Introduzione
Le vasculiti ANCA associate (AAV) sono malattie autoimmuni rare, caratterizzate da alterazioni infiammatorie e necrotizzanti della parete dei piccoli vasi, che causano ischemia dei tessuti1. Il coinvolgimento renale, particolarmente frequente, si manifesta clinicamente con un’insufficienza renale rapidamente progressiva e, sul piano istologico, con una glomerulonefrite extracapillare necrotizzante con pochi o assenti immunodepositi all’immunofluorescenza e alla microscopia elettronica. Le AAV includono la granulomatosi con poliangioite (GPA), la granulomatosi eosinofilica con poliangioite (EGPA) e la poliangioite microscopica (MPA)2 . Il coinvolgimento renale nelle AAV è associato ad una prognosi peggiore non solo renale ma del paziente, e nella MPA in particolare il rischio di morte ha un hazard ratio di 3.7 nei pazienti con insufficienza renale alla diagnosi3 . Le recenti linee guida KDIGO del 2024 suggeriscono di effettuare una terapia di induzione con rituximab o ciclofosfamide in associazione a terapia steroidea ad alte dosi da scalare progressivamente, o in alternativa in associazione con avacopan. All’induzione, i pazienti vengono solitamente trattati con metilprednisolone (dose complessiva 1–3 g) e, a seguire, prednisone orale fino a 1mg/kg/die 4. In ADVOCATE – un trial di fase III randomizzato, controllato, in doppio cieco, pubblicato sul New England Journal of Medicine nel 2021 – avacopan in associazione a ciclofosfamide o rituximab si è dimostrato più efficace rispetto al gruppo trattato con steroide ad alte dosi nel raggiungere la remissione alla settimana 52. I pazienti trattati con avacopan, un antagonista del recettore C5a del complemento, hanno
utilizzato un dosaggio nettamente inferiore di steroide, hanno raggiunto la remissione in percentuale più elevata e hanno avuto un maggior recupero della funzione renale in termini di filtrato glomerulare rispetto ai pazienti del gruppo placebo5 .
Nel giugno 2024 l’Agenzia Italiana del Farmaco (AIFA) ha approvato l’impiego in regime di rimborsabilità del medicinale avacopan. Ora sono attesi gli studi in real life che devono confermare gli ottimi risultati del trial.
Il caso clinico descritto è molto peculiare, perché si tratta di una paziente affetta da una diversa patologia autoimmune e quindi già in terapia immunosoppressiva, in cui il riscontro istologico di glomerulonefrite extracapillare necrotizzante è stato inatteso.
Presentazione del caso
Il caso clinico riguarda una donna di 56 anni che, già nel 1982, era stata ricoverata presso il reparto di Nefrologia per anomalie urinarie (proteinuria di 1,2 g/24h ed ematuria) in presenza di funzione renale normale. La biopsia renale eseguita in quell’occasione aveva evidenziato una glomerulonefrite a depositi di IgA. Non era stata avviata alcuna terapia specifica, e la proteinuria era andata remissione spontanea. La paziente ha quindi proseguito il follow-up nefrologico fino al 1996. Nel 2015 la paziente iniziava terapia con rivaroxaban per la comparsa di un’embolia polmonare idiopatica. Nello stesso anno veniva riscontrata diagnosi di connettivite indifferenziata e, successivamente, di sindrome di Sjögren (sindrome sicca-artralgie; SSA+ SSB+ ANA 1/320, FR/antiCCP/antidsDNA negativi). La paziente veniva quindi presa in carico dai colleghi reumatologi, che impostavano una terapia di fondo con metotrexato 10 mg/sett, idrossiclorochina 200 mg e
steroidi (dosaggio di mantenimento metilprednisolone 2 mg/die).
Nel novembre 2024 la paziente veniva nuovamente inviata all’attenzione del nefrologo per insufficienza renale acuta (creatininemia da 0,9 a 1,6 fino a 2 mg/dl), azotemia 80 mg/dl, proteinuria di circa 1 g/24h e sedimento urinario attivo con microematuria e cilindri eritrocitari. Gli esami ematochimici documentavano inoltre anemizzazione (Hb 9,4 g/dl rispetto a 12,8 g/dl del precedente controllo) e confermavano la nota positività per ENA e ANA, con anticorpi anti-dsDNA negativi.
Nel dicembre 2024 la paziente veniva ricoverata nel reparto di Nefrologia per eseguire una biopsia renale. Gli accertamenti confermavano un’insufficienza renale a rapida progressione e rilevavano la presenza di anticorpi ANCA anti-mieloperossidasi (MPO) ad alto titolo (205 UI/ml). Durante la degenza si verificavano due episodi di emottisi, risolti da terapia con aerosol medicati e broncodilatatori. La TAC torace evidenziava addensamenti polmonari di non univoca interpretazione (flogosi vs emorragia alveolare), motivo per cui veniva avviata terapia antibiotica con amoxicillina/acido clavulanico. Considerata la rapida progressione dell’insufficienza renale con sedimento nefritico e in attesa del referto istologico, la paziente veniva trattata per tre giorni consecutivi con boli di metilprednisolone da 500 mg, seguiti da prednisone 0,5 mg/kg/die.
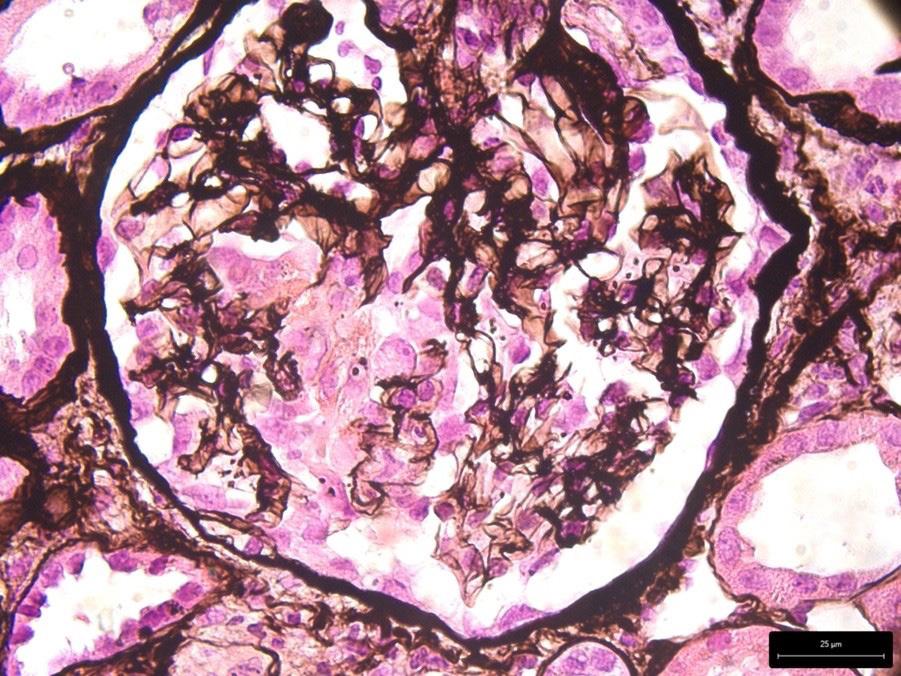
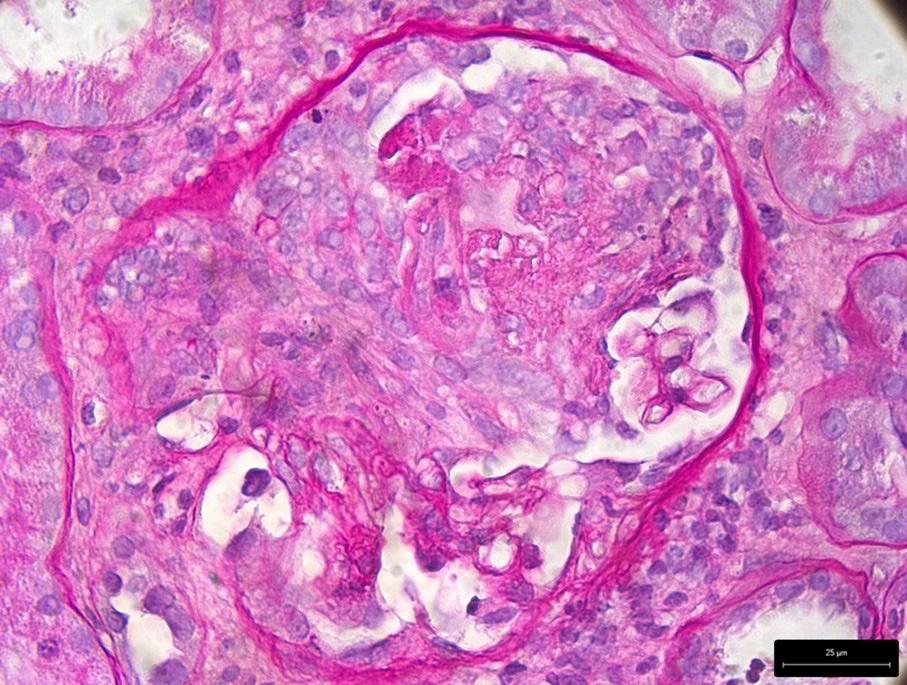
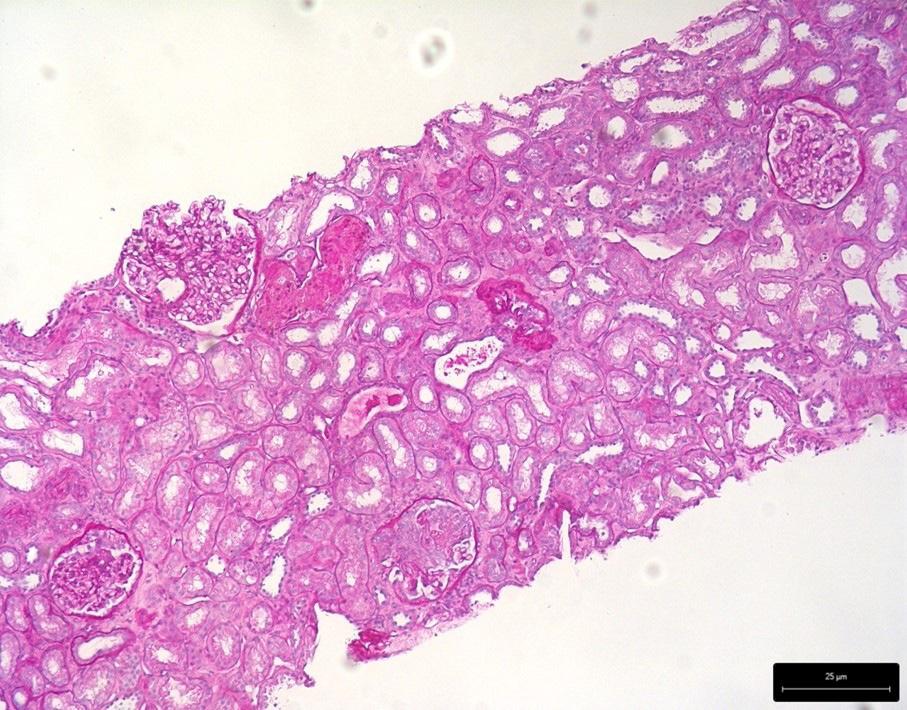
La biopsia renale consentiva la diagnosi di AAV, evidenziando una glomerulonefrite proliferativa extracapillare necrotizzante (Figura 1), una glomerulosclerosi segmentaria e globale focali (Figura 2), un diffuso danno tubulare acuto (Figura 3) e un focale fibroedema ed infiltrato interstiziale. L’immunofluorescenza mostrava sfumati depositi e aspecifici di IgM (+), due glomeruli contenenti semilune risultavano positivi per fibrinogeno (2–3+), in un glomerulo era presente necrosi fibrinoide. Non si rilevavano depositi di IgG, IgA,
Figura 1. Glomerulonefrite proliferativa extracapillare necrotizzante.
Figura 2. Glomerulosclerosi segmentaria e globale focali.
Figura 3. Diffuso danno tubulare acuto.
C3 o C1q. Alla terapia steroidea venivano associati rituximab (1 g per due somministrazioni a distanza di 15 giorni) e avacopan 30 mg due volte al giorno, introdotto già in fase di ricovero a scopo steroido-risparmiatore e con l’obiettivo di favorire una rapida remissione di malattia e un recupero della funzione renale. In accordo con la reumatologa curante, il metotrexato veniva temporaneamente sospeso.
Durante il follow up ambulatoriale la terapia steroidea veniva progressivamente scalata, arrivando a raggiungere un dosaggio di mantenimento di 5 mg/die dopo 14 settimane. Tuttavia, la paziente lamentava ricomparsa di artralgie localizzate a gomiti e anche; per questo motivo si decideva di aumentare temporaneamente il prednisone a 7,5 mg/die e l’idrossiclorochina a 400mg/ die, con beneficio clinico. Sempre in accordo con la reumatologa, il metotrexato rimaneva sospeso. Gli esami ematici effettuati mensilmente documentavano un rapido miglioramento dal punto di vista della risposta della vasculite, con progressiva riduzione del titolo degli ANCA e degli indici infiammatori, accompagnato da un graduale e progressivo recupero della funzione renale. Gli esami di maggio 2025 mostravano: creatinina 1,44 mg/dl, proteinuria 0,61 gr/24h, Hb 12,4 g/dl, anticorpi anti-MPO 20,9 UI/ml, CD20 0%, persistenza di microematuria ma assenza di cilindri, VES 29 mm/h e PCR 2,1 mg/L (v.n. <5). La clearance della creatinina risultava migliorata, da 28 a 52ml/min in sei mesi di terapia. Dopo circa tre mesi di terapia con avacopan si osservava un incremento significativo degli indici di sintesi epatici (GOT 54, GPT 306, gammaGT 146) con valori di bilirubina e leucociti nei limiti. La terapia veniva quindi temporaneamente sospesa. Si effettuavano controlli ematici settimanali, che mostravano rapida normalizzazione dei valori epatici dopo entro due settimane dalla sospensione, consentendo la reintroduzione graduale del
farmaco senza ulteriori segni di tossicità ai successivi monitoraggi. La paziente rimaneva asintomatica e non presentava altri eventi avversi.
Discussione
Questo caso clinico evidenzia diversi aspetti rilevanti nella gestione delle AAV in pazienti con comorbidità autoimmuni preesistenti. La paziente era già in trattamento con metotrexato e basse dosi di corticosteroidi per una sindrome di Sjögren, regime considerato adeguato anche come terapia di mantenimento nelle AAV6 . L’andamento clinico sottolinea quindi l’importanza di considerare diagnosi alternative anche in presenza di una nefropatia nota, qualora il quadro clinico e laboratoristico mostri modificazioni significative. Un secondo elemento di interesse riguarda l’impiego di avacopan in associazione a rituximab, che in questo caso si è dimostrato un approccio terapeutico efficace e sicuro. Tale combinazione ha consentito una rapida riduzione della terapia corticosteroidea, limitando così il rischio di tossicità da steroidi. Di rilievo anche il recupero rapido e sostenuto della funzione renale, documentato dal miglioramento del filtrato glomerulare. L’ottimo profilo di tollerabilità del farmaco, anche in presenza di alterazioni transitorie della funzionalità epatica, rappresenta un ulteriore elemento a favore dell’integrazione di avacopan nel trattamento delle AAV. Un attento monitoraggio clinico-laboratoristico si conferma cruciale per garantire la gestione sicura degli eventi avversi e la continuità terapeutica.
Conclusioni
Nonostante i significativi progressi terapeutici compiuti negli ultimi anni, le AAV restano patolo -
gie a potenziale esito fatale, che richiedono un trattamento immunosoppressivo prolungato nel tempo. L’introduzione di nuovi agenti terapeutici rappresenta un’importante innovazione nella gestione di queste malattie, consentendo un controllo rapido e sostenuto dell’attività di malattia e, al contempo, una significativa riduzione dell’esposizione cumulativa ai corticosteroidi. Sarà tuttavia necessario attendere i risultati di studi osservazionali condotti in contesti di real life per confermare se questa nuova strategia di induzione sia effettivamente in grado di migliorare i tassi di remissione e la sopravvivenza a lungo termine nei pazienti con AAV.
Bibliografia
1. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512–23.
2. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
3. Mukhtyar C, Flossmann O, Hellmich B, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis. 2008;67(7):1004–10.
4. KDIGO. KDIGO 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Kidney Int. 2024 Mar;105(3S):S71–S116.
5. Jayne DRW, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021 Feb 18;384(7):599–609.
6. Maritati F, Alberici F, Oliva E, et al. Methotrexate versus cyclophosphamide for remission maintenance in ANCA-associated vasculitis: A randomised trial. PLoS One. 2017 Oct 10;12(10):e0185880.
Il presente caso clinico è stato realizzato in collaborazione con