum Umbau der Muskulatur in Fett- und Bindegewebe führen [1,2]. Die Hemmung der HDACAktivität kann dazu beitragen, Entzündungen zu reduzieren und die Vernarbung und Verdickung des Gewebes zu verringern. Aufgrund seiner überzeugenden Behandlungserfolge bei einem breiten Patientenspektrum erhielt der HDAC-Inhibitor Givinistat (Duvyzat®) vor Kurzem die bedingte Marktzulassung von der Europäischen Kommission. Der oral verabreichbare HDAC-Inhibitor ist in Kombination mit einer Kortikosteroid-Therapie indiziert zur Behandlung der Duchenne-Muskeldystrophie bei gehfähigen Patienten ab 6 Jahren – unabhängig von der spezifischen DystrophinGenmutation, die die Krankheit verursacht [7]. Givinistat reguliert die für DMD-Muskeln charakteristische übermäßige HDAC-Aktivität und trägt so dazu bei, die Expression von Schlüsselgenen und
biologischen Prozessen wiederherzustellen, die für den Erhalt und die Wiederherstellung der Muskeln wichtig sind. Indem sein Wirkmechanismus auf krankheitsrelevante Prozesse abzielt, kann Givinistat unabhängig von der Genmutation die Progression der DMD verzögern und die Muskelfunktion erhalten [8], was die Ergebnisse der zulassungsrelevanten Studie EPIDYS eindrucksvoll belegen [9]. In der randomisierten, doppelblinden, placebokontrollierten PhaseIII-Studie erhielten insgesamt 179 gehfähige Jungen ab 6 Jahren zweimal täglich Givinistat oder Placebo zusätzlich zur Kortikosteroid-Therapie. Die EPIDYSStudie erreichte ihren primären Endpunkt: eine statistisch signifikante und klinisch relevante Änderung der Zeit zur Absolvierung des 4-Stufen-Tests. Außerdem zeigte Givinistat Vorteile bei wichtigen sekundären Endpunkten, darunter das North Star Ambulatory Assessment (NSAA) sowie die Beurteilung der Fettinfiltration mittels Magnetresonanzspektroskopie. Die Behandlung mit Givinistat war mit einem um 40 % geringeren kumulativen Verlust an NSAA-Items assoziiert, was auf das Potenzial des HDAC-Inhibitors hinweist, die Progression der Erkrankung zu verzögern.
Dies unterstützt auch der Vergleich von Langzeitdaten der laufenden EPIDYS-Verlängerungsstudie mit natürlichen Verlaufsdaten mittels Propensity-Score-Matching, denn es zeigte sich, dass das mediane Alter bei Verlust der Gehfähigkeit in der Givinistat-Gruppe bei 18,1 Jahren lag, während es in der Kontrollgruppe 15,2 Jahre waren [10]. Givinistat wurde von den Kindern gut vertragen, die meisten unter der Therapie beobachteten unerwünschten Ereignisse waren leicht bis moderat.
Brigitte Söllner, Erlangen
Literatur
1 Sandonà M et al. Int J Mol Sci 2023;24: 4306
2 Consalvi S et al. Mol Med 2011;17:457465
3 Bez Batti Angulski A et al. Front Physiol 2023;14:1183101
4 Giuliani G et al. FEBS J. 2022;289:64846517
5 Walter MC et al. J Cachexia Sarcopenia Muscle 2017;8:681-685
6 Crisafulli S et al. Orphanet J Rare Dis 2020;15:141
7 www.ema.europa.eu/en/medicines/human/EPAR/duvyzat
8 Licandro SA et al. Skelet Muscle 2021; 11:19
9 Mercuri E et al. Lancet Neurol 2024; 23:393-403
10 Vandenborne K et al. Givinostat in Duchenne muscular dystrophy: effect on disease milestones. Poster presentation. MDA Clinical &Scientific Conference, Dallas, March 2025
Hoffnung für neurogenetische Krankheiten: „Small Molecules“ wirken bereits, bevor Symptome auftreten
Bei Neugeborenen mit spinaler Muskelatrophie (SMA) kann Risdiplam, ein „small molecule“, sicher und wirksam mRNA editieren und so den Krankheitsprogress aufhalten. In einer aktuellen „Proof-of-Principle“-Studie zu Risdiplam zeigte sich, dass das kleinmolekulare Medikament bereits vor dem Auftreten der ersten Symptome wirkt. Angesichts der Tatsache, dass bei verschiedenen
neurologischen Erkrankungen Genmutationen zu fehlerhafter, krankheitsverursachender mRNA führen, z.B. bei einigen Formen von ALS, Parkinson oder Demenzerkrankungen, ist dieses Studienergebnis von hoher Relevanz, und zwar weit über das beschriebene Krankheitsbild hinaus.
Spinale Muskelatrophie –eine infauste Diagnose
Die spinale Muskelatrophie (SMA) ist eine seltene, autosomal-rezessive neuromuskuläre Erkrankung. Zurückzuführen ist sie auf Mutationen des Survival-of-MotorNeuron-Protein 1 (SMN)-Gens. Der Schweregrad der Erkrankung variiert. SMA Typ 1 betrifft schon Neugeborene, die Säuglinge erlernen meistens keine normale Kopfkontrolle oder freies Sitzen und können wegen Schluckbeschwerden nicht trinken. Die Lebenserwartung beträgt ohne Therapie maximal 13 Monate, meistens versterben die Kinder in den ersten 2 Lebensjahren an respiratorischen Komplikationen bei Versagen der Atemmuskulatur. Die anderen Typen der Erkrankung haben eine etwas bessere Prognose, stellen aber bis auf Typ IV ebenfalls infauste Diagnosen dar.
Ursache der SMA ist in ca. 90 % der Fälle eine „Loss-of-function“Genmutation auf dem Chromosom 5 im „Survival-Motor-Neuron-1“ (SMN-1)-Gen. Dadurch kann kein funktionsfähiges SMN-Protein gebildet werden, das Motoneuronzellen zum Überleben benötigen. Vom SMN-1-Gen gibt es eine fast identische Kopie im Erbgut, das SMN-2-Gen, welches den Funktionsverlust des SMN-1-Gens zumindest abmildern sollte. SMN2 ist jedoch bei der Erkrankung nicht voll funktionsfähig und kann lediglich kurzes und instabiles SMN-Protein bilden.
Mit Risdiplam die Ablesbarkeit des SMN-2-Gens verbessern
Aktuell gibt es neben der Genersatztherapie, bei der das SMN1Gen mit Hilfe von Viren als int-
ravenöse Infusion eingeschleust wird, den Therapieansatz, die Ablesbarkeit des SMN-2-Gens zu verbessern, sodass der Körper wieder selbst funktionsfähiges SMNProtein bilden kann. Das leistet das Antisense-Oligonukleotid Nusinersen, das via Lumbalpunktion in die Zerebrospinalflüssigkeit injiziert werden muss. Doch auch das „small molecule“ Risdiplam kann das Splicing der SMN2-prämRNA optimieren, so die Ablesbarkeit des SMN-2-Gens erhöhen und damit den Krankheitsprozess aufhalten. In einer aktuellen Studie wurde der Nachweis erbracht, dass Risdiplam bereits effektiv wirkt, bevor Symptome entstehen. Insgesamt wurden 26 Säuglinge mit genetisch bestätigter SMA und 2, 3, 4 oder mehr Kopien von SMN-2 in die Studie aufgenommen und erhielten Risdiplam (0,2 mg pro kg Körpergewicht), und zwar bevor sie symptomatisch wurden. Nach 12 Monaten konnten 21 Säuglinge (81 %) 30 Sekunden lang ohne Unterstützung sitzen, 14 (54 %) konnten alleine stehen und 11 (42 %) alleine gehen. Drei Säuglinge wurden nach 12 Monaten von einem Elternteil aus der Studie genommen. Von den 23 Säuglingen, die die 24-monatige Behandlung abgeschlossen hatten, lebten alle ohne permanente Beatmung oder künstlicher Ernährung. Es traten keine schwerwiegenden Nebenwirkungen auf. „Das Ergebnis ist ein Durchbruch in der Therapie der SMA, die damit behandelbar wird, bevor Symptome entstehen“, erklärt Professor Tim Hagenacker, Sprecher der DGN-Kommission Motoneuron- und neuromuskuläre Erkrankungen. „Ein weiterer Vorteil von small molecules liegt in ihrer Einfachheit. Viele können die BlutHirn-Schranke überwinden und
sind oral verfügbar, während ASOTherapien nur intrathekal, in der Regel unter Vollnarkose, verabreicht werden können.“ Auch seien sie generell sehr gut verträglich. Wie Prof. Hagenacker betont, können small molecules so konzipiert werden, dass sie nur auf bestimmte Zelltypen oder Signalwege innerhalb des Nervensystems abzielen, wodurch die Präzision der Therapie erhöht wird.
„Die aktuelle Studie zeigt, dass das Therapieprinzip funktioniert und small molecules auch erfolgreich vor der symptomatischen Erkrankung eingesetzt werden können. Wir werden in den nächsten Jahren sicher viele solcher wirksamen small molecules für monogenetisch bedingte neurologische Krankheiten wie Chorea Huntington, das Rett-Syndrom, die Charcot-Marie-Tooth-Erkrankung, Neurofibromatose oder auch genetische Formen der ALS, Parkinson- oder Alzheimer-Erkrankung sehen. Zu den drei letztgenannten wurden bereits Bluttests für die Frühdiagnose entwickelt. Wenn diese klinisch validiert sind, könnte die Therapie bereits beginnen, bevor die Betroffenen Symptome entwickeln. Gerade bei neurodegenerativen Erkrankungen mit langer Prodromalphase wie Alzheimer oder Parkinson wäre das von großer Relevanz“, kommentierte Professor Peter Berlit, Generalsekretär der DGN.
Quellen
1 Finkel RS et al. RAINBOWFISH Study Group. Risdiplam in presymptomatic spinal muscular atrophy. N Engl J Med 2025;393:671-682
2 Devinsky O et al. Gene therapies for neurogenetic disorders. Trends Mol Med 2025;31:814-826
10 Jahre Mepolizumab: IL5Inhibition bewährt sich in der Modulation von Typ2Inflammationen
Vor knapp 10 Jahren wurde Mepolizumab (Nucala) zur Behandlung des schweren eosinophilen Asthmas (SEA) zugelassen. Als erster Interleukin (IL)-5-Inhibitor stellte Mepolizumab einen Durchbruch in der Asthmatherapie dar, da erstmals die eosinophile Entzündungskaskade effizient gehemmt werden konnte. Diese therapeutische Innovation ist von hoher Relevanz, da bis zu 84 % der erwachsenen Asthma-Patienten eine Typ-2-Inflammation mit eosinophilem Phänotyp aufweisen. Heute, ein Jahrzehnt später, zeigt sich: Die gezielte IL5-Inhibition ist nicht nur wirksam und gut verträglich, sondern bietet auch krankheitsmodifizierende Potenziale bei SEA sowie verschiedenen weiteren Typ-2-entzündlichen Erkrankungen – darunter auch die chronische Rhinosinusitis mit Nasenpolypen (CRSwNP).
Im Rahmen einer Presseveranstaltung von GSK beleuchteten PD Dr. Christian Geßner, Leipzig, und Prof. Dr. Chia-Jung Busch, Greifswald, die breite Datenlage und berichteten von eigenen Erfahrungen mit dem monoklonalen Antikörper.
Krankheitsmodifizierende Wirkung
„Die IL-5-Inhibition hat sich bei schwerem eosinophilem Asthma als echter Gamechanger erwiesen. Wir sehen nicht nur eine Reduktion der Exazerbationen, sondern auch eine Stabilisierung der Lungenfunktion und eine Re-
Mepolizumab (Nucala) ist ein humanisierter monoklonaler Antikörper (IgG1, Kappa), der mit hoher Affinität und Spezifität an humanes Interleukin-5 (IL-5) bindet. IL-5 ist das wichtigste Zytokin für Wachstum, Differenzierung, Rekrutierung, Aktivierung und Überleben von Eosinophilen, die wichtige Mediatoren einer Typ2-Inflammation sind. Durch die Bindung an an IL-5 verhindert Mepolizumab dessen Bindung an die Alpha-Kette des IL-5-Rezeptorkomplexes auf der Zelloberfläche von Eosinophilen. Dadurch wird die IL-5-Signaltransduktion blockiert und die Produktion sowie das Überleben der Eosinophilen vermindert.
Der Anti-IL-5-Wirkansatz zeigt auch positive Effekte und krankheitsmodifizierende Potenziale jenseits der Wirkung auf Eosinophile, wie unter anderem eine verbesserte epitheliale Integrität, die Wiederherstellung des Gleichgewichts der Immunantwort, die Herunterregulierung von Transkripten für die Schleimproduktion, das Remodelling der Atemwege sowie die Verbesserung der Atemwegsparameter.
duktion der oralen Kortikosteroidgabe – das ist ein klarer Hinweis auf eine krankheitsmodifizierende Wirkung – und das über Jahre hinweg bei guter Verträglichkeit“, erklärte Geßner. Damit deckt sich die Praxiserfahrung mit den Studiendaten, in denen die Wirksamkeit von Mepolizumab unabhängig von Komorbidität oder BiomarkerLevel wie FeNO (fraktioniertes ausgeatmetes Stickstoffmonoxid) oder Immunglobulin E beobachtet wurde. So konnte in der REALITI-A-Studie mehr als jeder zweite Patient (57 %) die Dosis oraler Kortikosteroide (OCS) auf 0 mg reduzieren und es traten etwa 80 % weniger Exazerbationen auf, und das auch nach 2 Jahren.
Darüber hinaus konnte das Sicherheitsprofil von Mepolizumab im Rahmen einer Open-Label-Extension-Studie (OLE) bestätigt werden, in der Patienten mit SEA bis zu 10 Jahre lang mit Mepolizumab
behandelt wurden. Bei einer kumulativen Exposition von über 1.500 Patientenjahren, einem medianen Follow-up von 2,03 Jahren sowie bei 27 % der Teilnehmenden einem Follow-up von sogar länger als 4,5 Jahren blieb die Rate der schwerwiegenden therapieassoziierten unerwünschten Ereignisse konstant niedrig.
Alternative zur OP bei chronischer Rhinosinusitis mit Nasenpolypen
Da auch bei CRSwNP häufig eine Typ-2-Inflammation zugrunde liegt, sind die Daten insbesondere bezüglich der Sicherheit von Mepolizumab bei SEA auch für diese Indikation von Interesse. Darüber hinaus bestätigten klinische Studien und Real-World-Daten die Wirksamkeit des monoklonalen Antikörpers bei CRSwNP anhand einer signifikanten Reduktion der Polypenlast nach 6 Monaten (median: 4–1, p < 0,001), einer Verbesserung der Lebensqualität anhand des SNOT-22 Scores (um bis zu 63
Punkte) sowie einer Reduktion der systemischen Kortikosteroidgabe um bis zu 64 % (p < 0,001). Darüber hinaus konnte die Notwendigkeit für eine Operation um bis zu 100 % gesenkt werden. „Für viele Patienten mit schwerer CRSwNP bedeutet Mepolizumab eine echte Alternative zur wiederholten Operation. Die gezielte IL5-Inhibition adressiert die zugrunde liegende Entzündung, was eine Symptomlinderung bei gleichzeitig gutem Sicherheitsprofil mit sich bringt“, betonte Chia-Jung Busch. Mit Mepolizumab steht Patienten eine wirksame Therapieoption mit gutem Sicherheitsprofil zur Verfügung, die es ermöglicht die Krankheitslast langfristig zu reduzieren. Darüber hinaus konnte das Verständnis von IL-5 als therapeutischem Ansatz seit Zulassung von Mepolizumab durch weitere Forschung weiter vertieft sowie seine Bedeutung unterstrichen werden.
Elisabeth Wilhelmi, München
7 Jahre CARTZelltherapie: AxiCel und die Chance auf Heilung bei r/r (D)LBCL
Vor 7 Jahren wurden erste Chimäre Antigen-Rezeptor (CAR)T-Zelltherapien wie Axicabtagen ciloleucel (Axi-Cel, Yescarta®) in Deutschland eingeführt. Mit Zulassung der innovativen Therapieform kam es zu einer Zeitenwende, unter anderem in der Behandlung des rezidivierten/refraktären großzelligen B-Zell-Lymphoms (r/r LBCL): „Nach ca. 30 Jahren relativen Stillstands konnten mit der einmaligen Gabe von Axi-Cel überzeugende Ergebnisse in der Drittlinie erreicht werden, ehe in
Die CAR-T-Zelltherapie stellt eine innovative Therapieoption im Kampf gegen bestimmte rezidivierte/refraktäre hämatologische Malignitäten, wie Non-Hodgkin-Lymphome und Leukämien, dar. Sie basiert auf der genetischen Modifikation körpereigener T-Zellen der Erkrankten, wodurch diese synthetische Immunrezeptoren (CAR) exprimieren – wie eine maßgeschneiderte Immuntherapie. Über die extrazelluläre Domäne dieser CAR können die CAR-TZellen nach der Reinfusion tumorspezifische Oberflächenproteine erkennen und die Krebszellen eliminieren.
Durchführung
Nach der Indikationsstellung für eine CAR-T-Zelltherapie in einem interdisziplinären Tumorboard wird die Therapie ausschließlich in qualifizierten Behandlungszentren durchgeführt, die in der Verabreichung des jeweiligen CAR-T-Zellproduktes sowie dessen Therapie- und Nebenwirkungsmanagement geschult sind. In Deutschland nimmt die Zahl dieser qualifizierten CAR-T-Zentren stetig zu, derzeit gibt es bereits über 40 solcher Zentren. In der Europäischen Union sind aktuell 6 CAR-T-Zellprodukte zur Therapie bestimmter Patienten mit verschiedenen hämatologischen oder lymphatischen Malignitäten zugelassen, darunter Axicabtagen ciloleucel (Axi-Cel, Yescarta®) und Brexucabtagen autoleucel (Brexu-Cel, Tecartus®). Zu den akuten Nebenwirkungen, die im Nachgang einer Therapie mit CAR-T-Zellen auftreten können, zählen u.a. das Zytokinfreisetzungssyndrom (CRS) und das Immuneffektor-assoziierte Neurotoxizitätssyndrom (ICANS). Bei der Wahl des Behandlungszentrums gilt es daher zu berücksichtigen, dass sich die Patienten poststationär für 4 Wochen in räumlicher Nähe zur Einrichtung (Fahrtzeit <2 Stunden) aufhalten sollen, um bei einem potenziellen Auftreten der akute Nebenwirkungen schnell reagieren zu können. Zur weiteren Nachsorge können Patienten in die Praxis der Primärbehandelnden zurückkehren.
Herstellungsprozess
Die derzeitige Herstellung von CAR-T-Zellen ist ein auf den Patienten zugeschnittener Prozess: Im qualifizierten Behandlungszentrum werden mittels Leukapherese zunächst Leukozyten des Patienten gewonnen und die T-Zellen isoliert. Diese werden dann genetisch so modifiziert, dass sie CAR exprimieren, die spezifisch gegen ein auf den Zielzellen exprimiertes Oberflächenprotein, z.B. CD19, gerichtet sind. Anschließend erfolgt die T-Zell-Expansion. Nach der Qualitätssicherung werden die kryokonservierten CAR-TZellen an das qualifizierte Behandlungszentrum ausgeliefert und dort dem Patienten im Anschluss an eine konditionierende Chemotherapie als Einmalinfusion reinfundiert. Die Herstellungszeit der einzelnen Produkte kann variieren. Für Axi-Cel beispielsweise liegt die Herstellungsdauer für Europa im Median bei 19 Tagen.
der Folge auch erstmals ein signifikanter Vorteil beim Gesamtüberleben (OS) im Vergleich zum bisherigen Standard of Care (SOC)* in der Zweitlinie nachgewiesen wurde“, berichtete Professor Peter Borchmann, Köln, auf einer Pressekonferenz von Kite, ein Unternehmen von Gilead. Langzeit-Follow-ups der pivotalen Studien zu Axi-Cel (ZUMA-1 für die 3. Linie und ZUMA-7 für die 2. Linie) zeigen, dass die OS-Kurve bei etwa 50 % der Patienten ein Plateau ausbildet. „Das legt nahe, dass die CAR-T-Zelltherapie mit Axi-Cel tatsächlich die Chance auf Heilung eröffnet“, ergänzte Professor Björn Chapuy, Berlin.
Das CAR-T-Wirkprinzip als „Game Changer“ in der Zweitlinie
Die Erstlinien-Therapie des (D) LBCL mit R-CHOP** hat kuratives Potenzial, versagt aber bei jedem vierten Patienten. „Die Aussichten für diese Betroffenen mit r/r (D)LBCL waren dann lange Zeit sehr schlecht: Ab der Zweitlinie sind vor nicht langer Zeit etwa 80 – 90 % der Patienten innerhalb von 5 Jahren verstorben“, erklärte Borchmann. „Erst das neue Wirkprinzip der CAR-T-Zelltherapien hat es ermöglicht, viele vormals hoffnungslose Fälle effektiv therapieren zu können.“
Die gegen CD19 gerichtete CAR-TZelltherapie Axi-Cel ist zugelassen zur Behandlung von erwachsenen
Patienten mit diffus großzelligem B-Zell-Lymphom (DLBCL) und hochmalignem B-Zell-Lymphom (HGBL), das innerhalb von 12 Monaten nach Abschluss einer Erstlinien-Chemoimmuntherapie rezidiviert oder gegenüber dieser refraktär ist, sowie für die Therapie von erwachsenen Patienten mit r/r DLBCL und primär mediastinalem großzelligem B-Zell-Lymphom (PMBCL) nach 2 oder mehr systemischen Therapien***.
Basis der Erstzulassung 2018 bei r/r DLBCL in der 3. Linie war die Studie ZUMA-1, die Erwachsene mit einem refraktären LBCL und mindestens 2 Vortherapien einschloss. Eine Analyse nach 5 Jahren zeigt das kurative Potenzial: „Zwei von drei Patienten, die vollständig auf Axi-Cel angesprochen haben, sind nach 5 Jahren noch am Leben“, betonte Chapuy. Damit hat sich das CAR-T-Wirkprinzip als „Game Changer in der Therapie“ erwiesen.
Die Daten unterstreichen laut Chapuy die deutliche Überlegenheit der Therapie mit Axi-Cel in der Zweitlinie gegenüber dem vormaligen Standard. „Relevant ist dabei auch, dass der Unterschied zwischen Axi-Cel und SOC signifikant war, obwohl 57 % der Patienten im SOC-Arm progredient waren und eine zelluläre Immuntherapie – davon 78 % Axi-Cel – erhielten.“
Heilung als Therapieziel
* Platinbasierte Salvage-Kombinationschemotherapie, gefolgt von Hochdosischemotherapie und autologer Stammzelltransplantation bei Patienten, die auf die SalvageChemotherapie ansprechen.
** R-CHOP: Rituximab; Cyclophosphamid, Hydroxydaunorubicin, Vinchristin, Prednison
Die Phase-III-Studie ZUMA-7 verglich die Wirksamkeit und Sicherheit von Axi-Cel in der Zweitlinie mit dem damaligen SOC bei 359 Erwachsenen mit einem LBCL, das innerhalb von 12 Monaten nach Abschluss einer Erstlinientherapie rezidivierte oder gegenüber dieser refraktär war. Die Auswertung ergab nach 4 Jahren ein signifikant längeres geschätztes OS nach einer Einmalbehandlung mit AxiCel im Vergleich zum SOC (HR: 0,73; 95%-KI: 0,54 – 0,98; strat. Log-Rank p-Wert = 0,03). Eine Subgruppenanalyse von ZUMA-7 bestätigte einen numerischen OSVorteil auch für ältere Patienten über 65 Jahre.
*** Axi-Cel wird außerdem angewendet zur Behandlung von erwachsenen Patienten mit r/r follikulärem Lymphom (FL) nach 3 oder mehr systemischen Therapien.
Um bei einer CAR-T-Zelltherapie beim r/r DLBCL von Heilung sprechen zu können, nannte Chapuy 3 Anforderungen: Die einmalige Anwendung muss zu einem Plateau bei der OS-Kurve führen, das Follow-up muss eine ausreichende Zeitspanne erfassen und das Überleben der Patienten muss vergleichbar mit dem der Allgemeinbevölkerung sein. Diese Anforderungen werden laut Chapuy von einer Therapie mit Axi-Cel erfüllt: Das Langzeit-Follow-up der ZUMA-7-Studie zeigte nach etwa 24 Monaten ein klares Plateau bei ca. 55 % der mit Axi-Cel behandelten Patienten und weist darauf hin, dass das weitere Überleben vergleichbar mit dem der Allgemeinbevölkerung ist. Auch in der SOCGruppe bildete sich, wegen des späteren Einsatzes von Axi-Cel in der Drittlinie, im Laufe der Zeit ein Plateau aus – allerdings auf niedrigerem Niveau (46 % OS-Rate).
„Das spricht für einen möglichst frühen Einsatz von Axi-Cel beim r/r DLBCL in der Zweitlinie“, folgerte Chapuy.
Für Borchmann ist angesichts der gewonnenen Erkenntnisse „die Heilung immer das Ziel bei r/r DLBCL, auch bei Patienten im höheren Alter.“
Das Nebenwirkungsprofil von CAR-T-Zelltherapien ist mittlerweile gut bekannt und handhabbar. Die bedeutendsten, häufigsten unerwünschten Ereignisse unter Axi-Cel in ZUMA-1 bzw. ZUMA7 waren das Zytokinfreisetzungssyndrom (CRS, 93 % bzw. 92 %), Enzephalopathie (60 % bzw. 49 %) und Infektionen (40 % bzw. 45 %). Schwerwiegende Nebenwirkungen traten bei 51 % bzw. 54 % der Patienten auf. „Als Behandelnde
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
verstehen wir die spezifischen Nebenwirkungen immer besser und können gut damit umgehen“, sagte Chapuy und Borchmann ergänzte: „Auch das Management von CRS und ICANS – also dem Immuneffektorzell-assoziierten Neurotoxizitätssyndrom – kann sehr gut trainiert werden.“
CAR-T-Zelltherapie ist heute Standard in der Zweitlinie
Die Ergebnisse von ZUMA-7 haben sich auch in den Leitlinien nie-
dergeschlagen: Eine gegen CD19 gerichtete CAR-T-Zelltherapie wie Axi-Cel ist heute Zweitlinienstandard beim früh rezidivierten/primär refraktären DLBCL. Der Onkopedia-Leitlinie zufolge sollte eine CAR-T-Zelltherapie allen CAR-T-fähigen Patienten angeboten werden, auch älteren, ggf. nicht Hochdosis-fähigen Patienten.
Fabian Sandner, Nürnberg
Titelbild: Bronchialsystem (Quelle: magicmime, iStock).
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R.
Roland Welker Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de

Langanhaltend
Langzeitdaten aus Klinik und Praxis4,5
Behandeln Sie das Sichtbare. Verhindern Sie das Unsichtbare.
auf Haut & Gelenke1–3 Wirkstark
günstiges Sicherheitsprofil für Ihre Patient*innen6,7 Bewährt
a STEPIn: Randomisierte Open-Label-Studie zum Vergleich der Standardtherapie (Schmalband-UVB) mit Cosentyx® über einen Behandlungszeitraum von 52 Wochen bei Patient*innen mit neu aufgetretener (≤ 12 Monate) mittelschwerer bis schwerer Plaque-Psoriasis. 9 von 10 Patient*innen erreichten mit Cosentyx® 300 mg nach 1 Jahr ein PASI90-Ansprechen, fast 7 von 10 ein PASI100-Ansprechen.1 b FUTURE 5: In die randomisierte, placebokontrollierte Phase-III-Studie waren TNFi-naive Patient*innen mit aktiver PsA und solche, die unzureichend auf eine Therapie mit TNFi angesprochen haben, eingeschlossen. Cosentyx® konnte ab Woche 52 von 150 mg auf 300 mg erhöht werden, wenn aktive Krankheitssymptome in einer ärztlichen Beurteilung beobachtet wurden. Unter Cosentyx® 300 mg waren in Woche 104 89,5 % der PsA-Patient*innen ohne röntgenologische Progression. In der 150-mg-Dosierung waren es 82,3 %.2 c IVEPSA: Einarmige, prospektive, explorative Open-Label-Studie bei Psoriasis-Patient*innen mit Beteiligung der Kopfhaut oder Nägel oder einem PASI > 6 sowie entzündlichen oder erosiven Veränderungen im MRT oder CT. Nach 24 Wochen Behandlung mit Cosentyx® 300 mg konnte eine Reduktion der subklinischen Entzündung festgestellt werden.3 CT Computertomographie. IVEPSA Interception in very early PsA. MRT Magnetresonanztomographie. PASI Psoriasis Area and Severity Index. PsA Psoriasis-Arthritis. TNFi Tumornekrosefaktor-Inhibitor. UVB Ultraviolett B. 1. Iversen L, et al. J Eur Acad Dermatol Venereol. 2023;37(5):1004-1016. 2. Mease PJ, et al. RMD Open. 2021;7(2):e001600. 3. Kampylafka E, et al. Arthritis Res Ther. 2019;21(1):178. 4. Bissonnette R, et al. J Eur Acad Dermatol Venereol. 2018;32(9):1507-1514. 5. Augustin M, et al. EADV 2024. Poster 3355. 6. Fachinformation Cosentyx. 7. Sun R, et al. Dermatol Ther. 2024;14:729-743. Cosentyx® ist angezeigt für die Behandlung von Erwachsenen sowie Kindern und Jugendlichen ab 6 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis,die für eine systemische Therapie in Frage kommen. Cosentyx®, allein oder in Kombination mit Methotrexat (MTX), ist angezeigt für die Behandlung erwachsener Patient*innen mit aktiver Psoriasis-Arthritis, wenn das Ansprechen auf eine vorhergehende Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureichend gewesen ist. Cosentyx® 75 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einem Fertigpen, Cosentyx® 300 mg Injektionslösung in einer Fertigspritze, Cosentyx® 300 mg Injektionslösung in einem Fertigpen. Wirkstoff: Secukinumab (in Ovarialzellen d. chines. Hamsters [CHO-Zellen] produzierter, gg. Interleukin-17A gerichteter, rekombinanter, vollständig humaner monoklonaler Antikörper d. IgG1/κ-Klasse). Zus.-setz.: Arzneil. wirks. Bestandt.: 1 Fertigspritze enthält 75 mg Secukinumab in 0,5 ml bzw. 1 Fertigspritze/Fertigpen enthält 150 mg Secukinumab in 1 ml bzw. 300 mg Secukinumab in 2 ml. Sonst. Bestandt.: Trehalose-Dihydrat, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80, Wasser f. Inj.-zwecke. Anwend.: Behandl. v. Kindern u. Jugendl. ab 6 J. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. v. Kindern u. Jugendl. ab 6 J. mit Enthesitis-assoziierter Arthritis od. juveniler Psoriasis-Arthritis, allein od. in Kombination mit Methotrexat (MTX), wenn Erkrankung unzureich. auf eine konventionelle Therapie angesprochen hat od. d. diese nicht vertragen. 150/300 mg Injektionslösung zusätzl.: Behandl. erw. Pat. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. erw. Pat. mit mittelschwerer bis schwerer aktiver Hidradenitis suppurativa (Acne inversa), d. auf eine konventionelle system. HS-Therapie unzureichend angesprochen haben. Behandl. erw. Pat. mit aktiver Psoriasis-Arthritis, allein od. in Kombination mit MTX, wenn d. Ansprechen auf eine vorhergeh. Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureich. gewesen ist. Behandl. erw. Pat. mit aktiver ankylosierender Spondylitis, d. auf eine konventionelle Therapie unzureich. angesprochen haben. Behandl. erw. Pat. mit aktiver nichtröntgenolog. axialer Spondyloarthritis mit objektiven Anzeichen d. Entzündung, angez. durch erhöhtes C-reaktives Protein (CRP) u./od. Nachweis durch Magnetresonanztomographie (MRT), d. unzureich. auf nichtsteroid. Antirheumatika (NSAR) angesprochen haben. Gegenanz.: Überempfindlichkeit gg. d. Wirkstoff od. einen d. sonst. Bestandt. Klinisch relevante, aktive Infekt. (z. B. aktive Tuberkulose). Nebenw.: Sehr häufig: Infekt. d. oberen Atemwege. Häufig: Oraler Herpes. Kopfschmerzen. Rhinorrhö. Diarrhö, Übelkeit. Ekzem. Ermüdung. Gelegentl.: Orale Candidose, Otitis externa, Infekt. d. unteren Atemwege, Tinea pedis. Neutropenie. Konjunktivitis. Entzündl. Darmerkrankungen. Dyshidrot. Ekzem. Urtikaria. Selten: Anaphylakt. Reakt., Angioödem. Exfoliative Dermatitis, Hypersensitivitätsvaskulitis. Häufigkeit nicht bekannt: Mukokutane Candidose (einschl. ösophageale Candidose). Pyoderma gangraenosum. Verschreibungspflichtig. Weit. Angaben: S. Fachinformationen. Stand: Februar 2025 (MS 04/25.25). Novartis Pharma GmbH, Sophie-Germain-Str. 10, 90443 Nürnberg. Tel.: (09 11) 273-0. www.novartis.de