Alle Artikel, die mit “In Zusammenarbeit mit“ gekennzeichnet sind, sind keine neutrale Redaktion der Mediaplanet Verlag Deutschland GmbH. Aus Gründen der besseren Lesbarkeit wird auf die gleichzeitige Verwendung der Sprachformen männlich, weiblich & divers (m/w/d) verzichtet. Alle Personenbezeichnungen gelten gleichermaßen für alle Geschlechter.
Mediaplanet-Kontakt: de.redaktion@mediaplanet.com
iebe Leserinnen, liebe Leser, der Sommer ist vorüber und von Sommerloch keine Spur! Knapp 150 Tage ist die Bundesregierung nun im Amt. Im Mai hat sie ihren Koalitionsvertrag veröffentlicht. Die Ankündigung, Maßnahmen zu ergreifen, um die gesundheitliche Situation von Betroffenen Seltener Erkrankungen zu verbessern, werten wir als Lichtblick. Den Satz im Koalitionsvertrag zu platzieren, war ein dickes Brett. Nun müssen viele Akteure gemeinsam für die Konkretisierung sorgen. Wir, die ACHSE, bringen unsere Expertise ein, damit im Sinne der Betroffenen gehandelt wird.
Ein Thema, das mich gerade besonders umtreibt, ist die geplante Primärarztversorgung. Den Primärversorger als Lotsen im System zu etablieren, begrüßen wir. Auch, dass durch die Einführung bürokratische Hürden abgebaut werden sollen. Um die Bedarfe chronisch Kranker zu adressieren, könnten auch Fachärztinnen oder -ärzte als Primärversorger fungieren, heißt es in dem Vorhaben.
Menschen mit Seltenen Erkrankungen benötigen in jedem Fall zusätzliche Regelungen: Seltene Erkrankungen müssen in den Blick genommen werden, gerade im Behandlungsalltag.
Geske Wehr
Vorsitzende der ACHSE e. V.
in den Blick genommen werden, gerade im Behandlungsalltag. Wissen muss frühzeitig und stetig vermittelt werden. Es muss gewährleistet sein, dass Ärztin oder Arzt wissen, an welche Versorgungseinrichtung sie Betroffene überweisen können, um eine gerechte Versorgung sicherzustellen. Und überhaupt: Das System kann nur funktionieren, wenn die Kapazitäten gründlich aufgestockt werden.
Die Bretter, die wir bohren, werden dicker. Als Dachverband mit 140 Patientenorganisationen stehen wir mit konkreten Handlungsempfehlungen und der Expertise unseres Netzwerkes zur Verfügung, nicht nur bei der Ausgestaltung der Primärarztversorgung. Die Themen sind vielfältig. Doch es gibt 4 Millionen Gründe für eine bessere Gesundheitspolitik.
Für Menschen mit Seltenen Erkrankungen ist das nicht nur wichtig, sondern überlebenswichtig. Das Vorhaben ist zum jetzigen Zeitpunkt noch nicht ausgestaltet. Menschen mit Seltenen Erkrankungen benötigen in jedem Fall zusätzliche Regelungen: Seltene Erkrankungen müssen
Einige Stimmen aus unserem Netzwerk, lernen Sie in dieser Ausgabe kennen. Viel Freude beim Lesen.
Weitere Informationen finden Sie unter: www.achse-online.de
„Was ich mir wünsche? Akzeptanz statt dummer Sprüche!“
Jannis ist 26 Jahre alt, besucht leidenschaftlich gern Festivals, interessiert sich für Design und Streetwear. Dass er aber nicht so ist wie andere junge Menschen, sieht man bereits an seinem Rollstuhl. Jannis hat Friedreich-Ataxie (FA): eine seltene, erbliche Erkrankung des Nervensystems. Aber Jannis möchte seine Zeit genießen und setzt sich dafür ein, die Friedreich-Ataxie bekannter zu machen.
Text Hanna Sinnecker
Lieber Jannis, du hast eine seltene Erberkrankung: Friedreich-Ataxie. Wie hast du bemerkt, dass etwas gesundheitlich nicht stimmt? Es fing an, als ich elf Jahre alt war. Ich war mit meiner Mutter spazieren und bin beim Laufen immer nach rechts abgedriftet. Da ich zu der Zeit sehr schnell gewachsen bin, dachten wir, dass es daran liegt. Da es aber nicht besser wurde, gingen wir zum Hausarzt. Der meinte, dass er schon mal von einer Krankheit gehört hätte, die solche Symptome auslösen kann. Er nahm das Ganze sehr ernst und hat mich in eine Klinik überwiesen, die auf neuromuskuläre Erkrankungen spezialisiert ist. Dort wurde mir Rückenmarksflüssigkeit entnommen und untersucht. Danach stand fest: Ich habe Friedreich-Ataxie.
Wie alt warst du, als die Diagnose gestellt wurde und was hat das mit dir und deiner Familie gemacht? Bei der Diagnose war ich zwölf Jahre alt. Ich selbst habe damals noch gar nicht gecheckt, was das für mich und mein Leben bedeutet. Für meine Mutter war die Diagnose ein Schock. Sie hat verstanden, dass die Erkrankung große Auswirkungen auf meine Zukunft haben würde. Früher oder später sind die meisten Betroffenen auf einen Rollstuhl angewiesen, bei mir ist das seit meinem 16. Lebensjahr der Fall. In der Schule wurde es im Laufe der Zeit immer schwieriger für mich. Ich wurde extrem gemobbt, weil mein Gang immer wackeliger wurde und ich langsamer sprach. Ich wurde häufig rumgeschubst, meine Mitschüler haben sich ständig über mich lustig gemacht. Klar, irgendwann habe ich dann auch feststellen müssen, dass die Symptome schlimmer werden und mich immer mehr einschränken. Daran habe ich mich aber schnell gewöhnt, selbst, als ich einen Rollstuhl brauchte. Das Schlimmste für mich war die geballte Ignoranz meiner Mitschüler.
Schaut man sich deine Social MediaKanäle an, sieht man deutlich: du bist
immer mittendrin, trotz deiner Erkrankung und den damit verbundenen Einschränkungen. Wie schaffst du das?
Zu einem gewissen Teil dachte ich mir irgendwann: „Jetzt erst recht!“ Klar habe ich eine krasse Erkrankung, aber ich bin ja trotzdem ein normaler junger Typ, mein Körper funktioniert nur nicht so, wie er sollte. Meine Tattoos haben mir dabei geholfen, mich von anderen abzuheben und zu zeigen, dass ich so viel mehr bin als meine Krankheit!
Mittlerweile habe ich gute Freunde, die sich wirklich für mich interessieren. Diese Unterstützung fühlt sich natürlich gut an und ist toll für mich. Mitleid brauche ich nicht. Was ich brauche, sind Menschen, die sich die Zeit nehmen, den Typen kennenzulernen, der ich bin.
Nehmt euch die Zeit, die Person hinter der Erkrankung kennenzulernen und lernt, was es bedeutet, mit einer chronischen Krankheit leben zu müssen.
Jannis
Friedreich-Ataxie-Patient
Du sprichst offen über deine Erkrankung und nimmst dabei kein Blatt vor den Mund. Fiel dir das schon immer leicht?
Ich war schon immer ein Mensch, der Dinge gern offen angesprochen hat. Aber Social Media ist natürlich ein besonderer Weg. Angefangen habe ich auf TikTok, da war ich über den Zeitraum von etwa einem Jahr täglich live. So habe ich mir die erste kleine Reichweite aufgebaut. Aber eins muss ich sagen: Ich bekomme auf TikTok unfassbar viele eklige Nachrichten… Das härtet ab. Das zeigt aber noch mal mehr, wie wichtig es ist, Aufklärungsarbeit zu leisten.
Was ist dein Antrieb für deinen Einsatz rund um die Aufklärung über deine Erkrankung und was wünschst du dir für dich und andere Betroffene?
Kein kranker Mensch ist weniger wert! Nehmt euch die Zeit, die Person hinter der Erkrankung kennenzulernen und lernt, was es bedeutet, mit einer chronischen Krankheit leben zu müssen.
Die meisten Betroffenen sind super offen und erklären gern alles! Aber man muss nachfragen und echtes Interesse zeigen. Also bitte: Akzeptanz statt dummer Sprüche!
Mehr zu Jannis‘ Alltag mit Friedreich Ataxie finden Sie auf seinem Instagram-Profil:
Foto:
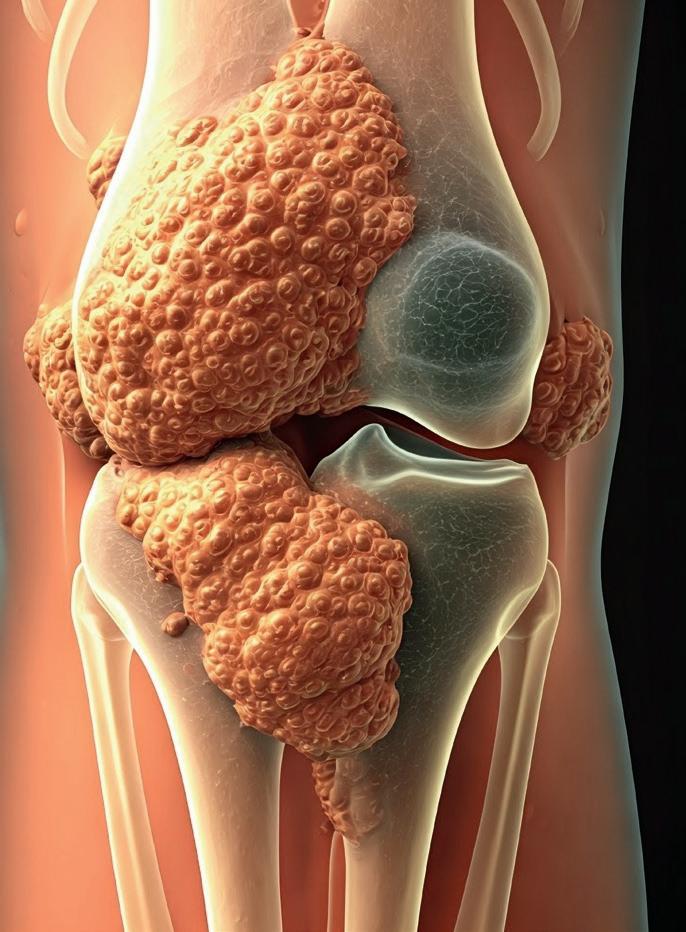
TENOSYNOVIALE RIESENZELLTUMOREN (TGCT):
„TGCT können fundamentale Konsequenzen für die Lebensqualität Betroffener haben.“
Hört man das Wort Tumor, denkt man sofort an Krebserkrankungen. Es gibt aber auch gutartige Tumore, die nicht lebensbedrohlich sind. Zu diesen zählen auch die sogenannten Tenosynovialen Riesenzelltumoren (kurz TGCT). Doch auch, wenn es sich um gutartige Tumore handelt, können sie die Lebensqualität Betroffener erheblich beeinträchtigen.
Herr Prof. Bauer, was passiert bei TGCT-Betroffenen im Körper? Wo treten diese Tumoren auf und welche Symptome können sie auslösen?
Hinter der Erkrankung steckt eine durch einen Tumor ausgelöste Entzündung der Gelenkhaut, die als Synovialitis bezeichnet wird. Die entzündlichen Veränderungen der Gelenkhaut kann man deutlich in Form von farblichen Veränderungen sehen. Lange dachte man, dass es sich um eine Erkrankung handelt, die eher mit Rheuma vergleichbar ist. Stattdessen ist es eine klonale Erkrankung, für die es eine definierte genetische Ursache gibt. In diesem Fall ist das eine nicht erblich bedingte Gen-Umlagerung, die dazu führt, dass ein Lokal-Hormon („Zytokin“) vermehrt ausgeschüttet wird. Das führt zu einer Wucherung Gewebe-ständiger Zellen im Bereich der Gelenkhaut.
Hinter der Erkrankung steckt eine durch einen Tumor ausgelöste Entzündung der Gelenkhaut, die als Synovialitis bezeichnet wird.
Die Erkrankung betrifft beim Großteil der Betroffenen die Knie-, Hüft- oder manchmal die Fußgelenke, seltener die Schulteroder Fingergelenke. Bei TGCT handelt es sich nicht um bösartige Tumoren, somit sind sie nicht lebensgefährdend. Sie können aber fundamentale Konsequenzen für die Lebensqualität Betroffener haben, denn durch die übermäßigen Wucherungen in der Gelenkhaut können Gelenke komplett
zerstört werden. Wir unterscheiden bei TGCT zwei Formen: Die erste Form ist klein und knotig und wird auch als lokalisierte Form bezeichnet. Es ist eine deutliche Abgrenzung zum umliegenden, gesunden Gewebe möglich. Diese Form ist in den meisten Fällen durch eine OP behandelbar. Schwieriger wird es bei der zweiten Form, den sogenannten diffusen TGCT. Diese verteilen sich im Gelenk, sind meist nicht gut abgegrenzt und daher auch operativ schwer behandelbar. Zudem besteht hier ein hohes Risiko, dass die Erkrankung erneut auftritt (sog. Rezidiv).
Wo liegen die Herausforderungen, wenn es um die Diagnose geht und wie kann ein TGCT zweifelsfrei festgestellt werden?
Die Erkrankung verursacht Symptome, die sehr häufig auch bei anderen Erkrankungen auftreten. Das ist die klassische Suche nach der Nadel im Heuhaufen. Erfahrene Radiologen können die Erkrankung durchaus erkennen, indem sie die typische Ausprägung auf den medizinischen Aufnahmen erkennen. Der ultimative Beweis kann durch eine Gewebeprobe erbracht werden, je nach betroffenem Gelenk mittels Gelenkspiegelung oder direkter Biopsie erreichbar. Wir gehen daher von einer hohen Dunkelziffer aus.
Wichtig ist: Die Erkrankung betrifft häufig jüngere Menschen, die in vielen Fällen keine medizinische Vorgeschichte haben. Die Patienten hatten in der Regel keine Gelenktraumata oder vorherige Verletzungen am betroffenen Gelenk. Für jüngere Menschen, die ohne medizinische Vorgeschichte solche Beschwerden haben und bereits eine Synovialitis diagnostiziert
bekommen haben, ist eine Nachfrage beim Arzt, ob es sich auch um TGCT handeln könnte, durchaus sinnvoll.
Warum ist eine möglichst frühe Diagnose so wichtig?
Es gibt unterschiedliche Verläufe von TGCT. Manche wachsen schnell, andere sehr langsam, oder auch spontan nicht mehr. Auch hier ist wieder die Unterscheidung zwischen der lokalisierten und der diffusen Form wichtig. Denn die lokale Form greift nicht auf umliegendes Gewebe wie z. B. Knorpel über.
Bei der diffusen Form kann es dazu kommen, dass der Tumor das Gelenk zerstört. Wenn das der Fall ist und der Tumor z. B. auch den Knorpel infiltriert, ist eine schnelle Diagnose absolut entscheidend. Denn Knorpel, der einmal zerstört ist, kann auch nicht wieder repariert werden. Im schlimmsten Fall muss ein Gelenk ersetzt werden, z. B. durch eine metallische Gelenkprothese.
Wie sehen die derzeitigen Behandlungsmöglichkeiten aus, um Betroffenen wieder zu mehr Lebensqualität zu verhelfen?
Als man noch glaubte, dass es sich um eine entzündliche Erkrankung handelt, wurde auch entsprechend behandelt: z. B. spritzte man eine Art Strahlentherapie in das Gelenk ein, wenn große Gelenke betroffen waren. Für TGCT ist die Wirkung dieser Art der Therapie nicht sicher belegt. Auch eine Strahlentherapie, wie sie z. B. bei bösartigen Tumoren angewandt wird, sollte nach Meinung von Experten nicht eingesetzt werden.
Text Levi Müller
Jetzt, wo wir wissen, was bei der Erkrankung passiert und welche Mechanismen ihr zugrunde liegen, können wir auch entsprechend therapieren. Die lokale Form kann operativ behandelt werden. Da bei der diffusen Form die Veränderungen an der Gelenkinnenhaut an mehreren Stellen auftreten können, sind auch die Übergänge zwischen betroffenen und nicht betroffenen Arealen schwer abgrenzbar. Bei dieser Form ist es also sehr schwer vorhersagbar, ob man bei einer OP auch alle betroffenen Stellen entfernen kann oder ob sogar schon andere Gewebearten wie der Knorpel betroffen sind. Das heißt nicht, dass bei der diffusen Form nicht operiert wird – eine Heilung kann auch hier durch eine alleinige Operation möglich sein.
In den letzten Jahren sind auch Medikamente zum Einsatz gekommen, die bei Patienten geprüft wurden, bei denen eine (radikale) OP zu starken OP-Langzeitfolgen führen würde. Mittlerweile gibt es tablettenbasierte Medikamente, die wir einsetzen können. Diese spielen besonders bei der diffusen Form und bei Rezidiven eine entscheidende Rolle.
Patienten mit
Diese adressieren zielgerichtet den Krankheitsmechanismus des Tumors, haben eine hohe Ansprechwahrscheinlichkeit und bewirken eine deutliche Symptomverbesserung. Unter dieser Therapie gibt es eine hohe Chance, dass die Tumoren sich zurückbilden. Diese Medika mente müssen dann allerdings dauerhaft eingesetzt werden.
Jetzt, wo wir wissen, was bei der Erkrankung passiert, können wir auch entsprechend therapieren.
Prof. Dr. med. Sebastian Bauer
Leiter des Sarkomzentrums am Westdeutschen Tumorzentrum der Universitätsklinik Essen und Vorstandsmitglied der Deutschen Sarkom-Stiftung
Es kann zwar Phasen der Remission geben, in denen man die Medikamente absetzen kann. Tritt ein erneutes Tumorwachstum auf, setzt man die medikamentöse Therapie fort. Es ist meine große Hoffnung für die Patientencommunity, dass wir diese Behandlungsalternativen bald auch in Deutschland
TGCT werden häufig aufgrund unspezifischer Symptome verspätet diagnostiziert1,2
Median 18-20
Monate vom ersten Arztbesuch bis zur Diagnose
Subtypen*:
N-TGCT – noduläre/lokalisierte Form
D-TGCT – diffuse Form
* Größe der farbigen Waben zeigt Häufigkeit der betroffenen Gelenke an
1. Xie GP, et al., PLoS One. 2015;10(3):e0121451.
2. De Ponti A, et al., Arthroscopy. 2003;19(6):602–7.
4. Brahmi M, et al., Curr Treat Options Oncol. 2016;17(2):10.
5. Wang C, et al., Oncol Lett. 2017;13(6):4459–62.
6. Stacchiotti S, et al., Cancer Treat Rev. 2023;112:102491.
einsetzen können. Das therapeutische Gebot lautet aktuell: So viel wie nötig, so wenig wie möglich. Das betrifft sowohl die Operationen als auch die Medikamentengabe. Man sollte sich auf jeden Fall überlegen, ob die Behandlung in einem spezialisierten Zentrum sinnvoll ist. Bei Patienten, die an der diffusen Form leiden, sollten meiner Meinung nach Experten die Behandlung übernehmen, da sie auch über etwaige medikamentöse Therapieoptionen entscheiden können und besonders auch beim Wiederauftreten der Erkrankung die nötige Erfahrung mitbringen, um die individuell richtige Behandlung für jeden Patienten in die Wege zu leiten.
Weitere Informationen zur Arbeit der Deutschen Sarkom-Stiftung finden Sie unter: . www.sarkome.de
ANZEIGE
Schneller zur Diagnose
Im MRT mit Kontrastmittel lassen sich wichtige Merkmale erkennen:3-6
Gelenkergüsse
Synoviale Ausdehnung
Zystische Erosionen
Hämosiderinablagerungen
Die WHO hat 2013 die Pigmentierte villonoduläre Synovialitis (PVNS) und den Riesenzelltumor der Sehnenscheide (GCT-TS) unter dem Übergriff Tenosynovialer Riesenzelltumor (TGCT) zusammengefasst.
„Die
Lebensqualität der PV-Patienten sollte bei jeder Therapieentscheidung im Vordergrund stehen“
Myeloproliferative Neoplasien (MPN) sind eine Gruppe von seltenen Erkrankungen des Knochenmarkes. Charakteristisch für diese Krankheitsbilder ist eine gesteigerte Produktion von Blutzellen, was sich in einer Vielzahl von Symptomen äußern kann. Diese Beschwerden können die Lebensqualität Betroffener enorm einschränken. Wir sprachen mit Frau Prof. Dr. med. Haifa Kathrin Al-Ali über die Symptome und Behandlungsmöglichkeiten der Polycythaemia Vera (PV), die zu den MPN zählt.
Frau Prof. Al-Ali, die PV ist eine seltene Erkrankung, die sich durch oft unspezifische Symptome bemerkbar macht. Wie sehen diese aus und wie kann man der Erkrankung auf die Spur kommen? Die Beschwerden der PV sind sehr unspezifisch. Das Hauptsymptom ist eine bleierne Müdigkeit (sog. Fatigue). Aber wenn wir ehrlich sind, sind wir natürlich alle mal erschöpft, allerdings nicht dauerhaft. Kommen weitere, spezifische Symptome wie z. B. ein Kribbeln in Händen und Füßen oder ein Juckreiz ohne Hautausschlag hinzu, dann sollte man hellhörig werden. Der Juckreiz ist besonders nach Kontakt mit Wasser sehr ausgeprägt und kann Betroffene schier in den Wahnsinn treiben, manche berichten gar über Suizidgedanken. Viele Patienten durchlaufen einen langen Leidensweg, bis die Krankheit korrekt diagnostiziert wird. Manche kämpfen jahrzehntelang mit den Symptomen.
Viele Patienten durchlaufen einen langen Leidensweg, bis die Krankheit korrekt diagnostiziert wird.
Prof. Dr. Haifa Kathrin Al-Ali Direktorin des Krukenberg-Krebszentrums Halle, Universitätsmedizin Halle, Fachärztin für Innere Medizin, Hämatologie, Onkologie
Was die Diagnose zusätzlich erschwert, ist, dass Betroffene aufgrund der erhöhten Anzahl der roten Blutkörperchen im Körper äußerlich gesund und geradezu rosig aussehen, sich aber sehr schlecht fühlen.
Dies hat Auswirkungen auf die psychische Verfassung, da viele von ihnen im persönlichen und beruflichen Umfeld nicht ernst genommen werden. Das Blutbild kann bereits einen ersten Hinweis geben: Erhöhte Werte von Hämoglobin und Hämatokrit sind dabei ein deutlicher Hinweis. Eine PCR-Analyse des Blutes kann zusätzlich die JAK2-Mutation nachweisen, die die Diagnose PV bekräftigt und eine Untersuchung des Knochenmarks rundet das diagnostische Vorgehen ab. Es ist aber auch möglich, dass eine MPN ohne auffällige Blutwerte vorliegt. Insbesondere bei jungen Menschen können plötzliche und ungewöhnliche Thrombosen auf eine vorhandene JAK2Mutation hinweisen, die bei nahezu allen PV-Patienten vorliegt. Es können Jahre zwischen den Thrombosen und den PV-typischen Blutbildveränderungen liegen.
i zi n H a l el
Ist die Diagnose PV gestellt, dann gibt es verschiedene Behandlungsansätze. Wie sehen diese aus? Bei jungen Betroffenen mit niedrigem Risiko für Thrombosen reicht ein Aderlass von Zeit zu Zeit und eine Behandlung mit Medikamenten zur Primärprophylaxe von Thrombosen meist aus.
Ist ein Patient über 60 Jahre alt oder hatte bereits eine oder mehrere Thrombosen, dann sprechen wir von einem Hochrisikopatienten für weitere Thrombosen. Dann kommt die sogenannte Zytoreduktion zum Einsatz, die darauf abzielt, die übermäßige Produktion von Blutzellen im Knochenmark zu reduzieren.
Eine wichtige Rolle bei der passenden Behandlung spielt also die Information, ob eine Hochrisiko-PV vorliegt. Wie sollten solche Hochrisiko-Patienten versorgt werden?
Dazu muss man sich erst noch einmal anschauen, was wir unter einem Hochrisikopatienten verstehen. Der Patient ist jung und hatte noch keine Thrombose? Somit gilt er als Niedrigrisikopatient. Der Patient hatte bereits eine oder mehrere Thrombosen oder/und ist über 60 Jahre alt? Damit wird er als Hochrisikopatient eingestuft. Diese Klassifizierung bezieht aber die weiteren Beschwerden wie z. B. den starken Juckreiz, Konzentrationsprobleme, Fatigue nicht mit ein, was hochproblematisch ist. Deswegen sollte man besonders bei den jüngeren Patienten ganz stark darauf achten, wie die Lebensqualität aussieht und eine Therapieentscheidung auch davon abhängig machen. Zudem sollten auch weitere Faktoren wie erhöhte Cholesterinwerte oder kardiovaskuläre Risiken mit einbezogen werden.
Wie sieht die derzeitige Versorgungsrealität aus: Werden Hochrisiko-Patienten heute auch entsprechend versorgt? Welche Auswirkungen kann es auf die Krankheitsentwicklung haben, wenn die Behandlung zu spät oder gar nicht dahingehend angepasst wird? Leider nein. Laut aktuellen Daten bekommen 40% der Hochrisikopatienten gar keine Zytoreduktion. Das ist schon bitter.
Text Levi Müller
Die Auswirkungen dieser bitteren Versorgungsrealität bekommen in erster Linie die Patienten zu spüren, deren Lebensqualität extrem leidet. Und bei einer lebenslangen chronischen Erkrankung wie der PV sollte die Lebensqualität der Betroffenen, neben dem Alter und dem Thromboserisiko, auch ein ausschlaggebendes Kriterium bei der Therapieentscheidung sein.
Ein informierter Patient, der sich mit anderen Betroffenen vernetzt, kann sich selbst am besten schützen und Einfluss auf seinen Krankheitsverlauf nehmen.
Welche Auswirkungen kann es wiederum auf die Lebensqualität Betroffener haben, wenn diese Faktoren berücksichtig werden und sie eine angepasste Therapie erhalten?
Sie können in der Regel ein normales Leben führen, das nicht durch die
Symptomlast bestimmt wird. Sie können zuversichtlich in die Zukunft blicken, was einen ungemeinen Zugewinn an Lebensqualität bedeutet. Außerdem gibt es Hinweise, dass Komplikationen wie Folgeerkrankungen oder Knochenvernarbungen seltener auftreten. Zudem wird das Risiko für thrombotische Ereignisse gesenkt. Diese Aspekte müssen bei behandelnden Ärzten unbedingt in den Fokus rücken.
Wie wichtig ist Ihrer Meinung nach der Austausch mit anderen Betroffenen, etwa in Selbsthilfegruppen, und wie finden Patienten Zugang zu solchen Netzwerken? Welche Rolle spielen Patientenschulungen oder Workshops bei der Bewältigung der Erkrankung? Der Austausch mit anderen Betroffenen ist von unschätzbarem Wert! Das Erste, was ein neuer Patient nach der Begrüßung von mir bekommt, ist ein Flyer des MPN-Netzwerkes. Denn ein informierter Patient, der sich mit anderen Betroffenen vernetzt, kann sich selbst am besten schützen und Einfluss auf seinen Krankheitsverlauf nehmen. Aber auch Ärzte müssen dranbleiben, sich informieren und weiterbilden.
Leben mit MPN –
Umfassende Hilfe für
Betroffene
Das forschende Pharmaunternehmen Novartis denkt Medizin neu, um besonders auch Menschen mit seltenen Erkrankungen mit innovativen Wirkansätzen und Informationsangeboten mehr Lebensqualität zu ermöglichen.
Speziell für Menschen, die an einer Myeloproliferativen Neoplasie (MPN) wie der Myelofibrose, der Polycythaemia Vera oder der Chronischen Myeloischen Leukämie leiden, hat Novartis eine umfangreiche Informationsinitiative ins Leben gerufen, die wissenschaftlich fundiertes Wissen zur Erkrankung und zum Umgang damit zur Verfügung stellt.
Über die Website www.leben-mit-blutkrankheiten.de können sich Betroffene über alle Facetten der verschiedenen Erkrankungen informieren. Hier finden sich auch Patienten-Erfahrungsberichte und Expertenbeiträge zu verschiedenen krankheitsrelevanten Schwerpunkten. Zudem finden Patient:innen ausführliche Checklisten, die ihnen die Gespräche mit dem Behandlungsteam erleichtern können. Dazu kann auch eine Anpassung der bestehenden Therapie gehören, wenn die bisherige Behandlung nicht den gewünschten Erfolg erzielt oder Nebenwirkungen auftreten, welche die Lebensqualität stark beeinträchtigen.
Dabei kann auch der MPN-Tracker unter de.mpn.your-symptom-questionnaire.com helfen, der Patient:innen in Form eines Therapietagebuches bei der Dokumentation zur Entwicklung ihrer Erkrankung unterstützt.
Die Möglichkeiten dafür sind in Form von Patientenveranstaltungen und Weiterbildungen da, sie müssen aber genutzt werden.
PCR-Analyse
Die PCR-Analyse (Polymerase-Kettenreaktion) ist eine molekulargenetische Methode, die es ermöglicht, spezifische DNA-Sequenzen, wie die JAK2-Mutation, die bei fast allen PV-Patienten vorhanden ist, in einer Blutprobe nachzuweisen.
Thrombose
Unter einer Thrombose versteht man ein Blutgerinnsel, auch Thrombus genannt, welches ein Blutgefäß verengt oder ganz verschließt. Die erhöhte Blutdicke und die veränderte Funktion der Blutplättchen bei PV erhöhen das Risiko für die Bildung von solchen Blutgerinnseln.
Weitere Informationen finden Sie unter: . www.mpn-netzwerk.de
ANZEIGE
Zusammen stärker
Auch der Austausch mit anderen Betroffenen, Selbsthilfeorganisationen und Fachärzt:innen stärkt Patient:innen und ihre Angehörigen im Umgang mit der Erkrankung. Seit 2016 können MPN-Betroffene einen bundesweit etablierten Treffpunkt nutzen: die MPN-Patient:innentage. Die Teilnahme an den MPN Veranstaltungen ist kostenlos. Auf www.leben-mit-blutkrankheiten.de/mpn-patiententage findet man die Anmeldung für den nächsten Patient:innentag sowie weitere Informationen und einen kleinen Rückblick auf vergangene Veranstaltungen. Zudem können Betroffene und ihre Angehörigen sich für die Initiative „Erfahrungsschatz“ anmelden, um andere Patient:innen, Angehörige und Interessierte an wichtigen Themen rund um ihre Erkrankung teilhaben zu lassen. Erzählen Sie Ihre Geschichte!
Scannen Sie den QR-Code und lesen Sie mehr zu uns auf unserer Webseite unter www.leben-mit-blutkrankheiten.de
GENTHERAPIE BEI HÄMOPHILIE
Innovative Therapien müssen zu den Patienten gelangen
Hämophilie ist eine seltene, erbliche Blutgerinnungsstörung. Ursache ist ein Mangel an Gerinnungsfaktor VIII (Hämophilie A) oder IX (Hämophilie B). Ohne diese Proteine gerinnt das Blut nicht richtig, was zu gefährlichen Blutungen in Gelenken, Muskeln oder Organen führen kann. Jahrzehntelang bestand die Standardtherapie darin, die fehlenden Faktoren regelmäßig zuzuführen. Dank dieser Prophylaxe können Patienten heute relativ normal leben. Doch die Belastung bleibt hoch. Gentherapien eröffnen nun eine deutlich höhere Lebensqualität.
Wir sprachen mit Christian Schepperle, Geschäftsführer der Interessengemeinschaft Hämophiler (IGH) e. V., warum die Gentherapie für die Hämophilie eine wichtige Therapieoption ist, für welche Patienten sie in Frage kommt, aber auch über die Hürden beim Zugang zu dieser wichtigen Behandlungsoption.
Text Hanna Sinnecker
Herr Schepperle, was unterscheidet die Gentherapie von der klassischen Behandlung?
Die klassische Faktortherapie erfordert regelmäßige Infusionen, oft zweibis dreimal pro Woche. Das ist aufwendig und schränkt den Alltag ein.
Wie funktioniert die Gentherapie stattdessen?
Sie wird einmalig verabreicht. Über sogenannte AAV-Viren gelangt der Bauplan für den fehlenden Gerinnungsfaktor in die Leberzellen. Diese können dann selbst den Faktor herstellen.
Was bedeutet das für Betroffene?
Sie sind besser vor Blutungen geschützt, auch vor kaum spürbaren Mikroblutungen. Gleichzeitig entfällt die ständige Faktorgabe. Reisen, Arbeit und Freizeit lassen sich viel spontaner gestalten.
Für wen ist die Therapie geeignet?
Derzeit für Erwachsene ab 18 Jahren mit schwerer Hämophilie und stabiler Leberfunktion. Sie dürfen keine Antikörper gegen Faktoren oder die eingesetzten Viren haben.
Wie groß ist das Interesse?
Sehr groß. Viele informieren sich bei Veranstaltungen, in Fachzentren oder über Medienberichte.
Warum wurden bisher so wenige Patienten behandelt?
Obwohl seit über zwei Jahren zugelassen, sind es in Deutschland weniger als zehn. Grund sind Verzögerungen bei Zulassungsschritten, Preisverhandlungen und organisatorische Unsicherheiten.
Gab es auch Probleme mit den Krankenkassen?
Ja. Manche Kassen erklärten die Therapie für unwirtschaftlich und verzögerten Anträge. Das Bundesamt für Soziale Sicherung (BAS) hat in ihrem gerade veröffentlichten Jahresbericht klargestellt, dass solche Ablehnungen nicht zulässig sind. Demnach ist unser Verständnis, dass der Arzt diese Gentherapien einfach verordnen kann, und die Krankenkassen nicht vorher informieren oder um Genehmigung bitten muss.
Und trotzdem kommt es immer noch zu Verzögerungen bei der TherapieGenehmigung?
Ja. Einige Krankenkassen versuchen nach Antragsstellung durch den Arzt ein langwieriges Prüfverfahren anzustoßen und auf Zeit zu spielen.
Wir konnten über die letzten zwei Jahre feststellen, dass besonders die Allgemeinen Ortskrankenkassen und die Techniker Krankenkasse sehr zurückhaltend sind, wenn es um die Genehmigung der Anträge geht.
Wie haben Ärzte darauf reagiert?
Einige zögerten, Patienten zu informieren oder überhaupt Anträge zu stellen. Nach der BAS-Entscheidung sind sie aber abgesichert. Dennoch fehlt vielerorts die aktive Aufklärung.
Welche Rolle spielen die Kosten?
Auf den ersten Blick sind sie hoch. Doch eine Faktortherapie verursacht jährlich 150.000 bis 250.000 Euro.
Über ein Leben summiert sich das auf Millionen. Gentherapie kann langfristig sogar günstiger sein – und Folgeschäden vermeiden.
Welche Folgen haben Verzögerungen für Patienten?
Vor der Therapie muss eine AAV-Testung erfolgen. Liegt zu viel Zeit dazwischen, können Antikörper entstehen. Dann ist eine Behandlung nicht mehr möglich. Wir kennen bereits tragische Fälle.
Welche Belastungen bestehen bei der bisherigen Therapie?
Regelmäßige Infusionen sind ein erheblicher Eingriff in den Alltag – sowohl für Erwachsene als auch für Eltern betroffener Kinder.
Welche nächsten Schritte sind nötig?
Die Gentherapie ist zugelassen und ihr Zusatznutzen ist erwiesen. Ärzte müssen Patienten jetzt aktiv darüber informieren. Patienten brauchen neutrale Beratung. Außerdem müssen Strukturen für Durchführung und Nachsorge geschaffen werden.
Welche Rolle spielt der Erfahrungsaustausch?
Eine große. Wir haben eine Fokusgruppe gegründet, in der bereits behandelte Patienten ihre Erfahrungen weitergeben. So können Interessierte realistisch abwägen – ohne Druck, sondern durch echte Information.
Entscheidend ist, dass Patienten Zugang erhalten – ohne Verzögerungen, mit klarer Unterstützung durch Ärzte und Krankenkassen.
Christian Schepperle Geschäftsführer der IGH e. V.
Die einmalige Gentherapie bedeutet hier einen enormen Zugewinn an persönlicher Freiheit.
Was raten Sie Patienten, die auf Hindernisse stoßen?
Sie sollten Anträge konsequent stellen lassen und sich nicht entmutigen lassen. Bei Ablehnung ist ein Eilantrag beim Sozialgericht möglich. Wir unterstützen dabei.
Wie wichtig ist die Lebensqualität in der Bewertung neuer Therapien?
Sehr wichtig. Doch in den alten Bewertungsbögen des G-BA wird sie kaum berücksichtigt. Wir fordern, dass dieser Faktor stärker in Entscheidungen einfließt.
Wie lautet Ihr aktuelles Fazit?
Die Gentherapie ist ein Meilenstein in der Behandlung von Hämophilie B. Sie bietet medizinischen Fortschritt und neue Lebensqualität. Entscheidend ist, dass Patienten Zugang erhalten –ohne Verzögerungen, mit klarer Unterstützung durch Ärzte und Krankenkassen.
Die Interessengemeinschaft Hämophiler e. V. ist ein bundesweit tätiger Patientenverband, der die Interessen der an einer angeborenen Blutungskrankheit leidenden Menschen und ihrer Angehörigen vertritt.
Informieren Sie sich weiter auf der Webseite der Interessengemeinschaft Hämophiler e. V. unter: www.igh.info
LEBEN MIT XLH:
„Eine kontinuierliche Behandlung ermöglicht ein enormes Plus an Lebensqualität und Teilhabe“
Die x-chromosomale Hypophosphatämie (kurz XLH) ist eine seltene Störung des Knochenstoffwechsels, die durch eine Mutation vererbt wird, welche auf dem X-Chromosom liegt und zu einem Phosphatmangel führt. Wir sprachen mit dem Experten Dr. Felix Reschke über die Herausforderungen für Betroffene, besonders beim Übergang ins Erwachsenenalter.
Text Hanna Sinnecker
Herr Dr. Reschke, was passiert bei XLH im Körper Betroffener und wie äußert sich die Erkrankung?
Die X-chromosomale Hypophosphatämie (kurz XLH) ist eine genetische Veränderung, die dazu führt, dass der Körper über den Urin zu viel Phosphat ausscheidet. Phosphat ist in Kombination mit Calcium entscheidend für den Aufbau und die Stabilität unserer Knochen und Zähne. Da dieser Kreislauf bei XLH-Betroffenen nicht richtig funktioniert, entsteht ein chronischer Phosphatmangel, was den Knochen- und Zahnaufbau stört. Bei Kindern, die sich im Wachstum befinden, äußert sich das häufig durch Beinfehlstellungen bzw. Verformungen der unteren Extremitäten (X- und/oder O-Beine), damit einhergehenden Wachstumsverzögerungen und Schmerzen in den Knochen und Gelenken.
In vielen Fällen haben betroffene Kinder Zahnprobleme, da der Zahnschmelz durch das fehlende Phosphat nicht richtig ausgebildet wird. Im Jugendlichen- und Erwachsenenalter gehen die Wachstumsverzögerungen in einen manifesten Kleinwuchs über, es kann zu Gelenksteifigkeit und frühen Arthrosen kommen. Zudem sind sie besonders anfällig für Knochenbrüche auch bei nur kleinen Unfällen.
Was sind die Herausforderungen bei der Diagnose und warum sollte sie möglichst früh erfolgen?
Durch die Seltenheit der Erkrankung kommen Ärzte nicht häufig mit ihr in Kontakt, statistisch gesehen sieht ein niedergelassener Kinderarzt in seiner Berufslaufbahn wahrscheinlich einen XLH-Patienten. Zudem können die Symptome auch andere Ursachen haben oder unterschiedlich ausgeprägt sein, weshalb es noch häufig zu Fehldiagnosen kommt. Viele Patienten erleben eine Odyssee, bis es
zur richtigen Diagnose kommt. Wir können die Erkrankung zwar nicht heilen, aber mittlerweile gut behandeln. Wir haben seit einigen Jahren eine kausale Therapie zur Verfügung, die an der Ursache der Krankheitsentstehung ansetzt und den Phosphatstoffwechsel wieder ins Gleichgewicht bringen kann. Der Behandlungserfolg ist aber absolut abhängig vom Diagnosezeitpunkt, da es sich um eine fortschreitende Stoffwechselerkrankung handelt.
Wir brauchen strukturierte Programme für den Übergang in die Erwachsenenmedizin.
Dr. Felix Reschke
Oberarzt am Kinder- und Jugendkrankenhaus „Auf der Bult“ in Hannover
Je früher die Erkrankung erkannt und behandelt wird, umso besser können wir in das Krankheitsgeschehen eingreifen und Folgeschäden verhindern. Wenn wir die Diagnose also bereits im frühen Kindesalter stellen, können wir z. B. Schmerzen und Fehlstellungen sehr gut vorbeugen, da noch ein großes Wachstumspotenzial besteht. Sind die Knochen einmal ausgewachsen und verkrümmt, dann wird es schwierig, das buchstäblich wieder therapeutisch „geradezubiegen“. Manche Schäden können dann nicht mehr rückgängig gemacht werden.
Betroffene Kinder werden nach der Diagnose im Regelfall gut betreut und überwacht. Wie sieht die Versorgungsrealität aus, wenn Betroffene heranwachsen?
Es ist wichtig zu verstehen, dass eine
Therapie ein Leben lang fortgesetzt werden muss, da es sich um eine chronische Erkrankung handelt. Die Wachstumsphase ist eine ganz entscheidende Phase, in der Eltern ihre Kinder in vielen Fällen gewissenhaft durch die Therapie begleiten und dafür sorgen, dass Therapieund Kontrolltermine wahrgenommen werden. Die Versorgung erfolgt in Deutschland in fast allen Fällen an Zentren, die sich auf Knochenstoffwechselerkrankungen im Kindesalter spezialisiert haben. Meist sind dabei maßgeblich Nephrologen oder Endokrinologen beteiligt, die dann Kollegen aus weiteren Fachbereichen hinzuziehen.
Fot o : P r i v a t
Wir bieten also eine auf den jungen Patienten bezogene Betreuung an, die einen ganzheitlichen Ansatz verfolgt und einen stark umsorgenden Charakter hat. Dieser enge Rahmen verändert sich beim Übergang in die Erwachsenenmedizin. Erwachsene finden seltener spezialisierte Anlaufstellen, zudem steigt die Eigenverantwortung. Klar, mit 18 hat man vielleicht auch einfach andere Dinge im Kopf, manche verlieren in dieser Zeit den Kontakt zu Spezialisten. Das führt dann aber dazu, dass wieder vermehrt Schmerzen und Bewegungseinschränkungen auftreten können, die vielleicht zu spät mit der XLH in Verbindung gebracht werden.
Diese Abbrüche wollen wir verhindern, weshalb wir in der Begleitung unserer Patienten ab einem gewissen Alter einen Fokus auf die Wissensvermittlung rund um ihre Erkrankung und das Therapiemanagement setzen und davor warnen, solche Lücken entstehen zu lassen.
Wir versuchen also, die Patienten schon vorausschauend aus der Fürsorge der Eltern herauszunehmen und die Eigenverantwortung zu schulen. Idealerweise finden wir dann gemeinsam mit dem Patienten ein nachsorgendes Zentrum und leiten einen „Übergabeprozess“ in die Wege, damit sie auch als Erwachsene gut versorgt bleiben. Man muss aber sagen, dass es kein strukturiertes Programm gibt, das den Übergang für XLH-Patienten in die Erwachsenenmedizin regelt. Wir bräuchten das aber dringend, um Versorgungslücken gar nicht erst entstehen zu lassen.
Warum ist es so wichtig, dass Betroffene auch im Erwachsenenalter weiter gut versorgt werden, und wie kann das gelingen?
Das ist eine spannende Frage, hier hole ich mal etwas weiter aus. Es passiert nämlich nicht selten, dass wir von XLH betroffene Kinder behandeln und in diesem Zuge auch die Eltern auf XLH (oder eine Trägerschaft) positiv getestet werden, da es sich ja um eine genetische Erkrankung handelt. Als pädiatrisches Zentrum können wir hier keine Behandlung anbieten, aber versuchen
dann an Experten zu vermitteln, die sich um die Versorgung der erwachsenen Patienten kümmern können. Hier wäre es wünschenswert, eine Art Register oder Landkarte zu haben, um diese Patienten dann einem Wohnort-nahen Experten zuführen zu können. Solche Register können auch helfen, die Versorgung von Patienten im Erwachsenenalter zu sichern, die bereits als Kinder diagnostiziert wurden.
Es ist unglaublich toll, was mir jugendliche Patienten berichten, die mit der kausalen Therapie behandelt werden. Ich hatte z. B. einen jungen Patienten, der mir freudestrahlend erzählte, dass er eines morgens zum Bus rennen musste und es noch geschafft hat, ihn zu erreichen. Er sagte, dass das ohne die Therapie nicht möglich gewesen wäre, weil er sonst starke Schmerzen gehabt hätte und sich gar nicht so schnell hätte bewegen können. Ein anderer Patient erzählte mir mit leuchtenden Augen, dass er ohne Angst bei der Eisdisco zum Schlittschuhlaufen war.
Die Therapie ermöglicht also ein enormes Plus an Lebensqualität und Teilhabe!
EINSATZ FÜR MENSCHEN MIT SELTENEN ERKRANKUNGEN
„Meine Hautprobleme begannen 2004“, berichtet eine Patientin. Sie litt zu dieser Zeit unter wiederkehrenden Hautausschlägen und Schmerzen. Erst Jahre später wurde bei ihr eine Mycosis fungoides diagnostiziert, eine Krebserkrankung, die in Europa weniger als einen von 110.000 Menschen betrifft.1
„Ich befand mich fast zehn Jahre lang in einer Grauzone“, erinnert sie sich. Ihre anfänglichen Symptome wurden zunächst als Ekzem erkannt. Erst ein Zufallsbefund führte zur richtigen Diagnose. Diese Geschichte ist kein Einzelfall: Der Weg bis zum Befund dauert bei diesem Krankheitsbild durchschnittlich zwei bis sieben Jahre.2
Kyowa Kirin ist ein global tätiges biopharmazeutisches Unternehmen, das die Versorgung von Menschen mit seltenen Erkrankungen verbessern möchte. Es wurde 1949 in Japan gegründet und entwickelt seit dieser Zeit
innovative Therapien in den Bereichen Nephrologie, Neurologie, Onkologie und Immunologie. Die Forschung, Entwicklung und Wirkstoffproduktion stützen sich auf Verfahren der Spitzenbiotechnologie aus eigenem Hause. So gilt das Unternehmen als Pionier in der Behandlung des nur selten auftretenden Phosphatdiabetes.
Kyowa Kirin ist ein global tätiges Unternehmen, das die Versorgung von Menschen mit seltenen Erkrankungen verbessern möchte.
Ein weiterer Schwerpunkt ist die Behandlung seltener Krebserkrankungen wie der Mycosis fungoides und des Sézary-Syndroms – beides Unterformen des kutanen T-Zell-Lymphoms (CTCL).
Die Patienten müssen daher verstehen, dass ihre Therapietreue ihnen genau das ermöglicht, aber umgekehrt ein Abbruch der Versorgung zwangsläufig negative Folgen haben wird. Zu diesen negativen Folgen im Erwachsenenalter zählen ständige chronische Knochenschmerzen, frühzeitige Gelenkveränderungen (z. B. Arthrose), Gelenksteifigkeit und Bewegungseinschränkungen.
Zudem können schmerzhafte Verkalkungen der Sehnenansätze entstehen. Des Weiteren besteht ein erhöhtes Frakturrisiko und es können Zahnprobleme auftreten (Abszesse, frühzeitiger Zahnverlust). Diese belastenden Symptome können zudem zu einer ständigen Müdigkeit und Erschöpfung führen. Aus diesem Grund ist es extrem wichtig, strukturierte Programme für den Übergang in die Erwachsenenmedizin zu schaffen, damit kein Patient durchs Raster fällt. Denn im Erwachsenenalter ergeben sich andere Behandlungsziele, aber ein gutes Management und eine gezielte Behandlung der Erkrankung können auch hier in vielen Fällen positive Auswirkungen auf die körperliche Situation und Lebensqualität haben.
Kyowa Kirin möchte sämtlichen Menschen, mit denen es sich im Austausch befindet, ein Lächeln schenken – nicht nur durch die Bereitstellung neuer Wirkstoffe, sondern auch durch gelebte Partnerschaften. Das Unternehmen sucht weltweit den Austausch mit Betroffenen und Beteiligten, um gemeinsam bessere Antworten auf Patientenbedürfnisse zu finden, getrieben von dem Ansporn „Making people smile“.
1 – Orphanet https://tinyurl.com/4vr9ar9v 2 – CL Foundation https://tinyurl.com/mvk67utw
Weitere Informationen finden Sie unter: www.kyowakirininternational.com
Text Katharina Lassmann
Dieser Artikel ist in Zusammenarbeit mit der Kyowa Kirin GmbH entstanden.
„Ich hätte viel früher die Reißleine ziehen sollen“
Nicht-dystrophe Myotonien (kurz NDM) sind seltene, genetisch bedingte neuromuskuläre Erkrankungen. Das charakteristische Merkmal: Betroffene sind aufgrund der Krankheit nicht fähig, die der körperlichen Bewegung dienenden Muskeln (Skelettmuskulatur) nach der Anspannung sofort wieder zu entspannen. Susanne ist eine von ihnen und gab uns im Gespräch Einblicke in ihren Alltag mit der Erkrankung.
Text Miriam Hähnel
Susanne, Sie leben mit der seltenen Erkrankung Myotonia congenita Thomsen, die zu den NDM zählt. Wann haben Sie bemerkt, dass etwas nicht stimmt?
Rückblickend hatte ich schon in der Kindheit Symptome. Sport war für mich immer der Horror, weil ich mich nicht so bewegen konnte wie die anderen, ich wurde aber einfach als unsportlich abgetan. Mit Anfang 20 fing ich an, in der Firma meines damaligen Mannes zu arbeiten. Da das eine körperlich harte Tätigkeit war, schob man von da an meine Beschwerden auf den Arbeitsstress. Ich bekam ein Reizdarmsyndrom, das mich sehr belastete, zudem kamen immer stärkere Schmerzen vor allem im Bereich des Nackens, Rückens und Kiefers dazu. Mir war häufig schwindelig, ich bekam einen Tinnitus. Heute weiß ich, dass all das von der ständigen Verkrampfung meiner Muskeln durch die NDM kam, aber damals wurde das als orthopädisches Problem gesehen. Jede Treppe war ein Problem für mich, ich kam die Stufen einfach nicht hoch und runter, weil meine Muskeln blockierten.
Die Orthopäden sagten, ich müsse mehr Sport treiben und an meiner Kondition arbeiten. Über Jahrzehnte wurde ich nicht ernst genommen und lief von Arzt zu Arzt, lebte mit Dauerschmerzen. Mit Mitte 30 kam die Fatigue hinzu. Mit Mitte 40 bekam ich Orthesen, um die Sprunggelenke zu schonen, die schon seit 20 Jahren chronisch entzündet waren: das schränkte mich in der Beweglichkeit
noch weiter ein. Es wurden verschiedene Schmerzmedikamente ausprobiert, nichts half. Irgendwann wurde ich in die psychosomatische Schublade gesteckt. Da wir selbstständig waren, konnte ich aber nie pausieren, ich powerte weiter durch.
Wie sah Ihr Weg bis zur richtigen Diagnose aus und gibt es in Ihrer Familie weitere Betroffene? NDM sind ja erblich bedingt.
Erst nachdem es eine NDM-Diagnose in meiner Familie gab, wurde ich mit einem Gentest daraufhin untersucht. Mitte 2022 hatte ich das Ergebnis: Ich habe Myotonia congenita Thomsen. Da war ich 54 Jahre alt. Alle anderen Ärzte, bei denen ich bis dahin war, haben die Symptome nicht in Verbindung gebracht und zusammenhängend betrachtet.
Mit 34 bekam ich meinen Sohn: Ich hatte also die Doppelbelastung Arbeit und Haushalt, war dabei ständig erschöpft und hatte chronische Schmerzen. Das machte auch mental etwas mit mir.
Ich bin abends oft beim Geschichte vorlesen eingeschlafen, weil ich so müde war. Viele Dinge konnte ich nicht mit ihm unternehmen, und es tut mir unglaublich leid, wieviel dabei auf der Strecke geblieben ist. Das war für mich das Schlimmste.
Jede Treppe war ein Problem für mich, ich kam die Stufen einfach nicht hoch und runter, weil meine Muskeln blockierten.
Was waren für Sie die größten Herausforderungen im Alltag und war es für Sie wichtig, sich selbst mit der Erkrankung auseinanderzusetzen?
Die größte Herausforderung für mich war, meiner Familie gerecht zu werden.
Ich habe aber zum Glück ein tolles Verhältnis zu meinem Sohn. Mein damaliger Mann zeigte für meine Beschwerden gar kein Verständnis. Ich fühlte mich immer wie die, die alle aufhält. Das wurde mir im Urlaub z. B. beim Wandern oder Fahrradfahren immer bewusster: Ich brauchte häufig Pausen, konnte beim Fahrradfahren nicht mehr ordentlich absteigen und verlor die Freude an solchen Aktivitäten. Das war eine schwere Zeit für mich, ich hatte ständig ein schlechtes Gewissen, wurde hochgradig depressiv. Mit dem Wissen um die Erkrankung kann ich nun gnädiger mit mir sein.
Was wäre in Ihrem Leben anders verlaufen, wenn Sie früher diagnostiziert und therapiert worden wären? Ich hätte viel früher die Reißleine ziehen sollen. Ich bereue sehr, dass ich mich stets der Arbeit untergeordnet habe. Denn wenn die Krankheit früher erkannt worden wäre, hätte man früher mit der Behandlung starten und gewisse Schäden verhindern können: Körperlich und mental. Ich musste also lernen, Grenzen zu setzen und auf meinen Körper zu hören.
Seit meiner Diagnose muss ich mir die Physiotherapie nicht mehr erbetteln, das ist schon mal ein Fortschritt! Ich bin beim Rehasport, und zwar im Wasser: Das funktioniert für mich am besten. Anfang 2023 war ich das erste Mal in einer Spezialklinik für neuromuskuläre Erkrankungen und habe viel über meine Erkrankung gelernt. Dort habe ich auch verstehen müssen, dass ich nicht mehr die „Alte“ werde, da ich jahrelang über meine Grenzen gegangen bin. Die einmal verlorene Muskelkraft bekomme ich leider nicht mehr zurück. Leider ist ein geregelter Alltag so nicht mehr möglich, da meine Kräfte manchmal schon am Mittag ausgeschöpft sind.
Jeder Tag ist eine neue Herausforderung: Termine und Aktivitäten müssen gut geplant werden, da die Kraft oft nicht reicht. Deswegen bin ich auf Gehhilfen und einen Rollator angewiesen. Aber ich sehe nach vorn und versuche, der Verbitterung keinen Raum zu geben.
Ich musste lernen, Grenzen zu setzen und auf meinen Körper zu hören.
Wie sieht Ihr Leben nun nach der richtigen Diagnosestellung und unter Therapie aus?
Ich taste mich langsam wieder an bestimmte Sachen heran: Ich versuche, mich mehr zu bewegen und gebe dem Fahrradfahren wieder eine Chance. Ich habe einen neuen Partner, der sehr verständnisvoll ist und sich großartig um mich kümmert: Eine tolle Erfahrung! Und eine Sache ist eine ganz große Erleichterung für mich:
Seit ich medikamentös gut eingestellt bin, ist mein Reizdarmsyndrom weg! Vom ersten Tag an! Auch muskulär bemerke ich eine deutliche Verbesserung, ich kann mich morgens z. B. einfach strecken, kann leichter aufstehen. Dafür bin ich sehr dankbar.
Welche Rolle spielt für Sie der Kontakt mit anderen Betroffenen, z. B. über die Patientenorganisation „Mensch und Myotonie e. V.“?
Ich wurde nach der Diagnose im Klinikum darauf hingewiesen, dass es Patientenvereinigungen gibt. Ich habe mich dann direkt bei „Mensch und Myotonie“ gemeldet. Das war eine riesige Hilfe für mich, um die Krankheit zu verstehen und zu sehen, dass ich nicht allein bin. Ich kann jedem nur empfehlen, das in Anspruch zu nehmen!
Weitere Informationen zur Patientenorganisation Mensch und Myotonie finden Sie unter: www.menschundmyotonie.de
ANZEIGE
WENN DER SCHMERZ DAS LEBEN DIKTIERT:
und potenzielle Trigger zu meiden, die bei jedem Patienten unterschiedlich aussehen können. Dennoch bleibt die Erkrankung unberechenbar.
Clusterkopfschmerz zählt zu den eher seltenen, aber extrem schmerzhaften primären Kopfschmerzerkrankungen. Die Attacken treten in bestimmten Zeiträumen (engl. „Cluster“) auf, die sich über Wochen oder Monate erstrecken können und mit mehreren Anfällen täglich verbunden sein können. Meist folgen darauf längere schmerzfreie Phasen, aber bei etwa 10 bis 15% der Betroffenen nimmt die Erkrankung einen chronischen Verlauf ohne nennenswerte Pausen zwischen den Attacken. Die Schmerzen sind so intensiv, dass der Clusterkopfschmerz auch als "Selbstmord-Kopfschmerz" bezeichnet wird.
Leben mit starken Schmerzattacken
Tokio-Hotel-Gitarrist Tom Kaulitz (35) lebt seit Jahren mit Clusterkopfschmerz und spricht offen über die quälenden Kopfschmerzattacken. Er berichtet, dass er schlimme Clusterkopfschmerz-Perioden hat und dann nur unter Einsatz seiner Medikamente funktionieren kann. Die Attacken kommen heftig und plötzlich, und kündigen sich durch ganz bestimmte Symptome an. Sein Bruder Bill weiß inzwischen, wie das aussieht: „Deine Augen hängen dann herunter, oder deine eine Gesichtshälfte. Du kannst dann gar nicht mehr richtig gucken und muss dich sofort zurückziehen.“ Tom Kaulitz erlebt somit ganz typische
ClusterkopfschmerzEpisoden, die sich durch einen intensiven einseitigen Augen- und Schläfenschmerz, begleitet von Augentränen und Gesichtshautveränderungen äußern. Die Schmerzattacken sind nicht nur körperlich, sondern auch mental sehr belastend – so stark, dass Tom bereits in die Notaufnahme musste. Für einen Musiker wie Tom sind solche Attacken verheerend und die Angst vor dem nächsten Schmerzschub wird zum ständigen Begleiter – auch für die Menschen, die ihm nahestehen. Um trotzdem auftreten zu können, ist er auf Medikamente angewiesen.
Therapie und Bewältigungsstrategien Klassische Schmerzmittel zeigen beim Clusterkopfschmerz keine Wirkung – stattdessen können Sauerstoff und sogenannte Triptane helfen, die auch als Injektion verabreicht werden können und so schneller ihre Wirkung entfalten. Tom berichtet, dass einzig diese Maßnahmen ihm Linderung verschaffen.
Zusätzlich versucht er, Strategien zur Entspannung in seinen Alltag zu integrieren
Offene Kommunikation schafft
Awareness
Indem Tom Kaulitz seine Erkrankung öffentlich macht, schafft er Bewusstsein für eine wenig bekannte, aber äußerst belastende Krankheit. Er gibt Einblick in seine schmerzgeplagte Welt – und betont gleichzeitig: Clusterkopfschmerz ist mehr als nur Kopfschmerz. Es ist eine Erkrankung, die das Leben massiv beeinträchtigen kann: Und dabei ist es der Erkrankung egal, ob man Rockstar ist oder einen ganz normalen Alltag bestreitet. Eine frühe Diagnose und eine gezielte Therapie sind entscheidend, um weiteren Schaden –körperlich und psychisch – zu verhindern.
Clusterkopfschmerz: So sehen die Symptome aus
• einseitige, extremste Schmerzattacken im Bereich von Schläfe oder Auge
• Unruhe und Bewegungsdrang während der Attacke
• Häufig zusätzlich auf der Schmerzseite: tränendes Auge, hängendes und/oder geschwollenes Augenlid, verkleinerte Pupille, laufende Nase, schwitzige und gerötete Haut (mind. eines dieser Begleitsymptome kann auf Clusterkopfschmerz hindeuten)
• Attackendauer: zwischen 15 Minuten und drei Stunden
Text Hanna Sinnecker
kcotsrettuhs
Orphan Drugs –es bleibt viel zu tun
Derzeit sind etwa 8.000 Seltene Erkrankungen bekannt, doch es gibt nur ca. 200 Arzneimittel zur Behandlung. Der therapeutische Bedarf an Arzneimitteln gegen Seltene Erkrankungen (Orphan Drugs) ist also groß. Oftmals bieten sie erstmalig eine Therapieoption und tragen erheblich dazu bei, die Lebensqualität von Patientinnen und Patienten zu verbessern.
Neue Therapien stehen hierzulande so schnell und umfassend zur Verfügung wie in keinem anderen europäischen Land.
Dr. Matthias Wilken BPI-Geschäftsführer Market Access, Märkte und Versorgung
Das Besondere an der Forschung zu Seltenen Erkrankungen ist, dass auch Menschen mit anderen Erkrankungen von den Ergebnissen profitieren können. Denn das gewonnene Wissen über das Krankheitsgeschehen einer Seltenen Erkrankung lässt sich oft auch auf andere Krankheitsbilder übertragen. Umso mehr Faktoren eines Krankheitsverlaufs bekannt sind, desto eher können pharmazeutische Unternehmen spezifische Therapien entwickeln.
Die Entwicklung eines neuen Orphan Drugs ist jedoch sehr herausfordernd. Sie kann bis zu 15 Jahre dauern und mehrere hundert Millionen bis Milliarden Euro kosten. Bereits die Vorbereitung und Durch-
führung von Studien (Datengenerierung) ist aufwendig: Will ein Unternehmen Therapieansätze für eine Seltene Erkrankung erforschen, sucht es nach der Stecknadel im Heuhaufen. Zudem sind die Patientenpopulationen klein, heterogen und geografisch oft weit verteilt.
Die Rekrutierung von Probanden ist daher sehr schwierig und kostenintensiv, was wiederum mit einem hohen Investitionsrisiko für pharmazeutische Unternehmen einhergeht. Da Hersteller diese Kosten refinanzieren müssen und der Absatzmarkt durch die kleinen Patientenpopulationen stark begrenzt ist, sind höhere Preise die Folge.
Trotz dieser Herausforderungen können Patientinnen und Patienten in Deutschland positiv gestimmt sein: Neue Therapien stehen hierzulande so schnell und umfassend zur Verfügung wie in keinem anderen europäischen Land.
Es ist jedoch wichtig, dass der Arzneimittelmarkt preispolitisch so aufgestellt bleibt, dass Unternehmen in der Lage sind, auch zukünftig Therapien für nur wenige Patientinnen und Patienten zu entwickeln. Stabile politische Rahmenbedingungen sind dafür entscheidend.
Leider weisen aktuelle Entwicklungen in eine andere Richtung: Auf europäischer Ebene werden Einschnitte im Anreizsystem zur Entwicklung von Orphan Drugs diskutiert. Und auch auf nationaler Ebene wird der sozialrechtliche Sonderstatus immer wieder in Frage gestellt.
Die aktuelle Bundesregierung sieht hier Handlungsbedarf: Im Koalitionsvertrag kündigt sie an, die gesundheitliche Situation von Betroffenen Seltener Erkrankungen, zum Beispiel durch Ausbau und Stärkung von digital vernetzten Zentren, zu verbessern.
Als Bundesverband der Pharmazeutischen Industrie (BPI) begrüßen wir dieses Signal und fordern weiterhin verbindliche Rahmenbedingungen. Gerade vor dem Hintergrund der angespannten Finanzlage der Gesetzlichen Krankenversicherung dürfen Patientinnen und Patienten mit Seltenen Erkrankungen nicht ins Hintertreffen geraten.
Um diesen Prozess ganzheitlich zu gestalten, engagieren wir uns im Nationalen Aktionsbündnis für Menschen mit Seltenen Erkrankungen (NAMSE). Gemeinsam mit Bundesministerien und knapp 30 Bündnispartnern setzen wir uns dafür ein, dass Patientinnen und Patienten mit einer Seltenen Erkrankung eine zeitnahe Diagnose und Therapie erhalten.
Über den QR-Code direkt zum Video “Seltene Leiden –Vorurteile und Fakten“
Bei seltenen Erkrankungen handelt es sich oft um genetisch bedingte Erkrankungen, oft sind Kinder betroffen.
Für viele schwerkranke Patientinnen und Patienten, bei denen andere Therapien oft ausgeschöpft sind, können mitunter Arzneimittel für neuartige Therapien (ATMP) zum Einsatz kommen.
Weitere Informationen zum Thema finden Sie im BPI-Themendienst „ATMP“ und im BPI-Themendienst „Seltene Erkrankungen“:
Weitere Informationen finden Sie unter: www.bpi.de
Text Dr. Matthias Wilken, Bundesverband der Pharmazeutischen Industrie (BPI)
„Die Behandlung der Mastozytose war noch nie so wirksam möglich wie heute“
Die systemische Mastozytose ist eine seltene, oft unterschätzte Erkrankung, die das Leben der Betroffenen stark beeinflussen kann. Die Symptome sind vielfältig – was die Diagnose oft erschwert. Wir sprachen mit dem Experten PD Dr. Frank Siebenhaar: Im Interview erklärt er, wie sich die Erkrankung zeigt, warum eine frühe Diagnose so entscheidend ist – und warum man trotz allem nicht in Panik verfallen sollte, wenn einzelne Symptome auftreten.
Text Miriam Hähnel
D ie systemische Mastozytose ist eine seltene Erkrankung, die durch die unkontrollierte Vermehrung von Mastzellen verursacht wird. Was passiert dabei im Körper Betroffener?
Mastzellen sind Teil unseres Immunsystems und entstehen im Knochenmark. Sie kommen in bestimmten Gewebearten vor, die uns zu unserer Außenwelt abgrenzen: In der Haut, im Magen-Darm-Trakt, in den Atemwegen. Wir gehen davon aus, dass sie eine Rolle in der Abwehr von Krankheitserregern und der Regulation von Entzündungsprozessen spielen, aber auch bei der Entstehung von allergischen Reaktionen. Bei Allergikern schütten diese Mastzellen Allergie- und Entzündungsstoffe aus, die dann die allergische Reaktion befeuern. Menschen mit Mastozytose haben zu viele dieser Mastzellen im Körper, da sie eine erworbene genetische Veränderung haben. Mastozytose wird im Regelfall nicht vererbt, es handelt sich um eine klonale Erkrankung: Betroffene bilden einen sog. „Stammzell-Klon“, sodass im Knochenmark vermehrt Mastzell-Vorläufer gebildet werden, die in den genannten Gewebearten zu vollständigen Mastzellen heranreifen. Diese Genmutation sitzt meist im KIT-Rezeptor. Bei gesunden Mastzellen funktioniert der KIT-Rezeptor wie ein Schloss, das nur durch den passenden Schlüssel aktiviert wird. Bei Mastozytose ist dieses Schloss durch die Mutation dauerhaft geöffnet. Dadurch teilen sich die Mastzellen unkontrolliert und sammeln sich vermehrt im Körper an. Mastozytosen der Haut können auch im Kindesalter auftreten, sind dann aber harmlos und verschwinden meist von selbst. Im Erwachsenenalter sind sie fast immer systemisch und werden klinisch unterteilt in aggressive und nicht aggressive Formen.
Welche Symptome können auf eine systemische Mastozytose hindeuten – und wie kann sich dies im Alltag der Betroffenen äußern?
Das Hauptsymptom sind Veränderungen der Haut: kleine rot-bräunliche Flecken, die überall am Körper auftreten können. Wenn auf diesen Flecken Reibung geschieht, werden die Mastzellen aktiviert: Es entstehen Rötungen, Quaddeln, die Stellen beginnen, stark zu jucken. Betroffene finden nachts keinen Schlaf, haben Konzentrationsprobleme und sind im Alltag aufgrund der sichtbaren Hautveränderungen mit Stigmatisierung konfrontiert. Es können aber auch Beschwerden im Magen-Darm-Trakt wie Bauchkrämpfe und chronische Durchfälle auftreten. Betroffene müssen dann bis zu 15mal am Tag zur Toilette und halten unterwegs immer nach der nächsten Toilette Ausschau. Da häufig auch junge Patienten betroffen sind, richten sie teils ihre Berufswahl nach der Erkrankung aus, manche sind nicht voll erwerbstätig. Auch atypische Osteoporosen im eher jüngeren Alter können auftreten, da die zu große Anzahl der Mastzellen auch die Knochenbildung stört. Zudem sollte bei Patienten mit schwersten Anaphylaxien aufgrund von Allergien immer auch an eine Mastozytose gedacht werden. Interessanterweise haben Menschen mit Mastozytose sehr häufig eine Insektengiftallergie, was sie zu „Super-Allergikern“ macht, sodass eine Anaphylaxie schlimmstenfalls bis zum Kreislaufkollaps und Tod führen kann. Sie leben mit der ständigen Angst, dass die nächste allergische Reaktion sie das Leben kosten könnte.
Das alles sind massive Einschränkungen der Lebensqualität, da sich oft der gesamte Alltag den Beschwerden unterordnet.
Die meisten Betroffenen haben eine nicht aggressive Mastozytose, deren Symptome aber trotzdem extrem belastend sein können. Zehn bis 15 Prozent haben eine aggressive Form, bei der die Mastzellen auch weitere Organe infiltrieren und deren Funktion beeinträchtigen. Diese aggressiven Formen können lebensbedrohlich werden. Die Erkrankung hat also ein breites Spektrum, daher vermuten wir eine hohe Dunkelziffer. Es ist also sehr wichtig, dass das Krankheitsbild Medizinern aller Fachbereiche bekannt ist.
Seit dem vorvergangenen Jahr können wir die Ursache der Erkrankung nun endlich therapeutisch am Schopfe packen.
PD Dr. Frank Siebenhaar Oberarzt am Interdisziplinären Mastozytose Centrum des Instituts für Allergieforschung an der Charité –Universitätsmedizin Berlin
Eine Diagnose ist also oft nicht leicht zu stellen. Wie wird die systemische Mastozytose diagnostiziert? Bei den typischen Veränderungen der Haut ist eine Diagnose recht einfach zu stellen. Wenn keine Hautveränderungen zu sehen sind, können verschiedene Biomarker auf die Mastozytose hinweisen.
Das ist z. B. der Tryptasewert im Blut, den auch ein Hausarzt untersuchen lassen kann. Bei 85% der Betroffenen ist dieser Wert deutlich erhöht. Wir kennen aber auch Patienten, die niedrige Werte aufweisen und trotzdem eine Mastozytose haben, daher ist dieser Wert allein oft nicht aussagekräftig. Wenn also in Kombination mit erhöhten Mastzellwerten solche unklaren Symptome oder Beschwerden auftreten, die ich vorab erwähnt habe, dann sollte eine genetische Analyse der KIT-Mutationslast erfolgen. Diese Testung ist so sensibel, dass die Diagnose mit über 98%iger Sicherheit gestellt werden kann. Bei all dem sollte man die Seltenheit der Mastozytose aber nie vergessen: beim Großteil der Patienten, bei denen eine Mastzellerkrankung vermutet wird, liegt mit großer Wahrscheinlichkeit keine vor. Und die Patienten, die tatsächlich eine systemische Mastozytose haben, können wir behandeln.
Wie wird die Krankheit behandelt und lässt es sich damit gut leben?
Bisher konnte man auf Antiallergika, Antihistaminika, Cortison oder Adrenalin im absoluten Notfall zurückgreifen. Zudem gab es für schwer betroffene Patienten sogenannte
Tyrosinkinasehemmer, die aber zu unspezifisch wirkten und starke Nebenwirkungen auslösten. Seit dem vorvergangenen Jahr können wir die Ursache der Erkrankung endlich therapeutisch am Schopfe packen, da wir nun eine Möglichkeit haben, das spezifische KIT der mutierten Mastzellen zu hemmen.
Wir führen dem Körper über ein Medikament also den fehlenden Schlüssel für das Schloss zu, das sonst permanent geöffnet war. Dadurch nehmen wir dem Feuer, das die Mastzellen dazu bringt, sich zu vermehren, das Brennmaterial. Die überzähligen Mastzellen „verhungern“, der Tryptasewert und die Mutationslast sinken. Damit erleben Patienten eine deutliche Verbesserung der Lebensqualität und können wieder ein normales Leben führen. Und den Patienten mit einer aggressiven Form rettet diese Behandlung das Leben. Die Behandlung der Mastozytose war noch nie so wirksam möglich wie heute.
Wo finden Betroffene Spezialisten, die mit dieser seltenen Erkrankung vertraut sind?
Entweder kennt der diagnostizierende Arzt
bereits eine Anlaufstelle, oder der Patient kann sich selbst informieren. Es gibt das „Kompetenznetzwerk Mastozytose e. V.“, das auf der Website über die größten Zentren informiert. Zudem kann man sich über die zwei Patientennetzwerke „Mastozytose Selbsthilfe Netzwerk e. V.“ und „Selbsthilfeverein Mastozytose e. V.“ informieren, die ebenfalls eine Auflistung der Experten zusammengestellt haben. Aber auch die allergologischen, dermatologischen und hämatologischen Zentren der Universitätsklinika sind geeignete Anlaufstellen.
Weitere Informationen finden Sie hier!
Kompetenznetzwerk Mastozytose e. V.: www.mastozytose.net
Mastozytose Selbsthilfe Netzwerk e. V.: www.mastozytose-info.de
Selbsthilfeverein Mastozytose e. V.: www.mastozytose.de
LEBEN MIT SYSTEMISCHER MASTOZYTOSE VERSTEHEN UND GESTALTEN
Die Systemische Mastozytose (SM) ist eine seltene, chronische Erkrankung, die zu ganz unterschiedlichen Beschwerden führen kann: Hautrötungen, Juckreiz, Magen-Darm-Beschwerden, Kreislaufprobleme bis hin zu anaphylaktischen Reaktionen. Viele dieser Symptome können auch bei anderen, harmloseren Erkrankungen auftreten, was die Diagnose erschwert und für Betroffene oft eine lange Zeit der Unsicherheit bis zur Diagnosestellung bedeuten kann.
Fundierte Informationen für Betroffene und ihre Angehörigen Navigating SM ist eine digitale Plattform, die Menschen mit Systemischer Mastozytose und ihre Angehörigen durch diese komplexe Erkrankung begleiten möchte. Das Ziel ist es, verlässliche, medizinisch fundierte Informationen bereitzustellen und dabei stets den Menschen im Fokus zu behalten. Denn jede Erkrankung ist individuell, und genauso individuell ist der Umgang damit.
Auf der Website finden Betroffene und ihre Angehörigen verständlich aufbereitete Informationen zu den Ursachen und unterschiedlichen Formen der Systemischen Mastozytose sowie zu typischen Symptomen. Es wird erklärt, wie die Diagnose gestellt werden kann, und warum sie ausschließlich durch spezialisierte Fachärzte erfolgen sollte.
Zudem finden Betroffene hier auch Informationen zu den Therapiemöglichkeiten. Besprechen Sie mit Ihrem Behandlungsteam, welche Optionen es gibt und wie sie individuell angepasst werden können.
Für mehr Orientierung im Alltag mit SM Leben mit SM bedeutet aber nicht nur, Fakten zu verstehen und den Alltag mit der Erkrankung zu organisieren, sondern auch, mit Ängsten umzugehen, Arbeit und Familie zu meistern und sich mit anderen auszutauschen. Deshalb bietet die Website auch praxisnahe Tipps für den Umgang mit Triggern, zur Ernährung, zur Notfallvorsorge oder zu psychischer Unterstützung. Außerdem sind dort Informationen zu Selbsthilfegruppen zu finden, um sich mit anderen Betroffenen zu vernetzen.
Navigating SM versteht sich als Wegbegleiter – informierend, stärkend und unterstützend. Denn auch wenn die Systemische Mastozytose eine Herausforderung ist, so ist sie kein Stillstand. Informationen über die Erkrankung und ein guter Kontakt zum Behandlungsteam ermöglichen eine individuelle Selbstfürsorge, mit der Betroffene ein erfülltes Leben führen können.
Informieren Sie sich weiter unter www.navigatingsm.com/de
Dieser Artikel ist in Zusammenarbeit mit dem Sächsischen Staatsministerium für Wissenschaft, Kultur und Tourismus entstanden.
„Bei Seltenen Erkrankungen ist detektivischer Spürsinn gefragt“
Zentren für Seltene Erkrankungen (ZSEs) spielen bei der Diagnostik und der Erforschung Seltener Erkrankungen eine tragende Rolle. Wir sprachen mit Prof. Dr. Reinhard Berner vom USE Dresden, welche Schwerpunkte dort eine besondere Rolle spielen und wie bereits Medizinstudierende in Dresden an dieses Thema herangeführt werden.
Herr Prof. Berner, die Diagnose von Seltenen Erkrankungen stellt auch versierte Mediziner oft noch vor Herausforderungen. Woran liegt das Ihrer Meinung nach, und was ist die größte Herausforderung dabei?
Dabei spielen unterschiedliche Faktoren eine Rolle. Zum einen ist das die Seltenheit an sich, so dass ein Hausarzt, Kinderarzt oder Internist in seinem Alltag nicht häufig mit solchen Krankheitsbildern konfrontiert ist, manche treten seltener als 1 zu 1 Million auf; bei mittlerweile 6.000 bis 8.000 bekannten Seltenen Erkrankungen ist es schlechterdings unmöglich, alle davon zu kennen. Zum anderen sind die Symptome oft unspezifisch oder schwer in den richtigen Zusammenhang zu bringen, denn oft weisen einzelne Symptome erst einmal auf weitaus häufiger auftretende Erkrankungen hin. Es ist also detektivischer Spürsinn gefragt, um die spezifischen Symptome oder Symptomkonstellationen ausfindig zu machen, die dann wirklich auf eine Seltene Erkrankung hindeuten.
Zentren für Seltene Erkrankungen wie das UniversitätsCentrum für Seltene Erkrankungen (USE) in Dresden sind daher eine wichtige Anlaufstelle für Betroffene und darüber hinaus auch für Angehörige. Was macht Ihre Arbeit an solchen Zentren so besonders? Eine wichtige Aufgabe dieser Zentren ist es, für Patienten mit unklarer oder fehlender Diagnose eine Anlaufstelle zu sein, wo sie mit ihren Beschwerden und ihrer oft langen, odyseehaft verlaufenden Krankheitsgeschichte aufgefangen und ggf. auch weitervermittelt werden können, wenn sich der Verdacht auf eine Seltene Erkrankung erhärtet. Wir besprechen solche Fälle in einer interdisziplinären Fallkonferenz:
Dabei sind Mediziner unterschiedlicher Fachbereiche involviert, die einen ganzheitlichen Blick auf den Patienten mit all seinen Symptomen und Befunden werfen. Wenn sich in einer solchen Fallkonferenz herausstellt, dass es sich z. B. um eine seltenere Form von Epilepsie handelt, verweisen wir den Patienten an neurologische Kollegen, die sich darauf spezialisiert haben. Vermuten wir eine seltene Herzerkrankung, verweisen wir an die entsprechenden spezialisierten Kardiologen etc. Wenn die Fallkonferenz zu dem Ergebnis kommt, dass die Diagnose weiterhin unklar ist, aber man übereinstimmend zu der Einschätzung gelangt, dass eine Seltene Erkrankung wahrscheinlich ist, dann kann eine genetische Diagnostik sinnvoll sein, die dann direkt über das Zentrum in die Wege geleitet wird. Das verkürzt den Weg zur richtigen Diagnosestellung oft enorm. Diese geballte Expertise, verknüpft mit den Zugängen zu diagnostischen Möglichkeiten, machen die Zentren für Seltene Erkrankungen unverzichtbar, um die bestmögliche Versorgung von Menschen mit Seltenen Erkrankungen anbieten zu können.
Fakt ist, dass jeder Mediziner in seinem Berufsleben mit Menschen konfrontiert sein wird, die eine Seltene Erkrankung haben.
Seit dem Sommersemester gibt es in der Medizin-Ausbildung in Dresden ein neues Wahlfach für Seltene Erkrankungen. Sie setzen damit bewusst auch einen zusätzlichen inhaltlichen Ausbildungsschwerpunkt. Warum ist das aus Ihrer Sicht so wichtig? In den verschiedenen medizinischen
Fächern, die gelehrt werden, wird das Thema der Seltenen Erkrankungen natürlich immer mal wieder gestreift. Wir haben aber festgestellt, dass das Grundproblem und auch die Möglichkeiten der Diagnostik, die es heute gibt, dabei oft nicht ausreichend adressiert werden. Dabei sollten bereits Studierende lernen, wann man an eine Seltene Erkrankung denken sollte, wie mögliche Leitsymptome aussehen können und wie dann vor allem auch der diagnostische Weg aussehen kann. Das möchten wir den Studierenden der Medizin in einer strukturierten Weise nahebringen. Die Anmeldeliste war sehr schnell gefüllt, das Interesse an diesem Wahlpflichtfach war sehr groß. Nach der Veranstaltung waren die Studierenden begeistert von dem, was sie gelernt hatten.
Da Seltene Erkrankungen ja grundsätzlich einem bestimmten medizinischen Fach zuzuordnen sind, haben wir verschiedene Experten unseres Klinikums, die sich regelmäßig auch zu den oben erwähnten Fallkonferenzen zusammenfinden, gebeten, je eine Doppelstunde zu einer Seltenen Erkrankung ihres Fachbereiches zu halten, aber immer unter dem Blickwinkel, wie nähere ich mich der Diagnose, wie komme ich zum Ziel. So bleibt es abwechslungsreich und wird der Bandbreite der Seltenen Erkrankungen gerecht. Zudem haben wir auch Betroffenen- und Selbsthilfeverbände einbezogen, um gerade die so wichtige Patientensicht mit einfließen zu lassen. Fakt ist, dass jeder Mediziner in seinem Berufsleben mit Menschen konfrontiert sein wird, die eine Seltene Erkrankung haben. Deshalb finden wir es ungemein wichtig, unseren Studierenden eine Art Leitfaden an die Hand zu geben, wie ein sinnvolles Vorgehen aussehen kann, um für diese Patienten die richtige Diagnose zu stellen und damit angemessen versorgen zu können.
Text Levi Müller
Um auch bei den angehenden Medizinern besondere Aufmerksamkeit für Ihr neues Angebot zu erzielen, haben Sie dem Vorhaben einen Untertitel gegeben: Stichwort: „Dr. House“. Warum? Wir wollten natürlich einen gewissen Anreiz schaffen, sich mit diesem Thema zu beschäftigen, und direkt an den detektivischen Spürsinn unserer Studierenden appellieren. Denn am Ende ist die Suche nach der richtigen Diagnose unglaublich spannend, es ist eine Suche nach Indizien und Bausteinen, die in der Gesamtheit dann idealerweise auf die richtige Spur und damit zur Diagnose führen.
Uin ev r is t ä t s k l in i kum Dresden
Die Suche nach der richtigen Diagnose ist unglaublich spannend. Es ist eine Suche nach Indizien und Bausteinen, die in der Gesamtheit dann idealerweise auf die richtige Spur und damit zur Diagnose führen.
Prof. Dr. med. Reinhard Berner
Sprecher des UniversitätsCentrums für Seltene Erkrankungen (USE) am Universitätsklinikum Carl Gustav Carus Dresden
Sie verstehen sich als vernetztes Zentrum, das seine interdisziplinäre Expertise auch für die Forschung nutzt. Wie muss man sich diese vernetzte Forschungsarbeit praktisch vorstellen, und gibt es Bereiche oder Krankheitsbilder, die Sie am dabei am USE besonders im Blick haben?
In Dresden haben wir hier schon seit vielen
Jahren ganz verschiedene Projekte erfolgreich umgesetzt, allen voran vielleicht das Projekt Translate NAMSE, in dem es exemplarisch darum ging zu zeigen, wie interdisziplinäre Fallkonferenzen und die genetische Diagnostik dazu beitragen, Diagnosewege zu verkürzen und Betroffenen eine Behandlung zu ermöglichen, wenn es für die Erkrankung zielgerichtete Therapien gibt und welche Voraussetzungen gegeben sein sollten, um eine Empfehlung für eine genetische Diagnostik abzuleiten. Denn nur dann erreicht man dabei auch eine Zielgenauigkeit von 30 bis 40 Prozent bei der Diagnosestellung. Und tatsächlich hat dieses Projekt dazu geführt, dass wir heute den Patienten, die dafür in Frage kommen, z. B. im Rahmen des Modellvorhabens Genomsequenzierung in der Initiative genom.de eine entsprechende genetische Diagnostik anbieten können. Zudem haben wir natürlich an unserem Uniklinikum, und im speziellen an der Kinderklinik, bestimmte Schwerpunkte der klinischen Forschung, aber auch der Grundlagenforschung. Das sind bei uns insbesondere Erkrankungen des Immunsystems, aber auch Erkrankungen der Nebenniere und neurologische und neuromuskuläre Erkrankungen. Außerdem beschäftigen wir uns intensiv mit dem Neugeborenenscreening, das es bereits seit Ende der 1960er Jahre und damit mittlerweile seit über 50 Jahren gibt und in gewisser Weise den Anfang der systematischen Beschäftigung mit den Seltenen Erkrankungen darstellt. Dabei wird ein Blutstropfen des Neugeborenen über eine Trockenblutkarte auf bestimmte Stoffwechselerkrankungen, Hormon- oder Bluterkrankungen und Immundefekte untersucht. Hier arbeiten wir mit anderen nationalen und internationalen Zentren zusammen u.a. zu der Frage, ob ein genetisches Neugeborenenscreening sinnvoll sein könnte; hier gibt es aber noch sehr viele offene Fragen, die es vorab zu klären gilt, weshalb wir uns auch damit beschäftigen, welche Voraussetzungen gegeben sein müssen und in welcher Form man Aufklärungsarbeit betreiben müsste, um die notwendige Akzeptanz für eine solch neue Screeningform zu schaffen. Insofern kann man zusammenfassen, dass es bei den Seltenen Erkrankungen immer sehr komplexe Zusammenhänge und Sachverhalte sind, mit denen man sich auseinandersetzen muss.
Ein Gastbeitrag von Sachsens Wissenschaftsminister Sebastian Gemkow
TECHNOLOGIE
IN KOMBINATION MIT SENSIBILISIERUNG HILFT BETROFFENEN
KONKRET
Eine der größten Herausforderungen im Umgang mit Seltenen Erkrankungen bleibt ihre Diagnose. Trotz enormer Fortschritte vergeht für den einzelnen Patienten in der Regel noch zu viel Zeit, bis er oder sie Gewissheit hat. Diese Zeit ist geprägt von Einschränkungen im Alltag, Beschwerden und der bohrenden Frage: „Was ist mit mir?“ Künstliche Intelligenz birgt ein ungeheures Potential, die Antwort auf diese Frage in Zukunft viel schneller zu geben.
Trotz enormer Fortschritte vergeht für den einzelnen Patienten in der Regel noch zu viel Zeit, bis er oder sie Gewissheit hat.
Der Freistaat Sachsen unterstützt deshalb gemeinsam mit weiteren Partnern beispielsweise die Forschung an KI-Modellen, die mit tausenden medizinischen Datensätzen trainiert werden und in der Lage sind, von der Analyse auf eine Seltene Erkrankung zu schließen. Die Universitätsmedizin in Leipzig und Dresden gehört hier klar zu den Spitzenzentren in Deutschland, im engen Netzwerk mit weiteren forschungsstarken Hochschulen im Bereich KI und außeruniversitären Einrichtungen.
Hinzu kommt die Expertise der beiden Zentren für Seltene Erkrankungen in Sachsen, als Anlaufstelle für Ärztinnen und Ärzte, Patienten und Mediziner in Ausbildung. Sie sind auch Bindeglied zwischen Biotechnologie, IT und Medizin und erreichen eine 360 Grad-Sensibilisierung für Seltene Erkrankungen, was dazu beiträgt, Betroffenen schneller helfen zu können.
Nothilfe
weltweit
Die weltweiten Katastrophen nehmen zu. Aktion Deutschland Hilft steht Menschen in ihrer größten Not bei, versorgt sie medizinisch und mit Trinkwasser und Lebensmitteln. In sicheren Unterkünften finden Betroffene Schutz.
Helfen Sie uns Leben zu retten – mit Ihrer Spende. Herzlichen Dank! Aktion-Deutschland-Hilft.de