Terry Taylor, BSc DipMLS MNZIMLS, Southern Community Laboratories, Dunedin
Sharon Tozer, DipBis Stud, AT CAANZ, NZIMLS, Rangiora
Robyn Wells, BApllSci(MT) GradCert Haem, Milton, Australia
Formatting
Sharon Tozer, AT DipBusStud, Executive Office NZIMLS, Rangiora
About the Journal
The New Zealand Journal of Medical Laboratory Science (the Journal) is the official publication of the New Zealand Institute of Medical Laboratory Science (NZIMLS). The Journal is peer reviewed and publishes original and review articles, case studies, technical communications, and letters to the Editor on all subjects pertaining to the practice of medical laboratory science. The Journal is open access (www.nzimls. org.nz/nzimls-journal) and is published three times per year in March, July, and November. Hard copies are circulated to all NZIMLS members and universities and research units in New Zealand and overseas. Current circulation is about 2,800 copies per issue. Printing is by Blueprint Ltd, Christchurch on environmentally responsible paper using elemental chlorine free third party certified pulp sourced from well managed and legally harvested forests and manufactured under the strict ISO14001 Environmental Management System. The Journal is indexed by CINAHL, EMBASE, SCOPUS, Informit, Thomson Gale, EBSCO and Biosis Citation Index, and the Journal Editors are members of the World Association of Medical Editors (www.wame.org).
Brief instructions to authors
The Journal accepts original submissions from anyone and anywhere. Comprehensive instructions can be found on the NZIMLS website (www.nzimls.org.nz/instructions-to-authors. html). All submissions will undergo single-blind peer review and possibly plagiarism checking with iThenticate™ software. If accepted for publication, copyright is vested in the author(s) under terms of the Creative Commons Attribution License (www. creativecommons.org/licenses/by/2.5/legalcode). The authors are responsible for the scientific content and views. Opinions expressed in the Journal are not necessarily those of the Editors, Editorial Board, or Council of the NZIMLS.
LDL receptor gene associated with familial hypercholesterolemia in a cohort of Egyptian children
Khalda S Amr, Miral M Refeat, Hala T El-Bassyouni, Nesma M Elaraby, Angie MS Tosson, Faten Mohamed Abdel Aziz and Shrouk M. Abdalla 49-55
Case Studies
A curious case of haemolytic disease of the newborn caused by cold-reacting Anti-M: a New Zealand case report Savannah C Young, Ben G Paterson and Dhana S Gounder 57-60
A lethal synergy: lymphoma associated haemophagocytic lymphohistiocytosis: a case report
Hari Priya Raghvan, Indhira Subbiah, Wee Shiang Yui, Nor Ashikin Azizan, Caroline Ho Siew Ling and Ehram Jamian 61-64
Pyroglutamic acidosis: an under-recognised cause of high anion gap metabolic acidosis with multi-factorial aetiology
Yassar Alamri, Polly Davison, Charlotte Reay, John Geddes and Christopher Florkowski ..............................................65-66
Book Reviews
How Life works, a user’s guide to the new biology by Philip Ball Reviewed by Michael Legge.................................................. 67
A fatal inheritance: how a family misfortune revealed a deadly medical mystery by Lawrence Ingrassia Reviewed by Michael Legge 67
In memorium
Sue Warrington, Supervising Scientist, New Zealand Blood Service, Christchurch Contributed by Nicole Crampton 87
Helen Beatrix Robertshawe, past Secretary of the Medical Laboratory Technologists Board Contributed by Rob Siebers 87
Regular features Advertisers
Otago student research project abstracts: semester 2, 2024 68-77
Advertising and subscription
Advertisement bookings and enquiries should be addressed to the NZIMLS Executive Officer, Sharon Tozer: sharon@nzimls.org.nz. Phone +64 3 313 4761.
The Editors have put their heads together in this issue to discuss Point of Care Testing (POCT) in New Zealand, their use and disparities in control and regulation. How do we educate and stem the tide of unregulated and uncalibrated tests and their potentially risky and ill-considered use by the worried-well?
Familial hypercholesterolemia (FH) is a genetic disorder distinguished by elevated levels of low-density lipoprotein cholesterol (LDL-C), leading to early cardiovascular disease (CVD). Most FH cases result from mutations in the lowdensity lipoprotein receptor (LDLR) gene encoding the LDLR protein. In a study conducted by Professor El-Bassyouni and colleagues at the National Research Center in Cairo, clinical manifestations, biochemical profiles, and patterns of LDLR gene mutations were investigated in 50 Egyptian children with Familial hypercholesterolemia. Fifty-eight percent of the study patients were offspring of consanguineous marriages, similarly, affected family member were reported in 52% of cases. In this report 24% of FH patients had a family history of sudden death compared to 39% in other investigations. Using Sanger DNA sequencing, the forward and reverse strands of the LDLR gene were sequenced from these patients, identifying eleven known pathogenic variants, including one rare homozygous variant and a novel pathogenetic variant in three patients. Early detection can enhance early genetic diagnosis of FH and next generation sequencing could initiate early treatment therefore delaying or reducing life-threatening cardiovascular complications.
Haemolytic disease of the fetus and newborn (HDFN) occurs when maternal IgG alloantibodies cross the placenta, resulting in destruction of fetal or neonatal red blood cells (RBC) carrying the corresponding paternally inherited antigen, in this case, M antigen. New Zealand case study, Savannah Young and colleagues at the NZ Blood Service, NZ Blood Service Reference Lab and the Wellington Blood and Cancer Centre report this rare late onset haemolytic disease case, which without clinical intervention, can be fatal. Typically anti-M is a naturally occurring IgM antibody reacting to temperatures below 37°C and normally considered clinically insignificant and rarely responsible for HDFN as the anti-M antibody is unable to cross the placenta due to its structure. Biochemistry and serological testing was conducted between days 2 and 35. Their findings were consistent with passively-acquired cold-reacting IgG anti-M as the cause of haemolysis and recommend this significance should not be discounted in determining HDFN, where determination of an IgG component could aid in risk assessment of alloimmunised pregnancies, alongside thermal amplitude and titre.
In our second case study, Dr Raghavin and colleagues from the Haematology Unit in the Department of Pathology at the Hospital Sultanah Aminah, in Malaysia, report the lethal synergy of lymphoma associated haemophagocytic lymphohistiocytosis (HLH). A challenging case of a 65-year-old male diagnosed as High-grade B-cell lymphoma with secondary HLH, who was initially investigated for HLH and recovered, but was re-admitted again with a similar presentation, leading to the eventual diagnosis of lymphoma and the patient succumbing to the illness before treatment could be initiated. Diagnostic criteria
for HLH in the 2004 guidelines were established as 5 out of 8 of the criteria needing to be met, The patient, met four criteria but the team were unable to test two of the variables as they were not offered (NK cell activity and soluble CD25). Studies have revealed that treatment of LA-HLH need to be centred towards both the lymphoma and HLH. They have extremely poor prognosis with survival of <1 month without lymphomaspecific treatment or with solely HLH-directed therapy. This case of LA-HLH highlights the critical need for early recognition and intervention. The dual pathology poses significant diagnostic and therapeutic challenges, necessitating a high index of suspicion and a multidisciplinary approach to improve patient outcome.
Pyroglutamic acidosis (PGA; also known as 5-oxoproline acidaemia) is a rare, mostly acquired, cause of high-anion gap metabolic acidosis (HAGMA) and remains under-recognised among practising clinicals. Dr Yassar Alamri, Medical Registrar at Christchurch Hospital in New Zealand and associates present our third case study. This recent case of a 85-year old female admitted to the Hospital, presented with an ankle fracture her admission was complicated by delirium, hospital-acquired pneumonia and an acute kidney injury (AKI). On day 8 her urinary metabolic screen returned positive increase in PGA. The pathogenesis of pyroglutamic acidosis (PGA) is complex and multi-factorial involving several clinico-biochemical “hits” before significant acidosis develops. There are a multitude of risk factors, including regular use of paracetamol, use of flucloxacillin/ vigabatrin, malnutrition and AKI) and untreatable factors as female sex, advancing age, and multi-morbidity- including renal and hepatic failure. The patient’s condition deteriorated, and she died on day 13 of her admission. Clinicians should be aware of the possibility of PGA, especially in the setting of otherwise unexplained HAGMA whereby more common causes such as lactic acidosis and ketoacidosis have been excluded. The probability of PGA is further increased if the clinical context also includes regular paracetamol usage, sepsis, renal impairment, and concomitant use of antibiotics and/or AED.
Dr Michael Legge reviews two interesting books, How Life Works: a user’s guide to biology, by Phillip Ball and A fatal inheritance: how are family misfortune revealed a deadly mystery, by Lawrence Ingrassia.
The Journal presents two “In Memorial” articles of valued NZIMLS members who passed away in 2024, including Sue Warrington, from the NZ Blood Service in Christchurch and Helen Robertshaw a past Secretary of the Medical Technologists Board. There are also a three long-servicing member retirement notices; Ailsa Bunker, Madhu Nahna and Tony Marcinkowski. The Journal and the NZIMLS thank them for their services to the profession and wish them all the very best.
Our other regular features include the University of Otago student research project abstracts from Semester 2, 2024, an update from the Pacific Pathology Training Centre (PPTC) in the Pacific Way, Science Digest, Recent Reviews and this issue’s all important CPD questionnaire.
Lisa Cambridge, Editor
EDITORIAL
Point of care testing: the elephant in the room
Michael Legge and Lisa Cambridge
Point of Care Testing (POCT) or near-patient testing is one of the most rapidly growing areas in healthcare globally. POCT devices are in-vitro diagnostic devices (IVDs), touted for their easy use, rapid results and accessibility. In the United Kingdom there are currently at least 27 different healthcare sectors identified as using POCT. By 2026 it is predicted to have an international POCT market in the region of £GBP34 billion ($NZ75 billion), with a growth rate of 8.4%. The principal areas for growth are seen in Clinical Biochemistry, Microbiology and Haematology testing. This does not include POCT areas such as blood pressure monitoring, pregnancy tests, blood-glucose and other non-invasive procedures. In addition to determining ‘traditional’ analytes, there is a rapidly developing market for their use as “wellness tests”. A recent review from Australia showed that more than 40% of direct-to-consumer tests were being sold as “health checks” including hormones, nutritional profiles and some haematology with pharmacies being the main provider with some, offering options for treatment. The spread of and increasing sophistication with POCT is clearly shown with a UK company developing POCT molecular home testing under the guise of rapid testing for COVID-19 and other viral infections.
As instrumentation becomes more sophisticated, portable POCT has become readily available to the consumer outside the more traditional and controlled routes for diagnostic testing and primary care in hospitals, general practice and community laboratories, and are now in the hands of the unqualified and untrained user and outstrips any legislation developed for their controlled use. For the more informed professions, this raises alarm-bells, highlights the importance of patient safety and recognises the need for a structured approach to establishing POCT guidance and training.
New Zealand medical laboratories must adhere to the specific requirements of ISO15189:2022, where POCT has been integrated throughout the Standard. Laboratory-supported POCT must be in the laboratory’s management systems and quality control and included in their laboratory’s Scope of Accreditation, including specific testing locations, what associated testing the laboratory performs and be part of mandatory onsite assessments conducted by IANZ (1). In addition, New Zealand POCT Advisory Group Guidelines (NZPOCTAG) (2) must be adhered to.
Currently in New Zealand, POCTs can be purchased, used and sold by any client or patient as long as they comply with the Medicines Act (1981) enforced by MedSafe. It is interesting to point out that medical devices must be entered into the Web-Assisted Notification of Devices (WAND) database (3) administered by MedSafe, but IVDs and therefore POCTs are exempt from this requirement, with no risk classification. In addition, regulatory guidance by MedSafe on IVDs have not been updated since 2014. Therefore, POCT devices have no analytical specifications or method validation requirements and no mandatory reporting of adverse events, including incorrect results, inaccuracy of labelling or instructions and no reporting of serious health threats through their use.
Because New Zealand has an unregulated POCT system, there is no oversight and with largely unskilled public workforce providing the testing, this raises three important points. First, is the equipment used, well-understood? What quality assessment is undertaken? And what understanding of the principles underpinning the tests are there? Secondly, the interpretation of the results, role of instrument calibration/quality control and the skill base of the person undertaking the tests. Third, Registered Pathology Scientists and Technicians are not supposed to interpret results, so why are non-qualified persons allowed to do this?
Given the rapid (and uncontrolled) development of POCT in New Zealand, the following must be considered:
• Can sufficient accuracy, validity and interpretation in a clinical context be achieved?
• Can sufficient oversight be maintained for the devices and reagent kits?
• Can consistency be achieved across different devices?
• What would be the outcomes and impact be across the care pathways?
• All POCT devices should be validated for suitable and appropriate use.
• What would be the training for any end user?
• How would any data/results be captured into the ‘digital health’ system?
• What would be the future relating to the use of ‘smart devices’ be in relation to the ‘digital health’ system e.g. blood glucose monitors, insulin pumps, blood pressure monitors etc?
• There would be an increasing development of Pharmacy based non-prescription treatments with no basis of efficacy.
• Do medical centres and GPs have the capacity to monitor any electronically transmitted patient data from individual devices?
Another significant factor in this unregulated POCT market relates to generating results in isolation of patient records and careful consideration relating to privacy, rights of data access, trust in the ‘testers’ and data storage must be given. Should, or more likely when, POCT increase in their complexity and develop genomic technologies, then POCT moves to a more significant level, therefore it is essential that governance, training and certification be required to protect public safety as currently none of the publicly available POCT providers come under the Health Practitioners Competency Assurance Act.
Author Information:
Michael Legge, PhD, MRSB, FIBMS, FNZIMLS, FFSc (RCPA), University of Otago.
Lisa Cambridge, BApplManagement, MNZIMLS, Editor, NZIMLS
Correspondence email: michael.legge@otago.ac.nz
REFERENCES
1. IANZ Specific Criteria for Accreditation. Medical Testing ISO15189-2022. AS LAB C7. 2024; 5th edition IANZ, Auckland New Zealand
2. New Zealand Point-of Care- Testing Advisory Group. New Zealand best practice guidelines for point-of care testing. 2022
3. MedSafe New Zealand [accessed June 2025], www. medsafe.govt.nz/regulatory/devicesnew/7InVitro.asp
receptor gene associated with familial hypercholesterolemia in a cohort of Egyptian children
Khalda S Amr, Miral M Refeat, Hala T El-Bassyouni, Nesma M Elaraby, Angie MS Tosson, Faten
Mohamed Abdel Aziz and Shrouk M. Abdalla
ABSTRACT
Aim: Familial hypercholesterolemia (FH) is a genetic disorder distinguished by elevated levels of low-density lipoprotein cholesterol (LDL-C), leading to early cardiovascular disease (CVD). Most FH cases result from mutations in the low-density lipoprotein receptor (LDLR) gene, which encodes the LDLR protein. This study intended to identify the clinical manifestations, biochemical profiles, and patterns of LDLR gene mutations in Egyptian children with Familial hypercholesterolemia.
Methods: Fifty children aged 5 to 15.5 years were clinically and biochemically assessed and diagnosed with Familial hypercholesterolemia. LDLR gene mutations were detected using Sanger sequencing for both the forward and reverse strands. The pathogenicity of a newly identified variant was examined through in silico analysis. All participating children (29 females and 21 males) exhibited xanthomas and atheromatous plaques as detected by echocardiography
Results: The biochemical analysis revealed high lipid levels among the enrolled cases, with total cholesterol levels and LDL-C measuring ≥5.18 and ≥3.37mmol/L, respectively DNA sequencing of the LDLR gene identified eleven known pathogenic variants (ten heterozygous and one homozygous). The most common genetic variants included the heterozygous types: c.502G>A, c.1217G>A, c.1721G>A, and c.2552A>G.
Conclusion: Identifying the prevalent LDLR gene variants can enhance definitive early genetic diagnosis of Familial hypercholesterolemia and facilitate early initiation of therapy to delay or reduce cardiovascular complications.
Familial hypercholesterolemia (FH, MIM: #143890) is a disorder of lipid metabolism that impairs LDL-C clearance, thereby increasing the risk of premature cardiovascular disease (CVD) (1, 2). FH is primarily inherited in an autosomal dominant pattern (3, 4), arising from mutations in the LDL receptor gene (LDLR, MIM: #606945), the apolipoprotein B gene (APOB; MIM: #107730), or the proprotein convertase subtilisin/kexin 9 gene (PCSK 9; MIM: # 607786), either individually or in combination (5, 6). Over 2, 000 genomic variants have been documented in the LDLR gene (7). In ClinVar, 580 variants in the APOB gene and 350 variants in the PCSK 9 gene have been identified (8). LDLR gene mutations are the most prevalent cause of FH (9). FH patients are classified into two clinical types: homozygous (HoFH) and heterozygous (HeFH). The prevalence of HoFH is approximately 1 in 1,000,000, while HeFH occurs in fewer than 1 in 500 individuals, depending on the population (10). Clinically, HeFH patients are characterized by tendon xanthomas, corneal arcus, and CVD complications (11), with total cholesterol levels and LDLC concentrations of ≥5.18 and ≥3.37mmol/L, respectively (12). In contrast, HoFH patients are typically diagnosed earlier, within the first two decades of life, exhibiting severe manifestations such as extremely premature atherosclerosis and elevated total cholesterol levels (>12.95mmol/L), with untreated individuals at risk of dying by age 20 (1). LDLR is a cell surface glycoprotein that mediates the specific binding and uptake of apoB-100 lipoproteins through receptor-mediated endocytosis (13). The human gene encoding LDLR is located on chromosome 19 (p13.1 - 13.3) and consists of 18 exons and 17 introns (14). The length of human LDLR mRNA is 5. 3 kb, encoding a glycoprotein of 860 amino acids crucial for cholesterol homeostasis. Identifying mutations in the LDLR gene within families enables thorough genetic counselling and facilitates follow-up for affected individuals (15). Once the genetic diagnosis is confirmed, family members at risk for cardiovascular disease can be identified through cascade screening and treated promptly (1). Here, we report the clinical, biochemical, and molecular findings in 50 familial hypercholesterolemia patients.
MATERIALS AND METHODS
The study included 50 familial hypercholesterolemia patients aged 5 to 15.5. They were referred from Cairo University
Children’s Hospital (Abu El Rish) or the Clinical Genetics
Outpatient Clinic of the National Research Centre. Before the study's enrolment, all patients and their guardians provided informed consent, which was approved by Cairo University's Research Ethics Committee (number N-479-2023) following the principles of the Declaration of Helsinki.
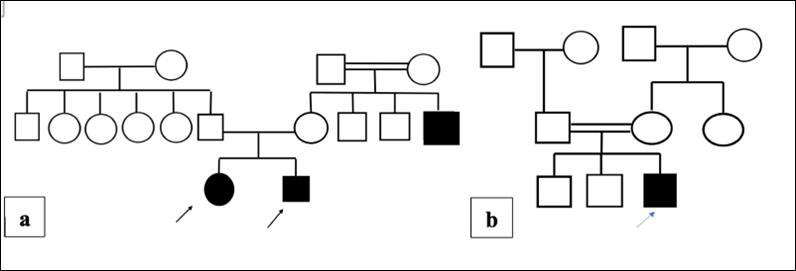
The following data were documented: detailed personal history, family history of dyslipidaemia, 3-generations pedigree analysis (Figure 1), meticulous clinical examination, and echocardiography.
Laboratory assessment
Collection of blood samples
Peripheral blood samples (5 mL) were withdrawn from all participants and available family members after 10 hours of fasting except for water to analyse TC, LDL, TG, and HDL concentrations. Then samples were centrifuged and analysed using Cobas® pro integrated solutions (ROCH diagnostics).
Biochemical investigations
Serum total cholesterol (TC), triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) were assessed. The lipid profile reference range for children is as follows: Total cholesterol: less than 4.40 mmo/L; LDL cholesterol: less than 2.85 mmol/L; HDL cholesterol: more than1.43 mmol/L; and Triglycerides: less than 1.70 mmol/L. (16).
Molecular Genetic Analysis
Genomic DNA extraction
Genomic DNA was extracted from peripheral blood samples using a commercial kit according to the manufacturer’s recommendations (QIAamp DNA Mini Kit, Qiagen, USA). The DNA concentration and purity were determined using the NanoDrop Spectrophotometer (Thermo Scientific NanoDrop™ 2000c) at an A260/A280 ratio between 1.8 and 2.0 followed by storage at −20°C.
LDLR gene sequence analysis
The whole length of the LDLR gene including coding exons and corresponding intron/exon boundaries was amplified by 18 forward and reverse primer sets using the primer designing tool (http://www.primer3.com). The primers were then checked
for specificity against the LDLR gene using the NCBI Blast tool (http://www.ncbi.nlm.nih.gov). Primer sequences and PCR cycling conditions will be shared upon request from the corresponding author. The PCR products were purified using the Exo-SAP PCR Cleanup Kit (Fermentas, Germany). Then PCR fragments were subjected to Sanger sequencing using the Big-Dye Terminator v3.1Cycle sequencing kit and analysed on the ABI Prism 3500 Genetic Analyzer (Applied Biosystems) according to the manufacturer's instructions. The outcomes were then compared to the human LDLR reference sequence using the FinchTV application. (NM_000527.5).
Bioinformatics prediction of variant pathogenicity
In-silico bioinformatics tools were used to identify the pathogenicity and frequency of the variants and predict the potentially damaging effect of the detected mutations on the stability and the expression of LDLR protein which will affect its function. These tools included Sorting Intolerant From Tolerant (SIFT) (HYPERLINK "http://sift.jcvi.org" a score less than 0.05 indicates it is deleterious, polymorphism phenotyping version 2 (Polyphen V2) (HYPERLINK http://genetics.bwh.harvard.edu/ pph2) where a SNP with a score ranging between 0.85 and 1.0 predicts a damaging effect on the encoded amino acid whereas a SNP with a score ranging between 0.0 and 0.15 is verified to be benign and a SNP with a score ranging between 0.15 and 1.0 predicts a possibly damaging function, Mutation Taster (http:// mutationtaster.org) with a score close to 1 indicates a prediction of the given variant to be disease-causing, MetaLR logistic regression (LR) based ensemble prediction score, ranges from 0 to 1 when higher values are more likely of being deleterious. Aggregated Prediction tool relies on ensemble methods such as REVEL, MetaLR which assigns different weights to the different in-silico tools, and REVEL ensemble method for predicting the pathogenicity of missense variants ranges from 0 to 1. A score above 0.932 indicates strong evidence of pathogenicity, a score between 0.773 and 0.932 indicates moderate significance, and a score between 0.644 and 0.773 supports mild pathogenicity. While a score between 0.183 and 0.29 supports a benign result, a score between 0.016 and 0.183 indicates a moderate benign, a score between 0.003 and 0.016 indicates a strong benign, and a score below 0.003 indicates a very strong benign. These tools rely on conservation scores across different species, and physiochemical differences between the wildtype and mutant amino acid residues.
RESULTS
Clinical and biochemical results
The children’s ages ranged from 5-15.5 years (6.51 ±3.45), they were 29 females and 21 males (1.4:1). Twenty-nine patients had positive parental consanguinity (58%), and 26 reported a similarly affected family member (52%). A history of sudden death in the family was reported in 12 patients (24%). Stroke or angina was found in 16 patients (32%). Furthermore, tendon xanthomas were present in 48 cases (96%) while 24 cases (48%) had atheromatous plaques detected by echocardiography. A summary of the patients' clinical features and lipid profile is provided in (Tables 1 and 2).
The serum total cholesterol and LDL-C levels in heterozygous FH ranged from 9.10 – 14.25mmol/L respectively. In homozygous
Table 1. The clinical characteristics of patients
FH, the total cholesterol level was 21.18mmol/L, and the LDL-C level was 20.38mmol/L respectively (Table 3.). Additionally, the parents of the patients had serum total cholesterol and LDL-C levels between 4.40 – 7.50 mmol/L and 3.18-6.73 mmol/L, respectively (Table 3). All patients were prescribed statins in the form of atorvastatin with doses ranging from 10 to 40 mg combined with 10 mg of ezetimibe as described by the treating physicians. Atorvastatin-treated patients had their cholesterol levels measured before and after therapy during routine follow-ups. Initial values were established by measuring lipid profiles, which revealed LDL cholesterol levels between 8.0 and 14.48mmol/L.
Following the initiation of atorvastatin therapy, follow-up lipid profiles were performed at 4- to 12-week intervals to assess treatment response. Three patients (2, 10, and 12, as shown in Table 3) demonstrated a statistically significant reduction in LDL levels after receiving medical treatment, with p-values of 21.19 and 20.38 mmol/L, respectively. Their pretreatment LDL cholesterol levels were 15.26, 15.0 and 16.58mmol/L. None of the other patients showed a favourable response to the medical treatment they received, highlighting the need for adjustments in subsequent treatments.
Molecular genetic results
Direct Sanger sequencing results of the whole coding exons, with their flanking intron sequences, of the LDLR gene revealed 11 reported variants. Ten FH patients comprised heterozygous LDLR gene variants and one patient (P15) carried a homozygous variant as presented in (Table 4) Missense substitution was detected in 9 variants of 14 patients descending from unrelated families, and a nonsense type was detected in two probands (P9 and P14) from two unrelated families; namely (c.1659C>G p.(Tyr553*) in exon 11 and c.1757C>G p.(Ser586*)) in exon 12, in which the amino acid tyrosine (Tyr) and serine (Ser) were replaced by the stop codon, respectively. The LDLR c.1659C>G and c.1757C>G variants were identified as stop gain variants, categorized as the most deleterious type of variants. A missense variant (c.2389G>A) was identified with a homozygous state in a 6-year-old proband (P15) with tendon xanthomas who had high total cholesterol and high LDL-C levels of 21.10 and 20.38 mmol/L respectively.
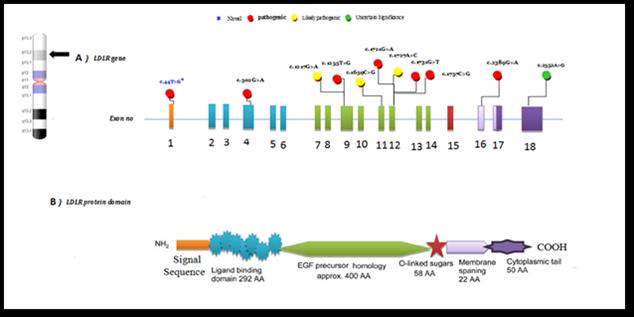
The result of the patient’s echocardiography revealed trivial mitral and aortic regurgitations. Both parents were heterozygous for the (c.2389G>A) LDLR mutation. This variant led to a change of valine, present in position 797, to methionine in exon 16 (P. Val797Met). Among the variants, eight different heterozygous missense variants were observed in different exons (1, 4, 9, 12, and 18) of the LDLR gene and they encoded in the ligandbinding domain, the epidermal growth factor (EGF) - precursor domain of LDLR, and the cytoplasmic domain (Figure 2).
Functional analysis of variants using bioinformatics tools
The current study included 11 disease causing variants, confirmed using mutation taster, nine of them were predicted to have a damaging effect on the encoded amino acid using the SIFT tool. The pathogenicity of identified variants varied between strong to moderate pathogenic scores predicted with the REVEL ensemble method. According to the Aggregated Prediction tool, most variants were deleterious with score ranges from 0.7 to 0.9.
Table 2. Clinical features of the enrolled FH patients
Atorvastatin 40mg one-ezitimibe 10mg onecholestyramine 4g bida clinical trial of evolocumab (ongoing for both)
Under normal physiological circumstances, cells typically attain cholesterol by absorbing plasma lipoprotein through the LDLR route. Therefore, any defect in the LDLR function can lead to FH (17).
Fifty-eight percent were the offspring of consanguineous marriage, which aligns with the elevated prevalence of consanguinity observed among Egyptians (18). Similarly, affected family members were reported in 52% of cases, which agrees with previous studies (19, 20). In our report, 24% of FH patients had a family history of sudden death, compared to 39% in other investigations (21).
In 32% of our cohort, a stroke or angina was reported. However, former research claimed that in 20% of their population, this may be due to different ethnicities (22). When xanthomas are present, a good diagnosis of familial hypercholesterolemia can be made. However, they are not always seen in children and adolescents, xanthomas were found in 96% of the cases in our study (23).
Aortic root and valve involvement are uncommon in heterozygotes and only occur in severe cases, according to echocardiography, atheromatous plaques in the aortic root were found in 48% of cases. In a recent study, 53% of FH subjects exhibited aortic root atheroma (24). In the present study, 11 variants were identified in 16 patients of unrelated families; nine were heterozygous FH, and one was homozygous, whose phenotype was more severe and experiencing an earlier onset. It was previously reported that the heterozygous FH phenotype is more common, while the rarely encountered homozygous FH has more severe symptoms (25). In cases with homozygous FH, genetic screening would be a crucial diagnostic tool to initiate early treatment (26). Several LDLR gene mutations have been identified in patients suffering from FH in several countries (27,28). Thus, we undertook the present study to detect the mutations of the LDLR gene in Egyptians. A previous study reported that p. Leu15pro disrupted the α-helical arrangement of the signal sequence, which has a pathogenic effect on LDLR protein folding in the endoplasmic reticulum (29). The missense variants detected in the current study have been previously described in different FH cases from diverse ethnic backgrounds: Russian, United Kingdom, Iranian, Brazilian, Saudi, Taiwan, French, and Japanese (30). In the current study, another vital finding was the homozygous FH genetic variant c. 2389G>A in exon 16, located in the EGF-like repeats of LDLR protein. The HoFH patient (P15) with this mutation had high blood LDL-C levels. This patient had an earlier disease onset than the other studied patients (2 years of age). However, there were no other notable differences in the phenotype. The variant has been published before in other FH patients. It was previously reported in two Chinese studies (31,32). This variant is associated with disturbing mRNA cleavage and subsequent protein retention in the Golgi apparatus, reducing the expression of LDLR on the
cellular membrane and the ultimate uptake ability of the LDLR protein for LDL cholesterol (33).
A different variant was previously found in the same position (c.2389G>T), causing in-frame skipping of exon 16, which led to the translocation of the entire L799R-LDLR into the lumen of the endoplasmic reticulum (34). Moreover, an Egyptian patient was previously reported to have a homozygous LINE-1 insertion in exon 7 of LDLR (35). Another heterozygous missense variant was found in one proband aged nine years (P16) with a serum LDL cholesterol level of 17.1mmol/L. The patient had mild stunting (height for age z-score < -2 standard deviation) despite normal weight for age. The c.2552A>G variant is in exon eighteen of the LDLR gene. This variant has not been detected in an individual affected with familial hypercholesterolemia in the previous literature but was reported in the ClinVar database. The function of this variant was also verified by in silico prediction tools as being a variant of uncertain significance (VUS), suggesting the need for further functional analysis. The remaining six missense variants were reported in the ClinVar database. Two nonsense mutations were observed in exon 11 (c.1659C>G p. Tyr553Ter) and exon 12 (1757C>G p.Ser586Ter), which code for the EGF-like domain. These variants were found in P9 and P14. Both variants led to a premature stop codon that produced a deleterious effect on the function of the LDLR protein products either through the synthesis of a truncated protein or through nonsense-mediated decay of LDLR mRNA. The p. Tyr553Ter variant was reported in the ClinVar database, whereas the p.Ser586Ter variant was reported for the first time in China in one proband (36). We did not find any notable differences in their biochemical or clinical status among the patients regarding their type of LDLR mutations.
CONCLUSION
To our knowledge, this is the first research on LDLR gene variants in Egyptian FH. Among the detected nucleotide variations were a rare homozygous FH in one patient and a novel pathogenic variant in three patients. Early genetic diagnosis of FH in children would lead to the early initiation of therapy thus delaying/reducing life-threatening cardiovascular complications. Additional research should be conducted on a larger cohort of FH cases to delineate the mutation spectrum and the phenotypegenotype correlation among FH Egyptian patients using nextgeneration sequencing (NGS) and Multiplex ligation-dependent probe amplification (MLPA).
ACKNOWLEDGMENTS
We want to express our gratitude to Prof. Dr. Mervat El Ansary for performing the lipid profile. Also, thanks to Athar Marawan for assistance in arranging patients’ data.
Figure 1. A three-generation family pedigree of patients 1 (a) and 15 (b)
Figure 2. LDLR gene and protein domain structure. a) Mutations reported in the present study in the literature are in black. b)The domain structure of LDLR protein corresponds to LDLR coding from exon 1 to exon 18
Table 3: The lipid profile of the patients
Table 4. Variants detected in LDLR gene among studied patients
The data will be available upon request from the corresponding author.
AUTHOR INFORMATION
Khalda S Amr, PhD, Professor of Molecular Genetics and Enzymology1
Miral M Refeat, PhD, Assistant Professor of Medical Molecular Genetics1
Hala T El-Bassyouni, MD, Professor of Clinical Genetics 2
Nesma M Elaraby, PhD, Assistant Professor of Medical Molecular Genetics1
Angie MS Tosson, MD, Professor of Pediatrics3
Faten Mohamed Abdel Aziz, MD, Professor of Pediatrics3
Shrouk M Abdallah, MD, Assistant Professon of Pediatrics3
1Medical Molecular Genetics Department, National Research Center, Cairo, Egypt
2Clinical Genetics Department, National Research Centre, Cairo, Egypt
3Pediatrics Department, Faculty of Medicine, Cairo University, Cairo, Egypt
Corresponding Author: Dr Hala T El-Bassyouni, Professor of Clinical Genetics, National Research Centre, Cairo, Egypt
Email: halabassyouni@yahoo.com
REFERENCES
1. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34: 3478–390a. doi.org/10.1093/eurheartj/ ehaa166.
2. Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society consensus panel. Eur Heart J 2017; 38: 2459-2472. doi.org/10.1093/eurheartj/ehx144.
3. Meshkov A, Ershova A, Kiseleva A, et al The LDLR, APOB, and PCSK9 variants of index patients with familial hypercholesterolemia in Russia. Genes (Basel) 2021; 12, 66.doi.org/10.3390/genes12010066.
4. Sawhney JPS, Madan K. Familial hypercholesterolemia. Indian Heart J 2024; 76(Suppl1):S108-S112. doi: 10.1016/j. ihj.2023.12.002
5. Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia Nat Clin Pract Cardiovasc Med 2007; 4: 214-225. doi:10.1038/ ncpcardio0836.
6. Kassner U, WÜhle-Demuth M, Missala I, et al. Clinical utility gene card for hyperlipoproteinemia, TYPE II. Eur J Hum. Genet 2014; 22(7): doi:10.1038/ejhg.2013.271.
7.
8. Chemello K, Garcia-Nafria J, Gallo A, et al. Lipoprotein metabolism in familial hypercholesterolemia. J Lipid Res 2021; 100062. doi.org/10.1016/j.jlr.2021.100062.
Lacocca MA, Chora JR, Carrié A, Freiberger T, et al. linVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat 2018; 39: 1631–1640. doi. org/10.1002/humu.23634.
9. Benn M, Watts GF, Tybjaerg-Hansen A, et al. Mutations causative of familial hypercholesterolemia: screening of 98098 individuals from the Copenhagen general population study estimated a prevalence of 1 in 217. Eur Heart J 2016; 37:1384-1394.
10.
11. Defesche JC, Gidding SS, Harada-Shiba M, et al. Familial hypercholesterolemia. Nat Rev Dis Primers 2017; 3, 17093. doi: 10.1038/nrdp.2017.93.
Santos RD, Wiegman A, Caprio S, et al. Alirocumab in pediatric patients with heterozygous familial hypercholesterolemia: a randomized clinical trial. JAMA Pediatr 2024; 178: 283-293.
12. Singh S, Bittner V. Familial hypercholesterolemiaepidemiology, diagnosis, and screening. Curr. Atheroscler Rep 2015; 17, 482. doi: 10.1007/s11883-014-0482-5.
13.Jeenduang N, Promptmas C, Pongrapeeporn S, et al. Molecular modeling of D151Y and M391T mutations in the LDL receptor. Biochem. Biophys. Res. Commun 2008; 377: 355-360.
14. Cuchel M, Raal FJ, Hegele RA, et al. Homozygous familial hypercholesterolemia: new insights and guidance for clinicians to improve detection and clinical management. a position paper from the consensus panel on familial hypercholesterolemia of the European Atherosclerosis Society. Eur Heart J. 2014; 35: 2146-2157.
15. Ward AJ, O’Kane M, Nicholls DP, et al. A novel single base deletion in the LDLR gene (211delG): effect on serum lipid profiles and the influence of other genetic polymorphism in the ACE gene, apoE and apoB genes. Atherosclerosis 1996; 120: 83–91. doi: 10.1016/0021-9150(95)05685-8
16. Magnan RA, Murphy T, Rosenthal L, et al. Improved adherence to lipid screening in the pediatric cardiology clinic: a quality improvement project. Pediatr Qual Saf 2024;10: e781. doi: 10.1097/pq9.0000000000000781.
17. Van der Graaf A, Avis HJ, Kusters DM, et al. Molecular basis of autosomal dominant hypercholesterolemia: Assessment in a large cohort of hypercholesterolemia children. Circulation 2011; 123: 1167-1173.
18.Afifi HH, El-Ruby MO, El-Bassyouni HT, et al. The most encountered groups of genetic disorders in Giza Governorate, Egypt. Bratisl Lek Listy 2010; 111: 62–69.
19. Harada-Shiba M, Ohtake A, Sugiyama D, et al. Guidelines for the diagnosis and treatment of pediatric familial hypercholesterolemia. J Atheroscler Thromb 2022; 30: 531557.
The combination of MosaiQ™ and LumiQ™ platforms provides a unique, automated, seamlessly integrated solution to cover your needs for screening and identi cation of autoantibodies fo r a wide range of diseases.
for autoimmune diseases, allergy and
A curious case of haemolytic disease of the newborn caused by cold-reacting Anti-M: a New Zealand case report
Savannah C Young, Ben G Paterson and Dhana S Gounder
ABSTRACT
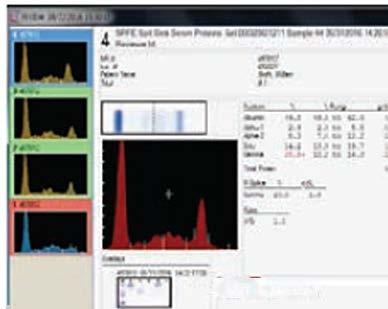
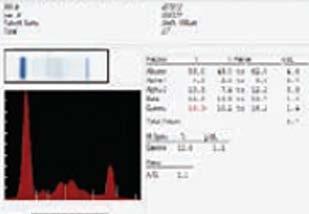
Maternal alloimmunisation with anti-M is rarely associated with haemolytic disease of the fetus and newborn (HDFN), as demonstrated in the current case study The infant exhibited poor reticulocyte response and low birthweight. Interesting findings included the late onset of prolonged anaemia, low maternal antibody titre of one and particularly, the cold-reactivity of Immunoglobulin GAnti-M (IgG anti-M) demonstrated by indirect anti-globulin testing (IAT). With a haemoglobin (Hb) drop to 69 g/L on day 15 of life, intervention via top-up transfusion was required, with further transfusion at day 29. Alongside the literature, this case suggests current laboratory testing may provide poor predictability of the clinical significance of some anti-M alloantibodies in causing severe HDFN.
Key words: Anti-M, haemolytic disease of the fetus and newborn, cold-reacting alloantibody, paediatric anaemia
NZ J Med Lab Sci 2025; 79(2): 57:60
INTRODUCTION
Haemolytic disease of the fetus and newborn (HDFN) occurs when maternal IgG alloantibodies cross the placenta, resulting in destruction of fetal or neonatal red blood cells (RBC) carrying the corresponding paternally inherited antigen, in this case M antigen. Anaemia ensues due to fetal RBC haemolysis, with potentially severe sequalae such as hepatosplenomegaly, hydrops fetalis and kernicterus (1). Without clinical intervention, HDFN can be fatal. Historically, anti-D accounted for a large proportion of cases, however the use of prophylactic RhD immunoglobulin has significantly reduced this burden (1,2).
Fetomaternal haemorrhage, previous pregnancy, or transfusion can act as sensitising events for alloantibody production (1,3).
Antithetical M and N antigens were first discovered in 1927 by Landsteiner and Levine, corresponding to glycophorin A (GPA) on the RBC membrane (1). Typically, anti-M is a naturally occurring IgM antibody reacting at temperatures below 37°C and is considered clinically insignificant as it is rarely responsible for HDFN or haemolytic transfusion reactions (HTR) (1). However, the frequency of reported HDFN case studies is high in Asian countries (3-6). While anti-M is predominantly an IgM antibody which is unable to cross the placenta due to its pentameric structure, IgG isotypes are present in 50-80% of cases (1). Laboratory identification of anti-M is aided by demonstration of dosage and susceptibility to enzymes ficin, bromelain, and papain in antibody identification panels (1). Around 10% of pregnancies with a positive red cell antibody screen (RCAS) are identified as anti-M, however subsequent antibody titre has a low predictive value for HDFN risk and severity (3, 7). We report a rare case of maternal anti-M causing late-onset HDFN, despite early identification and cold-reactivity in routine antenatal screening.
CASE REPORT
The male New Zealand (NZ) Māori-Samoan infant was born via uncomplicated birth at 38/40. APGAR scores were 10, however the infant was of low birth weight (2.68kg, 1.8th centile). Fetal ultrasound performed at 25/40 for low fundal height had demonstrated normal anatomy, normal umbilical artery Doppler velocity, and estimated fetal weight (EFW) of 819g. Repeat ultrasound at 35 weeks showed normal interval growth and EFW of 2681g.
On day two, the neonate was admitted for jaundice. Biochemistry results indicated abnormally elevated bilirubin of 350 µmol/L (reported by laboratory as above high normal paediatric bilirubin); however, haemoglobin (Hb) was normal at 144 g/L (135-215g/L). Serological testing confirmed the infant was AB RhD positive, with a positive direct antiglobulin test (DAT) of 2+ for IgG (Grifols DG Gel DC Scan). Insufficient sample was provided for elution. The mother was G3P3 and had no history of transfusion. The maternal blood group was A RhD positive, with a history of anti-M alloimmunisation first detected in antenatal
screening four and a half years prior.
Phototherapy was initiated, with a good response. Intravenous antibiotics were given over 24 hours on account of raised C-reactive protein (CRP) and suspicion of sepsis. Blood cultures yielded no growth. Blood films exhibited polychromasia and above normal levels of immature granulocytes. Reticulocyte count was normal, except for a slight raise on day three to 255 x 109/L (0-250 x 109/L). The neonate was discharged on day five with an abnormal, but decreasing, neonatal bilirubin of 184 µmol/L. The Hb had declined from 144 g/L (135-215 g/L) on admission to 119 g/L. Follow-up assessment was indicated.
On return at 15 days the haematology was markedly abnormal. The peripheral blood film showed polychromasia, occasional nucleated RBCs and a single example of erythrophagocytosis (Figure 1). A Hb of 69 g/L (125-205g/L), RBC count of 2.0 x 1012/L (3.5-6.0 x 1012/L) and low packed cell volume (PCV) indicated severe anaemia. Bilirubin had decreased further but was still above normal at 173 µmol/L. A neonatal pre-transfusion sample collected for top-up transfusion showed an IgG positive DAT (1+) and positive RCAS (Grifols Perfect Screen 3 Cell in Grifols DG Gel Coombs card). The sample was insufficient for elution and antibody identification.
A maternal sample was collected, the RCAS was positive and antibody identification (Grifols 11 cell Perfect Panel in Grifols DG Gel Coombs card) identified Anti-M. Clinically significant antibodies were excluded and dosage was not seen for the Anti-M. Phenotyping confirmed the mother was negative for the M antigen. Pre-warmed 37°C saline immediate spin tube technique resolved Anti-M interference with Grifols Perfect Reverse A1 cells (M-positive) in reverse grouping. Pre-warmed 37°C tube indirect antiglobulin test (IAT) RCAS (Grifols Perfect Screen 3 Cell) showed no reactivity at 37°C, consistent with previous identifications of cold-reacting anti-M at 35/40 and historically. Anti-M titre of one had been demonstrated at 9/40 and 22/40.
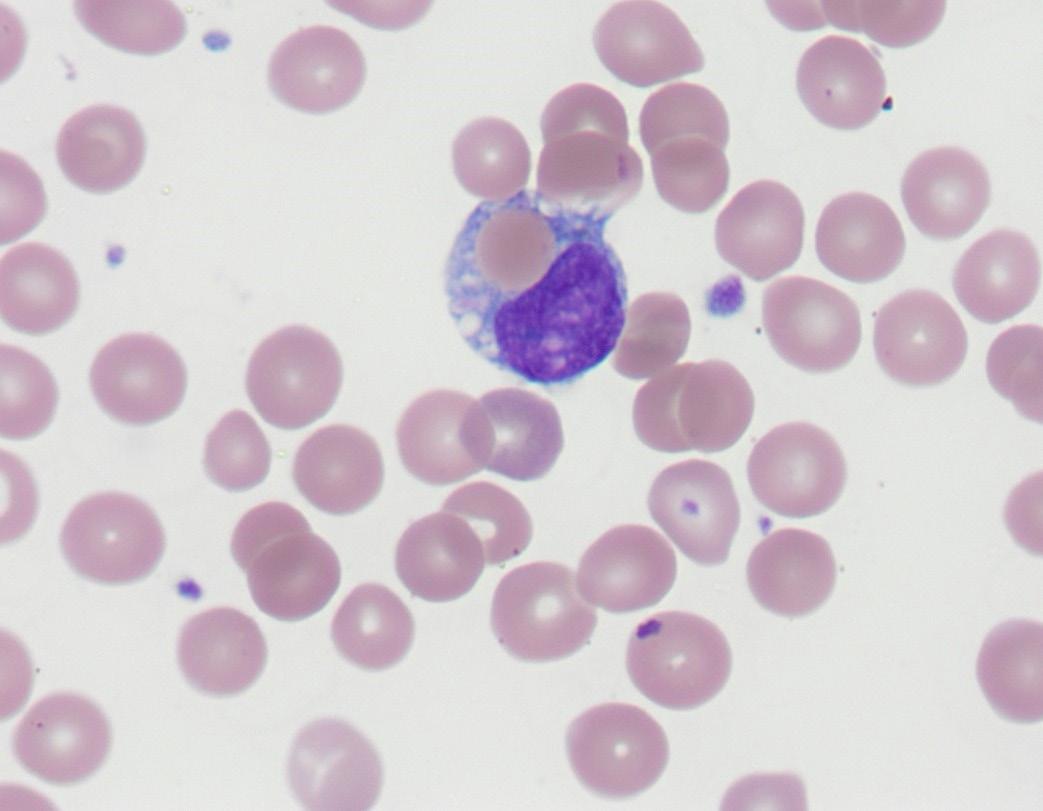
Figure 1. Erythrophagocytosis seen on day 15 in the peripheral blood film of the neonate
The antibody identification on baby confirmed passivelyacquired maternal anti-M present in the plasma and eluate (Table 1), exhibiting dosage (which was not seen in the maternal sample). The phenotype of the neonate was confirmed as M+N+. A pre-warmed 37°C tube IAT screen performed on stored neonatal plasma was non-reactive. Differential diagnosis of ABO HDFN via maternal anti-B was excluded by testing the eluate against M-negative B RhD positive donor cells, as Grifols Perfect Reverse B cells typed positive for M antigen. No Anti-B was eluted. O RhD-negative, M-negative, haemolysin-negative, CMV-negative, Kell-negative, fresh (<7days) neonatal red cell units were compatible by IAT crossmatch against a new neonatal sample on day 16, for subsequent transfusion of 20 mL/kg (60mL) on day 17.
The complete blood count (CBC) on day 23 showed Hb of 79 g/L (100-180g/L). Another Hb drop to 68 g/L on day 29 led to further transfusion of 80 mL of compatible M-negative red cells,
and folic acid supplementation. Further testing showed gradual resolution of hyperbilirubinemia and a negative DAT, despite anti-M persistence in the neonatal plasma, now reacting weakly with homozygote M-positive cells only. At three months of age the infant was discharged from paediatric care with a stable Hb of 107 g/L (90-130g/L) and was in good health at four months of age.
Of note, the mother’s previous (second) child had also experienced anti-M HDFN: term delivery with low birth weight and mild anaemia, with subsequent development of neonatal jaundice requiring phototherapy and moderate-severe anaemia. The blood group was A RhD positive and DAT IgG 3+. Anti-M was identified by elution; however, M phenotyping was not performed. The Hb reached a nadir of 67 g/L at day 35 of life, although transfusion was not pursued. The antibody titre was also one during this pregnancy and non-reactive by pre-warmed 37°C IAT.
Table 1. Antibody identification results (Grifols Perfect Panel 11-cell) showing Anti-M with dosage in neonatal plasma and eluate on day 16.
DISCUSSION
Throughout the literature, anti-M HDFN does not fit the typical presentation of non-ABO HDFN by demonstrating late-onset, prolonged anaemia with a negative DAT (2, 3, 8-11). Titres are not always predictive of severity, nor is reactivity at 37°C (12). Similar to anti-Kell HDFN, the proposed mechanism of prolonged anaemia is suppression of neonatal erythroid progenitor cells by anti-M, as M antigen on RBC is developed by nine weeks (3, 10, 13). This is supported by a study whereby maternal and infant sera containing anti-M suppressed proliferation of cultured M-positive erythroid precursor cells (13).
Anaemia promotes feedback to the bone marrow, stimulating erythropoiesis and early release of immature reticulocytes (10). Hence suppression of erythropoiesis by the alloantibody exacerbates anaemia and results in lower reticulocyte counts (3, 10, 11). Lack of correlation between haematocrit and reticulocyte percentage occurred in anti-M HDFN versus matched antiRhD HDFN cases which did show correlation, reiterating poor compensation for anaemia (4). Of reported anti-M HDFN cases 60-88% were DAT negative, hypothesised to be due to rapid intravascular destruction of DAT positive RBC and erythroid precursors (3, 6, 14).
Antenatal anti-M titres prove disproportionate to HDFN severity (3, 7). This has been attributed to either suppression of erythroid progenitor cells, or variation in laboratory titration methods (3, 7, 12, 15, 16). Antibody-dependent cell mediated cytotoxicity assay (ADCC) and monocyte monolayer assay (MMA) better mimic antibody reactivity in vivo but require specialised testing facilities (4). Despite a titre of one across three reported pregnancies in the literature, severe HDFN arose in three siblings: the first died in-utero of hydrops fetalis, the second premature with the same outcome, and the third treated with IUT and exchange transfusion (7).
Naturally occurring cold-reacting IgM forms of anti-M are considered clinically insignificant and are excluded using 37°C pre-warming techniques (14, 16). The literature explores the possibility that cold reactive anti-M HDFN is explained by either cold-reacting IgG Anti-M in vitro that may cause haemolysis in vivo, or IgM anti-M crossing the placenta (12). Often, both IgM and IgG are present in maternal plasma, but infant plasma demonstrates IgG only, consistent with the understanding that IgM anti-M does not cross the placenta (11, 17).
To our knowledge, ours is the first severe reported case of anti-M mediated HDFN in New Zealand. A retrospective 15-year United States study found no anti-M HDFN and after a 50-year lookback, no further cases (14). Anti-M HDFN was also rare in Iceland, despite high maternal alloimmunisation rates of 19.4% (18). Of reported anti-M HDFN cases in the literature, 88% were in Asian populations, who are postulated to be more susceptible (3-6). In the context of our case, antenatal alloantibody profiles in NZ Māori and Samoan populations are not reported; therefore, it cannot be ascertained whether these populations are susceptible to anti-M HDFN.
The present case exhibited late-onset prolonged haemolysis and anaemia requiring RBC transfusions. The mother had no transfusion history, though the previous pregnancy was also complicated by HDFN. Anti-M allo-immunised mothers with HDFN affected babies frequently had a history of complicated pregnancy (2, 6, 19). In our case, the haemoglobin dropped on day 15 and again on day 29, replicating prolonged anaemia observed in the literature (3, 11, 13, 15). In matched cases anti-M HDFN showed lowest Hb at 9.76 days (+/- 7.37) versus anti-RhD HDFN at 4.2 days (+/- 4.04) and bilirubin peaked later, in contrast to our case where phototherapy was initiated early to address bilirubinaemia (15). In over half of Japanese cases, anaemia was late onset despite early intervention and anti-M persistent
in neonatal plasma has been reported up to seven weeks postbirth (3, 11).
The current case supports the mechanism of erythroid progenitor suppression; reticulocytes were within normal ranges for the most part despite prolonged anaemia. The mother presented with a stable titre of one, supporting a lack of predictability of disease severity of antenatal anti-M titres (3,12,15). In Japanese cases with titres below 16, 80% developed severe HDFN (3). Fortunately, DAT positivity, anti-M in the neonatal plasma and elution aided rapid HDFN diagnosis in the present case, whereas cases in the literature frequently presented DAT negative (15). Our infant had a low birth weight, a feature specific to anti-M HDFN in a recent publication, however the mechanism is undetermined (15).
The absence of reactivity by pre-warmed 37°C tube IAT RCAS was at odds with the clinical picture and strikingly similar to a case reported by Crispin et al (2019) which also demonstrated late onset anaemia (day 20) requiring two transfusions (12). While a negative DAT differed, a single erythrophagocytic monocyte was reported in both cases, a rare feature found in HDFN, following RBC injury. Cold reactive Anti-M was reactive in IAT phase using card technique in both cases (12). In five reported HDFN cases dithiothreitol (DTT) treatment discerned IgG from IgM isotypes of anti-M and found higher maternal IgG titres at 4°C than 37°C (15). Cases of growth retardation, termination, and anaemia taking months to resolve, have been associated with cold-reacting IgG anti-M (5, 20).
Pre-warmed 37°C tube IAT RCAS performed in the current case was on thawed, stored neonatal plasma and weak reactions in card IAT may have been less sensitive in tube. If mixed with IgM in maternal plasma testing, IgG alone in neonatal plasma could be below detection thresholds. Plasma volume was a limitation to further investigation and due to anaemia, further neonatal samples were not requested for titration or DTT treatment, however the DAT indicates IgG. A paternal sample was unavailable.
Clinical management of fetal anaemia in utero is assisted by Doppler assessment of middle cerebral artery peak systolic velocity (MCA-PSV), cordocentesis and alloantibody titration (1, 21). Late onset anaemia in this case meant intravenous immunoglobulin (IVIg), in utero transfusion and exchange transfusion were not indicated. Postnatal treatment consisted of neonatal top-up transfusions of M-negative RBC and phototherapy for hyperbilirubinaemia.
Hospital stays in anti-M HDFN are longer and the likelihood of stillbirths higher than matched anti-RhD HDFN cases, thus accurate antenatal diagnosis is paramount (11, 15, 19). Coldreacting, low titre, IgG anti-M in the current case is deemed to be clinically insignificant based on current antenatal testing guidelines. Neonates with anti-M allo-immunised mothers should be observed for late haemoglobin level changes, even when maternal anti-M is cold-reacting, low titre, or DAT is negative (3, 22). Determination of an IgG component of anti-M during pregnancy may provide greater HDFN predictability and requires further investigation. Additionally, historical pregnancy outcomes should be reviewed. Given this case had two affected pregnancies, close neonatal monitoring is prudent for subsequent pregnancies.
CONCLUSION
Findings in the current case are consistent with passivelyacquired cold-reacting IgG anti-M as the cause of haemolysis. Alongside others in the literature, the current case challenges the notion that cold-reacting anti-M in vitro is not clinically significant in causing HDFN in vivo and its significance should not be discounted. Determination of an IgG component could aid in risk assessment of allo-immunised pregnancies, alongside thermal amplitude and titre. While anti-M HDFN is rare, the stakes are high with potentially severe outcomes. Further local data is required to assess the significance of antenatal anti-M alloimmunisation in the New Zealand population and to aid
implementation of further diagnostic guidelines.
DECLARATIONS OF INTEREST
The authors have no conflicts of interest or funding to declare.
CONSENT
Verbal consent obtained from the mother.
ACKNOWLEDGEMENTS
Special thanks to Steve Johnson of Medlab Central for assistance with blood film imaging and to Janine Gundersen of New Zealand Blood Service for reviewing the manuscript and providing technical advice.
AUTHOR INFORMATION
Savannah C Young, MMLSc, Medical Laboratory Scientist1
Ben G Paterson, MBChB, Haematology Registrar2
Dhana S Gounder, DSM, FRCPA, Transfusion Medicine Specialist3
1 New Zealand Blood Service, Palmerston North, New Zealand
2 Wellington Blood and Cancer Centre, Wellington, New Zealand
3 Reference Laboratory, New Zealand Blood Service, Auckland, New Zealand
Correspondence: Savannah Young, New Zealand Blood Service, Palmerston North, New Zealand email: savannah.young@nzblood.co.nz
REFERENCES
1. D Harmening (Editor). Modern blood banking & transfusion practices. 2012; 7th ed. F.A. Davis Company, Philadelphia, USA.
2. Li L, Huang L, Luo G, et al. Prenatal treatment of severe fetal hemolytic disease due to anti-M alloimmunization by serial intrauterine transfusions. Taiwan J Obstet Gynecol 2017; 56(3): 379-381.
3. Yasuda H, Ohto H, Nollet KE, et al. Hemolytic disease of the fetus and newborn with late-onset anemia due to anti-M: a case report and review of the Japanese literature. Transfus Med Rev 2014; 28(1): 1-6.
4. Li S, He Z, Mo C, et al. Hyporegenerative anemia in anti-Massociated hemolytic disease of the fetus. Transfusion 2021; 61(6): 1908-1915.
5. Liang YL, Shi Y, Su YQ, et al. Maternal Cold-Reacting Immunoglobulin G Anti-M of MNS Blood Group System Causing Hemolytic Disease of the Fetus. Iran J Immunol 2023; 20(1): 129-134.
6. Li S, Mo C, Huang L, et al. Hemolytic disease of the fetus and newborn due to alloanti-M: three Chinese case reports and a review of the literature. Transfusion 2019; 59(1): 385-395.
7. Wikman A, Edner A, Gryfelt G, et al. Fetal hemolytic anemia and intrauterine death caused by anti-M immunization. Transfusion 2007; 47(5): 911-917.
8. Hasani RD, Abdurazak G, Pribadi A. Serial intrauterine transfusion for severe fetal anemia due to anti-M alloimmunization. Asian J Transfus Sci 2023; 17(1): 1-4.
9. Hassan NM, Mohd Noor NH, Mohammed Yusoff S, et al. Anti-M induced severe haemolytic disease of foetus and newborn in a Malay woman with recurrent pregnancy loss. Malays J Pathol 2017; 39(1): 73-76.
10. HitoshiO,DenommeGA,ShoichiI,etal.Threenon-classical mechanisms for anemic disease of the fetus and newborn, based on maternal anti-Kell, anti-Ge3, anti-M, and anti-Jr (a) cases. Transfus Apher Sci 2020; 59(5): 102949.
11. Arora S, Doda V, Maria A, Kotwal U, Goyal S. Maternal anti-M induced hemolytic disease of newborn followed by prolonged anemia in newborn twins. Asian J Transfus Sci 2015; 9(1): 98-101.
New Zealand Journal of Medical Laboratory Science
12. Crispin P, Sliwinski K, Wilson C, et al. Cold reacting anti-M causing delayed hemolytic disease of the newborn. Transfusion 2019; 59(12): 3575-3579.
13. Ishida A, Ohto H, Yasuda H, et al. Anti-M antibody induced prolonged anemia following hemolytic disease of the newborn due to erythropoietic suppression in 2 siblings. J Pediatr Hematol Oncol 2015; 37(6): e375-377.
14. Stetson B, Scrape S, Markham KB.Anti-M alloimmunization: management and outcome at a single institution. AJP Rep 2017; 7(4): e205-210.
15. He Y, Gao W, Li Y, et al. A single-center, retrospective analysis of 17 cases of hemolytic disease of the fetus and newborn caused by anti-M antibodies. Transfusion 2023; 63(3): 494-506.
16. Bajpayee A, Dubey A, Sonker A, Chaudhary RK. A case of severe foetal anaemia due to anti-M isoimmunisation salvaged by intrauterine transfusions. Blood Transfus 2014; 12(S1): s302-s304.
17. Mathew AM, Shah S, Bhatnagar N, et al. Maternal allo anti-M antibody-induced hemolytic disease of newborn. Asian J Transfus Sci 2022; 16(1): 144-147.
18. Bollason G, Hjartardottir H, Jonsson T, et al. Red blood cell alloimmunization in pregnancy during the years 1996-2015 in Iceland: a nation-wide population study. Transfusion 2017; 57(11): 2578-2585.
19. Yu M, Graham K, Pasalic L, Alahakoon TI. Recurrent fetal hydrops with maternal M alloimmunisation: not a benign condition. BMJ Case Rep 2019; 12(7): e230552.
20. Andersen LH, Jacob EK, McThenia SS, et al. Hemolytic disease and reticulocytopenia of the newborn attributable to maternal immunoglobulin G anti-M reacting optimally at cold temperatures. Transfusion 2021; 61(3): 974-978.
21. Golshahi F, Sharbaf FR, Shirazi M, et al. Severe fetal hemolytic disease due to anti-M alloimmunization: A case report and literature. Case Rep Womens Health 2024; 42: e00620.
22. Sharma D, Murki A, Murki S, Pratap T Anti-M antibodies as a cause of intrauterine fetal death and neonatal hyperbilirubinaemia. BMJ Case Rep 2014; 203534
Was the pregnancy test the first Point of Care Test (POCT)?
Contributed
by Michael Legge
The oldest recorded pregnancy test was recorded in Ancient Egypt about 4,000 years ago in the Kahun Medical Papyrus and subsequently in the Greek Magni Hippocratis Opera Omnia. These must be the oldest recordings of a ‘point of care test’ although the result did take some time. Both cultures used a similar technique whereby seeds of wheat and spelt were planted and watered with the women’s urine daily. No growth indicated no pregnancy and if the wheat grows it would be a boy, and spelt growth indicated a girl. The Greeks added confusion by replacing wheat with barley as it was associated with males and wheat was associated with females. Later, Arab physicians ‘refined’ the test by adding seven beans and growth in any or all of these indicated fertility or pregnancy. Other ‘POCT” for pregnancy in ancient times were a recipe for pounded watermelon, mixed with milk and if the woman was sick, she was pregnant. Other tests involved change of smell and being sick after various potions. The latter continued to the middle-ages with a further refinement of the ancient method by incubating two pots of bran, one with male urine and the other with female urine then sealed for nine days (a controlled experiment perhaps!) If the bran with the female urine was preserved, then she was pregnant; refinements were adding beans, barley or wheat. Progressively refinements were undertaken such as putting a copper pot with clean water and nettles under the woman’s bed and if the nettles had red spots, then she was pregnant. One throwback to Aristotle was
for placing the women’s urine in a stoppered glass vile for three days then straining it through a cloth and if there were small living creatures in it, she was pregnant being part of her ‘own substance’.
Moving into more ‘modern times’ it was discovered that a pregnant woman’s urine if injected into female frogs induced spawning the next morning. This was 93% reliable after three weeks of the last missed period and became the mainstay for pregnancy testing as it offered several advantages over the historical methods e.g. being accurate and reliable. Except for an instance in one New Zealand laboratory. The frogs could be stored in a refrigerator prior to use where they would go into hibernation. Unfortunately, after completion of days testing the fridge door was not closed properly and the next morning the laboratory was greeted with frogs making a break for freedom. The discovery and isolation human chorionic gonadotrophin (hCG) removed the ‘romance’ of pregnancy testing and from the 1970’s onwards women could conduct pregnancy testing from their own homes using immunological based kits, with the current ‘stick tests’ available from the 1980’s now being 99% accurate. The development of the immunological pregnancy test was for the first time that a woman did not have to consult another person to get a pregnancy test. The pregnancy POCT was not initially supported by many in the medical profession as it was considered that ‘expert advice’ was required for pregnancy testing. However, the pregnancy test in its various forms must retain the title of the first known POCT.
A lethal synergy: lymphoma associated haemophagocytic lymphohistiocytosis: a case report
Hari Priya Raghvan, Indhira Subbiah, Wee Shiang Yui, Nor Ashikin Azizan, Caroline Ho Siew Ling and Ehram Jamian
ABSTRACT
Haemophagocytic Lymphohistiocytosis (HLH) is a serious hyperinflammatory syndrome caused by abnormal activity in cytotoxic T-lymphocytes and macrophages, leading to a cytokine storm and potential organ damage if not treated effectively In adults, HLH can be triggered by infections, inflammatory disorders, malignancies, and immunodeficiency syndromes. Among these, malignancy associated HLH has the poorest prognosis. We describe a complex case involving a middle-aged man who underwent extensive investigation for HLH initially, yet no trigger was identified. It was only during his second admission that the underlying lymphoma was discovered.
Haemophagocytic Lymphohistiocytosis (HLH) is a severe clinical syndrome caused by immune overactivity, resulting in uncontrolled inflammation and an increased risk of mortality (1). They can be either primary or secondary. Primary HLH is seen mostly in paediatric population and occurs mostly due to chromosomal/genetic alteration. Secondary HLH occurs mostly in adults and common triggers would be infection, malignancies, and autoimmune conditions. Among reported neoplasm associated HLH cases in adults, lymphoma comprises about 67% (2).
HLH can mask the underlying lymphoma. The majority of cases involving Lymphoma-associated Haemophagocytic Lymphohistiocytosis (LA-HLH) are linked to T-cells/NK cells lymphoma (2). LA-HLH secondary to B-cell lymphoma is quite frequent in Asia (1). The outcome is generally poor with survival less than 1-2 months. Defining treatment for LA-HLH remains challenging due to its low incidence rate, diagnostic complexities, rapid progression and the often-compromised physical condition of affected individuals.
We report a case of a 65-year-old male who was diagnosed as High-grade B-cell lymphoma with secondary HLH. This case had posed a challenge to us as the patient was initially investigated for HLH and recovered, but was re-admitted again with a similar presentation, leading to the eventual diagnosis.
CASE REPORT
A sixty-five-year-old gentleman, initially presented with spiking fever (≥38.5°C) and shortness of breath. No bleeding tendencies noted. On examination, no organomegalies/lymphadenopathies
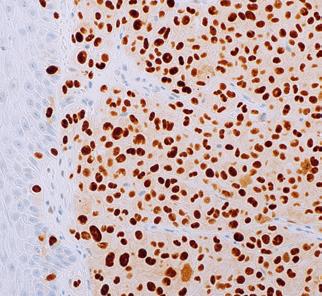
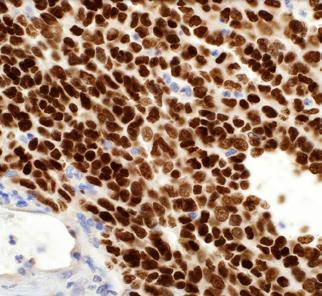
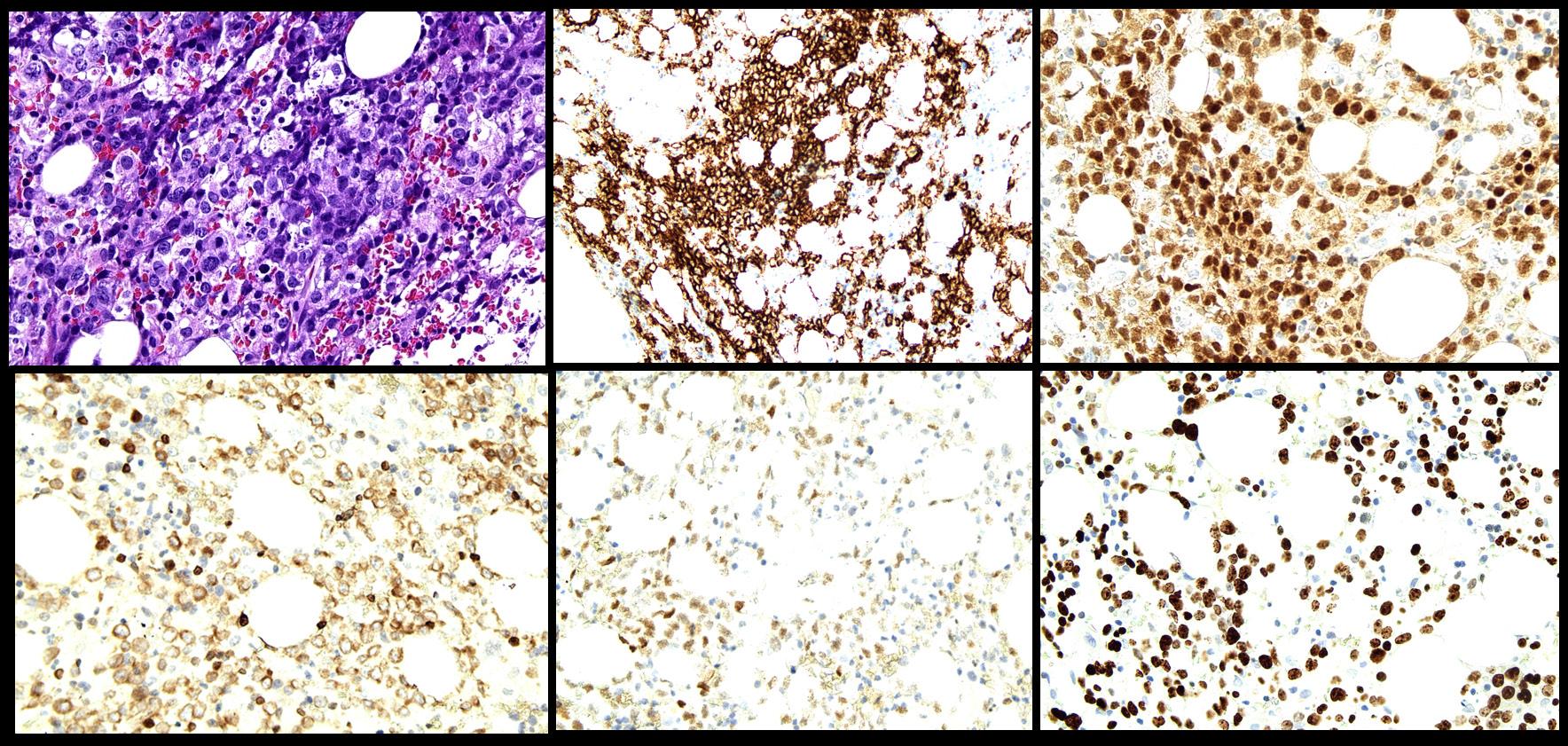
were noted, and no skin lesions were detected. A full blood count revealed normochromic normocytic anaemia and thrombocytopenia. Peripheral blood film did not reveal any blasts/ abnormal lymphoid cells. Ferritin and lactate dehydrogenase (LDH) were markedly raised. The patient then underwent a bone marrow aspirate and trephine biopsy (BMAT) procedure to rule out HLH. However, only occasional haemophagocytic activity was noted. No increase in blasts or evidence of lymphomatous infiltration were detected. He was also extensively investigated for the trigger of HLH (Table 1). A PET scan was done, however, there were no significant findings. He was then empirically treated with dexamethasone. During follow-up, his thrombocytopenia recovered and was responding to the treatment. However, 2-3 months after the initial admission, the patient presented again with similar symptoms. A full blood count during the second admission showed mild normochromic normocytic anaemia with thrombocytopenia. A repeat BMAT was done. Bone marrow aspirate revealed normocellularity with presence of 8% abnormal mononuclear cells which were medium in size, moderate elongated agranular basophilic cytoplasm, round nuclei, condensed chromatin pattern, and inconspicuous nucleoli (Figure 1.). However, immunophenotyping analysis failed to capture any abnormal lymphoid or significant blasts population. Trephine biopsy revealed presence of abnormal lymphoid cells which were positive for CD20, PAX5, CD79a, MUM1, BCL6, and high Ki-67 proliferative index, suggestive of High-grade B-cell lymphoma, favouring Diffuse Large B-Cell Lymphoma, non-GCB subtype (Figure 2.). Unfortunately, the patient succumbed to his illness before any treatment could be initiated.
Figure 1. Bone marrow aspirate showing A) presence of abnormal mononuclear cells (red arrow) (X400 and B) histiocytes ingesting a neutrophil (x 400)
Figure 2. Trephine biopsy showing A) presence of abnormal lymphoid cells (X400) which are positive for B) CD20 (X200), C) MUM1 (X400), D) BCL2 (X400), E) BCL6 (X400), and F) high Ki-67 (X400).
Table 1 Lists of tests that were done during 1st and 2nd admission
Test
Eosinophils (0.0-0.5x109/L)
Basophils (0.0-0.1x109/L)
Blood film malarial parasite No malarial parasite seen
Haemophagocytosis on bone marrow aspirate No: 0 Yes: +35
DISCUSSION
In adults, the majority of HLH cases stem from secondary triggers such as infection, immunodeficiency, or malignancy. Lymphoma is linked to a notably grim prognosis (3). Initial diagnosis may be delayed due to the resemblance between HLH and other inflammatory disorders. Histological diagnosis might be obstructed by tissue infiltration with activated lymphocytes and macrophages, which enables tumour cells to conceal themselves within the inflammatory infiltrates. A previous study has showed that in Asia, the distribution of B- and T-Non-Hodgkin Lymphoma (NHL) are almost equal (B-NHL: 47.4% and T-NHL: 50.8%) (1). Only one such similar case has been registered in Malaysia (1).
In B-cell lymphoma, the development of HLH may be linked to an abnormal rise in inflammatory cytokines produced by malignant cells. Both resting and activated T cells express the membrane bound IL2 receptors. Elevated levels of sIL2 receptors are associated with increased inflammatory activity in various disease conditions (4).
Diffuse Large B-Cell Lymphoma (DLBCL) are ranked the highest among the most common reported HLH-triggering B-cell lymphomas, followed by intravascular B-cell lymphoma (1). Studies indicate that over half of the patients are diagnosed at Stage III-IV (1).
The diagnostic criteria for HLH in the 2004 guidelines were established as 5 out of 8 of the criteria needing to be met (5). In our patient, 4 criteria were met but we were unable to test 2 of the variables as we do not offer those tests (NK cell activity and soluble CD25) (Table 2). However, diagnosing HLH in adults is often challenging due to the condition typically presenting in severely ill individuals, complicating early detection efforts. To address this, the H-score was developed, incorporating 9 variables with probability of HLH and our patient has a chance of HLH of 70-80% (Table 3) (6). Additionally, the MD Anderson Center introduced a scoring system for malignancy associated HLH (M-HLH), utilizing 18 variables to improve early diagnosis and treatment outcomes (7).
Studies have revealed that treatment of LA-HLH need to be centred towards both the lymphoma and HLH. They have extremely poor prognosis with survival of <1 month without lymphoma-specific treatment or with solely HLH-directed therapy (1). Thus, imaging and repeated biopsy of suspicious tissue or excision of lymph nodes is needed in a case of HLH without any obvious signs of a triggering disease/suspected but not proven yet as lymphoma (1). As in our case, we could only detect the lymphoma during the second presentation.
Cytotoxic chemotherapy continues to be the primary treatment approach for HLH associated with malignancy. However, they could be challenging in a severely ill patients with multi-organ failure. Many agents such as etoposide-based therapy together with corticosteroids has been used for malignancy associated HLH with stem cell transplant as the final option (8).
CONCLUSION
This case of LA-HLH highlights the critical need for early recognition and intervention. The dual pathology poses significant diagnostic and therapeutic challenges, necessitating a high index of suspicion and a multidisciplinary approach to improve patient outcomes.
In conclusion, managing HLH associated with B-cell lymphoma poses a distinctive challenge in clinical diagnosis and treatment. The presence of non-specific symptoms upon presentation and the absence of a definitive test underscores the critical need for a high level of suspicion and prompt initiation of therapy once a diagnosis is confirmed.
DECLARATION
Authors declare no conflict of interest in this publication.
AUTHOR INFORMATION
Hari Priya Raghvan, MD (UCSI), DrPath, Haematology, (UKM)1
Indhira Subbiah, MBBS, MPath, Haematology1
Wee Shiang Yui, MBBS, DrPath, Haematology1
Nor Ashikin Azizan, MD, DrPath, Haematology1
Caroline Ho Siew Ling, MBBS, DrPath, Haematology1
Ehram Jamian, MD, MPath, Haematology1
1 Haematology Unit, Department of Pathology, Hospital Sultanah Aminah, Johor Bahru, Johor, Ministry of Health, Malaysia.
Corresponding Author: Hari Priya Raghvan, Haematology Unit, Department of Pathology, Hospital Sultanah Aminah, Johor Bahru, Ministry of Health, Malaysia.
Email: priya_hari88@yahoo.com
REFERENCES
1. Knauft J, Schenk T, Ernst T, et al. Lymphoma-associated hemophagocytic lymphohistiocytosis (LA-HLH): a scoping review unveils clinical and diagnostic patterns of a lymphoma subgroup with poor prognosis. Leukemia 2024; 38(2): 235249.
2. Li N, Jiang M, Wu WC, et al. Lymphoma-associated hemophagocytic syndrome: a retrospective study from a single center. Hematology. 2022; 27(1): 909-916. doi:10.10 80/16078454.2022.2113600.
3. Li F, Li P, Zhang R, et al. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): an analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol 2014; 31(4) :1-7. doi: 10.1007/s12032-014-0902-y.
4. Ojo, A.S., Asemota, J., Ojukwu, S., et al. B-cell lymphoma-associated hemophagocytic lymphohistiocytosis: a case report. Oncol Lett 2022; 24(2): 246 doi.org/10.3892/ ol.2022.13365.
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48(2):124131.
6. Debaugnies F, Mahadeb B, Ferster A, Meuleman N, Rozen L, Demulder A, Corazza F. Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol 2016; 145(6): 862870.
7. Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017; 123(17): 3229-3240.
8. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019; 133(23): 2465-2477. doi: 10.1182/ blood.2018894618.
Pyroglutamic acidosis: an under-recognised cause of high anion gap metabolic acidosis with multi-factorial aetiology
Yassar Alamri, Polly Davison, Charlotte Reay, John Geddes and Christopher Florkowski
ABSTRACT
Pyroglutamic acidosis (PGA) remains under-recognised among practising clinicians. Predisposing factors are common. We present a recent case of pyroglutamic acidosis and highlight the role of drug-drug/drug-body interactions, as well as the necessity of good communication between clinicians at the bedside, and scientists at the bench.
Abbreviations: Anti-epileptic drug (AED), cytochrome P450 (CYP450), high-anion gap metabolic acidosis (HAGMA), N-acetylcysteine (NAC), pyroglutamic acidosis (PGA).
NZ J Med Lab Sci 2025, 79(2): 65:66
INTRODUCTION
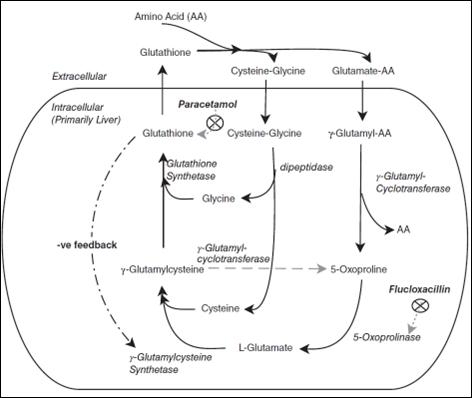
Pyroglutamic acidosis (PGA; also known as 5-oxoproline acidaemia) is a rare, mostly acquired, cause of high-anion gap metabolic acidosis (HAGMA). It is most often seen in female patients with multiple co-morbidities. Sustained use of therapeutic-dose paracetamol (acetaminophen) is a major risk factor (1). Other medications have also been implicated, most notably flucloxacillin (β-lactam antibiotic), and vigabatrin (antiepileptic drug; AED). However, other agents (including other AED) may also contribute to the development of PGA by, for example, inducing hepatic metabolic enzymes (e.g., CYP2E1) (2). We present a case of PGA which required inter-disciplinary team collaboration in caring for a multi-morbid female patient.
CASE REPORT
An 85-year-old female was admitted following a fall at her resthome. She had sustained a right-sided bi-malleolar ankle fracture (which was surgically treated on her third day of admission). Other past medical history included well-controlled epilepsy, hypertension, and stable chronic kidney disease (baseline eGFR = 25 mL/min/1.73m2). She received medication over-sight at her care facility, and regular medications included paracetamol (4 g/ day), carbamazepine, phenytoin, phenobarbital, and perindopril. Her admission was complicated by delirium, hospitalacquired pneumonia (for which she received an 8-day-course of intravenous cefuroxime), and an acute kidney injury (nadir eGFR = 14 mL/min/1.73m2) which was treated with cessation of perindopril, and receipt of intravenous balanced crystalloids.
Throughout her admission, she was noted to have an unexplained HAGMA on several arterial and venous blood gas samples. Further blood tests did not demonstrate significant rises in lactate, or β-hydroxybutyrate. Trough concentrations of her three AED were low-normal, and she had no detectable concentration of ethanol, methanol, paracetamol, or salicylic acid. She had an acellular urinary sediment without crystals; further samples for urine tandem mass spectrometry screen were sent.
Discussions about her HAGMA were had among multiple clinical teams, including surgical/geriatric medicine, clinical pharmacology, nephrology, and chemical pathology. Owing to the presence of multiple PGA risk factors, her paracetamol was ceased, and she was empirically commenced on N-acetylcysteine (NAC).
On day 8 her admission, her urinary metabolic screen returned positive for a marked increase in pyroglutamic acid. Despite ongoing supportive cares (including NAC infusions), her condition continued to deteriorate, and she died on day 13 of her admission.
DISCUSSION
Remarks on the patho-physiology of pyroglutamic acidosis
The pathogenesis of PGA is complex and multi-factorial involving several clinico-biochemical “hits” before significant acidosis develops. There are a multitude of risk factors (3, 4), some of which are modifiable (e.g., regular use of paracetamol, use of flucloxacillin/vigabatrin, malnutrition, and AKI), while others are not (e.g., female sex, advancing age, and multi-morbidity— including renal and hepatic failure).
PGA develops due to interference with glutathione metabolism, relative deficiency of cysteine/glycine, and/or reduced clearance of pyroglutamate (1, 3). Glutathione is an intermediary product required in multiple redox reactions, and its intra-cellular concentration is kept within a narrow homeostatic range. Glutathione is synthesised by the sequential addition of three non-essential amino acids: Glutamate, cysteine, and glycine. Glutamate is ubiquitous in the body, and without the two other amino acids, it can spontaneously cyclise to form pyroglutamate. Therefore, conditions that limit the availability of cysteine (e.g., chronic paracetamol use), and glycine (e.g., malnutrition, or the demands of pregnancy) can increase the rate of pyroglutamate formation (refer to Figure 1 for a schema of the metabolic pathway (5)). Glutathione depletion also reduces negative feedback inhibition of the gamma-glutamyl cycle, resulting in increased production of gamma-glutamyl cysteine, which is then converted to pyroglutamate (3, 5).
Under physiological conditions, pyroglutamate is immediately broken down back to glutamate by 5-oxoprolinase (a functional “lactamase” enzyme). If this enzyme is inhibited (e.g., by flucloxacillin which is an inhibitor of β-lactamases), then accumulation of pyroglutamate ensues. Additionally, the presence of renal dysfunction further limits its urinary excretion (3, 4).
Role of AED
In a recent review of 100 published cases of PGA, epilepsy and AED use featured in 8 patients. It is well-documented, however, that patients with epilepsy are at high-risk of other medical comorbidities, including malnutrition and poly-pharmacy. Therefore, the observation of 8% of PGA patients havingepilepsy could be coincidental (3).
Regardless, mechanistic causation (i.e., AED contributing to PGA) remains a possibility. It has been postulated that medications, such as many AEDs, that induce hepatic cytochrome P450 (CYP450) enzymes may interfere with the metabolism of other medications (including paracetamol) (2). This would further deplete glutathione (and cysteine/glycine) stores, and thus favour the formation of pyroglutamate.
Vigabatrin is an AED which inhibits the breakdown of GABA. It is often used as an adjunct agent for patients with resistant epilepsy. Its contribution to the development of PGA is unrelated to effects on the CYP450 system. The exact mechanism remains elusive, although it is thought to be related to disruption of the cysteine-taurine metabolic pathway (3).
Multi-factorial aetiology
Our patient, therefore, had at least had a “triple-whammy” that may have led to the development of PGA. Her three AED were known CYP450 inducers, and could have plausibly competed with paracetamol metabolism which she regularly consumed (first hit with cysteine and glutathione depletion). She also had malnutrition, and developed an inter-current sepsis (second hit with further glycineand glutathione depletion). She also developed an AKI on her CKD (third hit with reducedclearance of pyroglutamate). It is unclear whether her cefuroxime course had contributed to the PGA (e.g., by interferingwith 5-oxoprolinase activity), although at least one previous case
report has implicated cefuroxime in the development of PGA (6). Good inter-disciplinarcommunication in our case enabled a presumptive diagnosis of PGA, and empiric treatment with NAC.
Access to a validated pyroglutamate assay in our laboratory confirmed the diagnosis.
Figure 1. Multiple ‘hits’ may affect the gamma-glutamyl cycle, and predispose to the accumulation of pyroglutamic acid (Reproduced with permission (5)).
CONCLUSION
Clinicians should be aware of the possibility of PGA, especially in the setting of otherwise unexplained HAGMA whereby more common causes such as lactic acidosis and ketoacidosis have been excluded. The probability of PGA is further increased if the clinical context also includes regular paracetamol usage, sepsis, renal impairment, and concomitant use of antibiotics and/or AED.
DECLARATIONS
Participant consented for anonymised data publication.
AUTHOR INFORMATION
Yassar Alamri, MBChB, PhD, Medical Registrar, Department of Medicine1,2
Polly Davison, MBChB, Medical Registrar, Department of Medicine1
Charlotte Reay, MBChB, House Officer1
John Geddes, FRACP, Consultant Geriatrician3,4
Christopher Florkowski, BA, MB BS, MD(Lond), MRCP(UK), FRCPA, FRACP, Chemical Pathologist5
1Department of Medicine, Christchurch Hospital, Christchurch, New Zealand
2Department of Medicine, University of Otago, Christchurch, New Zealand
3Department of Older Persons’ Health, Christchurch Hospital, Christchurch, New Zealand
4Department of Medicine, University of Otago, Christchurch, New Zealand
5Department of Clinical Biochemistry and Genetics, Canterbury Health Laboratories, Christchurch, New Zealand
3. Tailor P, Raman T, Garganta CL, et al. Recurrent high anion gap metabolic acidosis secondary to 5-oxoproline (pyroglutamic acid). Am J Kidney Dis 2005; 46(1):e4-10. Peter JV, Rogers N, Murty S, et al. An unusual cause of severe metabolic acidosis. Med J Aust 2006; 185(4): 223225.
Stewart GW. Pyroglutamate acidosis 2023: a review of 100 cases. Clin Med (Lond) 2024; 24(2): 100030.
4. Crisp T, Sizeland P, Du Toit S, Chan LW. Pyroglutamic acidosis: an under-recognised cause of high anion gap metabolic acidosis. NZ Med J 2021; 134(1546): 117-121.
5. Mo L, Liang D, Madden A, Aung AK. A case of delayed onset pyroglutamic acidosis in the sub-acute setting. Intern Med J 2016; 46(6): 747-749.
6. Patel S, Parikh F, Kulkarni N. High anion gap metabolic acidosis as a result of 5-oxoproline. Indian J Med Spec 2022; 13: 198-200.
The author is a freelance science writer, broadcaster and a previous editor for the science journal Nature. In this book the author considers whether the slow processes of DNA changes over time, is correct or are there other molecular mechanisms contributing to change. The first four chapters start with the basic biology of DNA, protein synthesis and cell biology. These chapters are essential for an understanding of the concepts discussed later in the book. The acknowledged concept of the central dogma is well considered but the author raises the question, does our understanding with the accepted wisdom, fit with the discoveries and changes found in the new biology? While DNA is essential, does it “build a cell” i.e. is the genome enough? This consideration is continued throughout the book, examining the roles of RNA which we now know is a cell signalling system as well as making proteins. As the book progresses the author considers how do the cells of the embryo ‘know’ where to go to form separate body organs with incredible precision. What is the role of complex carbohydrates and epigenetics? Why is only two percent of our DNA used for coding proteins? What are the roles in human biology for the remaining 98 percent of our DNA? The title of this book accurately reflects the conventional wisdom of DNA but challenges the reader to think about “how life works”. The book is very well researched and written. It can challenge conventional wisdom at times, but the author writes with exceptional clarity. Definitely a good read for those that wonder whether there is more than just DNA
Reviewed by: Michael Legge, PhD
A fatal inheritance: how a family misfortune revealed a deadly medical mystery
Author: Lawrence Ingrassia, Henry Holt & Company, 2024
ISBN: 9781250837226
The author is a multiple Pulitzer Prize editor having worked with the Wall Street Journal and the Los Angeles Times and he writes about not only his personal story of living in a family with Li-Fraumeni syndrome, but he expertly traces the history of the discovery of the mutation that causes the rare cancercausing syndrome. The author’s personal story starts with the deaths of his mother, two sisters a brother and a nephew to cancer and a second unrelated family who had fatal early onset cancer. He traces the early family histories of several families all of whom had cancer dominating in their families and the curiosity of two young clinicians Li and Fraumeni in the early 1960s who initiated familial cancer pedigree collections. At the time cancer was firmly
believed to be caused be cancer causing viruses and for a few years it was the only accepted dogma. By 1982 Li and Fraumeni had collected extensive family pedigrees demonstrating that there must be an underlying genetic factor involved in the cancers and United Kingdom (UK) cancer researchers named the syndrome the Li-Fraumeni Syndrome. The dogma still considered a mix of viruses and specific cancer-causing genes. However, a joint discovery by scientists in the USA and the UK identified a normal gene that was transformed in cancer, the p53 gene and subsequently the gene implicated in retinoblastoma (Rb gene). This led to the term onco-gene and opened the way for studying the genetic basis of cancer. It soon became evident that mutations in the p53 gene were the cause of various cancers and in particular the families Li and Fraumeni had so carefully curated. The inheritance was established as autosomal dominant, which now accounted why certain family members did not develop cancer, and this fitted with earlier theoretical genetic calculations.
The book is an easy read, and the author cleverly integrates his own family story with the tragedy of cancer in numerous other families that were studied by Li and Fraumeni. It covers the numerous scientists and clinicians who worked on the p53 and Rb gene discoveries. It also highlights the competitive matters in the early days of gene discovery to be the first to publish.
Reviewed by: Michael Legge, PhD
Otago BMLSc student research project abstracts: semester 2, 2024
Evaluation of Colotect™ 1.0 faecal DNA stability for colorectal cancer screening
Pallak Bhatnagar1 and Amanda Dixon-McIver2
1University Of Otago, Dunedin and 2IGENZ Laboratory, Auckland
Objectives: Colorectal cancer is the second leading cause of cancer in New Zealand but has high cure rates when identified early. In June 2024, IGENZ launched Colotect™ 1.0, a privately funded bowel screening test. Colotect™ 1.0 requires selfcollection of a faecal sample which is then transported to the laboratory. The manufacturer states samples are stable for up to 7 days in the provided buffer. We aimed to determine whether the faecal sample remains suitable for analysis 10 days postcollection in case of transportation delays. Methods: Six individuals were selected, each providing two faecal samples. The initial sample was frozen on day 1, whilst second remained at room temperature until frozen at day 10. Following DNA extraction, bisulfite conversion and purification took place in accordance to the manufacturer's instructions. This assay uses qRT-PCR to measure the methylation status of four genes. Three genes are used to infer risk status (ADHFE1, SDC2, PPP2R5C), while GAPDH, a common house-keeping gene, is used as a control to validate methylation status findings. Results: Day one samples yielded reduced cycle threshold values for GAPDH compared to day 10 samples indicating nucleic acid degradation. However, all day ten samples remained within acceptable cycle threshold limits (<38). Detectable cycle threshold values were found for ADHFE1 and SDC2 in samples one and six initially but were absent in day ten samples.
Conclusion: Colotect™ 1.0 demonstrates potential for identifying colorectal cancer markers ten days post collection. The cycle threshold for GAPDH, although elevated, remains within the manufacturers acceptable range. The deterioration of ADHFE1 and SDC2 highlights the necessity for additional research utilising a larger sample size and known positive samples.
Evaluating the DFS70 IgG testing protocol at Pathlab Waikato
Khrystine Carullo1 and Andrew Soepnel2
1University of Otago, Dunedin and 2Pathlab Waikato, Hamilton
Objectives: The current protocol for the detection of anti-dense fine speckled 70 (DFS70) IgG antibodies at Pathlab Waikato involves adding DFS70 IgG testing to antinuclear antibody (ANA) samples with a DFS-like pattern and titre > 1:80. This approach may miss isolated DFS70 IgG positive cases that do not exhibit DFS-like pattern during initial screening. This study aimed to evaluate whether expanding the DFS70 IgG testing protocol to include homogenous patterns with a titre > 1:80 is warranted. Methods: Routine ANA samples were analysed using QUANTALyser 3000 and interpreted through NOVA-View automation and manual immunofluorescence reading. In the first half of the study, single well titre was used; in the second half, endpoint dilution testing (ET) was implemented. Samples displaying homogenous patterns with titre > 1:80 underwent DFS70 IgG testing on the BIO-FLASH. A total of 201 samples were included. A cost-benefit analysis assessed whether the proposed protocol change could be justified as a permanent change.
Results: Nine additional isolated DFS70 IgG cases were identified, including one that displayed a DFS-like pattern upon ET. The estimated monthly cost for the additional DFS70 IgG testing was $2,400 in the first half. For the second half, the estimated monthly cost was $1,620 for the additional DFS70 IgG tests and $608 for the extra wells required for ET.
Conclusion: The proposed change in the DFS70 IgG testing protocol was not implemented. Although additional isolated
DFS70 IgG cases were identified, the local immunopathologist deemed them not clinically significant. Therefore, the extra costs associated with the protocol change could not be justified.
Validation of Cepheid GeneXpert MRSA/SA blood culture test
Claire Chan1 and Rosie Greenlees2
1University of Otago, Dunedin and 2Canterbury Health Laboratories, Christchurch
Objectives: This study was to validate results from the Cepheid GeneXpert MRSA/SA blood culture test against traditional culture methods, considered to be the gold standard. The study was done to see if, after gram-positive cocci (GPC) resembling staphylococci were seen on gram stain from a blood culture bottle, GeneXpert may be used alongside culture methods, for a faster time to result. This may avoid unnecessary antibiotics or patient callbacks or delay in appropriate antibiotic treatment. GeneXpert produces results in about an hour, whereas culture takes 12-24 hrs, the coagulase test takes a minimum of 4 hrs, and sensitivity testing with a cefoxitin disc to determine methicillin resistance can take up to 18 hrs.
Methods: Nineteen positive blood culture bottles from ED (aerobic and anaerobic), resembling Staphylococcus species (GPC) from gram stain, were run on the GeneXpert over a period of three weeks. Sample preparation included sterilising the blood culture bottle with 70% ethanol, adding 50uL of blood to the elution reagent vial and vortexing on high speed, transferring the entire contents of the vial to the cartridge and analysing the sample with the GeneXpert system. Due to low numbers of MRSA samples, a blood culture bottle spiked with MRSA has been included in the study.
Results: For GeneXpert, the sensitivity and specificity were both 100% for Staphylococcus aureus and MRSA when compared to the gold standard, which is culture. For the coagulase test, sensitivity was 67% and specificity was 100% when compared to GeneXpert.
Conclusion: Based on these results, the Cepheid GeneXpert system could be used instead of the coagulase test in the lab to provide rapid, accurate results.
The relationship between hypogranular neutrophils and the [Ne-SSC(ch)] of Sysmex XN-series analysers
Tristan Cherrill1, Katherine Christie2 and Richard Parker1,2 1 University of Otago, Dunedin and 2 Awanui Labs, Dunedin
Objectives: To establish a reference range for the Ne-SSC measurement from the Dunedin Hospital population, investigate the relationship between this measurement and neutrophil hypogranularity and identify a cut-off value for generating blood films to assist detection of dysplastic cases, such as Myelodysplastic Syndrome.
Methods: The reference range was established using 62,000 patient specimens refined from a larger sample collected over a 4-month period on the Dunedin Hospital Haematology Analysers. Thirty-eight specimens from patients with Myelodysplastic Syndrome, Acute Myeloid Leukaemia, or Chronic Myelomonocytic Leukaemia and an extra specimen from a patient Polycythaemia Vera were investigated by microscopy for hypogranular neutrophils. These specimens’ Ne-SSC values were extracted from the Dunedin Haematology XN-20 analysers. Potential cutoff values were identified by the changes in sensitivity based on the collected specimens.
Results: A reference range of 139.6 - 156.7 was established, containing 95% of the ‘healthy’ Dunedin hospital patients in the sample. Specimens with hypogranular neutrophils, except
for two, consistently exhibited Ne-SSC values below this reference range with a mean value of 128.49. A cut-off value of approximately 137.2 was suggested as the most appropriate. This cut-off identified almost all hypogranular specimens in the sample with only a minor increase in the generation of false negative blood films.
Conclusion: Implementation of the cut-off will assist detection of Myelodysplastic Syndrome due to the correlation of hypogranular neutrophils and Myelodysplastic Syndrome. The cut-off can be adjusted by ±1.0 value to ensure a sustainable change in workload while keeping an almost consistent sensitivity.
Validation of a NOTCH2NLC GGC repeat primed PCR assay for diagnostic purposes
Sooin Choi1, Pei-Chun (Lisa) Hsu2, Hannah Wyatt2 and Kylie Drake2
1University of Otago, Dunedin and 2Canterbury Health Laboratories, Christchurch
Objectives: Neuronal intranuclear inclusion disease (NIID) is a rare neurodegenerative condition caused by a GGC repeat expansion in the 5′-untranslated region of NOTCH2NLC. This repeat was identified in patients of East Asian ancestry, however recent evidence suggests a high prevalence of NIID among New Zealand Māori/Polynesian individuals. A validated diagnostic assay for detecting NOTCH2NLC is unavailable in an accredited clinical laboratory within Australasia and patients are currently diagnosed as part of research protocols via sequencing of the repeat expansion. The objective of this study was to validate a repeat-primed PCR assay to detect the NOTCH2NLC repeat expansion at Canterbury Health Laboratories (CHL).
Methods: Six NOTCH2NLC GGC repeat positive control (sequenced overseas), and 2 negative control samples were used to validate the assay. Primer sequences were sourced from a previous publication, and testing was performed using FastStart Taq (Roche), RepliQA (Quantabio), Platinum Taq (Invitrogen), and Qiagen multiplex master mix (Qiagen) to amplify the triplet repeat expansion of NOTCH2NLC. PCR fragments were run on an ABI-3500 Genetic Analyser and analysed using GeneMapper. Various amplification conditions and DNA concentrations were utilised.
Results: The available polymerases at CHL displayed variable amplification of the repeat. However, amplification with FastStart Taq and Platinum Taq polymerase resulted in best saw-tooth pattern/expansion peaks and amplicon length analysis.
Conclusion: Amplification of the repeat expansion of NOTCH2NLC was successful. However, the PCR is not yet reproducible and requires further adjustment. This project represents the first step towards an accredited diagnostic NOTCH2NLC assay in Australasia. Completing the validation of this assay will be critical for the diagnosis of patients with NIID in New Zealand but is outside the scope of this project.
Development and validation of a next-generation sequencing cancer panel
Francis Costas 1 and Vivienne Bickley2
1University of Otago, Dunedin and 2Canterbury Health Laboratories, Christchurch
Objectives: Molecular oncology has a crucial role in cancer diagnosis, treatment and prognosis through identification of genetic alterations in genes that drive cancer. The Illumina TruSight Tumour 15 (TS15) next generation sequencing panel targets 15 genes associated with lung, melanoma, breast, colon, ovarian and prostate cancers. Treatment advances and updates in molecular classifications, such as the 2021 WHO Classification of central nervous system tumours and 2023 FIGO staging system for endometrial cancers, demonstrated the need for expanded genetic testing. The objective was to optimise and validate a new custom-designed 35-gene cancer panel
encompassing the current TS15 content, as well as additional CNS and endometrial cancer genes.
Methods: Validation was conducted with eighteen formalin-fixed paraffin-embedded tissue (FFPET) samples extracted with the QIAamp kit (Qiagen) through silica-gel membrane purification; and twenty samples previously tested on the TS15 panel. The custom panel (Twist Bioscience) utilises hybridisation-based target enrichment, which simplifies future customisation for more adaptable testing provision. Secondary analysis was performed using the DRAGEN enrichment app v4.3.6 (Illumina), while tertiary analysis was facilitated by Alissa software (Agilent).
Results: Variable DNA yield and quality was achieved from FFPET samples, resulting in inconsistent sequencing metrics despite optimisation efforts. High off-target hybridisation was observed, despite this, 19/19 of the known variants tested were correctly identified, validating variant detection in 19 of the 35 genes on the panel to date.
Conclusion: Validation of this panel illustrated the challenges associated with testing DNA derived from FFPET, particularly the variable quality and high fragmentation, while off-target probe hybridisation suggested re-design of some panel probes was needed. Following re-design further optimisation of the library preparation method and validation of the panel content will continue.
ABO blood group confirmation: Comparison of methods, Paragon Care Immulab and Grifols Erytra Eflexis
Maria Costas1, Ruth Brookes2 and Janine Gunderson2
1University of Otago, Dunedin, and 2New Zealand Blood Service, Palmerston North
Objectives: New Zealand Blood Service Palmerston North Blood Bank currently uses Paragon Care Immulab reagents for manual blood group confirmation of red blood cell. This study aimed to compare the efficiency and cost-effectiveness of manual tube testing utilising Paragon Care Immulab reagents against automated Grifols Erytra Eflexis with Grifols DG Gel Confirm P cards.
Methods: Medical Laboratory Scientists were observed performing two methods: manual tube testing with Paragon Care Immulab reagents and automated testing using Grifols DG Gel Confirm P cards. Twenty-eight batches were observed with a total of three hundred and eighty-two red cell units confirmed. Stopwatch timing ensured consistency by starting and stopping at the same points for both methods. Consumable costs per test were calculated for each method. All data was recorded and analysed on an Excel spreadsheet.
Results: The average time for manual testing was 27 minutes and 08 seconds, while automated testing 27 minutes and 27 seconds. The cost per test was $1.52 NZD for manual testing and $2.85 NZD for automated testing.
Conclusions: The study concluded that automation did not offer a significant time or cost advantage for the Palmerston North Blood Bank. However, automation may provide workflow efficiency benefits for smaller or understaffed blood banks, where reducing manual labour could have a more substantial impact.
Determining the suitability of testing anti-Jka antisera using Grifols AHG cards
Matthew Cresswell1 and Kate Anderson2
1University of Otago, Dunedin and 2New Zealand Blood Service, Dunedin
Objectives: Delivery of safe, compatible blood is incredibly important to a Blood Bank, and as such, phenotyping red cell units is paramount in providing compatible blood to patients. The Kidd blood group system is commonly associated with delayed haemolytic transfusion reactions, meaning units for a patient with an anti-Jka must receive Jka antigen-negative blood. However,
the current Jka phenotyping method has an unstable endpoint, as well as requiring more antisera and direct staff attention compared to card methods. The aim of this study was to determine the suitability of phenotyping for the Jka antigen using Grifols AHG card technology compared to the current standard tube test method.
Methods: Twenty red cell unit segments with unknown antigen status were subjected to three cycles of washing. Tube phenotyping was performed on every unit’s cells for both the Jka and Jkb antigens to determine their antigen status. Nineteen of the 20 units were phenotyped for Jka using Grifols AHG cards. Positive and negative controls were performed once daily, with the addition of washed controls on the second day of testing.
Results: Results indicate Grifols AHG cards could not reliably phenotype for Jka compared to the current standard phenotyping procedure. Of the 13 units positive for the Jka antigen, 9 had a weaker result compared to the positive control and were all Jk antigen heterozygotes. Interestingly, the four valid Jka positive units were a mixture of heterozygous (2) and homozygous (2).
Conclusion: Preliminary results show that Grifols AHG cards are unsuitable for Jka phenotyping. However, further research with a larger sample size, as well as investigating the incorporation of different pre-testing techniques should be explored before a conclusion is made.
A method comparison of the Axis Shields IgG anti-CCP manual ELISA, and the BioFlash platform for IgG anti-CCP
Alice Doney1, Jonathan Slade2 and Sarah Beck2 University of Otago, Dunedin1 and Canterbury Health Laboratories, Christchurch2
Objectives: Anti-CCP Antibodies are important markers in the diagnosis of Rheumatoid Arthritis (RA), a chronic inflammatory disease characterised by joint swelling, tenderness, and destruction of synovial joints. Anti-CCP antibodies are considered superior to Rheumatoid Factor (RF) due to greater clinical sensitivity, and earlier presence in disease. The manual ELISA was replaced in routine diagnostics for several reasons, including the time-consuming manual methodology, inability to automate with current laboratory instrumentation, and test not meeting stated turnaround time for results. The project aim was to compare the performance of an Anti-Cyclic Citrullinated Peptide (CCP) antibody assay by QuantaFlash chemiluminescent immunoassay (CLIA) on the BioFlash platform to the previously used enzyme-linked immunoassay using the Axis Shields IgG anti-CCP commercial kit.
Methods: Seventy-eight patient samples run routinely on the BioFlash were repeated by manual ELISA. Results were evaluated for sensitivity and specificity, and kappa agreement against clinical diagnosis of RA.
Results: Sensitivity and specificity of the BioFlash and Manual ELISA methods were calculated from a subset of samples with available clinical details, as summarised below. BioFlash Sensitivity = 86%, Specificity = 96%, Kappa = 0.83, ELISA Sensitivity = 86%, Specificity = 88%, Kappa = 0.73
Conclusion: These results show that the CLIA method has greater specificity for RA than manual ELISA. The BioFlash is faster and easier to automate, resulting in a major improvement in turnaround time and has other benefits to laboratory performance such as improved reagent traceability, sample traceability and no manual transcription of results.
Case study of an invasive breast cancer of no special type, with specific focus on the histopathological grossing technique
Jessica Evans1 and Karen Murcott2
1University of Otago, Dunedin and 2Awanui Labs, Dunedin
Objectives: Breast cancer affects approximately 3,500 Aotearoa women each year. This statistic is strongly influenced by the quality of histological grossing, which sets the foundation for
downstream reporting. This case-study involves a female patient who had multiple specimens submitted to the Dunedin Grossing Laboratory following the identification of a suspicious lump within the right breast. This case-study highlights the importance of standardised histological grossing guidelines which endorses correct identification, description and specimen grossing, maximizing the quality of histopathological results for optimal patient care.
Methods: Histological grossing of the breast cores and lumpectomy surgery was performed following formalin fixation using standardised protocols outlined by the Royal College of Pathologists of Australasia. A total of 23 cassettes were submitted for histological testing. A team of pathologists viewed the tissue under microscope, using the macroscopic dictation and standardised diagnostic guidelines to form a histopathological diagnosis.
Results: The histological results coherently showed evidence of invasive breast (ductal) carcinoma of no special type (WHO 5th edition), G3 pathological staging (American Joint Committee on Cancer (AJCC) 8th edition). The margins and sentinel lymph nodes were both clear, thus no additional surgical procedures were required.
Conclusion: This case-study examined the grossing of several breast specimens to highlight the direct link between correct grossing and confident diagnostic reporting. Furthermore, underpinning the importance of standardised histological grossing guidelines to comply with quality assurance policies which protects laboratory accreditation.
Retrospective analysis of emergency O RhD negative red blood cell units issued at Rotorua Hospital between 01 January 2022 and 31 December 2023
Felicity George1 and Raewyn Cameron2
1University of Otago, Dunedin and 2Pathlab, Rotorua
Objective: To retrospectively analyse emergency O RhD negative red blood cell units issued at Rotorua Hospital between 01 January 2022 and 31 December 2023.
Methods: The information was collected from 80 patient eTraceline profiles and included the status of the emergency O RhD negative red blood cell units, laboratory response times and patient demographics that may influence protocols for future blood products issued.
Results: There were 156 emergency O RhD negative red blood cell units issued between 01 January 2022 and 31 December 2023. Nine Emergency O RhD negative red blood cell units issued were returned to blood bank (7 units were able to be returned to stock and 2 units were required to be discarded). Average time taken for laboratory staff to provide group and screen results was 58 minutes. Average time taken for laboratory staff to crossmatch compatible units for patients with a positive antibody screen was 7 hours and 13 minutes. Fifty-nine patients required subsequent ABO compatible red blood cell units to be issued after emergency O RhD negative red blood cell units were issued and included 23 patients that were escalated to massive haemorrhage pathway activation. The emergency department requested most emergency O RhD negative red blood cell units and reasons for the requests were varied, the most common being gastrointestinal bleeding and motor vehicle accidents.
Conclusion: In most cases, the request for emergency O RhD negative units in Rotorua Hospital between 01 January 2022 and 31 December 2023 was for patient treatment, as their conditions did not allow sufficient timing for standard testing to be completed for ABO compatible red blood cells to be issued.
Evaluating the transition to automated urine analysis: A comparative study of Sysmex UF-5000 and UD-10 against manual microscopy
Yirui Gong1 and Koen Van Der Werff2
1University of Otago, Dunedin and 2Awanui Labs, Wellington
Objectives: Urine analysis is vital for diagnosing urinary tract infections (UTIs) and monitoring patient health. This study evaluates the transition at Awanui Lab in Wellington to automated urine analysis systems, Sysmex UF-5000 and UD-10, comparing their performance with traditional manual microscopy.
Methods: A total of 64 random fresh urine samples were analysed using the UF-5000, UD-10, and manual microscopy. White blood cell counts were the primary focus as indicators of UTIs. Statistical analyses, including the Friedman test and Kendall's W test, were employed to evaluate systematic differences and reproducibility.
Results: The UF-5000 and UD-10 demonstrated sensitivity at 87.18% and specificity at 76.19%. Statistical tests revealed systematic differences between automated methods, particularly in white blood cell readings. Advantages included reduced turnaround times and minimized human error, although sample characteristics affected analysis accuracy.
Conclusion: Automated systems, UF-5000 and UD-10, are viable replacements for traditional urine analysis methods, promoting efficiency and accuracy. Future work should focus on addressing sample variability and enhancing parameter analysis for further optimisation.
Comparative evaluation of Theradiag ez-Tracker for therapeutic drug monitoring of Infliximab
Ambryme Guergoua1, Sian Horan2, Christian Christian2 and Jason Wong2
1University of Otago, Dunedin and 2Awanui Labs, Dunedin.
Objectives: Infliximab is a chimeric monoclonal antibody targeting TNF-α, revolutionizing treatment for chronic inflammatory diseases. The Theradiag ez-Tracker employs a fluorescence immunoassay to quantitatively measure infliximab levels. This project aims to validate the ez-Tracker assay for integration into the IANZ scope of practice at Dunedin Awanui Laboratory.
Methods: Twenty-nine Infliximab samples were tested on the ez-Tracker and compared with current Canterbury Health Laboratories results. Quality control material was run before each batch of samples. Due to limited availability of test cartridges, precision testing was run three times on one random sample. Pooled serum was spiked with known concentrations of Remicade infliximab for internal validation. Analyse-it for Microsoft Excel was used to run statistical analyses such as Passing Bablok regression and Bland-Altman plotting.
Results: Statistical analysis of the ELISA and ez-Tracker assays showed a mean difference of 0.793 ug/mL. The BlandAltman plot indicated a wide range of limits of agreement (95%) from -13.483 to 11.896, suggesting significant variability in the differences between the two methods. Passing Bablok regression produced a fit equation of y = 0.02242 + 0.8014 x, indicating a slight positive bias and a 19.86% negative proportional bias in the new assay. For our spiked dilutions, Passing Bablok regression produced a fit equation of y = 0.232 +0.8964 x with a negative proportional bias of 10.36%.
Conclusion: The Theradiag ez-Tracker immunoassay showed poor comparative results, making it unviable for integration. Further investigation into external validation as well as potential causes, is necessary.
Immunohistochemistry versus fluorescence in-situ hybridization for anaplastic lymphoma kinase fusion protein expression status determination in non-small cell lung cancer
Senumi Gunawickrama1 and Shannon McCroskery2 1University of Otago, Dunedin and 2Te Whatu Ora Waikato Laboratory, Hamilton
Objectives: An innovative genomics (IGENZ) based qualitative florescence in-situ hybridization (FISH) method is utilised by Waikato histology laboratory for anaplastic lymphoma kinase (ALK) fusion protein expression status determination in non-small cell lung cancer cases. This method is costly and time consuming as the specimens need to be transported and tested by a nonfunded organisation. The aim of this study was to determine if an in-house qualitative Immunohistochemistry (IHC) based method using the ready-to-use primary antibody Anaplastic Lymphoma Kinase 54A (1° antibody ALK 54A) provided by Leica Biosystems can be used for ALK fusion protein status detection in non-small cell lung cancer cases.
Methods: Twenty formalin-fixed paraffin-embedded lung tissue specimens ALK testing performed initially by IGENZ via the FISH method, were tested using the 1° antibody ALK 54A reagent on the IHC BOND-PRIME device.
Results: Ninety percent (18/20) of the samples tested using the two methods had qualitative agreement. Ten percent (2/20) of the samples had an uninformative FISH result, while IHC provided an accurate negative result for these samples.
Conclusion: 1° antibody ALK 54A based IHC method can be used for ALK fusion protein status determination and can be considered as a more reliable and specific method in comparison to the reference FISH method for ALK fusion status determination in non-small cell lung cancer cases.
Validation of the MICROSED-SYSTEM® analyser for erythrocyte sedimentation rate.
Olivia Hurnard1 and Miyuki Neufeld2
1University of Otago, Dunedin and 2Health NZ, Te Whatu Ora Waikato Laboratories, Thames
Objectives: Erythrocyte sedimentation rate (ESR) can be used as biomarker of inflammation in diseases like lymphoma, arthritis, or infections. The EliTechGroup MICROSED-SYSTEM® is an analyser that employs the modified Westergren method to provide automated ESR results. The objective of this study was to validate the MICROSED analyser for use at Thames Hospital Laboratory by evaluating the agreement with the MixRate ESR analyser and the modified manual Westergren methods.
Methods: Patient samples (101) were run on both analysers, 21 of which were also tested via the manual method. QC data from both analysers was included to establish inter-run precision, and five replicates of two patient samples were run on the MICROSED to determine intra-run precision at both low and high ESRs. Ten samples had an additional ESR run 24 hours after collection to confirm the sample stability. Comparisons were performed using Bland-Altman and linear regression statistical analyses.
Results: When comparing the MICROSED to the MixRate, a bias of 1.356 was seen with 1.98% of samples falling outside the limits of error, R2 was 0.99. When comparing the MICROSED to the manual method, a bias of -1.714 was seen with 4.76% of samples falling outside the limits of error, R2 was 0.99. The MICROSED uncertainty of measurement (UoM) was 46.57% (low QC) and 11.52% (high QC), while the MixRate revealed an UoM of 89.26% and 11.69% respectively.
Conclusion: Evaluation of the MICROSED analyser proved it as a valid method. Results were comparable to the MixRate analyser and the manual methods with overall improved precision.
Evaluation of Hgb-O method for determination of haemoglobin in lipaemic samples
Jan Janerol1, Catherine Cahill2 and Karen Elizabeth2
1University of Otago, Dunedin and 2Awanui Labs, Invercargill.
Objectives: Haemoglobin is a basic parameter routinely tested in haematology. This is tested via SLS-Haemoglobin on XN-10 in Awanui Invercargill. However, this method produces falsely high results in lipaemic samples, thus requiring plasma replacement to correct this error. Hgb-O is a proposed alternative, using the RET channel to reduce time and operator error. This research aims to evaluate Hgb-O measurements in comparison to plasma replacement in lipaemic samples.
Methods: Forty-three patient samples were collected consisting of seven normal samples that fell within the CBC reference intervals and thirty-six according to MCHC (>365) and or lipemia index (>50). All samples were tested via SLS-Haemoglobin after plasma replacement, and through the RET channel. Analyze-It Excel plug-in was then used for statistical analysis, generating Bland-Altman, Passing Bablok, and Student’s T test.
Results: Between both methods, limits of agreement (95%) in Bland-Altman plot were –9.9 -10.8, with a mean difference of +0.5g/L. Passing Bablok regression generated a strong positive correlation slope equation of y=0 + 1x demonstrating an ideal linear relationship. A p value of 0.5659 was generated by the student’s T test.
Conclusion: Evaluation shows that statistical results were not significant. However, they are clinically significant as they fall out of the Royal Pathologists of Australia’s analytical performance specifications allowable limits (Hb ±5 >100, Hb ±5% > 100). Further investigations towards potential causes of significance, and the method’s limits of quantification and detection should be carried out before implementing Hgb-O as an alternative method for lipaemic samples in Awanui Invercargill.
Retrospective analysis of the Sysmex XN-10 body fluid eosinophil counts
Mameaw Jungsakul1 and Kevin Fomiatti2
1University of Otago, Dunedin and 2Awanui Labs, Hutt Valley
Objectives: Assess the impact of changing the Sysmex XN10 body fluid eosinophil percentage (EO-BF%) reflex testing requirement for a manual WBC differential count from the current 2% to 6%.
Methods: Body fluid results were downloaded from the Sysmex XN-10 analyser with body fluid mode activated. All samples meeting inclusion criteria and with EO-BF% >2 were categorised into testing outcome at both the 2% and 6% levels and compared in terms of 1) reflex testing requirement/workload and 2) impact of identifying clinical eosinophilia of >10% if a 6% rule was adopted. The WDF scattergrams were examined for five discrepant samples where EO-BF% was between 2-6 and the manual counts were ≥10%.
Results: Of 909 samples, 328 had an EO-BF% >2. For 31 samples, reflex testing was required at 2% but not at 6%. Twenty-six of these had manual eosinophil counts <10% and therefore were not indicative of eosinophilia. Of the five samples with manual counts >=10%, three manual counts were probably incorrect due to the absence of an eosinophil cloud on the WDF scattergrams, and two had distinct eosinophil clouds requiring manual counts. Changing the EO-BF% reflex testing limit from 2 to 6 reduced reflex testing requirements by 66%, from 328 to 116.
Conclusion: The importance of staff manually reviewing all WDF scattergrams rather than simply accepting the XN counts is highlighted by the three apparent false positive clinical eosinophilia’s based on manual counts. Occasional clinical eosinophilia’s may be missed if the 6% reflex rule is applied. Adding a second rule requiring staff to check the WDF scattergram if the EO-BF% is between 2 and 6% would mitigate this risk.
Introducing magnesium testing in the Oamaru laboratory
Patrick Kane1 and Kay Jones2
1University of Otago, Dunedin and 2Awanui Lab s, Oamaru
Objectives: Magnesium is the fourth most abundant cation in the human body. Most of the body’s magnesium is found in bone. Less than 1% is found in serum where it is either free, bound to proteins, or complexed with anions. Magnesium is a cofactor in over 300 enzymic reactions and supports glucose utilisation, muscle contraction, and neurological function. It also plays a role in regulating heart rhythm, vascular tone thrombosis, and bone formation. The objective of this study was to investigate if magnesium testing could be performed successfully in the Oamaru laboratory.
Methods: Analysis was performed on the Roche Cobas c 501 analyser, which uses a colourimetric endpoint reaction to measure serum magnesium. In an alkaline solution, magnesium forms a purple complex with xylidyl blue. The decrease in xylidyl blue absorbance is measured photometrically to determine the magnesium concentration. Thirty-six Patient samples were run in both the Oamaru and Dunedin laboratories and the results were compared.
Results: It was found that our analyser tended to report the magnesium concentration lower on average compared to the Dunedin laboratory. The mean difference was -0.027mmol/L. The 95% confidence interval is -0.0342 to -0.0203mmol/L. This difference was not clinically significant as the interval sits entirely within the RCPAQAP limit of a 0.10mmol/L deviation. The largest deviation between the two analysers on one sample was 0.07mmol/L.
Conclusion: Although the analyser tended to run lower than that of the reference laboratory, magnesium testing can be performed successfully in the Oamaru laboratory and meet RCPAQAP performance requirements.
A method comparison between Sediplast Autozero and Microvette CB 200 for the analysis of erythrocyte sedimentation rate
Elizabeth Laven1, Sonette Kruger2 and Stephen Johnson2 1University of Otago, Dunedin and 2MedLab Central, Palmerston North
Objectives: Erythrocyte sedimentation rate (ESR) testing is a long-standing haematological test for non-specific assessment of inflammation. CRP testing has largely replaced ESR at MedLab Central Palmerston North, however valid testing indications remain. The project aimed to evaluate the performance of the Microvette CB 200 ESR test kit against the original Sediplast Autozero method which has been discontinued.
Methods: Forty-nine EDTA samples were evaluated using both testing methods. Selected samples were <12 hours old with >1mL blood volume. Results for both ESR methods were read at 1 hour and analysed using the laboratory’s allowable limits of performance for ESR testing. Bias was evaluated using BlandAltman plots. The sensitivity and specificity of the Microvette method compared to the Sediplast was calculated using the reference interval of <15mm/hr.
Results: Allowable limits of performance were not met in 12/49 samples. A Bland-Altman plot of the raw results indicated that as the mean of the paired results increased, so did the variance.
Log transformation of observations in the Bland-Altman plot removed heteroscedasticity, revealing that Microvette results are 0.3 to 3x greater than Sediplast results, 95% of the time. Compared to the performance of the Sediplast method, the sensitivity and specificity of the Microvette method was 0.90 and 0.80, respectively.
Conclusion: ESR testing is understood to have poor performance compared to more modern testing techniques. However, under the current result interpretation the preliminary results are sufficient to support use of the new method in the laboratory.
Development of an immunohistochemistry protocol for Oct-2 Used by VENTANA ULTRA IHC Stainer
Chongyuan Li1 and Mandy Fisher2
1University of Otago, Dunedin and 2Awanui Labs, Dunedin
Objectives: This project aims to work up an immunohistochemistry (IHC) protocol for Oct-2 identification, which will fit for an effective lymphoma screening panel, with high sensitivity, specificity, and reproducibility, using the VENTANA BenchMark ULTRA IHC stainer.
Methods: Based on the existing Oct-4 protocol, variables were optimised to create an available Oct-2 protocol. Formalin-fixed, paraffin-embedded tissue sections were prepared and loaded onto the VENTANA ULTRA. After automatic deparaffinization by heating the slide, antigen retrieval was optimised using Cell Conditioning 1 (CC1). The anti-Oct-2 primary antibody was received in set dilution, therefore, no optimisation on the concentration of the primary antibody. The DAB (3, 3'-diaminobenzidine) was used for chromogenic development following the detection of Oct-2. Counterstaining was performed using Haematoxylin. Tonsil tissue sections were utilised as positive controls, whereas testis tissue sections were negative controls. Three tissue sections from patients previously stained with Oct-2 by different laboratories were also used in comparison. Finally, the stained slides were analysed and compared through digital pathology.
Results: An available immunohistochemistry (IHC) protocol for Oct-2 detection was successfully developed and optimized for use on the BenchMark ULTRA. The final protocol showed robust performance, with optimal antigen retrieval using Cell Conditioning 1 (CC1) for 48 minutes, the antibody incubation time for 32 minutes, and applying amplification.
Conclusion: The Oct-2 immunohistochemistry protocol developed for the VENTANA BenchMark ULTRA is reliable and efficient for routine lymphoma screening panels in our lab.
Comparison of GeneXpert Carba-R and NG-Test CARBA-5 kits for carbapenemase detection
Elinor McAuley1 and Vicki Raeder2
1University of Otago, Dunedin and 2Te Nikau Hospital Laboratory, Greymouth
Objective: Carbapenemase producing organisms are on the rise worldwide, causing the need for accurate and prompt carbapenemase detection in clinical laboratories. This includes Te Nikau, a multidisciplinary lab where due to the range of expertise, as well as limited space and resources there is a need for carbapenemase detection to be quick and easy. This study was a comparison of the Cepheid GeneXpert Carba-R (XpertCarba-R) and Biotech NG-Test CARBA-5 (NG-CARBA-5) kits to determine which was better suited for Te Nikau.
Methods: Isolates from Canterbury Health Laboratories with known carbapenemases were used for analysis, including 4 IMP, 1 VIM, 4 NDM, 2 KPC, and 4 OXA-48 families, one with both an NDM and OXA-48. Cultures from nutrient-rich slants were streaked on blood-agar plates for single colonies and used to run the Xpert-Carba-R and NG-CARBA-5 kits. The XpertCarba-R methodology was real time-PCR, and the NG-CARBA-5 methodology was immunochromatographic. After sample preparation the Xpert-Carba-R runtime was 48 minutes, while NG-CARBA-5 took 15 minutes. Results were then analysed by their sensitivity, specificity, negative (NPV) and positive predictive values (PPV).
Results: The NG-CARBA-5 correctly detected all the carbapenemases from the 15 isolates. In comparison, the XpertCarba-R did not detect two IMP variants that resulted in two false negatives. The NG-CARBA-5 provided a higher sensitivity at 100% against 86% of the Xpert-Carba-R. While the NPV followed the same trend, it was most likely underestimated by the limited true negative samples.
Conclusion: The comparison between the Xpert-Carba-R and NG-CARBA-5 kit revealed the NG-CARBA-5 was better suited for Te Nikau. It was quicker and had fewer steps while demonstrating a higher sensitivity.
Changes in the titre of passive anti-D over time following administration of RhD immunoglobulin-VF in pregnant women
Ella Mycock1 and Dan Gyles2
1University of Otago, Dunedin and 2New Zealand Blood Service, Wellington
Objectives: Haemolytic disease of the fetus and newborn (HDFN) is a complication of fetal-maternal blood group incompatible pregnancies. Maternal antibodies to blood group antigens can cross the placenta and destroy fetal red cells. RhD incompatibility is the cardinal cause of HDFN. The introduction of RhD immunoglobulin-VF to RhD-negative mothers has exceptionally reduced isoimmunization and consequently the incidence of HDFN. This study aimed to investigate how the titre of passive anti-D changes over time in RhD-negative pregnant women.
Methods: The antibody titration procedure involved preparing master dilutions of the participant’s plasma by performing a two-fold serial dilution up to a titre of 32. 50uL of 0.8% group O R1R1 reagent red cells and 25uL of each master dilution for each participant were pipetted into the columns of Grifols DG Gel Coombs cards. The Grifols DG Gel Coombs cards were incubated and centrifuged, then the reaction strength was graded using the 0-4+ scale.
Results: There was no relationship between the titre of passive anti-D and the duration of time since administration of the RhD immunoglobulin-VF dose.
Conclusion: The lack of association between the titre of passive anti-D from RhD immunoglobulin-VF and duration since the last dose in this cohort suggests that other factors likely influence the titre of passive anti-D acquired in the 3 months following administration. These factors include silent fetal-maternal haemorrhage or differing rates of clearance of IgG antibodies. Further research with more participants is needed to confidently report the trend between passive anti-D titre and time.
Evaluation of interfering factors in plasma methotrexate (MTX) assay after high dose methotrexate administration
Garima Panta1 and Campbell Heron2 1University of Otago, Dunedin and 2LabPLUS, Auckland
Objectives: This research aimed to evaluate an interference observed in a patient’s methotrexate (MTX) levels. The plasma MTX levels did not become undetectable (> 0.04 µmol/L) as expected by day 4 and unexpectedly increased from day 5. The patient’s symptoms did not correlate with the laboratory findings, and her urea and creatinine levels were normal, suggesting interference in the ARK MTX assay used. To confirm this, QTOF was performed, supporting the interference hypothesis. Identifying the interferent would allow the laboratory to inform clinicians about potential assay interference.
Methods: Lithium-heparin samples were reanalysed to determine MTX plasma concentration. Heterophile blocking tubes were used to test for heterophile antibodies, and IgM assay was performed to check for precipitation. MTX assay involves the reduction of NAD to NADH, assays involving similar reduction steps, including valproate, tobramycin, ethanol, and Creatine Kinase (CK) were analysed. Urea assay, involving an oxidation step, was analysed to determine if the interferent influenced the reaction towards NADH. Samples from days 5 to 12 were analysed for ethanol, urea and CK; the day 8 sample for IgM; and day 9 and 11 for heterophile antibodies, valproate and tobramycin using the Cobas 502 and 702 analysers.
Results: The concentration difference exceeded the MTX test’s imprecision following heterophile antibody testing; however, MTX remained above the quantitation limit. IgM concentration was within the reference range. Valproate, tobramycin, and ethanol concentrations were undetectable. As MTX concentration rose, CK concentration trended up, whereas urea concentration trended down.
Conclusion: The exact interferent in the ARK MTX assay was not identified, but the research ruled out several potential causes. A relationship between CK, urea, and MTX was observed. This report provides a foundation for further studies.
Method evaluation of the HemosIL AcuStar ADAMTS13 activity assay for rapid diagnosis of thrombotic thrombocytopenic purpura
Divya Patel1, Nick Heffernan2, Renee Tietjens2 and Rebecca O’Toole2
1 University of Otago, Dunedin and 2 Awanui Labs, Wellington
Objectives: Thrombotic thrombocytopenic purpura (TTP) is a rapidly fatal microangiopathic haemolytic anaemia that leads to a prothrombotic state due to reduced activity of the vonWillebrand factor-cleaving protease ADAMTS13. The formation of microthrombi can cause end organ damage and lead to a diverse range of clinical sequelae. The first line treatment for patients suspected to have TTP is pre-emptive plasma exchange, an invasive procedure which carries a substantial financial burden for the healthcare system. The clinical overlap between TTP and other conditions makes diagnosis challenging. The availability of the ADAMTS13 test adds further complications, being only available at two referral laboratories in New Zealand (NZ). The aim of this project was to evaluate the analytical accuracy of the fully automated, chemiluminescent HemosIL AcuStar ADAMTS13 activity (Hemosil). Having this test available on demand at Awanui Labs, Wellington would have a significant health-economic impact by eliminating the need for prolonged and potentially unnecessary plasma exchange.
Methods: This study involved 13 paired samples from nine patients suspected to have TTP. This included two lyophilised patient samples obtained from an External QA provider. ADAMTS13 levels were measured on the HemosIL assay and compared retrospectively to results obtained using the ‘goldstandard’ FRETS-VWF73 assay. Agreement between methods was assessed using Bland-Altman plots and Passing-Bablok regression. An ADAMTS of <10%, the threshold diagnostic of TTP, was used for sensitivity and specificity calculations.
Results: Overall, the HemosIL assay gave partial agreement to the FRETS-VWF73 assay. The Pearson correlation coefficient value was r =0.951, Bland-Altman analysis found a mean difference of -4.148 (95% limits of agreement, -27.869 to 19.573). No false negatives or false positives were recorded (sensitivity 100% and specificity 100%).
Conclusions: The HemosIL assay produced generally concordant results when compared with the FRETS-VWF73 assay indicating its potential to rapidly aid TTP diagnosis. However, a larger sample size with a broader range of patient conditions should be tested in a validation study to support these initial findings.
Note: This abstract replaces the previous version of the abstract printed in NZ J Med Lab Sci 2024; 78(3):150-160, by mistake and due to an author communication error.
Syphilis non-treponemal testing: Evaluation of the automated Mediace RPR against the manual Arlington Scientific RPR
Divya Patel1 and Paul Tustin2
1University of Otago, Dunedin and 2Awanui Labs, Wellington
Objectives: Syphilis is a notifiable sexually transmitted infection caused by Treponema pallidum. Diagnosis is based on clinical indications, initial treponemal testing, and subsequent nontreponemal testing for assessing active infection status. The study aimed to evaluate the analytical accuracy of the fully automated Mediace RPR assay in comparison to the manual Arlington Scientific RPR assay to reduce workload and increase throughput. The correlation between the quantitative values reported using the Mediace RPR and the titre reporting system used with the Arlington Scientific RPR was assessed alongside the ability of the Mediace RPR to recognise notifiable infections, seroreversion, and early infections.
Method: patient samples (8999) were screened for syphilis using treponemal testing (Roche EIA). The 619 samples (6.9%) derived from 396 patients required confirmatory treponemal (Mediace TPLA) and non-treponemal testing. The Mediace RPR was performed on the Roche Cobas c502 analyser in the Biochemistry Department, and the results were compared
to those performed by the Immunology Department using the Arlington Scientific RPR.
Results: Overall, the Mediace RPR gave comparable results to the Arlington Scientific RPR assay with a categorical agreement of 93.5%. The specificity was 98.1%, the negative predictive value was 92.9%, the positive predictive value was 95.3%, but the sensitivity was 83.5%. The Mediace RPR demonstrated poor sensitivity at low titres, a faster decline in patients with a history of syphilis, and a slower rise in titre when detecting early infections.
Conclusion: The quantitative values reported with the Mediace RPR did not clearly correlate to the titre reporting system used traditionally making assessment of reinfection and therapeutic monitoring difficult. Poor sensitivity at low titres may limit clinical use as there is a potential to miss active infections.
A PIEZO the puzzle: a preliminary investigation into PIEZO1 variants and clinical features of hereditary xerocytosis in a cohort from New Zealand
Kimberly J. Pinto1, Ian M. Morison1,2, Jacqueline L. Ludgate1 and Catherine L. Ronayne1
1University of Otago, Dunedin and 2Awanui Labs, Dunedin
Objectives: Ever enigmatic, hereditary xerocytosis (xerocytosis) is a disorder of dehydrated erythrocytes. It is largely caused by gain-of-function variants in the mechanosensitive ion channel PIEZO1. Identifying these is considered diagnostic. Despite notable ramifications for obstetric and diabetic monitoring, xerocytosis is not currently diagnosed in New Zealand. Recent evidence suggests it may be more common than previously thought, imploring diagnostic pathways be implemented. This study aimed to sequence PIEZO1 in adults from Otago and Southland with suspected hereditary xerocytosis, and secondarily to analyse clinical features of the cohort to clarify indicators of xerocytosis.
Methods: Eight participants with suspected xerocytosis were recruited. RNA was extracted from leukocytes and reverse transcribed. HotStart PCR was used to amplify PIEZO1 variant hotspots from single-stranded cDNA. Amplification products were sequenced by Sanger using both forward and reverse primers, and sequence mismatches identified by creating alignments in Benchling. To analyse clinical features, a review of participants’ medical histories was conducted with informed consent and peripheral blood films prepared.
Results: 24 PIEZO1 sequence variations (11 unique) were identified across the cohort. Of these, three were missense variants previously described in xerocytosis, leading to four probable diagnoses of xerocytosis. Predominant traits in these participants were elevated mean cell haemoglobin concentration, reticulocytosis and low HbA1c.
Conclusion: This study provides the first genetic confirmation of hereditary xerocytosis in New Zealand and highlights the polymorphic nature of PIEZO1. Further study is required to optimise sequencing and construct appropriate selection criteria for diagnostic PIEZO1 testing. However, this research has provided a critical first ‘PIEZO’ the puzzle that is hereditary xerocytosis.
Small cell neuroendocrine carcinoma of the vagina: a rare case study
Karla Rades1, Liz Pringle2 and Tamaki Inoue2
1University of Otago, Dunedin and 2Lab-PLUS, Auckland
Objectives: This Case Study followed a 65-year-old woman presenting with the symptoms, vaginal pain and abnormal discharge. The patient was also previously under screened. As she was under screened and had suspicious symptoms, the patient immediately received cytology and HPV co-testing with a cervical sample.
Methods: Methods used include HPV testing, cytology testing, vaginal biopsy, histology, immunohistochemistry and medical imaging via PET/CT scan.
Results: HPV Testing revealed Human-Papilloma-Virus-Type 18, via COBAS 6800, in a ThinPrep PreservCyt Solution,
therefore the patient received colposcopy recommendation. Cytology testing small cells ~3x size of mature lymphocyte in singles, chains, loose groups with scant delicate cytoplasm. Because of the high mitotic rate (102/2mm2), marked necrosis, apoptosis and diathesis, marked hyperchromatic nuclei, “saltand-pepper” coarse chromatin, and nuclear molding, the cytology was suspicious of malignancy, specifically small cell carcinoma. A vaginal biopsy, revealed multiple small (10-1) fragments of a pearly white mass in the vagina, showed, via histology; nested, trabecular, sheet-like growth patterns bellow the attenuated squamous mucosa. Immunohistochemistry of Synaptophysin and INSM1 and p16 were positive, confirming small cell carcinoma from an HPV infected site. P40, CK5/6, LCA, CDX2, were all negative, negating squamous a gastrointestinal origin. Medical Imaging via PET/CT scan showed no abnormalities in the abdomen or chest, apart from iliac and inguinal lymphadenopathy, confirming the vagina as the primary tumour site.
Conclusion: The results found the patient was positive for a small cell neuroendocrine carcinoma of the vagina and was positive with HPV-18. These findings have an extremely poor prognosis, and thought could be given to the importance of regular screening and vaccination.
The effect of centrifugation time on the platelet-poor plasma of citrate samples
Tegan Rutherford1 and Josef Fourie2
1University of Otago, Dunedin and 2Whangarei Hospital Automation Laboratory
Objectives: The objective of this investigation was to assess the ability of different centrifugation times to produce platelet-poor plasma in citrated blood samples used for coagulation testing. The Whangarei laboratory considers platelet-poor plasma to have a platelet count of less than 10 x109/L, and the citrate samples are centrifuged for five minutes using the Cobas p471 automated centrifuge.
Methods: The centrifugation times that were used were five, ten, and fifteen minutes. Twenty samples were centrifuged for each of these times. The five-minute samples were centrifuged at 3500rpm, and the ten- and fifteen-minute samples were centrifuged at 2000g. Approximately 0.5 mL of plasma was then removed from each sample post-centrifugation and pipetted into a 1.5mL microcentrifuge tube, after which the platelet count was manually tested using the Sysmex XN-10 analyser.
Results: All the samples that were centrifuged for ten and fifteen minutes produced platelet-poor plasma. However, only 40% of the samples centrifuged for five minutes produced platelet-poor plasma. The mean platelet counts of the plasma from five, ten, and fifteen minute centrifugation times were 9.9, 2.4, and 1.5 x109/L respectively, and the ranges were 0-36 x109/L, 1-7 x109/L, and 0-7 x109/L, respectively.
Conclusion: With the method used in this investigation, the ten and fifteen minute centrifugation times reliably produced plateletpoor plasma. The five-minute centrifugation time, however, was not always reliable in producing platelet-poor plasma. Although this does not indicate the laboratory’s automated centrifugation method is unsatisfactory.
Method comparison of reticulin stains: Titration vs. volumetric silver solutions
Amelia Scott1 and Rowena Hunter2
1University of Otago, Dunedin and 2Awanui Labs, Christchurch
Objectives: The current reticulin stain at Awanui Labs, Christchurch uses a titration method to prepare the silver solution. The method involves mixing ammonia drop by drop until the precipitate just disappears. A new method, introduced following a Histopathology conference, aims to optimise the staining process using a volumetric silver solution. This study evaluated the efficacy, efficiency and reproducibility of the new volumetric method for routine reticulin stain use.
Methods: Formalin fixed, paraffin embedded liver biopsies (n=3) were selected for comparative staining using the titration and volumetric methods. Sections were cut at standard 4μm
onto adhesive slides, and two rounds of 5μm sections as per the volumetric method instructions. H&E slides and the titration method were performed once, the volumetric method three times (twice with 5μm sections and once with 4μm sections).
Results: Staining quality was assessed on a 1-4 scale of 1=poor (25%), 2=acceptable (50%), 3=good (75%) and 4=excellent (100%). After refining silver impregnation and reduction steps, the volumetric method at 5μm averaged 79.2%, 4μm averaged 54.2%. The titration method at 4μm averaged 75%, compared to all volumetric method stains which averaged 70.8%. The volumetric method was easier to formulate and reduced preparation time from around 45 minutes to 30 minutes. Conclusion: Initial findings suggest that the volumetric method is a viable alternative. Further standardisation of the volumetric process, particularly from silver impregnation through formalin reduction, is recommended to ensure consistent staining quality across different laboratory workers. Further evaluation of the stains needs to be completed and a pathologist to verify the quality.
Method comparison: Advanced Instruments Osmo1 MicroOsmometer vs. Advanced Instruments Model 3250 SingleSample Osmometer
Abbie Sommerville1 and Maho Nakajima2
1University of Otago and 2 Awanui Labs, Wellington
Objectives: At Awanui Labs Wellington, the Advanced Instruments Osmo1 Micro-Osmometer has been introduced as a replacement for the discontinued Advanced Instruments Model3250 Single-Sample Osmometer. This study aimed to verify the linearity, accuracy, and precision of the Advanced Instruments Osmo1 Micro-Osmometer and determine if it exhibits a significant performance difference compared to the Advanced Instruments Model3250 Single-Sample Osmometer. Methods: Linearity testing involved eight levels of reference material, with three replicate measurements at each level. Precision testing used three levels of quality control material. For inter-run precision each of these were measured once a day for 20 days, while intra-run precision was determined by performing ten consecutive measurements in one day. Accuracy testing required ten levels of quality control material. To compare the performance of the two analysers, 36 biological samples (19 serum and 17 urine) were tested once on each analyser. Statistical analysis of this data was carried out using GraphPad Prism 10, and Analyse-it software.
Results: A proportional relationship between the response of the Advanced Instruments Osmo1 Micro-Osmometer and the osmolality of reference material was established across the measuring range (r = 1.0, p < 0.0001). The Advanced Instruments Osmo1 Micro-Osmometer met the acceptance criteria for precision, with coefficients of variation below 1%, and for accuracy with a correlation coefficient of r = 0.999 (p < 0.0001). There were no significant biases between the two analysers based on regression analysis.
Conclusion: The Advanced Instruments Osmo1 MicroOsmometer met acceptance criteria for linearity, precision, and accuracy. There was no significant performance difference compared to the Advanced Instruments Model3250 SingleSample Osmometer.
Investigation of the use of the Siemen’s INNOVANCE D-Dimer assay past the recommended onboard stability time
Imogen Taylor1 and Shona Brougham2 1University of Otago, Dunedin and 2Awanui Labs, Nelson
Objectives: Reduction of cost and waste is a major goal in every clinical medical laboratory. This study aimed to evaluate the accuracy of the Siemen’s INNOVANCE D-dimer assay kit as a negative predictor of venous thromboembolism after the recommended onboard stability of 120 hours. An extension of the onboard stability of this kit would reduce waste and reduce costs.
Methods: Sensitivity of the INNOVANCE D-Dimer assay was tested by repeated testing of 50 patient samples. Samples were analysed on the Sysmex CS-2500 coagulation analyser one hour after the reagent stability had expired, 25 hours after the reagent stability had expired and 49 hours after the reagent stability had expired. Samples were frozen after the initial testing had been run and results released. Samples were then thawed 15 minutes prior to testing, retested and refrozen. This process was repeated on three consecutive days.
Results: Overall accuracy after the 49-hour extension was 95.9%. Three of the samples included in the study were initially identified as positive but dropped below cutoff values and became negative after the reagent stability expiry. This may be due to sample degradation due to the change only occurring in samples with original results slightly above the cutoff values.
Conclusion: The INNOVANCE D-Dimer assay maintained 100% accuracy as a negative predictor of venous thromboembolism for 49 hours after the recommended onboard stability period.
Evaluation of the TriniCHROMTM Factor VIII:C kit assay for Factor VIII testing
Alan Uthup1, Brigid Carroll2 and Grace Lim2
1University of Otago, Dunedin and 2Hawkes Bay District Health Board Laboratory, Hastings
Objectives: Factor VIII (FVIII) testing can be performed by the one-stage clotting assay (OSA) and the chromogenic substrate assay (CSA). Although aPTT-based OSAs are relatively inexpensive and simple to-perform, CSAs such as the TriniCHROMTM Factor VIII:C show less variability by measuring FVIII cofactor activity (FVIII:C). This study aimed to compare the TriniCHROMTM assay against the OSA and evaluate the practicality of CSAs for routine FVIII testing.
Methods: Data from 20 citrated plasma samples (with normal INR and aPTT results) were collected from both HBDHB laboratory staff and patients not on anticoagulants. The laboratory’s Stago Compact Max® analyser tested the FVIII concentrate (IU/dL) in all samples by the OSA and CSA. Comparisons were made between samples with normal and high FVIII and C-reactive protein (CRP) levels.
Results: The FVIII difference between assays was statistically significant in samples with normal FVIII or CRP, with P-values <5%. However, 82-85% of these samples lacked clinical significance as their FVIII differences were within the acceptable variation limits of 0-30 IU/dL, while only 60-71% of samples with high FVIII or high CRP levels were clinically insignificant. Deming regression plots assessed the correlation (with R2 values between 0.569 and 0.915 for each comparison group), while Bland-Altman plots determined the bias and agreement between assays.
Conclusion: The higher FVIII levels detected by the CSA are clinically less significant for samples with normal FVIII or CRP. However, further testing is required to confirm the clinical significance of the FVIII difference between assays for samples with high FVIII and CRP. The expensive and technically complex nature of the TriniCHROMTM assay makes it impractical to implement for routine FVIII testing.
Optimisation, validation, and method comparison of LCMS against Liaison immunoassay for plasma aldosterone quantification
Ziqi Wang1, Ian Philips2, Josh Penese2 and Anne ZychlinskiKleffmann2
1University of Otago, Dunedin and 2Awanui Labs, Dunedin
Objectives: Diagnosing primary aldosteronism (PA) requires accurate quantification of plasma aldosterone. Due to the limitations associated with immunoassay-based diagnosis of PA, many laboratories are transitioning to liquid chromatographmass spectrometry (LCMS) methods. This study aimed to optimise the solid phase extraction (SPE) component of the LCMS-SPE method previously developed by Awanui Dunedin, then validate and compare it to the current Liaison immunoassay used at Awanui Dunedin.
Methods: SPE was optimized by comparing aldosterone recovery from a plasma pool, using modifications of the LCMSSPE and literature methods. The LCMS-SPE method was updated with the optimised SPE and validated for matrix effect, recovery, efficiency, and inter- and intraday precision using a plasma pool, with inter-day precision assessed using quality controls. Method comparison was conducted with 25 patient plasma samples using statistical analysis including the Wilcoxon signed-rank test, Passing-Bablok regression, and Bland-Altman plot for LCMS-SPE and Liaison results.
Results: The optimised LCMS-SPE method was identified, showing a matrix effect of -40.26%, recovery of -33.78%, efficiency of -56.87%, and precision with intraday of 5.37% and inter-day of 5.68%-6.92%. LCMS results demonstrated a Pearson correlation coefficient of r=0.999 and were significantly lower than Liaison results (p=0.007), with a mean difference, systematic and proportional bias of -48.60pM, -35.70%, and -14.82% respectively.
Conclusion: The optimised LCMS-SPE method demonstrated favourable matrix effect, recovery, efficiency, precision, and high correlation with the Liaison immunoassay. However, it reports lower aldosterone concentrations that are statistically and clinically significant. Further studies should expand on this method validation and increase sample size to confirm these findings.
Homogenization of sputum with mucolytic agent: Sputagest Selectavial
Anna Welch1 and Neil Campbell2
1University of Otago, Dunedin and 2Hawke’s Bay District Health Board Laboratory, Hastings
Objectives: Gold standard sampling procedures for investigation of lung infections are highly reliable for definitive diagnosis but require invasive methods that place heavy burdens upon the patient and hospital. To avoid this, routine laboratory diagnosis instead typically uses expectorated sputum, a non-invasive method. Its accuracy is often contested due to contamination by oropharyngeal microflora, uneven organism distribution and difficulty handling the specimens. To address these issues, chemical agents such as Sputagest Selectavial, which reduce disulphide bonds in mucus, have been developed to liquefy samples and aid in distribution and isolation of predominant organisms. This study evaluated pathogen recovery and ease of handling when using Sputagest Selectavial treated expectorated sputum compared to the standard laboratory procedure.
Method: Fifty-two sputum samples with previously reported pathogens were diluted 1:1 with a 1% Sputagest solution, then incubated, homogenised, inoculated onto standard media and grown for 48 hours. Pathogens were identified and quantified by laboratory standards and compared to reported pathogens from non-homogenized cultures.
Results: Homogenisation with Sputagest Selectavial resulted in a 94% recovery rate, with 32% of pathogens exhibiting increased growth. However, for Haemophilus influenzae samples, 24% of samples had reduced or no growth. Gram stains were generally easier to interpret compared to non-homogenised samples, though distinguishing small Gram-negative organisms from lysed mucus strands became challenging.
Conclusion: Treating expectorated sputum samples with Sputagest Selectavial improved the recovery of some pathogens, eased handling and simplified bacterial identification. However, it led to reduced growth of major pathogens and therefore will not be implemented into routine laboratory procedures.
Evaluating the stability of Quidel TriageTrue high sensitivity troponin I controls after shipment to a rural New Zealand hospital
Theresa Winckler, Rory Miller and Garry Nixon University of Otago, New Zealand
Objectives: The aim of the study was to assess whether shipment of the temperature sensitive Quidel TriageTrue High Sensitivity Troponin I control samples to a rural hospital site in
NZ affected their integrity.
Methods: Two shipments of quality control materials were analysed across three runs, a total of 15 level 1 (low) and 5 level 2 (high) controls. Samples were compared to expected values, manufacturer recommendations, and Royal College of Pathologists of Australia (RCPA) performance specifications. The percentage differences from each run were also compared to the last three quality control results from the Abbot i-STAT troponin I (i-STAT).
Results: The first run produced a mean of 21.7 ng/L (SD: 1.6 ng/L) compared to the expected 29 ng/L, with a CV of 7.2%; 20% of samples met RCPA guidelines, while 80% met manufacturer recommendations. In the second run, the mean was 431.8 ng/L (SD: 25.7 ng/L) versus the expected 583 ng/L, with a CV of 5.9%; none met RCPA guidelines, but 80% met manufacturer recommendations. The third run yielded a mean of 19.9 ng/L (CV: 7.2%) compared to 29 ng/L, with 0% meeting RCPA and 50% meeting manufacturer guidelines. The mean percentage difference from expected values was 13.5% for the I-STAT and 31.9% for the Quidel Triage MeterPro.
Conclusion: The findings raise concerns about the stability of Quidel TriageTrue High Sensitivity Troponin I control samples under the current shipping conditions, with observed values consistently lower than expected across all runs, with few samples meeting RCPA standards. In contrast, the i-STAT controls were consistently within RCPA limits. However, due to the small sample size, these results should be interpreted cautiously.
Comparison and validation of haemoglobin concentration measurements: assessing the transition from Varian Cary 50 to Cary 60 spectrophotometers
Ruoling Xiao1 and Ramesh Tiwari2
1University of Otago, Dunedin and 2Cantebury Health Laboratories, Christchurch
Objectives: To validate the accuracy and compare the results of haemoglobin concentration measurements from the old Varian Cary 50 Conc UV-visible spectrophotometer to the new Cary 60 UV/Vis spectrophotometer. Specifically, haemolysate absorbance was measured using both the old and new spectrophotometers, and the results were converted to Hb concentrations using the same standard curve. This study aims to validate the feasibility of replacing the old spectrophotometer with the new one.
Methods: Sixty blood EDTA samples were collected and stored for up to a week. Random samples, as well as those requested for thalassaemia screening, were included.
The Specialist Haematology Haemoglobinopathy Manual was followed for the preparation of 100 g/L haemolysate using the cyanmethaemoglobin method. However, instead of collecting as much haemolysate as possible for further procedures as suggested by the manual, only 200µL of haemolysate was collected from each sample for the purpose of comparing methods in this assay.
Results: The results suggest that there was a relatively strong positive linear correlation between the two methods, with a correlation coefficient of 0.934 and an r² value of 0.907, representing strong correlation between the measurements from the two methods. Ninety-five percent of the differences (3 out of 60) between the two populations are expected to lie within two standard deviations, ranging from 16.04 to -5.92 g/L, with a mean difference of 5.06 g/L.
Conclusion: Statistically, the measurements from the two methods demonstrated good correlation and agreement, providing evidence that replacing the old spectrophotometer with the new one is feasible. Further studies and possible adjustments may be needed to improve the consistency and accuracy of the results.
Assessment of the Elecsys PTH assay updates for the Roche Cobas e801 analyser
Xintong Zhang1, Dave Coles2 and Dale Coburn2
1University of Otago, Dunedin and 2Medlab Central, Palmerston North
Objectives In April 2024, Roche informed Medlab Central of the launch of an updated Elecsys parathyroid hormone (PTH) assay, which has increased tolerance against biotin compared to the current PTH assay. The aim of this project was to assess whether the new assay could replace the current assay without inducing a change in the measurement of PTH concentration.
Methods Thirty-eight retrospectively collected patient samples were firstly tested using the current PTH assay on Cobas Pro e801 analyser A and then tested using the new PTH assay on Cobas Pro e801 analyser C immediately afterwards. Two sets of results were compared and analysed using Passing-Bablok regression and Bland-Altman plot to assess the degree of agreement between two assays, both performed using MedCalc statistical software. Additionally, two levels of Roche Quality Control (QC) material and three levels of Bio-Rad QC material were run daily over 2-6 weeks to assess between-run precision of two assays.
Results The Passing-Bablok regression equation had an intercept of -0.261, with a 95% Confidence Interval (CI) of -0.457 to -0.102, and a slope of 1.078 (95% CI 1.053 to 1.110). The Bland-Altman plot identified a mean ratio estimate of 1.04 (95% CI 1.02 to 1.06) and limits of agreement of 0.95 to 1.14. The two assays have a similar level of precision, but the differences in running means are significant.
Conclusions Analysis revealed that the new PTH assay has significant positive bias compared to the current PTH assay. The trend was seen in both patient samples and QC samples. With pathologist review of the comparison study results, the reference range of PTH changed from 1.60-7.00 pmol/L to 1.8-7.9 pmol/L.
NZIMLS Annual Scientific Meeting Social Functions
Jim le Grice Welcome Function, Hugh Bloore Poster Session & Exhibition Opening
Join us on Wednesday, 27 August from 5pm - 7.30pm at the Distinction Hotel Hamilton for the Jim le Grice Welcome Function, Hugh Bloore Poster Session & Official Exhibition Opening. The Best Exhibit will also be announced during this session. Drinks & Canapes provided.
NZIMLS ASM 2025 Gala Dinner
Join us on Thursday, 28 August from 6.30pm - 11.30pm at the Distinction Hotel Hamilton for the Gala Dinner. The theme for this dinner is Garden Party.
Dress code: Garden Party Chic. Time to bloom!
Entertainment provided by Black & Gold. They are guaranteed to get you on your feet!
Dinner, drinks and entertainment provided. Tickets required - $145 pp (incl GST).
Biodiversity in the bathroom
SCIENCE DIGEST
Contributed by Michael Legge
There is a general tendency to associate biodiversity with conservation around the world. However, in a publication late last year from the USA, the authors reported an extensive phage biodiversity identified in toothbrushes and shower heads (1). Their research initially started as a project to try and identify what bacteria existed on surfaces such as tables, walls, shower heads, toothbrushes etc. However, they turned their attention to focusing on the formation of biofilms on shower heads and toothbrushes and the presence of viruses. They worked on an established knowledge that viruses were detectable in the bacterial biofilms and that bacteriophages infect bacteria with a high host specificity. Using a metagenomics approach they analysed 34 toothbrushes and 92 shower head metagenomes. Their results identified 22 complete, 232 high quality and 362 medium quality viral Operational Taxonomic Units (OTU). They identified that of the viral OUT quality 532 were connected with 32 bacterial families of which only Sphingmonadaceae, Burkholderiaceae and Caulobactereraceaae were found on both the shower heads and the toothbrushes otherwise there were diverse ’virus islands’ on both. Overall, the toothbrushes demonstrated genera such as Klebsiella, Streptococcus, and Veillonella and the shower heads demonstrated Mycolicibacterium and Mycobacteriodes as well as Sphingopyxis, Sphingobium and Aquabacterium which the authors considered would be normally found in drinking water. The authors conclude that the viruses they detected that were associated with bacteria did not raise concern for human health e.g. antibiotic resistance or virulence and proposed further work on bacterial-viral interactions on other areas of the built environment. They also indicated that simply washing the shower head in hot soapy water and regularly changing toothbrush heads would be better than using bleach.
Differentiating neonatal late onset sepsis with pneumonia
Clinically, differentiating neonates with sepsis poses a significant threat and represents the third leading cause of neonatal mortality. However, differentiating between neonatal pneumonia and neonatal sepsis is challenging as many of the symptoms overlap. In this research from China the authors investigated whether there were suitable biomarkers to distinguish between the two clinical conditions using conventional and readily available biomarkers: CRP, platelets, ALT, AST, CRN, Urea, procalcitonin and WBC as well as clinical parameters (2). All the neonates included in this research met the criteria for neonatal pneumonia as assessed by independent clinicians. A total of 1385 neonates met the inclusion criteria. Of these, 174 had positive blood cultures and assigned to the sepsis cohort the remaining neonates (1211) were assigned to the pneumonia cohort. The biochemical and white cell results were significantly higher in the neonatal sepsis group compared to the neonatal pneumonia group as was the CRP. In addition to the elevated CRP, procalcitonin was also significantly elevated which were strongly associated with neonatal sepsis. The authors also identified a highly significant correlation with neonatal sepsis and the CRP: platelet ratio and concluded that using either or both would significantly speed-up the diagnosis of neonatal sepsis being faster than blood culture results, although they did not recommend replacing the blood culture.
Genomic healthcare and ethical preparedness
Developments in genetic and genomic technologies will and are changing the delivery of healthcare and the diagnosis of diseases often earlier than previously. However, the integration of the new technologies creates problems over and above the standard considerations such as turnaround time and informed communication. In a publication from the United Kingdom (3), the authors explore the notion of preparedness for the new
technologies from an ethical perspective. The authors maintain that while the technical and infrastructure consideration are important, an ethical preparedness should run alongside these. Preparedness in the conventional sense is introduced and based on the COVID-19 pandemic and the unwanted upheaval of momentous social and work changes. They consider that healthcare innovation places new responsibilities on professionals working in health and the results transfer to the patient. Genomic medicine is changing rapidly and is being incorporated into routine health care. It does however, cross into both social and genetic relationships raising more complex issues. They quote a survey by the Wellcome Trust that indicated health practitioners’ “disquiet” over being prepared for routine genetic screening and testing. The authors consider that ethical preparedness inhabits the space between enthusiasts and pessimists, but to make it work, the ethical concerns and preparedness need to be identified and caution that an unprepared workforce would be the Achilles heel of health care professionals.
Frequent blood donors and clonal haematopoiesis
There is no doubt the blood donation and the use of blood products saves many lives each year and there many people who become regular blood donors. The frequency of individual blood donations is limited for both males and females to avoid any complications associated with blood donation. In a multinational research project, the issue of clonal haematopoiesis expansion was investigated (4). The authors indicate that each donation represents approximately 10% of the total blood volume. They considered that all red blood cells are replaced three times a year and that each donated unit of blood adds 2.5.to 4% excess haematopoiesis for that year. Taking this into consideration they hypothesised that blood donation may contribute to producing haematopoietic clones with functionally distinct mutations. In this research they screened 217 male older donors with >100 blood donations compared to 212 male sporadic donors (<10 donations), aged matched. Nextgeneration sequencing was performed on all donors as well as the measurement of erythropoietin as an indicator of bone marrow stimulus. The latter was found to be elevated for 120 days post-donation and was taken as stimulating bone marrow erythropoiesis. From the sequencing data the authors identified certain DNMT3A mutations that are involved in DNA methylation in the regular donors and correlated this with erythropoietin being a novel factor that promotes haematopoietic stem cells carrying the DNMT3A mutations. The significance of the mutations is not known at present but do not correlate with known diseasecausing mutations previously identified in DNMT3A.
Highly pathogenic H5N1 virus infections in humans
The H5N1 virus was first identified as causing illness in humans in 2024 in Hong Kong. To date 900 human cases in 24 countries have been reported with a cumulative fatality of 50%. In the USA, only one case of H5N1 was reported in a poultry worker who had mild symptoms. After 2024 there were reports of cow to human transmission. The research published this year investigates the transmission of H5N1 to humans from both cows and poultry in the USA (5). Data archived at the Centre for Disease Control (CDC)from identified infection using the A/H5 subtyping kit was analysed. A total of 46 cases were identified and 20 were poultry-related and 25 presumed to be from infected dairy cows. One case had no known animal contacts. All patients were hospitalised with mild symptoms and none died. No cases were detected in their respective households. It was concluded that to date, H5N1infections in humans appear to be mild and respond to anti-viral treatment. There was no evidence of human-tohuman transmission. The authors also comment on the poor use of PPE amongst the health care workers where only 71% wore
gloves, 60% eye protection and 47% face masks.
How good is GPT-4 for colorectal adenoma detection?
Colorectal adenomas are benign growths occurring in the inner lining of the colon or rectum. They are considered to be the precursor of colorectal cancer and early detection helps to prevent the progression to cancer and provide for better treatment outcomes. The gold standard for diagnosis is histopathological analysis. In recent research from Thailand (6), the authors investigated the use of GPT-4 for the image analysis of photomicrographs of previously identified colonic polyps at a x4 magnification. A total of 100 photomicrographs were used which included 50 adenomas (plus sub-categories), and 50 nonadenomas (plus sub-categories). The CPT-4 demonstrated a median sensitivity of 74% and specificity of 36% for adenoma detection. The median accuracy for polyp detection was 16% for non-specific changes and 36% for tubular adenomas. The authors conclude that the diagnostic consistency was low for GPT-4, however the diagnostic tool acknowledged its limitations and emphasised the need for a pathologist’s expertise.
Couple-based genetic carrier screening: a nationwide survey
Often couples who have a risk of children with a genetic condition are only aware of this once a child is born and their carrier status is discovered. Currently there are over 2500 genes linked to childhood onset autosomal recessive and X-linked conditions, and carrier status is only revealed with the birth of an affected child. To have reproductive carrier screening before a conception would help provide reliable information to inform reproductive decisions. This has been limited by the availability of any provider and the cost through commercial providers. In a recent publication (7) the results of an Australian Reproductive Genetic Carrier Screening Project called; Makenzie’s Mission are presented. The Australian government provided $A20 million for reproductive genetic screening. This enabled 10,000 couples contemplating a pregnancy or had a pregnancy at less than 10 weeks be screened for 1281 genes for more that 750 autosomal and X-linked conditions. Of the 10,038 reproductive couples enrolled in the study, 9107 completed screening and 175 were identified as at risk of having a child with a genetic condition which involved pathogenic variants in 90 different genes of which 74.3% were autosomal recessive. Within three months of receiving the results 76.6% of couples who received the results had used or planned to use reproductive interventions to avoid having an affected child. The authors conclude that couple based genetic carrier screening was largely acceptable and helped to make informed reproductive decisions.
What is the best time to treat bloodstream infections?
There is substantial morbidity and mortality resulting from bloodstream infections. Although early antibiotic intervention is essential, the duration of the treatment is uncertain. Currently there is an absence of evidence to guide clinical practice for treatment including the duration of antibiotics. Typically, a median duration of treatment is 14 days but there is little evidence that this duration is appropriate. In a multicentre clinical trial involving Canada, Australia and New Zealand, 74 hospitals and 3608 patients underwent randomisation and 1814 had 7-days of antibiotic treatment and 1794 had 14-days of antibiotic treatment (8). Infections were from a number of sources. Despite the source of the infection the authors reported that the treatment of 7-days antibiotics was non-inferior to that of 14-days of treatment of infections resulting from a wide range of bloodstream infections and conclude that more research is needed on shorter antibiotic treatment periods.
REFERENCES
1. Huttelmaier S, Shuai W, Sumner JT, Hartman EM. Phage communities in household-related biofilms correlate with bacterial hosts. Front Microbiomes 2024; doi:10.3389/ frmbi.2024.1396560. [Open Access]
2. Li X, Li T, Fu H, Lin F. et al. C-reactive protein to platelet ratio as an early marker in differentiating neonatal late-onset sepsis in neonates with pneumonia. Sci Rep 2025; 15(1): 10769. doi: 10.1038/s42598-025-94845-x. [Open Access]
3. Farsides B, Lucassen AM. Ethical preparedness and developments in genomic healthcare. J Med Ethics 2025; 51: 213-218. doi:10.1136/jme-2022-108528.
4. Karpova D, Encabo HH, Donato E, Calderazzo S. et al. Clonal hematopoiesis landscape in frequent blood donors. Blood 2025; doi:10.1182/blood.2024027999
5. Garg S, Reinhart K, Couture A, Knis K. et al. Highly pathogenic avian influenza A (H5N1) virus infections in humans. N Eng J Med 2025; 392(9): 843-854. doi:10.56/ NEJMoa2414610.
6. Laohawetwanit T, Namboolue C, Apornvirat S. Accuracy of GPT-4 in histological image detection and classification of colorectal adenomas. J Clin Pathol 2025; 78(3): 202-207. doi:1136/jcp-2023-209304.
7. Kirk EP, Delatycki MB, Tutty E, et al Nationwide, couplebased genetic carrier screening. N Eng J Med 2024; 391(20): 1877-1889. doi:10.1056/NEJMoa2314768.
8. The Balance Investigators (Multiple authorship). Antibiotic treatment for 7 versus 14 days in patients with blood stream infections. N Eng J Med 2025; 392: 1065-1078. doi: 10.1056/ NEJM0a2404991. NZIMLS Annual Scientific Meeting Member Registration Fees
The
Pacific Way
Warm Pacific greetings to you all from the PPTC
UPDATES
2025 New Zealand short term centre-based courses
Advanced Laboratory Quality Management - 7th April – 17th April 2025
This training course was delivered over a duration of 2 weeks at the Pacific Pathology Training Centre, based at the Wellington Hospital campus in Wellington, New Zealand to medical laboratory Quality Officers working in hospital laboratories within the South Pacific Region. Students who attended this course included:
• Eteuini Elika -Samoa (Apia)
• Tuimala Aleamotu -Tonga (Nuku’alofa)
• Nesia Rafaela -Fiji (Suva)
• Christopher Siga -Fiji (Labasa)
• Tauli Apearaamo -Samoa (Savai’i)
Course aims
The PPTC is committed to advancing and developing Pacific Island laboratories to be fully compliant to the ISO 15189 Medical Laboratory Standard guidelines and attain standards of practice consistent with International Accreditation status. The requirements of ISO15189 guidelines are a globally recognised mark of efficiency and quality in the scientific profession.
The advanced Laboratory Quality Management course is an opportunity for Quality Managers and senior laboratory personnel to discuss with PPTC Consultants their laboratory’s future plans and developments in light of recent quality management system influences, technology, directives, industry changes, etc.
This 2-week course aims to enhance and equip students with both skills and knowledge with reference to Laboratory Quality Management Systems and to formulate workplans focused towards ISO15189 accreditation requirements and operational management essentials.
A comprehensive theoretical component and a series of practical case studies are provided to students in the administration and management of medical laboratories. The purpose of this training is to also equip students with sufficient knowledge to be able to work confidently in their home laboratories and be able to provide and improve quality medical services.
Course content and objectives
This course provides students with the following:
• Leadership management skills.
• Training on the ISO15189:2022 standard and related IANZ /SLIPTA revised documents. Training includes impartiality, governance & structural requirements, response to complaints, corrective actions, non-conformances, risk management, management reviews and information management.
• Explore national plans and strategies to enhance and strengthen and build laboratory capacity and expectations for the laboratories to progress quality.
• Discuss quality improvement project activities in the countries, specifically long-term issues with procurement,
personnel management and CPD, internal process control and quality assurance monitoring, laboratory health and safety, information management and laboratory information systems
• Develop country specific quality improvement projects for addressing risk management, method, validation procedures, quality indicators, management review meetings, service feedback, equipment selection.
Medical Microbiology & Introduction to Molecular Diagnostics, 5th May – 30th May 2025
This training course was delivered over a duration of four weeks at the Pacific Pathology Training Centre, based at the Wellington Hospital campus in Wellington, New Zealand to medical laboratory personnel working in hospital laboratories within the South Pacific Region. Students who attended this course included:
• Folole Bartley -Samoa
• Silivelio Saumani -Tokelau
• Telehia Manuele -Tokelau
• Seleni Peleni -Tokelau
Course aims
A comprehensive theoretical component and a series of practical workshops are provided to students in the diagnostic medical field of Medical Microbiology including procedures and Molecular Testing Platforms.
The purpose of this training is to equip students with sufficient knowledge to be able to work confidently and competently in their home laboratories and be able to provide quality diagnostic test results to clinicians using the medical laboratory services for patient management and better health outcomes.
Course Content and Objectives
This course provides students with the following
• Understanding of major bacterial groups and identification characteristics of pathogens and commensal flora.
• Sample processing methods used in Microbiology
Figure 1 Students and Consultants: Advanced Laboratory Quality Management 2025
and molecular laboratories. This includes equipment, consumables and supplies used in for microscopy, culture and analysis.
• The theoretical and practical aspects of current methods used in the isolation and identification of bacterial pathogens isolated from various Microbiology specimens. PCR molecular testing theory and methodology will also be addressed.
• Emergence of automation and equipment used in Microbiology and molecular PCR laboratories including maintenance of instruments used, in-house media production for bacterial isolation, and choosing the right resources to carry out microbiological testing. Suppliers may also present on instruments currently available in the world market.
• Antimicrobial Susceptibility Testing of micro-organism pathogens using the CLSI and EUCAST guidelines.
• Monitoring of resistance patterns for multi drug resistant bacterial pathogens
• Discussions on emerging and re-emerging bacterial and viral organisms likely to cause infectious diseases.
• Identification and diagnosis of pathogenic parasites
• Health and Safety in a molecular and Microbiology laboratory, including disposing of infectious waste and rubbish.
• Reporting and presenting Microbiology results to clinicians and reporting requirements and guidelines for PCR testing.
• The role of the Microbiology laboratory in the surveillance of nosocomial infections and identification of infections of public health importance
• Serological and other rapid methods for the identification of bacterial and viral diseases including Hepatitis A, B, and C, HIV and other STIs.
• Standard Operating Procedures and written documents in Microbiology and molecular labs. Applying Laboratory Quality Management to diagnostic processes, including the management of Quality Controls, documentation, training records, stock management etc.
• Understanding UN/ IATA shipping requirements for infectious substances affecting humans and animals, biological substances category B.
Clinical Biochemistry, 23rd June- 18th July 2025 (4 weeks)
Foundations of Haematology, 4th August- 12th September 2025 (6 weeks)
Blood Transfusion, 22nd September – 17th October 2025 (4 weeks)
For course enrolment, course content and course objectives in relation to the above please contact: Emmanuel Marshall, Medical Laboratory Consultant and Education Manager, Pacific Pathology Training Centre (PPTC), WHO Collaborating Centre for External Quality Assessment in Health Laboratory Services.
E-mail: emmanuel.marshall@pptc.org.nz
Post: PPTC, PO Box 7013 Wellington, New Zealand Telephone: +64 4 389 6294, Mobile: +64 0272985326, E-mail: pptc@pptc.org.nz
OVERSEAS TRAVEL
The PPTC plays a vital role in strengthening the capacity of medical laboratories throughout the Pacific region through medical laboratory education, practical technical training and quality improvement initiatives. Its contribution to education is extensive, a significant portion of which is carried out in- country within the home laboratory. Each year, PPTC consultants travel extensively to selected countries in order to carry out teaching and training programmes in the disciplines of Clinical Microbiology, Haematology, Blood Transfusion Science, Biochemistry, Molecular Diagnostics and Laboratory Quality Management. The number of training visits to Pacific countries per year total approximately 25 to 32 the majority of which are carried out between June and December.
PPTC Consultants who have carried out in country teaching, workshops and assessments for the first 5 months of 2025 include:
INTERNATIONAL CONFERENCES/ PROFESSIONAL DEVELOPMENT
Blood cell morphology course, Sydney (28th – 29th March 2025)
Both Phil Wakem and Emmanuel Marshall attended a “Blood Cell Morphology Workshop” in Sydney conducted by Gillian Rozenberg. This workshop was excellent in its presentation and covered in detail, updates within each of the following sections:
• Red Cell Nomenclature
• WHO Classification of Myeloproliferative Neoplasms
• WHO Classification of Myelodysplastic Syndromes
• WHO Classification of Acute Myeloid Leukaemia
• Malaria
• Lymphocytes: Reactive / Neoplastic
• Paediatric Haematology
• Case Studies
Figure 2. Students & PPTC Consultants, Microbiology, 2025
Figure 3 Emmanuel Marshall, Gillian Rozenberg, Phil Wakem
Australasian Society of Infectious Diseases, Canberra (2nd6th April 2025)
Russell Cole attended the ASID Annual Scientific Conference held in Canberra on April 2 - 6th 2025.
The theme of the 'changing face of infectious disease’ explored what disease will look like into the futureincluding discussions on new technology in the diagnosis and management of infectious disease, public health and the newly formed Australian CDC, and regional infectious disease in Australia and the Pacific. This year's meeting saw 202 abstract submissions with 133 posters, 18 proffered paper presentations, and a new addition of 20 poster walks. With a program of 79 speakers over 17 sessions, the ASID welcomed over 450 delegates from across the world.
RCPA visit, Sydney (13th - 17th April 2025)
Filipo Faiga (PPTC EQAP Programme Manager) visited RCPA in Sydney to discuss shipment of samples across the Pacific
Region. The main purpose of the visit was to observe and understand how the RCPA QAP manages shipment of EQA samples outside of Australia about the logistic requirements and dispatch processes, including packaging and shipping requirements. This visit also involved the observation of RCPA’s processes and procedures with regards to sample preparation, packaging, and transportation criteria/requirements e.g., documentation and permits.
Please contact: Phil Wakem
Chief Executive Officer
Pacific Pathology Training Centre
Wellington, New Zealand
Email: pptc@pptc.org.nz or phil@pptc.org.nz
Tel: 64-4-389 6294 or 027 2305483
REVIEWS OF INTEREST
The reviews below can be accessed for their Abstracts and “Open Access” is indicated where applicable. Unfortunately, the NZIMLS cannot provide full access to the articles due to copyright restrictions, but full access may be available through various institution arrangements. Any feedback on this can be sent to: editor@nzimls.org.nz
1. Alwahaibl N, Alwahaibl MA. Mini review on skin biopsy: traditional and modern techniques. Front Med 2025; 5:12: 1476685. doi:10.3389/fmed.2025.1476685. [Open Access]
2. Garg S, Reinhart K, Couture A, Kniss K et al. Highly pathogenic avian influenza A (H5N1) virus infections in humans. N Eng J Med 2025; 392(9): 843-854.
3. MacIntyre RC, Chughtai AA, Kunasekaran M et al. The role of masks and respirators in preventing respiratory infections in healthcare and the community. BMJ 2025; 27:388e078573 doi:10.1136/bmj-2023-078573.
4. Sibira J, Soriano MJ, del Castillo LM, de Los Santos MJ. Mitochondrial replacement techniques to resolve mitochondrial dysfunction and ooplasmic deficiencies: where are we now? Hum Reprod 2025; 40(4): 585-600. doi. org/10.1093/humrep/deaf034.
5. Yung Y, Zhang J, Hong X, Yuan W et al. Comprehensive annotation of complete ABO alleles and resolution of ABO variants by improved full-length ABO haplotype sequencing. Clin Chem 2025; 71(4): 510-519. doi/10.1093/clinchem/ hvaf015/8058681. [Open Access]
6.Kim YJK, Bunyavanich S. Microbial influencers: the airway microbiome’s role in asthma. J Clin Invest (2025); 135(4) e184316. doi.org/10.1172/JCI184316. [Open Access]
7. Zhang Y, Li N, Ge Z, Li F. Blood component therapy for dry eye disease: a systematic review and network metaanalysis. Front Med 2024; 11:1500160. doi:103389/ fmed.2024.1500160. [Open Access]
8. Nolan BJ, Cheung AS. Laboratory monitoring in transgender and gender-diverse individuals. Clin Chem 2025; 71(3): 358-377. doi/10.1093/clinchem/hvaf001/8006615. [Open Access]
9. Weber PM, Michelson C, Kerou M. What’s in our bin: Labs kick off and demand the transition towards a circular economy for lab plastics. EMBO Rep 2025; 26(2): 297-302. doi.org/10.1038/s44319-024-00360-x. [Open Access]
10. Orchanian SB, Hsiao EY. The microbiome as a modulator of neurological health across the maternal-offspring interface. J Clin Invest 2025; 135(4): e184314. doi.org/101172/ JCI1184314. [Open Access]
11. Ng XJK, Mohd Khairuddin AS, Liu H et al. Artificial intelligence-assisted point-of-care devices for lung cancer. Clin Chim Acta 2025; 15:570:120191.doi.org/10.1016/j. cca.2025.120191.
12. Li Y, Sun S. RNA dysregulation in neurodegenerative diseases. EMBO J 2025; 44(3): 613-638. doi.org/10.1038/ s44318-024-00352-6. [Open Access]
13. Daily JP, Parikh SP. Malaria. New Eng J Med 2025; 392(13): 1320-1333. doi:10.1056/NEJMra2405313.
14.Wiewiorska-Krata N, Foroncewicz B, Mucha K, Zagozdzon R.Cell therapies for immune-mediated disorders. Front Med 2025; 26:12:1550527. doi:10.3389/fmed.2025.1550527. [Open Access]
15. Ran Q, Zhang J, Zhong J, Zang S et al. Organ preservation: current limitations and optimization. Front Med 2025; 26:12:1566080. doi:3389/fmed.2025.1566080. [Open Access]
Ailsa began her career at Middlemore Hospital Laboratory in 1974, qualifying with Part 2 subjects in Microbiology and Haematology. In 1980, she completed Part 3 in Haematology. In 2003, she became a Fellow of the New Zealand Institute of Medical Laboratory Science (NZIMLS) and completed a Diploma in Communication Studies from MIT that same year.
Ailsa has always been passionate about knowledge and education, both her own and others. She has been happy to step in as a lecturer in Haematology at Auckland University of Technology (AUT) and its predecessor institutions, the Auckland Technological Institute (ATI) and the Auckland Institute of Technology (AIT).
Her career was further shaped by her work in the mid-1980s when she and her family moved to Palmerston North. Ailsa worked at Massey University’s Department of Microbiology and Genetics as the curator of the stock culture collection, preparing cultures for classes. During this time, she also worked in the Haematology department at Palmerston North Hospital.
Upon returning to Auckland, Ailsa worked part-time at Middlemore Hospital Laboratory in the Haematology Laboratory and as a Phlebotomist. Her experience in Phlebotomy has deepened her understanding of, and appreciation for, the work performed by Phlebotomists. This inspired her to establish the Pre-Analytical Special Interest Group (PASSIG) of the New Zealand Institute of Medical Laboratory Science (NZIMLS) in 2001. Ailsa served as the convenor for the first six years. During this time, Phlebotomists and Specimen Reception staff became eligible for registration with the Medical Sciences Council. The group also developed the first NZIMLS syllabi for Phlebotomy and Specimen Services examinations, which led to the creation of a registerable qualification for these areas. Ailsa has since contributed to writing and moderating Medical Laboratory Preanalytical Technician examinations. She remains an active member of the PASSIG committee.
Ailsa served as the NZIMLS Region 1 Representative from 2014 to 2015. She has also contributed to the profession by
Madhu Nahna Charge Scientist, Te Kuiti Hospital, Waikato
Madhu Nahna began her training in Medical Laboratory Science on January 11th, 1971, at Te Kuiti Hospital. At that time, the hospital was a thriving 100-bed facility offering full services, including a nursing school where nurses were both trained and lived on site. “We were lucky in that we had onsite training and were paid at the same time,” she recalls, “but I had to move to Hamilton to complete my two ‘O’ levels.”
Madhu chose to specialise in Microbiology and Immunohematology (blood bank wasn’t a separate exam in those days). As part of her Immunohaematology training, she also spent time working at the Auckland Blood Transfusion Centre. After qualifying, Madhu spent a year travelling overseas with her younger sister a wonderful experience, as she describes it, before the days of crowded airports, intense security checks, and long queues at popular tourist attractions Upon returning home, she resumed working in the laboratory but soon felt the need for change. She began working part-time in a variety of roles: as a doctor’s
organising and speaking at both national and international conferences. She has helped convene numerous pre-analytical streams at the South Pacific Congress, including the 2007 and 2015 congresses in Auckland, as well as the 2019 event at the Gold Coast, Australia. In 2023, she took on the role of Convenor for the South Pacific Congress, held at the Viaduct Convention Centre, Auckland, coordinating all the various streams with the help of a dedicated team. Ailsa finds that being part of the pre-analytical area of the laboratory is both challenging and rewarding. Medical Laboratory Science has been her life for 51 years.
Ailsa retired from her role as Charge Scientist for Specimen Services at Middlemore Hospital Laboratory in Auckland, New Zealand, a position she held since 2004. For 15 years, Ailsa also managed the Phlebotomy Department, overseeing both Specimen Services and Phlebotomy during that time. In recognition of her outstanding service to the profession, NZIMLS awarded Ailsa Life Membership in 2023.
receptionist, a laboratory technician at the local milk treatment station, selling fruit and vegetables in her father’s shop, and even working as a DJ at the local club on Saturday nights.
In 1986, Madhu returned to work at the hospital laboratory and was later offered the Charge Scientist position, which she held until her retirement this year. Madhu has always enjoyed working in a small rural laboratory and being part of the Te Kuiti community where she grew up. In retirement, she now leads a popular Senior Stretch class in town, free of charge, to keep older adults moving and maintaining their balance and strength. She is also a qualified Iyengar Yoga teacher and runs classes every Thursday night.
Madhu lives on a five-acre lifestyle block about 9 km from Te Kuiti, which keeps her very busy. Coming from a large family of five brothers and five sisters, there is always plenty happening— she never has a chance to be bored! Reflecting on her career, Madhu says, “There are scientists of my generation who have retired or are planning to, but we have a new wave of young scientific minds entering the workforce. They are ready to embrace the exciting technological changes being introduced.
Over her 54-year career, Madhu has witnessed enormous changes in both the services provided by rural hospitals and in laboratory technology. Today, the hospital lab remains busy, supporting community testing, clinic work, emergency department services, and inpatient testing.
Life membership Presented by The Rt Hon Dame Cindy Kiro, Govenor General of New Zealand.
Tony Marcinkowski
Technical Specialist, Te Toka Tumai, Auckland
Congratulations to Tony Marcinkowski who recently retired after over 53 years of amazing service to Te Toka Tumai (and its predecessors ADHB/Auckland Healthcare Services).
Tony started his journey at Auckland Hospital in the January 1972 intake (just 2 weeks after another momentous occasion, his wedding!) Longevity is in the blood with his marriage also well past the golden anniversary stage. Back in those days, training was on the job with classes done in work time and in the staff’s own time. Three years was general laboratory technology, followed by 2 years specialising with either O levels in 2 different disciplines or and O and A level in the same discipline. Luckily for us Tony chose Biochemistry (aka Chemical Pathology) as his discipline of choice. Qualifying after 5 years he then continued gaining experience and moved into a graded position (now known as Senior Scientist/Technical Specialist) and then a higher graded position at Greenlane Hospital. Here he oversaw a diverse area covering Specimen Reception, Proteins, Blood Gases, Lipids, in fact everything bar the mainframe analyser.
With various lab mergers in the Auckland region over the coming years there were moves to National Women’s Hospital and then in 1999 the central labs merged to form LabPlus,
based on the Auckland Hospital site. Here Tony’s position was a Technical Specialist in charge of Special Techniques – a combination of manual and automated technologies on a variety of specimen types and including collections for sweat testing and breath hydrogen. He was involved prior to the National Bowel Screening programme starting up with selection and evaluation of 4 different analytical systems before we landed on the Eiken system (Diana OC, now Pledia). The programme to this day continues to go from strength to strength.
Apart from the wealth of experience built up in over 50 years of lab work and his huge knowledge of Biochemistry in general, but more particularly Special Biochemistry, Tony was the staff member we all wish for. He really enjoyed training younger scientists and was very knowledgeable and patient, the ultimate mentor. He got great satisfaction from seeing these younger scientists develop in their careers and progress to higher positions. It made it difficult for us in rostering as staff always wanted to work in his area. More than that he was very supportive of his workmates, empathetic, and took a genuine interest in each staff member as a person and in their lives. And if something needed fixing he was your man – the ultimate Mr Fixit.
Tony leaves with all our best wishes for a long and happy retirement, definitely very well deserved and earned. The department will certainly be a lot quieter without Tony around but his legacy lives on in all the budding scientists he has trained. We will miss him and his sense of humour but his time has come to spend precious time with his family and pursue his hobbies.
Thank you, Tony, for over 53 years of incredible service and for sharing your great wisdom, leaving the medical laboratory science profession in the capable hands of those you have trained.
Read the articles carefully as most questions require more than one answer. Answers are to be submitted through the NZIMLS website. Make sure you supply your correct email address and membership number, it is recommended that you write your answers in a word document and then cut and paste your answers on the website. You are reminded that to claim valid CPD points for successfully completing the journal questionnaire you must submit an individual entry. It must not be part of a consultative or group process. In addition, members who have successfully completed the journal questionnaire cannot then claim additional CPD points for reading the articles from which the questions were derived. The site will remain open until Friday 17 October 2025. You must get a minimum of eight questions correct per questionnaire to obtain 5 CPD points.
The Editor sets the questions but the CPD Co-Ordinator, Jillian Broadbent, marks the answers. Direct any queries to her at cpd@nzimls.org.nz.
JULY 2025 QUESTIONNAIRE
1. What is familial hypercholesterolemia (FML)? What is considered as the disease risk of this disorder? What three genes have mutations shown to give rise to FML?
2. FH patients are classified into two clinical types: homozygous (HoFH) and heterozygous (HeFH). What is the prevalence of each type in a population? How is each type characterised?
3. Where is Low-density lipoprotein receptor (LDLR) located? What does it encode for? And why is it crucial?
4. Why is antithetical M antigen typically considered as clinically insignificant and rarely responsible for Haemolytic disease of the fetus and newborn (HDFN) or haemolytic transfusion reactions (HTR)?
5. How is the identification of anti-M antigen aided in the laboratory?
6. What is the severe clinical syndrome Haemophagocytic Lymphohistiocytosis (HLH) caused by? What differences in presentation of the syndrome are seen between paediatric and adult populations?
7. In reported neoplasm-associated HLH cases in adults, lymphoma accounts for 67%, what are the majority of cases linked to? What is the survival rate? What are the challenges in defining treatment?
8. Why can the initial diagnosis of HLH be delayed? And how?
9. What medications are implicated in Pyroglutamic acidosis (PGA)?
10. How does PGA develop? Other than the medications above, what other factors contribute to the risk of PGA?
ANSWERS MARCH 2025 QUESTIONNAIRE
1. 70% of all diagnoses are based on a laboratory test result, what can happen when results do not meet their stated levels of sensitivity and specificity? What did an FDA report of twenty cases highlight in 2015? Potential for misdiagnosis, causing public and patient harm. Inaccurate testing led to false positive and negative results, inappropriate testing, and delayed treatment.
2. What three examples of Laboratory Developed Tests are available in Aotearoa NZ? Where? Embryonic screening by preimplantation genetic testing for rare monogenic disorders performed at Canterbury Health Laboratories. Comprehensive genetic testing for immunodeficiency disorders at LabPLUS laboratory in Auckland. CHL toxicology laboratory has applied inhouse developed mass spectrometry testing.
3. What cancers have reported overexpression of Aurora kinases? What does overexpression cause? What is/ are the most overexpressed members or subtypes of Aurora Aurora kinases found in liver cancer tissues?
Kidney, lung, and mesothelioma and particularly liver. Aurora kinases overexpression causes chromosomal instability that could lead into liver cancer development. The most overexpressed Aurora kinases in liver cancer tissues are AURKA and AURKB
4. What 3 findings in the Zywea study into AURKA expression in liver cancer were revealed? What was high mRNA expression of AURKA associated with? AKURA expression was up modulated in liver cancer tissues compared to normal tissues and was associated with patients’ poor survival. AURKA has the potential to be a prognostic marker for early prognosis of liver cancer. AURKA may serve as a novel therapeutic target for the treatment of liver cancer. A poorer or worst survival and two times high risk of death than those with a low mRNA expression.
5. Mutations in the glucose-6-phosphate dehydrogenase (G6PDH) gene manifest as a deficiency in G6PDH enzyme activity. What are the three clinical manifestations of this deficiency? How are these conditions caused? Neonatal jaundice, chronic nonspherocytic haemolytic anaemia (CNSHA), and acute haemolytic anaemia. These conditions are caused by factors such as certain foods, antibiotics, antimalarials, or infections that cause reactive oxygen species (ROS) to accumulate.
6. G6PDH deficiency and type of G6PDH variant present determines the probability and severity of haemolysis, what other abnormalities are caused by a G6PDH deficit? Why is the prevalence of G6PDH deficiency higher in males, than in females? Multiple abnormalities in the amounts of various vitamins, lipid profiles, blood proteins, trace elements, uric acid, and malondialdehyde Females receive a copy of the X chromosome containing the gene from each of their parents, whereas males only receive one copy from their mothers. Since females have two sources of the enzyme, females are less likely than males to be G6PDH deficient.
7. What is DiGeorge syndrome? What are the two forms and how are they characterised? It is a primary immunodeficiency caused by chromosome 22q11.2 deletion and is the greatest common microdeletion (1:4000 live births). There are two types: Complete DiGeorge syndrome where there is a dysregulation of T-B cell interactions and patients suffer from life-threatening severe T-cell lymphoma. Partial DiGeorge syndrome is characterized by decreased thymic output show and patients have a T-cell lymphopenia with a wide variability of phenotypic features presented.
8. What should be defined during an immunologic evaluation of DiGeorge syndrome? What assessments are required for immunological diagnosis? Immunologic assessment of affected subjects with 22q11.2del and any other developmental thymus deformity (DTD) is crucial to define immune status and assess infection susceptibility. T cell markers (CD3, CD4, CD8), B cell markers (CD19 or CD20), and natural killer cell markers (CD16 or CD56) should be included in the primary immunologic evaluation required for diagnosis. Plus, IgG, IgM, and IgA estimation.
9. Implementation of novel and unfamiliar technologies into a busy laboratory is complex and challenging. What does a traditional review of implementation entail? What different methodology did ESR use to review the implementation of next-generation sequencing (NGS) using the Oxford Nanopore Technologies platform? What were the purpose of using these systems? Traditional reports and reviews focus on implementation of new laboratory technologies focus on feasibility, technical challenges and solutions, comparison between similar technologies, and comments on cost-effectiveness. A social systems methodology to investigate the perceptions of those involved in the implementation, and a look at the wider context of laboratory policies and practices, both formal and informal. To understand the barriers and enablers from the point of view of the people involved.
10. What is one limitation of the greater use of nanopore sequencing? What solution is proposed and what would this scenario require? The lack of in-depth bioinformatic expertise. To leverage the advantages of the relatively small medical laboratory system in Aotearoa New Zealand and to promote collective working in order to decentralize sequencing carried out in routine diagnostic laboratories and create a centralised bioinformatics reference laboratory service. This would require development of appropriate data infrastructure, including data storage, transfer mechanisms, security, governance and sovereignty
IN MEMORIUM
Sue Warrington
Supervising Scientist, New Zealand Blood Service, Christchurch
I am honoured to be able to share a little bit about who Sue was to us. To us, Sue was a Medical Laboratory Scientist and our Supervising Scientist, with a special interest and aptitude for Red Cell Serology.
Sue graduated with a diploma in Medical Laboratory Technology, specialising in Haematology and Immunohaematology in 1986. She was entered into the Register of Medical Laboratory Technologists on the 4th of February 1987. She was number 1551 on the Register.
Sue was the very first Supervising Scientist at the New Zealand Blood Service and since becoming a Supervising Scientist 14 years ago, Sue worked to craft the position into the supportive, inspiring, educating role that it is today. “Let’s check with Sue”, “leave it for Sue”, “Sue will be able to help”, “thank goodness Sue is here”, “Sue will know what to do”, are phrases heard daily in our laboratory.
Sue had a hand in training every staff member, university placement student and registrar that has spent time in Christchurch Blood Bank. A little bit of Sue can be seen in every scientist who was lucky enough to have her as a mentor. She had time for everyone, was approachable for any question and always had the answer. Who needs the SOP when you have Sue! Sue worked so hard to share her knowledge and we had so much more to learn from her.
One of Sue’s great passions was supporting and educating regional blood banks. She was very focussed on doing her best for them and she loved every moment. Her knowledge of the science and requirements of a transfusion laboratory were phenomenal. This made her the perfect person to support these labs, and what a wonderful support she was! They had absolute faith in her and relied on her almost daily for advice.
Sue presented the “Supervising Scientist Forum” at our Regional Blood Bank Meetings. Sue made amazing PowerPoint presentations and spoke with such confidence – you wouldn’t know how nervous she was – I didn’t believe her when she told me!
Sue authored research papers and has presented at international conferences. In 2019 she presented at BLOOD in
Perth on the Blood Bank participation in the Mosque Shootings. I was living in Brisbane at the time and was lucky to be able to attend this conference and see Sue speak.
Just last year, Sue was accepted to present a case study at the BLOOD conference in Brisbane. Sue was the only scientist within NZBS who was approved to attend the conference with full funding.
Sue had an amazing work ethic and would strive for perfection, always. She wanted to do the very best for every patient. She inspired our team to have high standards. She loved to share her knowledge; she loved to help.
On a personal level – Sue trained me, initially as a university student and then when I had my very first Medical Laboratory Scientist job. I was completely in awe of her knowledge and skill. Sue was kind to me. I did not have my driver’s licence, and she used to drop me off at home after working a 4 to midnight shift with her. Sue has been such a wonderful support to me as a Team Leader. I am now in awe of not only her knowledge and skill, but her as a person, as a friend, as a wife, mother, grandmother.
Sue was very passionate about what she loved and cared about most, her family were at the very top of the list, she shared so many photos, videos and thoughts. She was immensely proud of her family. We heard about every Irish Dancing competition, followed every Christmas Grotto in preparation and watched as wool was lovingly knitted into clothing for her grandchildren. Without Sue, there is a gigantic hole in our Blood Bank and in our hearts.
On Friday 24th September 2010, Sue wrote the first edition of “Sue Says” – a newsletter, where she shared advice, insight and helped to ensure all staff were up to date with any changes or interesting cases. There are 276 editions of “Sue Says”. When looking for the answer to a technical question, staff turn to “Sue Says”. What “Sue Says” is gift that we will treasure always. There is something for everyone in “Sue Says”, and while I will not enlighten you on how to improve your blood banking skills, I would like to end by sharing some of Sue’s “Thoughts of the Week”:
• “Always laugh when you can. It is cheaper than medicine!”
• “If you don’t have time to do it right, when will you have time to do it over?”
• “Keep your face always toward the sunshine - and shadows will fall behind you.”
Sue passed away 4 December 2024.
Prepared by: Nicola Crampton, Team Leader, New Zealand Blood Service, Christchurch
IN MEMORIUM
Helen Beatrix Robertshawe, 1940-2024
Helen passed away on the 4th of November 2024. She is remembered as the Secretary of the Medical Laboratory Technologists Board, now the Medical Sciences Council, from 1974 to 1984. She was passionate about our profession and its members, attending many of our scientific meetings.
Helen was born in 1940 and attended the Nga Tawa Diocesan School in Wellington, New Zealand.
She moved to England where she worked in the office of the All England LawnTennis Club fortheWimbledonTennisTournament in 1968.
After her stint in the Medical Laboratory Technologists Board, she worked for the Legal Services Agency in Wellington before retiring to Waikanae in 2011 where she passed away in 2024. Helen was very musical, being a choral singer, pianist and organist. She was a member of many church groups and loved showing visitors around Wellington Cathedral.
Helen was a true friend to our members and profession and in 1978 she was made an Honorary Member of our Institute for her services to the profession.
Contributed by: Rob Siebers, Emeritus Editor
Saturday �
Startsat8:30am
JetParkAuckland
Airport Hotel & Conference Centre
63 Westney Road, Mongere,.
Auckland
CSPIFE NEXUS) ►
REVOLUTIONARY ANTISERA APPLICATIONSYSTEM
• Automated antisera for up to 15 profiles per gel application
• No crossover contamination or antisera templates
• Ready-to-use, color-coded antisera in temperature controlled on-board storage
INTEGRATED REPORTING
• QuickScanTouch Plus on-board with linking to other Helena gel and VS Nexus CE data
• Myeloma Module provides linking to historic data for rapid review and reporting
• NEW! Composite Key Identifier feature creates a unique identifier from two demographic fields for tracking outside samples.