ESTABLISHING CLINICALLY RELEVANT DISSOLUTION SPECIFICATIONS

Andre Hermans Merck & Co., Inc. 20 Sep 2022


The current practice in establishing specifications as defined in ICH specification guidelines is essentially based solely on clinical experience. This can result in overly tight specifications that are not necessarily relevant to safety or efficacy and thus can lead to rejection of what is actually good product and limit the ability to continuously improve the manufacturing process
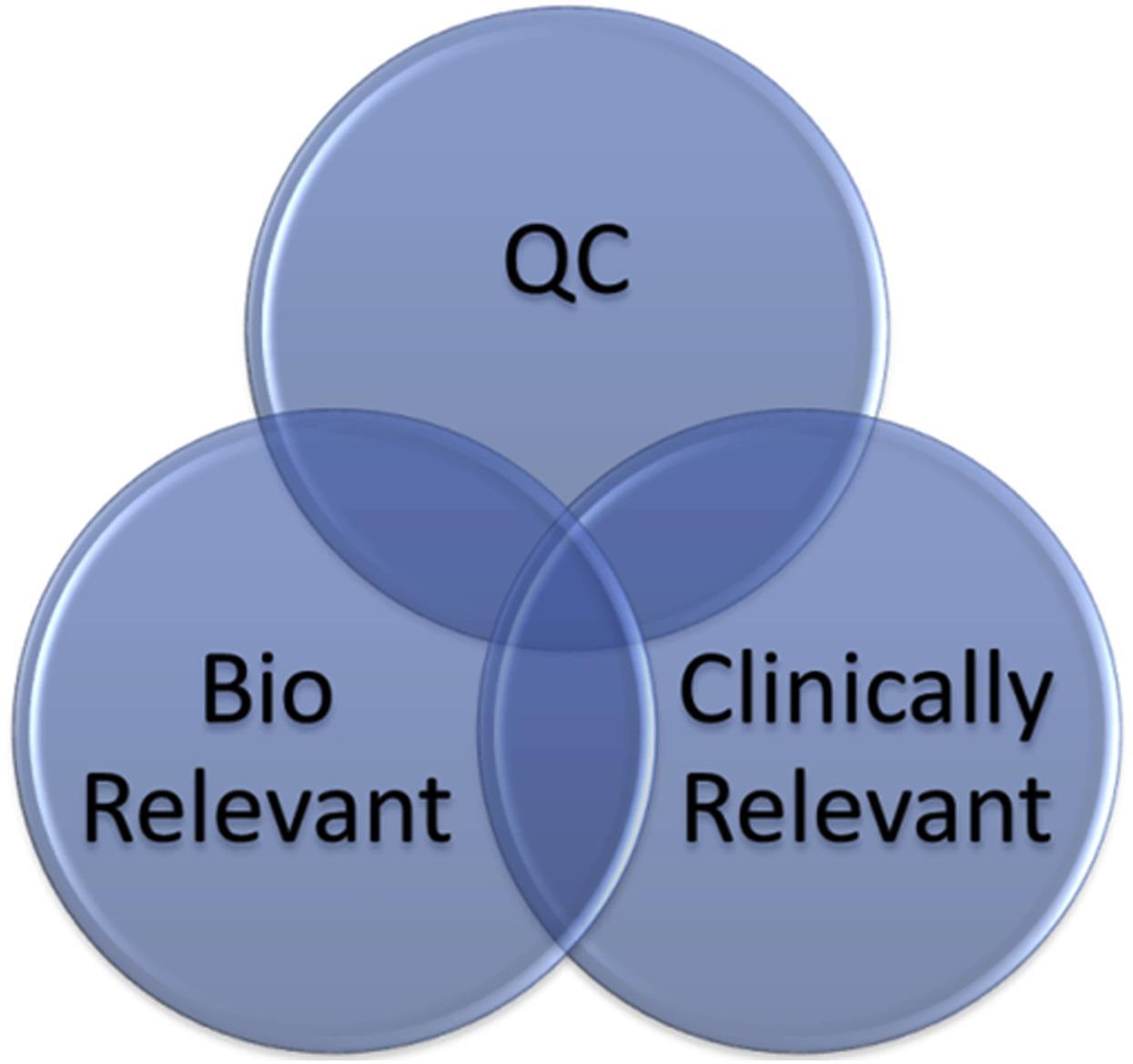
Patient-centric quality standards (PCQS) are a set of patient relevant attributes and their associated acceptance ranges to which drug product should conform within the expected patient exposure range to deliver the therapeutic benefit indicated in the label.
Patient-centricity in the development of specifications has additional focus on the impact to patient but using data beyond that simply what material was included in clinical studies.
The term “patient centric” and “patient relevant” are currently preferred as these phrases emphasize the link to the patient and attempt to break the misconception that “clinical relevance” equates to clinical trial experience alone. The
PCQS approach can benefit manufacturers who have an understanding of their process, product design and CQAs and their relation to patient impact.
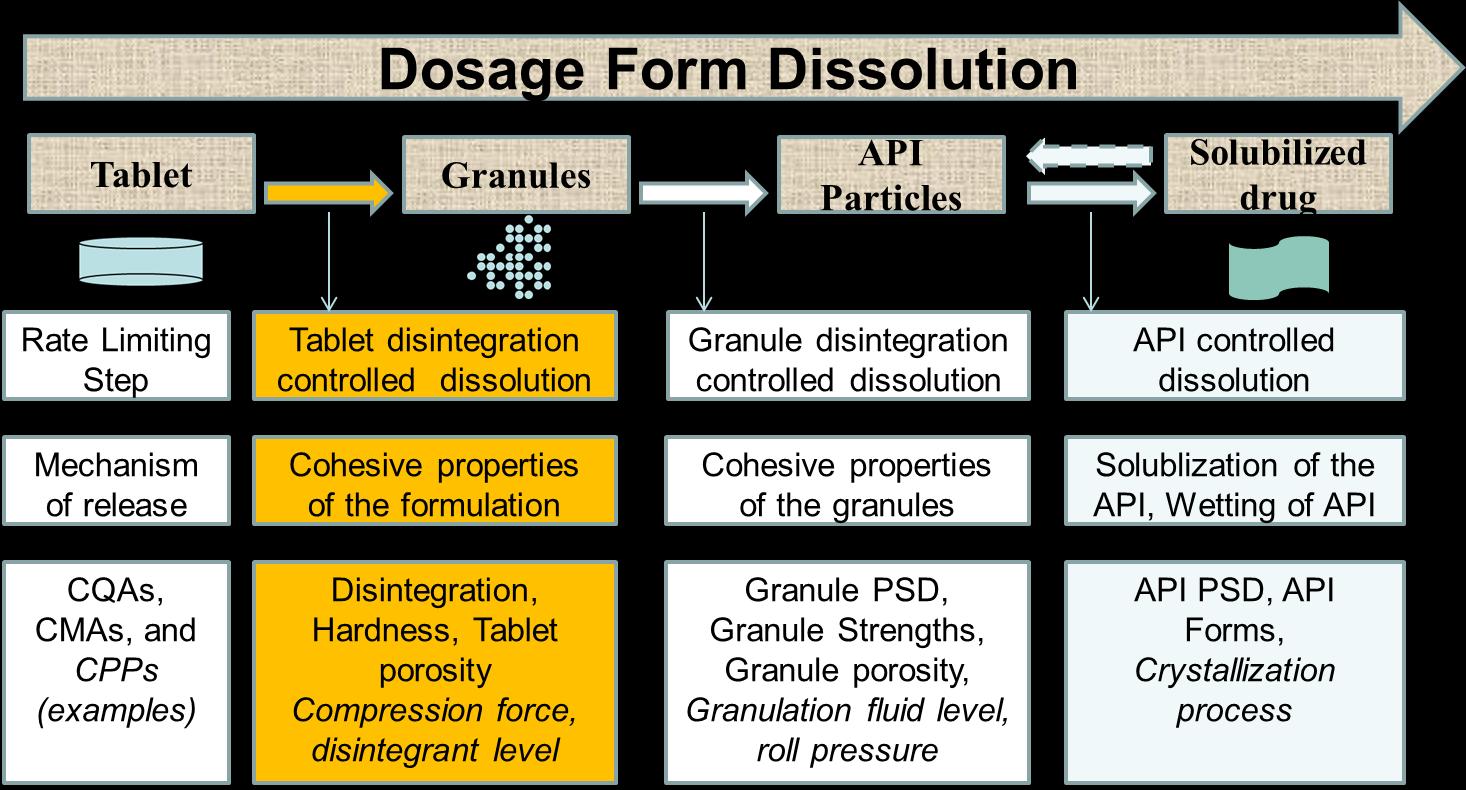
Clinically Relevant Dissolution Specifications (CRDS) Method conditions and acceptance criteria based on clinical performance (PK or PK/PD).
Goal is to reliably accept batches which are bioequivalent with pivotal batches
Clinical safe space—or bioequivalent space Boundaries defined by in vitro specifications within which drug product batches are anticipated to be bioequivalent to one another, or less optimally, but still possible, bioequivalent to the pivotal clinical batch(es).
• Abend A, Curran D, Kuiper J, Lu X, Li H, Hermans A, et al. Dissolution Testing in Drug Product Development: Workshop Summary Report, AAPS Journal, 2019. (21)
• Suarez Sharp S, Cohen M, Kesisoglou F, Abend A, Marroum P, Delvadia P, et al. Applications of Clinically Relevant Dissolution Testing: Workshop Summary Report. AAPS Journal. 2018;20(6):93.
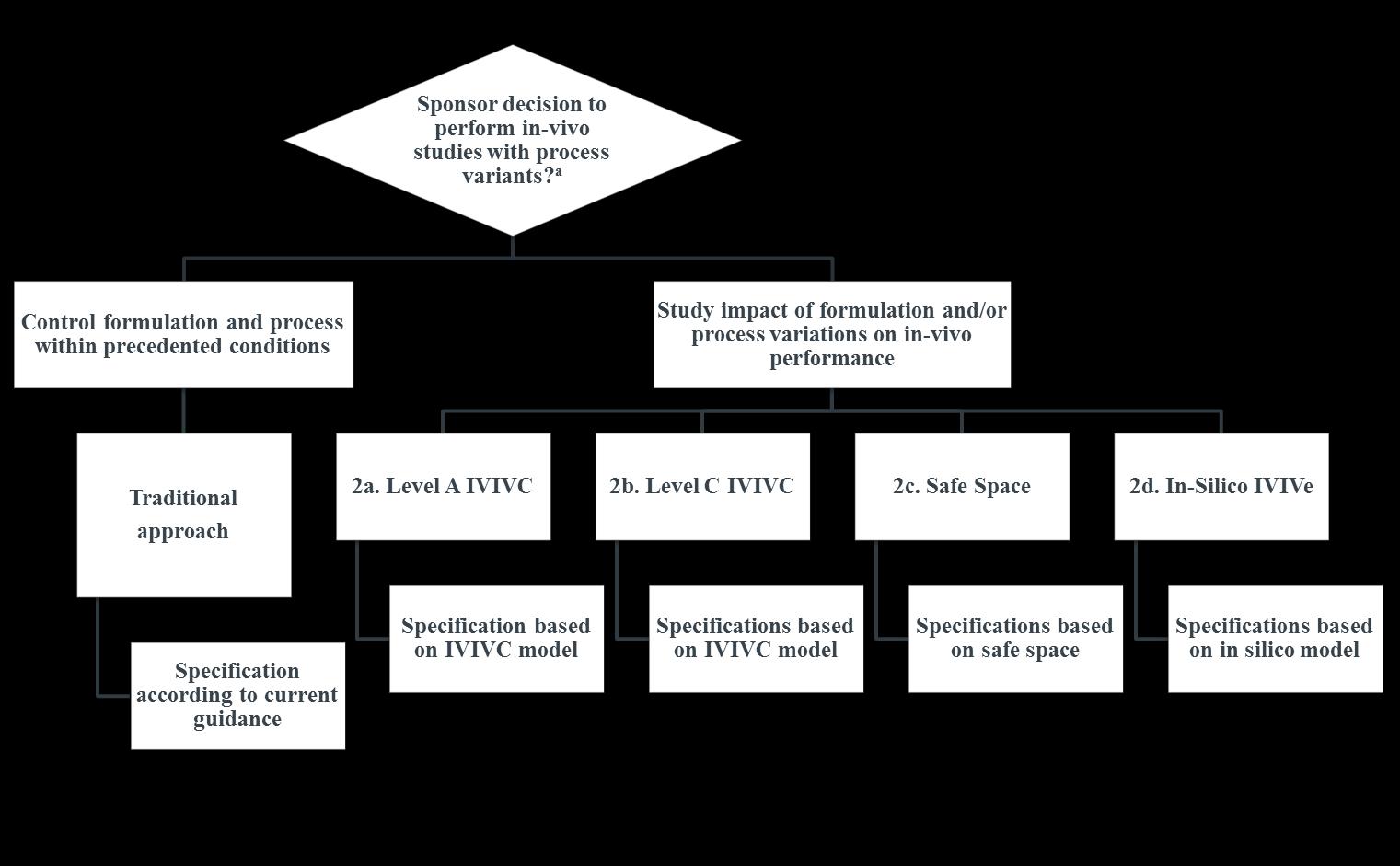
Approaches for Establishing Clinically Relevant Dissolution Specifications during Drug development The AAPS Journal, August 2017
The Evolving Roles of the Dissolution Test: Industry’s View on Using Quality Control, Biorelevant and Clinically Relevant Dissolution Tests for Pharmaceutical Development, Registration and Commercialization J. Pharm. Sci, January 2018
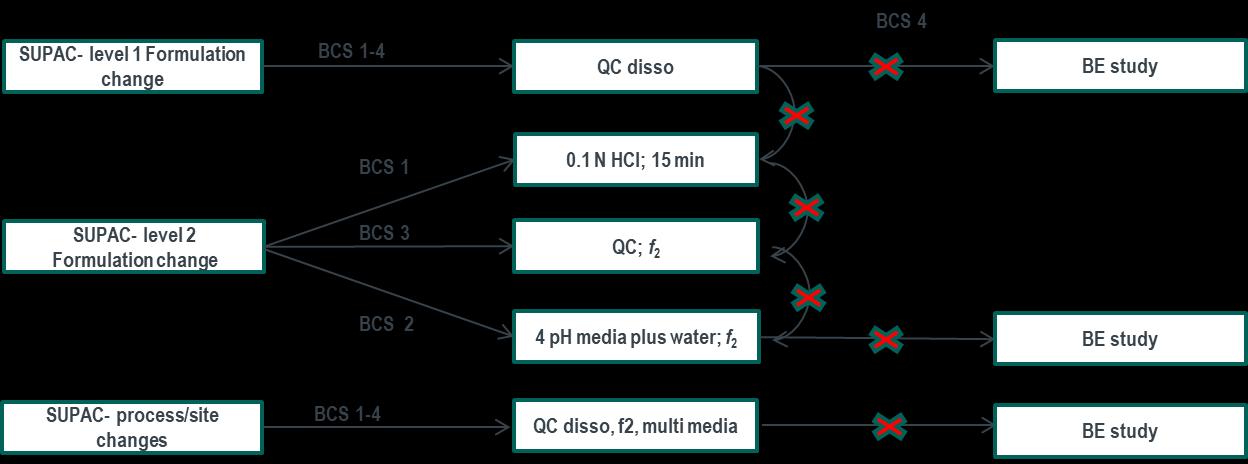
Setting specifications based on clinical experience (BCS 2/4) FDA Dissolution Guidance for BCS 1 and 3
Standardized method and acceptance criteria for BCS
IR dosage forms:
Guidance is aligned in principle with ICH M9 Method:
Basket Method (USP I) or Paddle Method (USP II)
Stirring rate = 100 RPM for USP I and 50 RPM for
Q=80% in 30 minutes. These specification can be considered clinically
specifications to ensure consistent
of the
1.31 (1.02,1.69) 1.21 (0.94,1.57) 1.00 (0.78,1.29) 1.04 (0.89,1.23)
1.04 (0.89,1.22) 1.03 (0.90,1.18) 0.974 (0.84,1.13) 1.00 (0.89,1.13)
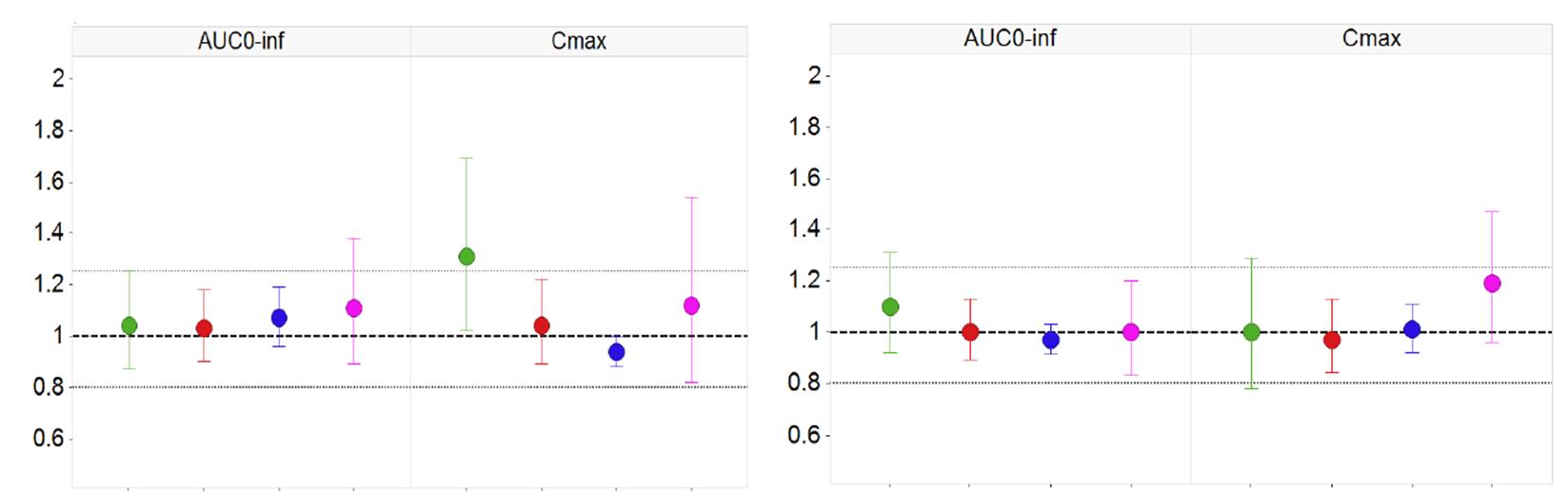
1.12 (0.82,1.54) 1.10 (0.89,1.38) 1.19 (0.96,1.47) 0.998 (0.83,1.20)
0.938 (0.88,1.00) 1.07 (0.96,1.19) 1.01 (0.92,1.11) 0.972 (0.91,1.03)
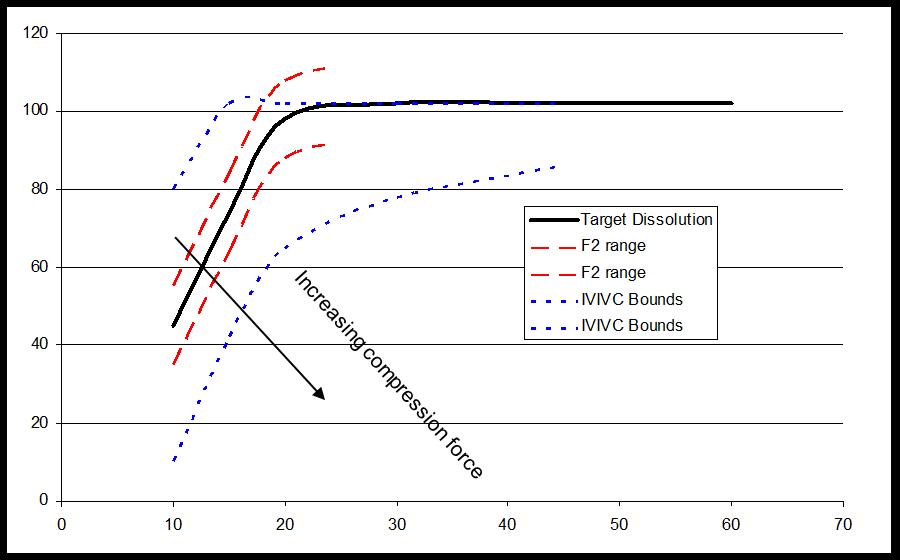
Relative Bioavailability Study on formulations with F2<50 that include desired compression range
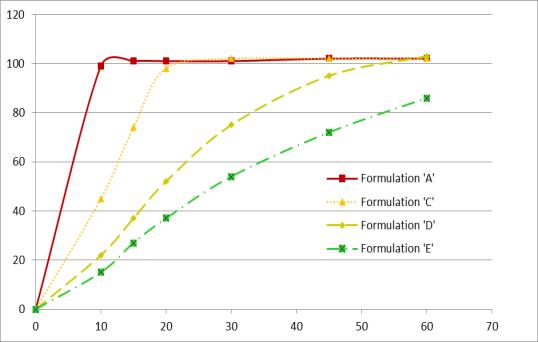
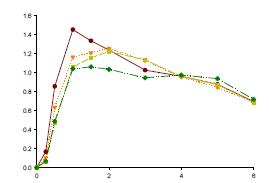

Multiple Level C IVIVC on Dissolution vs. Cmax (AUC not sensitive)

F, Hermans
Vivo

for


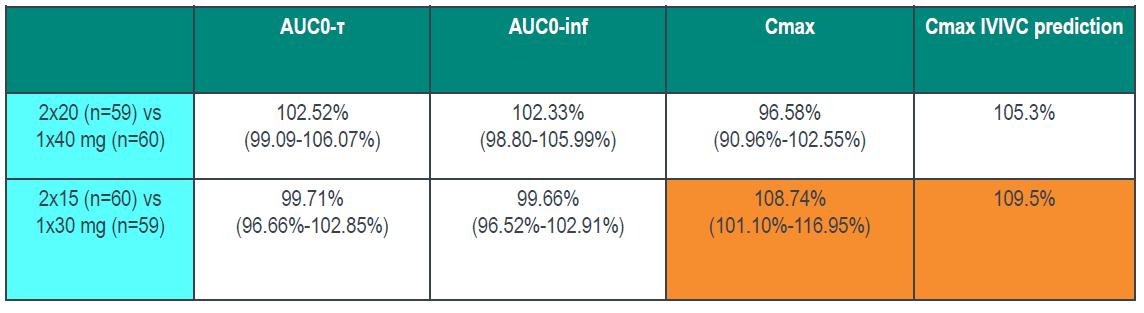
Bioequivalence study between strengths to support interchangeability (much faster dissolution for 15 vs 30 mg and 20 vs 40 mg tablets) IVIVC used to inform POS and power study (maximum 9.5% difference predicted based on 20 min dissolution)
Establishing the Bioequivalence Safe Space for Immediate-Release Oral Dosage Forms using Physiologically Based
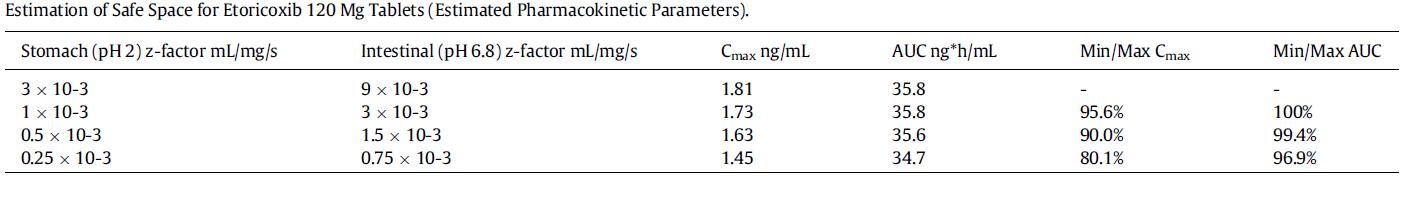
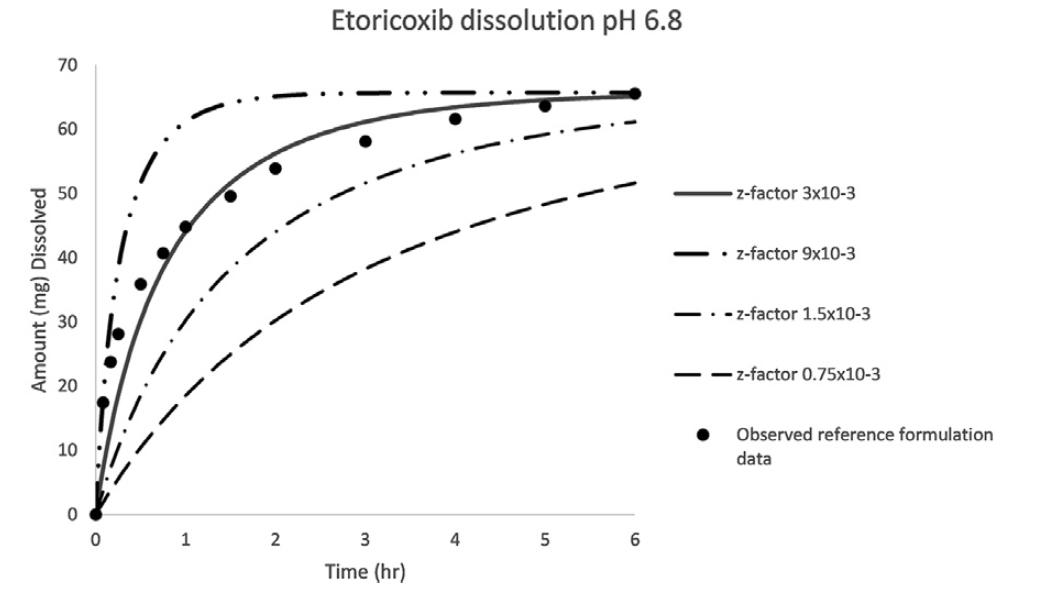
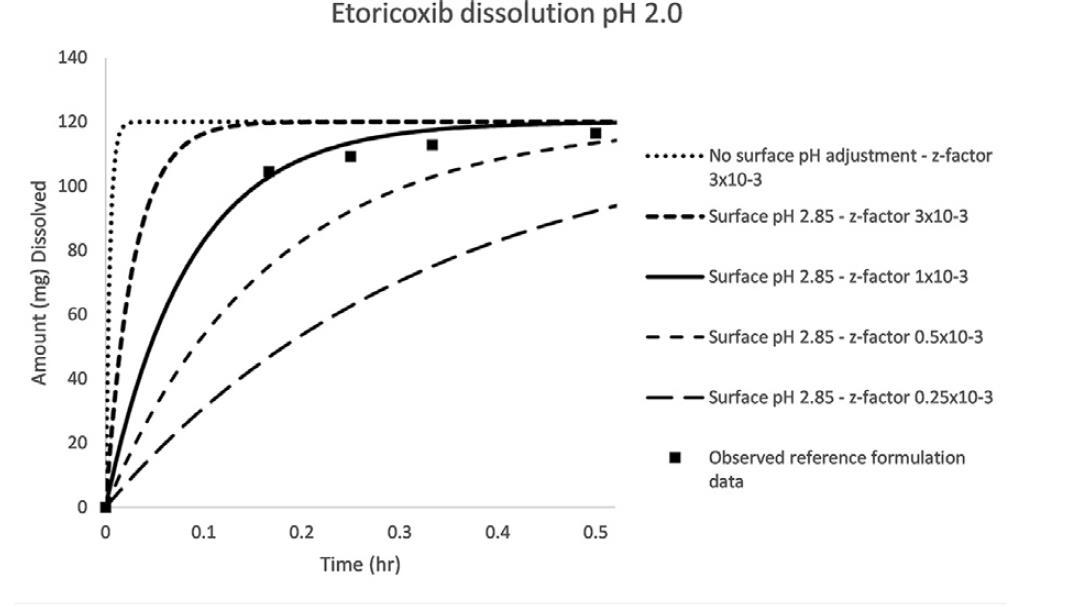
Biopharmaceutics Modeling (PBBM): Case Studies, J. Pharm Sci, 2021
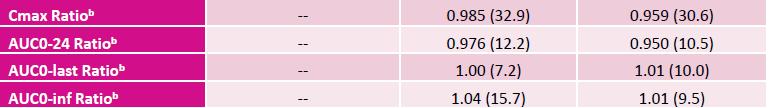
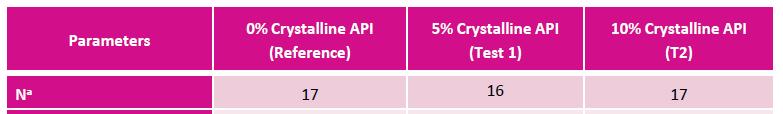
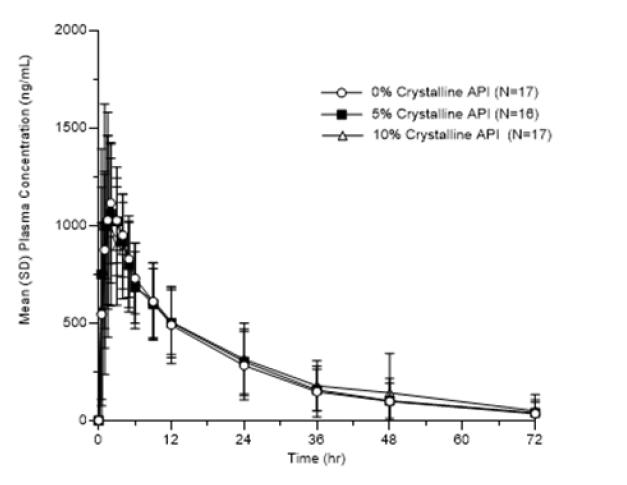
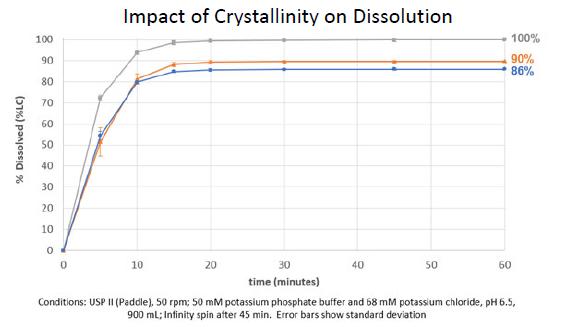
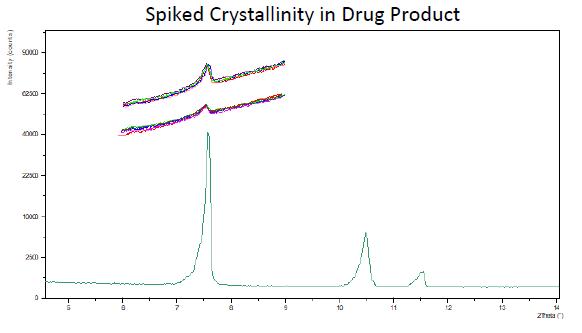
Excerpt from EMA Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action
The discriminatory power is the ability of a test procedure to discriminate between batches manufactured with different critical process parameters and /or critical material attributes which may have an impact on the bioavailability. Ideally all nonbioequivalent batches should be detected by the in vitro dissolution test results.
Batches representing different in vitro dissolution profiles, derived from the defined manufacturing process by setting process parameters within the range of maximum variability expected from process validation studies, are so-called “sidebatches”. The dissolution profiles of the side-batch can be used to set a suitable dissolution specification, when bioequivalence with the reference product is demonstrated.
Considering the approach given to the dissolution test in this guide, the main objective of the method development is to establish a test capable of failing batches with undesirable in vitro performance that, eventually, may be correlated with in vivo performance.
Therefore, the evaluation of batches whose pharmacokinetic results have been studied is the preferred approach to prove the discriminative capacity of the dissolution method (SCHEUBEL, 2010).
For this approach, it must be demonstrated that the dissolution method is able to distinguish batches with acceptable in vivo performance from those with unsatisfactory in vivo results. Results obtained in clinical studies or results of drug bioequivalence/bioavailability studies may be used; vitro:
When the company has a history of batches with known in vivo performance, the amplitude of the acceptance criteria can be established through the extreme values that were able to achieve satisfactory in vivo performance (EMA, 2014). That is, dissolution profiles of batches with deliberate variations and which have achieved satisfactory in vivo performance can be used to establish clinically relevant specifications.











• Filippos Kesisoglou
• Andreas Abend
• Jim DiNunzio
Tycho Heimbach
Jessica Miller
David Johnson
