
Progressione nella sclerosi multipla:
evidenze ed esperienze
La progressione
nella sclerosi multipla 2
Profilo del paziente: caratteristiche cliniche e strumenti di valutazione 9
Trattamento della progressione: evidenze e criticità 16
Conclusioni e prospettive future 20
Bibliografia 21
La progressione nella sclerosi multipla
Negli ultimi anni, la comprensione della sclerosi multipla (SM) si è evoluta oltre la tradizionale distinzione tra una fase precoce recidivante infiammatoria – caratterizzata dal riscontro di lesioni rilevabili strumentalmente – e una fase progressiva – caratterizzata da fenomeni neurodegenerativi –, verso una concezione che vede l’evoluzione della patologia come un continuum biologico-clinico la cui manifestazione fenotipica dipende dall’interazione tra fattori genetici, fisiologici e ambientali. In questa nuova visione, la progressione rappresenta un processo biologico e clinico già attivo nelle fasi iniziali di malattia, che può manifestarsi e procedere senza eventi infiammatori evidenti.
Questo quaderno rappresenta la seconda tappa di un progetto scientifico dedicato all’approfondimento condiviso dei temi emergenti nella gestione della sclerosi multipla. Dopo aver affrontato il concetto di “smoldering neuroinflammation”, il presente documento si concentra sulle sfide cliniche legate alla rilevazione precoce e alla gestione della progressione di malattia.1
Le tematiche presenti nel quaderno sono state trattate in occasione del progetto La rivelazione dell’invisibile: i sottili cambiamenti nel paziente SM e i segnali di progressione, sviluppatosi attraverso quattro incontri – organizzati a Milano, Firenze e Napoli – che hanno coinvolto neurologi ospedalieri, ricercatori e clinici esperti nella diagnosi e nel trattamento della SM.
Anche in ambito clinico l’evoluzione della SM è stata tradizionalmente suddivisa in due fasi: pre-diagnostica e diagnostica. Alla prima appartengono le sindromi radiologicamente isolate (radiologically isolated syndrome, RIS) e le manifestazioni prodromiche, caratterizzate da alterazioni subcliniche rilevabili tramite imaging, mentre quella diagnostica comprende le sindromi clinicamente isolate (clinically isolated syndrome, CIS) e le forme recidivanteremittente (SM-RR) e secondariamente progressiva (SM-SP). A queste si affianca la forma primariamente progressiva (SM-PP), che si distingue, sin dall’esordio, per un peggioramento continuo e precoce della funzione neurologica, in assenza di ricadute e fasi di remissione.
Uno dei temi più rilevanti emersi durante gli incontri del progetto La rivelazione dell’invisibile: i sottili cambiamenti nel paziente SM e i segnali di progressione è la necessità di una definizione operativa condivisa della progressione di disabilità. Durante il confronto, infatti, è emersa una forte disomogeneità nella modalità con cui viene definita la progressione nella
pratica quotidiana, con il risultato che la formalizzazione della diagnosi di SM-SP non sempre è esplicitata nei referti o viene comunicata al paziente solo su sua richiesta. Nella maggior parte dei casi, le cartelle cliniche riportano una sintomatologia in peggioramento (fatica, disequilibrio, difficoltà nelle attività lavorative), ma la visita specialistica si limita a confermare la stabilità clinica e radiologica, con conseguente prosecuzione della terapia in corso. Negli ultimi anni, la disponibilità di trattamenti ad alta efficacia in grado di agire sull’attività infiammatoria, sia clinica sia radiologica, ha contribuito a mettere in luce una progressione silente o indipendente, non attribuibile a recidive o alla comparsa di nuove lesioni demielinizzanti. Infatti, oltre al peggioramento associato alle ricadute (relapse-associated worsening, RAW), esiste una progressione indipendente dall’attività di ricaduta (progression independent of relapse activity, PIRA) già evidente nelle fasi precoci della SM-RR e responsabile di almeno il 50% dell’aggravamento complessivo della disabilità.2,3,4,5,6,7 Questo suggerisce che la malattia sia caratterizzata da un continuum biologico in cui meccanismi patologici differenti coesistono e si intrecciano fin dalle prime fasi. Tuttavia, la definizione attuale di PIRA, basata su scale che dipendono prevalentemente dalla componente motoria come l’EDSS (Expanded Disability Status Scale), risulta poco sensibile ad altre dimensioni cliniche rilevanti. In tale contesto si inserisce il concetto di smoldering associated worsening (SAW), che include un peggioramento subdolo, spesso riferito dai pazienti, legato a sintomi fisici e cognitivi attribuibili a meccanismi infiammatori cronici non acuti.8 Il monitoraggio del SAW richiede un approccio multidimensionale, con l’impiego di biomarcatori clinici, radiologici e liquorali o sierici per una valutazione più sensibile e precoce della progressione di disabilità [Figura 1].
Già prima della concettualizzazione della PIRA, i clinici osservavano un peggioramento graduale nei pazienti in trattamento con terapie modificanti la
malattia (disease-modifying therapy, DMT) ad alta efficacia. In diversi casi, tale peggioramento non seguiva un decorso lento e impercettibile, ma si manifestava con evidenza clinica nel giro di pochi mesi. Queste osservazioni precoci hanno rappresentato un primo indicatore della presenza di meccanismi patogenetici non pienamente controllati dalla modulazione dell’infiammazione acuta, suggerendo l’esistenza di processi neurodegenerativi autonomi, attivi anche in fasi apparentemente “silenti” della malattia.
Contestualmente, si è assistito a un proliferare di marcatori – PRL, SEL, CAL, PIRMA, PISMA – che mirano a fornire misure oggettive della progressione. I primi marker fanno riferimento all’esame di risonanza magnetica e includono le lesioni con bordo paramagnetico (paramagnetic rim lesions, PRL) e le lesioni a lenta espansione/evoluzione (slowly expanding/evolving lesions, SEL). Queste lesioni croniche attive (chronic active lesions, CAL) sono espressione di un’infiammazione persistente e rappresentano una delle componenti “smoldering” della SM, caratterizzata da una progressione indipendente dalle ricadute. Il concetto di PIRMA (progression independent of relapse and MRI activity) estende la definizione di PIRA escludendo ogni segno di attività infiammatoria, sia clinica che radiologica, mentre quello di PISMA (progression independent of symptomatic or MRI activity) – non ancora formalmente validato in letteratura – mira a isolare una progressione clinica del tutto svincolata da fenomeni infiammatori o peggioramenti transitori.
Abbandonata quindi la tradizionale visione dicotomica della malattia, nella comunità degli esperti di sclerosi multipla si discute oggi della necessità di ridefinire il concetto di progressione di malattia. Per tutti i partecipanti agli incontri di Milano, Firenze e Napoli, ad esempio, il modo di intendere il viraggio verso la fase progressiva della malattia è attualmente troppo legato all’aspetto motorio. In particolare, è stato criticato l’utilizzo quasi esclusivo dell’EDSS
come metrica di disabilità. Lo score, infatti, pur essendo lo strumento standard nella valutazione del peggioramento nella SM, presenta una sensibilità ridotta per sintomi non motori come la fatica e i disturbi cognitivi, sessuali e sfinterici [Figura 2].
Inoltre, un’analisi condotta su soggetti con più di cinquantacinque anni e privi di patologie neurologiche mostra che l’EDSS risulta essere, in questa fascia di età, poco affidabile: lo studio ha rilevato un punteggio medio dell’EDSS pari a 3.0, con valori
considerati anomali in tutti i partecipanti. Questo dato suggerisce che lo score possa essere influenzato dall’invecchiamento fisiologico e non esclusivamente dalla progressione della sclerosi multipla, sollevando dubbi sull’utilizzo del valore soglia di 4.0 come indicatore del passaggio alla SM-SP.9 L’EDSS è quindi percepito come un criterio di valutazione limitante, seppur consolidato: nel contesto ambulatoriale, infatti, il tempo a disposizione e la disponibilità limitata di strumenti rendono complessa l’iden-
Figura 1. Confronto tra i meccanismi patogenetici e clinici di RAW e SAW, che evidenzia modalità di presentazione, sintomi, biomarcatori e misure di esito. Fonte: Scalfari A. et al, Ann Neurol 2024.
Substrati patologici
Modalità di presentazione dei sintomi
Tipo di sintomi
Misure di esito clinico
Biomarcatori (MRI/CSF/siero)
infiammatoria focale (immunità adattativa)
Demielinizzazione acuta e perdita assonale
Ricaduta clinica: sintomi neurologici acuti/subacuti seguiti da deficit residuo
Perdita visiva
Deficit motorio
Atassia
Deficit sensoriale
Disfunzione sfinterica
EDSS
MIRI: lesioni T2/GAD+ nuove o in crescita
Siero/CSF: aumento transitorio dei livelli di NfL
di malattia a lenta evoluzione (immunità innata)
Neurodegenerazione progressiva e diffusa, neuroinfiammazione cronica
Accumulo graduale della disabilità non correlato a ricadute
PIRA (peggioramento principale nella funzione motoria, arti superiori/ inferiori)
EDSS e EDSS+ (EDSS/ T25FW/9HPT)
Ridotta resistenza (distanza minore nella camminata/corsa)
Sintomi indotti dall’esercizio
Peggioramento cognitivo
Peggioramento della fatica e delle funzioni sfinteriche
Peggioramento generale (“sentirsi peggio”)
Test neurologici da stress
PASAT/MMSE/BICAMS
Scale per fatica/umore/ dispositivi indossabili PROs (MSIS-29-Phys/HUl3)
MRI: SEL, CL, atrofia cerebrale/spinale, attivazione microgliale (TSPO, PRL)
Siero/CSF: aumento dei livelli di NfL, GFAP, CXCL13, CHl3L1
Perdita permanente della funzione
SAW
RAW
Attività
Attività
Figura 2. L’Expanded disability status scale (EDSS). Fonte: Brissart H et al. Eur Neurol 2010;64:345-350.
Nessuna disabilità
Disabilità minima
Sintomi minimi
Disabilità moderata
Disabilità rilevante
La disabilità limita le funzioni giornaliere È richiesto un suppor to alla deambulazione
Necessità della sedia a rotelle
Necessità di assistenza per la sedia a rotelle
Impossibilità di lasciare il letto Morte per SM
tificazione di una progressione di malattia che non coinvolga primariamente la funzione motoria. Secondo le evidenze, l’evento PIRA potrebbe essere considerato il determinante principale dell’accumulo di disabilità a lungo termine: il primo evento potrebbe infatti coincidere con la transizione e l’inizio della fase secondariamente progressiva, ma la variabilità nella sua definizione operativa in diversi studi clinici e osservazionali ha finora limitato la comparabilità dei risultati.3
I criteri più determinanti nel definire gli eventi PIRA risultano essere due – il metodo di aggiornamento del punteggio EDSS di riferimento (re-baselining) e la durata del periodo di conferma del peggioramento. Nel complesso, una recente analisi ha indicato come definizione più efficace per identificare la PIRA la seguente: un peggioramento significativo della disabilità rispetto al valore di riferimento – ricalibrato dopo ogni evento, ricaduta o miglioramento dell’EDSS – in assenza di ricadute dall’ultima valutazione EDSS. Tale peggioramento deve essere confermato da punteggi EDSS non preceduti da ricadute nei trenta giorni precedenti e mantenersi al di sopra della soglia di significatività clinica per almeno dodici mesi.10
Caso clinico 1
Profilo del paziente: Impiegato, 55 anni, cinque anni trascorsi dall’ultima relapse.
Tempo trascorso dalla diagnosi: quindici anni. Trattamento in corso: da due anni in trattamento con anti-CD20 (in precedenza natalizumab).
Sintomi: graduale peggioramento della deambulazione con peggioramento confermato della disabilità e passaggio da EDSS 3.0 a 6.0 dall’estate 2022 a oggi. Il paziente riferisce minor autonomia motoria, minor destrezza manuale e ipostenia dell’arto superiore sinistro, cessazione delle attività di hobby. Il medico curante sottolinea una spasticità dell’arto inferiore sinistro e maggior faticabilità.
Discussione: Negli ultimi anni è progressivamente cresciuta l’attenzione verso l’identificazione precoce di segni di progressione. In quest’ottica, il team specialistico che ha in carico il paziente ha scelto di valorizzare, durante le visite di follow-up, l’ascolto della quotidianità del paziente, considerandola una fonte clinicamente rilevante per cogliere manifestazioni sottili ma indicative di un’evoluzione progressiva della malattia.
Grazie alla recente acquisizione di una neuropsicologa, si sta implementando un tipo di valutazione che comprende batterie di test cognitivi tarati a seconda dell’età, scolarizzazione e sesso e standardizzati secondo le linee guida nazionali e internazionali. È stata valutata la possibilità di utilizzare dei questionari multiitems per analizzare sia gli aspetti fisici che cognitivo-comportamentali (come stati emotivi e faticabilità).
La progressione, anche senza infiammazione acuta, è presente sin dalle prime fasi di malattia oltre a essere il principale driver di disabilità. Inoltre, una volta manifestata, sembra procedere indipendentemente dall’infiammazione pre-esistente [Figura 3]. 3,4,11,12 Può però succedere che pazienti incontrati in setting ambulatoriale presentino comunque
recidive nel corso della malattia, a dimostrazione che l’assenza di progressione clinica significativa non è incompatibile con un’attività infiammatoria episodica.
Negli anni, sono stati proposti diversi criteri per identificare con maggiore precisione l’esordio della fase progressiva della SM, riflettendo una pratica ormai comune nella comunità scientifica e clinica: quella di ricorrere a definizioni operative, spesso non univoche, per descrivere una transizione che raramente si manifesta in modo netto. Tra queste, una delle più utilizzate si basa sul giudizio clinico soggettivo del neurologo, in accordo con quanto scritto da Lublin e colleghi: 13
“Nella maggior parte dei contesti clinici, la SM-SP viene diagnosticata retrospettivamente in base a un’anamnesi di graduale peggioramento dopo un iniziale decorso recidivante della malattia, con o
Figura 3. Il diagramma mostra traiettorie distinte di malattia, con variazioni nei processi infiammatori, neurodegenerativi e demielinizzanti che contribuiscono alla progressione.
Fonte: Krieger S. et al, Curr. Op. Neurol. 2024.
Paziente 1Paziente 2
In ammazione acuta della sostanza bianca ( WM)
Demielinizzazione della sostanza bianca
In ammazione parenchimale cronica
Degenerazione assonale
Fallimento della rimielinizzazione
Degenerazione neuronale
Demielinizzazione cor ticale
In ammazione interstiziale cronica
In ammazione acuta della sostanza bianca ( WM)
Demielinizzazione della sostanza bianca
In ammazione parenchimale cronica
Degenerazione assonale
Fallimento della rimielinizzazione
Degenerazione neuronale
Demielinizzazione cor ticale
In ammazione interstiziale cronica
senza esacerbazioni acute durante il decorso progressivo. Ad oggi non esistono criteri clinici, di imaging, immunologici o patologici chiari per determinare il punto di transizione tra SM-RR e SM-SP; la transizione è solitamente graduale. Ciò ha limitato la nostra capacità di studiare le caratteristiche di imaging e biomarcatori che possono distinguere questo decorso”.
Per identificare la progressione di malattia nei pazienti con SM sono stati poi proposti anche criteri operativi differenziati che affiancano il giudizio del neurologo: tra questi i criteri di MSBase – database che comprende più di 92.000 pazienti – e i criteri utilizzati in alcuni trial, come EXPAND – che ha valutato la sicurezza e l’efficacia di siponimod.14,15
Secondo l’algoritmo MSBase, la progressione viene definita in presenza di:
un punteggio EDSS minimo pari a 4.0 e un punteggio funzionale piramidale (FS) di almeno 2;
una variazione documentata dell’EDSS maggiore o uguale a un punto rispetto al basale per valori
≤5.5, oppure maggiore o uguale a mezzo punto per valori ≥ 6.0;
l’assenza di ricadute nei trenta giorni precedenti alla visita di follow-up;
la conferma del peggioramento a distanza di almeno tre mesi dal follow-up.
Per l’algoritmo non era previsto alcun intervallo minimo obbligatorio tra la valutazione basale e quella di controllo.16 Diversamente, i criteri di inclusione del trial EXPAND prevedevano: 14
un punteggio EDSS compreso tra 3.0 e 6.5 al momento dello screening;
una storia documentata di SM-RR;
l’assenza di ricadute nei tre mesi precedenti allo screening;
una progressione confermata dell’EDSS nei due anni antecedenti.
Definizioni anche solo lievemente diverse tra loro possono portare a stime significativamente differenti: secondo un’analisi basata sui dati del Registro
italiano sclerosi multipla si va da circa il 12% dei pazienti quando la valutazione si basa sul giudizio clinico soggettivo del neurologo, a quasi il 18% quando il passaggio dalla forma RR a quella SP viene identificato tramite criteri data driven, basati su una versione modificata dell’algoritmo di Lorscheider (DDA) [Figura 4].16,17
Nella comunità degli esperti di SM si è discusso anche dei limiti dell’applicazione in ambito ambulatoriale del concetto di PIRA: a differenza di quanto si fa nell’ambito degli studi clinici multicentrici o monocentrici, nella pratica clinica l’adozione del Neurostatus completo risulta rara nella valutazione dell’EDSS e dei sistemi funzionali, e la conferma del peggioramento degli score a tre o sei mesi è difficilmente sostenibile per ragioni organizzative e pratiche. Inoltre, la comunità di esperti concorda sulla necessità di identificare la corretta finestra tempo -
Figura 4. Proporzione di pazienti con SM-SP identificati secondo una definizione data-driven rispetto al giudizio clinico del neurologo.
Fonte: Iaffaldano et al. Mult Scler 2022.
Pazienti del Registro Italiano Sclerosi Multipla
Pazienti con forma SM-SP:
12% 18% secondo il giudizio del neurologo secondo i criteri DDA
Figura 5. Flusso decisionale che individua pazienti con SM “truly benign”. Fonte: Tallantyre et al. JNNP 2019.
Durata di malattia <15 anni
n = 899
Con dati su EDSS insufficienti
n = 77
EDSS più recente ≥4
n = 803
EDSS <4
Ha ricevuto DMT
n = 46
Pazienti con SM
n = 2025
Durata di malattia >15 anni
n = 1126
Con dati su EDSS associati
n = 1049
Ultimo EDSS <4
n = 246
Ultimo EDSS <4 No DMT
n = 200
Valutati clinicamente
n = 60
rale per un opportuno sequencing terapeutico, in pazienti già in trattamento con terapie ad alta efficacia. Questa pratica, che consiste nella scelta e successione dei trattamenti modificanti la malattia in base all’evoluzione clinica e radiologica del paziente, è stata analizzata in particolare nei soggetti in peggioramento lieve ma progressivo e in assenza di attività infiammatoria evidente.
Il monitoraggio longitudinale da parte dello specialista è un ulteriore fattore critico: quando il paziente è seguito da più professionisti, viene perduta la continuità necessaria per cogliere segnali di deterioramento graduale. Per superare questi limiti, alcuni clinici riferiscono di affidarsi anche a elementi funzionali riferiti dal paziente – la ridotta tolleranza all’attività lavorativa o sportiva – come indici precoci di progressione, soprattutto nei soggetti ancora in trattamento con DMT a bassa efficacia.
Uno studio recente ha proposto anche una definizione di SM “benigna” (truly benign), basata su quattro criteri principali: un punteggio EDSS inferiore a 3.0 per almeno quindici anni, assenza di impatto sulla vita lavorativa, assenza di sintomi depressivi o affaticamento rilevante e assenza di compromissione cognitiva.18 Applicando questa definizione a una coorte iniziale di più di duemila pazienti con un follow-up superiore a quindici anni, solo sessanta soggetti soddisfacevano i criteri clinici preliminari [Figura 5]. Di questi, dopo rivalutazione approfondita, appena nove pazienti presentavano effettivamente tutte le condizioni richieste. Questo dato sottolinea come il fenotipo clinico truly benign sulla carta esista, ma sia estremamente raro. Tuttavia, secondo molti clinici, il concetto di SM benigna appare utile solo retrospettivamente, dal momento che non esistono marcatori affidabili in grado di prevedere in fase precoce un decorso clinico favorevole. Ne consegue che il termine, sebbene storicamente radicato, abbia oggi un’utilità limitata nella pratica clinica, dove l’approccio precoce e proattivo alla gestione della malattia resta imprescindibile.
Profilo del paziente: caratteristiche
cliniche e strumenti di valutazione
La risonanza magnetica (MRI) rappresenta ancora oggi il principale strumento diagnostico e prognostico nella SM, in particolare nelle fasi iniziali della malattia. Se applicati correttamente, i criteri più recenti consentono una diagnosi anche con una singola scansione e la frequenza di riscontro di nuove lesioni supera spesso quella delle recidive cliniche.19,20,21
Oltre a rilevare l’attività infiammatoria acuta, l’MRI ha un ruolo anche nella rilevazione della progressione, quantificando l’atrofia cerebrale – espressione del danno neurodegenerativo cumulativo – e permettendo di rilevare le CAL. La degenerazione delle aree motorie e cognitive può contribuire in modo sostanziale all’accumulo di disabilità, e diversi studi hanno dimostrato che le variazioni del volume cerebrale rappresentano un predittore affidabile della progressione di malattia in tutte le fasi della SM.22 Inoltre, è stato osservato che fenomeni neurodegenerativi più marcati a livello del midollo spinale cervicale si associano all’accumulo di disabilità in modo indipendente dalla presenza di lesioni focali. 23 La presenza di atrofia del sistema nervoso centrale è stata documentata in modo significativo in studi di coorte, anche prima della comparsa delle manifestazioni cliniche.24,25
Un tema emerso nel confronto tra i partecipanti del progetto La rivelazione dell’invisibile riguarda l’eterogeneità della refertazione neuroradiologica nelle MRI di follow-up per pazienti con SM. Alcuni clinici segnalano che, nonostante l’elevata specializzazione dei diversi centri – che siano pubblici o privati – nei quali i pazienti sono inviati a svolgere l’esame diagnostico, i referti ricevuti risultano spesso eccessivamente sintetici, con formulazioni generiche come
“quadro invariato” e senza dettagli utili alla valutazione longitudinale della progressione. Sebbene l’impiego sistematico della MRI per monitorare la progressione silente possa sembrare ancora disomogeneo, la MRI rimane uno strumento centrale nella diagnosi e prognosi della SM. Marcatori avanzati come le PRL sono stati identificati come potenziali indicatori di infiammazione cronica attiva e stanno progressivamente guadagnando attenzione nella pratica clinica, anche se la loro identificazione richiede competenze specifiche e non è ancora standardizzata. Nonostante persistano differenze tra centri in termini di protocolli e risorse, si sta osservando una crescente tendenza verso l’adozione di tecniche di imaging più raffinate e clinicamente orientate. È stato discusso inoltre se l’MRI debba essere impiegata come strumento di monitoraggio continuo oppure come marcatore prognostico iniziale. In quest’ottica, un utilizzo mirato nelle fasi precoci della malattia – ad esempio nel corso dei tre anni successivi alla diagnosi – potrebbe fornire indicazioni utili sull’andamento futuro, evitando la ripetizione frequente di esami poco informativi. Per implementare l’uso di marcatori come le PRL, sarebbe necessario coinvolgere attivamente i radiologi, definire nuovi protocolli di acquisizione e standardizzare le letture attraverso linee guida condivise. Per rispondere all’esigenza di una maggiore precisione sui referti, in alcune regioni si stanno diffondendo modelli di referto predefiniti, adottati anche dalle strutture convenzionate: queste valutazioni includono informazioni dettagliate sul numero (ad esempio <10, 10–20, >20) e tipo di lesioni (T1, T2, lesioni captanti gadolinio), sulla presenza di black holes, sull’evoluzione dimensionale. Una prima iniziativa di questo tipo, implementata in Veneto, si sta estendendo anche ad altre realtà regionali: in Lombardia, la proposta di adottare un formato condiviso per la refertazione è stata presentata alla Tavola Tecnica per la Rete Sclerosi Multipla.
Oltre alla MRI, uno standard diagnostico molto utilizzato nei contesti ospedalieri è il già citato EDSS.
6.
EDSS in persone con o senza diagnosi di SM. Fonte: Lynch s. et al, MSRD 2021.
Questo score, per quanto semplice da usare, cattura solo una parte della disabilità e può risultare poco attendibile o non abbastanza sensibile a cambiamenti clinici sottili e progressivi, portando a una sottostima del deterioramento complessivo [Figura 6]. 26,27,28
Caso clinico 2
Profilo del paziente: impiegato presso Azienda dei trasporti, 45 anni, tre anni dall’ultima relapse.
Tempo trascorso dalla diagnosi: ventisei anni.
Trattamento in corso: ozanimod da luglio 2022 (prima interferone, teriflunomide).
Sintomi: nel 2022 l’EDSS era 4.0. L’EDSS passa da 5.0 a 6.0 in sei/otto mesi, da inizio 2024. Il paziente riferisce un aumento della fatica negli spostamenti e una diminuzione dell’equilibrio, una comparsa di parestesie persistenti agli arti inferiori, con estensione anche alla gamba destra – precedentemente asintomatica – e una progressione dei disturbi sfinterici, in particolare con accentuazione dell’urgenza minzionale. Il fisiatra comunica un peggioramento negli
esercizi di equilibrio. Viene prescritto un ciclo di fisioterapia.
Discussione: Nel percorso clinico è cruciale un approccio precoce e multidisciplinare, che coinvolga fisiatri, fisioterapisti, terapisti occupazionali, logopedisti, psicologi. Il supporto integrato può consentire una presa in carico più efficace, soprattutto nei casi in cui la progressione clinica è subdola e sottostimata. Fondamentale è anche il “racconto” del paziente: spesso è proprio dalla sua narrazione – più che da una variazione dell’EDSS – che emergono i primi segnali di transizione. I segnali possono essere disturbi sfinterici, disfagia, disfonia, alterazioni cognitive e peggioramento funzionale degli arti superiori. Inoltre, secondo lo specialista che ha presentato il caso, si sarebbe dovuto intervenire precocemente. Riferimenti espliciti a una fase secondariamente progressiva risalgono solo a luglio 2024, sulla base di una visita effettuata qualche mese prima. Tuttavia, considerando retrospettivamente la traiettoria clinica e i sintomi riferiti, è plausibile che il paziente si trovasse già da diversi anni in fase SP.
Figura
Punteggi
La crescente consapevolezza della complessità clinica della progressione nella SM ha messo in evidenza la necessità di affiancare ai tradizionali parametri motori anche valutazioni più ampie, come quelle cognitive e biologiche: in alcuni contesti si è affermato l’uso di scale composite come l’EDSS-plus, che integra i risultati dell’EDSS standard con i risultati del Timed 25-Foot Walk (T25FW) e del 9-Hole Peg Test (9HPT), consentendo una valutazione più approfondita della progressione che altrimenti sfuggirebbero all’esame neurologico di routine [Figura 7].29 Sebbene questi strumenti possano essere integrati in modo più agile nella pratica clinica, la loro applicazione sistematica resta limitata. Per questo motivo, diversi clinici hanno indicato la funzionalità cognitiva come un possibile indicatore utile nella definizione della progressione, seppur con un ruolo supplementare.30 Tuttavia, l’eterogeneità dei test utilizzati e la complessità nell’interpretazione dei risultati –soprattutto in assenza di una formazione specifica
– rendono difficile una valutazione coerente tra centri diversi e nel tempo. Strumenti di valutazione rapidi, come la Brief International Cognitive Assessment for MS (BICAMS) o il Symbol Digit Modalities Test (SDMT), sono ampiamente utilizzati e possono fornire indicazioni clinicamente rilevanti se somministrati a intervalli regolari, ad esempio ogni sei o dodici mesi.31,32 Tuttavia, diversi specialisti lamentano l’assenza di criteri condivisi e una sensibilità talvolta insufficiente nel rilevare variazioni cognitive di lieve entità. È opinione condivisa che nella pratica clinica siano pochi i centri a utilizzare in modo sistematico test e scale differenti: il BICAMS o l’SDMT vengono talvolta somministrati alla baseline ma raramente ripetuti nel follow-up, e la valutazione cognitiva è spesso eseguita su richiesta e non integrata nella routine. In parallelo, sta emergendo una nuova generazione di strumenti valutativi che integrano diversi indicatori in un’unica piattaforma di raccolta dati: sono i
Figura 7. Descrizione dei test T25FW, 9HPT ed EDSS-Plus per il monitoraggio della disabilità.
Fonte: slide di Repice.
Valuta la funzione deambulatoria misurando quanto velocemente una persona può camminare per 25 piedi (7,62 metri).
Un peggioramento >20% rispetto al valore iniziale è considerato clinicamente significativo.
Il punteggio EDSS-Plus combina EDSS, T25FW e 9HPT, creando un endpoint composito per valutare in modo più completo la progressione della disabilità.
Misura la funzione degli arti superiori cronometrando quanto tempo impiega una persona a inserire e rimuovere nove pioli da nove fori, usando una mano alla volta.
Un incremento >20% rispetto al baseline rappresenta un peggioramento significativo.
Analisi del cammino e dell’esercizio fisico
Il peggioramento dell’andatura indotto dall’esercizio può rivelare deficit neurologici sottili già nelle fasi precoci della SM.
T25FW
9HPT
dispositivi indossabili (motion wearable devices, MWD) e le applicazioni per smartphone.33,34 Lo sviluppo di queste soluzioni, in grado di monitorare in modo continuo l’attività motoria e le funzioni cognitive nella vita quotidiana, rappresenta potenzialmente un ulteriore passo avanti verso una valutazione personalizzata della progressione di malattia. Anche i Patient reported outcomes (PROs) sono utilizzati nell’ambito della SM per identificare tempestivamente eventuali segni di progressione, come raccomandato dalla Food and Drug Administration (FDA) e l’European Medicines Agency (EMA).35,36,37,38 I diversi strumenti utili per valutare la progressione di malattia – che siano scale, questionari o dispositivi digitali – possono però paradossalmente portare a un’eccessiva disponibilità di dati che lo specialista potrebbe non avere il tempo e i mezzi per gestire. Molti clinici hanno sottolineato due criticità: da una parte l’impossibilità, durante il tempo dedicato alla visita, di sottoporre ai pazienti le diverse scale di valutazione; dall’altra le difficoltà nell’analisi della mole di dati raccolti tramite app, questionari o MWD.
Caso clinico 3
Profilo del paziente: casalinga, 41 anni, quattro anni trascorsi dall’ultima relapse.
Tempo trascorso dalla diagnosi: dodici anni.
Trattamento in corso: anti-CD20 da quattro anni.
Sintomi: La paziente presenta EDSS stabile a 5.5 sin dall’inizio della terapia, sebbene con progressivo peggioramento della performance funzionale e della qualità di vita. Negli ultimi dodicidiciotto mesi, pur in assenza di nuove ricadute o di peggioramenti evidenti all’imaging, la paziente riferisce un complesso sintomatologico in progressivo aggravamento: spasticità severa e refrattaria (trattata senza successo anche con associazioni off-label di cannabinoidi), sindrome
dolorosa importante, incontinenza urinaria, disfagia e frequenti episodi di “testa cadente”. Riferisce inoltre una faticabilità invalidante che la costringe a trascorrere la maggior parte del tempo in sedia a rotelle, oltre a una marcata compromissione cognitiva e relazionale che si manifesta anche durante le visite cliniche.
Discussione: Il carico lesionale alla MRI è da tempo elevato, ma stabile rispetto all’ultima ricaduta. Secondo i partecipanti al gruppo di miglioramento, il quadro clinico è compatibile con una progressione silente che sfugge agli strumenti di monitoraggio standard e pone l’interrogativo sull’efficacia attuale della terapia in corso. Il caso solleva anche dubbi sull’opportunità e il timing di un eventuale passaggio a farmaci con diverso meccanismo d’azione.
Sullo sfondo rimane il nodo del coinvolgimento attivo dei pazienti, che raramente partecipano in modo strutturato alla raccolta dei dati necessari per valutare il decorso della malattia e l’eventuale progressione. Molti dei clinici coinvolti nei gruppi di miglioramento hanno concordato sull’utilità di disporre di report – eventualmente redatti dagli specializzandi – o, più in generale, di informazioni raccolte dai pazienti stessi a domicilio, dedicando pochi minuti a settimana a test o questionari. In questo modo, il tempo della visita potrebbe essere impiegato per l’analisi condivisa dei dati, rendendo il monitoraggio più efficiente. Alcuni suggeriscono anche la possibilità di proporre brevi questionari da compilare in sala d’attesa, prima della visita specialistica [Figura 8].
In mancanza di una procedura strutturata che regoli l’utilizzo dei diversi strumenti diagnostici, porre ai pazienti alcune domande sulla loro vita quotidiana è una soluzione pratica e attuabile. Domande mirate, ad esempio su eventuali difficoltà recenti nel salire o scendere le scale, sulla comparsa di disturbi della deglutizione o su alterazioni della funzione sfinterica o sessuale, possono rappresentare un uti-
Figura 8. Sintomatologia potenzialmente indicativa di una progressione della sclerosi multipla. Per ogni sistema e sintomo, in Tabella 1 vengono proposte diverse domande da sottoporre al paziente.
Autonomia motoria e coordinazione
Fatica
Fatica in caso di sforzo Fatica
Funzione sfinterica
Problemi vescicali
Problemi intestinali
Percezione sensoriale
Difficoltà nella deambulazione,
Spasticità
Disturbi della coordinazione
Funzioni cognitive
Aspetti emotivi
Intorpidimento, e formicolio
Problemi di vista
Dolore
le strumento per il neurologo, contribuendo ad accendere segnali d’allerta utili a ipotizzare una possibile progressione di malattia [Tabella 1]. 39,40 Nella pratica clinica, infatti, è spesso l’osservazione dei cambiamenti funzionali nella quotidianità del paziente a suggerire i primi segnali di progressione. Informazioni apparentemente semplici possono fornire indicazioni preziose e precoci, soprattutto nei pazienti con un basso grado di disabilità. Viene segnalato anche di osservare modifiche nella scrittura o nella firma, cambiamenti della voce, disfagia: sintomi spesso sottostimati, ma che impattano sulla qualità di vita e sono allarmi di un possibile viraggio a una fase prevalentemente progressiva.
In questo senso, si è anche ipotizzata inoltre la possibile introduzione di un questionario breve, da ripetere ogni sei mesi, composto da due versioni differenziate: una per i pazienti senza disabilità evidenti, mirata a cogliere i segni più precoci e subclinici, e una per i pazienti già con disturbi motori o progres-
Difficoltà di memoria e nella concentrazione
Ridotta velocità di elaborazione
Difficoltà nel problem solving e nella capacità decisionale
Ansia/depressione
sione nota. Il questionario dovrebbe contenere poche domande, personalizzabili, idealmente somministrate con la mediazione del clinico o dell’infermiere così da evitare l’effetto di un’autovalutazione separata dal contesto clinico.
Diversi studi internazionali condotti in contesti geografici eterogenei – come Stati Uniti, Sud America, Germania e Italia – mostrano una convergenza significativa: i pazienti con SM percepiscono l’inizio della fase progressiva della malattia circa tre anni prima rispetto a quando essa viene formalmente diagnosticata dal clinico.41,42,43 Questo ritardo – variabile tra medie di 2,6 e 3,3 anni a seconda delle valutazioni dei gruppi di ricerca – appare imputabile a molteplici fattori. Da un lato, la comunicazione della progressione è vissuta dal neurologo come un passaggio particolarmente delicato, privo di quelle “buone notizie” – come la disponibilità di trattamenti efficaci – che spesso accompagnano la diagnosi iniziale di SM. Dall’altro, la scarsità di opzioni
Tabella 1. Sintomatologia potenzialmente indicativa di una progressione della sclerosi multipla e esempi di possibili domande da rivolgere al paziente.
Sistema Sintomo Domande da rivolgere al paziente
Difficoltà nella deambulazione
Autonomia motoria e coordinazione
Spasticità
Disturbi della coordinazione
Difficoltà di memoria e nella concentrazione
Funzioni cognitive
Ridotta velocità di elaborazione
Difficoltà nel problem solving e nella capacità decisionale
Aspetti emotivi Ansia/depressione
Intorpidimento e formicolio
Percezione sensoriale
Problemi di vista
Ha notato un peggioramento della sua capacità di camminare?
Ha bisogno di un supporto per camminare, come un bastone o un deambulatore?
Ha difficoltà a mantenere l’equilibrio o cade più spesso?
Riesce a salire le scale senza difficoltà?
Avverte rigidità o spasmi muscolari?
In quali parti del corpo avverte questi sintomi?
La rigidità influisce sulla sua capacità di muoversi o svolgere attività quotidiane?
I sintomi sono peggiorati nel tempo?
Ha difficoltà con movimenti che richiedono coordinazione, come abbottonarsi i vestiti o scrivere?
Ha problemi a raggiungere oggetti o a maneggiarli?
Avverte tremori o movimenti involontari?
Ha difficoltà a ricordare cose, come appuntamenti o conversazioni?
Ha problemi a concentrarsi su compiti o attività?
Si distrae facilmente?
Ha difficoltà a seguire le istruzioni?
Si sente più lento nel pensare o nel reagire alle cose?
Ha difficoltà a tenere il passo con le conversazioni o a elaborare informazioni rapidamente?
Ha difficoltà a prendere decisioni o a risolvere problemi?
Si sente sopraffatto quando deve affrontare situazioni complesse?
Si sente spesso triste, ansioso o irritabile?
Ha notato cambiamenti nel suo umore?
Ha difficoltà a gestire le emozioni?
Ha pensieri negativi o di autolesionismo?
Avverte intorpidimento o formicolio in alcune parti del corpo?
Dove si localizzano queste sensazioni?
Sono costanti o intermittenti?
Sono peggiorate nel tempo?
Ha notato cambiamenti nella sua vista?
Vede doppio o la sua vista è offuscata?
Ha difficoltà a distinguere i colori o a vedere in condizioni di scarsa illuminazione?
Ha dolore agli occhi o mal di testa?
Soffre di dolore cronico?
Dove si localizza il dolore?
Dolore
Funzione sfinterica
Problemi vescicali
Problemi intestinali
Fatica Fatica
Fatica in caso di sforzo
Che tipo di dolore è (bruciore, lancinante, sordo)?
Il dolore è peggiorato nel tempo?
Cosa fa peggiorare o migliorare il dolore?
Ha difficoltà a controllare la vescica?
Sente spesso il bisogno di urinare urgentemente?
Ha difficoltà a svuotare completamente la vescica?
Ha episodi di incontinenza urinaria?
Soffre di stitichezza o diarrea?
Ha difficoltà a controllare l’intestino?
Ha episodi di incontinenza fecale?
Si sente spesso stanco o esausto, anche dopo aver riposato a sufficienza?
La fatica influisce sulla sua capacità di svolgere attività quotidiane?
La fatica è peggiorata nel tempo?
Si sente molto stanco dopo aver svolto attività fisiche o mentali?
Anche piccoli sforzi la fanno sentire esausto?
terapeutiche per la fase progressiva può alimentare una certa inerzia clinica e amministrativa nel modificare lo status del paziente, mantenendolo formalmente in una fase che consenta di proseguire la terapia già in corso.
Oltre agli indicatori già descritti, un altro insieme di fattori discussi nella comunità scientifica per l’individuazione precoce della progressione silente sono i biomarcatori plasmatici e strumentali. Tra questi, la catena leggera dei neurofilamenti (neurofilament light chain, NfL) – proteina dosabile sia nel liquor che nel sangue e le cui variazioni sono espressione del danno assonale – rappresenta uno dei marcatori più promettenti in quanto riflette l’attività infiammatoria e neurodegenerativa ed è potenzialmente utile sia nella predizione di una malattia smoldering sia nel monitoraggio della risposta alle DMT ad alta efficacia.44,45 Sebbene al momento l’uso sistematico della NfL non sia sostenibile nella pratica ambulatoriale, la sua introduzione – insieme ad altri marcatori di danno neuroassonale – potrebbe rappresentare un valido complemento al monitoraggio clinico e radiologico, a condizione che vengano consolidate evidenze più robuste a supporto. In questo contesto si inserisce anche la tomografia a coerenza ottica (optical coherence tomography, OCT), tecnica non invasiva che consente di monitorare la neurodegenerazione retinica in vivo, correlando l’assottigliamento di specifici strati retinici con la progressione clinica della malattia.46,47,48
Un aspetto rilevante è il potenziale ruolo dell’attività fisica come strumento di automonitoraggio. I pazienti che hanno contezza della propria routine sportiva possono essere facilitati nella “misurazione” delle proprie capacità: ad esempio, sulla distanza percorsa o sui tempi, possono cogliere precocemente segnali di deterioramento o peggioramento. Il momento in cui il paziente percepisce un cambiamento rispetto alla propria condizione iniziale può rappresentare un segnale chiave dell’avvio di una fase progressiva della malattia. Non si tratta neces-
sariamente di un peggioramento oggettivabile a livello motorio, ma spesso di una riduzione della capacità funzionale nelle attività quotidiane – lavorative o sociali – rispetto al periodo immediatamente successivo alla diagnosi. Per cogliere questa transizione è essenziale che il clinico adotti un approccio di ascolto attivo indagando esplicitamente, anche in assenza di una lamentela spontanea, il rendimento dei pazienti nei contesti relazionali.
Il riconoscimento dei fattori confondenti rappresenta infatti una delle sfide più complesse nella gestione della SM: è spesso difficile distinguere tra un sintomo realmente attribuibile alla malattia e una componente funzionale o secondaria. Nel valutare il versante cognitivo, ad esempio, è importante ricordare che condizioni come la depressione o un tono dell’umore deflesso possono influire in modo significativo sulla performance, indipendentemente da un reale peggioramento clinico. In assenza di una correlazione oggettiva con parametri neurologici o radiologici, emerge il quesito se sintomi come la ridotta tolleranza a eventi di lavoro – ad esempio un infermiere a cui vengono assegnati turni consecutivi di notte – debbano essere interpretati come manifestazione di una possibile infiammazione smoldering, ovvero un segnale clinico precoce potenzialmente rilevante, oppure come una normale variazione funzionale attribuibile all’età o al contesto lavorativo.
Caso clinico 4
Profilo del paziente: donna, 38 anni, dieci anni trascorsi dall’ultima relapse.
Tempo trascorso dalla diagnosi: undici anni.
Trattamento in corso: teriflunomide.
Sintomi: La paziente al momento della diagnosi aveva un EDSS di 1.5, arrivato ora a 3.0. Le ultime risonanze non mostrano nuove attività infiammatorie, se non una lesione frontale nel 2019. Tuttavia, la paziente lamenta da uno-due anni
una riduzione della resistenza fisica (da diversi chilometri, ora riesce a percorrerne meno di uno), difficoltà cognitive e urgenza sfinterica, in un contesto di sindrome depressiva in trattamento.
Discussione: La discussione si concentra su due temi centrali: la paziente è in progressione? È raccomandabile un cambio di terapia dopo più di dieci anni di stabilità apparente?
I pareri sono articolati e riflettono una varietà di approcci clinici. Alcuni specialisti sottolineano i fattori prognostici sfavorevoli come l’esordio con lesioni spinali e sottotentoriali e la giovane età, che giustificherebbero un’intensificazione terapeutica indipendentemente dalla presenza di nuove lesioni. Altri ribadiscono che l’attuale assenza di attività infiammatoria non rende automatica l’escalation terapeutica e non si fidano delle sensazioni della paziente, pensando che possano dipendere dalla sua condizione depressiva. In conclusione, non emerge una risposta univoca, ma si delinea un consenso di fondo sul fatto che la paziente sia verosimilmente in una fase di progressione latente. Il cambio di terapia viene considerato giustificabile anche solo in un’ottica di protezione prospettica. Per ora, l’unica strategia è rappresentata dall’introduzione di un antiCD20.
La possibilità di riconoscere precocemente segnali subclinici di progressione dipende, infine, anche dalla continuità del rapporto tra medico e paziente. Nei centri ad alta affluenza, dove più professionisti si alternano nella gestione clinica, il rischio di una frammentazione del monitoraggio longitudinale è concreto, specie se le visite non sono organizzate all’interno di un sistema che garantisca coerenza nella presa in carico. La presenza di una segreteria dedicata o di un’organizzazione interna che permetta di mantenere un riferimento clinico stabile può aiutare a mitigare questi limiti, facilitando l’intercettazione tempestiva dei cambiamenti soggettivi che spesso precedono quelli neurologici obiettivi.
Trattamento della progressione: evidenze e criticità
I dati più recenti relativi al trattamento della SM-SP, provenienti da studi clinici randomizzati e da registri osservazionali, stanno gradualmente ridefinendo le strategie terapeutiche per questa fase della malattia.
Nella pratica clinica attuale, la gestione terapeutica dei pazienti con SM in viraggio verso la fase progressiva rimane una delle sfide più complesse, non solo per la mancanza di criteri diagnostici univoci, ma anche per l’assenza di trattamenti approvati specificamente per una forma progressiva senza attività infiammatoria.
Come è stato già accennato, questa mancanza ha delle ripercussioni su un altro punto emerso dal confronto tra esperienze cliniche: la flessibilità nella classificazione della malattia. Alcuni neurologi scelgono di non etichettare rigidamente il fenotipo e distinguere tra RR e SP, preferendo descrivere il quadro clinico e radiologico in termini funzionali: presenza di attività infiammatoria, sospetti segni di progressione, comorbidità rilevanti. Questo approccio, considerato più “narrativo” e talvolta riportato direttamente nei referti clinici, può agevolare decisioni terapeutiche più personalizzate e la continuità nella presa in carico tra specialisti.
A livello terapeutico, l’utilizzo precoce di farmaci ad alta efficacia è ormai prassi diffusa. Una recente review ha però evidenziato come le terapie attualmente disponibili per la SM, pur avendo significativamente ridotto la quota di disabilità legata alle ricadute, mostrino una limitata efficacia nel contenere la PIRA.49 I dati disponibili delineano un quadro che ancora non lascia soddisfatti gli specialisti: in termini di efficacia generale delle DMT sul rischio di progressione emergono alcuni segnali positivi, ma a prevalere sono evidenze non del tutto incoraggianti. Un primo dato rilevante è che i pazienti con forme clinicamente più aggressive trattati fin dall’esordio con
farmaci come natalizumab mostrano comunque una maggiore probabilità di PIRA rispetto a pazienti con decorso meno aggressivo avviati a DMT meno potenti.50 D’altro canto una maggiore esposizione cumulativa ai DMT – indipendentemente dalla loro efficacia nominale – è associata a un minor rischio di progressione, suggerendo un possibile effetto protettivo legato alla durata della terapia.51 Nel confronto tra adulti e pazienti con esordio pediatrico emerge una differenza importante: la progressione indipendente dalle ricadute è molto più rara nei bambini, probabilmente per un’attività infiammatoria predominante e un intervallo più breve tra l’esordio biologico e quello clinico della malattia. Questo potrebbe spiegare il motivo della minore componente smoldering nei quadri pediatrici.52 Infine, uno studio comparativo su pazienti trattati con natalizumab o ocrelizumab (195 per ciascun gruppo) ha mostrato una lieve riduzione del rischio di PIRA con entrambi i farmaci, senza differenze significative.53 Nella pratica clinica, tuttavia, resta la percezione che alcuni pazienti siano destinati a evolvere verso una forma progressiva indipendentemente dal trattamento in corso.
Caso clinico 5
Profilo della paziente: ricercatrice presso la Facoltà di Lettere e filosofia, 36 anni.
Tempo trascorso dalla diagnosi: quattro anni. Trattamento in corso: trattata da quattro anni con anti-CD20 (ocrelizumab).
Sintomi: l’EDSS è passato da 2.0 a 3.0 negli ultimi dodici mesi. La paziente lamenta un aumento della fatica nello svolgimento delle attività di vita quotidiana e una ridotta attenzione.
Discussione: si discute se il peggioramento clinico sia espressione di reale progressione secondaria o se vi siano componenti funzionali reattive (es. disturbo dell’umore). Tuttavia, i test
cognitivi obiettivi sembrerebbero confermare la progressione. La paziente è in trattamento con anti-CD20 dal momento della diagnosi, e il controllo della malattia appare insoddisfacente. Si apre il dibattito – al momento puramente teorico – sull’eventualità di interrompere la terapia attuale in favore di un trattamento differente. Si esprime prudenza su uno switch in una paziente giovane con carico midollare e atrofia documentata. Il rischio di rebound infiammatorio è basso, ma permane la preoccupazione per una lesione midollare invalidante.
Rimangono alcune domande aperte. Il peggioramento è legato a progressione neurodegenerativa precoce o a fallimento terapeutico? Quanto incide la riserva cognitiva nella percezione soggettiva di peggioramento? La paziente è troppo avanti nella progressione per trarre beneficio da un nuovo trattamento o è questo il momento ideale per intervenire? Il caso evidenzia una tensione clinica tra il rischio di non agire prontamente e quello di introdurre una terapia nuova in assenza di dati consolidati per pazienti con progressione rapida e precoce. Si sottolinea la necessità di criteri più chiari per definire il momento opportuno per abbandonare gli anti-CD20 e l’eventuale indicazione a un trattamento differente.
Nel trattamento della PIRA, con particolare riferimento alla SM-SP, uno dei meccanismi patogenetici ritenuti centrali è l’infiammazione compartimentalizzata a livello del sistema nervoso centrale. Un ruolo importante viene attribuito in questo contesto alle cellule B, in particolare per la formazione di aggregati linfoidi negli spazi subaracnoidei, e alla microglia, coinvolta in una forma di infiammazione più diffusa e meno visibile. Un recente studio ha dimostrato attività microgliale in quasi il 60% delle lesioni altrimenti considerate stabili (ovvero non captanti gadolinio e non in crescita), suggerendo che anche in assenza di eviden-
Figura 9. Meccanismo d’azione degli inibitori di BTK e inibitori di BTK brain-penetrant nell’ambito della sclerosi multipla. Fonte: Krämer et al. Nat Rev Neurol 2023.
Inibitori di BTK
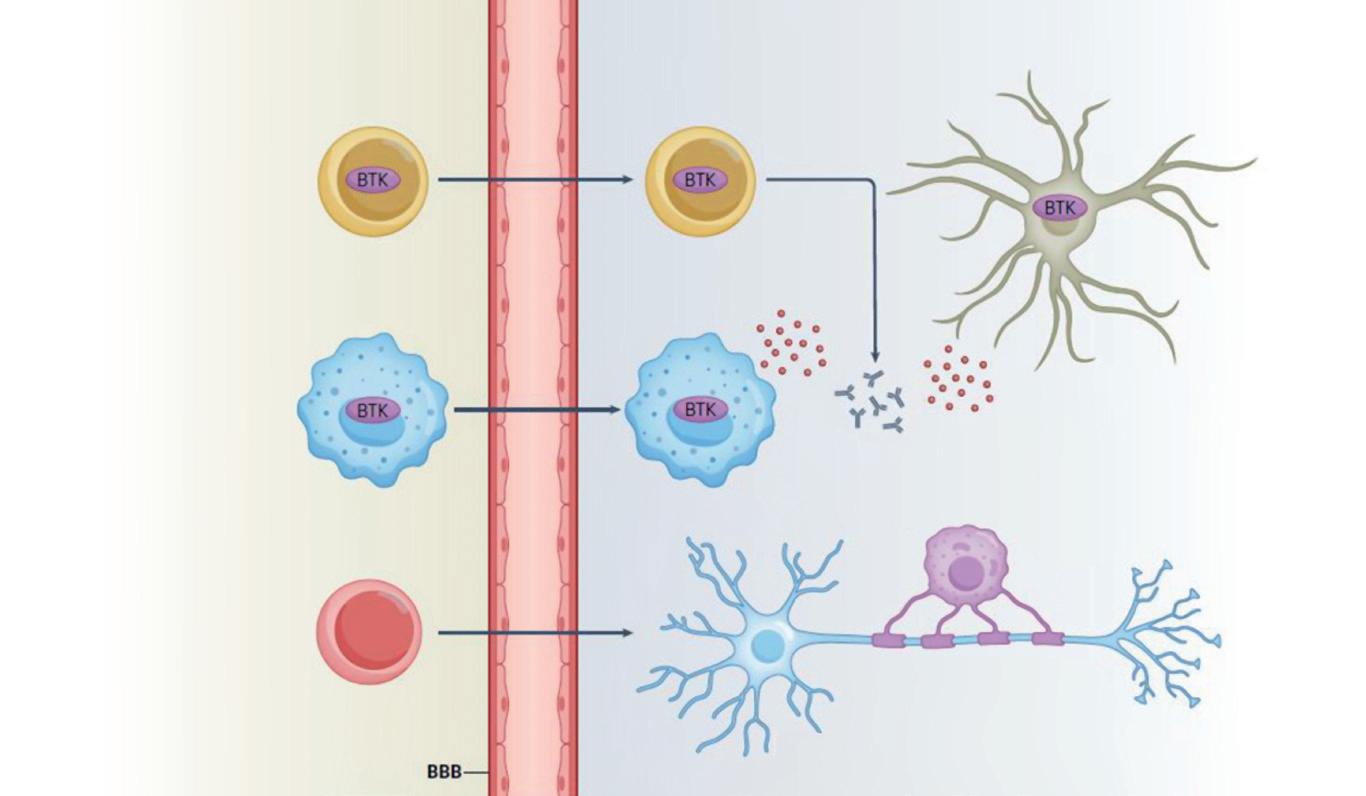
Inibitori di BTK brain-penetrant
Modulazione delle cellule B nel SNC
Inibizione dell’attivazione e della proliferazione delle cellule B
Inibizione dell’attivazione dei macrofagi
Inibizione della microglia e/o attivazione dei macrofagi
Demielinizzazione Degrado assonale
za radiologica tradizionale, possa persistere un’infiammazione cronica “sotto traccia”, associata alla progressione silente della malattia.54 Ulteriori conferme provengono da dati che evidenziano una correlazione tra la presenza ed evoluzione di lesioni caratterizzate da un bordo paramagnetico che riflette una persistente attivazione microgliale – e un aumentato rischio di progressione indipendente dalle ricadute.55 Una nuova prospettiva in questo ambito è rappresentata dall’inibizione della tirosina chinasi di Bruton (Bruton’s tyrosine kinase, BTK), una tirosina chinasi non recettoriale espressa nella maggior parte delle cellule ematopoietiche. La BTK funge da nodo di connessione tra i recettori specifici di superficie e le vie di segnalazione intracellulari, traducendo lo stimolo immunitario in attivazione cellulare. Il razionale dell’inibizione della BTK risiede nella capacità di
agire sia sulle cellule B sia sulla microglia, intervenendo simultaneamente sui due principali bersagli dell’infiammazione cronica attiva.15,56
Considerato il ruolo centrale di BTK nella segnalazione delle cellule B e della microglia a livello del sistema nervoso centrale, è stato ipotizzato che la sua inibizione possa non solo ridurre l’infiammazione acuta associata alle lesioni, ma anche esercitare effetti favorevoli sulle lesioni croniche attive e sugli infiltrati meningei [Figura 9].
La loro importanza, al momento, risiede nel fatto che queste riflettono l’evoluzione del ragionamento clinico: superare la dicotomia tra recidivante e progressiva per gestire in modo precoce quei pazienti in transizione fenotipica, spesso sottorappresentati nei trial ma che vengono naturalmente incontrati nella pratica reale.
Microglia
Citochine
Oligodendrocita
Neurone
Cellula T
Cellula B
Macrofago
Caso clinico 6
Profilo del paziente: Cacciatore, 55 anni.
Tempo trascorso dalla diagnosi: dodici anni.
Trattamento in corso: Fingolimod. Interferon beta-1b (fino a novembre 2017), poi teriflunomide (fino a ottobre 2018).
Sintomi: Il decorso nei primi tre anni risulta stabile, ma nel 2017 si rileva attività radiologica di malattia. Nel 2018 l’obiettività neurologica rileva una deambulazione tallonata e un incremento dell’EDSS. Si opta per un nuovo switch terapeutico, con miglioramento parziale post-steroide e apparente stabilizzazione clinica e radiologica nell’arco del primo anno. Tuttavia, l’andatura si rivela progressivamente scoordinata, si rileva una lieve atassia all’arto inferiore sinistro e aumenta la faticabilità. Emerge una diagnosi di diabete, non ben controllato.
Nel 2024, il paziente lamenta astenia marcata dell’arto inferiore e difficoltà nella marcia, con riduzione significativa dell’autonomia. Viene intrapreso un ciclo intensivo di fisioterapia, con beneficio soggettivo ma senza variazioni significative dell’EDSS. Le attività lavorative e cognitive restano integre, così come il livello di attività fisica. A maggio 2024 compaiono segni subclinici agli arti superiori (tremore e peggioramento della coordinazione fine) in assenza di sintomi riferiti.
Discussione: Alla luce di questi dati, si solleva l’ipotesi di un decorso progressivo presente già da anni, precedentemente mascherato da ricadute isolate o da un quadro clinico sfumato. La questione centrale riguarda la tempistica di un possibile switch terapeutico: il paziente presenta attualmente progressione clinica senza attività radiologica o nuove ricadute, ma non si ritiene ideale l’avvio di un anti-CD20 per età, comorbilità e rischio infettivo.
Il profilo immunologico e l’assenza di opzioni mirate alla progressione impongono una riflessione sull’appropriatezza e sulle tempistiche di un trattamento differente.
Nella loro pratica clinica, infine, molti neurologi hanno iniziato a introdurre anche indicazioni terapeutiche che fanno riferimento a interventi non farmacologici. Accanto alla raccomandazione – talvolta ancora percepita come controindicata da pazienti e caregiver – di mantenere una regolare attività fisica, alcuni professionisti propongono, ad esempio, interventi integrativi basati sulla fitoterapia e su principi della medicina tradizionale ayurvedica. È stata ricordata, inoltre, l’importanza di associare lo stretching e l’allungamento, svolti magari in occasione di attività come yoga o pilates, a sforzi aerobici per prevenire l’ipertono o la spasticità nel lungo termine. Assume un ruolo sempre più centrale, infine, la cura dell’alimentazione. In questa prospettiva, al momento della diagnosi vengono spesso fornite prime indicazioni pratiche sull’organizzazione della dieta quotidiana e viene incentivata l’assunzione di probiotici. Tra i suggerimenti più comuni quelli di organizzare al meglio i macronutrienti: ridurre il consumo di alimenti ultra-processati, limitare la carne rossa una volta alla settimana, evitare eccessi di sodio. Sebbene queste indicazioni non costituiscano ancora un protocollo formalizzato e non siano esclusive per la SM, riflettono un crescente interesse per strategie preventive non farmacologiche da integrare nella presa in carico globale del paziente [Figura 10]. Anche in questo caso, tuttavia, molti esperti lamentano l’assenza di linee guida riconosciute che regolamentino in modo chiaro un approccio strutturato a questo tipo di intervento. Alcuni operatori riferiscono di non sentirsi culturalmente preparati su alimentazione, attività fisica e integratori, sottolineando l’assenza di formazione specifica durante il percorso medico. In tal senso, è emersa la proposta di elaborare materiali condivisi da fornire al paziente al momento della diagnosi, con indicazioni pratiche e validate, in grado di conferire maggiore autorevolezza e concretezza al messaggio terapeutico. Sul piano educativo, è emerso un progetto che prevede colloqui infermieristici post-diagnosi focalizzati
Figura 10. Rappresentazione dell’integrazione tra interventi farmacologici e non farmacologici per una gestione multidisciplinare della malattia.
Neurologo
Assistente sociale
Interventi farmacologici
Le DMTs possono ridurre il numero di ricadute e rallentare Ia progressione della disabiIità
Caregiver
Gestione ottimale della SM + =
I trattamenti sintomatici aiutano ad alleviare i nuovi sintomi
I corticosteroidi vengono utilizzati nelle fasi acute di malattia
Interventi non farmacologici
Esempi
Riabilitazione cognitiva
Fisioterapia
Terapia occupazionale
Medicina complementare e/o alternativa
su alimentazione e movimento, integrati da un supporto psicologico iniziale offerto in collaborazione con l’Associazione Italiana Sclerosi Multipla (AISM).57
Conclusioni e prospettive future
Un elemento chiave emerso nel corso degli incontri del progetto La rivelazione dell’invisibile: i sottili cambiamenti nel paziente SM e i segnali di progressione è la speranza che nuovi interventi farmaceutici potranno rappresentare una strategia terapeutica per pazienti che non mostrano più segni di attività infiammatoria ma che continuano a progredire nella disabilità. Si tratta spesso di pazienti con una lunga storia di malattia, un EDSS compreso tra 3.5 e 6.0, già trattati con DMT a bassa o moderata efficacia, il cui trattamento è ancora in corso senza evidenti benefici oppure è stato progressivamente sospeso. Ma non solo: intercettare e trattare la PIRA in modo precoce aggiungerebbe un›opzione terapeutica in di-
Fisioterapista
Terapista occupazionale
Infermiere
Psicologo
In definitiva, un approccio multidisciplinare che coinvolga diversi professionisti sanitari è iI modo ottimale per gestire la SM. Non tutti i pazienti con SM, però, hanno accesso a una presa in carico di questo tipo.
verse classi di pazienti, in una fase in cui l’infiammazione compartimentalizzata resta l’unico target potenzialmente aggredibile.
Al termine degli incontri è stata infine sottolineata la necessità di ampliare l’attenzione clinica verso le manifestazioni invisibili della SM, soprattutto perché capaci di influenzare significativamente la qualità di vita dei pazienti.
La fatica, uno dei sintomi più frequenti e invalidanti della SM, può essere caratterizzata come centrale e periferica, primaria e secondaria, e può precedere o segnalare l’insorgenza di una progressione clinica.58 Nel contesto della malattia, questo sintomo emerge come uno tra i più comuni e debilitanti, con una prevalenza stimata tra il 53% e il 90% dei pazienti; è spesso riferito come uno dei più difficili da gestire nella quotidianità, sia per la sua natura soggettiva che per il suo impatto funzionale e psicologico.58,59,60
Negli incontri si è discusso anche dei limiti intrinseci delle scale per la valutazione della fatica – come la Fatigue Severity Scale (FSS), la Modified Fatigue Impact Scale (MFIS) e la Visual Analogue Scale (VAS) –
nel cogliere appieno l’impatto soggettivo di questo sintomo nei pazienti.61,62,63,64 Un’altra manifestazione chiave è quella dei disturbi cognitivi, che nei pazienti con SM possono comparire precocemente anche in presenza di un EDSS relativamente basso. Secondo molti specialisti la compromissione cognitiva – in particolare nei domini dell’attenzione, della memoria e delle funzioni esecutive – non è sempre adeguatamente indagata, sebbene possa costituire un indice precoce di progressione e contribuire alla disabilità percepita dal paziente. La componente emozionale, in particolare sotto forma di depressione e ansia, rappresenta poi un ulteriore livello di complessità nella gestione del paziente con SM: queste condizioni, spesso croniche e scarsamente riconosciute, possono alterare la percezione soggettiva dei sintomi e compromettere l’aderenza terapeutica. Altri segni spesso trascurati nelle fasi iniziali della progressione sono la compromissione degli arti superiori, i disturbi sfinterici e sessuali, la disfagia o la disfonia.65,66 Questi elementi, sebbene poco visibili o riferibili a scale di disabilità, contribuiscono in modo rilevante all’aggravamento complessivo della condizione funzionale. Il riconoscimento di un viraggio verso una fase secondariamente progressiva nella sclerosi multipla rappresenta oggi una delle principali sfide, sia nella ricerca che nella pratica clinica. Restano però dei nodi aperti che la comunità scientifica è chiamata ad affrontare. Tra questi, la necessità di una definizione condivisa che consenta di riconoscere precocemente la progressione, anche in assenza di segni radiologici evidenti; l’integrazione tra strumenti clinici oggettivi e test soggettivi o questionari, per ottenere una valutazione più completa e centrata sulla persona; l’adozione di un approccio multidisciplinare che coinvolga psicologi, terapisti occupazionali, logopedisti, fisioterapisti e neuroradiologi; la promozione di linee guida condivise e uniformi per il follow-up dei pazienti con SM, per garantire una gestione strutturata e coerente nel tempo.
Bibliografia
1. Smoldering neuroinflammation: una nuova prospettiva nella gestione clinica della sclerosi multipla.
2. Coles AJ, Cox A, Le Page E, et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol 2006; 253: 98-108.
3. Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol 2020; 77: 1132-1140.
4. Lublin FD, Häring DA, Ganjgahi H, et al. How patients with multiple sclerosis acquire disability. Brain 2022; 14; 145(9): 3147-3161.
5. Portaccio E, Bellinvia A, Fonderico M, et al. Progression is independent of relapse activity in early multiple sclerosis: a real-life cohort study. Brain 2022;145(8):2796-2805.
6. Müller J, Cagol A, Lorscheider J, et al. Harmonizing definitions for progression independent of relapse activity in multiple sclerosis: a systematic review. JAMA Neurol. 2023;80(11):1232-1245.
7. Prosperini L, Ruggieri S, Haggiag S, et al. Prognostic accuracy of NEDA-3 in long-term outcomes of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1059.
8. Scalfari A, Traboulsee A, Oh J et al. SmoulderingAssociated Worsening in Multiple Sclerosis: An International Consensus Statement on Definition, Biology, Clinical Implications, and Future Directions. Ann Neurol (2024); 96: 826-45.
9. Lynch S, Baker S, Nashatizadeh M, et al. Disability measurement in Multiple Sclerosis patients 55 years and older: What is the Expanded Disability Status Scale really telling clinicians. Mult Scler Relat Disord. 2021;49:102724.
10. Müller J, Sharmin S, Lorscheider J et al. Standardized Definition of Progression Independent of Relapse Activity (PIRA) in Relapsing-Remitting Multiple Sclerosis. JAMA Neurol 2025; 82;(6):614-625.
11. Kappos L, Butzkueven H, Wiendl H, et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Mult Scler 2018; 24(7): 963-973.
12. Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol 2019; 85(5): 653-666.
13. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014;83(3):278 -286.
14. Kappos L, Bar- Or A, Cree BA, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double -blind, randomised, phase 3 study. Lancet 2018;391(10127):1263-73.
15. Registro MSBase: https://www.msbase.org/.
16. Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain 2016;139(9):2395-2405.
17. Iaffaldano P, Lucisano G, Guerra T, et al. Towards a validated definition of the clinical transition to secondary progressive multiple sclerosis: A study from the Italian MS Register. Mult Scler 2022;28(14):2243-52.
18. Tallantyre EC, Major PC, Atherton MJ, et al. How common is truly benign MS in a UK population? J Neurol Neurosurg Psychiatry 2019;90:522-528.
19. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Eng J MEd 2018; 378: 169-180.
20. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology 2016; 87: 1393-9.
21. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17(2): 162-173.
22. Sastre-Garriga J, Pareto D, Battaglini M, et al. MAGNIMS consensus recommendations on the use of brain and spinal cord atrophy measures in clinical practice. Nat Rev Neurol 2020; 16: 171-182.
23. Sastre-Garriga J, Ingle GT, Rovaris M, et al. Long-term clinical outcome of primary progressive MS: predictive value of clinical and MRI data. Neurology 2005; 65: 633-635.
24. De Stefano N, Giorgio A, Battaglini M, et al. Assessing brain atrophy rates in a large population of untreated multiple sclerosis subtypes. Neurology 2010; 74: 1868-76.
25. Azevedo CJ, Overton E, Khadka S, et al. Early CNS neurodegeneration in radiologically isolated syndrome. Neurol Neuroimmunol Neuroinflamm 2015; 2(3): e102.
26. Giovannoni G, Hawkes CH, Lechner-Scott J, et al. Multiple sclerosis is one disease. Mult Scler Relat Disord. 2022; 63:103961.
27. Thompson AJ, Hobart JC. Multiple sclerosis: assessment of disability and disability scales. J Neurol 1998; 245: 189-196.
28. Kobelt G, Thompson A, Berg J, et al. New insights into the burden and costs of multiple sclerosis in Europe. Mult Scler J 2017; 23(8): 1123-1136.
29. Cadavid D, Cohen JA, Freedman MS, et al. The EDSSPlus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler 2017; 23(1): 94-105.
30. Goldman MD, LaRocca NG, Rudick RA, et al. Evaluation of multiple sclerosis disability outcome measures using pooled clinical trial data. Neurology 2019; 93: e1921-31.
31. Benedict RH, Amato MP, Boringa J, et al. Brief International Cognitive Assessment for MS (BICAMS): international standards for validation. BMC Neurol 2012; 12: 55.
32. Benedict RH, DeLuca J, Phillips G, et al. Validity of the Symbol Digit Modalities Test as a cognition performance outcome measure for multiple sclerosis. Mult Scler 2017; 23(5): 721-733.
33. Midaglia L, Mulero P, Montalban X, et al. Adherence and Satisfaction of Smartphone- and SmartwatchBased Remote Active Testing and Passive Monitoring in People With Multiple Sclerosis: Nonrandomized Interventional Feasibility Study J Med Internet Res 2019; 21(8): e14863.
34. Wendrich K, Van Oirschot P, Martens MB, et al. Toward digital self-monitoring of multiple sclerosis: investigating first experiences, needs, and wishes of people with MS. Int J MS Care 2019; 21: 282-291.
35. Venkatesan P. New European guidance on patientreported outcomes. Lancet Oncol 2016; 17: e226.
36. Kluzek S, Dean B, Wartolowska KA. Patient-reported outcome measures (PROMs) as proof of treatment efficacy. BMJ Evid Based Med 2021; 27: 153-155.
37. Retzer A, Aiyegbusi OL, Rowe A, et al. The value of patient-reported outcomes in early-phase clinical trials. Nat Med 2022; 28: 18-20.
38. D’Amico E, Haase R, Ziemssen T. Review: patientreported outcomes in multiple sclerosis care. Mult Scler Relat Disord 2019; 33: 61-66.
39. Meca-Lallana JE, Casanova B, Rodríguez-Antigüedad A, et al. Consensus on early detection of disease progression in patients with multiple sclerosis. Front Neurol 2022; 13: 931014.
40. Kaplan TB et al. Challenges to longitudinal characterization of lower urinary tract dysfunction in multiple sclerosis. Mult Scler Relat Disord 2022; 62: 103793.
41. Katz Sand I, Krieger S, Farrell C, et al. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Multiple Sclerosis Journal. 2014;20(12):1654-1657.
42. Rojas JI, Patrucco L, Alonso R, et al. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis: Multicenter study in Argentina. Multiple Sclerosis Journal. 2020;27(4): 579-584.
43. Solari A, Giovannetti AM, Giordano A, et al. Conversion to Secondary Progressive Multiple Sclerosis: Patient Awareness and Needs. Results From an Online Survey in Italy and Germany. Front Neurol 2019;10:916.
44. Olsson B, Portelius E, Cullen NC, et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol 2018: 1-8.
45. Gaetani L, Paolini Paoletti F, et al. CSF and blood biomarkers in neuroinflammatory and neurodegenerative diseases: implications for treatment. Trends Pharmacol Sci 2020; 41(12): 1023-1037.
46. Nolan-Kenney RC, Liu M, Akhand O, et al. Optimal intereye difference thresholds by optical coherence tomography in multiple sclerosis: An international study. Ann Neurol 2019; 85(5): 618-629.
47. Saidha S, Al-Louzi O, Ratchford JN, et al. Optical coherence tomography reflects brain atrophy in multiple sclerosis: a fouryear study. Ann Neurol 2015; 78: 801-13.
48. Martínez-Lapiscina EH, Arnow S, Wilson JA, et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol 2016; 15: 574-84.
49. Guerra T, Iaffaldano P. A Window into New Insights on Progression Independent of Relapse Activity in Multiple Sclerosis: Role of Therapies and Current Perspective. Int J Mol Sci 2025;26(3):884.
50. Graf J, Leussink VI, Soncin G, et al. Relapseindependent multiple sclerosis progression under natalizumab. Brain Commun 2021;3(4):fcab229.
51. Puthenparampil M, Gaggiola M, Ponzano M, et al. High NEDA and No PIRA in Natalizumab -Treated Patients With Pediatric- Onset Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm 2024;11(5):e200303.
52. Chisari CG, Aguglia U, Amato MP, et al. Long-term effectiveness of natalizumab in secondary progressive multiple sclerosis: A propensity-matched study. Neurotherapeutics 2024;21(4):e00363.
53. Iaffaldano P. A comparison of natalizumab and ocrelizumab on disease progression in multiple sclerosis. Ann Clin Transl Neurol 2024;11(8):2008-15.
54. Hamzaoui M, Garcia J, Boffa G, et al. Positron emission tomography with [^18F]-DPA-714 unveils a smoldering component in most multiple sclerosis lesions which drives disease progression. Ann Neurol 2023;94(2):366 -383.
55. Reeves JA, Bartnik A, Jakimovski D, et al. Associations Between Paramagnetic Rim Lesion Evolution and Clinical and Radiologic Disease Progression in Persons With Multiple Sclerosis. Neurology 2024;103(10):e210004.
56. Krämer J, Bar-Or A, Turner TJ, et al. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat Rev Neurol 2023;19(5):289-304.
57. https://www.aism.it/tipologie_di_attivit%C3%A0_di_ supporto_del_territorio
58. Penner IK. Evaluation of cognition and fatigue in multiple sclerosis: daily practice and future directions. Acta Neurol Scand 2016;134:19-23.
59. Thompson A, Kobelt G, Berg J, et al. New insights into the burden and costs of multiple sclerosis in Europe:
Results for the United Kingdom. Mult Scler 2017;23(2_ suppl):204 -216.;
60. Kister I, Bacon TE, Chamot E, et al. Natural history of multiple sclerosis symptoms. Int J MS Care 2013;15(3):146 -58.
61. Krupp LB, LaRocca NG, Muir-Nash J. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 1989;46(10):1121-3.
62. Fisk JD, Pontefract A, Ritvo PG. The impact of fatigue on patients with multiple sclerosis. Can J Neurol Sci 1994;21(1):9 -14.
63. National Multiple Sclerosis Society. Modified Fatigue Impact Scale. Available at: www.nationalmssociety. org/For-Professionals/Researchers/Resources-forResearchers/Clinical-Study-Measures/ModifiedFatigue-Impact-Scale-(MFIS);
64. Kos D, Nagels G, D’Hooghe MB. A rapid screening tool for fatigue impact in multiple sclerosis. BMC Neurol 2006;6:27.
65. Carotenuto A, De Giglio L, Chiodi A, et al. Validation of the Italian version of the Multiple Sclerosis Intimacy and Sexuality Questionnaire-19. Neurol Sci 2021;42(7):2903-2910.
66. Lakin L, Davis BE, Binns CC. Comprehensive approach to management of multiple sclerosis: addressing invisible symptoms—a narrative review. Neurol Ther 2021;10(1):75 -98.
Una pubblicazione de Il Pensiero Scientifico Editore e Think2it
Tutti i diritti riservati Neuroinfo – Anno II, settembre 2025
Rassegna realizzata da NeuroInfo in collaborazione con
