


India Sisler, MD


• Most common monogenic disease1
• Disproportionately affects marginalized communities1
• Pervasive misunderstanding surrounding disease1
– Provider bias and stigma
• Paucity of available resources1
– Underfunding for care, research, and surveillance
– Lack of access to specialty care
– Systemic racism within health care
– Less than half of patients receive recommended


80% to 100% of Hb concentration is HbS in HbSS


Heart
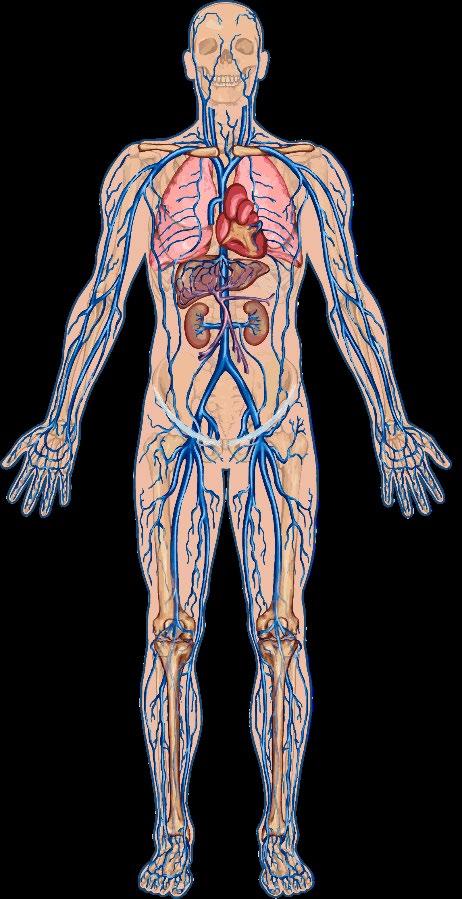
Pulmonary
Acute
Patients experience ~6 VOCs annually requiring hospitalization, miss ~10 hours of work/week, with an overall work productivity loss of 53.4%,1 and indirect economic burden of ~$9290 annually.2 Stroke
Acute

India Sisler, MD
Prophylactic penicillin, until age 5 Pneumococcal immunizations and other scheduled vaccines Routine screening for early signs of organ damage
Other clinical routine preventive services
Stroke
Pulmonary
Hypertension Retinopathy
Nephropathy
National Heart, Lung, and Blood Institute (NHLBI). Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014. https://www.nhlbi.nih.gov/health-topics/evidence-basedmanagement-sickle-cell-disease.
Not a One-Size-Fits-All Approach
Chronic Red Cell Transfusion (CRCT) can prevent:1,2
✓ Ischemic stroke in high-risk children
✓ Secondary stroke across the lifespan
When to initiate:1,2
✓ Recurrent acute chest syndrome
✓ Complications caused by pregnancy
Children with transcranial doppler (TCD) reading >200 cm/s High Strong
Adults and children with previous clinically overt stroke Moderate Moderate
Risks and limitations:3,4 % IRON OVERLOAD3
• Alloimmunization—up to 47%
• Delayed hemolytic transfusion reactions—up to 11%
• Hyperviscosity
• Accessibility in resource poor areas
Stroke recurs in approximately 20% of patients receiving CRCT therapy despite maintenance of a sickled hemoglobin <30%.3
1. NHLBI. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014. https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease; 2. Alan S, Kanter J. Expert Opin Pharmacother. 2024;25(10):1325-1334; 3. Udeze C, et al. Clinicoecon Outcomes Res. 2025:17:303-313; 4. Chou ST, et al. Blood. 2013;122(6):1062-1071.
A potent ribonucleotide reductase inhibitor (required for DNA synthesis and repair)

Vascular Effects Cellular Effects
Decreased Endothelial Activation and Adhesion
Decreased Thrombosis and Microparticle Formation
Decreased Vasoconstriction
Increased HbF Synthesis

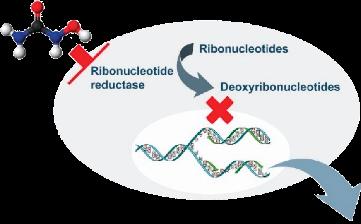

Decreased HbS Polymerization
Decreased RBC Membrane Damage
Decreased Hemolysis
Decreased Neutrophil Count
Increased Hb Synthesis
Initially approved in 1998 for the treatment of severe SCD in adults, indication expanded for use in children aged ≥2 years in 2017 and ≥6 months in 2024 to reduce painful crises and need for blood transfusions.
Follow-up, observational study (1996-2001)2
• Patients could continue, stop, or start HU
• N=75 died
• Cumulative mortality at 9 years, 28% with HbF <0.5 g/dL vs 15% with HbF ≥0.5 g/dL – ACS, 32% mortality vs 18% in those without
HU initiation recommended in patients aged 9 to 12 months.
% of patients hospitalized or receiving emergency care for VOC were NOT RECEIVING HU >75
In a study of Medicaid claims data (N=1035), only 20.9% of patients were adherent to HU; this is often due to drug intolerance.
Kang HA, et al. Am J Hematol. 2023;98(1):90-101; Stettler N, et al. JAMA. 2015;313(16):1671-1672.
• Significant contributors to the psychosocial distress:

Sleep Disturbances Interpersonal Relationship Challenges Stigmatization Workplace Discrimination Anxiety Reduced QOL Functional Impairment
Financial Strain Body Dissatisfaction
• Early identification and targeted interventions
– Integration of mental health into clinical care
– Comprehensive support programs
• School-based and community support groups
• Research and advocacy
– Patient and family education
– Enhanced pain management strategies
QOL, quality of life.
Essien EA, et al. Medicine (Baltimore). 2023;102(47):e36147; NHLBI. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014. https://www.nhlbi.nih.gov/health-topics/evidencebased-management-sickle-cell-disease.
“As patients with SCD become adolescents and young adults, they have more acute care presentations, have higher health care costs, and experience increased mortality rates.”1

15- to 19-year age group, 1.7 per 1,000,000
20- to 24-year age group, 0.7 per 1,000,000
A PROPOSED STANDARDIZED METRIC FOR SUCCESSFUL TRANSFER OF CARE
Standard 1
Standard 2
A successful transfer of care is defined as 2 visits with a comprehensive adult sickle cell program in the first year. Visits can be in person or via telemedicine.
A successful integration into adult care—an essential component of transition from pediatric to adult-centered health care—is defined as completion of ≥50% of scheduled outpatient visits in the 5-year period after transfer of care and the patient identification of the adult center as their sickle cell medical home. • Mortality rates:2
1. Pollock G, Curtis S. Blood Adv. 2025;9(18):4622-4623; 2. Karkoska KA, McGann PT. Pediatrics. 2024;154(6):e2024067341.

Modupe Idowu, MD
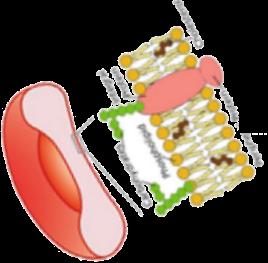
• L-glutamine is an oral precursor for NADH, the reduced form of nicotinamide adenine
dinucleotide
• Results in decreased oxidative stress within sRBCs

FDA-approved in 2017 to reduce acute complications of SCD in patients aged ≥5 years.
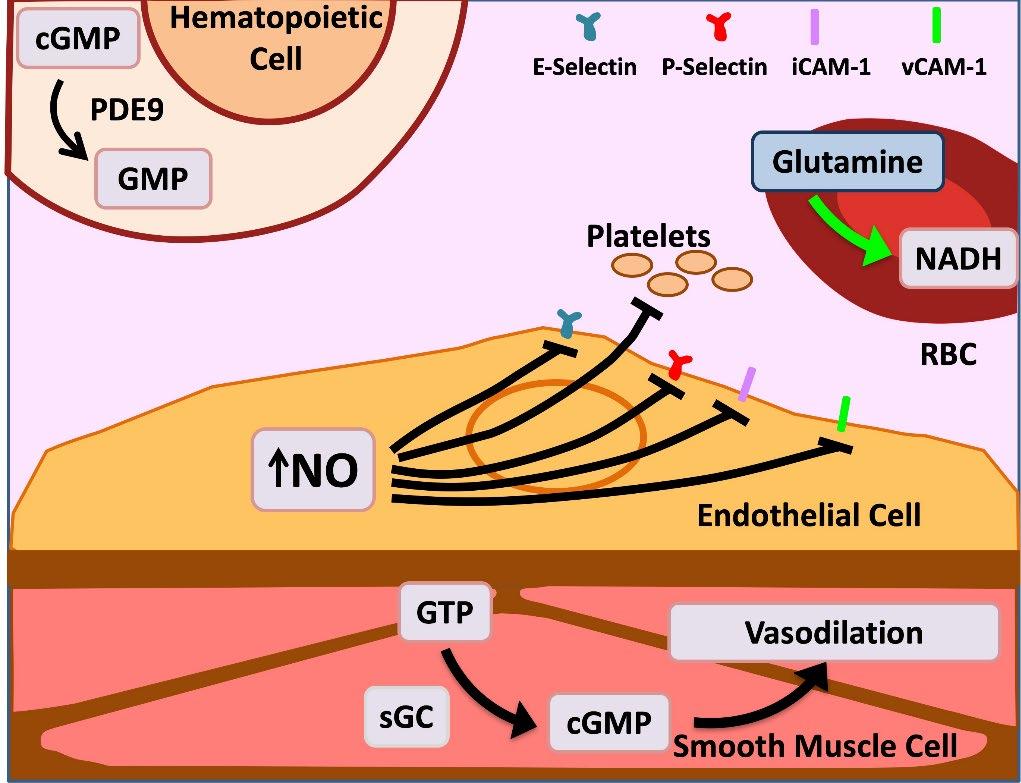
cGMP, cyclic guanosine monophosphate; iCAM, intercellular adhesion molecule-1; NADH, nicotinamide adenine dinucleotide plus hydrogen; sGC, soluble guanylate cyclase; sRBCs, sickle RBCs; vCAM, vascular cell adhesion molecule-1. Sadaf A, et al. Exp Biol Med (Maywood). 2020;245(2):146-154; Tang MS, Shan H. Vox Sang. 2024;119(6):521-528. Image adapted from . Morrone K, et al. Semin Hematol. 2018;55(2):68-75
• 25% reduction in number of pain crises (3.0 vs 4.0; P=0.005) • 33% lower hospitalization rates (2.0 vs 3.0; P=0.005)
• Reduced number of episodes of acute chest syndrome (8.6% vs 23.1%; P=0.003)

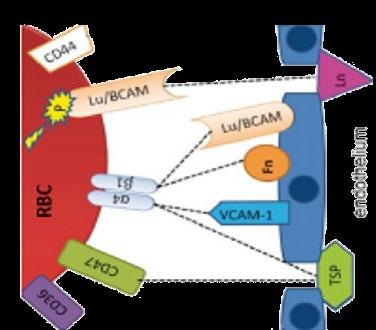
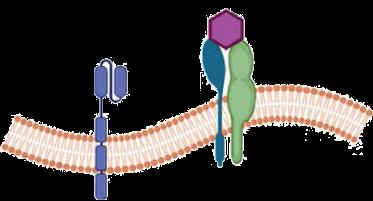
• Upregulation of P-selectin in endothelial cells and platelets contributes to the cell–cell interactions involved in the pathogenesis of vaso-occlusion and sickle cell–related pain crises
• P-selectin is the primary selectin that binds to sickled RBCs and reticulocytes, mediating their adhesion to other cells and the endothelium—an important step in initiating and propagating VOCs
• Interference with selectins represents a potential therapeutic strategy for the clinical management of SCD
• The safety and efficacy of selectin inhibitors have been evaluated in patients with SCD
• Humanized IgG2 monoclonal antibody to P-selectin
– Expression of P-selectin on endothelium mediates adherence of sickle cells and development of VOC
• Clinical effects— reduces frequency of VOCs
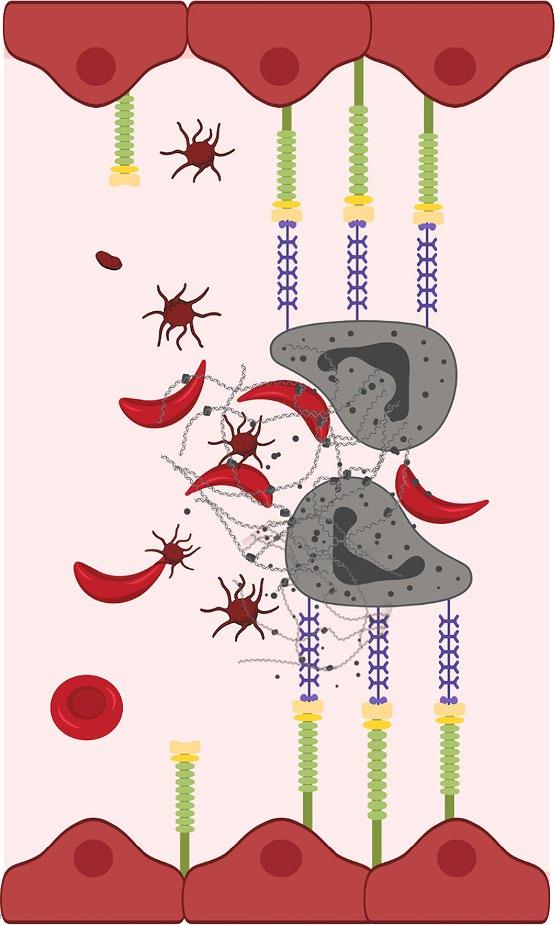
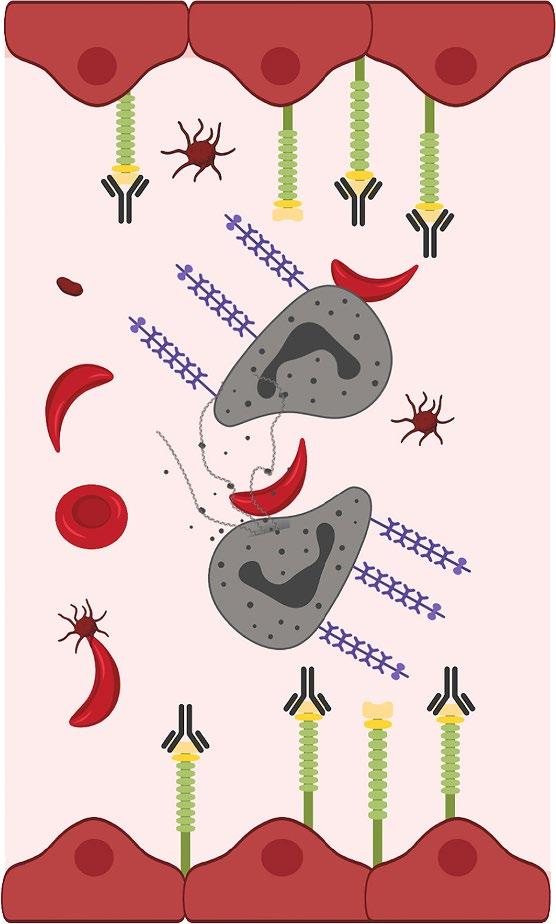






Efficacy of Crizanlizumab, 5.0 mg/kg and 7.5 mg/kg
Annualized Adjusted Rates of VOC, 1 Year Postrandomization
While the safety profile was consistent among the STAND and SUSTAIN studies, no significant reductions in VOC for crizanlizumab vs placebo were demonstrated in STAND.
Phase 3 study, SPARKLE, is currently recruiting to reinvestigate outcomes.
N=252 patients aged ≥12 years with SCD randomly assigned (1:1:1) to receive either 5.0 mg/kg of crizanlizumab, 7.5 mg/kg of crizanlizumab, or placebo, in addition to standard of care, for 1 year. The primary endpoint was the annualized rate of VOCs leading to a health care visit over the first-year postrandomization. Key secondary endpoint was the annualized rate of all VOCs managed at
and leading to a health care visit after 1 year postrandomization. Abboud MR, et al. Lancet Haematol. 2025;12(4):e248-e257.
Summary of Reduction of Priapic Events
≥60 Minutes in the Study Population
Percentage Change From Baseline in Cumulative No. Priapic Events by Week 26

Modupe Idowu, MD, and Santosh Saraf, MD
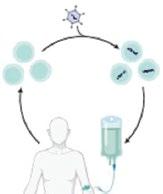
• Overview: autologous hematopoietic stem cells are collected from patients with SCD and genetically modified ex vivo to reduce sickling tendency before reinfusion1



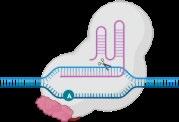
• Hemopoietic stem and progenitor cells are transduced with the BB305 lentiviral vector encoding modified β-globin gene, HbAT87Q
• HbAT87Q has a similar oxygen-binding affinity to wild-type HbA and sterically prevents RBC sickling1,2
Working copies of modified hemoglobin gene delivered into stem cells through a vector Cells express the


• HGB-206 Group C study + HGB-210 studiesa
– As of Feb 2024, N=55 received lovo-cel
– Among 34 VOE-evaluable participants
• 88.2% achieved VOE-CR
• 94.1% achieved sVOE-CR
– 6 to 18 months post infusion
– Among 46 evaluable participants
• 89.1% achieved GR
– Median peripheral blood HbAT87Q 6 months post infusion was 49%
GR, gene replacement; PB, peripheral blood; sVOE-CR, complete resolution of severe vaso-occlusive event. aNonrandomized, open-label, single-dose trials; primary outcome was resolution of VOC. 1. Tang MS, Shan H. Vox Sang. 2024;119(6):521-528; 2. Alan S, Kanter J. Expert Opin Pharmacother. 2024;25(10):1325-1334; 3. Gupta AO, et al. Transplant Cell Ther. 2025;31(2):S256-S257; 4. Lyfgenia. Prescribing information. bluebird bio, Inc; 2023. https://www.genetixbiotx.com/-/media/bluebirdbio/Corporate%20COM/Files/Lyfgenia/LYFGENIA_Prescribing_Information.pdf. Image adapted from Gene Therapy in Sickle Cell. https://www.sparksicklecellchange.com/treatment/sickle-cell-gene-therapy. Approved for patients aged ≥12 years with SCD and history of VOCs.4


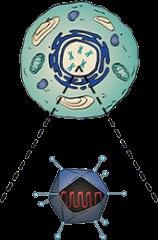
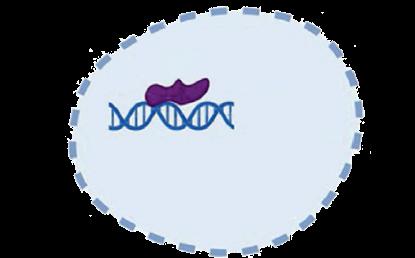
• Exa-cel uses CRISPR/Cas9 gene editing technology to disrupt the BCL11A erythroid enhancer on chromosome 2, leading to increased expression of fetal hemoglobin1,2
• BCL11A is a key regulator of the switch from fetal to adult Hb1,2
CRISPR/Cas9 delivered via electroporation "Switch" made to a different gene Working type of hemoglobin is produced to compensate for sickle hemoglobin Functioning red blood cells developed


Study2,3
• CLIMB SCD-121
– N=30 with sufficient duration of follow-up
– VF12: 97% (freedom from severe VOCs ≥12 consecutive months)
– HF12: 100% (freedom from VOC–related hospitalizations ≥12 months)
– Total Hb: improved to 12.5 g/dL at 6 months
– HbF: increased to 36.9% at 3 months and 43.9% at 6 months
– Neutrophil engraftment, median 27 days
– Platelet engraftment, median 35 days
CRISPR, clustered regulatory interspaced palindromic repeats.


1. Tang MS, Shan H. Vox Sang. 2024;119(6):521-528; 2. Alan S, Kanter J. Expert Opin Pharmacother. 2024;25(10):1325-1334; 3. Frangoul H, et al. N Engl J Med. 2024;390(18):1649-1662; 4. Casgevy. Prescribing information. Vertex Pharmaceuticals Inc; 2024. https://pi.vrtx.com/files/uspi_exagamglogene_autotemcel.pdf Image adapted from Gene Therapy in Sickle Cell. https://www.sparksicklecellchange.com/treatment/sickle-cell-gene-therapy.
• In the US, ~60% of patients with SCD rely on Medicare or Medicaid, limiting access to specialized health care
• Lack of access and scarcity of SCD specialists and specialized transplant centers
• Many people aren’t eligible because of the requirement for high-dose chemotherapy
• Gene therapy products cost between $1.4 and $2.1 million US dollars
• Gene therapy for SCD currently not available in low-income countries
INSURANCE SPECIALIZED TRANSPLANT CENTERS

Only ONE patient received gene therapy in 2024. Health care inequity must be addressed for gene therapy to bring benefit among patients with severe SCD.
. 2024;119(6):521-528.
• Once limited to full sibling donors of patients with SCD1
– 25% chance of being HLA-identical (fully matched)
• >90% EFS; <20% of patients have this donor




Donor Collection Processing




Patient Reinfection

Cryopreservation
– 50% chance of being haploidentical
• Risks include GF, GVHD, infections, infertility, vital organ injury, and death2


• Systematic review/meta-analysis (pooled data from 33 studies published between 1986 and 2017, N=2853 patients)3,a




• Advancements in donor selection and conditioning regimens continue to improve1
aGVHD, acute graft-versus-host disease; cGVHD, chronic GVHD; DFS, disease-free survival; EFS, event-free survival; GF, graft failure; HSCT, hematopoietic stem cell transplantation; OS, overall survival. aWhile MRD was the most common donor source, the analysis also included MUD, haploidentical, and umbilical cord blood.
1. Alan S, Kanter J. Expert Opin Pharmacother. 2024;25(10):1325-1334; 2. Kanter J, et al. Blood Adv. 2021;5(18):3668-3689; 3. Iqbal M, et al. Transplant Cell Ther. 2021;27(2):167.e1-167.e12.
Haploidentical or “half-matched” donor may include parent, sibling, child, niece, nephew, aunt, uncle, or cousin of the patient
• N=42 patients, age 15.5 to 43.2 years, received transplantation with RICa and GVHD prophylaxisb
• Primary outcome: 2-year EFS rate, 88.0%
• Secondary outcome: OS, 95.0%
How does this compare with other HSCT regimens?
Consider HLA–matched related HSCT in
1. Patients who have experienced an overt stroke or have an abnormal TCD
2. Patients with frequent pain episodes requiring medical attention
3. Patients with recurrent episodes of ACS
aRIC that contains melphalan/fludarabine regimens. ASH, American Society of Hematology. Kanter J, et al. Blood Adv. 2021;5(18):3668-3689.
For patients with an indication:
4. If no matched sibling donor (MSD), use alternative donors in the context of a clinical trial
5. Use either total body irradiation ≤400 cGy or chemotherapy–based conditioning regimens
a) For children with an MSD, use myeloablative conditioning over RICa
b) For adults with an MSD, use nonmyeloablative conditioning over RICa
6. Use transplantation at an earlier age rather than an older age
7. Use HLA-identical sibling cord blood when available over bone marrow

Santosh L. Saraf, MD
Let’s recap:
SCD is the most common monogenic disease It disproportionately affects marginalized communities
The average life expectancy is 52.6 years
Paucity of available and accessible resources
The few FDA-approved therapies are insufficient











2023;7(24):7539-7550.

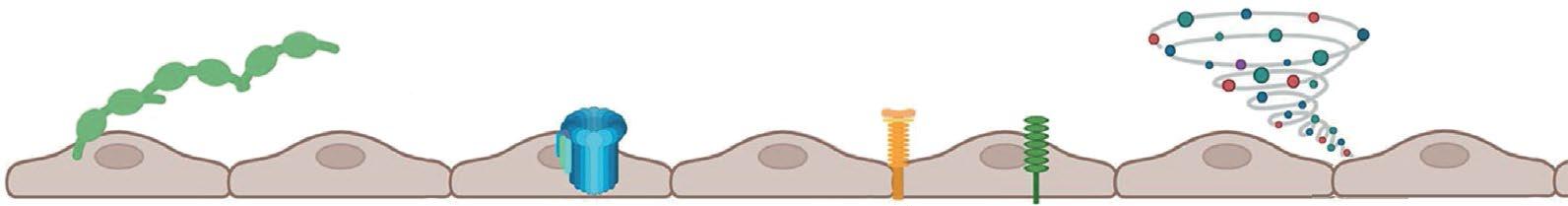
• Hemolysis
• RBC sickling
• Vaso-occlusive events
• Hb level
• Hb-oxygen affinity
The phase 2 dose-finding period included N=79 patients aged ≥16 years with SCD were randomly assigned 1:1:1 to mitapivat 50 mg orally (PO), mitapivat 100 mg PO, or matched placebo twice a day (BID) for 12 weeks. Patients who completed the 12-week double-blind period and did not have ongoing grade 3 or worse treatment-related adverse events could receive mitapivat in the 216-week openlabel extension period. Primary efficacy and safety endpoints were Hb response (≥1.0 g/dL increase from baseline in average Hb concentration from week 10 through week 12), and type, severity, and relationship to study drug of adverse and serious adverse events.
Idowu M, et al. Lancet Haematol. 2025;12(1):e35-e44.
• Annualized rates of pain crisis
– 0.83, mitapivat 50 mg
– 0.51, mitapivat 100 mg
– 1.71, placebo
• % reduction in annualized rate vs placebo – 51.6%, mitapivat 50 mg – 70.0%, mitapivat 100 mg

Endpoint (ITT population)
All hemolysis biomarkers decreased from BL in both etavopivat groups
adapted from Paikari A, Sheehan VA. Br J Haematol. 2017;180(2):189-200 and Pinto VM, et al. Blood. 2024;144(8):853-866.
Targeting
Endothelial
Ab
Ab


Ab, antibody; BMP, bone morphogenic protein; E-MP, erythroid microparticles; FPN, ferroportin; HAMP, hepcidin; HJV, hemojuvelin; IL, interleukin; MAC, macrophage; NO, nitric oxide; PS, phosphatidylserine; ROS, reactive oxygen species; Smad, suppressor of mothers against decapentaplegic; TMPRSS, transmembrane protease serine; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule; vWF, von Willebrand factor. Pinto VM, et al. Blood. 2024;144(8):853-866.
• SCD requires the early application of guideline-based management to reduce complications and improve outcomes.
• Holistic care is essential, addressing not only physical complications but also psychosocial distress and stigma.
• While disease modifying drugs are available, a crucial need persists for the development of novel agents, which are both effective and accessible.
• Emerging innovations such as pyruvate kinase activators are being investigated and may be promising new additions to the treatment armamentarium.
• Equitable access remains a challenge—closing gaps in specialty care, resources, and advanced therapies is necessary to ensure all patients with SCD can benefit from scientific progress.