EDITO : BIOTHÉRAPIES, LE NOUVEAU QUOTIDIEN DES DERMATOLOGUES
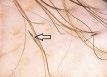
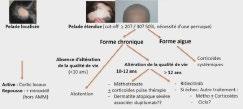
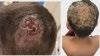
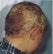
JDP 2024 : ALOPÉCIE CHEZ L’ENFANT : PAS À PAS DIAGNOSTIQUE ET THÉRAPEUTIQUE
DERMATITE ATOPIQUE ET AFFECTIONS ASSOCIÉES
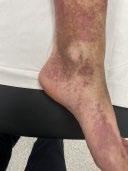
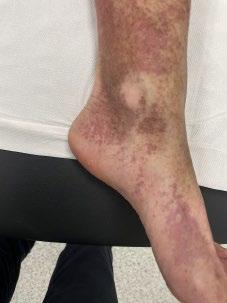
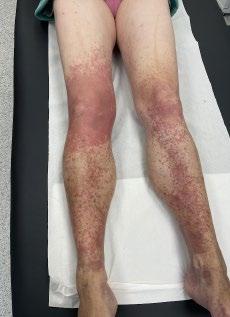
CAS CLINIQUE : PORPORA TÉLÉANGIECTASIQUE DE MAJOCCHI
DERMATOPOROSE : UN MARQUEUR DE FRAGILITÉ
COMMENT PRÉPARER MON PATIENT
Bimestriel | Bureau de dépôt : Leuven | P705015
EDITO : BIOTHÉRAPIES, LE NOUVEAU QUOTIDIEN DES DERMATOLOGUES
JDP 2024 : ALOPÉCIE CHEZ L’ENFANT : PAS À PAS DIAGNOSTIQUE ET THÉRAPEUTIQUE
DERMATITE ATOPIQUE ET AFFECTIONS ASSOCIÉES
CAS CLINIQUE : PORPORA TÉLÉANGIECTASIQUE DE MAJOCCHI
DERMATOPOROSE : UN MARQUEUR DE FRAGILITÉ
COMMENT PRÉPARER MON PATIENT
Bimestriel | Bureau de dépôt : Leuven | P705015
Otezla® 10 mg, 20 mg and 30 mg film-coated tablets 1. NAME OF THE MEDICINAL PRODUCT: Otezla 10 mg film-coated tablets. Otezla 20 mg film-coated tablets. Otezla 30 mg film-coated tablets. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION: Otezla 10 mg film-coated tablets: Each film- coated tablet contains 10 mg of apremilast. Excipient(s) with known effect: Each film-coated tablet contains 57 mg of lactose (as lactose monohydrate). Otezla 20 mg film-coated tablets: Each film-coated tablet contains 20 mg of apremilast. Excipient(s) with known effect: Each film-coated tablet contains 114 mg of lactose (as lactose monohydrate). Otezla 30 mg film-coated tablets: Each film-coated tablet contains 30 mg of apremilast. Excipient(s) with known effect: Each film-coated tablet contains 171 mg of lactose (as lactose monohydrate). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM: Film-coated tablet (tablet). Otezla 10 mg film-coated tablets: Pink, diamond shaped 10 mg film-coated tablet of 8 mm length with “APR” engraved on one side and “10” on the opposite side. Otezla 20 mg film-coated tablets: Brown, diamond shaped 20 mg film-coated tablet of 10 mm length with “APR” engraved on one side and “20” on the opposite side. Otezla 30 mg film-coated tablets: Beige, diamond shaped 30 mg film-coated tablet of 12 mm length with “APR” engraved on one side and “30” on the opposite side. 4. CLINICAL PARTICULARS: 4.1
Therapeutic indications: Psoriatic arthritis: Otezla, alone or in combination with Disease Modifying Antirheumatic Drugs (DMARDs), is indicated for the treatment of active psoriatic arthritis (PsA) in adult patients who have had an inadequate response or who have been intolerant to a prior DMARD therapy (see section 5.1). Psoriasis: Otezla is indicated for the treatment of moderate to severe chronic plaque psoriasis in adult patients who failed to respond to or who have a contraindication to, or are intolerant to other systemic therapy including cyclosporine, methotrexate or psoralen and ultraviolet-A light (PUVA). Behçet’s disease: Otezla is indicated for the treatment of adult patients with oral ulcers associated with Behçet’s disease (BD) who are candidates for systemic therapy.
4.2 Posology and method of administration: Treatment with Otezla should be initiated by specialists experienced in the diagnosis and treatment of psoriasis, psoriatic arthritis or Behçet’s disease.
Posology: The recommended dose of apremilast is 30 mg taken orally twice daily, approximately 12 hours apart (morning and evening), with no food restrictions. An initial titration schedule is required as shown below in Table 1. No re-titration is required after initial titration. Table 1.
Dose titration schedule:
Day 1: AM: 10 mg; Day 2: AM:
Otezla®, the first and only oral PDE4-inhibitor on the market for patients with PSO or PsA, is well tolerated and requires no dose adjustment (except in severe renal impairment), monitoring or analytical screening.1-3,a
CHOOSE EFFICACY & SIMPLICITY1-6
APREMILAST IN PATIENTS WITH PSO or PsA with or without CARDIOMETABOLIC COMORBIDITIES1-6 in the treatment of years of EXPERIENCE9 >8
Experts consider apremilast a suitable option in the treatment of the psoriatic patient*# and presence of comorbidities1,7,8
PM: 30 mg; Day 6 & thereafter: AM: 30 mg. PM: 30 mg. If patients miss a dose, the next dose should be taken as soon as possible. If it is close to the time for their next dose, the missed dose should not be taken and the next dose should be taken at the regular time. During pivotal trials the greatest improvement was observed within the first 24 weeks of treatment for PsA and PSOR and within the first 12 weeks of treatment for BD. If a patient shows no evidence of therapeutic benefit after this time period, treatment should be reconsidered. The patient’s response to treatment should be evaluated on a regular basis. Special populations: Elderly patients: No dose adjustment is required for this patient population (see sections 4.8 and 5.2). Patients with renal impairment: No dose adjustment is needed in patients with mild and moderate renal impairment. The dose of apremilast should be reduced to 30 mg once daily in patients with severe renal impairment (creatinine clearance of less than 30 mL per minute estimated by the Cockcroft-Gault equation). For initial dose titration in this group, it is recommended that apremilast be titrated using only the AM schedule listed in Table 1 and the PM doses be skipped (see section 5.2). Patients with hepatic impairment: No dose adjustment is necessary for patients with hepatic impairment (see section 5.2). Paediatric population: The safety and efficacy of apremilast in children aged 0 to 17 years have not been established. No data are available. Method of administration: Otezla is for oral use. The film-coated tablets should be swallowed whole, and can be taken either with or without food. 4.3 Contraindications: Hypersensitivity to the active substance(s) or to any of the excipients listed in section 6.1. Pregnancy (see section 4.6). 4.8 Undesirable effects: Summary of the safety profile: The most commonly reported adverse reactions with apremilast in PsA and PSOR are gastrointestinal (GI) disorders including diarrhoea (15.7%) and nausea (13.9%). The other most commonly reported adverse reactions include upper respiratory tract infections (8.4%), headache (7.9%), and tension headache (7.2%) and are mostly mild to moderate in severity. The most commonly reported adverse drug reactions with apremilast in BD are diarrhoea (41.3%), nausea (19.2%), headache (14.4%), upper respiratory tract infection (11.5%), upper abdominal pain (8.7%), vomiting (8.7%) and back pain (7.7%) and are mostly mild to moderate in severity. The gastrointestinal adverse reactions generally occurred within the first 2 weeks of treatment and usually resolved within 4 weeks. Hypersensitivity reactions are uncommonly observed (see section 4.3). Tabulated list of adverse reactions: The adverse reactions observed in patients treated with apremilast are listed below by system organ class (SOC) and frequency for all adverse reactions. Within each SOC and frequency grouping, adverse
reactions are presented in order of decreasing seriousness. The adverse drug reactions were determined based on data from the apremilast clinical development programme and post-marketing experience. The frequencies of adverse drug reactions are those reported in the apremilast arms of the four Phase III studies in PsA (n = 1,945) or the two Phase III studies in PSOR (n = 1184), and in the phase III study in BD (n = 207) (the highest frequency from either data pool is represented in table 2). Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to <1/10); uncommon (≥ 1/1,000 to <1/100); rare (≥ 1/10,000 to <1/1,000); not known (cannot be estimated from the available data). Table 2. Summary of adverse reactions in psoriatic arthritis (PsA), psoriasis (PSOR) and Behçet’s disease (BD) : System Organ Class, Frequency, Adverse reaction. Infections and infestations: Very Common: Upper respiratory tract infectiona; Common: Bronchitis, Nasopharyngitis*. Immune system disorders: Uncommon: Hypersensitivity. Metabolism and nutrition disorders: Common: Decreased appetite*. Psychiatric disorders: Common: Insomnia, Depression; Uncommon: Suicidal ideation and behaviour. Nervous system disorders: Very common: Headache*,a; Common: Migraine*, Tension headache*. Respiratory, thoracic, and mediastinal disorders: Common: Cough. Gastrointestinal disorders: Very Common: Diarrhoea*, Nausea*; Common: Vomiting*, Dyspepsia, Frequent bowel movements, Upper abdominal pain*, Gastroesophageal reflux disease; Uncommon: Gastrointestinal haemorrhage. Skin and subcutaneous tissue disorders: Uncommon: Rash, Urticaria; Not known: Angioedema. Musculoskeletal and connective tissue disorders: Common: Back pain*. General disorders and administration site conditions: Common: Fatigue. Investigations: Uncommon: Weight decrease. *At least one of these adverse reactions was reported as serious. aFrequency reported as common in PSA and PSOR. Description of selected adverse reactions: Psychiatric disorders: In clinical studies and post-marketing experience, uncommon cases of suicidal ideation and behaviour, were reported, while completed suicide was reported post-marketing. Patients and caregivers should be instructed to notify the prescriber of any suicidal ideation (see section 4.4). Body weight loss: Patient weight was measured routinely in clinical studies. The mean observed weight loss in PsA and PSOR patients treated for up to 52 weeks with apremilast was 1.99 kg. A total of 14.3% of patients receiving apremilast had observed weight loss between 5-10% while 5.7% of the patients receiving apremilast had observed weight loss greater than 10%. None of these patients had overt clinical consequences resulting from weight loss. A total of 0.1% of patients treated with apremilast discontinued due to adverse reaction of weight decreased. The mean observed weight loss in BD patients treated with apremilast for 52 weeks was 0.52 kg. A total of 11.8% of patients receving apremilast had observed weight loss between 5-10% while 3.8% of the patients receiving apremilast had observed weight loss greater than 10%. None of these patients had overt clinical consequences from weight loss. None of the patients discontinued the study due to adverse reaction of weight decreased. Please see additional warning in section 4.4 for patients who are underweight at beginning of treatment. Special populations: Elderly patients: From post-marketing experience, elderly patients ≥ 65 years of age may be at a higher risk of
complications of severe diarrhoea, nausea and vomiting (see section 4.4). Patients with hepatic impairment: The safety of apremilast was not evaluated in PsA, PSOR or BD patients with hepatic impairment. Patients with renal impairment: In the PsA, PSOR or BD clinical studies, the safety profile observed in patients with mild renal impairment was comparable to patients with normal renal function. The safety of apremilast was not evaluated in PsA, PSOR or BD patients with moderate or severe renal impairment in the clinical studies. Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system: Belgium: Federal agency for medicines and health products -Vigilance Division; EUROSTATION II; Victor Hortaplein, 40/40; B-1060 Brussels; www. famhp.be; adversedrugreactions@fagg-afmps.be Luxembourg: Centre Régional de Pharmacovigilance de Nancy ; crpv@chru-nancy.fr ; Tel : +33 3 83 65 60 85 / 87 ; Fax : +33 3 83 65 61 33 or Division de la Pharmacie et des Médicaments ; Direction de la Santé à Luxembourg ; pharmacovigilance@ms.etat. lu ; Tel : +352 247 85592 ; Fax : +352 247 95615. 7. MARKETING AUTHORISATION HOLDER: Amgen Europe B.V., Minervum 7061, 4817 ZK Breda, The Netherlands. Local representative: s.a. Amgen n.v., Telecomlaan 5-7, B-1831 Diegem, phone: 02/775.27.11. 8. MARKETING AUTHORISATION NUMBER(S): EU/1/14/981/001-002. Classification of the medicine: Medicinal product subject to medical prescription. Date of revision of the short SmPC: April 2020.
have
to respond to, or have a contraindication to or intolerance of, other systemic therapy, including cyclosporine, methotrexate or psoralene and ultraviolet A light (PUVA) and for the treatment of adult patients with oral ulcers associated with Behçet’s disease (BD) who are candidates for systemic therapy. #Otezla® alone or in combination with Disease Modifying Antirheumatic Drugs (DMARDs) is indicated for the treatment of active psoriatic arthritis (PsA) in adult patients who have failed to respond adequately to previous treatment with a DMARD or do not tolerate this treatment. 1. Otezla®SmPC, Summary of Product Characteristics, latest version. 2. http://riziv.be - last consultation 1st of February 2024. 3. Kavanaugh A, et al. Long-term experience with apremilast in patients with psoriatic arthritis: 5-year results from a PALACE
FRANÇOISE GUIOT
Dermatologue, Grez Doiceau
ÉDITORIAL LES BIOTHÉRAPIES
DEVIENNENT LE QUOTIDIEN
DES DERMATOLOGUES
Qu’il s’agisse du psoriasis, du mélanome, de la maladie de Verneuil, du vitiligo ou de l’urticaire chronique, l’arrivée massive des biothérapies occupent l’actualité en dermatologie depuis déjà quelques années. C’est en comprenant mieux la physiopathologie et l’évolution des pathologies inflammatoires cutanées, que ces traitements ciblés innovants ont pu être développés. A l’inverse, leur utilisation et l’analyse détaillée des résultats obtenus permettent en retour de mieux comprendre la physiopathologie. Un cercle vertueux qui amène de nouvelles solutions aux patients et améliore considérablement leur qualité de vie. La dermatite atopique et le psoriasis dominent bien évidemment les indications potentielles des biothérapies en dermatologie.
Lors de l’Académie européenne de dermatologie et de vénéréologie (EADV) qui s’est déroulée à Amsterdam, du 25 au 28 septembre 2024, de nombreux exposés ont été consacrés à ces nouveaux traitements porteurs d’espoir dans ces mêmes indications mais aussi à l’extension de ces mêmes traitements innovants à de nouvelles indications.
Ainsi par exemple, dans la dermatite atopique, le lébrikizumab, un anti-IL-13 administré par voie sous-cutanée, devrait arriver sur le marché en 2025. En effet, les données à trois ans des études ADvocate 1 et 2 ont été présentées et ont montré que la réponse à 52 semaines était maintenue à long terme chez les sujets de 12 ans et plus, que l’administration soit faite toutes les deux ou toutes les quatre semaines. Ces données permettent d’envisager une épargne thérapeutique, en envisageant de basculer vers un schéma mensuel lorsque les patients présentent une réponse satisfaisante prolongée sous schéma bimensuel. Ce médicament offre donc un nouveau traitement biologique de
première intention pour la dermatite atopique modérée à sévère mal contrôlée avec des traitements topiques et peut représenter une option supplémentaire utile dans la pratique clinique.
Le némolizumab en sous-cutané, un anti IL-31 développé dans le prurigo et qui a fait l’objet d’une étude spécifique dans le prurit associé à la dermatite atopique modérée à sévère, confirme son intérêt à 56 semaines, avec des scores d’efficacité supérieur à 75 % pour 80 % des patients inclus, âgés de 12 ans et plus. Les résultats décrivent, en outre, un renforcement de son efficacité avec le temps. L’une des dernières familles qui a émergé pour traiter la dermatite atopique concerne celles ciblant l’enzyme TYK2, une tyrosine kinase intracellulaire qui fait partie de la famille des JAK et qui est associée à la réponse de plusieurs cytokines inflammatoires impliquées dans le psoriasis, comme l’IL-12 ou l’interféron alpha.
Dans le psoriasis, le deucravacitinib est approuvé pour les adultes atteints de psoriasis en plaques modéré à sévère considérés comme candidats pour un traitement systémique. L’efficacité et la sécurité d’une étude de phase IIIb/IV, multicentrique, randomisée, en double aveugle et contrôlée contre placebo (L’étude PSORIATYK SCALP) a confirmé que le deucravacitinib était efficace et bien toléré dans le traitement du psoriasis du cuir chevelu modéré à sévère.
Actuellement d’autres pathologies semblent pouvoir bénéficier de thérapies ciblées. Ainsi, le baricitinib et le ritlécitinib ont été approuvés ces derniers mois dans la pelade et devraient être rejoints par d’autres molécules, comme le deuruxolitinib qui a montré son efficacité dans les études de phase III Thrive-AA1 et AA2. Dans le vitiligo, le ruxolitinib topique, récemment en-
« La pharmacogénétique peut contribuer à trouver des biomarqueurs permettant de prédire la réponse à des traitements spécifiques et ouvre la voie d’un traitement personnalisé pour nos patients souffrant de pathologies chroniques ».
registré, confirme son intérêt selon l’étude d’extension qui a été présentée et va voir d’autres anti-JAK lui succéder (povorcitinib, upadacitinib, baricitinib). Rappelons que les inhibiteurs de JAK topiques comprennent le delgocitinib, approuvé au Japon pour la dermatite atopique et actuellement testé en Europe pour l’eczéma chronique des mains, ainsi que le ruxolitinib, seul anti JAK topique disponible en Europe approuvé pour le vitiligo mais malheureusement non disponible en Belgique.
L’hidradénite suppurée est une maladie inflammatoire chronique dans laquelle l’interleukine 17A est impliquée. L’izokibep est une petite molécule protéique thérapeutique conçue pour inhiber sélectivement l’IL-17A. Dans une étude de phase III, cette molécule administrée par voie sous-cutanée montre une réponse supérieure à celle du placebo après 12 semaines. Un tiers des patients atteints d’hidradénite suppurée modérée à sévère ont atteint l’HiSCR75 à la semaine 12. L’izokibep a également apporté des améliorations précoces concernant des mesures clés de la maladie, y compris l’HiSCR90, l’HiSCR100, la douleur et la qualité de vie. Le ruxolitinib topique a aussi été décrit comme favorable dans les formes de la maladie légères à modérées, avec une diminution significative du nombre de nodules inflammatoires, mais des études de phase III sont attendues pour évaluer son efficacité sur les formes sévères.
La liste des indications des biothérapie ne cesse de s’allonger parmi les maladies dermatologiques et auto-immunes. Il est vrai que ces traitements ont pour cible les multiples « voies de passage » des cytokines avec la possibilité de moduler à la fois les réponses immunitaires innées et adaptatives. Dans un exposé, le Dr Wollenberg a rappelé qu’il est néanmoins très important de tenir compte des variations des profils de sécurité entre les différents traitements et prenant l’exemple des inhibiteurs de JAK, il a souligné l’importance d’une sélection minutieuse des patients et d’une évaluation individualisée des risques lors de la prescription de ces traitements. Les lignes directrices en matière de sécurité et de surveillance lors de la prescription des inhibiteurs de JAK, mettent l’accent sur les risques de MACE
(Major Adverse Cardiac Events) de thrombose et de cancer. En effet, si les inhibiteurs de JAK constituent une classe de médicaments rapidement efficaces et polyvalents, des problèmes de sécurité importants persistent chez les patients présentant des facteurs de risque particuliers. Face à ce dénombrement de molécules, il reste également à définir celle qui sera optimale pour son patient. La découverte de biomarqueurs permettant de prédire la réponse au traitement pourrait contribuer à prédire l’efficacité du traitement et à personnaliser la prise en charge. La pharmacogénétique est un domaine en plein essor qui cherche à définir ces liens. Les études s’intéressent particulièrement aux polymorphismes à nucléotide simple de gènes connus pour coder pour des protéines impliquées dans la pharmacodynamie et la pharmacocinétique de certains médicaments. Une revue de la littérature à partir des bases de données MEDLINE et Cochrane Library a mis en évidence de nombreux variants génétiques associées à la réponse au traitement chez les personnes atteintes de psoriasis. Parmi ces variants génétiques figurent le transporteur ABC, le DNMT3b, le MTHFR, l’ANKLE1, l’IL12B, l’IL-23R, le MALT1, le CDKAL1, l’IL17RA, l’IL1B, le LY96, et le TLR2. Des associations ont été trouvées pour divers médicaments utilisés dans la prise en charge du psoriasis, notamment le méthotrexate, la ciclosporine, l’acitrétine, les anti-TNF ou encore les agents anti-IL-12/23, anti-IL-17 et anti-PDE4. Dans cette étude, de nombreuses variants génétiques ont été associés à la réponse au traitement dans un certain nombre de classes de médicaments différents. La pharmacogénétique peut contribuer à trouver des biomarqueurs permettant de prédire la réponse à des traitements spécifiques et ouvre la voie d’un traitement personnalisé pour nos patients souffrant de pathologies chroniques. Un test permettant de trouver le médicament le plus efficace pour chaque patient pourrait contribuer à améliorer la qualité de vie ainsi qu’à réduire les coûts de santé et les effets indésirables.
RÉF :
33e congrès de l’Académie européenne de dermatologie et de vénéréologie (EADV), Amsterdam, du 25 au 28 septembre 2024.
45 mg vial or prefilled syringe or
130 mg vial
90 mg prefilled syringe packaging example, may di er per country
▼This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions. 1. NAME OF THE MEDICINAL PRODUCT: WEZENLA 45 mg solution for injection. WEZENLA 45 mg solution for injection in pre-filled syringe. WEZENLA 90 mg solution for injection in pre-filled syringe. WEZENLA 130 mg concentrate for solution for infusion. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION: WEZENLA 45 mg solution for injection: Each vial contains 45 mg ustekinumab in 0.5 mL. WEZENLA 45 mg solution for injection in pre-filled syringe: Each pre-filled syringe contains 45 mg ustekinumab in 0.5 mL. WEZENLA 90 mg solution for injection in pre-filled syringe: Each pre-filled syringe contains 90 mg ustekinumab in 1 mL. WEZENLA 130 mg concentrate for solution for infusion: Each vial contains 130 mg ustekinumab in 26 mL (5 mg/mL). Ustekinumab is a fully human IgG1κ monoclonal antibody to interleukin (IL)-12/23 produced in a Chinese hamster ovary cell line using recombinant DNA technology. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM: WEZENLA 45 mg solution for injection: Solution for injection. WEZENLA 45 mg solution for injection in pre-filled syringe: Solution for injection. WEZENLA 90 mg solution for injection in pre-filled syringe: Solution for injection. WEZENLA 130 mg concentrate for solution for infusion: Concentrate for solution for infusion. The solution is clear to opalescent, colourless to light yellow. The pH of the solution is 6.0. 4. CLINICAL PARTICULARS: 4.1 Therapeutic indications: Plaque psoriasis: WEZENLA is indicated for the treatment of moderate to severe plaque psoriasis in adults who failed to respond to, or who have a contraindication to, or are intolerant to other systemic therapies including ciclosporin, methotrexate (MTX) or PUVA (psoralen and ultraviolet A) (see section 5.1). Paediatric plaque psoriasis: WEZENLA is indicated for the treatment of moderate to severe plaque psoriasis in children and adolescent patients from the age of 6 years and older, who are inadequately controlled by, or are intolerant to, other systemic therapies or phototherapies (see section 5.1). Psoriatic arthritis (PsA): WEZENLA, alone or in combination with MTX, is indicated for the treatment of active psoriatic arthritis in adult patients when the response to previous non-biological diseasemodifying antirheumatic drug (DMARD) therapy has been inadequate (see section 5.1). Crohn’s disease: WEZENLA is indicated for the treatment of adult patients with moderately to severely active Crohn’s disease who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a TNFα antagonist or have medical contraindications to such therapies. 4.2 Posology and method of administration: WEZENLA is intended for use under the guidance and supervision of physicians experienced in the diagnosis and treatment of conditions for which WEZENLA is indicated. WEZENLA concentrate for solution for infusion should only be used for the intravenous induction dose in Crohn’s disease. Posology: Plaque psoriasis: The recommended posology of WEZENLA is an initial dose of 45 mg administered subcutaneously, followed by a 45 mg dose 4 weeks later, and then every 12 weeks thereafter. Consideration should be given to discontinuing treatment in patients who have shown no response up to 28 weeks of treatment. Patients with body weight > 100 kg: For patients with a body weight > 100 kg the initial dose is 90 mg administered subcutaneously, followed by a 90 mg dose 4 weeks later, and then every 12 weeks thereafter. In these patients, 45 mg was also shown to be efficacious. However, 90 mg resulted in greater efficacy (see section 5.1, table 4). Psoriatic arthritis (PsA): The recommended posology of WEZENLA is an initial dose of 45 mg administered subcutaneously, followed by a 45 mg dose 4 weeks later, and then every 12 weeks thereafter. Alternatively, 90 mg may be used in patients with a body weight > 100 kg. Consideration should be given to discontinuing treatment in patients who have shown no response up to 28 weeks of treatment. Elderly (≥ 65 years): No dose adjustment is needed for elderly patients (see section 4.4). Renal and hepatic impairment: Ustekinumab has not been studied in these patient populations. No dose recommendations can be made. Paediatric population: The safety and efficacy of ustekinumab in children with psoriasis less than 6 years of age or in children with psoriatic arthritis less than 18 years of age have not yet been established. Paediatric plaque psoriasis (6 years and older): The recommended dose of WEZENLA based on body weight is shown below (see tables 1 and 2). WEZENLA should be administered subcutaneously at weeks 0 and 4, then every 12 weeks thereafter. Table 1 Recommended dose of WEZENLA for paediatric psoriasis (body weight at the time of dosing and recommended dose): < 60 kg: 0.75 mg/kg; ≥ 60-≤ 100 kg: 45 mg; > 100 kg: 90 mg. To calculate the volume of injection (mL) for patients < 60 kg, use the following formula: body weight (kg) x 0.0083 (mL/kg) or see table 2. The calculated volume should be rounded to the nearest 0.01 mL and administered using a 1 mL graduated syringe. A 45 mg vial is available for paediatric patients who need to receive less than the full 45 mg dose. Table 2 Injection volumes of WEZENLA for paediatric psoriasis patients < 60 kg (body weight at time of dosing and dose in injection volume): 15 kg: 11.3 mg in 0.12 ml. 16 kg: 12.0 mg in 0.13 ml. 17 kg: 12.8 mg in 0.14 ml. 18 kg: 13.5 mg in 0.15 ml. 19 kg: 14.3 mg in 0.16 ml. 20 kg: 15.0 mg in 0.17 ml. 21 kg: 15.8 mg in 0.17 ml. 22 kg: 16.5 mg in 0.18 ml. 23 kg: 17.3 mg in 0.19 ml. 24 kg: 18.0 mg in 0.20 ml. 25 kg: 18.8 mg in 0.21 ml. 26 kg: 19.5 mg in 0.22 ml. 27 kg: 20.3 mg in 0.22 ml. 28 kg: 21.0 mg in 0.23 ml. 29 kg: 21.8 mg in 0.24 ml. 30 kg: 22.5 mg in 0.25 ml. 31 kg: 23.3 mg in 0.26 ml. 32 kg: 24.0 mg in 0.27 ml. 33 kg: 24.8 mg in 0.27 ml. 34 kg: 25.5 mg in 0.28 ml. 35 kg: 26.3 mg in 0.29 ml. 36 kg: 27.0 mg in 0.30 ml. 37 kg: 27.8 mg in 0.31 ml. 38 kg: 28.5 mg in 0.32 ml. 39 kg: 29.3 mg in 0.32 ml. 40 kg: 30.0 mg in 0.33 ml. 41 kg: 30.8 mg in 0.34 ml. 42 kg: 31.5 mg in 0.35 ml. 43 kg: 32.3 mg in 0.36 ml. 44 kg: 33.0 mg in 0.37 ml. 45 kg: 33.8 mg in 0.37 ml. 46 kg: 34.5 mg in 0.38 ml. 47 kg: 35.3 mg in 0.39 ml. 48 kg: 36.0 mg in 0.40 ml. 49 kg: 36.8 mg in 0.41 ml. 50 kg: 37.5 mg in 0.42 ml. 51 kg: 38.3 mg in 0.42 ml. 52 kg: 39.0 mg in 0.43 ml. 53 kg: 39.8 mg in 0.44 ml. 54 kg: 40.5 mg in 0.45 ml. 55 kg: 41.3 mg in 0.46 ml. 56 kg: 42.0 mg in 0.46 ml. 57 kg: 42.8 mg in 0.47 ml. 58 kg: 43.5 mg in 0.48 ml. 59 kg: 44.3 mg in 0.49 ml. Consideration should be given to discontinuing treatment in patients who have shown no response up to 28 weeks of treatment. Crohn’s disease: In the treatment regimen, the first dose of WEZENLA is administered intravenously. WEZENLA treatment is to be initiated with a single intravenous dose based on body weight. The infusion solution is to be composed of the number of vials of WEZENLA 130 mg as specified in table 1bis (see section 6.6 for preparation). Table 1bis Initial intravenous dosing of WEZENLA: Body weight of patient at the time of dosing ≤ 55 kg: recommended dosea of 260 mg, number of 130 mg WEZENLA vials: 2. Body weight of patient at the time of dosing > 55 kg to ≤ 85 kg: recommended dosea of 390 mg, number of 130 mg WEZENLA vials: 3. Body weight of patient at the time of dosing > 85 kg: recommended dosea of 520 mg, number of WEZENLA vials: 4. aApproximately 6 mg/kg. The first subcutaneous administration of 90 mg WEZENLA should take place at week 8 after the intravenous dose. After this, dosing every 12 weeks is recommended. Patients who have not shown adequate response at 8 weeks after the first subcutaneous dose, may receive a second subcutaneous dose at this time (see section 5.1). Patients who lose response on dosing every 12 weeks may benefit from an increase in dosing frequency to every 8 weeks (see sections 5.1 and 5.2). Patients may subsequently be dosed every 8 weeks or every 12 weeks according to clinical judgement (see section 5.1). Consideration should be given to discontinuing treatment in patients who show no evidence of therapeutic benefit 16 weeks after the IV induction dose or 16 weeks after switching to the 8weekly maintenance dose. Immunomodulators and/ or corticosteroids may be continued during treatment with ustekinumab. In patients who have responded to treatment with ustekinumab, corticosteroids may be reduced or discontinued in accordance with standard of care. In Crohn’s disease, if therapy is interrupted, resumption of treatment with subcutaneous dosing every 8 weeks is safe and effective. Elderly (≥ 65 years): No dose adjustment is needed for elderly patients (see section 4.4). Renal and hepatic impairment: Ustekinumab has not been studied in these patient populations. No dose recommendations can be made. Paediatric population: The safety and efficacy of ustekinumab in treatment of Crohn’s disease in children less than 18 years have not yet been established. No data are available. Method of administration: WEZENLA 45 mg vials or 45 mg and 90 mg pre-filled syringes are for subcutaneous injection only. If possible, areas of the skin that show psoriasis should be avoided as injection sites. After proper training in subcutaneous injection technique, patients or their caregivers may inject WEZENLA if a physician determines that it is appropriate. However, the physician should ensure appropriate follow-up of patients. Patients or their caregivers should be instructed to inject the prescribed amount of WEZENLA according to the directions provided in the package leaflet. Comprehensive instructions for administration are given in the package leaflet. For further instructions on preparation and special precautions for handling, see section 6.6. WEZENLA 130 mg is for intravenous use only. It should be administered over at least one hour. For instructions on dilution of the medicinal product before administration, see section 6.6. 4.3 Contraindications: Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. Clinically important, active infection (e.g. active tuberculosis; see section 4.4). 4.8 Undesirable effects: Summary of the safety profile: The most common adverse reactions (> 5%) in controlled periods of the adult psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis clinical studies with ustekinumab were nasopharyngitis and headache. Most were considered to be mild and did not necessitate discontinuation of study treatment. The most serious adverse reaction that has been reported for ustekinumab is serious hypersensitivity reactions including anaphylaxis (see section 4.4). The overall safety profile was similar for patients with psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis. Tabulated list of adverse reactions: The safety data described below reflect exposure in adults to ustekinumab in 14 phase 2 and phase 3 studies in 6 709 patients (4 135 with psoriasis and/or psoriatic arthritis, 1 749 with Crohn’s disease and 825 patients with ulcerative colitis). This includes exposure to WEZENLA in the controlled and non-controlled periods of the clinical studies for at least 6 months or 1 year (4 577 and 3 253 patients, respectively with psoriasis, psoriatic arthritis, Crohn’s disease or ulcerative colitis) and exposure for at least 4 or 5 years (1 482 and 838 patients with psoriasis, respectively). Table 3 provides a list of adverse reactions from adult psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis clinical studies as well as adverse reactions reported from post-marketing experience. The adverse reactions are classified by system organ class and frequency, using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1 000 to < 1/100), rare (≥ 1/10 000 to < 1/1 000), very rare (< 1/10 000), not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. Table 3 List of adverse reactions: Infections and infestations: Common: upper respiratory tract infection, nasopharyngitis, sinusitis; Uncommon: cellulitis, dental infections, herpes zoster, lower respiratory tract infection, viral upper respiratory tract infection, vulvovaginal mycotic infection. Immune system disorders: Uncommon: hypersensitivity reactions (including rash, urticaria); Rare: serious hypersensitivity reactions (including anaphylaxis, angioedema). Psychiatric disorders: Uncommon: depression. Nervous system disorders: Common: dizziness, headache; Uncommon: facial palsy. Respiratory, thoracic and mediastinal disorders: Common: oropharyngeal pain; Uncommon: nasal congestion; Rare: allergic alveolitis, eosinophilic pneumonia; Very rare: organising pneumonia*. Gastrointestinal disorders: Common: diarrhoea, nausea, vomiting. Skin and subcutaneous tissue disorders: Common: pruritus; Uncommon: pustular psoriasis, skin exfoliation, acne; Rare: exfoliative dermatitis, hypersensitivity vasculitis; Very rare: bullous pemphigoid, cutaneous lupus erythematosus. Musculoskeletal and connective tissue disorders: Common: back pain, myalgia, arthralgia; Very rare: lupus-like syndrome. general disorders and administration site conditions: Common: fatigue, injection site erythema, injection site pain; Uncommon: injection site reactions (including haemorrhage, haematoma, induration, swelling and pruritus), asthenia. * See section 4.4, Systemic and respiratory hypersensitivity reactions. Description of selected adverse reactions: Infections: In the placebo-controlled studies of patients with psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis, the rates of infection or serious infection were similar between ustekinumab-treated patients and those treated with placebo. In the placebo-controlled period of these clinical studies, the rate of infection was 1.36 per patient-year of follow-up in ustekinumab-treated patients, and 1.34 in placebo-treated patients. Serious infections occurred at the rate of 0.03 per patient-year of follow-up in ustekinumab-treated patients (30 serious infections in 930 patientyears of follow-up) and 0.03 in placebo-treated patients (15 serious infections in 434 patient-years of follow-up) (see section 4.4). In the controlled and non-controlled periods of psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis clinical studies, representing 11 581 patient-years of exposure in 6 709 patients, the median followup was 1.0 years; 1.1 years for psoriatic disease studies, 0.6 year for Crohn’s disease studies, and 1.0 years for ulcerative colitis studies. The rate of infection was 0.91 per patient-year of follow-up in ustekinumab-treated patients, and the rate of serious infections was 0.02 per patient-year of follow-up in ustekinumab-treated patients (199 serious infections in 11 581 patient-years of followup) and serious infections reported included pneumonia, anal abscess, cellulitis, diverticulitis, gastroenteritis and viral infections. In clinical studies, patients with latent tuberculosis who were concurrently treated with isoniazid did not develop tuberculosis. Malignancies: In the placebo-controlled period of the psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis clinical studies, the incidence of malignancies excluding non-melanoma skin cancer was 0.11 per 100 patient-years of follow-up for ustekinumab-treated patients (1 patient in 929 patientyears of follow-up) compared with 0.23 for placebo-treated patients (1 patient in 434 patient-years of followup). The incidence of non-melanoma skin cancer was 0.43 per 100 patient-years of follow-up for ustekinumab-treated patients (4 patients in 929 patient-years of follow-up) compared to 0.46 for placebo-treated patients (2 patients in 433 patient-years of follow-up). In the controlled and non-controlled periods of psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis clinical studies, representing 11 561 patient-years of exposure in 6 709 patients, the median followup was 1.0 years; 1.1 years for psoriatic disease studies, 0.6 year for Crohn’s disease studies and 1.0 years for ulcerative colitis studies. Malignancies excluding non-melanoma skin cancers were reported in 62 patients in 11 561 patient-years of follow-up (incidence of 0.54 per 100 patient- years of follow-up for ustekinumab-treated patients). The incidence of malignancies reported in ustekinumab-treated patients was comparable to the incidence expected in the general population (standardised incidence ratio = 0.93 [95% confidence interval: 0.71, 1.20], adjusted for age, gender and race). The most frequently observed malignancies, other than non-melanoma skin cancer, were prostate, colorectal, melanoma and breast cancers. The incidence of non-melanoma skin cancer was 0.49 per 100 patient-years of follow-up for ustekinumab-treated patients (56 patients in 11 545 patientyears of follow-up). The ratio of patients with basal versus squamous cell skin cancers (3:1) is comparable with the ratio expected in the general population (see section 4.4). Hypersensitivity and infusion reactions: During the controlled periods of the psoriasis and psoriatic arthritis clinical studies of ustekinumab, rash and urticaria have each been observed in < 1% of patients (see section 4.4). In Crohn’s disease and ulcerative colitis intravenous induction studies, no events of anaphylaxis or other serious infusion reactions were reported following the single intravenous dose. In these studies, 2.2% of 785 placebo-treated patients and 1.9% of 790 patients treated with the recommended dose of ustekinumab reported adverse events occurring during or within an hour of the infusion. Serious infusion-related reactions including anaphylactic reactions to the infusion have been reported in the post-marketing setting (see section 4.4). Paediatric population: Paediatric patients 6 years and older with plaque psoriasis: The safety of ustekinumab has been studied in two phase 3 studies of paediatric patients with moderate to severe plaque psoriasis. The first study was in 110 patients from 12 to 17 years of age treated for up to 60 weeks and the second study was in 44 patients from 6 to 11 years of age treated for up to 56 weeks. In general, the adverse events reported in these two studies with safety data up to 1 year were similar to those seen in previous studies in adults with plaque psoriasis. Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system: Belgium: Federal agency for medicines and health products; www.famhp.be; Vigilance Division; Website: www.notifieruneffetindesirable.be; email: adr@fagg-afmps.be Luxembourg: Centre Régional de Pharmacovigilance de Nancy or Division de la pharmacie et des médicaments de la Direction de la santé; Website: www.guichet.lu/pharmacovigilance 7. MARKETING AUTHORISATION HOLDER: Amgen Technology (Ireland) UC, Pottery Road, Dun Laoghaire, Co Dublin, Ireland. Local representative: s.a. Amgen n.v., Telecomlaan 5-7, B-1831 Diegem; phone: 02/775.27.11. 8. MARKETING AUTHORISATION NUMBER(S): WEZENLA 45 mg solution for injection: EU/1/24/1823/001. WEZENLA 45 mg solution for injection in pre-filled syringe: EU/1/24/1823/002. WEZENLA 90 mg solution for injection in pre-filled syringe: EU/1/24/1823/003. WEZENLA 130 mg concentrate for solution for infusion: EU/1/24/1823/004. Classification of the medicine: Medicinal product subject to medical prescription. Consult the price details on www.e-compendium.be Date of revision of the short SPC: June 2024. BEL-654-1024-80004 – V1.0 – creation date 29 Oct 2024
SOMMAIRE
Les biothérapies deviennent le quotidien des dermatologues
Françoise GUIOT
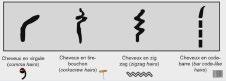
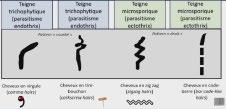
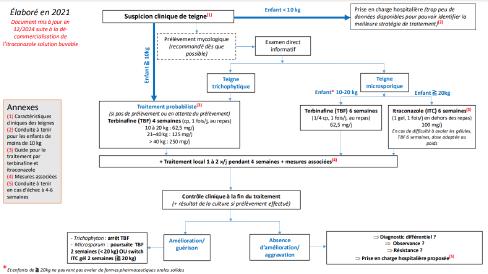
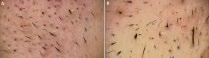
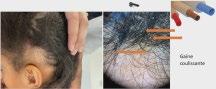
L’alopécie chez l’enfant ne sea plus en myst « hair » pour vous : Pas à pas diagnostique et thérapeutique – partie1
Sophie LEDUCQ, Stéphanie MALLET, Juliette MAZEREEUW- HAUTIER, Fanny MORICE-PICARD
Immunosuppression
Comment préparer mon patient à l’immunosuppression/modulation en dermatologie ?
Nicolas DAUBY
Dermatite atopique
Dermatite atopique et affections associées
Dr Hannah DE LEYE, Julie SAERENS, Pauline LUTTGENS, Liesl MATTON, Prof Jan GUTERMUTH
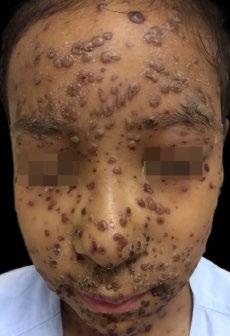
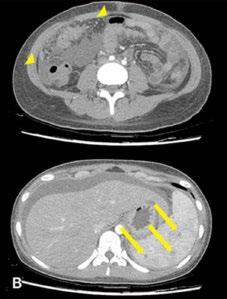
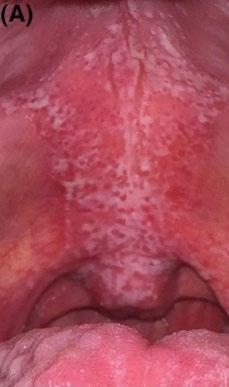
Cas clinique
La connexion suédoise
Stefan KERRE
SOMMAIRE
Cas clinique
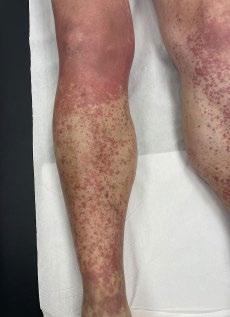
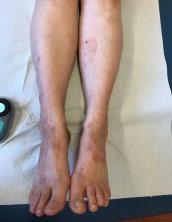
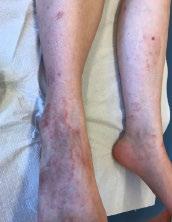
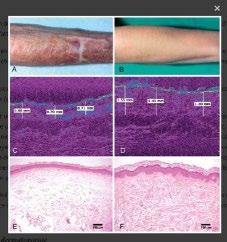
Cas clinique de porpora téléangiectasique de majocchi et approfondissement des dermatoses purpuriques pigmentées
Les articles signés, publiés dans Dermatologie Actualité sont écrits sous la responsabilité de leurs auteurs. En aucun cas le journal ne peut être tenu responsable du contenu rédactionnel.
Aucun article publié dans Dermatologie Actualité ne peut être reproduit en tout ou en partie sans l’autorisation de l’éditeur.
DÉNOMINATION DU MÉDICAMENT: Tremfya 100 mg solution injectable en seringue préremplie. Tremfya 100 mg solution injectable en stylo prérempli. COMPOSITION QUALITATIVE ET
QUANTITATIVE: Tremfya 100 mg solution injectable en seringue préremplie: Chaque seringue préremplie contient 100 mg de guselkumab dans 1 ml de solution. Tremfya 100 mg solution injectable en stylo prérempli: Chaque stylo prérempli contient 100 mg de guselkumab dans 1 ml de solution. Le guselkumab est un anticorps monoclonal (AcMo) entièrement humain, de type immunoglobuline G1 lambda (IgG1λ) produit par des cellules ovariennes de hamster chinois (CHO) par la technologie de l’ADN recombinant. FORME PHARMACEUTIQUE: Solution injectable. La solution est limpide et incolore à jaune clair. Indications thérapeutiques: Psoriasis en plaques: Tremfya est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez l’adulte qui nécessite un traitement systémique. Rhumatisme psoriasique: Tremfya, seul ou en association avec le méthotrexate (MTX), est indiqué dans le traitement du rhumatisme psoriasique actif chez les patients adultes ayant présenté une réponse inadéquate ou une intolérance à un traitement de fond antirhumatismal (DMARD) antérieur. Posologie et mode d’administration: Ce médicament est destiné à être utilisé sous la conduite et la surveillance d’un médecin expérimenté dans le diagnostic et le traitement des pathologies pour lesquelles il est indiqué. Posologie: Psoriasis en plaques: La dose recommandée est de 100 mg en injection sous-cutanée aux semaines 0 et 4, suivie d’une dose d’entretien toutes les 8 semaines. L’arrêt du traitement doit être envisagé chez les patients ne présentant pas de réponse au bout de 16 semaines de traitement. Rhumatisme psoriasique: La dose recommandée est de 100 mg en injection sous-cutanée aux semaines 0 et 4, suivie d’une dose d’entretien toutes les 8 semaines. Pour les patients présentant un risque élevé de lésion articulaire selon l’avis clinique, une dose de 100 mg toutes les 4 semaines peut être envisagée. L’arrêt du traitement doit être envisagé chez les patients ne présentant pas de réponse au bout de 24 semaines de traitement. Populations particulières: Personnes âgées (≥ 65 ans): Aucun ajustement posologique n’est nécessaire. Les données chez les sujets âgés de 65 ans et plus sont limitées, et elles sont très limitées chez les sujets âgés de 75 ans et plus. Insuffisance rénale ou hépatique: Tremfya n’a pas été étudié chez ces populations de patients. Aucune recommandation posologique ne peut être faite. Pour plus d’informations sur l’élimination du guselkumab, voir résumé des caractéristiques du produit. Population pédiatrique: La sécurité et l’efficacité de Tremfya chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible. Mode d’administration: Voie sous-cutanée. Dans la mesure du possible, les sites où la peau présente du psoriasis ne doivent pas être utilisés comme sites d’injection. Après une formation adaptée à la technique d’injection sous-cutanée, les patients peuvent s’injecter Tremfya si le médecin estime cela approprié. Cependant, le médecin doit assurer un suivi médical adéquat des patients. Les patients doivent être informés de la nécessité d’injecter la dose complète de solution conformément aux « Instructions d’utilisation » fournies dans la boîte. Pour les instructions concernant la préparation du médicament avant administration, voir résumé des caractéristiques du produit. Contre-indications: Hypersensibilité grave à la substance active ou à l’un des excipients. Infection active et cliniquement importante (par exemple tuberculose active). Effets indésirables: Résumé du profil de sécurité: L’effet indésirable le plus fréquent était les infections des voies respiratoires chez environ 14 % des patients dans les études cliniques sur le psoriasis et le rhumatisme psoriasique. Tableau récapitulatif des effets indésirables: Le tableau 1 fournit une liste des effets indésirables observés dans les études cliniques sur le psoriasis et le rhumatisme psoriasique, ainsi que depuis la mise sur le marché du produit. Les effets indésirables sont présentés par classe de système d’organes MedDRA et par fréquence, selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Tableau 1 : Liste des effets indésirables: Classe de système d’organes : Fréquence : Effets indésirables. Infections et infestations Très fréquent : Infections des voies respiratoires. Peu fréquent : Infections à Herpes simplex. Dermatophytoses. Gastro-entérite. Affections du système immunitaire : Peu fréquent : Hypersensibilité. Anaphylaxie. Affections du système nerveux : Fréquent : Céphalée. Affections gastro-intestinales : Fréquent : Diarrhée. Affections de la peau et du tissus sous-cutané : Peu fréquent : Urticaire. Rash. Affections musculo-squelettiques et systémiques Fréquent : Arthralgie. Troubles généraux et anomalies au site d’administration : Fréquent : Réactions au site d’injection. Investigations : Fréquent : Transaminases augmentées. Peu fréquent : Neutrophiles diminués. Description de certains effets indésirables : Transaminases augmentées: Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le rhumatisme psoriasique, les événements indésirables de type augmentation des transaminases (comprenant ALAT augmentée, ASAT augmentée, enzymes hépatiques augmentées, transamina ses augmentées, test hépatique anormal, hypertransaminasémie) ont été rapportés plus fréquemment dans les groupes traité par guselkumab (8,6 % dans le groupe toutes les 4 semaines et 8,3 % dans le groupe toutes les 8 semaines) que dans le groupe placebo (4,6 %). En un an, les événements indésirables de type augmentation des transaminases (ci-dessus) ont été rapportés chez 12,9 % des patients dans le groupe toutes les 4 semaines et 11,7 % des patients dans le groupe toutes les 8 semaines. Sur la base des analyses biologiques, la plupart des augmentations des transaminases (ALAT et ASAT) étaient ≤ 3 x la limite supérieure de la normale (LSN). Les augmentations des transaminases situées entre > 3 et ≤ 5 x LSN et > 5 x LSN étaient peu fréquentes, survenant plus souvent dans le groupe guselkumab toutes les 4 semaines que dans le groupe guselkumab toutes les 8 semaines (tableau 2). Une fréquence similaire a été observée quels que soit la sévérité et le bras de traitement à la fin de l’étude clinique de phase III de 2 ans sur le rhumatisme psoriasique. Tableau 2 Fréquence de patients présentant une augmentation des transaminases post-inclusion dans les études cliniques de phase III sur le rhumatisme psoriasique. A: Jusqu’à la semaine 24a : A1 : Placebo N : 370c – A2 : guselkumab 100 mg toutes les 8 semaines N = 373c – A3 : guselkumab 100 mg toutes les 4 semaines N = 371c – B: Jusqu’à 1 anb : B1 : guselkumab 100 mg toutes les 8 semaines N = 373c – B2 guselkumab 100 mg toutes les 4 semaines N = 371c ALAT : > 1 à ≤3 x LSN A1 : 30,0% ; A2 28,2% ; A3 : 35,0% ; B1 : 33,5% ; B2 41,2%. > 3 à ≤5 x LSN A1 : 1,4% A2 : 1,1% ; A3 : 2,7% ; B1 : 1,6% B2 : 4,6%. > 5 x LSN A1 : 0,8% A2 : 0,8% ; A3 1,1% ; B1 : 1,1% ; B2 : 1,1%. ASAT : > 1 à ≤3 x LSN A1 : 20,0% ; A2 : 18,8% ; A3 : 21,6% ; B1 : 22,8% B2 : 27,8%. > 3 à ≤5 x LSN A1 : 0,5% ; A2 : 1,6% A3 : 1,6% ; B1 2,9% ; B2 : 3,8%. > 5 x LSN A1 : 1,1% ; A2 0,5% ; A3 : 1,6% B1 : 0,5% ; B2 1,6%. a période contrôlée versus placebo. b les patients randomisés sous placebo à l’inclusion puis traités par guselkumab ne sont pas pris en compte. c nombre de patients ayant fait l’objet d’au moins une évaluation post-inclusion pour l’analyse spécifique au cours de la période. Dans les études cliniques sur le psoriasis, avec une dose de guselkumab toutes les 8 semaines, la fréquence des augmentations des transaminases (ALAT et
Remboursé pour le psoriasis et le rhumatisme psoriasique
Treat
Peau sans plaques 1
Early with Tremfya
Effet sur les symptômes articulaires 2
Tremfya est le premier et le seul inhibiteur de l’IL-23 à être administré manuellement par le biais d’un stylo injecteur, permettant aux patients de contrôler eux-mêmes la vitesse d’administration.4
ASAT), évaluée sur une période d’un an, a été similaire à celle observée dans les études cliniques sur le rhumatisme psoriasique avec une dose de guselkumab toutes les 8 semaines. Sur une période de 5 ans, l’incidence de l’augmentation des transaminases n’a pas augmenté par année de traitement sous guselkumab. La plupart des augmentations de transaminase étaient ≤ 3 x LSN. Dans la plupart des cas, l’augmentation des transaminases était transitoire et n’a pas entraîné l’arrêt du traitement. Diminution du nombre de neutrophiles: Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le rhumatisme psoriasique, l’événement indésirable de type diminution du nombre de neutrophiles a été rapporté plus fréquemment dans le groupe traité par guselkumab (0,9 %) que dans le groupe placebo (0 %). En un an, l’événement indésirable de type diminution du nombre de neutrophiles a été rapporté chez 0,9 % des patients traités par guselkumab. Dans la plupart des cas, la diminution du nombre de neutrophiles sanguins a été légère, transitoire, non associée à une infection et n’a pas entraîné d’arrêt du traitement. Gastro-entérite: Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le psoriasis, des gastro-entérites sont survenues plus fréquemment dans le groupe traité par guselkumab (1,1 %) que dans le groupe placebo (0,7 %). Jusqu’à la semaine 264, 5,8 % de tous les patients traités par guselkumab ont rapporté une gastro-entérite. Ces gastro-entérites étaient non graves et n’ont pas conduit à l’arrêt du traitement par guselkumab jusqu’à la semaine 264. Les taux de gastro-entérite observés pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique étaient similaires à ceux observés dans les études cliniques sur le psoriasis. Réactions au site d’injection: Lors de deux études cliniques de Phase III sur le psoriasis, 0,7 % des injections de guselkumab et 0,3 % des injections de placebo ont été associées à des réactions au site d’injection jusqu’à la semaine 48. Jusqu’à la semaine 264, 0,4 % des injections de guselkumab ont été associées à des réactions au site d’injection. Ces réactions au site d’injection étaient généralement de sévérité légère à modérée ; aucune n’était grave, et une seule a conduit à l’arrêt du traitement par guselkumab. Lors de deux études cliniques de Phase III sur le rhumatisme psoriasique jusqu’à la semaine 24, le nombre de patients pour lesquels une ou plusieurs réactions au site d’injection ont été rapportées était faible et légèrement plus élevé dans les groupes guselkumab que dans le groupe placebo ; 5 patients (1,3 %) dans le groupe guselkumab toutes les 8 semaines, 4 patients (1,1 %) dans le groupe guselkumab toutes les 4 semaines et 1 patient (0,3 %) dans le groupe placebo. Un patient a arrêté le guselkumab en raison d’une réaction au site d’injection pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique. En un an, la proportion de patients ayant présenté 1 réaction au site d’injection ou plus était de 1,6 % et de 2,4 % dans les groupes guselkumab toutes les 8 semaines et toutes les 4 semaines, respectivement. Dans l’ensemble, le taux d’injections associées à des réactions au site d’injection observé pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique était similaire aux taux observés dans les études cliniques sur le psoriasis. Immunogénicité: L’immunogénicité du guselkumab a été évaluée à l’aide d’une méthode sensible de dosage immunologique, tolérante au biomédicament. D’après les analyses des études poolées de Phase II et de Phase III menées auprès de patients atteints de psoriasis et de rhumatisme psoriasique, 5 % (n = 145) des patients traités par guselkumab ont développé des anticorps anti-médicament sur une durée de traitement allant jusqu’à 52 semaines. Parmi les patients ayant développé des anticorps antimédicament, environ 8 % (n = 12) présentaient des anticorps catégorisés comme neutralisants, soit 0,4 % de l’ensemble des patients traités par guselkumab. Dans les analyses poolées de Phase III, parmi les patients atteints de psoriasis, environ 15 % des patients traités par guselkumab ont développé des anticorps anti-médicament sur une durée de traitement allant jusqu’à 264 semaines. Parmi les patients ayant développé des anticorps anti-médicament, environ 5 % présentaient des anticorps qualifiés de neutralisants, soit 0,76 % de l’ensemble des patients traités par guselkumab. La présence d’anticorps anti-médicament n’a pas été associée à une réduction de l’efficacité ou à la survenue de réactions au site d’injection. Déclaration des effets indésirables suspectés: La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/ risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via : Belgique : Agence fédérale des médicaments et des produits de santé - Division Vigilance, Boîte Postale 97, 1000 BRUXELLES Madou. Site internet : www.notifieruneffetindesirable.be, e-mail : adr@afmps.be Luxembourg : Centre Régional de Pharmacovigilance de Nancy, Bâtiment de Biologie Moléculaire et de Biopathologie (BBB), CHRU de Nancy – Hôpitaux de Brabois, Rue du Morvan, 54 511 VANDOEUVRE LES NANCY CEDEX, tél (+33) 3 83 65 60 85 / 87, e-mail crpv@chru-nancy.fr. Ou Direction de la Santé, Division de la Pharmacie et des Médicaments, 20, rue de Bitbourg, L-1273 Luxembourg-Hamm, tél. : (+352) 2478 5592, e-mail pharmacovigilance@ms.etat.lu. Link pour le formulaire https:// guichet.public.lu/fr/entreprises/sectoriel/sante/medecins/notification-effets-indesirables-medicaments.html TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ : Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgique. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ EU/1/17/1234/001 1 seringue préremplie EU/1/17/1234/002 1 stylo prérempli. EU/1/17/1234/003 2 stylos préremplis. EU/1/17/1234/004 2 seringues préremplies. MODE DE DELIVRANCE : Médicament soumis à prescription médicale. DATE DE LA DERNIERE APPROBATION DU TEXTE : 15/07/2022. Toute information complémentaire peut être obtenue sur demande. Téléphone : 0800 93 377 (BE) – 800 29 504 (LUX) • E-mail: janssen@jacbe.jnj.com • Internet: www.janssen.com/belgium
Tolérance prouvée1-3
Tremfya® (guselkumab) est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les adultes éligibles à un traitement systémique 3
Tremfya, seul ou en association avec le méthotrexate, est indiqué dans le traitement du rhumatisme psoriasique actif chez les patients adultes ayant présenté une réponse inadéquate à un traitement antérieur par un antirhumatismal modi cateur de la maladie (ARMM) ou qui ne tolèrent pas ce traitement 3
1. Reich K et al, Br J Dermatol 2021; 185(6):1146-1159
2. McInnes IB et al, Arthritis & Rheumatology 2022; 74:475-485
3. Tremfya SmPC 2022
4. Ferris LK, et al. J Dermatolog Treat 2020;31:152–159.
Le fabricant de la spécialité Amgevita® a procédé au remplacement des dosages disponibles. Les nouveaux dosages sont deux fois plus concentrés que les anciens. Il est important d’informer les patients puisque ce médicament peut être auto-administré (injection sous-cutanée). Ces nouveaux dosages correspondent aux dosages déjà existants des autres spécialités à base d’adalimumab.
L’adalimumab est utilisé dans de nombreuses pathologies inflammatoires telles certaines formes d’arthrites, psoriasis, hidrosadénite suppurée, maladie de Crohn, colite ulcéreuse ou uvéite.
Réf. : www.cbip.be
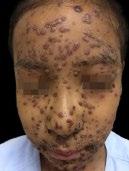
Évaluation du taux de benzène et du risque associé dans les produits formulés avec du peroxyde de benzoyle
Une équipe étasunienne a étudié la potentielle carcinogénicité du peroxyde de benzoyle (POB). En effet, le benzène, carcinogène connu et puissant est un produit de dégradation du POB. Du benzène se forme lorsque des produits contenant du POB sont incubés à la température du corps et plus généralement aux températures élevées comme lors du transport ou du stockage. Les auteurs de ce travail ont examiné en chromatographie gazeuse couplée à une spectrométrie de masse (CG-MS) 111 produits en vente libre contenant du peroxyde de benzoyle, conservés à température ambiante. Les concentrations de benzène mesurées allaient de 0,16 ppm à 33,30 ppm. Des études de stabilité à froid (2°C) et à température très élevée (50°C) sur un produit à base de PBO sur ordonnance montrent qu’il n’y a pas de formation de benzène à 2°C mais qu’il y en a à 50 °C. Ce qui est en faveur d’un stockage à froid des produits renfermant du POB. La micronisation ou l’encapsulation ne semblent pas réduire le risque de formation de benzène. En cas d’exposition aux UV (y compris à des niveaux moindres que l’ensoleillement maximal) après application d’un produit à base de POB, il y a aussi formation de benzène. Le risque lié à ces produits médicamenteux n’a pas été souligné dans la littérature avant 2024. Or, selon la FDA, il n’y a pas de niveau d’exposition au benzène sans risque on ne peut permettre une telle exposition que pour les produits pharmaceutiques qui constituent un progrès thérapeutique significatif, en limitant alors les concentrations de benzène à 2 ppm. Des études épidémiologiques ont rapporté une association entre cancer et exposition persistante au benzène à un niveau aussi bas que 0,8 ppm. Les résultats de cette étude suggèrent que l’utilisation de nombreux produits à base de peroxyde de benzoyle proposés dans le traitement de l’acné pourrait comporter des risques liés à l’exposition au benzène d’autant plus que ces produits sont employés le plus souvent sur de longues périodes.
Réf. : Kucera K, Zenzola N, Hudspeth A, et al. Evaluation of Benzene Presence and Formation in Benzoyl Peroxide Drug Products. J Invest Dermatol. 2024 Sep 27:S0022-202X(24)02155-9. doi: 10.1016/j. jid.2024.09.009.
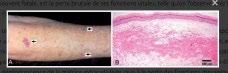
Dermatite atopique légère à modérée : agir sur le microbiote cutané
Une étude préclinique belge menée sur des patients atteints de dermatite atopique non grave met en avant une diversité du microbiote cutané jamais étudiée jusque-là. Dans cette étude, Lize Delanghe et coll. (Anvers) ont cherché à caractériser le microbiote cutané d’une cohorte belge de 38 patients atteints de DA en les comparant à 49 sujets sains, et ce, afin d’identifier les biomarqueurs du microbiote cutané, en particulier pour la DA légère, forme actuellement sous-explorée dans les études. Les patients ont été recrutés lors de consultations de suivi standard à l’hôpital universitaire d’Anvers. Ont été inclus des patients atteints de DA légère (sévérité déterminée selon l’échelle EASI n = 28) ou de lésions modérées à sévères (n = 6) sur n’importe quel site du corps. Ils ont été regroupés en différentes tranches d’âge : 0-3 ans, 3-6 ans, 6-12 ans, 12-18 ans et 18 ans et plus. Les auteurs ont constaté que la diversité globale du microbiote cutané est corrélée au degré de sévérité de la lésion de DA et à l’âge du patient. Cette analyse a indiqué que le nombre d’espèces sur la peau était similaire pour les patients atteints de DA légère à modérée et les témoins sains. Seul l’âge (inférieur ou supérieur à 12 ans) influait sur la diversité des espèces. Et contrairement à la DA grave, dans les formes moins prononcées il n’y a pas de S. aureus en cause. Lize Delanghe et coll. soulignent aussi la diversité des espèces retrouvées, puisque 11 espèces communes aux patients analysés et aux témoins ont été individualisées :la classe Actinomycetia (32 %), et les genres Acinetobacter (3,5 %), Corynebacterium (2,7 %),Cutibacterium (13,5 %), Micrococcus (0,9 %), Moraxella (1,9 %), Mycobacterium (1,9%), Paracoccus (1,1 %), Staphylococcus (19,4 %), Streptococcus (5,3 %),Streptomyces (3,6 %). L’analyse a mis en évidence que les genres Mycobacterium (p = 0,0012) et Streptomyces (p= 2,2 × 10-5) sont une cible thérapeutique possible, car leur abondance relative est majorée de 2,2 à 2,9 fois respectivement dans la DA légère par rapport aux témoins sains. Dans les cas modérés à sévères, ces genres étaient également significativement augmentés de respectivement 2,2 ( p = 0,0097) et 4,2 fois ( p = 2,5 × 10-4) Les présences de Moraxella, de Micrococcus, d’Acinetobacter et de Paracoccus étaient significativement diminuées dans les deux groupes de la DA par rapport aux échantillons sains. De plus, Corynebacterium (p = 0,04) et Cutibacterium (p = 0,034) étaient significativement diminués, en particulier dans les échantillons de patients modérés à sévères par rapport aux échantillons sains. Reste maintenant à imaginer des stratégies thérapeutiques permettant de cibler le microbiote cutané en prenant en compte le degré de gravité de la maladie.
Réf. : Mild atopic dermatitis is characterized by increase in non-staphylococcus pathobionts and loss of specific species. Scientific reports - ISSN 2045-2322-14:1 (2024) p. 1-10. Lize Delanghe, Ilke De Boeck, Joke Van Malderen, Camille Allonsius, Tim Van Rillaer, Peter A. Bron, Ingmar Claes, Margo Hagendorens, Sarah Lebeer, Julie Leysen
EBGLYSS® (lebrikizumab), remboursé le 1er janvier en dermatite atopique
Cher(e) Docteur,
Nous avons le plaisir de vous informer du remboursement d’ EBGLYSS® (lebrikizumab) pour le traitement de la dermatite atopique modérée à sévère chez les adultes et les adolescents âgés de 12 ans et plus pesant au moins 40 kg et qui sont éligibles pour un traitement systémique.
Précision1-5,10 : EBGLYSS® est un anticorps monoclonal qui se lie avec une haute affinité à l’IL-13, inhibant sélectivement sa signalisation en empêchant la formation du complexe IL-13Rα1/IL-4Rα
Contrôle6-10 : EBGLYSS® a démontré une efficacité clinique précoce et un maintien de la réponse tant en monothérapie qu’en association avec des corticostéroïdes topiques. Près de 90% des répondeurs* à la semaine 16 ont maintenu une réponse EASI-90 à 3 ans avec une injection toutes les 4 semaines.
Les effets indésirables les plus fréquents sont des conjonctivites (6,9 %), des réactions au site d’injection (2,6 %), des conjonctivites allergiques (1,8 %) et une sécheresse oculaire (1,4 %). Le profil de sécurité observé à 3 ans est consistent avec le profil de sécurité précédemment rapporté, sans nouveau signal de sécurité.
Dosage unique10 :
La dose recommandée est de 2 injections à la semaine 0 et à la semaine 2, suivie d’une injection toutes les deux semaines jusqu’à la semaine 16. La dose d’entretien** est ensuite d’une injection d’EBGLYSS® toutes les quatre semaines.
EBGLYSS® est disponible en stylo pré-rempli et en seringue pré-remplie, en boîtes de 2 et en boîtes de 3 unités.
Pour toute question ou pour convenir d’un rendez-vous afin d’obtenir de plus amples informations concernant EBGLYSS® et ses conditions de remboursement, n’hésitez pas à nous contacter.
Nous sommes à votre disposition pour vous fournir toutes les informations nécessaires.
Également disponible en seringe préremplie (250 mg)
Dosage pour adultes et adolescents ≥12 ans avec un poids corporel ≥ 40 kg
Précision
Inhibe la signalisation IL-13 avec précision et haute affinité1-5
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT EBGLYSS®
Contrôle
Efficacité élevée à 16 semaines, maintenue jusqu’à 3 ans* avec un profil de sécurité favorable6-10
2 x EBGLYSS® 250mg seringues/stylos préremplis € 2,009.44
3 x EBGLYSS® 250mg seringues/stylos préremplis € 3,008.87
Toutes les 4 semaines en phase de maintenance10 Prix public (TVAC)
Scannez pour plus d’infos sur EBGLYSS®
Dosage unique
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. DÉNOMINATION DU MÉDICAMENT : Ebglyss 250 mg, solution injectable en seringue préremplie. Ebglyss 250 mg, solution injectable en stylo prérempli. COMPOSITION QUALITATIVE ET QUANTITATIVE : Chaque seringue/stylo préremplie à usage unique contient 250 mg de lébrikizumab dans 2 mL de solution (125 mg/mL). FORME PHARMACEUTIQUE : Solution injectable. CLASSE PHARMACOTHÉRAPEUTIQUE : Autres préparations dermatologiques, traitements de la dermatite, à l’exclusion des corticostéroïdes. INDICATIONS THÉRAPEUTIQUES : Ebglyss est indiqué dans le traitement de la dermatite atopique modérée à sévère de l’adulte et de l’adolescent âgé de 12 ans et plus, pesant au moins 40 kg, qui nécessitent un traitement systémique. POSOLOGIE ET MODE D’ADMINISTRATION : Le traitement doit être initié par des professionnels de santé expérimentés dans le diagnostic et le traitement de la dermatite atopique. Posologie : La dose recommandée de lébrikizumab est de 500 mg (deux injections de 250 mg) à la semaine 0 et à la semaine 2, suivie de 250 mg administrés par injection sous-cutanée toutes les deux semaines jusqu’à la semaine 16. L’arrêt du traitement devra être envisagé chez les patients qui ne présentent aucune réponse clinique après 16 semaines de traitement. Certains patients présentant initialement une réponse partielle peuvent bénéficier d’une amélioration de la réponse en poursuivant le traitement toutes les deux semaines jusqu’à la semaine 24. Une fois la réponse clinique obtenue, la dose d’entretien de lébrikizumab recommandée est de 250 mg toutes les quatre semaines. Le lébrikizumab peut être utilisé avec ou sans dermocorticoïdes (DC). Les inhibiteurs de la calcineurine topiques (ICT) peuvent être utilisés, mais ils doivent être limités aux zones sensibles, telles que le visage, le cou, les zones intertrigineuses et les parties génitales. Dose manquée : En cas d’oubli d’une dose, celle-ci devra être administrée dès que possible. Par la suite, le schéma d’administration devra être repris à la date prévue habituellement. Populations particulières : Personnes âgées (≥ 65 ans) : Aucun ajustement posologique n’est recommandé chez les patients âgés (voir RCP). Insuffisance rénale ou hépatique : Aucun ajustement posologique n’est recommandé chez les patients présentant une insuffisance rénale ou hépatique (voir RCP). Poids corporel : Aucun ajustement posologique n’est recommandé en fonction du poids corporel (voir RCP). Population pédiatrique : La sécurité et l’efficacité du lébrikizumab chez les enfants âgés de 6 mois à moins de 12 ans ou chez les adolescents âgés de 12 à 17 ans et pesant moins de 40 kg n’ont pas encore été établies. Aucune donnée n’est disponible. Mode d’administration : Voie sous-cutanée. Le lébrikizumab est administré par injection sous-cutanée dans la cuisse ou l’abdomen, à l’exception de la zone de 5 centimètres autour du nombril. L’injection peut également être effectuée dans le haut du bras si elle n’est pas réalisée par le patient lui-même. Pour la dose initiale de 500 mg, deux injections de 250 mg doivent être réalisées consécutivement en choisissant des sites d’injection différents. Il est recommandé de changer de site d’injection à chaque injection. Le lébrikizumab ne doit pas être injecté dans les zones où la peau est sensible, endommagée ou présente des hématomes ou des cicatrices. Un patient peut s’auto-injecter le lébrikizumab ou un soignant peut le lui administrer si le professionnel de santé juge que cela est approprié. Une formation adéquate des patients et/ou des soignants à l’administration du lébrikizumab devra être assurée avant utilisation. Des instructions d’utilisation détaillées sont incluses à la fin de la notice. CONTRE-INDICATIONS : Hypersensibilité à la substance active ou à l’un des excipients. MISES EN GARDE SPÉCIALES ET PRÉCAUTIONS D’EMPLOI : Traçabilité : Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés. Hypersensibilité : En cas de survenue d’une réaction d’hypersensibilité systémique (immédiate ou retardée), l’administration du lébrikizumab doit être interrompue et un traitement approprié doit être instauré. Conjonctivite : Les patients traités par lébrikizumab qui développent une conjonctivite non résolue avec un traitement standard doivent effectuer un examen ophtalmologique (voir Effets indésirables). Infection par des helminthes : Les patients présentant des infections connues par des helminthes ont été exclus des études cliniques. L’influence du lébrikizumab sur la réponse immunitaire contre les infections dues à des helminthes, par inhibition de la voie de signalisation de l’IL-13, n’est pas connue. Les patients présentant des infections pré-existantes par des helminthes doivent être traités avant de commencer le traitement par lébrikizumab. Si des patients sont infectés au cours du traitement par lébrikizumab et ne répondent pas au traitement anti-helminthique, le traitement par lébrikizumab doit être interrompu jusqu’à la guérison de l’infection. Vaccinations : Avant l’instauration du traitement par lébrikizumab, il est recommandé de s’assurer que les patients sont à jour de toutes les vaccinations appropriées selon l’âge, conformément aux recommandations vaccinales en vigueur. Les vaccins vivants et les vaccins vivants atténués ne doivent pas être administrés au cours du traitement par lébrikizumab car la sécurité et l’efficacité cliniques n’ont pas été établies. Les réponses immunitaires aux vaccins non vivants ont été évaluées dans un vaccin combiné contre le tétanos, la diphtérie et la coqueluche acellulaire (dTCa) et un vaccin polysaccharidique anti-méningococcique (voir RCP). EFFETS INDÉSIRABLES : Résumé du profil de sécurité : Les effets indésirables les plus fréquents sont des conjonctivites (6,9 %), des réactions au site d’injection (2,6 %), des conjonctivites allergiques (1,8 %) et une sécheresse oculaire (1,4 %). Récapitulatif des effets indésirables : Dans toutes les études cliniques menées sur la dermatite atopique, 1 720 patients au total ont reçu du lébrikizumab, parmi lesquels 891 ont été exposés au lébrikizumab pendant au moins un an. Sauf indication contraire, les fréquences sont basées sur un regroupement de 4 études randomisées, en double aveugle, menées chez des patients atteints de dermatite atopique modérée à sévère, au cours desquelles 783 patients ont été traités par lébrikizumab par voie sous-cutanée pendant la phase contrôlée par placebo (16 premières semaines de traitement). Les effets indésirables observés dans les essais cliniques sont présentés ci-dessous et sont présentés par classe de système d’organe et par fréquence dans les catégories suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Infections et infestations : Fréquent : Conjonctivite. Peu fréquent : Zona. Affections hématologiques et du système lymphatique : Peu fréquent : Éosinophilie. Affections oculaires : Fréquent : Conjonctivite allergique, Sécheresse oculaire. Peu fréquent : Kératite, Blépharite. Troubles généraux et anomalies au site d’administration : Fréquent : Réaction au site d’injection. Description de certains effets indésirables : Conjonctivite et événements associés : Au cours des 16 premières semaines de traitement, des cas de conjonctivite, de conjonctivite allergique, de blépharite et de kératite ont été rapportés plus fréquemment chez les patients traités par lébrikizumab (respectivement, 6,9 %, 1,8 %, 0,8 % et 0,6 %) comparé aux patients sous placebo (1,8 %, 0,7 %, 0,2 % et 0,3 %). Au cours de la période de traitement d’entretien (16 à 52 semaines), l’incidence des conjonctivites et des conjonctivites allergiques observée avec le lébrikizumab était de 5,0 % et 5,9 % respectivement. Dans toutes les études cliniques, chez les patients traités par lébrikizumab, un arrêt du traitement dû à une conjonctivite et une conjonctivite allergique est survenu dans 0,7 % et 0,3 % des cas respectivement. Des cas sévères de conjonctivite et de conjonctivite allergique sont survenus dans 0,1 % et 0,2 % des cas, respectivement ; 72 % des patients ont récupéré, dont 57 % dans les 90 jours. Éosinophilie : Les patients traités par lébrikizumab ont présenté une augmentation moyenne de leur taux à l’inclusion d’éosinophiles, plus importante que les patients ayant reçu le placebo. Chez les patients traités par lébrikizumab, 20,3 % ont présenté une augmentation du taux d’éosinophiles, contre 11,7 % dans le groupe placebo. En général, cette augmentation chez les patients traités par lébrikizumab était d’intensité légère à modérée et transitoire. Une éosinophilie (> 5 000 cellules/µL) a été observée chez 0,4 % des patients traités par lébrikizumab et chez aucun de ceux ayant reçu le placebo. Des cas d’éosinophilie ont été rapportés chez 0,6 % des patients traités par lébrikizumab et avec un taux similaire chez les patients ayant reçu le placebo au cours de la période de traitement initiale. L’éosinophilie n’a pas entraîné d’arrêt du traitement, et aucun trouble lié aux éosinophiles n’a été rapporté. Réactions au site d’injection : Des réactions au site d’injection (y compris douleur et érythème) ont été rapportées plus fréquemment chez les patients ayant reçu le lébrikizumab (2,6 %) que chez ceux ayant reçu le placebo (1,5 %). La majorité (95 %) des réactions au site d’injection étaient d’intensité légère ou modérée, et peu de patients (< 0,5 %) ont arrêté le traitement par lébrikizumab. Zona :Un zona (herpès zoster) a été rapporté chez 0,6 % des patients traités par lébrikizumab et chez aucun des patients du groupe placebo. Tous les événements de zona rapportés étaient d’intensité légère ou modérée, et aucun n’a entraîné l’arrêt définitif du traitement. Tolérance à long terme : La tolérance à long terme du lébrikizumab a été évaluée dans 5 études cliniques : dans deux études en monothérapie (ADvocate 1, ADvocate 2) pendant une durée maximale de 52 semaines, dans une étude en association avec les DC (ADhere), puis dans une étude d’extension à long terme (ADjoin) pendant 56 semaines au total et dans l’étude ADore en monothérapie chez des adolescents pendant également une durée maximale de 52 semaines. Le profil de tolérance du lébrikizumab en monothérapie jusqu’à la semaine 52 ou en association avec un DC jusqu’à la semaine 56 est cohérent avec le profil de tolérance observé jusqu’à la semaine 16. Population pédiatrique : Adolescents âgés de 12 à 17 ans : La tolérance du lébrikizumab a été évaluée chez 372 patients âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère, dont 270 patients exposés pendant au moins un an. Le profil de tolérance du lébrikizumab chez ces patients était similaire à celui des adultes atteints de dermatite atopique. DÉCLARATION DES EFFETS INDÉSIRABLES SUSPECTÉS : La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration. Belgique : Agence fédérale des médicaments et des produits de santé, www.afmps.be, Division Vigilance, Site internet: www.notifieruneffetindesirable.be, e-mail: adr@afmps.be Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet : www.guichet.lu/pharmacovigilance TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ : Almirall S.A., Ronda General Mitre 151, 08022 Barcelona, Espagne. FABRICANT : Industrias Farmacéuticas Almirall, S.A., Barcelona, Espagne. NUMÉRO D’AUTORISATION DE MISE SUR LE MARCHÉ : EU/1/23/1765/001-012. MODE DE DÉLIVRANCE : Sur prescription médicale. DATE DE MISE À JOUR DU TEXTE : 02/2024. BE-EBG-2400037 11/2024 * chez les répondeurs (patients ayant atteint un EASI-75 ou un IGA 0/1 avec une réduction de ≥2 points sans médication de secours) ; p<0,001 vs. placebo à la semaine 16.10
** Certains patients présentant une réponse initiale partielle peuvent bénéficier d’une amélioration de la réponse en poursuivant le traitement toutes les deux semaines jusqu’à la semaine 24.10 Références : 1. Moyle M, et al. Exp Dermatol. 2019 Jul;28(7):756–768 ; 2. Gonçalves F, et al. Drugs Context. 2021 Mar 30;10:2021-1-7 ; 3. Ultsch M, et al. J Mol Biol. 2013 Apr 26;425(8):1330–1339 ; 4. Okragly AJ, et al. Dermatol Ther (Heidelb). 2023 Jul;13(7):1535–1547 ; 5. Silverberg JI, et al. N Engl J Med. 2023 Mar 23;388(12):1080-1091 ; 6. Thaçi D, et al. EADV, Amsterdam, Netherlands; 25-28 September 2024, poster D1T01.2 ; 7. Simpson EL, et al. JAMA Dermatol. 2023 Feb 1;159(2):182–191 ; 8. Paller AS, et al. Dermatol Ther (Heidelb). 2023 Jul;13(7):1517–1534 ; 9. Guttman-Yassky E, et al. SKIN The Journal of Cutaneous Medicine, 7(6), s271. 10. SmPC EBGLYSS® (lebrikizumab), dernière version disponible.
SOPHIE LEDUCQ, STÉPHANIE MALLET, JULIETTE MAZEREEUW-HAUTIER, FANNY MORICE-PICARD
Retranscription par Sophie Golstein
JOURNÉES DERMATOLOGIQUES DE PARIS 2024
L’ALOPÉCIE CHEZ
L’ENFANT NE SERA PLUS
UN MYST « HAIR » POUR VOUS : PAS À PAS DIAGNOSTIQUE ET THÉRAPEUTIQUE - PARTIE 1
1) ALOPÉCIE
A. ALOPÉCIE CONGÉNITALE/PRÉCOCE
i. Localisée
1. Aplasie cutanée congénitale du cuir chevelu (ACC)
Cliniquement, il peut s’agir d’une érosion ou d’une ulcération plus ou moins profonde.
La peau a parfois entièrement cicatrisé à la naissance et l’on peut avoir tout simplement une lésion cicatricielle. La question à se poser devant ces lésions est la suivante : y a-t-il des anomalies de fermeture plus profonde ?
Il faudra réaliser une IRM cérébrale dans les situations suivantes :
• ACC de grande taille
• Localisation médiane
• Défect osseux sous-jacent
• Signe du « collier de cheveux » (qui a une très bonne valeur prédictive négative)
• Signe de la touffe de cheveux
Il faut idéalement demander cette IRM cérébrale précocement, afin d’éviter de devoir faire une anesthésie générale.
Lorsque l’aplasie cutanée est de petite taille et isolée, on peut se limiter à l’échographie transfontanellaire.
2. Alopécie occipitale congénitale
L’alopécie occipitale congénitale ou néonatale est extrêmement fréquente ; elle atteint 20% des nouveau-nés. En
général, les parents pensent qu’elle est liée à la friction, mais ce sont en fait des fausses croyances. Il s’agit en réalité d’une chute physiologique des cheveux, due à la chute synchronisée des poils télogènes qui est initiée in utéro. Elle commence au niveau frontal, se poursuit sur le vertex et elle se finit au niveau occipital. Il faut absolument l’expliquer aux parents car leur réflexe pourrait être : « je ne veux pas que mon enfant perde ses cheveux donc je vais le mettre sur le ventre », et l’on sait que sur le ventre il y a un risque de mort subite du nourisson.
3. Alopécie « en auréole » L’alopécie en auréole est un diagnostic différentiel de l’alopécie occipitale congénitale. Il s’agit d’une pathologie beaucoup plus rare, liée à une ischémie locale et prolongée due à la pression sur l’orifice cervical. Les alopécies sont en général transitoires mais, de façon exceptionnelle, il existe des alopécies cicatricielles.
4. Alopécie triangulaire congénitale
Les parents ne le remarquent que tardivement mais elle est présente dès la naissance. Elle est bilatérale dans 20% des cas. Au niveau du cuir chevelu, on retrouve une petite plaque duveteuse
qui va persister toute la vie. Il ne faut pas proposer de traitement type Minoxidil, qui ne marchera pas. Il faut rassurer les parents.
ii. Diffus/hypotrichose
1. Dysplasie ectodermique
Dans les dysplasies ectodermiques, on retrouve l’atteinte d’au moins deux dérivés du tissu ectodermiques suivants : dents, ongles, glandes sudorales, follicule pilaire et glande mammaire. Ce sont des pathologies hétérogènes sur le plan moléculaire mais les gènes responsables sont principalement des gènes du développement.
Il peut y avoir l’association d’une hypotrichose généralisée plus ou moins marquée, associée à des anomalies du développement dentaire.
Le diagnostic clinique peut être posé grâce à une hypotrichose, avec souvent des cheveux fins et clairsemés, une pilosité très modérée. On retrouve parfois de l’eczéma dans ces pathologies.
2. Syndrome des cheveux anagènes caducs
Dans cette pathologie, il y a un défaut du cycle pilaire, dans la mesure ou les cheveux vont se détacher pendant la phase anagène. Si on réalise un trichogramme, on observe >50% de cheveux en phase anagène, alors que l’on s’attendrait à plutôt voir des cheveux en phase télogène.
En microscopie, on observe un bulbe anagène déformé, une absence de gaine interne et une cuticule ébouriffée.
Il s’agit d’une pathologie assez fréquente. L’évolution est plutôt favorable avec le temps ; il faut rassurer les parents.
On peut proposer un traitement par Minoxidil.
3. Hypotrichoses simplex
Sur le plan génétique, il y a un grand nombre de gènes impliqués dans les anomalies des cheveux. Certaines hypotrichoses sont complètement isolées (gènes impliqués dans le cycle pilaire ou dans la morphogénèse du cheveu).
L’important est de différencier une forme isolée ou syndromique, d’où l’intérêt d’un diagnostic moléculaire précis.
B. ACQUISE
i. Inflammatoire
1. Pelade
Il s’agit d’une pathologie qui n’est pas rare.
Le diagnostic de pelade est habituellement simple, mais il peut parfois être plus complexe.
Il est important de ne pas oublier de regarder les sourcils et les ongles (ponctuations, trachyonychie, leuconychie) lorsqu’un enfant est atteint de pelade.
La fréquence de l’atteinte unguéale est plus faible que celle de la perte de cheveux. Cette atteinte est le plus souvent simultanée à la chute des cheveux, mais elle peut parfois survenir avant ou après la chute.
La trichoscopie va aider énormément au diagnostic :
• Les points jaunes (yellow dots)
• Les points noirs (black dots)
• Les rétrécissements à la base (tapered hair)
• Les cheveux cassés (broken hairs)
• Les cheveux en points d’exclamation (exclamation mark hairs)
• Les cheveux à repousse verticale (upright regrowing hairs)
• Les cheveux en duvet (vellus hairs)
• Les cheveux en tire-bouchon (pigtail hairs)
• Les constrictions (constrictions de Pohl-Pinkus)
Une fois le diagnostic posé, quelle est la conduite à tenir devant un enfant atteint d’une pelade ?
1) Évaluer la sévérité/gravité. Comment ?
• Le SALT score :
S0 : pas de perte de cheveux
S1 : <25 % de perte de cheveux
S2 : 20-49 %
S3 : 50-74 %
S4 : 75-99 %
S5 : 100 % perte totale
• Test de traction : anomale si >5 cheveux
• Éléments trichoscopiques corrélés à la sévérité ?
Éléments corrélés positivement à la sévérité (c’est à dire que si on les observe, on est en présence d’une pelade active) : Yellow dots, black dots. Exclamation hairs ? Controversé.
Éléments corrélés négativement à la sévérité (ce sont des signes plutôt positifs) : Short vellus hairs ; qui correspondent à le repousse initiale et qui seront remplacés par des cheveux complètement pigmentés.
2) Faut-il réaliser un bilan devant une pelade chez l’enfant?
Bilan de « chute » : NFS, ferritine (elle doit être supérieure au moins à 40), 25-OH D3, zinc, vitamine B12, vitamine B9, TSH (à renouveler tous les 2 ans).
3) Traitement
Il n’existe pas d’algorithme de traitement ou de recommandations claires.
A. Forme localisées
Corticoïdes locaux
• Corticoïdes topiques forts en première intention
• Considérés comme efficaces et surs (effets secondaires : atrophie cutanée, télangiectasie, folliculite)
• Injections intra-lésionnelles ?
• Efficaces, douloureuses chez l’enfant, phobie des piqûres
Peu de données chez l’enfant dans la pelade : efficacité ?
Effet secondaire principal : hypertrichose, effets secondaires généraux possibles si mésusages (par exemple lors de l’applications 2x/j par exemple)
➞ Pas en première ligne, utile si repousse ? (à repérer en dermoscopie) (avis d’expert)
Application du Minoxidil 5 % possible, 3j/semaine, et prévoir un rinçage après 30 minutes
(Inhibiteurs de la calcineurine : peu de données, semblent inefficaces)
(Laser/photothérapie : laser excimer ou UBV, peu de données ou inefficaces)
B. Forme étendue (>50 %/port perruque)
Que faut-il prendre en compte avant de faire son choix thérapeutique ?
Il faut savoir, et bien dire au patient et à la famille :
• Il s’agit d’une maladie chronique, avec une histoire naturelle faite de poussées et de rémissions.
• Tous les traitements sont suspensifs, quels qu’ils soient.
• Certains paramètres influencent l’efficacité du traitement ; par exemple, la pelade ophiasique (de la nuque) est réputée pour répondre moins bien aux traitements, comme la pelade totale, universelle (qui s’associe à la chute des poils)
• La durée d’évolution semble importante : plus la pelade est ancienne moins, semble t-il, le traitement va être efficace
• Chez l’enfant, il semble particulièrement important d’évaluer le retentissement psychologique
• Il y a peu « d’evidence based medicine », peu d’études bien conduites. Il semble donc raisonnable d’utiliser des médicaments à faible risque et de réserver les traitements systémiques aux enfants avec des formes extensives et réfractaires, qui ont une altération significative de la qualité de vie.
Traitements systémiques :
a. Traitement « anciens »
i. Méthotrexate
8 études ont été identifiées ; incluant 42 enfants. Le plus souvent, le Méthotrexate était donné en association avec les corticoïdes systémiques.
Les auteurs ont estimé qu’il y avait une réponse moins bonne chez l’enfant que chez l’adulte (11,6% vs 44,7%) et des rechutes moins fréquentes que chez l’adulte (31,7% vs 52%).
Le principal effet secondaire sont les nausées, qui surviennent chez environ 1 enfant sur 2 (Méthotrexate prescrit par voie orale ou injectable).
La perturbation des tests hépatiques est possible mais assez rare.
ii. Corticoïdes systémiques
Dans les formes de pelade chronique, ils ne faut les utiliser en monothérapie ; il faut les associer au Méthotrexate au départ et on peut les utiliser en pulse thérapie.
b. Traitements nouveaux/ciblés – JAK inhibiteurs
La voie orale semble plus efficace que la voie locale.
Les effets indésirables sont d’intensité modérée dans la majorité des cas et les plus fréquents sont : élévation des transaminases, infections des voies respiratoires supérieures et hyperéosinophilie.
A ce jour, en France, le Baricitinib (OlumniantR) à l’AMM et le remboursement chez l’adulte uniquement.
Depuis quelques semaines, il existe une AMM et le remboursement en Europe pour le Ritlecitinib, à partir de l’âge de 12 ans.
Il s’agit d’un domaine en pleine expansion, avec des essais cliniques à venir.
Le Riltecitinib est donc le seul traitement ayant à ce jour une AMM pour la pelade de l’enfant à partir de 12 ans.
Proposition d’algorithme thérapeutique dans la pelade (pas de consensus d’experts)
En général, avant 10 ans, les enfants n’ont pas d’altération de la qualité de la vie. C’est en général les parents que ça gène. Si les enfants ne sont pas demandeurs, il ne faut pas les traiter de façon systémique.
2. Teigne
Les teignes sont des infections fréquentes chez les enfants. Elles sont dues à 2 germes : Tricophyton ou Microsporum.
Classiquement, on observe des cheveux qui seront cassés courts, avec des plaques alopéciques.
Il y a différents types cliniques de teigne.
• La teigne tricophytique : petites plaques d’alopécie sèches
• La teigne microsporique : grandes plaques d’alopécie
• Le kérion : forme très inflammatoire, purulente
• La teigne favique : « godet favique », alopécie définitive. Il s’agit d’une forme très exceptionnelle.
Pourquoi utiliser la lampe de Wood ?
Il ne faut pas hésiter à l’utiliser lorsque l’on a une suspicion clinique de teigne. Si le germe responsable est le microsporum, l’examen à la lampe de Wood va nous aider car on va retrouver une fluorescence jaune-verte qui va permette de conforter notre diagnostique de teigne et nous orienter vers le germe responsable.
Pourquoi utiliser son dermatoscope ?
Le dermatoscope est un outil indispensable. Il va nous permettre : 1) D’éliminer un diagnostic différentiel
Par exemple : la pelade. Lorsque l’on se trouve devant une plaque alopécique, il ne faut pas hésiter à poser son dermatoscope.
Les 4 signes trichoscopiques importants, les plus spécifiques dans la teigne :
2) Orienter sur le germe en cause : Il a été montré que les teignes tricophytiques, qui ont un parasitisme endothrix, vont plutôt avoir un pattern courbé (cheveux en virgule et cheveux en tire-bouchon) ; tandis que les teignes microsporiques auront un pattern droit (cheveux en zigzag ou cheveux en code-barres).
3) Suivre le patient et objectiver une guérison À 12 semaines, lorsque la culture est négative, les signes trichoscopiques persistants sont les squames, l’érythème et les points noirs. Il s’agit de signes peu spécifiques.
La régression des 4 signes spécifiques (cheveux en virgule, cheveux en tire-bouchon, cheveux en zigzag et cheveux en code-barres) permet d’assurer que le patient est guéri cliniquement.
Traitement
Depuis fin novembre 2024, le sirop d’itraconazole n’est plus commercialisé. Pour traiter les teignes microsporiques chez les enfants de moins de 20kg (ou pour les enfants ne savants pas avaler des gélules), il est recommandé de prescrire la terbinafine durant 6 semaines, à adapter en fonction du poids.
ii. Mécaniques
Trichotillomanie
La trichotillomanie est une pathologie fréquente. L’alopécie est classiquement unique, plutôt de contours irréguliers, et d’apparition rapide/brutale.
On la retrouve principalement sur le vertex, mais on peut également la retrouver au niveau des sourcils et des cils.
Cliniquement, le test de traction sera négatif. Il est important de le faire car un des diagnostics différentiels peut être la pelade. Un autre diagnostic différentiel est la teigne ; mais dans la trichotillomanie le cuir chevelu ne sera pas inflammatoire et la trichoscopie pourra nous aider à exclure des signes spécifiques de teigne.
Il y a 4 signes trichoscopiques de la trichotillomanie :
• Cheveux cassés courts, de longueur différente (vert)
• Black dots (rouge)
• Cheveux en tulipe (jaune)
• V sign (mauve)
Prise en charge
Le geste n’est souvent pas admis par les enfants ni par les parents.
Il faut donc être certain de notre diagnostic et on pourra alors proposer un suivi psychologique avec éventuellement une thérapie cognitivo-comportementale (avec le renversement d’une habitude).
Alopécie de traction
L’alopécie de traction est une pathologie fréquente.
En trichoscopie, on observe des gaines coulissantes.
Prise en charge
La prise en charge est assez simple ; le message à faire passer aux parents est l’arrêt de tout facteur traumatisant, de toute coiffure serrante.
Les dermocorticoïdes peuvent être prescrits en cas de phase inflammatoire ; éventuellement du Minoxidil topique. Pour les formes adultes, à distance, on peut discuter des greffes capillaires.
iii. Autre
Alopécie androgéno-génétique (AAG)
Cliniquement, comme chez l’adulte, on observe un recul symétrique des golfes temporaux et l’atteinte du vertex. Il y a une raréfaction et une miniaturisation des follicules pileux.
Les signes trichoscopiques de l’AAG sont :
• Anisotrichie (disparité du calibre des poils)
• Un seul poil par unité folliculaire (au lieu de 2-4)
• Cheveux duveteux (miniaturisation)
• Points jaunes d’aspect huileux
• Orifices pileux vides
L’AAG atteint plus les adolescentes que les adolescents et est probablement due à une hyperandrogénie fonctionnelle périphérique et/ou à un syndrome des ovaires micro-polykystiques (SOPK).
Le bilan hormonal est normal. Si on a un doute sur un SOPK, on peut demander une échographie ovarienne.
Si l’on a d’autres points d’appels cliniques de virilisation, il faudra éliminer une hyperandrogénie organique (bloc surrénalien, tumeur hormonosécrétante, …).
Prise en charge
On peut proposer du Minoxidil 2%.
Effluvium télogène
Il s’agit de la synchronisation brutale en phase terminale d’un grand nombre de follicules.
Les facteurs déclenchants peuvent être, entre autres, le stress physique (ex : fièvre élevée et prolongée), une intervention chirurgicale, un amaigrissement rapide.
L’effluvium télogène survient 2-4 mois après le facteur déclenchant.
Il est important de rassurer les patients et les parents car la repousse est constante.
L’effluvium télogène chronique
Si l’effluvium télogène est chronique, il faudra réaliser un bilan afin d’éliminer des carences isolées ou combinées (dans le cadre d’une dénutrition) : fer +++, oligoéléments, vitamines, protéines. D’autres causes peuvent être évoquées : des troubles hormonaux (dysthyroïdies +++), la prise de médicaments (les rétinoïdes dont l’isotrétinoïne).
Pelade diffuse
Le diagnostic est clinique et l’on peut s’aider du dermatoscope, comme dans la pelade en plaques.
La prise en charge repose sur les traitements systémiques.
Résumé des alopécies acquises diffuses
La dermoscopie aide vraiment au diagnostic.
COSENTYX®,
� 1st IL-17A antagonist
� 1st line biologic in the treatment of HS
Trust in a well-studied clinical profile1
*Observed prespecifi ed exploratory data—no clinical or statistical conclusions can be drawn.4 †Data presented are the results of an FDA-requested analysis of the SUNSHINE and SUNRISE trials.1 SUNSHINE and SUNRISE were identical, multicenter, randomized, placebo-controlled, doubleblind phase 3 trials. In both trials, patients were randomly assigned to receive COSENTYX® 300 mg Q2W or Q4W or placebo. SUNSHINE evaluated 541adult patients: 181 received COSENTYX® 300 mg Q2W, 180 received COSENTYX 300 mg Q4W, and 180 received placebo. SUNRISE evaluated 543 adult patients: 180 received COSENTYX® 300 mg Q2W, 180 received COSENTYX® 300 mg Q4W, and 183 received placebo. All patients were adults with moderate to severe HS (defined as a total of ≥5 inflammatory lesions affecting ≥2 distinct anatomical areas) for ≥1 year. The primary end point was the proportion of patients with an HS clinical response, defi ned as a decrease in AN count by ≥50% with no increase in the number of abscesses or in the number of draining fistulae at Week 16 compared with baseline. Co-secondary end points were AN50, flares, and NRS30 at Week 16.1,3
FDA, US Food and Drug Administration; IL, interleukin; NRS, numeric rating scale; Q2W, every 2 weeks; Q4W, every 4 weeks; QOL, quality of life
Name: Cosentyx 75 mg solution for injection in pre-filled syringe. Cosentyx 150 mg solution for injection in pre-filled syringe. Cosentyx 300 mg solution for injection in pre-filled syringe. Cosentyx 150 mg solution for injection in pre-filled pen. Cosentyx 300 mg solution for injection in pre-filled pen. Composition: Cosentyx 75 mg solution for injection in pre-filled syringe. Each pre-filled syringe contains 75 mg secukinumab in 0.5 ml. Cosentyx 150 mg solution for injection in pre- filled syringe. Each pre-filled syringe contains 150 mg secukinumab in 1 ml.Cosentyx 300 mg solution for injection in pre- filled syringe. Each pre-filled syringe contains 300 mg secukinumab in 2 ml. Cosentyx 150 mg solution for injection in pre- filled pen. Each pre-filled pen contains 150 mg secukinumab in 1 ml. Cosentyx 300 mg solution for injection in pre-filled pen. Each pre-filled pen contains 300 mg secukinumab in 2 ml.* Secukinumab is a recombinant fully human monoclonal antibody selective for interleukin-17A. Secukinumab is of the IgG1/κ-class produced in Chinese Hamster Ovary (CHO) cells. For the full list of excipients, see full leaflet. Pharmaceutical form: Solution for injection (injection). The solution is clear and colourless to slightly yellow.Therapeutic indications: Adult plaque psoriasis. Cosentyx is indicated for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy. Paediatric plaque psoriasis. Cosentyx is indicated for the treatment of moderate to severe plaque psoriasis in children and adolescents from the age of 6 years who are candidates for systemic therapy. Hidradenitis suppurativa (HS). Cosentyx is indicated for the treatment of active moderate to severe hidradenitis suppurativa (acne inversa) in adults with an inadequate response to conventional systemic HS therapy (see full leaflet).Psoriatic arthritis. Cosentyx, alone or in combination with methotrexate (MTX), is indicated for the treatment of active psoriatic arthritis in adult patients when the response to previous disease-modifying anti-rheumatic drug (DMARD) therapy has been inadequate. Axial spondyloarthritis (axSpA). Ankylosing spondylitis (AS, radiographic axial spondyloarthritis) Cosentyx is indicated for the treatment of active ankylosing spondylitis in adults who have responded inadequately to conventional therapy. Non-radiographic axial spondyloarthritis (nr-axSpA) Cosentyx is indicated for the treatment of active non-radiographic axial spondyloarthritis with objective signs of inflammation as indicated by elevated C- reactive protein (CRP) and/or magnetic resonance imaging (MRI) evidence in adults who have responded inadequately to non-steroidal anti-inflammatory drugs (NSAIDs). Juvenile idiopathic arthritis (JIA). Enthesitis-related arthritis (ERA) Cosentyx, alone or in combination with methotrexate (MTX), is indicated for the treatment of active enthesitis-related arthritis in patients 6 years and older whose disease has responded inadequately to, or who cannot tolerate, conventional therapy (see full leaflet). Juvenile psoriatic arthritis (JPsA) Cosentyx, alone or in combination with methotrexate (MTX), is indicated for the treatment of active juvenile psoriatic arthritis in patients 6 years and older whose disease has responded inadequately to, or who cannot tolerate, conventional therapy (see full leaflet). Posology and method of administration: Cosentyx is intended for use under the guidance and supervision of a physician experienced in the diagnosis and treatment of conditions for which Cosentyx is indicated. Adult plaque psoriasis The recommended dose is 300 mg of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Based on clinical response, a maintenance dose of 300 mg every 2 weeks may provide additional benefit for patients with a body weight of 90 kg or higher. Each 300 mg dose is given as one subcutaneous injection of 300 mg or as two subcutaneous injections of 150 mg. Paediatric plaque psoriasis (adolescents and children from the age of 6 years). The recommended dose is based on body weight (Table 1) and administered by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Each 75 mg dose is given as one subcutaneous injection of 75 mg. Each 150 mg dose is given as one subcutaneous injection of 150 mg. Each 300 mg dose is given as one subcutaneous injection of 300 mg or as two subcutaneous injections of 150 mg. Table 1. Recommended dose for paediatric plaque psoriasis : Body weight at time of dosing: Recommended Dose; <25 kg : 75 mg; 25 to <50 kg: 75 mg; ≥50 kg : 150 mg (*may be increased to 300 mg). *Some patients may derive additional benefit from the higher dose. The 150 mg and 300 mg solution for injection in pre-filled syringe and in pre-filled pen are not indicated for administration to paediatric patients with a weight <50 kg. Cosentyx may be available in other strengths and/or presentations depending on the individual treatment needs. Hidradenitis suppurativa (HS) The recommended dose is 300 mg of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing. Based on clinical response, the maintenance dose can be increased to 300 mg every 2 weeks. Each 300 mg dose is given as one subcutaneous injection of 300 mg or as two subcutaneous injections of 150 mg. Psoriatic arthritis For patients with concomitant moderate to severe plaque psoriasis, please refer to adult plaque psoriasis recommendation. For patients who are anti-TNFα inadequate responders (IR), the recommended dose is 300 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Each 300 mg dose is given as one subcutaneous injection of 300 mg or as two subcutaneous injections of 150 mg. . For other patients, the recommended dose is 150 mg by subcutaneous injection with initial dosing at Weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Based on clinical response, the dose can be increased to 300 mg. Axial spondyloarthritis (axSpA). Ankylosing spondylitis (AS, radiographic axial spondyloarthritis).The recommended dose is 150 mg by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Based on clinical response, the dose can be increased to 300 mg. Each 300 mg dose is given as one subcutaneous injection of 300 mg or as two subcutaneous injections of 150 mg. Non-radiographic axial spondyloarthritis (nr-axSpA). The recommended dose is 150 mg by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3 and 4, followed by monthly maintenance dosing. Juvenile idiopathic arthritis (JIA). Enthesitis-related arthritis (ERA) and juvenile psoriatic arthritis (JPsA). The recommended dose is based on body weight (Table 2) and administered by subcutaneous injection at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing. Each 75 mg dose is given as one subcutaneous injection of 75 mg. Each 150 mg dose is given as one subcutaneous injection of 150 mg. Table 2. Recommended dose for juvenile idiopathic arthritis : Body weight at time of dosing : Recommended dose; <50 kg : 75 mg; ≥50 kg: 150 mg. The 150 mg and 300 mg solution for injection in pre-filled syringe and in pre-filled pen are not indicated for administration to paediatric patients with a weight <50 kg. Cosentyx may be available in other strengths and/or presentations depending on the individual treatment needs. For all of the above indications, available data suggest that a clinical response is usually achieved within 16 weeks of treatment. Consideration should be given to discontinuing treatment in patients who have shown no response by 16 weeks of treatment. Some patients with an initial partial response may subsequently improve with continued treatment beyond 16 weeks. Special populations: Elderly patients (aged 65 years and over): No dose adjustment is required. Renal impairment / hepatic impairment: Cosentyx has not been studied in these patient populations. No dose recommendations can be made. Paediatric population The safety and efficacy of Cosentyx in children with plaque psoriasis and in the juvenile idiopathic arthritis (JIA) categories of ERA and JPsA below the age of 6 years have not been established. The safety and efficacy of Cosentyx in children below the age of 18 years in other indications have not yet been established. No data are available. Method of administration. Cosentyx is to be administered by subcutaneous injection. If possible, areas of the skin that show psoriasis should be avoided as injection sites. The solution in the syringe or pen must not be shaken. After proper training in subcutaneous injection technique, patients may self-inject Cosentyx if a physician determines that this is appropriate. However, the physician should ensure appropriate follow-up of patients. Patients should be instructed to inject the full amount of Cosentyx according to the instructions provided in the package leaflet. Comprehensive instructions for administration are given in the package leaflet. Contraindications: Severe hypersensitivity reactions to the active substance or to any of the excipients. Clinically important, active infection (e.g. active tuberculosis). Undesirable effects: Summary of the safety profile: see full leaflet. List of adverse reactions: ADRs from psoriasis, psoriatic arthritis and ankylosing spondylitis clinical studies as well as from post-marketing experience are listed by MedDRA system organ class. Within each system organ class, the ADRs are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); and not known (cannot be estimated from the available data).List of adverse reactions in clinical studies1) and post-marketing experience: Infections and infestations: Very common: Upper respiratory tract infections; Common: Oral herpes; Uncommon: Oral candidiasis; Uncommon: Otitis externa. Uncommon: Lower respiratory tract infections. Uncommon: Tinea pedis. Not known: Mucosal and cutaneous candidiasis (including oesophageal candidiasis). Blood and lymphatic system disorders: Uncommon: Neutropenia. Immune system disorders: Rare: Anaphylactic reactions. Rare:Angioedema. Nervous system disorders: Common: Headache. Eye disorders: Uncommon: Conjunctivitis. Respiratory, thoracic and mediastinal disorders: Common: Rhinorrhoea. Gastrointestinal disorders: Common: Diarrhoea. Common: Nausea. Uncommon: Inflammatory bowel disease. Skin and subcutaneous tissue disorders: Common: Eczema. Uncommon: Urticaria. Uncommon: Dyshidrotic eczema. Rare: Exfoliative dermatitis2). Rare: Hypersensitivity vasculitis. Not known: Pyoderma gangrenosum. General disorders and administration site conditions: Common: Fatigue. 1) Placebo-controlled clinical studies (phase III) in plaque psoriasis, PsA, AS, nr-axSpA and HS patients exposed to 300 mg, 150 mg, 75 mg or placebo up to 12 weeks (psoriasis) or 16 weeks (PsA, AS, nr-axSpA and HS) treatment duration. 2) Cases were reported in patients with psoriasis diagnosis. Description of selected adverse reactions: see full leaflet. Paediatric population. Undesirable effects in paediatric patients from the age of 6 years with plaque psoriasis. The safety of secukinumab was assessed in two phase III studies in paediatric patients with plaque psoriasis. The first study (paediatric study 1) was a double-blind, placebo-controlled study of 162 patients from 6 to less than 18 years of age with severe plaque psoriasis. The second study (paediatric study 2) is an open-label study of 84 patients from 6 to less than 18 years of age with moderate to severe plaque psoriasis. The safety profile reported in these two studies was consistent with the safety profile reported in adult plaque psoriasis patients. Undesirable effects in paediatric patients with JIA. The safety of secukinumab was also assessed in a phase III study in 86 juvenile idiopathic arthritis patients with ERA and JPsA from 2 to less than 18 years of age. The safety profile reported in this study was consistent with the safety profile reported in adult patients. Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system. Mode of delivery: Medicinal product subject to medical prescription. Marketing authorisation holder and numbers: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland. Cosentyx 75 mg solution for injection in pre-filled syringe. EU/1/14/980/012-013. Cosentyx 150 mg solution for injection in pre-filled syringe. EU/1/14/980/002, EU/1/14/980/003, EU/1/14/980/006. Cosentyx 300 mg solution for injection in pre-filled syringe. EU/1/14/980/008-009. Cosentyx 150 mg solution for injection in pre-filled pen. EU/1/14/980/004, EU/1/14/980/005, EU/1/14/980/007. Cosentyx 300 mg solution for injection in pre-filled pen. EU/1/14/980/010-011. Date of revision of the text: 19.12.2024
1. Cosentyx. Prescribing information. Novartis Pharmaceuticals Corp. 2. FDA approves Novartis Cosentyx® as the fi rst new biologic treatment option for hidradenitis suppurativa patients in nearly a decade [press release]. Basel, Switzerland: Novartis: October 31, 2023. Accessed November 1, 2023. https://www.novartis.com/news/media-releases/fda-approves-novartis-cosentyx-fi
PROF. NICOLAS DAUBY
Service des Maladies Infectieuses, CHU Saint-Pierre, Université Libre de Bruxelles (ULB)
COMMENT PRÉPARER MON PATIENT À L’IMMUNOSUPPRESSION/ MODULATION EN DERMATOLOGIE ?
INTRODUCTION
La prise en charge de maladies inflammatoires ou auto-immunes en dermatologie nécessite souvent l’utilisation de médicaments immunosuppresseurs ou immunomodulateurs. Ces dernières années, une augmentation de l’utilisation de ces médicaments, notamment les biologiques, avec un mécanisme d’action ciblé, est observée en médecine avec une amélioration substantielle du pronostic de certaines pathologies et une amélioration de la qualité de vie des patients.
Néanmoins, l’utilisation de ces médicaments peut entraîner des conséquences au niveau du risque infectieux, que ce soit en augmentant l’incidence ou la sévérité de certaines infections ou en augmentant le risque de réactivation de certaines infections latentes.
Avant l’initiation de ces médicaments, il est recommandé de procéder, dans la mesure du possible à un bilan pré-immunosuppression.
Nous reprendrons ici les grandes lignes d’un tel bilan. Cet article n’a pas l’objectif de se substituer à une consultation médicale spécialisée mais plutôt de sensibiliser le prescripteur afin qu’il informe de manière adéquate le patient avant de le référer à une telle consultation spécialisée. Au CHU Saint-Pierre, cette consultation est basée à la Travel & Vaccine Clinic et les patients sont en général référés par leur spécialiste traitant (rhumatologue, dermatologue, etc)
VACCINATION PRÉ-IMMUNOSUPPRESSION
Deux aspects sont primordiaux avant de commencer un traitement immunosuppresseurs :
• Vérifier le statut sérologique vis-à-vis de différentes maladies à prévention vaccinale. En effet, le traitement
Figure 1 : Varicelle sévère chez une enfant de 12 ans non-vaccinée et traitée par méthotrexate depuis 1 ans pour un psoriasis. D’après Galvao, 2022(2)
pourrait augmenter la sévérité d’une MPV (par exemple la varicelle, voir figure 1) ou diminuer l’efficacité de la vaccination
• Proposer la vaccination avec des vaccins vivants qui ne pourraient être administrés sous traitement immunosuppresseurs. Le cas exemplatif est celui du vaccin contre la fièvre jaune. Même si le patient n’a pas l’intention de voyager dans un délai court dans un pays endémique, il vaut mieux recevoir le vaccin avant le début du traitement. En effet, pour certains traitements (corticoïdes hautes doses, anti-TNF ou méthotrexate haute dose), ce vaccin est contre-indiqué et nécessité une période de “wash-out” du traitement immunosuppresseurs.
Les MPV dont nous vérifions le statut sérologique sont : rougeole, rubéole (pour les femmes en âge de procréer), les virus des hépatites A et B et la varicelle. Une dose de rappel du vaccin tétanos-dipthérie-coqueluche est proposé si le dernier rappel date de plus de 10 ans. Enfin, la vaccination contre les infections respiratoires ‘saisonnières’ (grippe, COVID-19, VRS, pneumocoque) est recommandée, d’autant plus si le patient souffre de comorbidités augmentant le risque de ces infections (diabète, obésité, etc.).
DÉPISTAGE DE LA TUBERCULOSE LATENTE
Certains traitements utilisés en dermatologie (surtout les anti-TNF ou les corticoïdes hautes doses) vont augmenter grandement le risque de réactivation d’une tuberculose latente. En Belgique, entre 5-10% des cas de tuberculose sont diagnostiqués chez des patients recevant un traitement immunosuppresseurs(3). Les anti-TNF vont surtout être associée à des tuberculoses extra-pulmonaires (Figure 2)
Le dépistage de la tuberculose latente doit se faire idéalement avant le début du traitement par immunosuppresseurs. Il faut être particulièrement vigilant chez les patients issus de zones endémiques (plus de 60% des cas de tuberculose diagnostiqués en Belgique sont chez personnes nées à l’étranger). En Belgique, on utilise une stratégie combinée : intra-dermo réaction et test immunologique de type IGRA (le plus souvent le Quantiferron ®). Une Radiographie de thorax est également demandée afin d’exclure une tuberculose active. L’interprétation des résultats se fera en consultation spécialisée d’infectiologie ou pneumologie. Si un traitement de la tuberculose latente est nécessaire, on choisira entre la rifampicine (avec risque d’interactions médicamenteuses mais traitement court de 4 mois) ou la nicotibine(traitement plus long et avec plus de risque d’effets indésirables). A noter, que la prophylaxie pour la tuberculose latente n’abolit pas complètement le risque de développer une tuberculose active (voir Figure 2)
LE CAS DES ANTI-JAK
Comme discuté dans un numéro précédent de Dermactu, les inhibiteurs de JAK sont associés à un risque particulièrement élevé de réactivation du virus de la varicelle et du zona (VZV)(5).
Figure 2 : Tuberculose péritonéale et splénique (flèches jaunes) chez une patiente coréenne de 27 ans traitée par adalimumab (Humira ®) ) pour un psoriasis depuis 45 jours (d’après Hong Ho 2021(4)). La tuberculose s’est manifestée malgré une prophylaxie par nicotibine
Il est recommandé par le conseil supérieur de la santé de vacciner contre le zona avec le vaccin recombinant avec l’initiation de ce traitement (2 doses du vaccin Shingrix ® avec au moins 2 mois d’intervalle)(6). Le tableau 1 reprend les inhibiteurs de JAK disponibles en Belgique et leurs indications respectives en dermatologie. Malheureusement, le remboursement en Belgique n’est pas encore disponible pour cette indication.
Tableau 1 : liste des inhibiteurs de JAK utilisés en dermatologie. Ces molécules sont associées à un risque accru de zona et il est recommandé de vacciner avant le début du traitement.
Molécule
Indication en dermatologie
Abrocitinib (Cibinqo ® ) Dermatite atopique
Baricitinib (Olumiant ®) Pelade
Tofacitinib (Xeljanz ®) Psoriasis articulaire
Upadacitinib (Rinvoq ®) Dermatite atopique
LE CAS DU PSORIASIS ET DES ANTI-IL17
Différents agents bloquant la voie de l’IL-17 sont maintenant approuvés en Belgique et principalement utilisés dans le traitemnt du psoriasis (bimékizumab, le brodalumab, l’ixékizumab et le sécukinumab).
« Attention, l’utilisation de ces médicaments peut entraîner des conséquences au niveau du risque infectieux, aussi, avant l’initiation de ces médicaments, il est recommandé de procéder, dans la mesure du possible, à un bilan pré-immunosuppression. »
La voie de l’interleukine 17 est également impliquée dans l’immunité anti-fongique. Les anti-IL17 sont ainsi associés à un risque accru d’infection à Candida par rapport au placebo ou aux anti-TNF. La majorité de ces infections sont bénignes et mucocutanées. Elles peuvent être traitées en topique. Il n’y a pas d’indication de dépistage du Candida avant initiation d’un traitement par IL-17 ; il faudra bien informer le patient néanmoins(7).
Il est important de souligner que les patients avec psoriasis ont un risque accru d’infections sévères (pneumonie par exemple) indépendamment du traitement reçu(8). Il est donc important que ces patients soient à jour pour leurs vaccins (grippe, pneumocoque notamment). Une vaccination en cocon pourra être proposée aux membres du foyer (grippe, pneumocoque, COVID-19). Enfin, on rappellera qu’en cas de diagnostic d’un psoriasis, il est indiqué de réaliser une sérologie VIH.
CONCLUSIONS
L’utilisation de traitements immunomodulateur ou immunosuppresseur a révolutionné le pronostic de certaines pathologies dermatologiques. Les infections peuvent constituer des complications graves de ces traitements et une approche systématique permet de mitiger ce risque en combinant bilan sérologique, dépistage de la tuberculose latente et vaccinations ciblées avant l’initiation du traitement. Une collaboration entre spécialistes (dermatologue et infectiologue ou pneumologue pour la tuberculose latente) est indispensable, au bénéfice du patient et du système de santé.
RÉFÉRENCES
1. Martin C, Muls V, Brasseur C, Meric de Bellefon L, Lam Hoai XL, Vanderhilst J, et al. ImmunoStart: preparing patients for immunosuppression. Rheumatol Adv Pract. 31 août 2021;5(3):rkab092.
2. Galvão LO, Reis CMS, Alves NL, Maciel ES. Severe varicella in a child immunosuppressed with methotrexate. An Bras Dermatol. 1 mars 2022;97(2):184-8.
3. Fond des affections respiratoires ASBL. Registre belge de la tuberculose, 2022. Bruxelles; 2024 mars.
4. Oh JH, Ham SP, Park HJ. Disseminated Tuberculosis in a Psoriasis Patient under Adalimumab Treatment despite the Chemoprophylaxis of Latent Tuberculosis: A Case Report. Ann Dermatol. 1 févr 2021;33(1):77-81.
5. Mansilla-Polo M, Morgado-Carrasco D. Biologics Versus JAK Inhibitors. Part II: Risk of Infections. A Narrative Review. Dermatol Ther. 1 août 2024;14(8):1983-2038.
6. Conseil Supérieur de la Santé. Vaccination contre l’herpès zoster - CSS n°9684. Bruxelles; 2022. Report No.: 9684.
7. Yeung J, Bunce PE, Lynde CW, Turchin I, Vender RB. Review and Practical Guidance on Managing Fungal Infections in Patients With Psoriasis Receiving Anti-IL-17 Therapies. J Cutan Med Surg. 1 juill 2022;26(1_suppl):3S-23S.
8. Rademaker M, Agnew K, Anagnostou N, Andrews M, Armour K, Baker C, et al.
Figure 3 : Candidose pseudomembraneuse orale chez un patient recevant un anti-IL17A pour le traitement d’un psoriasis en plaque (d’après Armstrong(9))
Psoriasis and infection. A clinical practice narrative. Australas J Dermatol. mai 2019;60(2):91-8.
9. Armstrong AW, Blauvelt A, Mrowietz U, Strober B, Gisondi P, Merola JF, et al. A Practical Guide to the Management of Oral Candidiasis in Patients with Plaque Psoriasis Receiving Treatments That Target Interleukin-17. Dermatol Ther. 1 mars 2022;12(3):787-800.
DR DE LEYE HANNAH 1 , DR SAERENS JULIE 1, DR LUTTGENS PAULINE 1 , DR MATTON LIESL 2, PROF GUTERMUTH JAN 3
1 Assistante en formation en dermatologie, UZ Brussel
2 Médecin généraliste en formation, cabinet de Keerbergen
3 Chef du département de dermatologie, UZ Brussel
DERMATITE ATOPIQUE ET AFFECTIONS ASSOCIÉES
INTRODUCTION
Plusieurs comorbidités sont associées à la dermatite atopique (DA). Les comorbidités atopiques et non atopiques sont décrites avec une prévalence et une incidence différente dans le groupe de la DA.
COMORBIDITÉS ATOPIQUES
Le concept de « la marche atopique » est désormais bien connu. La marche atopique décrit la progression de la DA vers l’allergie alimentaire (AAL), l’asthme allergique (AA), la rhinite allergique (RA) et la conjonctivite allergique (CA). Selon cette théorie, la rupture de la barrière épidermique dans la DA entraîne une exposition accrue aux allergènes épicutanés, ce qui provoque une inflammation. Cette inflammation et cette sensibilisation se transmettent ensuite à d’autres épithéliums, y compris le tractus gastro-intestinal, les voies respiratoires supérieures (conduisant à la RA) et les voies respiratoires inférieures (contribuant à l’AA). (1, 19)
Cependant, une étude récente a remis en question ce concept en montrant que seuls 3 % des enfants correspondent à la marche atopique traditionnelle, ce qui suggère que les facteurs génétiques et l’exposition environnementale, entre autres, sont importants, en plus de la perturbation de la barrière comme événement primaire. Il existe donc de multiples sous-groupes et profils de développement pour les comorbidités allergiques de la DA. (2)
La prévalence des comorbidités atopiques dans la DA, quel que soit l’âge, est plus élevée que dans la population générale (tableau 1). (3, 4, 5) Il existe un terrain génétique commun entre la DA et les comorbidités atopiques. La gravité de la DA est également associée à un risque accru de développer une AA, entre autres. (6, 7)
Les symptômes ophtalmologiques constituent un aspect souvent sous-étudié de l’atopie. Les maladies de la peau ont un impact local sur l’œil et vice versa. Le larmoiement et les démangeaisons dus à la conjonctivite allergique peuvent amener le patient à se gratter et à irriter la peau autour des yeux, provoquant un eczéma irritatif ou aggravant la dermatite atopique. Un examen systématique réalisé en 2023 a révélé que la conjonctivite a une prévalence de 31,7 % chez les personnes atteintes de la DA, contre 13,3 % dans le groupe témoin, la conjonctivite allergique étant le sous-type le plus courant chez les patients atteints de la DA. Les affections telles que la blépharite, la sécheresse oculaire et la kératite sont fréquentes chez les personnes atteintes de la DA, avec respectivement 22 %, 9,1 % et 1,4 %. (7) S’ils ne sont pas traités, ces symptômes ophtalmiques peuvent entraîner des lésions irréversibles des yeux (kératocône et cataracte, par exemple). Il est important de procéder à un dépistage et d’adresser le patient à un ophtalmologiste si nécessaire.
ALLERGIQUE 25.7% 8.1%
ALIMENTAIRE
Tableau 1. Comorbidités chez les personnes atteintes de la DA par rapport à celles qui n’en sont pas atteintes (3, 4, 5).
Plus les patients sont jeunes au moment de l’apparition de la DA, plus ils sont susceptibles de développer une ou plusieurs comorbidités (tableau 2). La DA qui n’apparaît qu’à l’âge adulte est deux fois plus susceptible de développer une rhinite allergique que la DA apparue dans l’enfance. Cependant, la DA qui
débute dans l’enfance entraîne trois fois plus de cas d’allergie alimentaire que la DA qui ne débute qu’à l’âge adulte. (8)
ONSET DA ≥ 1 COMORBIDITÉS ATOPIQUES
NOURISSON (0-2J) 90.8%
ENFANT (2-18J) 87.9%
JEUNE ADULTE (18-41J) 72.4%
ÂGE MUR (41-61J) 63%
MATURITÉ TARDIVE (61+J) 50%
Tableau 2 : Comorbidités atopiques en relation avec l’âge d’apparition de la maladie atopique. (8)
En 2022, une vaste cohorte longitudinale de nourrissons atteints de la DA et âgés de moins de 2 ans a étudié la marche atopique chez les nourrissons atteints de la DA selon la couleur de peau. Cette étude a clairement montré que les patients à la peau noire atteints de la DA sont plus susceptibles d’évoluer vers l’AA, qui présente alors un niveau de gravité plus important. Les enfants avec la peau blanche atteints de la DA, quant à eux, étaient plus susceptibles de souffrir d’allergie alimentaire, de rhinite allergique et de sensibilisation aux allergènes alimentaires et respiratoires sans risque élevé d’asthme. Il a donc été clairement démontré que le paradigme classique de la marche atopique n’est pas universellement applicable et qu’il est sujet à des différences entre les types de peau. (9) Une faiblesse de cette étude est que tous les facteurs environnementaux pertinents, en particulier l’exposition à la pollution environnementale, n’ont pas été inclus dans l’analyse.
COMORBIDITÉS INFECTIEUSES
Dans la DA, les infections récidivantes sont la conséquence de plusieurs facteurs tels qu’un défaut de la barrière cutanée, la colonisation par Staphylococcus aureus, la suppression de l’immunité innée cutanée due à l’inflammation de type 2 et la dysbiose cutanée. (7) La prévalence du SARM est nettement plus fréquente chez les patients atteints de DA que dans la population générale. La virulence élevée de ces bactéries génère une abondance de superantigènes (exotoxines staphylococciques), augmentant le potentiel d’infections et exacerbant l’inflammation cutanée chez les patients atteints de la DA. (10)
Ces infections peuvent évoluer en des infections cutanées bactériennes et virales aux complications graves telles que la bactériémie et l’endocardite. (11) L’impétigo, une infection cutanée bactérienne souvent causée par S. aureus, est plus fréquent chez les personnes atteintes de la DA (OR = 1,6-8,7. (1) Bien qu’il n’y ait pas d’augmentation claire du risque d’infection par l’herpès simplex, les personnes atteintes de dermatite atopique (DA) ont un risque accru d’infection fulminante avec un tableau d’eczéma herpétique (OR = 11,7). Cette infection par le virus de l’herpès simplex, potentiellement mortelle, survient principalement chez les patients atteints de DA sévère, les patients atteints de DA et ayant des antécédents d’infection cutanée à Staphylococcus aureus, des antécédents d’allergie
alimentaire, ou une DA à début précoce. Les patients infectés par le virus coxsackie, en particulier le coxsackie A6, sont plus susceptibles de développer un « eczéma coxsackium ». Cette affection est associée à une atteinte cutanée étendue, similaire à l’eczéma herpétique. (12) La question de savoir si les personnes atteintes de la DA sont plus sensibles à l’infection par le molluscum contagiosum est controversée. Cependant, la dermatite à molluscum et les signes plus graves d’infection à molluscum contagiosum (y compris l’accentuation des lésions dans la DA) sont plus fréquents chez les enfants atteints de la DA que chez les témoins (OR = 1,6). (12) On manque de données précises sur les comorbidités infectieuses chez les adultes, mais on observe que les enfants atteints de la DA sont plus sensibles à l’eczéma herpétique et aux infections bactériennes que les adultes atteints de la DA et que les deux groupes sont plus vulnérables aux infections bactériennes et mycosiques que les témoins. (8)
COMORBIDITÉS PSYCHOSOCIALES
Une étude systématique réalisée en 2024 a montré que les personnes atteintes de la DA étaient plus susceptibles de souffrir de TDAH (rapport de cotes de 1,28) et de troubles du spectre autistique (rapport de cotes de 1,87).(20) La prévalence des troubles du comportement est également plus élevée chez les enfants de 5, 9 et 15 ans atteints de la DA. (21)
Une revue systématique et une méta-analyse ont montré que la DA à l’âge adulte était significativement associée à la dépression, aux troubles anxieux et aux pensées suicidaires. Pour la DA chez l’enfant, un lien (bien que plus faible) avec la dépression a été trouvé en particulier (13). Une méta-analyse a montré que 20,1 % des personnes atteintes de la DA souffraient de dépression, contre 14,8 % dans le groupe témoin. Chez les adultes atteints de la DA, on observe des taux de dépression clinique nettement plus élevés (14,9 % contre 12,6 %), une augmentation de la consommation d’antidépresseurs (29,3 % contre 20,3 %) et des taux de suicide nettement plus élevés (12,2 % contre 6,4 %), par rapport aux adultes non atteints de la DA (7). Les preuves d’un lien entre la gravité de la DA et l’apparition de comorbidités psychiatriques sont insuffisantes. Il est donc important d’y prêter attention chez tous les patients atteints de la DA. La cause exacte de l’apparition de comorbidités psychiatriques n’est pas tout à fait claire. On suppose que les symptômes purement physiques, tels que les démangeaisons, peuvent avoir une influence négative, mais l’isolement social et la stigmatisation, le manque de sommeil et la nature chronique de la maladie jouent certainement (et peut-être même davantage) un rôle. (14)
COMORBIDITÉS CARDIOVASCULAIRES ET HÉMATOLOGIQUES
Par analogie avec le psoriasis, la question de savoir si les personnes atteintes de la DA présentent également un risque accru de maladie cardiovasculaire est aujourd’hui un sujet d’actualité. Une revue systématique a observé un lien entre la DA, le tabagisme (actif et passif) et l’hypertension. (15,16) En outre, les patients atteints de DA originaires d’Asie et d’Amérique du Nord sont plus susceptibles d’être en surpoids ou obèses, mais cette corrélation n’est pas observée en Europe. (17) Dans
« Une étude systématique réalisée en 2024 a montré que les personnes atteintes de la DA étaient plus susceptibles de souffrir de TDAH et de troubles du spectre autistique. »
une vaste étude de cohorte, les événements cardiovasculaires non mortels étaient plus fréquents chez les patients atteints de la DA, en particulier dans le sous-groupe des patients les plus gravement atteints. Il s’agit notamment des AVC, de l`angine de poitrine instable, de l`infarctus du myocarde, de la fibrillation auriculaire et de l`insuffisance cardiaque, après ajustement des facteurs de risque cardiovasculaire distincts (tels que l`IMC, le tabagisme, la consommation d`alcool, l`hyperlipidémie, l`hypertension et le diabète). (18)
CONCLUSION
La dermatite atopique est une affection fréquente dans nos consultations. La littérature conclut que cette population de patients est plus « à risque » de certaines comorbidités que la population générale. Le dermatologue est souvent le premier professionnel de santé consulté. Pendant l’enfance, le risque est significativement plus élevé de développer une rhinite allergique, une allergie alimentaire, un asthme allergique, des infections cutanées récurrentes et des pathologies ophtalmiques. L’éducation des patients est importante pour les sensibiliser aux symptômes d’alarme possibles, afin qu’une aide puisse être proposées rapidement et ainsi permettre le contrôle des symptômes.
Il est nécessaire d’informer les patients adultes des risques cardiovasculaires et des conséquences psychosociales de la DA ; cela ne demande pas nécessairement beaucoup de temps, mais peut se traduire par le simple conseil de se faire suivre par son médecin généraliste (contrôle de la tension artérielle par exemple).
LISTE D’ABRÉVIATIONS
AA asthme allergique
CA conjonctivite allergique
DA dermatite atopique
TDAH trouble du déficit de l’attention/hyperactivité
RA rhinite allergique
SARM S. aureus résistant à la méthicilline
OR odds ratio
ALA allergie alimentaire
RÉFÉRENCES
1. Davis DMR, Drucker AM, Alikhan A, Bercovitch L, Cohen DE, Darr JM, et al. American Academy of Dermatology Guidelines: Awareness of comorbidities associated with atopic dermatitis in adults. J Am Acad Dermatol. 2022 Jun;86(6):1335-1336.e18.
2. Roduit C, Frei R, Depner M, Karvonen AM, Renz H, Braun-Fahrländer C, et al. Phenotypes of Atopic Dermatitis Depending on the Timing of Onset and Progression in Childhood. JAMA Pediatr. 2017 Jul 1;171(7):655-662.
3. Ravnborg N, Ambikaibalan D, Agnihotri G, Price S, Rastogi S, Patel KR, et al. Prevalence of asthma in patients with atopic dermatitis: A systematic review and meta-analysis. J Am Acad Dermatol. 2021 Feb;84(2):471-478.
4. Knudgaard MH, Andreasen TH, Ravnborg N, Bieber T, Silverberg JI, Egeberg A, Halling AS, Thyssen JP. Rhinitis prevalence and association with atopic dermatitis: A systematic review and meta-analysis. Ann Allergy Asthma Immunol. 2021 Jul;127(1):49-56.e1.
5. Spolidoro GCI, Amera YT, Ali MM, Nyassi S, Lisik D, Ioannidou A, et al. Frequency of food allergy in Europe: An updated systematic review and metaanalysis. Allergy. 2023 Feb;78(2):351-368.
6. Ferreira MA, Vonk JM, Baurecht H, Marenholz I, Tian C, Hoffman JD, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet. 2017 Dec;49(12):1752-1757.
7. Thyssen JP, Halling AS, Schmid-Grendelmeier P, Guttman-Yassky E, Silverberg JI. Comorbidities of atopic dermatitis-what does the evidence say? J Allergy Clin Immunol. 2023 May;151(5):1155-1162.
8. Maintz L, Schmitz MT, Herrmann N, Müller S, Havenith R, Brauer J, et al. Atopic dermatitis: Correlation of distinct risk factors with age of onset in adulthood compared to childhood. Allergy. 2023 Aug;78(8):2181-2201.
9. Biagini JM, Kroner JW, Baatyrbek Kyzy A, Gonzales A, He H, Stevens M et al. Longitudinal atopic dermatitis endotypes: An atopic march paradigm that includes Black children. J Allergy Clin Immunol. 2022 May;149(5):1702-1710.e4.
10. Ong PY, Leung DY. Bacterial and Viral Infections in Atopic Dermatitis: a Comprehensive Review. Clin Rev Allergy Immunol. 2016 Dec;51(3):329-337.
11. Wang V, Boguniewicz J, Boguniewicz M, Ong PY. The infectious complications of atopic dermatitis. Ann Allergy Asthma Immunol. 2021 Jan;126(1):3-12.
12. Paller A, Jaworski JC, Simpson EL, Boguniewicz M, Russell JJ, Block JK, et al. Major Comorbidities of Atopic Dermatitis: Beyond Allergic Disorders. Am J Clin Dermatol. 2018 Dec;19(6):821-838.
13. Rønnstad ATM, Halling-Overgaard AS, Hamann CR, Skov L, Egeberg A, Thyssen JP. Association of atopic dermatitis with depression, anxiety, and suicidal ideation in children and adults: A systematic review and meta-analysis. J Am Acad Dermatol. 2018 Sep;79(3):448-456.e30. doi: 10.1016/j.jaad.2018.03.017. PMID: 30119868.
14. Matthews T, Danese A, Wertz J, Odgers CL, Ambler A, Moffitt TE, Arseneault L. Social isolation, loneliness and depression in young adulthood: a behavioural genetic analysis. Soc Psychiatry Psychiatr Epidemiol. 2016 Mar;51(3):339-48. doi: 10.1007/s00127-016-1178-7. Epub 2016 Feb 3. PMID: 26843197; PMCID: PMC4819590.
15. Kantor R, Kim A, Thyssen JP, Silverberg JI. Association of atopic dermatitis with smoking: A systematic review and meta-analysis. J Am Acad Dermatol. 2016 Dec;75(6):1119-1125.e1. doi: 10.1016/j.jaad.2016.07.017. Epub 2016 Aug 16. PMID: 27542586; PMCID: PMC5216172.
16. Yousaf M, Ayasse M, Ahmed A, Gwillim EC, Janmohamed SR, Yousaf A, Patel KR, Thyssen JP, Silverberg JI. Association between atopic dermatitis and hypertension: a systematic review and meta-analysis. Br J Dermatol. 2022 Feb;186(2):227-235. doi: 10.1111/bjd.20661. Epub 2021 Sep 30. PMID: 34319589.
17. Zhang A, Silverberg JI. Association of atopic dermatitis with being overweight and obese: a systematic review and metaanalysis. J Am Acad Dermatol. 2015 Apr;72(4):606-16.e4. doi: 10.1016/j.jaad.2014.12.013. PMID: 25773409.
18. Silverwood RJ, Forbes HJ, Abuabara K, Ascott A, Schmidt M, Schmidt SAJ, Smeeth L, Langan SM. Severe and predominantly active atopic eczema in adulthood and long term risk of cardiovascular disease: population based cohort study. BMJ. 2018 May 23;361:k1786. doi: 10.1136/bmj.k1786. PMID: 29792314; PMCID: PMC6190010.
20. Cheng Y, Lu JW, Wang JH, Loh CH, Chen TL. Associations of Atopic Dermatitis with Attention Deficit/Hyperactivity Disorder and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Dermatology. 2024;240(1):13-25. doi: 10.1159/000533366. Epub 2023 Nov 8. PMID: 37939694.
21. Manjunath J, Silverberg JI. Atopic Dermatitis Is Associated With Multiple Behavioral Problems in US Children and Adolescents. Dermatitis. 2022 NovDec 01;33(6S):S52-S60. doi: 10.1097/DER.0000000000000749. Epub 2021 Apr 9. PMID: 33840782.
MARQUE RECOMMANDÉE PAR PLUS DE 70.000 DERMATOLOGUES1
ÉLIMINE 100%
DES PELLICULES VISIBLES2
ACTION ANTIRÉCIDIVE 6 SEMAINES 3
JOUR 0 JOUR 31
APRÈS 4 SEMAINES DE TRAITEMENT
LE MICROBIOME DU CUIR CHEVELU EST RÉÉQUILIBRÉ
DS 1% ACIDE SALICYLIQUE L’EFFET KÉRATOLYTIQUE
1% DISULFURE DE SÉLÉNIUM L’EFFET ANTIFONGIQUE À UTILISER EN MONOTHÉRAPIE OU EN COMPLÉMENT D’UN MÉDICAMENT AVANT/APRÈS CLINIQUEMENT PROUVÉ
1 Enquête menée au sein du marché de la dermocosmétique réalisée par AplusA et d’autres partenaires entre janvier 2023 et mai 2023, auprès de dermatologues de 34 pays, représentant plus de 80% du PIB mondial.
2 Test de consommateur auprès de 262 sujets – utilisation régulière après 2 semaines – Italie.
3 Étude clinique sur 45 sujets après 4 semaines de traitement et 6 semaines traitement d’entretien.
4 étude clinique sur 45 sujets avec 4 applications par semaine, images correspondantes à un résultat équivalent de scorage de pellicules adhérentes.
5 Étude clinique avec suivi des principaux micro-organismes impliqués dans la formation des pellicules, sur 56 sujets avec 3 traitements par semaine pendant 4 semaines.
STEFAN KERRE
Dermatologue, Aarschot - Hôpital Imelda Bonheiden
LA CONNEXION SUÉDOISE
La vascularite est souvent une pathologie difficile en clinique dermatologique, tant en termes de diagnostic que de traitement.
À l’aide d’une étude de cas, une forme rare de vascularite, la vascularite maculaire, également appelée purpura hypergammaglobulémique de Waldenström, est décrite.
Le lien avec la maladie de Sjögren est remarquable, de sorte que l’on peut parler d’une connexion suédoise Sjögren, Waldenström.
CASUS
Une femme de 42 ans est vue en consultation pour l’apparition soudaine et très rapide d’un purpura sur les deux jambes. La patiente présente également un gonflement du genou droit. Elle se plaint également des sueurs nocturnes, mais elle les décrit depuis un certain temps. Elle tousse occasionnellement, il n’y a pas d’arguments clairs en faveur d’un épisode infectieux.
La patiente a comme traitement : befact forte, mirtazapine, serlain, atarax, ibuprofène et pilocarpine en collyre pour les yeux. La patiente souffre depuis un an d’un syndrome de Sjögren qui a débuté par une parotidite et s’est ensuite traduit par une xérostomie et une xérophtalmie classiques.
Sur le plan biochimique, elle présente une hypergammaglobulinémie, un ANA ou ANF positif et une positivité pour le SSA/ Ro 60, le SSA/Ro 52 et le SSB/La.
Il y a quelques semaines, la patiente a présenté un épisode similaire et bref de purpura sur les deux jambes.
L’examen clinique a montré de multiples lésions purpuriques plates sur les deux jambes inférieures et, sur le genou droit, une large zone érythémateuse plate.
Sur le plan biochimique, il n’y a pas d’anomalies évidentes, le coefficient de coagulation est normal, il n’y a qu’une augmentation marginale de la CRP et une légère augmentation de la sédimentation. Les fonctions hépatiques et rénales sont normales. Il y a les ANF positifs déjà connus avec les anticorps de Sjögren et un facteur rhumatoïde de 435 UI/ml (c’est-à-dire <14).
Une biopsie a montré un infiltrat périvasculaire et interstitiel important avec de nombreux neutrophiles et des débris nucléaires. Le DIF (Direct immunofluorescence) était négatif.
Aucun traitement n’a été mis en place et le patient nous a rapporté une disparition complète après 3 jours.
Diagnostic : vascularite maculaire récurrente dans le contexte d’une hypergammaglobulinémie, également appelée purpura hypergammaglobulémique de Waldenström.
DESCRIPTION
Le purpura hypergammaglobulémique de Waldenström se caractérise par des épisodes récurrents et de courte durée de
vascularite d’apparence bénigne (c’est-à-dire avec peu d’inflammation, donnant plutôt une image de purpura plat). Les lésions sont également limitées aux membres dans la grande majorité des cas. Sur le plan biochimique, on retrouve une hypergammaglobulinémie, avec un facteur rhumatoïde positif.
L’affection peut être primaire ou secondaire et elle est alors souvent associée à un syndrome de Sjögren mais aussi à d’autres affections associées à une hypogammaglobulinémie comme une hépatite auto-immune, une hépatite C, un lupus systémique.
L’hypergammaglobulinémie peut créer des complexes immuns qui peuvent déclencher une vascularite. L’évolution positive (inflammation moins prononcée, rémission rapide, présentation histologique classique moins prononcée) est probablement liée au type de complexes immuns formés (plutôt igG et igA qu’IgM, ils sont donc plus petits, activent moins le complément et se résorbent plus rapidement).
Il est également à noter que cette pathologie survient plus facilement en association avec des périodes de position debout prolongée, de marche et de consommation d’alcool, comme c’est le cas pour la vascularite du golfeur.
Le diagnostic est posé sur la base de cette présentation classique complétée par un bilan sanguin, une électrophorèse des protéines (mise en évidence d’une hypogammaglobulinémie), un facteur rhumatoïde, un dosage AFN, un bilan hépatique, un bilan rénal et une analyse d’urine. Une biopsie ne montre pas toujours l’image complète d’une vascularite.
Le traitement n’est généralement pas nécessaire étant donné la rémission rapide des lésions.
Il est particulièrement important d’envisager cette forme bénigne particulière de purpura chez un patient déjà connu pour être atteint du syndrome de Sjögren, ce qui donne lieu à un lien suédois (Sjögren-Waldenström). Des formes plus sévères comme par exemple la vascularite cryoglobulinémique peuvent se produire avec cette pathologie.
« Cette pathologie survient plus facilement en association avec des périodes de position debout prolongée, de marche et de consommation d’alcool, comme c’est le cas pour la vascularite du golfeur. »
FRANCESCO MESSINA
CAS CLINIQUE DE PORPORA TÉLÉANGIECTASIQUE DE MAJOCCHI ET APPROFONDISSEMENT DES DERMATOSES PURPURIQUES PIGMENTÉES
CAS CLINIQUE
Une patiente de 35 ans s’est présentée à la consultation avec des lésions cutanées pigmentées et asymptomatiques, principalement situées sur les pieds et les jambes, apparues quelques mois avant (fig. 1-2). Les lésions, de forme annulaire et non prurigineuses, n’étaient associées à aucun symptôme inflammatoire. La patiente n’a pas d’antécédents familiaux de maladies dermatologiques, mais mentionne des éruptions similaires à l’enfance et à l’adolescence, alors diagnostiquées comme des mycoses.
La patiente avait été initialement traitée avec du mométasone furoate, sans amélioration significative. Un examen mycologique a été effectué et s’est révélé négatif pour la présence de champignons sur les lésions cutanées, bien que des cultures positives aient été obtenues dans les espaces interdigitaux.
L’examen histopathologique d’un fragment cutané incluant l’hypoderme a révélé un épiderme orthokératosique et papillomateux. Le derme superficiel montrait un infiltrat lymphocytaire discret, avec des signes de dermatite de l’interface focale et une vasculopathie sans leucocytoclasie. Une incontinence pigmentaire dans le derme était également observée, tandis que le réseau d’élastine restait intact. La recherche de mycoses par coloration PAS était négative. Ces observations histologiques ainsi que l’aspect clinique des lésions ont conduit au diagnostic de porpora téléangiectasique de Majocchi, une forme rare de capillarite purpurique caractérisée par une altération vasculaire associée à une hyperpigmentation.
Suite à ce diagnostic, un traitement comprenant de la vitamine C (1 g/jour), du rutoside, et un corticostéroïde topique à base de bétaméthasone a été initié. Après six semaines, les lésions cutanées avaient régressé de façon marquée, avec une nette diminution des signes de purpura.
Dans la suite de cet article, nous allons examiner les caractéristiques de la porpora téléangiectasique de Majocchi en les comparant aux autres capillarites purpuriques. Nous explo -
rerons les caractéristiques cliniques et histopathologiques de ces différentes formes de capillarites, en abordant les options diagnostiques et thérapeutiques pour offrir un cadre de référence aux cliniciens
REVUE DE LA LITTERATURE
Les dermatoses purpuriques pigmentées (DPP) sont un groupe de maladies cutanées caractérisées par des hémorragies petechiales dues à une capillarite [1]. Cela entraîne l’extravasation des érythrocytes, provoquant une purpura, et les lésions plus anciennes prennent une teinte rouge-brun en raison des macrophages chargés d’hémosidérine. Les DPP se manifestent généralement par une purpura plate, non palpable, avec une distribution bilatérale sur les jambes, souvent chez les personnes âgées, et suivent un cours de type remittent-relapsing. Bien que les adultes soient plus fréquemment affectés, des cas de DPP chez les enfants sont également bien documentés dans la littérature [2].
Bien que les DPP n’aient pas de symptômes systémiques associés, leur présentation clinique peut parfois susciter des inquiétudes, entraînant une évaluation pour des conditions telles que la thrombocytopénie ou la vascularite. En effet, la nature purpurique (généralement petechiale) des lésions peut conduire à un mauvais diagnostic. En fonction de leur apparence clinique, les DPP sont classées en cinq types principaux : 1) dermatoses pigmentaires progressives (maladie de Schamberg), la plus courante ; 2) dermatoses purpuriques lichénoïdes pigmentées (Gougerot et Blum) ; 3) purpura annulaire télangiectodique (Majocchi) ; 4) purpura de type eczématide (Doucas et Kapetanakis) ; et 5) lichen aureus. D’autres formes plus rares de DPP peuvent se présenter de manière atypique [1-2]
La pathogénie des dermatoses purpuriques pigmentées n’est pas complètement comprise et peut varier selon les sous-types. Cependant, des éléments communs incluent l’inflammation et l’hémorragie des vaisseaux dermiques papillaires superficiels, principalement des capillaires. Le mécanisme inflammatoire sous-jacent à ces conditions est inconnu, mais il semble pro-
venir d’un événement immunologique médié par les cellules [3]. Certaines études suggèrent une régulation à la hausse des molécules d’adhésion cellulaire (CAM), telles que LFA-1 (antigène de fonction des lymphocytes-1) et ICAM-1 (molécule d’adhésion intercellulaire-1) dans les cellules inflammatoires, ainsi que ICAM-1 et ELAM-1 (molécule d’adhésion leucocytaire endothéliale-1) dans les cellules endothéliales [4]. Cette réaction pourrait représenter une réaction d’hypersensibilité médiée par les cellules.
De plus, l’infiltrat inflammatoire périvasculaire inclut des cellules T CD4+ et des cellules dendritiques CD1a+ [5]. Les cytokines produites par les leucocytes peuvent induire l’expression des CAM, influençant l’activité fibrinolytique et provoquant la déposition intravasculaire de fibrine, observée dans les DPP [5,6]. La dilatation et la fragilité des capillaires, peutêtre attribuées à une altération de la fonction des cellules de la structure vasculaire comme les fibroblastes et l’endothélium, peuvent entraîner une fuite de globules rouges, déclenchant une réponse immunitaire.
Certains médicaments, tels que le paracétamol, les anti-inflammatoires non stéroïdiens, les antibiotiques et les antidiabétiques oraux, peuvent constituer des facteurs déclenchants, en particulier dans la maladie de Schamberg [5]. Bien que la plupart des DPP soient idiopathiques, des associations avec des maladies systémiques telles que le diabète, la polyarthrite rhumatoïde, le lupus érythémateux systémique, les dysfonctionnements thyroïdiens, la mycose fongoïde, les néoplasies hématologiques et solides, les maladies hépatiques et l’hyperlipidémie ont été rapportées.
Une autre théorie émergente suggère que les DPP pourraient représenter une altération épithélitropique des cellules T, soutenue par des observations d’épidermotropisme ou par un motif monoclonal dans l’infiltrat inflammatoire. Il a été rapporté que les DPP progressaient vers la mycose fongoïde, soulignant la nécessité d’évaluations cliniques, moléculaires et histopathologiques intégrées pour un diagnostic et une prise en charge précis [4,7].
HISTOLOGIE
Malgré les variations cliniques, toutes les DPP partagent des caractéristiques histologiques communes. Les quatre éléments caractéristiques principaux sont : la dilatation des vaisseaux sanguins avec un gonflement des cellules endothéliales, les dépôts d’hémosidérine, l’extravasation des globules rouges et l’infiltrat périvasculaire de lymphocytes et de macrophages. Ces caractéristiques sont observées dans toutes les DPP, avec des différences d’intensité notées dans certains sous-types. Les dépôts d’hémosidérine dans le derme superficiel, visibles grâce aux colorations de Perl et Fontana-Masson, permettent de différencier les DPP de la dermatite de stase. L’infiltrat est principalement constitué de lymphocytes CD4+, de cellules dendritiques CD1a+ occasionnelles et de macrophages. Les plasmocytes et les neutrophiles peuvent être présents, ces derniers étant observés dans les lésions purpuriques prurigineuses, comme dans la purpura eczématiforme de Doucas et Kapetanakis [6,8].
L’infiltrat typique du lichen aureus se caractérise par un infiltrat dermique en bande séparé de l’épiderme par une zone de collagène non impliquée (zone de Grenz). Cet infiltrat est également présent dans la dermatosis purpurique lichénoïde de Gougerot et Blum. Une forme rare granulomateuse des DPP, avec un infiltrat granulomateux hémorragique superposé, a également été décrite. La spongiose épidermique légère et l’exocytose lymphocytaire sont courantes dans toutes les variantes, à l’exception du lichen aureus [9].
Une autre caractéristique diagnostique importante est la présence d’une fluorescence immunologique directe positive, qui peut révéler le dépôt de fibrinogène, d’IgM et/ou de C3 dans les vaisseaux dermiques superficiels. En plus de confirmer le diagnostic d’éruption purpurique pigmentée, une biopsie cutanée constitue un outil précieux pour exclure le lymphome cutané à cellules T, en particulier dans ses stades précoces, car il peut imiter à la fois cliniquement et histologiquement les DPP [10].
VARIANTES CLINIQUES
Maladie de Schamberg ou Dermatosis Purpurique
Pigmentée Progressive
La maladie de Schamberg, décrite pour la première fois en 1901, est la forme la plus courante de DPP [1]. Elle peut affecter toutes les tranches d’âge, mais survient principalement chez les hommes d’âge moyen à avancé, et plus rarement chez les enfants [2]. Les localisations typiques sont les membres inférieurs, mais elle peut également toucher les cuisses, les fesses, le tronc et les bras. L’examen clinique montre généralement des pétéchies ponctiformes, comparables à des “poivres de Cayenne”, qui se présentent sous forme de taches jaune-orange-brun avec des contours ovales ou irréguliers. Il s’agit d’une dermatosis chronique et persistante qui, dans certains cas, peut s’étendre aux zones proximales. La plupart des patients sont asymptomatiques, bien que certains signalent des démangeaisons légères. L’insuffisance veineuse pourrait jouer un rôle dans leur localisation.
FIG 1
De plus, une consommation chronique d’alcool semble être liée au développement de la purpura et de la maladie de Schamberg, surtout lorsqu’il y a des dommages au foie [1, 4, 11].
Purpura Annulaire Télangiectodique de Majocchi
Décrite pour la première fois en 1896, la purpura annulaire télangiectodique de Majocchi est une forme rare de DPP qui touche principalement les adolescents et les jeunes adultes, en particulier les femmes [12]. Les lésions initiales sont des macules annulares bleu-rouge, avec des points télangiectatiques rouges foncés ; par la suite, elles s’étendent centrifuge avec une résolution progressive centrale et une légère atrophie, leur conférant une configuration annulaire . L’éruption débute bilatéralement sur les membres inférieurs, puis s’étend aux membres supérieurs, mais une forme unilatérale rare a également été décrite [4]. Les lésions peuvent varier de quelques-unes à innombrables ; les patients peuvent être asymptomatiques ou se plaindre de démangeaisons ou de brûlures légères. Parmi les facteurs déclenchants, en plus de ceux déjà mentionnés, on trouve la sclérothérapie, qui entraîne des dommages endothéliaux directs avec extravasation des globules rouges et dépôt d’hémosidérine [4, 12].
Dermatite Lichénoïde Pigmentée de Gougerot et Blum
Cette entité a été définie par Gougerot et Blum en 1925 . Il s’agit d’une forme rare caractérisée par deux types de lésions : des papules lichénoïdes rouge-brun purpurique et une purpura semblable à celle de Schamberg. Les lésions se situent généralement sur les membres et touchent parfois la muqueuse buccale. Cette maladie touche généralement les hommes d’âge moyen ou âgés, a un cours chronique et se manifeste parfois par des démangeaisons [13].
Lichen Aureus
Décrit pour la première fois comme lichen purpuricus en 1958, il a été renommé lichen aureus par Calnan en 1960. Il s’agit d’une éruption plus localisée, persistante et intensément purpurique, rarement douloureuse. Elle se présente sous forme de lésion purpurique associée à une papule lichénoïde, généralement localisée sur les membres inférieurs, souvent au-dessus d’une veine perforante, mais parfois également sur le tronc, les membres supérieurs, les fesses ou le visage. La couleur varie du doré au rouille au marron-violet, justifiant le terme “lichen aureus” ainsi que le pattern pathologique de dermatite lichénoïde. Contrairement aux autres formes, les jeunes hommes sont les plus affectés [14].
Purpura Eczématiforme de Doucas et Kapetanakis
Cette forme est considérée comme une variété inflammatoire et plus étendue de la maladie de Schamberg, présentée pour la première fois en 1953 . En effet, elle tend à se propager cranialement des membres inférieurs jusqu’à l’abdomen, et se caractérise par des démangeaisons intenses et des macules, papules et plaques purpuriques ou pétéchiales squameuses [15]. À la confirmation, l’histologie montre une spongiose et un infiltrat lymphocytaire périvasculaire intense. La résolution est toujours spontanée, mais les récidives sont fréquentes. Une
association avec une dermatite de contact allergique due au caoutchouc et aux vêtements a été décrite.
AUTRES FORMES
La purpura pigmentée granulomateuse est une forme rare de DPP qui semble toucher plus fréquemment les personnes asiatiques que les caucasiennes. Elle a été décrite pour la première fois en 1997 par Saito et al. [16]. Elle survient généralement chez les patients d’âge moyen ou âgé et suit souvent un cours chronique avec des phases alternées. [17]. L’examen clinique montre souvent des taches brunes avec des papules hémorragiques superposées sur les jambes. L’échantillon histologique est caractérisé par un infiltrat granulomateux hémorragique, comprenant des histiocytes, situé dans le derme superficiel.
La purpura pigmentée linéaire est une forme rare qui peut survenir chez les enfants et les adolescents. Les termes “capillaropathie quadrantique” et “capillarite linéaire unilatérale” ont été utilisés par le passé pour décrire cette entité [4]. Elle se caractérise par des lésions similaires à celles du lichen aureus ou de la maladie de Schamberg, mais unilatérales et linéaires.
La purpura prurigineuse de Loewenthal doit être considérée comme une variante symptomatique de la maladie de Schamberg qui touche presque exclusivement les adultes [4]. Elle se caractérise par une apparition brutale et des démangeaisons sévères sur les membres inférieurs.
DIAGNOSTIC
Le diagnostic est clinique dans la majorité des cas : les macules orange-rouge-brun avec des taches purpuriques, associées aux caractéristiques spécifiques des variantes mentionnées ci-dessus, sont décisives.
L’examen des dermatoses purpuriques pigmentées par dermoscopie révèle des caractéristiques uniques facilitant leur diagnostic. La coloration de base, allant du brun-rouge à l’orange, reflète le dépôt sous-jacent d’hémosidérine. Les points et globules rouges, qui correspondent aux capillaires dilatés et à l’extravasation des globules rouges, sont des observations courantes, contribuant à l’apparence purpurique caractéristique de ces dermatoses. En outre, un motif en réseau de teintes jaunes-brunes, évoquant des vaisseaux ramifiés, peut être discerné, soulignant la nature vasculaire de ces lésions [18]. D’autres caractéristiques moins fréquentes incluent des cercles bruns ou rouges, des points gris, des vaisseaux serpentinés ou épais, ainsi que des structures en rosette. Les points et globules bruns résultent de la présence de mélanocytes ou de mélanophages à la jonction dermo-épidermique [19].
Les tests de numération sanguine et de frottis sanguin périphérique sont nécessaires pour exclure la thrombocytopénie ; un bilan de coagulation peut aider à exclure d’autres causes possibles de purpura. Parfois, des tests pour les facteurs rhumatoïdes, les anticorps antinucléaires, les anticorps anti-virus de l’hépatite B et les anticorps anti-virus de l’hépatite C peuvent être utiles pour identifier les purpuras associés à des maladies auto-immunes ou secondaires à une hépatite.
La DPP doit être différenciée de la purpura causée par une altération des plaquettes et de la purpura non-thrombocytopénique, incluant la “purpura induite par l’exercice”, qui survient sur les jambes après une activité musculaire inhabituelle ou importante [4].
Les éruptions purpuriques pigmentées sur les jambes doivent également être différenciées des hémorragies dermiques secondaires à l’hypertension veineuse, qui se manifestent par des pétéchies superposées à une hémosidérose diffuse, en particulier chez les personnes âgées présentant une dermatite de stase.
Enfin, la dermatite purpurique causée par les vêtements, la purpura hyperglobulinémique, la mycose fongoïde précoce, le scorbut, la vascularite leucocytoclastique et les réactions d’hypersensibilité médicamenteuses peuvent imiter la maladie de Schamberg.
OPTIONS DE TRAITEMENT
Le traitement des dermatoses purpuriques pigmentées présente des défis, car les options disponibles offrent souvent des résultats insatisfaisants. Diverses approches, notamment les agents topiques, les médicaments oraux, la photothérapie et le traitement au laser, ont été utilisées, mais avec un succès limité.
Traitement topique
Les corticostéroïdes topiques sont couramment utilisés, mais les résultats sont souvent insatisfaisants, même avec une combinaison de stéroïdes oraux et topiques [6]. Les traitements topiques combinés avec la vitamine C ou d’autres agents comme le rutoside n’ont pas montré de résultats probants [6]. Les inhibiteurs de la calcineurine (pimécrolimus, tacrolimus) peuvent être envisagés, étant plus efficaces dans certaines formes de DPP, mais pas dans la dermatose lichénoïde pigmentée de Gougerot-Blum [20]. Les crèmes à la vitamine K et les antibiotiques topiques se sont avérés inefficaces pour traiter la maladie de Schamberg.
Pentoxifylline (PTX)
La pentoxifylline (PTX) est un médicament qui a suscité un intérêt croissant dans le traitement des DPP en raison de ses effets bénéfiques sur la microcirculation et l’inflammation. Dans plusieurs études, la PTX a montré des résultats positifs chez des patients atteints de la maladie de Schamberg et d’autres formes de DPP, particulièrement à des doses élevées. Les doses utilisées varient généralement entre 200 et 1200 mg par jour. Kano et al. ont observé des améliorations notables après seulement 2 à 3 semaines d’utilisation de doses faibles (200–300 mg/jour), bien que des doses plus élevées, comme 1200 mg/jour, aient montré une efficacité plus prononcée [21].
L’un des principaux mécanismes d’action de la pentoxifylline réside dans sa capacité à améliorer la fluidité sanguine et à inhiber l’agrégation plaquettaire. Cela est dû à son action sur les phosphodiestérases, en particulier la PDE-4, ce qui entraîne une élévation du cAMP intracellulaire. Cette action contribue à la relaxation des cellules musculaires lisses vasculaires et améliore ainsi la circulation sanguine. En outre, elle joue un rôle clé dans
l’inhibition de l’infiltration des leucocytes, la réduction de l’expression des cytokines pro-inflammatoires (comme TNF-alpha, IL-1, et IL-6) et la suppression de la voie de signalisation NFκB, ce qui conduit à une diminution de l’inflammation et à une amélioration clinique des lésions cutanées [6].
Dans une étude comparative entre la pentoxifylline et les corticostéroïdes topiques, il a été observé que, bien que l’avantage de la pentoxifylline s’estompe après 6 mois de traitement, elle a montré une efficacité supérieure après 2 mois, avec un nombre significativement plus faible de récurrences par rapport aux stéroïdes [21]. De plus, PTX a un profil d’effets secondaires plus favorable par rapport aux corticostéroïdes, ce qui en fait une option préférable pour de nombreux patients, en particulier ceux qui présentent une résistance aux traitements conventionnels.
Photothérapie
La photothérapie à base de PUVA et de lumière UVB-NB a montré des résultats positifs dans le traitement des DPP, en particulier dans la maladie de Schamberg. Ces traitements, qui possèdent des effets immunomodulateurs et anti-inflammatoires, ont été efficaces même en cas de lésions étendues et résistantes aux autres traitements. Un protocole de traitement de maintenance peut permettre une rémission à long terme [22].
Vitamine C, Flavonoïdes et Calcium Dobesilate
L’utilisation de la vitamine C dans le traitement des DPP repose sur ses propriétés antioxydantes et son rôle dans la synthèse du collagène. La vitamine C améliore la résistance des parois vasculaires et réduit la perméabilité endothéliale, ce qui diminue les fuites de sang dans les tissus environnants et atténue les manifestations cliniques des DPP. Dans plusieurs études, des doses de 500 mg d’acide ascorbique deux fois par jour, associées à 50 mg de rutoside deux fois par jour, ont montré une amélioration des lésions cutanées ou même une rémission
FIG 2
complète des symptômes [6, 23]. Les bioflavonoïdes, comme le rutoside, ainsi que le calcium dobesilate, ont également montré une action bénéfique en réduisant les radicaux libres, en inhibant la production de prostaglandines et en diminuant l’expression des facteurs de croissance vasculaire.
En plus de leurs effets antioxydants, la vitamine C et les flavonoïdes ont une action protectrice sur les vaisseaux sanguins. Ils réduisent l’activation des cellules endothéliales, par exemple en inhibant l’expression des molécules d’adhésion cellulaires comme ICAM-1 et VCAM-1, et ils diminuent les métalloprotéinases (MMP), qui dégradent les composants de la matrice extracellulaire. De plus, le calcium dobesilate contribue à la relaxation des vaisseaux en favorisant la production d’oxyde nitrique (NO), ce qui diminue les lésions cellulaires endothéliales et améliore la circulation [23, 24].
Ces traitements sont généralement bien tolérés par les patients et n’ont pas d’effets secondaires majeurs. En raison de leur efficacité et de leur bonne tolérance, ils représentent une option thérapeutique viable, en particulier lorsqu’ils sont utilisés dans des thérapies combinées avec d’autres médicaments, comme les corticostéroïdes ou les agents topiques.
Médicaments Immunomodulateurs et Immunosuppresseurs
Des rapports anecdotiques indiquent l’utilisation de médicaments immunomodulateurs, tels que la dapsone et la colchicine, dans les DPP résistantes aux stéroïdes. La dapsone et la colchicine ont montré une réduction significative des lésions cutanées chez certains patients, bien que ces traitements ne soient pas systématiquement utilisés [25,26]. L’efficacité de ces thérapies soutient une étiologie immunologique pour les DPP.
Dispositifs Basés sur l’Énergie
Le traitement au laser, notamment au laser vasculaire 595 nm et à la lumière pulsée intense (IPL), a montré des résultats positifs dans le traitement de la maladie de Schamberg et d’autres DPP. Ces thérapies, qui visent à éliminer les dépôts d’hémosidérine et à remodeler le collagène, sont prometteuses pour les lésions localisées résistantes aux traitements topiques [27].
Approche de Surveillance Active
Dans de nombreux cas, les DPP sont asymptomatiques et ont une évolution auto-limitante. Par conséquent, une approche de surveillance active sans traitement immédiat peut être envisagée [28]. Plusieurs études ont rapporté une amélioration spontanée dans les DPP, et certains patients peuvent préférer cette approche au traitement actif, en particulier lorsque l’impact psychologique et physique est faible. Toutefois, pour les patients présentant des formes plus sévères ou persistantes, un traitement actif est recommandé.
En conclusion, la diversité des options thérapeutiques, qu’il s’agisse de traitements topiques, systémiques ou de photothérapie, reflète la complexité des mécanismes sous-jacents à ces dermatoses. L’approche personnalisée et la surveillance
active, notamment pour les formes asymptomatiques, restent essentielles pour garantir le bien-être des patients tout en minimisant l’intervention médicale inutile.
REFERENCES
1. Torrelo, A.; Requena, C.; Mediero, I.G.; Zambrano, A. Schamberg’s purpura in children: A review of 13 cases. J. Am. Acad. Dermatol. 2003, 48, 31–33.
2. Coulombe, J.; Jean, S.E.; Hatami, A.; Powell, J.; Marcoux, D.; Kokta, V.; McCuaig, C. Pigmented purpuric dermatosis: Clinicopathologic characterization in a pediatric series. Pediatr. Dermatol. 2015, 32, 358–362.
3. Smoller, B.R.; Kamel, O.W. Pigmented Purpuric Eruptions: Immunopathologic Studies Supportive of a Common Immunophenotype. J. Cutan. Pathol. 1991 18, 423–427
4. Spigariolo, C.B.; Giacalone, S.; Nazzaro, G. Pigmented Purpuric Dermatoses: A Complete Narrative Review. J. Clin. Med. 2021, 10, 2283. https://doi. org/10.3390/jcm10112283
5. Ghersetich, I.; Lotti, T.; Bacci, S.; Comacchi, C.; Campanile, G.; Romagnoli, P. Cell infiltrate in progressive pigmented purpura (Schamberg’s disease): Immunophenotype, adhesion receptors, and intercellular relationships. Int. J. Dermatol. 1995, 34, 846–850.
6. Kimak, A.; Z˙ ebrowska, A. Therapeutic Approach in Pigmented Purpuric Dermatoses—A Scoping Review. Int. J. Mol. Sci. 2024, 25, 2644. https://doi. org/10.3390/ ijms25052644
7. Magro, C.M.; Schaefer, J.T.; Crowson, A.N.; Li, J.; Morrison, C. Pigmented purpuric dermatosis: Classification by phenotypic and molecular profiles. Am. J. Clin. Pathol. 2007, 128, 218–229.
8. Huang, Y.-K.; Lin, C.-K.; Wu, Y.-H. The Pathological Spectrum and Clinical Correlation of Pigmented Purpuric Dermatosis—A Retrospective Review of 107 Cases. J. Cutan. Pathol. 2018, 45, 325–332.
9. Zeng, Y.P.; Fang, K.; Ma, D.L. Lichen aureus: Clinicopathological features in a Chinese series. Eur. J. Dermatol. 2016, 26, 290–294.
10. Ratnam, K.V.; Su, W.P.D.; Peters, M.S. Purpura simplex (inflammatory purpura without vasculitis): A clinicopathologic study of 174 cases. J. Am. Acad. Dermatol. 1991, 25, 642–647
11. Torrelo, A.; Requena, C.; Mediero, I.G.; Zambrano, A. Schamberg’s Purpura in Children: A Review of 13 Cases. J. Am. Acad. Dermatol. 2003, 48, 31–33.
12. Hoesly, F.J.; Huerter, C.J.; Shehan, J.M. Purpura Annularis Telangiectodes of Majocchi: Case Report and Review of the Literature. Int. J. Dermatol. 2009, 48, 1129–1133.
13. Park, M.-Y.; Shim, W.-H.; Kim, J.-M.; Kim, G.-W.; Kim, H.-S.; Ko, H.-C.; Kim, M.-B.; Kim, B.-S. Dermoscopic Finding in Pigmented Purpuric Lichenoid Dermatosis of Gougerot-Blum: A Useful Tool for Clinical Diagnosis. Ann. Dermatol. 2018, 30, 245–247.
14. Mahajan, V.K.; Chauhan, P. Lichen Aureus. Indian J. Pediatr. 2014, 81, 420–421.
16. Saito, R.; Matsuoka, Y. Granulomatous Pigmented Purpuric Dermatosis. J. Dermatol. 1996, 23, 551–555.
17. Allan, A.; Altman, D.A.; Su, W. Granulomatous Pigmented Purpuric Dermatosis. Cutis 2017, 100, 256–258.
18. Ozkaya, D.B.; Su, O.; Bahali, A.G.; Demirkesen, C.; Emiroglu, N.; Cengiz, F.P.; Yildiz, P.; Onsun, N. Dermatoscopic findings of pigmented purpuric dermatosis. An. Bras. Dermatol. 2016, 91, 584.
19. Metin, M.S.; Elmas, O.F. Dermoscopic profile of pigmented purpuric dermatosis: New observations. Adv. Dermatol.Allergol./Poste,py Dermatol. I Alergol. 2019, 36, 687.
20. Murota, H.; Katayama, I. Lichen aureus responding to topical tacrolimus treatment. J. Dermatol. 2011, 38, 823–825
21. Panda, S.; Malahar, S.; Lahiri, K. Oral Pentoxifylline vs Topical Betamethasone in Schamberg Disease: A Comparative Randomized Investigator-Blinded Parallel-Group Trial. Arch. Dermatol. 2004, 140, 491–493.
22. Fathy, H.; Abdelgaber, S. Treatment of pigmented purpuric dermatoses with Narrow-band UVB: A report of six cases. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 603–606.
23. Schober, S.M.; Peitsch, W.K.; Bonsmann, G.; Metze, D.; Thomas, K.; Goerge, T.; Luger, T.A.; Schneider, S.W. Early treatment with rutoside and ascorbic acid is highly effective for progressive pigmented purpuric dermatosis | Die frühzeitige Therapie der Purpura pigmentosa progressiva mit Rutosid und Ascorbinsäure ist hocheffektiv. JDDG J. Ger. Soc. Dermatol. 2014, 12, 1112–1119.
24. Gupta, A.; Sardana, K.; Kishan Gautam, R. Venoprotective drugs in pigmented purpuric dermatoses: A case report. J. Cosmet. Dermatol. 2019, 18, 1580–1583.
25. Geller, M. Benefit of colchicine in the treatment of Schamberg’s disease. Ann. Allergy Asthma Immunol. 2000, 85, 246.
26. Cavalcante, M.L.L.L.; Masuda, P.Y.; de Brito, F.F.; Pinto, A.C.V.D.; Itimura, G.; Nunes, A.J.F. Schamberg’s disease: Case report with therapeutic success by using colchicine. An. Bras. Dermatol. 2017, 92, 246–248.
27. Hilerowicz, Y.; Sprecher, E.; Gat, A.; Artzi, O. Successful treatment of Schamberg’s disease with fractional non-ablative 1540 nm erbium: Glass laser. J. Cosmet. Laser Ther. 2018, 20, 265–268.
28. Ollech, A.; Paller, A.S.; Kruse, L.; Kenner-Bell, B.; Chamlin, S.; Wagner, A.; Shen, L.; Yousif, R.; Balmert, L.C.; Mancini, A.J. Pigmented purpuric dermatosis in children: A retrospective cohort with emphasis on treatment and outcomes. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2402–2408.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identi cation rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir «Effets indésirables» pour les modalités de déclaration des effets indésirables. DENOMINATION DU MEDICAMENT Adtralza 150 mg solution injectable en seringue préremplie ; Adtralza 300 mg solution injectable en stylo prérempli. COMPOSITION QUALITATIVE ET QUANTITATIVE Adtralza 150 mg solution injectable en seringue préremplie : Chaque seringue préremplie contient 150 mg de tralokinumab dans 1 ml de solution (150 mg/ml). Adtralza 300 mg solution injectable en stylo prérempli : Chaque stylo prérempli contient 300 mg de tralokinumab dans 2 ml de solution (150 mg/ml). Le tralokinumab est produit dans des cellules de myélome de souris grâce à la technologie de l’ADN recombinant. FORME PHARMACEUTIQUE Solution injectable (injection). Solution limpide à opalescente, incolore à jaune pâle, avec un pH de 5,5 et une osmolarité d’environ 280 mOsm/l. INDICA-
TIONS THERAPEUTIQUES Adtralza est indiqué dans le traitement de la dermatite atopique modérée à sévère de l’adulte et de l’adolescent à partir de 12 ans qui nécessitent un traitement systémique. POSOLOGIE ET MODE D’ADMINISTRATION Le traitement doit être instauré par des professionnels de santé expérimentés dans le diagnostic et le traitement de la dermatite atopique. Posologie : La dose recommandée de tralokinumab pour les patients adultes et les adolescents à partir de 12 ans est une dose initiale de 600 mg de la manière suivante: quatre injections de 150 mg en seringues préremplies ou deux injections de 300 mg en stylos préremplis. Cette dose initiale est suivie d’une injection de 300 mg administrée toutes les 2 semaines de la manière suivante: deux injections de 150 mg en seringues préremplies ou une injection de 300 mg en stylo prérempli. Une administration toutes les quatre semaines peut être envisagée par le prescripteur pour les patients dont la peau est blanchie ou presque blanchie après 16 semaines de traitement. La probabilité de maintenir une peau blanchie ou presque blanchie peut être diminuée avec une administration toutes les 4 semaines (voir rubrique 5.1 du RCP complet). Un arrêt du traitement doit être envisagé chez les patients n’ayant pas présenté de réponse après 16 semaines de traitement. Certains patients présentant initialement une réponse partielle peuvent béné cier d’une amélioration de la réponse par la suite en poursuivant le traitement toutes les deux semaines au-delà de 16 semaines. Le tralokinumab peut être utilisé avec ou sans corticostéroïdes topiques. L’utilisation de corticostéroïdes topiques, lorsqu’elle est appropriée, peut apporter un effet complémentaire à l’efcacité globale du tralokinumab (voir rubrique 5.1 du RCP complet). Les inhibiteurs de la calcineurine topiques peuvent être utilisés, mais ils doivent être limités aux zones sensibles, telles que le visage, le cou, les zones intertrigineuses et les parties génitales. Dose oubliée : En cas d’oubli d’une dose, celle-ci devra être administrée dès que possible. Par la suite, le schéma d’administration devra être repris à la date prévue habituellement. Populations particulières : Sujets âgés : Aucun ajustement posologique n’est recommandé chez les patients âgés (voir rubrique 5.2 du RCP complet). Les données disponibles chez les patients de plus de 75 ans sont limitées. Insufsance rénale Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuf sance rénale. Les données disponibles chez les patients présentant une insuf sance rénale sévère sont très limitées (voir rubrique 5.2 du RCP complet). Insu sance hépatique : Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuf sance hépatique. Les données disponibles chez les patients présentant une insuf sance hépatique modérée ou sévère sont très limitées (voir rubrique 5.2 du RCP complet). Poids corporel élevé : Chez les patients de poids corporel élevé (> 100 kg), dont la peau est blanchie ou presque blanchie après 16 semaines de traitement, la réduction de la posologie à une administration toutes les 4 semaines peut ne pas être appropriée (voir rubrique 5.2 du RCP complet). Population pédiatrique : La sécurité et l’ef cacité du tralokinumab chez les enfants âgés de moins de 12 ans n’ont pas encore été établies. Aucune donnée n’est disponible. Mode d’administration : Voie sous-cutanée. La seringue préremplie ou le stylo prérempli ne doivent pas être secoués. Après avoir sorti les seringues préremplies ou les stylos préremplis du réfrigérateur, il est conseillé d’attendre qu’elles/ils reviennent à température ambiante en patientant 30 minutes avant de réaliser l’injection avec la seringue préremplie, 45 minutes avant de réaliser l’injection avec le stylo prérempli. Le tralokinumab est administré par injection souscutanée dans la cuisse ou l’abdomen, à l’exception d’une zone de 5 centimètres autour du nombril. L’injection peut également être effectuée dans le haut du bras si elle n’est pas réalisée par le patient lui-même. Pour la dose initiale de 600 mg, quatre seringues préremplies de 150 mg ou deux stylos préremplis de 300 mg doivent être administrés consécutivement en choisissant des sites d’injection différents dans la même zone du corps. Il est recommandé de changer de site d’injection à chaque injection. Le tralokinumab ne doit pas être injecté dans des zones où la peau est sensible, endommagée, ou présente des contusions ou des cicatrices. Un patient peut s’injecter le tralokinumab lui-même ou un soignant peut lui administrer le tralokinumab si le professionnel de santé juge que cela est approprié. Une formation adéquate des patients et/ou des soignants à l’administration du tralokinumab devra être assurée avant utilisation. Des Instructions d’utilisation détaillées sont présentées à la n de la notice. CONTRE-INDICATIONS Hypersensibilité à la substance active ou à l’un des excipients. EFFETS INDESIRABLESRésumé du pro l de sécurité : Les effets indésirables les plus fréquents sont des infections des voies respiratoires supérieures (23,4% ; principalement signalées comme des rhumes ordinaires), des réactions au site d’injection (7,2%), des conjonctivites (5,4%) et des conjonctivites allergiques (2,0%). Tableau des e ets indésirables : Les effets indésirables observés dans les essais cliniques sont présentés par classe de systèmes d’organes et par fréquence, selon les catégories suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. Ces fréquences sont basées sur la période de traitement initiale de 16 semaines dans l’ensemble des 5 études portant sur des patients atteints de dermatite atopique. Liste des effets indésirables : Infections et infestations : Très fréquent : Infections des voies respiratoires supérieures. Fréquent : Conjonctivite. Affections hématologiques et du système lymphatique : Fréquent : Éosinophilie. Affections oculaires : Fréquent : Conjonctivite allergique. Peu fréquent : Kératite. Troubles généraux et anomalies au site d’administration : Fréquent : Réactions au site d’injection. La sécurité à long terme du tralokinumab a été évaluée dans 2 études en monothérapie jusqu’à 52 semaines et dans 1 étude en association aux corticostéroïdes topiques jusqu’à 32 semaines. Le pro l de sécurité du tralokinumab à 52 semaines et à 32 semaines, respectivement, correspondait au pro l de sécurité observé jusqu’à
la semaine 16. Description de certains e ets indésirables : Conjonctivite et événements liés :
Une conjonctivite est survenue plus fréquemment chez les patients atteints de dermatite atopique traités par tralokinumab (5,4%) que chez ceux ayant reçu le placebo (1,9%), au cours de la période de traitement initiale de 16 semaines dans l’ensemble des 5 études. Une conjonctivite a été rapportée plus fréquemment chez les patients présentant une dermatite atopique sévère que chez les patients présentant une dermatite atopique modérée, à la fois dans le groupe de patients traités par tralokinumab (6,0% contre 3,3% ; période de traitement initiale) et dans le groupe de patients ayant reçu le placebo (2,2% contre 0,8% ; période de traitement initiale). La plupart des patients ont guéri ou étaient en cours de guérison pendant la période de traitement. Une kératite a été signalée chez 0,5% des patients traités par tralokinumab, au cours de la période de traitement initiale. Parmi ces cas, la moitié était classée comme kératoconjonctivites, toutes étaient non graves et de sévérité légère à modérée, et aucune n’a entraîné l’arrêt du traitement. Éosinophilie : Des effets indésirables tels que des éosinophilies ont été signalés chez 1,3% des patients traités par tralokinumab et 0,3% des patients ayant reçu le placebo, au cours de la période de traitement initiale de 16 semaines dans l’ensemble des 5 études. Les patients traités par tralokinumab ont présenté une augmentation initiale moyenne de leur taux d’éosinophiles par rapport à l’inclusion, plus importante que les patients ayant reçu le placebo. Une éosinophilie (≥ 5 000 cellules/µl) a été mesurée chez 1,2% des patients traités par tralokinumab et 0,3% des patients ayant reçu le placebo, au cours de la période de traitement initiale. Toutefois, cette augmentation chez les patients traités par tralokinumab était transitoire et le taux moyen d’éosinophiles est revenu à sa valeur à l’inclusion lors de la poursuite du traitement. Le pro l de sécurité pour les patients présentant une éosinophilie était comparable au pro l de sécurité de tous les patients. Eczéma herpétiforme : Des cas d’eczéma herpétiforme ont été signalés chez 0,3% des patients traités par tralokinumab et 1,5% des patients du groupe placebo, au cours de la période de traitement initiale de 16 semaines dans l’ensemble des 5 études portant sur la dermatite atopique. Quelle que soit la durée de traitement dans ces 5 études, tous les événements d’eczéma herpétiforme signalés dans le groupe tralokinumab étaient non-graves, aucun n’était grave, et un seul événement chez un patient a conduit à l’arrêt dé nitif du traitement. Immunogénicité : Comme avec toutes les protéines thérapeutiques, il existe un risque d’immunogénicité avec le tralokinumab. Les réponses en anticorps anti-médicament (Anti-drug-antibody, ADA) n’ont pas eu d’impact sur l’exposition, la sécurité ou l’efcacité du tralokinumab. Dans les études ECZTRA 1, ECZTRA 2, ECZTRA 3 et dans l’étude sur la réponse vaccinale, l’incidence des ADA jusqu’à 16 semaines était de 1,4% chez les patients traités par tralokinumab et de 1,3% chez les patients ayant reçu le placebo ; des anticorps neutralisants ont été observés chez 0,1% des patients traités par tralokinumab et 0,2% des patients ayant reçu le placebo. Quelles que soient les durées des études, l’incidence des ADA chez les patients traités par tralokinumab était de 4,6% ; 0,9% des patients a présenté des ADA persistants et 1,0% des patients a présenté des anticorps neutralisants. Réactions au site d’injection : Des réactions au site d’injection (incluant douleur et rougeur) sont apparues plus fréquemment chez les patients ayant reçu le tralokinumab (7,2%) que ceux ayant reçu le placebo (3,0%) au cours de la période de traitement initiale de 16 semaines dans l’ensemble des 5 études. Quelle que soit la durée de traitement dans ces 5 études portant sur la dermatite atopique, la grande majorité (99%) des réactions au site d’injection étaient d’intensité légère à modérée, et peu de patients (< 1%) ont interrompu le traitement par tralokinumab. La plupart des réactions au site d’injection signalées étaient de courte durée et approximativement 76% d’entre elles se sont résolues après 1 à 5 jours. Population pédiatrique La sécurité du tralokinumab a été évaluée dans une étude chez 289 patients âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère (ECZTRA 6). Le pro l de sécurité du tralokinumab chez ces patients suivis pendant la période de traitement initiale de 16 semaines et la période à long terme de 52 semaines était semblable au pro l de sécurité constaté dans les études chez les adultes. Cependant, une fréquence plus faible de sujets avec des conjonctivites a été observée avec le tralokinumab chez les adolescents (1,0%) que chez les adultes (5,4%), et contrairement à chez l’adulte, la fréquence des conjonctivites allergiques était similaire pour le tralokinumab et le placebo chez les patients adolescents. Déclaration des e ets indésirables suspectés : La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle per met une surveillance continue du rapport bénéfi ce/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via : Belgique : Agence fédérale des médicaments et des produits de santé, Division Vigilance, Avenue Galilée 5/03, 1210 Bruxelles ou Boîte Postale 97, B-1000 Bruxelles, Madou, Site internet: www.notifi eruneffetindesirable.be, e-mail: adr@af-mps.be. Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la Pharmacie et des Médicaments de la Direction de la santé, site internet: www.guichet.lu/ pharmacovigilance. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE LEO Pharma A/S. NUMERO D’AUTORISATION DE MISE SUR LE MARCHE EU/1/21/1554/001-005. DELIVRANCE Sur prescription médicale. DATE DE MISE A JOUR DU TEXTE 09/2023. Des informations détaillées sur ce médicament sont dispo-nibles sur le site internet de l’Agence européenne des médicaments
LA DERMATOPOROSE : UN MARQUEUR DE FRAGILITÉ
La dermatoporose s’associe à d’autres pathologies liées au vieillissement d’organes et s’inscrit en tant que telle comme insuffisance d’organe par sa fragilité « extrême » cutanée. Avec l’augmentation de l’espérance de vie, l’approche du vieillissement cutané prend une nouvelle dimension, qui n’est plus seulement d’ordre cosmétique, mais aussi fonctionnelle. Les dermatologues sont des acteurs-clés de sa prévention et de son traitement.
N. Evrard, d’après une présentation de Françoise Guiot, Dermatologue, Grez-Doiceau
SUR LE PLAN CLINIQUE
Le terme de dermatoporose a été proposé par analogie avec l’ostéoporose pour désigner le vieillissement cutané. La dermatoporose est caractérisée par une atrophie cutanée, un purpura plan, des cicatrices blanches. La peau atrophique est plus fragile, la cicatrisation est ralentie laissant des marques définitives. Des hémorragies superficielles sont fréquentes avec parfois des hémorragies profondes sévères disséquantes. En conséquence, la répercussion fonctionnelle et esthétique de la dermatoporose impacte très significativement la qualité de vie quotidienne. Chez de nombreuses personnes âgées de 50 ans et plus, de nombreux facteurs peuvent accélérer la fragilité vasculaire cutanée liée a l’âge : la prise d’antiagrégants plaquettaires (aspirine, clopidrogel), d’anticoagulants oraux (antivitamine K, nouveaux anticoagulants oraux), une thrombopénie (taux < 40 000 éléments par mm3 ), les corticoïdes pris au long cours (les corticostéroïdes topiques et/ou systémiques, lesquels sont connus pour moduler l’expression des gènes des collagènes I, III, IV et V, de l’élastine, des métalloprotéinases 1, 2 et 3, de la ténascine, ainsi que des inhibiteurs de métalloprotéinases 1, 2 et 6), un diabète, une carence en vitamine C, mais aussi un mécanisme d’héliodermie sur les parties découvertes, ainsi qu’un déficit vasculaire veineux des jambes.
– Les manifestations cliniques de la dermatoporose comprennent des marqueurs morphologiques de fragilité (atrophie cutanée, purpura sénile, pseudo-cicatrices stellaires), ainsi que l’expression fonctionnelle de la fragilité cutanée (lacérations cutanées, mauvaise cicatrisation, hémorragies sous-cutanées). (figure1) –
PHYSIOPATHOLOGIE
Plusieurs mécanismes potentiels expliquent la fragilité cutanée dans la dermatoporose. Le stress oxydant se manifeste par la production de formes réactives de l’oxygène et de radicaux libres, lesquels provoquent entre autres des altérations de l’ADN nucléaire et mitochondrial et l’activation de diverses métalloprotéinases matricielles, avec de nombreuses conséquences
Figure 1 : Aspects cliniques, échographiques et histologiques de la dermatoporose
Avant-bras (A), examens échographique (C) et histologique (E) d’un patient présentant une atrophie cutanée, un purpura sénile et des pseudo-cicatrices stellaires.
Avantbras (B), examens échographique (D) et histologique (F) d’un sujet sain.
biologiques. Ainsi les UV activent des récepteurs de facteurs de croissance situés à la surface des kératinocytes, et il en résulte un signal mettant en jeu l’activation de facteurs de transcription de la famille AP-1 dans le noyau, ce qui interfère avec la production de collagène, en se liant et séquestrant des facteurs nécessaires à la transcription des procollagènes de types I et III (TGFb). La signalisation dépendant de l’acide hyaluronique et du CD44 est également réprimée par les UV.Les altérations de
Les 4 stades de la dermatoporose
• Stade I. Ce stade est caractérisé par un fort amincissement de la peau avec purpura sénile et pseudo-cicatrices stellaires. L’évaluation clinique de la visco-élasticité de la peau est un bon paramètre de mesure.
Stade II. En plus des lésions observées dans le stade I, on observe de petites lacérations cutanées localisées, lesquelles résultent d’un clivage entre le derme et l’épiderme.
• Stade III. Les lacérations cutanées sont plus nombreuses et plus grandes, et peuvent impliquer toute la surface des extrémités. Il y a clairement un retard de cicatrisation.
• Stade IV. La progression de ces lésions conduit à l’apparition d’hématomes cutanés disséquants, constituant une urgence médicale et nécessitant une hospitalisation.
Aspects clinique (A) et histologique (B) du purpura sénile
Pseudo-cicatrices : linéaire (A), en forme d’étoile (B) ou en plaques (C)
la visco-élasticité cutanée provoquent des fractures des fibres de collagène et une diminution de l’acide hyaluronique, dont la principale conséquence est une perte progressive de cette propriété essentielle de la peau qu’est sa visco-élasticité. L’acide hyaluronique, principal constituant de la matrice extracellulaire, diminue progressivement au cours du vieillissement. D’autre part, l’acide hyaluronique, en dehors de ses propriétés physiques participant à la visco-élasticité de la peau, est une molécule biologiquement active, induisant une signalisation intracellulaire suite à sa liaison avec un récepteur membranaire appelé CD44. Des études indiquent que la voie de signalisation dépendant de l’acide hyaluronique et du CD44 est affectée dans la dermatoporose et constitue une bonne cible pour le développement de stratégies thérapeutiques pour cette pathologie dermatologique.
PRISE EN CHARGE
La pathologie évolue en 4 stades, et au stade initial, les différentes manifestations cliniques doivent éveiller la vigilance des soignants et des personnes concernées. Le traitement repose avant tout sur la prévention. « Ne pas trop s’exposer au soleil et se protéger à chaque exposition est indispensable. Je recommande une protection d’indice élevé avec un spectre de protection incluant les UVA qui pénètrent le derme. Se protéger des UVB pour éviter les coups de soleil n’est pas suffisant. Bien hydrater sa peau est aussi un incontournable pour ralentir le vieillissement cutané. Pour cela, je recommande de privilégier des soins à base d’acide hyaluronique car les patients atteints de dermatoporose présentent une importante réduction de la production d’acide hyaluronique. C’est un actif hydratant reconnu. Son effet réparateur sur les différents manifestations cliniques de la dermatoporose, en association avec d’autres actifs tels que l’acide boswellique, la vitamine B5 et la centella asiatica, a été prouvé » explique Françoise Guiot.
LES ACIDES BOSWELLIQUES, UN ACTIF INNOVANT EN DERMO-COSMETIQUE
La recherche moderne s’est intéressée aux propriétés dermatologiques de la gomme-résine du Boswellia serrata, et plus globalement à l’ensemble des gommes-résines exsudées de Boswellia serrata. En effet, la médecine traditionnelle lui attribue des propriétés anti-inflammatoires pour le traitement de diverses affections cutanées comme par exemple dans l’accompagnement du traitement du psoriasis, de la dermatite atopique, de la dermatite séborrhéique, et de la dermatoporose. Les acides boswelliques inhibent le facteur nucléaire kappa-B (NF-κB), un facteur de transcription impliqué dans la réponse inflammatoire. Ils inhibent également l’élastase leucocytaire.
Les acides boswelliques provoquent une diminution des cytokines pro-inflammatoires et une diminution du blocage des niveaux d’interleukines. Ils entraînent, outre une activité antiallergique associée à une dégradation des mastocytes, une diminution de la production d’histamine. Les acides boswelliques ont également une action désinfectante (antibactérienne et antivirale). Ils boostent également la production d’acide hyaluronique. Les acides boswelliques en topique ont été étudiés dans le traitement du psoriasis et de l’eczéma érythémateux. Le traitement topique, en se localisant préférentiellement dans les structures protéiques de la couche cornée, a amélioré les desquamations (70%), les démangeaisons (60%) et l’érythème (50%) sans aucun cas d’aggravation .
RÉFÉRENCES
Kaya G, Saurat J-H. Dermatoporosis: A Chronic Cutaneous Insufficiency/ Fragility Syndrome. Dermatology. 18 octobre 2007; 215(4):28494
Kaya G, Tran C, Sorg O, Hotz R, Grand D, Carraux P, et al. Hyaluronate Fragments Reverse Skin Atrophy by a CD44-Dependent Mechanism. PLOS Medicine. 19 décembre 2006; 3(12):e493
Baumann L. Skin ageing and its treatment. Journal of Pathology. Janvier 2007; 211(2):24151
• Schoepe S, Schäcke H, May E, Asadullah K. Glucocorticoid therapy-induced skin atrophy. Experimental Dermatology Journal. 2006; 15(6):406–420
Kaya G. New therapeutic targets in dermatoporosis. Journal of Nutrition, Health and Aging. 2012; 16(4):285–288
• Togni S, Maramaldi G, Di Pierro F, Biondi M. A cosmeceutical formulation based on boswellic acids for the treatment of erythematous eczema and psoriasis. Clin Cosmet Investig Dermatol. 2014 Nov 11;7:321-7. doi: 10.2147/ CCID.S69240. PMID: 25419153; PMCID: PMC4235203.
Evolution des conjonctivites survenant sous dupilumab pour le traitement de la dermatite atopique après remplacement du dupilumab par du tralokinumab ou un inhibiteur de Janus kinase
Des auteurs ont mené une étude rétrospective sur des données recueillies à l’arrêt du dupilumab, trois à six mois après, puis lors d’une dernière visite. Parmi 1 109 patients inclus dans 12 centres, 83 ont développé une affection oculaire nécessitant l’arrêt du dupilumab (7,5 %) et un changement de traitement. L’analyse a porté sur 71 patients (35 ans en moyenne, 55 % d’hommes) qui ont été vus ensuite au moins une fois. Lorsque ces patients ont arrêté le dupilumab, leur atteinte cutanée avait diminué. Le délai moyen entre le début du traitement par DU et l’apparition de l’atteinte était de 4,5 mois : blépharoconjonctivite dans 70 % des cas, kératoconjonctivite dans 24 % et sècheresse oculaire dans 4 %. La maladie oculaire était sévère pour 30 %, modérée dans 63 % et légère dans 6 %.
Parmi ces 71 patients, 25 sont passés au tralokinumab et 46 à un inhibiteur de JAK dont 19 au baricitinib, 24 à l’upadacitinib et 3 à l’abrocitinib. Lors de la deuxième visite, à trois-six mois après l’arrêt du dupilumab, deux patients étaient repassés au dupilumab, 21 étaient sous tralokinumab, 11 sous baricitinib, 34 sous upadacitinib et 3 sous abrocitinib.
La maladie oculaire était alors améliorée chez 77 % des patients, de manière complète chez 59 % (dans un délai de 0,7 mois) et partielle chez 18 % ; il n’y avait pas de changement pour 18 % et une aggravation pour 3 %. Une amélioration était observée pour 52 % des patients passés au tralokinumab, complète dans 12 %, et une aggravation pour deux patients, contre une amélioration complète pour 87,5 % de ceux passés à l’upadacitinib, 79 % au baricitinib et 100 % à l’abrocitinib.
Lors de la dernière visite, à 10,9 mois en moyenne, cinq patients étaient revenus au traitement par dupilumab, 16 étaient encore sous tralokinumab, 12 sous baricitinib, 29 sous upadacitinib et 7 sous abrocitinib. Les trois quarts des patients ont obtenu une résolution totale de leur maladie oculaire, dans un délai de 2,5 mois par rapport à la précédente visite, et 10 % une amélioration partielle. La situation était inchangée dans 14 % et aggravée chez un patient. Enfin, tous les patients qui sont passés à un inhibiteur de JAK ont obtenu une amélioration de leur maladie oculaire, 96 % complète et 4 % partielle, contre une amélioration complète chez 45,5 % de ceux passés au tralokinumab et 22,7 % partielle (31,8 % sans changement).
Source : Reguiai Z. Evolution des conjonctivites survenant sous dupilumab pour le traitement de la dermatite atopique après remplacement du dupilumab par du tralokinumab ou un inhibiteur de Janus Kinase.
Erenumab dans le traitement de la rosacée
Des chercheurs ont évalué l’efficacité et la tolérance d’un Ac monoclonal ciblant le récepteur du CGRP sur l’érythème persistant et le flush de la rosacée. Il s’agissait d’une étude ouverte, non randomisée, prospective, réalisée dans plusieurs centres au Danemark. Les patients éligibles étaient des adultes atteints de rosacée présentant dans les 4 semaines avant le début du traitement au moins 15 jours d’érythème modéré à sévère et/ou de flushs modérés à extrêmes. Tous les traitements topiques ou oraux de la rosacée devaient avoir été arrêtés depuis 1 mois ou 5 demi-vies. Après une période d’observation de 4 semaines, les patients recevaient 140 mg d’érénumab par voie sous- cutanée toutes les 4 semaines pendant 12 semaines. Une visite de suivi pour la tolérance était réalisée à la semaine 20. Le critère de jugement principal était la variation moyenne du nombre de jours avec un flush modéré à extrême pendant les semaines 9 à 12, comparativement à la période d’observation initiale de 4 semaines. La variation moyenne du nombre de jours avec un érythème modéré à sévère était un critère secondaire, ainsi que la qualité de vie et la tolérance. Au total, 30 patients (âge moyen [SD], 38,8 [13,1] ans), majoritairement des femmes (n = 23 femmes [77 %]), ont été inclus. Vingt-sept de ces patients ont terminé l’étude de 12 semaines. Parmi eux, 87 % avaient déjà reçu un traitement local ou systémique pour la rosacée et 43 % au moins trois traitements. En ce qui concerne le critère de jugement principal, le nombre moyen de jours avec flushs modérés à extrêmes, qui était de 23,6 jours (écart type 5,8) au début de l’étude, était réduit de 6,9 jours (IC à 95 %, –10,4 à –3,4 jours ; P < 0,001), et 26 % des patients ont eu une réduction de plus de 50 % du nombre de jours avec flush. De façon similaire, le nombre moyen de jours avec un érythème modéré à sévère, qui était de 15,2 jours (9,1) au début de l’étude, a été réduit de 8,1 jours (IC à 95 %, –12,5 à –3,7 jours ; P < 0,001). Il existait une amélioration significative de la qualité de vie évaluée par le DLQI et le RosaQoL, un score de la qualité de vie de la rosacée, mais pas d’amélioration des scores d’anxiété et de dépression. Les événements indésirables comprenaient une constipation transitoire légère à modérée et des infections des voies respiratoires supérieures (3 patients [10 %]), ce qui était attendu par rapport aux données sur la migraine. En conclusion, ces résultats suggèrent que l’érénumab pourrait être efficace dans la réduction des flushs et de l’érythème associés à la rosacée avec un profil de tolérance acceptable.
Réf. : Wienholtz NKF, Christensen CE, Do TP et al. Erenumab for treatment of persistent erythema and flushing in rosacea : a nonrandomized controlled trial. JAMA Dermatol 2024 ; 160(6) : 612-9.
L’URÉE, L’INGRÉDIENT UNIQUE DE LA PEAU SÈCHE
Àchaque inconfort de peau, des soins spécifiques doivent être recommandés aux patients.
« Les patients souffrant de sécheresse cutanée sévère peuvent ressentir un impact majeur sur leur qualité de vie. L’un des effets secondaires les plus courants d’une peau très sèche est la démangeaison. En tant que dermatologue, je recommande à mes patients ayant la peau très sèche des produits qui nourrissent et aident à restaurer la couche protectrice naturelle de la peau » note la dermatologue Heather Woolery-Lloyd. En matière de réparation des peaux sèches, l’urée est un actif incontournable que l’on retrouve à des concentrations et formulations différentes dans de nombreux soins dermatologiques.
NATURE ET ORIGINE DE L’URÉE
DANS LA PEAU
L’urée, encore appelé carbamide est le diamide de l’acide carbonique. Cette petite molécule est présente naturellement dans l’organisme et résulte de la dégradation des protéines.
Au niveau de la peau, l’urée entre dans la composition du N.M.F. (Natural Moisturizing Factor) où elle représente 7% de ce N.M.F.
Le N.M.F est présent dans les cornéocytes où il joue un rôle d’hydratation de ces cellules.
Il regroupe en fait un ensemble de facteurs hygroscopiques, capables de capter l’eau dont: des acides aminés libres, du PCA (acide pyrrolidone carboxylique), de l’urée, des lactates, des sels minéraux, des sucres (fructose, mannose, galactose, glucose). L’urée présente dans les NMFs des cornéocytes provient de deux sources: la transpiration et la dégradation de l’arginine par l’arginase durant le processus de kératinisation. L’hydrolyse de l’arginine en ornithine et urée est catalysée par l’arginase. Le contenu total en urée dans une peau normale est de 1%.
PRINCIPE D’ACTION DE L’URÉE
DANS
LA PEAU
Lors de l’ultime phase de kératinisation, la filaggrine se dégrade progressivement et libère des acides aminés qui composent le N.M.F. Cette dégradation est régulée par des protéases (enzymes), elles-mêmes sous la dépendance du pourcentage d’eau dans le stratumcorneum. Parmi les acides aminés formés, il en est un en particulier qui va fournir l’urée que l’on trouve dans le N.M.F., c’est l’arginine.
La dégradation de l’arginine va conduire à la formation de citrulline (l’un des acides aminés le plus importants du N.M.F.) et d’urée qui va se fixer à l’eau et donc réguler les enzymes de dégradation. L’urée est donc un véritable régulateur de l’hydratation.
DEUX ACTIONS DIFFÉRENTES
EN FONCTION DE LA DOSE
Par sa capacité à fixer l’eau, l’urée va donc être un véritable régulateur des protéases.
Les protéases vont jouer sur 2 sites d’action:
• Celles qui interviennent sur la dégradation de la filaggrine et donc sur la formation du N.M.F.
• Celles qui interviennent sur la dégradation de la cornéodesmosine (protéine de jonctions cellulaires entre kératinocyte)
Par sa capacité à activer les protéases, l’urée peut donc être pro-hydratante ou pro-kératolytique en fonction de la dose et de l’environnement.
• Les propriétés hydratantes de l’urée se manifestent préférentiellement à faible dose (0.5 à 4 %), surtout en présence d’humectant tels que la glycérine, …
• Les propriétés kératolytiques ou kérato-régulatrices de l’urée se manifestent à plus forte concentration, à savoir 10% et plus.
LIPIKAR GEL UREA 30%:
LA SOLUTION INNOVANTE
DES LABORATOIRES LA ROCHE-POSAY POUR L’HYPERKÉRATOSE
Pour répondre aux défis de l’hyperkératose, les laboratoires La RochePosay ont formulé Lipikar Gel Urée 30%, un nouveau soin qui complète la gamme Lipikar. Lipikar Gel Urée 30% est actuellement sur le marché le seul produit à base d’urée à une concentration de 30% formulé avec de la niacinamide.
➢ Ce soin dermatologique intensif transforme visiblement les zones d’hyperkératose sur le corps dès la première utilisation. Il se caractérise par sa tolérance optimale, et ce même pour les peaux sensibles, grâce à l’inclusion dans la formule de niacinamide.
➢ Les études cliniques démontrent l’efficacité impressionnante du Lipikar Gel Urée 30%.
• Réduction de 88% de la sécheresse et une amélioration de 34% de la douceur de la peau après seulement 30 minutes.
• Galénique agréable: Lipikar Gel Urée 30% a une texture de gel transparent qui fond immédiatement sur la peau, ce qui permet une application facile et rapide, tout particulièrement pour les personnes âgées.
• Ces résultats se traduisent par des expériences réelles pour les patients, puisque 71 % d’entre eux ont constaté une amélioration immédiate de la rugosité de la peau. Cette amélioration rapide et perceptible peut améliorer de manière significative l’observance et la satisfaction des patients.
➢ A appliquer 1 à 2 fois par jour.
CERAVE, LAIT HYDRATANT INTENSIF POUR UNE HYDRATATION LONGUE DURÉE ET APAISEMENT
DES DÉMANGEAISONS CAUSÉES
PAR LA XÉROSE
Grâce à sa formule unique, le lait hydratant Intensif de Cerave est une lotion riche à absorption rapide qui aide à restaurer la barrière cutanée:
➢ INTRODUCTION DE L’HYDRO-UREA™
Formulé avec 5 % d’Hydro-Urea™, un puissant cocktail de substances hydratantes: de l’urée hydroxyéthyl, des acides aminés et de l’acide pyrrolidonecarboxylique, qui favorise l’hydratation de la peau à un pH équilibré.
➢ TROIS CÉRAMIDES ESSENTIELS
Formulée avec le mélange signature de CeraVe de trois céramides essentiels, cette lotion aide à restaurer la barrière cutanée.
➢ LA TECHNOLOGIE MVE®
La technologie d’émulsion multivésiculaire (MVE®), un système unique de libération multicouches, encapsule les ingrédients hydratants et les libère progressivement dans le temps, les diffusant en continu à la surface de la peau pour une hydratation longue durée.
➢ BEURRE DE KARITÉ
Cet ingrédient occlusif et hydratant recouvre la peau d’une couche protectrice qui scellera l’hydratation.
« Le Lait Hydratant Intensif de CeraVe est une excellente option pour les peaux très sèches et répond efficacement à deux problèmes clés. Il enrichit la peau en céramides pour aider à restaurer et à renforcer sa barrière naturelle, en retenant l’humidité et en prévenant la déshydratation. En outre, l’ingrédient Hydro-Urea™ agit comme un aimant qui attire et retient l’eau pour une hydratation durable » souligne la dermatologue Heather Woolery-Lloyd.
• Testé sous contrôle dermatologique.
• Un lait pour le corps qui prend en charge les divers signes de peau sèche, rugueuse, squameuse, qui tiraille et qui démange.
• Cliniquement prouvé pour apaiser des principaux signes de sécheresse.
Après une semaine d’utilisation:
➢ réduit de 99 % les démangeaisons de la peau irritée, inconfortable et très sèche (1)
➢ réduit la desquamation de 98 %(1)
Après 4 semaines d’utilisation
➢ réduit la TEWL (perte d’eau transépidermique) de 45 %(2)
Après une application
➢ 90 % des consommateurs ont constaté que la peau tiraillait moins pendant 72 heures (3).
1. Étude clinique, 90 sujets avec des types de peau Fitzpatrick, de 19 à 75 ans, 2x/ jour, pendant 4 semaines, sur les jambes uniquement.
2. Évaluation instrumentale, 80 personnes, utilisation sur les jambes uniquement, 2 fois par jour
3. Auto-évaluation, 29 femmes 55-63ans), après 72 heurs sur les jambes uniquement.
Les recommandations de la prise en charge de la peau sèche soulignent l’importance d’une routine de soins cohérente et hydratante pour les peaux très sèches, avec un nettoyage quotidien, une hydratation et une protection solaire. Avec les différents soins formulés pour les peaux sèches, CeraVe répond parfaitement à ces recommandations pour accompagner le patient.
Pas d’association entre dose cumulée de méthotrexate et fibrose hépatique chez les patients psoriasiques
Une étude transversale monocentrique, incluant tous des patients adultes atteints de psoriasis modéré à sévère a été menée afin de déterminer la prévalence de la fibrose hépatique chez les patients psoriasiques (par fibroscan), d’évaluer l’association entre la dose cumulée de méthotrexate et la fibrose hépatique, d’évaluer la relation entre les autres facteurs de risque connus de fibrose hépatique et la fibrose hépatique, et d’évaluer la pertinence d’utiliser des facteurs de risque cliniques et le score FIB4 pour prédire le risque de fibrose hépatique afin de rationaliser l’utilisation du Fibroscan. Deux-centquatre patients étaient inclus, d’âge moyen 48 ans, 51% de femmes, 56% d’obèses, tour de taille médian 103 cm. Le PASI médian était de 2,1 (pire PASI médian 12,8). 21% des patients étaient diabétiques, la plupart des patients avaient une consommation modérée d’alcool, mais 23% avaient une consommation excessive et 13% avaient eu antérieurement une consommation excessive d’alcool. 92% des patients avaient reçu du méthotrexate (durée médiane 36 mois, dose cumulée moyenne 2,16gr). 38% des patients étaient traités par biothérapie. La prévalence de la fibrose hépatique était respectivement de 36, 25 et 17% en utilisant les seuils de 7kPA, 7,9kPA et 9,5kPa. 17% des patients avaient un score FIB-4 >1,3. Il y avait 35 patients un Fibroscan ≥7,9kPa et 23 avec un Fibroscan ≥9,5kPa mais avec un score FIB-4 normal. Il n’y avait pas d’association entre le FIB-4 et les résultats du Fibroscan. Il n’y avait pas d’association entre la dose cumulée de méthotrexate et les résultats du Fibroscan, et ce même après ajustement sur les facteurs confondants de fibrose hépatique. L’association entre les facteurs de risque de fibrose hépatique (IMC, tour de taille, syndrome métabolique, diabète et consommation excessive d’alcool) était retrouvée. Dans cette étude, un IMC ≥28 était un facteur associé à une valeur augmentée du Fibroscan. Un IMC ≥28, un diabète et un syndrome métabolique étaient plus performant que le score FIB-4 pour identifier les patients à risque de fibrose hépatique modérée à sévère. Ces résultats sont rassurants sur l’association dose cumulée de méthotrexate et risque de fibrose hépatique, cependant l’impact du méthotrexate chez les patients avec une fibrose hépatique débutante ou déjà existante n’est pas correctement identifié. L’effet d’une dose cumulée supérieure de méthotrexate sur le risque de fibrose hépatique ne peut pas être exclu. Les auteurs proposent de réaliser un Fibroscan chez les patients psoriasiques avec IMC >28, un syndrome métabolique ou un diabète et de ne plus utiliser de méthotrexate chez les patients avec un IMC ≥40.
Réf. : Cumulative methotrexate dose is not associated with liver fibrosis in patients with a history of moderate to severe psoriasis. Babakinejad P, Lapsley R, Forster L et al. Br J Dermatol. 2024 Feb 17:ljae069.
Aucun impact des prébiotiques sur la dermatite atopique
Actuellement aucun traitement curatif de la dermatite atopique n’a été identifié. Dans ce contexte, les stratégies de prévention primaire de cette maladie représentent un enjeu de santé majeur. Parmi ces stratégies, les interventions visant à modifier le microbiote intestinal par les probiotiques ou les prébiotiques en période périnatale semblent les plus prometteuses. L’essai clinique PREGRALL a permis de tester pour la toute première fois l’impact de prébiotiques sur la survenue de la dermatite atopique chez des nouveau-nés à risque de développer cette maladie. Pour cette étude multicentrique PREGRALL, 376 femmes enceintes ont été incluses à 20 semaines d’aménorrhée puis randomisées en double aveugle entre une supplémentation quotidienne en prébiotiques (des galacto-oligosaccharides et de l’inuline dans un rapport de 9 pour 1) et un placebo jusqu’à la fin de la grossesse. Le critère principal d’évaluation - la prévention de la survenue de la dermatite atopique chez les enfants à 12 mois - n’a pas été atteint, avec une prévalence de 20 % environ dans les deux groupes. Les résultats étaient similaires après exclusion des femmes ayant pris des antibiotiques au cours de leur grossesse, que l’accouchement soit par voie basse ou par césarienne, que l’enfant soit allaité ou non et quelle que soit la durée de l’allaitement.
Les objectifs secondaires de l’étude n’ont pas non plus été atteints : que ce soit la prévalence de la dermatite atopique à 6 mois, la sévérité de la dermatite atopique ou la qualité de vie. Il n’y avait pas non plus de différence entre les deux groupes concernant la sensibilisation aux principaux allergènes (lait, œuf, arachide, chat, pollens de graminées, acariens) et les allergies alimentaires.
Pourtant, l’analyse des selles des mères qui ont pris la supplémentation en prébiotiques montre bel et bien une modification du microbiote, qui a été ensuite transmise à leur enfant en début de vie.
Malheureusement, ce transfert du microbiote de la mère à l’enfant ne permet pas de prévenir la survenue de dermatite atopique chez l’enfant à un an. Ces résultats s’ajoutent à ceux de la méta-analyse des études réalisées pendant les premiers mois de vie.
Les enfants de la cohorte PREGRALL vont être suivis jusqu’à leurs 5 ans pour évaluer un éventuel effet des prébiotiques maternels sur l’asthme.
Réf. : Bodinier M, Aubert H, Boivin M et coll. La supplémentation maternelle en prébiotiques pendant la grossesse régule la colonisation du microbiote des enfants à haut risque, mais ne permet pas de prévenir la dermatite atopique à l’âge d’un an. Revue Française d’Allergologie Volume 64, Supplement, April 2024, 104035. https:// doi.org/10.1016/j.reval.2024.104035
Accélère le processus de régénération cutanée
❙ APAISE ET RÉPARE
la peau très sèche à extrêmement sèche, craquelée ou irritée
❙ ACCÉLÈRE LE PROCESSUS DE RÉGÉNÉRATION
de la peau abîmée tout en protégeant la zone concernée
❙ CRÉE UNE BARRIÈRE PROTECTRICE semi-occlusive
❙ RÉSULTAT :
La peau est visiblement régénérée, protégée et réparée
Convient pour le visage et le corps des : Bébés Enfants Adultes
DRY, CRACKED SKIN
PEAU SÈCHE, CRAQUELÉE
PIEDS ET TALONS CRAQUELÉS
DRY HANDS & CUTICLES
SENSITIVE BABY SKIN
MAINS SÈCHES & CUTICULES PEAU SENSIBLE DU BÉBÉ
PEAU AQUAPHOR
PEAU CRAQUELÉE
DES SOLUTIONS INNOVANTES POUR LES PEAUX SÈCHES
En raison de l’altération de la barrière cutanée, la peau sèche est très sensible aux agressions extérieures, ce qui peut induire des sensations de démangeaisons, une dermatite de contact et aggraver la dermatite atopique (1). Un adulte sur trois présente une sécheresse cutanée (2), huit patients sur dix présentant une peau sèche déclarent subir une altération de leur qualité de vie (3) et un patient sur deux d’entre eux n’utilise pas des soins adaptés (4). Les experts s’accordent à dire que les émollients et les soins d’hygiène formulés pour la xérose cutanée améliorent l’hydratation de la peau et améliorent la fonction globale de la barrière cutanée (1). Pourtant, de nombreuses personnes touchées n’ont pas encore n’ont pas encore trouvé le produit adapté à leur mode de vie.
PHYSIOPATHOLOGIE
La capacité de la peau à emmagasiner de l’eau dépend en grande partie de la formation de la barrière lipidique dans la couche cornée. Provenant des corps d’Odland, les lipides épidermiques de l’espace extracellulaire, représentent 10 à 30 % du volume total du SC et sont constitués de Céramides d’acides gras libres et de cholestérol. Ils forment le ciment intercellulaire et assurent la régulation de la teneur en eau et en fluide ; fonction cruciale puisque l’élasticité et la fermeté de la couche cornée dépendent de son niveau d’hydratation. Cette fonction barrière minimise la perte insensible en eau (PIE) et protège ainsi le corps de la déshydratation. Les NMF, ou facteurs naturels d’hydratation, sont naturellement synthétisés lors du processus de cornification des kératinocytes. Parmi ses constituants, l’acide pyrrolidone carboxylique, l’urée et les lactates possèdent un fort pouvoir hydratant par leur action hygroscopique (capables de retenir jusqu’à 70 % de leur poids en eau) permettant ainsi de fixer l’eau dans la couche cornée. La structure protéique des cellules cornées influence par conséquent la capacité de la peau à fixer l’eau. Le film hydrolipidique de surface est constitué pour sa partie hydrosoluble de la sueur (provenant de la perspiration insensible en eau et de la sécrétion sudorale) et pour sa partie liposoluble de sébum et de lipides libérés par l’épiderme lors de la kératinisation. La fonction principale de ce film est de retenir l’eau à la surface de la peau
La sécheresse cutanée est due à différents facteurs corrélés les uns aux autres et induisant un cercle vicieux:
• une fragilisation de la barrière cutanée est due en une déficience en lipides épidermiques
• cette fragilisation entraîne une plus grande évaporation de l’eau épidermique ainsi qu’un déficit en facteurs hygroscopiques capables de capter cette eau libre épidermique.
• un déficit intrinsèque en facteurs hygroscopiques, entraînant une sécheresse épidermique, l’absence ou le
manque d’eau étant à l’origine d’une mauvaise circulation et synthèse des protéines de desquamation épidermique et donc d’une altération de leur activité.
La desquamation du Stratum Corneum se fait alors par amas de cornéocytes de surface, avec une fragilisation de la barrière cutanée.
L’ÉTIOLOGIE DE LA PEAU SÈCHE
Plusieurs facteurs peuvent être responsables de la peau sèche :
• une prédisposition génétique : la xérose désigne une peau sèche, l’ichtyose est une peau très sèche, écailleuse sur de grandes surfaces, difficile à traiter, pathologique et qui correspond à une dermatose caractérisée par un déficit génétique de synthèse de profilaggrine.
• des expositions solaires trop fréquentes et trop prolongées
• des facteurs environnementaux : froid, vent, chauffage extrême
• la prise de certains médicaments : dérivés de la vitamine A (acitrétine, isotrétinoïne), diurétiques, statines, traitements oncologiques, …
• le vieillissement : la xérose sénile affecte plus de 50 % des personnes âgées de plus de 65 ans, le plus souvent au niveau des jambes et du tronc, mais peut être présente sur tout le corps.
• une dermatose comme le psoriasis, la dermatite atopique,
• la kératose pilaire (KP) est une affection cutanée génétique fréquente et bénigne qui provoque des bouchons kératiniques dans l’orifice folliculaire, principalement sur les bras et les jambes, associés à des degrés variables d’érythème périfolliculaire, de sécheresse et de rugosité. Elle touche environ 40 % des adultes et 50 à 80 % des adolescents.
QUELLE PRISE EN CHARGE ?
De simples gestes permettent de prévenir ou de limiter la peau sèche : hygiène douce avec des soins adaptés à la xérose (savons de type surgras, syndets, huile, …), hydratation interne suffisante, alimentation équilibrée, … Il est important d’appliquer des soins (idéalement lorsque la peau est encore humide, pour retenir l’humidité) pour préserver ou rétablir la barrière cutanée.
Sur la base des données scientifiques disponibles, l’urée est le gold standard pour le traitement de la xérose cutanée. Non seulement l’urée hydrate efficacement la peau, mais elle améliore également la fonction barrière et les mécanismes de défense et d’hydratation de la peau. Elle augmente la pénétration des substances actives dans la peau, soulage les sensations de dé-
mangeaisons et, à des concentrations plus élevées, a un effet kératolytique. Sa concentration et sa galénique doivent être choisies en fonction de l’état individuel de la peau, de l’âge du patient et de l’environnement. Le produit ne doit pas être utilisé sur des zones inflammatoires ou ouvertes. En association avec les Céramides et les facteurs naturels d’hydratation (NMF), l’efficacité hydratante de l’urée est supérieure à celle d’un soin contenant uniquement de l’urée. Rappelons qu’une réduction des NMF dans l’épiderme a été observée dans la xérose cutanée, l’eczéma atopique, l’ichtyose vulgaire et chez les personnes âgés.
EUCERIN : L’EXPERTISE DES PEAUX SÈCHES
Deux gammes importantes et complémentaires accompagnent les signes de xérose.
1. UreaRepair, Hydratation immédiate et durable
L’émollient 10% d’urée UreaRepair : Hydratation intensive pendant 72 heures et renforcement de la barrière cutanée (5,6)
UreaRepair d’Eucerin offre une gamme complète de produits pour les peaux sèches à très sèches, comprenant des émollients, des crèmes hydratantes pour le visage et le corps, un nettoyant et des produits de soins pour les mains et les pieds. Leur formule avec de l’Urée (ingrédient clé de la gamme) et de divers facteurs d’hydratation naturels (NMFs) améliore la capacité de la peau à retenir l’eau et augmente ainsi son taux d’humidité. Les Céramides et d’autres lipides renforcent la barrière cutanée et la protègent contre la perte d’humidité. Les produits de la gamme sont disponibles en plusieurs concentrations d’Urée (5 % à 30 %), selon le degré de sécheresse cutanée.
L’Émollient 10% d’Urée UreaRepair est nos solution cliniquement prouvée avec des ingrédients identiques à ceux de la peau (10 % Urée, 17 autres NMFs, Céramides, …) (6), il est recommandé pour les peaux sèches à très sèches.
➢ Hydratation intense : 7x plus d’hydratation après première application (5)
➢ Hydratation en profondeur : Hydratation supérieure dans les couches profondes de la couche cornée
➢ Amélioration de la fonction barrière : Réduction supérieure de la perte d’eau trans-épidermique
➢ Hydratation supérieure comparée à d’autres hydratants avec et sans urée (7)
2 Atopi Control, Une peau apaisée et une barrière cutanée renforcée
Eucerin AtopiControl offre des soins adaptés aux différentes phases dont la peau à tendance atopique a besoin.
➢ Nos solutions cliniquement prouvées pour un soulagement durable des sensations de démangeaisons : Licochacone A + Omega + Céramides + NMF’s & Glycérine (8)
➢ Les sensations de démangeaisons sont atténuées (9)
➢ Augmentation de l’hydratation 24h après la première application et après 2 semaines d’utilisation régulière, mesurée par cornéométrie (8)
➢ Amélioration de la qualité de vie : 91 % confirment un apaisement durable des sensations de démangeaisons (11)
RÉFÉRENCES
1. Augustin M et al. (2019)., J Dtsch Dermatol Ges;17(S7):3:33
3. von Stülpnagel et al. (2021), Journal of Dermatological Treatment, 33:5, 24822487
4. Multi-centre study in seven countries (Coratia, France, Germany, Hungary, Italy, Serbia, Slovenia) with 1555 patients with dry skin. BDF data on file.
5. Clinical study with 44 women with very dry skin, Measurement of skin moisturization using SkiCon® 30 minutes after single application of UreaRepair 10% Lotion. BDF Data on file.
6. Schuster et al (2023). “Intelligent” combination of skin identical moisturizers and lipids in emollients boosts moisturization and barrier repair in patients with Xerosis cutis. Poster presented at 32nd EADV Congress Berlin.
8. Permuted, controlled, blind study with 31 male and female patients and five assessments over a 2-week period. Comparison with baseline and untreated control.
9. Permuted, controlled, blind study with 31 male and female patients and five assessments over a 2-week period. Comparison with baseline and untreated control.
10. In-home study with 160 patients with mild to moderately severe atopic dermatitis using AtopiControl Calming Balm over a 4-week period.
11. Test consommateurs, 160 volontaires souffrant de dermatite atopique légère à modérée. Résultats après 4 semaines d'utilisation.
* Cet article est sous la responsabilité rédactionnelle de Dermactu.
Une avancée scientifique contre l'hyperpigmentation
La Roche-Posay a développé le Sérum Mela B3, une innovation dermatologique ciblant l'hyperpigmentation cutanée. Ce sérum associe deux actifs majeurs : le Melasyl™, une molécule multibrevetée issue de 18 ans de recherche, et la niacinamide à une concentration de 10%.
Le Melasyl™ agit en interceptant sélectivement les précurseurs de la mélanine, empêchant ainsi leur transformation en pigments responsables des taches brunes, tout en préservant l'intégrité des mélanocytes.
La niacinamide (vitamine B3) réduit les micro-inflammations à l'origine des taches brunes et renforce la barrière cutanée en apportant des lipides essentiels à son bon fonctionnement.
Des études cliniques ont démontré l'efficacité de Mela B3, avec des résultats visibles dès une semaine d'utilisation. Après huit semaines, 80% des femmes ont constaté une correction des taches persistantes, 98% ont observé une diminution du nombre de taches, et 96% ont noté une réduction de leur taille.
Des soins experts pour une peau saine
durant la saison froide
La gamme CeraVe, développée avec des dermatologues, propose des soins adaptés pour maintenir une hydratation optimale pendant l'hiver.
L'Huile Lavante Moussante Hydratante est conçue pour nettoyer en douceur les peaux normales à très sèches, y compris celles à tendance atopique. Sa formule associe des triglycérides et du squalane, à une base lavante douce dérivée d'acides aminés. Enrichie en trois céramides essentiels et en acide hyaluronique, sa texture huile se transforme en une mousse enveloppante au contact de l'eau, nettoyant sans laisser de film gras et apaisant immédiatement la peau.
Pour une hydratation prolongée, le Baume Hydratant de CeraVe hydrate les peaux sèches à très sèches pendant 48 heures. Enrichi en céramides et en acide hyaluronique, il nourrit et aide à restaurer la barrière cutanée grâce à la technologie MVE, qui diffuse les actifs tout au long de la journée.
Un sérum pour accompagner les injections de neurotoxines
P-TIOX est un sérum anti-rides avancé aux peptides, cliniquement prouvé pour atténuer l'apparence de 9 types de rides d'expression. Formulé avec un complexe peptidique avancé comprenant hexapeptide et dipeptide, il s'inspire de l'action des neurotoxines injectables pour améliorer l'apparence des principales rides. Enrichi d'un complexe d'actifs puissants: 5 % de PHA, 5 % de niacinamide et extrait de Laminaria, P-TIOX améliore aussi visiblement la texture et l'éclat de la peau dès la 1ère semaine pour un effet glass skin. P-TIOX de SkinCeuticals a été testé par des médecins comme complément aux injections de neurotoxines pour traiter les rides dans toutes les zones du visage, y compris les zones difficiles à injecter, et améliorer la qualité de la surface de la peau. Combiné aux injections de neurotoxines, P-TIOX assure une correction visible des signes du vieillissement cutané avec des résultats immédiats et durables, même après 3 mois.
"P-TIOX est idéal utilisé en complément d'injections de toxine botulique, ou intégré à votre routine de soins. Son efficacité et sa bonne tolérance sont cliniquement prouvées pour aider à réduire l'apparence des principales rides de contraction et améliorer l'éclat de la peau grâce à une synergie unique de peptides et de PHA." souligne le Docteur Van Riet, dermatologue, Carpe Clinic, Anvers.
ET SI RÊVER VOUS FAISAIT RAJEUNIR RETINAL INTENSE
Nouveau sérum de nuit à base de retinaldehyde pour tous types de peau*
Une gamme de produits nettoyants et de soins
journaliers pour les peaux acnéiques au Thiamidol breveté
Pour faire face aux imperfections de la peau ainsi qu’aux marques post-acné, Eucerin a développé DermoPure, une gamme de produits nettoyants et de soins journaliers adaptés à ce type de peau aux formules très ciblées.
Soin phare de la gamme Dermopure, le Sérum DermoPure Triple Action permet d’offrir un soin continu et efficace pour les peaux sujettes aux marques post-acné grâce au Thiamidol breveté. Il prévient et réduit les marques post-acné dès 2 semaines.
Pour davantage de résultats, l’idéal est d’intégrer le Sérum DermoPure Triple Action dans une routine bien-être parfaitement adaptée aux peaux à imperfections, par exemple :
• le Gel Nettoyant DermoPure, un soin nettoyant ultra-efficace,
• le Fluide Matifiant DermoPure, qui offre un effet anti-brillance durant 8h
• le soin solaire Sun Oil Control SPF 50+, mais aussi
• le Gommage DermoPure qui désobstrue les pores,
• le K10 Soin Rénovateur Cutané DermoPure, un soin qui réduit visiblement les imperfections et aide à prévenir leur réapparition et enfin
• la Crème Compensatrice Apaisante DermoPure, soin hydratant intense spécialement formulé pour apaiser la peau fragilisée par les traitements dermatologiques asséchants et atténuer leurs effets secondaires (inconfort, rougeurs, sécheresse).
Traiter l'acné du corps
Une étude américaine a montré qu'environ 50 % des patients souffrant d'acné présentent aussi de l'acné corporelle ou tronculaire. La plupart des régions du corps ciblées sont le dos, la poitrine et les épaules.
Jusqu'à 87 % des personnes touchées par l'acné présentent également des marques post-acné, qui peuvent persister pendant plusieurs années, voire une décennie, le soleil aggravant les symptômes.
La Crème Corps Triple Action DermoPure des Laboratoires Eucerin associe des ingrédients actifs puissants pour réduire les imperfections et les marques post-acné tout en apaisant et en hydratant la peau :
• Anti-imperfections : L'Acide Salicylique réduit efficacement les imperfections persistantes
• Anti-marques : Le Thiamidol® breveté réduit visiblement les marques post-acné persistantes et prévient leur apparition.
• Action apaisante et hydratante : La Licochalcone A apaise les sensations d'irritations et aide à réduire les rougeurs. La Glycérine hydrate intensément.
Pour une peau instantanément apaisée et hydratée, et une peau visiblement plus nette et plus uniforme après deux semaines.
Traitements des poux : la société française de dermatologie fait le point
La Société française de dermatologie (SFD) a fait le point, dans un communiqué, sur les traitements à utiliser pour lutter contre les poux de tête qui sévissent souvent chez les enfants d’âge scolaire, et entrainent un prurit et parfois des surinfections bactériennes. “Depuis les années 80, les poux ont développé des résistances et les traitements ont perdu en efficacité”, rappelle la SFD. Les spécialistes conseillent donc d’utiliser en priorité des traitements mécaniques comme le peignage régulier sur cheveux humides et le délentage manuel. Les produits qui peuvent être utilisés sont une lotion à base de diméticone, ou une huile de silicone. Ils agissent par action physique et non chimique, et permettent de tuer les parasites en les étouffant. Concernant les huiles essentielles, la SFD reconnait qu’elles “peuvent se montrer efficaces” ; mais elles sont contre indiquées chez les enfants et chez les femmes enceintes. En revanche, “les shampooings n’ont pas montré leur efficacité et ne doivent pas être utilisés”, affirment les dermatologues. Enfin, une désinfection des objets qui ont été en contact avec le cuir chevelu doit être réalisée. Et ce, “soit par isolation une dizaine de jours dans un sac fermé soit par lavage à une température de plus de 50°C”, ajoute la SFD, qui insiste sur la nécessité d’”examiner la tête des enfants en collectivité régulièrement et à leur apprendre à ne pas se prêter brosses et bonnets !”.
MAGIC : une étude prospective multicentrique britannique pour améliorer la prise en charge des abcès sous cutanés
Cette étude anglaise concernait tous les hôpitaux du NHS disposant d’un service de soins chirurgicaux d’urgence. Les patients inclus étaient âgés de plus de 18 ans et consultaient pour un abcès aigu sous-cutané de toute localisation à l’exclusion de ceux siégeant dans les régions péri-anale, mammaire, des mains et des doigts, et ceux de la face qui sont le plus souvent pris en charge par des services spécialisés. Etaient également exclus les abcès post-opératoires, ceux avec fistules et ayant une origine vasculaire. Le principal critère de jugement était la récidive de l’abcès et la nécessité de reconsulter à l’hôpital. Les données étaient recueillies 60 jours après la présentation initiale. Cette étude montre une grande variation dans les pratiques en Grande Bretagne concernant le traitement des abcès sous-cutanés. Le bénéfice de l’anesthésie locale est souligné par les auteurs notamment en termes de réduction de morbidité induite. Ses autres avantages sont connus : diminution des admissions hospitalières, réduction des durées de séjour, optimisation des flux au bloc opératoire, reprise du travail accélérée, diminution des coûts financiers et environnementaux. Pour ces patients vus le plus souvent en urgence, l’accessibilité à un plateau de type ambulatoire en dehors des horaires habituels est un plus. Le bénéfice de l’antibiothérapie post-drainage est également discuté par les auteurs. En Grande Bretagne, l’antibiothérapie n’est pas prescrite en routine. Cependant un travail collaboratif a montré, après lecture de 14 essais randomisés, un bénéfice modeste sur la réduction du risque de récidive. Ainsi un panel d’experts a émis une recommandation faible en faveur du trimethoprime-sulfaméthoxazole ou de la clindamycine (avec une suggestion plutôt en faveur du premier qui induit moins de diarrhée). Les experts soulignaient néanmoins que ce bénéfice faible était contrebalancé d’un point de vue sociétal par une possible induction d’antibiorésistance. De façon pragmatique, une antibiothérapie est justifiée en présence de facteurs de risque (obésité, diabète, immunosuppression…) ou de présentation particulière (fièvre, aspect cellulitique…).
l’eczéma chronique des mains (ECM)
Intérêt du dupilumab dans le traitement de
L’eczéma chronique de la main est une maladie dermatologique handicapante et récurrente, et il existe peu de traitements efficaces. Le dupilumab, inhibiteur des cytokines IL-4/13 a démontré largement son efficacité clinique au cours de la dermatite atopique (DA) et est actuellement en essai pour l’eczéma chronique de la main. La base de données PubMed a été utilisée pour rechercher les termes dupilumab et eczéma/ dermatite chronique de la main, y compris les sous-types d’ECM (dyshidrotique, hyperkératotique, allergique, contact, atopique, professionnel, vésiculaire). Vingt-deux publications d’études cliniques ont satisfait aux critères d’inclusion et d’exclusion pour l’évaluation. Deux études prospectives multicentriques, un essai contrôlé randomisé, deux études observationnelles prospectives, une étude rétrospective, neuf séries de cas et sept rapports de cas ont été inclus dans l’analyse avec 374 patients au total. Une réponse partielle ou une résolution complète de l’ECM est survenue chez 80,3 % des patients traités par dupilumab dans les 4 à 16 semaines du traitement. La réponse des patients a eu tendance à augmenter tout au long de ces études pour un pourcentage important de patients et, pour beaucoup, elle a duré au-delà de la durée de l’étude. Il a été noté une amélioration significative des scores de sévérité de l’eczéma de la main et des scores de qualité de vie. L’efficacité a été rapportée dans plus de la moitié des cas d’eczéma hyperkératotique de la main ; cependant, l’efficacité était diminuée par rapport à une efficacité relativement élevée et constante pour les autres sous-types d’ECM tels que dyshidrotique, vésiculaire, atopique et de contact. Les patients ayant une DA actuelle ou antérieure avaient des scores d’amélioration plus élevés que ceux qui n’en avaient pas. Les effets indésirables les plus fréquents étaient la conjonctivite (9,9%), l’infection virale herpètique (1,4%) et l’érythème/dermatite faciale (1,3%). Les auteurs concluent que le dupilumab a démontré son efficacité thérapeutique dans le traitement de l’ECM. Il faut mener d’autres études contrôlées randomisées sur l’utilisation du dupilumab dans les sous-types d’ECM.
Réf. : Dupilumab for Chronic Hand Eczema: A Systematic Review and Meta-Analysis.
HR Riva, T Yoon, AJ Hendricks et al. Dermatitis. 2024 Nov 6. doi:10.1089/derm.2024.0186.
Méthotrexate: photosensibilité et interaction médicamenteuse
Dans le cadre de l’évaluation périodique des données de sécurité pour le méthotrexate, le PRAC a conclu qu’une mise à jour des RCP et des notices des médicaments contenant du méthotrexate était nécessaire. Le risque de photosensibilité doit désormais y être mentionné. En effet, une photosensibilité se manifestant par une réaction « coup de soleil accrue » a été observée chez certaines personnes prenant du méthotrexate. L’exposition à la lumière solaire intense ou aux rayons UV doit être évitée, sauf indication médicale. Les patients doivent utiliser une protection solaire adéquate pour se protéger de la lumière intense du soleil. D’autre part, le PRAC a estimé que les RCP des médicaments contenant du méthotrexate doivent mentionner que l’administration simultanée de métamizole et de méthotrexate peut augmenter l’effet hématotoxique du méthotrexate, en particulier chez les patients âgés.
Chlorhexidine à usage cutané : lésion cornéenne persistante et déficience visuelle significative
Le PRAC a conclu que le RCP et la notice des médicaments contenant de la chlorhexidine devaient être mis à jour afin d’y mentionner les effets indésirables suivants : érosion cornéenne, lésion épithéliale/lésion cornéenne, déficience visuelle significative permanente. Ces médicaments ne doivent pas entrer en contact avec l’œil. Des cas graves de lésion cornéenne persistante, pouvant nécessiter une greffe cornéenne, ont été signalés à la suite d’une exposition oculaire accidentelle à des médicaments contenant de la chlorhexidine malgré la prise de mesures de protection des yeux en raison de la migration de la solution au-delà de la zone de préparation chirurgicale prévue. Lors de l’application, il convient de faire preuve d’une extrême prudence afin de s’assurer que la solution ne migre pas au-delà du site d’applicationprévu dans les yeux. Des précautions particulières doivent être prises chez les patients anesthésiés qui ne sont pas en mesure de signaler immédiatement une exposition oculaire. Si de la solution de chlorhexidine entre en contact avec les yeux, il faut rincer rapidement et soigneusement avec de l’eau. Il convient de demander conseil à un ophtalmologue.
Traitement de l’acné : quelle place pour les lasers et les dispositifs à base d’énergie
Une revue systématique a analysé les publications qui ont mesurées et étudiées l’action des lasers et dispositifs à base d’énergie (EBD) sur le niveau de sébum dans l’acné vulgaire.
Les résultats retrouvent pour la réduction du niveau sébum par les différentes techniques :
• PDT : 30-40 %. Des temps d’incubation plus courts pour la PDT (ALA ou autres photosensibilisants) peuvent être tout aussi efficaces, simplifiant ainsi les protocoles.
Les différents dispositifs par radiofréquence : 30 à 35 %,
• Le traitement par laser était généralement moins efficace, sauf pour le laser à Diode 1450 nm qui a permis des réductions de 40 à 48 %.
Les publications comparent entre elles l’efficacité des traitements :
• LED : les résultats sont très variables et moins fiables dans la réduction du sébum, mais plus efficace quand les LED sont combinées avec d’autres modalités comme la PDT ou le laser.
• Laser 1064 nm Nd:YAG est moins efficace que le laser Diode 1450.
• Les traitements combinant la radiofréquence fractionnée avec microneedling (FMR) avec les lasers ont été plus efficaces que les lasers seuls.
• La combinaison de RF et de RF avec microneedling permet une meilleure pénétration et destruction des glandes sébacées, souvent plus efficace comparé au seul laser.
Le choix de la zone de mesure du sébum est important : il est recommandé de mesurer le sébum sur les joues (zone U), plutôt que la zone T (front, nez), pour plus de stabilité dans les résultats. Les effets de la réduction de sébum sur l’acné sont visibles généralement après 12 semaines, le suivi prolongé est essentiel. Les auteurs citent également les premières études de nouveaux dispositifs comme le laser 1726 nm, ciblant spécifiquement le sébum comme chromophore, qui ouvrirait la voie à des traitements plus ciblés.
Réf. : The impact of energy-based devices on sebum in acne vulgaris: A systematic review. Jaalouk D, Pulumati A, Algarin YA, Humeda J, Goldberg DJ. J Cosmet Dermatol. 2024 Oct;23(10):3066-3077.
INDICATION
Formes sévères d’acné résistantes à des cures appropriées de traitement standard avec des antibactériens systémiques et un traitement topique.
Disponible en boîtes de 30, 60 et 100 capsules
DÉNOMINATION DU MÉDICAMENT : Isotiorga 10 mg et 20 mg capsules molles. COMPOSITION QUALITATIVE ET QUANTITATIVE : Isotiorga 10 mg : chaque capsule molle contient 10 mg d’isotrétinoïne. Isotiorga 20 mg : chaque capsule molle contient 20 mg d’isotrétinoïne. Excipients à effet notoire : Chaque capsule molle de 10 mg contient : environ 147 mg d’huile de soja (huile de soja raffinée, huile de soja hydrogénée et huile végétale hydrogénée [huile de soja de type II]), jusqu’à 5 mg de sorbitol, moins de 1 mg de rouge ponceau 4R (E-124). Chaque capsule molle de 20 mg contient : environ 294 mg d’huile de soja (huile de soja raffinée, huile de soja hydrogénée et huile végétale hydrogénée [huile de soja de type II]), jusqu’à 7 mg de sorbitol, moins de 1 mg de jaune soleil FCF (E-110). FORME PHARMACEUTIQUE : Capsule molle. Isotiorga 10 mg : capsules molles ovales, violettes et de taille 3. La longueur de la capsule est d’environ 11,1 mm et sa largeur est d’environ 6,8 mm. Isotiorga 20 mg : capsules molles ovales, de couleur blanc cassé à crème et de taille 6. La longueur de la capsule est d’environ 13,8 mm et sa largeur est d’environ 8,1 mm. INDICATIONS THÉRAPEUTIQUES : Formes sévères d’acné (telles que l’acné nodulaire ou l’acné conglobata, ou l’acné à risque de cicatrices permanentes) résistantes à des cures appropriées de traitement standard avec des antibactériens systémiques et un traitement topique. Ce médicament est indiqué chez les adultes, y compris les adolescents et les personnes âgées. POSOLOGIE ET MODE D’ADMINISTRATION : Posologie : Isotiorga ne doit être prescrit que par ou sous la supervision d’un médecin spécialiste de l’utilisation des rétinoïdes systémiques dans le traitement de l’acné sévère et comprenant parfaitement les risques liés au traitement par isotrétinoïne et les exigences en matière de surveillance. Les capsules doivent être prises au cours d’un repas une ou deux fois par jour. Population pédiatrique : Isotiorga ne doit pas être utilisé dans le traitement de l’acné prépubertaire et n’est pas recommandé chez les enfants de moins de 12 ans en raison de l’absence de données relatives à son efficacité et à sa sécurité. Adultes, dont adolescents et personnes âgées : Le traitement par isotrétinoïne doit être instauré à la dose de 0,5 mg/kg par jour. La réponse thérapeutique à l’isotrétinoïne et certains des effets indésirables sont dose-dépendants et varient d’un patient à l’autre. Cela nécessite un ajustement individuel de la posologie au cours du traitement. Pour la plupart des patients, la dose est comprise entre 0,5 et 1,0 mg/kg par jour. Les taux de rémission à long terme et de rechute sont plus étroitement liés à la dose totale administrée qu’à la durée du traitement ou à la dose quotidienne. Il a été démontré qu’aucun bénéfice supplémentaire important n’est attendu au-delà d’une dose de traitement cumulée de 120 à 150 mg/kg. La durée du traitement dépendra de la dose quotidienne individuelle. Une cure de traitement de 16 à 24 semaines suffit normalement à atteindre la rémission. Chez la majorité des patients, une seule cure de traitement permet d’arriver à une disparition complète de l’acné. En cas de rechute confirmée, une nouvelle cure de traitement par isotrétinoïne peut être envisagée en utilisant la même dose quotidienne et la même dose de traitement cumulée. Dans la mesure où l’amélioration de l’acné peut se poursuivre jusqu’à 8 semaines après l’arrêt du traitement, une nouvelle cure ne peut être envisagée qu’après la fin de ce délai. Patients présentant une insuffisance rénale : Chez les patients présentant une insuffisance rénale sévère, le traitement doit être instauré à une dose plus faible (p. ex. 10 mg/jour). La dose doit ensuite être augmentée jusqu’à 1 mg/kg/jour ou jusqu’à ce que le patient reçoive la dose maximale tolérée (voir rubrique 4.4 du RCP complet). Patients présentant une intolérance : Chez les patients présentant une intolérance sévère à la dose recommandée, le traitement peut être poursuivi à une dose plus faible, exposant le patient à un traitement plus long et à un risque accru de récidive. Afin d’arriver à la meilleure efficacité possible chez ces patients, le traitement doit normalement être poursuivi à la dose maximale tolérée. Mode d’administration : Voie orale. CONTRE-INDICATIONS : Isotiorga est contre-indiqué chez les femmes enceintes ou qui allaitent (voir rubrique 4.6 du RCP complet). Isotiorga est contre-indiqué chez les femmes en âge de procréer, sauf si toutes les conditions du Programme de prévention de la grossesse sont remplies (voir rubrique 4.4 du RCP complet). Isotiorga est également contre-indiqué chez les patients présentant une hypersensibilité à la substance active ou à l’un des excipients. Isotiorga contient de l’huile de soja raffinée, de l’huile végétale hydrogénée et de l’huile de soja hydrogénée. Par conséquent, Isotiorga est contre-indiqué chez les patients allergiques à l’arachide ou au soja. Isotiorga est également contre-indiqué chez les patients présentant une insuffisance hépatique, présentant une lipidémie excessivement élevée, présentant une hypervitaminose A, recevant un traitement concomitant par tétracyclines (voir rubrique 4.5 du RCP complet). EFFETS INDÉSIRABLES : Certains des effets indésirables associés à l’utilisation de l’isotrétinoïne sont dose-dépendants. Les effets indésirables sont généralement réversibles après la modification de la dose ou l’arrêt du traitement, mais certains peuvent persister après l’arrêt du traitement. Les symptômes suivants sont les effets indésirables les plus fréquemment rapportés avec l’isotrétinoïne : sécheresse de la peau, sécheresse des muqueuses, par ex. des lèvres (chéilite), de la muqueuse nasale (épistaxis) et des yeux (conjonctivite). Liste tabulée des effets indésirables : L’incidence des effets indésirables calculée à partir des données groupées d’essais cliniques impliquant 824 patients et des données post-commercialisation est présentée dans le tableau ci-dessous. Les effets indésirables sont listés ci-dessous par classe de systèmes d’organes (SOC) de MedDRA et par catégories de fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque regroupement de fréquences et SOC, les effets indésirables sont présentés par ordre décroissant de gravité. Infections et infestations : Très rare : Infection bactérienne (cutanéo-muqueuse) gram positif. Affections hématologiques et du système lymphatique : Très fréquent : Thrombopénie, anémie, thrombocytose, vitesse de sédimentation augmentée ; Fréquent : Neutropénie ; Très rare : Lymphadénopathie. Affections du système immunitaire : Rare : Réactions anaphylactiques, hypersensibilité, réaction allergique cutanée. Troubles du métabolisme et de la nutrition : Très rare : Diabète, hyperuricémie. Affections psychiatriques : Rare : Dépression, dépression aggravée, anxiété, tendances agressives, changements de l’humeur ; Très rare : Suicide, tentative de suicide, idées suicidaires, trouble psychotique, comportement anormal. Affections du système nerveux : Fréquent : Céphalée ; Très rare : Hypertension intracrânienne bénigne, convulsions, somnolence, sensation vertigineuse. Affections oculaires : Très fréquent : Blépharite, conjonctivite, sécheresse oculaire, irritation oculaire ; Très rare : Œdème papillaire (en tant que signe d’hypertension intracrânienne bénigne), cataracte, daltonisme (altération de la vision des couleurs), intolérance aux lentilles de contact, opacité cornéenne, vision nocturne diminuée, kératite, photophobie, troubles de la vision, vision trouble. Affections de l’oreille et du labyrinthe : Très rare : Altération de l’audition. Affections vasculaires : Très rare : Vasculite (par exemple granulomatose de Wegener, vascularite allergique). Affections respiratoires, thoraciques et médiastinales : Fréquent : Rhinopharyngite, épistaxis, sécheresse nasale ; Très rare : Bronchospasme (en particulier chez les patients asthmatiques), enrouement. Affections gastro-intestinales : Très rare : Maladie entérique inflammatoire, colite, iléite, pancréatite, hémorragie gastro-intestinale, diarrhée hémorragique, nausées, gorge sèche (voir rubrique 4.4 du RCP complet). Affections hépatobiliaires : Très fréquent : Transaminases augmentées (voir rubrique 4.4 du RCP complet) ; Très rare : Hépatite. Affections de la peau et du tissu sous-cutané : Très fréquent : Prurit, rash érythémateux, dermatite, chéilite, sécheresse cutanée, exfoliation localisée, fragilité cutanée (risque de traumatisme frictionnel) ; Rare : Alopécie ; Très rare : Acné fulminans, acné aggravée (poussée d’acné), érythème (facial), exanthème, troubles pileux, hirsutisme, dystrophie unguéale, paronychie, réaction de photosensibilité, botryomycome, hyperpigmentation cutanée, transpiration augmentée ; Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) : Érythème polymorphe, syndrome de Stevens-Johnson, nécrolyse épidermique toxique. Affections musculo-squelettiques et du tissu conjonctif : Très fréquent : Arthralgie, myalgie, dorsalgie (en particulier chez les enfants et les adolescents) ; Très rare : Arthrite, calcinose (calcification des ligaments et des tendons), soudure prématurée des cartilages de croissance, exostose, (hyperostose), densité osseuse diminuée, tendinite ; Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) : Rhabdomyolyse, Sacro-iliite. Affections du rein et des voies urinaires : Très rare : Glomérulonéphrite ; Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) : Urétrite. Affections des organes de reproduction et du sein : Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles) : Dysfonction sexuelle, y compris dysérection et diminution de la libido, gynécomastie, sécheresse vulvovaginale. Troubles généraux et anomalies au site d’administration : Très rare : Granulation tissulaire (augmentation de la formation de), malaise. Investigations : Très fréquent : Triglycérides sanguins augmentés, lipoprotéines de haute densité diminuées ; Fréquent : Cholestérol sanguin augmenté, glucose sanguin augmenté, hématurie, protéinurie ; Très rare : Créatine phosphokinase sanguine augmentée. Déclaration des effets indésirables suspectés : La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/ risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via Belgique : Agence fédérale des médicaments et des produits de santé - www.afmps.be - Division Vigilance : Site internet : www.notifieruneffetindesirable.be - e-mail : adr@fagg-afmps.be Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé - Site internet : www.guichet.lu/pharmacovigilance TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ : Laboratoires Bailleul S.A., 14-16 Avenue Pasteur, L-2310 Luxembourg, Luxembourg NUMÉROS D’AUTORISATION DE MISE SUR LE MARCHÉ : Belgique : Isotiorga 10 mg capsules molles: BE596071 ; Isotiorga 20 mg capsules molles: BE596080 Luxembourg : Isotiorga 20 mg capsules molles:
X plus dès la 1re application1
Peau très sèche, rugueuse
Formule intelligente pour soulager les signes d’une peau sèche - instantanément et durablement
HYDRATATION SUPÉRIEURE3
Hydratation intense 72h1
Lisse la peau et soulage les sensations de démangeaison
en semaines
❙ URÉE: 7x plus d’hydratation dès la 1re application1 ❙ LES FACTEURS NATURELS D’HYDRATATION + GLUCO-GLYCÉROL : hydratation intense ❙ CÉRAMIDES: amélioration de la fonction barrière de la peau
| CNK: 2816-049
| CNK: 2816-056
(8):29-39
1. Test instrumental, 42 volontaires. | 2. Mesures cornéométriques, augmentation de 92% des valeurs obtenues après 2 semaines d’application régulière en comparaison à une zone sans soin, 44 participants. | 3. Weber TM et al. Treatment of Xerosis with a