Granulomatosi con poliangioite a interessamento meningeo
Francesco Delle Monache
UOC
di Medicina Interna
Servizio di Reumatologia, Ospedale “Giuseppe Mazzini” – ASL Teramo

Francesco Delle Monache
UOC
di Medicina Interna
Servizio di Reumatologia, Ospedale “Giuseppe Mazzini” – ASL Teramo
Le vasculiti associate ad autoanticorpi anti-citoplasma dei neutrofili (ANCA) sono un gruppo di disturbi che includono la granulomatosi con poliangioite (GPA), la poliangioite microscopica (MPA) e la granulomatosi eosinofila con poliangioite (EGPA). La granulomatosi con poliangioite (GPA) e la poliangioite microscopica (MPA) sono entrambe vasculiti necrotizzanti che colpiscono prevalentemente arterie di piccole dimensioni e si presentano in modo variabile in termini di manifestazioni organiche e gravità della malattia. Gli organi più comunemente e gravemente colpiti includono le vie respiratorie superiori e inferiori e i reni. A causa della loro forte associazione con ANCA, sono anche chiamate vasculiti associate ad ANCA (AAV). In Europa la prevalenza della granulomatosi con poliangioite (GPA) varia da 24 a 157 casi per milione di persone 1 . I pazienti con AAV possono sviluppare manifestazioni cliniche che coinvolgono il sistema nervoso, tra cui mononeuropatia multipla (chiamata anche mononeurite multipla), neuropatia sensoriale, anomalie dei nervi cranici, lesioni di massa del sistema nervoso centrale, oftalmoplegia esterna e perdita dell’udito neurosensoriale. La malattia meningea è una manifestazione rara che più comunemente si associa a un interessamento granulomatoso del sistema nervoso centrale.
Il caso clinico descritto è quello di una paziente, infermiera professionale nata nel 1972, che viene ricoverata presso il reparto di Medicina interna del Presidio Ospedaliero di Teramo per una diar-
rea profusa resistente alla terapia domiciliare, nell’ aprile del 2016. Nel corso degli anni il quadro clinico è variato, ponendo dubbi sia sull’aspetto clinico che terapeutico.
La storia clinica della paziente ha inizio nel 2007, a quattro mesi dall’espletamento del parto, con artralgie migranti a piccole e grandi articolazioni trattate con farmaci antinfiammatori non steroidei (FANS) con temporaneo beneficio. Nel 2009, il primo consulto con uno specialista reumatologo a seguito della comparsa di manifestazioni artritiche a carico delle caviglie. Dal consulto risultava un incremento degli indici di flogosi e positività dei c-ANCA (65 U/ml) mentre erano negativi tutti gli altri marcatori bioumorali autoimmuni. In relazione al quadro clinico, alla familiarità per psoriasi e al riscontro di aplotipo HLA-Cw6, viene posta diagnosi di oligoartrite sieronegativa, subset psoriasico. Un’ulteriore valutazione del caso in altri due centri di reumatologia confermava diagnosi e terapia. Nell’agosto del 2009, la paziente è sottoposta a nuovo ricovero presso la UOC di Reumatologia in altra regione, dove agli esami di laboratorio si osservavano ancora un lieve incremento degli indici di flogosi e la positività dei c-ANCA a medio titolo (62 U/ml). La paziente non riceve diagnosi specifiche ma le viene consigliata una terapia con metilprednisolone 4 mg/die in associazione con metotrexato (MTX). La paziente ha successivamente presentato episodi di artro-mialgie migranti senza eseguire terapia, fino all’inizio di una nuova gravidanza nel 2010, caratterizzata da pericarditi recidivanti trattate con cicli di steroide. Nel marzo 2013, un nuovo consulto con uno specialista reumatologo conferma la diagnosi di artrite psoriasica con indicazione per una progressiva diminuzione dello steroide e introduzione di etanercept 50 mg. Nel marzo 2013, causa episclerite
OS trattata con terapia topica e prednisone al dosaggio di 1 mg/kg, la terapia con etanercept è sospesa e sostituita con terapia con golimumab. Nell’ottobre 2013 la paziente è nuovamente ricoverata in ambiente reumatologico fuori regione, dove l’episclerite OS è confermata e si evidenzia ipoacusia neurosensoriale lieve. Posta diagnosi per episclerite OS, ipoacusia neurosensoriale e artrite simmetrica e bilaterale in paziente con positività agli c-ANCA, la paziente viene trattata con rituximab (RTX, un ciclo completo secondo quanto già previsto per l’artrite reumatoide) con beneficio per la salute oculistica. Segue un periodo di benessere senza alcuna terapia che si protrae fino al 2016, anno in cui compare una intensa sintomatologia caratterizzata da dolore con “senso di costrizione e riduzione della sensibilità” localizzato a livello di entrambe le gambe e i piedi. A fine marzo 2016, il quadro clinico si complica ulteriormente con la comparsa di fastidi all’OD, addominalgie intense in regione dell’ipocondrio sinistro e ematochezia auto-risolta dopo circa una settimana. In data 03/04/2016 la paziente è ricoverata presso l’Ospedale di Teramo.
Vengono eseguiti esami ematici che documentano un marcato incremento degli indici di flogosi (VES 71 mm/h, PCR 103 mg/dL) e una lieve anemia normocromico-microcitica (Hgb 11,4 g/dL), una TAC all’addome con mezzo di contrasto (04/04/2016) che evidenzia una “milza con multiple, ubiquitarie ed estese aree ipodense prive di vascolarizzazione in sede intra-parenchimale, molto confluenti e da riferire a infarti splenici su base vasculitica” e una consulenza oculistica che evidenzia “cheratite limbica in OD”. Poco prima delle dimissioni viene formulata una diagnosi per “infarti splenici su base vasculitica in paziente con artrite psoriasica e granulomatosi di Wege -
Figura 1. Esteso ispessimento e potenziamento leptomeningeo in sede frontale e parietale destra, specie al vertice con ispessimento durale diffuso (circa 6 mm) che attualmente si estende a interessare anche la grande falce cerebrale (con spessore max di circa 4 mm in sede anteriore). Permangono gli ispessimenti focali a carattere pseudonodulariforme in particolare a livello del giro frontale medio (diam. di 11 mm) nonché in sede occipitale destra.
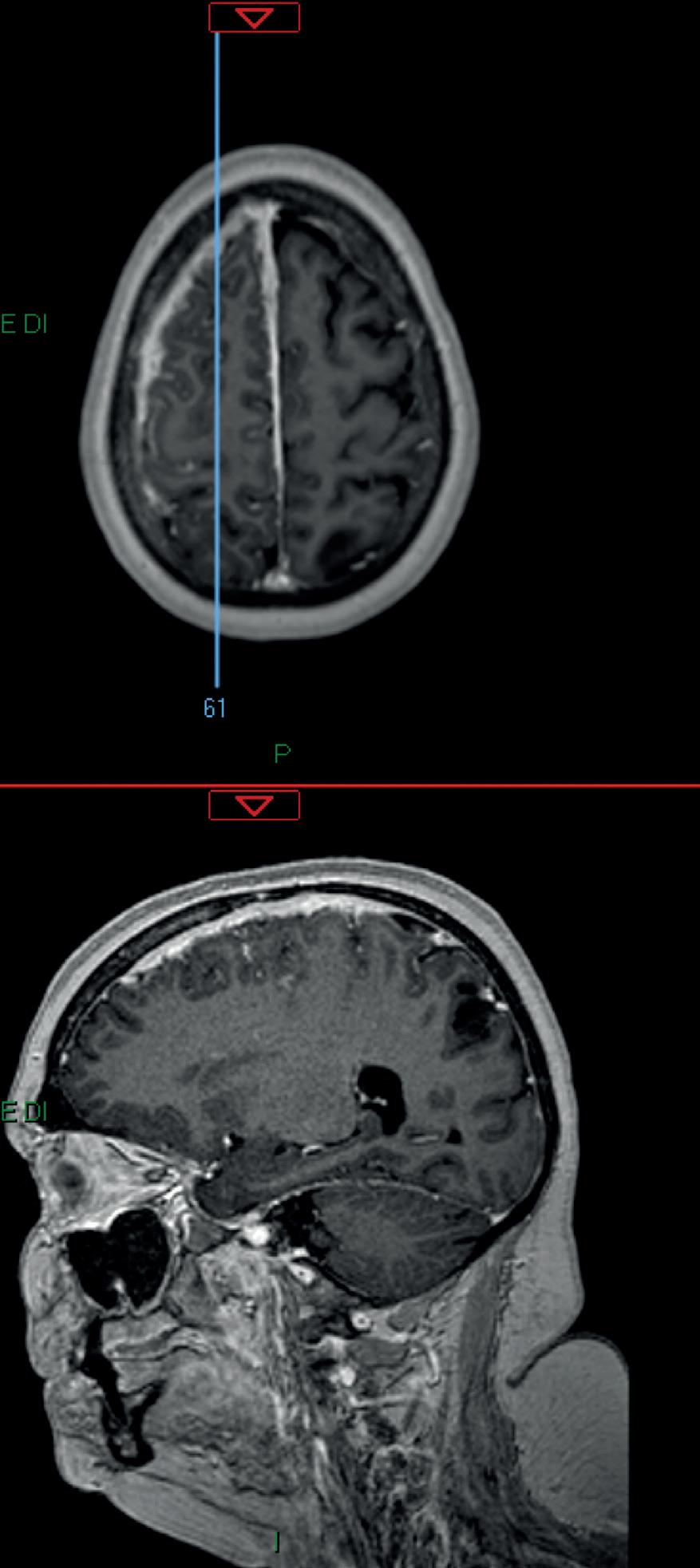
ner”, cui segue la prescrizione di una terapia con enoxaparina sodica (4000 UI/die per un mese), golimumab (50 mg) e MTX (15 mg/sett), senza beneficio. Agli inizi di maggio 2016, il quadro si complica ulteriormente, con la comparsa di un’intensa, persistente e diffusa cefalea in regione sopraorbitaria e al movimento dei bulbi, che costringe la paziente a un ricovero in ambiente internistico. Sospesa la terapia farmacologica, durante la degenza vengono ripetuti gli esami di laboratorio e confermati nuovamente sia l’incremento degli indici di flogosi sia la presenza di lieve anemia. Viene anche eseguita RMN dell’encefalo (20/05/2016), con riscontro di piccole aree di alterato segnale a carico della sostanza bianca fronto-parietale, corone radiate bilateralmente in regione periventricolare frontale sinistra a distribuzione sottocorticale (da riferire a gliosi) e una piccola alterazione dell’intensità di segnale in regione temporale profonda sinistra (da riferire a cisti della fessura corioidea). Somministrato un mezzo di contrasto, si documenta anche una diffusa e discreta pachimeningite in assenza di aree nodulari e un sistema ventricolare simmetrico, in asse e normoampliato (Figura 1)
Alla luce dei suddetti dati e al riferimento da parte della paziente della comparsa, da circa un mese, di lesioni al cavo orale e pregresse lesioni genitali, viene diagnosticata una Sindrome neuroBehçet da trattare con azatioprina (100 mg/die) in associazione a prednisone (50 mg/die). Il trattamento non dà beneficio e la paziente viene nuovamente ricoverata. Si assiste alla comparsa di croste nasali sanguinanti, specie al mattino, con associata congestione nasale e si riscontrano in più determinazioni positività ad alto titolo dei cANCA (dovuta a storia pregressa di oligoartrite non erosiva), decorso cronico-recidivante, sclerite con associata intensa cefalea, pachimeningite in assenza di aree nodulari, infarti splenici, rettorragia con riscontro endoscopico di colite ischemi-
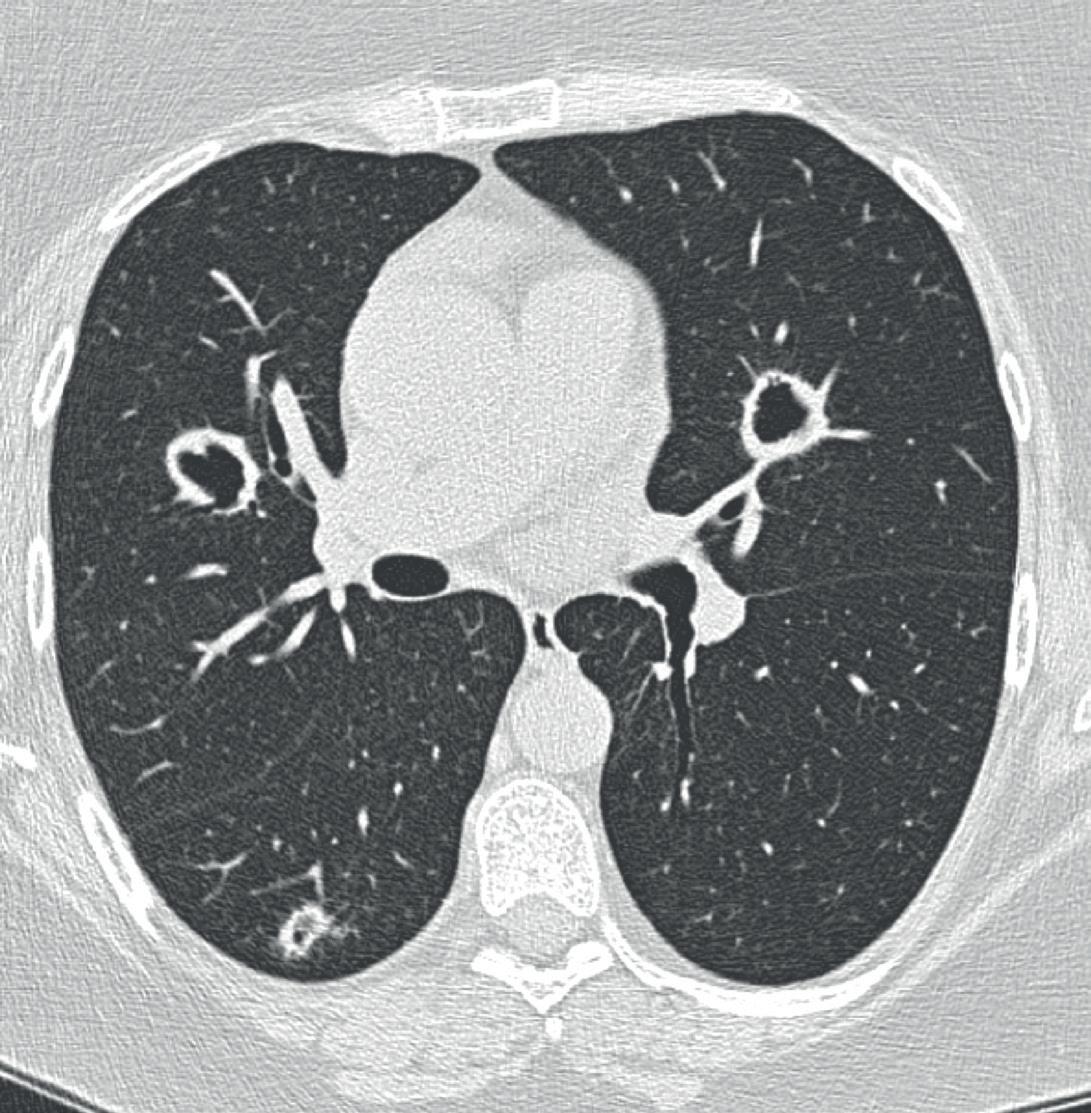
ca e multiple aree di consolidamento pseudonodulare del polmone, alcune delle quali escavate (Quantiferon e broncoscopia con studio BL per BK + BAL per esami colturali, nei limiti) (Figura 2). A fronte di quanto riscontrato, si sospende la terapia con azatioprina e si avvia una progressiva diminuzione del prednisone (1 mg/Kg), in previsione di iniziare una terapia con RTX secondo lo schema previsto per AAV.
Alla luce delle variazioni cliniche, della comparsa di lesioni polmonari escavate e della confermata positività dei c-ANCA, viene posta diagnosi di granulomatosi con poliangioite (GPA) o “granulomatosi di Wegener”.
Dato il progressivo peggioramento clinico e l’interessamento multiorgano, a seguito di una biopsia cerebrale per escludere una patologia linfoproliferativa e/o infettiva, la paziente viene sottoposta a terapia di induzione con ciclofosfamide (12 boli endovenosi da 1,2 g)1,3 e RTX (1000 mg al T0 e al T15) con successiva introduzione di micofenolato mofetile (2 g/die) più medi dosaggi di steroide (metilprednisolone 8 mg/die)2,3 . Tuttavia, il mancato miglioramento ha spinto a optare per una terapia di emergenza con immunoglobuline e.v. al dosaggio di 0,5 g/Kg/die per quattro giorni consecutivi e progressiva riduzione dello steroide, mantenendo il solo micofenolato mofetile al dosaggio di 2 g/die. Ha quindi proseguito le infusioni di immunoglobuline e.v. al dosaggio di 0,5 g/ kg/die per quattro giorni consecutivi al mese con riduzione dello steroide fino a 8 mg/die. Nel gennaio 2020, a seguito di una riacutizzazione della malattia a livello meningeo e oculare, si è deciso di intraprendere una terapia ricombinata con RTX 1000 mg al T0 e T15 (16/01/2020) e ciclofosfamide e.v. 500 mg ogni 15 giorni per sei
volte, associata a un’unica infusione di glucocorticoidi da 500 mg e.v., seguita da metilprednisolone 16 mg a scalare (1 cp alternata a ¾ di cp per 20 giorni, quindi ¾ di cp per 20 giorni, quindi ¾ di cp alternata a ½ cp per 20 giorni, quindi ½ cp per 20 giorni, quindi ½ cp alternata a ¼ di cp per 20 giorni, quindi ¼ di cp fino al successivo controllo clinico). Veniva quindi prescritta anche una terapia con antibiotico a base di sulfametoxazolo e trimetopim per tre giorni alla settimana. Già in seguito alla prima infusione, la paziente ha riferito un discreto miglioramento, con riduzione della diplopia, dell’esoftalmo OD e della cefalea. La paziente ha ripetuto RTX 1000 mg e ciclofosfamide 500 mg a febbraio dello stesso anno, per ulteriori cinque somministrazioni, tutte ben tollerate (dosaggio cumulativo di ciclofosfamide raggiunto: 17,4 g).
Nel marzo 2023, in un momento di apparente discreto controllo della malattia, si documenta la comparsa di un nuovo interessamento polmonare, caratterizzato da lesioni escavate multiple. Considerati anche l’incremento di ANCA anti-PR3, le rilevanze della tipizzazione linfocitaria, la storia clinica e il coinvolgimento polmonare in corso, la paziente inizia a essere trattata con metilprednisolone 500 mg in mono-somministrazione, infuso in vena al dosaggio di 0,5 mg/Kg per quattro giorni. Assume inoltre RTX 375 mg/m2 SC (dosaggio 675 mg), somministrato secondo uno schema settimanale per un totale di quattro somministrazioni e intraprende un trattamento con antibiotico battericida a base di sulfametoxazolo e trimetopim per la profilassi da Pneumocystis jirovecii. La paziente prosegue la terapia con prednisone (17,5 mg/die), con intento di progressiva riduzione. Dopo la terza somministrazione di RTX (21/04/2023), a causa dell’accentuazione delle parestesie e del dolore nella loggia posteriore della gamba, si esegue una EMG (24/04/2023) dalla quale emergono polineuropatia sensitiva cronica, segni di sofferenza del nervo peroneo di recente insorgenza e una lenta riduzione dei parametri di flogosi all’ultima infusione di RTX (28/04/2023: PCR 67 mg/dL).
Naturalmente, il complesso iter terapeutico associato a una persistente terapia steroidea a medio-alto dosaggio ha comportato una serie di complicanze legate alla terapia stessa, come una grave osteoporosi fratturativa con necrosi asettica delle teste omerali.
Nell’impossibilità di ridurre tale terapia steroidea (prednisone 17,5 mg/die) si è chiesto di poter aggiungere, in uso compassionevole, terapia con avacopan4,5,6 .
Dopo poche settimane dall’introduzione di avacopan, la paziente ha presentato un graduale ma costante miglioramento, con riduzione della te -
rapia steroidea (prednisone fino a 2,5 mg/die). Al momento, la malattia è in remissione sia clinica che laboratoristica e a ogni controllo si segnala la riduzione di tutti i noti effetti collaterali dello steroide.
Le patologie autoimmuni tendono spesso ad assumere aspetti diversi e a modificare la propria espressione clinica e laboratoristica. Il caso descritto è stato valutato dai migliori centri di reumatologia nazionali che, interpretando i segni e i sintomi, hanno effettuato diagnosi che dall’artrite psoriasica sono passate al Behçet e alla granulomatosi di Wegener (GPA). Di fatto, tutte le patologie elencate avevano i criteri diagnostici per giustificare le ipotesi formulate. Anche gli organi interessati sono stati diversi (occhio, articolazioni, meningi, massiccio facciale, polmone). In questi casi, per poter eseguire una diagnosi corretta ed elaborare il piano terapeutico più efficace, l’approccio multidisciplinare rappresenta l’unica possibilità di successo.
Negli corso degli anni è emerso che una terapia steroidea protratta per anni a una determinata dose cumulativa, incide sulla qualità della vita e sulla prognosi del paziente in percentuale pari a quella legata all’aggressività della patologia. Per tale ragione, è fondamentale ragionare e attuare tutto quanto in nostro potere per ridurre la quantità di cortisone somministrato, per quanto, soprattutto in pazienti giovani con malattie che pongono in pericolo di vita, siamo portati a sottovalutare l’effetto iatrogeno dello steroide. Questo, ad oggi, merita un’attenzione di primo rilievo.
1. Chung SA, Langford CA, Maz M, et al. American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis 2021. Arthritis Rheumatol 2021; 73(8):1366.
2. Cortazar FB, Muhsin SA, Pendergraft WFIII, et al. Combination Therapy With Rituximab and Cyclophosphamide for Remission Induction in ANCA Vasculitis. Kidney Int Rep 2018; 3(2): 394–402.
3. Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. European Vasculitis Study Group N Engl J Med 2010; 363(3):211.
4. Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis 2024; 83(1):30-47.
5. Chandwar K. Avacopan for the Treatment of ANCA-Associated Vasculitis. N Engl J Med 2021; 384(21):e81.
6. Geetha D, Dua A, Yue H, et al. Efficacy and safety of avacopan in patients with ANCA-associated vasculitis receiving rituximab in a randomised trial. ADVOCATE Study Group. Ann Rheum Dis 2024; 83(2):223-232.
Ilpresentecasoclinicoèstatorealizzatoin collaborazionecon
