SELTENE ERKRANKUNGEN
DIE WAISEN DER MEDIZIN.


„Meine Erkrankung hat mich stark gemacht“

Nadine Großmann ist BiochemieDoktorandin und erforscht ihre eigene Erkrankung FOP.
NICHT VERPASSEN: Spinale Muskelatrophie Patientin Carolin über ein Leben zwischen Einschränkung und Selbstbestimmung.
Seite 12

Hypoparathyreoidismus Ursachen und Folgen fehlender Nebenschilddrüsen.
Seite 20
EINE UNABHÄNGIGE KAMPAGNE VON MEDIAPLANET
Lesen Sie mehr auf www.seltenekrankheiten.de
VERANTWORTLICH FÜR DEN INHALT IN DIESER AUSGABE: MIRIAM HÄHNEL

Vier Millionen Menschen in Deutschland leben mit einer seltenen Erkrankung. Vier Millionen Gründe, um die Forschung beständig voranzutreiben!

IN DIESER AUSGABE
08
Plötzlich (fast) blind Andreas verlor mit 33 Jahren fast vollständig seine Sehfähigkeit.
online

Kristina Mayer hat akute hepatische Porphyrie und erzählt ihre persönliche Geschichte.
Industry Manager Health: Miriam Hähnel
Geschäftsführung: Richard Båge (CEO), Philipp Colaço (Managing Director), Franziska Manske (Head of Editorial & Production), Henriette Schröder (Sales Director) Designer: Elias Karberg
Mediaplanet-Kontakt: redaktion.de@ mediaplanet.com Coverbild: Privat
Artikel, die mit mit Unterstützung gekennzeichnet sind, sind keine neutrale Mediaplanet-Redaktion.
facebook.com/ MediaplanetStories
@Mediaplanet_germany
Please recycle
DANKE,
dass Sie dem Thema Seltene Erkrankungen und vor allem den vier Millionen betroffenen Kindern und Erwachsenen in Deutschland Ihre Aufmerksamkeit schenken. Die letzten Monate waren vor allem durch die Corona-Pandemie geprägt, die die „Waisen der Medizin“ einmal mehr in den Schatten gestellt hat. Es wird Zeit, den vielen chronisch kranken Kindern und Erwachsenen, die in der Pandemie vor zusätzlichen Herausforderungen gestanden haben, wieder mehr Gehör zu schenken! Dabei möchte ich gern die Patientenselbsthilfe stärker in Ihr Blickfeld rücken.

Geske Wehr Vorsitzende der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V.
Gerade bei den Seltenen Erkrankungen, wo es an Wissen und Expertise mangelt, es nur wenige Therapien und Behandlungsmethoden gibt, ist die Patientenselbsthilfe eine stützende Säule! Ehrenamtliche Kräfte, zumeist Eltern kranker Kinder oder selbst erkrankte Menschen, übernehmen Aufgaben, die unser Gesundheitssystem nicht stemmt oder stemmen kann. Ich berichte Ihnen kurz aus meinem Verein, der Selbsthilfe Ichthyose. Erst kürzlich konnten wir einer Familie helfen, die uns nur per Zufall gefunden hat, nachdem sie bereits mehr als ein Jahr lang Untersuchungen über ihr Kind ergehen lassen musste, ohne Ergebnis. Als betroffene Mutter kenne ich die verzweifelte Suche nach Antworten. Der kleine Junge leidet an einer schweren Form von Ichthyose. Diese angeborene Verhornungsstörung erfordert mehrmals täglich sehr aufwendige Pflege der Haut, damit es nicht zu Verhärtungen bis hin zur Unbeweglichkeit kommt. Die Selbsthilfe hätte Antworten zum Umgang mit der Erkrankung gehabt und darauf, wie sich die Erkrankung weiterentwickelt, dass mit der richtigen Pflege ein mehr oder weniger normales Leben des Kindes möglich
ist. Der Familie wäre viel Leid erspart geblieben.
So wie unsere Selbsthilfe stehen auch andere Verbände ratsuchenden Betroffenen zur Seite. Viele Verbände haben sich weltweit vernetzt, weil so noch mehr Wissen ausgetauscht werden kann und Kräfte gebündelt werden können. Sie arbeiten an Leitlinien mit, damit das Wissen rund um die Behandlung der Erkrankung erweitert wird, denn sie verfügen über ein enormes Erfahrungswissen. Selbsthilfeverbände unterstützen und beraten, geben seelischen Halt, tragen dazu bei, dass richtige Diagnosen gestellt werden und Forschung vorangetrieben wird. Es ist an der Zeit, dass diese Leistung anerkannt wird – öffentlich und mit struktureller Förderung.
Was darüber hinaus in Politik und Gesundheitswesen getan werden sollte, damit Menschen mit chronischen seltenen Erkrankungen länger und besser leben können, erfahren Sie auf Seite 3.
Ich wünsche Ihnen viele „AhaErlebnisse“ beim Lesen dieser Ausgabe - und bitte erzählen Sie es weiter. Danke!
Lesen Sie mehr auf seltenekrankheiten.de 2
Unsere
Liste mit konkreten
Forderungen ist lang. Sie können sie hier
nachlesen: www.achseonline.de/ de/was_tut_
ACHSE/4MillionenGruendejetzt-zuhandeln. php
Vier Millionen Gründe, jetzt zu handeln
In Deutschland leben vier Millionen Menschen, die von einer der etwa 8.000 verschiedenen und oftmals sehr komplexen Seltenen Erkrankungen betroffen sind, darunter vor allem viele Kinder. Die Erkrankungen gehen mit schwerwiegenden körperlichen und geistigen Einschränkungen einher, viele Menschen sind ihr Leben lang auf pflegerische Unterstützung angewiesen.
Text Mirjam Mann
Aufgrund der Vielzahl der Erkrankungen, die für sich genommen in geringen Zahlen vorkommen, ist das Interesse, daran zu forschen, gering oder mit hohem Aufwand und Kosten verbunden, sodass es kaum Therapien und Behandlungsmöglichkeiten gibt. Das wenige Wissen zu den einzelnen Erkrankungen liegt in nur wenigen Händen. Betroffene sind oft Jahre auf der Suche nach einer richtigen Diagnose und erleben dabei wahre Odysseen. In Beruf, Schule oder Gesellschaft finden sie oft keine Anerkennung.
Um die Waisen der Medizin – wie die Betroffenen auch genannt werden – in den Fokus der Politik, Wissenschaft, Forschung und Medizin zu rücken und die Lebenslage der Menschen zu verbessern – gar Leben zu retten –, hat sich vor 17 Jahren die ACHSE gegründet. Unter deren Dach kommen heute über 130 Patientenorganisationen zusammen und teilen ihr
Erfahrungswissen. Mit ihrem Netzwerk aus Vertreterinnen und Vertretern aus den Bereichen Medizin, Wissenschaft, Forschung sowie Kontakten im Gesundheitswesen ist die ACHSE zugleich Anker und starke Stimme der Menschen mit Seltenen Erkrankungen, die in Deutschland leben. Vier Millionen Menschen sind vier Millionen Gründe für unseren Einsatz. Aus Anlass der Bundestagswahl hat die ACHSE ein umfassendes Positionspapier für die kommende Legislaturperiode verfasst. Es enthält nicht nur Forderungen, sondern konkrete Maßnahmen, die dazu beitragen sollen, die Leben von Menschen mit chronischen seltenen Erkrankungen zu verbessern. Es gibt darin zwei Kernforderungen: Zum einen die nach strukturierten Patientenpfaden. Diese bilden die Behandlung und Pflege eines Patienten mit einer definierten Erkrankung ab. Wir fordern gut beschriebene und öffentlich zugängliche

Mirjam Mann Geschäftsführerin der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V.
Patientenpfade mit konkreten Handlungsanweisungen. Sie können für den Arzt verdeutlichen, wie der ideale Weg eines Patienten ist, denn Menschen mit Seltenen Erkrankungen irren oft lange durch unser Gesundheitssystem. Auf diese Weise sollen auch Ärzte, die nicht auf diese Erkrankungen spezialisiert sind, wissen, wie sie weiter vorgehen können. Denn auch wenn es mittlerweile 35 Zentren für Seltene Erkrankungen gibt, in denen Ärzte vernetzt arbeiten –Patienten mit unerklärlichen Symptomen gehen zuerst zum Hausarzt, vielleicht noch zum Facharzt. Der weitere Weg des Patienten sollte nicht vom einzelnen Engagement des jeweiligen Arztes abhängen müssen. Sein Weg in das Zentrum und zu einer richtigen Diagnose oder Behandlung sollte sichergestellt werden. Unsere zweite Forderung knüpft daran an: Wir möchten Betroffenen einen Case Manager auf Rezept ermöglichen, was eine Gesetzesänderung erforderlich macht. Wir möchten, dass alle Menschen mit einer chronischen Erkrankung jemanden an die Seite gestellt bekommen, der ihnen hilft, alle notwendigen Schritte einzuleiten: zum Beispiel Pflegeleistungen oder HartzIV-Zusatzleistungen zu beantragen. Nicht jeder Mensch hat treusorgende Angehörige um sich und das Gesundheitssystem ist so komplex, dass es für Laien schwer verständlich ist. Die Betroffenen benötigen eine Art Bauleiter, der sich um alles kümmert. Denn sie selbst haben genug mit ihrer Erkrankung zu tun.
Wir freuen uns außerdem über Ihre Unterstützung unserer Social-Media-Kampagne #4MillionenGründe.
3 Lesen Sie mehr auf seltenekrankheiten.de
Leben mit ITP
In Deutschland leben derzeit rund 16.000 Patienten mit Immunthrombozytopenie (ITP). Bei dieser Erkrankung erkennt das Abwehrsystem des Körpers die eigenen Blutplättchen (Thrombozyten) fälschlicherweise als Fremdkörper und baut diese vermehrt ab. Im Interview spricht Prof. Dr. Axel Matzdorff, Chefarzt der Abteilung für Innere Medizin II an der Asklepios Klinik Uckermark, über die seltene Autoimmunerkrankung.
Text Franziska Manske
 Prof.
Prof.
Dr.
Axel Matzdorff
Chefarzt der Abteilung für Innere Medizin II an der Asklepios Klinik Uckermark
ITP gehört ja zu den seltenen Erkrankungen: Wie äußert sich die Erkrankung und wie wird sie diagnostiziert?
Man unterscheidet die ITP bei Erwachsenen und bei Kindern. Kinder haben häufig einen akuteren und schwereren Verlauf, haben häufiger Blutungen. Das Erfreuliche ist, dass 90 Prozent der Kinder spontan ausheilen, die Erkrankung geht nach wenigen Wochen vorbei. Bei den Erwachsenen ist es umgekehrt – rund 90 Prozent entwickeln einen chronischen Verlauf und nur rund zehn Prozent eine spontane Heilung.
Bei einem Drittel der Patienten treten stecknadelkopfgroße Einblutungen (Petechien) der Haut und Schleimhäute sowie blaue Flecken auf. Schwere Blutungen in den Kopf, die Augen, andere Organe oder dass man eine Transfusion braucht, sind zum Glück selten. Bei zwei Drittel der Patienten ist die ITP eine Zufallsdiagnose, die durch eine Blutuntersuchung beim Arzt auffällt.
Über welche Belastungen im Alltag berichten Ihre ITPPatienten am häufigsten?
Neben den genannten Symptomen belasten vor allem Fatigue, eine bleierne Müdigkeit und anhaltende Erschöpfung, und die Angst vor schweren Blutungen die Betroffenen sehr. Hinzu kommen die engmaschigen Kontrollen beim Arzt, die Zeit kosten und gerade für ältere Menschen sehr aufwendig sind. Denn um den Erfolg der ITP-Behandlung beurteilen zu können, müssen die Blutplättchenwerte zu Therapiebeginn besonders engmaschig, d. h. mitunter alle paar Tage kontrolliert werden. Wenn sich die Thrombozytenzahl erholt und stabilisiert hat, können die Kontrollintervalle immer weiter ausgedehnt werden. Dann sind nur noch alle 2–4 Wochen oder gar Monate ein Arztbesuch und eine Überprüfung der Blutplättchenzahl notwendig.
Welche Therapieoptionen gibt es, und können Pati-
enten mit der passenden Therapie wieder ein „normales“ Leben führen? Ein weitestgehend normales Leben ist möglich, wenn die Therapie bei Betroffenen anschlägt, was zum Glück zu 90 Prozent der Fall ist. Nach der Diagnose der ITP erfolgt die Behandlung in aufeinanderfolgenden Schritten.
1 Erstlinientherapie
Patienten werden standardmäßig mit Kortikosteroiden (Nichtmediziner sagen häufig „Kortison“) in hoher Dosierung behandelt. Bei Bedarf kommen zusätzlich Immunglobuline und Thrombozytenkonzentrate zum Einsatz.
2 Zweitlinientherapie
Wenn mit der Erstlinientherapie keine ausreichende oder anhaltende Steigerung der Blutplättchenzahl erreicht wird oder die Mittel vom Patienten schlecht vertragen werden, kann der Einsatz von Thrombopoetin-RezeptorAgonisten oder eines für
Lesen Sie mehr auf seltenekrankheiten.de 4
die ITP zugelassenen neuen Milz-Tyrosinkinase-Hemmers ins Auge gefasst werden. Auch die Entfernung der Milz (Splenektomie) stellt eine Therapieoption dar, wird heute aber nur noch selten angeboten.
die ITP zugelassenen neuen Milz-Tyrosinkinase-Hemmers ins Auge gefasst werden. Auch die Entfernung der Milz (Splenektomie) stellt eine Therapieoption dar, wird heute aber nur noch selten angeboten.
3 Drittlinientherapie Sprechen behandlungsbedürftige Patienten auch auf die Zweitlinientherapie nicht an oder erleiden sie immer wieder einen Rückfall, dann kann auf verschiedene Medikamente, sogenannte Immunsuppressiva, zurückgegriffen werden, die normalerweise in der Transplantations- oder Krebsmedizin eingesetzt werden, um das körpereigene Abwehrsystem zu unterdrücken. Ziel ist auch hier, die Bildung von
3 Drittlinientherapie Sprechen behandlungsbedürftige Patienten auch auf die Zweitlinientherapie nicht an oder erleiden sie immer wieder einen Rückfall, dann kann auf verschiedene Medikamente, sogenannte Immunsuppressiva, zurückgegriffen werden, die normalerweise in der Transplantations- oder Krebsmedizin eingesetzt werden, um das körpereigene Abwehrsystem zu unterdrücken. Ziel ist auch hier, die Bildung von
Autoantikörpern und somit den übermäßigen Blutplättchenabbau zu verhindern. In den späteren Therapielinien ist die Milzentfernung eine häufigere Therapieoption.
Autoantikörpern und somit den übermäßigen Blutplättchenabbau zu verhindern. In den späteren Therapielinien ist die Milzentfernung eine häufigere Therapieoption.
Warum ist es so wichtig, dass ITP-Patienten ihre Erkrankung kennen und sich mit dem Arzt austauschen können?
Warum ist es so wichtig, dass ITP-Patienten ihre Erkrankung kennen und sich mit dem Arzt austauschen können?
Die ITP ist eine seltene Erkrankung und der Arzt sieht in seiner Praxis viele andere Erkrankungen, die häufiger sind und mit denen er sich logischerweise besser auskennt. Mit einer seltenen Erkrankung auf dem Laufenden zu bleiben, ist schon schwierig. Der ITP-Patient, der den ganzen Tag mit seiner Erkrankung konfrontiert ist, weiß schon
Die ITP ist eine seltene Erkrankung und der Arzt sieht in seiner Praxis viele andere Erkrankungen, die häufiger sind und mit denen er sich logischerweise besser auskennt. Mit einer seltenen Erkrankung auf dem Laufenden zu bleiben, ist schon schwierig. Der ITP-Patient, der den ganzen Tag mit seiner Erkrankung konfrontiert ist, weiß schon
innerhalb kürzester Zeit sehr viel darüber – manchmal sogar mehr als der Arzt. Das liegt daran, dass sich Betroffene viel mehr damit auseinandersetzen und informieren. Sie sind in Selbsthilfegruppen, tauschen sich mit anderen aus, wissen, wo Experten sitzen, holen sich Zweitmeinungen ein. Das ist sehr wichtig und trägt sehr positiv zur ArztPatienten-Kommunikation bei.
innerhalb kürzester Zeit sehr viel darüber – manchmal sogar mehr als der Arzt. Das liegt daran, dass sich Betroffene viel mehr damit auseinandersetzen und informieren. Sie sind in Selbsthilfegruppen, tauschen sich mit anderen aus, wissen, wo Experten sitzen, holen sich Zweitmeinungen ein. Das ist sehr wichtig und trägt sehr positiv zur ArztPatienten-Kommunikation bei.
Am 25.09. findet der erste nationale ITP-Patiententag als virtuelle Veranstaltung im Internet statt und Prof. Matzdorff hofft, dass noch viele weitere folgen – denn Aufklärung ist das A und O, bei jeder seltenen Erkrankung.
Am 25.09. findet der erste nationale ITP-Patiententag als virtuelle Veranstaltung im Internet statt und Prof. Matzdorff hofft, dass noch viele weitere folgen – denn Aufklärung ist das A und O, bei jeder seltenen Erkrankung.
Aufklärung ist das A und O – bei jeder seltenen Erkrankung.
Aufklärung ist das A und O – bei jeder seltenen Erkrankung.
ITP Immunthrombozytopenie –eine Autoimmunkrankheit nimmt Lebensqualität
Häufiges Nasen- oder Zahnfleischbluten, eine Neigung zu blauen Flecken oder kleine Einblutungen unter der Haut (Petechien) können Anzeichen für eine Immunthrombozytopenie, kurz ITP, sein. ITP ist eine Erkrankung des blutbildenden Systems, von deren chronischer Form in Deutschland etwa 16.000 Menschen betroffen sind.1 Das körpereigene Abwehrsystem erkennt die eigenen Blutplättchen (Thrombozyten) und ihre Vorläuferzellen fälschlicherweise als Fremdkörper und beginnt sie abzubauen. So entsteht ein Thrombozytenmangel,
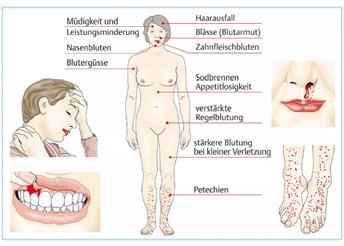
durch den es zu Störungen der Blutgerinnung kommt. Dies kann wiederum zu einer erhöhten Blutungsneigung mit den oben aufgeführten Folgen führen. Hinzu kommen für viele Patienten nicht sichtbare Symptome, wie Müdigkeit und starke Erschöpfungszustände (Fatigue). Der Erfolg einer ITP-Therapie sollte daher nicht allein an der Thrombozytenzahl gemessen werden, sondern auch nicht sichtbare Symptome beachten.
Mit der passenden ITP-Therapie die Blutplättchenzahl erhöhen und die Lebensqualität verbessern
Für die Behandlung der ITP stehen unterschiedliche Therapien zur Verfügung: Nach der Diagnose wird standardmäßig mit einer Kortisontherapie gestartet. Bei einem Großteil der Patienten kann damit nach kurzer Zeit ein Anstieg der Blutplättchen erzielt werden. Sollte dies nicht gelingen oder die Medikamente vom Patienten schlecht vertragen werden, kann in einem zweiten Schritt auf eine Therapie mit Thrombopoetin-RezeptorAgonisten (TPO-RAs) oder einen für die ITP
zugelassenen Milz-Tyrosinkinase-Hemmer umgestellt werden. TPO-RAs können die Produktion der Blutplättchen im Knochenmark anregen, Milz-Tyrosinkinase-Hemmer können den Abbau der Blutplättchen verringern. In Ausnahmefällen kann die operative Entfernung der Milz (Splenektomie) helfen. Die ITP verläuft oft individuell sehr unterschiedlich, ob und wie die ITP behandelt wird, sollte gemeinsam von Arzt und Patient getroffen werden.
Beim 1. Nationalen ITP-Patiententag stehen die Fragen der Patienten im Mittelpunkt. Auf großes Interesse im Vorfeld stießen die Workshops zum Umgang mit Erschöpfung und Müdigkeit sowie die Frage, ob eine Therapiefreiheit auch bei chronischer ITP möglich ist. In Kürze finden Sie die Vorträge auch auf www.leben-mit-itp.de
1) Onkopedia-Leitlinie Immunthrombozytopenie https:// www.onkopedia.com/de/onkopedia/guidelines/immunthrombozytopenie-itp/@@guideline/html/index.html, zuletzt aufgerufen am 23.06.2021
5 Lesen Sie mehr auf seltenekrankheiten.de
5 Lesen Sie mehr auf gesunder-koerper.info
ANZEIGE
©Novartis
Geerbte Attacken
Akute hepatische Porphyrien sind eine Gruppe seltener metabolischer Erkrankungen, die bei den Betroffenen starke Beschwerden hervorrufen und den Alltag stark beeinträchtigen können. Wie bei vielen seltenen Erkrankungen müssen Ärzte bei der Diagnosefindung medizinische Detektivarbeit leisten, um Patienten helfen zu können. Ein Gespräch mit Nils Wohmann und Dr. Ilja Kubisch vom Porphyriezentrum Chemnitz.
Text Hanna Sinnecker

Nils Wohmann Porphyriezentrum Chemnitz

Dr. Ilja Kubisch Porphyriezentrum Chemnitz
Von akuter hepatischer Porphyrie (AHP) hat Otto NormalBürger wahrscheinlich noch nie etwas gehört. Was passiert im Körper Betroffener?
Generell sind sogenannte Porphyrine lebenswichtige biochemische Bausteine für Pflanzen, Tiere und Menschen. Sie haben die Fähigkeit, elektromagnetische Strahlung im Lichtspektrum zu absorbieren und die aufgenommene Energie wieder abzugeben. Bei den tierischen und damit auch den menschlichen Zellen bilden sie zusammen mit Eisen Häm-Moleküle. Diese sind als Hämo- beziehungsweise Myoglobin essenziell für die Sauerstoffversorgung im Körper. Auch als Bestandteil vieler Enzyme sind Porphyrine von grundlegender Bedeutung für den geregelten Ablauf von Stoffwechselprozessen, besonders in der Leber.
Wie äußert sich eine AHP konkret?
Wie der Name schon sagt, leiden die Patienten oft unter akuten Attacken. Es kommt zu Übelkeit, Erbrechen, diffusen Bauchschmerzen, aber auch zur Ausstrahlung in den Rücken oder die Beine. Dazu kommen mögliche psychische Beschwerden, auch ein roter Urin zählt zu den Symptomen. Lähmungen in Armen oder Beinen, Krampfanfälle oder Atembeschwerden führen dann zur Aufnahme auf die Intensivstation. Atemlähmungen oder Herzrhythmusstörungen sind mögliche lebensbedrohliche Komplikationen.
Wo liegt die Schwierigkeit bei der Diagnosefindung, und gibt es Verwechslungsmöglichkeiten mit anderen Erkrankungen?
autosomal-dominant vererbt. Frauen im Alter zwischen 20 und 30 Jahren sind anfälliger dafür. Da spielen hormonelle Veränderungen im Rahmen des Monatszyklus eine entscheidende Rolle. Circa eine von 100.000 Personen kann daran erkranken. Sinnvoll ist nach einer Diagnose daher die genetische Untersuchung der gesamten Familie. Auf diese Weise lassen sich bei Betroffenen die auslösenden Faktoren leichter meiden und die Beratung verbessern.
Gibt es die Möglichkeit, AHP zu behandeln?
Das komplette Interview sowie einen Patientenbericht lesen Sie unter: seltenekrankheiten.de
Bei Porphyrien handelt es sich um acht verschiedene, meist angeborene Stoffwechselerkrankungen. Bei den Betroffenen gibt es jeweils einen Defekt eines Enzyms der Hämsynthese. In der Folge kommt es zur Anhäufung von Zwischenprodukten. Durch ihre Ablagerungen in Haut, Leber und Nervensystem treten verschiedene Symptome auf.
Die eben beschriebenen Symptome legen natürlich Verwechslungen nahe. Die größte Schwierigkeit liegt darin, dass ein Arzt nicht sofort an diese seltene Erkrankung denkt. Die Diagnose im Labor mit einer Urinprobe ist dann an sich nicht kompliziert.
Handelt es sich um eine erblich bedingte Erkrankung?
Ja, die akuten hepatischen Porphyrien werden
Entscheidend ist, wie gesagt, eine frühe Diagnose. Danach kann man die Faktoren beseitigen, die Attacken auslösen: Nikotin, Alkohol, Stress, Ernährung, Fasten oder Medikamente. Hier ist die Aufklärung wichtig. Idealerweise ist sogar der Patient besser informiert als der nächste behandelnde Arzt, gerade wenn es um die bisherige Behandlung mit Medikamenten geht.
Bei den Medikamenten gibt es seit 2020 ein innovatives Präparat. Es hemmt selektiv die messenger-RNA des ersten Enzyms der Hämsynthese in der Leber und senkt nachweislich chronische Symptome, die Schubhäufigkeit und den Bedarf an Hämarginat.
Lesen Sie mehr auf seltenekrankheiten.de 6
Gen-Stilllegung mit RNA
Die Ursache der meisten seltenen Erkrankungen liegt im Erbgut. Mit konventionellen Behandlungsmethoden lassen sich häufig nur die Symptome lindern. Das Prinzip der RNA-Interferenz ermöglicht einen neuen Ansatz, mit dessen Hilfe sich die Aktivität einzelner Gene gezielt regulieren lässt. Genetisch bedingte Erkrankungen können so ursächlich therapiert werden –ohne dabei das Erbgut zu verändern.
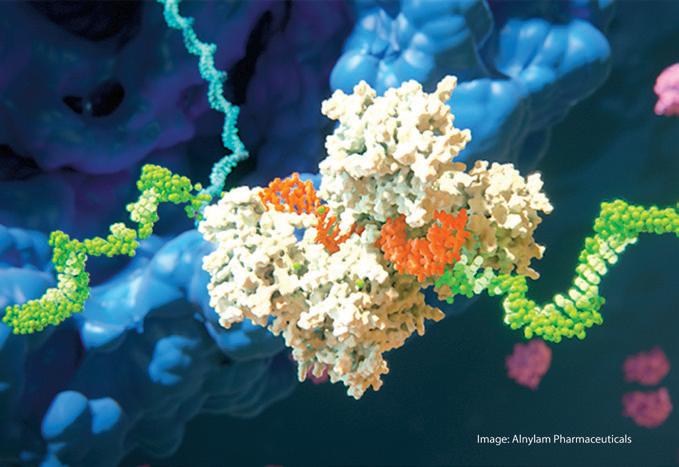
Im vergangenen Jahr hat eine neue Klasse von Impfstoffen auf Basis von Boten-RNA (Messenger-RNA, mRNA) ihren Durchbruch erlebt. Durch das Einbringen von mRNA in die Zellen erhalten diese den Bauplan für ein bestimmtes Virus-Protein, das sie dann selbst herstellen. Gegen diese Proteine erzeugt das Immunsystem anschließend eine Immunantwort. mRNA gibt es in jeder Zelle in Hülle und Fülle. Ihre natürliche Funktion ist es, die im Erbgut gespeicherten Protein-„Baupläne“ an die Protein-„Fabriken“, die Ribosomen, zu übermitteln. Diese Transportfunktion macht die mRNA zu einem Ziel für neue therapeutische Ansätze – weit über Impfstoffe hinaus.
Viele seltene Erkrankungen gehen zurück auf Mutationen im Erbgut. Dadurch können etwa die Baupläne für wichtige Proteine fehlerhaft sein. Diese „defekten“ Proteine können zu schweren Komplikationen im Stoffwechsel führen, zum Beispiel wenn sie toxisch wirken, wie bei der akuten hepatischen Porphyrie, oder aufgrund ihrer veränderten Struktur Ablagerungen (Amyloid) bilden, die wiederum die Funktionsfähigkeit der Organe beeinträchtigen können, zum Beispiel bei der ATTRvAmyloidose.
RNAi-Medizin: Eine neue Klasse von Arzneimitteln
Vor gut 20 Jahren entdeckten Forschende einen natürlichen Mechanismus, mit dem Zellen die Aktivität einzelner Gene steuern können. Dieser Mechanismus wird als RNA-Interferenz (RNAi) bezeichnet. Für ihre Forschung erhielten die US-Wissenschaftler Andrew Z. Fire und Craig C. Mello im Jahr 2006 den Medizin-Nobelpreis. Die Entdeckung der RNA-Interferenz legte den Grundstein für eine völlig neue Klasse von Arzneimitteln.
Die Grundidee ist simpel. Die Aktivität eines für eine Erkrankung ursächlichen Gens wird einfach herunterreguliert. Dies geschieht, indem der Informationsträger – die mRNA – abgebaut wird, bevor er die Ribosomen erreicht. Mittels des zelleigenen Mechanismus der RNA-Interferenz lässt sich präzise genau jene mRNA deaktivieren, die den fehlerhaften Bauplan überträgt. Um diesen Prozess zu aktivieren, wird eine kurze RNA-Sequenz in die Zellen eingebracht. Diese teilt der Zelle mit, welche mRNA abgebaut werden soll. Im Ergebnis wird die Produktion der krankheitsverursachenden Proteine erheblich reduziert. Ein Vorteil der RNA-Interferenz: Im Gegensatz zu einer Gentherapie wird nicht in das Erbgut einge-
griffen. Setzt man die Behandlung aus, wird das betreffende Protein wieder hergestellt. Das Potenzial der RNAi zum Wohle von Patienten nutzbar machen – mit dieser Vision wurde 2002 das biopharmazeutische Unternehmen Alnylam Pharmaceuticals gegründet. Seither hat Alnylam mehr als drei Milliarden US-Dollar in die Entwicklung von RNAi-Therapeutika investiert. Seit 2018 wurden bereits drei RNAi-Therapeutika zur Behandlung seltener, genetisch bedingter Erkrankungen in Europa zugelassen. Weitere sind in Entwicklung. Perspektivisch lassen sich mit RNAi-Therapeutika nicht nur genetische Erkrankungen behandeln, sondern potenziell auch Herz- und Stoffwechselkrankheiten, Infektionskrankheiten und Erkrankungen des zentralen Nervensystems. Dies ist ein gutes Beispiel, wie von der Forschung an seltenen Erkrankungen mittelfristig auch viele weitere Patienten profitieren können.
Erfahren Sie mehr über RNA-Interferenz und die Forschung von Alnylam unter alnylam.de
ANZEIGE
08.2021 PH1-DEU-00041
Kurze RNA-Stränge (orange) teilen der Zelle mit, welche mRNA (grün) gezielt abgebaut werden soll.
Plötzlich (fast) blind
Als Andreas 33 Jahre alt ist, bekommt er Probleme mit den Augen. Er geht zum Augenarzt und erhält den Verdacht Hirntumor. Dass eine seltene Erkrankung dahintersteckt, ahnte zu diesem Zeitpunkt niemand. Heute hat der Elektromeister ein Sehvermögen von einem Prozent. Um welche Erkrankung es sich handelt und wie sich sein Alltag verändert hat, erzählt er im Interview.
Andreas, welche Augenerkrankung haben Sie?
Ich habe die seltene Augenerkrankung Lebersche Hereditäre Optikus-Neuropathie (LHON). Das ist eine genetische Erkrankung der Nervenzellen des Auges, die vor allem junge Männer betrifft. Sie tritt in Deutschland nur circa 80-mal pro Jahr als Neuerkrankung auf.
Wie haben Sie gemerkt, dass etwas mit Ihren Augen nicht stimmt, und welche Beschwerden traten auf?
Im September 2019 traten Sehbeschwerden auf, ich sah teilweise verschwommen und ging zum Optiker, weil ich vermutete, dass ich eine neue Brille benötige. Beim Sehtest konnte ich kaum die Zahlen erkennen. Der Optiker reinigte extra das Gerät, da er nicht glauben konnte, dass ich kaum etwas sah. Doch mit dem Gerät war alles in Ordnung. Er schickte mich zum Augenarzt, der sämtliche Untersuchungen machte und mich dann per Notfallüberweisung ins Krankenhaus schickte wegen des Verdachts

auf Hirntumor. Zum Glück bestätigte sich die Diagnose nicht. Doch warum ich immer schlechter sehen konnte, wusste immer noch niemand.
Wie lange hat es gedauert, bis die Diagnose LHON gestellt wurde, und was hat die Diagnose für Ihr Leben bedeutet?
Es ging wochenlang hin und her und die ständig neuen Verdachtsdiagnosen machten mich wahnsinnig. Die ständige Angst und die immer größer werdende Unsicherheit haben mich sehr viel Kraft gekostet. Im Oktober kam dann der erste Hinweis auf LHON, was dann auch durch einen genetischen Test bestätigt wurde. Zu diesem Zeitpunkt hatte ich noch eine Sehkraft von vier bis fünf Prozent. Doch das ging weiter bergab. Heute habe ich eine Sehkraft von einem Prozent. Mein Leben war quasi von heute auf morgen nicht mehr das gleiche. Ich brauchte sehr lange, um mich mit meinem neuen Leben zu arrangieren. Lange wollte ich es nicht wahrhaben und habe mich immer gefragt: Warum ich?
Wie sieht Ihr Alltag mit der Erkrankung aus, und fühlen Sie sich als Patient mit einer seltenen Augenerkrankung gut versorgt?
Ich lebe in einer Kleinstadt im ländlichen Raum. Hier ist man auf das Auto angewiesen. Doch natürlich kann ich mich als fast blinder Mensch nicht mehr hinters Steuer setzen. Auch der Alltag mit der Familie hat sich natürlich verändert und auch die Arbeit. Doch ich habe das große Glück, dass sowohl meine Frau als auch mein Arbeitgeber, bei dem ich als Kalkulator arbeite, immer hinter mir standen und stehen. Zudem habe ich mich an die PRO RETINA gewandt, die mir sehr viele Hilfestellungen an die Hand gegeben hat und nach wie vor gibt. Nicht allein zu sein, ist ein gutes Gefühl. Ich muss vierteljährlich zum Arzt, der mir mein Medikament verschreibt. Zudem stehe ich mit sechs weiteren Patienten auf der Warteliste für eine derzeit noch nicht zugelassene Therapie. Ich hoffe täglich auf den Anruf.
Lesen Sie mehr auf seltenekrankheiten.de 8
Text Franziska Manske Foto privat
Das LHON-Patientenregister
Forschung und Medizin mit Patienten vernetzen.
Text Hanna Sinnecker
 Dr. Sandra Jansen
Dr. Sandra Jansen
Projektmanagerin und Leitung Patientenregister des PRO RETINA Deutschland e. V.
Was verbirgt sich hinter dem LHON-Patientenregister von PRO RETINA?
Das LHON-Patientenregister von PRO RETINA ist eine Datenbank, über die wir online Daten zur LHON erfassen, die Forscher, Augenärzte und Neurologen einsehen können. Sie wird angeschlossen an unser Patientenregister mit generellen Daten zu Netzhauterkrankungen. Aber wir bringen darüber auch Betroffene und Ausrichter klinischer Studien zusammen. Unser Ziel ist es, einen Standard für Diagnostik und Therapie bei LHON zu entwickeln. Und mein persönliches Anliegen ist es, den Leidensdruck der Patienten zu reduzieren.
Welche Vorteile hat ein Patient, der sich im Patientenregister eintragen lässt, und wie werden die Daten genutzt?
LHON ist eine komplexe Krankheit, die wir verstehen wollen. Es geht um Erkenntnisgewinn, der elementar zu einer Verbesserung von Diagnose und Therapie beiträgt. Gerade bei LHON dauert es oft lange bis zur Diagnose. Bevor die richtige Diagnose steht, ist das Augenlicht meist fort, denn die Sehfähigkeit sinkt im Schnitt in drei Monaten unter zehn Prozent. Hinzu kommt, dass die Daten bis jetzt nur dezentral vorliegen. Die Lösung ist das zentrale Register, das alle Informationen bündelt.
Der Datenschutz ist uns wichtig. Die Datensätze werden pseudonymisiert. Die Daten liegen auf geschützten Servern in Deutschland, die medizinischen auf einem anderen als die persönlichen.
Was sind die Herausforderungen bezüglich eines Patientenregisters, speziell wenn es sich um eine seltene Erkrankung wie LHON handelt?
Die Herausforderung liegt schon in der Seltenheit der Erkrankung. Freiwillige für das Patientenregister zu finden, gestaltet sich dann schon etwas schwieriger. Aber bei dem LHON-Patientenregister handelt es sich um eine multizentrische Registerstudie. D.h. momentan haben wir 14 Augenkliniken in Deutschland unter Vertrag, die für uns LHON-Patienten ins Register aufnehmen.
Weitere Infos unter: www.pro-retina.de

Sie sehen was, was sie nicht sehen.
9 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE
Menschen mit der seltenen Augenkrankheit Lebersche Hereditäre Optikusneuropathie leiden unter verschiedenen Einschränkungen ihres Sehvermögens – bis hin zur Erblindung. Chiesi setzt sich für die Betroffenen ein. Erfahren Sie mehr über unser Engagement im Bereich der seltenen Erkrankungen: www.chiesi.de/seltene-erkrankungen Chiesi setzt für www.chiesi.de/seltene-erkrankungen
Gentherapie bei LHON
Die seltene erbliche Augenkrankheit LHON soll Studien zufolge erstmals ursächlich behandelt werden können. Durchgeführt wurden die randomisierten, placebokontrollierten, doppelblinden Studien an sieben Zentren weltweit, darunter am LMU Klinikum München (Friedrich-Baur-Institut an der Neurologischen Klinik und Augenklinik) unter der Leitung von Prof. Thomas Klopstock. Im Interview spricht er über den aktuellen Stand der Forschung.
Text Paul Howe

Prof. Dr. med.
Klopstock Friedrich-BaurInstitut an der Neurologischen Klinik der Ludwig-Maximilians-Universität (LMU)
Können Sie uns kurz die Erkrankung LHON beschreiben?
Die Lebersche Hereditäre Optikus-Neuropathie (LHON) ist mit einer Häufigkeit von ca. 1:30.000 eine der häufigsten mitochondrialen Erkrankungen. Die Erkrankung kann in jedem Alter auftreten. In der akuten Phase beschreiben die Patienten eine schmerzlose subakute Verschlechterung des zentralen Sehens, häufig auch des Farbensehens, die i. A. zunächst monokulär beginnt und dann innerhalb weniger Wochen oder Monate auch das zweite Auge betrifft. In der Mehrzahl der Fälle bleibt eine hochgradige
Die Diagnosestellung gelingt oft nicht auf Anhieb. Entscheidend ist daher, an die Möglichkeit einer LHON zu denken.
permanente Sehverschlechterung, insbesondere des zentralen Sehens, zurück. Die klinisch-ophthalmologische Diagnosestellung gelingt oft nicht auf Anhieb, meist wird zunächst unter der Verdachtsdiagnose einer Optikusneuritis weitere Diagnostik und Therapie veranlasst. Entscheidend ist daher, an die Möglichkeit einer LHON zu denken, und möglichst schnell den einfachen und kostengünstigen Gentest aus dem Blut zu veranlassen.
Warum ist das Auge für eine Gentherapie bestens geeignet?
Die derzeit per Gentherapie adressierten Augen-Erkrankungen sind auf Netzhaut und Sehnerv beschränkt. Das heißt: Man kann die Gentherapie lokal in das Auge und somit direkt in die Nähe der Zellen injizieren, wo sich die Wirkung entfalten soll. Zudem ist das Auge ein „immun-privilegiertes“ Organ, d.h. es ist sehr unwahrscheinlich, dass die lokale Injektion in das Auge zu einer systemischen Immunreaktion führt.
Gentherapien wecken großes Interesse. Was können Sie zur Sicherheit einer solchen Therapie sagen?
Das größte Risiko besteht bei systemisch verabreichten Gentherapien in einer überschießenden Immunreaktion auf Bestandteile des Gentherapie-Vektors, meist auf den Trägervirus. Die inzwischen meist verwendeten Adeno-assoziierten VirusVektoren (AAV-Vektoren) sind diesbezüglich bereits viel weniger immunogen als früher verwendete Vektoren, doch auch bei AAVbasierten Gentherapien können Immunreaktionen auftreten. Bei lokaler Verabreichung in das Auge ist diese Gefahr deutlich geringer. Insgesamt gilt: Auch wenn die Gentherapien heutzutage relativ sicher sind, muss jeder Ansatz in Studien neu geprüft werden.
Da bei der Gentherapie defekte Gene ausgetauscht oder repariert werden, wird das Problem sozusagen an
Lesen Sie mehr auf seltenekrankheiten.de 10
Thomas
der Wurzel gepackt. Können Sie schon etwas zur Wirksamkeit der Therapie bei LHON sagen, und wie nachhaltig die einmal erzielten Verbesserungen sein könnten? Die Gentherapie wurde inzwischen bei LHON-Patienten mit der Mutation G11778A in mehreren klinischen Studien untersucht. Die einmalige, unilaterale, intravitreale Injektion des Gentherapie-Vektors (ND4-cDNA verpackt in rekombinanten Adenoassoziierten Virus 2, rAAV2) war in zwei Phase-3-Studien gut verträglich und wirksam. Bei 37 LHON-Patienten, die die Injektion 6-12 Monate nach Symptombeginn erhielten, fand sich nach 96 Wochen eine Verbesserung der Sehkraft des injizierten Auges um im Mittel 15 Buchstaben auf der Sehtafel und des kontralateralen Auges um 13 Buchstaben. Als Erklärung für den kontralateralen Effekt fand sich in Primatenversuchen ein Transfer des Gentherapie-Konstrukts über die Sehnervenkreuzung. Ähnlich positive Ergebnisse fanden sich bei weiteren 38 Patienten mit unilateraler Injektion weniger als sechs Monate nach




Symptombeginn. Auch nach mehrjähriger Nachverfolgung schneiden die behandelten Patienten deutlich besser ab als im natürlichen Verlauf der Erkrankung. Eine Zulassung des Gentherapeutikums ist beantragt.
Aktuell ist die Therapie in Deutschland noch nicht verfügbar. An wen können sich Patienten wenden, um möglichst frühzeitig behandelt zu werden?
Die randomisierten Studien sind erfolgreich beendet, eine Zulassung ist bei der EMA beantragt. Um die Zeit bis zur Zulassung zu überbrücken, planen wir ein sog. ExpandedAccess-Programm (EAP). Solche Programme bieten Patienten mit Krankheiten, für die es noch keine Behandlungsmöglichkeiten gibt, Zugang zu Präparaten außerhalb von klinischen Studien und vor der Einführung des Medikaments.
Patienten mit LHON und der Mutation 11778 können sich per E-Mail an fbi@med.uni-muenchen.de wenden.

INNOVATIVE THERAPIEANSÄTZE
BEI SELTENEN NETZHAUTERKRANKUNGEN
LHON betrifft hauptsächlich Jugendliche und junge Erwachsene, vorwiegend Männer. Die Erkrankung ist mit einem schmerzlosen, plötzlichen Verlust des zentralen Sehvermögens in einem Auge und einer rasch einsetzenden Beeinträchtigung des zweiten Auges verbunden

Gensight Biologics, ein Biopharma-Unternehmen aus Frankreich, hat sich auf die Forschungsarbeit an neurodegenerativen Augenerkrankungen und Erkrankungen des zentralen Nervensystems spezialisiert. Die innovativen Therapieansätze fokussieren sich dabei besonders auf Patienten mit Leberscher hereditärer Optikusneuropathie (LHON) und Retinis Pigmentosa.
International und auch unter Beteiligung deutscher Forscher wird derzeit eine neue, noch nicht zugelassene Gentherapie klinisch erprobt, die sich speziell auf ProbandInnen fokussiert, die an der schwersten klinischen Form der LHON (ND4-LHON) erkrankt waren. Die Ergebnisse sind vielversprechend und bilden die Grundlage für den Zulassungsantrag und die Freigabe für den Einsatz an qualifizierten Zentren für Seltene Erkrankungen.
11 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE
Meine Realität ist hier und jetzt
Carolin hat spinale Muskelatrophie (SMA). Seit ihrem ersten Lebensjahr hat das sowohl ihr Leben als auch das ihrer Familie komplett verändert. Im Interview gibt sie uns einen Einblick in ihren Alltag, der eigentlich ziemlich normal ist.
Text Hanna Sinnecker
Sie sind von der seltenen Erkrankung SMA betroffen. Wann wurde die Erkrankung bei Ihnen diagnostiziert?
Meine Eltern stellten erste Auffälligkeiten fest, als ich mit ca. sechs Monaten nicht anfing, zu krabbeln oder mich selbstständig zu drehen. Damals hieß es jedoch noch, ich sei „einfach etwas faul“, und so begann für meine Eltern eine regelrechte Odyssee. SMA war zu diesem Zeitpunkt noch kaum erforscht, konnte erst seit wenigen Jahren dem richtigen Chromosom zugeordnet werden. Bei mir wurden sehr viele Tests gemacht und anfangs wussten die Ärzt*innen wohl nicht so genau, wonach sie überhaupt suchen. Für meine Familie bedeutete das monatelange Ungewissheit. Mittels einer Muskelbiopsie konnte dann schließ-

lich die richtige Diagnose gestellt werden. Da war ich ein Jahr alt. Heute geht das viel einfacher. Es reicht eine kleine Blutentnahme und schon weiß man, was Sache ist.
Wie hat sich die Erkrankung auf Ihr Leben ausgewirkt?
Natürlich wirkt sich die SMA sehr stark auf mein Leben aus, da ich in jeder Lebenssituation auf die Unterstützung von anderen angewiesen bin. Ich sitze im Rollstuhl, kann mittlerweile nicht mal mehr einen Arm heben und brauche unterwegs beispielsweise immer eine Begleitung.
Gleichzeitig ist Selbstbestimmung ein wichtiges Thema für mich und ich versuche, mein Leben so unabhängig wie nur möglich zu gestalten. Oft ist
es aber auch gar nicht meine Behinderung, die mich davon abhält, sondern vielmehr die deutsche Bürokratie oder fehlende Inklusion.
Ich würde sagen, meine SMA hat mich darauf vorbereitet, dass man im Leben nicht immer das bekommt, was man gerade möchte. Meine Erkrankung hat mir beigebracht, mich mit Gegebenheiten zu arrangieren, auf die ich keinen Einfluss habe, und dennoch nach Möglichkeiten zu suchen, wie ich meine Ziele erreichen kann. Wer weiß, vielleicht hätte ich ohne diese Diagnose im Ausland studiert oder eine Weltreise gemacht. Alles Dinge, die sich als Rollstuhlfahrerin doch etwas schwieriger gestalten. Aber egal ob mit oder ohne Behinderung, man kann das Leben nie vorhersagen. Ich weiß nicht, wo
Lesen Sie mehr auf seltenekrankheiten.de 12
FOTO:
@CAROLIN.CORALINART
ich heute ohne SMA wäre. Allerdings interessiert es mich auch nicht (mehr). Meine Realität ist hier und jetzt.
Sie leben nun bereits seit Jahren mit der Erkrankung. Wie sieht Ihr Alltag derzeit aus, und fühlen Sie sich medizinisch gut versorgt?
Tatsächlich würde ich meinen Alltag als ziemlich normal beschreiben. Ich arbeite im Homeoffice und mache in meiner Freizeit all die Dinge, die für junge Frauen selbstverständlich dazugehören: Reisen, Konzerte besuchen, Freund*innen treffen oder shoppen gehen. Momentan wohne ich noch bei meinen Eltern, aber Ende nächsten Jahres möchte ich in eine eigene Wohnung ziehen.
Was die medizinische Versorgung betrifft, bin ich zufrieden und fühle mich in meiner lokalen Klinik stets gut aufgehoben. Dort bin ich bereits seit meiner Kindheit Patientin, und
weil mich das Personal so gut kennt, kann ich nun auch als Erwachsene noch dort behandelt werden. Allerdings lebe ich in einer ländlichen Gegend und deswegen mangelt es leider an Spezialist*innen, die bei einer seltenen Erkrankung weiterhelfen können. Die meisten Neurolog*innen verweisen einen nur an große Unikliniken, die meistens Hunderte von Kilometern entfernt sind. Wenn ich also beispielsweise ins Schlaflabor muss oder eine neue Therapie beginne, ist das mit viel Aufwand verbunden. Ich würde mir wünschen, dass die Versorgung auf dem Land genauso gut wäre, wie in der Stadt.
Welche Rolle spielt für Sie die Vernetzung mit anderen Betroffenen? Ich empfinde den Austausch mit anderen Betroffenen als sehr bereichernd! Mit meinen Freund*innen, die ebenfalls SMA haben, kann ich
Die Welt bewegen trotz
Spinaler Muskelatrophie
Unabhängig im Leben stehen – das wünschen sich alle Eltern für ihre Kinder. Doch was, wenn das Kind aufgrund einer seltenen Muskelerkrankung nicht stehen kann? „Wenn meine Tochter etwas erreichen will, dann gibt sie alles“ – davon ist Klaus, Vater einer SMA-Patientin, felsenfest überzeugt. Trotz SMA sind sie eine normale Familie – mit einer normalen, nur anders normalen, Tochter.
Menschen mit Spinaler Muskelatrophie (SMA), einer erblich bedingten neuromuskulären Krankheit, leben ihr Leben oft seit frühster Kindheit anders: Dinge, die selbstverständlich scheinen – laufen, gehen, stehen – sind für sie schwierig oder gar unmöglich. Wie bei einem komplexen Mechanismus, dem ein Zahnrad fehlt und der deswegen stillsteht, fehlt dem Körper
ein für Bewegung essenzielles Eiweiß. Ohne dieses Eiweiß sterben bestimmte Nervenzellen im Rückenmark – und damit bricht die Kommunikation zu den Muskeln ab.
Für Betroffene äußert sich dies in Muskelschwäche und Muskelschwund, viele sind auf einen Rollstuhl angewiesen. Bei man-
„Sie ist ein normales Kind – eines, das natürlich ab und an mal zusätzliche Unterstützung braucht.“
chen SMA-Patienten können die Beeinträchtigungen anfangs geringer sein – die Zahnräder drehen sich mit Mühe weiter. Unter der Oberfläche schreitet die Erkrankung jedoch immer weiter voran. Ein Lichtblick: Moderne Therapien können das Voranschreiten verlangsamen. Und die Betroffenen denken
Lebensqualität und Glück haben nichts mit Muskelkraft zu tun!
über Dinge sprechen, die sonst niemand versteht. Immerhin machen sie die gleichen Erfahrungen, werden mit den gleichen Problemen konfrontiert und können sich deswegen besser in meine Gefühlswelt hineinversetzen. Außerdem helfen wir uns häufig gegenseitig, wenn es um medizinische Themen, Hilfsmittel, Rechtliches etc. geht.
nicht daran aufzugeben: „Tomke ist ein ganz normales elfjähriges Mädchen – aufgeweckt und fröhlich“, so Klaus über seine Tochter, die seit dem ersten Lebensjahr Symptome von SMA aufweist.
Rund 1.600 Menschen in Deutschland trotzen täglich dieser schweren Erkrankung. Viele davon sind sehr jung und müssen ihren Weg durchs Leben mit SMA noch finden. „Es ist natürlich eine etwas andere Kindheit“, so Klaus. „Ich glaube aber, sie wachsen ganz normal auf – sie wachsen trotzdem sehr glücklich auf.“
Ihre Zukunft werden junge Menschen mit SMA eigenständig wählen: Im digitalen Zeitalter haben sie gute Chancen, erfolgreich zu sein, sich selbst zu verwirklichen und ihre Unabhängigkeit zu bewahren. Und ein wenig Unterstützung braucht jeder von uns ab und an.
Mehr Informationen zum Leben mit SMA, Alltagstipps oder News aus der Forschung gibt es auf www.FaceSMA.de.

13 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE
„Meine Krankheit hat mich stark gemacht“

Nadine Großmann ist 29 Jahre alt und Doktorandin für Biochemie an der FU Berlin. Sie forscht an einer sehr seltenen Erkrankung, von der die meisten wahrscheinlich noch nie gehört haben: Fibrodysplasia ossificans progressiva (kurz FOP). Das Besondere daran: Nadine Großmann ist selbst betroffen und erforscht damit ihre eigene Erkrankung. Wir sprachen mit ihr über den Weg bis zur Diagnose, über Behandlungsfehler und die so wichtige Forschungsarbeit im Bereich der seltenen Erkrankungen.
Text Franziska Manske
Sie leben mit der seltenen Erkrankung FOP. Was ist das für eine Krankheit?
Die FOP ist eine seltene, erbliche Erkrankung. Sie lässt Knochen an Stellen wachsen, wo sie nicht hingehören, sowie Muskeln, Binde- und Stützgewebe fortschreitend verknöchern, sodass Erkrankte häufig in ihrer Bewegung eingeschränkt sind.
Wann haben Sie das erste Mal bemerkt, dass etwas nicht stimmt? Mir ist das gar nicht aufgefallen, sondern meiner Mama. Als ich 13 Jahre alt war, hat sie bemerkt, dass ich vornübergebeugt laufe. Schmerzen oder andere Symptome hatte ich nicht. Wir sind erst zum Hausarzt und dann zum Orthopäden gegangen. Keiner konnte die Ursache für meinen humpelnden Gang herausfinden. Ich wurde dann zur Uniklinik geschickt, und durch Röntgenbilder, auf denen der zusätzliche Knochen zu sehen war, sowie histologische Untersuchungen wurde die FOP dann diagnostiziert. Das war circa ein halbes Jahr nach dem ersten Arztbesuch. Und das hat sich
dann auch später durch eine genetische Analyse bestätigt. Die Diagnosefindung ist für eine sehr seltene Erkrankung wie FOP sehr kurz. Durchschnittlich dauert es 18 Monate.
Wie ging es danach weiter: Haben Sie sich ärztlich gut betreut gefühlt?
Leider gar nicht, denn ich wurde komplett falsch behandelt.
Inwiefern?
Ich wurde dreimal operiert, was das Allerschlimmste ist, was man bei einer FOP machen kann, denn diese OPs können weitere Knochenschübe auslösen. Das ist ein Punkt, den ich bis heute nicht verstehe. Sie haben meine Erkrankung richtig diagnostiziert, aber komplett falsch behandelt. Das waren auch keine kleinen Operationen, sondern sehr komplizierte, die jeweils rund sieben Stunden gedauert haben und bei denen auch wichtige Nerven hätten irreparabel verletzt werden können.
Was wäre denn die richtige Therapie
gewesen?
Nur beobachten. Wenn ein akuter Schub ansteht, kann man versuchen, diesen mit Kortison zu unterdrücken. Es gibt zudem verschiedene Medikamente, die zum Stoppen der Entzündung führen können. Die Behandlung bei FOP ist sehr individuell und von Patient zu Patient unterschiedlich. Eine Therapie oder Heilung gibt es leider noch nicht.
Wie sind Sie in jungen Jahren mit der Erkrankung umgegangen?
Es war mir total egal (lacht). Ich habe mein Leben einfach weitergelebt, machte alles, worauf ich Lust hatte. Für meine Eltern war es viel schwerer, weil sie sich große Sorgen um die Folgen gemacht haben.
Ihre berufliche Laufbahn ist eng mit Ihrer Krankheitsgeschichte verbunden. Wann stand für Sie fest, dass Sie sich der Forschung an FOP verschreiben wollen?
Das kam durch meinen Bruder. Er ist Bioinformatiker und war für seine
Lesen Sie mehr auf seltenekrankheiten.de 14
FOTO: PRIVAT
Doktorarbeit in den USA. Dort hat er sich auf die Suche nach einem FOP-Experten gemacht und wurde auch fündig. Als ich ihn in den USA besuchte, hat er ein Treffen organisiert, und das war für mich ein Aha-Erlebnis. Seitdem wollte ich auch etwas zur Forschung an FOP beitragen, und das mache ich jetzt.
Die Diagnose ist 16 Jahre her. Wie geht es Ihnen heute?
Es hat sich zum Glück nur wenig verschlechtert, da ich einen sehr milden Verlauf habe. Ich bin sehr mobil, lebe allein, bin selbstständig und gehe arbeiten. Derzeit habe ich drei Gelenke, die betroffen sind, doch diese schränken mich in meinem alltäglichen Leben nicht zu sehr ein. Natürlich weiß ich nicht, wie meine
FOP in den nächsten Jahren voranschreitet. Aus diesem Grund genieße ich das Leben jetzt und denke nicht darüber nach, was irgendwann sein könnte.
Zudem muss ich sagen, dass mich die Erkrankung stärker gemacht hat. Wenn ich Rückschläge habe – beruflich oder privat –, kann ich das viel besser wegstecken als früher. Ich schaue immer nach vorn.
Wenn Sie auf Ihre eigene Geschichte zurückblicken: Was möchten Sie anderen Betroffenen mit auf den Weg geben?
Es lohnt sich immer zu kämpfen. Man sollte Ärzten nicht uneingeschränkt vertrauen. Ich weiß, das ist hart, aber es lohnt sich, auch selbst zu recherchieren und kritisch zu sein. Man muss sehr viel
Eigeninitiative zeigen, denn natürlich kann nicht jeder Arzt alle 7.000 seltenen Erkrankungen kennen. Aus diesem Grund ist es mir auch so wichtig, dass Patienten gehört werden und eine laute Stimme haben. Am wichtigsten ist mir jedoch, dass das größte Merkmal für eine FOP schon in die U1, also die erste Untersuchung nach der Geburt eines Babys, eingeführt wird. Denn ist der große Zeh verkürzt und/oder nach innen gebogen, kann das ein Hinweis auf eine FOP sein und sollte unbedingt genetisch abgeklärt werden.
Nadine Großmann ist stellvertretende Vorsitzende des FOP e. V. Weitere Informationen unter: fop-ev.de Das ganze Interview lesen Sie auf: seltenekrankheiten.de
FOP: Die unaufhaltsame Verknöcherung
Die Fibrodysplasia ossificans progressiva (FOP) ist eine der seltensten erblichen Erkrankungen. Laut aktueller Studienlage sind zwischen 0,881 und 1,362 Menschen pro 1 Mio. Einwohner betroffen, in Deutschland sind laut der Patientenvereinigung FOP e.V. derzeit 44 Patient*innen bekannt. Die limitierte Zahl der Patient*innen erschwert die Forschung enorm, da zur Durchführung der erforderlichen Studien eine ausreichend große Zahl an Patient*innen notwendig ist und die klinische Erprobung möglicher Therapien erhebliche Ressourcen erfordert. Ein Gespräch mit FOP-Experte Dr. med. Rolf Morhart.
Text Paul Howe
Dr. Morhart, wen trifft die Erkrankung, wie zeigt sie sich?
Schon bei Neugeborenen weisen verkürzte und zur Fußaußenseite abgeknickte große Zehen (sog. Hallux valgus) auf FOP hin. Im Kleinkindalter zeigen sich dann häufig faustgroße, gerötete, überwärmte Beulen vorwiegend am Kopf, Nacken und Rücken. Dies sind Entzündungsherde, die in manchen Fällen aber als Verhärtungen bestehen bleiben. Dort bildet sich dann fälschlicherweise zusätzliche Knochensubstanz. Mit fortschreitendem Alter breiten sich diese Entzündungsherde auf den ganzen Körper aus.
Warum ist eine möglichst frühe Diagnose so wichtig?
Da der Körper der Patient*innen mit
FOP mit jeder Entzündung unbeweglicher wird und schubweise oder schleichend in unregelmäßiger Form versteift, wird der Zustand irgendwann lebensbedrohlich: In einem schon im Kleinkindalter verknöcherten Brustkorb kann kein Herz auswachsen, keine Lunge sich entfalten. Je eher die Diagnose erfolgt, desto eher können die Patient*innen bekannte Auslöser von Entzündungen zu meiden versuchen. Dazu werden Schäden durch falsche therapeutische Maßnahmen bei Fehldiagnosen vermieden. Wir wissen heute genau, wo der Gendefekt der FOP liegt. Ein Gentest aller Neugeborenen mit Hallux valgus könnte sehr früh zur sicheren Diagnose führen. Leider ist diese Erkrankung aber bei Medizinern wie bei Laien weitgehend unbekannt.
Wie behandeln Sie FOP derzeit? Die Krankheit ist noch nicht heilbar. Wir behandeln vorwiegend mit Medikamenten, die Entzündungen hemmen. Inzwischen werden aber verschiedene FOP-Medikamente in klinischen Studien getestet. Ich sehe deshalb optimistisch in die Zukunft.
Die Entwicklung und Zulassung eines solchen Medikamentes ist medizinische Pionierarbeit, die stets die Betroffenen im Blick hat. Die Schwierigkeit liegt aber nach wie vor in der Bereitstellung und Bündelung der erforderlichen Ressourcen, um diese Arbeit schneller voranzutreiben und Patient*innen entsprechend versorgen zu können.
15 Lesen Sie mehr auf seltenekrankheiten.de
1 Pignolo et al. 2021; 2 Baujat et al. 2017 ALL-DE-000678
Dieser Beitrag entstand mit freundlicher Unterstützung der IPSEN PHARMA GmbH
Nicht-dystrophe Myotonien: Wenn plötzlich alles stillsteht
Nicht-dystrophe Myotonien (NDM) sind eine Gruppe seltener erblicher Erkrankungen. Das Hauptsymptom: Betroffene sind aufgrund der Krankheit nicht fähig, die der körperlichen Bewegung dienenden Muskeln (Skelettmuskulatur) nach der Kontraktion sofort wieder zu entspannen. Dies führt zu einer Blockade der Muskulatur, welche die Mobilität und Lebensqualität der Betroffenen negativ beeinflusst. Ein Gespräch mit dem Experten Prof. Dr. med. Benedikt Schoser.
Text Hanna Sinnecker

Prof. Dr. med. Benedikt Schoser Oberarzt
Friedrich-BaurInstitut an der Neurologischen Klinik und Poliklinik des LMU Klinikums München
Wie äußern sich NDM, wie wirken sie sich auf den Alltag Betroffener aus und was sind die Gefahren für Betroffene?
Zunächst ist vielen Ärzten, aber auch den Betroffenen unklar, was mit dem Patienten bzw. ihnen nicht stimmt. Sie haben gegebenenfalls eine recht gute Muskelkraft, sehen muskulös-sportlich aus, aber können weder schnell loslaufen, zum Beispiel an einer Ampel, im Sport oder beim Treppensteigen, noch schnell die Faust öffnen, Finger schnell bewegen und verkrampfen
NDM-Patienten wissen oft erst nach dem Start einer Therapie, wie sich ein Leben mit weniger oder gegebenenfalls ohne Myotonie wirklich anfühlt.
sich beim Schreiben oder Schneiden. Viele Betroffene erleben das bereits in früher Kindheit und werden damit erwachsen. Sie waren nie so richtig gut im Schulsport und anderen sportlichen Aktivitäten, ein Musikinstrument spielen war schwierig, und bei manch einem war das Schlucken, die Lidöffnung oder das Kauen „verkrampft“. Immer wieder gab es aber auch Phasen, in denen die bereits genannten Symptome weniger oder auch mal kurzfristig gar nicht vorhanden waren. Oft wissen die Betroffenen selbst ja erst nach Start einer antimyotonen Therapie, wie sich ein Leben mit weniger oder gegebenenfalls ohne Myotonie wirklich anfühlt und zu welchen körperlichen Leistungen sie dann ohne Probleme in der Lage sind.
Bei seltenen Erkrankungen ist die Zeit bis zur Diagnose oft lang, da sie selbst für erfahrene
Mediziner nicht leicht zu erkennen sind. Wie lange dauert es im Schnitt, bis NDM-Patienten diagnostiziert werden?
Bis vor wenigen Jahren waren es in der Regel drei bis zehn Jahre, bis die Diagnose gesichert war. Heute sind es immer noch zwei bis drei Jahre im Durchschnitt.
Die einzige Möglichkeit, diese immer noch sehr lange Zeit zu verkürzen, ist die kontinuierliche Weiterund Fortbildung. Denn um eine Diagnose stellen zu können, muss man die Symptome kennen und zuordnen können, um dann eine genetische Diagnostik einzuleiten.
Was sind die Herausforderungen bei der Diagnosestellung?
Die Myotonie stellt eine Dekontraktionshemmung der Skelettmuskulatur dar. Dieses Nicht-loslassenKönnen nach einer dynamischen oder statischen Muskelanspannung wird oft
Lesen Sie mehr auf seltenekrankheiten.de 16

weder von Ärzten noch dem Patienten selbst verstanden und adäquat als ein spezifisches Krankheitssymptom wahrgenommen und eingeordnet. Oft berichten Patienten nur von einem Muskelschmerz oder einem Verkrampfungsgefühl der Muskulatur nach Belastung. Erst ein Neurologe oder Neuropädiater wird in der Regel die Zuordnung der Symptome zu einem myotonen Syndrom leisten. Gegebenenfalls kann dann die Nadeluntersuchung der Muskulatur (Elektromyografie, EMG) bereits eine Diagnosesicherung liefern. Die endgültige Diagnosesicherung erfolgt heute über eine Genanalyse aus dem Blut.
Wie sehen die derzeitigen Therapieoptionen aus, und können Betroffene unter Therapie ein weitestgehend normales Leben führen?
Wir haben heute unterschiedliche antimyoton wirksame Medikamente, die erfolgreich in der Behandlung einer Myotonie eingesetzt werden können. Durch diese Medikamente wird der Spannungszustand der Muskulatur herabgesetzt und es kommt zu einer Verbesserung der Dekontraktion nach Muskelanspannung, sodass eine gebesserte dynamische Kraftentwicklung und Leistungsfähigkeit erzielt werden kann. Nicht allen, aber sehr vielen Betroffenen
hilft die täglich wiederholte Einnahme dieser Medikamente, um eine deutlich verbesserte Lebensqualität zu erreichen.
Was wünschen Sie sich bezüglich der Versorgung von Menschen mit NDM?
Das müssen Sie in erster Linie die Betroffenen selbst fragen. Ich wünsche mir, dass alle Betroffenen diagnostiziert werden, langfristig adäquat fachärztlich versorgt werden und ihnen eine medikamentöse Therapie angeboten wird. Die zusätzlichen Angebote der Selbsthilfeorganisationen wie „Mensch und Myotonie e. V.“ und der „Deutschen Gesellschaft für Muskelkranke e. V.“ sind hier sehr
wichtig und sollten allen Betroffenen bekannt sein.
Haben Sie die Hoffnung, dass es irgendwann eine ursächliche Therapie geben wird, die die Erkrankungen heilbar macht?
Es sind alles sehr seltene erbliche Erkrankungen, die zumindest schon eine ganz gut funktionierende symptomatische Therapie haben, das haben wir in dieser Form für viele andere Muskelerkrankungen nicht. Wünschenswert wäre natürlich auch hier eine spezifische Gentherapie, wenn wir die nötige Sicherheit dann einmal für diese Therapiemethode haben.


17 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE
Morbus Fabry – das Chamäleon unter den seltenen Krankheiten
Bei der Erkrankung Morbus Fabry kommt es zur übermäßigen Speicherung von Stoffwechselprodukten.
Im Interview erklärt Dr. med. Jessica Kaufeld, Nierenexpertin aus dem Fabry-Zentrum der Medizinischen Hochschule Hannover, warum das dazu führt, dass sich die Krankheit vielfältig wie ein Chamäleon zeigt.
Warum gilt Morbus Fabry als das Chamäleon unter den seltenen Erkrankungen?

Dr. med.
Jessica Kaufeld
Fachärztin für Innere Medizin und Nephrologie, Klinik für Nierenund Hochdruckerkrankungen, Medizinische Hochschule Hannover (MHH)
Die erblich bedingte Speichererkrankung Morbus Fabry führt zu Störungen beim Abbau bestimmter Fette (Lipide). Insbesondere das Globotriaosylceramid (kurz GB3) lagert sich übermäßig stark in einer Vielzahl von Organen ab. Das beeinträchtigt nach und nach deren Funktion. Je nachdem welches Organ betroffen ist, ergeben sich andere Symptome. Die Erkrankung erscheint daher so vielfältig wie ein Chamäleon.
Was passiert bei der Erkrankung im Körper?
Die Stoffwechselstörung beruht auf einem Mangel am Enzym Alpha-Galaktosidase A. Das sorgt normalerweise dafür, dass Fettstoffe aufgespalten und verarbeitet werden können. MorbusFabry-Patient*innen stellen das Enzym kaum bis gar nicht her. Dies führt unter anderem zu Herz-, Nieren- und Nervenproblemen. Daher spricht man im weiteren Verlauf auch von einer Multiorganerkrankung.
Was sind erste Anzeichen für einen Morbus Fabry?
Klassische Anzeichen sind beispielsweise Brennschmerzen in Händen und Füßen und ein spezieller Hautausschlag (stecknadelkopfgroße dunkelrotviolette Papeln). Häufig berichten Patienten von Herzproblemen wie Herzrasen, Magenproblemen, Müdigkeit und Erschöpfung.
Findet man keine gute Erklärung, sollte man an Morbus Fabry denken.
Viele Morbus-FabryPatient*innen leiden Jahre, bis sie endlich ihre Diagnose bekommen. Woran liegt das?
Die Vielfalt möglicher Symptome ist immens und viele davon könnten durchaus auch andere Ursachen haben. Meist kommt es erst zur Diagnose, wenn sich mit Fortschreiten der Erkrankung immer mehr Beschwerden zeigen und diese ganzheitlich und von Mediziner*innen verschiedener Disziplinen gemeinsam betrachtet werden. Bei unseren Patient*innen kann der Leidensweg bis dahin im Schnitt bis zu zwölf Jahre dauern.
Wie lässt sich der Leidensweg abkürzen?
Mit Aufklärung. Denn ein früher Verdacht könnte schneller zur sicheren Diagnose und damit zur Behandlung führen. Wir wissen längst, dass der Morbus Fabry von einem Gendefekt verursacht wird und dass das veränderte Gen auf dem X-Chromosom der Geschlechtschromosomen sitzt. Deshalb könnte auch der Hinweis eines Familienmitgliedes mit Symptomen dienlich sein. Oder das Wissen einer Ärztin oder eines Arztes darüber, dass zum Beispiel der Nachweis von Eiweiß im Urin nicht nur unnormal ist, sondern ein Anzeichen für Morbus Fabry sein kann. Wer mit einem solchen Befund bei uns im
Zentrum nachfragt, sei es der*die behandelnde Arzt*Ärztin oder der*die Betroffene selbst, kann sofort mit der Hilfe und Expertise eines multidisziplinären Teams rechnen.
Wie behandeln Sie Morbus Fabry?
Morbus Fabry lässt sich mit einer lebenslangen Enzymersatztherapie als Infusion behandeln. Eine alternative Therapie besteht in einer Kapsel zum Einnehmen, die die Enzymaktivität unterstützt, aber nur für spezielle Fabry-Patienten geeignet ist (sog. Chaperontherapie). Über die Indikation und die Art der Behandlung entscheidet das Fabry-Zentrum. Die Therapien haben möglicherweise auch Nebenwirkungen, die ebenfalls durch die Spezialisten überwacht werden müssen.
Was wünschen Sie MorbusFabry-Patient*innen für die Zukunft?
Ich wünsche mir schnellere Diagnosen und damit kürzere Leidenswege für die Patient*innen. Ganz weit oben auf meiner Wunschliste stehen zudem Therapieformen, die leichter oder seltener anzuwenden sind. Weniger Nebenwirkungen sind ebenso wünschenswert. Voller Hoffnung schaue ich derzeit auf die Arbeit der Kolleg*innen in der Forschung, denn neue Methoden in der Diagnostik und den Therapien für Morbus Fabry sind schon in der klinischen Erprobung.
Lesen Sie mehr auf seltenekrankheiten.de 18
Text Doreen Brumme
Wenn die Diagnose 18 Jahre dauert
Ein Gespräch mit Conny Rudolph, Morbus-Fabry-Patientin, über die jahrelange Odyssee bis zur Diagnose ihrer seltenen Erkrankung und ein neues Leben mit der Therapie.
Text Hanna Sinnecker

Sie sind Morbus-Fabry-Patientin und haben einen langen Leidensweg hinter sich. Wann wurde die Diagnose gestellt?
Das ganze Interview lesen Sie auf: seltenekrankheiten. de
Ich habe die Krankheit wohl seit meiner Kindheit. Die Diagnose habe ich aber erst Ende 2017 erhalten. Als Kind litt ich unter Schmerzen, mit Anfang 20 kamen Migräne und Schmerzkrisen an Händen und Füßen hinzu. 2003 hatte ich einen Schlaganfall, der zu spät erkannt wurde. Dann wurden die Schmerzen immer schlimmer. Man diagnostizierte eine Ablösung der Netzhaut, korrigierte den Befund aber wieder. 2015 war der berufliche und private Stress so groß, dass ich wegen Depressionen
medikamentös behandelt wurde. Allerdings merkte ich, dass die Antidepressiva meine Schmerzen minderten. Da wurde meine Neurologin hellhörig. Daraufhin diagnostizierten Ärzte jedoch fälschlicherweise erst MS und dann vaskuläre Demenz. Irgendwann entschied sich meine Neurologin für eine Genomuntersuchung. Sie teilte mir nach 18 Jahren meine Diagnose mit: Ich habe Morbus Fabry.
Wie ging es danach weiter?
Ich musste ein halbes Jahr auf einen Termin in einem medizinischen Zentrum warten. Dort teilten die Ärzte mir mit, dass mein Morbus Fabry nicht krank-
heitsrelevant sei, ich bekam keine Behandlung. 2019 schlug mir meine Neurologin einen zweiten Versuch in einem Zentrum in Dresden vor. Im Juli 2020 wurde ich dann endlich behandelt, sechsmal in der Klinik alle 14 Tage. Seit Oktober 2020 therapieren mich Krankenschwestern bei mir zu Hause. Diese Therapie mit Medikamenten erhalte ich nun ein Leben lang.
Wie sieht Ihr Leben nun aus? Ich bin äußerst zufrieden. Der Umgang mit meinen Schmerzen ist um Welten besser. Seit vielen Jahren kann ich endlich richtig schlafen.
Weitere Infos unter: www.fabry-shg.org
Morbus Fabry in der Familie?
Informationen für Betroffene und deren Angehörige
Morbus Fabry ist eine genetische Erkrankung, die über mehrere Generationen einer Familie vererbt werden kann. Das heißt: Wenn eine Person in einer Familie die Diagnose Morbus Fabry hat, können andere Familienangehörige ebenfalls betroffen sein1. Eine ausführliche Analyse des Familienstammbaums ist daher sehr wichtig für Betroffene und deren Angehörige.
Ich bin betroffen – Was nun?
Ist die Diagnose Morbus Fabry gestellt, dann ist es für Betroffene wichtig zu wissen, was die eigene Diagnose für Familienangehörige bedeuten kann und wer aufgrund des Vererbungsmusters ein erhöhtes Risiko für Morbus Fabry hat. Hier kommt die neue Website www. fabryfamilytree.de ins Spiel, die Betroffenen umfassende Informationen und Hilfestellungen an die Hand geben möchte. Dazu gehören grundlegenden Informationen, wie die Erkrankung vererbt wird und wer in der Familie ein erhöhtes Risiko hat. Über ein Online StammbaumTool kann man zusammen mit seinem behandelnden Arzt seinen individuellen Fabry-Stammbaum erstellen und für die persönliche Nutzung herunterladen, um Angehörige mit erhöhtem Fabry-Risiko gezielt informieren zu können. Die Daten werden streng vertraulich behandelt. Die Website gibt professionelle Hilfestellung, wie man Angehörige mit erhöhtem Risiko dann darauf ansprechen und sie aufklären kann. Dazu gehört auch eine Briefvorlage, die man nutzen

kann, wenn eine direkte Ansprache sich schwierig gestalten sollte.
Informationen für Familienangehörige mit erhöhtem Fabry Risiko
Auf der Website gibt es aber auch für Angehörige von Morbus Fabry-Patienten detaillierte Informationen, die dabei helfen sollen, die Erkrankung zu verstehen und warum sie selbst ein erhöhtes Risiko haben. Dabei ist eines sehr wichtig: ein erhöhtes Risiko bedeutet nicht zwangsläufig, dass man tatsächlich auch betroffen ist. Daher sollten Angehörige, die laut Stammbaum ein erhöhtes Risiko haben, unbedingt einen Arzt ansprechen und eine genetische Analyse durchführen lassen. Das kann der eigene Hausarzt oder aber der Fabry-Spezialist des betroffenen Angehörigen sein.
Informationen für das Fachpersonal Aber auch medizinisches Fachpersonal findet auf der Website Materialien und Hilfestellungen, wenn es darum geht, Fabry-Patienten oder deren Angehörige zu beraten und aufzuklären. Dazu gehört ebenfalls die Nutzung des Online StammbaumTools in Zusammenarbeit mit dem Patienten, sowie weitere Broschüren, die beim Familienscreening unterstützen sollen.
Informieren Sie sich unter: fabryfamilytree.de und amicusrx.de
19 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE 1 GERMAIN DP. ORPHANET J RARE DIS. 2010;5:30
Conny Rudolph MorbusFabryPatientin
Darf es noch etwas mehr Calcium sein?
Wenn auch nach mehr als einem Jahr nach einer Schilddrüsenoperation der Calciumspiegel nicht im Normbereich liegt
Eine Schilddrüsenoperation gehört zu den häufigen Eingriffen in Deutschland, der überwiegende Teil der Betroffenen sind dabei Frauen. Dabei kann es (beabsichtigt oder unbeabsichtigt) auch zur Entfernung der Nebenschilddrüsen kommen. Da die linsengroßen Organe aber für die Produktion eines wichtigen Hormons, des Parathormons, zuständig sind, kann die Folge ihrer Entfernung eine seltene chronische Erkrankung mit dem Namen Hypoparathyreoidismus sein. Wir sprachen mit der Endokrinologin Priv.-Doz. Dr. med. Dorothee Maria Baur über Ursachen und Folgen fehlender Nebenschilddrüsen.
Text Hanna Sinnecker

Priv.-Doz. Dr. med. Dorothee
Maria Baur
Endokrinologin im Endokrinologikum Ulm
Frau Dr. Baur, Sie behandeln unter anderem auch Patienten, die an einem chronischen Hypoparathyreoidismus (kurz Hypopara) leiden. Wie entsteht diese Erkrankung?
In drei Viertel der Fälle entsteht diese Erkrankung als Folge einer Schilddrüsenoperation mit beabsichtigter oder versehentlicher Entfernung der Nebenschilddrüsen. Damit kann der Körper kein Parathormon mehr herstellen. Ist das Parathormon mehr als sechs Monate nach der Operation erniedrigt, dann spricht man von einem
Häufiger kommt es zu Ängstlichkeit und Depressionen und somit zu starken Einschränkungen im sozialen Leben der betroffenen Patienten.
chronischen Hypoparathyreoidismus. Da in Deutschland mehr Frauen als Männer an der Schilddrüse operiert werden, leiden auch mehr Frauen an dieser Form des chronischen Hypoparas.
Was passiert dabei im Körper der Betroffenen? Fehlt dem Körper Parathormon, dann kann die Niere kein aktives Vitamin D3 mehr herstellen. Somit kann der Körper nicht mehr genügend Calcium resorbieren und zur Verfügung stellen. Der niedrige Calciumspiegel führt in vielen Organsystemen zu Symptomen. Sehr häufig sind Muskelkrämpfe, Kribbeln in den Extremitäten und Spasmen (Tetanien). Das kann auch andere Muskelsysteme betreffen wie zum Beispiel im Darm oder in der Lunge (sogenannte Bronchospasmen, die sehr unangenehm sind). Dazu kommen Müdigkeit, Schlafstörungen und Konzentrationsstörungen (sogenannter „Brain Fog“). Zudem kommt
es häufiger zu Ängstlichkeit und Depressionen und somit zu starken Einschränkungen im sozialen Leben der betroffenen Patienten.
Wie wird der Hypopara behandelt?
Das wichtigste Ziel ist es, den Calciumspiegel anzuheben und möglichst im unteren Normbereich konstant zu halten. Dazu muss dem Körper das aktive Vitamin D3 zugeführt werden, da der Körper es aufgrund des Parathormonmangels nicht mehr selbst herstellen kann. Zudem wird Calcium gegeben, in verschiedenen oralen Applikationsformen, zum Beispiel als Brausetablette oder in Tablettenform, oder bei sehr schweren Tetanien und Beschwerden auch als intravenöse Gabe. Zudem hilft die Gabe von Magnesium und sogenanntem inaktivem Vitamin D3. Erwachsene Patienten mit chronischem Hypopara, die trotz maximaler konservativer Therapie nicht ausreichend
Lesen Sie mehr auf seltenekrankheiten.de 20
behandelt werden können und bestimmte Kriterien aufgrund der Schwere der Erkrankung erfüllen, können glücklicherweise seit wenigen Jahren auch eine Hormonersatztherapie mit Parathormon bekommen. Dieses Medikament müssen sich Betroffene dann einmal täglich spritzen. Das führt oft zu einer Verbesserung der Einstellung und damit Linderung der Symptomatik. In laufenden Studien wird evaluiert, ob diese Therapie auch die Entstehung von Langzeitkomplikationen der konservativen Therapie verhindern kann. Gezeigt werden konnten bereits die positiven Effekte auf die Lebensqualität und Reduktion der Einnahme von Vitamin D und Calcium unter
Hormonersatztherapie. Die Möglichkeit, Parathormon als Hormonersatztherapie einzusetzen, stimmt mich als Endokrinologin sehr hoffnungsfroh.
Warum kann man denn nicht einfach mehr Calcium nehmen?
Das ist aus zwei Gründen nicht unproblematisch. Erstens wird die orale Gabe von Calcium oft schlecht vertragen und führt zu gastrointestinalen Symptomen. Zweitens hat die Therapie mit Calcium und aktivem Vitamin D weitere Folgen, wie zum Beispiel den Anstieg des Phosphatspiegels, sodass es durch Entstehung von Calciumphosphatkristallen zu Ablagerungen in den Organsystemen kommen
kann, beispielsweise in den Nieren. Quälend kann somit die Entstehung von Nierensteinen sein – und auch die Verschlechterung der Nierenfunktion über die Zeit der Behandlung. Auch im Gehirn können sich Ablagerungen bilden, dies nennt man Morbus Fahr.
Welche Blutwerte sollten Patientinnen und Patienten von ihrem Arzt bestimmen lassen, um festzustellen, ob es sich um einen chronischen Hypoparathyreoidismus handelt, und was sind Normbereiche?
Die wichtigste Blutentnahme ist sicherlich die Bestimmung von Albumin-kontrolliertem Calciumspiegel, am besten direkt postoperativ und im weiteren Verlauf,
sowie die Bestimmung von Parathormon, anorganischem Phosphat, Magnesium sowie 25-OH-Vitamin D3. Idealerweise sollte unter Therapie auch die Bestimmung von Calcium im 24h-Urin erfolgen, um die Belastung der Nieren zu überprüfen. Bei der Bestimmung von Elektrolyten und Hormonspiegel müssen die präanalytischen Empfehlungen beachtet werden, dies ist von Labor zu Labor unterschiedlich. Zeigt sich ein unklar erniedrigter Calciumspiegel, ist in jedem Fall eine weitere Abklärung sinnvoll. In Zusammenhang mit einer stattgefundenen Schilddrüsenoperation ist der Verdacht auf einen Hypoparathyreoidismus naheliegend.
21 Lesen Sie mehr auf seltenekrankheiten.de
EXA/DE/HYPO/0096_07/2021 Takeda Pharma Vertrieb GmbH & Co. KG Jägerstr. 27, 10117 Berlin Erkennen Sie die Symptome von Hypopara rechtzeitig. NEBENSCHILDDRÜSEN.DE Wenn mit den Neben schilddrüsen die Lebensfreude entfernt wurde ANZEIGE EXA/DE/HYPO/0096_07/2021 Takeda Pharma Vertrieb GmbH & Co. KG Jägerstr. 27, 10117 Berlin
Sie die Symptome von Hypopara rechtzeitig. NEBENSCHILDDRÜSEN.DE Wenn mit den Neben schilddrüsen die Lebensfreude entfernt wurde
Erkennen
Mit 3D gegen die EoE
Ein Gespräch mit Prof. Dr. med. Joachim Labenz, Direktor der Inneren Medizin am Diakonie Klinikum Jung-Stilling in Siegen, über die eosinophile Ösophagitis, kurz EoE, eine seltene immunvermittelte Erkrankung, bei der die Speiseröhre chronisch entzündet ist.
Text Dominik Maaßen

Innere Medizin, Diakonie Klinikum JungStilling
Was passiert bei der EoE im Körper der Betroffenen und wer ist der „typische EoE-Patient“?
Betroffen sind meistens junge Männer im Alter von 30 bis 50 Jahren. Nicht selten leiden sie zum Beispiel schon unter Heuschnupfen oder Asthma. Bei Männern ist das Erkrankungsrisiko zwei- bis dreimal höher.
Aber auch Frauen, Kinder oder ältere Menschen können betroffen sein. Es gibt verschiedene Beschwerden wie Brennen hinter dem Brustbein, Schmerzen beim Schlucken oder die Nahrung bleibt sogar in der Speiseröhre stecken. Viele Betroffene vermeiden daher irgendwann bestimmte Nahrungsmittel, oft feste Nahrung wie Brot oder Fleisch. Die Symptome sind vielfältig und unspezifisch. Als Gründe vermuten Experten genetische Veranlagungen, aber auch Faktoren aus der Umwelt.
Was sind die Herausforderungen bei der Diagnosefindung, und besteht Verwechslungsgefahr zu anderen Erkrankungen?
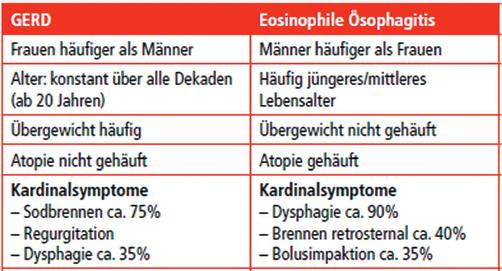
Die EoE wird häufiger mit anderen Erkrankungen verwechselt, als man glaubt. Auch bei der GERD, der Refluxkrankheit, klagen Patienten über ein Brennen hinter dem Brustbein oder Schluckbeschwerden. Die GERD tritt aber häufiger auf und ist deshalb bekannter. Allerdings muss der Arzt sie anders behandeln, ihr Verlauf ist ebenfalls anders. Verwechselt wird die EoE außerdem mit anderen Entzündungen der Speiseröhre, zum Beispiel aufgrund einer Pilzinfektion.
Zur Diagnose macht der Arzt, ein Gastroenterologe, eine Spiegelung und entnimmt mindestens sechs Gewebeproben entlang der Speiseröhre. Wichtig ist nur, dass der Patient 14 Tage vorher keinen Säureblocker genommen hat, der die Entzündung unterdrückt. Danach ist eine Diagnose recht einfach. Das Problem ist nur, dass diese relativ junge Krankheit selbst unter Ärzten vergleichsweise unbekannt ist. Beim Hausarzt fällt die Krankheit
daher oft nicht auf. Dann startet womöglich eine falsche Therapie. Mein Appell ist daher: Wenn ein Patient unter Schluckbeschwerden leidet, ein Brennen während der Mahlzeit spürt, Nahrung hängen bleibt und eine Behandlung bisher nicht zielführend war, dann sollte man auch an die EoE denken. Irgendwann fangen Patienten an, bei der Nahrungsaufnahme lange zu kauen oder viel nachzutrinken. Auch Ärzte können bei der Anamnese gezielt nach solchen Verhaltensweisen fragen.
Wie kann die EoE behandelt werden, und können Betroffene unter Therapie ein beschwerdefreies Leben führen?
Man fasst die drei Möglichkeiten unter den „3 D“ zusammen, also Diet, Drugs und Dilatation. Eine Diät ist recht kompliziert, weil man mit ihr potenzielle Allergieauslöser herausfinden möchte. Dafür werden erst Allergene vom Speiseplan gestrichen und dann wieder Schritt für Schritt zugeführt. Die erste Wahl ist daher eine
Lesen Sie mehr auf seltenekrankheiten.de 22
Prof. Dr. med. Joachim Labenz Fachabteilung
medikamentöse Behandlung. Man wählt ein lokal wirksames Kortison, ähnlich wie bei der Behandlung von Asthma. Rund ein Drittel der Patienten ist auch initial erfolgreich mit Säureblockern behandelbar, allerdings fehlen aussagekräftige Daten zum langfristigen Einsatz. Bei Patienten, bei denen eine medikamentöse Therapie nicht möglich oder ungenügend wirksam ist, kann der Arzt eine mechanische Aufweitung der Speiseröhre, die sogenannte Dilatation, durchführen. Da dieser Eingriff aber nicht die eigentliche Entzündung bekämpft, wird die Dilatation grundsätzlich mit entzündungshemmenden Medikamenten kombiniert,
um die Beschwerden dauerhaft zu lindern. Ziel der Therapie ist, dass die Patienten wieder einen geregelten Alltag aufnehmen können.
Warum ist es so wichtig, die EoE so früh wie möglich zu diagnostizieren, und welche Gefahren bestehen für Patienten, die falsch oder gar nicht diagnostiziert werden?
Wichtig ist für viele Betroffene sicher erst mal die Botschaft, dass es aufgrund dieser Erkrankung keine Krebsgefahr gibt. Kommt es jedoch zu einem chronischen Entzündungsprozess, kann die Speiseröhre vernarben und sie wird eng. Wie schwer das Schlucken
dann fällt, kann sich jeder vorstellen. Laut Untersuchungen in der Schweiz kommt es in 100 Prozent der Fälle zu solchen gravierenden Folgen, wenn die Krankheit über Jahrzehnte nicht behandelt wurde. Auch eine Therapie ist in dieser Art Endzustand nur
noch schwer möglich. Natürlich kommt hinzu, dass die Betroffenen im Alltag sehr unter den Folgen leiden. Deshalb sollte man die Krankheit frühzeitig und damit effektiv behandeln, um Langzeitschäden zu vermeiden.
Typische Patientenbilder GERD und EoE
Frank*, EoE-Patient, erzählt seine Geschichte
Frank ist im Außendienst tätig und viel im Auto unterwegs, sein Mittagessen verzehrt er daher oft „on the road“. Als er mal wieder ein Sandwich im Auto isst, verkrampft sich plötzlich seine Speiseröhre, er kann den Bissen einfach nicht herunterschlucken. Nach langen fünf Minuten löst sich der Krampf. Frank denkt, er habe einfach zu schnell gegessen oder nicht ordentlich gekaut. Aber die Krämpfe kommen wieder und passieren häufiger. Er geht zum Arzt, aber wird beschwichtigt: auch der Mediziner ist der Meinung, dass er sich einfach mehr Zeit beim Essen lassen solle.
Sein Bauchgefühl sagt ihm, dass hinter den Krämpfen etwas anderes stecken muss. Vielleicht eine Reflux-Erkrankung wie bei seinem Vater? 2015 besucht er daher den Arzt seines Vaters, aber die Diagnose ist eine ganz andere: Frank leidet an einer eosinophilen Ösophagitis (meist abgekürzt als EoE), einer seltenen entzündlichen Erkrankung der Speiseröhre. Sofort wird er medikamentös eingestellt, die verschriebenen Protonenpumpenhemmer, die für die EoE nicht zugelassen und für die Dauertherapie nicht getestet sind, nutzt er nach Bedarf. Solange er die Medikamente einnimmt, ist er beschwerdefrei. Setzt er sie ab, kommen die Beschwerden schon nach wenigen Tagen zurück und beeinträchtigen seinen Alltag enorm. Einmal kommt es so
weit, dass er beim Abendessen mit Freunden ein Stück Fleisch nicht schlucken kann. "Es fühlte sich an, als hätte mir jemand ein Messer in die Brust gestoßen", sagt er. Die Schmerzen halten zwei Stunden lang an und sind so stark, dass er sich übergeben muss.
Solche oder ähnliche Geschichten haben viele Betroffene erlebt, die an einer eosinophilen Ösophagitis leiden. Seit 2018 gibt es nun das erste, eigens für EoE-Patienten entwickelte und offiziell zugelassene Medikament, das die Beschwerden dauerhaft im Zaum hält. Es ist als Schmelztablette mit Brauseeigenschaften einfach einzunehmen und lokal in der Speiseröhre wirksam, dadurch ist es gut verträglich und nebenwirkungsarm. Als Frank von der Zulassung dieser Therapie hört, kamen ihm die Tränen, sagt er. Ein deutliches Zeichen, wie groß der Leidensdruck ist, unter dem Betroffene stehen und wie groß die Hoffnung, durch diese neue Behandlungsoption ein großes Stück Lebensqualität zurückzugewinnen.
Wenn Sie mehr zu dieser Erkrankung wissen möchten, finden Sie auf www.schluckbeschwerden.de umfangreiche Informationen zur EoE. Für Ärzte und medizinisches Fachpersonal gibt es zudem ein gesondertes Informationsportal für Fachkreise. *Name von der Redaktion geändert
23 Lesen Sie mehr auf seltenekrankheiten.de
ANZEIGE

Ein gutes Leben trotz Hämophilie
Jan (31) leidet seit seiner Geburt an Hämophilie A. Schon in seiner Kindheit bemerkt er außerdem, dass er Transgender ist: „Ich hatte immer zwei Lasten, die ich mit mir tragen musste.“ Beides wirft dunkle Schatten auf seine Kindheit und Jugend. Seit einem halben Jahr nimmt Jan moderne Medikamente gegen die Bluterkrankheit. Zum ersten Mal in seinem Leben fühlt er sich nun trotz allem frei und sagt: „Du musst versuchen, dich zu lieben, und mit anderen Menschen respektvoll umgehen.“
Text Paul Howe
Lesen Sie mehr auf seltenekrankheiten.de 24
FOTO: NINA SCHÖNER
Früher Schicksalsschlag
Jan ist ein aufgeschlossener, freundlicher und hilfsbereiter Mensch, der gern mit seinen beiden Hunden spazieren und zur Arbeit geht. Man sieht ihm nicht an, welches Schicksal er seit seiner Geburt mit sich trägt. Als Jan in den späten 80ern geboren wurde, war recht schnell klar, dass etwas mit ihm nicht stimmt. „Ich war gerade geboren, da haben sie festgestellt, dass ich Hämophilie habe“, sagt er. Der Säugling bekam einen Port in die rechte Brust gelegt. Alle zwei Tage wurde ihm darüber der Blutgerinnungsfaktor VIII gespritzt – denn dieser fehlt den Betroffenen.
Erbkrankheit ohne Heilung
Übertragen wird der zugrunde liegende Defekt auf dem X-Chromosom. Von Hämophilie A sind also meist nur Männer betroffen. Jans Eltern mussten früh lernen, mit ihrem kranken Baby umzugehen. Alle zwei Tage fuhren sie mit ihrem neugeborenen Sohn in die Uniklinik, um ihm über den Port Medikamente spritzen zu lassen. Dort betreut Dr. Carmen Escuriola-Ettingshausen Jan bis heute als Patient im Hämophilie-Zentrum Rhein-Main. „Wenn wir mit dem Auto fuhren, egal wohin, habe ich immer geschrien: 'Nicht auf die Autobahn, nicht auf die Autobahn!'“, erinnert sich Jan. Zu groß war die Angst, dass es wieder in die Klinik geht.
Belastung für Körper und Geist Bis heute ist Jan diese Angst geblieben –vor der Klinik und vor allen Dingen vor den Nadeln. Mit sechs Jahren lernt er dennoch, sich selbst zu spritzen. Er will unabhängiger sein. Auch wird ihm auf seinen Wunsch der Port entfernt, weil er sich für ihn schämt. Von da an erfolgen die Injektionen intravenös. Die psychischen Belastungen lassen Jan aggressiv und gewalttätig werden. „Wenn du klein bist, bist du sauer auf alle, weil du siehst, deine Freunde sind frei und müssen sich nicht spritzen.“ Zusätzlich fühlt er sich schon früh als Mädchen in einem Jungenkörper gefangen. Vielleicht, so sagt er, führte auch das damals zu seinem ablehnenden Verhalten, dass er sich immer falsch fühlte.
Ein Leben mit Schmerzen, Angst und langfristigen Folgen
So zieht Jan mit 14 schon von zu Hause aus. Er sehnt sich nach Freiheit, will ein normales Leben und die Hämophilie und das ständige Spritzen hinter sich lassen. Von da an spritzt er sich nur noch, wenn die Schmerzen kommen. Er beginnt, Drogen zu nehmen, um sich zu betäuben, und achtet nicht auf seine Krankheit. Raubbau am eigenen Körper und einen großen Fehler nennt er das heute. Was er damals nicht bedenkt: Die Folgen der unbehandelten Einblutungen in die Gelenke bleiben ein Leben lang. Heute kann Jan kaum rennen oder springen. Und auch die Angst vor den Injektionen begleitet ihn bis heute noch. Selbst seine Ausbildung als Tierpfleger und sieben Jahre Arbeit im Tierschutz schaffen es auf Dauer nicht, ihm ein ausgefülltes und ruhiges Leben zu geben. Seine Rastlosigkeit treibt ihn schließlich immer weiter von einem Beruf zum nächsten, die Schmerzen und die Hämophilie trägt er dabei stets mit sich.
Dank innovativer Therapien ein fast normales Leben
Im Oktober 2019 berichtet ihm Dr. Escuriola-Ettingshausen zum ersten Mal von modernen Medikamenten für Hämophilie-A-Erkrankte. Bereits ein Jahr zuvor hatte Jan sich für eine Testreihe neuer Medikamente angemeldet, die dann aber so nicht stattfinden konnte. „Ich hätte es gemacht, auch wenn ich gewusst hätte, dass man sterben kann. Hauptsache war für mich: nicht mehr dauernd in die Vene spritzen“, sagt er. Seine Ärztin stellt ihm die fortschrittlichen Therapiemöglichkeiten so vor: „Deutlich seltenere Injektionen als vorher reichen in der Regel aus, und das bei einem guten Blutungsschutz.“ Jan spürt die Erleichterung für sein Leben schnell.
Wie ein neues Leben
Die Angst vor den Spritzen ist nicht verschwunden. Besonders Blutabnehmen ist immer noch schlimm, das macht Jan nur selbst. Nachdenklich sagt er: „Das, was du erlebt hast, das
bleibt immer. Aber jetzt habe ich Lebensqualität geschenkt bekommen.“ Mit den fortschrittlichen Medikamenten fühlt der junge Mann sich stärker und sicherer. Er hat gelernt, mit der Hämophilie umzugehen. Schon nach nur wenigen Monaten mit den modernen Therapien kann er spürbar besser laufen und hat weniger Schmerzen beim Auftreten. Er arbeitet mittlerweile am Empfang eines Unternehmens, übernimmt Verantwortung und fühlt sich aufgehoben. Von den Drogen ist er lange los. Und auch das Verhältnis zu seinen Eltern hat Jan repariert. Heute ist es so gut wie nie zuvor. Dabei hatten sie jahrelang keinen Kontakt. Doch inzwischen weiß Jan, was es bedeutet, wenn jemand für ihn da ist. Und er lebt endlich eine Freiheit, nach der er sich lange gesehnt hat. Die neu gewonnene Lebensqualität lässt ihn entspannter sein. Er ist glücklicher.
Hoffnung für alle Betroffenen Auch für seine zweite Last hat Jan sich nach einer Erleichterung erkundigt. Tatsächlich gibt es in Deutschland Ärzte, die transsexuelle Bluterkranke operieren. Einerseits möchte Jan diese Möglichkeit nutzen. Dennoch sagt er: „Man muss sich vielleicht einfach mit manchem abfinden. Ich werde mein Leben so weiterleben, und im nächsten Leben wird vielleicht alles besser.“ Auf dem Weg der Besserung befindet Jan sich schon jetzt mit den modernen Therapien. „Wenn ich etwas bestimmen würde“, lächelt er, „würde ich sagen, man soll den Betroffenen nur noch diese Medikamente geben.“
Dieser Beitrag ist nur einer von vielen, die auf der Website www.mutmachseiten.de zu finden sind. Der Zweck dieser Website ist die für Laien verständlich formulierte Verbreitung positiver Erfahrungsberichte von erkrankten oder schwer beeinträchtigten Menschen über ihre ganz individuellen Therapiewege und die Förderung des Austausches von Patienten und Betroffenen über Therapie und Heilung untereinander. Instagram.com/mutmachseiten
25 Lesen Sie mehr auf seltenekrankheiten.de
Hoher Blutdruck und Gewichtszunahme? Denken Sie an Cushing!
Das Cushing-Syndrom gehört zu den seltenen endokrinologischen Erkrankungen und ist auch für erfahrene Mediziner nicht leicht zu diagnostizieren.
Im Schnitt warten Betroffene drei bis fünf Jahre bis zur Diagnose: eine Zeit der quälenden Ungewissheit, die zudem oft von starken Beschwerden geprägt ist und die Lebensqualität der Patient*innen stark einschränken kann. Wir sprachen mit Brigitte Martin, die selbst vom Cushing-Syndrom betroffen ist.
Text Benjamin Pank

Lesen Sie mehr auf seltenekrankheiten.de 26
FOTO: PRIVAT
Frau Martin, Sie sind betroffen vom Cushing-Syndrom. Woran haben Sie gemerkt, dass etwas mit Ihrer Gesundheit nicht stimmt?
Im Oktober 1999 habe ich das erste Mal bemerkt, dass etwas nicht stimmt. Ich habe stark zugenommen, besonders am Bauch, bekam ein sehr rundes Gesicht, Wassereinlagerungen in den Beinen und Haarausfall, hohen Blutdruck, Muskelund Gelenkschmerzen am ganzen Körper und hatte eine verminderte Leistungsfähigkeit.
Welche Auswirkungen hatten die Beschwerden auf Ihren Alltag?
Ich konnte nicht mehr in dem Umfang arbeiten wie früher. Auch im Haushalt konnte ich nicht mehr so zupacken. Fenster putzen oder volle Wäschekörbe tragen war mit Schmerzen und Schweißausbrüchen verbunden. Mein Alltag war definitiv nicht mehr derselbe.
Sicher sind Sie zum Arzt gegangen: Wie sah Ihr Weg bis zur Diagnose aus?
Zuerst bin ich zum Hausarzt gegangen, der hat mich zum Internisten geschickt. Der hatte dann den Verdacht auf Cushing, doch die Urinwerte belegten diesen Verdacht nicht. Man muss dazu sagen, dass man am Anfang der Erkrankung das Zuviel an Cortisol noch nicht nachweisen kann. Also ging der Arztmarathon weiter. Ich war beim Urologen, beim Radiologen, beim Gynäkologen. Durch Zufall kam ich in die Hormonsprechstunde der
Ich war kurz vor der Verzweiflung, hatte sogar einen Nervenzusammenbruch.
Inneren Organe. Der Arzt schaute mich dann an und sagte, dass ich entweder ein Adenom an der Hypophyse oder an der Nebenniere habe. Die Ergebnisse haben den Anfangsverdacht dann bestätigt: Cushing. Ich wurde dann ins Krankenhaus eingewiesen, wo viele Untersuchungen gemacht wurden. Das war dann ein richtiger Krankenhausmarathon. Mittlerweile war klar, dass auf der linken Seite ein Adenom sitzt, wo genau, war aber immer noch nicht klar. Das war sehr zermürbend. Ich war kurz vor der Verzweiflung, hatte sogar einen Nervenzusammenbruch. Obwohl nach wie vor nicht klar war, ob ich ein Hypophysenadenom habe, wurde ein OP-Termin geplant. Ein Vierteljahr später hatte ich dann endlich die OP. Das war im November 2000. Ich habe auf den besten Operateur gewartet, und der hat das Adenom an der Hypophyse ohne Komplikationen entfernt.
Wie sieht Ihr Leben jetzt aus, wo Sie eine entsprechende Behandlung bekommen?
Häufigkeit klinischer Merkmale des Cushing-Syndroms*
Häufigkeit klinischer Merkmale des Cushing-Syndroms*
Gerötetes Gesicht (70–90 %) Mondgesicht (81–90 %) Akne (20–35 %) männlicher Behaarungstyp / Haarausfall (75 %)
Gerötetes Gesicht (70–90 %) Mondgesicht (81–90 %) Akne (20–35 %) männlicher Behaarungstyp / Haarausfall (75 %)
Ich habe nur sechs Jahre Hydrokortison genommen. Das wurde dann schleichend abgesetzt. Heute nehme ich, bis auf Blutdruckmedikamente, nichts mehr ein. Und es geht mir sehr gut. Ich führe ein komplett normales Leben.
Sie sind selbst sehr aktiv in der Selbsthilfe. Was ist Ihr Antrieb für dieses Engagement, und welche Rolle spielt die Vernetzung in der Selbsthilfe für Sie persönlich?
Ich war damals komplett allein, habe jedoch durch meine Hartnäckigkeit innerhalb eines Jahres alles geregelt. Die meisten Betroffenen brauchen zehn Jahre und länger, um eine Diagnose zu bekommen. Das ist fatal, denn man stirbt nicht am Cushing selbst, sondern an Herzinfarkt oder Hirnschlag. Dass es mir heute so gut geht, liegt auch an dem professionellen Operateur. Ich möchte Menschen begleiten, ihnen Mut machen und ihnen den richtigen Weg weisen. Das ist mein Antrieb, mich immer wieder erneut für Menschen mit Cushing starkzumachen.
ANZEIGE

Bluthochdruck (70–85 %) Glukoseintoleranz (45–70 %) erhöhte Blutfettwerte (70 %)

* Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015;7:281–93.
* Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology
management. Clin Epidemiol 2015;7:281–93.
Bluthochdruck (70–85 %) Glukoseintoleranz (45–70 %) erhöhte Blutfettwerte (70 %) rote Dehnungsstreifen (Striae rubrae > 1 cm) (44–50 %) Zyklusstörungen (70–80 %) verringerte Libido (24–80 %)
rote Dehnungsstreifen (Striae rubrae > 1 cm) (44–50 %)
Zyklusstörungen (70–80 %) verringerte Libido (24–80 %)
Neigung zu Blutergüssen (35–65 %)
Neigung zu Blutergüssen (35–65 %)



Depression/emotionale Labilität, Psychose/Manie, kognitive Dysfunktion/ gestörtes Kurzzeitgedächtnis (70–85 %) Büffelnacken (50 %)
Depression/emotionale Labilität, Psychose/Manie, kognitive Dysfunktion/ gestörtes Kurzzeitgedächtnis (70–85 %) Büffelnacken (50 %)
Muskelschwund, Muskelschwäche (60–82 %)
Muskelschwund, Muskelschwäche (60–82 %)
Nierensteine (21–50 %)
Nierensteine (21–50 %)
Übergewicht oder Gewichtszunahme (70–95 %)
Übergewicht oder Gewichtszunahme (70–95 %)
verringerte Knochendichte oder Knochenbrüche(40–70 %)
verringerte Knochendichte oder Knochenbrüche(40–70 %)
27 Lesen Sie mehr auf seltenekrankheiten.de
0-21-08-2 Stand Aug. 2021
and developments in disease
0-21-08-2 Stand Aug. 2021


