
6 minute read
Una presentación atípica de neuritis óptica

Autores: Dra. Fiorella Dapper (residente de 4° año), Dr. Marcelo Rudzinski (especialista en uveítis) y Dr. Pablo Piccilli (jefe), Servicio de Oftamología, Hospital Escuela de Agudos Dr. Ramón Madariaga (HEADRM)
La neuritis óptica es una causa frecuente de pérdida visual súbita en adultos jóvenes y puede constituir la primera manifestación de enfermedades desmielinizantes del sistema nervioso central como la esclerosis múltiple (EM), la más prevalente dentro de ellas. Sin embargo, la neuromielitis óptica (NMO) representa una entidad menos frecuente, pero con implicaciones pronósticas y terapéuticas significativamente diferentes.
La NMO es un trastorno inflamatorio autoinmune caracterizado por afectación recurrente del nervio óptico y la médula espinal, con mayor riesgo de secuelas permanentes si no se instaura un tratamiento adecuado.
A diferencia de la esclerosis múltiple, la NMO se asocia a anticuerpos anti-aquaporina 4 (AQP4-IgG) positivos que permiten establecer un diagnóstico más preciso y diferenciarla de otras entidades desmielinizantes como MOGAD (trastorno asociado a anticuerpos anti-Glicoproteína oligodendrocitaria de mielina). La identificación temprana es esencial, dado que el manejo inmunosupresor oportuno reduce la incidencia de recaídas y mejora la recuperación visual y neurológica.
“Este caso subraya la relevancia de la detección temprana de NMO en pacientes con neuritis óptica atípica y síntomas neurológicos asociados, además de la importancia del tratamiento inmunosupresor precoz para reducir el riesgo de recaídas y mejorar el pronóstico funcional. También destaca la importancia del enfoque multidisciplinario, involucrando oftalmología, neurología y laboratorio especializado para optimizar el diagnóstico y manejo; así como la vigilancia clínica y la educación del paciente sobre signos de recaída son fundamentales para mejorar el pronóstico a largo plazo”.
Dra. Fiorella Dapper
Este reporte de caso tiene como objetivo presentar un ejemplo clínico de neuritis óptica atípica que condujo al diagnóstico de NMO, destacando la importancia del enfoque multidisciplinario y el inicio temprano de tratamiento inmunosupresor.
Palabras Claves
neuritis óptica neuromielitis óptica esclerosis múltiple diagnóstico diferencial tratamiento inmunosupresor
Caso Clínico
Paciente femenina de 41 años, con antecedente de toxoplasmosis ocular en ojo derecho, consulta por pérdida progresiva e indolora de agudeza visual en ojo izquierdo de una semana de evolución intensificada en las últimas 72 horas. Refiere episodios de vómitos y cefalea que aparecieron 48 horas después del inicio del cuadro visual, sin otros síntomas neurológicos asociados.
EXAMEN OFTALMOLÓGICO:
• Agudeza visual (AV) sin corrección: OD no percepción de luz (NPL); OI: percepción de luz.
• Pupilas arreactivas con midriasis media; motilidad ocular conservada.
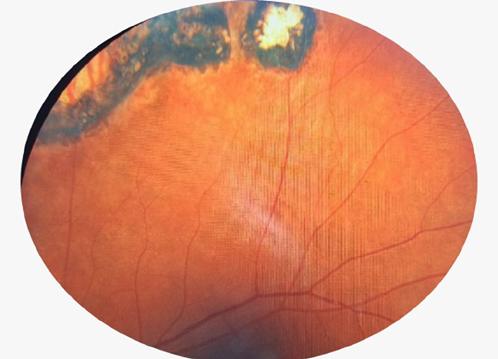
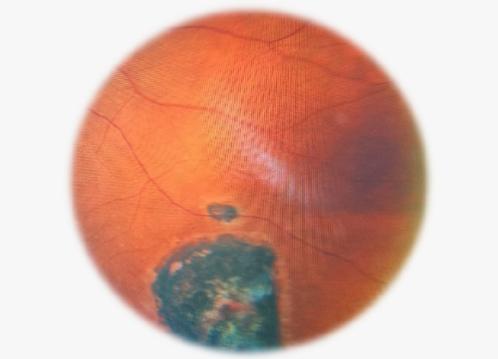
Fondo de ojo:
OD con nervio óptico pálido y cicatrices periféricas por toxoplasmosis


OI con papila de bordes netos y cicatrices periféricas, sin signos de edema activo.

Diagnósticos diferenciales considerados a partir de la presentación clínica de la paciente:
Neuritis óptica: NMO, EM, MOGAD. AION
Otras patologías autoinmunes: Lupus eritematoso sistémico, sarcoidosis, granulomatosis de Wegener. La presencia de síntomas sistémicos asociados (cefalea, vómitos) y la afectación monocular progresiva sugirieron un proceso inflamatorio/inmunológico más amplio que una neuritis óptica típica.
Tratamiento
Se indica internación inmediata e inicio de pulsoterapia con metilprednisolona intravenosa. Una vez confirmado el diagnóstico, se inicia tratamiento inmunosupresor con azatioprina, acompañado de corticoides orales en dosis descendente —de manera ambulatoria—, logrando estabilización de la agudeza visual y prevención de nuevas recaídas. Se inicia interconsulta con los servicios de Neurología, Clínica Médica e Infectología para manejo interdisciplinario. Se solicitan estudios complementarios:
Resonancia magnética (RMN) cerebral y de órbitas: el departamento de imágenes informa presencia de hiperintensidades en sustancia blanca y compromiso del quiasma óptico izquierdo, sin lesiones características de EM.


RMN cervical y torácica: se observan cambios degenerativos leves, sin lesiones desmielinizantes significativas en médula espinal.


Laboratorio: perfil inmunológico completo, anticuerpos anti-AQP4 y anti-MOG. El resultado fue positivo para anti-AQP4, compatible con NMO.
Discusión
La neuromielitis óptica es un trastorno inflamatorio autoinmune que afecta principalmente el nervio óptico y la médula espinal. Se caracteriza por recaídas graves que pueden conducir a discapacidad visual o motora permanente. Su diagnóstico se basa en la combinación de hallazgos clínicos, estudios de imagen y pruebas serológicas. La diferenciación de la esclerosis múltiple es crucial, ya que el manejo y pronóstico difieren significativamente. En este caso, la presencia de neuritis óptica unilateral atípica, pérdida visual progresiva, síntomas sistémicos y la serología positiva para anti-AQP4 orientaron al diagnóstico de NMO. La literatura señala que aproximadamente el 70 % de los pacientes con NMO presentan anticuerpos anti-AQP4, lo que permite un diagnóstico más seguro y la instauración temprana de terapia inmunosupresora. El tratamiento de primera línea incluye pulsoterapia con metilprednisolona y agentes inmunosupresores como azatioprina o rituximab, con el objetivo de prevenir nuevas recaídas y minimizar el daño neurológico. La terapia temprana se asocia con mejor recuperación visual y menor incidencia de discapacidad permanente. Este caso también destaca la importancia del enfoque multidisciplinario, involucrando Oftalmología, Neurología y Laboratorio especializado para optimizar el diagnóstico y manejo. La vigilancia clínica y la educación del paciente sobre signos de recaída son fundamentales para mejorar el pronóstico a largo plazo.
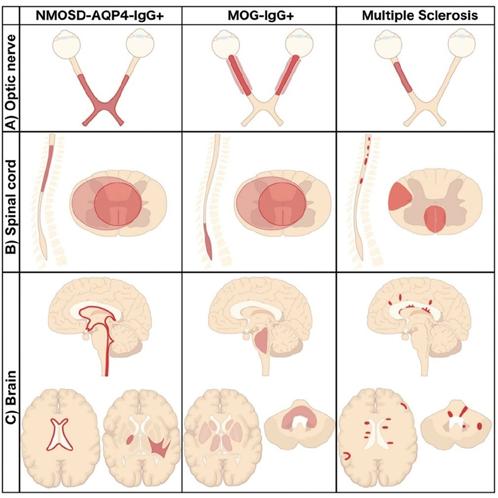
Además, se deben considerar los diagnósticos diferenciales de NMO, incluyendo MOGAD, esclerosis múltiple, neuropatías ópticas isquémicas y enfermedades autoinmunes sistémicas. Cada entidad presenta diferencias en edad de inicio, patrón de recaídas, hallazgos de imagen y respuesta al tratamiento, lo que subraya la necesidad de un enfoque individualizado y basado en evidencia (tabla 2)
Conclusión
La neuromielitis óptica debe sospecharse en pacientes con neuritis óptica atípica acompañada de síntomas sistémicos o neurológicos. La detección precoz, la confirmación mediante estudios de imagen y serología, y el inicio oportuno de tratamiento inmunosupresor permiten reducir el riesgo de recaídas, preservar la función visual y mejorar el pronóstico funcional.
El manejo multidisciplinario es clave para un seguimiento adecuado, asegurando la detección temprana de complicaciones y la optimización del tratamiento a largo plazo. Este caso clínico resalta la relevancia de considerar NMO como diagnóstico diferencial frente a neuritis óptica, especialmente en pacientes con antecedentes de enfermedad ocular previa y presentación clínica atípica.
El abordaje temprano y dirigido puede marcar la diferencia entre la recuperación visual parcial y la discapacidad irreversible, reafirmando la importancia de la educación continua del personal de salud y la colaboración entre especialidades.

Bibliografía
1. Wingerchuk DM, Banwell B, Bennett JL, Cabrera P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurólogo. 2015; 88: 177-189. Disponible en: https://pmc.ncbi.nlm.nih.gov/articles/PMC4515040/
2. Cacciaguerra L y Flanagan EP. A tale of Two Central Nervous System Autoinmune Inflammatory Disorders. Neurol Clin. 2024; 42: 77-144.Disponible en: https://doi.org/10.1016/j.ncl.2023.06.009
3. Rojas JI, González SJ, Patrucco L y Critiano E. Neuromielitis Óptica. Actualización de los conceptos clínicos y fisiopatología de la enfermedad. Rev HospItal B Aires. 2011; 31(1). Disponible en: http://revista.hospitalitaliano.org.ar





