RIAGEM NEONATAL

ANEMIA FALCIFORME

FENILCETONÚRIA
FIBROSE CÍSTICA
HIPOTIREOIDISMO
NO DIAGNÓSTICO PRECOCE DE DOENÇAS
IMPORTÂNCIA
DA

ANEMIA FALCIFORME
FENILCETONÚRIA
FIBROSE CÍSTICA
HIPOTIREOIDISMO
Montagem, edição e revisão: Amanda Pezzini Martins
Introdução: Joana D’Arc Souza Rodrigues
Professora orientadora: Cristina Souza
Colaboradores:
Amanda Pezzini Martins, Ana Luíza Nogueira Costa, Ana Luiza Sena Costa Brant, Bruno Vieira de Oliveira, Carla Rebeca Coelho Vieira, Esther Mara Maciel Oliveira, Gabriele Fernanda Batista Penido, Giovanna Melo, Helena Cristina dos Anjos Martins, Joana D’Arc Souza Rodrigues, Kaillany Silva dos Santos,
Lívia Vitória de Aguiar Oliveira, Lucas Vitorino Missiagia, Maria Laura Gonçalves dos Santos, Millena Faria Ribeiro, Morgana Alves Maia, Otilha Maria Alves Fernandes, Paulo Francisco Falcão Almeida, Quézia Emanuelle Ferreira Rocha, Rafaela Nayara Souza Silva, Samara Santana Peixoto, Sarah Pales de Pádua Pradino, Tainá Goulart dos Santos, Victor Matheus Costa Alves.
Vinícius Augustus Liberato Gomes Pedrosa
A triagem neonatal, mais conhecida como teste do pezinho, é um exame laboratorial simples, considerado a maior ação de saúde pública quanto à prevenção do agravo das doenças O exame de triagem neonatal apresenta papel fundamental desde os primeiros dias de vida do recém-nascido identificando diversas patologias precocemente como hipotireoidismo congênito, fibrose cística, anemia falciforme, fenilcetonúria, hiperplasia congênita da supra-renal, hemocistinúria, hiperfenilalaninemia, deficiência de globulina ligadora de tiroxina (TBG) entre outros e, assim, intervir na patologia detectada, possibilitando mais rapidamente o processo de cura

Essa triagem é dividida entre básica e ampliada, sendo a primeira responsável por detectar quatro tipos de doença: hipotireoidismo congênito, anemia falciforme, fibrose cística e fenilcetonúria e a segunda por detectar as doenças do teste básico e ainda outras quatro: hiperplasia congênita da suprarenal; deficiência de globulina ligadora de tiroxina (TBG); hemocistinúria; hiperfenilalaninemias
TRIAGEM NEONATAL A IMAGEM MOSTRA A REALIZAÇÃO DE UM TESTE DE TRIAGEM NEONATAL
O obgetivo deste trabalho é descrever as doenças do PNTN com ênfase na descrição genetica e bioquímica e descrever os exames diagnósticos utilizados. Foram estudadas seis doenças triadas tais como, fenilcetonúria, hipotireoidismo congênito, doença falciforme e outras hemoglobinopatias, fibrose cística, hiperplasia adrenal congenita e deficiência de biotinidase Cada capitulo sera dedicado a uma doença descrevendo os sintomas, dados epidemiológicos, etiologia, diagnostico detalhado e tratamento.

A anemia falciforme é uma doença genética autossômica de caráter recessivo que afeta, principalmente, indivíduos afrodescendentes. Indivíduos heterozigotos possuem o "traço falciforme", que na maioria dos casos é assintomático É de extrema importância que o parceiro do indivíduo falciforme ou com traço realize aconselhamento hematológico para verificar se há possibiliudiade do casal ter filhos falciformes

É considerada doença falciforme quando o indivíduo é homozigoto recessivo, apresentando hemácias no formato de foice que podem ser facilmente observadas no esfregaço sanguíneo. São consequência da doença crises de dor, vasoclusão, palidez, entre outras.
Não existe cura conhecida para a doença, contudo algumas práticas podem ser adotadas para melhorar a qualidade de vida do paciente falciforme: A hidroxiureia é hoje uma das drogas mais usadas no tratamento da anemia falciforme por ser capaz de aumentar a produção de um outro tipo de hemoglobina, conhecida como hemoglobina fetal (mais presente no período de vida uterina). Em caso de complicações, podem ser necessárias transfusões sanguíneas.
HERANÇA DO GENE HBS PELOS PAIS
A IMAGEM MOSTRA OS PAIS COM TRAÇOS FALCIFORMES (HBS EM HETEROZIGOSE)
PODENDO GERAR FILHOS COM TRAÇOS FALCIFORMES (AS) COM ANEMIA FALCIFORME (SS) OU SEM QUALQUER DOENÇA FALCIFORME (AA)
FONTE: HTTPS://BLOG VARSOMICS COM/DOENCA-FALCIFORME-AIMPORTANCIA-DA-CONSCIENTIZACAO/
Segundo estimativa da Organização Mundial da Saúde (OMS), 5% da população mundial é portadora do gene para Hemoglobinopatias, e a cada ano nascem 250 000 a 300 000 pessoas com a doença falciforme (DF) em todo o mundo Estima-se que essa hemoglobinopatia afeta aproximadamente 30 000 a 50 000 brasileiros, e que anualmente, de 1 000 a 3 500 crianças nascem com a doença
Dentre os estados brasileiros, os que possuem as maiores prevalências da doença são Bahia, Rio de Janeiro e Minas Gerais, respectivamente

A causa da doença falciforme é uma mutação pontual no gene β da hemoglobina, que acarreta a substituição do códon GAG para GTG, resultando na troca do Glutamato (Glu) por Valina (Val). Essa substituição produz uma molécula anormal chamada hemoglobina S (do inglês Sickle), que promove a modificação estrutural da hemácia para um formato de foice, podendo resultar em vasoclusão
A vasoclusão repercute no quadro clínico da doença falciforme, que inclui fortes crises de dor provocadas pela obstrução de pequenos vasos sanguíneos e afeta diretamente a vida do paciente falciforme
E FENÓTIPO DA HBS
A IMAGEM DESCREVE DA MUTAÇÃO NOS AMINOÁCIDOS À EXPRESSÃO DAS HEMÁCIAS O CICLO DA ANEMIA FALCIFORME
FONTE: HTTPS://SICKLE-CELL COM/CAUSES



Crise de dor: É o sintoma mais freqüente da doença falciforme, causado pela obstrução de pequenos vasos sanguíneos pelos glóbulos vermelhos em forma de foice. A dor é mais frequente nos ossos e nas articulações, podendo, porém atingir qualquer parte do corpo Essas crises têm duração variável e podem ocorrer várias vezes ao ano Geralmente são associadas ao tempo frio, infecções, período prémenstrual, problemas emocionais, gravidez ou desidratação;
Síndrome mão-pé: Nas crianças pequenas as crises de dor podem ocorrer nos pequenos vasos sanguíneos das mãos e dos pés, causando inchaço, dor e vermelhidão no local;
Infecções: As pessoas com doença falciforme têm maior propensão a infecções e, principalmente as crianças podem ter mais pneumonias e meningites
Úlcera de Perna: Ocorre mais frequentemente próximo aos tornozelos, a partir da adolescência As úlceras podem levar anos para a cicatrização completa, se não forem bem cuidadas no início do seu aparecimento. Para prevenir o aparecimento das úlceras, os pacientes devem usar meias grossas e sapatos;
Sequestro do Sangue no Baço: O baço é o órgão que filtra o sangue Em crianças com anemia falciforme, o baço pode aumentar rapidamente por seqüestrar todo o sangue e isso pode levar rapidamente à morte por falta de sangue para os outros órgãos, como o cérebro e o coração É uma complicação da doença que envolve risco de vida e exige tratamento emergencial
SÍNDROME MÃO-PÉ
HEMÁCIA NORMALO diagnóstico dessa hemoglobinopatia é bastante complexo, por isso um diagnóstico precoce é importante para um tratamento mais completo. Os exames mais utilizados para a determinação dessa patologia são: Hemograma, Teste de falcização, Teste de Solubilidade, Eletroforeses, Focalização Isoelétrica, Imunoensaio, Diagnóstico em Neonatos, dosagem de Hemoglobina Fetal e Hemoglobina A2.
Eletroforese: A eletroforese de hemoglobina é usada como diagnóstico pré-natal para identificar os diferentes tipos de hemoglobina que podem ser encontrados no sangue. A eletroforese baseia-se na migração de íons de acordo com um campo elétrico, e as proteínas são carregadas negativamente e migram para o eletrodo positivo por atração eletrostática
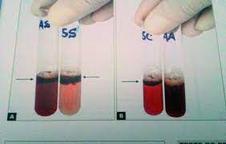

FONTE: HTTP //WWW HEMOGLOBINOPATIAS COM BR/HEMOGLOBINOPATIAS/ANALISES HTML
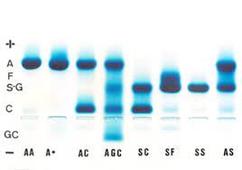
Teste de Falcização: Avaliação qualitativa para determinar a presença ou ausência de hemoglobina S (HbS) nas hemácias. Devido à baixa resolução, o teste falciforme é o menos indicativo, pois não consegue diferenciar genótipo de Hb S (Hb SS, Hb SF, Hb SC, Hb AS, Hb SD) quando positivo
FONTE: HTTPS://REPOSITORIO FAEMA EDU BR/BITSTREAM/12 3456789/2479/1/TCC%20EDELSON%20COSTA%20DE%20SOUZA PDF
Teste de Solubilidade: Um teste baseado na insolubilidade da desoxiemoglobina S, uma vez que a hemoglobina normal é solúvel. Muitas vezes é usado como teste de triagem ou confirmatório para HbS em emergências, mas não tem boa sensibilidade no diagnóstico de recém-nascidos prematuros porque suas hemácias ainda não são HBF (hemoglobina fetal)
FONTE: HTTPS //SISTEMAS UFT EDU BR/PERIODICOS/ INDEX PHP/PATOLOGIA/ARTICLE/DOWNLOAD/2245/9552/
TESTE DE ELETROFORESEReação em Cadeia da Polimerase (PCR): Um método de amplificação in vitro de seqüências específicas de DNA, permitindo a identificação de mutações presentes no material genético, antes mesmo do nascimento, e análise das variações dos produtos protéicos

FONTE: HTTPS://WWW BIOMEDICINAPADRAO COM BR/2011/10 /REACAO-EM-CADEIA-DA-POLIMERASE-PCR HTML

Cromatografia líquida de alta performace (HPLC): É uma técnica molecular que viabiliza a exclusão de anomalias da Hemoglobina. A HPLC tem a quantificação de Hb A2, Hb F, Hb A, Hb S, Hb C e triagem para variantes Na técnica, coloca-se um pequeno volume da amostra em uma coluna cromatográfica com partículas porosas (fase estacionária) Então um reagente é dispensado na coluna, fazendo com que os componentes da mistura desloquem-se através da coluna As substâncias com maior afinidade pelo reagente (solvente) movem-se mais lentamente; já as com menor afinidade movem-se mais rapidamente Ao sair da coluna, os componentes passam por um detector que emite um sinal elétrico o qual é registrado, constituindo um cromatograma.

O teste do pezinho surgiu nos anos 1960 nos Estados Unidos O Brasil possui registros de programas de aconselhamento genético desde a década de 1950, No entanto, os Programas de Triagem
Neonatal começaram somente em 1976. Em 1992, o Ministério da Saúde criou o Comitê de Hemoglobinopatias, responsável pelas primeiras medidas de divulgação e de normatização do tratamento dessas doenças em nosso país
RODRIGUES, D. DE OW, ET AL. "HISTÓRIA DA TRIAGEM NEONATAL PARA DOENÇA FALCIFORME NO BRASIL–CAPÍTULO DE MINAS GERAIS " REV MED MINAS GERAIS 22 1 (2012): 1-128.
Apesar de não ter cura, existem fatores que podem aumentar a qualidade de vida e longevidade do paciente falciforme Os tratamentos incluem medicamentos, transfusões de sangue e, raramente, transplante de medula óssea.
Medicamentos podem ser divididos em analgesiantes, sendo os principais opióides e antiinflamatórios não esteroidais Outro medicamento muito comum na rotina do falciforme é a Hidroxiuréia, um quimioterápico oral que é capaz de aumentar os níveis de hemoglobina fetal, quem em altos níveis de elevação provoca a diminuição da polimerização de hemácias defeituosas diminuindo o risco de vasooclusão Podem ser utilizados outros medicamentos como protocolo para evitar ou tratar complicações como ácido fólico, quelantes de ferro, dentre outros É importante salientar que somente médicos podem diagnosticar a doença e nenhum medicamento deve ser usado sem indicação e orientação do profissional habilitado.

Um dos testes realizados para detecção precoce, o Programa Nacional de Triagem Neonatal Biológica é extremamente benéfica para o paciente. Quando descoberta a doença, o bebê deve ter acompanhamento médico adequado baseado num programa de atenção integral realizado pelo SUS. Nesse programa, os pacientes devem ser acompanhados por toda a vida por uma equipe com vários profissionais treinados no tratamento da anemia falciforme para orientar a família e o doente a descobrir rapidamente os sinais de gravidade da doença, a tratar adequadamente as crises e a praticar medidas para sua prevenção
MÚLTIPLOS EFEITOS BENÉFICOS DA HIDROXIUREIA NA DOENÇA FALCIFORME
(1) INDUÇÃO DA HB FETAL POR MEIO DA ATIVAÇÃO DA GUANILATO CICLASE ALTERANDO A CINÉTICA DOS PRECURSORES ERITRÓIDES; (2) BAIXOS VALORES DE NEUTRÓFILOS E RETICULÓCITOS POR INIBIÇÃO DA RIBONUCLEOTÍDEO REDUTASE E CITOXICIDADE DA MEDULA; (3) DIMINUIÇÃO DA ADESIVIDADE E MELHORA DA REOLOGIA DOS NEUTRÓFILOS E RETICULÓCITOS CIRCULANTES; (4) REDUÇÃO DA HEMÓLISE PELA MELHOR HIDRATAÇÃO DOS ERITRÓCITOS, MACROCITOSE, E REDUÇÃO DA FALCIZAÇÃO; (5) O ÓXIDO NÍTRICO (NO) É LIBERADO E COMO POTENTE VASODILATADOR LOCAL, MELHORA A RESPOSTA VASCULAR
1 GALIZA NETO, Gentil Claudino de; PITOMBEIRA, Maria da Silva Aspectos moleculares da anemia falciforme Jornal Brasileiro de Patologia e Medicina Laboratorial, v 39, p 51-56, 2003
2. CANÇADO, Rodolfo D.; JESUS, Joice A. A doença falciforme no Brasil. Revista Brasileira de Hematologia e hemoterapia, v 29, p 204-206, 2007
3 DA SILVA, Neila Caroline Henrique et al Principais técnicas para o diagnóstico da anemia falciforme: uma revisão de literatura Caderno de Graduação-Ciências Biológicas e da Saúde-UNIT-PERNAMBUCO, v 3, n. 2, p. 33-33, 2017.
4 LOBO, Clarisse; MARRA, Vera Neves; SILVA, Regina Maria G Crises dolorosas na doença falciforme Revista Brasileira de hematologia e hemoterapia, v 29, p 247-258, 2007
5 https://portal fiocruz br/noticia/anemia-falciforme-hidroxiureia-diminui-ativacao-de-celulas-de-defesa
6. https://revista.abrale.org.br/hidroxiureia-como-funciona-e-quando-usar/
7 https://bvsms saude gov br/anemia-falciforme/
8 https://www gov br/saude/pt-br/composicao/saes/sangue/programa-nacional-da-triagem-neonatal
Amanda Pezzini Martins,
Ana Luíza Nogueira Costa,
Ana Luiza Sena Costa Brant,
Joana D’Arc Souza Rodrigues,
Helena Cristina dos Anjos Martins,
Otilha Maria Alves Fernandes,
Victor Matheus Costa Alves.
A fenilcetonúria (PKU - do inglês, phenilketonuria [OMIM: 261600]) é uma entre as 300 doenças hereditárias causadas pela desordem dos processos bioquímicos celulares É considerada um dos erros inatos do metabolismo, onde o defeito metabólico gerado geralmente da enzima fenilalanina hidroxilase (PAH) leva a não conversão do aminoácido fenilalanina (Phe) em tirosina (Tyr), importante na produção de neurotransmissores Com isso, provoca o acúmulo de fenilalanina no sangue e excreção urinária de ácido fenilpirúvico, que atribui odor desagradável característico.
É uma enfermidade caracterizada por padrão de herança autossômica recessiva, como exemplificado na Figura 1, sendo que os indivíduos afetados são homozigotos recessivos (genótipo aa), filhos de pais portadores obrigatórios do genótipo heterozigoto (Aa) Por se tratar de uma doença com alteração em cromossomos autossômicos, pode afetar ambos os sexos de maneira semelhante 97% dos casos de PKU são resultantes de mutações no gene PAH, sendo que os outros 3% são deficiências do cofator, a tetraidrobiopterina (BH4)

REFERÊNCIA: HTTPS://RETINABRASIL ORG BR/OS-TRESPRINCIPAIS-PADROES-DE-HERANCA/
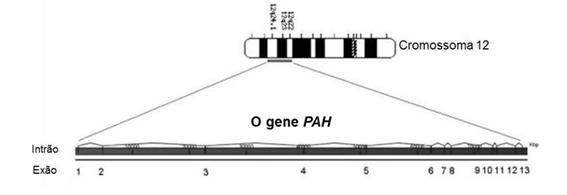
A fenilcetonúria é causada por mais de 1 180 mutações diferentes no gene (figura 2) que codifica a enzima fenilalanina hidroxilase, presente no braço longo (q) do cromossomo autossômico 12 (12q22–24 1)
Esse gene é composto por 13 exons e codifica um polipeptídeo que contém 452 aminoácidos
FIGURA 2: O GENE AFETADO REPRESENTAÇÃO DA ESTRUTURA DO GENE AFETADO (PAH) NO CROMOSSOMO 12Mundialmente, estima-se que 0 45 milhões de indivíduos possuam fenilcetonúria, com prevalência global de um afetado a cada 23 930 nascidos vivos Essa prevalência varia entre diferentes regiões e etnias
No continente europeu é encontrada uma maior prevalência, com representantes como Itália (1 caso a cada 4.000 nascidos vivos) e Irlanda (1 caso a cada 4.545 nascidos vivos). Em seguida, a região do Oriente Médio, onde encontram-se Irã e Jordânia (1:5000) As Américas do Norte e do Sul apresentam taxas menores: Canadá (1:15 000), Estados Unidos (1:25 000) e México (1:27 778), Argentina (1:15,715), Chile (1:19,231) e Brasil (1:25,000)
As menores taxas registradas são encontradas em países asiáticos, Tailândia (1:227,273), Japão (1:125,000), Filipinas (1:116,006) e Singapura (1:83,333) A figura 3 apresenta as taxas de prevalência no contexto mundial
REFERÊNCIA: THE GENETIC LANDSCAPE AND EPIDEMIOLOGY OF PHENYLKETONURIA AM J HUM GENET 2020 DISPONÍVEL EM: HTTPS://PUBMED NCBI NLM NIH GOV/32668217/

Na fenilcetonúria, o excesso de fenilalanina é desviado para uma via metabólica alternativa, na qual são produzidos compostos secundários, como os ácidos fenilpirúvico, feniláctico e fenilacético, que são considerados neurotóxicos, ou seja, provocam danos ao sistema nervoso A fenilalanina e seus metabólitos tóxicos são acumulados, principalmente, no sistema nervoso central, levando a PKU clássica, leve ou HPA não PKU (também chamada de HPA benigna), como demonstrado na Figura 4.
FIGURA 4: EFEITOS DO ACÚMULO DE FENILALANINA A IMAGEM DESCREVE POTENCIAIS MECANISMOS RESPONSÁVEIS PELA DEFICIÊNCIA NEUROCOGNITIVA OBSERVADAS NOS PORTADORES DE PKU (BBB) BARREIRA HEMATO ENCEFÁLICA (LAT1) TRANSPORTADOR DE AMINOÁCIDOS L DO TIPO I (LNAA) AMINOÁCIDOS NEUTROS DE MAIOR MASSA MOLECULAR
REFERÊNCIA: GOMES K ET AL NOVAS ABORDAGENS PARA O TRATAMENTO DA FENILCETONÚRIA: AVALIAÇÃO EM MODELOS CELULARES DE FORMULAÇÕES DA FENILCETONÚRIA HIDROXILASE HUMANA 2016 DISPONÍVEL EM HTTPS://REPOSITORIO UL PT/BITSTREAM/10451/26534/1/ULFC120723 TM K%C3%A1TIA GOMES PDF

Esse acúmulo de substâncias tóxicas prejudiciais ao SNC provoca sintomas como deficiência intelectual severa, epilepsia, problemas psicomotores (ataxia: dificuldade na função motora) e de conduta (irritabilidade, distúrbios de atenção e aprendizagem, hiperatividade), microcefalia (figura 5) Ainda, a fenilcetonúria pode provocar hipopigmentação de pele e cabelo (figura 6), eczema, odor desagradável na urina

FIGURA 5: MICROCEFALIA
DIMINUIÇÃO DO TAMANHO NORMAL DO CRÂNIO DEVIDO AO DESENVOLVIMENTO ANORMAL DO CÉREBRO

FIGURA 6: INDIVÍDUO COM FENILCETONÚRIA APRESENTANDO HIPOPGMENTAÇÃO DA PELE E CABELOS
REFERÊNCIA: HTTPS://DOUTORNUBERTO COM BR/MICROCEFALIA/
REFERÊNCIA: FENILCETONÚRIA - PKU | SAÚDE E A QUÍMICA DA CÉLULA HUMANA (SAUDECELULAHUMANA BLOGSPOT COM)
Para evitar sequelas irreversíveis, o diagnóstico da fenilcetonúria deve ser precoce, idealmente através de programas de Triagem Neonatal. A triagem é realizada através da dosagem quantitativa da Fenilalanina no sangue, obtida de amostras colhidas em papel filtro Para que o aumento desse aminoácido possa ser detectado, é fundamental que a criança tenha ingerido proteínas, portanto é recomendado que a coleta seja feita após 48 horas do nascimento Nesse momento, mesmo crianças de risco, que ainda não tiveram contato com leite materno, podem colher material desde que estejam sob dieta rica em aminoácidos essenciais
Os métodos laboratoriais utilizados para avaliar o nível de fenilalanina são: espectrometria de massa em tandem, cromatografia líquida de alto desempenho (HPLC), cromatografia gasosa e fluorimetria


Espectrometria de massa em Tandem (figura 7): as moléculas existentes na amostra biológica são convertidas em íons em fase gasosa, e posteriormente estes íons são separados no espectrômetro de massas de acordo com sua razão massa (m) sobre a carga (z), m/z, permitindo a identificação e quantificação do metabólito analisado, nesse caso, a fenilalanina
REFERÊNCIA: HTTPS://TESTEDABOCHECHINHA COM BR/TESTEDA-BOCHECHINHA-ESPECTROMETRIA-DE-MASSAS-EM-TANDEMMS/ Cromatografias (líquida de alto desempenho e gasosa): A cromatografia é uma técnica utilizada para a análise, identificação e separação dos componentes de uma mistura É definida pela separação dos componentes na interação desses com a fase estacionária e com a fase móvel; líquido-líquido (alto desempenho; figura 8) e gás-líquido (gasosa; figura 9)

Fluorimetria (figura 10): técnica de análise que determina a quantidade ou concentração de uma determinada substância na amostra medindo a intensidade da luz emitida por um fluoróforo em um aparelho chamado espectrofotômetro

É possível também diagnosticar por meio análise molecular, onde o gene PAH é sequenciado para detecção da origem da mutação, e essa genotipagem pode favorecer a correlação entre a gravidade clínica e a instituição do melhor tratamento à cada caso.
Não existe uma cura para a fenilcetonúria, mas quando iniciado após diagnóstico precoce, o tratamento, quando iniciado até os primeiros três meses de vida, pode garantir a ausência de sequelas mais graves e consequentemente, melhor qualidade de vida para o indivíduo É realizado em duas etapas: dieta e uso de fórmulas metabólicas A dieta deve conter baixo teor de fenilalanina, pois a Phe é um aminoácido essencial ao corpo humano, logo deve estar presente nem que seja em pequenas quantidades, com o consumo de alimentos pobres em proteínas (como vegetais e frutas) e uso de fórmula metabólica rica em aminoácidos.

O tratamento é multidisciplinar: necessita de supervisão por nutricionistas especializados e pediatras, com avaliações clínicas, bioquímicas, estado nutricional, mudanças fisiológicas e fisiopatológico que podem indicar o aumento ou redução da fenilalanina no sangue
REFERÊNCIA:HTTPS://CORDEIROPOLIS CORDEROVIRT UAL COM BR/NOTICIAS/14404/SAUDE/NO-DIANACIONAL-DO-TESTE-DO-PEZINHO-APAE-LIMEIRAALERTA-PARA-EXAME
107,2 (2020): 234-250 doi:101016/jajhg202006006
MAIS
A fenilcetonúria é, dentre os erros inatos do metabolismo, o mais comum, com mais de 1.180 variações no gene PAH já registradas.Hillert, Alicia et al. “The Genetic Landscape and Epidemiology of Phenylketonuria.” American journal of human genetics vol FIGURA 10: ANÁLISE FLUORIMÉTRICA FIGURA 11: COLETA DE SANGUE NO TESTE DO PEZINHO
1. HILLERT A. et al. The Genetic Landscape and Epidemiology of Phenylketonuria. American journal of human genetics v 107, p 234-250 2020
2 SANTOS, M ; HAACK, A Fenilcetonúria: diagnóstico e tratamento Com Ciências Saúde, v 23, n 4, p 263270, 2012
3. PAVIA, D. L.; LAMPMAN, G. M.; KRIZ, G. S.; ENGEL, R. G. Química orgânica experimental: técnicas de escala pequena 2 ed Porto Alegre: Bookman, 2009
4 VAN SPRONSEN, F et al Phenylketonuria. Nature reviews Disease primers v 71, p 36 2021
5 DE MARQUI, Alessandra Bernadete Trovó Fenilcetonúria: aspectos genéticos, diagnóstico e tratamento. Revista da Sociedade Brasileira de Clínica Médica, v 15, n 4, p 282-288, 2017
6 GOMES, Kátia Cristina Morais Soares et al Novas abordagens para o tratamento da fenilcetonúria: avaliação em modelos celulares de formulações da fenilcetonúria hidroxilase humana. 2016. Tese de Doutorado
7 MONTEIRO, Lenice Teresinha Bussolotto; CÂNDIDO, Lys Mary Bileski Fenilcetonúria no Brasil: evolução e casos. Revista de Nutrição, v 19, p 381- 387, 2006
8 COSTA, Roseli Divino, et al IDENTIFICATION OF MUTATIONS IN THE PAH GENE IN PKU PATIENTS IN THE STATE OF MATO GROSSO. Revista Paulista de Pediatria, v 38, 2020
9. RAMALHO, Antônio R. O; AGUIAR-OLIVEIRA, Manuel H. Avaliação da eficiência e desfecho da triagem e manejo da PKU no Estado de Sergipe, Brasil. Arquivos Brasileiros de Endocrinologia e Metabologia, v 58 2014
10 Programa Nacional de Triagem Neonatal, Ministério da Saúde 2021 Disponível em: https://www.gov.br/saude/pt-br/composicao/saes/sangue/programa-nacional-da-triagemneonatal/fenilcetonuria-pku
11 COSTA, Roseli Divino, et al IDENTIFICATION OF MUTATIONS IN THE PAH GENE IN PKU PATIENTS IN THE STATE OF MATO GROSSO Revista Paulista de Pediatria, v 38, 2020 Disponível em: https://www scielo br/j/rpp/a/xMFgpLvfzcmB7P6nzbN4zRK/?lang=en Acesso em: 18 set 2022
12 RAMALHO, Antônio R O; AGUIAR-OLIVEIRA, Manuel H Avaliação da eficiência e desfecho da triagem e manejo da PKU no Estado de Sergipe, Brasil Arquivos Brasileiros de Endocrinologia e Metabologia, v 58 2014 Disponível em: https://www.scielo.br/j/abem/a/7QPczgDYR87QhJnsfZSz6xj/?lang=en. Acesso em: 18 set. 2022.
Bruno Vieira de Oliveira,
Carla Rebeca Coelho Vieira,
Morgana Alves Maia,
Rafaela Nayara Souza Silva, Sarah Pales de Pádua Pradino, Vinícius Augustus Liberato Gomes Pedrosa.
A fibrose cística (FC) é a doença genética mais frequente em populações brancas (descendentes de Caucasianos), atingindo ambos os sexos É uma doença cujo problema está localizado em um dos cromossomos do DNA
Uma das menções mais antigas atribuídas a Fibrose Cística é no folclore europeu por volta dos séculos XVIII ou XIX (FIRMIDA, Mônica, LOPES, Agnaldo, 2011)
A lenda diz que crianças que, quando beijadas na fronte, tivessem sabor salgado morreriam precocemente. Esse gosto salgado na criança provavelmente era devido a alta concentração de sal no suor da mesma, uma das características principais para o diagnóstico da Fibrose Cística Mas somente há pouco mais de 70 anos a doença foi oficialmente reconhecida
Em 1936, Fanconi descreveu o caso de uma criança portadora de síndrome celíaca com alterações pancreáticas que diferia da doença celíaca clássica, pois apresentava sintomas pulmonares e intestinais. Ao longo dos anos, Fanconi formulou uma hipótese da etiologia da doença e propôs a normalização do seu tratamento

A fibrose cística é uma doença genética rara que se dá principalmente no pulmão, para a qual atualmente não há cura Também conhecida como mucormicose, a Fibrose Cística, causa secreções de muco no pulmão extremamente pegajoso, essas secreções são mais espessas que o normal e são difíceis de eliminar, podendo causar vários problemas nos pacientes afetados Além de afetar os pulmões, a FC afeta as secreções do pâncreas e do sistema digestivo
A Fibrose Cística é uma doença genética autossômica recessiva, assim, é passada de pais portadores, ou carregadores, para seus filhos. Não é uma doença infecciosa Apesar de rara, a fibrose cística é uma das doenças raras mais comuns no Brasil De acordo com o Registro Brasileiro de Fibrose Cística (REBRAFC), um a cada 10 mil nascidos vivos são afetados pela doença
A Fibrose Cística é um distúrbio genético monossômico com alteração no cromossomo 7. Dispondo de 1 872 mutações registradas nas bases de dados Tem como frequência populações de origem Caucasiana tais quais as da Europa, América do Norte e Austrália Afeta cerca de 70 000 pessoas em todo o mundo Na União Europeia, 1 em cada 2 000 a 3 000 recém-nascidos são afetados , nos EUA, esta frequência é de 1 em cada 3 500, no sul da África, 1 em cada 42 pessoas são carreadoras e a incidência é estimada em 1 a cada 7 056 indivíduos (FIRMIDA, LOPES, 2011) Em países com população heterogênea a incidência da Fibrose Cística é menor por consequência da grande mistura étnica
Os avanços das últimas décadas vêm mudando o cenário da doença, que anteriormente consideravam um distúrbio letal, atualmente os seus portadores já possuem uma realidade melhor podendo viver até os 50 ou 60 anos. Quanto mais cedo o paciente tem acesso a tratamento e acompanhamento em centros especializados, melhor tende a ser sua sobrevida e maior a chance de se promover qualidade de vida
A fibrose cística é uma doença monogênica autossômica recessiva. A etiologia dessa doença é determinada pelo gene CFTR (Cystic Fibrosis Transmembrane Regulator), localizada do braço longo do cromossomo 7, que é dividido em 27 exons representando cerca de 5% do DNA genômico (Reis FJC et al, 2018) Ele codifica um RNAm que é transcrito em uma proteína de 1480 aminoácidos, também determinada CFTR Esse gene pode ter mais de 1800 mutações, sendo a mais frequente a p Phe508del, também conhecida como DF508, ela está presente em 66% dos alelos em estatística mundial Esta é uma mutação que afeta o processamento da proteína CFTR, impedindo-a de chegar à membrana celular e atuar como um canal de Cl–.

A DF508 é uma deleção de 3 pb no éxon 10 do gene CFTR, que ocasiona a perda da fenilalanina na posição 508 da proteína A proteína CFTR com DF508 é produzida, mas seu transporte para a membrana celular não ocorre, pois ela apresenta dobramento incorreto (Saraiva-Pereira ML et al, 2011).

Teste do pezinho: Ao nascer, os pacientes fibrocísticos apresentam uma quantidade elevada de tripsina, e ela se mantém assim durante os 30 primeiros dias de vida do recém-nascido Na triagem neonatal, emprega-se uma dosagem de tripsina imunorreativa (IRT) e ela detecta a tripsina presente Em caso de exame alterado é necessário realizar uma nova coleta duas semanas após a primeira. Caso essa amostra também esteja alterada, o paciente é encaminhado para realizar o teste de suor pelo método de Gibson&Cooke


Teste do Suor: A análise da concentração dos íons Na e Cl20 no suor do paciente é considerada como um método ouro no diagnóstico da fibrose cística A produção de suor é estimulada por uma e iontoforese de pilocapina, processo que consiste na aplicação de uma corrente elétrica leve à pele, servindo para aumentar temporariamente sua permeabilidade, o que permite que a pilocapina passe através do que normalmente seria uma barreira Os resultados com uma concentração de cloro >60 mmol/L são considerados diagnóstico de fibrose cística até prova do contrário É necessário repetir o teste em ocasiões diferentes para confirmar o diagnóstico
Tanto os sintomas, quanto a gravidade, da Fibrose Cística podem variar de pessoa para pessoa, devido ao fato de que parte dos sintomas é baseado no tipo de mutação presente no gene, podendo haver mais de 1000 tipos diferentes de mutação para esse determinado gene.
O gene defeituoso acarreta disfunção de uma proteína que se situa na membrana apical das células epiteliais de muitos órgãos do nosso corpo, como as do trato respiratório, de glândulas submucosas e do pâncreas exócrino Essa proteína, conhecida como CFTR, tem como principal função ser um canal de transporte de cloro, regulando o balanço entre íons e água através do epitélio (FIRMIDA et al, 2011) Com o mau funcionamento da CFTR, não ocorre o balanço entre íons e água eficiente nas células do corpo, gerando alterações nas propriedades físico químicas do muco, desidratando-o e tornando-o mais espesso e viscoso Esse muco hiperconcentrado pode causar uma série de problemas, podendo obstruir ductos pancreáticos e ductos da árvore brônquica, causando graves infecções crônicas
Porém, a fibrose cística tem como sintomas mais comuns: pele ou suor de sabor muito salgado; íleo meconial e obstruções intestinais; insuficiência pancreática; tosse persistente, muitas vezes com catarro grosso; presença de infecções pulmonares frequentes, como a pneumonia e a bronquite; presença de chiados no peito ou falta de fôlego causado pelo acúmulo de muco nos pulmões; crescimento lento ou baixo ganho de peso, apesar de bom apetite; presença de diarreia; surgimento de pólipos nasais; infertilidade; e baqueteamento digital, que é o alongamento e arredondamento na ponta dos dedos

Apesar de não existir cura para a Fibrose Cística, o tratamento pode aliviar os sintomas e reduzir as complicações. O tratamento é realizado por uma equipe de profissionais da saúde de diversas áreas, como médicos (geralmente pneumologistas e gastroenterologistas), enfermeiras, psicólogos, fisioterapeutas e nutricionistas. Alguns dos métodos de tratamento são:
Drenagem postural - drenagem de secreções pulmonares sob a influência da gravitação Esse método é usado para tratar sintomas relacionados às secreções que normalmente se acumulam nos pulmões dos pacientes de Fibrose Cística;
Suplemento alimentar - preparações alimentares que se destinam a fornecer nutrientes como vitaminas, minerais, fibras, ácidos graxos ou aminoácidos, que podem estar ausentes ou não absorvidos em quantidades suficientes na dieta do paciente;
Medicamentos - o uso de medicamentos é muito utilizado no tratamento da FC. Os antibióticos matam as bactérias, ou impedem o seu crescimento excessivo. A penicilina, por exemplo, mata bactérias específicas que costumam causar infecções nos portadores da doença Os medicamentos para tosse bloqueiam o reflexo da tosse e podem afinar o muco e limpar as vias aéreas;
Terapia inalatória - a nebulização ajuda a melhorar a respiração, pois consegue fazer com que a medicação chegue diretamente nos pulmões, podendo alcançar um melhor efeito do medicamento no órgão;
Vibração torácica - tratamento com o uso de um dispositivo que vibra o corpo do paciente para a terapia de desobstrução das vias aéreas com o objetivo de remover o excesso de muco das vias aéreas pulmonares

No entanto, esses tratamentos não são a cura para a Fibrose Cística Porém, a substituição pulmonar, através de transplante de órgãos, poderia melhorar os sintomas relacionados aos pulmões. Essa cirurgia é possível de ser realizada quando disponibilizada pelo SUS.
O aprimoramento no conhecimento, nas últimas décadas é visivelmente significativo quando falamos de progresso nos tratamentos e diagnósticos diligentes dos pacientes, especialmente com relação às terapias gênicas em estudo, a inclusão da dosagem da tripsina imunorreativa (IRT) na triagem neonatal, possibilitando o diagnóstico precoce da doença e a introdução do tratamento multidisciplinar visando à prevenção da desnutrição e da deterioração da função pulmonar
Em suma, atualmente mesmo com o avanço nas pesquisas e nos diagnósticos, a fibrose cística ainda é conhecida como uma doença rara de pouca divulgação nos meios sociais E a partir disso, intensificar o estudo sobre possíveis novas perspectivas de tratamentos para que assim, os portadores da doença possam observar a possibilidade de melhores condições em sua vivência
Lançado em 2019, o filme “A cinco passos de você" conta a história de um casal de jovens com a doença genética Fibrose Cística Will, um dos protagonistas, é portador da bactéria B, Cepacia. Para evitar contaminação cruzada e outras complicações causadas pela bactéria, ele precisa manter pelo menos cinco passos de distância de outros pacientes, incluindo seu interesse amoroso Stella, devido a vulnerabilidade que a FC causa nas vias respiratórias dos afetados. A trama segue mostrando a vida dos adolescentes e as dificuldades que a doença os causa nos seus dia-a-dias Apesar de ter várias menções recentes da Fibrose Cística na mídia, como no filme, e em estudos, a doença já era mencionada em contos a mais de cinco séculos atrás.

1 FIRMIDA, Mônica; LOPES, Agnaldo Aspectos Epidemiológicos da Fibrose Cística Revista do Hospital Universitário Pedro Ernesto, Rio de Janeiro, v 10, n 4, p 12 Dezembro de 2011
2 ROSA, Fernanda; et al Fibrose cística: uma abordagem clínica e nutricional Revista Nutricional, Campinas, v. 21, n. 6, p. 725. Novembro/Dezembro de 2008.
3 REIS, Francisco; DAMACENO, Neiva Artigo de revisão: Fibrose cística Jornal de Pediatria, DOI: 10 2223/JPED 489, v 74, n S1, p 76 Novembro/Dezembro de 1998
4 FIRMIDA, Mônica; MARQUES, Bruna; COSTA, Cláudia Fisiopatologia e Manifestações Clínicas da Fibrose Cística. Revista do Hospital Universitário Pedro Ernesto, Rio de Janeiro, v. 10, n.4, p. 46. Dezembro de 2011.
5 CABELLO, Giselda Avanços da Genética na Fibrose Cística. Revista do Hospital Universitário Pedro Ernesto, Rio de Janeiro, v 10, n 4, p 36 Dezembro de 2011
6. PEREIRA, Maria Luiza; KIEHL, Mariana; SANSEVERINO, Maria Teresa. A GENÉTICA NA FIBROSE CÍSTICA. Clinical and Biomedical Research, v 31, n 2 Junho de 2011
Gabriele Fernanda Batista Penido
Giovanna Melo
Kaillany Silva dos Santos
Lucas Vitorino Missiagia
Millena Faria Ribeiro
Tainá Goulart dos Santos
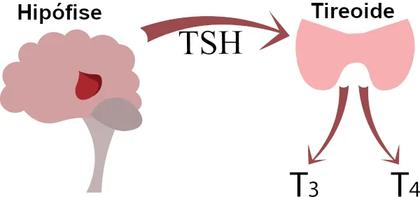
O hipotireoidismo congênito (HC) é uma síndrome de deficiência de hormônio tireoidiano em recémnascidos, caracterizada pela produção baixa ou mesmo nula do hormônio da glândula tireoide, ou, ocasionalmente, pela resistência à ação deste A tireoide é a primeira glândula endócrina que surge durante o desenvolvimento embrionário, é uma das glândulas mais importantes do sistema endócrino, produz os hormônios TSH, T3 e T4 (Figura 1), responsáveis por regular a função de órgãos essenciais como o cérebro, coração, fígado e os rins Nas crianças, principalmente durante os primeiros anos de vida, esses hormônios são fundamentais para o crescimento físico, o desenvolvimento do cérebro, além das várias funções do organismo.
Quando é originado por razões na própria glândula, é hipotireoidismo primário, por alterações na hipófise, hipotireoidismo secundário, e no hipotálamo, hipotireoidismo terciário (Figura 2) No hipotireoidismo primário encontramos o TSH elevado e o T4 baixo, pois quando a tiroide tem um problema e começa a produzir pouco hormônio, a hipófise aumenta progressivamente sua produção de TSH para tentar contornar esse déficit Se o problema for central, na hipófise, encontraremos um TSH baixo por falta de secreção e um T4 também baixo por falta de estímulo para sua produção
A doença pode ser esporádica ou ter causa genética Pode ser permanente ou transitória e, em alguns casos, sem causa definida.
T3 e T4, também chamados de triodotironina ou tiroxina, respectivamente, são dois hormônios produzidos pela tireoide, a função destes é manter o metabolismo em equilíbrio Hormônio tíreo-estimulante (TSH) da hipófise, aumenta a produção de hormônios tireóideos


Fonte: https://wwwmedicinanetcombr/conteudos/revisoes/560
Fonte: https://geglobocom/eu-atleta/saude/noticia/hipotireoidismo-ehipertireoidismo-afetam-desempenho-fisicoghtml
Mais informações em: https://youtu be/Vsw7YAdGynI
O hipotireoidismo congênito possui incidência de 1:2 000 a 1:4 000 crianças nascidas vivas em países com suficiência iódica, no Brasil varia de 1:2 595 a 1:4 795, tornando-se o distúrbio endócrino congênito mais frequente. Os grupos étnicos influenciam diretamente na prevalência de HC, sendo menor em negros americanos comparando com hispânicos, além de ser um distúrbio com maior prevalência entre as mulheres Crianças com Síndrome de Down possuem risco 35 vezes maior hipotireoidismo congênito
Em Minas Gerais o Nupad é responsável pela realização da triagem neonatal para hipotireoidismo congênito, entre 1994 e agosto de 2022 mais de 6,8 milhões de crianças foram triadas e 1651 fizeram o acompanhamento ambulatorial, com incidência de aproximadamente 1:3500 em nascidos vivos
As causas mais frequentes de hipotireoidismo congênito incluem a disgenesia e defeitos na produção hormonal (disormonogênese). O hipotireoidismo congênito (HC) é o distúrbio endócrino congênito mais frequente, é uma das principais causas de retardo mental que pode ser prevenida Resulta da deficiência dos hormônios tireoidianos.
O HC pode ser transitório e resulta de diversas causas, entre elas estão a ingestão excessiva (ou deficiente) de iodo pela mãe; ingestão materna de drogas antitireoidianas em mães portadoras de hipertireoidismo; passagem placentária de anticorpos maternos bloqueadores do receptor de TSH e esse diagnóstico deve ser considerado quando houver relato de mais de um filho com hipotireoidismo transitório detectado pela triagem neonatal geralmente perdura 1 a 3 meses até que os anticorpos desaparecem da circulação. Estudos mostram que a incidência do HC pode variar dependendo de etnias, presença de consangüinidade ou área geográfica A frequência é maior em neonatos brancos do que em neonatos negros sendo de três a quatro vezes mais freqüentes no sexo feminino comparativamente ao masculino
Fonte: https://www.nupad.medicina.ufmg.br/wp-content/uploads/2016/12/protocolo_hipo.pdf.
O HC pode ser classificado em 4 grupos principais: hipotireoidismo
permanente esporádico, permanente primário, permanente hipotalâmicohipofisário, e transitório (Tabela 2)

Fonte:https://www.teses.usp.br/teses/disponiveis/5/5135/tde-15042009140742/publico/Solangecneves.pdf.2008.

A maioria das formas genéticas do Hipotireoidismo

Congênito Primário possui um padrão de herança autossômica recessiva Nesses casos, para ter a doença, o bebê precisa herdar as duas cópias do gene alterado, uma do pai e outra da mãe Quando a criança herda apenas um gene alterado, ela é considerada "portadora”. Portadores não têm sintomas da doença, porém podem passar o gene alterado para seus filhos
Em outros casos, quando a doença é causada, por exemplo, por mutações nos genes PAX8, TSHR e DUOX2, o hipotireoidismo congênito apresenta um padrão de herança autossômica dominante
Isto é, herdar apenas uma cópia do gene alterado é suficiente para causar a doença
A alteração genética que causa a doença em um padrão “dominante” pode não ter sido herdada, mas ter surgido pela primeira vez na criança Isso explica os casos de hipotireoidismo congênito causados por alterações genéticas e sem histórico familiar.
O padrão de herança depende de qual gene está alterado
Vale ressaltar que cerca de 15-20% têm causa genética conhecida.
Os sintomas do hipotireoidismo congênito se manisfesta de forma diferente ao longo do tempo, nas primeiras semanas podem envolver dificuldade para se alimentar Do primeiro ao terceiro mês os sintomas incluem letargia e macroglossia, após o terceiro mês os sintomas se intensificam e ficam cada vez mais evidente O déficit de crescimento e problemas relacionados ao sistema nervoso são outros dos sintomas característicos do hipotireoidismo congênito.

A identificação do hipotireoidismo congênito (HC) é feita durante exames de triagem neonatal, normalmente por meio do teste do pezinho (figura 4), em que são coletadas algumas gotas de sangue do calcanhar do bebê, e que são encaminhadas para o laboratório para que possam ser analisadas

O teste de triagem neonatal de hipotireoidismo congênito analisa somente a concentração do hormônio tireoestimulante (TSH) no sangue dos bebês Quanto maior a elevação do hormônio TSH, indica que menor está sendo a quantidade de T4 produzido. Pois, o TSH é responsável por estimular e por regular a quantidade de hormônios produzidos pela glândula tireoide (T3 e T4), garantindo que esses hormônios não sejam insuficientes nem excessivos. (figura 5)
O teste do pezinho em si não é considerado um exame diagnóstico, por isso os bebês com o Teste do Pezinho alterado são convocados para uma consulta médica, onde é feito exames laboratoriais
Para a confirmação do diagnóstico é necessário realizar a dosagem do hormônio T4 (total e livre) e do TSH em amostra de sangue.

Os bebês identificados na triagem neonatal, e tratadas precocemente têm desenvolvimento físico e intelectual dentro do esperado para a idade.
Caso não seja diagnosticado e tratado precocemente, a partir da 2ª semana de vida a deficiência dos hormônios tireoidianos poderá causar sintomas neurológicos graves no bebê Pois o T4 é fundamental para o funcionamento do metabolismo
Aproximadamente cerca de 5%-10% dos casos de HC não são diagnosticados no período neonatal devido à elevação tardia do TSH
Por isso, caso o bebê apresente sintomas sugestivos de HC, recomenda-se realizar a dosagem hormonal de TSH e T4L, mesmo que tenha apresentado resultados normais no Teste do Pezinho.

A constatação de um TSH acima de 10µUI/mL, com T4 livre normal ou diminuído, confirma o diagnóstico da doença Bebês com TSH menor que 5,6µUI/ mL têm o diagnóstico de HC descartado , e são orientadas a suspender o tratamento e recebem alta do programa Já aquelas com elevação discreta do TSH (entre 5,6 e 10 µUI/mL) e T4 livre normal, são orientadas a suspender o tratamento, mas permanecem em acompanhamento clínicolaboratorial rigoroso, sem tratamento medicamentoso (Figura 6).
Não há cura para o HC, mas quando o diagnostico por precoce, e tendo o tratamento imediato, pode impedir a ocorrência do atraso mental
Saiba mais em: https://www gov br/saude/ptbr/composicao/saes/sangue/programa-nacional-da-triagemneonatal
CURIOSIDADE:
Há um exame de triagem neonatal feito para ajudar a identificar a ocorrência de HC, o Teste da Bochechinha É um exame de triagem neonatal genético, capaz de identificar mais de 340 doenças genéticas tratáveis que se manifestam na infância Além de complementar o tradicional teste do pezinho

 FIGURA 6: FLUXOGRAMA DA TRIAGEM NEONATAL PARA HC DO PROGRAMA DE TRIAGEM NEONATAL
FIGURA 6: FLUXOGRAMA DA TRIAGEM NEONATAL PARA HC DO PROGRAMA DE TRIAGEM NEONATAL
COMO INTERPRETAR UM DIAGNOSTICO DO TESTE DO PEZINHO?
INTERPRETAÇÃO DOS RESULTADOS DA TRIAGEM NEONATAL NA AVALIAÇÃO DO HIPOTIREOIDISMO CONGENITO

FONTE: HTTPS://WWW SBP COM BR/FILEADMIN/USER UPLOAD/ 21369CDC HIPOTIREOIDISMO CONGENITO PDF
Crianças com mais de 48 horas de vida, e valores de TSH neonatal menores que 10 mUI/l no sangue total, nenhum seguimento é realizado.
Resultados de TSH entre 10 e 20 mUI/l determinam a solicitação de uma segunda amostra do calcanhar e, na maioria das vezes, este segundo resultado virá normal.
Quando o resultado do TSH neonatal for maior que 20 mUI/l, solicita-se que a criança compareça para consulta clínica e realize testes de função tireoidiana em amostras de soro. A maioria das crianças com valores de TSH neonatal maior que 20 mUI/l apresentará a doença.
O hipotireoidismo congênito altera a produção do hormônio tireoidiano, por isso o tratamento consiste na reposição diária deste hormônio A avaliação sérica de TSH e T4livre, ATPO, TRAB e TG são determinantes para o estabelecimento da dosagem individual do medicamento. Levotiroxina(L-T4) é a medicação mais indicada para o tratamento, sendo ofertada já na primeira consulta pós diagnóstico e posteriormente pelos postos de saúde e farmácias credenciadas.
O monitoramento é realizado através de exame clínico de crescimento, desenvolvimento e dosagens séricas de T4livre e TSH, de 3 em 3 meses até os três anos e de 6 em 6 após Aos 3 anos o tratamento costuma ser interrompido por quatro semanas para aferição das taxas e determinação etiológica do HC
O objetivo do tratamento é a normalização do TSH e manutenção do T4livre superior a metade do valor de referência, com atenção especial para os seis primeiros meses O tratamento bem executado resulta em melhoras no desenvolvimento, o que não pode ocasionar a interrupção do tratamento, já que o mesmo é o responsável por esse bom desenvolvimento. O acompanhamento médico e o uso correto da medicação são promordiais para bons resultados no tratamento.
FONTE: HTTP://WWW RMMG ORG/ARTIGO/DETALHES/1855
APRESENTAÇÕES COMERCIAIS DE LIVOTIROXINA DISPONÍVEIS NO BRASIL
(1) EUTHYROX®; (2) LEVOID®; (3) LEVOTIROXINA; (4) PURAN-T4®; (5) SYNTHROID®

A levotiroxina atua no organismo como o hormônio tireoidiano tiroxina (T4), regularizando quantidade hormonal no organismo A ação de reposição hormonal da levotiroxina possibilita a continuidade da conversão de T4 em T3, aproximando as taxas hormonais as recomendadas

ALVES, Ana Lília Vieira, SAVASSI, Eric Aparecido, FERREIRA, Maria Luisa, SILVA, Nicole Cordeiro da, COSTA, Robson Antonio da. Hipotireoidismo, 2021. Trabalho de conclusão de curso (Curso Técnico em Farmácia) -- Etec Deputado Salim Sedeh, Leme, 2021
ANDRADE, Caio Leônidas de et al Prevalência de sintomas otoneurológicos em indivíduos com hipotireoidismo congênito: estudo piloto. Cadernos Saúde Coletiva, v 25, p 144-151, 2017
BENEVIDES, Alex Mota et al Perfil epidemiológico de portadores de hipotireoidismo congênito Revista Paraense de Medicina, v. 20, n. 3, p. 23-26, 2006.
CASTRO, Marcos de Paula Ramos; SOARES, João César Castro Hipotireoidismo RBM rev bras med, 2014
Ferreira, Ligia Oliva et al Manifestações fonoaudiológicas relatadas por pais de crianças com hipotireoidismo congênito Revista da Sociedade Brasileira de fonoaudiologia [online]. 2011, v.16, n. 3 [Acessado 15 Setembro 2022] , pp. 317-322.
La Franchi S Thyroid function in the preterm infant Thyroid; 1999;9:71-8
LIMA, F. E.; CABRAL, D. M.; FERREIRA, T. S.; CARDOSO, L. P.; ROMERO, A.; LEÃO, P. O. MACHADO, L. C. S. A importância do diagnóstico precoce e adesão terapêutica no hipotireoidismo congênito Curitiba, 2020
MACIEL, Léa Maria Zanini et al Hipotireoidismo congênito: recomendações do Departamento de Tireoide da Sociedade Brasileira de Endocrinologia e Metabologia Arquivos Brasileiros de Endocrinologia & Metabologia, v 57, p 184-192, 2013
NASCIMENTO, M L Situação atual da triagem neonatal para hipotireoidismo congênito: críticas e perspectivas Hospital Infantil Joana de Gusmão, Serviço de Endocrinologia Pediátrica; Universidade Federal de Santa Catarina (UFSC) Departamento de Pediatria, Florianópolis, SC, Brasil, 2011
Núcleo de Ações e Pesquisa em Apoio Diagnóstico da Faculdade de Medicina da Universidade Federal de Minas Gerais Nupad em números – Programa de Triagem Neonatal de Minas Gerais
PEDRO, I. G. A.; MAGALHÃES, P. DE S.; REIS, B. C. C. Hipotireoidismo congênito diagnóstico precoce e suas complicações: uma revisão de literatura Revista Eletrônica Acervo Médico, v 12, p e10365, 30 jun 2022
PEZZUTI, Isabela L ; LIMA, Patrícia P de; DIAS, Vera Hipotireoidismo congênito: perfil clínico dos recém nascidos identificados pelo Programa de Triagem Neonatal de Minas Gerais Jornal de Pediatria, v 85, p 72-79, 2009
Schoenmakers N The genetic basis of congenital hypothyroidism Endocrinology org
Esther Mara Maciel Oliveira
Lívia Vitória de Aguiar Oliveira
Maria Laura Gonçalves dos Santos
Paulo Francisco Falcão Almeida
Quézia Emanuelle Ferreira Rocha
Samara Santana Peixoto