JAHRGANG 34
HEFT 5/6
Dezember 2025

JAHRGANG 34
HEFT 5/6
Dezember 2025

Chronische Rhinosinusitis mit Nasenpolypen – mehr Lebensqualität mit Tezepelumab
Myelofibrose mit Anämie: DGHO-Leitlinie empfiehlt Momelotinib
Generalisierte tonisch-klonische Anfälle: Aktuelle Daten zum Praxiseinsatz von Perampanel als frühe Zusatztherapie
Akute myeloische Leukämie: Orales Decitabin bietet Vorteile bei der Therapie unfitter Patienten
Typ-2-Inflammation bei schwerem Asthma: Leitlinie empfiehlt zielgerichtete Therapie mit Biologika
Hereditäres Angioödem: Lanadelumab kann die Krankheitskontrolle auch bei niedriger
Attackenrate verbessern
Bronchitis-Behandlung in der realen Welt: Pflanzlicher Spezialextrakt EPs® 7630 kann mehr als nur Symptome lindern
Selumetinib zur Behandlung plexiformer Neurofibrome bei Erwachsenen mit Neurofibromatose
Typ 1
Durvalumab – das erste perioperative Immuntherapie-Regime für das muskelinvasive Blasenkarzinom
Mavenclad® bei schubförmiger MS: Anhaltende Wirksamkeit auch in der behandlungsfreien Zeit
Neue Studiendaten belegen die Wirksamkeit von Deucravacitinib bei Psoriasis-Arthritis und systemischem Lupus erythematode
Tim Eikermann, TSV Bayer Leverkusen, 60 & 100 m Hürden
Deutscher Hallenmeister 2023 & 2024 (60 m Hürden)


Vor etwa 30 Jahren befragten wir in Großbritannien fast 3500 Personen, die schriftlich Interesse an der Teilnahme einer Studie zur Untersuchung an einer komplementärmedizinischen Therapie bei rheumatischen Beschwerden bekundet hatten, zu ihren einschlägigen Erfahrungen [1]. Von den gut 1000 Befragten, die uns den Fragebogen ausgefüllt zurückschickten, antwortete auf die Frage, ob ihr Hausarzt wusste, dass sie auch bei einem nichtärztlichen komplementärmedizinischen Therapeuten in Behandlung waren, gerade mal ein Viertel mit Ja. Fehlschlüsse in Bezug auf Erfolg wie auf unerwünschte Entwicklungen sind da vorprogrammiert … Ob auch hierzulande und heutzutage Hausärzte in aller Regel nicht zuverlässig wissen, wo ihre Patienten sonst noch in Behandlung sind und was da alles gemacht wird? Ob sich diese Informationslücke nur auf auf Auspendler und Gesundbeter beschränkt oder auch auf (fach-) ärztliche Behandlungen? Da erscheint es intuitiv als Segen, dass es nach viele Jahre währendem Gezerre (und mehr oder weniger dilettantischen Umsetzungsversuchen) seit 1. Oktober 2025 endlich soweit ist, dass die elektronische Patientenakte (ePA) für die Mitglieder der Gesetzlichen Krankenversicherungen – und damit für das Gros aller Patienten – für die Praxen Pflicht geworden ist. Zahlen verschiedener Krankenkassen sprechen dafür, dass wohl nur wenige Prozent (zwischen 1 und 9 %) aller Versicherten grundsätzlich widersprechen.
Keine Frage, dass ein kleiner Teil der Versicherten mit der Zustimmung eine Öffnung der Büchse der Pandora befürchtet, dass Befürchtungen aufkeimen, die ePA schaffe den irreversibel gläsernen Patienten. Auf der anderen Seite werden auch viele
Praxen offensichtlich ein dumpfes Unbehagen der ePA gegenüber nicht los – allerdings wohl nicht so sehr wegen möglicher Probleme mit dem Datenschutz, sondern primär wegen der ganz offensichtlich nicht adäquat vergüteten Mehrarbeit.
Denn die ePA soll ja nicht die „hauseigene“ Dokumentation ersetzen, sondern wesentliche Teile davon in einen elektronischen Speicher spiegeln. Warum eigentlich nicht „statt“ anstelle von „sowohl als auch“? Digitalisierung ist doch allenthalben das Zauberwort, das alles einfacher, schneller und sicherer machen soll. Mit schöner Regelmäßigkeit werden uns notorisch skeptischen Deutschen leuchtende Vorbilder wie Finnland, Dänemark, Schweden und die Benelux-Staaten [2] angedient, bei denen „alles“ digital funktioniert. Wenn die Welt gut wäre, wäre das sicher eine gute Sache. Ist sie aber nicht. Immer häufiger stolpere ich über Meldungen, wonach ein „hybrider Krieg“ gegen die Länder der Europäischen Union, allen voran Deutschland, insbesondere seitens Russlands längst im Gange ist. Rezente erfolgreiche Hackerangriffe auf kleine Gemeindeverwaltungen über Unternehmen bis hin zum amerikanischen Verteidigungsministerium belegen klar, dass Cybersicherheit ein naiver Wunschtraum ist.
Solange meine Haustüre mit einem traditionellen, mechanischen Schloss versehen ist, muss sich ein potenzieller Einbrecher vor Ort physisch daran zu schaffen machen. Sobald

Prof. Dr. med. Karl-Ludwig Resch
statt eines mechanischen ein „smartes“ Schloss installiert wird, das in das häusliche WLAN eingebunden ist, kann jeder Erdenbewohner von jedem Winkel des Planeten aus jederzeit einen Angriff starten. Umgekehrt bleibt man selbst ausgesperrt, wenn z.B. das WLAN wegen eines Stromausfalls lahmgelegt oder wenn der Akku des Handys mit der Sesam-öffne-dich-App leer ist. Zu Ende gedacht bedeutet das, dass die gesamte digitale Infrastruktur in einer neuen, zusätzlichen Dimension unsicher ist. Unsicher, weil die Gefahr, dass Unbefugte sich Zugang verschaffen ungleich größer ist als bei einer analogen Variante. Unsicher auch, weil jede Störung der Energieversorgung den Zugang unmöglich macht. Zusätzliche Unsicherheit in Form einer zunehmenden politischen Unberechenbarkeit geht von den
am meisten genutzten Datenspeichern aus, die sich außerhalb Europas befinden. An den Schaltzentralen der politischen wie der wirtschaftlichen Macht sitzen zunehmend Persönlichkeiten, für die die traditionellen Spielregeln von Kooperation und Verlässlichkeit Makulatur sind, und die jederzeit (auch) den verwalteten Datenschatz für Erpressungsversuche einsetzen könnten.
Ich halte mich grundsätzlich für aufgeschlossen allem Neuen gegenüber – allerdings nur und solange die Vorteile überwiegen und die Nachteile überschaubar sind. Gerade deshalb halte ich es für unerlässlich, bei aller Sympathie für die Digitalisierung Essenzielles weiterhin in analoger Form und mechanisch gut gesichert im Tresor vor unbefugtem Zugriff zu sichern.
Karl-Ludwig
Resch, Nürnberg
Chronische Rhinosinusitis mit Nasenpolypen –mehr Lebensqualität mit Tezepelumab 132 Brigitte Söllner
Myelofibrose mit Anämie: DGHO-Leitlinie empfiehlt Momelotinib 136
Generalisierte tonisch-klonische Anfälle: Aktuelle Daten zum Praxiseinsatz von Perampanel als frühe Zusatztherapie 138
Akute myeloische Leukämie: Orales Decitabin bietet Vorteile bei der Therapie unfitter Patienten 140
Typ-2-Inflammation bei schwerem Asthma: Leitlinie empfiehlt zielgerichtete Therapie mit Biologika 142
Hereditäres Angioödem: Lanadelumab kann die Krankheitskontrolle auch bei niedriger Attackenrate verbessern 144
Bronchitis-Behandlung in der realen Welt: Pflanzlicher Spezialextrakt EPs® 7630 kann mehr als nur Symptome lindern 147
Selumetinib zur Behandlung plexiformer Neurofibrome bei Erwachsenen mit Neurofibromatose Typ 1 148
Durvalumab – das erste perioperative ImmuntherapieRegime für das muskelinvasive Blasenkarzinom 150
Mavenclad® bei schubförmiger MS: Anhaltende Wirksamkeit auch in der behandlungsfreien Zeit 152
Neue Studiendaten belegen die Wirksamkeit von Deucravacitinib bei Psoriasis-Arthritis und systemischem Lupus erythematodes 155
Quellen
1 Ernst E, Resch KL, Hill S. Referrals between GPs and complementary practitioners. Br J Gen Pract 1996;46(409):494. PMID: 8949337
2 https://atpinfo.de/wirtschaft-und-u nternehmen/digitalisierungsfortschritt-deutschland-im-eu-vergleichauf-platz-14/
Wissenswertes 146, 156 Kongresse 157



Im Einsatz für ärzte ohne grenzen : Basma al-Chajat, Anästhesistin aus dem Irak
Unterstützen Sie Ihre Kolleg*innen bei weltweiten Hilfseinsätzen mit einer Dauerspende und werden Sie so zur Partnerärzt*in von ärzte ohne grenzen . Erfahren Sie mehr über unser Programm ärzte für ärzte : www.msf.de/partner-aerzte
JETZT SPENDEN UND PARTNERÄRZT*IN WERDEN!
Spendenkonto:
Bank für Sozialwirtschaft
IBAN: DE72 3702 0500 0009 7097 00
BIC: BFSWDE33XXX
Die chronische Rhinosinusitis (CRS) ist eine chronische Erkrankung der oberen Atemwege, die die Lebensqualität der Betroffenen erheblich einschränkt [1]. Dabei unterscheidet man innerhalb des Krankheitsbildes zwischen einer CRS mit (CRSwNP) oder ohne (CRSsNP) Nasenpolypen [1]. Die CRSwNP ist gekennzeichnet durch eine anhaltende Entzündung der Nasenschleimhaut und der Nasennebenhöhlen, die mindestens 12 Wochen andauert, sowie durch gutartige Wucherungen, die Nasenpolypen [1]. Typische Symptome sind eine nasale Obstruktion oder Schwellung, nasale Sekretion, Schmerzen im Gesicht sowie Störungen oder Verlust des Riechvermögens [2]. Die CRSwNP betrifft etwa 2 – 4 % der europäischen Bevölkerung [3]. Das durchschnittliche Alter bei Krankheitsbeginn liegt bei etwa 50 Jahren [1]. Die CRSwNP ist mit einer höheren Morbidität verbunden als die CRSsNP, was sich in einem höheren Schweregrad der Krankheit und einer höheren Anzahl von Operationen oder Medikamentenexpositionen widerspiegelt [1]. Obwohl es Fortschritte in der Behandlung von CRSwNP gibt, besteht weiterhin ein erheblicher ungedeckter medizinischer Bedarf. Trotz medikamentöser Therapie und Operation treten Nasenpolypen bei vielen Patienten erneut auf: 80 % der Betroffenen berichten 3 – 5 Jahre nach der Operation über unzureichend kontrollierte Symptome [4] und 35 % der Patienten hatten 6 Monate nach ihrer endoskopischen Nasennebenhöhlenoperation ein NasenpolypenRezidiv [5]. Zudem verkürzt sich das Operationsintervall bei rezidivierenden Nasenpolypen mit zunehmender Operationszahl [6] – eine erhebliche Belastung für die
Brigitte
Söllner, Erlangen
Patienten. Hinzu kommt das Risiko für schwerwiegende systemische Nebenwirkungen durch den wiederholten Einsatz oraler Kortikosteroide.
Eine vielversprechende neue Option zur Behandlung der chronischen Rhinosinusitis ist Tezepelumab (Tezspire®), ein monoklonaler Antikörper, der spezifisch das Thymus-Stroma-Lymphopoietin (TSLP) blockiert. Tezepelumab ist bereits seit 2023 für die Behandlung des schweren unkontrollierten Asthmas zugelassen und hat im Oktober 2025 die Zulassungserweiterung für die Therapie des CRSwNP erhalten [7].
Alarm von der Nase bis in die Lunge –das Atemwegsepithel als Schlüsselakteur
Die oberen und unteren Atemwege sind anatomisch, histologisch und immunologisch miteinander verbunden und auch die zelluläre Zusammensetzung und Struktur des Epithels sind ähnlich [8]. Daher treten Asthma und CRSwNP häufig gemeinsam auf: In Studien hatten bis zu 56 % der Patienten mit CRSwNP ein komorbides Asthma [9] und bei Patienten mit schwerem, unkontrolliertem Asth-
ma hatten in Registern rund 41 % eine komorbide CRSwNP [10].
Dabei tragen Patienten mit einer CRSwNP-Diagnose und komorbidem Asthma im Vergleich zu Patienten ohne Asthma eine höhere Krankheitslast. Auch die Asthmakontrolle ist bei komorbiden Patienten verringert [11].
Es besteht demnach eine bidirektionale Beziehung zwischen Erkrankungen der oberen und der unteren Atemwege. Die Koexistenz von CRSwNP und Asthma deutet auf eine gemeinsame Pathophysiologie hin und hat zum Konzept der United Airway Disease (UAD) geführt [12].
Epitheliale Dysfunktion bestimmt die Pathogenese
Das gesunde Epithel ist eine Schutzbarriere und damit der erste Kontaktpunkt gegenüber pathogenen Einflüssen wie Viren, Bakterien und Umwelteinflüssen. Die Bildung von Nasenpolypen ist ein vom Epithel getriebener Prozess und beinhaltet folgende Reaktionen:
1. Epitheliale Barrierestörung: Es gibt Hinweise auf eine gestörte epitheliale Barrierefunktion bei CRSwNP-Patienten [1]. Schäden am Epithel der Nasenschleimhaut führen zu einer Proliferation der

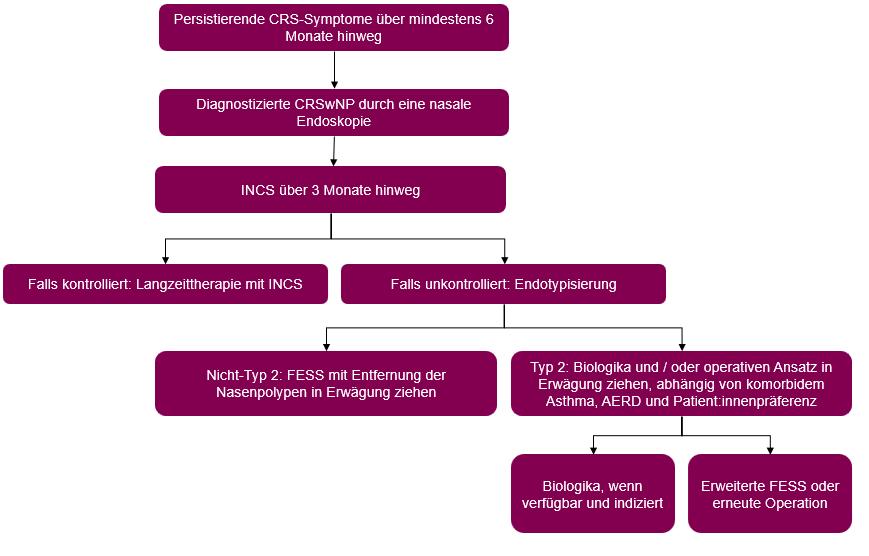
Abbildung 1: Therapieoptionen bei CRSwNP (mod. nach [1]). AERD: Analgetikaasthma; CRS: chronische Rhinosinusitis; CRSwNP: chronische Rhinosinusitis mit Nasenpolypen; FESS: funktionelle endoskopische Nasennebenhöhlenoperation; INCS: intranasale Kortikosteroide.
Epithelzellen, sodass diese in Mesenchym-Zellen übergehen. Dieser Prozess trägt zu persistierenden Entzündungsreaktionen und einem strukturellen Gewebeumbau bei, wie er für chronisch-entzündliche Erkrankungen der oberen Atemwege charakteristisch ist [13].
2. Epitheliale Entzündung: Als Reaktion auf Umweltreize oder sonstige Trigger setzt das Epithel zunächst Alarmine wie das Thymus-Stroma-Lymphopoietin (TSLP), Interleukin(IL)-25 und IL-33 frei, die mehrere nachgelagerte Entzündungswege aktivieren [14, 15]. In der Folge erhöhen sich die Konzentrationen von Zytokinen wie IL-4, IL-5 und IL-13 und die Mastzellen werden aktiviert [15]. Mastzellen und Basophile induzieren den Austritt von Plasma aus den Blutgefäßen der Nase, was zu Schleimhautödemen und letzt-
lich zur Bildung von Nasenpolypen führt [16, 17].
3. Remodelling: Langanhaltende Entzündungen können strukturelle Veränderungen von Zellen und Geweben in der Nasenschleimhaut verursachen [1, 18]:
• veränderte Epithelstruktur durch Hyperplasie oder Metaplasie
• Angiogenese, erhöhte Permeabilität der Gefäße sowie Ödeme
• Veränderungen in der extrazellulären Matrix wie Ablagerungen, Abbau oder Anreicherung von Plasmaproteinen
• Einwanderung von Granulozyten oder eine Aktivierung von Fibroblasten
Diese Remodelling-Prozesse können sich durch Schleimhautschwellungen, Nasenpolypen Pseudozysten oder fibrotisches Gewebe bemerkbar machen [18].
Der Krankheitsverlauf der CRSwNP ist oft rezidivierend [5]. Therapieziel ist daher, eine klinische Kontrolle zu erreichen und aufrechtzuerhalten. Zur Behandlung der CRSwNP stehen verschiedene Ansätze zur Verfügung (Abb. 1). Intranasale Kortikosteroide in Kombination mit Kochsalzlösung stellen die Erstlinientherapie dar und haben sich als wirksam zur Symptomreduktion und Verkleinerung der Polypen erwiesen [1]. Kurzzeitig verabreichte orale Kortikosteroide können bei moderaten bis schweren Symptomen zum Einsatz kommen. Wenn die medikamentöse Therapie nicht ausreichend wirksam ist, wird eine endoskopische Sinuschirurgie in Betracht gezogen [1]. Auch Biologika haben vielversprechende
Ergebnisse gezeigt [19]. Die bislang zugelassenen Wirkstoffe zur Behandlung der CRSwNP sind monoklonale Antikörper, die spezifisch gegen Typ-2-Entzündungsprozesse gerichtet sind [5].
Tezepelumab –ein Anti-TSLP-Antikörper zur zielgerichteten Therapie
Seit Oktober 2025 ist mit Tezepelumab (Tezspire®) ein weiterer Antikörper zur Behandlung der CRSwNP zugelassen*. Der Firstin-Class humaner monoklonaler Antikörper bindet spezifisch an das Thymus-Stroma-Lymphopoietin (TSLP), ein epitheliales Alarmin, das direkt aus dem Atemwegsepithel freigesetzt wird und als Signalstoff eine zentrale Rolle in der Entzündungskaskade spielt. Durch Blockade der Interaktion von TDLP mit seinem heterodimeren Rezeptor wirkt Tezepelumab direkt am Ursprung der Entzündung und beeinflusst eine Vielzahl von nachgeschalteten Signalwegen. Dabei verringert Tezepelumab die Aktivität von Zytokinen wie IL-5, IL-13 und IL-4, was eosinophile und neutrophile Entzündungen hemmt [20].
Bei Asthma werden durch die Therapie mit Tezepelumab gängige
* Tezepelumab ist angezeigt als Add-onTherapie mit intranasalen Kortikosteroiden zur Behandlung von Erwachsenen mit schwerer CRSwNP, die mit systemischen Kortikosteroiden und/oder chirurgischem Eingriff nicht ausreichend kontrolliert werden kann [7].
In beiden Indikationen wird Tezepelumab in der Dosierung 210 mg alle 4 Wochen verabreicht. Zur Verfügung stehen eine Fertigspritze oder ein Fertigpen für die Selbstapplikation zu Hause. Die Vorteile:
• eine Fixdosis von Anfang an
• keine Loading-Phase
• keine Anpassung der Dosierung
• keine Anpassung aufgrund des Gewichts
Bei einer überschießenden Immunreaktion und der daraus folgenden Entzündungskaskade spielen die epithelialen Alarmine als Signalstoffe eine zentrale Rolle. Einer dieser Schlüsselmediatoren ist das Thymus-Stroma-Lymphopoietin (TSLP) [25, 26]. TSLP wirkt früh im Entzündungsprozess und treibt die Immunreaktion weiter voran, indem es Mastzellen und Eosinophile aktiviert. Bei Asthma fördert dies die Wechselwirkung zwischen Mastzellen und der glatten Atemwegsmuskulatur, was zu einer Verdickung der Atemwegswand führt und zum strukturellen Umbau der Atemwege beiträgt. Bei CRSwNP kommt es durch die Freisetzung der Alarmine TSLP, IL25 und IL-33 zur Aktivierung von Mastzellen und Basophilen. Diese induzieren den Austritt von Plasma aus den Blutgefäßen der Nase, was zu Schleimhautödemen und letztlich zur Bildung von Nasenpolypen führt [15].
Biomarker wie FeNO, Eosinophile oder IgE reduziert [21, 22], auch die Hyperreagibilität der Atemwege nimmt ab [23]. Bei CRSwNP reduzierte Tezepelumab den Schweregrad der Nasenpolypen und die nasale Obstruktion [24].
Überzeugende Ergebnisse der WAYPOINT-Studie
Die WAYPOINT-Studie [25] untersuchte die Wirksamkeit und Verträglichkeit von Tezepelumab bei Erwachsenen mit schwerer, unkontrollierter chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP). In die randomisierte, doppelblinde, placebokontrollierte Phase-III-Studie wurden insgesamt 408 Patienten eingeschlossen, davon 203 Patienten in der Tezepelumab-Gruppe und 205 in der Placebo-Gruppe. Die Studienteilnehmer mussten seit mindestens 12 Monaten eine ärztlich diagnostizierte CRSwNP haben, deren Schweregrad mit der Notwendigkeit für eine Operation einherging. Dies wurde durch einen endoskopischen Nasenpolypen-Score (NPS) ≥5 definiert, mit einem Minimum von ≥2 für jedes
Nasenloch. Zusätzlich waren ein Nasal-Congestion-Score (NCS) von ≥2 und ein SNOT-22-Gesamtscore von ≥30 beim Screening erforderlich. Alle Teilnehmer erhielten eine Standardbehandlung mit intranasalen Glukokortikoiden sowie Tezepelumab 210 mg oder Placebo subkutan alle 4 Wochen für 52 Wochen.
Ko-primäre Endpunkte waren die Veränderungen des endoskopischen Gesamt-NPS und des mittleren NCS in Woche 52 im Vergleich zur Baseline. Wichtige sekundäre Endpunkte waren:
• Verlust des Riechvermögens
• Verbesserung der krankheitsspezifischen gesundheitsbezogenen Lebensqualität, gemessen anhand des SinoNasal Outcome Test 22 (SNOT-22)
• Lund-Mackay-Score
• Zeit bis zur Operationsentscheidung und/oder zur Gabe systemischer Kortikosteroide bei Nasenpolypen
• Gesamtsymptom-Score des Symptomtagebuchs für Nasenpolypen
In Woche 52 zeigten sich bei den mit Tezepelumab behandelten Patienten signifikante Verbesse-
rungen in beiden primären Endpunkten: Die Veränderung des Gesamt-NPS vs. Baseline war unter Tezepelumab signifikant größer als unter Placebo (mittlere LSDifferenz: –2,08; 95%-KI: –2,40 bis –1,76; p < 0,001). Auch der NCS verbesserte sich unter Tezepelumab signifikant stärker als unter Placebo (mittlere LS-Differenz: –1,04; 95%-KI: –1,21 bis –0,87; p < 0,001).
Die Verbesserungen des NPS und des NCS in der TezepelumabGruppe wurden bereits nach 4 Wochen beobachtet und blieben bis Woche 52 bestehen. Die Behandlung mit Tezepelumab führte zu konsistenten Verbesserungen in nahezu allen vordefinierten Subgruppen, einschließlich bei Patienten mit einer Bluteosinophilenzahl von weniger als 150 Zellen/µl zu Studienbeginn [25].
Auch hinsichtlich der sekundären Endpunkte war Tezepelumab der Placebobehandlung überlegen: Es verbesserte signifikant den Lossof-smell-Score zur Bewertung des Verlusts des Riechvermögens (mittlere LS-Differenz vs. Placebo: –1,01; 95%-KI: –1,18 bis –0,83; p < 0,001). Auch der SNOT22-Gesamtscore verbesserte sich deutlich gegenüber Placebo (mittlere LS-Differenz: –27,44; 95%KI: –32,51 bis –22,37; p < 0,001). Darüber hinaus konnte unter der Therapie mit Tezepelumab im Vergleich zu Placebo auf chirurgische Eingriffe im Studienzeitraum nahezu vollständig verzichtet werden (HR: 0,02; 95%-KI: 0,00 bis 0,09; p < 0,001) und der Bedarf an Kortikosteroiden war signifikant niedriger (HR: 0,11; 95%-KI: 0,04 bis 0,25; p < 0,001). Der LundMackay-Score verbesserte sich gegenüber Placebo um –5,70 (95%KI: –6,37 bis –5,03; p < 0,001) und der Gesamtsymptom-Score um
–6,96 (95 %-KI: –8,09 bis –-5,83; p < 0,001).
Das Sicherheitsprofil von Tezepelumab war vergleichbar mit dem von Placebo. Unerwünschte Ereignisse traten bei 78,3 % der Patienten in der Tezepelumab-Gruppe und bei 77,1 % der Patienten in der Placebo-Gruppe auf. Die häufigsten unerwünschten Ereignisse waren COVID-19, Nasopharyngitis, Infektionen der oberen Atemwege, Kopfschmerzen, Nasenbluten und eine Verschlechterung der chronischen Rhinosinusitis mit Nasenpolypen. Eine Verschlechterung der CRSwNP wurde häufiger in der Placebo-Gruppe als in der Tezepelumab-Gruppe beobachtet, nämlich bei 22,9 % gegenüber 5,4 % der Patienten. Asthma-Exazerbationen wurden ebenfalls häufiger unter Placebo als unter Tezepelumab berichtet (bei 5,9 % gegenüber 0,5%) [25]. Schwerwiegende unerwünschte Ereignisse traten bei 10 Patienten in der Tezepelumab-Gruppe (4,9 %) und bei 12 Patienten in der Placebo-Gruppe (5,9 %) auf. In der Placebo-Gruppe verstarb ein Teilnehmer an einer bakteriellen Sepsis. Die Behandlung wurde aufgrund unerwünschter Ereignisse von 0,5 % der Patienten in der Tezepelumab-Gruppe und von 1,5 % der Patienten in der Placebogruppe abgebrochen. Neutralisierende Antikörper wurden bei 3 Patienten unter Tezepelumab und bei 2 Patienten unter Placebo festgestellt [25].
Deutschen Gesellschaft für Allgemeinmedizin und Familienmedizin (DEGAM). Stand: April 2017
3 Bachert C et al. JAA 2021;14:127-134
4 van der Veen J et al. Allergy 2017;72: 282-290
5 DeConde AS et al. Laryngoscope 2017; 127:550-555
6 Smith KA et al. Int Forum Allergy Rhinol 2019;9:402-408
7 Fachinformation Tezspire®; Stand: Oktober 2025
8 Laulajainen-Hongisto A et al. Front Cell Dev Biol 2020;8:204
9 Chen S et al. Curr Med Res Opin 2020; 36:1897-1911
10 Caveney S et al. Proportion of patients with severe asthma and comorbid chronic rhinosinusitis with nasal polyps: literature review. Poster präsentiert auf dem Kongress des American College of Allergy, Asthma and Immunology (ACAAI); 24.–-27. Oktober 2024; Boston, MA, USA
11 Laidlaw TM et al. J Allergy Clin Immunol Pract 2021;9:1133-1141
12 De Corso E et al. JPM 2022;12:846
13 Siddiqui S et al. J Allergy Clin Immunol 2023;152:841-857
14 Menzies-Gow A et al. Respir Res 2020; 21:268
15 Kato A et al. J Allergy Clin Immunol 2022;149:1491-1503
16 Takabayashi T et al. J Allergy Clin Immunol 2020;145:740-750
17 Hulse KE et al. Clin Experiment Allergy 2015;45:328-346
18 Fokkens WJ et al. Rhinology 2024; 62: 287-298
19 Luk HG et al. Ear Nose Throat J 2025; 01455613251363018
20 Caminati M et al. Allergy 2024;79:11341145
21 Menzies-Gow A et al. N Engl J Med 2021;384:1800-1809 + Supplement
22 Menzies-Gow A et al. Lancet Respir Med 2023;11:425-438
23 Diver S et al. Lancet Respir Med 2021; 9:1299-1312
24 Lipworth B et al. N Engl J Med 2025; 392:1178-1188
25 Sun N et al. Allergy 2024;79:3192-3237
26 Menzies-Gow A et al. Respir Res 2020; 21:268
Literatur
1 Bachert C et al. Nat Rev Dis Primers 2020;6:86
2 Stuck BA et al. Rhinosinusitis: S2k-Leitlinie der Deutschen Gesellschaft für HalsNasen-Ohren-Heilkunde, Kopf- und Hals-Chirurgie (DGHNO-KHC) und der
Anschrift der Verfasserin: Brigitte Söllner Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen brigitte.soellner@online. de
Die Deutsche Gesellschaft für Hämatologie und Onkologie (DGHO) hat im September 2025 eine aktualisierte Version der Onkopedia-Leitlinie zur Diagnose und Therapie der Myelofibrose (MF) herausgegeben [1]. Darin wird Momelotinib (Omjjara®) im Therapiealgorithmus nunmehr als dritter zugelassener Januskinase (JAK)-Inhibitor aufgeführt. Der Einsatz von Momelotinib wird empfohlen bei krankheitsbedingter Splenomegalie oder bei Symptomen und Vorliegen einer klinisch symptomatischen, moderaten bis schweren Anämie und Thrombozytenwerten >25 Giga/l. Diese Empfehlung gilt sowohl für die Erstlinie als auch für die Zweitlinie nach Ruxolitinib-Versagen.
Der aktualisierten OnkopediaLeitlinie zufolge sollten alle Patienten mit MF und hohem oder sehr hohem Risiko initial auf ihre Eignung für eine allogene Stammzelltransplantation (SZT) hin untersucht werden. Auch für Patienten mit intermediärem Risiko kann dies sinnvoll sein.
Bei Patienten mit Splenomegalie oder Symptomen, für die eine allogene SZT nicht geeignet ist, empfiehlt die Leitlinie bei gleichzeitiger klinisch symptomatischer, moderater bis schwerer Anämie und Thrombozyten >25 Giga/l den Einsatz von Momelotinib. Liegt keine oder eine klinisch nicht relevante Anämie vor und beträgt der Thrombozytenwert >50 Giga/l, werden als Erstlini-
enoptionen unabhängig von der Risikoeinstufung die JAK-Inhibitoren Ruxolitinib oder Fedratinib empfohlen.
Bei Versagen der Ruxolitinib-Therapie empfiehlt die aktualisierte Leitlinie, bei klinisch symptomatischer, moderater bis schwerer Anämie und Thrombozyten >25 Giga/l Momelotinib als Folgetherapie einzusetzen, bei relevanter Splenomegalie und Thrombozyten >50 Giga/l Fedratinib.
Für die Bestimmung eines Ruxolitinib-Versagens wird z.B. der Einsatz des RR6 (Ansprechen auf Ruxolitinib nach 6 Monaten)-Prognosemodells empfohlen, um ein rechtzeitiges Umstellen auf eine Zweitlinientherapie zu ermöglichen. Alternativ können definierte klinische Kriterien zur Bestimmung einer Refraktärität, Progression oder Intoleranz herangezogen werden [1].
Momelotinib
Studien belegen Wirksamkeit und Sicherheit
Die Aufnahme von Momelotinib in die Leitlinie basiert auf der Zulassung durch die EMA im Januar 2024. Diese fußte auf den Ergebnissen der randomisierten, kontrollierten Studien MOMENTUM (JAK-Inhibitor-vorbehandelte Patienten, Vergleich von Momelotinib mit Danazol) und SIMPLIFY-1 (JAK-Inhibitor-naive Patienten, Vergleich von Momelotinib mit Ruxolitinib, Subgruppe der Patienten mit Anämie, Hämoglobin <10 g/dl) fußt. Diese Studien wiesen die Wirkung von Momelotinib auf die Reduktion des Milzvolumens sowie auf die beiden Anämieparameter, den Hämoglobinwert (Hb) und die Rate der Transfusionsabhängigkeit nach [2, 3, 4]. Außerdem bezieht sich die Onkopedia-Leitlinie bei ihrer
Momelotinib verfügt über einen differenzierten Wirkmechanismus, der die Hemmung von 3 wichtigen Signalwegen ermöglicht: JAK1, JAK2 und Aktivin-A-Rezeptor Typ I (ACVR1). Die Inhibition von JAK1 und JAK2 kann die konstitutionellen Symptome und die Splenomegalie verbessern. Zudem führt die Hemmung von ACVR1 zu einem Rückgang von zirkulierendem Hepcidin, was möglicherweise zu einer Verbesserung der Anämie beiträgt [2, 3, 8, 9].
Momelotinib ist zugelassen für die Behandlung der krankheitsbedingten Splenomegalie oder der Symptome bei erwachsenen Patienten mit moderater bis schwerer Anämie, die an primärer Myelofibrose (MF), Post-Polycythaemia Vera-MF oder post-essenzieller Thrombozythämie-MF erkrankt sind und nicht mit einem JAK-Inhibitor vorbehandelt sind oder mit Ruxolitinib behandelt wurden [6].
Empfehlung auf die retrospektive MoReLife-Analyse, bei der Momelotinib bei 60 Patienten unter realen Bedingungen und außerhalb einer klinischen Studie hinsichtlich Wirksamkeit und Sicherheit untersucht wurde [5]. Die Sicherheit und Verträglichkeit von Momelotinib wurden in den 3 Phase-III-Studien MOMENTUM, SIMPLIFY-1 und SIMPLIFY-2 untersucht (n = 448) [2, 3, 6]. Die häufigsten Nebenwirkungen waren Diarrhö (23 %), Thrombozytopenie (21 %), Übelkeit (17 %), Kopfschmerzen (13 %), Schwindelgefühl (13 %), Fatigue (12 %), Asthenie (11 %), Abdominalschmerzen (11 %) und Husten (10 %) [7]. Als häufigste schwere Nebenwirkung (≥Grad 3) unter
Momelotinib trat Thrombozytopenie auf (12 %) [7].
Weitere Änderungen der Onkopedia-Leitlinie umfassen neben der Aktualisierung des Therapiealgorithmus eine stärkere Gewichtung der Symptomerfassung, eine ausführlichere Empfehlung zur Diagnose und Therapie der präfibrotischen primären Myelofibrose (präPMF), deren Differenzierung von einer essenziellen Thrombozythämie (ET) und die Unterteilung in einen ET-ähnlichen und Overt-MF-ähnlichen Phänotyp sowie die Empfehlung zur Nutzung neuerer mutationsbasierter Risi-
koscores und zusätzliche Untersuchungen auf bestimmte Hochrisikomutationen, JAK2-Allel-Last und Zytogenetik mittels Next Generation Sequencing [1]. Fabian Sandner, Nürnberg
Literatur
1 Gießhammer M et al. Leitlinie Primäre Myelofibrose, Stand 09/2025
2 Verstovsek S et al. Lancet 2023;401:269280
3 Mesa RA et al. J Clin Oncol 2017; 35:3844-3850
4 Gupta V et al. Leuk Lymphoma 2024; 65:965-977
5 Jilg S et al. Ann Hematol 2024;103:40654077
6 Harrison CN, Lancet Hematol 2018; 5:e73-e81
7 Fachinformation Omjjara®; Stand: März 2025
8 Chifotides HT et al. J Hematol Oncol 2022; 15:7
9 Asshoff M et al. Blood 2017; 129:18231830






Machen Sie mit und melden Sie sich an!
Scannen Sie zur Registrierung den QR-Code oder geben Sie folgenden Link in Ihrem Webbrowser ein: https://www.eisaipro.eu/de-de/neurology/fortbildung/veranstaltungen/neuronar/anmeldung
Termine und Themen#:
10.12.25 Schlaglichter vom DGN 2025 –Kongresshighlights aus Berlin
T. Duning, Bremen
17.12.25 Schlaglichter vom DGPPN 2025 –Kongresshighlights aus Berlin
L. Hausner, Mannheim
14.01.26 Best Practice Sharing Lecanemab –Tipps zur Umsetzung in der Praxis
21.01.26
D. Saur, Leipzig / B. Kallmann, Bamberg
Demenz und Sexualität – ein Tabuthema?
M. Berner, Karlsruhe
28.01.26
Wissenswertes zur Bildgebungspraxis für die Anti-AmyloidTherapie bei früher Alzheimer-Krankheit (als radionar: Fokus-Webinar radiologische Alzheimer-Diagnostik)
J. Fiebach, Berlin
04.02.26 Amyloid, Tau und Mikroglia als therapeutische Targets zur Behandlung der Alzheimer-Krankheit
11.02.26
M. Pawlowski, Münster
Do‘s and Don‘ts bei der Epilepsietherapie
B. Steinhoff, Kehl-Kork
# Änderungen vorbehalten
Informationen zum Datenschutz bei Eisai erhalten Sie in unserer Datenschutzerklärung unter
Die Einladung für diese von der Eisai GmbH organisierte Online-Fortbildungsveranstaltung kann nur für fachliche Teilnehmer:innen ausgesprochen werden.
Der Praxiseinsatz von Perampanel (Fycompa®) in der Zusatztherapie bei unzureichend kontrollierten primär oder sekundär generalisierten tonischklonischen Anfälle wurde im Rahmen der deutschen, nicht-interventionellen Studie PERPRISE untersucht. Dabei zeigte sich nach 12 Monaten Perampanel-Therapie eine Retentionsrate von 66,7 % in der Gesamtpopulation. Die vollständigen Ergebnisse wurden kürzlich veröffentlicht und unterstreichen den klinischen Nutzen von Perampanel im Praxisalltag [1].
Alleinige Zusatztherapie bei aktiver Epilepsie
Bei der PERPRISE-Studie handelt es sich um eine 12-monatige, multizentrische, prospektive Beobachtungsstudie zur Wirksamkeit und Verträglichkeit von Perampanel als alleinige Zusatztherapie in der klinischen Praxis [1]. Eingeschlossen wurden erwachsene Patienten mit einer aktiven Epilepsie – hier definiert, als mindestens ein großer Anfall in den letzten 3 Monaten.
Von den 183 ausgewerteten Patienten erhielten 86 Perampanel zusätzlich zu einer bestehenden Monotherapie (Add-on-Gruppe).
Bei 96 Patienten wurde ein anfallssuppressives Medikament (ASM) einer Kombinationstherapie gegen Perampanel ausgetauscht (Substitutionsgruppe).
Die durchschnittliche Erkrankungsdauer in der Add-on-Gruppe war mit 12,5 Jahren niedriger als in der Substitutionsgruppe mit 20,4 Jahren. Dies spiegelte sich auch in der medikamentösen Vorbehandlung der Epilepsie wider. Während in der Substitutionsgruppe der größte Anteil (39,6 %) schon mindestens 5 unterschiedliche ASM
als Vortherapie bekamen, erhielten die meisten Patienten der Add-onGruppe (36,0 %) Perampanel als erste Zusatztherapie zur initialen Monotherapie. Die gängigsten Kombinationen zu Perampanel waren Lamotrigin (41,0 %) und Levetiracetam (33,3 %), gefolgt von Lacosamid (19,1 %) und Valproat (19,1 %) [1].
Hohe Retentions- und Anfallsfreiheitsraten
Nach 12 Monaten lag die Retentionsrate – ein Maß für die Wirksamkeit und Verträglichkeit – in der Gesamtpopulation bei 66,7 %
Perampanel
(Abb. 1A). Sowohl in der Add-on(67,4 %) als auch in der Substitutionsgruppe (66,7 %) hat sich die Zusatztherapie bei 2 von 3 der Studienteilnehmer nach einem Jahr als sehr erfolgreich gezeigt. Zudem wurde insgesamt mit 42,3 % eine hohe Anfallsfreiheitsrate über den gesamten Beobachtungszeitraum von 12 Monaten bei primär oder sekundär generalisierten Anfällen erzielt. In diesem Endpunkt zeigte sich ein höherer Wert bei der Addon- (51,7 %) gegenüber der Substitutionsgruppe (35,1 %) [1]. Die hohen Retentionsraten sind laut Studienleiter Professor Bernhard Steinhoff auch der guten Wirksamkeit schon in geringer
Perampanel (Fycompa®) ist ein hochselektiver, nicht-kompetitiver AMPA-Rezeptor-Antagonist (α-Amino-3-hydroxy-5-methyl-4isoxazolpropionsäure). AMPA-Rezeptoren, die in fast allen exzitatorischen Neuronen vorhanden sind, übertragen Signale, die vom Neurotransmitter Glutamat im Gehirn vermittelt werden. Es wird davon ausgegangen, dass sie eine Rolle bei Erkrankungen des zentralen Nervensystems spielen, die sich durch übermäßige exzitatorische Signalbildung auszeichnen, u.a. die Epilepsie [3].
Perampanel ist in Deutschland als Zusatztherapie für Patienten ab 4 Jahren mit fokalen Anfällen mit oder ohne sekundäre Generalisierung zugelassen. Außerdem ist es indiziert bei Patienten ab 7 Jahren zur Zusatzbehandlung primär generalisierter tonischklonischer Anfälle in Verbindung mit idiopathisch generalisierter Epilepsie [2].
Späte
Gesamt Frühe Gabe (2. oder 3. ASM) n = 53/74 n=122/183 n = 26/51 n=58/86 n = 43/57 n=64/96
Abbildung 1: Ergebnisse der PERPRISE-Studie:
A: Retentionsraten von Perampanel (Fycompa®) nach 12 Monaten, in denen Perampanel zusätzlich zu einer bestehenden Monotherapie verabreicht (Add-on) oder ein Wirkstoff einer Kombinationstherapie gegen Perampanel ausgetauscht wurde (Substitution).
B: Retentionsraten in Abhängigkeit von der Anzahl der vor Perampanel eingesetzten anfallssuppressiven Medikamente (ASM).
Abb. 1: Retentionsraten von Perampanel nach 12 Monaten in der PERPRISE-Studie in Abhängigkeit der Vortherapie als Addon-Gabe zu einer bestehenden Monotherapie bzw. Substitution eines Wirkstoffs einer Kombinationstherapie (A) und nach Anzahl der zuvor eingesetzten ASM (B). ASM, anfallssuppressives Medikament. A modifiziert nach: Steinhoff BJ et al. PERPRISE Study (PERampanel in patients with PRImary or SEcondarily generalized seizures): Final Analysis. Posterpräsentation anlässlich des 10. Kongress der European Academy of Neurology, 29. Juni - 02. Juli 2024, Helsinki. B: Data on file, Tabelle 8.1.2
Diagramm A modifiziert nach Steinhoff BJ et al. PERPRISE Study: Final Analysis. Posterpräsentation anlässlich des 10. Kongresses der European Academy of Neurology, 29. Juni – 02. Juli 2024, Helsinki. Diagramm B: Data on file, Tabelle 8.1.2
Dosierung zu verdanken. Im Mittel erhielten die Patienten der Add-onGruppe im Mittel 5,4 mg Perampanel/Tag und die Patienten der Substitutionsgruppe 6,4 mg/Tag [1].
Keine Beeinträchtigung der Kognition
In der Gesamtpopulation traten bei 44,0 % der Patienten unerwünschte Ereignisse auf und führten bei 16,5 % zu einem Abbruch der Behandlung. Am häufigsten wurden Schwindel (12,1 %), Fatigue (7,7 %) und Übelkeit (5,5 %) berichtet [1].
Ein Einfluss von Perampanel auf die Kognition (Messung mittels EpiTrack®) konnte im Vergleich zur Baseline bei der ersten Visite
(~6 Monate) nicht festgestellt werden. Folglich blieben die kognitiven Fähigkeiten der Patienten unter Perampanel erhalten [1].
In der Gesamtkohorte wurde Perampanel am häufigsten (40,7 %) früh – definiert als zweites oder drittes ASM – eingesetzt. Lediglich 28,0 % hatten vor Perampanel 5 oder mehr anfallssuppressive Medikamente eingenommen. Die Retentionsraten nach 12 Monaten waren bei früher und intermediärer Gabe (71,6 % bzw. 75,4 %) deutlich höher als bei später Gabe (51,0 %) (Abb. 1B). „Somit zeigt ein früher Einsatz ein besseres Outcome als eine späte
Kombination. Moderne ASM wie Perampanel sollten daher nicht deswegen spät eingesetzt werden, weil sie spät eingeführt wurden, sondern entsprechend der Behandlungssituation und des Arzneimittelprofils“, resümierte Steinhoff. Elisabeth Wilhelmi, München
Literatur
1 Steinhoff BJ et al. Epilepsia Open, 2025; doi: 10.1002/epi4.70117
2 Fachinformation Fycompa®, aktueller Stand
3 Hanada T et al. Epilepsia 2011;52:13311340
Die akute myeloische Leukämie (AML) ist die häufigste Form akuter Leukämien bei Erwachsenen [1] mit einem medianen Erkrankungsalter in Deutschland von etwa 73 Jahren. Die Zahl aller hier im Zeitraum von 2016 bis 2021 diagnostizierten Erkrankten betrug etwa 26.000 bei einer Neuerkrankungsrate von 4,72 Patienten pro 100.000 Einwohner [2]. Darüber hinaus gilt die AML als Erkrankung des Alters, denn die jährliche Inzidenz bei Patienten über 70 Jahre liegt im Vergleich zum gesamten Bundesdurchschnitt etwa 30-mal höher (ca. 100 Er-
Akute myeloische Leukämie
krankte pro 100.000 Einwohner) [1]. Da die Bevölkerung immer älter wird, geht man davon aus, dass sich die Inzidenz bis 2050 um weitere 14,6 % erhöhen wird.
Die akute myeloische Leukämie (AML) ist eine biologisch heterogene Erkrankung des blutbildenden Systems, die unbehandelt zum Tod führt. Denn infolge genetischer Veränderungen vermehren sich bei der AML myeloische Blutvorläuferzellen unkontrolliert und verdrängen dabei die gesunde Blutbildung. Die daraus resultierende hämatopoetische Insuffizienz hat zur Folge, dass die Zahl an ausdifferenzierten Blutzellen aller 3 Blutzelllinien im peripheren Blut stark abnimmt und es zu einer Leukozytopenie (v.a. Neutropenie), einer Anämie und Thrombozytopenie kommen kann, auf die sich die im Verlauf auftretenden Symptome zurückführen lassen [3]:
• Mögliche Folgen einer Neutropenie: Infektionen (u. a. bakterielle Pneumonien, systemische Mykosen)
• Mögliche Folgen einer Anämie: Müdigkeit, Blässe, Leistungsminderung/Schwäche, Dyspnoe
• Mögliche Folgen einer Thrombozytopenie: Blutungen (u.a. Nasenbluten, Petechien)
In einigen Fällen befallen leukämische Infiltrate auch andere Organe, wie Leber, Milz und Lymphknoten, die sich dann vergrößern und Beschwerden verursachen können [3]. Krankheitsdefinierend ist ein Blastenanteil von ≥20 % im peripheren Blut, im Knochenmark oder in anderen Geweben (Myelosarkom) bzw. der Nachweis AML-definierender, rekurrenter genetischer Veränderungen [3]:
• Nachweis von AML-definierenden Mutationen: Laut WHO-Klassifikation beispielsweise PML::RARA-Fusion, RUNX1::RUNX1T1Fusion, KMT2A-Umlagerung, NUP98-Umlagerung oder NPM1Mutation.
• Nachweis von Blasten: Die ICC-Klassifikation fordert beispielsweise einen Blastenanteil von ≥10 % sowie eine bestimmte Genetik/ Zytogenetik.
Die 5-Jahres-Überlebensrate liegt trotz vielversprechender Verbesserungen in Versorgung und Behandlung aktuell nur bei etwa 23,8 % [2].
Unterscheidung zwischen „fitten“ und „unfitten“ Patienten
Die Leitlinie teilt AML-Patienten hinsichtlich ihrer Therapieeignung in 2 Gruppen ein: zum einen in die für eine Standard-Induktionschemotherapie (SIC; z.B. mit Anthrazyklinen und Cytarabin) geeigneten „fitten“ Patienten und zum anderen in die für eine SIC ungeeigneten „unfitten“ Patienten. Das Verhältnis beträgt dabei etwa 50:50 [3].
Als „unfit“ gelten Patienten, wenn sie mindestens eines der folgenden Kriterien erfüllen:
• biologisches Alter ≥75 Jahre
• relevante Komorbiditäten
– diabetisches Spätsyndrom
– schwere Leber- oder Nierenerkrankungen
– Herzinsuffizienz (EF <30 %)
• ECOG* ≥3
• geringe Heilungschancen aufgrund bestimmter Mutationen (z. B. TP53**)
• hohes Risiko für eine Frühsterblichkeit unter einer SIC
* ECOPG = Eastern Cooperative Oncology Group
** TP53 = Tumorsuppressor-Gen 53
Während die Therapie „fitter“ Patienten die Heilung als Ziel hat, geht es bei den „unfitten“Patienten, bei denen eine Heilung unwahrscheinlich ist, um die Lebensverlängerung bei bestmöglicher Lebensqualität [3].
Orales Decitabin schließt die therapeutische Lücke für unfitte AML-Patienten
Die Therapie unfitter AML-Patienten ist besonders herausfordernd, da hierbei individuelle patientenund krankheitsbezogene Faktoren berücksichtigt werden müssen und die Betroffenen nur noch mit weniger intensiven Therapieregimen behandelt werden können. Medikamentös kommen hierzu hauptsächlich hypomethylierende Substanzen (HMA) infrage, die u.a. als Kombinationstherapie oder als Monotherapie eingesetzt werden können [3].
Herkömmliche HMA wie das intravenöse Decitabin oder das subkutane Azacitidin sind jedoch mit einem erheblichen Aufwand für die schwer kranken Patienten verbunden: Für i.v. Decitabin müssen sie an 5 und für s.c. Azacitidin an 7 Tagen jedes 28-tägigen Behandlungszyklus in der Klinik oder Praxis vorstellig werden [4, 5]. Dies geht für etwa 30 % der Patienten mit einer Fahrtzeit von über 2 Stunden pro Therapietag einher [6].
Daher ist es nicht verwunderlich, dass laut einer Umfrage (n = 21) über die Hälfte der Befragten die für die applikationsbedingten Anfahrts- und Wartezeiten als belastend empfindet [7] und sich sogar 95 % für eine orale HMA-Therapie entscheiden würden, die zu Hause einnehmbar ist – vorausgesetzt, sie ist verfügbar [7].
Bei Inaqovi® handelt es sich um eine oral einzunehmende Fixdosiskombination des bereits seit Jahren zugelassenen HMA Decitabin (35 mg) zusammen mit dem Cytidin-DesaminaseInhibitor Cedazuridin (100 mg) [8].
Die Hemmung der CytidinDesaminase in Darm und Leber ermöglicht es, dass Decitabin oral verabreicht werden kann, um eine vergleichbare systemische Exposition zu erreichen wie bei der i.v. Verabreichung mit demselben Dosierungsschema [8].
Mit Inaqovi® steht für neu diagnostizierte unfitte AML-Patienten seit eineinhalb Jahren eine orale Alternative für die HMA-Monotherapie zur Verfügung (Dosierung: 1 täglich eine Tablette an Tag 1 – 5 eines 28-tägigen Behandlungszyklus) [8]. Damit lässt sich nicht nur der Wunsch der Patienten nach einer einfachen oralen Therapieoption erfüllen, sondern sie können sich durch die Einnahme zu Hause auch die applikationsbedingt zeitaufwändigen Besuche im Behandlungszentrum ersparen, die viele auch aus Angst vor möglichen Infektionen vermeiden möchten.
Lebensqualität vor Lebensquantität –HMA-Venetoclax-Kombination oder (orale) HMA-Monotherapie
Der aktuelle Therapiestandard für unfitte AML-Patienten sieht eine Kombination aus dem BCL2-Inhibitorc Venetoclax und i.v. Decitabin oder s.c. Azacitidin vor, da hierdurch aktuell das beste
Gesamtüberleben erreicht werden kann [3]. Für IDH1-mutierte Patienten steht darüber hinaus die Kombination aus dem IDH1-Inhibitor Ivosidenib und s.c. Azacitidin in erster Priorität zur Verfügung [3].
Es gibt jedoch Gründe, die potenziell gegen eine Kombinationstherapie mit Venetoclax und für eine orale HMA-Monotherapie sprechen können. Zum einen sind dies medizinische Gründe, wie eine stärker ausgeprägte Zytopenie mit erhöhtem Risiko für Infektionen [3] durch Venetoclax oder der ausbleibende Überlebensvorteil durch die Hinzunahme von Venetoclax bei bestimmten genetischen Voraussetzungen (z. B. TP53-Mutation*** oder FLT3-ITD****) [9–12]. Zum anderen sind es persönliche Gründe, wie der Wunsch nach weniger therapiebedingtem Aufwand.
Orales und i.v. Decitabin zeigen vergleichbare Wirksamkeit und Verträglichkeit
Die Zulassung von Inaqovi® basiert im Wesentlichen auf der Phase-III-Studie ASCERTAIN, in der orales mit i.v. Decitabin verglichen wurde [13]. Dabei zeigte die orale Form nicht nur eine zu 99,64 % vergleichbare Bioverfügbarkeit mit der Infusion (kumulative 5-Tage-Decitabin-AUC-Äquivalenz; 90%-KI: 91,23 – 108,80 %), sondern auch eine vergleichbare Wirksamkeit und Verträglichkeit. Es wurden auch keine neuen Sicherheitssignale (z.B. spezifische gastrointestinale Nebenwirkungen) für Inaqovi® festgestellt [13].
*** BCL2 = Apoptoseregulatoren der BZell-Lymphom-2-Familie
**** FLT3-ITD = FMS-like Tyrosine Kinase-3-internal Tandem Duplications
Die Kombination von Cedazuridin mit Decitabin ermöglicht eine effiziente orale Verabreichung von Decitabin und erhöht die systemische Exposition von Decitabin. Das orale HMA bietet einen therapeutischen Fortschritt gegenüber der aktuellen Therapie bei älteren unfitten Patienten, die für eine intensive Therapie nicht geeignet sind, denn es ermöglicht eine Behandlung zu Hause und kann die Belastung durch monatliche, mehrfache intravenöse oder subkutane Behandlungen in einem Zentrum reduzieren. Da AML-Patienten nach der Diagnosestellung bisher
Bei den meisten Asthmaerkrankungen liegt den Symptomen wie Husten und Atemnot eine Typ-2-Inflammation zugrunde – ein immunologischer Mechanismus, der in rund 80 % der Fälle die Krankheitsentstehung wesentlich beeinflusst [1, 2, 3]. Diese Form der Immunreaktion ist nicht nur für die typischen Symptome verantwortlich, sondern auch für die hohe Krankheitslast vieler Patienten. Besonders herausfordernd wird es, wenn das Asthma trotz maximaler inhalativer Therapie nicht ausreichend kontrolliert werden kann: Dann spricht man von schwerem Asthma [4, 5]. Charakteristisch sind plötzliche, teils schwere Exazerbationen mit mindestens zwei leichten oder einer schweren Attacke pro Jahr, oft verbunden mit Krankenhausaufenthalten. Ursache sind Prozesse wie Bronchokonstriktion, Schleimbildung und Atemwegsödeme, die durch die zugrunde liegende Entzündung befeuert werden [4].
Doch schweres Asthma kommt selten allein: Viele Betroffene lei-
mehr als 40 % ihrer verbleibenden Lebenszeit entweder im Krankenhaus oder bei ambulanten Klinikterminen verbringen mussten [14], kann dies für viele einen Zugewinn an Lebensqualität bedeuten.
Brigitte Söllner, Erlangen
Literatur
1 De Kouchkovsky et al. Blood Cancer J 2016;6:e441
2 Baden D et al. The Lancet Regional Health–Europe 2025;59:101503
3 Röllig C. et al. Onkopedia Leitlinien: Akute Myeloische Leukämie (AML), Stand September 2025
4 Dacogen® 50 mg powder for concentrate for solution for infusion. Summary of product characteristics. February 2022
5 Vidaza® 25 mg/mL powder for suspen-
sion for injection. Summary of product characteristics. November 2023
6 Acute Leukemia Advocates Network (ALAN) Global Quality of Life Survey 2023
7 Eberhardt A et al. Exploring preferences of different modes of administration of hypomethylating Agent treatments among patients with acute myeloid leukaemia Poster #PCR8; presented at ISPOR, Vienna, Austria, 2022
8 Fachinformation Inaqovi®; Stand: März 2025
9 Döhner H et al. Blood 2024;144:22112222
10 DiNardo CD et al. Blood 2020;135:791803
11 Daver NG et al. Hematol Oncol 2023; 16:19
12 Konopleva M et al. Clin Cancer Res 2022;28:2744-2752
13 Geissler K et al. Br J Haematol 2024; 00:1734-1745
14 Potenza L et al. Cancers 2022;14:478
den zusätzlich unter Komorbiditäten wie einer chronisch obstruktiven Lungenerkrankung (COPD) oder einer chronischen Rhinosinusitis mit Nasenpolypen (CRSwNP) – Erkrankungen, die ebenfalls mit Typ-2-Inflammation in Verbindung stehen. Das zeigt, dass die Entzündung nicht lokal begrenzt, sondern systemisch relevant ist [6].
Biologika als Schlüssel zur Kontrolle der Typ-2-Inflammation
Die Typ-2-Inflammation rückt zunehmend in den Fokus der moder-
nen Asthmatherapie, insbesondere bei schwerem Asthma [4]. Charakterisiert durch T2-Helfer-Zellen, die entzündungsfördernde Mediatoren wie IL-4, IL-5 und IL-13 freisetzen, beeinflusst diese Immunantwort nicht nur die Symptomatik, sondern auch die Krankheitsprogression maßgeblich [7]. IL-5 gilt als ein zentrales Zytokin in der Regulation der Typ-2-Inflammation. Es aktiviert und moduliert eine Vielzahl immunologischer und struktureller Zellen, darunter eosinophile Granulozyten, Mastzellen, Th2-Zellen, Fibroblasten und glatte Muskelzellen [8–15]. Eine gestörte IL-5-Re-
gulation beeinträchtigt die Struktur und Funktion der Epithelzellen –jene Schutzbarriere der Atemwege, die eigentlich vor äußeren Reizen schützen soll. Wird diese Barriere durch die Entzündung geschwächt, verliert sie ihre Schutzfunktion [16].
Auch das Remodeling, die langfristige strukturelle Veränderung der Atemwege, wird durch IL-5 beeinflusst [14]. Die Dysregulation von IL-5 verändert zudem die Immunantwort [13, 17]. Darüber hinaus fördert IL-5 die Schleimproduktion, die zur Verschlechterung der Symptome beiträgt und spielt bei der Ausbildung von Nasenpolypen eine Rolle [18–20]. Als zentraler Treiber der Typ-2-Inflammation ist IL-5 ein vielversprechender Angriffspunkt für Biologika, die gezielt in diesen Prozess eingreifen und schweres Asthma effektiv therapieren können [4].
Versorgungslücke trotz klarer Empfehlung für die zielgerichtete Behandlung
Schlafstörungen, begrenzte Leistungsfähigkeit und psychische Belastungen sind häufige Begleiterscheinungen von schwerem Asthma – mit spürbaren Auswirkungen auf die Lebensqualität [6]. Umso wichtiger ist eine strukturierte und wirksame Therapie. Die nationale Versorgungsleitlinie Asthma orientiert sich dabei am Stufenschema der GINA-Asthmatherapie und bietet klare Empfehlungen für eine zielgerichtete Behandlung, insbesondere bei schwerem Asthma mit Typ-2-Inflammation [4, 6]. Aktuell stehen bei schwerem Asthma ab Stufe 5 Biologika mit verschiedenen Zielstrukturen zur Verfügung, auch gegen IL-5 [4, 6]. Trotz ihrer hohen Wirksamkeit kommen diese Medikamente bei vielen schwer
betroffenen Patienten jedoch gar nicht oder erst verspätet zum Einsatz: Nur 20 % der Patienten, für deren Krankheitslast diese Medikamente empfohlen werden, erhalten auch Biologika [21]. Dabei kann bei schwerem Asthma die unzureichende Kontrolle der Typ2-Inflammation zu unvorhersagbaren Exazerbationen und einer irreversiblen Krankheitsprogression führen [22–31].
Dosierungsschema beeinflusst Therapieadhärenz
Schwere Exazerbationen mit Hospitalisierung gelten als Marker für schweres Asthma – und betreffen in Deutschland bis zu 15 % der Patienten [32]. Sie können lebensbedrohlich sein und verursachen bei unzureichender Versorgung hohe Gesundheitskosten sowie Effizienzverluste, die durch eine bessere Versorgungsqualität vermeidbar wären [4, 6].
Eine US-amerikanische Studie mit Patienten mit schwerem Asthma, die von Fachärzten behandelt wurden, ergab, dass das Dosierungsschema der Biologika eine wichtige Rolle für die Therapieadhärenz spielt. 33 Patienten und Ärzte bevorzugen eine BiologikaTherapie mit weniger häufigen Verabreichung und somit weniger Injektionen [34]. Biologika mit längeren Dosierungsintervallen sind mit weniger Beeinträchtigungen im Alltag der Patienten und geringerem Verwaltungsaufwand für das medizinische Fachpersonal verbunden [34].
Fabian Sandner, Nürnberg
2 Seys SF et al. Respir Res 2017;18:39
3 Jackson DJ et al. Thorax. 2018;73(Suppl 4):A124-A125. Abstract P48
4 Global Initiative for Asthma (GINA). Global Strategy for Asthma Management and Prevention, Update Mai 2025. www. ginasthma.org
5 chen Diagnostik und Therapie von Asthma, Version 3.1, 2023; AWMF-RegisterNr. 020-009
6 AWMF. Nationale Versorgungsleitlinie Asthma – Langfassung. AWMF-RegisterNr. nvl-002. https://register.awmf.org/de/ leitlinien/detail/nvl002
7 Pelaia C et al. Front Physiol 2019;10: 1514
8 Sohail A et al. J Allergy Clin Immunol 2024;153:527-532
9 Galdiero MR et al. Front Med 2017;4:103
10 Lawrence MG et al. Ann Allergy Asthma Immunol 2022;128:53-60.e3
11 Borish L et al. Ann Allergy Asthma Immunol 2023;130:617-621.e1
12 Gorski SA et al. PLoS One 2019;14: e0221113
13 Bergantini L et al. Biomed Pharmacother 2023;166:115385
14 Bajbouj K et al. Allergy 2023;78:882-885
15 Khalfaoui L et al. Allergy 2022;77:29742986
16 Barretto KT et al. Allergy 2020;75:21272130
17 Malik B et al. Respirology 2023;28:758766
18 Dunican EM et al. J Clin Invest 2018; 128:997-1009
19 Tomomatsu K et al. Sci Rep 2023;13: 5468
20 Zielińska-Bliźniewska H et al. BMC Immunol 2022;23:33
21 Park J et al. CHEST. 2024;166:A4649A4650
22 Pascual RM et al. J Allergy Clin Immunol 2005;116:477-486
23 Bourdin A et al. Eur Respir J 2019;54: 1900900
24 Barnig C et al. Front Immunol 2019;10: 1699
25 Hough KP et al. Front Med (Lausanne) 2020;7:191
26 Tupper OD et al. Respir Res 2021;22:269
27 Ortega H et al. J Allergy Clin Immunol Pract 2018;6:980-986.e1
28 Engelkes M et al. Respir Med 2020;165: 105919
29 Sado AI et al. Cureus 2023;15:e25225
30 Mailhot-Larouche S et al. Ann Allergy Asthma Immunol 2025;134:31-45
31 Varricchi G et al. Allergy 2025;80:408422
32 Bergmann KC et al. J Asthma Allergy 2022;15:897-906
Literatur
1 Heaney LG et al. Chest. 2021;160:814830
33 Ledford DK et al. Annals of Allergy, Asthma & Immunology 2023;131:598605.e3
34 Gelhorn HL et al. Patient Prefer Adherence 2019;13:1253-1268
Das hereditäre Angioödem (HAE) ist eine genetisch bedingte, chronische Erkrankung, die durch rezidivierende Ödem-Attacken gekennzeichnet ist. Die Schwellungen können im Gesicht, an den Extremitäten oder Genitalien stark beeinträchtigend sein, im Gastrointestinaltrakt kolikartige Abdominalschmerzen verursachen und zu lebensbedrohlichen Kehlkopfschwellungen führen [1]. Durch die Attacken selbst und die Angst vor der nächsten Attacke ist die Krankheitslast enorm, Alltag, Ausbildung und Beruf der Patienten können daher erheblich eingeschränkt sein [2, 3], selbst wenn die Attacken nur ab und zu auftreten [4].
Therapieziele beim HAE sind eine vollständige Krankheitskontrolle und ein normales Leben für die Betroffenen [1]. Dies wird auch bei Patienten mit niedriger Attackenrate durch eine Langzeitprophylaxe mit Lanadelumab (Takhzyro®) ermöglicht, wie die Ergebnisse einer Interimsanalyse der PIQHAR-Studie zeigen, die auf dem Deutschen Allergiekongress 2025 präsentiert wurden [4].
Auch bei niedriger Attackenrate erheblich eingeschränkte Lebensqualität
Die multizentrische Phase-IVBeobachtungsstudie PIQHAR un-
Lanadelumab kann die Krankheitskontrolle auch bei niedriger Attackenrate verbessern
tersucht erstmals prospektiv im Real-World-Setting die Langzeitprophylaxe mit Lanadelumab bei HAE-Patienten, die trotz niedriger Attackenrate unter einer erheblichen Beeinträchtigung der Lebensqualität leiden. Eingeschlossen wurden bislang 22 Patienten mit HAE Typ 1 und Typ 2 ab 12 Jahren aus 8 deutschen Zentren. Sie litten unter weniger als 2 Attacken pro Monat im Jahr vor Studienbeginn, etwa 37 % davon hatten sogar weniger als eine Attacke pro Monat [4].
Zur Beurteilung des Patient Reported Outcome wurden 2 Fragebögen verwendet:
• der Angioedema Quality of Life (AE-QoL) Fragebogen, der die Beeinträchtigung der Lebensqualität ermittelt. Höhere Scores auf der Skala von 1 – 100 entsprechen einer höheren Krankheitslast [5, 6].
• der Angioedema Control Test (AECT), der die Krankheitskontrolle erfasst. Höhere Scores auf der Skala von 0 – 16 zeigen eine bessere Krankheitskontrolle an. Ein Score von ≥10 bedeutet eine gute bis sehr gute Krankheitskontrolle [7].
Die Lebensqualität war zu Studienbeginn bei ca. 74 % der Patienten stark bis sehr stark beeinträchtigt (AE-QoL-Score >50). Außerdem hatten ca. 84 % der Patienten vor Studieneinschluss einen AECT-
Score <10, was auf eine unzureichende Krankheitskontrolle hinweist.
Alle Studienteilnehmer erhielten den humanen monoklonalen Antikörper Lanadelumab subkutan gemäß der aktuellen Produktinformation (siehe auch Insert auf S. 145) [8].
Die Studie läuft von Q2/2022 bis Q4/2026, für die Interimsanalyse wurden die Daten nach einem Jahr ausgewertet [4].
Die Ergebnisse der Interimsanalyse der PIQHAR-Studie zeigen erstmals, dass auch Betroffene mit niedriger Attackenrate von einer Langzeitprophylaxe mit Lanadelumab profitieren können [4]. Wie die Daten der 13 ausgewerteten Patienten belegen, verbesserte sich bei ihnen die Lebensqualität signifikant: Der AE-QoL-Gesamtscore sank bei allen im Mittel um 33,26 Punkte nach 12 Monaten und lag im Durchschnitt bei 25,70 Punkten von Monat 1 bis 12, was einer nur noch geringen Beeinträchtigung der Lebensqualität entspricht [5, 6]. Auch beim AECT-Score zeigte sich eine Verbesserung hin zu einer guten bis sehr guten Krankheitskontrolle [7]: Er stieg im Mittel klinisch signifikant um 7 Punkte nach
Lanadelumab (Takhzyro®) ist ein vollständig humaner monoklonaler Antikörper (IgG1/κ-Leichtkette), der die proteolytische Aktivität von aktivem Plasmakallikrein hemmt. Eine erhöhte Plasmakallikrein-Aktivität führt bei Patienten mit einem hereditären Angioödem durch die Proteolyse von hochmolekularem Kininogen (HMWK), bei der gespaltenes HMWK und Bradykinin entstehen, zu Angioödem-Attacken. Lanadelumab ermöglicht eine anhaltende Kontrolle der Plasmakallikrein-Aktivität und beschränkt daher die Bradykinin-Bildung bei HAE-Patienten. Die empfohlene Anfangsdosis beträgt bei Patienten über 12 Jahren 300 mg Lanadelumab alle 2 Wochen. Bei Patienten, die unter Behandlung attackenfrei sind, kann eine Dosisreduzierung von 300 mg alle 4 Wochen in Betracht gezogen werden. Dosisanpassungen sind weder bei älteren Patienten, noch bei Patienten mit Leber- oder Nierenfunktionsstörungen erforderlich [8].
12 Monaten und lag bei 14,49 von 16 möglichen Punkten in Monat 6 bis 12 (Abb. 1a) [4].
Über 50 % der Patienten blieben im Behandlungszeitraum von einem Jahr ab der ersten Injektion vollständig attackenfrei. Die Attackenrate wurde um ca. 94 % reduziert (Abb. 1b). Lanadelumab war insgesamt gut verträglich, die meisten behandlungsbedingten Ereignisse waren mild ausgeprägt [4].
Ergebnisse der PIQHAR-Studie verdeutlichen zum einen, dass es bei der Behandlung des hereditären Angioödems nicht allein auf die Anzahl der Attacken ankommt – auch Patienten mit selteneren Schwellungen können im Alltag spürbare Einschränkungen erleben. Zum anderen belegen die Interimsergebnisse, dass gerade die-
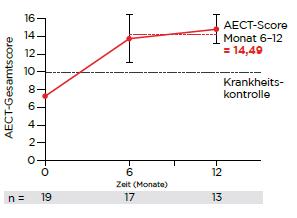
Abb. 1a
se Patienten mit Lanadelumab eine stabile Krankheitskontrolle und eine deutliche Lebensqualitätsverbesserung erreichen können. Besonders hervorzuheben ist, dass mehr als die Hälfte der Behandelten über ein Jahr hinweg attackenfrei blieb.
Brigitte Söllner, Erlangen
Literatur
1 Maurer M et al. Allergy 2022;77:19611990
2 Aygören-Pürsün E et al. Orphanet J Rare Dis 2014;9:99
3 Caballero T et al. Allergy Asthma Proc 2014;35:47-53
4 Lochbaum R et al. Propylaxis impact on quality of life impairment of HAE patients with lower annual baseline attack rates: 1-year interim results from the PIQHAR study. Poster #P8.13 präsentiert beim Deutschen Allergiekongress, 2.-4. Oktober 2025, Düsseldorf
5 Weller K et al. Allergy 2016;71:12031209
6 Kulthanan K et al. Health Qual Life Outcomes 2019;17:160
7 Weller K et al. J Allergy Clin Immunol Pract 2020;8:2050-2057.e4
8 Fachinformation Takhzyro®; Stand: April 2025
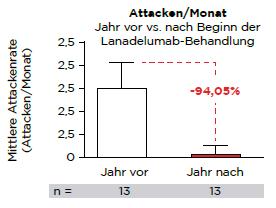
Abb. 1b
Abbildung 1: Die Ergebnisse der Interimsanalyse der PIQHAR-Studie zeigen nach 12-monatiger Langzeitprophylaxe mit Lanadelumab (Takhzyro®) eine signifikant verbesserte Krankheitskontrolle (a) und eine fast vollständige Attackenfreiheit (b) [mod. nach 4].
Frühe symptomatische AlzheimerKrankheit:
Donanemab in Deutschland verfügbar
Nur wenige Wochen nach Zulassung durch die Europäische Kommission ist Donanemab (Kisunla®) seit Anfang November auch in Deutschland erhältlich. Damit steht Patienten mit einer frühen symptomatischen AlzheimerKrankheit eine weitere dringend benötigte Behandlungsoption zur Verfügung. Erste Behandlungen mit dem Antikörper haben bereits begonnen.
Auch an der Uniklinik RWTH Aachen erhalten die ersten Patienten Donanemab. „Wir freuen uns, kurz nach der Einführung von Lecanemab mit Donanemab eine weitere kausale Therapie zur Behandlung der frühen symptomatischen Alzheimer-Krankheit anbieten zu können. Dies umfasst die Phasen der leichten kognitiven Störung und der leichten Alzheimer-Demenz bei positivem Biomarker Nachweis einer zugrundeliegenden Alzheimer Pathologie. Dazu sind Nervenwasser- oder PET-Untersuchungen notwendig“, erklärte Professor Jörg B. Schulz, Direktor der Klinik für Neurologie an der RWTH Aachen, und ergänzte: „Wir haben eine Reihe von Patienten, die wegen des vierwöchentlichen Infusionsintervalls entschieden haben, auf Donanemab zu warten. Die Therapie stellt keine Heilung dar, aber Studien mit Donanemab haben eindeutig eine Verlangsamung der Erkrankungsprogression gezeigt. In dem Studienzeitraum von 18 Monaten bedeutete dies einen Gewinn von 4 – 6 Monaten. Weitere Beobachtungsstudien le-
Donanemab, verabreicht als Infusion alle 4 Wochen, wird derzeit in den USA und anderen Ländern unter dem Markennamen Kisunla® vermarktet, darunter Japan, China, Großbritannien, die Vereinigten Arabischen Emirate, Katar, Kuwait, Bahrain, Singapur (hier wird Donanemab unter dem Markennamen Lormalzi vermarktet), Taiwan, Brasilien, Mexiko, Australien und der EU.
In den USA, Japan, China und vielen anderen Ländern ist Donanemab für Patienten unabhängig vom ApoE4-Status zugelassen. In der EU umfasst die Zulassung nur heterozygote ApoE4-Träger und -Nichtträger.
Donanemab ist die erste und einzige gegen Amyloid-Plaques gerichtete Therapie, die nachweislich ein Therapieende unterstützt, wenn Amyloid-Plaques ausreichend entfernt wurden. Dies kann zu niedrigeren Behandlungskosten und weniger Infusionen führen.
gen nahe, dass dieser Gewinn nach den ersten 18 Monaten nicht nur stabil ist, sondern weiter zunimmt. Damit können Patienten länger in frühen Stadien der Erkrankung verbleiben und länger selbständig bleiben.“
Die EU-Zulassung von Donanemab war im September 2025 erfolgt für die Behandlung von Erwachsenen mit einer leichten kognitiven Störung oder leichten Demenz infolge der AlzheimerKrankheit (frühe symptomatische Alzheimer-Krankheit), bei denen es sich um heterozygote Apolipoprotein E-ε4 (ApoE-ε4)-Träger oder -Nichtträger handelt und bei denen eine Amyloid-Pathologie bestätigt wurde. Der Wirkstoff kann den Abbau kognitiver und alltagsrelevanter Funktionen klinisch relevant verlangsamen. Donanemab ist die erste und einzige gegen Amyloid gerichtete The-
rapie, die beendet werden kann, wenn Amyloid-Plaques entfernt wurden.* Dies kann zu weniger Infusionen und dadurch zu niedrigeren Behandlungskosten führen als eine dauerhafte Therapie. Die Zulassung erfolgte unter Auflage eines kontrollierten Zugangsprogramms (Controlled Access Programme, CAP). Um die sichere und wirksame Anwendung von Donanemab zu fördern, ist der Beginn der Behandlung für alle Patienten über ein zentrales Registrierungssystem zu dokumentieren, das Teil des CAP ist. Die Therapie darf nur von Ärzten eingeleitet werden, die unter anderem Erfahrungen in der Diagnose und Behandlung der AlzheimerKrankheit und zeitnahen Zugang zur Magnetresonanztomographie (MRT) haben.
S. M.
* Die Entfernung von Amyloid-Plaques (Amyloid-Negativität) ist definiert als <24,1 Centiloids im Amyloid-PET-Scan. Die Behandlung sollte so lange fortgesetzt werden, bis die Amyloid-Plaques entfernt sind. Die Entfernung der Amyloid-Plaques ist durch einen validierten Test zu bestätigen. Die maximale Behandlungsdauer beträgt 18 Monate und sollte nicht überschritten werden, auch wenn die Plaques-Entfernung nicht bestätigt wird.
Eine neue, umfangreiche RealWorld-Analyse mit Daten von über 370.000 Patienten stellt die gängige Behandlungspraxis bei akuter Bronchitis infrage: Antibiotika werden häufig verordnet, obwohl die meisten Infekte viral bedingt sind. Demgegenüber zeigt der Wurzelextrakt EPs® 7630 aus Pelargonium sidoides (enthalten in Umckaloabo®) in mehreren klinisch relevanten Endpunkten klare Vorteile: Weniger Folgeverordnungen von Antibiotika, weniger Rückfälle, weniger Komplikationen und kürzere Krankschreibungen.
Dies belegen die Ergebnisse einer retrospektiven Auswertung der Verschreibungs- und Versorgungsdaten (Real-World-Daten) von über 370.000 Patienten mit akuter Bronchitis, die von Gillissen et al. anhand der IQVIA™ Disease Analyzer-Datenbank durchgeführt wurde. Diese Datenbank enthält Fallinformationen von fast 3.000 niedergelassenen Ärzten und repräsentiert etwa 3 – 5 % aller deutschen Praxen. Einbezogen wurden die Daten von Patienten ab 18 Jahren, die in Deutschland ambulant behandelt wurden (d.h. Hausarztpraxen besuchten) und bei denen zwischen Januar 2005 und Dezember 2022 mindestens einmal eine akute Bronchitis in der IQVIA™ Dis-
7630 kann mehr als

Der Wurzelextrakt EPs® 7630 aus Pelargonium sidoides überzeugt auch in der realen Behandlungswelt mit starker Evidenz (© Dr. Willmar Schwabe).
ease Analyzer-Datenbank diagnostiziert wurde. Als Vergleichstherapien wurden Acetylcystein, Ambroxol, Antibiotika (Initialverordnung) und EPs® 7630 untersucht. Endpunkte waren der Bedarf an nachfolgenden Antibiotika, Wiedererkrankungen innerhalb eines Folgejahres, Komplikationen wie chronische Bronchitis oder Lungenentzündung (CAP) und die Dauer der Arbeitsunfähigkeit.
Hier die Kernaussagen der Analyse:
• Antibiotika-Übergebrauch : Obwohl die akute Bronchitis
in rund 90 % der Fälle viral bedingt ist, erhielten 89 % der Patienten, die nur ein Medikament bekamen, ein Antibiotikum –ohne gesicherten Nutzen und mit dem Risiko, Resistenzen zu fördern.
• Weniger Bedarf an späteren Antibiotika: Initial mit EPs® 7630 behandelte Patienten benötigten seltener nachfolgend Antibiotika als unter Acetylcystein (p = 0,02). Gegenüber Ambroxol zeigte sich kein signifikanter Unterschied (p = 0,112). Im Vergleich zu einer initialen Antibiotikatherapie war das Risiko einer weiteren Antibiotika-
Verordnung nach 31 – 365 Tagen dagegen um 44 % geringer (p < 0,001).
• Langfristiger Schutz: Das Risiko für eine erneute Arztkonsultation wegen akuter Bronchitis innerhalb eines Jahres war mit EPs® 7630 immer signifikant niedriger als unter Ambroxol, Acetylcystein und Antibiotika.
• Weniger Komplikationen: Das Risiko einer chronischen Bronchitis war im Vergleich zu Ambroxol und Acetylcystein signifikant geringer (p < 0,001).
Das Risiko einer Lungenentzündung war gegenüber Acetylcystein und Antibiotika signifikant (p < 0,003 und p = 0,005) und gegenüber Ambroxol tendenziell geringer.
• Schnellere Rückkehr in den Alltag: Der Anteil längerer Arbeitsunfähigkeiten (>3, ≥7, ≥14 Tage) war in der EPs® 7630-Gruppe durchweg am niedrigsten.
Die Real-World-Daten zeigen zum einen eine deutliche Diskrepanz zwischen Evidenz und Verordnungspraxis: Patienten mit akuter, durch Viren verursachter Bronchitis erhalten häufig Antibiotika. Zum anderen belegen die Ergebnisse, dass der pflanzliche Spezialextrakt EPs® 7630 messbare Vorteile in der Versorgung bringt. Die Autoren diskutieren als Erklärung unter anderem immunmodulatorische Effekte wie das Anregen einer Interferon-Antwort, die zu einem besseren Schutz vor zukünftigen Virusinfektionen beitragen könnten. Zudem könnte der Extrakt auch das Anhaften von Bakterien an Schleimhäuten erschweren. Diese aus dem Labor bereits
bekannten Mechanismen bedürfen weiterer Forschung. Als retrospektive Real-WorldAnalyse kann die Studie kausale Zusammenhänge nicht abschließend belegen und erfasst nicht alle potenziellen Einflussfaktoren des Lebensstils. Dennoch sind die Ergebnisse aufgrund der großen Stichprobe und der Konsistenz der Effekte für die Versorgungspraxis relevant.
Die Daten sprechen für ein Umdenken bei der Behandlung der akuten Bronchitis: EPs® 7630 geht über eine reine Symptomlinderung hinaus, senkt den Bedarf an Antibiotika, reduziert Rückfälle und Komplikationen und verkürzt die Dauer der Arbeitsunfähigkeit. Auch vor dem Hintergrund zunehmender Antibiotikaresistenzen sollten bewährte pflanzliche Optionen in der Erstlinien-Therapie stärker berücksichtigt werden.
Elisabeth Wilhelmi, München
Die Neurofibromatose Typ 1 (NF1, Morbus Recklinghausen) ist eine seltene, progressive, genetisch bedingte Erkrankung, die meist schon in der frühen Kindheit diagnostiziert wird, aber bis ins Erwachsenenalter fortschreitet und jedes Organsystem beeinträchtigen kann [1, 2]. Bis zu 50 % der Betroffenen entwickeln plexiforme Neurofibrome (PN), primär gutartige Nervenscheidentumoren, die alle peripheren Nerven befallen können [2, 3]. PN können auch in einer späteren Lebensphase auftreten und sehr groß werden, was zu erheblichen Beeinträchtigungen wie Entstellungen, Schmerzen, sensiblen und motorischen Defiziten führen kann. Darüber hinaus besteht die Möglichkeit einer malignen Transformation [2, 3]. Bis 2025 gab es weltweit keine zugelassenen medikamentösen Therapien für Erwachsene mit NF1 und symptomatischen, inoperablen PN. Zur Behandlung im Kindesund Jugendalter ab 3 Jahren steht seit 2021 der oral einzunehmende Kinase-Inhibitor Selumetinib (Koselugo®) zur Verfügung, der inlabel über das 18. Lebensjahr weiter verordnet werden konnte. Die Zulassungserweiterung auf Erwachsene mit NF1 PN bietet Ärzten nun die Möglichkeit, auch Patienten im Erwachsenenalter auf Selumetinib einzustellen [4].
KOMET-Studie bestätigt signifikante Verringerung des Tumorvolumens auch bei Erwachsenen
Quelle: Gillissen A et al. Prescription of EPs 7630 is associated with short- and long-term benefits in acute bronchitis: a real-world data analysis. Front Pharmacol 2025;16:1652203. doi: 10.3389/fphar.2025.1652203
Die Zulassung von Selumetinib für Erwachsene folgte auf die positive Stellungnahme des Ausschusses für Humanarzneimittel (CHMP) und basiert auf den Ergebnissen der randomisierten, doppelblin-
den, placebokontrollierten KOMET-Studie, die die Wirksamkeit und Sicherheit von Selumetinib bei Erwachsenen mit NF1 und symptomatischen, inoperablen PN untersuchte [5]. An dieser bislang einzigen placebokontrollierten globalen Phase-III-Studie in dieser Patientengruppe nahmen 145 Erwachsene aus 13 Ländern in Nord- und Südamerika, Europa, Asien und Australien teil. Die Baseline-Charakteristika der Teilnehmer, einschließlich Geschlecht und Verteilung der PN, spiegelte die globale Population erwachsener NF1-Patienten wider. Voraussetzungen für die Aufnahme in die Studie waren die Diagnose NF1, mindestens ein symptomatisches, inoperables, mit volumetrischer MRT-Analyse messbares PN, ein während des Screenings
Selumetinib
dokumentierter chronischer PNSchmerz-Score, eine ausreichende Organ- und Knochenmarkfunktion sowie eine stabile chronische PNSchmerzmedikation [5, 6].
Die Patienten erhielten 1:1 randomisiert Selumetinib (n = 71) oder Placebo (n = 74) über 12 Zyklen von jeweils 28 Tagen. Nach 12 Zyklen wurden Patienten, die Placebo erhielten, auf Selumetinib umgestellt. Patienten, die Selumetinib erhielten, setzten diese Behandlung für weitere 12 Zyklen fort. Primärer Endpunkt war die bestätigte objektive Ansprechrate (ORR) bis Zyklus 16. Definiert wurde die ORR als der Prozentsatz der Patienten mit bestätigtem vollständigem Ansprechen (Rückbildung der PN) oder partiellem Ansprechen (mindestens 20%ige Verringerung des Tumorvolumens
Selumetinib (Koselugo®) ist ein Kinase-Inhibitor, der die Mitogenaktivierten Proteinkinase-Kinasen 1 und 2 (MEK1/2) selektiv blockiert, die an der Stimulierung des Zellwachstums beteiligt sind. Bei Neurofibromatose Typ 1 (NF1) sind diese Enzyme überaktiviert, was dazu führt, dass Tumorzellen unkontrolliert wachsen und plexiforme Neurofibrome (PN) bilden. Durch die Blockierung dieser Enzyme hemmt Selumetinib das Wachstum von Tumorzellen und kann somit zur Volumenreduktion der PN führen.
Die Selumetinib-Monotherapie ist fürKinder ab 3 Jahren, Jugendliche und Erwachsene zur Behandlung von symptomatischen, inoperablen PN bei NF1 zugelassen. Die empfohlene Dosis beträgt 25 mg/m2 Körperoberfläche, zweimal täglich (alle 12 Stunden) als Hartkapsel oral eingenommen [4].
gegenüber dem Ausgangswert).
Sekundäre Endpunkte waren u.a. eine Verbesserung der PN-bedingten Schmerzen und der gesundheitsbezogenen Lebensqualität (HRQoL) bis Zyklus 12 [5, 6].
In der Primäranalyse der Studie zeigte sich unter Selumetinib bis Zyklus 16 eine statistisch signifikante objektive Ansprechrate (ORR) von 20 % (n = 14/71, 95%KI: 11,2 – 30,9) im Vergleich zu 5 % unter Placebo (n = 4/74, 95%KI: 1,5 – 13,3; p = 0,01) [5].
Das Sicherheitsprofil entsprach dabei dem der etablierten Anwendung bei pädiatrischen Patienten. Die wichtigsten Nebenwirkungen waren Hautveränderungen und Durchfall [5].
In der KOMET-Studie konnte durch die zielgerichtete Therapie mit Selumetinib eine signifikante Verringerung des Tumorvolumens bei Erwachsenen erzielt werden. Dies bedeutet einen maßgeblichen therapeutischen Fortschritt in der Behandlung von plexiformen Neurofibromen bei Patienten mit Neurofibromatose Typ 1, denn nun können auch erwachsene Patienten mit einer Erstdiagnose vom klinischen Nutzen der Behandlung mit Selumetinib profitieren.
Brigitte Söllner, Erlangen
Literatur
1 Tamura R. Int J Mol Sci 2021;22:5850
2 Hirbe AC et al. Lancet Neurol 2014; 13:834-843
3 Bergqvist C et al. Orphanet J Rare Dis 2020;15:37
4 Fachinformation Koselugo®; Stand: Oktober 2025
5 Chen AP et al. Lancet. 2025;405: 2217-30
6 ClinicalTrials.gov. Efficacy and safety of selumetinib in adults with NF1 who have symptomatic, inoperable plexiform neurofibromas (KOMET). NCT Identifier: NCT04924608
Jährlich erkranken in Deutschland etwa 31.000 Menschen an einem Harnblasenkarzinom, das bei Männern die vierthäufigste Krebsneuerkrankung darstellt [1]. Bei etwa einem Viertel der Neuerkrankungen handelt es sich um einen Tumor, der in die Muskelwand der Blase eindringt [2]. Die derzeitige Standardbehandlung des muskelinvasiven Blasenkarzinoms (MIBC) ist eine radikale Zystektomie (RC) mit bilateraler Lymphadenektomie, bei Cisplatinfähigen Patienten in Kombination mit einer neoadjuvanten Chemotherapie (NAC) [3]. Doch selbst nach dieser kurativ intendierten Behandlung kommt es nach der Resektion häufig zu einem Rezidiv mit schlechter Prognose, sodass viele Patienten versterben [4]. Daher werden dringend Behandlungsoptionen benötigt, die einen Rückfall nach der Operation verhindern. Eine vielversprechende neue Therapieoption ist der monoklonale Antikörper Durvalumab (Imfinzi®), der im Juli 2025 von der Europäischen Kommission zur Behandlung erwachsener Patienten mit resezierbarem muskelinvasivem Blasenkarzinom zugelassen wurde. Die Zulassung gilt in Kombination mit Gemcitabin und Cisplatin (Gem-Cis) zur neoadjuvanten Behandlung, gefolgt von Durvalumab als Monotherapie zur
adjuvanten Behandlung nach radikaler Zystektomie [5].
Signifikante Verbesserung des ereignisfreien Überlebens und des Gesamtüberlebens
Die Zulassung von Durvalumab basiert auf den Ergebnissen der offenen, randomisierten Phase-IIIStudie NIAGARA, die erstmals zeigen, dass die zusätzliche Gabe von Durvalumab zur Chemotherapie vor der Zystektomie, gefolgt von Durvalumab als Monotherapie zur adjuvanten Behandlung, das Leben der Patienten im Vergleich zur alleinigen neoadjuvanten Chemotherapie signifikant verlängern kann [6].
Eingeschlossen in die NIAGARAStudie wurden 1063 Patienten mit MIBC. Sie erhielten randomisiert entweder das NIAGARA-Regime, bestehend aus 4 Zyklen Durvalumab plus einer neoadjuvanten Chemotherapie vor der Zystektomie, gefolgt von 8 Zyklen Durvalumab-Monotherapie alle 4 Wochen (n = 533), oder nur den bisherigen Standard, d.h. eine neoadjuvante Chemotherapie in Form von Gemcitabin-Cisplatin vor der Zystektomie, ohne weitere Behandlung nach der Operation (n = 530). Die dualen primären Endpunkte waren das ereignisfreie Überleben (EFS)
und die pathologische Komplettremission (pCR) [6].
In der geplanten Interimsanalyse mit einer medianen Nachbeobachtungszeit von 42,3 Monaten führte die perioperative Behandlung mit Durvalumab zu einer statistisch signifikanten relativen Risikoreduktion von 32 % beim EFS* (HR: 0,68; 95%-KI: 0,56 – 0,82; p < 0,0001) gegenüber dem Vergleichsarm [6]. Der geschätzte Median des EFS wurde in der Durvalumab-Gruppe noch nicht erreicht, während er in der Vergleichsgruppe 46,1 Monate betrug. Schätzungsweise 67,8 % der mit dem NIAGARA-Regime behandelten Patienten waren nach 2 Jahren ereignisfrei, verglichen mit 59,8 % in der Vergleichsgruppe [6].
Die Ergebnisse für den wichtigsten sekundären Endpunkt, das Gesamtüberleben (OS), zeigten, dass das perioperative Immuntherapieregime mit Durvalumab das Sterberisiko gegenüber der Vergleichsgruppe um 25 % verringerte (HR: 0,75; 95%-KI: 0,59 – 0,93; p = 0,0106) [6]. Die mediane Überlebenszeit wurde in beiden Grup-
* Risikoreduktion für Ereignisse des ereignisfreien Überlebens. Relevante Ereignisse sind: progressive Erkrankung, die eine radikale Zystektomie verhinderte; ein Rezidiv nach einer radikalen Zystektomie; das Datum der erwarteten Operation bei Patienten, die sich keiner radikalen Zystektomie unterzogen; Tod jeglicher Ursache.
Durvalumab (Imfinzi®) ist ein vollständig humaner, monoklonaler Immunglobulin-G1-kappa-(IgG1κ)-Antikörper, der selektiv die Interaktion von PD-L1 mit PD-1 und CD80 (B7.1) blockiert. Dadurch verbessert es die antitumorale Immunantwort und erhöht die TZellaktivierung [5].
Die Expression des programmed cell death ligand 1 (PD-L1)-Proteins ist eine adaptive Immunantwort, die Tumoren dabei hilft, sich der Erkennung und Elimination durch das Immunsystem zu entziehen. PD-L1 kann durch inflammatorische Signale induziert (z. B. IFN-gamma) und sowohl auf Tumorzellen als auch tumorassoziierten Immunzellen im Tumormikromilieu exprimiert werden. Durch Interaktion mit PD-1 und CD80 (B7.1) blockiert PD-L1 die T-Zellfunktion und -aktivierung. Durch Bindung an seine Rezeptoren verringert PD-L1 die zytotoxische T-Zellaktivität, Proliferation und Zytokinproduktion [5].
pen noch nicht erreicht. 82,2 % der mit dem NIAGARA-Regime behandelten Patienten waren nach 2 Jahren noch am Leben, verglichen mit 75,2% in der Vergleichsgruppe (Kaplan-Meier-Schätzung) [6].
Handhabbares Verträglichkeitsprofil
Unter der Behandlung mit dem NIAGARA-Regime traten keine neuen Sicherheitssignale bei der neoadjuvanten und adjuvanten Behandlung auf. Verglichen mit der alleinigen NAC führte die Zugabe von Durvalumab nicht zu einer erhöhten Abbruchrate aufgrund von Nebenwirkungen und beeinträchtigte auch nicht die Fähigkeit der Patienten, die radikale Zystektomie abzuschließen (88 % unter dem NIAGARA-Regime, 83 % im Vergleichsarm). Immunver-
mittelte unerwünschte Ereignisse betrafen 21 % der Patienten unter dem NIAGARA-Regime, was dem bekannten Profil von Durvalumab entsprach [6]. Die Mehrheit der immunvermittelten unerwünschten Ereignisse war niedriggradig [7].
Die Ergebnisse der NIAGARAStudie lassen den Schluss zu, dass die Immuntherapie mit Durvalumab (Imfinzi®) bereits in den frühen Stadien der Behandlung des muskelinvasiven Blasenkarzinoms zusätzlich zur Standardbehandlung eingesetzt werden sollte, da derzeit fast die Hälfte der Patienten trotz neoadjuvanter Chemotherapie und radikaler Zystektomie ein Rezidiv erleidet oder verstirbt. Das perioperative Immuntherapieregime mit Imfinzi® senkte in der NIA-
GARA-Studie das Rezidivrisiko um fast ein Drittel und führte zu einer signifikanten Verlängerung der Überlebenszeit. Damit hat das NIAGARA-Regime das Potenzial, die klinische Praxis im kurativen Setting zu verändern. Dies spiegelt sich auch in der Bewertung der Europäischen Gesellschaft für Medizinische Onkologie (ESMO) wider, die das NIAGARA-Regime anhand der Magnitude of Clinical Benefit Scale (MCBS) bewertet und ihm im kurativen Bereich die höchstmögliche Note „A“ verliehen hat [8].
Brigitte Söllner, Erlangen
Literatur
1 Robert Koch Institut. Krebs in Deutschland 2019/2020 (Stand: 2023). https:// www.krebsdaten.de/Krebs/DE/Content/ Publikationen/Krebs_in_Deutschland/ krebs_in_deutschland_2023.pdf?__ blob=publicationFile
2 Fletcher, A et al. ISRN Urol 2011;2011: 545241
3 S3-Leitlinie: Früherkennung, Diagnose, Therapie und Nachsorge des Harnblasenkarzinoms, Version 3.0 (Stand: März 2025). AWMF-Register-Nr. 032/038OL
4 Pfister C et al. Lancet Oncol 2024;25: 255-264
5 Fachinformation Imfinzi®; Stand: Juli 2025
6 Powles T et al. N Engl J Med 2024; 391:1773-1786
7 Galsky MD et al. J Clin Oncol 2025; 43:15-21
8 ESMO. Durvalumab NIAGARA. https:// www.esmo.org/guidelines/esmo-mcbs/esmo-mcbs-for-solid-tumours/esmo-mcbsscorecards?scorecard=498-2
Auf einem Symposium anlässlich des Kongresses der Deutschen Gesellschaft für Neurologie (DGN) präsentierte Merck aktuelle Langzeitdaten zur Therapie der schubförmigen Multiplen Sklerose mit CladribinTabletten (Mavenclad®*). Diese untermauern die anhaltenden Effekte der oralen Impulstherapie auf PIRA (Progression Independent of Relapse Activity), Behinderungsprogression und kognitive Fähigkeiten auch in der behandlungsfreien Zeit [1, 2, 3].
PIRA, definiert als schubunabhängige Behinderungsprogression (CDP, Confirmed Disability Progression) innerhalb von 90 Tagen über ≥6 Monate, gilt heute als etabliertes Maß für eine langfristige Behinderungszunahme bei MS, die nicht auf Schubaktivität zurückzuführen ist [1]. Unter der Therapie mit Cladribin-Tabletten waren ca. 90 % der Patienten 4 Jahre nach Therapiebeginn frei von PIRA; bei etwa 94 % war keine schubassoziierte Verschlechterung (RAW) und bei ca. 84 % keine bestätigte Behinderungsakkumulation (CDA, Confirmed Disability Accrual, definiert definiert als jegliches PIRA- oder RAW-Ereignis über ≥ 6 Monate) zu verzeichnen – so die Ergebnisse einer gepoolten Analyse der Studien MAGNIFY-MS-(EXT) und CLARIFY-MS-(EXT) mit insgesamt 752 Patienten (Abb. 1) [1]. Die niedrigsten Raten an
* Mavenclad® wird angewendet zur Behandlung von erwachsenen Patienten mit hochaktiver schubförmiger MS (RMS), definiert durch klinische oder bildgebende Befunde [12].
MS-Verlaufsformen
Bei Patienten mit Multipler Sklerose entwickelt sich eine Behinderung hauptsächlich durch 2 Mechanismen: zum einen durch die schrittweise Zunahme der Beeinträchtigung aufgrund einer unvollständigen Erholung von einem Schub, es kommt also zu einer schubassoziierten Verschlechterung (RAW, Relapse-Associated Worsening), zum anderen durch ein schubunabhängiges Fortschreiten (PIRA, Progression Independent of Relapse Activity). Beide Mechanismen sind bei den verschiedenen MS-Verlaufsformen unterschiedlich ausgeprägt:
• RRMS (Relapsing Remitting Multiple Sclerosis): Die schubförmige remittierende MS ist mit etwa 80 % die häufigste MS-Form; kennzeichnend sind die Schübe, bei denen neue Symptome auftreten oder sich bestehende verschlimmern können, gefolgt von einer Phase der teilweisen oder vollständigen Besserung (Remission). Zwischen den Schüben ist die Erkrankung stabil.
• SPMS (Secondary Progressive Multiple Sclerosis): Die sekundär progrediente MS entwickelt sich meist aus einer RRMS; hier nehmen die Beschwerden allmählich zu und die Symptome verschlechtern sich kontinuierlich, Schübe treten seltener auf.
• PPMS (Primary Progressive Multiple Sclerosis): Die primär progrediente MS ist mit ca. 10 % die seltenste MS-Form. Sie verläuft von Beginn an allmählich fortschreitend mit schleichender Verschlechterung. Schübe sind selten, die Symptome bilden sich nicht zurück.
Aufgrund dieser Erkenntnisse haben sich auch die Therapieziele geändert: Heute gilt es, nicht nur Schübe zu verhindern, sondern auch die schleichende Progression der MS früh mit einer hochwirksamen MS-Therapie zu bremsen.
PIRA, RAW und CDA wurden dabei bei jüngeren MS-Patienten (≤ 50 Jahre) beobachtet, was für einen frühen Behandlungsbeginn mit Cladribin-Tabletten spricht [1].
Etwa 83 % der Patienten waren frei von Behinderungsprogression
Eine weitere gepoolte Analyse zur Langzeitwirksamkeit von Cladribin-Tabletten konnte zeigen, dass
Abbildung 1: Ein Großteil der Patienten war nach 4 Jahren Therapie mit Cladribin-Tabletten (Mavenclad®) frei von PIRA, RAW und CDA (kumulative Wahrscheinlichkeiten) (mod. nach [1]).
Abbildung 2: Signifikant reduzierte Serum-NfL- und GFAP-Z-Scores nach 2 Jahren (mod. nach [4]).
nach 4 Jahren ca. 83 % der Patienten frei von bestätigter Behinderungsprogression geblieben waren; bei ca. 15 % konnte eine Verbesserung der Behinderung (CDI, Confirmed Disability Improvement) verzeichnet werden (n = 752) [2]. Jüngere, therapienaive Patienten zeigten dabei häufiger eine Behinderungsverbesserung als ältere oder vorbehandelte Personen [2].
Professor Ralf Gold, Bochum, ordnete diese Langzeitdaten in den Kontext aktueller Diskussionen in der Fachgemeinschaft ein: „Eine
‚therapiefreie Remission‘ galt lange nicht als realistisches Behandlungsziel bei MS. Die neuen Daten zu Cladribin-Tabletten auch jenseits der Behandlungszeit [1, 2] zeigen jedoch, dass es möglich sein kann, sich dieser Vision zu nähern.“
Signifikant gesenkte NfL- und GFAP-Serumspiegel
NfL (Neurofilament-Leichtketten) und GFAP (saures Gliafaserpro-
tein) sind wichtige Biomarker für neuroaxonale Schäden bzw. chronische Neuroinflammation bei MS [4]. In einer aktuellen Analyse konnte gezeigt werden, dass unter Cladribin-Tabletten 2 Jahre nach Therapiebeginn diese Biomarker im Serum der Patienten (n = 256) signifikant reduziert waren (Abb. 2) [4]. Darüber hinaus korrelierte die Reduktion beider Marker im Serum signifikant mit dem Erhalt von Hirnvolumend [4]. Außerdem konnte in früheren Untersuchungen unter Cladribin-Tabletten bereits eine signifikante Verringerung von NfL auch im Liquor der Patienten beobachtet werden [5].
„Die neuen Daten unterstreichen die potenziellen Vorteile der Therapie mit Cladribin-Tabletten bei der Verringerung von Neurodegeneration und Neuroinflammation über die klinische Wirksamkeit hinaus“, kommentierte Dr. Melanie Schütte, Düsseldorf, die aktuellen Ergebnisse.
Kognitive Fähigkeiten erhalten – bei hoher Behandlungszufriedenheit
Kognitive Symptome bei MS werden als „unsichtbares“ Symptom häufig unterschätzt [6, 7]. Dies könnte sich jedoch langfristig ändern: Bereits die Eröffnungsrede des ECTRIMS-Forums 2025** rückte die Kognition bei MS in den Fokus. Professorin Iris-Katharina Penner, Bern, betonte dabei nachdrücklich die klinische Relevanz während einer Veranstaltung auf dem DGN-Kongress: „Wir wissen, dass kognitive Einschränkungen
** ECTRIMS Lecture: The Cognitive Dimension of MS. ECTRIMS 2025, Plenary Session 1: Opening Session; 24.09.2025, Barcelona, Spain
als prädiktiver Marker für eine langfristige Behinderungsprogression bei MS fungieren können [8]. Umso wichtiger ist es demnach, die Symptome frühzeitig zu erkennen, zu quantifizieren und bei der Therapieentscheidung mit zu berücksichtigen.“
Bereits Daten aus dem Jahr 2024 konnten zeigen, dass unter Cladribin-Tabletten auch 4 Jahre nach Behandlungsbeginn die kognitiven Fähigkeiten bei ca. 88 % der Patienten erhalten geblieben waren (gemessen anhand der 8-PunkteDifferenz im Symbol Digit Modalities Test, SDMT) [9].
Diese Ergebnisse wurden nun durch die nicht-interventionelle CLADQoL-Studie bestätigt, die in Deutschland und Österreich durchgeführt und auf dem ECTRIMS 2025 präsentiert wurde: Auch darin waren nach 4 Jahren die kog-
nitiven Fähigkeiten der Patienten bei ca. 77 % stabil oder verbessert (8-Punkte-Differenz im SDMT) [3]. Gleichzeitig wurde eine hohe und anhaltende Behandlungszufriedenheit berichtet [3].
Die neuesten Daten bestätigen die anhaltende Wirksamkeit von Cladribin-Tabletten auch in den behandlungsfreien Jahren 3 und 4 bei einem großen Anteil der MSPatienten. In über 360.000 Patientenjahren und bei mehr als 130.000 behandelten Patienten weltweit wurden keine neuen Sicherheitssignale oder bestätigten Fälle von progressiver multifokaler Leukenzephalopathie (PML) beobachtet [9, 10]. Diese Ergebnisse untermauern das konsistent gute Sicher-
Literatur
Hyperexzitabilität:
Risikofaktor und Therapietarget auch jenseits der Epilepsie
Epileptische Anfälle treten nicht nur bei Menschen mit Epilepsie auf, sondern auch bei anderen neurologischen Erkrankungen. Sie sind oft nur subklinisch – und entsprechend schwer zu diagnostizieren. Doch sie sind klinisch relevant, weil sie signifikant die Prognose der Betroffenen beeinflussen. So können epileptische Anfälle bei Schlaganfall-Patienten die Krankenhaussterblichkeit verdoppeln und bei MS-Patienten mit einer schnelleren Progression, mehr Behinderungen und schlechteren kognitiven Leistungen einhergehen. Auch bei vielen anderen neuro-
logischen Krankheiten kommt es vermehrt zu epileptischen Anfällen, z.B. bei Enzephalitis, Meningitis, bei Intoxikationen, zerebraler Hypoxie oder Hirntumoren. So induzieren Gliome eine neuronale Übererregbarkeit, die häufig zu epileptischen Anfällen führt. Auch scheint die Hyperexzitabilität weit mehr als nur ein Krankheitssymptom bei Hirntumoren zu sein. Es gibt die Hypothese, dass die Synaptogenese zwischen glutamatergen Neuronen und Gliomen sowie die Übererregbarkeit das Tumorwachstum fördern. Ein neuer, vielversprechender Therapieansatz, der derzeit untersucht wird, ist die medikamentöse Reduktion der peritumoralen Hyperexzitabilität [1]. Die Hyperexzitabilität neuronaler Netzwerke ist außerdem auch ein
Brigitte Söllner, Erlangen
1 Montalban X et al. ECTRIMS 2025 [P1707]
2 Vermersch P et al. ECTRIMS 2025 [P335]
3 Penner IK et al. ECTRIMS 2025 [P1820]
4 Wiendl H et al. ECTRIMS 2025 [P1694]
5 Schmierer K et al. ACTRIMS 2025 [P143]
6 Penner IK. Acta Neurol Scand 2016; 134(Suppl 200):19-23
7 Penner IK et al. Eur J Neurol 2025; 32:e16234
8 Pitteri M et al. Mult Scler 2017;23:848854
9 De Stefano N et al. ECTRIMS 2024 [P349]
10 Glaser A et al. ECTRIMS 2025 [P856]
11 Leist T et al. MENACTRIMS 2024 [P347]
12 Fachinformation Mavenclad®; aktueller Stand
Begleitphänomen der AlzheimerErkrankung. Derzeit wird erforscht, inwieweit sie eine aktive Rolle in der Progression neurodegenerativer Krankheiten spielt – und auch hier Therapietarget sein könnte. Eine aktuelle Studie zeigte, dass es eine pathologische Wechselwirkung zwischen Aβ-Akkumulation und neuronaler Übererregbarkeit bei Alzheimer gibt; die Autorinnen und Autoren halten daher duale Therapien, die beide Targets adressieren, für aussichtsreicher als die alleinige Therapie mit Antikörpern gegen Aβ [2].
Quellen
DGN
1 Tobochnik S et al. Clin Cancer Res 2024; 30:5365-5373
2 Wang Y et al. ACS Nano. 2025 Oct 22. doi: 10.1021/acsnano.5c08317 heitsprofil von Cladribin-Tabletten in der MS-Therapie [11].
Auf dem Kongress des American College of Rheumatology (ACR) hat Bristol Myers Squibb aktuelle Ergebnisse aus 3 Studien zu Deucravacitinib (Sotyktu®) präsentiert, die die anhaltende Wirksamkeit und Sicherheit des oralen, selektiven Tyrosinkinase 2 (TYK2)-Inhibitors bei erwachsenen Patienten mit aktiver Psoriasis-Arthritis (PsA) sowie bei Patienten mit mittelschwerem bis schwerem systemischem Lupus erythematodes (SLE) erneut bestätigen [1, 2].
Phase-III-Studie POETYK PsA-1: Verbesserte Ansprechraten, keine Progression der PsA
Neue Daten aus Woche 52 dieser Studie bei erwachsenen Patienten mit aktiver Psoriasis-Arthritis (PsA) zeigen, dass sich bei Patienten, die kontinuierlich mit Deucravacitinib bis Woche 52 behandelt wurden, die ACR20-Ansprechraten (d.h. mindestens 20 % Verbesserung der Anzeichen und Symptome der Erkrankung), die in Woche 16 erreicht wurden (unter Deucravacitinib: 54,2 %; unter Placebo: 34,1 %; p < 0,0001) unter der Behandlung mit dem TYK2-Inhibitor weiter auf 63,1 % verbesserten und aufrechterhalten wurden [1].
Deucravacitinib
Patienten, die in Woche 16 von Placebo auf Deucravacitinib umgestellt wurden, wiesen nach 52 Wochen vergleichbare ACR20Ansprechraten von im Mittel 60,8 % auf, was mit den Ergebnissen aus der Studie POETYK PsA2 übereinstimmte [1].
Die bereits in Woche 16 beobachtete Hemmung der Progression struktureller Gelenkschäden blieb bis Woche 52 erhalten, gemessen als mittlere Veränderung im modifizierten Sharp-van der Heijde (mSvdH)-Score gegenüber Baseline. Außerdem zeigte ein höherer Anteil der mit Deucravacitinib behandelten Patienten im Vergleich zu Placebo keine Progression der Gelenkschädigung sowohl in Woche 16 (Deucravacitinib: 82,0 %; Placebo: 71,5 %) als auch in Woche 52 (bei kontinuierlicher Behandlung mit Deucravacitinib: 73,3 %;
Deucravacitinib (Sotyktu®) ist ein oraler, selektiver Tyrosinkinase 2 (TYK2)-Inhibitor und repräsentiert pharmakologisch eine neue Klasse innerhalb der niedermolekularen Wirkstoffe („Small Molecules“). Deucravacitinib hemmt u.a. die Signalkaskade von Interleukin (IL)-23, IL-12 und Typ-1-Interferon (IFN), die zu den zentralen Zytokinen der Pathogenese zahlreicher immunvermittelter Erkrankungen gehören.
Deucravacitinib ist in der EU derzeit für die Behandlung von Erwachsenen mit mittelschwerer bis schwerer Plaque-Psoriasis (PsO) zugelassen, die für eine systemische Therapie infrage kommen.
bei Umstellung in Woche 16 von Placebo auf Deucravacitinib: 66,5 %). Des Weiteren verbesserten sich unter der DeucravacitinibTherapie klinische Messgrößen der Krankheitsaktivität, die Patient-Reported Outcomes sowie die extraartikulären Manifestationen der PsA, die bis Woche 52 stabil blieben [1].
Bis dahin wurden zudem keine neuen Sicherheitssignale festgestellt. Die am häufigsten berichteten unerwünschten Ereignisse (UE) waren Infektionen der oberen Atemwege. Schwerwiegende UEs sowie UEs, die zum Abbruch der Behandlung führten, traten nur selten auf [1].
Phase-II-Studien PAISLEY-SLE und PAISLEY LTE: Anhaltende Wirksamkeit und konsistente Sicherheit bei SLE
Die integrierten Analysen der Phase-II-Studien PAISLEY-SLE und PAISLEY LTE (Langzeitverlängerungsstudie) zeigten bei einer Exposition von bis zu 4 Jahren eine anhaltende Wirksamkeit und ein konsistentes Sicherheitsprofil von Deucravacitinib bei Patienten mit mittelschwerem bis schwerem SLE. Trotz komplexer Hintergrundtherapien wurden keine neuen Sicherheitssignale beobachtet [2].
Die Ansprechraten blieben im zeitlichen Verlauf stabil, gemessen anhand des SLE Responder Index-4 (SRI-4), British Isles Lupus Assessment Group-based Composite Lupus Assessment (BICLA), Lupus Low Disease Activity State (LLDAS) und Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI-50) [2].
In Woche 72 zeigten Patienten, die initial im Placebo-Arm der Hauptstudie PAISLEY-SLE eingeschlossen wurden und dann in PAISLEY LTE auf Deucravacitinib umgestellt wurden, Verbesserungen, die mit denen der kontinuierlich behandelten Patienten vergleichbar waren. Schwerwiegende unerwünschte Ereignisse (UEs) traten jeweils bei 17,0 %, 14,9 % bzw. 21,8 % der Patienten auf, die Deucravacitinib in Dosierungen von 3 mg 2 täglich, 6 mg 2 täglich bzw. 12 mg 1x täglich erhielten. UEs, die zum Therapieabbruch führten, wurden bei jeweils 13,4 %, 11,4 % bzw. 17,3 % beobachtet [2].
Brigitte Söllner, Erlangen
Staloral® Birke ist auch bei Kindern und Jugendlichen wirksam und sicher
Unter einer allergischen Rhinokonjunktivitis leiden mehr als 500 Millionen Menschen. In Nordeuropa gehört die Birke zu den stärksten Allergenquellen. Auch die Allergenpotenz anderer Bäume aus der birkenverwandten Gruppe – wie Erle und Hasel – nimmt zu. Bei Kindern und Jugendlichen kann die allergische Rhinokonjunktivitis zu Schlafstörungen, Müdigkeit, Fehlzeiten in der Schule und Einschränkungen bei Freizeitaktivitäten führen. Eine frühe Sensibilisierung erhöht zudem das Risiko, Asthma zu entwickeln. Je jünger die Kinder sind, desto größer ist dieses Risiko – was die Bedeutung einer frühzeitigen kausalen Therapie in der Pädiatrie unterstreicht. Die Allergen-Immuntherapie (AIT) ist derzeit die einzige Behandlungsmöglichkeit, die den natürlichen Krankheitsverlauf nachhaltig beeinflussen kann. In flüssiger Form bietet die sublinguale AIT eine nadel- und stressfreie flexible Therapieoption für Kinder und Jugendliche.
Kausale Therapie von Birkenpollen-induzierten allergischen Erkrankungen
nischen Studien im Vergleich zu Placebo konsistent nachgewiesen wurde.
Dass die Therapie mit Staloral® Birke auch im Kindesalter wirksam und sicher ist, bestätigen die Ergebnisse der placebokontrollierten Phase-IIIb-Studie YOBI, in die 553 Kinder und Jugendliche im Alter von 5 – 17 Jahren mit moderater bis schwerer Birkenpollen-induzierter allergischer Rhinokonjunktivitis (ARC) – mit oder ohne Asthma – aus 12 europäischen Ländern eingeschlossen wurden. Untersucht wurden die Wirksamkeit und Sicherheit einer täglichen Erhaltungsdosis von 300 IR Staloral® Birke, die prä- und cosaisonal über 2 aufeinanderfolgende Birkenpollensaisons verabreicht wurde.
Literatur
1 Mease PJ et al. Oral presentation at American College of Rheumatology Convergence (ACR) Congress 2025 in Chicago, Abstract #LB20
2 Morand E et al Oral presentation at American College of Rheumatology Convergence (ACR) Congress 2025 in Chicago, Abstract #LB10
Staloral® ist eine sublinguale Lösung von Allergenextrakten für die AIT und deckt ein breites Spektrum relevanter Allergene ab. Für die kausale Behandlung speziell von Birkenpollen-induzierten allergischen Erkrankungen steht Staloral® Birke zur Verfügung, dessen klinische Wirksamkeit bei Erwachsenen in zahlreichen kli-
Die YOBI-Studie erreichte ihren primären Endpunkt: Staloral® Birke führte im Vergleich zu Placebo in der pädiatrischen Population während der zweiten Pollensaison zu einer 41%igen Reduktion des kombinierten ARC-Gesamtscores – eines kombinierten Gesamtscores aus Symptomen und Medikation – im Vergleich zu Placebo. Die Ergebnisse waren hochsignifikant (p < 0,0001) und klinisch relevant, mit einer Differenz zwischen den Behandlungsgruppen von –2,62 Punkten. Auch beim sekundären Endpunkt, dem von der European Academy of Allergy and Clinical Immunology (EAACI)1 empfohlenen kombinierten Symptom- und Medikationsscore, zeigte sich ein hochsignifikanter Unterschied (–0,41; p < 0,0001). Sicherheit und Verträglichkeit bestätigten das bekannte Sicherheitsprofil von Staloral® Birke.
B. S.
Fortgeschrittenes ALKpositives nicht-kleinzelliges Lungenkarzinom:
Effektive Krankheitskontrolle mit Lorlatinib
Die Fortschritte in der zielgerichteten Therapie des ALK-positiven nicht-kleinzelligen Lungenkarzinoms (ALK+ aNSCLC) ermöglichen Patienten heute eine längerfristige Krankheitskontrolle. Aktuelle Langzeitdaten belegen für Lorlatinib (Lorviqua®) in der Erstlinientherapie ein progressionsfreies Überleben (PFS) von über 5 Jahren – ein bisher unerreichtes Ergebnis in dieser Patientengruppe.
Im Rahmen einer virtuellen Fachpresseveranstaltung im Anschluss an den ESMO-Kongress ordneten Professor Christian Schulz (Regensburg) und Professor Christian Schumann (Kempten, Allgäu) diese und weitere Ergebnisse in den Kontext aktueller Leitlinienempfehlungen ein und diskutierten praxisrelevante Aspekte des evidenzbasierten Langzeitmanagements. Beide betonten: „Die aktuellen Daten unterstreichen nicht nur die hohe Wirksamkeit von Lorlatinib, sondern auch die Bedeutung eines strukturierten Langzeitmanagements.“
Ein adäquates Langzeitmanagement kann die Weichen für Therapieadhärenz, langanhaltendes Ansprechen und den Erhalt der Lebensqualität stellen. Nebenwirkungen treten unter Lorlatinib überwiegend in den ersten Monaten der Therapie auf und lassen sich meist gut kontrollieren. Dosisunterbrechungen oder -reduktionen beeinträchtigen dabei weder die systemische noch die intrakranielle Wirksamkeit.
Seit Beginn dieses Jahres wird Lorlatinib in den S3- und Onkopedia-Leitlinien bevorzugt empfohlen und ist damit der präferierte Erstlinienstandard beim ALK+ aNSCLC.
Lorlatinib in der Erstlinientherapie etabliert: über 5 Jahre systemische und intrakranielle Wirksamkeit
In der Follow-up-Analyse der Phase-III-Studie CROWN waren nach 5 Jahren noch 60 % der mit Lorlatinib behandelten Patienten mit ALK+ aNSCLC am Leben und progressionsfrei (HR: 0,19; 95%KI: 0,13 – 0,27). Das ist das längste PFS, das bislang in der Erstlinientherapie beim ALK+ aNSCLC beobachtet wurde. Die objektive Gesamtansprechrate (ORR) lag unter Lorlatinib bei 81 % (95%-KI: 73 – 87) im Vergleich zu 63 % unter Crizotinib (95%-KI: 54 – 70). Intrakraniell zeigte Lorlatinib ebenfalls eine hohe Wirksamkeit. 92 % der Patienten blieben nach 5 Jahren ohne ZNS-Progression (HR: 0,06; 95%-KI: 0,03 – 0,12), bei Vorliegen von Hirnmetastasen betrug die ORR 92 % (95%-KI: 62 – 100) im Vergleich zu 33 % unter Crizotinib (95%-KI: 4 – 78). Das erstmalige Auftreten von ZNS-Metastasen sowie die intrakranielle Progression bei bestehenden ZNS-Metastasen konnten mit Lorlatinib weitestgehend verhindert werden.
Das Sicherheitsprofil von Lorlatinib war dabei konsistent mit dem der primären Analyse. Neue Sicherheitssignale wurden nicht beobachtet. Basierend auf diesen Daten empfehlen seit diesem Jahr sowohl Onkopedia- als auch S3-Leitlinie bevorzugt Lorlatinib in der Erstlinientherapie beim ALK+
aNSCLC. Laut Schulz wurde Lorlatinib bereits auf dem ASCO 2024 als neuer Standard in der Erstlinientherapie diskutiert, vor allem im Hinblick auf die sich abzeichnende Chronifizierung der Erkrankung.
Lorlatinib versus Alectinib: überlegenes Langzeit-PFS bei vergleichbarem Sicherheitsprofil
Die beim ELCC 2025 vorgestellte MAIC-Analyse demonstrierte für Lorlatinib eine überlegene Wirksamkeit im Vergleich zu Alectinib. In der ITT-Gesamtpopulation reduzierte sich das Risiko für Progression oder Tod unter Lorlatinib um 45 % (HR: 0,55; 95%KI: 0,34 – 0,88). Das Sicherheitsprofil hinsichtlich unerwünschter Ereignisse (UE), die zu Behandlungsabbruch, Dosisreduktion oder -unterbrechung führten, war vergleichbar, auch wenn die Rate der Grad ≥3 UE unter Lorlatinib höher war.
„Die MAIC-Daten belegen einmal mehr, dass Lorlatinib eine hochwirksame Behandlungsoption mit verlängertem Therapieversprechen bietet“, betonte Schulz.
Lorlatinib-Therapie:
Tiefe des Ansprechens verbessert PFS
Post-hoc-Analysedaten der CROWN-Studie, präsentiert beim ASCO 2025, zeigten, dass eine größere Tiefe des Ansprechens (DepOR) bei Lorlatinib mit einem längeren PFS einhergeht. Patienten mit einer DepOR von >75 %–100 % hatten nach 5 Jahren eine Wahrscheinlichkeit von 75 %, progressionsfrei zu bleiben, verglichen mit 37 % bei Patienten mit einer DepOR von 0 %–50 %. Die
Daten belegen, dass die Tiefe des Tumoransprechens ein relevanter Prädiktor für den Langzeiterfolg der Therapie mit Lorlatinib ist.
Patientenorientiertes Langzeitmanagement unterstützt Therapieadhärenz
Das frühe Erkennen von Nebenwirkungen ermöglicht gezielte Interventionen wie z.B. Dosisanpassungen. Das Management von UE wie Hyperlipidämie, Gewichtszunahme, periphere Ödeme, Neuropathien oder ZNS-Effekte erfolgt nach spezifischen Empfehlungen. Schumann betonte: „Patientenzentrierte Betreuung und bedarfsgerechtes Langzeitmanagement von Nebenwirkungen sind entscheidend, um die Therapie langfristig aufrechtzuerhalten und die Adhärenz zu sichern.“
Eine aktuelle Post-hoc-Analyse der 5-Jahres-Follow-Up-Daten der CROWN-Studie bestätigt, dass Dosisreduktionen zum Handling von Nebenwirkungen gut geeignet sind.
Fabian Sandner, Nürnberg
Plädoyer für eine frühe Behandlung des Morbus Crohn
Beim Morbus Crohn (MC) mehren sich die Hinweise, dass ein möglichst früher Therapiebeginn sinnvoll ist, berichtete Professor Sebastian Zeißig, Greifswald, auf einem von Takeda veranstalteten Symposiums im Rahmen der Tagung „Viszeralmedizin 2025“ in Leipzig. Er verwies auf die Pro-
Vedolizumab (Entyvio®) ist ein humanisierter monoklonaler Antikörper, der selektiv im Gastrointestinaltrakt wirkt. Er bindet spezifisch an α4β7Integrin, das bevorzugt auf T-Helfer-Lymphozyten in der Darmwand exprimiert wird. Über mehrere Schritte wird
durch diese Bindung der Entzündungsprozess gebremst.
gression des MC in den ersten Jahren nach der Erstdiagnose: Während kurz nach Diagnosestellung unter 5 % der Erkrankten operiert werden, hat nach 5 Jahren bereits jeder vierte MC-Patient einen chirurgischen Eingriff hinter sich [1]. „Je länger man wartet, desto schwieriger wird es, den Patienten in Remission zu bringen“, sagte Zeißig.
Deutliche Argumente für einen frühen Therapiebeginn liefert die PROFILE-Studie, in der ein Topdown-Behandlungskonzept mit einem Step-up-Konzept verglichen wurde. Nach einer zweiwöchigen Steroidbehandlung zur Remissionsinduktion für alle 380 Teilnehmer ging es in 2 Gruppen weiter: Die Top-down-Gruppe wurde ab Tag 1 mit Infliximab + Immunmodulator behandelt. Bei gutem Ansprechen wurde die Dosis beibehalten, bei fehlendem Erfolg wurden Steroide dazugegeben. In der Step-up-Gruppe wurden nur die Steroide abgesetzt und dies beibehalten, solange die Remission anhielt. Bei Versagen wurden Steroide und ein Immunmodulator verabreicht, wenn keine Remission einsetzte, kam noch Infliximab hinzu. Nach 48 Wochen waren
67 % der Top-down-Gruppe und 44 % der Step-up-Gruppe in endoskopischer Remission; ohne Steroide und chirurgischen Eingriff waren 79 % der Top-down-Gruppe gegenüber 15 % der Step-up-Gruppe [2].
Auswahl des Medikaments entscheidet über den Erfolg
Ob eine Behandlung erfolgreich wird, hängt entscheidend von der Auswahl des Medikaments ab, das zudem die Akzeptanz des Patienten finden muss.
In der Real-World-Studie VEDOIBD erreichten nach 2 Jahren signifikant mehr Patienten unter Vedolizumab (Entyvio®) als unter TNFa-Antagonisten eine steroidfreie Remission (62,5 % vs. 41,6 %). Die Patienten der Vedolizumab-Gruppe zeigten zudem mit 82 % eine signifikant höhere Therapiepersistenz über 2 Jahre als die der TNFa-Antagonisten-Gruppe mit 57 % [3].
Am stärksten profitieren nach Zeißigs Erfahrung diejenigen Patienten von Vedolizumab, die keine perianale, fistulierende Erkrankung und keine extraintestinalen Manifestationen hatten, zuvor noch nicht mit einem TNF-a-Blocker behandelt und nicht operiert wurden sowie einen niedrigen Albuminwert und ein CRP <3 mg/l aufwiesen.
Dr. Barbara Voll, Bergisch Gladbach
Quellen
1 Burisch J et al. Gut 2019;68:423-433
2 Noor N et al. Lancet Gastroenterol Hepatol 2024;9:415-427
3 Bokemeyer B et al, Inflamm Bowel Dis 2024;30:746-756
Anti-B-Zelltherapien bei Kollagenosen: Ianalumab als Hoffnungsträger bei Sjögren-Syndrom, immunologischer Thrombozytopenie und systemischem Lupus erythematodes
Bei Kollagenosen wie der SjögrenErkrankung (SjE) und dem systemischen Lupus erythematodes (SLE) sind B- und Plasmazellen entscheidend in die Pathogenese involviert, wie aktuell die teils spektakulären Erfolge der CAR-TZelltherapien bei therapierefraktären Kollagenosen verdeutlichen. Da Letztere bis auf Weiteres nur für „austherapierte“ Betroffene infrage kommen, richtet sich der Blick jenseits etablierter Antikörper wie Rituximab oder Belimumab auf neue Therapieoptionen wie den dual wirksamen, B-Zell-depletierenden und gegen den BAFF (BCell Activating Factor)-Rezeptor gerichteten vollhumanen monoklonalen Antikörper Ianalumab, der derzeit in verschiedenen Indikationen geprüft wird.
Wie Dr. Johanna Mucke, Düsseldorf, Erstautorin der deutschen SLE-Leitlinie auf einem Satellitensymposium anlässlich des Deutschen Rheumatologiekongresses verdeutlichte, besteht bei SLE und Lupusnephritis (LN) noch ein großer „unmet medical need“. So weisen SLE-Patienten weiterhin eine um den Faktor 2,65 erhöhte Mortalität auf, 50 % entwickeln in den ersten 5 Jahren irreversible Organschäden – sei es durch die Erkrankung und/oder hohe Glukokortikoid-Dosen. In Studien werden Remissionsraten von nur 8 – 30 % erreicht, die Glukokortikoid-Zieldosis von 0 mg/Tag wird in der Mehrzahl der Fälle verfehlt.
Auch die Fatigue wird durch zugelassene Therapien nur selten adäquat adressiert.
Systemischer Lupus: Ein Blick in die Glaskugel
In der aktuellen deutschen SLELeitlinie wird bei extrarenalen Manifestationen nach dem Versagen auf Hydroxychloroquin und Glukokortikoid (oder wenn dessen Dosierung ≥5 mg/Tag beträgt) die Eskalation auf konventionelle DMARDs oder Biologika empfohlen; mit dem an BAFF (BLyS) ansetzenden Belimumab ist hier bereits eine Anti-B-Zelltherapie zugelassen.
Bei Lupusnephritis wird in Kürze mit dem Anti-CD20-Antikörper Obinutuzumab, quasi ein Rituximab 2.0, eine zweite Therapie hinzukommen – auch ein PhaseIII-Programm dazu läuft. Darüber hinaus befinden sich mit Dapirolizumab pegol, Telitacicept oder Ianalumab bei SLE noch weitere an der B-Zelle ansetzende Substanzen in Phase-III-Studien, erläuterte Mucke.
In Bezug auf Ianalumab wurde in einer Phase-II-Studie bei extrarenalem SLE ein im Vergleich zu Placebo (jeweils plus Standardtherapie) ein signifikant höheres SLE Responder Index (SRI)-4-Ansprechen mit anhaltender Steroidreduktion zu Woche 28 erreicht (49,4 % vs. 19,2 %; entsprechend einem Delta von 34,5 %). Auch über ein erweitertes Follow-up von 12 Monaten blieb die Response erhalten. Angesichts dieser überaus positiven Daten wird Ianalumab derzeit in gleich drei Phase-III-Studien bei SLE (SIRIUS-SLE 1 und 2) sowie LN (SIRIUS-LN) geprüft.
Sjögren-Syndrom: Erstmals Aussicht auf zugelassene Therapien
Noch wesentlich größere Probleme als beim SLE bestehen in puncto Therapie bei der ähnlich vielgestaltigen Sjögren-Erkrankung, betonte Professorin Diana Ernst, Hannover. Oft wird die Diagnose spät gestellt, ist die Symptomatik unspezifisch und das Erkrankungsbild heterogen. Auch hier weisen die Patienten oft eine hohe Krankheitslast mit Fatigue als wichtigem Symptom auf. Vor allem ist – wenn man von der Sicca-Symptomatik absieht – bei der SjE mit Organmanifestationen bislang keine einzige systemische Therapie zugelassen, was sich aber in Zukunft ändern dürfte.
Gemäß den EULAR-Empfehlungen mit jedoch durchweg geringer Evidenzbasis kommen off-label jenseits von Glukokortikoiden und konventionellen DMARDs noch am ehesten Abatacept oder mit Rituximab und Belimumab zwei Anti-B-Zell-Therapien (auch in Kombination) infrage, wie Ernst weiter ausführte.
Nach langen Jahren der Frustration gibt es derzeit ein „Wettrennen“ und die erste Zulassung beim SjE. Ein vielversprechender Kandidat ist das B-Zell-depletierende Ianalumab, das in einer Phase-IIb-Studie überzeugte. Nach 24 Wochen kam es unter 300 mg Ianalumab s.c. zu einer signifikanten Reduktion des EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI)Scores um ca. 8 Punkte, die sich bis Woche 52 fortsetzte. Im Gegensatz zu bisherigen Therapieformen fand sich u.a. auch eine deutliche Besserung der Sicca-Symptomatik. Derzeit laufen mit NEPTUNUS-1 und -2 gleich 2 Phase-III-Studien in dieser Indikation.
Ebenfalls in Phase-III-Studien untersucht werden Telitacicept und das indirekt an der B-Zelle angreifende Dazodalibep. Auch am neonatalen Fc-Rezeptor (FcRn) ansetzende Antikörper (Nipocalimab, Efgartigimod) werden bei SjE in Phase-III-Studien untersucht, ebenso der parallel bei SLE evaluierte orale Tyrosinkinase (TYK)2-Inhibitor Deucravacitinib.
Durchbruch in Sicht
Wie aus einer Vorab-Pressemitteilung von Novartis am 12. August 2025 hervorgeht, erreichte Ianalumab in beiden zulassungsrelevanten Phase III-Studien NEPTUNUS-1 und -2 mit 275 respektive 504 Teilnehmern eine statistisch signifikante Reduktion der Erkrankungsaktivität gemäß ESSDAI-Score – ein Novum für eine systemische Therapie bei SjE. Genauere Ergebnisse sollen in Kürze vorgestellt werden.
Dr. Michael Lohmann, Limburg
Ofatumumab als Erstlinientherapie bei aktiver RMS
Anlässlich des diesjährigen Kongresses der Deutschen Gesellschaft für Neurologie (DGN) stellte Novartis aktuelle Daten der AIOLOSStudie zum frühen Einsatz von Ofatumumab bei therapienaiven Patienten mit nicht-hochaktiver* schubförmiger Multipler Sklerose (RMS) im deutschen Behandlungsalltag vor. Die Daten stützen die positiven Ergebnisse der Zulassungsstudien ASCLEPIOS I und II sowie der offenen Verlängerungsstudie ALITHIOS und sprechen
Ofatumumab (Kesimpta®) ist ein vollhumaner Anti-CD20-Antikörper, der durch eine monatliche (ab Woche 4) subkutane Applikation via Fertigpen (Kesimpta® Sensoready® Pen) von den Patienten – nach Schulung durch entsprechend geschultes medizinisches Fachpersonal – selbst verabreicht werden kann. Ofatumumab zeigte ein vergleichbares Sicherheits- und Verträglichkeitsprofil zur Erstlinientherapie mit Teriflunomid.
Der Antikörper bindet zielgerichtet an das CD20-Molekül auf der Oberfläche von B-Zellen, wobei es auf einem anderen Epitop andockt als andere Anti-CD20-Antikörper und dadurch eine B-ZellLyse und Depletion induziert.
Der selektive Wirkmechanismus und die subkutane Verabreichung ermöglichen eine spezifische Wirkung auf die B-Zellen in den Lymphknoten, die in der MS-Pathologie eine entscheidende Rolle spielen. Ofatumumab ist zugelassen zur Behandlung erwachsener Patienten mit schubförmig verlaufender Multipler Sklerose (Relapsing Multiple Sclerosis; RMS) mit aktiver Erkrankung, definiert durch klinischen Befund oder Bildgebung.
für den Einsatz von Ofatumumab als Erstlinientherapie.
Anhaltende Vorteile gegenüber der Therapie mit Interferon β / Glatirameracetat
Die nicht-interventionelle RealWorld-Studie AIOLOS untersucht Ofatumumab als Erstlinientherapie bei RMS-Patienten ohne Merkmale für einen hochaktiven Krankheitsverlauf im Vergleich zur Erstlinientherapie mit Interferon β/ Glatirameracetat (IFNβ/GA) in der klinischen Routine in Deutschland. Die Ergebnisse der Interimsanalyse (n = 514) bestätigen das bekannte positive Nutzen-Risiko-Profil von Ofatumumab.
Patienten mit einer OfatumumabErstlinientherapie hatten im Vergleich mit IFNβ/GA über den gesamten Beobachtungszeitraum von 24 Monaten eine niedrigere jährliche Schubrate (0,14 vs. 0,20). Auch hinsichtlich der Therapietreue zeigte sich für Ofatumumab ein
anhaltender Vorteil: Sowohl nach 12 Monaten als auch nach 24 Monaten lag unter einer Therapie mit Ofatumumab die Therapietreue mit 96 % deutlich höher als unter einer Therapie mit IFNβ/GA (75 % bzw. 65 %). Zudem berichtete in der Ofatumumab-Gruppe ein geringerer Anteil von Patienten schwere Nebenwirkungen (4 % vs. 6,5 %) oder Nebenwirkungen, die zum Therapieabbruch führten (1,5 % vs. 12,9 %), im Vergleich zu Patienten in der IFNβ/GA-Gruppe. Die Immunglobulin G (IgG)-Spiegel blieben bei den Patienten, die eine Erstlinientherapie mit Ofatumumab erhielten, über den gesamten Zeitraum von 24 Monaten stabil im Normbereich.
Biomarker sNfL zur Überwachung des Therapieansprechens
Die Echtzeitüberwachung der klinischen und subklinischen Krankheitsaktivität bei MS gewinnt zunehmend an Bedeutung, wobei
der blutbasierte Biomarker Serum-Neurofilament-Leichtketten (sNfL) eine zentrale Rolle spielt. Die prospektive Datensammlung NeofiLos untersucht den Nutzen von sNfL im niedergelassenen neurologischen Bereich bei der Versorgung von RMS-Patienten in Deutschland. Über einen Zeitraum von 12 Monaten zeigten die vierteljährlichen sNfL-Messungen, die im Rahmen der NeofiLos-
Datenerhebung durchgeführt wurden, deutliche Unterschiede in den sNfL-Werten, abhängig von der Therapiesituation zu Beginn des Projekts. So waren die sNfL-Werte bei unbehandelten Patienten höher als bei jenen, die bereits eine Therapie mit Ofatumumab erhielten. Darüber hinaus sanken erhöhte (ZScore = 1,5 – 3) und stark erhöhte (Z-Score >3) sNfL-Werte nach Beginn einer Therapie mit Ofatu-

Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
mumab und erreichten Werte, die vergleichbar waren mit denen von bereits mit Ofatumumab behandelten Patienten.
Die Auswertungen aus NeofiLos verdeutlichen die hohe Relevanz von sNfL als zusätzlichem Parameter zur Überwachung von Krankheitsaktivität und Therapieansprechen in der klinischen Praxis.
Elisabeth Wilhelmi, München
Titelbild: Aus den Wurzeln der südafrikanischen Kapland-Pelargonie (Pelargonium sidoides) wird ein Auszug hergestellt, der zur Behandlung von Erkältungen eingesetzt wird, um Symptome wie Husten, Schleimbildung, Halsschmerzen und Abgeschlagenheit zu lindern (© Dr. Willmar Schwabe).
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz: HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R.
Roland Welker
Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de


Endlich mittendrin als PKU-Patient*in Phe senken. Möglichkeiten steigern.
• Senkung des Phe-Wertes im Blut um durchschnittlich 410 μmol / l (~ 7 mg / dl) / 63 % ggü. Placeboa,2
• 66 % Ansprechrate mit ≥ 30 % Reduktion des Phe-Wertes im Blut ggü. Baseline2
• Reduktion des Phe-Spiegels um durchschnittlich 524 μmol / l (~ 9 mg / dl) / 69 % ggü. Placebo bei klassischer PKUa,b,2
• Gut verträglich1,2
Phe = Phenylalanin; PKU = Phenylketonurie.
a Unter Einhaltung der Diät. b Klassische PKU wird definiert als Phe-Wert im Blut ≥ 1200 µmol / l (~ 20 mg / dl) bei Geburt oder entsprechend dokumentierte Werte in der medizinischen Vorgeschichte des Patienten.
1. SephienceTM (Sepiapterin) Fachinformation, aktueller Stand. 2. Muntau AC, et al. Lancet. 2024;404(10460):1333-1345.
Sephience 250 mg Pulver zum Einnehmen im Beutel / Sephience 1 000 mg Pulver zum Einnehmen im Beutel Wirkstoff: Sepiapterin.
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8 der Fachinformation. Qualitative und quantitative Zusammensetzung:
Sephience 250 mg bzw. 1 000 mg Pulver zum Einnehmen im Beutel: Jeder Beutel enthält 250 mg bzw. 1 000 mg Sepiapterin. Sonstige Bestandteile mit bekannter Wirkung: Sephience 250 mg bzw. 1 000 mg Pulver zum Einnehmen im Beutel: Jeder Beutel enthält 400 mg bzw. 1 600 mg Isomaltitol. Vollständige Auflistung der sonstigen Bestandteile: Mikrokristalline Cellulose (E460), Isomaltitol (E953), Mannitol (E421), Croscarmellose-Natrium (E468), Xanthangummi (E415), Kolloidales wasserfreies Siliciumdioxid oder kolloidales Siliciumdioxid (E551), Sucralose (E955), Magnesiumstearat (E470).
Anwendungsgebiete: Sephience wird angewendet für die Behandlung von Hyperphenylalaninämie (HPA) bei erwachsenen und pädiatrischen Patienten mit Phenylketonurie (PKU). Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der genannten sonstigen Bestandteile. Nebenwirkungen: sehr häufig (≥ 1 / 10): Infekt der oberen Atemwege, Kopfschmerzen, Diarrhö, Abdominalschmerz, Oberbauchschmerzen, Abdominalbeschwerden; häufig (≥ 1 / 100, < 1 / 10): Stuhlverfärbung, Hypophenylalaninämie. Verkaufsabgrenzung: Deutschland: Verschreibungspflichtig, Österreich: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. Pharmakotherapeutische Gruppe: Andere Mittel für das alimentäre System und den Stoffwechsel, sonstige Mittel für das alimentäre System und den Stoffwechsel, ATC-Code: A16AX28. Weitere Angaben: Ausführliche Informationen zu Warnhinweisen und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen, Schwangerschaft und Stillzeit, Nebenwirkungen, sowie Dosierung entnehmen Sie bitte der veröffentlichten Fachinformation (Zusammenfassung der Merkmale des Arzneimittels).
Pharmazeutischer Unternehmer / Inhaber der Zulassung: PTC Therapeutics International Limited, Unit 1, 52-55 Sir John Rogerson’s Quay, Dublin 2, D02 NA07, Irland. SEP-001