Contagem de Carboidratos no Diabetes Melito –Abordagem Teórica e Prática, 2a ed.
Débora Lopes Souto • Eliane Lopes Rosado
Doenças Exantemáticas em Pediatria, 2a Ed.
Carlos Eduardo Schettino
Emergências Endócrinas e Metabólicas
Rodrigo de Azeredo Siqueira
Endocrinologia Clínica no Dia a Dia, 2a Ed.
Alberto K. Arbex
Endocrinologia Pediátrica, 4a Ed.
Marilia Martins Guimarães • Maria Alice Neves Bordallo •
Kássie Regina Neves Cargnin • Honomar Ferreira de Souza • Cláudia Braga Monteiro
Manual de Terapêutica em Gastrenterologia e Hepatologia Pediátrica
Ana Daniela Izoton de Sadovsky • Vera Lúcia Ângelo Andrade
Neuropediatria no Dia a Dia
Flávia Nardes dos Santos • Giuseppe Pastura
Nutrição Clínica Aplicada à Pediatria
Patricia Padilha • Elizabeth Accioly
Pediatria no Dia a Dia, 2a Ed.
Giuseppe Pastura • Flávia Nardes dos Santos
Puericultura no Dia a Dia
Giuseppe Pastura • Flávia Nardes dos Santos
Seletividade Alimentar – Da Definição ao Tratamento
Mariana Catta-Preta
Semiologia Pediátrica, 3a Ed. Adauto Dutra
Saiba mais sobre estes e outros títulos em nosso site: www.rubio.com.br
A editora e os autores deste livro não mediram esforços para assegurar dados corretos e informações precisas. Entretanto, por ser a medicina uma ciência em permanente evolução, recomendamos aos nossos leitores recorrer à bula dos medicamentos e a outras fontes fidedignas, bem como avaliar, cuidadosamente, as recomendações contidas no livro em relação às condições clínicas de cada paciente.
Organizadoras
Ana Hermínia de Azevedo Ferreira
Endocrinologista Pediátrica com Título de Especialista em Pediatria com Área de Atuação em Endocrinologia Pediátrica pela Sociedade Brasileira de Pediatria (SBP).
Preceptora da Residência Médica em Endocrinologia Pediátrica do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Supervisora da Residência Médica em Endocrinologia Pediátrica do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Chefe do Serviço de Endocrinologia Pediátrica do Imip.
Claudia Andrade Coutinho
Endocrinologista Pediátrica com Título de Especialista em Pediatria com Área de Atuação em Endocrinologia Pediátrica pela Sociedade Brasileira de Pediatria (SBP).
Mestre em Ciências da Saúde pela Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Preceptora da Residência Médica em Endocrinologia Pediátrica do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Médica do Ambulatório de Triagem Neonatal de Hipotireoidismo Congênito no Hospital Barão de Lucena, PE.
Taciana de Andrade Schuler
Supervisora e Preceptora da Residência Médica em Endocrinologia Pediátrica do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Preceptora da Residência Médica em Endocrinologia Pediátrica do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
CIP-BRASIL. CATALOGAÇÃO NA PUBLICAÇÃO SINDICATO NACIONAL DOS EDITORES DE LIVROS, RJ)
E46
Endocrinologia pediátrica no dia a dia/organização Ana Hermínia de Azevedo Ferreira, Claudia Andrade Coutinho, Taciana de Andrade Schuler. –1. ed. – Rio de Janeiro: Rubio, 2025. 344 p. ; 24 cm.
Inclui bibliografia
ISBN 978-65-88340-93-6
1. Endocrinologia pediátrica. I. Ferreira, Ana Hermínia de Azevedo. II. Coutinho, Claudia Andrade. III. Schuler, Taciana de Andrade. III. Título.
CDD: 618.924 25-99353.0
CDU: 612.43-053.2
Carla Rosa Martins Gonçalves – Bibliotecária – CRB-7/4782
Editora Rubio Ltda.
Av. Franklin Roosevelt, 194 s/l. 204 – Castelo 20021-120 – Rio de Janeiro – RJ
Tel: 55(21) 2262-3779
E-mail: rubio@rubio.com.br www.rubio.com.br
Impresso no Brasil
Printed in Brazil
Colaboradores
Aline Lopes
Mestre em Ciências da Saúde pela Universidade de Pernambuco (UPE).
Aline Campitelli Fernandes
Residência Médica em Endocrinologia Pediátrica pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Ana Carla Lins Neves
Médica Pediatra pela Universidade de Pernambuco (UPE).
Especialização em Endocrinologia Pediátrica pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Mestre em Saúde da Criança e do Adolescente pela Universidade Federal de Pernambuco (UFPE).
Ana Carla Montenegro
Residência Médica em Endocrinologia pelo Hospital Agamenon Magalhães, PE.
Mestre em Tocoginecologia pela Universidade de Pernambuco (UPE).
Doutora em Neurociências pela Universidade Federal de Pernambuco (UFPE).
Ana Carolina Maués de Oliveira
Médica com Formação em Endocrinologia e Metabologia pelo Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (HC-FMRPUSP), Titulada pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Doutoranda do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Pesquisadora do Laboratório de Hormônios e Genética Molecular (LIM/42).
Ana Claudia Latronico
Professora Titular do Departamento de Clínica Médica, Disciplina de Endocrinologia da Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Ana Luiza Pessoa
Residência Médica em Endocrinologia Pediátrica pelo Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Ana Pinheiro Machado Canton
Médica Endocrinologista e Pesquisadora Assistente da Divisão de Endocrinologia e Metabologia da Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Doutorado e Pós-doutorado em Endocrinologia pela FMUSP.
Pós-doutorado em Endocrinologia Pediátrica pela Sorbonne Université (Paris).
Antonio Fernandes de Oliveira Filho
Título de Especialista em Endocrinologia pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM). Mestre em Tecnologia em Saúde Universidade Estadual da Paraíba (UEPB).
Doutorando do Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (Incor-HC-FMUSP).
Barbara Gomes
Médica Assistente e Preceptora da Residência Médica em Endocrinologia Pediátrica do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE). Doutora em Neuropsiquiatria e Ciências do Comportamento pela UFPE.
Bruna Silva de Oliveira
Graduada em Medicina pela Faculdade de Medicina Nova Esperança (Famene), João Pessoa – PB.
Residência Médica em Clínica Médica pela Universidade Federal de Campina Grande (UFCG), PB.
Residência Médica em Endocrinologia e Metabologia pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Carlos Alberto Longui
Chefe de Clínica Adjunto da Irmandade da Santa Casa de Misericórdia de São Paulo.
Professor Titular da Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Caroline Rosa Pellicciari
Residência Médica em Endocrinologia Pediátrica pela Irmandade da Santa Casa de Misericórdia de São Paulo.
Mestra em Ciências da Saúde pela Faculdade de Ciências
Médicas da Santa Casa de São Paulo.
Crésio de Aragão Dantas Alves
Professor Associado de Pediatria da Universidade Federal da Bahia (UFBA).
Chefe do Serviço de Endocrinologia Pediátrica do Complexo Hospitalar Universitário Professor Edgard Santos (Complexo HUPES) da Faculdade de Medicina da UFBA.
Cristiane Kochi
Professora Titular da Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Médica Assistente da Unidade de Endocrinologia Pediátrica da Santa Casa de São Paulo.
Cristiane Kopacek
Professora de Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS).
Preceptora de Endocrinologia Pediátrica da Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA)/ Santa Casa de Porto Alegre.
Coordenadora do Ambulatório de Triagem Neonatal das Endocrinopatias/Hiperplasia Adrenal Congênita do Hospital Materno-Infantil Presidente Vargas, RS.
Dalva Castro de Oliveira
Endocrinologista Pediátrica Formada pela Universidade Federal de São Paulo (Unifesp).
Médica Assistente do Ambulatório de Tecnologias em Diabetes da Unifesp.
Sócia Fundadora do Instituto Brasileiro de Tecnologia e Educação em Diabetes (IBTED).
Mestranda em Endocrinologia pela Unifesp.
Daniel Lins
Doutor em Medicina pela Universidade Federal de Pernambuco (UFPE).
Preceptor de Endocrinologia e Clínica Médica da Universidade de Pernambuco (UPE).
Eduardo Just
Residência Médica em Pediatria e Estágio Especializado em Diagnóstico por Imagem no Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Mestre e Doutor em Saúde da Criança e do Adolescente pela Universidade Federal de Pernambuco (UFPE).
Médico Radiologista do Imip e do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Érico Higino de Carvalho
Graduado em Medicina pela Universidade de Pernambuco (UPE).
Especialista em Endocrinologia e Metabologia pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Mestre em Ciências da Saúde pela UPE.
Doutor em Medicina Tropical pela Universidade Federal de Pernambuco (UFPE).
Professor Adjunto da Disciplina de Endocrinologia na UFPE.
Membro da SBEM.
Coordenador do Serviço de Endocrinologia do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Ex-presidente da SBEM PE – 2023/2024.
Coordenador da Liga Acadêmica de Endocrinologia (Liame) da UFPE.
Preceptor da Residência de Clínica Médica do Hospital
Santa Joana – Recife.
Erik Trovão Diniz
Médico Endocrinologista com Título de Especialista pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Preceptor da Residência Médica em Endocrinologia e Metabologia do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Fábio Moura
Especialista em Endocrinologia e Metabologia no Hospital Agamenon Magalhães, PE.
Preceptor da Residência em Endocrinologia no Instituto de Medicina Integral Professor Fernando Figueira (Imip). Endocrinologista do Hospital Universitário Osvaldo Cruz da Universidade de Pernambuco (UPE).
Gabriela Corrêa Lima Pereira
Endocrinologista Pediátrica Formada pelo Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Gabriela Ferraz Leal
Doutora em Genética pela Universidade Federal de Pernambuco (UFPE).
Especialista em Genética Médica pela Sociedade Brasileira de Genética Médica (SBGM).
Médica Geneticista da Universidade de Pernambuco (UPE) e do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Gil Guerra-Júnior
Professor Titular do Departamento de Pediatria da Faculdade de Ciências Médicas (FCM) da Universidade Estadual de Campinas (Unicamp).
Coordenador do Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo da Unicamp.
Jacqueline Araújo
Mestre em Saúde da Criança e do Adolescente pela Universidade Federal de Pernambuco (UFPE).
Doutora em Ciências Biológicas pela UFPE.
Chefe do Serviço de Endocrinologia Pediátrica do Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Jéssica Me Lin Ie
Médica com Formação em Endocrinologia e Metabologia pelo Instituto de Assistência Médica ao Servidor Público Estadual (Iamspe) de São Paulo, titulada pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Doutoranda do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP).
Pesquisadora do Laboratório de Lípides (LIM/42).
Júlia Constança C. Souza Fernandes
Endocrinologista Pediátrica do Complexo Hospitalar Universitário Professor Edgard Santos (Complexo HUPES) da Universidade Federal da Bahia (UFBA) e do Centro de Diabetes e Endocrinologia do Estado da Bahia (Cedeba).
Karla S. P. R. Vilar Calheiros
Residência Médica em Pediatria pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Residência Médica em Endocrinologia Pediátrica pelo Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Mestre em Saúde da Criança e do Adolescente pela UFPE. Médica Endocrinologista Pediátrica e Preceptora do Programa de Residência Médica em Pediatria no Hospital Universitário Osvaldo Cruz (HUOC) da Universidade de Pernambuco (UPE).
Professora de Pediatria da Faculdade de Ciências Médicas da UPE.
Latife Salomão Tyszler
Professora Colaboradora/Coordenadora do Curso de Endocrinologia da Pontifícia Universidade Católica do Rio de Janeiro (PUC-Rio)/Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione (Iede).
Diretoria da Associação de Ensino e Pesquisa (Assep) do Iede.
Presidente do Comitê de Endocrinologia Pediátrica da Sociedade de Pediatria do Estado do Rio de Janeiro (Soperj).
Chefe do Serviço de Endocrinologia Pediátrica do Iede. Membro da Sociedade Brasileira de Pediatria (SBP).
Membro do Comitê de Obesidade Infantil da Associação Brasileira para o Estudo da Obesidade e Síndrome Metabólica (Abeso).
Letícia Duarte de Carvalho Xavier do Nascimento
Médica pela Faculdade de Medicina Estácio de Juazeiro do Norte.
Especialista em Clínica Médica pelo Hospital Agamenon Magalhães da Secretaria de Saúde do Estado (SES-PE).
Especialista em Endocrinologia e Metabologia pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Lucas Moura
Estudante de Medicina da Faculdade Pernambucana de Saúde (FPS).
Lucian Batista de Oliveira
Mestre e Doutorando em Ciências da Saúde pela Universidade de Pernambuco (UPE).
Residência Médica em Endocrinologia e Metabologia pelo Hospital Agamenon Magalhães da Secretaria de Saúde do Estado de Pernambuco (SES-PE).
Professor Substituto de Endocrinologia da Universidade Federal de Campina Grande (UFCG).
Luciana Pimentel de Andrade Residência Médica em Endocrinologia Pediátrica no Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE).
Luciano Albuquerque
Mestre em Neurociências pela Universidade Federal de Pernambuco (UFPE).
Endocrinologista Titulado pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Preceptor do Serviço de Endocrinologia do Hospital das Clínicas da UFPE (HC-UFPE).
Chefe do Serviço de Clínica Médica do Hospital Otávio de Freitas da Secretaria de Saúde do Estado de Pernambuco (SES-PE).
Lucio Vilar
Doutor em Ciências da Saúde pela Universidade de Brasília (UnB).
Professor Titular da Disciplina de Endocrinologia da Universidade Federal de Pernambuco (UFPE).
Chefe do Serviço de Endocrinologia do Hospital das Clínicas da UFPE (HC-UFPE).
Luiz Claudio Gonçalves de Castro
Doutor em Ciências da Saúde pela Faculdade de Ciências da Saúde da Universidade de Brasília (UnB).
Pediatra com Área de Atuação em Endocrinologia Pediátrica pela Sociedade Brasileira de Pediatria (SBP).
Professor do Departamento de Pediatria da Faculdade de Medicina da UnB (FM/UnB).
Coordenador do Ambulatório Pediátrico de Metabolismo Ósseo e Mineral do Hospital Universitário de Brasília.
Manuella Galvão de Oliveira
Médica pela Universidade de Pernambuco (UPE) com Residência em Genética Médica pela Universidade Federal de São Paulo (Unifesp).
Médica Geneticista do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Marcela Barbosa
Mestre em Saúde da Criança e do Adolescente pela Universidade Federal de Pernambuco (UFPE).
Especialista em Endocrinologia pelo Hospital Agamenon Magalhães da Secretaria de Saúde do Estado de Pernambuco (SES-PE).
Médica Assistente do Centro Médico Senador José Ermírio de Moraes, PE.
Maria Paula Bandeira
Mestre em Ciências da Saúde pela Universidade de Pernambuco (UPE).
Preceptora da Residência Médica em Endocrinologia e Metabologia do Hospital Agamenon Magalhães da Secretaria de Saúde do Estado de Pernambuco (SES-PE).
Médica Endocrinologista Pediátrica das Provas Funcionais
A+, Grupo Fleury, PE.
Marilza Leal Nascimento
Mestre em Ciências Médicas pela Universidade Federal de Santa Catarina (UFSC).
Professora Adjunta do Departamento de Pediatria da UFSC.
Pediatra pela Secretaria de Estado da Saúde de Santa Catarina.
Especialista em Endocrinologia Pediátrica pelo Serviço de Endocrinologia Pediátrica da Universidade Federal do Paraná (UFPR).
Título de Especialista em Pediatria, conferido pela Associação Médica Brasileira (AMB) e Sociedade Brasileira de Pediatria (SBP).
Preceptora do Serviço de Residência Médica em Endocrinologia Pediátrica do Hospital Infantil Joana de Gusmão, SC.
Membro do Conselho Científico de Endocrinologia da SBP. Ex-coordenadora do Programa de Triagem Neonatal do Estado de Santa Catarina – 1991 a 2017.
Presidente do Departamento Científico de Endocrinologia da Sociedade Catarinense de Pediatria (SCP).
Mirela Costa de Miranda Médica Endocrinologista e Doutora em Endocrinologia pela Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Pós-doutoranda pela FMUSP. Linha de pesquisa, desde o doutorado, no diagnóstico, tratamento e seguimento da Hiperplasia Adrenal Congênita. Participa dos atendimentos do Ambulatório de Hiperplasia Adrenal Congênita da FMUSP desde 2015, no qual desenvolve atividade assistencial, de pesquisa e ensino.
Monica Gabbay
Pós-doutorado em Endocrinologia pela Universidade Federal de São Paulo (Unifesp).
Coordenadora do Ambulatório de Tecnologia em Diabetes do Centro Diabetes da Unifesp.
Mônica de Oliveira
Mestre em Medicina Interna da Universidade Federal de Pernambuco (UFPE).
Osmar Monte
Professor Emérito da Faculdade de Ciências Médicas da Santa Casa de São Paulo.
Patrícia Oliveira de Almeida Freire
Mestre em Saúde da Criança e do Adolescente pela Universidade Estadual de Campinas (Unicamp).
Residência Médica em Pediatria e em Endocrinologia Pediátrica na Unicamp.
Certificado de Área de Atuação em Endocrinologia Pediátrica (CAAEP) pela Sociedade Brasileira de Pediatria
(SBP) e pela Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Priscilla Mayara Padilha Ribeiro
Pediatra pelo Hospital das Clínicas da Universidade Federal de Pernambuco (HC-UFPE) – Residência Médica de 2014-2016.
Endocrinologista Pediátrica Formada pelo HC-UFPE –Residência Médica de 2016-2018.
Raphael Del Roio Liberatore Junior
Endocrinologista Pediátrico da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FMRP-USP). Livre-docente em Pediatria pela Universidade de São Paulo (USP).
Professor Associado do Departamento de Puericultura e Pediatria da FMRP-USP.
Rayana Maria de Melo Azedo Vieira
Médica pela Faculdade Pernambucana de Saúde (FPS).
Residências Médicas em Pediatria e Endocrinologia
Pediátrica pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Ricardo A. Machado e Silva
Coordenador da Residência em Medicina Nuclear do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Ricardo Fernando Arrais
Mestre em Pediatria e Ciências aplicadas a Pediatria pela Universidade Federal de São Paulo (Unifesp).
Doutor em Endocrinologia Clínica pela Escola Paulista de Medicina da Universidade Federal de São Paulo (EPM-Unifesp).
Coordenador da Unidade de Endocrinologia Pediátrica da Universidade Federal do Rio Grande do Norte (UFRN).
Professor Titular do Departamento de Pediatria da UFRN.
Taís Andrade Dantas
Pediatra pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Endocrinologista Pediátrica pela Universidade Federal de Pernambuco (UFPE).
Endocrinologista Pediátrica do Hospital Universitário Alcides Carneiro da Universidade Federal de Campina Grandes (UFCG).
Coordenadora da Residência de Pediatria do Município de Campina Grande, PB.
Tânia Aparecida Sartori Sanchez Bachega
Docente pela Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Credenciada no Serviço de Pós-graduação da FMUSP nas áreas de Concentração de Endocrinologia e Clínica Médica.
Presidente da Sociedade Brasileira de Triagem Neonatal e Erros Inatos do Metabolismo (SBTEIM).
Thereza Selma Soares Lins
Residência Médica em Pediatria no Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Curso de Especialização em Endocrinologia Pediátrica na Santa Casa de São Paulo.
Título de Especialista em Pediatria e Certificado de Atuação na Área de Endocrinologia Pediátrica.
Fundadora e Ex-chefe do Serviço de Endocrinologia Pediátrica do Imip – 1995-2023.
Endocrinologista Pediátrica Assistente do Serviço de Endocrinologia Pediátrica do Imip.
Thiago Cavalcanti de França Arruda
Preceptor do Curso de Medicina da Faculdade Pernambucana de Saúde (FPS).
Preceptor da Residência Médica de Pediatria e de Neonatologia do Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Vanessa Leão
Residência em Pediatria pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Especialista em Endocrinologia pelo Hospital Agamenon Magalhães, PE.
Fellow em Endocrinologia Pediátrica pela University of Florida, EUA.
Preceptora de Endocrinologia Pediátrica do Imip.
Victória Rodrigues Granja Alencar
Residências Médicas em Clínica Médica e Endocrinologia e Metabologia pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Mestranda do Programa de Mestrado Profissional do Imip.
Membro Titular da Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
Membro da Comissão de Valorização de Novas Lideranças da SBEM – 2023-2024.
Vinicius Nahime Brito
Professor Livre-docente da Faculdade de Medicina da Universidade de São Paulo (FMUSP).
Médico Pesquisador do Laboratório de Hormônios e Genética Molecular (LIM/42) do Hospital das Clínicas da FMUSP (HC-FMUSP).
Doutor Assistente da Unidade de Endocrinologia do Desenvolvimento da Divisão de Endocrinologia e Metabologia da FMUSP.
Responsável pelo Ambulatório de Puberdade Precoce do HC-FMUSP.
Wagner E. Nascimento
Médico pela Universidade Federal de Pernambuco (UFPE).
Residência Médica em Pediatria pelo Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Residência Médica em Endocrinologia Pediátrica pelo Hospital das Clínicas da UFPE (HC-UFPE).
Dedicatória
Este livro nasce do encontro entre o saber científico e o cuidado humano. Dedicamos, com afeto e respeito, esta primeira edição do livro Endocrinologia Pediátrica no Dia a Dia, do Instituto de Medicina Integral Professor Fernando Figueira (Imip), a todos que, direta ou indiretamente, contribuíram para a construção do nosso serviço, da nossa história e deste trabalho.
Aos nossos pacientes e suas famílias, que com coragem e esperança, nos ensinam diariamente o verdadeiro sentido da Medicina e são a razão maior do nosso compromisso diário, nossa mais profunda gratidão. Cada encontro, cada desafio, cada conquista compartilhada nos ensinam, nos transformam e nos impulsionam a sermos profissionais melhores.
Agradecemos especialmente à Dra. Thereza Selma, fundadora e chefe até 2023 do Serviço de Endocrinologia Pediátrica do Imip. Sua trajetória, marcada por dedicação, competência e humanidade, segue sendo inspiração para todos nós. O serviço que hoje mantemos e aprimoramos é fruto de sua visão e de seu trabalho incansável. Que esta dedicatória alcance, mesmo em silêncio, todos que ajudaram a construir este projeto. Este livro é também uma celebração do conhecimento construído em equipe, do cuidado que humaniza e da ciência que transforma. Que ele inspire novas gerações a seguirem esse legado com ética, empatia e excelência.
As Organizadoras
Agradecimentos
Agradecemos, com profunda admiração e reconhecimento, a todos os nossos colaboradores, cuja generosidade em compartilhar saberes tornou possível a realização deste livro.
Cada capítulo aqui apresentado reflete não apenas excelência técnica e científica, mas também o compromisso com a formação de novos profissionais e com a disseminação de um cuidado mais qualificado e humano para nossas crianças e adolescentes.
Sem a dedicação, o tempo e o entusiasmo de cada um dos autores convidados, esta obra não teria se concretizado. Que este trabalho conjunto, fruto da experiência e da colaboração, sirva como referência e inspiração para todos que buscam o conhecimento na área de Endocrinologia Pediátrica.
Nosso mais sincero agradecimento.
As Organizadoras
Apresentação
É com grande satisfação que apresentamos a primeira edição de Endocrinologia Pediátrica no Dia a Dia, fruto da colaboração entre três autoras que não só compartilham um profundo interesse pela Endocrinologia Pediátrica, mas também uma amizade duradoura. Como colegas e amigas, temos a honra de contribuir com este trabalho pioneiro, que representa além de um marco em nossas carreiras individuais, um testemunho do compromisso conjunto em proporcionar cuidados integrais às crianças e aos adolescentes que atendemos no Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Ao longo de nossas trajetórias profissionais no Imip, fomos inspiradas diariamente pela resiliência e determinação das crianças e das famílias que enfrentam desafios relacionados à saúde endócrina. Este livro reflete nosso compromisso em compartilhar conhecimento, experiência e insights clínicos adquiridos ao longo dos anos de prática clínica.
O Imip é reconhecido nacionalmente como referência no cuidado integral à criança e ao adolescente, e ao longo dos anos tem construído um legado sólido em ensino, assistência e pesquisa. As publicações anteriores tornaram-se ferramentas essenciais para a formação e a atuação de inúmeros profissionais da saúde em todo o país. Após a consolidação de importantes publicações, era natural que o Serviço de Endocrinologia Pediátrica também compartilhasse sua experiência por meio de uma obra própria.
Esta primeira edição reúne o conhecimento acumulado ao longo da história do serviço de Endocrinologia Pediátrica, fundado e liderado até 2023 por Dra. Thereza Selma – profissional exemplar, que nos orienta a termos uma cultura de ensino, ética e compromisso com o cuidado humanizado.
Desde os princípios básicos da Endocrinologia Pediátrica até as mais recentes abordagens diagnósticas e terapêuticas, cada capítulo foi elaborado com cuidado e atenção aos detalhes, abrangendo conteúdos fundamentais para o diagnóstico e o manejo das principais condições endócrinas na infância e na adolescência. A proposta é oferecer aos leitores – sejam estudantes, residentes, pediatras gerais ou subespecialistas – uma fonte prática, acessível e atualizada, alinhada às necessidades da Endocrinologia Pediátrica contemporânea.
Este livro também é uma celebração da colaboração interdisciplinar que caracteriza o trabalho no Imip. Reconhecemos e agradecemos a contribuição de nossos colegas endocrinopediatras e de diversas especialidades, tanto do Imip quanto de outras instituições de ensino do Recife e de diversas outras cidades de todo o Brasil, cujos apoio e parceria foram fundamentais para o desenvolvimento deste projeto.
Que esta obra sirva como uma fonte valiosa de conhecimento e inspiração, capacitando os leitores a fazerem uma diferença positiva na vida das crianças e dos adolescentes com doenças endócrinas.
As Organizadoras
Prefácio
O Instituto de Medicina Integral Professor Fernando Figueira (Imip) é um marco da Medicina Social e do ensino no Brasil, fundado sob a visão pioneira e inclusiva do professor Fernando Figueira, cuja trajetória inspira gerações de médicos comprometidos com a ética e a humanidade no cuidado à saúde.
É com grande honra e emoção que apresento Endocrinologia Pediátrica no Dia a Dia, fruto de trabalho, dedicação e excelência de um serviço que tive o privilégio de fundar no Imip. Ao longo dos anos, o serviço de Endocrinologia Pediátrica cresceu, consolidando-se como referência, especialmente pela oferta da residência médica em endocrinologia pediátrica, um espaço em que buscamos unir ciência, prática e cuidado com os pequenos pacientes que nos chegam.
Hoje, testemunho com orgulho o legado de excelência e compromisso mantido pelas profissionais que, atualmente, conduzem com competência e amor esta especialidade tão desafiadora. Minha gratidão à equipe médica que segue fortalecendo este trabalho com o esmero que ele merece.
Um agradecimento muito especial vai para os nossos pacientes e suas famílias, que nos confiam o cuidado do
que há de mais precioso. São eles que, com seus desafios e histórias, nos inspiram e nos ensinam diariamente, reafirmando a importância de nossa missão.
Que este livro seja um reflexo do compromisso do Imip com a formação de profissionais qualificados e do cuidado integral que marcam a sua história. Que ele também homenageie a memória de Fernando Figueira, cuja visão continua a guiar o trabalho que todos aqui realizamos com tanto afinco.
Com profundo respeito e admiração,
Thereza Selma Soares Lins de Freitas Residência Médica em Pediatria no Instituto de Medicina Integral Professor Fernando Figueira (Imip).
Curso de Especialização em Endocrinologia Pediátrica na Santa Casa de Misericórdia de São Paulo. Portadora de Título de Especialista em Pediatria e Certificado de Atuação na Área de Endocrinologia Pediátrica. Fundadora e Ex-chefe do Serviço de Endocrinologia Pediátrica do Imip no período 1995-2023.
Endocrinopediatra Assistente do Serviço de Endocrinologia Pediátrica do Imip.
Crescimento I
CAPÍTULOS
1 Baixa Estatura, 3
2 Deficiência do Hormônio do Crescimento, 15
3 Alta Estatura, 19
1 Baixa Estatura
Thereza Selma Soares Lins Ana Hermínia de Azevedo Ferreira
Introdução
O crescimento é um processo complexo e dinâmico e apesar de ser multifatorial, ocorre de maneira previsível em crianças e adolescentes normais, tornando-se um marcador sensível para saúde e bem-estar. Desvio do padrão de normalidade de crescimento pode ser a primeira manifestação de uma ampla variedade de doenças porque potencialmente qualquer doença pode afetar o crescimento normal.
A baixa estatura (BE) é um dos motivos mais frequentes de encaminhamento à endocrinologia pediátrica e causa grande ansiedade nos pacientes e em seus pais.
Definições
BE é definida como estatura abaixo de –2 desvios-padrão (DP) em relação à média da população de referência para sexo e idade.1 No Brasil, as curvas de crescimento adotadas são as da Organização Mundial da Saúde (OMS). Para o cálculo do escore Z, existem aplicativos disponíveis na Internet, como Anthro e Anthro Plus (https://www.who. int/tools/child-growth-standards/software).
Crescimento deficiente é definido quando ocorre redução da velocidade de crescimento (VC), que representa o número de centímetros que a criança/adolescente cresce em um ano e varia de acordo com sexo e idade. É considerado o principal critério de normalidade na avaliação do crescimento e deve ser calculada em período mínimo de seis meses, porém preferencialmente com cerca de um ano de intervalo.1 O retardo do crescimento é um sinal precoce de patologia subjacente em crianças. O gráfico de referência para avaliar VC é o de Tanner (Figuras 1.1 e 1.2).2
Critérios para Investigação de Crianças com Queixa de Baixa Estatura3
Estatura ou comprimento abaixo de −2DP (ou percentil 3) em relação à média populacional para sexo e idade.
Estatura abaixo de −1,5DP, em relação à estaturaalvo (EA).
Queda de mais de 0,5DP de estatura no período de um ano, ou seja, mudanças das linhas de percentis no gráfico de crescimento para um percentil inferior após a idade maior que 24 meses.
VC inferior a 2DP abaixo da média (percentil 3) em relação à sua faixa etária, independentemente da estatura.
VC inferior a 1,5DP abaixo da média (p10) por dois anos seguidos (para crianças sem BE).
VC inferior a 1DP abaixo da média (p25) em período de um ano (para crianças com BE).
Métodos de Avaliação do Crescimento
Estatura e Peso
É importante que a avaliação do peso e da estatura seja plotada nos gráficos de referência, e deve-se verificar se há maior comprometimento do peso em relação à estatura, o que ajuda a diferenciar entre causas endócrinas e não endócrinas.
A medição da estatura requer precisão, devendo ser realizada preferencialmente pelo mesmo observador.
Até os 2 anos de idade, a medição deve ser feita com a criança deitada. Nessa fase, a medida é denominada comprimento.
A partir dos 2 anos de idade, a medição deve ser feita com a criança em posição ereta, com o estadiômetro de parede. A medida obtida denomina-se altura ou estatura. Os valores obtidos devem ser registrados em centímetros e milímetros e plotados no gráfico correspondente para idade e sexo (https://www.who.int/tools/childgrowth-standards/standards).
Estatura-Alvo
Além da estatura e da VC, a estatura da criança também deve ser avaliada de acordo com seu padrão genético, a estrutura-alvo (EA), que deve ser plotada na mesma curva de crescimento estatural para ajudar a definir um padrão de crescimento esperado para uma determinada família.
5 Puberdade Precoce: Etiologia, Diagnóstico e Tratamento, 32
6 Atraso Puberal, 43
7 Síndrome dos Ovários Policísticos, 48
Puberdade
4 Avaliação da Puberdade Normal
Karla S. P. R. Vilar Calheiros
Introdução
A puberdade é uma fase crucial no desenvolvimento humano, marcada por mudanças físicas, hormonais, psicológicas e sociais significativas. É um processo de maturação biológica que resulta em desenvolvimento dos caracteres sexuais secundários e capacidade reprodutiva, além do crescimento somático com estirão puberal. De origem multifatorial, tem em seu processo a influência de fatores genéticos, epigenéticos, metabólicos, ambientais, étnicos, geográficos e socioeconômicos.1 Compreender esse processo é essencial para oferecer cuidados de saúde eficazes e apoiar, assim, os adolescentes na transição para a idade adulta.
Fisiologia da Puberdade
O desenvolvimento puberal é, na verdade, a finalização de um processo de maturação contínuo que acontece desde a concepção. O eixo hipotálamo-hipófise-gonadal (HHG) tem sua ativação inicial na vida fetal, passando por um período de quiescência durante a infância até atingir um processo de reativação durante a puberdade.2
O entendimento do conceito do “gonadostato” no desenvolvimento puberal é de fundamental importância para compreender esse processo ao longo das diversas fases da vida fetal, neonatal, infância e puberdade (Figura 4.1).3 Durante a maior parte da vida fetal e perinatal, o gonadostato encontra-se insensível ao feedback negativo dos esteroides sexuais no hipotálamo, acarretando níveis puberais desses hormônios. O gonadostato torna-se, então, cada vez mais sensível, ocasionando atividade mínima dos pulsos de hormônio liberador de gonadotrofinas (GnRH) na metade da infância. Perto do período puberal, começa a haver abandono dessa inibição, permitindo o início da puberdade, com o começo da pulsatilidade do GnRH no hipotálamo e o aumento da sensibilidade das células gonadotróficas hipofisárias, culminando com o estímulo gonadal e a produção dos hormônios sexuais, iniciando a cascata de eventos de maturação da puberdade.3
A produção de esteroides sexuais durante a vida fetal desempenha papel primordial na formação do embrião, especialmente no sexo masculino, sendo responsável
pela virilização da genitália masculina a partir da sétima semana de gestação, além de promover a descida testicular e o desenvolvimento genital no terceiro trimestre. A gonadotrofina coriônica humana (hCG) placentária liga-se ao receptor de LH provocando produção de esteroides sexuais, particularmente os andrógenos (testosterona e di-hidrotestosterona) no sexo masculino.2 Logo após o nascimento, com a ruptura da passagem transplacentária dos estrogênios maternos, os níveis de gonadotrofinas e esteroides sexuais atingem o ponto mais baixo, apenas voltando a aumentar em torno de de 1 a 3 meses de idade, em razão da imaturidade do gonadostato. Nessa fase, há aumento dos níveis de esteroides sexuais, configurando o que se denomina “minipuberdade”.2 Do ponto de vista fisiológico, esse evento tem relevância crucial sobretudo no sexo masculino, acarretando continuação do processo de descida testicular e a maturação adicional das células gonadais. Após esse período de atividade do eixo HHG, há a fase de quiescência relativa (mas não completa) até a puberdade.2
O momento exato da reativação do eixo HHG sofre grande variação em relação à faixa etária, estando sob o controle de fatores genéticos (herança poligênica) e epigenéticos (influências ambientais – dieta, estado nutricional, nível de atividade física, estresse, nível socioeconômico e disruptores endócrinos).4 A reativação desse eixo consiste no principal evento neuroendócrino e hormonal associado ao desenvolvimento puberal, porém os mecanismos exatos que provocam esse gatilho permanecem não completamente elucidados. Acredita-se que os fatores externos ambientais, por meio da epigenética, atuem como controladores da expressão gênica nesse início puberal.5 Há mudança no equilíbrio entre a expressão gênica estimulatória (KISS1, GnRH, GLS, TTF1) e a inibitória (MKRN3, GADG7, PDYN, GnIH) durante esse período de transição puberal.4,5 O gene KISS1, que codifica a kisspeptina, é um importante gene na ativação puberal do eixo HHG. Do ponto de vista clínico, mutações nos genes KISS1, MKRN3 e DLK1 desempenham papel crucial no desenvolvimento da puberdade precoce central.4,6
Os neurônios hipotalâmicos secretores de GnRH são ativados por meio de três neuropeptídios principais: kisspeptina, neuroquinina B e dinorfina, secretados
Neurônio GABAérgico
Neurônio KNDy
NKB DYN KISS
Neurônio glutamatérgico
GABA Glutamato
Neurônio
Hipotálamo
Neurônio kisspeptina
Feminino
Figura 4.2 Representação esquemática do eixo hipotálamo-hipófise-gonadal
Em resposta ao GnRH, as células gonadotróficas da hipófise anterior começam a síntese e a secreção das gonadotrofinas do hormônio luteinizante (LH) e do hormônio foliculoestimulante (FSH). O LH sérico primeiro aumenta desproporcionalmente em relação ao FSH; essa disparidade LH-FSH é particularmente evidente durante o sono.3 As gonadotrofinas hipofisárias estimulam as gônadas (ovários e testículos), com consequente produção dos hormônios esteroides. Nas meninas, os ovários irão produzir estrogênios, progestágenos e andrógenos, todos estimulados por LH e FSH e necessários para o ciclo menstrual e fertilidade normais. Nos meninos, o LH estimula as células de Leydig a produzirem testosterona e o FSH é responsável pela espermatogênese, nas células de Sertoli, com consequente fertilidade.3
Cronologia e Desenvolvimento Puberal Normal
O início normal do desenvolvimento puberal difere entre meninos e meninas, sendo entre 8 e 13 anos de idade
nas meninas e entre 9 e 14 anos de idade nos meninos.9 Algumas evidências recentes, porém, sugerem tendência à diminuição desse limite de normalidade nas meninas, recomendando que o início de puberdade entre 6 e 8 anos de idade poderia ser considerado dentro do normal, principalmente nas meninas de raça negra;13 porém, não há, até o momento, dados suficientes que justifiquem a alteração desse limite da normalidade. Dessa forma, o início de puberdade entre 6 e 8 anos de idade nas meninas deve ser considerado precoce, devendo ser criteriosamente avaliado e acompanhado pelo especialista, para verificar a necessidade de intervenção e tratamento. O aumento dos hormônios esteroides das gônadas é responsável pelo desenvolvimento dos caracteres sexuais primários e secundários. O desenvolvimento sexual na puberdade segue um padrão de normalidade em meninas e meninos, em média, dentro do período de 4,5 anos (variando de 1,5 a 6 anos), com as meninas começando a puberdade mais cedo que os meninos.9
Adrenal
CAPÍTULOS
12 Adrenarca Precoce, 75
13 Passo a Passo da Triagem Neonatal da Hiperplasia Adrenal Congênita por Deficiência de 21-Hidroxilase, 80
14 Diagnóstico Diferencial de Hiperplasia Adrenal Congênita, 88
15 Insuficiência Adrenal, 96
16 Retirada de Corticoterapia Crônica, 101
17 Síndrome de Cushing em Crianças, 106
18 Feocromocitoma em Crianças, 110
Passo a Passo da Triagem Neonatal da
Hiperplasia Adrenal Congênita por Deficiência de 21-Hidroxilase
Introdução
A hiperplasia adrenal congênita (HAC) representa uma condição patológica com considerável morbimortalidade que costumava ser frequentemente subdiagnosticada durante o período neonatal, até a sua inclusão em programas de triagem neonatal. A detecção precoce e o tratamento da doença são amplamente disponíveis e eficazes na prevenção de complicações pósnatais. Essas características, aliadas a um teste de triagem de execução simples, resultam na incorporação da HAC em programas públicos de triagem neonatal em vários países.1
Tendo em vista a importância desse diagnóstico precoce, este capítulo visa atualizar os profissionais de saúde nos diversos passos da triagem neonatal para a HAC.
Definição da Hiperplasia Adrenal Congênita e seus Defeitos Enzimáticos mais Frequentes
A HAC engloba um grupo de alterações genéticas, em sua maioria com herança autossômica recessiva, potencialmente letais, resultantes da deficiência de uma das enzimas e/ou proteínas responsáveis pela síntese de cortisol e/ou aldosterona.2
Em populações caucasianas, 90% a 95% dos casos ocorrem por redução da atividade da enzima 21hidroxilase (21 OH),24 em razão da presença de variantes patogênicas no gene que codifica essa enzima, gene CYP21A2.1,4 Essas mutações provocam diferentes graus de comprometimentos na atividade enzimática, resultando em um espectro de formas clínicas distintas (Figura 13.1).
Na maioria dos grupos étnicos, a deficiência de 11betahidroxilase corresponde à segunda causa mais comum (5% dos casos), mas, no Brasil, essa deficiência é muito rara e a deficiência da 17alfahidroxilase ocupa essa posição.5
A incidência da HAC21 OH varia de acordo com a população estudada e, com base nos resultados da triagem, é de 1:14.000 a 1:18.000 nascimentos, na maioria das populações, sendo mais prevalente em grupos pequenos e geneticamente isolados.1 Trabalhos realizados na população brasileira demonstraram que a HAC também possui frequência elevada no país, variando de 1:10.000 a 1:18.000 nascidos vivos.611
Formas Clínicas da Hiperplasia
Adrenal Congênita-21-Hidroxilase
As manifestações clínicas da deficiência da 21 OH estão correlacionadas ao grau de deficiência enzimática. Os sinais e sintomas podem ser decorrentes da deficiência de glicocorticosteroide, do excesso de andrógenos e/ou da deficiência de mineralocorticosteroide.
As formas clínicas são tradicionalmente divididas em dois grandes grupos:
1. Forma clássica: com manifestações ao nascimento e subdividida em formas perdedora de sal (PS) e virilizante simples (VS).
2. Forma não clássica: cujos sintomas se iniciam tardiamente, podendo ser na infância, puberdade ou vida adulta, mas com intensidades e complicações muito menos graves do que as observadas na forma clássica.1,3 Na infância, esta forma clínica pode ser suspeitada na presença de pubarca precoce em ambos os sexos, com ou sem avanço de idade óssea (IO). Na adolescência e na idade adulta, principalmente com manifestações semelhantes às da síndrome dos ovários policísticos (SOP), ou mesmo na presença de infertilidade isolada.3 Destacamos que esta forma clínica não é de interesse para identificação em triagem neonatal, por apresentar menor gravidade, porém alguns casos podem ser acidentalmente identificados.
Na forma clássica VS, os pacientes são carreadores de variantes patogênicas que resultam em 3% a 7% de atividade residual da 21 OH em homozigose ou em heterozigose composta, com mutações que causam total comprometimento da atividade enzimática.1 Caracterizase, no sexo feminino, por virilização prénatal da genitália externa, variando desde clitoromegalia isolada à fusão completa dos grandes lábios, podendo ter aparência semelhante à da genitália masculina normal. No sexo masculino, é descrita macrogenitossomia ao nascimento, não perceptível na maioria dos casos. Esta forma clínica também ocasiona virilização pósnatal em ambos os sexos, com aumento progressivo do clitóris ou pênis, surgimento precoce dos pelos pubianos, aumento da massa muscular, acne e engrossamento da voz. Ocorrem, também, incremento da velocidade de crescimento (VC)
Mirela Costa de Miranda Ana Carolina Maués de Oliveira Jéssica Me Lin Ie Cristiane Kopacek Tânia Aparecida Sartori Sanchez Bachega
Figura 13.1 Esteroidogênese adrenal. A enzima 21-hidroxilase (P450c21) catalisa a conversão da 17-OH-progesterona em 11-desoxicortisol e da progesterona em desoxicorticosterona. A diminuição de sua atividade causa prejuízo na síntese de cortisol, com consequente aumento compensatório na produção do hormônio adrenocorticotrófico (ACTH), hiperplasia do córtex adrenal e estímulo da produção excessiva dos precursores do cortisol, que são desviados para síntese de andrógenos
e avanço da IO, resultando em fechamento epifisário precoce e baixa estatura (BE) final.1,3,4
A forma clássica PS corresponde a 70% a 75% das formas clássicas em países desenvolvidos.1,4 Esses indivíduos são carreadores de variantes patogênicas que resultam em comprometimento total ou quase total da atividade enzimática (<2%), em homozigose ou heterozigose composta.12 Uma vez que a enzima 21 OH também é essencial para a síntese de mineralocorticosteroide, esse comprometimento enzimático na síntese de aldosterona acarreta perda de peso seguida de desidratação hiponatrêmica nas primeiras semanas de vida, com potencial evolução para choque hipovolêmico e óbito sem diagnóstico precoce. Essa manifestação, conhecida como crise de perda de sal, surge, em geral, entre o 8o a 23o dia de vida. Manifestações hiperandrogênicas semelhantes às da forma VS também acontecem na forma PS em ambos os sexos.3,4
Quando Suspeitar
do Diagnóstico da Hiperplasia Adrenal Congênita?
Todos os recémnascidos com atipia genital (AG) e sem gônadas palpáveis no períneo ou na região inguinal devem ser investigados para HAC, antes mesmo da alta do berçário e do resultado do cariótipo, pelo fato de esta ser a principal causa de AG e de a forma PS ser potencialmente letal.13 Tendo em conta o grande espaço de tempo para se ter o resultado do cariótipo, essa ferramenta diagnóstica pode ser substituída pela detecção de sequências do gene SRY por reação em cadeia da polimerase (PCR) em amostra de sangue periférico, cujo resultado pode ser obtido em poucos dias.
O diagnóstico clínico do recémnascido do sexo feminino com forma clássica da HAC21 OH, seja virilizante simples ou perdedora de sal, pode ser suspeitado pela presença da virilização da genitália externa. Já nos recém
Pâncreas
19 Diabetes Melito: Classificação e Características Clínicas, 117
20 Diabetes Melito Tipo 1: Diagnóstico e Tratamento, 124
21 Diabetes Melito Tipo 1: Rastreio de Doenças Autoimunes, 132
22 Complicações Agudas Relacionadas ao Diabetes Melito Tipo 1, 135
23 Complicações Crônicas do Diabetes Melito, 142
24 Diabetes Melito Tipo 2 na Infância e na Adolescência, 148
25 Hipoglicemia, 154
Pâncreas
Diagnóstico
Diabetes Melito Tipo 1: Diagnóstico e Tratamento
Monica Gabbay Dalva Castro de Oliveira
O diabetes melito tipo 1 (DM1) é o tipo mais frequente de diabetes melito (DM) em crianças e adolescentes. A cetoacidose ainda está presente em 40% a 60% no diagnóstico do DM11 e, por esse motivo, deve-se atentar aos sinais e sintomas e buscar prontamente os exames laboratoriais para confirmação do diagnóstico (Tabela 20.1).
Na ausência de sintomas, o diagnóstico requer dois resultados de testes de triagem anormais. Se apenas um exame for alterado, um segundo teste deve ser realizado, que pode ser o mesmo ou um diferente. Glicemia ao acaso >200mg/dL, acompanhada de sintomas clássicos, já confirma o diagnóstico.2,3
Peptídio C baixo (<0,6ng/dL) dosado 2h após refeição mista pode fortalecer o diagnóstico de DM1. Além disso, a presença de pelo menos um dos marcadores de autoimunidade relacionados ao DM1 (anticorpo antidescarboxilase do ácido glutâmico [anti-GAD], anticorpo antitirosina fosfatase [anti-IA2], anti-insulina, anticorpo antitransportador de zinco 8 [anti-ZnT8]) corrobora com esse diagnóstico, porém anticorpos negativos não afastam o diagnóstico.1 A presença persistente de dois ou mais autoanticorpos de ilhotas, mesmo antes dos sinais e sintomas do DM1, é preditor quase certo do desenvolvimento da doença.4
Tabela 20.1 Critérios diagnósticos de diabetes
Critério Normal Pré-diabetesDiabetes
Glicemia de jejum (8h – mg/dL) <100 100 a 125 ≥126
Glicemia ao acaso (mg/dL) ≥200
Teste oral de tolerância à glicose 2h com 75g dextrose ou 1,75g/kg de peso corporal, até o máximo de 75g (mg/dL) <140 140 a 199 ≥200
Hemoglobina glicada (%) <5,7 5,7 a 6,4 ≥6,5
Fonte: adaptada de Diretrizes ADA, 2022;2 Cobas et al., 2023,3 Libman et al., 2022.5
Tratamento
Bases da Insulinoterapia
A administração de insulina exógena deve replicar a secreção fisiológica da insulina pelas células beta pancreáticas, o padrão basal-bólus. Neste, a secreção de insulina basal ocorre de modo contínuo e em níveis baixos para diminuir a produção hepática de glicose em jejum e entre as refeições; já a secreção da insulina em bólus sucede durante as refeições, para controlar os níveis pósprandiais de glicose, conforme a Figura 20.1.
Recomenda-se o tratamento intensivo do diabetes com múltiplas aplicações diárias de insulina desde o diagnóstico para a prevenção das complicações crônicas micro- e macrovasculares.6,7
Atualmente, existem duas formas de tratamento intensivo da glicemia amplamente utilizadas na prática clínica baseadas no padrão basal-bólus (Figura 20.2).
Bólus Bólus Bólus
Concentração de insulina
Refeição
Basal
Tempo (0 a 24h)
RefeiçãoRefeição
Figura 20.1 Esquematização do padrão basal-bólus de secreção pancreática da insulina
Múltiplas injeções de insulina
Sistema de infusão contínuo de insulina (bombas)
Figura 20.2 Tipos de tratamento intensivo do diabetes melito tipo 1
No primeiro esquema, há aplicação de pelo menos
4 injeções/dia de dois tipos de insulina com diferentes tempos de ação:
1. Insulina de ação intermediária ou preferencialmente de longa ação para mimetizar a secreção basal.
2. Insulina de ação rápida ou ultrarrápida para a função bólus.
No segundo esquema, utiliza-se apenas a insulina de ação rápida ou ultrarrápida, para as funções basal e bólus. Mais detalhes serão apresentados adiante em Sistemas de Infusão Contínua de Insulina (Bomba de Infusão de Insulina).8,9
Tipos de Insulinas
As insulinas são classificadas de acordo com seu tempo de ação.
Existem insulinas de ação rápida, ultrarrápida, intermediária, longa e ultralonga. Na Tabela 20.2 e na Figura 20.3, é apresentado um resumo das insulinas disponíveis no Brasil e seu tempo de ação.
Tabela 20.2 Classificação das insulinas
Tipo de insulina Início de ação Pico Duração
Insulinas rápidas
Asparte 15min 1 a 3h3 a 5h
Glulisina 15min 1 a 3h3 a 5h
Lispro 15min 1 a 3h3 a 5h
Regular 30min 2 a 4h5 a 8h
Ação ultrarrápida
Faster Asparte 5 a 10min1 a 3h3 a 5h
Ação intermediária
NPH 2 a 4h 4 a 12h12 a 24h
Ação longa
Detemir 1 a 2h 4 a 7h20 a 24h
Glargina 2h – 22 a 24h
Glargina U300– – 30 a 36h
Ação ultralonga
Degludeca0,5 a 1,5h– >42h
NPH: neutral protamine hagedorn Fonte: adaptada de Cengiz et al., 2022.10
Início do Tratamento
As necessidades diárias totais de insulina podem ser estimadas com base no peso, com doses variando de acordo com a idade e tempo de diagnóstico. Quantidades maiores de insulina são necessárias durante puberdade, gravidez, infecções e após cetoacidose diabética (CAD).12,13
Na Tabela 20.3, visualiza-se um resumo das doses em UI/kg de acordo com a faixa etária.
Em pacientes sem cetoacidose na apresentação inicial, recomenda-se iniciar 0,5UI/kg/dia, com 50% administrados como insulina prandial, para controlar a glicemia após as refeições e os outros 50% como insulina basal, para controle da glicemia nos períodos entre refeições.14
A Figura 20.4 apresenta o fluxograma para prescrição da insulinoterapia no paciente recém-diagnosticado.
Prescrição e Ajuste de Dose Basal
A insulina neutral protamine hagedorn (NPH) geralmente é prescrita 3×/dia. Sugerimos aplicar de manhã cedo, pré-almoço e ao deitar-se. A aplicação da NPH pré-almoço, por seu início de ação de 2 a 4h após, pode ajudar na cobertura de lanches. A aplicação noturna deve ser o mais tarde possível a fim de evitar o término de ação da insulina na madrugada, por conseguinte, evitar hiperglicemia matinal.
A insulina glargina deve ser aplicada 1×/dia, no mesmo horário, podendo ser matinal ou noturna (em alguns casos pode ser necessário 2×/dia). A insulina detemir deve ser aplicada 2×/dia, com intervalo de 12h. A insu-
Tabela 20.3 Dose total diária de insulina recomendada para pessoas com diabetes melito tipo 1
Pacientes com DM1
Dose total diária de insulina (UI/kg/dia)
Diagnóstico recente (lua de mel) <0,5
Após a remissão parcial/adultos 0,7 a 1
Lactentes
0,2 a 0,4
Pré-púberes 0,5 a 0,8
Púberes 0,8 a 2
DM1: diabetes melito tipo 1. Fonte: adaptada de Silva Júnior et al., 2023.12
Figura 20.3 Uma ilustração do tempo de ação das insulinas basal e bólus
Fonte: adaptada da SBD, 2017.11
Diferenciação Sexual VII
30 Diferenciação Sexual Normal, 187
31 Distúrbios da Diferenciação do Sexo, 191
32 Criptorquidismo, 197
33 Micropênis, 200
34 Ginecomastia, 205
30 Diferenciação Sexual Normal
Gil Guerra-Júnior
Estado Sexualmente Neutro
Até cerca de sete semanas após a fertilização, o embrião humano é um organismo bissexual, contendo primórdios gonadais e genitais idênticos nos dois sexos, sem que seja possível fazer a distinção macro- ou microscópica entre embriões com predestinação masculina ou feminina. Até esse momento são encontrados gônadas indiferenciadas, primórdios dos condutos genitais internos masculinos e femininos (dois sistemas de canais bilaterais, os dutos de Wolff e os de Müller, respectivamente) e rudimentos genitais externos.1,2
A diferenciação para o sexo masculino ou feminino depende do sexo genético do embrião, que determina a diferenciação da gônada primordial em testículo ou ovário e o sexo fenotípico (diferenciação específica
dos dutos genitais internos, seio urogenital e genitália externa).1,2
O sexo genético é estabelecido pela fertilização do óvulo por um espermatozoide contendo um cromossomo X ou um Y, sendo o sexo heterogamético (XY) masculino e o homogamético (XX) feminino. O cromossomo Y é determinante da masculinidade, uma vez que no braço curto desse cromossomo encontra-se o gene SRY (sexdetermining region on the Y chromosome), responsável por liderar uma cascata de genes necessários para a formação dos testículos.1,2
Diferenciação Sexual Masculina
Em embriões 46,XY, na presença do SRY e dos demais genes necessários à diferenciação testicular (Figura 30.1)
Crista gonadal indiferenciada (6ª semana)
SOX9 FGF9 RSPO1 FOXL2
GADD45G
Crista gonadal XY (7ª semana)
MAP3K4 p38 GATA4 FOG2 SP1 SRY
WT1 SF1
CITED2
SOX3 SOX8 SOX10 SOX9
PGD2 FGF9
RSPO1
DAX1 FOXL2
WNT4 -catenin
AXIN1 GSK3 FST
VEGF-A PDGF-B Activins
Vasos celômicos
Testículos
Crista gonadal XX (7ª semana)
CITED2 WT1 SF1
SOX3 SOX8 SOX10
PGD2 FGF9 AXIN1 GSK3
RSPO1
DAX1 FOXL2
WNT4 -catenin SOX9 FST
VEGF-A
PDGF-B Activins
Sem vasos celômicos
Ovários
Figura 30.1 Esquema dos principais genes e fatores de transcrição envolvidos na determinação gonadal SRY: sex-determining region on the Y chromosome Fonte: adaptada de Rey et al., 2020.2
observa-se, por volta da sétima semana, a diferenciação de células epiteliais em células de Sertoli. Estas se agrupam formando cordões que englobam as células sexuais primitivas, que se tornam, assim, as espermatogônias. Esses cordões testiculares desenvolvem-se para formar os túbulos seminíferos e rede testis. Sob o epitélio surge uma espessa cápsula fibrosa, a túnica albugínea. As células de Leydig, derivadas do mesênquima, podem ser observadas entre os túbulos a partir da oitava semana. Uma vez iniciada a diferenciação do testículo, este determinará o restante da diferenciação masculina por meio dos hormônios por ele produzidos (Figuras 30.2 e 30.3).1-3
Diferenciação masculina
hCG <20s SRY
LH >20s
5-alfa-redutase
DHT
46,XY
Testículo
Leydig
Rud. embr.
Epidídimo/deferente v. seminal/d. ejacul.
Glande e corpo do pênis
Pênis em torno da uretra
Bolsa escrotal
Uretra peniana
As células de Sertoli secretam a partir da sétima semana o hormônio antimulleriano (HAM), glicoproteína de alto peso molecular que induz a regressão dos dutos de Müller, cujos resquícios são representados por utrículo prostático e apêndices testiculares. A ação do HAM tem diversas peculiaridades. Sua ação é parácrina, por difusão célula a célula, ligando-se ao seu receptor específico, de modo que cada testículo é responsável pela regressão do duto de Müller de seu lado. Além disso, esses dutos somente são sensíveis ao HAM até a oitava semana; a partir de então, sua diferenciação em genitais internos femininos ocorre mesmo em presença desse
Estado neutro
Gônada bissexual HAM
Diferenciação de Müller
Diferenciação de Wolff
Tubérculo genital Pregas genitais
Saliências labioescrotais
Seio urogenital
Figura 30.2 Esquema das diferenciações sexuais masculina e feminina normais
Diferenciação feminina
46,XX
Ovário
Útero/trompas
Porção sup. da vagina
Rud. embr. Clitóris
Pequenos lábios
Grandes lábios
Porção inferior da vagina
hCG: gonadotrofina coriônica humana; SRY: sex-determining region on the Y chromosome; LH: hormônio luteinizante; HAM: hormônio antimulleriano; DHT: di-hidrotestosterona.
Fonte: imagem elaborada pelo autor.
Migração das CGP
Ação das células de Sertoli
Ação das células de Leydig
Descida do testículo para a bolsa escrotal
Diferenciação da genitália externa Crescimento da genitália externa
Diferenciação dos dutos mesonéfricos (Wolff)
Regressão dos dutos paramesonéfricos (Müller)
Diferenciação testicular
Gestação (em semanas, a partir do dia da fecundação) 45 6 7 8910 11 1213 14 15
Figura 30.3 Linha do tempo ilustrando os eventos embriológicos da diferenciação masculina
CGP: células germinativas primordiais.
Fonte: elaborada pelo autor.
Osso VIII
CAPÍTULOS
35 Deficiência de Vitamina D, 213
36 Hipocalcemia, 219
37 Hipercalcemia, 227
38 Raquitismos, 236
39 Osteogênese Imperfeita, 245
Osso
38 Raquitismos
Luiz Claudio Gonçalves de Castro
Conceitos e Definições
O termo raquitismo engloba um conjunto de condições clínicas decorrentes da insuficiência da biomineralização da matriz osteoide da placa de crescimento óssea. Já a osteomalácia refere-se à insuficiência de biomineralização da matriz osteoide durante o processo de remodelamento do osso já formado. Assim, em situações de comprometimento do suprimento mineral (cálcio e/ou fósforo) para os processos de formação e remodelamento ósseo, o raquitismo e a osteomalácia podem coexistir em crianças e adolescentes em crescimento. Entretanto, apenas a osteomalácia pode-se instalar em adolescentes que já pararam de crescer e em adultos, uma vez que suas placas de crescimento se fecharam.1-3
Do ponto de vista histopatológico, o raquitismo é caracterizado pela excessiva expansão dos condrócitos hipertróficos e, consequentemente, da zona hipertrófica da placa de crescimento, associada à desorganização de sua estrutura. Esse fato decorre da diminuição da taxa de apoptose dos condrócitos hipertróficos da placa de crescimento. Fisiologicamente, a cascata de eventos que gera a apoptose dos condrócitos hipertróficos é deflagrada pelo fosfato extracelular, através da indução de fosforilação de moléculas componentes da via proteína quinase ativada por mitógeno (MAPK; do inglês, mitogen activated protein kinase) na cartilagem de crescimento. A deficiência de fosfato inibe o desencadeamento de apoptose dessas células. Esse processo de apoptose é essencial à mineralização da matriz osteoide da placa. Como resultado de apoptose, estabelece-se o suporte para a calcificação da matriz osteoide, por meio da liberação, pelos condrócitos apoptóticos, de substâncias que induzem à invasão de vasos sanguíneos e de células osteoprogenitoras provenientes do periósteo em direção à cartilagem de crescimento. Se a apoptose não ocorrer adequadamente, a biomineralização da matriz osteoide fica prejudicada. Dessa forma, compreende-se que o evento fisiopatológico comum a todos os tipos de raquitismo é a deficiência de fósforo.1-3 Entende-se também que pode haver raquitismo sem deficiência de cálcio. A hipofosfatemia no raquitismo pode decorrer da deficiência de absorção intestinal de fósforo, da redistribuição inadequada desse elemento entre os compartimentos do organismo e/ou
da diminuição da reabsorção tubular renal de fosfato. Nos raquitismos calciopênicos, a hipofosfatemia decorre da hiperfosfatúria consequente do hiperparatireoidismo secundário gerado por hipocalcemia e/ou hipovitaminose D e, em certo grau, da diminuição da absorção intestinal de fosfato e cálcio nos casos de deficiência de vitamina D.
Causas
Os raquitismos e a osteomalácia podem ser classificados, de acordo com a deficiência mineral desencadeadora do processo fisiopatológico. Tem-se, assim, os raquitismos calciopênicos ou fosfopênicos. Também podem ser classificados de acordo com a origem do evento fisiopatológico, em adquiridos (ambientais) ou genéticos.1-3
Sob um aspecto geral, as principais etiologias de raquitismo e osteomalácia estão sumarizadas na Tabela 38.1.4 Atualmente mais de 20 causas etiológicas de raquitismo e osteomalácia são descritas, com considerável superposição clínica, radiológica e bioquímica entre elas.
Diagnóstico
Características Clínicas
Embora a caracterização e as repercussões do raquitismo sejam primariamente esqueléticas, o quadro clínico é sistêmico, afeta diversos tecidos e órgãos e compromete a saúde global da criança e do adolescente.
A suspeição de raquitismo decorre da associação entre história clínica e exame físico do paciente, complementada por avaliações metabólicas e radiológicas e, quando necessário, confirmada por testes genéticos.
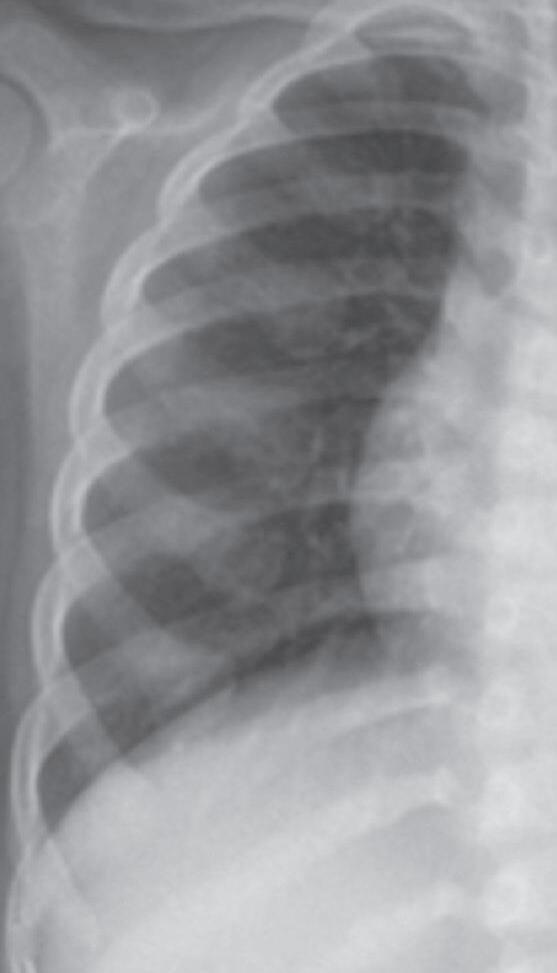
A história típica de uma criança ou adolescente com raquitismo traz relato de baixa velocidade de crescimento (VC) para a faixa etária, fraqueza muscular, dores ósseas, que podem ser demonstradas na forma de irritabilidade em crianças menores, fronte ampla, demora no fechamento das fontanelas em lactentes, alargamento das placas de crescimento em punhos, joelhos e tornozelos, deformidades em valgo e em varo nos membros inferiores. Podem-se observar, ainda, porém raramente e em condições clínico-laboratoriais mais graves, alargamento das junções costocondrais (rosário raquítico), retração do diafragma na altura do último arco costal (sulco de Har-
calcificação provisória na interface metáfise/epífise. Posteriormente, ocorre desorganização progressiva da placa metafisária, que se apresenta côncava, alargada, limites irregulares e franjeados, constituindo o sinal da taça (Figura 38.2).
Os achados típicos são rarefação óssea generalizada e alterações na placa de crescimento, que alcançam diferentes graus de gravidade em relação à perda dos limites, franjeamento das bordas metafisárias, concavidade e distanciamento entre a metáfise e a epífise. Na osteomalácia pode haver adelgaçamento da cortical dos ossos longos e pseudofraturas (zonas de Looser), mais frequentes em colo de fêmur, omoplata e púbis.5-7
A gravidade do raquitismo pode ser analisada a partir do escore de gravidade do raquitismo.8 Essa é uma escala de pontos (de 0 a 10 no total) obtida pela análise das alterações radiológicas observadas nas placas de crescimento dos punhos (rádio e ulna distais) e do joelho (fêmur distal e tíbia proximal), avaliando-se o grau de irregularidade, de concavidade e da extensão afetada da placa de crescimento.
Na osteomalácia, os sinais radiológicos característicos são as zonas de Looser ou fraturas de insuficiência. São áreas radiolucentes, compostas de osteoide não mineralizado, que se dispõem em linhas perpendiculares ao córtex ósseo e que não atravessam todo o diâmetro do osso afetado, com margens escleróticas e geralmente em regiões de submetidas a maior estresse mecânico ou áreas de suprimento vascular. São mais frequentes
no colo do fêmur, ossos do quadril, borda lateral da escápula e nos arcos costais.
No caso de suspeita de TIO, como os tumores expressam receptores 2A de somatostatina, o método imagenológico com maior sensibilidade e especificidade para detecção da lesão é a cintilografia de corpo inteiro com tomografia de emissão de pósitron/tomografia computadorizada empregando análogos da somatostatina marcado com gálio-68Ga-DOTA-DPhe1,Tyr3-octreotate (68Ga-DOTATATE PET/CT). Outros exames que podem ser utilizados nesses casos são:
Octreoscan (cintilografia de corpo inteiro com octreotida).
Cintilografia de corpo inteiro com tomografia de emissão de pósitron com 2-deoxi-2-[fluor-18]fluoro-Dglicose (18FDG-PET/CT).
Investigação Laboratorial
Como há significativa sobreposição entre as manifestações clínicas e radiográficas dos vários tipos de raquitismos, a avaliação laboratorial é essencial para diferenciá-los, ou ao menos direcionar o diagnóstico a um determinado grupo de raquitismos que partilham parte de seus mecanismos fisiopatológicos. Em determinadas situações, a confirmação diagnóstica exige teste molecular.2-4 Uma regra para adequada organização da investigação bioquímica do paciente com raquitismo é ter como objetivo encontrar a causa da hipofosfatemia, que é o
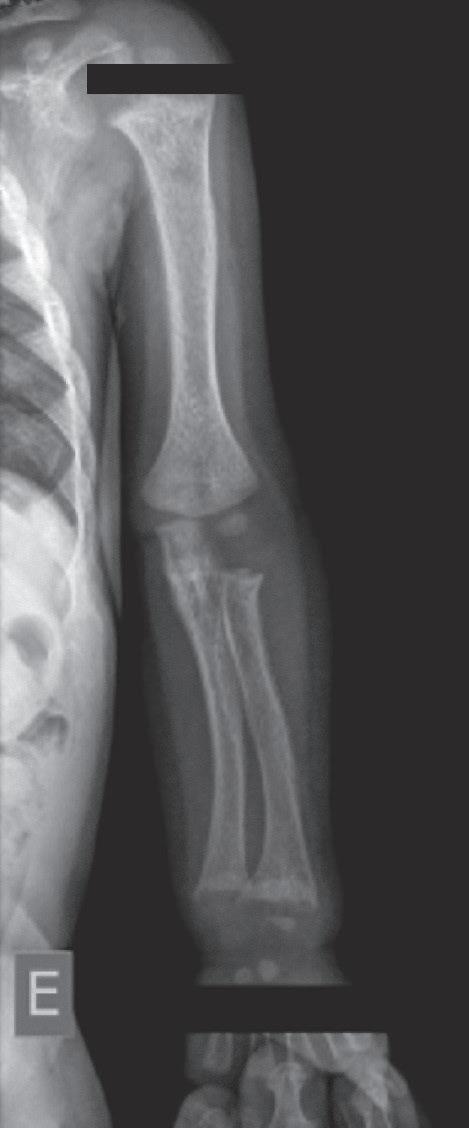
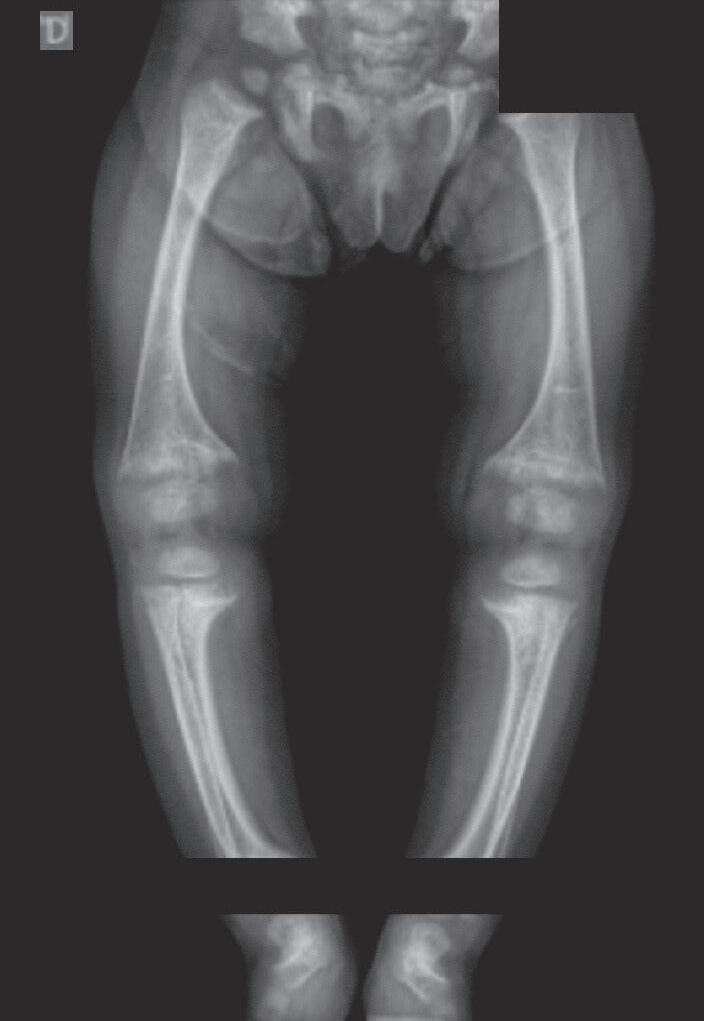
Figura 38.2 (A a C) Achados radiológicos característicos em pacientes com raquitismo. Sexo feminino, 8 anos de idade, raquitismo calciopênico por deficiência da 25-hidroxilase (mutação no gene CYP2R1), observa-se alargamento das junções costocondrais (rosário raquítico) (A ). Sexo feminino, 2 anos de idade, raquitismo hipofosfatêmico ligado ao X (mutação no gene PHEX), observam-se rarefação óssea difusa, placas de crescimento dos membros superiores e inferiores com limites mal definidos, alargadas; metáfises distais do rádio e ulna com bordas côncavas (sinal da taça) e maior distanciamento entre metáfise e epífise (B e C)
Fonte: imagens pertencentes ao autor.
A C B
ANEXO
I Memento, 315
I Memento
Nome farmacológico
Acetato de desmopressina
Acetato de gosserelina
Acetato de lanreotide
Acetato de leuprorrelina
Acetato de medroxiprogesterona
Acetato de octreotide
Acetilcisteína
ACTH, análogo (tetracosactido)
Nomes comerciais
DDAVP
Zoladex
Zoladex LA
Somatuline autogel
Eligard
Contracep
Octride
Acetilcisteína
Aires
Cisteil
Flucetil
Fluimucil
Fluiteína
Cortrosyn
Cosyntropin
Synacthen depot
Ácido zoledrônico (zolendronato)Ácido zoledrônico
Aclasta
Blaztere
Alendronato sódico
Alfacalcidol
Alfacoriogonadotrofina
Apresentações
Comprimido = 0,1 e 0,2mg
Solução nasal = 0,1mg/mL
Spray nasal = 0,1mg/mL
Seringa = 3,6mg
Seringa = 10,8mg
Seringas = 60, 90 e 120mg
FA = 7,5, 22,5 e 45mg
Ampola = 150mg/mL
FA = 0,05 e 0,1mg/mL
Envelopes = 200 e 600mg
Solução oral = 20 e 40mg/mL
Envelopes = 600mg
Solução oral = 20mg/mL
Envelopes = 200 e 600mg
Solução oral = 20 e 40mg/mL
Envelopes = 600mg
Envelopes = 200 e 600mg
Solução oral = 20 e 40mg/mL
Envelopes = 200 e 600mg
Solução oral = 20 e 40mg/mL
Ampola = 0,25mg (importado)
Ampola = 0,25mg (importado)
Ampola = 0,25mg (importado)
FA = 4mg
FA = 5mg
FA = 4mg
Densis FA = 5mg
Zolibbs
Zometa
Alendronato sódico
Bonagran
Bonalen
Minusorb
Ostenan
Osteoform
Sigmacalcidol
Ovidrel
FA = 4mg
FA = 4mg
Comprimido = 70mg
Comprimido = 70mg
Comprimido = 70mg
Comprimido = 70mg
Comprimido = 70mg
Comprimido = 70mg
Cápsula = 0,25 e 1µg
Caneta = 250µg
9-Alfafludrocortisona
Anastrozol
Atenolol
Atorvastatina
Bezafibrato
Bicalutamida
Bromocriptina
Burosumabe
Cabergolina
Carbonato de cálcio
Florinefe
Anastrozol
Anya
Arazabi
Arimidex
Anastrolibbs
Ablok
Angipress
Atenolab
Atenolol
Atenopress
Ateroma
Atorvastatina cálcica
Citalor
Lipistat
Lipitor
Vast
Bezafibrato
Cedur
Cedur retard
Bicalutamida
Casodex
Gepeprostin
Parlodel
Crysvita
Cabergolina
Caberedux
Cabertrix
Dostinex
Calciprev
Calsan
Os-CAL 500
PrevCal
Apresentações
Comprimido = 0,1mg
Comprimido = 1mg
Comprimido = 1mg
Comprimido = 1mg
Comprimido = 1mg
Comprimido = 1mg
Comprimido = 25, 50 e 100mg
Comprimido = 25 e 50mg
Comprimido = 25mg
Comprimido = 25, 50 e 100mg
Comprimido. = 50mg
Comprimido = 10, 20 e 40mg
Comprimido = 10, 20, 40 e 80mg
Comprimido = 10, 20, 40 e 80mg
Comprimido = 10, 20 e 40mg
Comprimido = 10, 20, 40 e 80mg
Comprimido = 10, 20 e 40mg
Comprimido = 200 e 400mg
Drágea = 200mg
Comprimido = 400mg
Comprimido = 50mg
Comprimido = 50mg
Comprimido = 50mg
Comprimido = 2,5mg
Frascos-ampola = 10, 20 e 30mg/mL
Comprimido = 0,5mg
Comprimido = 0,5mg
Comprimido = 0,5mg
Comprimido = 0,5mg
Comprimido= 500mg Ca elementar
Comprimido mastigável = 500mg Ca elementar
Comprimido = 500mg Ca elementar
Comprimido = 500mg Ca elementar
Calcitonina sintética de salmão
Calcitriol
Calsynar
Ostriol
Rocaltrol
Sigmatriol
Canagliflozina
Captopril
Cetoconazol
Invokana
Captopril
Captocrod
Captomed
Candoral
Cetoconazol
Izonax
Lozan
Solução nasal 100UI/mL
Comprimido = 0,25µg
Comprimido = 0,25µg
Comprimido = 0,25µg
Comprimido = 100 e 300mg
Comprimido = 25 e 50mg
Comprimido = 25 e 50mg
Comprimido = 25mg
Comprimido = 200mg
Comprimido = 200mg
Comprimido = 200mg
Comprimido = 200mg
A Endocrinologia Pediátrica é uma especialidade desafiadora, que exige do profissional conhecimento atualizado, raciocínio clínico apurado e sensibilidade no cuidado às crianças e aos adolescentes. Com esse propósito, apresentamos Endocrinologia Pediátrica no Dia a Dia , uma obra pensada para servir como um manual prático e objetivo, oferecendo suporte às decisões clínicas diante das diferentes patologias endócrinas que podem surgir na prática pediátrica.
O livro está organizado em seções temáticas que abrangem as principais áreas da especialidade: crescimento, puberdade, tireoide, adrenal, pâncreas, obesidade e dislipidemia, diferenciação sexual, metabolismo ósseo, outros distúrbios da hipófise e síndromes genéticas, bem como um capítulo dedicado à interpretação de exames de imagem aplicados à Endocrinologia Pediátrica.
Cada capítulo foi elaborado de forma didática, com enfoque em condutas práticas e baseadas em evidências, visando facilitar o manejo clínico no consultório, no ambulatório ou no hospital. Além de fornecer informações atualizadas, a obra busca transmitir segurança e clareza nas decisões, o que contribui para um atendimento mais preciso e humanizado.
Esperamos que este livro seja não apenas uma fonte de consulta rápida, mas também um guia de apoio contínuo, auxiliando médicos, residentes e demais profissionais de saúde a enfrentar, com segurança e confiança, os desafios do cuidado endocrinológico de crianças e adolescentes.