

Expert Review of Neurotherapeutics
ISSN: (Print) (Online) Journal homepage: www.tandfonline.com/journals/iern20
The management of neurofibromatosis type 1 (NF1) in children and adolescents
Nino Kerashvili & David H. GutmannTo cite this article: Nino Kerashvili & David H. Gutmann (27 Feb 2024): The management of neurofibromatosis type 1 (NF1) in children and adolescents, Expert Review of Neurotherapeutics, DOI: 10.1080/14737175.2024.2324117
To link to this article: https://doi.org/10.1080/14737175.2024.2324117

Published online: 27 Feb 2024.

Submit your article to this journal

Article views: 41


View related articles
View Crossmark data

Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=iern20

The management of neurofibromatosis type 1 (NF1) in children and adolescents
Nino
Kerashvilia and David H. Gutmann baDepartment of Neurology, University of Oklahoma Health Science Center, Oklahoma City, OK, USA; bDepartment of Neurology, Washington University School of Medicine, St. Louis, MO, USA
ABSTRACT
Introduction: Neurofibromatosis type 1 (NF1) is a rare neurogenetic disorder characterized by multiple organ system involvement and a predisposition to benign and malignant tumor development. With revised NF1 clinical criteria and the availability of germline genetic testing, there is now an opportunity to render an early diagnosis, expedite medical surveillance, and initiate treatment in a prompt and targeted manner.
Areas covered: The authors review the spectrum of medical problems associated with NF1, focusing specifically on children and young adults. The age-dependent appearance of NF1-associated features is highlighted, and the currently accepted medical treatments are discussed. Additionally, future directions for optimizing the care of this unique population of children are outlined.
Expert opinion: The appearance of NF1-related medical problems is age dependent, requiring surveillance for those features most likely to occur at any given age during childhood. As such, we advocate a life stage-focused screening approach beginning in infancy and continuing through the transition to adult care. With early detection, it becomes possible to promptly institute therapies and reduce patient morbidity. Importantly, with continued advancement in our understanding of disease pathogenesis, future improvements in the care of children with NF1 might incorporate improved risk assessments and more personalized molecularly targeted treatments.
1. Introduction
Neurofibromatosis comprises five distinct neurogenetic disorders caused by variants in different genes, including neurofibromatosis type 1 (NF1; NF1 gene), Legius syndrome (LGGS; SPRED1 gene), NF2-related schwannomatosis (NF2-SWN; NF2 gene), 22q-related schwannomatosis (SWN; SMARCB1 and LTZR1 genes), and schwannomatosis-not otherwise specified [1]. Of these conditions, the most common is NF1, affecting one in 3,000 people worldwide [2].
While NF1 was first reported by Frederich von Recklinghausen in 1882, formal diagnostic criteria were not established until 1987 [3]. At that time, a diagnosis of NF1 could be rendered for individuals who harbored two or more features from a list of seven diagnostic criteria. These clinical attributes included (1) six or more café-au-lait macules, (2) axillary or inguinal freckling, (3) iris hamartomas, called Lisch nodules, (4) distinctive bony lesions, like tibial pseudarthrosis or orbital dysplasia, (5) multiple cutaneous neurofibromas or one plexiform neurofibroma, (6) an optic pathway glioma, and (7) a first-degree relative with NF1. With the advent of molecular testing, these diagnostic criteria were updated in 2021 to now incorporate a positive genetic test (presence of a germline NF1 gene variant), broadening the bony abnormalities also to include anterolateral bowing of the long bones and the addition of choroidal abnormalities to Lisch nodules [4] (Table 1).
These advances, coupled with a better understanding of the molecular and cellular pathogenesis of NF1-associated medical problems, set the stage for more tailored surveillance and improved treatment options for affected children. In this review,
ARTICLE HISTORY
Received 5 January 2024
Accepted 23 February 2024
KEYWORDS
Neurofibromatosis; NF1; plexiform neurofibroma; optic pathway glioma; management
we discuss the genetics of NF1, the clinical features of the disorder, and an age-appropriate monitoring plan.
2. Literature search
A search strategy was developed using the terms ‘neurofibromatosis type 1,’ ‘NF1,’ ‘treatment,’ ‘management,’ focused specifically on clinical studies.
3. Establishing a diagnosis of NF1
Using the initial set of established clinical criteria, approximately 46% of children with sporadic (non-familial) NF1 met diagnostic criteria by 1 year of age, 97% by 8 years old, and nearly 100% by 20 years old [5]. As such, it is important to recognize that young children with NF1 May only manifest café-au-lait macules in the first couple years of life and do not develop a second feature (usually skinfold freckling) until the end of the first decade [6], leaving a large number of children undiagnosed in early childhood. This is particularly important for children in this age group, as many of the associated morbidities, like optic pathway glioma (OPG) [7,8], manifest prior to the emergence of skinfold freckling. For this reason, most NF specialists manage children with >5 café-au-lait macules as though they have already confirmed a diagnosis of NF1.
For those families desiring a definite diagnosis prior to the appearance of features necessary to establish a clinical
REVIEW
CONTACT David H. Gutmann gutmannd@wustl.edu Department of Neurology, Washington University, 660 S. Euclid Avenue, St. Louis, MO 63110, USA EXPERT REVIEW OF NEUROTHERAPEUTICS https://doi.org/10.1080/14737175.2024.2324117 © 2024 Informa UK Limited, trading as Taylor & Francis Group
Article highlights
● Neurofibromatosis type 1 (NF1) is a complex genetic disorder characterized by extreme clinical variability.
● Updated NF1 diagnostic criteria now incorporate genetic testing.
● The establishment of a clinical trials consortium has streamlined the evaluation of promising agents for the treatment of common medical problems arising in children and teenagers with NF1.
● The management of children and young adults with NF1 should entail age-specific monitoring.
● Future research should focus on adolescent/young adult transition to adult care.
diagnosis (e.g. skinfold freckling), genetic testing can also be offered in a child with only café-au-lait macules. Genetic testing can also distinguish NF1 from Legius syndrome and can be helpful for children with atypical presentations (e.g. isolated optic glioma or plexiform neurofibroma) [9]. Moreover, a providing a definitive confirmation of NF1 can guide future medical surveillance and the early incorporation of specialized academic services (e.g. individualized education plans).
4. Genetics
NF1 is inherited as an autosomal dominant disorder caused by pathogenic variants (mutations) in the NF1 gene encoding neurofibromin [10]. While NF1 can be inherited from an affected parent, ~50% of individuals have no clear family history and thus represent de novo NF1 variants. As an autosomal dominant disorder, there are no silent carriers of NF1, and everyone with a germline NF1 variant has the condition. In this regard, NF1 is completely penetrant; however, there is great phenotypic variability, even within families with the same germline NF1 variant [11]. This heterogeneity likely reflects the influence of other risk factors on the development and progression of the various features of NF1.
While NF1 largely manifests with clinical features involving the entire body, some people with NF1 have their disease limited to only one segment of their body (segmental or mosaic NF1), resulting from a spontaneous NF1 variant occurring after conception and only affecting a subset of fetal tissues [12,13]. These individuals will have NF1-related medical problems confined to that segment of their body.
For the most part, germline NF1 gene variants are distributed throughout the NF1 coding region [14]. However, there are individuals with missense variants (mutations) involving
Table
codon Arg1809 [15] or an in-frame deletion of a single methionine residue [16] who harbor pigmentary features of NF1, but do not develop brain or nerve tumors. Additionally, there are people who harbor a large genomic deletion of the NF1 locus who manifest with severe cognitive delays, an increased risk of cancer, and a greater overall tumor burden [17–19]. Similarly, those with missense mutations affecting codons 844–848 tend to have more severe disease manifestations, such as plexiform and spinal neurofibromas, optic gliomas, skeletal abnormalities, and malignancy [20]. Lastly, individuals with other missense variants are more likely to harbor facial dysmorphism, cardiovascular abnormalities, and spinal neurofibromas [21]. Further studies on the impact of germline NF1 mutations using genetically engineered mice and human induced pluripotent stem cells might reveal the underlying mechanisms responsible for this clinical heterogeneity [22,23].
5. Clinical features
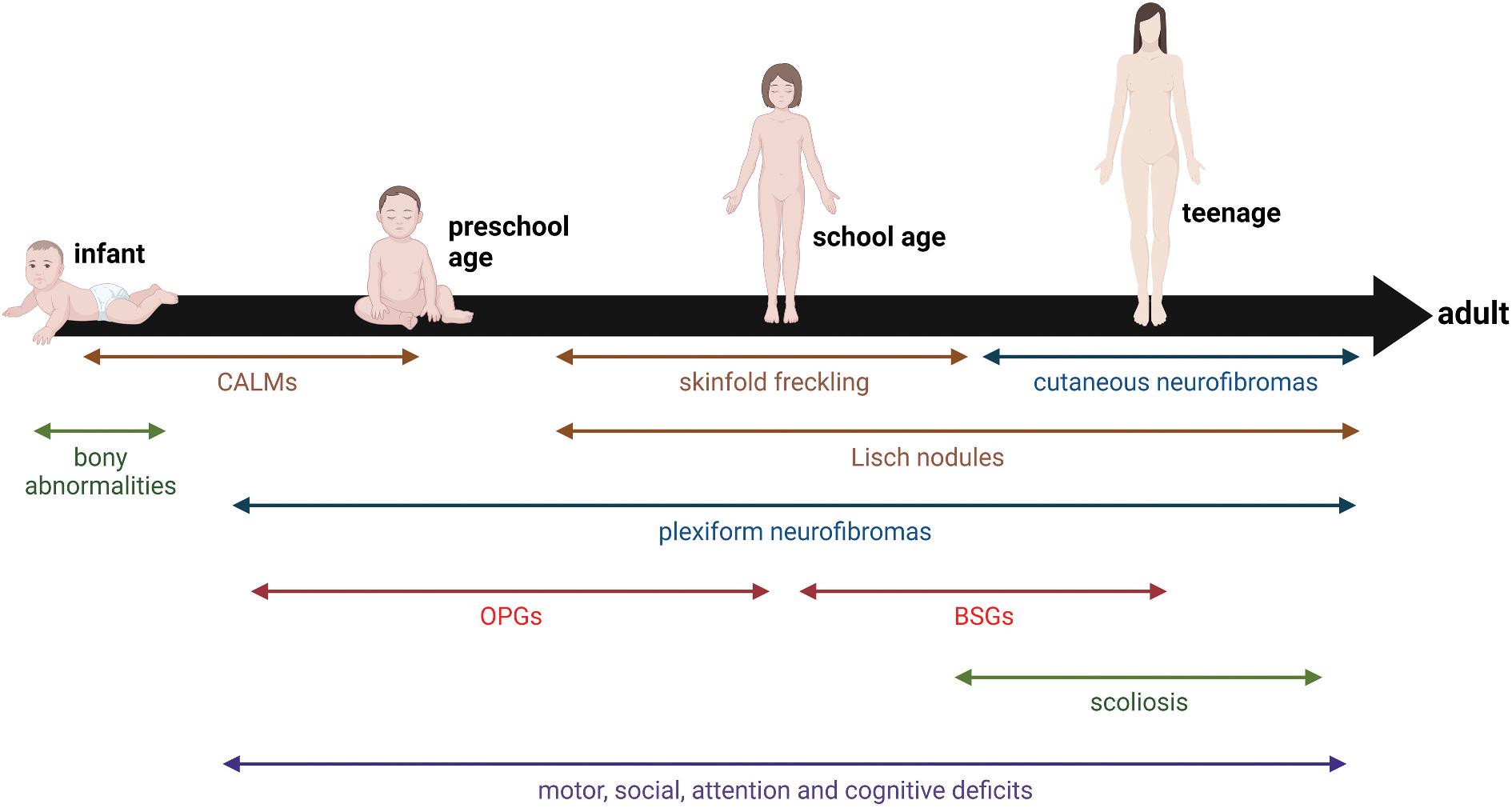
The clinical features of NF1 appear in an age-dependent fashion, requiring individualized monitoring for those medical problems most likely to occur at any given age (Figure 1). Herein, we will focus on the clinical manifestations that affect children and adolescents with NF1.
5.1. Café-au-lait macules (CALMs)
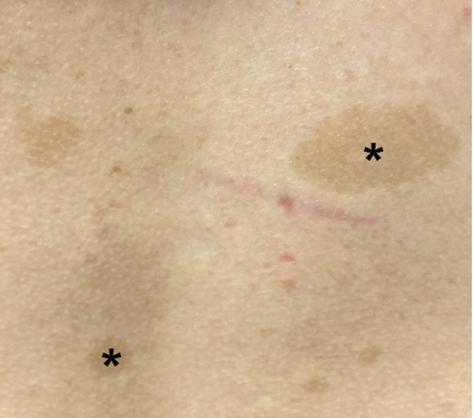
Cafe-au-lait macules are light to dark-brown pigmented spots that appear on the skin (Figure 2). A solitary CALM is a common finding in the general population, occurring in 3% of infants and 25% of healthy children. However, more than three CALMs are seen in only 0.2% to 0.3% of children with no known evidence of an underlying disorder. In striking contrast, children with NF1 typically manifest with numerous macules within the first year of life [24], representing the most common presenting feature of NF1. CALMs typically increase in size and number during early childhood. Where CALMs in the setting of NF1 usually have uniform pigmentation and distinct regular borders, atypical CALMs with irregular borders and inhomogeneous pigmentation are less likely to be associated with NF1 [25]. As such, in addition to the number of CALMs, the morphological appearance of CALMS may also be a useful predictive feature in the diagnosis of NF1 [6].
If an individual does not have a parent diagnosed with NF1, at least two of the following are required to establish a diagnosis of NF1:
(1) At least six café-au-lait macules over 5 mm in greatest diameter before puberty or over 15 mm after puberty
(2) Freckling in the axillary or inguinal region
(3) Two or more neurofibromas of any type or one plexiform neurofibroma
(4) Optic pathway glioma
(5) At least two Lisch nodules (identified by slit lamp examination) or at least two choroidal abnormalities (identified by ocular coherence tomography or nearinfrared reflectance imaging)
(6) Distinctive osseous lesion (sphenoid dysplasia, anterolateral tibial bowing, long bone pseudarthrosis)
(7) Heterozygous pathogenic germline NF1 variant with a variant allele fraction of 50%
If an individual has a parent diagnosed with NF1, only one of more of the above features are required to establish a diagnosis of NF1
1. Revised diagnostic criteria for NF1.2 N. KERASHVILI AND D. H. GUTMANN
5.2. Skinfold freckling
Freckling is denoted by the presence of 1–2 mm-sized pigmented lesions of light brown color that develop during childhood, usually after 6 years of age. Their appearance is similar to sun-induced freckling, but in NF1, they typically occur in areas with minimal to no sun exposure, such as the axillary and inguinal regions [26]. They can also occur in skinfold regions, like under the breasts in women or within fat folds.
5.3. Lisch nodules
Lisch nodules (LNs) are 1–2 mm-sized yellowish-brown domeshaped solid lesions on the surface of the iris [27]. The color of LNs varies with the pigmentation of the iris and the race/ ethnicity of the child. Their prevalence gradually increases to 50% at five years of age, 75% at 15 years of age, and 90–95% over the age of 30. They are never symptomatic, do not cause any visual disturbances, and do not require treatment.
5.4. Choroidal abnormalities
Choroid abnormalities (CAs) are ovoid bodies in the choroid consisting of hyperplastic Schwann cells, melanocytes, and ganglion cells [28]. CAs were recently added to the diagnostic criteria [4] based on their high specificity and sensitivity for a diagnosis of NF1 [29]. The appearance and dimensions of CAs can increase with age [30].
5.5. Optic pathway gliomas (OPGs)
The most common brain tumor is the optic pathway glioma, a low-grade (World Health Organization grade 1) pilocytic astrocytoma, seen in 15–20% of children with NF1 [31].
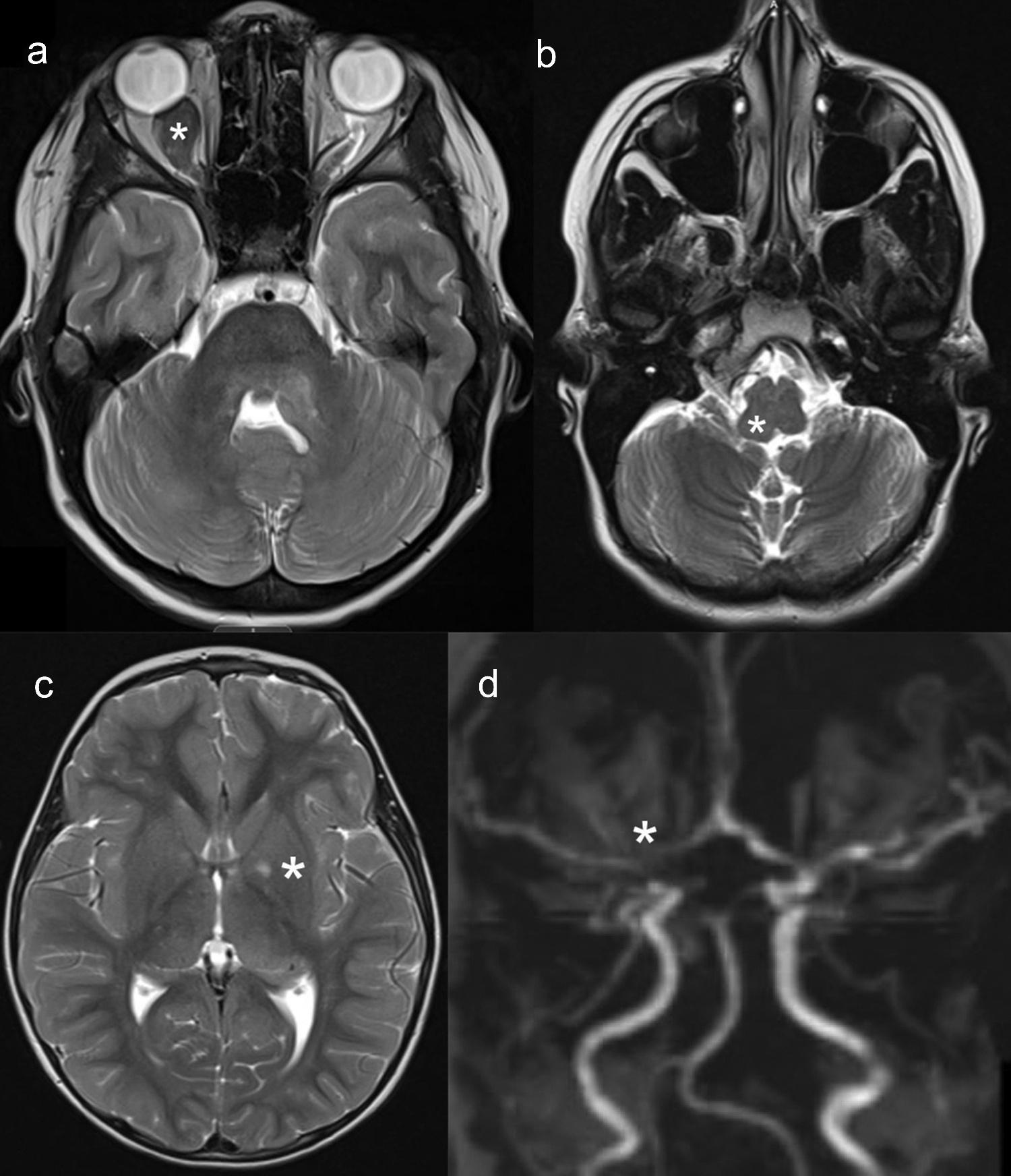
These tumors can arise anywhere along the optic pathway, including the prechiasmal optic nerves, optic chiasm, or postchiasmal tracts and radiations (Figure 3(a)). Over 75% of these tumors arise in the anterior optic pathway (optic nerves and chiasm) [31–34]. With optic nerve involvement, there can be unilateral proptosis, visual acuity loss, visual field defects, strabismus, relative afferent pupillary defect, and optic disc edema [7,35,36]. Children with chiasmal involvement can also present with precocious puberty; OPGs involving the hypothalamus can cause obstructive hydrocephalus and other neurological signs depending on their encroachment of adjacent structures [34,37].
NF1-OPGs can arise at any age in children, including after an otherwise normal neuroimaging study [38], but usually are detected between 2 and 5 years of age [35,38], with clinical progression less common after the first decade of life [38,39]. It is important to recognize that only fewer than 50% of children with NF1-OPG are symptomatic and require treatment [40].
There are several identified risk factors for OPG, such as Caucasian race [41], high birth weight [42], and some allergic conditions, like eczema and asthma [43]. In addition, the risk of
Figure 1. Age-dependent appearance of medical problems in children with NF1. Arrows denote the typical age of detection. Abbreviations: BSGs, brainstem glioma; CALMs, café-au-lait macules; OPGs, optic pathway gliomas. Created with BioRender.com.EXPERT REVIEW OF NEUROTHERAPEUTICS 3
Figure 2. Café-au-lait macules. Asterisks denote the macules.
vision loss secondary to an OPG includes involvement of the postchiasmal structures [39], age younger than 2 years or older than 8–10 years [39], and female sex [44]. Currently, these risk factors are not robust enough to use in clinical practice; however, the implementation of artificial intelligence-driven risk assessment algorithms using these and other disease modifiers may help identify individuals at greatest risk for specific NF1-associated medical problems [24].
5.6. Brainstem gliomas
Brainstem gliomas are also low-grade gliomas, mostly pilocytic astrocytomas, representing the second most common tumor in children with NF1 [45]. Occurring in fewer than 10% of individuals with NF1, they are more likely to be localized to the midbrain and medulla (Figure 3(b)). While the majority are asymptomatic and do not require medical or surgical intervention, symptoms can include headache, nausea/vomiting, cranial neuropathies (e.g. dysarthria), and
ataxia/gait instability. In contrast to NF1-OPGs, brainstem gliomas typically present later in childhood, with a mean age of 7.2 years [45,46].
5.7. Other brain tumors
In addition to OPGs and brainstem gliomas, children with NF1 can develop low-grade gliomas in other parts of the central nervous system, including the cerebral hemispheres, cerebellum, and spinal cord [47,48]. These gliomas can occur at any age, but are worrisome if they occur in adolescents or young adults, where close surveillance is recommended [49,50].
5.8. T2 hyperintensities
Previously referred to as unidentified bright objects (UBOs), these T2 hyperintense areas are now called focal areas of signal intensity (FASI) (Figure 3(c)). Differentiating FASI from
4 N. KERASHVILI AND D. H. GUTMANN
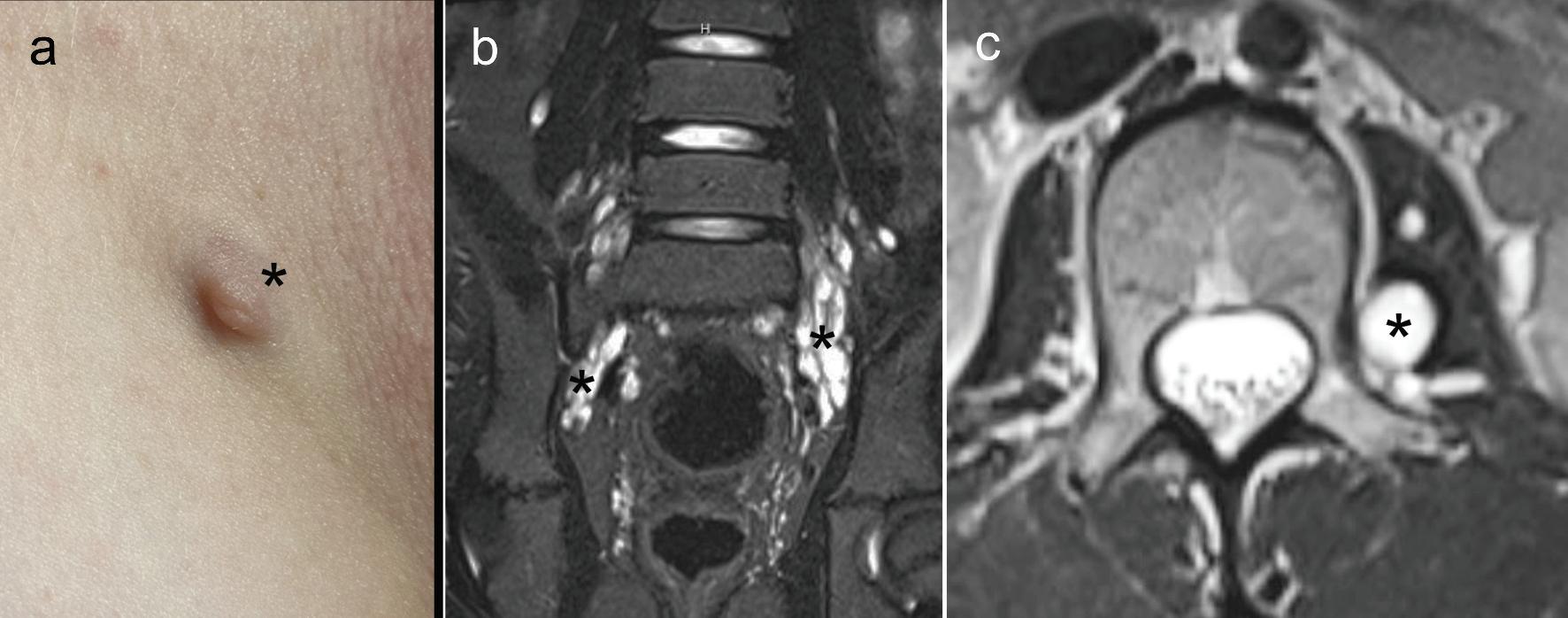
Figure 3. (a) optic pathway glioma with marked enlargement of the right optic nerve. (b) brainstem glioma. (c) basal ganglia foci of abnormal signal intensity (FASI). (d) narrowing of the cerebral blood vessels (M1 segment). Asterisks denote the lesions of interest.
a low-grade brain tumor on brain MRI can be challenging, as both appear as high-signal-intensity foci on T2-weighted images [51]. FASI most frequently occurs in the cerebellum, medial temporal lobes, and thalamus, whereas tumors are more commonly observed in the brainstem and basal ganglia. In addition, FASI are more likely to be bilateral and have nondiscrete borders, while tumors are typically characterized by mass effect or contrast enhancement on gadolinium MRI. The number of FASI is highest in early childhood and decreases with age [51]. While FASIs are common in children with NF1, they are not preneoplastic lesions and do not warrant treatment.
5.9. Neurofibromas
Neurofibromas are benign peripheral nerve sheath tumors that can arise as cutaneous, nodular or diffuse plexiform, spinal, or atypical neurofibromatous neoplasms of uncertain biologic potential. These tumors comprise a mixture of Schwann cells, perineurial-like cells, mast cells, macrophages, fibroblasts, blood vessels, and extracellular matrix.
Cutaneous neurofibromas (cNFs), one of the hallmarks of NF1, arising either within the dermis (intradermal; Figure 4(a)) or extending from the skin surface (exophytic), are nearly always in association with single nerves. Subcutaneous neurofibromas are located below the dermis and often feel like small, firm beads under the skin, while intradermal neurofibromas have a buttonhole appearance, often with a purplish central coloration. Exophytic neurofibromas are pedunculated and protrude from the surface of the skin. They may cause discomfort or bleeding when appearing in areas of constant friction (e.g. bra lines in women). cNFs tend to appear in late childhood or early adolescence, often heralding the onset of puberty [52]. Importantly, cNFs are invariably benign and do not progress to malignant tumors.
Plexiform neurofibromas (PN) have a highly infiltrative nature and can infiltrate into all three skin tissue layers [53]. They are usually associated with major nerve trunks and nerve plexi and can cause major disfigurement (Figure 4(b)). As such, PNs can erode neighboring bone, stimulate bone growth to result in leg length discrepancies, result in hemorrhage, or replace
muscle. Nearly one-third of patients with NF1 develop PNs, which can be congenital or develop in early childhood [54]. PNs exhibit the most rapid growth during childhood, although individual growth rates vary widely between individuals. In contrast to cNFs, PNs can undergo malignant transformation (Section 5.10 below).
Spinal neurofibromas (sNFs) arise from the proximal portion of the dorsal spinal nerves. They can appear as a discrete mass or part of a plexiform neurofibroma (Figure 4(c)), where they involve single or multiple nerve roots. The majority of sNFs present in individuals younger than 10 years of age and can be associated with both sensory and motor deficits.
Atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP) (previously termed ‘atypical neurofibromas’) are considered pre-malignant lesions lying on the spectrum between benign plexiform neurofibromas and malignant peripheral nerve sheath tumors (MPNST) [55]. These Schwann cell tumors exhibit at least two of the four following features: hypercellularity, atypical nuclei, loss of neurofibroma architecture, and few mitoses. ANNUBPs can exhibit increased FDG uptake on PET/CT, and their presence is strongly correlated with MPNST.
5.10. Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
MPNSTs are high-grade sarcomas with prominent nuclear atypia, high mitotic indices, and often areas of necrosis, affecting 12–15% of individuals with NF1 [56]. The median age of MPNST diagnosis in the setting of NF1 is 26 years [57]. These tumors can be associated with rapid growth, pain, neurologic deficits (e.g. weakness), and constitutional symptoms (e.g. weight loss, night sweats, fatigue). While more common in young adults, these cancers can arise in children and teenagers with NF1.
5.11. Learning disabilities
Nearly 20% of children with NF1 can have specific learning disorders [58], such as difficulties with reading, dyslexia,
EXPERT REVIEW OF NEUROTHERAPEUTICS 5
Figure 4. (a) cutaneous neurofibroma. (b) plexiform neurofibroma surrounding the spine. (c) paraspinal neurofibroma. Asterisks denote the lesions of interest.
mathematics, and writing/spelling [59,60]. In addition, issues with working memory and receptive language are also seen [60], along with impairments in visual processing [61]. Taken together, many children with NF1 will require some type of school accommodation.
In addition to specific learning disorders, children with NF1 can exhibit deficits in specific cognitive domains, namely executive function and visuospatial processing. These problems are related to more global measure functioning, including problem solving, planning, and taskswitching [ 62 ], and contribute to reading difficulties [ 61 ].
5.12. Attention problems/ADHD
Attention problems and ADHD are common in children with NF1, affecting as many as two-thirds of children with NF1 [63]. ADHD is rarely associated with hyperactivity/impulsive behavior and typically is of the inattentive type or combined type, which likely accounts for unrecognized ADHD in children with NF1. Elevated ADHD symptomatology mediates the relationship between executive functions and social skills problems, although it does not fully account for social dysfunction [64]. Overall, ADHD symptoms negatively affect daily functioning in children with NF1, underscoring the importance of interventions to minimize ADHD symptoms [65]. In this regard, ADHD is a significant risk factor for bullying [66].
5.13. Social perception deficits/autism spectrum disorder
Children with NF1 are prone to difficulties with social perception [67]. 40% of children with NF1 have poor social function, are less likely to be liked by their peers, are more socially isolated, and have fewer reciprocated friendships. As such, children with NF1 engage in fewer prosocial behaviors [68], exhibit deficits in perspective-taking [69], and have difficulties with facial and emotional recognition [70].
Autism spectrum disorder (ASD) is observed in 25% of individuals with NF1 [71,72]. NF1-associated-ASD symptomatology is most pronounced in the domains of social communication/interaction, with fewer stereotyped behaviors and greater engagement and interest in social interactions compared to children with idiopathic autism spectrum disease [73]. Interestingly, restrictive and repetitive behavior in children with NF1 may reflect the close association between ASD and ADHD in the NF1 population, leading to inflexibility, poor planning and organization, and limited self-starting/initiation [74].
5.14. Motor delays
Motor delays are one of the most common comorbid developmental problems in children with NF1, found in nearly 50% of affected children [75].
5.15. Scoliosis
Scoliosis is the most common musculoskeletal manifestation of NF1, affecting 10–30% of children with NF1, with a mean age at presentation of 9.4 years [76]. Children with NF1 can develop dystrophic or non-dystrophic scoliosis. While nondystrophic scoliosis resembles idiopathic scoliosis in the general population, dystrophic scoliosis is characterized by progressive, sharply angulated short segment curvature with severe wedging, rotation and scalloping of the apical vertebral bodies. Dystrophic spinal defects can be associated with paraspinal or other internal neurofibromas adjacent to the vertebrae, as well as vertebral scalloping, dural ectasia, and rib penciling [77,78]. Dystrophic scoliosis tends to evolve more rapidly, resulting in severe curvatures and often requiring complicated corrective surgery. Prompt recognition, orthopedic referral, and treatment are indicated.
5.16. Congenital bowing/dysplasia
Long bone dysplasia (without pseudarthrosis) is present in 3–4% of children with NF1, most commonly involving the long bones, such as the tibia, and more rarely, the fibula, radius, and ulna. It typically presents in early infancy, with unilateral anterolateral bowing of the lower leg [79] and can be associated with medullary canal narrowing and cortical thickening. Long bone dysplasia usually appears at birth, but in most cases, it is recognized months later. Pseudarthrosis occurs in 5% of children with NF1 [80], caused by abnormal bone formation and the appearance of a ‘false joint.’ Some of these children with poor bone healing will require an amputation and prosthetic devices for mobility.
5.17. Sphenoid wing dysplasia
Unilateral sphenoid wing dysplasia is found in 3–11% of children with NF1 [81]. While it can arise secondary to a plexiform neurofibroma, it can also result from a primary ossification defect with poor mesodermal development and bone formation. Abnormal growth of the skull associated with sphenoid wing dysplasia may also lead to progressive facial deformities and poor alignment of the eyes.
5.18. Cerebral vasculopathy
Cerebrovascular abnormalities are severe and often unrecognized complications in children with NF1 [82]. The most prevalent form of NF1-associated cerebral vasculopathy is moyamoya syndrome (MMS), with a predilection for anterior circulation vessels. Other cerebral arterial abnormalities include arterial stenosis (Figure 3(d)), occlusion, hypoplasia, fusiform dilation, ectasia, aneurysm, and arteriovenous fistula. Hypertension is also common in individuals with NF1, both with and without MMS, and could be a sign of renal artery stenosis [82].
5.19. Sleep disorders
The prevalence of obstructive sleep apnea (OSA) is high in individuals with NF1, requiring a detailed sleep evaluation
6 N. KERASHVILI AND D. H. GUTMANN
[83]. Early clinical intervention strategies to promote sleep hygiene are beneficial to optimize developmental outcomes.
5.20. GI symptomatology
Children with NF1 have more GI symptoms, such as functional dyspepsia, irritable bowel syndrome, or constipation [84]. This high prevalence of GI dysfunction is not ‘functional’ in nature, necessitating a work-up by an experienced pediatric gastroenterologist for underlying etiologies.
6. Expert opinion
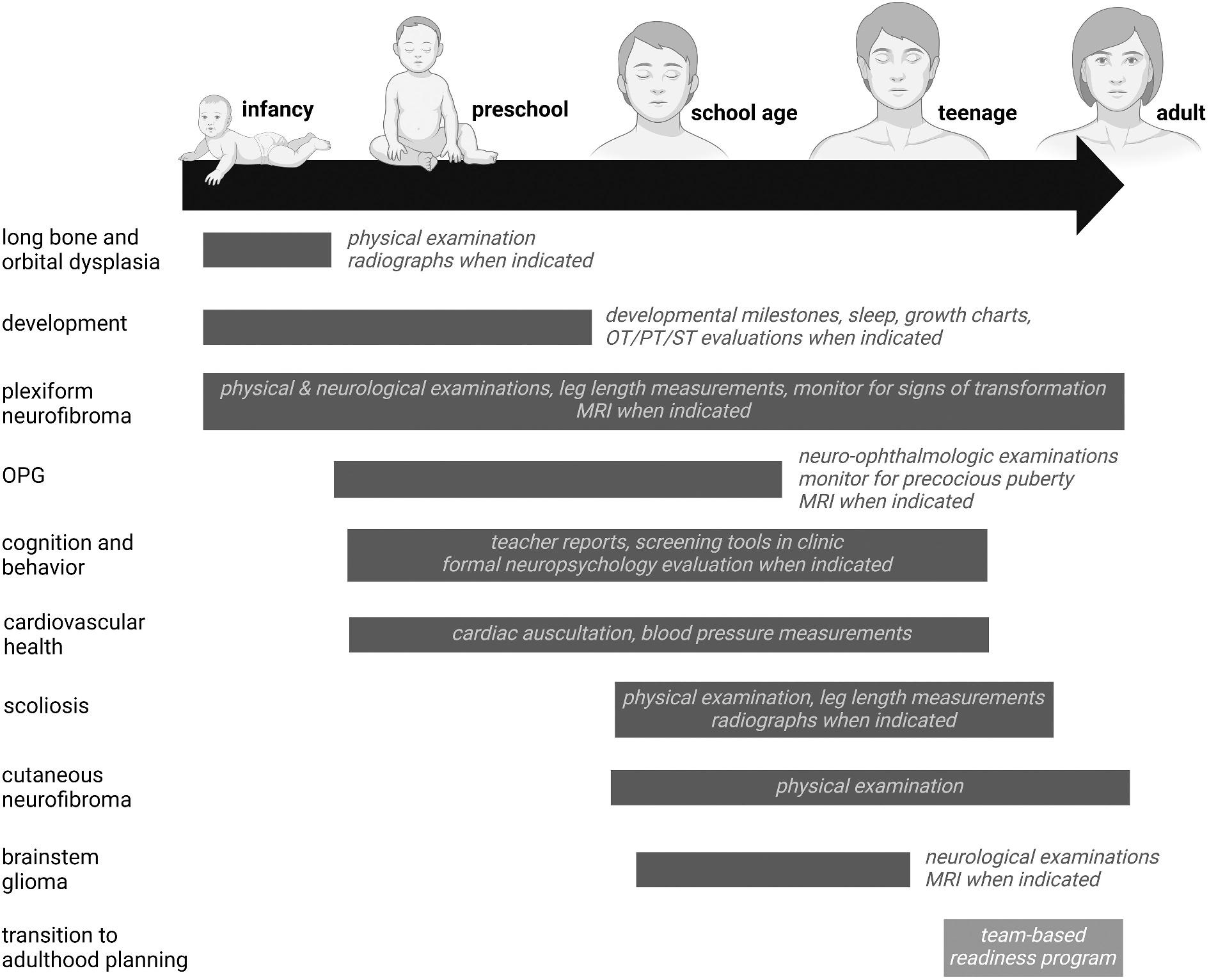
The appearance of NF1-related medical problems is agerelated, as previously noted in recent publications [85]. For this reason, we advocate surveillance screening that focuses on the clinical features most likely to appear at any given life stage (Figure 5). We recommend screening for NF1-related conditions as early as the neonatal period and continuing throughout life. Children are typically seen on an annual basis or more frequently to monitor specific medical complications of NF1.
6.1. Infancy
Early detection of bony abnormalities, including tibial and orbital dysplasia, is paramount. Physical examination, followed by long bone radiographs or orbital CT scans when indicated, is recommended, with prompt referral to orthopedic or craniofacial plastic surgery, respectively. Long bone dysplasia is initially managed with external bracing, although definitive treatment is largely surgical.
Examination of infants for body asymmetries, like hemihypertrophy, proptosis, and scoliosis, is important to identify plexiform neurofibromas. When suspected, MRI is indicated to define the location and extent of the tumor. The decision to treat depends on the location, rate of growth, and involvement of adjacent structures. Measurements of leg lengths can be helpful in identifying asymmetries requiring further investigation.
Neurodevelopmental monitoring is recommended to identify developmental delays and hypotonia. Using the American Academy of Pediatrics guidelines [86], age-appropriate milestones should be assessed. If delays are identified, prompt referrals to physical, occupational, or speech therapists should ensue. Head lag, slip-through when suspended under the arms, and increased hip abduction are predictors of hypotonia [87]. Given
EXPERT REVIEW OF NEUROTHERAPEUTICS 7
Figure 5. Age-dependent management of children and adolescents with NF1. Abbreviations: MRI, magnetic resonance imaging; OPG, optic pathway glioma; OT, occupational therapy; PT, physical therapy; ST, speech therapy. Created with BioRender.com.
an association between hypotonia and brain tumors [88], neuroimaging may be indicated to exclude an intracranial etiology.
Neuro-ophthalmological evaluations should commence before the first birthday using age-appropriate visual acuity measurements by an experienced neuro-ophthalmologist.
Visual acuity testing with Teller acuity cards is employed for children prior to 2 years of age [7]. Brain MRI for asymptomatic children is not recommended as a screening modality, as it does not predict visual outcome [89]. Should visual abnormalities be noted on the eye examination, neuroimaging is indicated. Treatment decisions are largely made based on changes in visual acuity [7,35]. In the setting of NF1-OPG, radiotherapy is not recommended due to an elevated risk of malignant transformation. The standard of therapy is chemotherapy with carboplatin and vincristine [90,91], which is more effective at controlling tumor growth than halting or reversing visual loss [92]. As a result, only one-third of children experience improved vision after treatment, while 30% continue to exhibit visual decline despite treatment and require additional treatment [93]. Risk factors associated with refractory/relapsed NF1-OPG and poor visual outcomes are young age (<24 months) and posterior tumor location [93]. Surgical decompression or cerebrospinal fluid diversion is usually reserved for tumors causing compression of the third ventricle with associated hydrocephalus.
6.2. Preschool-age children
Developmental monitoring should continue on a yearly basis along with annual ophthalmological evaluations using Lea symbols or HOTV cards [7]. Evaluation of retinal pathology by optic coherence tomography (OCT) is helpful for children with poor cooperation due to cognitive or behavioral comorbidities [7]. Questions about sleep and eating habits are asked and appropriate referrals made for further evaluation and treatment. Growth charts should be used to assess linear growth trajectories. In addition, concerns about social perception and social functioning should be discussed, and evaluation for early signs or features of autism spectrum disorder initiated using standardized tools (e.g. ADOS-2, CARS-2) [94–96]. Signs of attention deficit should be evaluated [97], and treatment initiated as appropriate [98–100]. In planning for school, children with cognitive or developmental delays often benefit from comprehensive neuropsychological testing, assessing function across multiple domains. This extensive evaluation helps guide further therapeutic interventions (e.g. school adaptations, speech and reading therapy).
For children with plexiform neurofibromas, monitoring by physical examination and MRI is necessary. The decision to treat depends on the age of the patient (younger children are more likely to have a faster growing PN), the risk of morbidity (e.g. secondary spinal cord compression), and progressive growth (rapidly growing vs. slowly growing) [101,102]. Surgery is sometimes indicated for debulking the tumor mass, but the risk of severe hemorrhage and nerve damage should be considered in surgical planning. Medical treatment for symptomatic PNs entails the use of selumetinib, an oral MEK1/2 inhibitor [103]. Partial clinical responses have been reported in two-thirds of children concomitant with
a reduction in pain intensity [104]. Common adverse reactions include vomiting, diarrhea, acneiform skin rash, nausea, abdominal pain, mucositis, and fatigue. Asymptomatic decreased left ventricular ejection fraction is encountered in 14% of individuals receiving MEK inhibitors, necessitating regular echocardiograms.
Monitoring blood pressure and cardiac auscultation continue throughout life, starting in this developmental period.
6.3. School-age children
Annual neuro-ophthalmological assessments for asymptomatic children with NF1 are recommended until 10 years of age [7], typically using Snellen acuity charts. NF1-OPG involvement of the chiasm can precipitate precocious puberty [105], requiring referral to pediatric endocrinology. Children in this age group may exhibit cutaneous neurofibromas. These do not typically require treatment, although surgical excision can be performed for those in problematic areas.
Regular discussions of school performance, social interactions, and behavior are important. Incorporating teacher reports provides additional complementary information. Following formal neuropsychological assessment, accommodations are typically incorporated into an individualized educational plan or 504 plan. Questions about restrictive eating behaviors or aversions to clothing tags and textures may be early signs of autism spectrum disorder.
A detailed neurological examination is required to identify gliomas involving the brainstem or other brain locations. The indications for treatment are tumor enlargement and neurologic deficits [45]. Medical treatment involves carboplatin/vincristine chemotherapy, whereas CSF diversion for secondary hydrocephalus or surgical debulking may be required.
Children may present with sleep disorders or epilepsy, necessitating referral to appropriate specialists for evaluation and treatment [83,106]. While the management of children with these medical problems in the setting of NF1 does not differ from those in the general population, intracranial structural etiologies (e.g. brain tumors) should be excluded for children with NF1 who present with seizures.
Scoliosis requires yearly screening with a physical examination, assessment of leg lengths, and evaluation of linear growth curves. Scoliosis evaluation entails complete spinal radiographs and orthopedic evaluation [107,108]. Bracing is usually the initial medical intervention, although definitive treatment requires internal fixation with the insertion of rods.
6.4. Teenagers
Complete physical and neurological examinations are performed with each annual visit. Monitoring the growth of plexiform neurofibromas, scoliosis, and intracranial neoplasms continues with radiologic assessments. The presence of a rapidly growing plexiform neurofibroma with associated pain or neurologic deficit should prompt immediate evaluation for malignant transformation by an experienced oncologist. Similarly, given the cognitive and behavioral problems seen in individuals with NF1, there is a continued need for
8 N. KERASHVILI AND D. H. GUTMANN
individualized education plans, school accommodations, and discussions about life skills to ensure future meaningful employment.
It is also recommended that discussions about the transition of care planning begin at 12–14 years of age to facilitate a smooth transition to adult care providers. There are several survey-based tools available to determine readiness [109,110]. We advocate a team-based approach involving therapists and other health care professionals, as well as regular meetings with pediatric and adult providers. A seamless transition to adult care avoids lapses in continuity of care, which are particularly problematic for young adults with NF1 in whom the risk of MPNST and breast cancer [111,112] is high.
6.5. Future directions
Over the past decade, there has been great progress in our understanding of the medical features of NF1, as well as the evaluation of promising drugs within a clinical trials consortium structure [ 113 ]. Future improvements in management and therapy will require a better appreciation of the risk factors for specific problems in NF1, such as sex, age, allergic conditions, maternal exposures, and specific germline NF1 variants. This will likely involve the deployment of risk assessment algorithms using machine learning and artificial intelligence methodologies [ 24 ]. Similarly, as a more complete dissection of the cellular and molecular determinants underlying NF1 disease become elucidated, other modalities, such as neuron or immune targeted therapies, might emerge as next-generation treatments for children with NF1 [ 114 , 115 ]. Finally, the development of NF1specific transition of care models is clearly needed, ensuring uninterrupted care for individuals with NF1 across the lifespan.
Funding
This paper was not funded.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
ORCID
David H. Gutmann http://orcid.org/0000-0002-3127-5045
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
1. Plotkin SR, Messiaen L, Legius E, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022 Sep;24(9):1967–1977.
2. Lee TJ, Chopra M, Kim RH, et al. Incidence and prevalence of neurofibromatosis type 1 and 2: a systematic review and meta-analysis. Orphanet J Rare Dis. 2023 Sep 14;18(1):292. doi: 10. 1186/s13023-023-02911-2
3. National institutes of health consensus development conference statement: neurofibromatosis. Bethesda, MD., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172–178.
4. Legius E, Messiaen L, Wolkenstein P, Huson SM, Evans DG, Plotkin SR, International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC), et al. Revised diagnostic criteria for neurofibromatosis type 1 and legius syndrome: an international consensus recommendation. Genet Med. 2021 Aug;238:1506–1513. doi: 10. 1038/s41436-021-01170-5
•• This international consensus group was convened to revise the diagnostic criteria to NF1 based on advances over the past thirty years.
5. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000 Mar;105(3):608–614. doi: 10.1542/peds.105.3.608
6. Nunley KS, Gao F, Albers AC, et al. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch Dermatol. 2009 Aug;145(8):883–7.
7. de Blank PMK, Fisher MJ, Liu GT, et al. Optic pathway gliomas in neurofibromatosis type 1: an update: surveillance, treatment indications, and biomarkers of vision. J Neuroophthalmol. 2017 Sep; Suppl 37(Suppl 1):S23–S32. doi: 10.1097/WNO.0000000000000550
• This consensus statement outlines the current recommendations for surveillance and treatment of optic pathway gliomas in children with NF1.
8. Listernick R, Louis DN, Packer RJ, et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 optic pathway glioma task force. Ann Neurol. 1997 Feb;41(2):143.
9. Miller DT, Freedenberg D, Schorry E, et al. Council of genetics: American College of Medical Genetics and Genomics. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019 May;143(5):e20190660. doi: 10.1542/peds.2019-0660
10. Gutmann DH, Ferner RE, Listernick RH, et al. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017 Feb 23;3(1):17004. doi: 10.1038/nrdp.2017.4
11. Napolitano F, Dell ‘Aquila M, Terracciano C, et al. Genotypephenotype correlations in neurofibromatosis type 1: identification of novel and recurrent NF1 gene variants and correlations with neurocognitive phenotype. Genes (Basel). 2022 Jun 23;13(7):1130. doi: 10.3390/genes13071130
12. Listernick R, Mancini AJ, Charrow J. Segmental neurofibromatosis in childhood. Am J Med Genet A. 2003 Aug 30;121A(2):132–5. doi: 10. 1002/ajmg.a.20183
13. Tinschert S, Naumann I, Stegmann E, et al. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet. 2000 Jun;8(6):455–9.
14. Messiaen LM, Callens T, Mortier G, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15 (6):541–55. doi: 10.1002/1098-1004(200006)15:6<541:AID-HUMU6>3.0. CO;2-N
•• This report describes the current state of NF1 genetic testing.
15. Rojnueangnit K, Xie J, Gomes A, et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat. 2015 Nov;36 (11):1052–1063. doi: 10.1002/humu.22832
• This manuscript details the features of individuals with a clear genotype-phenotype association, where affected patients do not develop optic pathway gliomas or neurofibromas.
16. Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically
EXPERT REVIEW OF NEUROTHERAPEUTICS 9
significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007 Jan;80(1):140–51.
17. De Raedt T, Brems H, Wolkenstein P, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003 May;72(5):1288–92.
18. Kluwe L, Nguyen R, Vogt J, et al. Internal tumor burden in neurofibromatosis Type I patients with large NF1 deletions. Genes Chromosomes Cancer. 2012 May;51(5):447–51.
19. Pacot L, Vidaud D, Sabbagh A, et al. Members of the NF France network, Wolkenstein P, pasmant E. Severe phenotype in patients with large deletions of NF1 Cancers (Basel). 2021 Jun 13;13 (12):2963. doi: 10.3390/cancers13122963
20. Koczkowska M, Chen Y, Callens T, et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am J Hum Genetic. 2018 Jan 4;102(1):69–87. doi: 10.1016/j.ajhg.2017.12.001
21. Koczkowska M, Callens T, Chen Y, et al. Clinical spectrum of individuals with pathogenic N F1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype–phenotype study in neurofibromatosis type 1. Hum Mutat. 2020 Jan;41(1):299–315.
22. Guo X, Pan Y, Gutmann DH. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell–specific chemokine recruitment of T cells and microglia. Neuro Oncol. 2019 Oct;21(10):1250–1262. doi: 10.1093/neuonc/ noz080
23. Anastasaki C, Wegscheid ML, Hartigan K, et al. Human iPSC-derived neurons and cerebral organoids establish differential effects of germline NF1 gene mutations. Stem Cell Rep. 2020 Apr;14 (4):541–550.
24. Morris SM, Gupta A, Kim S, et al. Predictive modeling for clinical features associated with neurofibromatosis type 1. Neurol Clin Pract. 2021 Dec;11(6):497–505.
25. Lalor L, Davies OMT, Basel D, et al. Café au lait spots: when and how to pursue their genetic origins. Clin Dermatol. 2020 Jul;38 (4):421–431.
26. Miraglia E, Moliterni E, Iacovino C, et al. Cutaneous manifestations in neurofibromatosis type 1. Clin Ter. 2020 Sep;171(5):e371–e377.
27. Ragge NK, Falk RE, Cohen WE, et al. Images of Lisch nodules across the spectrum. Eye (Lond). 1993;7(Pt 1):95–101. doi: 10.1038/eye. 1993.20
28. Kurosawa A, Kurosawa H. Ovoid bodies in choroidal neurofibromatosis. Arch Ophthalmol. 1982 Dec;100(12):1939–41. doi: 10.1001/archopht.1982.01030040919010
29. Parrozzani R, Clementi M, Frizziero L, et al. In vivo detection of choroidal abnormalities related to NF1: feasibility and comparison with standard nih diagnostic criteria in pediatric patients. Invest Ophthalmol Vis Sci. 2015 Sep;56(10):6036–42. doi: 10.1167/iovs.1416053
• This report describes choroidal abnormalities in individuals with NF1, prompting revision of the clinical diagnostic criteria for NF1.
30. Chilibeck CM, Shah S, Russell HC, et al. The presence and progression of choroidal neurofibromas in a predominantly pediatric population with neurofibromatosis type-1. Ophthalmic Genet. 2021 Jun;42(3):223–229.
31. Guillamo JS, Créange A, Kalifa C, et al. Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003 Jan;126(Pt 1):152–160.
32. Listernick R, Charrow J, Greenwald MJ, et al. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989 May;114 (5):788–92.
33. Listernick R, Charrow J, Greenwald M, et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994 Jul;125(1):63–6.
34. Listernick R, Ferner RE, Piersall L, et al. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology. 2004 Nov 23;63(10):1944–6. doi: 10.1212/01.WNL.0000144341. 16830.01
35. Listernick R, Ferner RE, Liu GT, et al. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007 Mar;61(3):189–98.
36. Fisher MJ, Avery RA, Allen JC, et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology. 2013 Nov 19;81(21 Suppl 1):S15–24. doi: 10.1212/01.wnl. 0000435745.95155.b8
37. Cambiaso P, Galassi S, Palmiero M, et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am J Med Genet A. 2017 Sep;173(9):2353–2358.
38. Listernick R, Charrow J, Greenwald M. Emergence of optic pathway gliomas in children with neurofibromatosis type 1 after normal neuroimaging results. J Pediatr. 1992 Oct;121(4):584–7. doi: 10. 1016/S0022-3476(05)81151-5
39. Fisher MJ, Loguidice M, Gutmann DH, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol. 2012 Jun;14(6):790–7. doi: 10.1093/neuonc/ nos076
• This report is one of the most comprehensive clinical studies on visual outcomes in children with NF1-associated optic gliomas.
40. Thirunavu VM, Mohammad LM, Kandula V, et al. Vision outcomes for pediatric patients with optic pathway gliomas associated with neurofibromatosis type I: a systematic review of the clinical evidence. J Pediatr Hematol Oncol. 2021 May 1;43(4):135–143. doi: 10.1097/MPH.0000000000002060
41. Abadin SS, Zoellner NL, Schaeffer M, et al. Racial/Ethnic differences in pediatric brain tumor diagnoses in patients with neurofibromatosis type 1. J Pediatr. 2015 Sep;167(3):613–20.e1–2.
42. Johnson KJ, Zoellner NL, Gutmann DH. Peri-gestational risk factors for pediatric brain tumors in neurofibromatosis type 1. Cancer Epidemiol. 2016 Jun;42:53–9. doi: 10.1016/j.canep.2016.03.005
43. Porcelli B, Zoellner NL, Abadin SS, et al. Associations between allergic conditions and pediatric brain tumors in neurofibromatosis type 1. Fam Cancer. 2016 Apr;15(2):301–8.
44. Fisher MJ, Loguidice M, Gutmann DH, et al. Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann Neurol. 2014 May;75(5):799–800.
45. Mahdi J, Shah AC, Sato A, et al. A multi-institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology. 2017 Apr 18;88(16):1584–1589. doi: 10.1212/WNL. 0000000000003881
•• This report is the most comprehensive analysis of brainstem gliomas in children with NF1.
46. Ullrich NJ, Raja AI, Irons MB, et al. Brainstem lesions in neurofibromatosis type 1. Neurosurgery. 2007 Oct;61(4):762–766.discussion 766-7. doi: 10.1227/01.NEU.0000298904.63635.2D
47. Mahdi J, Goyal MS, Griffith J, et al. Nonoptic pathway tumors in children with neurofibromatosis type 1. Neurology. 1059 [2020 Aug 25];95(8):e1052–e. doi: 10.1212/WNL.0000000000009458
48. Sellmer L, Farschtschi S, Marangoni M, et al. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J Rare Dis. 2017 Feb 15;12(1):34. doi: 10.1186/s13023-017-0588-2
49. Byrne S, Connor S, Lascelles K, et al. Clinical presentation and prognostic indicators in 100 adults and children with neurofibromatosis 1 associated non-optic pathway brain gliomas. J Neurooncol. 2017 Jul;133(3):609–614.
50. Gutmann DH, Rasmussen SA, Wolkenstein P, et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology. 2002 Sep 10;59(5):759–61. doi: 10.1212/WNL. 59.5.759
51. Griffith JL, Morris SM, Mahdi J, et al. Increased prevalence of brain tumors classified as T2 hyperintensities in neurofibromatosis 1. Neurol Clin Pract. 2018 Aug;8(4):283–291.
52. Poplausky D, Young JN, Tai H, et al. Dermatologic manifestations of neurofibromatosis type 1 and emerging treatments. Cancers (Basel). 2023 May 16;15(10):2770. doi: 10.3390/ cancers15102770
53. Waggoner DJ, Towbin J, Gottesman G, et al. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet. 2000 May 15;92(2):132–5. doi: 10.1002/(SICI)1096-8628(20000515)
92:2<132:AID-AJMG10>3.0.CO;2-6
10 N. KERASHVILI AND D. H. GUTMANN
54. Fisher MJ, Blakeley JO, Weiss BD, et al. Management of neurofibromatosis type 1-associated plexiform neurofibromas. Neuro Oncol. 2022 Nov 2;24(11):1827–1844. doi: 10.1093/neuonc/noac146
• This consensus report describes the current management recommendations for NF1-associated plexiform neurofibromas.
55. Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathological evaluation of atypical neurofibromatous tumours and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis type 1 – a consensus overview. Hum Pathol. 2017 Sep;67:1–10.
56. Uusitalo E, Leppavirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015 Mar;135(3):904–906.
57. Prudner BC, Ball T, Rathore R, et al. Diagnosis and management of malignant peripheral nerve tumors: Current practice and future perspectives. Neurooncol Adv. 2019 Nov 14;2(Suppl 1):i40–i49. doi: 10.1093/noajnl/vdz047
58. Vogel AC, Gutmann DH, Morris SM. Neurodevelopmental disorders in children with neurofibromatosis type 1. Dev Med Child Neurol. 2017 Nov;59(11):1112–1116. doi: 10.1111/dmcn.13526
59. Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005 Oct 11;65(7):1037–44. doi: 10.1212/01.wnl. 0000179303.72345.ce
60. Arnold SS, Payne JM, McArthur G, et al. Profiling the word reading abilities of school-age children with neurofibromatosis type 1. J Int Neuropsychol Soc. 2021 May;27(5):484–496.
61. Vernet M, Jover M, Bellocchi S, et al. Visual-processing deficits in children with neurofibromatosis type 1: a clinical marker of reading difficulties. Eur J Paediatr Neurol. 2022 May;38:25–32.
62. Beaussart-Corbat ML, Barbarot S, Farges D, et al. Executive functions in preschool-aged children with neurofibromatosis type 1: value for early assessment. J Clin Exp Neuropsychol. 2021 Mar;43 (2):163–175.
63. Lidzba K, Granström S, Lindenau J, et al. The adverse influence of attention-deficit disorder with or without hyperactivity on cognition in neurofibromatosis type 1. Dev Med Child Neurol. 2012 Oct;54(10):892–7.
64. Haebich KM, Dao DP, Pride NA, et al. The mediating role of ADHD symptoms between executive function and social skills in children with neurofibromatosis type 1. Child Neuropsychol. 2022 Apr;28 (3):318–336.
65. Payne JM, Haebich KM, MacKenzie R, et al. Cognition, ADHD symptoms, and functional impairment in children and adolescents with neurofibromatosis type 1. J Atten Disord. 2021 Jun;25 (8):1177–1186.
66. Stavinoha PL, Solesbee C, Swearer SM, et al. Risk factors for bullying victimization in children with neurofibromatosis type 1 (NF1). Children (Basel). 2021 Feb 15;8(2):145. doi: 10.3390/ children8020145
67. Barton B, North K. Social skills of children with neurofibromatosis type 1. Dev Med Child Neurol. 2004 Aug;46(8):553–63. doi: 10.1017/ S0012162204000921
68. Noll RB, Reiter-Purtill J, Moore BD, et al. Social, emotional, and behavioral functioning of children with NF1. Am J Med Genet A. 2007 Oct 1;143A(19):2261–73. doi: 10.1002/ajmg.a.31923
69. Payne JM, Porter M, Pride NA, et al. Theory of mind in children with neurofibromatosis type 1. Neuropsychology. 2016 May;30 (4):439–48.
70. Lewis AK, Porter MA, Williams TA, et al. Attention to faces in social context in children with neurofibromatosis type 1. Dev Med Child Neurol. 2019 Feb;61(2):174–180.
71. Garg S, Green J, Leadbitter K, et al. Neurofibromatosis type 1 and autism spectrum disorder. Pediatrics. 2013 Dec;132(6):e1642–8.
72. Morris SM, Acosta MT, Garg S, et al. Disease burden and symptom structure of autism in neurofibromatosis type 1: a study of the international NF1-ASD consortium team (INFACT). JAMA Psychiatry. 2016 Dec 1;73(12):1276–1284. doi: 10.1001/jamapsychia try.2016.2600
•• This international study is the largest and most comprehensive analysis of autism symptomatology in children with NF1.
73. Plasschaert E, Descheemaeker MJ, Van Eylen L, et al. Prevalence of autism spectrum disorder symptoms in children with neurofibromatosis type 1. Am J Med Genet B Neuropsychiatr Genet. 2015 Jan;168B(1):72–80.
74. Janusz JA, Klein-Tasman BP, Payne JM, et al. REiNS international collaboration. Recommendations for social skills end points for clinical trials in neurofibromatosis type 1. Neurology. 2021 Aug 17;97(7 Suppl 1):S73–S80. doi: 10.1212/WNL.0000000000012422
• This study outlines the recommendations for social skill endpoints for NF1 clinical trials.
75. Wessel LE, Gao F, Gutmann DH, et al. Longitudinal analysis of developmental delays in children with neurofibromatosis type 1. J Child Neurol. 2013 Dec;28(12):1689–93.
76. Toro G, Santoro C, Ambrosio D, et al. Natural history of scoliosis in children with NF1: an observation study. Healthcare. 2021 Jul 13;9 (7):881.10.3390/healthcare9070881
• This report describes the natural history of scoliosis in children with NF1.
77. Khong PL, Goh WHS, Wong VCN, et al. MR imaging of spinal tumors in children with neurofibromatosis 1. AJR Am J Roentgenol. 2003 Feb;180(2):413–417.
78. Ramachandran M, Tsirikos AI, Lee J, et al. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004 Dec;17(6):483–91.
79. Elefteriou F, Kolanczyk M, Schindeler A, et al. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A. 2009 Oct;149A(10):2327–38.
80. Brekelmans C, Hollants S, De Groote C, et al. Neurofibromatosis type 1-related pseudarthrosis: beyond the pseudarthrosis site. Hum Mutat. 2019 Oct;40(10):1760–1767.
81. Chauvel-Picard J, Lion-Francois L, Beuriat PA, et al. Craniofacial bone alterations in patients with neurofibromatosis type 1. Childs Nerv Syst. 2020 Oct;36(10):2391–2399.
82. Lehman LL, Ullrich NJ. Cerebral vasculopathy in children with neurofibromatosis type 1. Cancers (Basel). 2023 Oct 23;15 (20):5111. doi: 10.3390/cancers15205111
83. Licis AK, Vallorani A, Gao F, et al. Prevalence of sleep disturbances in children with neurofibromatosis type 1. J Child Neurol. 2013 Nov;28(11):1400–1405.
84. Ejerskov C, Krogh K, Ostergaard JR, et al. Gastrointestinal symptoms in children and adolescents with neurofibromatosis type 1. J Pediatr Gastroenterol Nutr. 2018 Jun;66(6):872–875.
85. Carton C, Evans DG, Blanco I, et al. ERN GENTURIS NF1 tumour management guideline team. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine. 2023 Jan;56:101818.
86. Lipkin PH, Macias MM, Norwood KW. Council on children with disabilities, section on developmental and behavioral pediatrics. Promoting optimal development: identifying infants and young children with developmental disorders through developmental surveillance and screening. Pediatrics. 2020 Jan;145(1):e20193449. doi: 10.1542/peds.2019-3449
87. Soucy EA, Wessel LE, Gao F, et al. A pilot study for evaluation of hypotonia in children with neurofibromatosis type 1. J Child Neurol. 2015 Mar;30(3):382–5.
88. Wessel LE, Albers AC, Gutmann DH, et al. The association between hypotonia and brain tumors in children with neurofibromatosis type 1. J Child Neurol. 2013 Dec;28(12):1664–7.
89. King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet A. 2003 Oct 1;122A(2):95–9. doi: 10. 1002/ajmg.a.20211
90. Packer RJ, Ater J, Allen J, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997 May;86(5):747–54.
91. Ater JL, Xia C, Mazewski CM, et al. Nonrandomized comparison of neurofibromatosis type 1 and non-neurofibromatosis type 1
EXPERT REVIEW OF NEUROTHERAPEUTICS 11
children who received carboplatin and vincristine for progressive low-grade glioma: a report from the children’s oncology group. Cancer. 2016 Jun 15;122(12):1928–1936. doi: 10.1002/cncr.29987
92. Rakotonjanahary J, Gravier N, Lambron J, et al. Long-term visual acuity in patients with optic pathway glioma treated during childhood with up-front BB-SFOP chemotherapy-analysis of a French pediatric historical cohort. PloS One. 2019 Mar 8;14(3):e0212107. doi: 10.1371/journal.pone.0212107
93. Kotch C, Avery R, Getz KD, et al. Risk factors for treatment-refractory and relapsed optic pathway glioma in children with neurofibromatosis type 1. Neuro Oncol. 2022 Aug 1;24 (8):1377–1386. doi: 10.1093/neuonc/noac013
94. Luyster R, Gotham K, Guthrie W, et al. The Autism Diagnostic Observation Schedule-toddler module: a new module of a standardized diagnostic measure for autism spectrum disorders. J Autism Dev Disord. 2009 Sep;39(9):1305–1320.
95. Chlebowski C, Green JA, Barton ML, et al. Using the childhood autism rating scale to diagnose autism spectrum disorders. J Autism Dev Disord. 2010 Jul;40(7):787–99.
96. Hyman SL, Levy SE, Myers SM. COUNCIL on CHILDREN with DISABILITIES, SECTION on DEVELOPMENTAL and BEHAVIORAL PEDIATRICS. Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics. 2020 Jan;145(1): e20193447. doi: 10.1542/peds.2019-3447
97. Wolraich ML, Hagan JF Jr, Allan C, et al. Subcommittee on children and adolescents with attention-deficit/hyperactive disorder. Clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/Hyperactivity disorder in children and adolescents. Pediatrics. 2019 Oct;144(4):e20192528.
98. Moscoso A, Julien A, Tanet A, et al. Clinical management of children and adolescents with neurofibromatosis type 1 like phenotypes and complex behavioural manifestations: a multidisciplinary and dimensional approach. Case Rep Psychiatry. 2019 Dec 31;2019:1–12.
99. Mautner VF, Kluwe L, Thakker SD, et al. Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol. 2002 Mar;44 (3):164–70.
100. Lion-François L, Gueyffier F, Mercier C, et al. The effect of methylphenidate on neurofibromatosis type 1: a randomised, double-blind, placebo-controlled, crossover trial. Orphanet J Rare Dis. 2014 Sep 10;9(1):142. doi: 10.1186/s13023-014-0142-4
101. Gross AM, Singh G, Akshintala S, et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol. 2018 Nov 12;20 (12):1643–1651. doi: 10.1093/neuonc/noy067
102. Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007 Feb 27;68(9):643–7. doi: 10.1212/01.wnl. 0000250332.89420.e6
103. Armstrong AE, Belzberg AJ, Crawford JR, et al. Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas. BMC Cancer. 2023 Jun 16;23(1):553. doi: 10.1186/s12885-023-10996-y
•• This review discusses the indications and uses of MEK inhibitors for treating children with NF1-associated plexiform neurofibromas.
104. Gross AM, Dombi E, Wolters PL, et al. Long-term safety and efficacy of selumetinib in children with neurofibromatosis type 1 on a phase 1/2 trial for inoperable plexiform neurofibromas. Neuro Oncol. 2023 Oct;25(10):1883–1894.
105. Habiby R, Silverman B, Listernick R, et al. Precocious puberty in children with neurofibromatosis type 1. J Pediatr. 1995 Mar;126 (3):364–7.
106. Ostendorf AP, Gutmann DH, Weisenberg JL. Epilepsy in individuals with neurofibromatosis type 1. Epilepsia. 2013 Oct;54 (10):1810–1814. doi: 10.1111/epi.12348
107. Crawford AH, Herrera-Soto J. Scoliosis associated with neurofibromatosis. Orthop Clin North Am. 2007 Oct;38(4):553–62. vii. doi: 10.1016/j.ocl.2007.03.008
108. Tsirikos AI, Saifuddin A, Noordeen MH. Spinal deformity in neurofibromatosis type-1: diagnosis and treatment. Eur Spine J. 2005 Jun;14(5):427–39. doi: 10.1007/s00586-004-0829-7
109. Radtke HB, Berger A, Skelton T, et al. Neurofibromatosis type 1 (NF1): addressing the transition from pediatric to adult care. Pediatric Health Med Ther. 2023 Feb 9;14:19–32. doi: 10.2147/ PHMT.S362679
• This review from the Children’s Tumor Foundation outlines recommendations for the transition to adult care in children with NF1.
110. Weisman AG, Haws T, Lee J, et al. Transition readiness assessment in adolescents and young adults with neurofibromatosis type 1 (NF1). Compr Child Adolesc Nurs. 2023 Sep;46(3):223–229.
111. Yan K, Gao Y, Heller SL. Breast cancer screening utilization and outcomes in women with neurofibromatosis type 1. Clin Breast Cancer. 2023 Jun;23(4):e200–205. doi: 10.1016/j.clbc.2023.02.005
112. Madanikia SA, Bergner A, Ye X, et al. Increased risk of breast cancer in women with NF1. Am J Med Genet. 2012 Dec;158A (12):3056–3060.
113. Gutmann DH, Blakeley JO, Korf BR, et al. Optimizing biologically targeted clinical trials for neurofibromatosis. Exp Opin Investig Drugs. 2013 Apr;22(4):443–462. doi: 10.1517/13543784.2013.772979
114. Hartman LLR, Crawford JR, Makale MT, et al. Pediatric phase II trials of poly-ICLC in the management of newly diagnosed and recurrent brain tumors. J Pediatr Hematol Oncol. 2014 Aug;36(6):451–457.
115. Anastasaki C, Mo J, Chen J-K, et al. Neuronal hyperexcitability drives central and peripheral nervous system tumor progression in models of neurofibromatosis-1. Nat Commun. 2022 May;13(1):2785.
12 N. KERASHVILI AND D. H. GUTMANN