treballs
de la Societat Catalana de Biologia









Volum 72 2022 · revista anual ISSN 0212-3037 (edició impresa) ISSN 2013-9802 (edició digital)
Treballs de la Societat Catalana de Biologia , revista anual de la SCB
Societat Catalana de Biologia, filial de l’Institut d’Estudis Catalans Carrer del Carme, 47. 08001 Barcelona scb@iec.cat
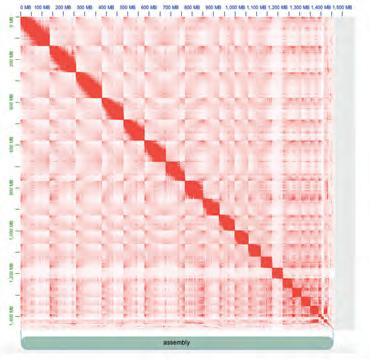
1) Mapa de contactes Hi-C (p. 31).
2) Paràsit de la malària (p. 45).
3) Xiphosura, Limulus polyphemus juvenil (p. 35).
4) Alga cocolitòfor (p. 45).
5) Pycnogonida (p. 35).
6) Ameba social (p. 45).

7) Paris polyphylla (p. 13).

8) Esquema de les possibles implicacions de la CBP en l’àmbit socioeconòmic (p. 6).
9) Ricinulei, Cryptocellus sp. (p. 35).




La propietat intel·lectual dels articles és dels autors respectius. La SCB està exempta de tota responsabilitat derivada de l’eventual vulneració de drets de propietat intel·lectual per part dels autors. Els continguts publicats a la revista estan subjectes —llevat que s’indiqui el contrari en el text o en el material gràfic— a una llicència Reconeixement - No comercialSense obres derivades 3.0 Espanya (by-nc-nd) de Creative Commons, el text complet de la qual es pot consultar a http://creativecommons.org/licenses/by-nc-nd/3.0/ es/deed.ca. Així doncs, s’autoritza el públic en general a reproduir, distribuir i comunicar l’obra sempre que se’n reconegui l’autoria i l’entitat que la publica i no se’n faci un ús comercial ni cap obra derivada. treballs de la societat catalana de biologia no es fa responsable de les idees i opinions exposades pels autors dels articles publicats.
© Societat Catalana de Biologia, filial de l’Institut d’Estudis Catalans, per a aquesta edició Dipòsit Legal B 12164-1963 ISSN 0212-3037 (ed. impresa) 2013-9802 (ed. digital) Imprès per Ediciones Gráficas Rey, SL Web de la versió digital: http://revistes.iec.cat/index.php.TSCB
COMITÈ DE PUBLICACIONS
Jordi Barquinero, VHIR Rafel Abós-Herràndiz, vocal, ICS Josep M. Espelta, vocal, UAB
EQUIP EDITORIAL
Jordi Barquinero, redacció editorial Unitat d’Edició del Servei Editorial, IEC, correcció
La Societat Catalana de Biologia (SCB) és una de les filials més antigues de l’Institut d’Estudis Catalans. Està regida per un Consell Directiu i organitzada en seccions especialitzades, que són les que organitzen les activitats principals que duu a terme la Societat.

CONSELL DIRECTIU DE LA SCB
Presidència: Marc Martí-Renom
Vicepresidència primera: Montserrat Corominas Vicepresidència segona: Maria Montserrat Sala
Secretaria general: Albert Jordan
Vicesecretaria: Oriol Cabré
Tresoreria: Marina Rigau
Vocalia d’Acció Territorial: Eduard Escrich
Vocalia de Comunicació: Toni Hermoso
Vocalia d’Ensenyament: Jordi Morral
Vocalia de Promoció: Sandra Acosta
Vocalies de Publicacions i Lexicografia: Jordi Barquinero

Vocalia de Seccions: Josep Saura

Vocalia d’Estudiants: Eva Coll
Delegat de l’IEC: Jaume Bertranpetit
SECCIONS
Vocalia de seccions temàtiques: Josep Saura
Aqüicultura: Nerea Roher Biofísica: Álex Perálvarez
Bioinformàtica i Genòmica: Roderic Guigó Biologia de la Reproducció: Rafael Oliva Biologia de Plantes: Anna Caño
Biologia del Càncer: Oriol Casanovas Biologia del Desenvolupament: Marta Morey
Biologia Evolutiva: Aurora Ruiz-Herrera Biologia i Societat: Laura Castarlenas Biologia Molecular i Cel·lular: Joan Roig Cromatina i Epigenètica: Albert Jordan Ecologia: Josep Maria Espelta Ensenyament: Jordi Morral Estudiants: Eva Coll Microbiologia: Eduard Torrents Neurobiologia Experimental: Carles Saura Neurociència Computacional i de Sistemes: Albert Compte i Gemma Huguet Proteòmica i Estructura de Proteïnes: Patrick Aloy i Eduard Sabidó
Senyalització Cel·lular i Metabolisme: Laura Herrero Virologia: Sílvia Bofill i Susana Guix Vocalia de seccions territorials: Eduard Escrich SCB a Alacant: Ivan Quesada i Sergi Soriano SCB a Andorra: Eros Alexandre Marín Millán SCB a Balears: Núria Marbà i Anna Traveset SCB a Castelló: Vicent Arbona i Ferran Martínez-Garcia SCB a Catalunya del Nord: Héctor Escrivà i Thierry Noguer SCB a Catalunya del Sud: Jaume Folch i Miguel Mulero SCB a Girona: Elisabeth Pinart i Enrique Verdú SCB a Lleida: Maria Laplana i Judit Ribas SCB a València: Ferran Palero SCB a Vic: Julita Oliveras
Volum
delaSocietatCatalanadeBiologia Volum72 2022 revistaanual ISSN0212-3037(edicióimpresa) ISSN2013-9802(ediciódigital) lestecnologiesdeseqüenciació DNA ElenaVila MònicaBayés Assemblatgedegenomesaescalacromosòmica redescobrir conservar biodiversitatcatalanaGómez-Garrido,FernandoCruz, PalmadaTylerAlioto Genòmica quelicerats:ladesconstrucciódelsaràcnids genòmica laseda, verins altres rellevànciabiològica Arnedo JulioRozas
eucariotes
SARS-CoV-2 exemple
JosepQuer CREU DE SANT JORDI 2012 PLACA NARCÍS MONTURIOL 2003 vols… seminariscursosijornades... ...perquènot’hiassocies? http://scb.iec.cat
treballs
Protists,laprincipalfontdediversitatgenòmica
RamonMassana,RamiroLogares,DavidLópez-Escardó Javier Campo Variabilitat seqüenciaciómassivadevirus.
CristinaAndrés,DavidTabernero,TomásPumarola, AndrésAntón
Editorial. Jaume Pellicer El racó de la SCB. Lisa Pokorny
rticles
3
La iniciativa catalana per a l’Earth BioGenome Project
Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas 10
Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
34
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica Miquel A. Arnedo i Julio Rozas
Protists, la principal font de diversitat genòmica en eucariotes
43
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
Del genoma als gens
Jaume Pellicer, Oriane Hidalgo, Joan Vallès i Teresa Garnatje 16
Ferriol Calvet i Roderic Guigó 21
Avenços en les tecnologies de seqüenciació del DNA
Berta Fusté, Elena Vila i Mònica Bayés 28
Assemblatge de genomes a escala cromosòmica per redescobrir i conservar la biodiversitat catalana Jèssica Gómez-Garrido, Fernando Cruz, Marc Palmada-Flores i Tyler Alioto
51
Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
72
Índex
A
2
La vida al nostre planeta ha anat evolucionant des de l’aparició dels organismes més senzills, sotmesa a un degoteig constant de canvis i innovacions, que han contribuït a la impressionant complexitat i diversitat que coneixem avui en dia. Mentrestant, la curiositat dels éssers humans per a resoldre els fonaments biològics que han permès l’aparició d’aquestes innovacions, així com el seu significat evolutiu, no han fet més que créixer. Per a entendre com s’organitza la vida, però, necessitem conèixer en profunditat com funciona l’engranatge molecular que ens pot donar accés al seu codi secret, el genoma.
Vivim en un present de grans avenços tecnològics en tots els àmbits i, específicament, en les ciències de la vida, entre els quals els dedicats al desenvolupament de noves tècniques de seqüenciació del DNA. Més que mai, ens podem permetre plantejar-nos estudiar detalladament els racons més inaccessibles del genoma eucariota, intractables fins ara. A més, necessitem conèixer, quantificar i catalogar la biodiversitat actual, ja que sols així podrem començar a pensar a mitigar l’emergència global que genera l’activitat humana, responsable de la pèrdua d’hàbitats i de moltes de les espècies que hi viuen.
La revolució genòmica actual ha propiciat la creació de consorcis internacionals com l’Earth BioGenome Project (EBP), que neix amb l’objectiu comú de seqüenciar aproximadament dos milions de plantes, animals, fongs i organismes unicel·lulars eucariotes. La creació d’una gran biblioteca digital de genomes de referència serà d’una importància enorme per a controlar i protegir els ecosistemes i els serveis que proveeixen, però també per a fer front a la propagació de patògens, entre altres funcions. La iniciativa catalana per a l’EBP (CBP), impulsada per l’Institut d’Estudis Catalans, té per objectiu contribuir a aquest gran catàleg internacional amb l’estudi d’espècies d’interès en els territoris de llengua catalana. En aquest volum, presentem un recull de treballs liderats per investigadors i investigadores associats a aquesta iniciativa, que ens mostren, entre d’altres, alguns dels avenços tecnològics més recents en la seqüenciació genòmica, contribucions a l’elaboració de catàlegs de recursos genòmics i l’estudi dels mecanismes moleculars responsables de la diversitat genòmica que coneixem actualment, tot posant en rellevància les seves implicacions a escala evolutiva i el seu impacte en la societat.
Jaume Pellicer Institut Botànic de Barcelona
El racó de la SCB
Les tècniques de seqüenciació massiva, tant de seqüències curtes (el màxim exponent de les quals són les plataformes Illumina) com de seqüències llargues (les plataformes de Pacific Biosciences —PacBio— i d’Oxford Nanopore Technologies —ONT— són les més conegudes), en combinació amb les tècniques de captura de la conformació dels cromosomes (Hi-C), han permès caracteritzar el genoma d’un organisme, cromosoma a cromosoma i de telòmer a telòmer. Durant molts anys, l’accés a aquest tipus de tecnologies s’ha centrat en l’estudi d’organismes model, en humans, atesa la gran inversió econòmica que portava associada aquesta recerca. Sens dubte, la proliferació de plataformes de seqüenciació i de tècniques moleculars ha abaratit els costos de producció de dades genòmiques considerablement, i l’ha fet molt més accessible a la comunitat científica i, consegüentment, ha obert un ventall incomparable d’oportunitats per entendre com mai abans hauríem pogut imaginar la biodiversitat que ens envolta. En paral·lel, la producció massiva d’un volum de dades genòmiques ha requerit una millora considerable de la logística i la manipulació d’aquestes dades. Els avenços, en aquest sentit, també han sigut força importants, amb el desenvolupament d’un gran nombre d’eines i de fluxos de treball bioinformàtics. Actualment, la computació d’alt rendiment en clústers i superordinadors està en evolució constant, cosa que permet processar de manera molt més eficient aquestes dades massives. Les condicions són, sens dubte, les ideals per expandir el nostre coneixement de la genòmica dels organismes, més enllà d’un grapat d’espècies, al conjunt de la biodiversitat.
Un dels projectes amb més repercussió mundial ha estat la seqüenciació del genoma humà, els resultats del qual ofereixen un marc incomparable per conèixer a fons com funciona el nostre organisme i que ha suposat grans avenços i aplicacions en els camps de la medicina i l’evolució, entre altres. Nosaltres, però, som solament una petita fracció de l’entorn, una peça més de l’engranatge global. No obstant això, hem influenciat en gran manera la dinàmica global del planeta, i ara vivim en un present en què cada vegada som més conscients de la influència del canvi climàtic a les nostres vides i de l’impacte devastador que té en el conjunt de la biodiversitat que ens envolta. S’han engegat multitud de projectes per a la seqüenciació de genomes complets de tots els organismes coneguts i els que encara ens queden per conèixer. Les repercussions d’aquestes iniciatives i d’aquests consorcis ja són massives. Més enllà del saber per saber, l’ús d’eines genòmiques té un potencial crític per a la comprensió dels mecanismes que governen l’origen i el manteniment de la biodiversitat, en aquest nostre món canviant, i podran proporcionar-nos les eines necessàries per mitigar els efectes nefastos del canvi global.
Lisa Pokorny Institut Botànic de Barcelona
Editorial
2
La iniciativa catalana per a l’Earth BioGenome Project
1
5
4
3
Resum
Els potents avenços en les tecnologies de seqüenciació de genomes, juntament amb la reducció de costos, permeten per primera vegada a la història utilitzar la genòmica per a ajudar a caracteritzar molecularment tant les espècies conegudes de la Terra com les que encara resten per identificar. L’Earth BioGenome Project (EBP) és una iniciativa internacional que aspira a seqüenciar, catalogar i caracteritzar els genomes de tota la biodiversitat eucariota de la Terra. Estructurada com una xarxa internacional de xarxes, vol crear una nova base per a la biologia, per a buscar les bases moleculars de molts caràcters únics a les espècies, per a trobar solucions per a preservar la biodiversitat i per a contribuir, finalment, al benestar i a la millora econòmica de les nostres societats. La iniciativa catalana per a l’Earth BioGenome Project és un dels nodes de la xarxa que té per objectiu la caracterització i la catalogació del genoma de les espècies eucariotes que viuen als territoris de parla i cultura catalanes.
Paraules clau: biodiversitat, genoma, genòmica, seqüenciació.
Introducció
Que la biodiversitat de la Terra està minvant és un fet. En els darrers quaranta anys, s’ha perdut el 60 % de la població d’espècies salvatges un 52 % en el cas dels vertebrats— i actualment hi ha més de 35.000 espècies en perill d’extinció ( https://nc.iucnredlist.org/redlist/ resources/files/1630480997-IUCN_RED_LIST
_QUADRENNIAL_REPORT_2017-2020.pdf).
El canvi climàtic, la destrucció d’hàbitats i l’explotació d’algunes espècies amb finalitats econòmiques són només alguns exemples de com l’activitat humana és un dels principals causants de la pèrdua d’espècies. Segons l’entomòleg Edward Osborne Wilson, reconegut mundialment pels seus estudis sobre la biodiversitat, ens trobem davant de la sisena gran extinció en la història de la Terra (Wilson, 1999). En aquest escenari, és necessari que actuem per a preservar i conservar els ecosistemes, tant per al futur de la Terra com també per a la supervivència de l’espècie humana. Per
DOI: 10.2436/20.1501.02.210
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 08/03/2022
Acceptat: 05/04/2022
The Catalan Initiative for the Earth BioGenome Project
Abstract
The major advances in genome sequencing technologies, together with the reduction of the costs involved, allow genomics to be used for the first time in history to help to characterise molecularly both the known species on Earth and those still remaining to be identified. The Earth BioGgenome Project (EBP) is an international initiative that aims to sequence, catalogue and characterise the genomes of all the Earth’s eukaryote biodiversity. Structured as an international network of networks, it seeks to create a new basis for biology in order to look for the molecular bases of many unique characters in species, to find solutions for preserving biodiversity, and to contribute to the welfare and the economic improvement of our societies. The Catalan Initiative for the Earth BioGenome Project forms one of the nodes of the network. Its goal is to characterise and catalogue the genome of the eukaryote species living in the territories of Catalan language and culture.
Keywords: biodiversity, genome, genomics, sequencing.
tal de donar resposta a aquesta emergència, primer cal conèixer les espècies que habiten la Terra, i una de les maneres més potents de ferho és estudiant-ne el genoma.
Malgrat els més de dos-cents cinquanta anys de classificació taxonòmica i més d’1,2 milions d’espècies catalogades, s’ha suggerit que un 86 % de les espècies existents en la totalitat de la Terra i el 91 % de les espècies de l’oceà encara esperen ser descrites (Mora et al., 2011). Els grans avenços en la tecnologia de seqüenciació de genomes, juntament amb la informàtica, l’automatització i la intel·ligència artificial, permeten per primera vegada a la història afrontar un dels reptes científics i socials més importants actualment: augmentar la nostra comprensió de la biodiversitat i millorar-ne sensiblement l’ordenació taxonòmica.
L’Earth BioGenome Project
L’Earth BioGenome Project (EBP; www.earth biogenome.org) és una iniciativa a escala mun-
Treballs de la Societat Catalana de Biologia, 72: 3-9
dial impulsada l’any 2017 per la Universitat de Califòrnia a Davis (UC Davis), la Smithsonian Institution i la Universitat d’Illinois a UrbanaChampaign, que té com a objectiu global seqüenciar, catalogar i caracteritzar el genoma de tota la biodiversitat eucariota de la Terra. L’abril de l’any 2018, la revista científica Proceedings of the National Academy of Science (PNAS) va publicar l’article original en què els coordinadors de l’EBP, juntament amb altres investigadors de set països, detallaven els objectius del projecte, així com les fases en què es desenvoluparia i el retorn a la societat que podria comportar (Lewin et al., 2018). El títol de l’article, que ha esdevingut el lema del projecte, és prou suggestiu de les implicacions d’aquesta iniciativa: «The Earth BioGenome Project: Sequencing life for the future of life», és a dir, seqüenciar la vida per al futur de la vida. Més específicament, els objectius de l’EBP es poden concretar a: 1) revisar i aportar una nova visió de la comprensió de la biologia,
3
Elisabet Tintó-Font,1 Helga Simon-Molas,2 Oriane Hidalgo,3 Roderic Guigó4 i Montserrat Corominas5
Institut de Salut Global de Barcelona (ISGlobal), Hospital Clínic - Universitat de Barcelona
Department of Experimental Immunology and Department of Hematology, Amsterdam UMC, Amsterdam, Països Baixos
Institut Botànic de Barcelona (IBB), CSIC - Ajuntament de Barcelona
Centre de Regulació Genòmica (CRG), Universitat Pompeu Fabra i Institut d’Estudis Catalans
Universitat de Barcelona i Institut d’Estudis Catalans
Correspondència: Montserrat Corominas. Departament de Genètica, Microbiologia i Estadística, Facultat de Biologia, Institut de Biomedicina de la Universitat de Barcelona (IBUB). Av. Diagonal, 643. 08028 Barcelona. Adreça electrònica: mcorominas@ub.edu
els ecosistemes, la filogènia i l’evolució; 2) permetre la conservació, la protecció i la regeneració de la biodiversitat; 3) maximitzar la rendibilitat per a la societat i el benestar humà.
L’EBP s’estructura com una xarxa internacional de xarxes, i fomenta la implicació d’institucions i centres d’arreu del món, indispensable per tal de complir els objectius del projecte. Els nodes d’aquesta xarxa estan inclosos en el Consell Coordinador de l’EBP i poden representar una localització geogràfica (Canadà, Àfrica, etc.) o incloure comunitats i organitzacions relacionades amb tàxons (vertebrats, insectes, plantes, etc.). Entre les funcions de l’EBP hi ha: desenvolupar i promoure estàndards per a la producció escalable de genomes de qualitat de referència; fer difusió de les millors pràctiques; coordinar les activitats de seqüenciació, anotació, anàlisi de dades i formació; facilitar l’accessibilitat pública de les dades i estimular la comunicació sobre el progrés del projecte (Lewin et al., 2022). El funcionament de l’EBP es vertebra en vuit comitès: recollida i processament de mostres; seqüenciació i assemblatge; anotació de gens; anàlisi de dades; tecnologies de la informació i informàtica; qüestions ètiques, legals i socials; justícia, equitat, diversitat i inclusió, i comunicació i assumptes públics.
Cal destacar que tota la informació generada per l’EBP és pública i accessible, des del primer moment, amb l’objectiu del benefici comú. Això implica un grau elevadíssim de coordinació i col·laboració, i fa absolutament indispensable que les dades siguin compatibles i comparables des del moment inicial en què es generen. Els nodes de la xarxa implicats en l’EBP han de respectar el Protocol de Nagoya sobre Accés i Intercanvi de Beneficis (ABS, de l’anglès access and benefit-sharing). Aquest protocol de 2010 és un acord complementari al Conveni sobre la Diversitat Biològica (CBD, de l’anglès Convention on Biological Diversity) de 1992 i promou la implementació d’un dels tres objectius del CBD: el repartiment just i equitatiu dels beneficis derivats de la utilització dels recursos genètics, per a contribuir així a la conservació i l’ús sostenible de la biodiversitat.
Una vegada recollides, les mostres s’han d’acompanyar de metadades robustes i completes. Això inclou identificar-ne l’espècie i fer el dipòsit en institucions públiques d’una mostra de cada exemplar, com a referència permanent i revisable del treball. Els materials frescs s’han de processar en tubs sobre gel sec i a partir d’aquest punt, mantenir-los a –80 °C o en
nitrogen líquid, malgrat que s’estan estudiant altres maneres de preservar-los en situacions en què aquest tipus de preservació no sigui possible. L’extracció de DNA d’alt pes molecular i de bona qualitat és absolutament necessari per a poder generar seqüències que segueixin els estàndards quantitatius establerts pel comitè corresponent. Les directrius actuals d’assemblatge consisteixen a generar una combinació de tipus de dades que inclouen lectura llarga (metodologia PacBio HiFi i/o ONT ultra long), de llarg abast (Hi-C) i RNAseq (lectures curtes d’Illumina, PacBio Iso-Seq o ONT cDNA-PCR) del mateix exemplar sempre que sigui possible. Un accés obert a les dades que respecti els principis ètics és imprescindible perquè es compleixi un dels objectius principals del projecte: contribuir al coneixement i a la ciència. Per tant, una vegada obtingudes, les seqüències dels genomes de referència s’han de dipositar en alguna de les bases de dades públiques següents: l’Arxiu Europeu de Nucleòtids, de l’Institut Europeu de Bioinformàtica del Laboratori Europeu de Biologia Molecular, ENA (EMBL-EBI) (www.ebi.ac.uk/ ena/); Genbank, del Centre Nacional per a la Informació Biotecnològica (NCBI) ( www. ncbi.nlm.nih.gov/genbank/), o DNA Databank of Japan (www.ddbj.nig.ac.jp). Aquestes dades de seqüència, conegudes com a informació de seqüència digital (DSI, de l’anglès digital sequence information ), són clau per al progrés científic i la innovació tecnològica en camps tan diversos com la medicina, la seguretat alimentària, la producció d’energia verda i la conservació de la biodiversitat. Les regles per a accedir a la DSI, però, generalment no són clares i per aquesta raó s’ha proposat recentment un marc per a l’ús de la DSI (Scholz et al., 2022) que es discutirà els propers mesos a la reunió de la CBD, on assistiran representants de l’EBP.
L’obtenció de la seqüència dels genomes de totes les espècies eucariotes tindrà una gran influència en molts aspectes de la biologia. Qüestions relacionades amb l’evolució o l’ecologia només seran abordables quan es disposi de dades de genomes sencers que permetin identificar divergències en totes les ramificacions de l’arbre de la vida o totes les espècies dels ecosistemes naturals. Tal com es conclou a l’article publicat recentment amb el títol «Why sequence all eukaryotes?» (Blaxter et al., 2022), aquesta biblioteca fonamental d’informació canviarà l’economia i el creixement social del futur, fomentant l’agricultura sostenible i noves bioeconomies, accedint a una
Treballs de la Societat Catalana de Biologia, 72: 3-9
farmacopea ampliada i promovent l’equitat i la diversitat de la societat a través de les ulleres d’una biodiversitat molt valorada.
El novembre de 2020 va fer dos anys del llançament de l’EBP i s’han fet avenços significatius en tots els aspectes del full de ruta (Lewin et al ., 2022). En aquests moments, l’EBP té quaranta-nou projectes afiliats, repartits per tots els continents i que cobreixen una diversitat de tàxons. Un dels més avançats és el Darwin Tree of Life (DToL; www.darwintreeof life.org/), que pretén seqüenciar els genomes de les setanta mil espècies d’organismes de la Gran Bretanya i Irlanda (The Darwin Tree of Life Project Consortium, 2022). Cal tenir en compte, però, que alguns dels països amb més representació de la biodiversitat de la Terra es troben en zones no industrialitzades. L’EBP contribuirà a destacar aquests països i a fernos a tots més conscients de la necessitat de treballar sense oblidar ningú.
La iniciativa catalana per a l’Earth BioGenome Project
La iniciativa catalana per a l’Earth BioGenome Project (CBP, de l’anglès Catalan Initiative for the Earth BioGenome Project; www.bioge noma.cat) és un projecte afiliat a l’EBP que té com a objectiu seqüenciar el genoma de les més de setanta mil espècies eucariotes que es calcula que viuen als Països Catalans. La CBP es va plantejar inicialment des de l’Institut d’Estudis Catalans (IEC), concretament des de dues de les seves societats filials: la Societat Catalana de Biologia (SCB) i la Institució Catalana d’Història Natural (ICHN). La iniciativa es va presentar en el marc del congrés Genomics for Biodiversity, que va tenir lloc a la seu de l’IEC el setembre de 2019. Actualment, la CBP és un projecte col·laboratiu que compta amb el suport i la participació directa d’una trentena d’institucions i d’una xarxa d’un centenar de membres d’arreu dels territoris de parla catalana. Es tracta d’un projecte ambiciós que, per tal d’implementar-se amb èxit, té dos pilars fonamentals: la rica i extensa tradició naturalista del nostre territori i les potents infraestructures tecnològiques de què disposem.
L’àmplia diversitat climàtica, topogràfica i geològica dels Països Catalans ha fet que s’hi concentri una biodiversitat molt rica. Des dels cims pirinencs de més de 3.000 metres d’altitud fins a les costes, illes mediterrànies i la mar que les envolta, passant per boscos atlàntics i per ambients subàrids, la diversitat de formes de vida hi adquireix un dels màxims exponents. Aquest territori es troba a la intersec-
4
Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas
ció de les plaques europees i africanes (CasasSainz i Vicente, 2009), i a la cruïlla entre les regions biogeogràfiques eurosiberiana i mediterrània. Els Països Catalans representen un punt calent (hotspot) de biodiversitat: tot i que cobreixen menys de l’1 % del territori europeu (70.520 km2, 2.500 km de vora del mar), són la llar d’aproximadament una quarta part de totes les espècies eucariotes europees conegudes. També es caracteritzen per un alt nivell d’endemisme. Per exemple, de les 7.500 plantes vasculars estimades en aquest territori, unes 5.500 són autòctones i més de 300 es consideren endèmiques (Peñuelas et al., 2019). Moltes espècies endèmiques estan amenaçades (fins al 40 % en alguns dels territoris, com les Illes Balears), una tendència que s’agreujarà en el futur, ja que el canvi climàtic afectarà especialment la conca mediterrània i les zones de muntanya (Cramer et al., 2018; Hoegh-Guldberg et al., 2019; Pepin et al., 2015; Tuel i Eltahir, 2020). A Catalunya, l’informe Estat de la natura a Catalunya 2020 (Brotons et al., 2020) conclou que les poblacions de vertebrats i invertebrats autòctons de les quals es tenen dades han perdut de mitjana el 25 % dels indivi-
dus en els darrers vint anys, i que els canvis en els usos del sòl són la principal causa de pèrdua de biodiversitat, malgrat que el canvi climàtic i les espècies invasores també hi tenen un paper important. Globalment, la problemàtica de la conservació de la biodiversitat a Catalunya és similar a la del conjunt d’Europa. L’informe Natura, ús o abús? (2018-2019) també destaca que la sobreexplotació de recursos té un gran impacte sobre el sòl, l’aire i l’aigua, i indica que els problemes de la conservació de la natura s’originen en el model econòmic actual, que és ambientalment insostenible (Peñuelas et al., 2019).
Pel que fa al funcionament de la CBP, s’han definit els diferents processos implicats en l’obtenció d’un catàleg detallat dels genomes (figura 1). Aquests processos, que segueixen els estàndards de l’EBP, es poden resumir en els punts següents: recollida de mostres, processament i catalogació; conservació de les mostres en bancs de materials biològics; extracció i seqüenciació del DNA; assemblatge i anotació del genoma;
anàlisi i adaptació de les dades per a la seva visualització; publicació de les dades en repositoris públics.

Implicacions i retorn social: més enllà de la biodiversitat
La CBP tindrà un impacte directe en el coneixement que tenim de les espècies que habiten els territoris de parla catalana. D’una banda, contribuirà a actualitzar, millorar i digitalitzar el catàleg de les espècies que hi viuen. De l’altra, entendre l’evolució genòmica d’aquestes espècies al llarg del temps i relacionar-la amb els efectes de l’activitat humana i el canvi climàtic ens farà ser més conscients de les nostres accions, dissenyar estratègies per a pal·liar-ne els efectes sobre els ecosistemes i millorar les estratègies de conservació que es duen a terme amb espècies amenaçades. Valorar la biodiversitat a través del desenvolupament d’aquesta iniciativa, i de la implicació i el compromís d’entitats tant públiques com privades—, tindrà un efecte de presa de consciència en la població. Els ciutadans i les ciutadanes entendran com les accions de l’espècie humana han afectat els ecosistemes al llarg dels anys i com podem actuar per a preservar la biodiversitat actual.
A més, el coneixement obtingut, que es posarà a l’abast de tothom, ha de comportar un retorn social en àmbits molt diversos, des de l’agricultura, l’alimentació i la salut fins al sector energètic i la indústria, sense oblidar l’impuls que suposarà per a la recerca, la indústria i l’economia. En aquest sentit, cal destacar que els objectius globals de la CBP s’emmarquen en el concepte one health (‘una sola salut’), definit per l’Organització Mundial de la Salut (OMS) el 2017 ( www.who.int/news -room/q-a-detail/one-health), i estan inclosos en l’Estratègia de Biodiversitat de la Unió Europea per al 2030 (https://ec.europa.eu/info/ sites/default/files/communication-annex -eu-biodiversity-strategy-2030_en.pdf ) i en l’Agenda 2030 per al Desenvolupament Sostenible de l’Organització de les Nacions Unides (ONU) (www.un.org/sustainabledevelopment/ development-agenda/).
Entre les implicacions principals de la CBP en l’àmbit socioeconòmic (figura 2), es pot destacar, en primer lloc, l’impacte en l’agricultura, la ramaderia, la pesca i l’alimentació. La identificació de noves variants genètiques permetrà millorar els cultius, reduir-ne les possibilitats d’infecció per plagues, augmentar-ne la productivitat o trobar noves maneres de conrear determinades espècies. Tam-
5
La iniciativa catalana per a l’Earth BioGenome Project
7
Figura 1. Diagrama de processos que se segueixen a la CBP. Elaboració pròpia.
Figura 2. Esquema de les possibles implicacions de la CBP en l’àmbit socioeconòmic. Elaboració pròpia.
bé es podran recuperar espècies de cultiu tradicionals que actualment s’han perdut o trobar noves aplicacions, com, per exemple, nous biocarburants. La resiliència de les espècies davant amenaces com el canvi climàtic prové de la diversitat genètica, i una font de variació genètica que ens podria ser molt útil es troba en els parents salvatges de les nostres espècies domesticades (Castañeda-Álvarez et al., 2016). La seqüència detallada del genoma facilitarà la selecció genòmica per a la producció i la conservació de races i espècies en perill d’extinció. A banda d’ampliar les possibilitats de generació de noves varietats de productes transgènics o genèticament modificats, saber més sobre les espècies conegudes actualment i descobrir-ne de noves permetrà obtenir aliments de fonts que ara mateix no s’aprofiten.
En segon lloc, cal destacar l’impacte de la CBP en la salut i la qualitat ambiental. Conèixer de manera completa el genoma de tots els eucariotes ajudarà, d’entrada, a comprendre millor moltes malalties i el sistema de transmissió entre espècies dels patògens infeccio-
sos, la qual cosa farà que el control de les pandèmies sigui més eficient. D’altra banda, es podran descobrir noves molècules terapèutiques per a millorar la salut. Cal destacar que, en el context actual, s’ha plantejat la necessitat urgent d’establir una plataforma global de biovigilància basada en la genòmica (un sistema d’intercepció de pandèmies), que afavoriria enormement la nostra comprensió del món natural i, per tant, tindria un gran valor per a la bioseguretat, la biodefensa i l’economia (Kress et al., 2020). Respecte a la qualitat ambiental, l’estudi de l’evolució dels genomes ens permetrà entendre quines accions humanes, inclòs el canvi climàtic, han tingut un impacte negatiu en la biodiversitat. Fruit d’això, podrem dissenyar estratègies per a millorar la qualitat del sòl, l’aire i l’aigua, i modificar i regular les activitats humanes que es duen a terme al nostre territori i que tenen un impacte directe en la salut de tota la població.
En tercer lloc, l’impacte en l’economia i la indústria. L’anomenat capital natural fa referència al conjunt de recursos naturals del pla-
neta, que inclouen geologia, sòls, aire, aigua i tots els organismes vius (https://www.unepfi. org/fileadmin/documents/ncd_booklet.pdf). La inversió en capital natural és un multiplicador de l’economia i permet retornar al medi més del que se n’extreu, cosa que posa l’economia al servei de la societat, perquè invertir en natura és invertir en la millora de les condicions de vida de les persones. A més, a la nostra societat, l’energia és precursora de l’activitat econòmica. La transició energètica cap a la descarbonització és la base d’un planeta més sa i saludable i, per això, cal apostar per energies renovables o més netes. El descobriment de noves espècies aportarà informació sobre formes alternatives d’energia fins ara desconegudes. A partir del genoma dels organismes seqüenciats es podran descobrir nous biomaterials o crear-ne a través de tècniques d’enginyeria genòmica i biologia sintètica, cosa que ha de contribuir al teixit industrial. Respecte a les tecnologies de la informació, l’EBP, i de la mateixa manera la CBP, representen un repte tant pel que fa a l’anàlisi com a l’emmagatzematge d’un nombre tan important de dades. Si bé amb les possibilitats actuals el projecte és factible, caldrà donar un impuls a noves tecnologies que facilitin la visualització, comparació i classificació de les dades, i això suposarà un revulsiu per a aquest sector.

En quart lloc, l’impacte en la recerca i la internacionalització. La implicació de la bioinformàtica i les ciències òmiques és clau en aquest projecte. Aquests sectors, que tenen una importància creixent al nostre país, oferiran noves possibilitats de projectes de recerca en l’àmbit de l’EBP. La iniciativa també contribuirà a la formació, donat que estudiants predoctorals, i també aquells que cursen estudis universitaris, podran participar activament en el projecte, tant en els treballs de camp de recollida de mostres com en les etapes de seqüenciació, anàlisi i classificació de les dades. També es dissenyaran activitats específiques per a escoles i instituts amb la finalitat de donar a conèixer i aprendre a respectar i estimar el nostre gran patrimoni natural. En el context internacional, la CBP és el node EBP a casa nostra i ens emmarca en un context internacional que afavorirà la presa de decisions en col·laboració i a través d’accions globals. Els projectes científics seran més competitius a causa de la seva projecció internacional.
Finalment, però no de menys importància, cal destacar l’impacte d’aquesta iniciativa en la comunicació científica i la ciència ciutadana. Al llarg de tot el projecte es faran dife-
6
Treballs de la Societat Catalana de Biologia, 72: 3-9
Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas
rents accions de divulgació per tal que la ciutadania conegui els objectius, la metodologia i l’estat dels resultats, que es comunicaran a través del web del projecte (www.biogenoma.cat) i de les xarxes socials de les institucions implicades. Els ciutadans que ho vulguin podran col·laborar en el projecte de manera activa a través d’iniciatives que podrien incloure, per exemple, participar en la tria de les espècies que cal seqüenciar o en l’obtenció de mostres.
Estat actual (març de 2022)
L’activitat principal de la CBP s’ha centrat a estructurar la xarxa en el territori i crear tres grups de treball i un grup de coordinació, i a establir reunions periòdiques on es fan presentacions científiques i es discuteix com va avançant la iniciativa. Es va iniciar amb una fase pilot, que ha servit com a prova de concepte.
Com a part d’aquesta fase, la CBP va obrir una convocatòria de projectes la tardor de 2020, que, juntament amb el finançament addicional dels mateixos investigadors i investigadores de la Generalitat de Catalunya i del Parc Zoològic de Barcelona, va permetre començar la seqüenciació dels genomes de trenta espècies sota el paraigua de la CBP (figura 3). En aquesta fase pilot, la CBP va seguir un enfocament de baix a dalt, responent als interessos de la comunitat investigadora local, i, després d’una avaluació internacional, es van seleccionar projectes que proposaven seqüenciar espècies d’interès i icòniques.
La tardor de 2021 es va obrir una segona convocatòria amb els mateixos criteris i sistema d’avaluació, amb la qual cosa el nombre d’espècies cofinançades des de la CBP són divuit avui en dia (taula 1). Entre aquestes espè-
cies hi ha tàxons poc explorats, espècies rares, endèmiques o difícils de catalogar, espècies model emergents, espècies en greu perill d’extinció o espècies medicinals. Actualment, el nombre de genomes de referència d’alta qualitat que s’estan generant és de quaranta-dos, repartits com es mostra a la figura 4. Cal destacar que el genoma de la baldriga balear (Puffinus mauretanicus) es troba ja disponible a la base de dades ENA (Cuevas-Caballé et al ., 2021). Com s’ha comentat, un aspecte crucial de l’EBP, i per tant també de la CBP, és la coordinació per tal d’evitar seqüenciar la mateixa espècie en llocs diferents. En el marc del DToL, s’ha creat l’aplicació GoaT ( https://goat.ge nomehubs.org), que utilitza la taxonomia de l’NCBI (https://www.ncbi.nlm.nih.gov) i serveix com a font centralitzada de metadades rellevants per al genoma per a la comunitat global. Funciona com a sistema de seguiment de seqüenciació per a la xarxa de l’EBP i conté, entre d’altres, la llista amb la informació de les espècies que se seqüencien sota el paraigua de la CBP.

La ICHN treballa, des de l’inici de la CBP, en l’actualització del catàleg de les espècies eucariotes que viuen als territoris catalans, que el desembre de 2021 inclou 26.433 tàxons. Aquest catàleg es digitalitzarà i servirà de referència per a prioritzar els genomes que cal seqüenciar durant les fases següents de la CBP. Entre els criteris de priorització s’inclouen la posició filogenètica i la novetat, l’interès per als grups de recerca locals, el grau d’endemisme i conservació, l’interès biomèdic, agrícola i industrial.
Dins la CBP també hi ha un gran interès a desenvolupar programari de codi obert. Així, el portal de dades que s’ha implementat https://dades.biogenoma.cat) conté tota la informació de les espècies, des del moment de la recol·lecció fins a l’obtenció de la seqüència. És un portal universal que importa automàticament totes les dades associades a projectes de seqüenciació genòmica que han estat dipositades a l’ENA. El portal és flexible i pot ser utilitzat per a mostrar les dades de qualsevol node de l’EBP. Altres desenvolupaments dins de la CBP inclouen mètodes eficients d’anotació de gens, d’alineament múltiple de gran nombre de seqüències (fins a milions) (Tommaso et al., 2017) i de filogenòmica.
Figura 3. Projectes de seqüenciació del genoma sota el paraigua de la fase pilot de la CBP. La figura captura la posició filogenètica de l’espècie, la mida del genoma quan es coneix, les tecnologies emprades per a la seqüenciació i la raó de la selecció d’espècies. A la part superior esquerra hi ha el logotip de la CBP. Elaboració pròpia.
Més enllà dels límits dels territoris catalans, i dins de la xarxa EBP de xarxes (superposades), la CBP pretén tenir un paper central en els projectes de genòmica de la biodiversitat a Europa, que ara s’organitzen sota el European
7
La iniciativa catalana per a l’Earth BioGenome Project
ANIMALS CORDATS P R O T I S T S PLANTES CNIDARIS MOLLUSCS CRUSTACIS
A R ÀCN I DS AMFIBIS PEIXOS RÈPTILS OCELLS Tecnologies Planificat Interès Endèmic Vulnerable / Amenaçat Invasiu Econòmic / Etnobotànic Icònic Filogenètic / Model Bionano HiC Illumina Omni-C ONT PacBio
ARTRÒPODES
Taula 1. Espècies en procés de seqüenciació del genoma que han rebut finançament de la CBP. Elaboració pròpia.
Animals
Esponges
Clathrina sp. Ctenòfors Mnemiopsis sp. Cnidaris
Cladocora caespitosa
Corallium rubrum
Equinoderms
Arbacia lixula Vertebrats
Diplodus puntazzo
Xyrichtys novacula
Iberolacerta aurelioi Podarcis lilfordi Artròpodes
Sensonator valentiensis
Tethysbaena scabra Cyprideis torosa Belisarius xambeui Anèl·lids Norana najaformis
Reference Genome Atlas (ERGA), una iniciativa europea per a crear un atles del genoma de la biodivertitat d’Europa ( https://www.erga -biodiversity.eu). Així mateix, la CBP vol ser també central en els projectes dirigits a les regions geogràfiques a les quals pertanyen naturalment els territoris catalans: la península
Plantes
Achillea ptarmica ssp. pyrenaica
Protists
Amastigomonas sp. Caecitellus Paraphysomonas
recerca, des de la història natural fins a la genòmica, que tradicionalment han estat aïllades les unes de les altres. Aquests sinergismes dotaran les institucions d’història natural de la infraestructura d’última generació i dels recursos humans necessaris per a garantir la documentació, la catalogació i la preservació d’exemplars, teixits i DNA per a les generacions futures. Caldrà, doncs, estructurar una xarxa de biobancs a tot el territori. La CBP reactivarà la investigació taxonòmica en un moment en què es troba en el seu mínim històric (p. ex. Crisci et al., 2020). A més, atesa l’heterogeneïtat dels paisatges catalans, la CBP necessitarà la contribució de l’àmplia xarxa d’associacions naturalistes que coneixen els ecosistemes locals per a facilitar l’accés a les mostres biològiques. Més enllà d’un esforç científic pur, veiem aquesta iniciativa com a part d’un moviment transformador mundial que augmenta la consciència social sobre l’amenaça que suposa la pèrdua de biodiversitat per al benestar humà i que implica la societat a escala mundial en una relació diferent i més equilibrada amb la natura.
Agraïments
Ibèrica i la conca mediterrània. La CBP és ja un projecte associat a ERGA i, dins d’aquesta xarxa, treballem en estreta coordinació i col·laboració amb ERGA Andorra, ERGA Espanya i ERGA França.
A escala local, la CBP representa una oportunitat excel·lent per a reunir comunitats de
La CBP ha rebut finançament de l’IEC (a través del llegat Asenjo i dels projectes propis de recerca PRO2019-SO2, PRO2020-SO2 i PRO2021-SO2), del Departament d’Agricultura, Ramaderia, Pesca i Alimentació de la Generalitat de Catalunya, del Parc Zoològic de Barcelona i d’Andorra Recerca + Innovació. Agraïm a Mercè Rocadembosch l’ajuda en l’elaboració de la figura 3 i a Teresa Garnatje, Josep Germain, Manel Niell i Tomàs Marquès-Bonet, la lectura crítica del manuscrit.
Figura 4. Resum de l’estat actual dels projectes de seqüenciació del genoma sota el paraigua de la CBP. Elaboració pròpia.
 Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas
Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas
Bibliografia
Blaxter, M. [et al.] (2022). «Why sequence all eukaryotes?». Proc. Natl. Acad. Sci. USA, 119 (4): 1-9.
Brotons, L. [et al.] (2020). Estat de la natura a Catalunya 2020. Barcelona: Generalitat de Catalunya: Departament de Territori i Sostenibilitat.
Camarasa, J. M.; Casassas, O. (2020). Cent anys de la Societat Catalana de Biologia, la primera societat filial de l’Institut d’Estudis Catalans. Barcelona: Institut d’Estudis Catalans. ISBN: 978-84-9965-556-7.
Casas-Sainz, A. M.; Vicente, G. de (2009). «On the tectonic origin of Iberian topography». Technophysics, 474: 214-235.
Castañeda-Álvarez, N. P. [et al.] (2016). «Global conservation priorities for crop wild relatives». Nat. Plants, 2: 1-6.
Cramer, W. [et al.] (2018). «Climate change and interconnected risks to sustainable development in the Mediterranean». Nat. Clim. Chang., 8: 972-980.
Crisci, J. V. [et al.] (2020). «The end of botany». Trends Plant Sci., 25: 1173-1176.
Cuevas-Caballé, C. [et al.] (2021). «The genome of the Balearic shearwater (Puffinus mauretanicus), a critically endangered seabird: a valuable resource for evo-
lutionary and conservation genomics». BioRxiv , 2021.12.17.473171.
Folch i Guillén, R. (dir.) (1984-2012). Història natural dels Països Catalans. Barcelona: Enciclopèdia Catalana. Hinchliff, C. E. [et al.] (2015). «Synthesis of phylogeny and taxonomy into a comprehensive tree of life». Proc. Natl. Acad. Sci. USA, 112: 12764-12769.
Hoegh-Guldberg, O. [et al.] (2019). «The human imperative of stabilizing global climate change at 1.5°C». Science, 365: aaw6974.
Kress, W. J. [et al.] (2020). «Opinion: Intercepting pandemics through genomics». Proc. Natl. Acad. Sci. USA, 117 (25): 13852-13855.
Lewin, H. A. [et al.] (2018). «The Earth BioGenome Project: Sequencing life for the future of life». Proc. Natl. Acad. Sci. USA, 115: 4325-4333. (2022). «The Earth BioGenome Project 2020: Starting the clock». Proc. Natl. Acad. Sci. USA , 119 (4): e2115635118.
Mora, C. [et al.] (2011). «How many species are there on Earth and in the ocean?». PLoS Biol., 9 (8): e1001127.
Peñuelas, J. [ et al .] (2019). Natura, ús o abús? (20182019): Ús i abús de la natura, impactes i propostes de
gestió. El cas de Catalunya com a paradigma. Barcelona: Institut d’Estudis Catalans. ISBN: 978-84-9965457-7.
Pepin, N. [et al.] (2015). «Elevation-dependent warming in mountain regions of the world». Nat. Clim. Chang., 5: 424-430.
Scholz, H. A. [et al.] (2022). «Multilateral benefit-sharing from digital sequence information will support both science and biodiversity conservation». Nat. Commun., 13 (1): 1086.
The Darwin Tree of Life Project Consortium (2021). «Sequence locally, think globally: The Darwin Tree of Life Project». Proc. Natl. Acad. Sci. USA , 119 (4): e2115642118.
Tommaso, P. di [et al.] (2017). «Nextflow enables reproducible computational workflows». Nat. Biotechnol., 35: 316-319.
Tuel, A.; Eltahir, E. A. B. (2020). «Why is the Mediterranean a climate change hot spot?». J. Clim., 33: 58295843.
Wilson, E. O. (1999). The diversity of life. Cambridge, Massachussetts: Harvard University Press. ISBN: 9780674058170.
Treballs de la Societat Catalana de Biologia, 72: 3-9
9
La iniciativa catalana per a l’Earth BioGenome
Project
1
Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
4
3
2
DOI: 10.2436/20.1501.02.211
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 16/01/2022
Acceptat: 12/03/2022
Resum
El coneixement dels mecanismes responsables de mantenir la biodiversitat requereix, entre d’altres, iniciatives que faciliten l’estudi en profunditat del genoma dels organismes eucariotes. D’aquesta manera, podrem assolir una visió crítica i robusta sobre la seua estructura i el seu funcionament. En el cas de les plantes, a més, es fa palesa la necessitat de continuar generant dades fonamentals referents al contingut de DNA nuclear i el nombre de cromosomes. Aquests trets són molt importants en l’evolució de les plantes, atesa la seua gran diversitat. A més, aporten informació bàsica, i alhora essencial, per a plantejar l’assemblatge d’un genoma. Aquests reptes, però, necessiten el suport d’estratègies d’investigació coordinades a escala global com l’Earth BioGenome Project (EBP) i les seues filials regionals, que faciliten l’accés a la tecnologia, a l’intercanvi d’informació i al desenvolupament de plans de formació. Prioritzar l’estudi comparatiu d’espècies amb un mostratge equilibrat i inclusiu, representatiu de la diversitat taxonòmica i geogràfica de les plantes, així com dels seus trets genòmics, morfològics i ecològics serà, per tant, un dels grans reptes en els pròxims anys.
Paraules clau: cromosoma, evolució, grandària del genoma, poliploïdia, seqüenciació.
Abstract
Understanding the mechanisms responsible for maintaining biodiversity requires, among other things, initiatives that facilitate in-depth genomic surveillance across eukaryote organisms. These initiatives support critical research programs for gaining robust insights into how genomes are structured and operate. In relation to land plants, there is also a clear need to continue generating fundamental data regarding both nuclear DNA contents and chromosome numbers. As a result of their great diversity, these traits are unquestionably important drivers of plant evolution. In addition, they provide basic yet essential information for the planning of a genome assembly. These challenges, however, need the support of globally coordinated research strategies such as the Earth BioGenome Project (EBP) and its regional subsidiaries, which facilitate access to technology, information exchange and the development of training plans. Prioritizing comparative studies across species with a balanced and inclusive taxonomic sampling that covers the geographical diversity of plants as well as their genomic, morphological and ecological features will consequently be one of our major challenges in the years to come.
Keywords: chromosome, evolution, genome size, polyploidy, sequencing.
Introducció
Vivim un present de grans projectes de seqüenciació de genomes, com l’Earth BioGenome Project (EBP, https://www.earthbioge nome.org), i alguns dels seus ramals com, per exemple, el Darwin Tree of Life (DToL, https:// www.darwintreeoflife.org) o la iniciativa catalana per a l’EBP (CBP, de l’anglès Catalan Initiative for the Earth BioGenome Project, https:// www.biogenoma.cat). Aquestes iniciatives, entre moltes altres, juntament amb el desenvolupament constant de les noves tècniques de seqüenciació d’alt rendiment, contribueixen de
manera significativa a accelerar el descobriment i el coneixement de la gran diversitat de genomes presents en el nostre planeta. Entre aquests genomes, les plantes (Viridiplantae) tenen un paper fonamental en la dinàmica dels ecosistemes, amb implicacions directes en el benestar dels éssers humans i amb un gran impacte en l’agroeconomia global (Schaal, 2019; Hidalgo et al., 2021). A mesura que l’activitat humana impacta de manera descontrolada sobre els ecosistemes, accelerant la pèrdua d’hàbitats, i consegüentment d’espècies, es fa més patent la necessitat de generar coneixement
Treballs de la Societat Catalana de Biologia, 72: 10-15
crític i evidències sòlides que faciliten la implementació d’estratègies de gestió i conservació per tal de mitigar l’emergència climàtica i de diversitat a la qual ens enfrontem actualment.
Ateses la gran diversitat i la rellevància de les plantes, són moltes les investigacions centrades en el seu estudi. Un punt clau és el coneixement del seu genoma, pel que fa a l’estructura i al funcionament, en un context tant aplicat com evolutiu. Si bé la majoria de treballs se centren en organismes model, d’interés econòmic i/o sanitari, l’accés a les grans
10
Jaume Pellicer,1, 2 Oriane Hidalgo,1, 2 Joan Vallès3, 4 i Teresa Garnatje1
Institut Botànic de Barcelona (IBB), CSIC - Ajuntament de Barcelona
Royal Botanic Gardens, Kew, Regne Unit
Laboratori de Botànica, Unitat Associada al CSIC, Facultat de Farmàcia i Ciències de l’Alimentació, Institut de la Biodiversitat IRBio, Universitat de Barcelona
Secció de Ciències Biològiques, Institut d’Estudis Catalans
Correspondència: Jaume Pellicer. Institut Botànic de Barcelona (IBB). Parc de Montjuïc. Passeig del Migdia, s. n. 08038 Barcelona. Adreça electrònica: jaume.pellicer@ibb.csic.es.
On the need to study genetic traits influencing the organization and structure of the genome in plant sequencing projects
plataformes de seqüenciació, que estan revolucionant el camp de la genòmica, ha facilitat en gran manera la possibilitat de plantejar i dur a terme iniciatives cada vegada més ambicioses, centrades en llinatges no estudiats fins al moment amb aquestes noves tècniques d’anàlisi genòmiques. De fet, actualment ja s’han publicat al voltant de 300 genomes de plantes assemblats a escala cromosòmica (Kress et al., 2022), tot i que encara representen una petita fracció (al voltant de 900 espècies) de les aproximadament 400.000 espècies de plantes terrestres que coneixem (Lughadha et al., 2016), però que, alhora, conformen un dipòsit global de coneixement sense precedents. L’esforç actual, tant tecnològic com científic, centralitzat a través de la creació de consorcis de recerca, té com a objectiu principal la coordinació i l’estandardització de metodologies, amb la finalitat de continuar millorant el procés d’assemblatge de genomes, resultat de la millora continuada de les eines de seqüenciació. L’obtenció de genomes de referència, però, actualment encara està esbiaixada, amb uns llinatges molt més explorats que d’altres, com per exemple les Poaceae, atesa la nombrosa presència d’espècies d’interés agronòmic com l’arròs (Oryza sativa). L’objectiu final ha de ser, per tant, intentar representar el màxim nombre de llinatges vegetals coneguts, la qual cosa ha d’implementar-se paral·lelament a la millora del coneixement sobre les relacions taxonòmiques entre les plantes. Certament, aquesta és una altra parcel·la de recerca que també està vivint una revolució substancial, a través d’iniciatives com el Plant and Fungal Trees of Life (PAFTOL, https://www.kew.org/ science/our-science/projects/plant-and-fungal -trees-of-life), i la creació de nous kits de seqüenciació dirigida d’alt rendiment, com l’Angiosperms353 (Johnson et al. , 2019) o el GoFlag451 (Breinholt et al. , 2021), que han suposat un revulsiu en la millora del coneixement de la història evolutiva de les plantes terrestres.
Sense cap mena de dubte, la revolució genòmica actual està sotmesa a les exigències tècniques d’una comunitat científica decidida a conéixer els secrets més íntims que s’amaguen en el genoma. L’arribada de tecnologies que permeten seqüenciar fragments de DNA cada vegada més llargs ha estat el punt de partida del que es coneix com l’era dels genomes de qualitat platí , amb assemblatges contigus de gran precisió a escala cromosòmica, que ens permetran estudiar en profunditat patrons de reestructuració i evolució a gran escala. Ini-
ciatives com el 10,000 Plant Genome Sequencing Project, també conegut com a 10KP (Twyford, 2018), representen una fita històrica en el progrés del coneixement del genoma vegetal, i ofereixen una plataforma incomparable per a abordar qüestions sobre els processos biològics al llarg de l’arbre de la vida. En aquest sentit, l’estudi de genomes de llinatges generalment poc investigats és un dels objectius d’aquest projecte. Les plantes, però, plantegen un gran repte, atesa l’extraordinària diversitat pel que fa a una sèrie de trets específics de la seua biologia, com són: a) la grandària del genoma, o valor C, que es refereix a la quantitat de DNA total present en un nucli cel·lular haploide no replicat; b) el nombre de cromosomes, és a dir, en quants cromosomes es distribueix la seqüència de DNA, i c) el nivell de ploïdia, que indica el nombre total de genomes (o còpies) presents en el nucli cel·lular. Aquests trets bàsics, però de gran rellevància en l’evolució de les plantes (Pellicer et al., 2018), s’han de tenir en compte a l’hora de plantejar-se seqüenciar un genoma, ja que poden representar una sèrie de reptes computacionals que creixen de manera exponencial en funció de la mida (Kelly et al., 2012).
Genomes i cromosomes: la diversitat cromosòmica en les plantes
Els cromosomes de les plantes presenten una gran variabilitat, ja siga en nombre com en forma i mida. Encara que puga semblar una informació menys important, el nombre cromosòmic és un paràmetre fonamental en la botànica sistemàtica i evolutiva (Stuessy, 2009 i 2011). El nombre de cromosomes més petit descobert fins avui és de 2n = 4, present tant en llinatges de monocotiledònies —per exemple, Zingeria biebersteiniana (Poaceae) o Ornithogalum tenuifolium ( Liliaceae )— com d’eudicotiledònies — Haplopapus gracilis i Brachyscome dichromosomatica ( Asteraceae )— (Castiglione i Cremonini, 2012). A l’altre extrem podem trobar nombres cromosòmics molt grans, com per exemple al gènere Ophioglossum (Ophioglossaceae, pteridòfits), en què s’han reportat nombres cromosòmics de més de 1.000 cromosomes, com en l’espècie O. reticulatum, amb 2n = 1.400 (Khandelwal, 1990). Aquest valor no només representa el nombre de cromosomes més gran entre les plantes, sinó que segurament és un rècord entre els organismes eucariotes, almenys entre tots els estudiats fins al moment present. Si el comparem amb el nostre propi genoma, amb
Treballs de la Societat Catalana de Biologia, 72: 10-15
una seqüència de DNA distribuïda en 46 cromosomes, aquesta falguera presenta trenta vegades més cromosomes en cada cèl·lula que l’ésser humà. La diversitat de nombres cromosòmics no només és evident entre espècies. De fet, es coneixen exemples en què una sola espècie pot presentar una gran diversitat de nombres cromosòmics, com per exemple Cardamine pratensis ( Brassicaceae ), amb aproximadament 65 nombres cromosòmics descrits, que varien des de 2n = 16 fins a 2n = 96. Si posem en perspectiva aquests nombres, en comparació amb la diversitat que s’ha trobat entre els animals, observem que les plantes són molt més variables. No obstant això, el nombre de cromosomes més petit que s’ha trobat entre els animals és inferior que en les plantes, 2n = 2, reportat a Myrmecia pilosula (Formicidae ), una espècie de formiga nativa d’Austràlia (Crosland i Crozier, 1986). En canvi, el més gran no supera els 500 cromosomes a l’espècie de papallona Polyommatus atlanticus (Lycaenidae) (Lesse, 1970), amb una tercera part de la dotació cromosòmica de la falguera abans esmentada.
En gran manera, la diversitat de nombres cromosòmics que trobem en les plantes es deu a fenòmens de multiplicació del genoma (o poliploïdia), que són extremament freqüents, i sense cap dubte estan considerats un dels principals motors de la seua diversificació (Wood et al., 2009; Soltis i Soltis, 2016; Clark et al., 2016). Els organismes poliploides s’originen en moltes ocasions a través d’un fenomen d’hibridació entre tàxons diferents, i que en molts casos dona lloc a la formació d’un híbrid estèril, com per exemple l’híbrid d’ Urospermum (Asteraceae), descobert recentment a Montjuïc, Barcelona (Pellicer et al., 2022). Si aquest organisme híbrid pateix una duplicació genòmica, a més de restaurar-ne la fertilitat, donarà lloc a la formació d’un individu poliploide (Hegarty i Hiscock, 2008). Els organismes poliploides originats arran de l’encreuament entre dues espècies són anomenats al·lopoliploides i són molt comuns entre els pteridòfits i les angiospermes. De fet, al llarg de l’evolució, i lligat als fenòmens de domesticació, els humans hem tret profit de la poliploïdia (atesos molts dels seus efectes en el fenotip) i actualment molts dels vegetals que utilitzem diàriament són d’origen poliploide (p. ex. blat, plàtan o cotó, entre molts altres). A banda de la gran diversitat de ploïdies descobertes en plantes actuals, el coneixement cada vegada més profund que tenim sobre les relacions evolutives entre les plantes, juntament amb la
11
Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
millora d’eines de seqüenciació i transcriptòmica, han permés descobrir evidències que indiquen una alta incidència d’episodis de multiplicació del genoma en el passat. Aquests fenòmens es coneixen com a episodis de paleopoliploïdia, i se n’han evidenciat més d’un centenar al llarg de la història evolutiva de les plantes terrestres (Landis et al., 2018).
La poliploïdia, però, no requereix sempre un fenomen d’hibridació, ja que aquesta duplicació del genoma pot tenir lloc en un mateix individu, per autofecundació, o per entrecreuament d’individus diferents de la mateixa espècie. Aquest fenomen rep el nom d’autopoliploïdia, com és el cas, per exemple, de Senecio carniolicus (Asteraceae, 2n = 2x = 90) (Sonnleitner et al., 2016). Molts autors suggereixen que el paper evolutiu de l’autopoliploïdia no es pot menysprear, atés el seu impacte en el genoma i la capacitat d’induir processos de diversificació (Parisod et al., 2010). De fet, al voltant d’un 13 % de les angiospermes presenta citotips poliploides, i les conseqüències en el genoma poden incloure canvis en l’expressió de gens, el metabolisme i la fisiologia (Panchy et al ., 2016). Per exemple, en el cas d’ Artemisia annua (Asteraceae), una herba de gran importància en el tractament de la malària, ja que se n’extrau l’artemisinina, s’ha comprovat que individus tetraploides produeixen 1,5 vegades més artemisinina que els individus diploides (Banyai et al., 2010), fet que podria tenir un impacte econòmic rellevant en la producció d’aquest compost químic d’una manera més sostenible amb una reducció de la biomassa necessària per a la seua producció.
L’impacte de la poliploïdia en la biodiversitat global és patent, ja que pot influenciar interaccions ecològiques, processos evolutius i patrons de distribució. De fet, hi ha estudis que demostren que la distribució de la freqüència de poliploides al planeta no és aleatòria, amb una incidència més elevada segons ens allunyem de l’equador i ens aproximem a zones properes als pols terrestres (Rice et al., 2019). Si comparem la incidència de la poliploïdia en les plantes amb la que trobem en els animals, és evident que és molt inferior entre aquests últims, tot i que, en els vertebrats, és present en rèptils, amfibis i peixos (Otto i Whitton, 2000). Pel que fa als invertebrats, alguns llinatges de mol·luscs, crustacis, anèl·lids o insectes també s’han reportat com a poliploides (Gregory i Mable, 2005). De fet, en aquest últim grup s’ha posat en evidència l’existència de fins a divuit esdeveniments de paleoploïdització al llarg de la seua història evolutiva (Li et al., 2018). Tota
aquesta diversitat impressionant de nombres cromosòmics i d’incidència de fenòmens de duplicació del genoma té una sèrie de conseqüències a l’hora de plantejar-se la seqüenciació d’un genoma, especialment si es vol obtenir un assemblatge del genoma d’alta qualitat. Bàsicament, i de manera rutinària, n’hauríem de saber, amb seguretat, el nombre de cromosomes i el nivell de ploïdia, ja que d’això dependrà la base de l’assemblatge que construirem de qualsevol organisme (i/o individu) del qual ens plantegem seqüenciar el genoma. Aquest punt és especialment rellevant en espècies per a les quals s’ha descrit un nombre variable de cromosomes i nivells de ploïdia, ja que poden tenir un impacte directe en la interpretació de les dades de seqüenciació.
La grandària del genoma en les plantes: una extraordinària diversitat que planteja grans reptes bioinformàtics
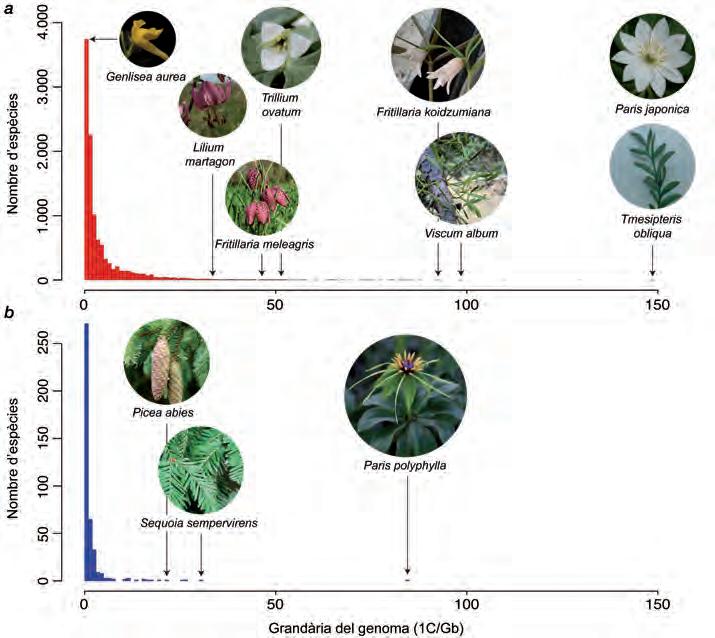
La quantitat de DNA nuclear en un nucli gametofític no replicat és el que es coneix com a mida del genoma o valor C (Greilhuber et al., 2005), i és un caràcter de gran diversitat, del qual disposem de dades per a aproximadament unes 20.000 espècies d’organismes eucariotes. Entre aquests organismes, les plantes han estat estudiades de manera extensa des de 1951, quan s’estimà, per primera vegada, la grandària del genoma en l’espècie Lilium longiflorum (Liliaceae ) (Ogur et al. , 1951). Des d’aleshores, i fins avui, s’han investigat al voltant de 12.000 espècies. Un esforç que s’ha dut a terme en paral·lel al desenvolupament de bases de dades en què la informació és recopilada i accessible al públic en general i a la comunitat investigadora en particular com, per exemple, la Plant DNA C-values Database ( https:// cvalues.science.kew.org ) (Pellicer i Leitch, 2020), que conté dades dels grans llinatges de plantes terrestres (angiospermes, gimnospermes, pteridòfits i briòfits), sense deixar de banda les algues. A més, la base inclou dades cariològiques que permeten interpretar millor la mida del genoma. Com s’ha indicat en apartats anteriors, aquest paràmetre és immensament divers entre els organismes eucariotes en general (> 64.000 ×), i particularment entre les plantes (aproximadament, 2.400 ×, vegeu la figura 1a) (Pellicer et al., 2018). Entre aquestes plantes, el genoma més petit reportat s’ha trobat en l’espècie Genlisea aurea (figura 1), una petita planta carnívora de la família de les Lentibulariaceae, amb un genoma de només 64 Mb (Fleischmann et al., 2014). En canvi, a
Treballs de la Societat Catalana de Biologia, 72: 10-15
l’altre extrem de l’espectre trobem el genoma gegantesc de l’espècie Paris japonica (figura 1), un lliri que pertany a la família de les Melanthiaceae, el qual presenta un genoma de 149 Gb (Pellicer et al., 2010). La seqüència del genoma d’aquesta planta és, en aquest cas, aproximadament cinquanta vegades més gran que la del genoma humà i la seua seqüenciació plantejaria un veritable repte tècnic i computacional, malgrat els avanços recents. De nou, les plantes representen un grup extraordinari pel que fa a la diversitat, fet que queda palés si comparem les dades de què disposem per a alguns grups d’animals, com per exemple els ocells, amb una variació de la grandària del genoma que no supera les 2,5 vegades (0,912,16 Gb/1C, https://www.genomesize.com ). Malgrat la gran diversitat de mides del genoma reportades en les plantes, la majoria de les angiospermes, per exemple, es caracteritzen pels genomes petits i/o molt petits (figura 1a), tot i l’elevada incidència d’episodis de poliploïdia. Aquest fet contrasta, per exemple, amb la distribució de valors en les gimnospermes, que presenten mides del genoma menys diverses, i relativament més grans que les angiospermes, un fet que s’il·lustra també amb una amplitud de nombres cromosòmics molt més discreta que en les angiospermes (Rastogi i Ohri, 2020). Aquesta tendència contrasta amb el que trobem, per exemple, entre els pteridòfits, en què l’amplitud de valors de quantitat de DNA no és tan elevada com en les angiospermes, tot i que observem que espècies amb genomes gegants de mida similar al d’algunes angiospermes presenten un nombre molt superior de cromosomes (p. ex. Tmesipteris obliqua [Psilotaceae], 1C = 149 Gb, 2n = 416, figura 1) (Hidalgo et al., 2017), enfront dels 2n = 40 de Paris japonica (Pellicer et al. , 2010). Aquestes diferències naixen del fet que aquests llinatges han mostrat patrons d’evolució diferents. Un genoma poliploide a les angiospermes està generalment sotmés a fenòmens de diploïdització, els quals provoquen una pèrdua parcial de DNA repetitiu i còpies de gens, entre d’altres (Wendel, 2015), que en general afavoreixen l’aparellament de cromosomes homòlegs; aquest fet generalitzat és el responsable de la falta de correlació entre la mida del genoma i el nombre de cromosomes (Pellicer et al. , 2018; Wang et al., 2021). En canvi, en els pteridòfits s’ha observat que la grandària del genoma està correlacionada amb el nombre de cromosomes (Clark et al. , 2016), a pesar de l’elevada incidència de poliploides (Wood et al., 2009). Les diferències, doncs, rauen en el
12
Jaume Pellicer, Oriane Hidalgo, Joan Vallès i Teresa Garnatje
Figura 1. a) Distribució de la grandària del genoma (valors 1C) en les plantes terrestres (dades extretes de la Plant DNA C-values Database, https://cvalues.science.kew.org/); b) distribució de la grandària del genoma de les espècies per a les quals s’ha seqüenciat el genoma (dades extretes de la base de dades PlaBiPD, https://plabipd.de/index.ep, i publicacions addicionals). Imatges: Jaume Pellicer (Gensilea aurea, Lilium martagon, Fritillaria koidzumiana, Fritillaria meleagris, Trillium ovatum, Paris polyphylla, Sequoia sempervirens, Picea abies, Viscum album, Tmesipteris obliqua) i Laurence Hill (Paris japonica).
procés de diploïdització, que en els pteridòfits impacta de manera prioritària silenciant gens, sense que hi haja associada una pèrdua significativa aparent de DNA repetitiu, i que, en definitiva, permet que al llarg de l’evolució es puguen mantenir nombres de cromosomes elevats (Haufler, 2014).
Quins són els principals mecanismes responsables de la gran diversitat de mides del genoma en les plantes?
Com s’ha indicat en apartats anteriors, un dels principals motors de l’evolució del canvi en la mida del genoma és la poliploïdia, que implica la presència de més de dues còpies del genoma i, per tant, l’increment de la quantitat de DNA (que pot ser proporcional o no) a mesura que ascendim en nivells de ploïdia. No obstant això, els canvis en la composició i l’abundància de seqüències repetitives de DNA també tenen un paper crític en l’estructura i en la mida del genoma. Aquestes seqüències es troben pre -
sents en el genoma en múltiples còpies, que varien en ordres de magnitud i que són principalment els retrotransposons, elements transposables de DNA i seqüències repetides en tàndem (o satèl·lits de DNA) (Bennetzen i Wang, 2014). Els transposons de DNA i els retrotransposons són elements mòbils del genoma que s’insereixen a través de mecanismes de tallar-i-enganxar o copiar-i-enganxar, respectivament, que els confereixen la capacitat de colonitzar nous espais de l’espectre genòmic. Entre aquests últims, els retrotransposons de repetició terminal llarga (LTR, de l’anglès long terminal repeat) són molt coneguts per la seua capacitat de dominar una fracció substancial del genoma repetitiu de les plantes. Comprenen diferents llinatges organitzats en superfamílies, en què els Ty1/Copia i Ty3/Gypsy són els més comuns entre les plantes (Wicker et al., 2007).
A mesura que creix el nostre coneixement sobre l’organització i el funcionament dels genomes, gràcies en part al desenvolupament de
tècniques de seqüenciació cada vegada més i més potents, podem observar que la fracció repetitiva del genoma augmenta a mesura que ho fa la mida (p. ex., el 80 % del genoma de la dacsa, Zea mays, és repetitiu [Sofi et al., 2007]).
A més, en genomes relativament petits, els canvis en la mida moltes vegades estan governats per l’acció d’un o pocs llinatges d’aquests elements, que arriben a tenir un elevadíssim nombre de còpies (Piegu et al., 2006; Macas et al., 2015). Aquests genomes mostren signatures d’un elevat dinamisme, atés que la majoria d’elements repetitius presenten evidències d’activitat d’amplificació i eliminació freqüent, amb una signatura evolutiva compatible amb un procés de vida recent, d’entre 3 i 5 milions d’anys. D’aquesta manera, les diferències en la capacitat dels elements repetitius per a amplificar-se és el que impacta directament en la grandària final del genoma. La pregunta és, però, si al llarg de l’amplitud de 2.400 × de variació de la grandària del genoma, l’escenari descrit anteriorment per a plantes amb genomes relativament petits continua present. La resposta és que no.
Un dels gèneres model per a l’estudi de l’obesitat genòmica (1C > 30 Gb) el trobem en les Liliaceae, en el gènere Fritillaria. Aquest gènere presenta una diversitat elevada de mides del genoma, que varia des dels 29 Gb/1C a F. degeniana fins als 87 Gb/1C a F. koidzumiana (Pellicer i Leitch, 2020). L’anàlisi d’alguns d’aquests genomes gegants ha posat de manifest una dinàmica totalment diferent de la que s’havia trobat en genomes petits. Per exemple, el seu genoma repetitiu és heterogeni i està compost per un gran nombre d’elements, amplificats a baixa freqüència, sense presentar evidències d’amplificació o eliminació recents (Kelly et al., 2015). Aquesta dinàmica també s’ha observat en algunes gimnospermes amb genomes grans com Picea abies ( Pinaceae ) (Nystedt et al., 2013), i en els genomes gegants d’algunes salamandres (Sun et al., 2012). En conseqüència, la hipòtesi més acceptada per a explicar com han arribat aquests genomes a proporcions gegantesques és la falta d’habilitat per a eliminar seqüències amplificades de DNA repetitiu, fet que n’afavoreix l’acumulació i la fossilització posterior en el genoma. Anàlisis comparatives més recents, incloent-hi una amplitud de diversitat de mides del genoma que va des de 64 Mb fins a 88,5 Gb, indiquen que la fracció repetitiva del genoma canvia en funció de la seva grandària (Novák et al., 2020). De fet, aquesta fracció s’incrementa amb relació a la grandària del genoma fins a arribar
13
Treballs de la Societat Catalana de Biologia, 72: 10-15
Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
aproximadament a uns 10 Gb/1C, mida a partir de la qual s’observa un canvi de dinàmica que suggereix una saturació de l’increment de DNA repetitiu, i fins i tot un declivi en algunes espècies. L’escenari anterior és compatible amb les evidències prèvies descrites en apartats anteriors, que apunten una reducció del dinamisme i una taxa baixa d’eliminació de DNA repetitiu.
Seqüenciant genomes: quins, com i per què és important tenir un coneixement bàsic dels trets genètics?
La publicació del primer genoma vegetal, seqüenciat de l’espècie model Arabidopsis thaliana (Brassicaceae) (Initiative, 2000), representà el punt de partida de més de dues dècades de recerca en aquest camp que han donat lloc a la publicació de centenars de genomes disponibles en repositoris públics i d’accés obert a la comunitat científica. No obstant això, com s’ha descrit en apartats anteriors, els genomes vegetals són particularment diversos pel que fa a la mida i a la complexitat citotípica, característiques que moltes vegades han descoratjat investigadors i investigadores, ateses la magnitud de la tasca d’assemblatge i les limitacions de la tecnologia disponible durant molts anys. És per això que la distribució taxonòmica dels genomes seqüenciats no ha estat equilibrada, amb una representació esbiaixada (Vallée et al., 2016). Els últims anys, però, han significat una revolució constant pel que fa a les tecnologies disponibles de seqüenciació i d’anàlisi computacional (Suzuki, 2020). Actualment podem analitzar cada vegada fragments més llargs de DNA amb tecnologies com PacBio Iso-Seq i HiFi, Oxford Nanopore i l’ancoratge Hi-C, que permeten assemblar cromosomes amb alta fidelitat i més contigüitat, la qual cosa d’alguna manera obri la possibilitat de poder començar a pensar en la viabilitat de projectes i iniciatives adreçats a seqüenciar qualsevol espècie vegetal independentment dels trets genètics que tinga (Kress et al., 2022). Tanmateix, aquests autors també fan una anàlisi sobre la qualitat dels assemblatges publicats fins al present i posen de rellevància que només una petita fracció compleix els estàndards mínims que s’exigeixen actualment (còntigs amb un N50 d’1 Mb i ancoratges amb un N50 de 10 Mb). Part d’aquestes limitacions rau en el fet que les noves tecnologies de seqüenciació requereixen extraccions de DNA d’alt pes molecular, i aquest punt pot resultar limitant en algunes
espècies, atesa la complexitat química del seu citosol, que pot dificultar el procés d’extracció amb els estàndards de qualitat requerits. En l’era dels grans consorcis i iniciatives de seqüenciació globals, una solució per a aquest problema hauria de raure en la creació de més fòrums de col·laboració, que facilitaren l’accés a protocols i a serveis tecnològics que ajudarien a superar aquests reptes d’una manera més eficient. Igualment, i com era esperable, aquesta «febre de l’or» per la seqüenciació no arriba de manera paritària arreu del planeta. De fet, ja són alguns els autors que alerten sobre la falta de vincles entre l’origen geogràfic de les espècies i dels equips investigadors que les estudien —que moltes vegades descuiden establir i mantenir col·laboracions amb equips de recerca i comunitats locals (Marks et al. , 2021)— i que apunten la necessitat d’encoratjar una implicació més gran per part de la comunitat científica.
Segons la publicació recent sobre l’estat de la qüestió de l’abast de la seqüenciació de genomes de Viridiplantae (Kress et al. , 2022), actualment s’han seqüenciat ja 812 espècies, incloent 543 angiospermes, 11 gimnospermes, 5 pteridòfits, 8 briòfits i 249 algues (figura 1b). De fet, entre aquestes últimes hi trobem alguns dels genomes seqüenciats més petits, com és el cas d’ Ostreococcus tauri ( Prasinophyceae ), amb un genoma de tan sols 12,6 Mb i que representa un dels organismes eucariotes de vida lliure més diminuts que coneixem (Derelle et al., 2006). Un altre exemple, de dimensions similars a l’anterior però que en aquest cas correspon a una alga paràsita, és Helicosporidium sp. (Chlorellaceae), amb un genoma compacte de només 12,3 Mb (Pombert et al., 2014). Entre els més grans, cal destacar el genoma de Pinus lambertiana (Pinaceae), que pertany a les gimnospermes i presenta un genoma de 27,6 Gb (Stevens et al., 2016). Més recentment, l’anunci de la seqüenciació del genoma de Paris polyphylla (Melanthiaceae, angiospermes) ha representat un gran salt quantitatiu, ja que presenta un assemblatge d’aproximadament 80 Gb (Li et al., 2020). Aquest projecte no ha estat exempt de debat, atesa la gran discordança que s’observa entre la mida del genoma assemblat i les dades de què disposem obtingudes amb citometria de flux, les quals indiquen que les espècies diploides del gènere Paris , si bé tenen genomes gegants, varien entre 30 i 55 Gb (Pellicer et al., 2014). Certament, la metodologia emprada per a estimar la grandària del genoma ha estat un punt conflictiu, per l’impacte que té en els resultats obtinguts. La
Treballs de la Societat Catalana de Biologia, 72: 10-15
citometria de flux s’ha consolidat durant molts anys com una tècnica altament efectiva i robusta per a estimar la quantitat de DNA (Doležel et al., 2007). Molts projectes de seqüenciació, però, basen estrictament l’estimació de la mida del genoma en l’anàlisi de distribució de k-mers o en la representació de seqüències en assemblatges contigus (Sun et al. , 2018; Pucker, 2019), que poden produir resultats diferents dels obtinguts amb citometria de flux segons algunes característiques inherents del genoma (p. ex., poliploïdia, nivell d’heterozigosi, contingut de DNA repetitiu). D’altres simplement usen valors previs publicats i que potser no són representatius de l’individu analitzat, atesa l’elevada taxa de poliploïdització dels genomes vegetals. L’impacte que pot tenir el mètode de quantificació sobre la mida estimada del genoma ha estat estudiat en llinatges d’organismes molt variats, com per exemple en alguns fongs (Kooij i Pellicer, 2020), insectes (Pflug et al., 2020) i en plantes (Al-Qurainy et al., 2021), entre d’altres. Molts d’aquests estudis han trobat diferències entre els valors obtinguts depenent de la metodologia emprada, tot i que no sembla que hi haja un patró comú clar que apunte la sobreestimació i/o infraestimació de la mida del genoma dels mètodes basats en l’anàlisi de seqüències. Per tot això, un punt clau a la base de qualsevol projecte de seqüenciació hauria d’incloure una caracterització citogenètica de l’individu que s’haja d’estudiar que com a mínim incloguera un recompte de cromosomes i la quantificació del contingut de DNA nuclear mitjançant citometria de flux.
Conclusions
El coneixement dels mecanismes responsables de mantenir la biodiversitat requereix, entre d’altres, iniciatives que faciliten l’estudi en profunditat del genoma dels organismes eucariotes, que permeten obtenir-ne una visió crítica i robusta sobre l’estructura i el funcionament. En el cas de les plantes, a més, volem posar de manifest la necessitat de continuar generant dades fonamentals referents al contingut de DNA nuclear i al nombre de cromosomes, que són molt rellevants i, a més a més, aporten informació bàsica, però essencial, a l’hora d’assemblar un genoma; una necessitat que es veu reforçada pel fet que aquests camps estan lluny d’haver estat explorats en profunditat per a molts dels grups taxonòmics (p. ex., només prop del 3 % de les espècies d’angiospermes tenen una mida del genoma coneguda [Pellicer et al., 2018]). Aconseguir
14
Jaume Pellicer, Oriane Hidalgo, Joan Vallès i Teresa Garnatje
Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
aquests reptes requereix estratègies de recerca coordinades a escala global com l’EBP i les seues filials regionals, que han de facilitar l’accés a la tecnologia, a l’intercanvi d’informació
Bibliografia
Al-Qurainy, F. [et al.] (2021). «Estimation of genome size in the endemic species Reseda pentagyna and the locally rare species Reseda lutea using comparative analyses of flow cytometry and k-mer approaches». Plants, 10: 1362.
Banyai, W. [et al.] (2010). «Overproduction of artemisinin in tetraploid Artemisia annua L.». Plant Biotechnol., 27: 427-433.
Bennetzen, J. L.; Wang, H. (2014). «The contributions of transposable elements to the structure, function, and evolution of plant genomes». Ann. Rev. Plant Biol., 65: 505-530.
Breinholt, J. W. [et al.] (2021). «A target enrichment probe set for resolving the flagellate land plant tree of life». App. Plant Sci., 9: e11406.
Castiglione, M.; Cremonini, R. (2012). «A fascinating island: 2n = 4». Plant Biosyst., 146: 711-726.
Clark, J. [et al.] (2016). «Genome evolution of ferns: Evidence for relative stasis of genome size across the fern phylogeny». New Phytol., 210: 1072-1082.
Crosland, M. W. J.; Crozier, R. H. (1986). «Myrmecia pilosula, an ant with only one pair of chromosomes». Science, 231: 1278.
Derelle, E. [et al.] (2006). «Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features». Proc. Nat. Acad. Sci. USA, 103: 11647-11652.
Doležel, J. [et al.] (2007). «Estimation of nuclear DNA content in plants using flow cytometry». Nat. Protoc., 2: 2233-2244.
Fleischmann, A. [et al.] (2014). «Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms». Ann. Bot., 114: 1651-1663.
Gregory, T. R.; Mable, B. K. (2005). «Polyploidy in animals». A: Gregory, T.; Ryan, B. T. (ed.). The evolution of the genome. Burlington (MA): Academic Press, 427-517.
Greilhuber, J. [et al.] (2005). «The origin, evolution and proposed stabilization of the terms “genome size” and “C-value” to describe nuclear DNA contents». Ann. Bot., 95: 255-260.
Haufler, C. H. [ et al.] (2014). «Ever since Klekowski: Testing a set of radical hypotheses revives the genetics of ferns and lycophytes». Am. J. Bot., 101: 2036-2042.
Hegarty, M. J.; Hiscock, S. J. (2008). «Genomic clues to the evolutionary success of polyploid plants». Curr. Biol., 18: R435-R444.
Hidalgo, O. [et al.] (2017). «Genomic gigantism in the whisk-fern family (Psilotaceae): Tmesipteris obliqua challenges record holder Paris japonica». Bot. J. Linn. Soc., 183: 509-514. (2021). «La botànica: entre art i ciència». A: Redondo, M.; Figueras, M. (ed.). HerbArt: Confluències entre art i ciència. Barcelona: Universitat de Barcelona, 226-243.
Initiative, T. A. G. (2000). «Analysis of the genome sequence of the flowering plant Arabidopsis thaliana». Nature, 408: 796-815.
Johnson, M. G. [et al.] (2019). «A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering». Syst. Biol., 68: 594-606.
Kelly, L. J. [et al.] (2012). «Why size really matters when sequencing plant genomes». Plant Ecol. Div., 5: 415425.
i al desenvolupament de plans de formació. Prioritzar l’estudi comparatiu d’espècies amb un mostratge equilibrat i inclusiu, representatiu de la diversitat taxonòmica i geogràfica de
les plantes, així com dels seus trets genòmics, morfològics i ecològics serà, llavors, el gran repte d’aquestes iniciatives en els pròxims anys.
(2015). «Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size». New Phytol., 208: 596-607.
Khandelwal, S. (1990). «Chromosome evolution in the genus Ophioglossum L.». Bot. J. Linn. Soc., 102: 205217.
Kooij, P. W.; Pellicer, J. (2020). «Genome size versus genome assemblies: Are the genomes truly expanded in polyploid fungal symbionts?». Genome Biol. Evol., 12: 2384-2390.
Kress, W. J. [et al.] (2022). «Green plant genomes: What we know in an era of rapidly expanding opportunities». Proc. Nat. Acad. Sci. USA, 119: e2115640118.
Landis, J. B. [et al.] (2018). «Impact of whole-genome duplication events on diversification rates in angiosperms». Amer. J. Bot., 105: 348-363.
Lesse, H. (1970). «Les nombres de chromosomes dans le groupe de Lysandra argester et leur incidence sur sa taxonomie [Lep. Lycaenidae ]». Bull. Soc. Entomol. France, 75 (3-4): 64-68.
Li, J. [et al.] (2020). «An enormous Paris polyphylla genome sheds light on genome size evolution and polyphyllin biogenesis». BioRxiv, 2020.06.01.126920.
Li, Z. [et al.] (2018). «Multiple large-scale gene and genome duplications during the evolution of hexapods». Proc. Nat. Acad. Sci. USA, 115: 4713-4718.
Lughadha, E. N. [et al.] (2016). «Counting counts: Revised estimates of numbers of accepted species of flowering plants, seed plants, vascular plants and land plants with a review of other recent estimates». Phytotaxa, 272: 82-88.
Macas, J. [et al.] (2015). «In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae». PLOS ONE, 10: 1-23.
Marks, R. A. [et al.] (2021). «Representation and participation across 20 years of plant genome sequencing». Nat. Plants, 7: 1571-1578.
Novák , P. [ et al .] (2020). «Repeat-sequence turnover shifts fundamentally in species with large genomes». Nat. Plants, 6: 1325-1329.
Nystedt, B. [et al.] (2013). «The Norway spruce genome sequence and conifer genome evolution». Nature , 497: 579-584.
Ogur, M. [et al.] (1951). «Nucleic acids in relation to cell division in Lilium longiflorum». Exp. Cell. Research, 2: 73-89.
Otto, S. P.; Whitton, J. (2000). «Polyploid incidence and evolution». Annu. Rev. Genet., 34: 401-437.
Panchy, N. [et al.] (2016). «Evolution of gene duplication in plants». Plant Physiol., 171: 2294-2316.
Parisod, C. [et al.] (2010). «Evolutionary consequences of autopolyploidy». New Phytol., 186: 5-17.
Pellicer, J. [et al.] (2010). «The largest eukaryotic genome of them all?». Bot. J. Linn. Soc., 164: 10-15. (2014). «A universe of dwarfs and giants: Genome size and chromosome evolution in the monocot family Melanthiaceae ». New Phytol ., 201: 14841497.
(2018). «Genome size diversity and its impact on the evolution of land plants». Genes, 9: 88. (2022). «Morphological and genome-wide evidence of homoploid hybridisation in Urospermum (Asteraceae)». Plants, 11: 182.
Pellicer, J.; Leitch, I. J. (2020). «The Plant DNA C-values Database (release 7.1): An updated online repository
of plant genome size data for comparative studies». New Phytol., 226: 301-305.
Pflug, J. M. [et al.] (2020). «Measuring genome sizes using read-depth, k-mers, and flow cytometry: Methodological comparisons in beetles ( Coleoptera )». G3 Genes|Genomes|Genetics, 10: 3047-3060.
Piegu, B. [et al.] (2006). «Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis , a wild relative of rice». Genome Research , 16: 1262-1269.
Pombert, J. F. [et al.] (2014). «A lack of parasitic reduction in the obligate parasitic green alga Helicosporidium». PLOS Genetics, 10: e1004355.
Pucker, B. (2019). «Mapping-based genome size estimation» BioRxiv, 607390.
Rastogi, S.; Ohri, D. (2020). «Chromosome numbers in Gymnosperms - an update». Silvae Genet., 69: 13-19.
Rice, A. [et al.] (2019). «The global biogeography of polyploid plants». Nat. Ecol. Evol., 3: 265-273.
Schaal, B. (2019). «Plants and people: Our shared history and future». Plants, People, Planet, 1: 14-19.
Sofi, P. [et al.] (2007). «Sequencing the maize genome: Rationale, current status and future prospects». Curr. Sci., 92: 1702-1708.
Soltis, P. S. [et al.] (2016). «Ancient WGD events as drivers of key innovations in angiosperms». Curr. Opin. Plant Biol., 30: 159-165.
Sonnleitner, M. [et al.] (2016). «Ecological differentiation of diploid and polyploid cytotypes of Senecio carniolicus sensu lato (Asteraceae) is stronger in areas of sympatry». Ann. Bot., 117: 269-276.
Stevens, K. A. [et al.] (2016). «Sequence of the sugar pine megagenome». Genetics, 204: 1613-1626.
Stuessy, T. F. (2009). Plant taxonomy : The systematic evaluation of comparative data. Nova York: Columbia University Press. (2011). «Multiple sources of comparative data for creative monography». A: Stuessy, T. F.; Lack, H. (ed.). Monographic plant systematics: Fundamental assessment of plant biodiversity . Ruggell: A. R. G. Gantner Verlag K. G., 33-47.
Sun, C. [et al.] (2012). «Slow DNA loss in the gigantic genomes of salamanders». Genome Biol. Evol., 4: 13401348.
Sun, H. [et al.] (2018). «findGSE: Estimating genome size variation within human and Arabidopsis using k-mer frequencies». Bioinformatics, 34: 550-557.
Suzuki, Y. (2020). «Advent of a new sequencing era: Longread and on-site sequencing». J. Hum. Genet., 65: 1. Twyford, A. D. (2018). «The road to 10,000 plant genomes». Nat. Plants, 4: 312-313.
Vallée, G. C. [et al.] (2016). «Economic importance, taxonomic representation and scientific priority as drivers of genome sequencing projects». BMC Genomics, 17: 782.
Wang, X. [et al.] (2021). «Genome downsizing after polyploidy: Mechanisms, rates and selection pressures». Plant J., 107: 1003-1015.
Wendel, J. F. (2015). «The wondrous cycles of polyploidy in plants». Amer. J. Bot., 102: 1753-1756.
Wicker, T. [et al.] (2007). «A unified classification system for eukaryotic transposable elements». Nature Rev. Genet., 8: 973-982.
Wood, T. E. [et al.] (2009). «The frequency of polyploid speciation in vascular plants». Proc. Nat. Acad. Sci. USA, 106: 13875-13879.
Treballs de la Societat Catalana de Biologia, 72: 10-15
15
Del genoma als gens
Resum
Durant el segle xx es va establir la naturalesa molecular dels gens —les unitats bàsiques de l’herència biològica— i dels processos involucrats en el flux d’informació dels gens a les proteïnes. La generalització de les tècniques de seqüenciació va facilitar l’obtenció de la seqüència dels genomes, però la seva utilitat és limitada sense un mapa dels gens. Per tal de construir-lo, hom utilitza una combinació de mètodes experimentals i computacionals. Les noves tecnologies que permeten l’obtenció de la seqüència completa dels RNA missatgers són les preferides, però no sempre es poden utilitzar. Per aquest motiu, els mètodes computacionals són essencials. Aquests mètodes inclouen la localització de regions en el genoma amb uns biaixos en la composició de la seqüència característics de les regions codificants de proteïnes. Els projectes en marxa que seqüenciaran el genoma de totes les espècies eucariotes faran necessari el desenvolupament de mètodes computacionals cada cop més acurats i eficients.
Paraules clau: anotació, seqüenciació, gens, predictors ab initio.
La naturalesa molecular dels gens
El terme gen va ser introduït l’any 1905 per Wilhem Johansen, per referir-se als «factors» hereditaris que Gregor Mendel havia proposat, mig segle abans, com a responsables dels trets observables dels individus. A principis del segle xx, es va determinar que els gens estaven situats als cromosomes, i el primer mapa genètic es va construir l’any 1913. Aquest avenç va ser seguit pel desenvolupament de mapes de més resolució i el càlcul de distàncies entre gens. Tots aquests models es van construir sense conèixer la naturalesa química del material hereditari, però dos grups de científics, Oswald Avery, Colin MacLeod i Maclyn McCarty (Avery et al. 1944), primer, i Alfred Hershey i Martha Chase (Hershey i Chase, 1952), després, van demostrar que es tractava d’àcid desoxiribonucleic (DNA). El pas final en la caracterització d’aquesta molècula va ser el descobriment de la seva naturalesa polimèrica fet per Watson i Crick l’any 1953 (Watson i Crick, 1953). Al mateix temps, Frederick Sanger desenvolupava metodologies de seqüenciació que van permetre caracteritzar la seqüència de proteïnes i, dues dècades més tard, la seqüència del DNA (Sanger i Tuppy,
DOI: 10.2436/20.1501.02.212
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 02/05/2022
Acceptat: 14/06/2022
From genome to genes
Abstract
In the 20th century, scientists established the molecular nature of genes – the basic units of biological inheritance – and of the processes involved in the flow of information from genes to proteins. The generalization of sequencing techniques facilitated the determination of the sequence of genomes, but their usability was limited without a map of all the genes involved. In order to build it, a combination of experimental and computational methods are used. The new sequencing technologies that allow the determination of the complete sequence of messenger RNAs are preferred, but they cannot always be used and this is why computational methods are essential. These methods include the identification of regions in the genome with sequence composition biases similar to those in the regions known to code for proteins. The ongoing projects that will sequence the genome of all the eukaryote species will make it necessary to develop more accurate and efficient computational methods.
Keywords: annotation, sequencing, genes, ab initio predictors.
1951; Sanger et al., 1977). Sanger va ser guardonat amb un Premi Nobel per cadascun d’aquests descobriments.
Paral·lelament a aquests avenços en l’estructura molecular del material hereditari, George Beadle i Edward Tatum van establir l’equivalència entre gens i proteïnes el 1941 (Beadle i Tatum, 1941). Poc després, George Gamow va establir la relació entre les seqüències d’aquestes dues biomolècules (Gamow et al., 1956) i el 1961 Sidney Brenner, François Jacob i Matthew Melenson van descobrir l’àcid desoxiribonucleic (RNA) (Brenner et al ., 1961). En concret, van descobrir l’RNA missatger (mRNA), que era la biomolècula que va permetre explicar com la informació codificada en el DNA, que es trobava dins el nucli, podia arribar al citoplasma, on es produeixen les proteïnes. Se sabia que hi havia d’intervenir una tercera molècula, i aquests tres científics la van identificar. Un mes després, Jacob i Jaques Monod van descriure l’mRNA en profunditat i en van destacar la possible funció reguladora (Jacob i Monod, 1961); i poc després d’aquest descobriment, van definir la correspondència entre els triplets de nucleòtids de les seqüències de DNA i mRNA (codons), i els ami-
Treballs de la Societat Catalana de Biologia, 72: 16-20
noàcids, que formen la seqüència de les proteïnes. El 1970, recopilant els avenços fets des de principis de segle, es va establir el dogma central de la biologia molecular (Crick, 1970), el qual estableix que «el DNA que resideix al nucli es transcriu a mRNA, que es desplaça al citoplasma, on successivament es tradueix a proteïna».
A partir d’aquests treballs i d’altres, i un cop establerta la naturalesa molecular dels gens, es va anar configurant una visió de com la informació genètica és configurada en la seqüència del DNA dels cromosomes d’acord amb la qual els gens correspondrien a regions (loci) d’aquesta seqüència, separades les unes de les altres, cada una de les quals especificaria la seqüència d’un mRNA, el qual, al seu torn, especificaria la seqüència d’aminoàcids d’una proteïna.
Anotació gènica
Entenem per anotació gènica el procés mitjançant el qual s’identifiquen els gens codificats en la seqüència genòmica. A part de la seva ubicació al genoma, cada gen té una estructura interna. Aquesta estructura es compon d’exons i introns i és determinada pels proces-
16
Ferriol Calvet1 i Roderic Guigó1, 2
1 Centre de Regulació Genòmica (CRG), Barcelona Institute of Science and Technology (BIST) 2 Universitat Pompeu Fabra (UPF)
Correspondència: Roderic Guigó. Centre de Regulació Genòmica. C. del Dr. Aiguader, 88. 08003 Barcelona. Adreça electrònica: roderic.guigo@crg.cat. Ferriol Calvet. Institut de Recerca Biomèdica. C. de Baldiri Reixac, 10. 08028 Barcelona. Adreça electrònica: ferriol. calvet@irbbarcelona.org
sos d’empalmament (splicing, en anglès) alternatiu i maduració als quals se sotmet l’mRNA després de produir-se amb la transcripció. El procés d’empalmament consisteix a eliminar les regions intròniques de l’mRNA i concatenar les regions restants conegudes com a exons. Aquest procés està mediat pel complex de tall i unió (spliceosome, en anglès), i és responsable que hi hagi una diversitat més gran d’RNA missatgers que de gens. Utilitzant les estadístiques de l’anotació de GENCODEv39 (Frankish et al., 2021) per al genoma humà, hi ha 3,98 vegades més transcrits que gens (244.939 i 61.533, respectivament).
L’objectiu del procés d’anotació d’un genoma és identificar tots els gens presents en aquell genoma, determinar les coordenades exactes de tots els exons i introns (figura 1) i assignar la funció o funcions corresponents a cada gen (Brent, 2005; Harrow et al., 2009). La identificació de la posició de tots els elements d’un gen en el genoma l’anomenem anotació estructural, mentre que el procés d’assignar la funció corresponent a cada gen és l’anotació funcional.
Centrant-nos en l’anotació estructural, tenint en compte la naturalesa de l’estructura interna dels gens, es poden utilitzar diferents
enfocaments per arribar a una anotació gènica precisa. L’obtenció experimental de la seqüència d’mRNA és la via més directa, i es va començar a implementar a finals del segle anterior amb el desenvolupament dels primers mètodes de seqüenciació. Una manera alternativa d’identificar gens és fer prediccions mitjançant eines computacionals que poden calcular mètriques sobre la seqüència del genoma i determinar quines regions tenen més probabilitat de codificar proteïnes. Aquest segon grup de mètodes es van poder començar a desenvolupar i aplicar un cop es van veure les característiques distintives de les regions amb gens respecte de les regions en què no n’hi havia.
Tots dos enfocaments han evolucionat des de les primeres implementacions, i en la majoria de protocols d’anotació es complementen amb l’objectiu d’identificar l’estructura exacta de tots els gens en el genoma de qualsevol espècie seqüenciada.
Les següents seccions d’aquesta revisió se centraran en els avenços fets en les eines experimentals i computacionals per obtenir l’estructura dels gens, i en els passos seguits per la majoria de mètodes d’anotació gènica ( pipelines, en anglès).
Obtenció experimental de l’estructura gènica
Amb els grans avenços en el camp de la biologia molecular durant les dècades de 1970 i 1980, els investigadors van poder estudiar regions específiques del genoma amb un detall sense precedents. L’interès principal se centrava en aquells loci relacionats amb malalties que havien estat prèviament cartografiats en el genoma. Tanmateix, un cop disponible la seqüència de nucleòtids de la regió, la seqüència exacta del gen codificat en aquella regió no era fàcil d’aconseguir, ja que les seqüències dels exons i dels introns no es poden distingir fàcilment, i això és imprescindible per poder trobar marcs de lectura oberta (ORF, de l’anglès open reading frame). Aquestes són les regions dels gens que contenen la seqüència que codifica la proteïna final.
Com que la seqüència genòmica no era suficient, es van desenvolupar estratègies per obtenir la seqüència de l’mRNA, on es poden buscar directament els codons d’inici i aturada que defineixen l’ORF. La seqüenciació directa de l’mRNA no era possible, i la millor alternativa era utilitzar un pas intermedi per retrotranscriure la molècula d’mRNA a DNA complementari (cDNA), ja que aquesta sí que es
Treballs de la Societat Catalana de Biologia, 72: 16-20
17 Del genoma als gens
Figura 1. Visualització de la posició i l’estructura interna d’un gen. a) Posició, també anomenada loci, de 8 gens a la regió p22.2 del cromosoma X d’humà. Representació dels gens reduïda a un únic transcrit per gen. b) Descripció de l’estructura interna del gen FANCB. S’observen tots els exons i els introns d’aquest gen i també els llocs d’acoblament. El canvi exó-intró l’anomenem lloc donador i l’intró-exó, lloc acceptor. Figura extreta d’http://genome.ucsc.edu (Lee et al., 2022), amb les anotacions de Morales et al. (2022) [consulta: 15 març 2022].
Ferriol Calvet i Roderic Guigó
podia seqüenciar amb els protocols de seqüenciació de DNA ja desenvolupats. Aquesta encara és actualment una de les tècniques més utilitzades per obtenir la seqüència d’mRNA. Havent trobat aquesta alternativa, hi havia un altre obstacle. Les lectures de seqüenciació més llargues produïdes per les diverses tècniques de seqüenciació disponibles fins al moment no eren suficients per cobrir tota la molècula d’mRNA. A causa d’això, els mRNA s’havien de retrotranscriure a cDNA i fragmentar-se en segments prou petits que sí que es podien seqüenciar de manera completa. Finalment, es necessitava un procés d’assemblatge per reconstruir les molècules d’mRNA a partir de tots els fragments seqüenciats. Aquest procés era especialment complex perquè no es podien aïllar només les molècules d’mRNA d’interès, tret que es tingués coneixement a priori de la seqüència.
Els mètodes experimentals per determinar l’estructura de l’mRNA s’han actualitzat des de les primeres versions (Wang et al., 2009). El volum de dades generat és més gran i les dades són de millor qualitat, i els mètodes per analitzar-les també són millors i més eficients. Aquests canvis es tradueixen en la capacitat de determinar l’estructura dels gens de manera molt més precisa.
Entre les millores més rellevants, en l’àmbit experimental, que faciliten el procés d’anotació gènica podem trobar el desenvolupament de tècniques de seqüenciació d’RNA (RNA-seq) de lectura llarga i tècniques de seqüenciació directa. En aquests moments, hi ha dues tecnologies principals per RNA-seq de lectura llarga. La primera es basa en la capacitat de detectar la incorporació d’un únic nucleòtid en una cadena de DNA; aquesta incorporació està catalitzada per una DNApolimerasa unida a la molècula de DNA que es vol seqüenciar (Schadt et al., 2010). La segona fa ús de la capacitat de detectar canvis en un camp elèctric que s’indueixen quan cada nucleòtid d’una molècula de DNA passa per un porus (Clarke et al. , 2009). Un avantatge d’aquesta segona tecnologia és que fa possible seqüenciar directament les molècules d’RNA, sense necessitat de copiar-les a cDNA.
Pipelines d’anotació
d’anotació dels principals centres que generen i publiquen més anotacions (Aken et al., 2016; Thibaud-Nissen et al., 2013).
El primer pas abans d’iniciar el procediment d’anotació ha de ser comprovar la qualitat de la seqüència del genoma. Idealment, hauríem de disposar d’una única seqüència per a cada cromosoma, però, ateses les limitacions tecnològiques, això sovint és difícil, i molts cops les seqüències dels cromosomes estan fragmentades. Mirant les estadístiques del genoma, centrant-nos en el nombre de seqüències i la seva llargada, i comparant-ho amb el nombre de cromosomes esperats, es pot veure com és de fragmentat el genoma. Això influirà en l’anotació, ja que alguns gens poden caure a les unions entre seqüències i es dividiran en dos o més gens diferents. Un altre procés més elaborat consisteix a utilitzar el mètode BUSCO (de l’anglès benchmarking universal single-copy orthologs) (Simão et al., 2015) per avaluar la integritat del genoma. Aquest mètode es basa en la recerca d’un conjunt de gens conservats en totes les espècies d’un grup taxonòmic concret, i que haurien d’existir en el genoma que es vol anotar; informa, aleshores, de la presència o absència d’aquests gens, així com de possibles duplicacions o fragmentacions. Manni et al. (2021) indica els protocols que cal seguir per avaluar la qualitat de les seqüències genòmiques.
Un cop s’ha comprovat que la qualitat del genoma és suficient per poder iniciar el procés d’anotació, la disponibilitat de dades experimentals determinarà quins protocols es poden seguir. Quan hi ha dades transcriptòmiques de l’espècie que es vol anotar o d’una espècie propera, s’utilitzen per aconseguir una identificació més precisa dels gens en el genoma. En cas contrari, quan no hi ha dades transcriptòmiques disponibles de l’espècie en estudi ni de cap espècie propera, es poden usar procediments alternatius, però amb l’inconvenient que la qualitat de l’anotació final és pitjor.
Mètodes dependents de la disponibilitat de dades transcriptòmiques
la qual es vol anotar el genoma. Si és possible, és encara millor tenir les dades transcriptòmiques del mateix individu del qual es va obtenir el genoma.
Després de la comprovació de la qualitat inicial de les dades d’RNA-seq, s’han d’alinear amb el genoma. Aquest pas és clau per poder construir després els models dels gens i els seus mRNA. L’alineació de seqüències d’RNA-seq amb el genoma permet identificar els llocs d’acoblament (splice sites, en anglès) i les posicions dels introns, les quals són indicades per interrupcions en alineament (figura 2). També permet identificar parcialment les regions exòniques en què s’alineen lectures senceres. Aquesta informació pot ser utilitzada per diversos programes que construeixen els models de gens i transcrits més probables. Si fem servir RNA-seq de lectura llarga, el procés d’assemblatge del model de transcripció és més senzill, ja que les cadenes contínues d’exons i introns són capturades de manera completa per les seqüències. Les dades generades amb les primeres versions d’RNA-seq de lectura llarga eren propenses a errors i el procés d’alineació tampoc no era trivial. Actualment, amb els avenços de les tecnologies de seqüenciació, s’obtenen dades de més qualitat i aquest problema s’ha minimitzat.
gènica
El procés d’anotació no té uns requeriments estrictes i els investigadors que volen produir una anotació ho fan seguint els mètodes que els semblin més adequats en cada cas. Tot i això, hi ha un conjunt de passos que són els més habituals i els utilitzats per les pipelines
El millor enfocament per determinar la ubicació i l’estructura dels gens és alinear l’evidència transcriptòmica amb el genoma en estudi. Aquesta evidència transcriptòmica pot tenir la forma de dades d’RNA-seq de lectura curta o de lectura llarga procedents de la mateixa espècie o d’una espècie evolutivament propera. El cas ideal és utilitzar dades transcriptòmiques de lectura llarga de la mateixa espècie de
Treballs de la Societat Catalana de Biologia, 72: 16-20
Mètodes independents de la disponibilitat de dades transcriptòmiques Després d’utilitzar les dades experimentals per generar el primer conjunt de transcrits altament fiable, es pot combinar l’ús d’altres mètodes per afegir nous models de gens. Aquests es basen en l’alineació de la informació disponible en bases de dades públiques o analitzen les periodicitats i els biaixos de les regions codificants del genoma per suggerir gens potencials que codifiquin proteïnes, alguns dels quals podrien no haver estat detectats en el procés d’alineació de les dades d’RNA-seq. La majoria d’aquests mètodes són sensibles a les regions repetitives o de baixa complexitat i, per aquest motiu, necessiten un fitxer amb la seqüència completa on aquestes regions que se sap que no acostumen a tenir gens estiguin senyalades d’una manera diferent de la resta. Els genomes que estan en aquest format s’anomenen genomes emmascarats (masked genomes, en anglès).
El procés d’emmascarament del genoma consisteix a identificar regions de DNA de baixa complexitat i regions amb elements repetitius (Hancock, 2002). Es pot fer ab initio o basant-se en biblioteques de regions repetitives.
18
Figura 2. Representació simplificada del procés d’RNA-seq. En primer lloc, s’obtenen els mRNA cel·lulars que s’han produït en el procés de transcripció i la maduració posterior, durant la qual els introns són eliminats. Al laboratori, aquests mRNA es retrotranscriuen a cDNA. Les molècules resultants es fragmenten en trossos més petits, els quals poden ser seqüenciats de manera completa. Un cop s’obtenen les seqüències d’aquests fragments, s’alineen amb el genoma de l’espècie que es vol anotar. En funció de si els fragments seqüenciats provenien d’una regió que corresponia a un mateix exó o es trobava a la intersecció entre dos exons, ens donarà informació de la localització dels introns i dels llocs d’acoblament o només de la localització dels exons. Elaboració pròpia.
Si no s’ha emmascarat prèviament el genoma de cap espècie estretament relacionada, el millor enfocament és utilitzar un mètode ab initio. Això requereix més temps, però analitza la seqüència en detall i troba regions que es repeteixen en diferents regions del genoma, així com aquelles amb una composició esbiaixada a causa de les repeticions de seqüències curtes. El segon enfocament, basat en una biblioteca de regions repetitives de referència, és més ràpid, ja que busca regions del genoma que tinguin la mateixa seqüència que les proporcionades d’entrada, però també pot ser inexacte en cas que les regions repetitives del genoma que es vol emmascarar siguin molt diferents de les descrites per la biblioteca de regions repetitives utilitzada (Storer et al., 2021).
Entre els mètodes que requereixen el genoma emmascarat i que aprofiten les bases de dades públiques, es troben els mètodes que alineen seqüències de proteïnes directament a la seqüència de DNA (Iwata i Gotoh, 2012). De manera similar a les dades d’RNA-seq, aquests alineaments són informatius per identificar les regions exòniques, però també els llocs d’acoblament potencials. Aquest és un mètode basat en l’homologia, i els models d’mRNA construïts amb aquestes dades poden ser fiables, encara que menys que els generats a partir de dades d’RNA-seq.
L’altra font de models de gens i d’mRNA són els predictors de gens ab initio (Scalzitti et al., 2020). Es tracta de programes que, utilitzant només el conjunt del genoma emmascarat i un fitxer de paràmetres, prediuen les es-
tructures gèniques d’aquesta seqüència. Aquests mètodes es basen en el biaix en l’ús de codons que exhibeixen els exons, és a dir, en el fet que en les regions que codifiquen proteïnes, a diferència de les no codificants, els diferents codons no apareixen amb freqüència idèntica (figura 3). El biaix en l’ús de codons en les seqüències codificants és conseqüència, en primer lloc, de l’ús desigual dels aminoàcids en les proteïnes. És a dir, en les proteïnes hi ha aminoàcids que apareixen amb molta
més freqüència que d’altres (per exemple, en les proteïnes dels vertebrats, més del 8 % dels aminoàcids són lisines, mentre que només l’1 % són triptòfans). En segon lloc, el biaix és conseqüència de l’ús desigual de codons sinònims per un mateix aminoàcid. S’han desenvolupat nombrosos mètodes per mesurar aquests biaixos (Guigó, 1997).
Cadascun dels programes té les seves particularitats, però els fitxers de paràmetres requerits contenen informació similar per modelar, principalment, les regions de codificants, els llocs d’acoblament i els introns (figura 3). Idealment, aquests fitxers de paràmetres es generen amb un procés d’entrenament basat en l’ús d’mRNA reals, obtinguts de la mateixa espècie que es vol anotar, i ajustant els paràmetres progressivament fins a arribar a una bona predicció d’aquests gens coneguts. Un inconvenient d’aquest tipus de mètodes és que aquest entrenament només és possible quan hi ha dades experimentals disponibles per a aquella espècie. Hi ha diverses maneres d’abordar aquest problema. Un és desenvolupar mètodes d’autoentrenament que no requereixin cap conjunt de gens coneguts per generar el fitxer de paràmetres, però la solució més habitual és utilitzar fitxers de paràmetres entrenats en espècies taxonòmicament properes.
Durant els pròxims deu anys, gràcies a projectes com l’Earth BioGenome Project (EBP) (Lewin et al., 2022) i els seus projectes afiliats, com la iniciativa catalana per a l’EBP (CBP, de
Figura 3. Representació del biaix en l’ús dels codons i dels llocs d’acoblament en una seqüència. Utilitzant la seqüència d’un gen humà, veiem com el biaix en l’ús dels codons és més gran a les regions que es corresponen als exons del gen. Les línies verticals indiquen les puntuacions dels llocs d’acoblament, calculades utilitzant matrius de pesos posicionals (PWM, de l’anglès position weight matrix). Aquestes es calculen a partir de seqüències reals dels donadors i dels acceptors i després s’utilitzen per puntuar la probabilitat que una posició en concret sigui un donador o un acceptor, respectivament. Figura adaptada de la memòria La codificació de la informació biològica en el genoma: Memòria llegida per l’acadèmic electe Dr. Roderic Guigó i Serra a l’acte de la seva recepció del dia 28 d’octubre de 2021, núm. 1064 de les Memòries de la Reial Acadèmia de Ciències i Arts de Barcelona [en línia], <https://www.racab.cat/publicacions/memories/1064> [consulta: 15 març 2022].
Treballs de la Societat Catalana de Biologia, 72: 16-20
19 Del genoma als gens
Ferriol Calvet i Roderic Guigó
l’anglès Catalan Initiative for the Earth BioGenome Project), seran desxifrats els genomes de centenars de milers d’espècies eucariotes. La seqüenciació, catalogació i caracterització d’aquests genomes proporcionarà per primera vegada una visió global de la vida a la Terra, i contribuirà a la identificació dels esdeveniments genòmics subjacents a les principals transicions durant la història de la vida: l’emergència dels eucariotes, de la pluricel·lularitat, de l’especialització de cèl·lules i d’òrgans, de la reproducció sexual, etc. També contribuirà a la comprensió de fenòmens biològics fonamentals, com ara el desenvolupament, la diferenciació (inclosa la regeneració), o aquells que estan involucrats en malalties i altres condicions humanes, com ara el càncer i l’envelliment.
Bibliografia
Aken, B. L. [et al.] (2016). «The Ensembl gene annotation system». Database: J. Biol. Databases Curation (2016).
Alexandersson, M. [et al.] (2003). «SLAM: Cross-species gene finding and alignment with a generalized pair hidden Markov model». Genome Res., 13 (3): 496-502.
Avery, O. T. [et al.] (1944). «Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Inductions of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III». J. Exp. Med., 79 (2): 137-158.
Beadle, G. W.; Tatum, E. L. (1941). «Genetic control of biochemical reactions in neurospora». Proc. Natl. Acad. Sci. USA, 27 (11): 499-506.
Brenner, S. [et al.] (1961). «An unstable intermediate carrying information from genes to ribosomes for protein synthesis». Nature, 190 (4776): 576-581.
Brent, M. R. (2005). «Genome annotation past, present, and future: How to define an ORF at each locus». Genome Res., 15 (12): 1777-1786.
Clarke, J. [et al.] (2009). «Continuous base identification for single-molecule nanopore DNA sequencing». Nat. Nanotechnol., 4 (4): 265-270.
Crick, F. (1970). «Central dogma of molecular biology». Nature, 227 (5258): 561-563.
Frankish, A. [et al.] (2021). «GENCODE 2021». Nucleic Acids Res., 49 (D1): D916-923.
Gamow, G. [et al.] (1956). «The problem of information transfer from the nucleic acids to proteins». Adv. Biol. Med. Phys., 4: 23-68.
Guigó, R. (1997). «DNA composition, codon usage and exon prediction». A: Genetic databases. Cambridge (RU): Elsevier, 53-80.
Aquest coneixement, però, no pot ser inferit directament de la seqüència dels genomes, si abans els gens codificats en aquestes seqüències no han estat identificats. En aquest sentit, els mètodes eficients per a la identificació de gens són prerequisits per a projectes com l’EBP; tanmateix, alhora, són també la conseqüència d’aquests projectes. Efectivament, la continuïtat històrica que lliga totes les espècies que habiten el planeta queda reflectida en la semblança de la seva seqüència genòmica, una semblança que és molt més acusada en les regions que codifiquen proteïnes, és a dir, en els gens, els quals estan sotmesos a processos de selecció més forts que no pas les regions no gèniques. La conservació de regions funcionals ens proporciona una manera general d’identi-
ficar gens en seqüències de DNA: mitjançant la comparació de seqüències genòmiques d’espècies diferents, fins i tot en el cas que les seqüències que comparem siguin completament anònimes (és a dir, que en totes les seqüències que comparem desconeixem els gens que eventualment puguin estar-hi codificats). La identificació de regions conservades entre seqüències genòmiques de dues o més espècies ens pot revelar la presència de gens homòlegs en aquestes seqüències (Alexandersson et al., 2003; Korf et al., 2001; Wiehe et al., 2001). En conseqüència, a mesura que el nombre de genomes coneguts creixi, també ho farà la nostra capacitat per identificar-ne els gens en cadascun i per entendre, en conseqüència, com la biologia de les espècies està codificada en els seus genomes.
Hancock, J. M. (2002). «Genome size and the accumulation of simple sequence repeats: Implications of new data from genome sequencing projects». Genetica, 115 (1): 93-103.
Harrow, J. [et al.] (2009). «Identifying protein-coding genes in genomic sequences». Genome Biol., 10 (1): 201.
Hershey, A. D.; Chase, M. (1952). «Independent functions of viral protein and nucleic acid in growth of bacteriophage». J. Gen. Physiol., 36 (1): 39-56.
Iwata, H.; Gotoh, O. (2012). «Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features». Nucleic Acids Res., 40 (20): e161.
Jacob, F.; Monod, J. (1961). «Genetic regulatory mechanisms in the synthesis of proteins». J. Mol. Biol., 3: 318-356.
Korf, I. [et al.] (2001). «Integrating genomic homology into gene structure prediction». Bioinformatics, 17, supl. 1: S140-148.
Lee, B. T. [et al.] (2022). «The UCSC Genome Browser database: 2022 update». Nucleic Acids Res . [en línia], 50, D1: D1115-D1122. <https://doi.org/10.1093/nar/ gkab959>.
Lewin, H. A. [et al.] (2022). «The Earth BioGenome Project 2020: Starting the clock». Proc. Natl. Acad. Sci. USA, 119 (4): e2115635118.
Manni, M. [et al.] (2021). «BUSCO: Assessing genomic data quality and beyond». Curr. Protoc., 1 (12): e323.
Morales, J. [et al.] (2022). «A joint NCBI and EMBL-EBI transcript set for clinical genomics and research». Nature, 604: 310-315
Sanger, F. [et al.] (1977). «DNA sequencing with chainterminating inhibitors». Proc. Natl. Acad. Sci. USA, 74 (12): 5463-5467.
Sanger, F.; Tuppy, H. (1951). «The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates». Biochem. J., 49 (4): 481-490.
Scalzitti, N. [et al.] (2020). «A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms». BMC Genomics, 21 (1): 293.
Schadt, E. E. [et al.] (2010). «A window into third-generation sequencing». Hum. Mol. Genet., 19 (R2): R227240.
Simão , F. A. [ et al .] (2015). «BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs». Bioinformatics, 31 (19): 32103212.
Storer, J. [et al.] (2021). «The Dfam community resource of transposable element families, sequence models, and genome annotations». Mob. DNA, 12 (1): 2.
Thibaud-Nissen, F. [et al.] (2013). «Eukaryotic genome annotation pipeline» A: The NCBI Handbook [en línia]. < https://www.ncbi.nlm.nih.gov/books/ NBK169439/> [Consulta: 30 abril 2022].
Wang, Z. [et al.] (2009). «RNA-seq: A revolutionary tool for transcriptomics». Nat. Rev. Genet., 10 (1): 57-63.
Watson, J. D.; Crick, F. H. (1953). «Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid». Nature, 171 (4356): 737-738.
Wiehe, T. [et al.] (2001). «SGP-1: Prediction and validation of homologous genes based on sequence alignments». Genome Res., 11 (9): 1574-1583.
Treballs de la Societat Catalana de Biologia, 72: 16-20
20
Avenços en les tecnologies de seqüenciació del DNA
Berta Fusté,1 Elena Vila1 i Mònica Bayés1, 2
1 Centre Nacional d’Anàlisi Genòmica - Centre de Regulació Genòmica (CNAG-CRG), Barcelona Institute of Science and Technology (BIST)
2 Universitat Pompeu Fabra (UPF)
Correspondència: Berta Fusté. Centre Nacional d’Anàlisi Genòmica. C. de Baldiri Reixac, 4. 08028 Barcelona. Tel.: +34 934 037 289. Adreça electrònica: berta.fuste@cnag.crg.eu.
Resum
L’any 1977 Frederick Sanger va desenvolupar un mètode per a determinar l’ordre de les bases dels fragments de DNA. Aquesta tecnologia encara s’utilitza actualment i ha sigut clau per a aconseguir fites tan importants com la primera seqüenciació completa del genoma humà. L’aparició d’una nova generació de tecnologies de seqüenciació del DNA, tecnologies d’NGS (de l’anglès next generation sequencing), més la gran explosió d’eines computacionals per a analitzar-lo, ha permès seqüenciar de manera ràpida, econòmica i amb una elevada precisió genomes de microbis, plantes i animals. Durant l’última dècada, hi ha hagut una gran expansió de plataformes d’NGS: primer van sorgir les tecnologies d’NGS de cadena curta i, més endavant, les de cadena llarga. Tot i que les tecnologies d’NGS de lectures llargues prometien grans avenços per a resoldre genomes complexos, les freqüències d’error d’aquestes tecnologies són elevades. Això ha fet que, en els últims anys, hagin aparegut una sèrie de mètodes de seqüenciació complementaris per a resoldre les deficiències de les tecnologies d’NGS de lectures curtes i llargues.
Paraules clau: genoma, seqüenciació de nova generació (NGS), seqüenciació per síntesi (SBS), seqüenciació de molècules úniques en temps real (SMRTseq), seqüenciació per nanoporus (ONT).
Introducció
a la genòmica
El genoma és el conjunt del material genètic de les cèl·lules d’un organisme que s’emmagatzema en forma d’àcid desoxiribonucleic o DNA i que conté tota la informació per al seu desenvolupament i funcionament correctes. Anomenem gens els segments de DNA que confereixen instruccions específiques a la cèl·lula, sovint mitjançant la síntesi de proteïnes a partir de molècules intermèdies anomenades ARN missatgers. Les proteïnes són les que formen els òrgans i teixits del cos, i controlen les reaccions químiques i la comunicació entre les cèl·lules.
Tal com van descriure James Watson i Francis Crick l’any 1953 gràcies a les observacions prèvies de Rosalind Franklin, la molècula de DNA està formada per dues cadenes d’unes unitats químiques que anomenem nucleòtids, enrotllades al voltant d’un eix comú formant una doble hèlix (Watson i Crick, 1953). Hi ha quatre tipus de nucleòtids, que identifiquem amb les lletres A, T, G i C (adeni-
DOI: 10.2436/20.1501.02.213
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 24/01/2022
Acceptat: 16/03/2022
Advances in DNA sequencing technology
Abstract
In 1977, Frederick Sanger developed a method for determining the order of the bases of DNA fragments. This technology still works today and has been crucial in achieving such important milestones as the first complete sequencing of the human genome. The emergence of the new generation of DNA sequencing technologies (NGS) plus the great explosion of computer tools for their analysis has become a matter of routine and allows the sequencing of the genomes of microbes, plants and animals in a way that is quick, relatively cheap and highly precise. There has been a great expansion of NGS sequencing platforms over the last decade, first involving short-read and later longread NGS sequencing technologies. Although long-read NGS sequencing promised great advances in solving complex genomes, the error rates of these technologies are high. This has led to the appearance in recent years of a number of complementary sequencing methods to address the shortcomings of NGS sequencing of short and long readings.
Keywords: genome, next-generation sequencing (NGS), sequencing by synthesis (SBS), single-molecule real-time sequencing (SMRT-seq), nanopore sequencing (ONT).
na, timina, guanina i citosina, respectivament). Les dues cadenes queden unides per ponts d’hidrogen formats entre la base d’una cadena i la de l’altra cadena, amb la qual queda enfrontada. Els aparellaments sempre són entre A-T i G-C. El DNA dels individus d’una mateixa espècie varia en un petit percentatge (<1 %); hi trobem canvis d’un únic nucleòtid (single nucleotide variants o SNV), però també reordenaments genòmics o variants estructurals (structural variants o SV) que afecten centenars o milers de bases, com ara inversions, delecions o duplicacions. Aquestes variacions en el DNA són responsables de les diferències entre les persones, i en alguns casos també poden donar lloc a malalties diverses.
El genoma humà té uns tres mil milions de nucleòtids i al voltant de vint-i-cinc mil gens, cada un dels quals dona lloc a tres proteïnes diferents de mitjana. La primera seqüència del genoma humà, és a dir, l’ordre de pràcticament tots els nucleòtids en la cadena del DNA, es va completar l’any 2003, en el marc d’una col·la-
Treballs de la Societat Catalana de Biologia, 72: 21-27
boració internacional, el Projecte Genoma Humà (http://www.genome.gov/10001772). Es tracta d’un dels assoliments més importants de la biologia; el projecte es va dur a terme en tretze anys i es calcula que va tenir un cost d’uns tres mil milions de dòlars (NHGRI, 2020).
L’any 1977, un investigador de la Universitat de Cambridge, Frederick Sanger, va desenvolupar un mètode per a determinar l’ordre dels nucleòtids i obtenir-ne la seqüència de DNA (Sanger et al., 1977). Es basa en l’ús d’un enzim, la DNA polimerasa, per a generar noves cadenes a partir de la cadena que es vol seqüenciar. En aquesta síntesi, es generen fragments que acaben en les quatre possibles bases del DNA, cadascuna marcada amb una molècula fluorescent diferent. Aquests fragments se separen després segons la seva mida en una matriu porosa i es detecten mitjançant el senyal fluorescent que emeten. L’any 1980 Sanger va ser guardonat amb el Premi Nobel de Química per aquest descobriment. Actualment, el mètode de Sanger continua sent el
21
Berta Fusté, Elena Vila i Mònica Bayés
més adequat per a seqüenciar petites regions de DNA en moltes mostres o, fins i tot, per a validar resultats obtinguts amb tecnologies més noves (Hert et al., 2008).
Evolució de la nova generació de tecnologies de seqüenciació
L’any 2004 hi va haver un canvi de paradigma en el camp de la seqüenciació del DNA i de la genòmica, amb l’aparició d’una nova generació de tecnologies de seqüenciació (tecnologies d’NGS, de l’anglès next negeneration sequencing) o seqüenciació massiva. Les tecnologies d’NGS combinen l’ús de tècniques d’enginyeria genètica, la nanotecnologia i la generació de milions de dades basades en la imatge. A diferència de la seqüenciació pel mètode de Sanger, que es basa en l’anàlisi individual de fragments de DNA, els seqüenciadors d’NGS són capaços d’analitzar milions de fragments de DNA en paral·lel i, en conseqüència, de seqüenciar un genoma humà sencer en pocs dies. Generen una quantitat de dades genòmiques impensable fa vint anys, de qualsevol organisme, en coneguem o no el genoma prèviament, i de manera sòlida. Les tecnologies d’NGS, juntament amb la gran explosió d’eines computacionals a finals de la primera dècada del segle xxi, com ara els programes informàtics per a alinear milions de lectures curtes en genomes de referència o per a detectar variants genètiques, han provocat un increment sense precedents de les dades de seqüenciació, a una velocitat superior a la llei de Moore, segons la qual la capacitat dels ordinadors es dobla cada any.
Amb l’ús d’aquestes noves tecnologies, el 2005 es van seqüenciar dos genomes bac -
terians sencers (Shendure et al., 2005; Margulies et al., 2005), i el mateix any l’empresa biotecnològica 454 Life Sciences va comercialitzar el primer equip de seqüenciació massiva, el 454 Genome Sequencer, capaç de produir lectures d’uns 110 parells de bases (pb) i un total de 20 megabases (Mb) en cada carrera (unitat de funcionament de cada seqüenciador). Roche Diagnostics va adquirir 454 Life Sciences i va comercialitzar-ne els instruments de seqüenciació durant uns anys, però actualment aquesta plataforma ja no està en funcionament.
L’any 2006, l’empresa Solexa, una empresa derivada de la Universitat de Cambridge, va llançar al mercat el seqüenciador Genome Analyzer 1G, que permetia seqüenciar 1 gigabase (Gb) de seqüència en una única carrera. La multinacional Illumina va adquirir Solexa poc després i des d’aleshores s’han seqüenciat una gran quantitat de genomes de microbis, plantes i animals amb aquesta tecnologia, que és avui en dia la principal tecnologia d’NGS de cadena curta. El primer genoma humà seqüenciat per NGS va ser el de James Watson el 2008, que ho va fer utilitzant els equips Genome Analyzer 1G, amb un cost d’un milió de dòlars i en un període de temps de dos mesos. Des del seu inici, el 2005, fins avui, Illumina ha dedicat tots els seus esforços a incrementar la producció dels seus seqüenciadors al mateix temps que es reduïa el cost per gigabase de dades generades. El NovaSeq6000 és, ara mateix, el seqüenciador més potent del mercat, capaç de produir 6 terabases (Tb) de seqüència i 20 bilions de lectures en menys de 48 h, l’equivalent a 48 genomes humans a una cobertura 30 ×.
El 2009 van sorgir dues tècniques d’NGS de cadena llarga que utilitzen aproximacions molt diferents: la seqüenciació de molècules úniques en temps real (SMRT), de Pacific Biosciences (PacBio), i la seqüenciació basada en nanoporus, d’Oxford Nanopore Technologies (ONT). PacBio va comercialitzar el primer instrument el 2015. En l’actualitat, PacBIO opera amb tres seqüenciadors de tecnologia d’NGS de lectures llargues, els Sequel Systems. Els seqüenciadors més grans són els Sequel II i Sequel IIe, amb una capacitat de seqüenciar 8 milions de molècules alhora. Pel que fa a ONT, el 2014 va comercialitzar el primer seqüenciador, el MinION, capaç de produir entre 5 i 10 Gb de seqüència per carrera. Gràcies a les millores continuades en els mètodes de preparació de biblioteques i en la química del MinION, el 2018 es va seqüenciar el primer genoma humà amb lectures ultrallargues (>100 kb). Avui dia, l’empresa comercialitza dos seqüenciadors més, que són més potents: el GridION, que pot generar entre 50 i 250 Gb de seqüència per carrera, i el més potent dels instruments, el PromethION, capaç de generar entre 1.000 i 2.000 Gb de seqüència en una sola carrera.
Durant l’última dècada hem vist una gran expansió de plataformes de seqüenciació, i seqüenciar el genoma d’un vertebrat ja és una cosa rutinària. Tot i així, l’assemblatge de quasi tots els genomes diploides continua sent incomplet i altament fragmentat. En els últims anys, juntament amb les tecnologies d’NGS, han aparegut mètodes de seqüenciació complementaris, com el Hi-C (Bonev i Cavalli, 2016) o el mapatge òptic (Giani et al., 2019) per a resol-
Treballs de la Societat Catalana de Biologia, 72: 21-27
22
1953 Descobriment de l’estructura del DNA (Watson iCrick) 1977 Seqüenciacióde Sanger 2005 2009 2014 Primer seqüenciador amb tecnologia de nanoporusMinION (OxfordNanoporeTechnologies) Primer seqüenciadord’NGS: 454 Genome Sequencer (454 LifeScience) 2017 1990 Inici del Projecte Genoma Humà (19902001) 2008 SeqüenciadorGenome Analyzer 1G (Solexa/Illumina) Primers articles publicats sobre tecnologia d’NGS de cadena llarga 2006 Seqüenciadors NovaSeq6000 (Illumina) Seqüenciació dels primers genomes humans amb tecnologia d’NGS 2018 Primer genoma humà assemblat de novo amb tecnologia d’ONT 2020 Assemblatge del cromosoma X humà de telòmer a telòmer 2015 Instruments Sequel System (PacificBiosciences) 2010 Primera anàlisi del metilomahumà seqüenciat Seqüenciador PromethION (OxfordNanopore Technologies)
Fites clau en les tecnologies de seqüenciació del DNA.
Figura 1.
Elaboració pròpia.
dre les deficiències de l’NGS de lectures curtes i llargues. De fet, s’ha acabat demostrant que la combinació d’aquestes tècniques pot solucionar les limitacions de cada una per separat. El 2020 es va publicar per primera vegada l’assemblatge d’un cromosoma humà, concretament el cromosoma X, de telòmer a telòmer (T2T) sense haver-hi cap buit per resoldre (Miga et al., 2020) (vegeu la figura 1).
Principis bàsics de les tecnologies d’NGS
Seqüenciació per síntesi (SBS), d’Illumina
Avui dia, quan parlem de tecnologies d’NGS de lectures curtes o NGS de cadena curta, parlem de seqüenciació de DNA amb instruments d’Illumina (www.Illumina.com). Més del 90 % de les dades de seqüenciació al món es generen mitjançant aquesta plataforma. Es parla de seqüenciació de cadena curta quan els fragments que se seqüencien van entre 50 i 300 pb.
La tecnologia d’Illumina sintetitza la nova cadena de DNA usant el mètode de seqüenciació per síntesi (SBS), que consisteix en la fabricació d’una cadena de DNA complementària a una cadena motlle mitjançant la DNA polimerasa. Tot i que la tecnologia d’Illumina està basada en l’aproximació de Sanger, difereix en la longitud de les lectures, que són més curtes, però, sobretot, en la capacitat d’analitzar milions de fragments de DNA de manera massiva i en paral·lel. La taxa d’error amb aquest mètode és més alta que amb el de Sanger, però es veu compensada per la gran capacitat de generar moltíssimes dades de seqüència alhora i, per tant, incrementar la cobertura de cada base seqüenciada. Per aquesta raó, la química d’Illumina és considerada seqüenciació d’alta resolució i precisió (precisió del 99,9 %). El 80 % de les bases seqüenciades per Illumina presenten un valor de qualitat Phred quality score igual o més alt de Q30, és a dir, que la probabilitat d’identificar erròniament una base és d’una entre un miler. Aquest nivell de fiabilitat és ideal per a abordar tot el ventall d’aplicacions d’NGS, incloent-hi les de diagnòstic clínic.
En general, l’NGS implica quatre passos bàsics, que es divideixen en: 1) la preparació de la biblioteca, que consisteix en la lligació de seqüències curtes conegudes, anomenades adaptadors; 2) la generació de milers de molècules de DNA idèntiques, o sigui la immobilització i clonació de les molècules de DNA que es vol
seqüenciar; 3) la seqüenciació, i 4) l’anàlisi de les dades.
El primer pas en la seqüenciació d’Illumina consisteix a trencar el DNA en fragments més manejables d’entre 200 i 600 pb. Als fragments de DNA se’ls uneixen unes seqüències curtes conegudes, anomenades adaptadors . Aquests adaptadors tenen tres funcions, que són la clau en els mètodes d’NGS de cadena curta. En primer lloc, serveixen per a immobilitzar les seqüències de DNA en una superfície sòlida ( flowcells o FC) que contenen nanopous, on s’amplifica i se seqüencia el DNA. En segon lloc, s’empren per a replicar les seqüències ancorades i produir milers de molècules de DNA idèntiques. Aquest procés es coneix amb el nom de bridge PCR amplification i és necessari per a després emetre un senyal prou fort per a ser detectat per una càmera. I en tercer lloc, aquests adaptadors són la seqüència complementària a l’encebador que, juntament amb la DNA polimerasa, elongaran la cadena i, per tant, generaran la seqüència que després llegirem (Goodwin et al., 2016; Barton et al., 2018).
Igual que el mètode de Sanger, Illumina utilitza la incorporació de nucleòtids modificats (dNTP, de l’anglès deoxynucleotide triphosphates) i reversibles durant diferents cicles consecutius, de manera que, una vegada incorporats, impedeixen l’elongació de la cadena de DNA. Els nucleòtids modificats són marcats amb un fluorocrom diferent que en ser excitat per un làser dona diferents longituds d’ona. L’emissió del senyal generat és capturada per una càmera i emmagatzemada en un ordinador. El procés de seqüenciació d’Illumina és un procés cíclic. A cada cicle de seqüenciació s’incorpora un únic dNTP a la molècula de DNA. La incorporació d’aquest dNTP, juntament amb el senyal emès pel fluoròfor, queda enregistrat per la càmera i, per tant, guardat a l’ordinador. Al final de cada cicle hi ha un trencament del grup que bloqueja a 3’ del nucleòtid i de l’etiqueta fluorescent que permetrà la incorporació del següent nucleòtid en el cicle posterior, i així successivament. El nombre de cicles es repeteix n vegades i és equivalent a la longitud de lectures seqüenciades, lectures de n bases de longitud. La seqüència de DNA s’analitza base a base durant la seqüenciació d’Illumina, per la qual cosa és un mètode molt precís. Una vegada acabat el procés de seqüenciació, la seqüència generada es pot alinear amb una seqüència de referència per a buscar coincidències o canvis en el DNA seqüenciat.
Treballs de la Societat Catalana de Biologia, 72: 21-27
Seqüenciació de molècules úniques en temps real (SMRT), de PacBio
La seqüenciació de molècules úniques en temps real (SMRT, de l’anglès single-molecule real-time ) és una tècnica d’NGS de lectures llargues que permet seqüenciar milions de molècules llargues de DNA al mateix temps utilitzant el procés natural de la replicació del DNA.
La tecnologia SMRT genera seqüències molt llargues que poden ser de 30-50 quilobases (kb) o inclús més llargues. A més, la tecnologia SMRT, tal com n’indica el nom, sintetitza una única molècula de DNA i ho fa en temps real. A diferència de la tècnica d’NGS de cadena curta, SMRT no cal que amplifiqui fragments de DNA ni de cicles químics repetitius per a elongar la cadena, fet que afavoreix l’eliminació de tots els errors sistemàtics induïts per la mateixa amplificació del DNA.
La tecnologia SMRT parteix de DNA circular de cadena simple. Al DNA se li uneixen uns adaptadors de cadena doble a cada extrem que en permeten la circularització i també l’ancoratge de les molècules a la DNA polimerasa. La reacció de seqüenciació es produeix sobre un suport sòlid (flowcell, SMRT FC) que conté milions de nanopous (pous ZMW, zeromode waveguide). El pou ZMW és una cavitat de desenes de nanòmetres de diàmetre que es fabrica en una pel·lícula metàl·lica de 100 nm dipositada sobre un substrat de vidre i on la llum no pot entrar. A la superfície de vidre inferior de cada pou ZMW hi ha ancorada una DNA polimerasa. En el procés de seqüenciació s’afegeix a cada pou ZMW una única molècula de DNA circular, més els quatre nucleòtids marcats amb fluorescència. A mesura que la DNA polimerasa incorpora nucleòtids a la cadena, s’alliberen els fluoròfors que, juntament amb l’excitació per làser, emeten diferents longituds d’ona. Cada longitud d’ona s’identifica amb una de les bases. El procés es fa en temps real i és enregistrat pel mateix instrument en format de vídeo (Eid et al., 2009; Ansorge et al., 2017).
Un dels avantatges de la tecnologia d’NGS de cadena llarga SMRT és la capacitat de sintetitzar seqüències de diverses kilobases (1025 kb) amb la resolució necessària per a resoldre de manera senzilla zones del genoma amb un alt nombre d’elements repetitius o identificar les diferents isoformes d’un mateix gen. Per contra, un dels principals desavantatges que té és la taxa d’error, que és força més alta que el de les tecnologies d’NGS de cadena curta. En aquest sentit, PacBio ha desenvolupat dues es-
23
Avenços en les tecnologies de seqüenciació del DNA
tratègies per a resoldre aquest problema: una d’enfocada a obtenir lectures molt llargues (CLR, de l’anglès continous long reads) i un nou mètode anomenat d’alta fidelitat (HiFi), en què la mateixa molècula de DNA és seqüenciada diverses vegades, de manera que la seqüència final és un consens d’alta qualitat autocorregit (CCS, de l’anglès circular consensus sequencing ) que aconsegueix lectures molt precises (99,8 %) (Taishan et al., 2021).


Seqüenciació per nanoporus, d’Oxford Nanopore Technologies (ONT) Oxford Nanopore Technologies (ONT, www. nanoporetech.com) ha desenvolupat una tec -
Genoma que es vol seqüenciar Fragmentació del gDNA
Lligació de seqüències adaptadores als extrems dels fragments de DNA
dispositius de seqüenciació d’ONT utilitzen uns suports sòlids (FC) que contenen centenars d’aquests nanoporus, l’un al costat de l’altre. Cada nanoporus conté el seu propi elèctrode connectat a un sensor que mesura el corrent elèctric que flueix a través del nanoporus. Quan una molècula passa a través d’un nanoporus, el corrent canvia i produeix un senyal elèctric característic per cada nucleòtid o grup de nucleòtids. Aquest senyal elèctric es descodifica utilitzant algoritmes bioinformàtics i determina l’ordre de les bases seqüenciades (Jain et al., 2018; Lin et al., 2021).
Ancoratge dels fragments de DNA en un suport sòlid
Amplificació dels fragments de DNA (bridge PCR)
Seqüenciació dels fragments de DNA u�litzant nucleò�ds marcats amb fluorescència

Captació de la fluorescència emesa pels nucleò�ds incorporats amb una càmera d’alta resolució

Transformació de les imatges fluorescents en seqüències de nucleò�ds
La seqüenciació d’ONT analitza tota la cadena de DNA i RNA que passa pel nanoporus; és a dir, la longitud de les seqüències és equivalent a la longitud de la mostra inicial que volem processar. La longitud de les lectures, doncs, és condicionada pel protocol que s’utilitza per a extreure el DNA o l’RNA, però també pel protocol utilitzat en la preparació de les biblioteques. En treballar amb molècules de DNA o RNA sense manipular, el procés de preparació de biblioteques és molt simple i ràpid. A les molècules de DNA o RNA se’ls afegeix un adaptador de cadena senzilla i una proteïna motora. Aquesta cadena senzilla permet l’entrada d’una de les dues cadenes de la molècula de DNA dins del porus amb l’ajut de la proteïna motora que comença el procés d’elongació de la seqüència.
nologia d’NGS de cadena llarga que permet seqüenciar molècules de DNA i RNA en temps real. De la mateixa manera que la tecnologia SMRT, la seqüenciació per nanoporus (ONT) també pot seqüenciar molècules úniques de DNA i RNA sense passos d’amplificació. La seqüenciació d’ONT és el mètode d’NGS que aconsegueix les lectures de seqüència més llargues: pot generar seqüències de més d’1 Mb de longitud i arribar a les 2 Mb per fragment amb ajuda computacional.
La seqüenciació d’ONT es basa a passar una molècula de DNA o RNA per uns petits forats, anomenats nanoporus , que es troben incrustats en una membrana sintètica. Tots els
Treballs de la Societat Catalana de Biologia, 72: 21-27
La seqüenciació d’ONT es produeix en temps real, ja que, a mesura que se seqüencia, es pot anar llegint la seqüència generada en un ordinador. Aquest pas és un avantatge respecte d’altres sistemes de seqüenciació perquè la qualitat de la seqüència pot ser validada en el moment precís i no cal esperar que l’experiment finalitzi per a veure’n els resultats. Un altre avantatge és la mida reduïda dels instruments utilitzats. Un MinION és més petit que un telèfon mòbil, la qual cosa fa que sigui totalment transportable i que pugui connectar-se directament a qualsevol ordinador per mitjà d’un port USB. Ara bé, igual que la tecnologia SMRT, la seqüenciació d’ONT, tot i produir seqüències molt llargues, presenta valors de qualitat de seqüència inferiors a les plataformes d’NGS de cadena curta. Això es pot resoldre mitjançant estratègies de combinació de tecnologies d’NGS de lectures llargues i curtes, tot i que recentment ONT està desenvolupant reactius d’alta fiabilitat per a la seva tecnologia, Q20+, que incrementa la fiabilitat de la seqüenciació i, en conseqüència, la precisió de les lectures s’aproxima a nivells similars als de l’NGS de cadena curta (Taishan et al., 2021).
24
Berta Fusté, Elena Vila i Mònica Bayés
ATTGCGACATCAGCGAGGG
ATTGCG
Encebador
Polimerasa T C A
A G G
1 2 6 3 4 5 7 8
Figura 2. Flux de seqüenciació per síntesi (SBS), d’Illumina. Elaboració pròpia.
Molècules de DNA d’alt pes molecular (HMW DNA)
Lligació de seqüències adaptadores als extrems dels fragments de DNA. Les biblioteques són DNA circular
Ancoratge de la DNA polimerasa al fons dels pous ZMW. A cada pou hi ha una molècula de DNA única que cal seqüenciar
Seqüenciació dels fragments de DNA u�litzant nucleò�ds marcats amb fluorescència. La fluorescència és alliberada a mesura que s’hi van incorporant nucleò�ds
Captació de la fluorescència emesa pels nucleò�ds incorporats amb una càmera de vídeo a temps real
l’RNA total (coding RNA i long-noncoding RNA [lncRNA]), l’RNA missatger (mRNA) i els RNA de mida petita (small RNA) (Stark et al., 2019). La caracterització de modificacions epigenètiques, com la detecció de la metilació de les citosines, també es fa actualment per NGS. El mètode més utilitzat es basa en un tractament amb bisulfit sòdic que converteix les citosines no metilades en uracils, però deixa les citosines metilades intactes. Quan se seqüencia el DNA tractat d’aquesta manera (WGBS, de l’anglès whole-genome bisulfite sequencing), s’obtenen dades precises sobre la freqüència de la metilació de totes les citosines del genoma. També podem identificar les interaccions entre determinades proteïnes i el DNA utilitzant tècniques d’immunoprecipitació seguides d’NGS (ChIP-seq), o alternativament estudiar les regions no protegides pels nucleosomes mitjançant l’ús de transposases o d’altres estratègies que permeten enriquir els fragments de DNA accessibles de la cromatina (ATAC-seq, FAIRE-seq). Finalment, s’han desenvolupat protocols específics per a explorar l’organització tridimensional de la cromatina al nucli, com ara el Hi-C. En la seqüenciació Hi-C s’estabilitzen els complexos DNA-proteïna amb formaldehid, s’enriqueixen aquells fragments que es troben en un espai proper en el nucli de la cèl·lula i se seqüencien, de manera que s’obté un mapa precís de les interaccions de la cromatina.
Principals aplicacions de les tecnologies d’NGS
Les tecnologies d’NGS possibiliten l’estudi del material genètic a un nivell de resolució sense precedents i amb protocols molt diversos segons l’objectiu de l’estudi (Buermans et al. , 2014). Actualment, i gràcies a la gran disminució del seu cost, la seqüenciació de genomes sencers (WGS, de l’anglès whole genome sequencing) és una de les aplicacions d’NGS més utilitzades. Permet obtenir una visió completa de tot el genoma, comparar-la amb el genoma de referència de l’espècie i identificar-ne les variants de seqüència (SNV, SV i CNV), que en determinen les característiques fenotípiques, incloent-hi les responsables d’algunes malalties. La seqüenciació del genoma d’espècies d’interès s’utilitza també en agrigenòmica per a accelerar els processos de millora genètica en plantes i animals.
En l’entorn clínic, la seqüenciació d’exomes sencers (WES, de l’anglès whole exome sequencing) o de panells de gens (panel-seq) d’interès per a una malaltia són les aplicacions d’NGS més comunes (Manolio et al. , 2019).
S’utilitzen mètodes basats en la hibridació de fragments de DNA per a capturar regions genòmiques específiques, com ara el conjunt de regions codificants de tot el genoma o l’exoma, usant sondes de DNA que són complementàries a les regions dianes. Aquests fragments de DNA enriquits en les regions d’interès després se seqüencien amb tècniques d’NGS. En comparació amb la seqüenciació de genomes sencers, la seqüenciació d’exomes o de panells de gens genera una quantitat de dades més manejable i permet obtenir més lectures de les regions codificants, que són les que concentren la gran majoria de mutacions que causen malalties, i tot a un preu més reduït.
Les tecnologies d’NGS permeten també estudiar els mecanismes de regulació del genoma. L’ús de microarrays per a l’estudi de l’expressió gènica ha donat pas a la seqüenciació de transcriptomes sencers (RNA-seq) per a quantificar de manera precisa l’expressió dels gens en una mostra i identificar-ne els fenòmens de splicing alternatiu i preservar-ne la informació sobre la cadena que es transcriu. Hi ha mètodes específics per a seqüenciar
Treballs de la Societat Catalana de Biologia, 72: 21-27
Hi ha hagut avenços en els darrers deu anys que permeten seqüenciar el DNA, el transcriptoma o l’epigenoma de cèl·lules individuals (scDNA-seq, de l’anglès single cell DNA sequencing; scRNA-seq, de l’anglès single cell RNA sequencing , i scWGBS, de l’anglès single cell whole genome bisulfite sequencing), i que han revelat una elevada heterogeneïtat de tipus cel·lulars en molts teixits i noves poblacions de cèl·lules, la qual cosa afegeix una nova capa de complexitat en molts processos biològics i malalties (Anaparthy et al., 2019). El primer pas consisteix a aïllar les cèl·lules individuals utilitzant mètodes de separació de les cèl·lules per citometria de flux (FACS, de l’anglès fluorescence activated cell sorting), micromanipulació, microdissecció per captura làser (LCM, de l’anglès laser capture microdissection) o sistemes de microfluids. Hi ha equips, com ara els desenvolupats per l’empresa 10X Genomics, que permeten analitzar desenes de milers de cèl·lules individuals en paral·lel, que queden encapsulades en petites gotes en les quals tenen lloc les primeres reaccions: lisi cellular, transcripció inversa en el cas de l’RNA i
25
Avenços en les tecnologies de seqüenciació
del DNA
1 2 4
5
3
Polimerasa
T
C A G G llum Excitació llum
Figura 3. Flux de seqüenciació de molècules úniques en temps real (SMRT), de PacBio. Elaboració pròpia.
A les molècules de DNA se’ls uneix un adaptador de cadena senzilla i una proteïna motora
noves tecnologies o modificacions de les existents en desenvolupament.
La proteïna motora s’uneix al porus i promou el pas de la molècula de DNA o RNA
Un exemple en són els avenços que s’estan fent al voltant de la seqüenciació per nanoporus. La tecnologia de nanoporus, actualment, utilitza nanoporus basats en proteïnes. Amb la finalitat d’augmentar la resolució i la velocitat de seqüenciació, s’estan investigant nous tipus de nanoporus fabricats a partir de l’ús de materials sintètics com són el grafè o el carboni (Wang et al., 2015). També, altres millores van encaminades a fer els instruments cada vegada més petits per tal de fer-los encara més portables; un exemple en seria el nou instrument SmidgION (Oxford Nanopore Technologies), el funcionament del qual aniria lligat a un telèfon mòbil (Kumar et al., 2019).
La seqüència es llegeix aprofitant el perfil de corrent elèctric que genera cada molècula en passar a través del nanoporus
Durant els últims anys, els progressos en la resolució de la microscòpia òptica, juntament amb l’aparició dels mètodes de seqüenciació de RNA de cèl·lula única, han unit forces i han desencadenat l’aparició de tot un ventall de tècniques que tenen l’objectiu d’estudiar l’expressió gènica en el seu context en l’espai, seqüenciació in situ (ISS, de l’anglès in situ sequencing). El marcatge de l’expressió gènica es fa directament sobre els teixits i s’utilitza microscòpia òptica per a detectar-la. El marcatge de l’expressió gènica pot ser via hibridació in situ (seqFISH), que implica l’ús de múltiples oligonucleòtids amb etiquetes fluorescents que s’uneixen a les molècules d’RNA, o via ISS, on l’RNAm se seqüencia directament en una secció del teixit (Marx et al., 2021).
el marcatge de tots els fragments de material genètic que provenen d’una mateixa cèl·lula utilitzant oligonucleòtids específics. A partir d’aquí, se n’extreuen els àcids nucleics, s’amplifica tot el genoma, les regions d’interès o el transcriptoma, es preparen les biblioteques i se seqüencien per NGS. Es tracta d’un camp en evolució contínua, en el qual cada mes es publiquen protocols nous que permeten analitzar moltes més cèl·lules i que minimitzen els biaixos causats per l’ínfim material de partida.
Més del 90 % de les dades de seqüenciació es generen amb la tecnologia i els instruments de l’empresa Illumina. La seva elevada capacitat de producció, precisió i el baix cost que té per base fan que sigui la tecnologia més adequada per a dur a terme la major part de les aplicacions descrites anteriorment. Les lectures relativament curtes dels instruments d’Illumina (50-150 pb) són, però, insuficients per a resoldre zones complexes del genoma. Per aquest
motiu, cal recórrer a les plataformes de lectures llargues (10-40 kb de mitjana) quan no coneixem el genoma de referència d’una espècie (de novo WGS), o per a identificar variants estructurals o les diverses isoformes d’un mateix gen. Tot i així, per a obtenir el genoma de referència d’una espècie diploide normalment es combinen la seqüenciació genòmica amb lectures curtes i llargues juntament amb altres mètodes complementaris com el Hi-C o el mapatge òptic (Giani et al., 2019; Graham et al., 2020).
Darrers desenvolupaments en tecnologies de seqüenciació
El gran potencial de les tecnologies d’NGS i l’impacte que han generat en tots els camps de la biologia han desencadenat un gran interès en l’ampliació de noves tecnologies i aplicacions enfocades a augmentar el rendiment, la velocitat i la precisió de la seqüenciació. En aquest sentit, avui dia hi ha un gran ventall de
Treballs de la Societat Catalana de Biologia, 72: 21-27
Tot i així, els darrers avenços en seqüenciació i en microscòpia no només van encaminats a entendre millor el paper de les cèl·lules i la seva ubicació en els processos biològics, sinó que també ens han proporcionat eines per a sondejar l’estructura del mateix genoma. Les tecnologies actuals de microscòpia òptica d’alta resolució estan limitades a observar un grapat de gens o, en el millor dels casos, un 1 % del genoma. Tanmateix, recentment s’ha publicat una nova tecnologia, OligoFISSEQ, que combina la microscòpia d’alta resolució, l’NGS i la hibridació amb oligonucleòtids marcats amb fluorescència, que permetrà visualitzar amb una resolució molecular el genoma sencer (Huy et al., 2020). Continuant en aquesta línia, el Centre Nacional d’Anàlisi GenòmicaCentre de Regulació Genòmica (CNAG-CRG) formarà part del Center for Genome Imaging, una nova infraestructura amb seu a la Universitat de Harvard, amb l’objectiu de desenvolupar tecnologies que permetin la visualització, l’anàlisi i la modelització de tot el genoma humà en 3D i a un nivell extremament d’alta resolució.
26
Berta Fusté, Elena Vila i Mònica Bayés
+
1 Adaptador Membrana Nanoporus
Proteïna motora
2+
Adaptador Membrana Nanoporus Proteïna motora
3
+
Adaptador Nanoporus
Proteïna motora Membrana
Figura 4. Flux de seqüenciació per nanoporus, d’Oxford Nanopore Technologies (ONT). Elaboració pròpia.
Les tecnologies de seqüenciació de DNA a Catalunya: el Centre Nacional d’Anàlisi Genòmica Catalunya té una reconeguda tradició en l’àmbit de la genòmica i la bioinformàtica. Els principals hospitals, centres de recerca en ciències de la vida i universitats del territori tenen unitats de genòmica amb tecnologies de seqüenciació massiva, generalment de petita o mitjana escala.
A més a més, des de 2009, a Catalunya hi trobem un dels centres europeus de referència en seqüenciació i anàlisi de dades genòmiques, el CNAG-CRG. Gràcies a un equip multidisciplinari d’investigadors, tècnics de laboratori, bioinformàtics i enginyers, el CNAG-CRG ofereix serveis de seqüenciació massiva amb tecnologies de segona i tercera generació i d’anàlisi bioinformàtica de les dades.
Actualment, la plataforma del CNAGCRG té tres unitats dels seqüenciadors de lectures curtes més potents del mercat, cinc equips de seqüenciació massiva mitjançant nanopo-
Bibliografia
Anaparthy, N. [et al.] (2019). «Single-cell applications of next-generation sequencing». Cold Spring Harb. Perspect. Med., 9 (10): a026898.
Ansorge, W. J. [et al.] (2017). «Prespectives for future DNA sequencing techniques and applications». A: Molecular diagnostics, cap. 8: 141-153.
Barton, E. [et al.] (2018). «Overview of next generation sequencing technologies». Curr. Protoc. Mol. Biol., 122 (1): e59.
Bonev, B.; Cavalli, G. (2016). «Organization and function of the 3D genome». Nat. Rev. Genet., 17 (11): 661-678.
Buermans, H. P. J.; Dunnen, J. T. [et al.] (2014). «Next generation sequencing technology: Advances and applications». Biochim. Biophys. Acta, 1842 (10): 19321941.
Eid, J. [et al.] (2009). «Real-time DNA sequencing from single polymerase molecules». Science, 323 (5910): 133-138.
Giani, A. M. [et al.] (2019). «Long walk to genomics: History and current approaches to genome sequencing and assembly». Comput. Struct. Biotechnol. J., 18: 9-19.
Goodwin, S. [et al.] (2016). «Coming of age: Ten years of next-generation sequencing technologies». Nat. Rev. Genet., 17: 333-351.
rus, diverses plataformes per a fer experiments de seqüenciació a partir de cèl·lules individuals, així com d’altres equips de laboratori complementaris. Aquest parc de seqüenciadors pot generar més de 10.000 Gb de seqüència cada 24 hores, o el que és el mateix, permet seqüenciar cada dia 100 genomes humans sencers amb una cobertura suficient per a identificar-ne de manera fiable les variants en el 95 % del genoma. Per tal de processar aquesta gran quantitat de dades, el centre té un superordinador amb més de 3.500 nodes de computació, 8 petabytes d’espai de disc per a emmagatzemament i una xarxa interna de 56 GB per segon. Tots els processos del centre es fan sota controls de qualitat estrictes, queden enregistrats en el LIMS (laboratory management information system) i disposen de la certificació ISO 9001:2015 i l’acreditació ISO 17025:2017. El 2020, el CNAG-CRG va seqüenciar més de 12.000 mostres de DNA o RNA en el marc de 521 projectes de 190 investigadors d’hospitals, universitats i centres de recerca.
Les activitats de recerca i suport a la recerca del CNAG-CRG s’articulen al voltant de sis àrees: la medicina personalitzada, les malalties rares, la genòmica del càncer, la genòmica de cèl·lules individuals, la genòmica funcional i la biodiversitat. Els investigadors del centre participen activament en algunes de les principals iniciatives nacionals i internacionals en aquestes àrees, com ara els projectes IMPaCT de medicina personalitzada de l’Instituto de Salud Carlos III (www.isciii.es/QueHacemos/Finan ciacion/IMPaCT/Paginas/default.aspx), l’International Rare Diseases Research Consortium (irdirc.org), el consorci del Human Cell Atlas (www.humancellatlas.org) i el European Reference Genome Atlas (www.erga-biodiversity.eu).
En resum, la seqüenciació de DNA i RNA ha esdevingut una eina imprescindible per a la recerca bàsica i aplicada, i la missió del CNAGCRG és facilitar-ne la implementació al país, oferint equipaments de darrera generació, preus competitius i el saber fer d’experts en diverses disciplines genòmiques.
Graham, J. E. [et al.] (2020). «Sequencing smart: De novo sequencing and assembly approaches for non-model mammal». GigaScience, 9: 1-14.
Hert, D. G. [et al.] (2008). «Advantages and limitations of next-generation sequencing technologies: A comparison of electrophoresis and non-electrophoresis methods». Electrophoresis, 23: 4618-4626.
Huy , Q. [ et al .] (2020). «3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing». Nat. Methods, 17: 822-832.
Jain, M. [et al.]. (2018). «Nanopore sequencing and assembly of a human genome with ultra-long reads». Nat. Biotechnol., 36 (4): 338-345.
Kumar, K. R. [et al.] (2019). «Next-generation sequencing and emerging technologies». Semin. Thromb. Hemost., 45 (7): 661-673.
Lin, B. [et al.] (2021). «Nanopore technology and its applications in gene sequencing». Biosensors, 11: 214.
Manolio, T. A. [et al.] (2019). «Genomic medicine year in review: 2019». Am. J. Hum. Genet., 105 (6): 1072-1075.
Margulies, M. [et al.] (2005). «Genome sequencing in microfabricated high-density picolitre reactors». Nature, 437: 376-380.
Marx, V. [et al.] (2021). «Method of the year: Spatially resolved transcriptomics». Nat. Methods, 18: 9-14.
Miga, K. H. [et al.] (2020). «Telomere-to-telomere assembly of a complete human X chromosome». Nature, 585: 79-84.
National Human Genome Research Institute (NHGRI) (2020). «Human Genome Project completion: Frequently asked questions» [en línia]. < www. genome.gov/about-genomics/educational-resources/ fact-sheets/human-genome-project> [Consulta: 11 desembre 2022].
Sanger, F. [et al.] (1977). «DNA sequencing with chainterminating inhibidors». Proc. Natl. Acad. Sci. USA, 74 (12): 5463-5467.
Shendure, J. [et al.] (2005). «Accurate multiplex polony sequencing of an evolved bacterial genome». Science, 309 (5741): 1728-1732.
Stark, R. [et al.] (2019). «RNA sequencing: The teenage years». Nat. Rev. Genet., 20 (11): 631-656.
Taishan, H. [et al.] (2021). «Next-generation sequencing tecnologies: An overview». Hum. Immunol., 82: 801811.
Wang, Y. [et al.] (2015). «The evolution of nanopore sequencing». Front. Genet., 5: 449.
Watson, J.; Crick, F. (1953). «Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid». Nature, 171: 737-738.
Treballs de la Societat Catalana de Biologia, 72: 21-27
27
Avenços en les tecnologies de seqüenciació del DNA
Assemblatge de genomes a escala cromosòmica per redescobrir i
conservar la biodiversitat catalana
DOI: 10.2436/20.1501.02.214
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 31/01/2022
Acceptat: 24/03/2022
Resum
Conèixer el genoma de les espècies que ens envolten és crucial per preservar la biodiversitat del territori. Assemblar un genoma consisteix a reconvertir les lectures fragmentades produïdes pels seqüenciadors en una seqüència contigua que representa el genoma complet de l’individu seqüenciat. Abans de l’arribada de les tecnologies de seqüenciació de lectura llarga, la majoria d’assemblatges produïts eren molt fragmentats, cosa que en limitava algunes de les utilitats. La incorporació de les noves tecnologies al camp de l’assemblatge de genomes ha permès una simplificació del procés i una millora de la qualitat dels assemblatges produïts. Els passos per obtenir un genoma de referència són: elaboració de blocs de seqüències consensuades, correcció de la seqüència, reconstrucció de cromosomes i perfeccionament de l’assemblatge. Un assemblatge de referència ens permet fer moltes anàlisis posteriors, com descobrir trets únics d’una espècie, que poden beneficiar les estratègies de conservació.
Paraules clau: assemblatge de genomes, refinament, reconstrucció de cromosomes, seqüenciació, assemblatges a escala cromosòmica.
Chromosome-level genome assemblies to rediscover and conserve Catalonia’s biodiversity
Abstract
It is very important to have a knowledge of the genomes of the species around us in order to preserve a region’s biodiversity. Assembling a genome involves combining the fragmented reads produced by sequencers into a contiguous sequence that represents the complete genome of the sequenced individual. Before the incorporation of long-read sequencing technologies, most of the genome assemblies that were produced were highly fragmented, limiting their utility for many downstream genomic analyses. The appearance of new technologies in the field of genome assembly has simplified the process and improved the quality of the resulting assemblies. The steps for producing a reference genome include contig assembly, sequence polishing, chromosome-level scaffolding and manual curation of the final assembly. A reference genome assembly allows multiple genomic analyses, which can greatly benefit the design of conservation plans.
Keywords: genome assembly, polishing, chromosome-level scaffolding, sequencing, chromosome-level assemblies.
Introducció
Conèixer el genoma de les espècies que ens envolten pot aportar molt a l’hora de desenvolupar estratègies per conservar-les i protegir-les. A més, ens pot ajudar a saber com interactuen entre elles, potenciar-ne l’aplicació en ramaderia i agricultura o ajudar-nos a entendre com s’han originat determinats trets i com responen al canvi climàtic, entre d’altres. Projectes com la iniciativa catalana de l’Earth BioGenome Project (EBP), la CBP (de l’anglès Catalan Initiative for the Earth BioGenome Project), que té com a objectiu seqüenciar i assemblar el genoma de totes les espècies eucariotes dels territoris de parla catalana, són crucials per tal de garantir l’existència de dades genòmiques d’alta qualitat de les espècies que habiten la regió. La qualitat de l’assemblatge final és molt important perquè en depenen, en gran manera, la robustesa i fiabilitat de les anàlisis posteriors.
Com que la seqüenciació d’un genoma produeix lectures fragmentades (més o menys llargues segons la tecnologia de seqüenciació), és necessari un procés d’assemblatge capaç de transformar aquests fragments en una seqüència contigua que representi el genoma complet (com a mínim, el genoma nuclear i els orgànuls) de l’individu seqüenciat. L’assemblatge ideal tindria tantes seqüències contínues (també anomenades còntigs) com cromosomes contingui el genoma, i tots els nucleòtids d’aquests cromosomes serien coneguts. Desafortunadament, aconseguir el genoma perfecte és molt complicat i el que normalment obtenim són assemblatges fragmentats i incomplets.
El procés de seqüenciació i assemblatge de genomes ha canviat molt al llarg dels anys. Es va trigar més d’una dècada a produir els primers assemblatges del genoma humà (International Human Genome Sequencing Con -
Treballs de la Societat Catalana de Biologia, 72: 28-33
sortium et al ., 2001; Venter et al ., 2001) i d’altres espècies model, com el ratolí (Mouse Genome Sequencing Consortium et al., 2002). Per obtenir-los, calia seqüenciar múltiples clons mitjançant el mètode de Sanger (500-1.000 nucleòtids per lectura) i van ser necessaris molts esforços manuals, així com el desenvolupament de programes informàtics. Més endavant, la seqüenciació del genoma complet amb lectures curtes d’Illumina (30-150 nucleòtids) va donar pas a un procés d’assemblatge més assequible, que va conduir a una revolució genòmica en la qual es va publicar l’assemblatge de moltes espècies, com per exemple el del panda gegant (Li et al., 2010). Desafortunadament, l’ús de lectures tan curtes portava a l’obtenció d’assemblatges força fragmentats, amb problemes en certes zones del genoma, sobretot en les més repetitives, la qual cosa provocava l’absència o fragmentació d’alguns gens. Recentment,
Jèssica Gómez-Garrido,1 Fernando Cruz,1 Marc Palmada-Flores2 i Tyler Alioto1, 3
1 Centre Nacional d’Anàlisi Genòmica - Centre de Regulació Genòmica (CNAG-CRG), Barcelona Institute of Science and Technology (BIST)
2 Department of Medicine and Life Sciences (MELIS), Institut de Biologia Evolutiva, Universitat Pompeu Fabra ‐ CSIC
3 Universitat Pompeu Fabra (UPF)
28
Correspondència: Tyler Alioto. Centre Nacional d’Anàlisi Genòmica. C. de Baldiri Reixac, 4. 08028 Barcelona. Tel.: +34 934 037 098. Adreça electrònica: tyler.alioto@cnag.crg.eu
l’aparició de les tecnologies de seqüenciació de lectura llarga ha fet que l’obtenció d’un assemblatge a escala cromosòmica d’una espècie qualsevol sigui possible en només unes quantes setmanes, la qual cosa ha canviat les normes del joc (Rhie et al., 2021). Per a més detall sobre les tecnologies de seqüenciació, convidem el lector a llegir l’article «Avenços en les tecnologies de seqüenciació del DNA», de Fusté et al., present en aquest mateix monogràfic.
Avaluació d’un assemblatge
Abans d’entrar en la descripció dels passos que hem de seguir durant el procés d’assemblatge, definirem les diferents maneres que tenim per avaluar la qualitat d’un assemblatge de novo (utilitzant únicament dades de seqüenciació, sense fer servir cap altra referència prèviament coneguda). Però com podem avaluar la qualitat del nostre assemblatge si desconeixem com és realment el genoma? Els dos aspectes bàsics que cal tenir en compte són la contigüitat i la integritat (proporció del genoma que s’ha inclòs) de la seqüència assemblada.
Hem dit que l’assemblatge ideal tindria tantes seqüències com cromosomes tingui el genoma; així doncs, per avaluar-ne la contigüitat es té en compte el nombre total de seqüències assemblades i la longitud. Per a això es fan servir mètriques com el L50 i el N50 (Earl et al., 2011). El L50 és el menor nombre de fragments la suma de longituds dels quals conforma el 50 % del total de nucleòtids assemblats. El N50 es defineix com la longitud del fragment més petit d’aquest mateix conjunt. Per calcular-los, s’ordenen tots els fragments segons les longituds, de més gran a més petit, i es van comptabilitzant fins que la suma acumulada és més gran o igual que la meitat de l’assemblatge (figura 1a).
a b
Un altre aspecte clau per determinar la contigüitat de l’assemblatge és el nombre de regions buides d’informació que conté. De vegades, es pot determinar que dues seqüències formen part del mateix fragment genòmic, però desconeixem la seqüència de nucleòtids que les uneix; en aquests casos, es construeix un pont entre elles afegint-hi unes quantes «N» entremig. Aquests ponts de seqüència desconeguda s’anomenen buits (gaps). Les seqüències assemblades que no contenen cap N s’anomenen còntigs i les formades per diversos còntigs connectats per buits són conegudes com a scaffolds (figura 1b).
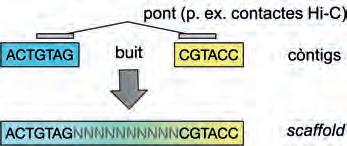
Pel que fa a la integritat de l’assemblatge, es pot avaluar buscant quants gens d’entre un conjunt de gens coneguts i que, en principi, hauríem de trobar al genoma, són presents a l’assemblatge, o bé calculant la fracció de lectures del seqüenciador que han estat assemblades. CEGMA ( core eukaryotic genes mapping approach) (Parra et al., 2007) i BUSCO (benchmarking universal single-copy orthologs) (Simão et al., 2015) són dos exemples de mètodes basats en la primera estratègia. CEGMA va ser el primer mètode d’aquest tipus que es va desenvolupar i utilitza com a referència 456 gens altament conservats en tots els eucariotes. D’altra banda, BUSCO fa servir bases de dades d’OrthoDB (Zdobnov et al., 2021) que contenen grups de gens conservats i presents una sola vegada en una branca filogenètica concreta (ortòlegs de còpia única). Aquests programes reporten quants dels gens presents a la base de dades es troben a l’assemblatge i quants estan duplicats o fragmentats. Totes aquestes dades són crucials per determinar la qualitat del genoma assemblat, ja que si falten molts gens que en teoria haurien d’estar presents o apareixen fragmentats, això pot voler
dir que hi ha una part del genoma que no es troba en l’assemblatge, que no l’hem assemblat correctament o que la qualitat de la seqüència no és gaire bona, fet que provoca que els gens continguin errors de seqüència i no es puguin identificar. D’altra banda, si trobem molts gens duplicats que en teoria haurien de ser presents únicament una vegada, pot ser indicatiu de la presència de regions duplicades artificialment en el nostre assemblatge, tot i que també podria ser degut a altres raons biològiques, com, per exemple, una duplicació recent del genoma.
La segona estratègia es basa a calcular quina part de la seqüència present a les lectures ha acabat continguda a l’assemblatge. Aquesta estratègia és més universal, ja que no se centra només en unes regions concretes del genoma, sinó que n’analitza totes les posicions. Durant aquest procés s’acostumen a dividir les lectures en subseqüències de longitud k (sovint al voltant de 21 nucleòtids), anomenades k-mers Així doncs, extraiem totes les subseqüències de longitud k de les lectures i les busquem a l’assemblatge per tal de determinar el percentatge de k-mers que comparteixen i saber, així, com és de complet el nostre assemblatge. A més, comparant els k-mers presents en les seqüències assemblades i en les lectures, també podem obtenir un valor de qualitat (QV), que ens proporciona informació sobre la quantitat d’errors produïts durant la seqüenciació que es traslladen a l’assemblatge. La mètrica QV (Rhie et al., 2020) reflecteix de manera logarítmica la precisió de la seqüència assemblada, de manera que com més alt sigui el valor, més precisa serà la seqüència. Per exemple, un QV de 30 correspon a una precisió del 99,9 %, és a dir, un error cada 1.000 nucleòtids; un QV de 40, a una precisió del 99,99 %, etc.
El procés d’assemblatge
Ara que ja coneixem alguns dels conceptes més importants darrere dels assemblatges genòmics, ens endinsarem en el procés d’elaboració. Podem dividir el procés bàsic d’assemblatge en quatre passos principals:
1. obtenció de blocs de seqüències contigües (còntigs);
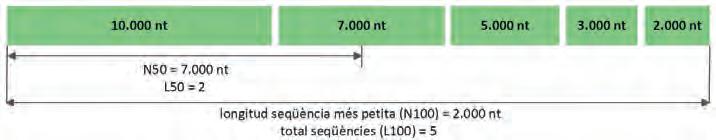
2. correcció de la seqüència de cada un dels còntigs (refinament);
3. reconstrucció de cromosomes ( chromosome scaffolding);
4. perfeccionament de l’assemblatge.
La figura 2 mostra un exemple d’esquema d’un procés d’assemblatge real: Figura 1. a) Representació gràfica de les mètriques. b) Representació gràfica de còntigs, scaffolds, buits. Elaboració pròpia.
Treballs de la Societat Catalana de Biologia, 72: 28-33
29
Assemblatge de genomes a escala cromosòmica per redescobrir i conservar la biodiversitat catalana
Figura 2. Esquema del procés d’assemblatge que mostra els passos que es podrien seguir per assemblar el genoma de qualsevol organisme amb dades de seqüenciació d’ONT de lectura llarga, Illumina de lectura curta i dades de contacte de Hi-C. Elaboració pròpia.
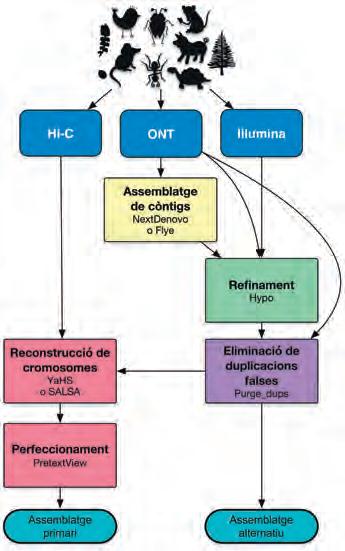
1. Obtenció de blocs de seqüències contigües (còntigs)
Un cop s’han eliminat adaptadors (seqüències tècniques utilitzades en el procés de seqüenciació) i seleccionat les lectures de seqüenciació amb millor qualitat, aquestes es fan servir per generar blocs de seqüències contigües, que, com hem dit anteriorment, s’anomenen còntigs
Fins fa pocs anys, el més freqüent era construir els còntigs a partir de lectures curtes mitjançant programes d’assemblatge que primer extreien tots els k-mers presents en les lectures i després construïen un graf de Bruijn connectant els diferents k-mers extrets (Pevzner et al ., 2001) en funció de les seves seqüències superposades. Entre els programes d’assemblatge que es van dissenyar per assemblar genomes amb lectures curtes i que es basen en grafs de Bruijn destacaríem Velvet (Zerbino i Birney, 2008), ABySS (Simpson et al., 2009), SPAdes (Bankevich et al., 2012) i SOAPdenovo (Li et al., 2010). Com s’ha comentat, aquest procés produïa uns assemblatges molt fragmentats. Amb l’avenç en les tecnologies de seqüenciació de lectura llarga, el procés d’assemblatge no només ha millorat notablement, sinó que
s’ha simplificat considerablement. Com que les lectures de tercera generació cobreixen regions molt més llargues del genoma en una sola lectura (hem passat d’aproximadament 150-200 nucleòtids a milers de nucleòtids), és molt més senzill resoldre el trencaclosques i saber on col·locar cada peça, fins i tot, les procedents de zones repetitives del genoma. En contraposició, l’alta taxa d’error de les tecnologies de lectura llarga provoca que hi hagi errors a les seqüències assemblades i fa necessari un pas posterior de correcció d’aquestes seqüències per millorar-ne la precisió.
Com es comenta a l’article sobre tecnologies de seqüenciació present en aquest monogràfic, les dues principals companyies amb seqüenciació de lectura llarga són Oxford Nanopore Technologies (ONT) i Pacific Biosciences (PacBio). El fet que construir grafs de Bruijn a partir de lectures llargues doni lloc a estructures molt enrevessades ha provocat que s’hagin desenvolupat programes d’assemblatge específics per a lectures llargues basats en altres mètodes. En alguns casos, s’ha mantingut la idea del graf de Bruijn, però s’ha adaptat a les característiques i peculiaritats de les lectures llargues; un exemple en seria el programa Flye (Freire et al., 2021), que es basa en grafs A-Bruijn, una modificació dels grafs de Bruijn que té en compte els encavalcaments entre les lectures. En altres casos, s’apliquen mètodes jeràrquics (hierarchical genome assembly process) (Al-Okaily, 2016), en els quals inicialment es produeixen diversos assemblatges amb les lectures de més qualitat i longitud i després es corregeixen i s’allarguen aquests miniassemblatges amb la resta de lectures que pertanyen a cada regió. Els programes Canu (Koren et al., 2017), Falcon-Unzip (Chin et al., 2016) i Hifiasm (Cheng et al., 2021) es basen en aquesta idea.
Atès que les lectures llargues poden contenir molts errors, per tal de simplificar els grafs que es construeixen durant el procés d’assemblatge, alguns programes fan un pas de correcció de les lectures llargues (p. ex., Nextdenovo, https://github.com/Nextomics/NextDenovo ).
Alternativament, hi ha la possibilitat de corregir les lectures prèviament i després donar-les als programes d’assemblatge. Quina és la millor opció varia en funció de les dades de les quals disposem i de l’organisme que estiguem assemblant.
2. Correcció de la seqüència de cada un dels còntigs (refinament)
Els còntigs obtinguts a partir de lectures llargues sovint contenen errors en forma de peti-
tes insercions, delecions o canvis de nucleòtids. Abans de poder fer servir aquests blocs de seqüència en anàlisis posteriors, com, per exemple, en l’anotació de gens, cal corregir aquests errors. De fet, si intentéssim anotar gens directament amb els assemblatges no corregits, no podríem definir correctament les seqüències codificants, ja que en molts casos trobaríem errors en la codificació dels aminoàcids i codons de terminació enmig de la seqüència.
El procés de correcció de la seqüència conegut com a refinament o polishing consisteix a determinar un consens per cada posició a partir de totes les lectures que cobreixen els còntigs. Les lectures emprades en aquest pas poden ser les mateixes amb les quals s’ha fet l’assemblatge base o diferents. En assemblatges obtinguts amb ONT o PacBio SMRT és freqüent fer diverses rondes de correcció amb les mateixes lectures llargues i, després, afegir-hi unes quantes rondes de correcció amb un altre tipus de lectures més precises, generalment, lectures curtes d’Illumina (que tenen una taxa d’error molt més baixa). També és possible fer servir programes com Hypo (Kundu et al., 2019), que poden corregir amb diversos tipus de lectures al mateix temps.
En assemblatges obtinguts a partir de lectures de tipus HiFi de PacBio no és necessari corregir amb altres tecnologies, ja que elles mateixes ja són fruit d’un procés de consens. De fet, s’obtenen a partir de múltiples lectures sobre la mateixa molècula de DNA, fet que fa que la seva taxa d’error sigui força baixa (<1 %). Per produir assemblatges a partir d’aquestes lectures s’acostuma a fer servir el programa HiFiasm (Cheng et al., 2021), que utilitza les mateixes lectures per corregir els possibles errors i produeix assemblatges contigus que, en principi, no cal corregir.
Un cop obtingudes les seqüències consensuades corregides, encara ens queda un pas de correcció addicional: eliminar duplicacions falses i separar els haplotips. Tot i que la majoria d’organismes contenen més d’una còpia del genoma, els assemblatges de referència idealment en contenen una sola còpia, és a dir, són una representació haploide del genoma. No obstant això, atès que ambdues còpies d’un mateix individu no són idèntiques, és possible que els programes d’assemblatge no detectin tots els casos i sovint més d’una còpia de certes regions acaba introduïda a l’assemblatge inicial. Per eliminar aquestes duplicacions, es tornen a alinear les lectures llargues i, basant-se en els alineaments, s’intenta detectar quines parts assemblades corresponen a la mateixa
30
Treballs de la Societat Catalana de Biologia, 72: 28-33
Jèssica Gómez-Garrido, Fernando Cruz, Marc Palmada-Flores i Tyler Alioto
regió del genoma. Com a resultat del procés de detecció de duplicats (per exemple, amb el programa Purge_dups (Guan et al ., 2020), s’obté un assemblatge primari que hauria de correspondre a una de les còpies del genoma. A més, si les lectures són prou acurades, és possible obtenir assemblatges secundaris amb tots els haplotips alternatius.
3. Reconstrucció de cromosomes (chromosome scaffolding)
Tot i que amb les lectures llargues podem reconstruir blocs molt llargs, és poc freqüent aconseguir assemblar cromosomes sencers sense ajuda d’informació suplementària. Per tant, un cop el nostre assemblatge és prou contigu i complet (per exemple, amb diverses megabases [Mb] de N50, un QV de més de 40 i amb més d’un 90 % de gens BUSCO i k-mers presents), podem procedir a emplaçar els blocs seqüència en estructures més llargues, idealment de la mida dels cromosomes. És el moment, doncs, d’ordenar i orientar les peces per crear superestructures (super-scaffolds) amb diversos blocs de seqüència connectats per un nombre arbitrari de n (sovint un valor fix, p. ex. 100). Hi ha diferents aproximacions que permeten aquest pas: mapes genètics, mapes òptics (p. ex. Bionano), mapes de contacte (p. ex. Hi-C), etc.
Durant dècades, els biòlegs han estudiat àmpliament nombroses espècies, de les quals, tot i no conèixer-ne amb exactitud la totalitat del genoma, han estat capaços d’extreure molta informació. Gràcies als esforços de molts investigadors, disposem de mapes genètics per a moltes d’aquestes espècies, que consisteixen en llistes de marcadors dels quals coneixem la seqüència i localització en el genoma. Aquests mapes es poden fer servir per ordenar i orientar les seqüències assemblades i, si aquestes són prou llargues, ens permeten reconstruir els cromosomes (p. ex. Guerrero-Cózar et al., 2021).
Com que no hi ha mapes genètics per a totes les espècies, és important poder reconstruir els cromosomes amb altres mètodes. Amb aquesta finalitat, podem seqüenciar amb tecnologies com Bionano ( https://bionanogeno mics.com/) o Hi-C (Belton et al., 2012).
Bionano Genomics ha desenvolupat una tecnologia que permet fer «fotografies» d’una molècula de DNA. El procés consisteix a marcar el DNA en determinades seqüències, capturar la imatge amb un instrument especialitzat a detectar-ne el senyal i fer-ne mapes òptics. A continuació, podem comparar els mapes òptics i les seqüències que hem assem-
blat prèviament per tal de detectar llocs d’unió entre els nostres blocs de seqüència i així crear superestructures.
L’altra opció àmpliament emprada per a la reconstrucció de cromosomes són els mapes de contacte. Tècniques com el Hi-C permeten detectar les interaccions de la cromatina al nucli cel·lular i construir matrius de distància que reflecteixen la conformació en tres dimensions del genoma. Aquestes tècniques es basen en el fet que la probabilitat d’interacció disminueix ràpidament amb l’augment de la distància genòmica. L’anàlisi d’aquestes matrius de distància fa possible detectar tant zones properes com regions separades per diversos centenars de megabases en el mateix cromosoma. Un cop detectades les interaccions entre els fragments assemblats, podem unir els que interactuen i crear una macroestructura que corres-
pon al cromosoma (figura 3). Aquest procés es coneix com a Hi-C scaffolding i els millors programes per dur-lo a terme són 3Ddna (Dudchenko et al., 2017), SALSA2 (Ghurye et al., 2019) i YaHS (Chenxi Zhou et al ., 2022). Aquest darrer, que s’ha desenvolupat recentment, incorpora noves maneres de netejar el graf que donen lloc a superestructures de molta qualitat.

Les tècniques descrites anteriorment són complementàries, de manera que és freqüent combinar diversos mètodes per tal d’arribar a obtenir el millor assemblatge possible, és a dir, el més contigu, complet i amb menys errors.
4. Perfeccionament de l’assemblatge Un cop finalitzat el pas de scaffolding, és possible que encara quedin algunes seqüències curtes sense col·locar o que algunes de les peces no
Figura 3. Mapa de contactes Hi-C que mostra com la intensitat dels contactes es correlaciona amb la proximitat al llarg de cada seqüència. Aquest cas en concret mostra el genoma d’1,5 Gb d’un vertebrat heterogamètic. S’observa una reducció del nombre de contactes a la seqüència 14 (s14), que correspon al cromosoma sexual més llarg, degut a la presència d’una sola còpia d’aquest cromosoma al genoma. Per conveniència, s’han marcat només les 17 seqüències més llargues. El nombre total de seqüències assemblades és 258.066, la gran majoria de les quals són petites porcions del genoma que romanen sense col·locar pel seu alt contingut en repeticions. Elaboració pròpia.
Treballs de la Societat Catalana de Biologia, 72: 28-33
31
Assemblatge de genomes a escala cromosòmica per redescobrir i conservar la biodiversitat catalana
es trobin correctament orientades en el cromosoma. Generalment, es tracta de blocs massa curts que no han pogut ser emplaçats correctament en les superestructures per manca d’informació en els mapes que els relacioni amb la resta de l’assemblatge. Per tal d’intentar col·locar aquestes peces, podem fer una revisió del mapa de contactes i reorganitzar els blocs de seqüència, assegurant-nos que els contactes siguin més freqüents en regions veïnes. A més, una altra manera d’intentar trobar-los el seu lloc és tornant a recórrer a les lectures llargues. Si tornem a buscar les lectures a l’assemblatge on tenim els cromosomes i els fragments restants, podem trobar lectures que continguin parts de la seqüència present a les superestructures i parts als còntigs o scaffolds deslocalitzats. Programes com Dentist (Ludwig et al., 2021) o RagTag (Alonge et al., 2019) permeten omplir els buits que s’han generat durant el procés de scaffolding amb els fragments curts que no s’havien pogut col·locar. Aquest procés es coneix com a emplenament de buits ( gap filling, en anglès).
Un altre motiu pel qual alguns fragments no poden ser col·locats als cromosomes és perquè no pertanyen al genoma que estem assemblant. En alguns casos, és possible trobar contaminació, ja sigui de genomes d’orgànuls cel·lulars, d’organismes que viuen dins del ma-
teix individu (endosimbionts) o bé seqüències contaminants d’altres organismes. Quan detectem la presència d’aquests fragments exògens, els podem eliminar de l’assemblatge final durant el procés de descontaminació. Una bona eina per a la detecció d’aquests contaminants és Blobtools (Challis et al., 2020).
Després de tots aquests passos, és possible que encara quedin algunes peces sense col·locar. Aquestes seqüències, que sovint són curtes, altament repetitives i pobres en gens, es poden mantenir al final del fitxer amb l’assemblatge, ja que poden ser útils per a determinats tipus d’anàlisis.
Selecció de l’assemblatge final Durant tot el procés d’assemblatge d’un genoma es produeixen assemblatges intermedis que poden ser avaluats amb les tècniques descrites a l’apartat «Avaluació d’un assemblatge». A més, com que les característiques del genoma de diferents organismes poden ser molt variades (per exemple, aspectes com la mida, l’heterozigositat, la ploïdia, les regions repetitives, la manera de determinar el sexe, etc.), és difícil seleccionar una única estratègia universal que sigui la millor en tots els casos. Per tant, sovint es proven diferents tècniques d’assemblatge i es comparen entre elles per tal de determinar la combinació òptima en cada cas.
A la figura 4 es mostren, a tall d’exemple, els resultats obtinguts en assemblar el genoma d’1,5 × 109 nucleòtids (Gb) d’un vertebrat. La primera figura (a) recull les mètriques després del primer pas del procés d’assemblatge, és a dir, després de la construcció dels còntigs (figura 2). A la segona figura ( b) s’observen els estadístics obtinguts durant l’assemblatge del mateix vertebrat, però després de tot el procés, en concret, després de la reconstrucció de cromosomes amb dades de Hi-C. En comparar ambdues imatges veiem que el primer assemblatge conté moltes més seqüències i que són molt més curtes. A més, el percentatge de gens BUSCO complets és molt més baix en el primer cas perquè el pas de refinament o correcció de la seqüència encara no ha tingut lloc.
L’Earth BioGenome Project (Lawniczak et al., 2022), que té com a objectiu seqüenciar i assemblar el genoma de totes les espècies del planeta, ha definit uns estàndards de qualitat mínims per als assemblatges obtinguts (Lewin et al., 2022). En resum, es considera un genoma d’alta qualitat si compleix els requisits següents:

més d’un 90 % de l’assemblatge emplaçat en cromosomes; N50 dels còntigs més gran d’una megabase;

32
Treballs de la Societat Catalana de Biologia, 72: 28-33
Jèssica Gómez-Garrido, Fernando Cruz, Marc Palmada-Flores i Tyler Alioto
a b
Figura 4. Gràfics de cargol (snail plots) (Challis et al., 2020) que mostren les mètriques obtingudes durant l’assemblatge d’un vertebrat. a) Mostra de l’assemblatge inicial generat a partir de les lectures llargues i b) mostra de l’assemblatge final generat després de tot el procés exposat a la figura 2. Elaboració pròpia.
QV superior a 40; més d’un 90 % de gens BUSCO complets; més d’un 90 % de k-mers presents.
Un cop finalitzat el procés i seleccionat el nostre assemblatge final, ja podem anotar-lo i fer-lo servir per a múltiples tipus d’estudis i anàlisis. Mitjançant aquest procés podem descobrir aspectes únics de la nostra espècie d’interès, comparar expressions gèniques entre diferents condicions, analitzar poblacions o, fins i tot, comparar el genoma de la nostra espècie amb el d’altres espècies. Gràcies a la presència de genomes de referència de bona qualitat és possible estudiar l’espectre complet de la variació genètica a la natura, fet que millora molt la nostra capacitat a l’hora de desenvolupar estratègies de conservació i també d’estudiar interaccions ecològiques dins d’un mateix ecosistema (Formenti et al., 2022). Aquesta informació serà clau per mantenir la diversitat a escala global.
Bibliografia
Abascal, F. [et al.] (2016). «Extreme genomic erosion after recurrent demographic bottlenecks in the highly endangered Iberian lynx». Genome Biol., 17: 251.
Al-Okaily, A. A. (2016). «HGA: De novo genome assembly method for bacterial genomes using high coverage short sequencing reads». BMC Genomics , 17: 193.
Alioto, T. [et al.] (2020). «Transposons played a major role in the diversification between the closely related almond and peach genomes: Results from the almond genome sequence». Plant J., 101: 455-472.
Almudi, I. [et al.] (2020). «Genomic adaptations to aquatic and aerial life in mayflies and the origin of insect wings». Nat. Commun., 11: 2631.
Alonge, M. [et al.] (2019). «RaGOO: fast and accurate reference-guided scaffolding of draft genomes». Genome Biol., 20: 224.
Bankevich, A. [et al.] (2012). «SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing». J. Comput. Biol., 19: 455-477.
Belton, J. M. [ et al .] (2012). «Hi-C: A comprehensive technique to capture the conformation of genomes». Methods, 58: 268-276.
Challis, R. [et al .] (2020). «BlobToolKit - Interactive Quality Assessment of Genome Assemblies». G3, 10: 1361-1374.
Cheng, H. [et al.] (2021). «Haplotype-resolved de novo assembly using phased assembly graphs with Hifiasm». Nat. Methods, 18: 170-175.
Chin, C. S. [et al.] (2016). «Phased diploid genome assembly with single-molecule real-time sequencing». Nat. Methods, 13: 1050-1054.
Cruz, F. [et al.] (2016). «Genome sequence of the olive tree, Olea europaea». Gigascience, 5: 29.
Dudchenko, O. [et al.] (2017). «De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds». Science, 356: 92-95.
Earl, D. [et al.] (2011). «Assemblathon 1: A competitive assessment of de novo short read assembly methods». Genome Res., 21: 2224-2241.
Figueras, A. [et al.] (2016). «Whole genome sequencing of turbot ( Scophthalmus maximus ; Pleuronecti -
Genomes de referència assemblats a Catalunya
Molts grups catalans han assemblat i publicat els genomes de diverses espècies al llarg dels anys. El Centre Nacional d’Anàlisi GenòmicaCentre de Regulació Genòmica (CNAG-CRG) de Barcelona, un dels centres europeus de referència en seqüenciació i anàlisi de dades genòmiques, té un equip que es dedica a produir assemblatges i anotacions de bona qualitat de tot tipus d’organismes ( https://denovo.cnag. cat /). Entre els genomes publicats en destaquen els d’algunes plantes com l’olivera (Cruz et al., 2016; Julca et al., 2020) i l’ametller (Alioto et al., 2020); vertebrats com el turbot (Figueras et al., 2016) i el linx ibèric (Abascal et al., 2016); insectes com la mosca Drosophila guanche (Puerma et al., 2018) i l’efemeròpter Cloeon dipterum (Almudi et al., 2020), i bivalves com el musclo (Gerdol et al., 2020).
En els darrers anys han aparegut múltiples iniciatives per generar genomes de refe -
rència de les espècies del planeta. Alguns d’aquests projectes se centren en branques taxonòmiques concretes, com el Vertebrate Genomes Project ( https://vertebrategenomes project.org/) o el Bird 10K (https://b10k.geno mics.cn / ), que assemblen genomes de vertebrats i d’ocells, respectivament. Per una altra banda, trobem els projectes enfocats a determinades zones geogràfiques, com el Darwin Tree of Life ( https://www.darwintreeoflife. org / ), l’African BioGenome Project ( https:// africanbiogenome.org/ ) o la iniciativa catalana per a l’Earth BioGenome Project (https:// www.biogenoma.cat/). Tots aquests projectes es troben emmarcats dins de l’Earth BioGenome Project, que té l’objectiu ambiciós de generar almenys un genoma de referència per cada espècie eucariota de la Terra. Científics que desenvolupen la seva tasca professional al llarg de tots els territoris de parla catalana estan contribuint que aquesta fita sigui possible.
formes): A fish adapted to demersal life». DNA Res., 23: 181-192.
Formenti, G. [et al.] (2022). «The era of reference genomes in conservation genomics». Trends Ecol. Evol. [en línia], 37 (3): 197-202. <https://doi.org/10.1016/ j.tree.2021.11.008>.
Freire, B. [et al.] (2021). «Memory-efficient assembly using flye». IEEE/ACM Trans. Comput. Biol. Bioinform.
G erdol, M. [ et al .] (2020). «Massive gene presenceabsence variation shapes an open pan-genome in the Mediterranean mussel». Genome Biol., 21: 275.
Ghurye, J. [et al.] (2019). «Integrating Hi-C links with assembly graphs for chromosome-scale assembly». PLoS Comput. Biol., 15: e1007273.
Guan, D. [et al.] (2020). «Identifying and removing haplotypic duplication in primary genome assemblies». Bioinformatics, 36: 2896-2898.
Guerrero-Cózar, I. [et al.] (2021). «Chromosome anchoring in Senegalese sole (Solea senegalensis) reveals sex-associated markers and genome rearrangements in flatfish». Sci. Rep., 11: 13460.
International Human Genome Sequencing Consortium [et al.] (2001). «Initial sequencing and analysis of the human genome». Nature, 409: 860-921.
Julca, I. [et al.] (2020). «Genomic evidence for recurrent genetic admixture during the domestication of Mediterranean olive trees (Olea europaea L.)». BMC Biol., 18: 148.
Koren, S. [ et al.] (2017). «Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation». Genome Res., 27: 722-736.
Kundu, R. [et al.] (2019). «Hypo: super fast & accurate polisher for long read genome assemblies». BioRxiv <https://doi.org/10.1101/2019.12.19.882506>.
Lawniczak, M. K. N. [et al.] (2022). «Standards recommendations for the Earth BioGenome Project». Proc. Natl. Acad. Sci. USA, 119
Lewin, H. A. [et al.] (2022). «The Earth BioGenome Project 2020: Starting the clock». Proc. Natl. Acad. Sci. USA, 119 (4): e2115635118.
Li, R. [et al.] (2010). «De novo assembly of human genomes with massively parallel short read sequencing». Genome Res., 20: 265-272.
Ludwig, A. [et al.] (2021). «DENTIST – using long reads to close assembly gaps at high accuracy». BioRxiv <https://doi.org/10.1101/2021.02.26.432990>.
Mouse Genome Sequencing Consortium [ et al .] (2002). «Initial sequencing and comparative analysis of the mouse genome». Nature, 420: 520-562.
Parra, G. [et al.] (2007). «CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes». Bioinformatics, 23: 1061-1067.
Pevzner, P. A. [et al.] (2001). «An Eulerian path approach to DNA fragment assembly». Proc. Natl. Acad. Sci. USA, 98: 9748-9753.
Puerma, E. [et al.] (2018). «The high-quality genome sequence of the oceanic island endemic species Drosophila guanche reveals signals of adaptive evolution in genes related to flight and genome stability». Genome Biol. Evol., 10: 1956-1969.
Rhie, A. [et al.] (2020). «Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies». Genome Biol., 21: 245. (2021). «Towards complete and error-free genome assemblies of all vertebrate species». Nature, 592: 737746.
S imão, F. A. [ et al .] (2015). «BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs». Bioinformatics , 31: 32103212.
Simpson, J. T. [et al.] (2009). «ABySS: A parallel assembler for short read sequence data». Genome Res., 19: 11171123.
Venter, J. C. [et al.] (2001). «The sequence of the human genome». Science, 291: 1304-1351.
Zdobnov, E. M. [et al.] (2021). «OrthoDB in 2020: Evolutionary and functional annotations of orthologs». Nucleic Acids Res., 49: D389-D393.
Zerbino, D. R.; Birney, E. (2008). «Velvet: Algorithms for de novo short read assembly using de Bruijn graphs». Genome Res., 18: 821-829.
Zhou, C. [et al.] (2022). «YaHS: yet another Hi-C scaffolding tool». BioRxiv [en línia], 495093. <https://doi.org/ 10.1101/2022.06.09.495093>.
Treballs de la Societat Catalana de Biologia, 72: 28-33
33
Assemblatge de genomes a escala cromosòmica per redescobrir i conservar la biodiversitat catalana
3
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
1
Resum
Des de sempre, les aranyes, els escorpins i els seus parents han fascinat i horroritzat els humans per igual. Tot i que es van originar als mars del precambrià, els quelicerats són uns dels organismes més abundants i diversos dels ecosistemes terrestres, on tenen un paper cabdal en les xarxes tròfiques com uns dels depredadors dominants. L’anàlisi genòmica comparativa dels quelicerats és encara a les beceroles: hi ha pocs genomes complets i la seva distribució taxonòmica és força esbiaixada, cosa que en compromet la representativitat. Tot i així, la informació disponible ha contribuït molt a millorar el nostre coneixement sobre l’origen i l’evolució d’aquest grup d’organismes, i l’arquitectura genòmica de trets de rellevància biològica, econòmica i mèdica com ara la seda, els verins, les famílies gèniques implicades en l’olfacte o el gust (sistema quimiosensorial), o l’adaptació a diferents dietes, incloent-hi el parasitisme. L’obtenció de nous genomes d’alta qualitat representatius de l’arbre de la vida dels quelicerats promet futurs descobriments clau, tant per a comprendre la gran diversificació i les extraordinàries adaptacions d’aquests animals fascinants, com per a aplicar-ho en conservació, biomedicina, control sostenible de plagues i obtenció de nous materials biològics.
Paraules clau: duplicació del genoma (WGD), terrestrialització, evolució, diversitat biològica, sistema quimiosensorial.
Introducció
Pocs organismes desperten sentiments tan contraposats entre els humans com les aranyes, les paparres o els escorpins. Des d’invocar les fòbies i pors més atàviques amb la seva sola menció, fins a fascinar-se contemplant el miracle d’enginyeria que és una teranyina, les aranyes i els seus parents —els quelicerats— formen part del nostre dia a dia i de la cultura popular. Convivim amb les aranyes, que són un dels organismes més abundants en els ambients urbans, els escorpins figuren en la mitologia de molts pobles i els àcars són el malson de pagesos i metges.
Els artròpodes són els organismes més diversos del planeta i constitueixen el principal component de la biomassa animal (Bar-On et al., 2018). Un dels seus trets característics és l’existència d’una cutícula externa segregada
DOI: 10.2436/20.1501.02.215
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 10/02/2022
Acceptat: 23/03/2022
Chelicerate genomics: The deconstruction of arachnids and the genomic basis of silk, venoms and other traits of biological importance
Abstract
Spiders, scorpions, and their kin have always fascinated and horrified humans alike. Although they originated in the pre-Cambrian seas, chelicerates are one of the most abundant and diverse organisms in terrestrial ecosystems, where they play a key role in food webs as one of the dominant predators. Comparative genomic analysis of chelicerates is still in its infancy: there are few complete genomes, unevenly distributed taxonomically, which compromises their representativeness. However, the available information has greatly contributed to improving our current knowledge about the origin and evolution of the group and the genomic architecture of traits of biological, economic and medical relevance such as the synthesis of silk and venoms, the gene families involved in smell and taste (chemosensory system) or the adaptation to different diets, including parasitism. The acquisition of new high-quality genomes throughout the tree of life of chelicerates, promises key future discoveries for the understanding of the great diversification and extraordinary adaptations of these fascinating animals, but also for their applications in conservation, biomedicine, sustainable pest management and the development of new biological materials.
Keywords: whole genome duplication (WGD), terrestralization, evolution, biological diversity, chemosensory system.
per l’epidermis i formada principalment de quitina, que embolcalla i protegeix l’animal. Aquest exoesquelet ha de ser mudat regularment per tal de permetre el creixement dels individus, mitjançant un procés anomenat ècdisi, que està regulat per una hormona esteroide, l’ecdisona. La regulació hormonal dels cicles d’ècdisi és un tret que els artròpodes comparteixen amb altres fílums, com ara els nematodes, els nematomorfs, els cinorrincs, els loricífers o els priapúlids, que col·lectivament formen el llinatge evolutiu conegut com a Ecdysozoa. Els artròpodes es diferencien dels altres ecdisozous perquè presenten apèndixs articulats, generalment dos per cadascun dels segments en què es divideix el cos. Aquest tret el comparteixen amb dos petits fílums, els tardígrads, que formen part de la meiofauna, i els onicòfors, que viuen en am-
Treballs de la Societat Catalana de Biologia, 72: 34-42
bients tropicals terrestres. Tots tres grups constitueixen els panartròpodes (figura 1). Tanmateix, els artròpodes són els únics que han patit un procés de regionalització anatòmica i funcional dels segments del cos i els seus apèndixs, la tagmosi. El nombre i la distribució d’aquestes regions corporals, anomenades tagmes, són característics de cadascun dels principals llinatges evolutius d’aquest grup divers.
Dins els artròpodes, en els insectes (com ara les paneroles, les mosques o les papallones), els crustacis (les puces d’aigua, els escamarlans o els porquets de Sant Antoni) i els miriàpodes (els centpeus i els milpeus), el primer parell d’apèndixs del cos són antenes, generalment amb funció sensitiva. Hi ha un seguit d’artròpodes, però, que en el lloc d’aquestes antenes hi tenen unes pinces o uns ullals, generalment
Miquel A. Arnedo1, 2 i Julio Rozas2, 3
Departament de Biologia Evolutiva, Ecologia i Ciències Ambientals, Universitat de Barcelona
2 Institut de Recerca de la Biodiversitat (IRBio), Universitat de Barcelona
Departament de Genètica, Microbiologia i Estadística, Universitat de Barcelona
34
Correspondència: Miquel A. Arnedo. Departament de Biologia Evolutiva, Ecologia i Ciències Ambientals, Universitat de Barcelona. Av. Diagonal, 643. 08028 Barcelona. Adreça electrònica: marnedo@ub.edu.
Figura 1. Filogènia de consens actual dels fílums de panartròpodes i subfílums d’artròpodes. Imatges de siluetes descarregades sota llicència de domini públic o CC d’http:// phylopic-org: Onycophora (imatge de Mali’o Kodis, fotografia de Bruno Vellutini, publicada sota una llicència Creative Commons AttributionShareAlike 3.0 Unported (CC BY-SA 3.0), https:// creativecommons.org/licenses/by-sa/3.0/),
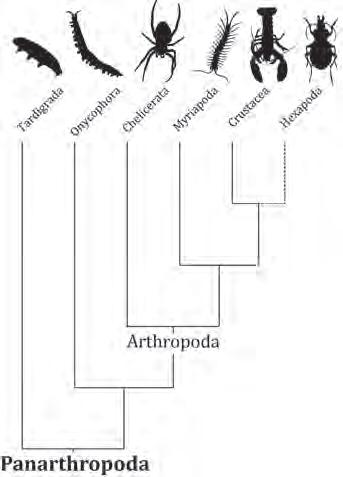
Chelicerata (imatge de Birgit Lang publicada sota una llicència Creative Commons AttributionShareAlike 3.0 Unported (CC BY-SA 3.0), https:// creativecommons.org/licenses/by-sa/3.0/),
Hexapoda (imatge de Michael Keesey [vectorització], Thorsten Assmann, Jörn Buse, Claudia Drees, Ariel-Leib-Leonid Friedman, Tal Levanony, Andrea Matern, Anika Timm i David W. Wrase [fotografia], publicada sota una llicència Creative Commons Attribution 3.0 Unported (CC BY 3.0), https://creativecommons. org/licenses/by/3.0/).
amb funció alimentària, anomenats quelícers Aquest grup el coneixem com a quelicerats
Morfologia
En els quelicerats, el cos està generalment dividit en dos tagmes. El prosoma, o tagma anterior, conté els ulls i els principals apèndixs que, juntament amb els quelícers, són propis del grup: un parell de pedipalps, la forma i funció dels quals varia en funció del grup de quelicerats, i quatre parells de potes o apèndixs locomotors. L’opistosoma, el tagma posterior, inclou l’orifici genital, que se situa en posició ventral anterior, i apèndixs molt modificats. Exemples d’apèndixs opistosòmics són les brànquies i els opercles de
a) Pycnogonida; b) Xiphosura, Limulus polyphemus juvenil; c) Solifugae, Gluvia dorsalis; d) Opiliones, Odiellus cf. troguloides; e) Opilioacarida, Opilioacarus baeticus; f ) Mesostigmata sp.; g) Holothryda; h) Ixodida, Ixodes vespertilionis; i ) Trombidiformes, Penthaleidae sp.; j ) Sarcoptiformes, Damaeus onustusi; k) Ricinulei, Cryptocellus sp.; l ) Palpigradi, Eukoenenia strinatii; m) Scorpiones, Belisarius xambeui; n) Pseudoscorpiones, Chthoniidae sp.; o) Araneae, Dysdera catalonica; p) Amblypygi, Damon diadema; q) Uropygi, Mastigoproctus giganteus; r) Schizomida, Stenochrus portoricensis. Fotografies: a, e: Eduardo Mateos; b, i, k: Gonzalo Giribet; c, d, f, h, m-q : Marc Domènech; g: Damien Brouste; j: Donald Hobern, reproduït sota llicència CC BY 2.0; l: Alberto Chiarle; r: Pedro Oromí.

protecció de l’orifici genital dels xifosurs (figura 2), les pectines sensorials dels escorpins, o les fileres, els òrgans per on segreguen la seda les aranyes. En alguns grups de quelicerats hi ha una subdivisió dels dos tagmes principals: en els escorpins, per exemple, l’opistosoma està subdividit en un mesosoma, més gruixut i on hi ha cinc parells de pulmons o fil·lotràquees, i uns
Treballs de la Societat Catalana de Biologia, 72: 34-42
segments terminals més prims que formen el metasoma, la «cua» dels escorpins (figura 2), al final de la qual hi ha el fibló, connectat a una glàndula interna del verí. En el cas dels àcars, els pedipalps i els quelícers formen una estructura individualitzada, el gnatosoma, mentre que en solífugs els segments de les potes 3 i 4 no estan fusionats a la resta del prosoma (figura 2).
35
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
Figura 2. Fotografies dels ordres de quelicerats:
Figura 3. Filogènia de consens actual dels ordres de quelicerats. Les capses de color indiquen agrupacions taxonòmiques superiors. Les xifres en els extrems de les branques terminals indiquen el nombre de genomes actualment disponibles de cadascun dels ordres. Les capses en les branques fan referència a l’existència de fenòmens de duplicació completa del genoma (WGD), els colors blancs i negres indiquen la possibilitat que aquests fenòmens no siguin homòlegs (vegeu el text). La línia de punts indica la posició alternativa dels xifosurs segons estudis genòmics recents (Ballesteros et al., 2022). Cal esmentar que, en aquest cas, els ricinulis apareixen també com a grup germà dels xifosurs. Imatges de siluetes pròpies o descarregades sota llicència de domini públic o CC d’http://phylopic-org: Xiphosura (imatge de Noah Schlottman publicada sota una llicència Creative Commons Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0), https://creativecommons.org/licenses/by-sa/3.0/), Opiliones i Pseudoscorpiones (imatges de Gareth Monger publicades sota una llicència Creative Commons Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0), https:// creativecommons.org/licenses/by-sa/3.0/), Ixodida i Trombidiformes (imatges de Mathilde Cordellier publicades sota una llicència Creative Commons AttributionShareAlike 3.0 Unported (CC BY-SA 3.0), https://creativecommons.org/licenses/by-sa/3.0/), Sarcoptiformes (imatge de Birgit Lang publicada sota una llicència Creative Commons Attribution-ShareAlike 3.0 Unported (CC BY-SA 3.0), https://creativecommons.org/licenses/by-sa/3.0/)
Taxonomia i diversitat
Els quelicerats inclouen vora de cent vint mil espècies, i ocupen el segon lloc darrere els insectes entre els grups d’animals més diversos. Des d’un punt de vista taxonòmic, els quelicerats inclouen tres classes (figura 3): els picnogònids, amb un únic ordre, els pantòpodes o aranyes d’aigua; els merostomats, els únics representants actuals dels quals són l’ordre dels xifosurs, els límuls o cassoles de les Moluques, i els aràcnids, dels quals hi ha fins a setze ordres (figura 2, taula 1). Cal fer una menció especial als àcars, que inclouen espècies d’interès mèdic, com les paparres ( Ixodida ), que són vectors de la malaltia de Lyme, i d’interès agropecuari, com les aranyes vermelles (Tetranychus), que són una de les principals plagues de cítrics, o les Varroa , que parasiten bresques
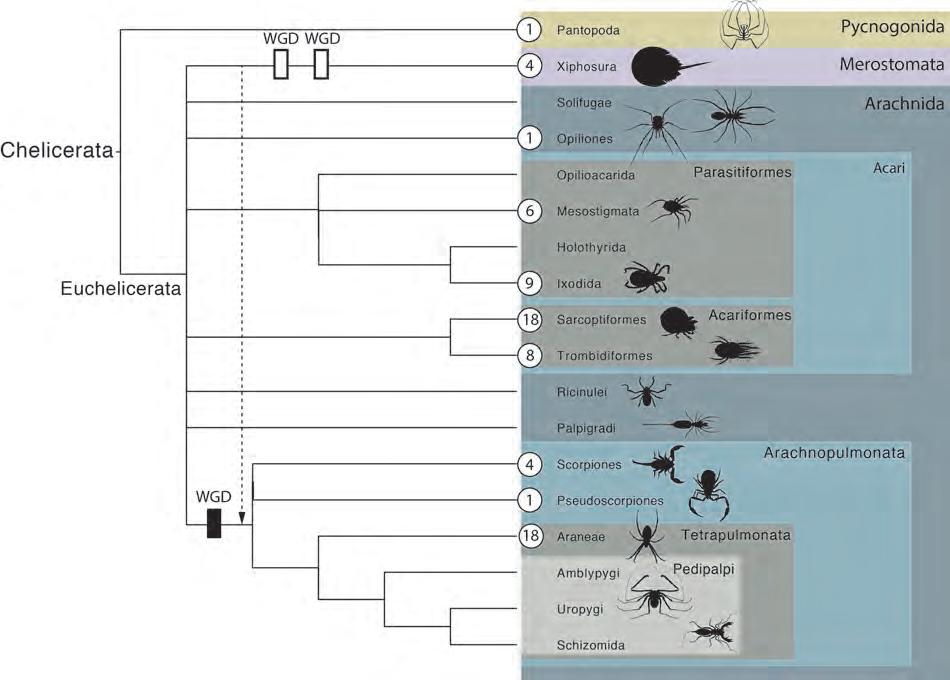
d’abelles, la taxonomia de les quals ha estat objecte de nombroses revisions en els últims anys (Dunlop i Alberti, 2008). Actualment, es consideren una superclasse que inclouria dos llinatges principals, els Acariformes i els Parasitiformes, la monofília dels quals ha estat qüestionada per nombrosos estudis recents de filogènica molecular.
Història evolutiva
Els quelicerats tenen una llarga història evolutiva: l’evidència més antiga correspon a una larva de picnogònid del cambrià mitjà, datat fa uns cinc-cents milions d’anys (Waloszek i Dunlop, 2002). Tot i els seus orígens marins i que dues de les classes actuals, els picnogònids i els límuls, viuen en aquest ambient, la major part de la diversitat actual habita ecosistemes
Treballs de la Societat Catalana de Biologia, 72: 34-42
terrestres, on constitueixen un dels grups de depredadors més diversos i abundants. L’aràcnid més antic conegut és un escorpí fòssil del silurià inferior de Wisconsin (Estats Units), datat fa 437,5-436,5 milions d’anys (Wendruff et al., 2020), que, tot i que va ser trobat en sediments marins, probablement corresponents a una zona d’intermareal —la seva anatomia és molt semblant a la dels escorpins actuals—, tot suggereix que ja era capaç de passar llargs períodes de temps en aquests ambients.
L’establiment de les relacions filogenètiques dels ordres de quelicerats ha estat una carrera llarga i plena d’obstacles, conflictiva i, a voltes, exasperant. Des de la formulació de les primeres hipòtesis explícites sobre les seves relacions a finals del segle xix, fins a la irrupció de les dades genòmiques en la segona dèca-
36
Miquel A. Arnedo i Julio Rozas
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
Taula 1. Llista dels genomes de quelicerats disponibles en bases de dades públiques a desembre de 2021. Genome ID: codi d’accés a base de dades del Centre Nacional per a la Informació Biotecnològica (NCBI); Contingut GC: percentatge de guanina i citosina en el genoma. Elaboració pròpia.
Classe Ordre
Família
Espècie
Genome ID Grandària (Mb) Contingut GC (%)
Pycnogonida Pantopoda Nymphon Nymphon striatum 97840 744,79 35,4
Merostomata Xiphosura Limulidae Carcinoscorpius rotundicauda 15673 1.697,14 33,5
Merostomata Xiphosura Limulidae Limulus polyphemus 787 1.828,26 34,5
Merostomata Xiphosura Limulidae Tachypleus gigas 93827 1.830,81 33,4
Merostomata Xiphosura Limulidae Tachypleus tridentatus 22334 1.974,11 32,85
Arachnida Acariformes
Arachnida Acariformes
Arachnida Acariformes
Sarcoptiformes Achipteria coleoptrata 37199 88,44 29,8
Sarcoptiformes Archegozetes longisetosus 103391 198,02 30,9
Sarcoptiformes Dermatophagoides farinae 9138 58,78 30,4
Arachnida Acariformes Sarcoptiformes Dermatophagoides pteronyssinus 8901 113,61 29,2
Arachnida Acariformes Sarcoptiformes Euroglyphus maynei 54478 43,43 26,2
Arachnida Acariformes
Arachnida Acariformes
Sarcoptiformes Hypochthonius rufulus 37200 172,4 27,9
Sarcoptiformes Medioppia subpectinata 95392 213,17 30,8
Arachnida Acariformes Sarcoptiformes Oppiella nova 95393 196,73 30,4
Arachnida Acariformes Sarcoptiformes Platynothrus peltifer 37201 100,58 28,9
Arachnida Acariformes Sarcoptiformes Psoroptes ovis 67302 63,21 28,2
Arachnida Acariformes Sarcoptiformes Sarcoptes scabiei 36095 56,58 33,2
Arachnida Acariformes Sarcoptiformes Tyrophagus putrescentiae 35692 95,13 29,1
Arachnida Acariformes Trombodiformes Aculops lycopersici 96667 32,54 45,2
Arachnida Acariformes Trombodiformes Brevipalpus yothersi 74771 71,16 36,6
Arachnida Acariformes Trombodiformes Dinothrombium tinctorium 73047 180,4 32,3
Arachnida Acariformes Trombodiformes Fragariocoptes setiger 105974 40,89 43,2
Arachnida Acariformes Trombodiformes Leptotrombidium deliense 7830 117,32 33,6
Arachnida Acariformes Trombodiformes Leptotrombidium pallidum 7646 170,87 35,1
Arachnida Acariformes Trombodiformes Panonychus citri 12054 83,97 31,3
Arachnida Acariformes Trombodiformes Tetranychus urticae 2710 90,82 32,5
Arachnida Araneae Araneidae Araneus ventricosus 34620 3.656,62 31,9
Arachnida Araneae Araneidae Argiope bruennichi 33398 1.670,29 29,3
Arachnida Araneae Araneidae Nephila pilipes 17580 2.694,57 29,7
Arachnida Araneae Araneidae Trichonephila antipodiana no disponible 2.2900 29,5
Arachnida Araneae Araneidae Trichonephila clavata 93228 2.497,9 31
Arachnida Araneae Araneidae Trichonephila clavipes 80095 2.874,35 34,2
Arachnida Araneae Araneidae Trichonephila inaurata 105982 2.507,04 29,5
Arachnida Araneae Dysderidae Dysdera silvatica 82280 1.365,69 34,7
Arachnida Araneae Eresidae Stegodyphus dumicola 87353 2.551,87 33,3
Arachnida Araneae Eresidae Stegodyphus mimosarum 12925 2.738,7 33,8
Arachnida Araneae Linyphiidae Oedothorax gibbosus 104832 821,43 32,08
Arachnida Araneae Lycosidae Pardosa pseudoannulata 83627 4.207,95 30,4
Arachnida Araneae Pisauridae Dolomedes plantarius 101471 2.580,75 33,49
Arachnida Araneae Sicariidae Loxosceles reclusa 14028 1.793,25 39,4
Arachnida Araneae Theraphosidae Acanthoscurria geniculata 22960 5.102,16 40,2
Arachnida Araneae Theridiidae Anelosimus studiosus 83943 2.033,43 21,2
Arachnida Araneae Theridiidae Latrodectus hesperus 14107 781,83 27,9 Arachnida Araneae Theridiidae Parasteatoda tepidariorum 13270 1.228,97 29,7
Arachnida Opiliones Palpatores Phalangium opilio 24136 576,9 37,2
Arachnida Parasitiformes Ixodida Dermacentor silvarum 36355 2.474,31 46,9
Arachnida Parasitiformes Ixodida Haemaphysalis longicornis 69202 4.958,66 47,4
Arachnida Parasitiformes Ixodida Hyalomma asiaticum 92161 1.713,63 46,6
Arachnida Parasitiformes Ixodida Ixodes persulcatus 7207 1.901,64 45,9
Arachnida Parasitiformes Ixodida Ixodes ricinus 16267 514,51 45
Arachnida Parasitiformes Ixodida Ixodes scapularis 523 2.226,88 46,2
Arachnida Parasitiformes Ixodida Rhipicephalus annulatus 93261 2.762,43 45,7
Arachnida Parasitiformes Ixodida Rhipicephalus microplus 2797 2.269,15 45,45
Arachnida Parasitiformes Ixodida Rhipicephalus sanguineus 2716 2.365,54 46,8
Arachnida Parasitiformes Mesostigmata Dermanyssus gallinae 75128 959,01 44,7
Arachnida Parasitiformes Mesostigmata Galendromus occidentalis 3487 151,7 51,6
Arachnida Parasitiformes Mesostigmata Stratiolaelaps scimitus 104968 426,5 45,9
Arachnida Parasitiformes Mesostigmata Tropilaelaps mercedesae 53919 352,54 40,9
Arachnida Parasitiformes Mesostigmata Varroa destructor 937 368,93 40,9
Arachnida Parasitiformes Mesostigmata Varroa jacobsoni 62339 365,59 40,9
Arachnida Pseudoscorpiones Cheliferoides Cordylochernes scorpioides 69337 2.807,11 30,1
Arachnida Scorpiones Buthida Androctonus mauritanicus 88051 1.459,99 34,9
Arachnida Scorpiones Buthida Centruroides sculpturatus 14105 925,48 31,4
Arachnida Scorpiones Buthida Mesobuthus martensii 14571 925,54 29,3
Treballs de la Societat Catalana de Biologia, 72: 34-42
37
Miquel A. Arnedo i Julio Rozas
da del segle xxi (vegeu Giribet, 2018, per a una síntesi històrica de les dades i hipòtesis proposades de la filogènia de quelicerats), el consens actual és que l’arbre dels quelicerats està poc resolt i el seu gran nombre de politomies formen una gran pinta (figura 3).
L’era genòmica
Des de la incorporació de les dades moleculars als estudis de la biodiversitat en la dècada de 1980, bona part de la informació disponible havia estat basada en la seqüenciació de pocs fragments seleccionats del genoma mitjançant la tècnica de seqüenciació de Sanger. L’aparició de les tècniques de seqüenciació de nova generació a inicis del segle xxi va permetre l’obtenció massiva de seqüències de DNA a una escala sense precedents i a uns costos raonables, cosa que va tenir un gran impacte en diferents aspectes de la variabilitat genètica, especialment en organismes no model. A finals de la primera dècada del segle, apareixen les tècniques de seqüenciació massiva de tercera generació (seqüenciació de lectura llarga), que, en combinació amb altres mètodes, com l’assemblatge basat en tecnologia de seqüenciació Hi-C i els avenços en computació i anàlisi, van obrir la porta a la generació de genomes d’alta qualitat i continuïtat, que cobreixen gairebé tots els cromosomes. Ara bé, de quina manera la informació a escala genòmica millora el nostre coneixement sobre la biodiversitat respecte de tècniques anteriors?
Tot i que hi ha multitud de camps en què el fet de disposar de genomes de referència contribueix de manera important a entendre les bases genètiques de trets rellevants des d’un punt de vista evolutiu, ecològic o fins i tot aplicat, hi ha aspectes relacionats amb l’adaptació, especiació i conservació en els quals la seqüenciació de tot el genoma (WGS, de l’anglès whole-genome sequencing) en lloc de biblioteques de representació reduïda (RRL, de l’anglès reduced representation library) és cabdal. Aspectes com la continuïtat, el lligament i la densitat de marcadors milloren les nostres estimacions de processos rellevants des del punt de vista ecològic i evolutiu, com són les bases genètiques de l’adaptació, la història demogràfica i l’estructuració poblacional, amb implicacions en la conservació, la introgressió, la variació estructural i el desenvolupament metodològics per a l’anàlisi de dades WGS (Taylor et al., 2021).
Què sabem del genoma dels quelicerats?
Actualment (29 de desembre de 2021), hi ha disponibles a les bases de dades públiques
63 genomes de diferents grups de quelicerats, amb un ampli espectre de qualitat pel que fa a la cobertura i continuïtat, que va des de genomes molt fragmentats obtinguts mitjançant piroseqüenciació —p. ex. l’escorpí Mesobuthus martensii (Cao et al., 2013)— fins a genomes de nivell de qualitat d’assemblatge d’escala cromosòmica —p. ex. l’aranya Dysdera silvatica (Escuer et al., 2022). Tot i que hi ha representants de les tres classes principals de quelicerats, només la meitat dels ordres (vuit de setze) tenen almenys un genoma de referència disponible. Els grups millor representats són les aranyes (18 genomes) i els àcars parasitiformes i acariformes (17 i 20, respectivament).
En el cas de les aranyes, un aspecte destacat de l’estudi de genoma és que ha revelat una gran variabilitat en la seva grandària, de fins a vuit cops. El genoma més petit reportat són els 0,75 Gb de l’aranya orbicular de quelícers llargs Tetragnatha elongata, i el més gran els 5,6 Gb de l’aranya saltadora Habronattus (Gregory i Shorthouse, 2003). L’estudi de genòmica comparada en la publicació recent del genoma complet d’una espècie d’aranya orbicular de quelícers llargs endèmica de les illes Hawaii ( Tetragnatha kauaiensis ) suggereix que bona part de la variabilitat en la grandària del genoma es deu a la presència de transposons i elements repetitius (Cerca et al., 2021).
Tot i que la quantitat de genomes de quelicerats actualment disponibles representen una ínfima part de la diversitat total del grup (taula 1), han aportat informació important en aspectes clau de l’evolució i biologia d’aquest important grup d’organismes. A continuació, destaquem i comentem algunes de les principals fites.
La desconstrucció dels aràcnids i el procés de terrestrialització Un dels punts més controvertits de la història evolutiva dels quelicerats és el que té a veure amb el procés de terrestrialització, més específicament, amb el fet de determinar quants cops els quelicerats van colonitzar independentment el medi terrestre a partir dels seus ancestres marins.
Un repte fonamental en la transició de la vida en els ambients aquàtics a la vida en un medi aeri és obtenir l’oxigen de l’aire i evitar la dessecació (Vieira i Rozas, 2011). Els quelicerats marins, els picnogònids i els límuls han desenvolupat diferents estratègies per a respirar dins de l’aigua: mentre que els primers no tenen estructures especialitzades i obtenen l’oxigen per difusió a través de la cutícula, transportant-lo al llarg del cos mitjançant moviments peristàltics dels intestins que activen
Treballs de la Societat Catalana de Biologia, 72: 34-42
l’hemolimfa (Woods et al., 2017), els segons disposen de sis parells de brànquies laminars (brànquies en llibre) formades pels exopodis d’apèndixs opistosòmics (el primer constitueix l’opercle genital) (Sharma, 2017) i d’un sistema circulatori tancat i força desenvolupat que inclou un cor dorsal i pigments respiratoris de tipus hemocianina (una proteïna que conté el metall de coure en lloc del ferro de l’hemoglobina per a transportar l’oxigen als teixits). Un aspecte interessant és que els limúlids són capaços d’aventurar-se fora de l’aigua per a pondre els ous a platges sorrenques, tal com fan les tortugues marines. Pel que fa als ordres terrestres, trobem dos sistemes de respiració internalitzats per a controlar la pèrdua d’aigua (taula 1): els pulmons en llibre (o fil·lotràquees), que consisteixen en invaginacions en forma de bosses en els segments anteriors de l’opistosoma, a l’interior de les quals hi ha una sèrie de làmines on es produeix el bescanvi d’oxigen amb l’hemolimfa, i les tràquees, invaginacions tegumentàries que es ramifiquen internament, i distribueixen l’oxigen directament a l’interior del cos. En certs grups, generalment associats a ambients molt humits, com els palpígrads i alguns acariformes, no hi ha estructures respiratòries especialitzades i la respiració es fa mitjançant el tegument (Dunlop, 2019). Tot i que el nombre i la posició dels pulmons en llibre en escorpins (quatre parells en els segments opistosòmics 4-7) difereixen dels d’aranyes i pedipalps (caracteritzats per la presència de dos parells de pulmons en llibre en els segments 2-3, raó per la qual hom es refereix a aquest grup com tetrapulmonata), la seva homologia ha estat confirmada per dades ultraestructurals (Scholtz i Kamenz, 2006) i per dades genòmiques (Giribet, 2018).
Tradicionalment, s’havia considerat que els aràcnids, els quelicerats terrestres, formaven un grup monofilètic, és a dir, amb un ancestre comú exclusiu i que, per tant, haurien colonitzat el medi terrestre un sol cop. Estimacions recents basades en rellotge molecular amb dades transcriptòmiques i informació d’un grapat de fòssils indiquen que els quelicerats haurien colonitzat els ambients terrestres al límit entre el cambrià i l’ordovicià, fa al voltant de quatre-cents vuitanta milions d’anys (Lozano-Fernández et al., 2020). Tanmateix, tal com s’ha esmentat, aquestes estimacions es basen en l’assumpció d’un origen únic dels ordres de quelicerats terrestres, quelcom que ha estat qüestionat recentment per un seguit d’estudis filogenòmics (Ballesteros et al., 2022; Ballesteros i Sharma, 2019; Noah et al., 2020; On-
38
tano et al., 2021a; Sharma et al., 2021; Sharma et al., 2014). Aquests estudis revelen que, contràriament a la visió clàssica: 1) els escorpins no serien el grup germà de la resta d’aràcnids, sinó que són més propers a les aranyes i a ordres col·lectivament coneguts com a Pedipalpi (amblipigis, uropigis i esquizòmids), amb els quals formarien el grup anomenat Arachnopulmonata degut a la presència de pulmons en llibre, quelcom que ja havia estat apuntat per estudis anteriors basats en seqüenciació de Sanger (Giribet, 2018), i 2) els limúlids podrien estar evolutivament més relacionats amb aquest grup que no pas amb la resta d’ordres d’aràcnids (a excepció dels ricinulis) (figura 3).
Una de les principals fonts de conflicte per tal de resoldre les relacions intraordinals dels quelicerats és deguda a l’existència d’organismes amb taxes d’evolució molecular molt per sobre de la mitjana de la resta. Aquests organismes ocupen branques llargues en els arbres filogenètics i poden generar errors metodològics durant la reconstrucció filogenètica. Els principals responsables d’això semblen ser els pseudoescorpins, els àcars i els palpígrads (Ontano et al., 2021b). L’anàlisi de genomes complets d’aranyes i escorpins ha contribuït a resoldre part del conflicte, ja que ha revelat que ambdós grups van patir al llarg de la seva evolució la duplicació completa del seu genoma (WGD, de l’anglès whole genome duplication) (Schwager et al., 2017). L’escenari més probable és que la WGD es donés en l’ancestre comú d’ambdós grups, cosa que confirmaria la seva proximitat filogenètica i, per tant, l’homologia dels pulmons en llibre d’ambdós grups. Sorprenentment, la publicació recent del primer genoma complet d’un pseudoescorpí, un dels grups conflictius filogenèticament, ha evidenciat que aquest grup també va experimentar una WGD (Ontano et al., 2021a) i que, per tant, probablement formi part també del llinatge evolutiu dels aracnopulmonats, tal com havien suggerit estudis anteriors (Benavides et al., 2019; Howard et al., 2020; Lozano-Fernández et al., 2019; Sharma et al ., 2015) (figura 3). El més notable d’aquests resultats és que, a diferència dels altres ordres d’aquest llinatge, els pseudoescorpins respiren mitjançant tràquees en lloc de pulmons en llibre, cosa que suggereix que les tràquees haurien evolucionat independentment més d’un cop. En aquest sentit, convé indicar que s’ha proposat que el procés de miniaturització sofert per diferents grups d’aràcnids per tal d’adaptar-se a nínxols nous, especialment els acariformes i els parasitiformes (àcars), però també els pseudoescorpins, podria explicar en
part el reemplaçament dels pulmons per tràquees o fins i tot la pèrdua dels òrgans respiratoris (Dunlop, 2019).
Més difícil d’entomar resulta la posició dels limúlids com a grup germà dels quelicerats terrestres, com els aracnopulmonata o ricinulis, ja que això suposaria la recolonització secundària dels ambients marins per part d’ancestres terrestres (figura 3), o bé una colonització de la terra múltiple i independent per part de diversos grups d’aràcnids. S’ha adduït, en aquest sentit, que bona part dels trets morfològics utilitzats tradicionalment per a demostrar l’origen únic dels aràcnids poden, de fet, explicar-se com el resultat de fenòmens de convergència adaptativa associats a les fortes pressions de seleccions imposades per la vida en ambients terrestres (Ballesteros et al., 2022). En aquest sentit, convé recordar que l’estreta relació que s’havia establert clàssicament entre miriàpodes i insectes va saltar pels aires amb l’aparició de les primeres dades moleculars que apuntaven, tal com s’ha confirmat posteriorment amb dades de seqüenciació massiva, una evolució dels insectes a partir d’un ancestre crustaci. D’altra banda, també és cert que altres hipòtesis primerenques basades exclusivament en dades moleculars, com ara que els quelicerats eren un grup germà dels miriàpodes, van demostrar ser producte d’artefactes metodològics. L’anàlisi genòmica no ha aportat, en aquest cas, gaire llum a la qüestió, ja que, tot i que els genomes obtinguts de les quatre espècies actuals de limúlids apunten l’existència de dos o fins i tot tres fenòmens de duplicació completa del genoma (Nong et al., 2021), els patrons de relacions inferits de les còpies múltiples de gens homeòtics no confirmen que cap estigui relacionada amb la WGD dels Arachnopulmonata (Kenny et al., 2016).
Haurem d’esperar la seqüenciació dels genomes d’altres ordres d’aràcnids per tal de poder determinar exactament la distribució de les WGD, la seva homologia i polaritat, així com l’existència d’altres canvis genòmics rars, per exemple la col·linealitat de gens en els cromosomes (sintènia) (Simakov et al., 2022), que permetin obtenir una filogènia completament resolta i ben fonamentada, i poder així reconstruir unívocament les principals fites evolutives dels quelicerats. En aquest sentit, un estudi recent basat en la sintènia dels fragments HOX entre limúlids, aranyes i escorpins no descarta completament la possibilitat de l’existència d’un esdeveniment únic de WGD en l’avantpassat comú de tots ells (Nong et al ., 2021).
Treballs de la Societat Catalana de Biologia, 72: 34-42
L’arquitectura genòmica de trets biològics de rellevància
La seda
Des de la publicació dels primers genomes complets d’aranyes l’any 2014 —l’aranya social Stegodyphus mimosarum i l’aranya migalomorfa Acanthoscurria geniculata— (Sanggaard et al., 2014), aquestes, juntament amb els àcars, han esdevingut el grup de quelicerats del qual disposem de més informació genòmica. Tanmateix, la distribució filogenètica és força esbiaixada, i més de la meitat dels 18 genomes disponibles (13 de publicats) pertanyen al grup d’aranyes Araneoidea, que inclou la major part de famílies que teixeixen teranyines aèries, incloses les teranyines orbiculars. Donada la representativitat encara força limitada de genomes d’aranya disponibles, la principal contribució dels estudis publicats fins al moment és proveir d’un recurs genòmic per a aportar llum a temes com l’arquitectura genòmica de certs trets rellevants, com ara els mecanismes genòmics darrere de l’evolució de la seda, els verins o el sistema quimiosensorial (olfacte i gust).
La capacitat de fabricar i manipular la seda és un dels trets distintius de les aranyes. Tot i que no són els únics animals capaços de sintetitzar seda, les aranyes han desenvolupat un grau de sofisticació sense comparació tant en la producció com en l’ús d’aquest material. La seda se sintetitza a les glàndules sericígenes, situades al final de l’opistosoma, i se segrega a través de les fileres, uns apèndixs opistosòmics molt modificats. Tot i que generalment hi ha tres parells de fileres, el seu nombre i posició a l’opistosoma pot canviar segons la família. Els grups d’aranyes que mostren més complexitat de seda són els araneoids, en els quals s’han descrit fins a set parells de glàndules sericígenes que segreguen sedes amb diferents propietats mecàniques i usos (p. ex. trampes de cacera, immobilització de preses, fils d’ancoratge, construcció de nius, protecció dels ous, senyalitzadors de feromones, etc.). Tot i l’homologia seriada de les diferents glàndules sericígenes, les duplicacions gèniques han tingut un paper clau en la diferenciació dels diferents tipus de sedes segregades per cadascuna. Tanmateix, s’ha vist que els canvis en els patrons d’expressió gènica no estan necessàriament relacionats amb aquestes duplicacions (Clarke et al ., 2017). La seda està formada principalment per les espidroïnes, unes proteïnes relacionades amb la queratina de les ungles, els pèls i la pell. Aquestes proteïnes s’emmagatzemen a les
39
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
Miquel A. Arnedo i Julio Rozas
glàndules en altes concentracions, i són processades posteriorment durant el seu transport a través dels conductes fins que les connecten a les fileres, mitjançant canvis en el gradient de pH, la concentració d’ions i tensions de tall, que acaben transformant-les en làmines beta (Andersson et al., 2016). La seda és un biomaterial amb una combinació única de resistència i extensibilitat, motiu pel qual hi ha força interès per a la seva síntesi a escala industrial. No és estrany, doncs, que molts estudis genòmics s’hagin dirigit especialment a identificar la diversitat de gens de la seda, la seva arquitectura i expressió (Babb et al., 2017; Kono et al., 2019). Els gens de la seda havien estat tradicionalment difícils de caracteritzar, donada la seva gran llargada i la presència de motius repetitius que caracteritzen les espidroïnes. L’obtenció de genomes complets ha permès catalogar bona part de les espridroïnes, caracteritzar-ne les variants i descobrir noves proteïnes en la seda (SpiCE) que confereixen a determinats tipus de seda les seves particularitats mecàniques.
Els verins
El verí és un dels aspectes que més ha contribuït a la mala reputació de les aranyes. Les glàndules productores del verí es localitzen a la zona anterior del prosoma, i l’injecten a la víctima mitjançant un canal que les connecta amb els quelícers i que s’obre a l’extrem dels ullals. Les aranyes no són els únics aràcnids verinosos, com és ben sabut; també ho són els escorpins, que injecten el verí mitjançant el fibló que es troba al final del metasoma, i, potser menys coneguts, les paparres (Cabezas-Cruz i Valdés, 2014) i els pseudoescorpins (Santibáñez-López et al., 2018), aquests últims amb les glàndules del verí als pedipalps. El verí dels aràcnids és un còctel de diferents components, que inclouen toxines que activen els canals iònics, inhibidors de proteases per a protegir-les, enzims amb propietats fosfolipases i hialuronidasa, i defensines (Santibáñez-López et al., 2018). L’evolució dels verins dels aràcnids ha estat tan complexa com la seva composició, i ha involucrat processos de duplicacions gèniques, reclutament i neofuncionalització, i, fins i tot, episodis de transmissió horitzontal des de bacteris (Luddecke et al., 2022). Encara que molt parcial, la seqüenciació de genomes complets ha contribuït de manera important a millorar el nostre coneixement sobre el nombre, la diversitat i la composició dels gens responsables dels diferents components del verí (Garb et al., 2018). És interessant destacar, en aquest sentit, que s’ha observat que en aranyes
errants, que no construeixen teles per a caçar sinó que persegueixen activament i immobilitzen les preses mitjançant el verí, hi ha un increment substancial del nombre de neurotoxines codificades, mentre que, d’altra banda, es redueix la quantitat de gens de la seda (Yu et al., 2019).
El sistema quimiosensorial
El sistema quimiosensorial és probablement un dels més primitius en els éssers vius. Tots els animals tenen òrgans especialitzats que els permeten detectar estímuls de l’ambient extern i respondre-hi, i que són els responsables dels sentits de l’olfacte i el gust. Tot i que la línia que separa aquests sentits en alguns organismes és molt difusa, com a regla general l’olfacte permet el reconeixement de molècules volàtils que confereixen a l’organisme la capacitat de detectar aliments, predadors o parelles, mentre que el gust sovint intervé en la detecció de substàncies solubles, que, a més d’induir comportaments associats amb l’alimentació, pot provocar respostes relacionades amb l’aparellament i la reproducció. La quimiorecepció és, per tant, una capacitat biològica crítica per a la supervivència i la reproducció dels organismes (Sánchez-Gracia et al., 2009; Vieira i Rozas, 2011). En els artròpodes, el primer pas en la recepció del senyal químic de l’exterior s’efectua a les sensílies, unes estructures cuticulars poroses presents a diverses parts del cos (en Drosophila, per exemple, es troben a les antenes, el bulb maxil·lar, la probòscide i les potes). En el cas de l’olfacte, les molècules que actuen com a odorants entren a través dels porus, travessen la limfa de la sensília (directament o ajudades per proteïnes solubles) i activen els receptors transmembrana, unes proteïnes molt especialitzades situades a les dendrites de les neurones olfactives. El procés desencadena una sèrie de senyals elèctrics que es processen i s’interpreten en el cervell (Pelosi, 1996).
Tant les proteïnes solubles com els receptors de membrana són codificats per famílies multigèniques enormes, que poden variar entre unes desenes i uns milers de còpies per genoma (Vizueta et al., 2020a; Vizueta et al., 2018). En artròpodes, les famílies multigèniques que codifiquen les principals proteïnes solubles són les de les proteïnes d’unió a odorants (OBP, odorant binding protein), les proteïnes quimiosensorials (CSP, chemosensory protein) i NPC2 (Niemann-Pick C2). Per la part dels receptors de membrana, tenim les famílies dels receptors olfactius (OR, odorant receptors; presents únicament a insectes), receptors gustatius (GR,
Treballs de la Societat Catalana de Biologia, 72: 34-42
gustatory receptors) i receptors ionotròpics (IR, ionotropic receptors). En els quelicerats amb genoma seqüenciat, la família més nombrosa és la dels GR, que en l’escorpí Centruroides exilicauda inclou 832 membres. El gran nombre de gens ha impossibilitat, fins ara, la identificació correcta i l’estudi rigorós d’aquestes famílies amb les metodologies de seqüenciació de fragments curts (short-reads). Tanmateix, la ràpida generalització de les tècniques de seqüenciació genòmica de tercera generació (de fragments llargs; long-reads), combinada amb el desenvolupament de metodologies bioinformàtiques que permeten obtenir un assemblatge genòmic a escala cromosòmica, està possibilitant fer estudis en genòmica comparada de famílies multigèniques i variants estructurals, inabordables fins ara.
La diversificació tròfica
A diferència de la resta de quelicerats que són depredadors, els àcars mostren una gran diversitat de preferències tròfiques, que inclouen, a més, espècies herbívores i formes ectoparasítiques, tant hematòfagues (p. ex. les paparres) com especialistes a alimentar-se de greixos (p. ex. Varroa, que ataca abelles). Un estudi de genòmica comparada ha identificat els principals canvis genòmics associats als diferents tipus de dieta en àcars (Liu et al., 2021). Mitjançant la comparació de quinze genomes complets d’àcars, s’ha observat que determinats gens associats a aspectes de l’obtenció, de la preparació i del metabolisme de nutrients estan sota diferents pressions de selecció depenent del tipus d’alimentació. Un aspecte important d’aquest estudi és la possibilitat d’identificar possibles dianes gèniques per al desenvolupament de nous pesticides per al control d’espècies nocives, principalment plagues agrícoles i dels ruscs d’abelles. En aquest sentit, s’està investigant activament en les bases genètiques dels canals KIR (de l’anglès killer-cell immunoglobulin-like receptor), els quals tenen un paper important en el funcionament de les glàndules salivals i els sistemes excretors d’aquests organismes. Estudis recents han identificat els diferents complements d’aquests canals transmembranals, cosa que permetrà estudiar-los in vitro per al desenvolupament de bloquejadors (Saelao et al., 2021).
La recerca en genòmica de quelicerats en els territoris de parla catalana
Els equips liderats pels doctors Julio Rozas i Àlex Sánchez, de la secció de Genètica, i el
40
doctor Miquel A. Arnedo, de la secció de Zoologia, tots membres de l’Institut de Recerca de la Biodiversitat (IRBio) de la Universitat de Barcelona (UB), col·laboren activament en la seqüenciació, l’anàlisi i la interpretació de genomes d’alta qualitat de diferents grups d’aràcnids. En aquest sentit, s’ha publicat recentment el genoma d’una espècie d’aranya dimoni vermella, endèmica de les Illes Canàries, Dysdera silvatica, l’únic representant disponible fins ara d’un dels principals llinatges d’aranyes, les sinespermiata, el qual ha ofert informació important sobre els processos associats a la diversificació insular i a l’evolució de l’especialització tròfica (Escuer et al., 2022; Sánchez-Herrero et al ., 2019). L’anàlisi exhaustiva del seu genoma ha permès quantificar i estudiar amb un detall sense precedents els gens codificats per les grans famílies multigèniques del sistema quimiosensorial en un quelicerat. En particular, en total s’han identificat 570 receptors de membrana codificats al genoma d’aquesta espècie, amb una infrarepresentació notòria al cromosoma X. A més, hem determinat que molts d’aquests gens dels receptors (un 54 %) es troben agrupats en clústers cromosòmics (fins a 83), i molts tenen un origen molt recent. Per a aquesta tasca es va desenvolupar una nova eina bioinformàtica: BITACORA (Vizueta et al., 2020b). Aquests resultats tenen repercussions i implicacions importants tant per al coneixement de la bio-
Bibliografia
Andersson, M. [et al.] (2016). «Silk spinning in silkworms and spiders». Int. J. Mol. Sci. [en línia], 17 (8). <https://doi.org/10.3390/ijms17081290>.
Babb, P. L. [et al.] (2017). «The Nephila clavipes genome highlights the diversity of spider silk genes and their complex expression». Nat. Genet. [en línia], 49 (6): 895-903. <https://doi.org/10.1038/ng.3852>.
Ballesteros, J. A. [et al.] (2022). «Comprehensive species sampling and sophisticated algorithmic approaches refute the monophyly of Arachnida». Mol. Biol. Evol. [en línia], 39 (2). <https://doi.org/10.1093/molbev/ msac021>.
Ballesteros, J. A.; Sharma, P. P. (2019). «A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error». Syst. Biol. [en línia], 68 (6): 1-62. <https://doi. org/10.1093/sysbio/syz011>.
Bar-On, Y. M. [et al.] (2018). «The biomass distribution on Earth». Proc. Natl. Acad. Sci. USA [en línia], 115 (25): 6506-6511. < https://doi.org/10.1073/pnas. 1711842115>.
Benavides, L. R. [et al.] (2019). «Phylogenomic interrogation resolves the backbone of the Pseudoscorpiones tree of life». Mol. Phylogenet. Evol. [en línia], 139: 106509. < https://doi.org/10.1016/j.ympev.2019. 05.023>.
Cabezas-Cruz, A.; Valdés, J. J. (2014). «Are ticks venomous animals?». Front. Zool. [en línia], 11 (1): 47. <https://doi.org/10.1186/1742-9994-11-47>.
Cao, Z. [et al.] (2013). «The genome of Mesobuthus martensii reveals a unique adaptation model of arthro-
logia quimiosensorial dels diferents grups d’artròpodes, com en la nostra comprensió de la biodiversitat a escala molecular.
Un segon genoma d’alta qualitat d’aquest mateix gènere, el de l’espècie Dysdera catalonica (figura 2o), la distribució de la qual se circumscriu al terç nord-oriental de Catalunya, està sent analitzat actualment. Curiosament, la mida del genoma de l’espècie continental (~3,2 Gb) és el doble que el de l’espècie insular (1,7 Gb), i les dades preliminars obtingudes pel grup suggereixen que són les espècies insulars les que han reduït la mida del genoma, i han proporcionat així un model únic per a estudiar com les pèrdues d’informació genòmica poden estar associades a la colonització dels ambients insulars i quines d’aquestes pèrdues són més preponderants. D’altra banda, cal esmentar que en el context dels ajuts de la iniciativa catalana per a l’Earth BioGenome Project (EBP), la CBP (de l’anglès Catalan Initiative for the Earth BioGenome Project), i amb finançament de la Societat Catalana de Biologia (SCB) i de la Institució Catalana d’Història Natural (ICHN) de l’Institut d’Estudis Catalans (IEC), aquest grup està actualment en la fase d’anotació del genoma de l’escorpí cec (Belisarius xambeui) (figura 2m), una espècie endèmica del Pirineu i Prepirineu oriental, de distribució relicta i força localitzada, amb preferència per llocs ombrívols i humits, i de relacions evolutives incertes. El genoma d’aquesta
espècie de morfologia aberrant, d’aproximadament 4 Gb, aportarà llum sobre l’adaptació als ambients troglòfils i l’evolució dels verins en els escorpins, ja que serà la primera espècie amb genoma disponible dels Iurida, un dels dos llinatges principals (parvordres) d’escorpins (Santibáñez-López et al., 2020).
D’altra banda, Jesús Lozano-Fernández, professor lector de la secció de Genètica de la UB i membre de l’IRBio, porta una línia de recerca on estudia les relacions filogenètiques de quelicerats fent servir dades a escala genòmica. Lidera estudis on integra fonts d’evidència alternatives per establir la filogènia d’aquest grup i on tracta d’inferir una escala de temps per a datar els processos de colonització de la Terra.
Agraïments
Volem agrair a Jaume Pellicer l’oportunitat que ens va oferir de participar en aquest número de la revista. Els comentaris de Jesús Lozano-Fernández van contribuir a millorar la qualitat final del manuscrit. Un agraïment especial a Marc Domènech, Gonzalo Giribet, Pere Oromí i Eduardo Mateos per compartir les seves fotografies per tal de confeccionar la figura 2. Aquest treball ha estat parcialment finançat pel Ministeri de Ciència i Innovació (PID2019-105794GB, PID2019-103947GB) i l’Agència de Gestió d’Ajuts Universitaris i Recerca (2017SGR83, 2017SGR1287).
pods». Nat. Commun. [en línia], 4: 2602. <https://doi. org/10.1038/ncomms3602>.
Cerca, J. [et al.] (2021). «The Tetragnatha kauaiensis genome sheds light on the origins of genomic novelty in spiders». Genome Biol. Evol. [en línia], 13 (12). <https://doi.org/10.1093/gbe/evab262>.
Clarke, T. H. [et al.] (2017). «Evolutionary shifts in gene expression decoupled from gene duplication across functionally distinct spider silk glands». Sci. Rep. [en línia], 7 (1): 8393. <https://doi.org/10.1038/s41598017-07388-1>.
Dunlop, J. A. (2019). «Miniaturisation in Chelicerata». Arthropod. Struct. Dev. [en línia], 48: 20-34. <https:// doi.org/10.1016/j.asd.2018.10.002>.
Dunlop, J. A.; Alberti, G. (2008). «The affinities of mites and ticks: A review». J. Zoolog. Syst. Evol. Res . [en línia], 46 (1): 1-18. < https://doi.org/10.1111/ j.1439-0469.2007.00429.x>.
Escuer, P. [et al.] (2022). «The chromosome-scale assembly of the Canary Islands endemic spider Dysdera silvatica (Arachnida, Araneae) sheds light on the origin and genome structure of chemoreceptor gene families in chelicerates». Mol. Ecol. Resour. [en línia], 22 (1): 375-390. < https://doi.org/10.1111/17550998.13471>.
Garb, J. E. [et al.] (2018). «Recent progress and prospects for advancing arachnid genomics». Curr. Opin. Insect. Sci. [en línia], 25: 51-57. < https://doi.org/ 10.1016/j.cois.2017.11.005>.
Giribet, G. (2018). «Current views on chelicerate phylogeny—A tribute to Peter Weygoldt». Zool. Anz. [en
Treballs de la Societat Catalana de Biologia, 72: 34-42
línia], 273: 7-13. < https://doi.org/10.1016/j.jcz. 2018.01.004>.
Gregory, T. R.; Shorthouse, T. R. (2003). «Genome sizes of spiders». J. Hered. [en línia], 94: 285-290. < https://doi.org/papers://B79588E5-2CC1-4C57 -804E-28592FC88A1E/Paper/p5767>.
Howard, R. J. [et al.] (2020). «Arachnid monophyly: Morphological, palaeontological and molecular support for a single terrestrialization within Chelicerata». Arthropod. Struct. Dev. [en línia], 59: 100997. <https:// doi.org/10.1016/j.asd.2020.100997>.
Kenny, N. J. [et al.] (2016). «Ancestral whole-genome duplication in the marine chelicerate horseshoe crabs». Heredity [en línia], 116 (2): 190-199. < https://doi. org/10.1038/hdy.2015.89>.
Kono, N. [et al.] (2019). «Orb-weaving spider Araneus ventricosus genome elucidates the spidroin gene catalogue». Sci. Rep. [en línia], 9 (1): 8380. <https://doi. org/10.1038/s41598-019-44775-2>.
Liu, Q. [et al.] (2021). «Comparative analysis of mite genomes reveals positive selection for diet adaptation». Commun. Biol. [en línia], 4 (1): 668. <https://doi. org/10.1038/s42003-021-02173-3>.
Lozano-Fernández, J. [et al.] (2019). «Increasing species sampling in chelicerate genomic-scale datasets provides support for monophyly of Acari and Arachnida». Nat. Comm. [en línia], 10 (1): 2295. <https://doi. org/10.1038/s41467-019-10244-7>. (2020). «A Cambrian–Ordovician terrestrialization of arachnids». Front. Genet. [en línia], 11: 896. <https://doi.org/10.3389/fgene.2020.00182>.
41
Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
Miquel A. Arnedo i Julio Rozas
Luddecke, T. [et al.] (2022). «The biology and evolution of spider venoms». Biol. Rev. Camb. Philos. Soc. [en línia], 97 (1): 163-178. < https://doi.org/10.1111/ brv.12793>.
Noah, K. E. [et al.] (2020). «Major revisions in arthropod phylogeny through improved supermatrix, with support for two possible waves of land invasion by Chelicerates». Evol. Bioinform. [en línia], 16: 117693432 090373. <https://doi.org/10.1177/1176934320903735>.
Nong, W. [et al.] (2021). «Horseshoe crab genomes reveal the evolution of genes and microRNAs after three rounds of whole genome duplication». Commun. Biol. [en línia], 4 (1): 83. <https://doi.org/10.1038/ s42003-020-01637-2>.
Ontano, A. Z. [et al.] (2021a). «Taxonomic sampling and rare genomic changes overcome long-branch attraction in the phylogenetic placement of Pseudoscorpions». Mol. Biol. Evol. [en línia], 38 (6): 2446-2467. <https://doi.org/10.1093/molbev/msab038>. (2021 b). «How many long branch orders occur in Chelicerata? Opposing effects of Palpigradi and Opilioacariformes on phylogenetic stability». Mol. Phylogenet. Evol. [en línia], 168: 107378. <https://doi. org/10.1016/j.ympev.2021.107378>.
Pelosi, P. (1996). «Perireceptor events in olfaction». J. Neurobiol . [en línia], 30 (1): 3-19. < https://doi. org/10.1002/(sici)1097-4695(199605)30:1<3::Aid -neu2>3.0.Co;2-a>.
Saelao, P. [et al.] (2021). «Phylogenomics of tick inward rectifier potassium channels and their potential as targets to innovate control technologies». Front. Cell. Infect. Microbiol. [en línia], 11: 647020. <https://doi. org/10.3389/fcimb.2021.647020>.
Sánchez-Gracia, A. [et al.] (2009). «Molecular evolution of the major chemosensory gene families in insects». Heredity [en línia], 103 (3): 208-216. <https://doi.org/ 10.1038/hdy.2009.55>.
Sánchez-Herrero, J. F. [et al.] (2019). «The draft genome sequence of the spider Dysdera silvatica (Araneae, Dysderidae): A valuable resource for functional and evolutionary genomic studies in chelicerates». Gigascience [en línia], 8(8): 1-9. <https://doi.org/ 10.1093/gigascience/giz099>.
Sanggaard, K. W. [et al.] (2014). «Spider genomes provide insight into composition and evolution of venom and silk». Nat. Commun. [en línia], 5: 3765. <https:// doi.org/10.1038/ncomms4765>.
Santibáñez-López, C. [et al.] (2018). «Transcriptomic analysis of pseudoscorpion venom reveals a unique cocktail dominated by enzymes and protease inhibitors». Toxins [en línia], 10 (5): 207. <https://doi.org/ 10.3390/toxins10050207>. (2020). «Phylogenomics of scorpions reveal a co-diversification of scorpion mammalian predators and mammal-specific sodium channel toxins». BioRxiv [en línia]. <https://doi.org/10.1101/2020.11.06.372045>.
Scholtz, G.; Kamenz, C. (2006). «The book lungs of Scorpiones and Tetrapulmonata (Chelicerata, Arachnida): Evidence for homology and a single terrestrialisation event of a common arachnid ancestor». Zoology, 109 (1): 2-13.
Schwager, E. E. [et al.] (2017). «The house spider genome reveals an ancient whole-genome duplication during arachnid evolution». BMC Biol. [en línia], 15 (1): 62. <https://doi.org/10.1186/s12915-017-0399-x>.
Sharma, P. P. (2017). «Chelicerates and the conquest of land: A view of arachnid origins through an evo-devo spyglass». Integr. Comp. Biol. [en línia], 57 (3): 510522. <https://doi.org/10.1093/icb/icx078>.
Sharma, P. P. [et al.] (2014). «Phylogenomic interrogation of Arachnida reveals systemic conflicts in phylogenetic signal». Mol. Biol. Evol. [en línia], 31 (11): 29632984. <https://doi.org/10.1093/molbev/msu235>. (2015). «Phylogenomic resolution of scorpions reveals multilevel discordance with morphological phylogenetic signal». Proc. R. Soc. B. Biol. Sci. [en línia], 282 (1804): 20142953. <https://doi.org/10.1098/rspb.2014. 2953>. (2021). «What is an “arachnid”? Consensus, consilience, and confirmation bias in the phylogenetics of Chelicerata». Diversity [en línia], 13 (11). <https:// doi.org/10.3390/d13110568>.
Shingate, P. [et al.] (2020). «Chromosome‐level genome assembly of the coastal horseshoe crab (Tachypleus gigas)». Mol. Ecol. Resour. [en línia], 20 (6): 17481760. <https://doi.org/10.1111/1755-0998.13233>.
Simakov, O. [et al.] (2022). «Deeply conserved synteny and the evolution of metazoan chromosomes». Sci. Adv. [en línia], 8 (5): eabi5884. < https://doi.org/ 10.1126/sciadv.abi5884>.
Taylor, R. S. [et al.] (2021). «Seeing the whole picture: What molecular ecology is gaining from whole genomes». Mol. Ecol. [en línia], 30 (23): 5917-5922. <https://doi.org/10.1111/mec.16282>.
Vieira, F. G.; Rozas, J. (2011). «Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: Origin and evolutionary history of the chemosensory system». Genome Biol. Evol. [en línia], 3: 476-490. <https://doi.org/ 10.1093/gbe/evr033>.
Vizueta, J. [et al.] (2018). «Comparative genomics reveals thousands of novel chemosensory genes and massive changes in chemoreceptor repertories across Chelicerates». Genome Biol. Evol. [en línia], 10 (5): 12211236. <https://doi.org/10.1093/gbe/evy081>. (2020a). «Evolutionary history of major chemosensory gene families across Panarthropoda». Mol. Biol. Evol. [en línia], 37 (12): 3601-3615. <https://doi.org/ 10.1093/molbev/msaa197>.
(2020b). «bitacora: A comprehensive tool for the identification and annotation of gene families in genome assemblies». Mol. Ecol. Resour. [en línia], 20 (5): 14451452. <https://doi.org/10.1111/1755-0998.13202>.
Waloszek, D.; Dunlop, J. A. (2002). «A larval sea spider (Arthropoda: Pycnogonida) from the Upper Cambrian of Sweden, and the phylogenetic position of pycnogonids». Palaeontology, 45 (3): 421-446.
Wendruff, A. J. [et al.] (2020). «A Silurian ancestral scorpion with fossilised internal anatomy illustrating a pathway to arachnid terrestrialisation». Sci. Rep. [en línia], 10 (1): 14-16. <https://doi.org/10.1038/s41598 -019-56010-z>.
Woods, H. A. [et al.] (2017). «Respiratory gut peristalsis by sea spiders». Curr. Biol. [en línia], 27 (13): R638-R639. <https://doi.org/10.1016/j.cub.2017.05.062>.
Yu, N. [et al.] (2019). «Genome sequencing and neurotoxin diversity of a wandering spider Pardosa pseudoannulata (pond wolf spider)». BioRxiv [en línia]. <https://doi.org/10.1101/747147>.
Treballs de la Societat Catalana de Biologia, 72: 34-42
42
2
Protists, la principal font de diversitat genòmica en eucariotes
Resum
La genòmica, la determinació de la seqüència de DNA d’una espècie, avui dia està en expansió gràcies a noves eines de seqüenciació. Les iniciatives per a obtenir els genomes de totes les espècies vives sovint obliden els protists, eucariotes unicel·lulars cabdals per a entendre l’evolució de la vida i el funcionament dels ecosistemes. En aquest treball repassarem el que se sap de genòmica de protists, sovint a partir d’organismes cultivats, i presentarem com es pot obtenir el genoma d’espècies no cultivades que conformen una part enorme de la biodiversitat. Després explicarem exemples de com la genòmica de protists permet abordar qüestions com l’origen del llinatge eucariota, l’adquisició i expansió del cloroplast, la multicel·lularitat, els processos de simbiosi, l’estudi d’activitats biogeoquímiques a partir de gens funcionals o la genètica de poblacions. Aquesta contribució vol donar a conèixer l’extraordinària diversitat de protists que hi ha i promoure la consideració que mereixen en els estudis genòmics.
Paraules clau: biodiversitat, ecologia, evolució, genòmica, protists.
Introducció
Un dels grans misteris desxifrats al segle passat va ser el mecanisme de manteniment i transmissió de la informació que codifica els éssers vius. El dogma central de la biologia estableix que la informació està escrita en la seqüència (ordre) de les quatre bases que formen el DNA, i que les unitats d’aquesta informació (els gens) es tradueixen a proteïnes (fent servir RNA com a missatger), les quals formen les estructures cel·lulars i controlen les reaccions químiques de les rutes metabòliques. En aquest context, esbrinar la seqüència del DNA d’una espècie podrà donar respostes a la seva ecofisiologia i història evolutiva. En contrast amb la genètica, que estudia el paper de gens concrets, la genòmica pretén caracteritzar conjuntament tots els gens de l’espècie, així com les regions no codificants, la seva posició relativa i la seva estructura tridimensional. En procariotes, el DNA acostuma a trobar-se en una sola cadena circular, un sol cromosoma, mentre que en eucariotes hi sol haver un nombre concret de cromosomes lineals embolcallats dins del nucli que varia segons l’espècie.
DOI: 10.2436/20.1501.02.216
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 24/03/2022
Acceptat: 17/04/2022
Protists, the primary source of genomic diversity in eukaryotes
Abstract
Genomics, which unravels the DNA sequence of species, is an expanding discipline thanks to new sequencing tools. Initiatives aimed at obtaining the genomes of all living species often overlook protists, which are unicellular eukaryotes that are crucial for understanding the evolution of life and the functioning of ecosystems. In this paper we review what is known about protist genomics, often on the basis of cultured species, and present new ways to obtain the genomes of uncultured species, which represent a large part of eukaryotic diversity. We then present some examples of how protist ge nomics is crucial to address key issues in evolution and ecology, such as the origin of the eukaryotes, the acquisition and expansion of chloroplasts, the emergence of multicellularity, symbiotic events, the study of biogeochemical activities on the basis of functional genes, and population genetics. This paper seeks to highlight the vast diversity of protists and to underscore the fact that they should be given greater consideration in genomic studies.
Keywords: biodiversity, ecology, evolution, genomics, protists.
La genòmica tractaria, doncs, d’obtenir la seqüència completa del DNA de tots els cromosomes i la seva interpretació funcional.
Vist el gran potencial científic de la informació genòmica en pràcticament tots els camps de la biologia (evolució, ecologia, biomedicina, biotecnologia), hi ha hagut moltes iniciatives per a obtenir els genomes de les espècies de la biosfera. Aquesta informació serà fonamental per a esbrinar la història natural i els patrons evolutius de cada espècie, permetrà conèixer-ne les potencialitats genètiques i, en alguns casos, identificar mutacions en gens concrets responsables de disfuncions o malalties. Aquest esforç és encara més crític en la situació actual de canvi climàtic, que podria acabar derivant en la sisena extinció massiva degut a les activitats humanes. És en aquest context en què s’emmarca l’Earth BioGenome Project (EBP), començat el 2018 i que té l’ambició de seqüenciar, catalogar i caracteritzar el genoma de totes les espècies eucariotes existents en un termini de deu anys (Lewin et al., 2022). El projecte està estructurat en diverses iniciatives, i els autors d’aquest treball en són
Treballs de la Societat Catalana de Biologia, 72: 43-50
part d’una: la iniciativa catalana per a l’EBP (CBP, de l’anglès Catalan Initiative for the Earth BioGenome Project, https://www.bioge noma.cat). Tot i que l’EBP proposa caracteritzar exhaustivament la biodiversitat d’eucariotes, és evident que fa un èmfasi especial en animals, plantes i fongs, i que posa poca atenció en els eucariotes unicel·lulars, els protists. Enlloc no es diu que no siguin importants, però fàcilment s’obvien. Gran part de la comunitat implicada en aquests projectes genòmics passa per alt que els protists representen la major part de la diversitat eucariota i que són clau per a entendre algunes de les principals transicions evolutives, com l’origen de la fotosíntesi, que permetrà l’aparició de les plantes superiors, o l’origen de la multicel·lularitat, clau per a l’aparició d’animals i plantes. Així, aquests organismes microscòpics i estructuralment senzills queden desenfocats del lloc que els pertoca en l’evolució de la vida, el funcionament dels ecosistemes i la seva enorme contribució a la biodiversitat. Amb aquest article volem posar una mica de llum a la foscor, i explicar en quina situació es troben els eucariotes unicel·lulars.
43
Ramon Massana,1 Ramiro Logares,1 David López-Escardó1 i Javier del Campo2
1 Institut de Ciències del Mar (CSIC)
Institut de Biologia Evolutiva (CSIC - Universitat Pompeu Fabra)
Correspondència: Ramon Massana. Institut de Ciències del Mar (CSIC). Passeig Marítim de la Barceloneta, 37-49. 08003 Barcelona. Tel.: +34 932 309 500. Adreça electrònica: ramonm@icm.csic.es
Projectes genòmics, de virus a tota la biodiversitat
La capacitat d’obtenir dades genòmiques ha anat lligada al desenvolupament de mètodes per a seqüenciar el DNA. Amb l’aparició de la tècnica de Sanger, el 1977 es va obtenir el primer genoma, el del bacteriòfag φX174 (5,4 kb). Així es va iniciar la cursa per a dur a terme projectes cada cop més complexos i ambiciosos: el genoma del mitocondri humà el 1981 (16,6 kb), el de cloroplasts de plantes el 1986 (155,8 kb), un cromosoma de fong el 1992 (315 kb) i, finalment, els primers genomes d’un organisme, el bacteri Haemophilus influenzae (1,8 Mb) el 1995 i el llevat Saccharomyces cerevisiae el 1996 (12,1 Mb). Poc després, la comunitat científica es va plantejar seriosament el Projecte Genoma Humà, que va esdevenir una altra cursa que va acabar amb la publicació d’una primera versió incompleta el 2001, una versió acabada el 2003 i la versió final corregida (menys d’un error en 20 kb) el 2007.
Paral·lelament, es van anar triant espècies concretes que servien com a model per a representar grans grups d’organismes. Dins dels mamífers es van completar els genomes del gos (Canis familiaris), la rata (Rattus norvegicus), el ratolí (Mus musculus) i el ximpanzé (Pan troglodytes), tots utilitzats en recerca biomèdica. Respecte d’altres animals emprats com a organismes model, el primer genoma obtingut va ser el del nematode Caenorhabditis elegans , després el de la mosca del vinagre Drosophila melanogaster i, finalment, el genoma del peix zebra, Danio rerio. Com a model de planta amb flor, es va triar Arabidopsis thaliana, i de fong, el llevat Saccharomyces cerevisiae. Amb relació a protists, els genomes complets disponibles estan esbiaixats cap a espècies d’interès biomèdic, com l’agent causant de la malària, l’apicomplex Plasmodium falciparum, o el responsable de la leishmaniosi, el cinetoplàstid Leishmania major. L’objectiu dels projectes genòmics amb espècies model era determinar el nombre de cromosomes, seqüenciar-los de punta a punta, identificar-ne els gens i predir-ne la funció. Donat que cada individu de la mateixa població té genomes lleugerament diferents, la seqüència de referència per a l’espècie és la del component haploide d’un sol individu, i aquesta referència serveix per a cartografiar la variabilitat intrínseca dins la població.
Un salt crític en la història dels projectes genoma ha estat l’aparició de les tècniques de seqüenciació massiva (Reuter et al., 2015). El rendiment en lectures de DNA d’aquestes noves màquines ha anat evolucionant amb el temps, inicialment produint fragments curts (primer la
tecnologia 454 i avui dia Illumina), i més endavant, fragments llargs (tecnologies com PacBio o ONT). Aquest increment espectacular de rendiment ha anat acompanyat d’avenços igualment trepidants en el maquinari computacional, amb ordinadors cada cop més potents que permeten emmagatzemar i processar l’allau d’informació generada, i en les eines bioinformàtiques per a gestionar i analitzar les dades. La seqüenciació massiva s’ha aplicat ràpidament a moltes disciplines, també per a obtenir el genoma d’una gran varietat d’espècies. Tot i que sovint ha anat acompanyada d’una qualitat inferior de les seqüències i de menys ambició a l’hora de finalitzar els projectes genòmics, és innegable que la seqüenciació massiva ha promogut una explosió en el camp de la genòmica. A més, ha permès avançar en el coneixement dels microorganismes dominants als ecosistemes, molts dels quals encara no cultivats. És en aquest nou escenari de capacitat enorme de seqüenciació que s’emmarca l’Earth BioGenome Project.
Els protists, els grans oblidats
Diversitat dels protists i la seva importància ecològica L’obtenció de dades genòmiques d’un ampli espectre d’espècies de macro- i microorganismes, juntament amb la millora d’algorismes de reconstrucció filogenètica, han permès arribar a una visió realista de la biodiversitat d’eucariotes (Burki et al., 2020). L’arbre resultant mostra com els animals, els fongs i les plantes formen una petitíssima part dels llinatges existents (vegeu la figura 1) i evidencia la descompensació en el coneixement que tenim dels protists respecte a la tríada multicel·lular, que representa al vol-
tant del 95 % de les espècies descrites (Pawlowski et al., 2012). Aquest biaix té arrels històriques i metodològiques. Des d’abans dels treballs de Darwin, els protists eren un calaix de sastre on es posaven tots els eucariotes unicel·lulars, equiparats com a regne al mateix nivell que animals, fongs i plantes, i obviant que darrere aquesta simplicitat aparent hi havia diferents orígens evolutius i dissenys cel·lulars. La informació genòmica ha permès caracteritzar aquesta gran diversitat i fer-li justícia. A escala evolutiva, és clar que l’últim ancestre comú de tots els eucariotes va ser un organisme unicel·lular i que durant una gran part del temps geològic només hi havia protists, que anaven diferenciant-se i ocupant nous nínxols ecològics. Vista aquesta presència seminal, no és estrany que siguin molt importants en tots els ecosistemes i en els cicles biogeoquímics com a productors primaris, depredadors, paràsits o descomponedors. Conèixer, doncs, l’enorme diversitat de protists és fonamental per a entendre la diversitat eucariota i el funcionament de la biosfera.
L’arbre de la vida eucariota està estructurat en uns quants supergrups, cadascun dels quals inclou força llinatges rellevants i multitud d’espècies. Un dels supergrups més importants i diversos és el dels TSAR, format per telonèmids, estramenòpils, alveolats i rizaris (vegeu la figura 1). Tots ells contenen grups de flagellats heterotròfics que tenen impacte en els ecosistemes com a depredadors de bacteris i eucariotes microbians. A més, també s’hi troben llinatges fotosintètics amb una gran rellevància ecològica, com les diatomees dins dels estramenòpils i els dinoflagel·lats dins dels alveolats, o importants paràsits, com els apicomplexos. Un segon supergrup és el dels Amorphea, que in-
Figura 1. Esquema de l’arbre de la vida eucariota en forma de supergrups i els seus principals components. L’arbre mostra com els animals i fongs (dintre d’Opisthokonta) i les plantes (dintre de Chloroplastida) representen una petita fracció del disseny evolutiu dels eucariotes. Elaboració pròpia a partir de Burki et al. (2020).
Treballs de la Societat Catalana de Biologia, 72: 43-50
44
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
clou els opistoconts, amb animals, fongs i els seus parents unicel·lulars, i els amebozous, que engloben la majoria d’amebes. Altres supergrups són els arqueplàstids, que contenen les plantes i les algues vermelles i verdes, els Cryptista i Haptista, que, al seu torn, inclouen algues unicel·lulars importants a escala ecològica i geològica, i els excavats, que contenen majoritàriament protists heterotròfics, molts dels quals paràsits. Finalment, hi ha una sèrie de llinatges orfes que tenen una posició filogenètica poc clara a l’arbre, generalment formats per poques espècies molt distants entre si.
Accés als genomes de protists i cobertura dins de l’arbre de la vida El coneixement de la gran biodiversitat d’eucariotes microbians, present en cultius de laboratori, en situacions de parasitisme o en estudis de biodiversitat ambiental, no ha anat acompanyat d’esforços genòmics equivalents. Això es reflecteix en les llistes de genomes disponibles, on estan clarament subrepresentats, i on hi ha un biaix claríssim cap a espècies d’interès econòmic o mèdic. Un bon lloc de referència amb genomes de bona qualitat és la plataforma ENSEMBL (www.ensembl.org), inicialment plantejada per a genomes de vertebrats, però que s’ha ampliat per a incloure tots els éssers vius. En aquest repositori, hi trobem 311 genomes d’espècies de vertebrats, 123 d’altres animals, 1.506 de fongs, 119 de plantes i 237 de protists. Dins d’aquests darrers, el 75 % són de patògens humans (100 genomes d’apicomplexos i 25 de cinetoplàstids) o de plantes (53 d’oomicets). Tot i així, ENSEMBL té també representants d’un ampli ventall de grups de protists, i recull aspectes cabdals de les mides de genoma, nombre de cromosomes i nombre de gens (vegeu la figura 2). Amb tot, és evident que la representació de la veritable biodiversitat d’eucariotes, sobretot els microbians, encara queda lluny.
En els darrers anys hi ha hagut iniciatives que han intentat omplir aquest buit de coneixement genòmic. Una de molt rellevant és el projecte MMETSP, The Marine Microbial Eukaryotic Transcriptome Sequencing Project (Keeling et al., 2014), que va fer una crida a la comunitat científica internacional per a aconseguir el transcriptoma de tants protists marins cultivats com fos possible. D’aquesta manera, es va afegir informació de més de tres-centes espècies d’un gran ventall de grups marins. Aquesta iniciativa no resolia, però, un dels grans reptes que va aparèixer en els estudis ambientals: la gran presència d’espècies no cultivades en les comunitats naturals. En els darrers anys s’han
Figura 2. Selecció de protists amb genomes seqüenciats. A dalt, taula amb un resum de les dades genòmiques disponibles al repositori ENSEMBL (elaboració pròpia). Cal destacar que per a alguns gèneres hi ha múltiples soques seqüenciades: Plasmodium spp., amb seixanta soques; Phytophtora spp., amb vint-i-tres, i Leishmania spp., amb quatre. A baix, microfotografies de les espècies anteriors, seguint l’ordre en què apareixen en la taula. Imatges descarregades sota llicència de domini públic o de CC de Wikimedia Commons: Dictyostelium discoideum (imatge extreta d’https://commons.wikimedia.org/wiki/File:Dictyostelium _discoideum_fb_2.jpg [autor: Tyler Larsen] i publicada sota una llicència Attribution-ShareAlike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/), Chlamydomonas reinhardtii (fragment d’una imatge extreta d’https://commons.wikimedia.org/wiki/File:Biohybrid_Chlamydomonas_reinhardtii _microswimmers_2.jpg [autors: Mukrime Birgul Akolpoglu, Nihal Olcay Dogan, Ugur Bozuyuk, Hakan Ceylan, Seda Kizilel i Metin Sitti; doi:10.1002/advs.202001256] i publicada sota una llicència Attribution-ShareAlike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/), Guillardia theta (fragment d’una imatge extreta d’https://commons.wikimedia.org/wiki/File:41598_2017_2668_Fig2d_HTML.jpg [autors: Ryo Onuma, Neha Mishra, Shin-ya Miyagishima; doi:10.1038/s41598-017-02668-2] i publicada sota una llicència Attribution-Share Alike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/), Leishmania major (fragment d’una imatge extreta d’https://commons.wikimedia.org/wiki/File:Leishmania _spp._-_promastigote.jpg [autor: Stefan Walkowski] i publicada sota una llicència Attribution-ShareAlike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/), Emiliania huxleyi (fragment d’una imatge extreta d’https://commons.wikimedia.org/wiki/File:Emiliania_huxleyi_coccosphere_and _coccolith.jpg [autors: Griet Neukermans i Georges Fournier (Dr. Jeremy Young, University College London, London, amb permís); doi: 10.3389/fmars.2018.00146] i publicada sota una llicència Attribution-ShareAlike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/), Monosiga_Brevicollis (imatge extreta d’https://commons.wikimedia.org/wiki/File:Monosiga_Brevicollis_Phase.jpg [autor: Stephen Fairclough] i publicada sota una llicència Attribution-ShareAlike 2.5 Generic (CC BY-SA 2.5), https:// creativecommons.org/licenses/by-sa/2.5/), Nannochloropsis gaditana (fragment d’una imatge extreta d’https://commons.wikimedia.org/wiki/File:CSIRO_ScienceImage_10697_Microalgae.jpg [autor: CSIRO, http:// www.scienceimage.csiro.au/pages/about/] i publicada sota una llicència Creative Commons Attribution 3.0 Unported (CC BY 3.0), https://creativecommons.org/licenses/by/3.0/), Phytophthora infestans (imatge extreta d’https://commons.wikimedia.org/wiki/File:Oospore_of_Phytophthora_infestans.jpg i publicada sota una llicència Attribution-ShareAlike 2.5 Generic (CC BY-SA 2.5), https://creativecommons.org/licenses/by-sa/2.5/).
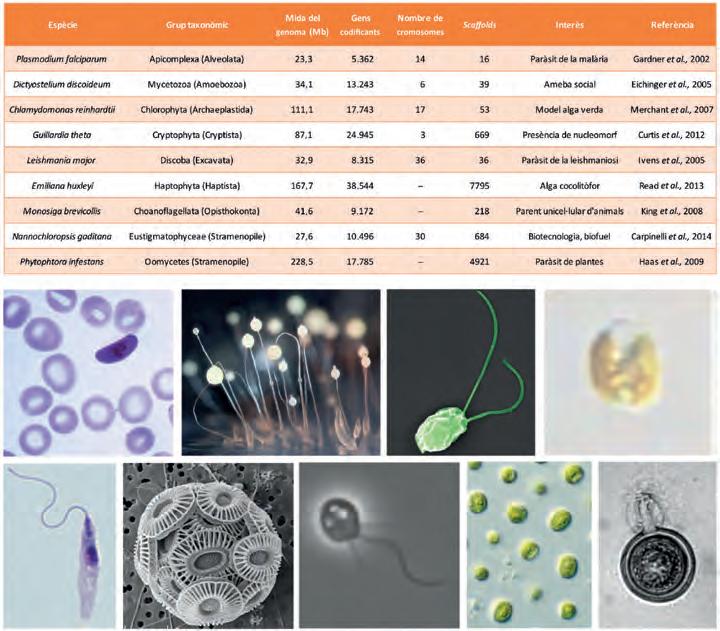
desenvolupat aproximacions per a aconseguir els genomes d’espècies no cultivades; potser la més rellevant és la basada en la separació de cèllules de la comunitat per citometria de flux, se-
Treballs de la Societat Catalana de Biologia, 72: 43-50
guida per l’amplificació i seqüenciació del DNA de cada cèl·lula (Labarre et al., 2021), que genera SAG (single amplified genomes). La genòmica de cèl·lules individuals permet, doncs, accedir a
45
Protists, la principal font de diversitat genòmica en eucariotes
Figura 3. Nombre d’estudis existents, separats en genomes, transcriptomes i -omes de cèl·lules individuals, en els grups principals de l’arbre de la vida eucariota. En els anells concèntrics s’indica també si el grup és fotosintètic o multicel·lular, amb color sòlid si aquesta capacitat és prevalent dins del grup, i en cercle obert si només ho és per a alguns dels membres. Elaboració pròpia a partir de dades del repositori EukProt (per tant, animals, plantes i fongs hi estan subrepresentats).
la diversitat natural i pot produir genomes quasi complets combinant diverses cèl·lules de la població (Latorre et al., 2021). El repositori genòmic de protists més complet el trobem avui a EukProt (www.github.com/beaplab/EukProt), que inclou genomes estàndard, transcriptomes i dades de cèl·lules individuals (vegeu la figura 3). Finalment, s’estan obtenint nous genomes a partir dels metagenomes de comunitats naturals, els anomenats MAG (metagenome assembled genomes), una aproximació d’èxit en procariotes i que comença a donar fruits en eucariotes, sobretot gràcies als esforços massius de seqüenciació, com els fets pel consorci TaraOceans (Delmont et al., 2022).
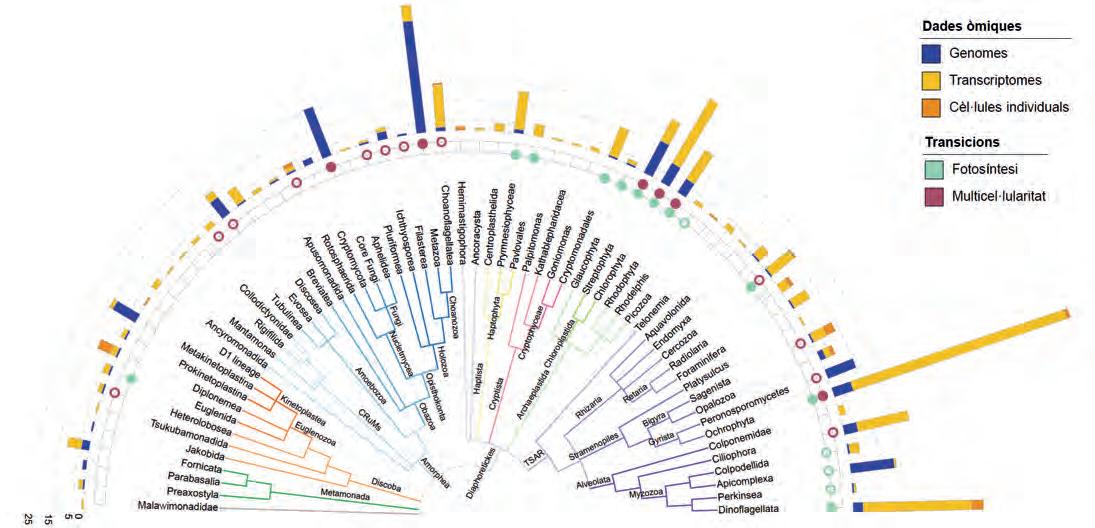
Genomes de protists en qüestions evolutives
Les dades genòmiques permeten aprofundir en el coneixement de la biologia dels protists i obren una finestra per a entendre les grans transicions evolutives que han donat forma a l’arbre de la vida eucariota. D’aquestes transicions, en destaquem tres per la gran transcendència que tenen: l’origen dels eucariotes, l’adquisició de la fotosíntesi i la multicel·lularitat.
L’origen de la cèl·lula eucariota
La cèl·lula eucariota és una quimera, fruit d’un procés d’endosimbiosi entre almenys un arqueu i un bacteri fa al voltant de dos milers de
milions d’anys. De manera molt resumida, l’arqueu va proporcionar la paret i el sistema de membranes que va permetre la generació del nucli, mentre que el bacteri va esdevenir el mitocondri. Val a dir, però, que hi ha un debat obert sobre si el primer eucariota tenia mitocondri, o bé si l’adquisició d’aquest mitocondri va ser posterior (Martin et al., 2017). Els primers estudis sobre l’origen de la cèl·lula eucariota es basaven en dades bioquímiques i d’ultraestructura cel·lular, i darrerament la genòmica els ha donat un gran impuls, ja que ha permès identificar les espècies procariotes properes a les que haurien participat en l’eucariogènesi. A partir de MAG de sistemes extrems, es va caracteritzar i posicionar a l’arbre de la vida l’arqueu més proper al que va participar en l’establiment de la cèl·lula eucariota, els asgardarqueus (Spang et al., 2015). Aquesta troballa també ha tingut un impacte directe en com entenem l’arbre de la vida, i s’ha proposat passar d’una estructura de tres dominis (bacteris, arqueus, eucariotes) a una altra de dos dominis (bacteris i arqueus), en la qual els eucariotes serien una branca dins dels arqueus i no pas un llinatge independent. Fent servir genòmica comparada, s’han trobat gens que prèviament es creien exclusius d’eucariotes, com gens del citoesquelet, en els asgardarqueus, la qual cosa suggereix que, a més de donar origen al nucli, també van donar lloc al citosquelet
eucariota. En relació amb l’altre component de l’eucariogènesi, les dades genòmiques han permès identificar les rickèttsies, que són alfaproteobacteris paràsits intracel·lulars, com el grup bacterià més proper al que va formar el mitocondri (Andersson et al ., 1998). A més d’aquests dos components, que estan prou ben documentats, hi ha anàlisis massives de genòmica comparada que suggereixen la participació en l’eucariogènesi d’altres procariotes (Pittis i Gabaldon, 2016), tot i que encara queda molta feina per a poder integrar aquesta informació, que tot sovint és desendreçada i complexa. Les tècniques genòmiques han permès reconstruir amb més detall l’eucariogènesi, desenvolupar noves hipòtesis i revolucionar completament com entenem l’arbre de la vida.
L’adquisició de la fotosíntesi
Segons el consens actual, el darrer ancestre comú eucariota era un organisme fagòtrof sense capacitat fotosintètica. Tanmateix, avui dia trobem aquesta capacitat a moltes branques de l’arbre dels eucariotes (vegeu la figura 3), cosa que duu a plantejar-se com va aparèixer i es va expandir aquesta capacitat (Keeling et al., 2013). El primer cop que la fotosíntesi va entrar dins el llinatge eucariota va ser a través de l’endosimbiosi amb un cianobacteri, bacteris fotosintètics que es troben a tots els ambients aquàtics. Gràcies a estudis de
46
Treballs de la Societat Catalana de Biologia, 72: 43-50
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
genòmica comparada i filogenòmica, s’ha identificat Gloeomargarita lithophora, aïllat fa pocs anys i relacionat amb Synechococcus, com el cianobacteri actual més proper al que va donar lloc al primer cloroplast (Ponce-Toledo et al., 2017). Aquesta endosimbiosi s’anomena primària i va originar els arqueplàstids, que inclouen glaucòfits, rodòfits i cloròfits, i dintre d’aquests darrers, les plantes. Posteriorment, els cloroplasts d’altres llinatges, com estramenòpils, alveolats, rizaris, euglènids, haptistes i criptistes, es van originar per endosimbiosis secundàries. Aquí estem parlant d’una relació entre dos eucariotes, on un organisme fagòtrof n’incorpora un de fotosintètic amb cloroplast primari. Les endosimbiosis primàries i secundàries es poden diferenciar pel nombre de membranes que embolcallen el cloroplast, dues o quatre, respectivament, o per genòmica comparada, ja que hi ha una transferència constant de gens entre els diferents genomes implicats en la simbiosi (Archibald, 2015). En l’endosimbiosi primària veiem senyals provinents del nucli de l’hoste i del cianobacteri inicial, mentre que en la secundària la imatge és més complicada, ja que inclou el senyal del nucli de l’hoste, i del nucli i el cloroplast del fotòtrof eucariota incorporat. Normalment en les endosimbiosis secundàries el nucli de la cèl·lula integrada desapareix amb el temps (només en uns pocs llinatges roman com a nucleomorf), però hi ha parts del seu genoma que s’integren dins del nucli de la cèl·lula hoste, cosa que permet esbrinar quins organismes hi han estat implicats. En la majoria de casos, l’alga implicada en l’endosimbiosi secundària és un rodòfit (alga vermella), mentre que en pocs casos, com els cloraracniòfits (dins els rizaris) i els euglenòfits (dins d’excavats), és un cloròfit (alga verda). L’endosimbiosi secundària no és, però, el darrer estadi, i en grups com els dinoflagel·lats hi ha força casos d’endosimbiosis terciàries, on el cloroplast deriva d’una alga amb endosimbiosi secundària com diatomees, haptòfits o criptomonadals. La major part de la fotosíntesi al planeta, i per tant bona part de la captura de CO2 atmosfèric, és duta a terme per eucariotes, tant plantes com protists fotosintètics. Sense aquest procés d’endosimbiosi que va incorporar la fototròfia dins el llinatge eucariota no podríem entendre el planeta ni els seus cicles biogeoquímics tal com avui els coneixem.
La multicel·lularitat
Després de la fotosíntesi, una de les transicions evolutives més significatives és l’origen de la multicel·lularitat. De multicel·lularitats n’hi ha
de molts tipus, i de fet no és exclusiva dels eucariotes; se’n troben els primers rastres en els estromatòlits, apareguts fa uns 3.500 milions d’anys, per agregació de cianobacteris filamentosos. En eucariotes, la multicel·lularitat ha aparegut independentment en almenys vint-i-cinc llinatges (Grosberg i Strathmann, 2007), en sis dels quals amb multicel·lularitat complexa, que dona lloc als organismes pluricel·lulars: animals, fongs, plantes i macroalgues verdes, vermelles i marrons. La resta pertanyen a esdeveniments de clonalitat reversible simple o multicel·lularitat agregativa. Un dels exemples paradigmàtics el trobem en l’ameba Dictyostelium discoideum, en què diferents cèl·lules s’ajunten per a formar un cos fructífer. Així doncs, mentre que la multicel·lularitat agregativa ha succeït força vegades al llarg de l’evolució dels eucariotes, la pluricel·lularitat és un fet més singular i relativament recent per als grups més emblemàtics (fa 600 milions d’anys en animals, 470 en plantes i 300 en fongs; per a algues vermelles, les primeres, fa 1.000 milions d’anys). La genòmica permet comparar les seqüències del DNA dels organismes pluricel·lulars i dels seus parents unicel·lulars propers, la qual cosa dona com a resultat una comprensió millor dels mecanismes de transició cap a la multicel·lularitat. Segurament el cas més significatiu és l’estudi de l’origen de la multicel·lularitat en animals. Des que es va publicar el genoma del coanoflagel·lat Monosiga brevicollis (King et al., 2008),
s’han publicat molts altres genomes o transcriptomes d’aquest grup unicel·lular, el més proper als animals, i també d’altres grups relacionats, com els filasteris, ictiosporis i pluriformeus (Ros-Rocher et al., 2021). A més de situar aquests nous llinatges a l’arbre de la vida, la informació genòmica ha revelat que alguns gens involucrats en funcions multicel·lulars i que es pensava que eren exclusius d’animals ja estaven presents al seu ancestre unicel·lular. Aquest ancestre, doncs, tindria una composició genètica relativament complexa i inclouria gens implicats en l’adhesió cel·lular (integrines), en la comunicació cel·lular (receptors tirosina-quinasa, receptors dels sistemes nerviós i immunitari), i factors transcripcionals per a regular la divisió cel·lular, el desenvolupament embrionari o la resposta immunitària (vegeu la figura 4). A més, en els quatre llinatges unicel·lulars parents dels animals hi ha espècies que presenten multicel·lularitat simple: el coanoflagel·lat Salpingoeca rosetta pot fer colònies, el filasteri Capsaspora owczarzaki presenta un estadi agregatiu en el cicle vital, i els ictiosporis i els pluriformeus poden formar cèl·lules multinucleades prèvies a la seva divisió. El pas següent ha estat, doncs, estudiar en detall els gens responsables d’aquests estadis multicel·lulars. Per exemple, les integrines, que són proteïnes d’unió a la matriu extracel·lular en els teixits animals, estan expressades en els agregats de C. owczarzaki i implicades en la seva adhesió al substrat (Parra-Acero
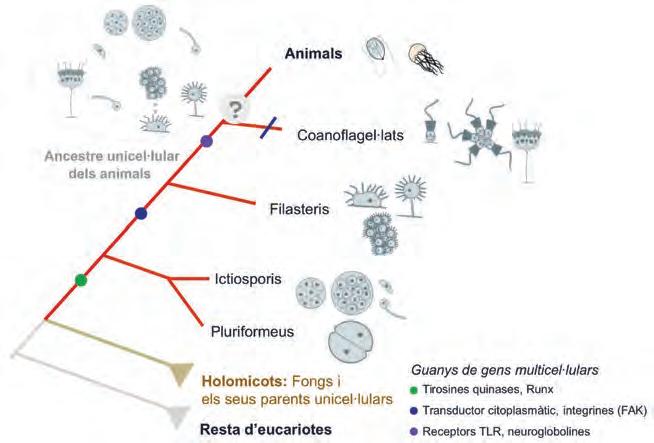
Figura 4. Representació filogenètica dels animals i els seus parents unicel·lulars que mostra les morfologies i els estadis de multicel·lularitat simple dels parents unicel·lulars, juntament amb el possible cicle de vida de l’ancestre unicel·lular dels animals. També s’hi inclouen exemples de gens relacionats amb funcions multicel·lulars adquirits en diferents moments evolutius previs als animals. Els guanys estan indicats amb punts de colors i les pèrdues secundàries, amb una ratlla (FAK = focal adhesion kinase [quinasa d’adhesió focal]; TLR = toll-like receptor [receptor de tipus Toll]). Elaboració pròpia.
Treballs de la Societat Catalana de Biologia, 72: 43-50
47
Protists, la principal font de diversitat genòmica en eucariotes
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
et al., 2020). Conèixer els genomes d’aquestes espècies ha donat peu a una sèrie d’estudis funcionals que han portat a una visió única de l’ancestre unicel·lular dels animals: una espècie amb un cicle de vida complex, amb regulació temporal de l’expressió gènica i que hauria evolucionat cap a integrar els estadis temporals dintre d’un estadi multicel·lular amb regulació espacial dins el propi organisme (Ros-Rocher et al., 2021). Les transformacions genòmiques clau que van acompanyar aquesta transició foren l’adquisició de nous gens formats a còpia de reordenar dominis proteics existents (López-Escardó et al., 2019), la duplicació de factors de transcripció i senyalització cel·lular, i el guany d’introns (Grau-Bové et al., 2017).
Genomes de protists en qüestions ecològiques
Les dades genòmiques també permeten aprofundir en el coneixement de l’ecofisiologia de les diferents espècies i expliquen fenòmens de diversificació i de la seva participació en els cicles biogeoquímics. A continuació en presentem alguns casos particularment interessants.
Les simbiosis
La simbiosi, individus de diferents espècies que estableixen una relació que els duu a viure junts, és molt freqüent a la natura. L’espectre de relacions simbiòtiques va des del parasitisme, on un dels dos membres de la relació es veu perjudicat, fins al mutualisme, on els dos membres en treuen un benefici. Els protists han establert relacions simbiòtiques entre ells, amb bacteris, i amb plantes i animals, i han participat en tot l’espectre de simbiosis. La relació que ha cridat més l’atenció ha estat el parasitisme, que no deixa de ser un cas extrem de simbiosi, i gran part de la informació que tenim de protists prové de l’interès històric en espècies paràsites d’humans i de bestiar. L’especificitat del parasitisme feia pensar que hi havia d’haver una empremta en el genoma del paràsit, i trobem, doncs, que la majoria de genomes de protists pertanyen a paràsits, tant d’humans (com Plasmodium, Toxoplasma, Cryptosporidium o Leishmania) com de plantes (com Phythophtora). Tanmateix, tot i que si ens basem en la literatura disponible podria semblar que el parasitisme és la forma dominant de simbiosi, l’exploració incipient de l’eucarioma d’animals, la fracció eucariota del microbioma, suggereix que la immensa majoria de relacions no són més que comensals (J. del Campo, J. et al., 2019); és a dir, que els hostes on viuen ni es veuen beneficiats ni perjudicats pels simbionts. Hi ha, doncs, un gran ventall de rela-
cions simbiòtiques que involucren protists que esperen ser estudiades.
Un aspecte rellevant és que la simbiosi no és un estadi fix. Per dir-ho d’una manera entenedora, un protist no és paràsit o mutualista, més aviat està paràsit o mutualista en el moment en què fem l’observació. A més, la posició dins de l’espectre de la simbiosi depèn de l’estat fisiològic de l’hoste i del simbiont i de les condicions ambientals, i un organisme pot tenir més d’un paper al llarg del seu cicle vital. Un cas paradigmàtic és el de l’alga verda Ostreobium queckettii, que viu a l’esquelet dels coralls formadors d’esculls (J. del Campo et al., 2017). Durant la major part de la seva vida, O. queckettii és comensal, però, com a resultat de canvis fisiològics en el corall, pot passar a actuar com a mutualista i en qüestió de setmanes esdevenir un paràsit. Aquesta gradació d’estats queda també reflectida a escala genòmica. S’havia assumit que el parasitisme comportava una sèrie d’innovacions a escala genòmica que permetien la transició de la vida lliure al parasitisme. En comparar, però, els genomes de protists paràsits amb els d’espècies més properes de vida lliure s’ha vist que la major part de mecanismes associats al parasitisme ja estan presents en les formes de vida lliure (Janouskovec i Keeling, 2016). Pel que fa al mutualisme, en sabem molt menys, degut sobretot a la manca d’informació genòmica, però el que s’intueix és que la situació és similar a la dels paràsits.
Diversitat funcional i adaptacions ecològiques
És fascinant pensar com la informació continguda en el DNA és cabdal per a entendre com és i com es comporta un organisme. Així, una de les tasques òbvies ha estat buscar gens de rellevància ecològica que expliquin la capacitat d’adaptació i funcionament de l’espècie. Aquesta cerca és molt profitosa en procariotes, en els quals es poden traçar papers biogeoquímics dins dels cicles del nitrogen, fòsfor o sofre a partir de gens funcionals (Ferrera et al., 2015).
Tanmateix, fer el mateix a partir de genomes eucariotes no és trivial, ja que moltes funcions eucariotes depenen de la mida, de l’estructura i del comportament cel·lular, propietats que involucren molts gens sovint també implicats en altres funcions. Potser l’excepció en seria la fotosíntesi, basada en complexos proteics especialitzats en aquesta funció. Una primera aproximació en eucariotes ha estat fer genòmica comparada entre espècies amb modes tròfics coneguts com fototròfia i fagotròfia i des-
Treballs de la Societat Catalana de Biologia, 72: 43-50
Figura 5. Predicció del mode tròfic d’espècies no cultivades MAST utilitzant genòmica comparada. Cada espècie és representada per un punt, que se situa dins d’un espai bidimensional definit per models de predicció fotòtrofa (a dalt) i fagòtrofa (a baix). La zona verda del panell superior posiciona les espècies fotòtrofes (els MAST no hi són) i la zona carbassa del panell inferior inclou les fagòtrofes (on se situen la majoria de MAST). Figura modificada de Labarre et al. (2021) i publicada sota una llicència AttributionShareAlike 4.0 International (CC BY-SA 4.0), https:// creativecommons.org/licenses/by-sa/4.0/.
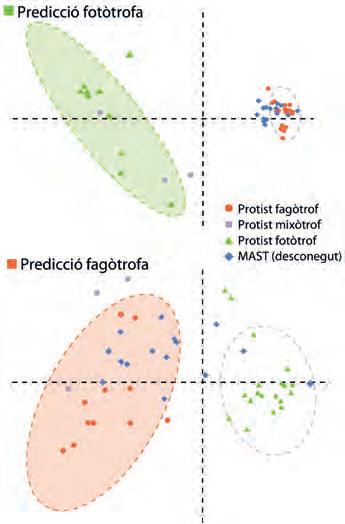
envolupar un model estadístic per a aplicar a organismes desconeguts (Burns et al., 2018). Certament, avui dia podem tenir el genoma d’una espècie de la qual ni tan sols coneixem el mode tròfic a partir d’una única cèl·lula (SAG) o de la comunitat (MAG). Aquest model prediu que diverses espècies marines no cultivades dels grups MAST (marine stramenopiles) (Labarre et al., 2021) són fagòtrofes i no fotòtrofes (vegeu la figura 5), fet ja observat empíricament en algunes espècies, però no en totes.
També s’han fet estudis per a esbrinar la relació entre famílies gèniques i un mode tròfic concret, com seria el cas de les peptidases i la fagotròfia sobre bacteris. Les peptidases són enzims importants durant la digestió de les preses ingerides, ja que més de la meitat del pes sec dels bacteris és proteïna, però també es troben en espècies no fagòtrofes que les fan servir en el reciclatge de les seves proteïnes. Es podria pensar que les espècies fagòtrofes tindrien molta més varietat i un nombre més ele-
48
vat de gens de peptidases, però l’anàlisi de genomes d’estramenòpils no va detectar una diferència clara entre espècies fotòtrofes, fagòtrofes i osmòtrofes (Labarre et al., 2021). Per tant, la simple presència d’aquests gens no és suficient per a explicar la fagotròfia. El que sí que ha resultat prometedor ha estat l’estudi de la seva expressió en condicions fisiològiques conegudes. Així, s’ha vist que els gens de peptidases estan més expressats en la fase exponencial d’un bacterívor quan creix digerint bacteris, que en la fase estacionària (Massana et al., 2021). També s’han fet estudis amb organismes mixòtrofs per a avaluar canvis en la seva expressió en funció de la llum i de la disponibilitat de nutrients (Kang et al., 2021). A més, disposar de genomes d’espècies dominants a la natura permet estudiar-ne l’activitat a partir de metatranscriptomes, l’expressió gènica de la comunitat. Per exemple, s’ha vist que un altre grup d’enzims digestius, les glucosidases, presenten diferències rellevants en la seva expressió a l’oceà global entre espècies properes de protists no cultivats (Latorre et al., 2021).
La genòmica també pot servir per a establir noves hipòtesis a partir de la troballa de gens no esperats. Un exemple interessant n’és el gen de la rodopsina, que codifica una proteïna descrita
inicialment a la retina dels mamífers, i que va agafar protagonisme a l’ecologia microbiana en trobar-se dins d’un bacteri marí abundant i no cultivat. Es va veure que la rodopsina formava part d’un sistema pigmentari que funcionava com a bomba de protons fent servir la llum solar, cosa que generava ATP independentment de la respiració. Des del seu descobriment, la presència de rodopsina no ha parat de créixer entre llinatges de bacteris, d’arqueus i també de protists (Pinhassi et al., 2016). Recentment s’ha trobat també en els genomes de bacterívors marins no cultivats (Labarre et al., 2021). De fet, es pensava que aquestes espècies vivien indiferents a la llum, però aquesta troballa podria qüestionar-ho i explicar la seva tendència a viure a les capes il·luminades dels oceans. Tenim, doncs, un cas interessant per a investigar i desenvolupar, aparegut a partir de l’observació i anàlisi dels nous genomes.
Els genomes de la comunitat microbiana
Seqüenciar els genomes del conjunt de microorganismes de la comunitat, el metagenoma, va ser un repte per als pioners en ecologia microbiana i cada cop és més factible gràcies als avenços en seqüenciació. La metagenòmica
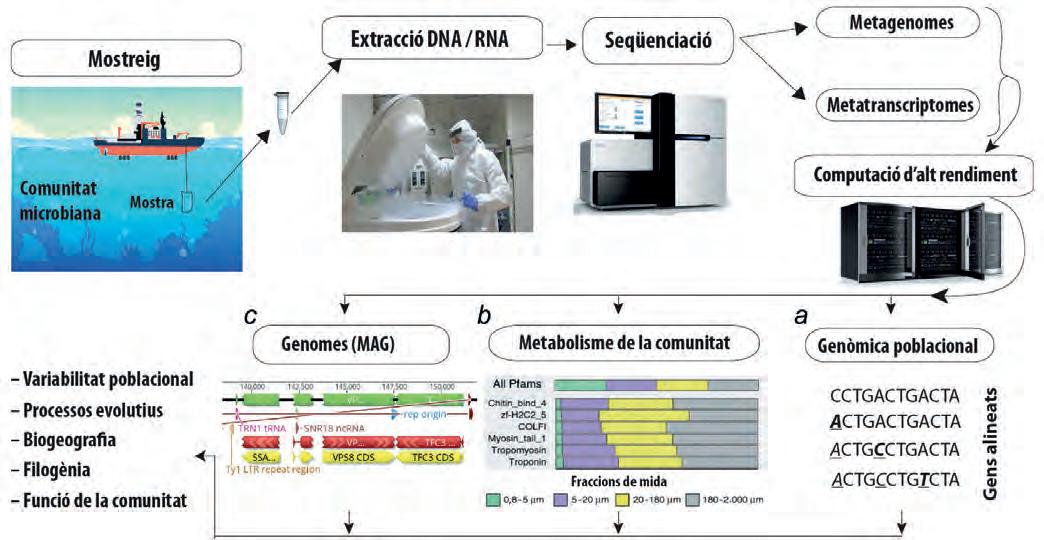
de protists ha avançat més lentament que la de procariotes, perquè tenen uns genomes més grans i complexos, però avui dia està esdevenint un camp molt prometedor. Així, pot ajudar a entendre la distribució geogràfica i la variabilitat poblacional, els processos evolutius a diverses escales temporals i el potencial metabòlic de les comunitats (vegeu la figura 6). També, com hem dit anteriorment, els metagenomes permeten reconstruir genomes d’espècies no cultivades, els MAG. L’aplicació simultània de la metagenòmica, la metatranscriptòmica i la genòmica de cèl·lules individuals ofereix un ventall enorme de possibilitats per a respondre preguntes ecològiques, de manera que esdevé una caixa d’eines molecular de gran utilitat.
La genòmica poblacional investiga la variabilitat genètica de les poblacions microbianes, les adaptacions de les poblacions a nínxols ecològics i la diversificació intraespecífica que pot derivar en especiació (Logares, 2011). A més, permet relacionar la variabilitat genètica amb el potencial d’adaptació de cada espècie als canvis ambientals. Cal destacar que, fins fa pocs anys, identificar poblacions de protists naturals era una tasca laboriosa i costosa, i que ara s’ha tornat accessible gràcies a la seqüenciació massiva. El mapatge de metagenomes a
Figura 6. Flux de treball en genòmica ambiental de protists marins. La comunitat de protists es mostreja de l’ambient, es fa una extracció del DNA o RNA ambiental i se seqüencia per a produir metagenomes i metatranscriptomes. Aquests són analitzats bioinformàticament en ordinadors d’alt rendiment amb l’objectiu d’estudiar la genòmica poblacional, el metabolisme de la comunitat o d’obtenir nous genomes. En el panell a cada seqüència correspon a un gen alineat de diferents genomes de la mateixa espècie, que indica els SNP (elaboració pròpia); el panell b mostra l’expressió relativa de diversos dominis Pfam en fraccions de mida del plàncton eucariota marí (fragment d’imatge de Carradec et al., 2018, publicada sota una llicència Attribution-ShareAlike 4.0 International (CC BY-SA 4.0), https://creativecommons.org/licenses/by-sa/4.0/); el panell c mostra una regió anotada d’un genoma eucariota (elaboració pròpia).
Treballs de la Societat Catalana de Biologia, 72: 43-50
49
Protists, la principal font de diversitat genòmica en eucariotes
genomes de referència permet detectar el polimorfisme de nucleòtids simples (SNP, single nucleotide polimorphism) que representen les variants genòmiques de la mostra analitzada. La detecció i quantificació dels SNP permet determinar poblacions i el grau de diferenciació genètica (Sjöqvist et al., 2021). Les anàlisis d’SNP possibiliten, a més, detectar processos selectius. La proporció de substitucions no sinònimes (dN) enfront de les sinònimes (dS) per a gens de la mateixa espècie pot servir per a detectar selecció positiva (dN/dS > 1) o selecció estabilitzadora (dN/dS < 1). Així, els gens que han experimentat una alta selecció positiva serien els que explicarien les millores adaptatives. En aplicar aquesta aproximació metagenòmica a sèries temporals, hi pot haver canvis en la freqüència dels SNP, cosa que suggereix processos evolutius en directe.
La metagenòmica i la metatranscriptòmica poden ajudar a comprendre el metabolisme de les comunitats de protists. Per exemple, un estudi de l’oceà global fet pel consorci TaraOceans va produir prop de 400 metatranscriptomes de les zones epi- i mesopelàgiques, cosa
Bibliografia
Andersson, S. G. E. [et al.] (1998). «The genome sequence of Rickettsia prowazekii and the origin of mitochondria». Nature, 396: 133-140.
Archibald, J. M. (2015). «Genomic perspectives on the birth and spread of plastids». Proc. Natl. Acad. Sci. USA, 112: 10147-10153.
Burki, F. [et al.] (2020). «The new tree of eukaryotes». Trends Ecol. Evol., 35: 43-55.
Burns, J. A. [et al.] (2018). «Gene-based predictive models of trophic modes suggest Asgard archae are not phagocytic». Nature Ecol. Evol., 2: 697-704.
Carpinelli, E. C. [et al.] (2014). «Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion». Mol. Plant, 7: 325-335.
Carradec, Q. [et al.] (2018). «A global ocean atlas of eukaryotic genes». Nat. Commun., 9: 373.
Colot, V.; Rossignol , J. L. (1999). «Eukaryotic DNA methylation as an evolutionary device». BioEssays, 21: 402- 411.
Curtis, B. A. [et al.] (2012). «Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs». Nature, 492: 59-65.
Del Campo, J. [et al.] (2017). «The “other” coral symbiont: Ostreobium diversity and distribution». ISME J., 11: 296-299.
(2019). «The eukaryome: Diversity and role of microeukaryotic organisms associated with animal hosts». Funct. Ecol., 34: 2045-2054.
Delmont, T. O. [et al.] (2022). «Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean». Cell Genom., 2: 100123.
Eichinger, L. [et al.] (2005). «The genome of the social amoeba Dictyostelium discoideum». Nature, 435: 4357.
Ferrera, I. [et al.] (2015). «Prokaryotic functional gene diversity in the sunlit ocean: Stumbling in the dark». Curr. Opin. Microbiol., 25: 33-39.
Gardner, M. J. [et al.] (2002). «Genome sequence of the human malaria parasite Plasmodium falciparum». Nature, 419: 498-511.
que va generar un catàleg de ~116 milions de gens no redundants (Carradec et al ., 2018). Aquest catàleg va permetre tenir una visió global dels metabolismes eucariotes, sobretot unicel·lulars, actius a l’oceà, a més de constituir un recurs de gran valor per a estudiar l’expressió d’espècies determinades. La reconstrucció de genomes de protists a partir de metagenomes (MAG) continua sent un desafiament, tot i que hi ha dades recents que utilitzen mostres enriquides en protists i seqüenciacions massives que estan donant bons resultats (Delmont et al., 2022). Probablement aquests MAG milloraran quan s’analitzin metagenomes obtinguts a partir de lectures llargues (PacBio, ONT). Aquesta seqüenciació també pot servir per a analitzar els canvis epigenètics en els protists associats a l’aclimatació, per exemple, com la metilació en algunes bases pot afectar l’expressió gènica (Colot i Rossignol, 1999). En aquest sentit, diferenciar els canvis epigenètics dels canvis evolutius és important a l’hora d’entendre els mecanismes emprats pels protists per a adaptar-se als canvis ambientals.
Cloenda
La vida inclou una multitud de formes i llinatges evolutius diferents, i per la seva visibilitat, el seu impacte i la seva proximitat, els animals, les plantes i els fongs han captivat l’atenció de científics i naturalistes. Tanmateix, aquesta diversitat eucariota ha evolucionat a partir d’un substrat previ extremament divers d’eucariotes unicel·lulars, i han seguit coevolucionant plegats. Els protists, doncs, representen baules fonamentals en el coneixement biològic, i mereixen igualment ser inclosos en la revolució genòmica actual. En aquesta contribució hem presentat, a grans trets, què sabem sobre genòmica de protists i el que es pot saber a curt termini gràcies a les noves tècniques disponibles, com la genòmica de cèl·lules individuals i la metagenòmica. També expliquem alguns exemples en què els genomes dels protists han sigut clau per a abordar diferents qüestions biològiques fonamentals i avançar en el seu estudi. Esperem, doncs, que aquest treball contribueixi a posar de nou els protists sota el focus i que tinguin la consideració que es mereixen.
Grau-Bové, X. [et al.] (2017). «Dynamics of genomic innovation in the unicellular ancestry of animals». ELife, 6: e26036.
Grosberg, R. K.; Strathmann, R. R. (2007). «The evolution of multicellularity: A minor major transition?». Annu. Rev. Ecol. Evol. Syst., 38: 621-654.
Haas, B. J. [et al.] (2009). «Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans». Nature, 461: 393-398.
Ivens, A. C. [et al.] (2005). «The genome of the kinetoplastid parasite, Leishmania major». Science, 309: 436-442.
Janouskovec, J.; Keeling, P. J. (2016). «Evolution: Causality and the origin of parasitism». Curr. Biol., 26: R174-177.
Kang, Y. [et al.] (2021). «Transcriptomic responses of four pelagophytes to nutrient (N, P) and light stress». Front. Mar. Sci., 8: 636699.
Keeling, P. J. [et al.] (2013). «The number, speed, and impact of plastid endosymbioses in eukaryotic evolution». Annu. Rev. Plant Biol., 64: 583-607. (2014). «The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing». PLoS Biol., 12: e1001889.
King, N. [et al.] (2008). «The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans». Nature, 451: 783-788.
Labarre, A. [et al.] (2021). «Comparative genomics reveals new functional insights in uncultured MAST species». ISME J., 15: 1767-1781.
Latorre, F. [et al.] (2021). «Niche adaptation promoted the evolutionary diversification of tiny ocean predators». Proc. Natl. Acad. Sci. USA, 118: e2020955118.
Lewin, H. A. [et al.] (2022). «The Earth BioGenome Project 2020: Starting the clock». Proc. Natl. Acad. Sci. USA, 119: e2115635118.
Logares, R. (2011). «Population genetics: the next stop for microbial ecologists?». Open Life Sci., 6: 887-892.
López-Escardó, D. [et al.] (2019). «Reconstruction of protein domain evolution using single-cell amplified genomes of uncultured choanoflagellates sheds light
Treballs de la Societat Catalana de Biologia, 72: 43-50
on the origin of animals». Phil. Trans. R. Soc. B., 374: 20190088.
Martin, W. F. [et al.] (2017). «The physiology of phagocytosis in the context of mitochondrial origin». Microb. Molec. Biol. Rev., 81: e00008-17.
Massana, R. [et al.] (2021). «Gene expression during bacterivorous growth of a widespread marine heterotrophic flagellate». ISME J., 15: 154-167.
Merchant, S. S. [et al.] (2007). «The Chlamydomonas genome reveals the evolution of key animal and plant functions». Science, 318: 245-251.
Parra-Acero, H. [et al.] (2020). «Integrin-mediated adhesion in the unicellular holozoan Capsaspora owczarzaki». Curr. Biol., 30: 4270-4275.
Pawlowski, J. [et al.] (2012). «CBOL Protist Working Group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms». PLoS Biol ., 10: e1001419.
Pinhassi, J. [et al.] (2016). «Marine bacterial and archaeal ion-pumping rhodopsins: Genetic diversity, physiology, and ecology». Microbiol. Mol. Biol. Rev., 80: 929954.
Pittis, A. A.; Gabaldón, T. (2016). «Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry». Nature, 531: 101-104.
Ponce-Toledo, R. I. [et al.] (2017). «An early-branching freshwater cyanobacterium at the origin of plastids». Curr. Biol., 27: 386-391.
Read, R. A. [et al.] (2013). «Pan genome of the phytoplankton Emiliania underpins its global distribution». Nature, 499: 209-213.
Reuter, J. A. [et al.] (2015). «High-throughput sequencing technologies». Mol. Cell., 58: 586-597.
Ros-Rocher, N. [et al.] (2021). «The origin of animals: An ancestral reconstruction of the unicellular-to-multicellular transition». Open Biol., 11: 200359.
Sjöqvist, C. [et al.] (2021). «Ecologically coherent population structure of uncultivated bacterioplankton». ISME J., 15: 3034-3049.
Spang, A. [et al.] (2015). «Complex archaea that bridge the gap between prokaryotes and eukaryotes». Nature, 521: 173-179.
50
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
1
Variabilitat i seqüenciació massiva de virus.
El SARS-CoV-2 com a exemple
Cristina Andrés,1 David Tabernero,2, 3 Tomás Pumarola,1, 5 Andrés Antón1, 5 i Josep Quer2, 4, 6
2 Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Instituto de Salud Carlos III
3 Unitat de Patologia Hepàtica, Departaments de Bioquímica i Microbiologia, Vall d’Hebron Barcelona Hospital Campus
4 Malalties Hepàtiques - Hepatitis Virals, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Barcelona Hospital Campus
5 Departament de Microbiologia, Universitat Autònoma de Barcelona (UAB)
6 Departament de Bioquímica i Biologia Molecular, Universitat Autònoma
Correspondència:
Josep
Resum
Una de les coses que hem après en els darrers dos anys, concretament amb la pandèmia del SARS-CoV-2, és que no podem emmurallar els virus per a evitar-ne la propagació, i la manera més eficaç de lluitar-hi en contra és amb una detecció i una caracterització genètica ràpides. Les pandèmies causades per virus han ocorregut, n’estem patint una i tard o d’hora en tornarem a patir. En els darrers anys, el risc de noves pandèmies s’ha incrementat degut a factors que les afavoreixen, com ara la destrucció d’espais naturals, la facilitat per a fer viatges de llarga distància en qüestió d’hores, el canvi climàtic i l’augment de concentracions humanes i activitats socials en un món cada vegada més globalitzat. En aquest sentit, la seqüenciació massiva o seqüenciació de nova generació (NGS, next generation sequencing) ha demostrat ser una eina molt poderosa que ha ajudat en el diagnòstic, en el desenvolupament de vacunes, en la identificació i el seguiment de variants i a conèixer millor la biologia del virus. La pandèmia del SARS-CoV-2 ha permès demostrar la utilitat de l’NGS i la necessitat de reforçar la xarxa de laboratoris de seqüenciació. Com més preparats estiguem, més ràpida i eficaç serà la resposta contra una possible amenaça.
Paraules clau: variabilitat, virus, seqüenciació massiva, NGS, diagnòstic.
Abstract
DOI: 10.2436/20.1501.02.217
ISSN (ed. impresa): 0212-3037
ISSN (ed. digital): 2013-9802
http://revistes.iec.cat/index.php/TSCB
Rebut: 01/02/2022
Acceptat: 14/03/2022
One of the things that we have learned in the last two years, specifically from the SARS-CoV-2 pandemic, is that we cannot wall in viruses to prevent their propagation, and that the most effective way to combat them is to ensure their rapid detection and genetic characterisation. Pandemics caused by viruses have broken out in the past, we are currently experiencing such a pandemic, and sooner or later we will surely be struck by another one. The risk of new pandemics has grown in recent years, due to factors favouring their appearance such as the destruction of natural areas, the ease of long-distance travel in a matter of hours, climate change, and the increase of human concentrations and social activities in a constantly more globalised world. In this respect, massive or next-generation sequencing (NGS) has proven to be a very powerful tool that has been of help in diagnosis and in the development of vaccines, the identification and monitoring of variants, and the study of the biology of the virus. The SARS-CoV-2 pandemic has allowed to demonstrate the utility of NGS as well as the need to strengthen the network of sequencing laboratories. The better we are prepared, the quicker and more effective will be our response to a possible threat.
Keywords: variability, virus, massive sequencing, NGS, diagnosis.
Introducció
Les pandèmies causades per virus han ocorregut, n’estem patint ara mateix i tard o d’hora en tornarem a patir. La raó principal la trobem en la gran capacitat que tenen els virus de generar variabilitat i seleccionar mutacions que, per exemple, els permeten saltar d’una espècie a una altra i causar una zoonosi (transmissió d’un animal a un humà). Els virus amb més capacitat de variar són els que tenen el genoma en forma d’RNA, com els virus de les hepatitis A, C i E; el virus del Zika (ZIKV); el virus del chikungunya; el virus de la immunodeficiència humana (HIV); el virus de Crimea-Congo; el virus de l’Ebola; el virus de Marburg (EVM); el virus de la influença; els virus entè-
rics; el virus de la febre groga; el virus del xarampió; el virus de les galteres o els enterovirus, i els coronavirus (CoV) estacionals (NL63, OC43, HKU1 o 229E), així com el SARS-CoV, el MERS-CoV i el SARS-CoV-2. Dins el grup de virus altament variables s’inclouen els que durant el cicle de replicació passen per una fase d’RNA, com el virus de l’hepatitis B (Domingo, 2020).
qüestió d’hores; el canvi climàtic, que ha facilitat la distribució de vectors virals on abans no estaven presents, i, a més a més, l’augment de concentracions humanes i activitats socials en un món cada vegada més globalitzat (Wearn et al., 2012; Gibb et al., 2020; Tollefson, 2020).
Una de les coses que hem après en els darrers dos anys, concretament amb la pandèmia del SARS-CoV-2, és que no podem emmurallar els virus per a evitar-ne la propagació, i que la manera més eficaç de lluitar-hi en contra és una detecció i una caracterització genètica ràpides. En aquest sentit, la seqüenciació massiva o seqüenciació de nova generació (NGS, next generation sequencing) ha demostrat ser Treballs de la Societat Catalana de Biologia,
En els darrers anys, el risc de noves pandèmies s’ha incrementat degut a factors que les afavoreixen, com la destrucció d’espais naturals, que ha facilitat el contacte directe amb animals salvatges, i que moltes vegades són reservoris de virus fins ara desconeguts; la facilitat per a fer viatges de llarga distància en
Variability and massive sequencing of viruses. SARS-CoV-2 as an example
Departament de Microbiologia, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Barcelona Hospital Campus
de Barcelona (UAB)
Andrés Antón. Departament de Microbiologia, Vall d’Hebron Institut de Recerca, Vall d’Hebron Barcelona Hospital Campus. Passeig de la Vall d’Hebron, 119-129. 08035 Barcelona. Adreça electrònica: aanton@ vhebron.net
Quer. Malalties Hepàtiques. Vall d’Hebron Institut de Recerca, Vall d’Hebron Barcelona Hospital Campus. Passeig de la Vall d’Hebron, 119-129. 08035 Barcelona. Tel.: +34 934 894 034. Adreça electrònica: josep.quer@vhir.org
51
72: 51-59
una eina molt poderosa, ja que ha permès desvelar en temps rècord la seqüència del genoma viral. A partir de les primeres seqüències, s’han pogut desenvolupar assajos moleculars basats en amplificació d’àcids nucleics (per exemple, per al disseny dels encebadors i sondes) per a la detecció del virus; caracteritzar la regió més immunògena (l’espícula o spike) per a desenvolupar vacunes basades en mRNA o de DNA (Amanat i Krammer, 2020; Ruiz-Rodriguez et al., 2021; Wouters et al., 2021); identificar i fer el seguiment de les noves variants identificant les mutacions associades, i estudiar-ne l’evolució i la variabilitat; detectar i monitorar brots nosocomials i a la comunitat, i conèixer millor la biologia del virus (identificar-ne les debilitats) per al disseny de compostos amb activitat antiviral.
Un tret comú entre els virus amb aquesta gran capacitat per a generar variabilitat és que no podem parlar d’un virus com una única seqüència, sinó com una població de genomes. I és en aquesta població de genomes o quasiespècies on podem descriure un reservori de genomes minoritaris que podrien ser seleccionats davant una pressió selectiva, perquè són aquestes variants minoritàries portadores d’una característica que li permeten un avantatge biològic. Per a descriure en detall aquesta població de genomes, és a dir, la seva variabilitat, l’eina més adient és, sens dubte, la tecnologia d’NGS.
1. Conceptes bàsics per a entendre la variabilitat dels virus RNA
Els virus amb el genoma d’RNA i els que durant el seu cicle de replicació passen per una fase d’RNA basen el seu èxit evolutiu en la gran capacitat que tenen de generar variabilitat. Això és degut al fet que tots fan servir RNA-polimerases RNA-dependents que, en la gran majoria de casos, no poden corregir aquells errors que de manera natural s’acumularien durant la còpia de l’RNA genòmic. Una excepció en són els CoV, que, com veurem més endavant, presenten una proteïna amb propietats correctores d’error.
Per a poder entendre la variabilitat viral, ens cal definir dos conceptes (taula 1): la taxa de mutació (MR, de l’anglès mutation rate) i la taxa d’evolució (ER, de l’anglès evolution rate), també coneguda com a taxa de fixació de mutacions (rate of accumulation [fixation] of mutations), a més a més del concepte de quasiespècies (taula 2).
La majoria de virus RNA tenen una gran capacitat de variar degut a la manca de proteïnes de lectura i correcció d’errors (proofread-
Taula 1. Definició de conceptes clau per a entendre la variabilitat viral. Elaboració pròpia.
Concepte Definició Exemples
1a. Taxa de mutació (mutation rate, MR)
Quantifica el nombre d’errors (substitucions) que ha introduït l’RNA-polimerasa RNA-dependent per cada nucleòtid copiat i en cada cicle de replicació (substitucions / nucleòtid / cicle de replicació).
Un virus com el de l’hepatitis C (VHC), que consta d’un genoma d’RNA de 10.000 nucleòtids amb una MR de 10–3-10–4, vol dir que cada cop que un genoma és copiat, el nou genoma que es genera tindrà entre 1 i 10 mutacions aleatòries al llarg del nou genoma, respectivament.
Aquesta taxa és un valor que depèn del procés de còpia de l’RNA viral, de la presència o absència de mecanismes de lectura i correcció d’errors, i del fet que hi hagi mancança o desequilibri en els quatre nucleòtids ATGC, entre altres necessitats bioquímiques. Per tant, la taxa de mutació (MR) és la conseqüència d’un procés bioquímic.
Virus RNA: 10–3-10–5
Retrovirus: 10–3-10–6
Virus DNA: 10–3-10–8
DNA cel·lular: 10–9-10–11
Un SARS-CoV-2 amb un genoma de 29.903 nucleòtids i una MR estimada de 10–6-10–7 vol dir que de cada 300 genomes (virions) que analitzem, un tindrà una mutació situada a l’atzar en el genoma.
1b. Taxa d’evolució (evolution rate, ER) o taxa de fixació de mutacions (rate of accumulation [fixation] of mutations)
Mesura del nombre de substitucions per nucleòtid i per any (substitucions/nt/any). L’ER mesura com el virus està evolucionant en la natura, i està directament afectada per l’eficàcia de transmissió, o facilitat que té el virus per a infectar i generar noves partícules virals. És a dir, un virus amb una taxa de mutació molt alta, però que no es pugui transmetre (per exemple, perquè la persona infectada estigui confinada), tindrà una taxa d’evolució nul·la; en canvi, un virus amb una taxa de mutació baixa, però que es transmeti molt fàcilment i amb capacitat per a generar un gran nombre de partícules virals (càrrega viral elevada), tindrà una taxa d’evolució alta.
Virus respiratoris: 10–3-10–4
Virus de l’hepatitis: 10–3- 7,9-10–5
HCV: 1,5 · 10–3 subst/nt/any
El SARS-CoV-2 té una ER estimada de 8,4 · 10–4 molt semblant als altres virus respiratoris i altres virus RNA.
Taula 2. Definició de quasiespècies. Elaboració pròpia.
Concepte Definició Rellevància clínica
Quasiespècies
Els virus RNA amb les taxes de mutació més elevades són els organismes amb més capacitat de generar diversitat. La conseqüència d’aquesta alta capacitat de variació és l’aparició continuada de variants. Per tant, si seqüenciem el genoma dels virus (població viral) que detectem en un òrgan o teixit d’un pacient infectat, no trobarem una sola seqüència, sinó una barreja complexa de genomes que s’assemblen molt, però presenten diferències. Tota aquesta població viral l’anomenem quasiespècies.
Una quasiespècie està composta per una barreja de genomes diferents, però que s’assemblen molt (tenen unes poques diferències a escala de seqüència l’una respecte de l’altra). La composició d’una quasiespècie està sotmesa a canvis continuats (és una estructura molt dinàmica) mentre el virus està replicant i depèn dels principis de l’evolució darwiniana:
Cada partícula viral pot alhora replicar i, per tant, generar noves variants.
Els virus que componen aquesta quasiespècie estan sotmesos a selecció competitiva.
Els membres de la quasiespècie poden interaccionar i cooperar.
ing activity en anglès), però, en canvi, els virus de la família Coronaviridae, entre els quals el SARS-CoV-2, tenen un mecanisme de correcció d’errors que fa que tinguin taxes de mutació mil vegades inferiors que altres virus RNA. Aquest mecanisme de correcció d’errors està basat en l’expressió de la proteïna no estructural nsp14 (non-structural protein 14 en anglès),
Treballs de la Societat Catalana de Biologia, 72: 51-59
Quins avantatges biològics té per al virus aquesta gran variabilitat?
Li facilita:
Adquirir canvis antigènics i així escapar de la resposta adquirida per vacunació o infecció natural.
Seleccionar variants amb mutacions associades a resistència a antivirals, que a més poden ser transmeses.
Adquirir un mecanisme d’evasió de la resposta immunitària induint fenòmens de tolerància perifèrica.
Afavorir una virulència o transmissibilitat superiors.
que té la funció de lectura de l’RNA que sintetitza l’RNA-polimerasa RNA-dependent (RNA-dependent RNA polymerase o nsp12) i, comparant-la amb la seqüència motlle, detecta si hi ha un error i el corregeix amb la seva activitat 3’ → 5’ exoribonucleasa (ExoN). Això fa que la còpia d’un genoma del SARS-CoV-2 tingui alta fidelitat i en redueix la taxa de mu-
52
Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple
tacions, o el que és el mateix, el nombre de substitucions en el nou genoma resultant (Eckerle et al. , 2010; Smith i Denison, 2013; Gribble et al., 2020; Rambaut et al., 2020). Ara bé, des de la seva circulació inicial entre la població humana el desembre de 2019, el virus no ha deixat d’evolucionar i en van apareixent variants noves dia rere dia, algunes amb impacte a escala mundial, com són les variants de preocupació (VOC, de l’anglès variants of concern ). Es defineixen com a VOC perquè les mutacions que incorporen poden suposar canvis que afectin significativament la salut pública de manera global, com, per exemple, un increment de la transmissibilitat, la virulència i la gravetat de la malaltia, o una reducció de la capacitat de detecció de la variant, l’efectivitat de vacunes o els tractaments antivirals. El febrer de 2022 es consideren VOC les variants alfa (B.1.1.7), beta (B.1.351), gamma (P.1), delta (B.1.617.2) i ara òmicron (B.1.1.529).
Com s’explica que el SARS-CoV-2 tingui una taxa de mutació baixa, mil vegades inferior a altres virus RNA, i en canvi s’observi una aparició continuada de variants noves arreu del món?
És obvi que el SARS-CoV-2 està evolucionant, i alguns dels seus canvis permeten que s’adapti cada cop millor a la població humana, amb una aparició continuada de variants. El febrer de 2022, s’havien comptabilitzat 22 clades (19A,B. 20A-J, 21A,C,D,F-J,L-M) segons la classificació establerta per Nextstrain (Nextstrain, 2021), i 1.728 llinatges (O’Toole et al., 2022) seguint la classificació PANGOLIN (Phylogenetic Assignment of Named Global Outbreak Lineages) (Rambaut et al. , 2020). Bàsicament, estem parlant d’una nomenclatura dinàmica basada en l’anàlisi de similitud de seqüències per inferència filogenètica (Rambaut et al., 2020).
Però, tal com hem comentat abans, la baixa taxa de mutació del SARS-CoV-2 es veu compensada per una elevada grandària de la població viral que cada persona infectada pot presentar, especialment en el pic de màxima càrrega viral, de fins a un total de 109 a 1011 virions, i que a més es veu afavorida per una gran capacitat de transmissió entre persones. Això permet que milions de persones s’estiguin infectant on el virus està replicant i, al mateix temps, canviant de manera continuada. Tot i el gran nombre de morts, el SARS-CoV-2 és causa d’una d’infecció de baixa gravetat de -
mostrada per un nombre elevat d’individus infectats de manera asimptomàtica o lleu, fet que ajuda a facilitar-ne la transmissió, ja que com més silenciós és (asimptomàtic), més fàcil és que una persona infectada pugui transmetre el virus a una persona no infectada, en no adoptar mesures per a evitar aquesta infecció, com és l’ús de mascaretes o reduir el contacte social, entre altres mesures no farmacològiques.
A l’inici de les pandèmies, amb l’absència d’immunitat preexistent a la població humana, el virus es va estendre sense límits i sense necessitat de canviar, fet que es va traduir en una similaritat de seqüència molt alta (99,9 %) entre els virus detectats i caracteritzats per seqüenciació arreu del món (Rambaut et al ., 2020). A mesura que el virus s’ha anat distribuint mundialment i la pandèmia ha anat progressant, amb una incidència elevada degut a la seva gran capacitat per a la transmissió de persona a persona, el virus ha anat canviant, de manera que hem pogut observar cocirculació de diferents variants, tot i que només unes poques s’han seleccionat, segurament pel fet de presentar un avantatge biològic. Aquest avantatge biològic pot entendre’s com una capacitat més gran de transmissió o d’infecció en una població cada vegada més immunitzada, sobretot si parlem dels països rics, que, a hores d’ara, presenten taxes elevades de cobertura vacunal. En la majoria de pandèmies prèvies, en les quals no es tenien recursos sanitaris, de diagnòstic ni de control (Rodríguez-Frías et al., 2021a), s’observa que, a mesura que avança la pandèmia, es van seleccionant noves variants més transmissibles però menys agressives i el virus pot esdevenir un virus endèmic i estacional en el cas de virus respiratoris i entèrics. Una variant que causi una malaltia greu es detecta més ràpidament, fet que permet adoptar mesures d’aïllament per al pacient i evitar, així, que infecti altres persones. Contràriament, un virus silenciós, és a dir, una variant que causi una infecció asimptomàtica, no és detectat perquè els malalts no necessiten atenció sanitària i, per tant, els infectats poden transmetre el virus sense ser-ne conscients, cosa que en facilita l’expansió i dona més probabilitats al virus de persistir en la població i seguir canviant.
Els virus tenen una capacitat elevada de generar variabilitat, però també és cert que tenen constrenyiments, és a dir, limitacions en la variació sense perdre capacitat de transmissió o d’infecció. La seqüenciació massiva o NGS no sols ens permet detectar mutacions minori-
Treballs de la Societat Catalana de Biologia, 72: 51-59
tàries, sinó també les regions genòmiques més conservades. En el cas del SARS-CoV-2, si som capaços d’identificar quins són aquests límits que té el virus per a variar, quines són les proteïnes més sensibles a aquests canvis aleatoris (regions del genoma més conservades) i quines són les mutacions que li donen un avantatge selectiu, podrem desenvolupar eines de vacunació i tractament eficaces que puguin veure’s menys compromeses en el cas de virus capaços de generar variants d’escapament, i que ens permetran a mitjà termini arribar al control de la pandèmia. Aquesta informació de variabilitat i evolució de seqüència completa del genoma viral i de diversitat només la pot aportar l’NGS.
De totes maneres, hem d’ésser conscients que el virus s’està transmetent a escala mundial, i que les variants estan emergint arreu, però les capacitats de seqüenciació no són les mateixes a tots els continents. Cal reforçar aquestes capacitats de seqüenciació a tots els països per a detectar-ne variants noves, que, tot i tenir un origen llunyà, tard o d’hora ens poden arribar i ser causa de problemes al nostre territori. Així ha passat amb la variant delta (amb origen a l’Índia) i òmicron (amb origen a Sud-àfrica i Botswana).
2. Origen i evolució de les variants del SARS-CoV-2 a Catalunya
2.1. Variants dominants de la primera a la sisena onada pandèmica L’NGS del genoma complet del virus ha estat clau per a fer la seqüenciació completa del genoma viral i així fer un seguiment setmana a setmana de les variants del SARS-CoV-2 en cocirculació a partir de mostrejos aleatoris entre els virus detectats, per a detectar possibles variants que eren importades d’altres països, o bé per a fer estudis selectius, especialment, dels casos sospitosos de reinfecció o fallades vacunals (infeccions irruptives). En aquest sentit, a Catalunya la variant predominant (figura 1) durant la primera onada de la pandèmia va ser la B.1.5 (51 %), que va dominar des de març fins a maig de 2020, quan la variant B.1.1 (33 %) va augmentar significativament, coincidint amb l’inici de la desescalada. Aquest virus, junt amb altres virus pertanyents també al clade G de GISAID, es caracteritzaven per ser portadors de la mutació D614G (canvi de l’aa àcid aspàrtic/D per Glicina/G a la posició 614) a l’espícula, la qual n’afavoreix la transmissibilitat (Korber et al., 2020) i es va fixar en el genoma viral, que encara és present en la majoria
53
Figura 1. Distribució setmana a setmana dels llinatges des de març de 2020. S’han encerclat els llinatges (variants) predominants en cada onada pandèmica. Els valors numèrics indiquen el nombre de casos positius (incidència) atesos a l’atenció primària de la ciutat de Barcelona més els ingressats a l’hospital (planta i UCI. Elaboració pròpia.
de virus en circulació actualment. No obstant això, el final del confinament i la flexibilització de la mobilitat i l’organització de les activitats d’estiu van coincidir amb la detecció de la variant B.1.177, que primer es va detectar a Casp (Saragossa), Alcanyís (Terol) i en una localitat holandesa el juny de 2020, i que va ser escampada pels temporers d’Osca i Lleida (López et al., 2021). Més tard, els anglesos la van anomenar variant de les vacances, ja que va esdevenir predominant durant tota la segona onada fins a finals de 2020 a mig Europa (Alm et al., 2020; Hodcroft et al., 2021). Aquesta variant tenia com a mutació principal la substitució de l’aminoàcid alanina per valina a la posició 222 (A222V) de la proteïna de l’espícula.
El 14 de desembre de 2020, el Regne Unit va declarar l’augment d’incidència d’una nova variant a l’est i al sud-est d’Anglaterra i a l’àrea metropolitana de Londres. Aquesta variant es va caracteritzar per tenir vint-i-tres mutacions diferents, vuit de les quals en el gen de l’espícula. De les vint-i-tres mutacions, catorze donen lloc a canvi d’aminoàcid (mutacions no sinònimes), tres delecions (del) i sis mutacions sinònimes (no canvia l’aminoàcid). Les mutacions més importants a l’espícula, i que definien aquesta variant, eren deleció H69-V70 (del69/70), delY144, N501Y, A570D, D614G, P681H, T716I, S982A i D1118H. Aquesta variant, pertanyent al llinatge PANGOLIN B.1.1.7, a principis de 2021 va ser considerada per l’Organització Mundial de la Salut (OMS) variant de preocupació (VOC-202012/01), anomenada alfa, i a Catalunya i a la resta de països va substituir progressivament els virus pertanyents al llinatge B.1.177, que s’havia detectat a Europa amb la mutació A222V (Hodcroft et al., 2021), de manera que es va convertir en dominant durant el final de la tercera onada (55 %). A principis de la quarta onada
pandèmica la seva prevalença ja era del 86 % (figura 1).
La selecció de noves variants amb més capacitat de replicació i transmissió (més fitness biològic) pot haver sorgit de manera independent en diferents parts del món i per diferents mecanismes, encara que a vegades hi pot haver semblança entre elles. Una de les variants que ha tingut més impacte i que ha desplaçat completament la variant alfa és la variant delta, pertanyent al llinatge B.1.617.2, que es va detectar per primer cop a l’Índia, i que va ser considerada VOC el 31 de maig de 2021. Aquesta variant estava definida per les mutacions T19R, E156G, delF157/R158, L452R, T478K, D614G, P681R i D950N a l’espícula, a més de múltiples canvis distribuïts en el seu genoma. Tal com va passar abans, aquesta variant va entrar a la nostra població substituint completament les altres variants fins a arribar a estar present en el 100 % de les mostres seqüenciades (Andrés et al. , 2022) i dominant completament la cinquena onada, però va ser desplaçada per la variant òmicron a mitjan desembre de 2021, a partir del començament de la sisena onada, de la qual és, en gran manera, responsable. A mesura que s’ha anat distribuint arreu del món, fins a finals de 2021, aquesta variant delta s’ha anat diferenciant en més de 200 llinatges i subllinatges designats com a AY, adquirint noves mutacions i delecions, no només a l’espícula, sinó també a altres gens del virus (Shu i McCauley, 2017).
2.2. Òmicron
El 9 de novembre de 2021 es va confirmar el primer cas d’una variant que, més tard, es va classificar com a pertanyent al llinatge B.1.1.529 en el Lancet Laboratories de Johannesburg, Sud-àfrica. Aquesta variant va ser batejada per l’OMS com a variant òmicron i en
Treballs de la Societat Catalana de Biologia, 72: 51-59
pocs dies va passar a ser considerada VOC pel Centre Europeu de Prevenció i Control de Malalties (European Centre for Disease Prevention and Control, ECDC) (ECDC, 2022). Encara que els primers casos van ser detectats a Botswana i després a Sud-àfrica, els experts suggereixen que la variant va sorgir cap a principis d’octubre en algun lloc que es desconeix. Però és interessant esmentar que es va observar un fort augment de la càrrega viral en aigües residuals a l’àrea de Pretòria cap a finals d’octubre o principis de novembre, i ja se sap que l’anàlisi en aigües residuals és un bon indicador predictiu i primerenc d’alerta (Carcereny et al., 2021; NICD, 2022). La variant òmicron ha estat la causa d’una nova onada pandèmica amb una elevada taxa d’incidència, primer als països del sud d’Àfrica i més tard a tot el món, encara que la població hagués estat prèviament molt exposada a aquest virus i hagués adquirit una bona immunitat natural, o bé hagués adquirit aquesta immunitat per campanyes de vacunació massiva que han permès arribar a taxes elevades de cobertura vacunal. Durant aquesta darrera onada, la variant òmicron ha estat capaç de desplaçar la delta com a variant dominant, tal com la delta va fer amb la variant alfa. És evident que aquesta variant presenta avantatges selectius respecte a les altres. La seqüenciació del seu genoma complet ha permès identificar que només en el gen de l’espícula, aquesta variant té seixanta mutacions respecte de la de Wuhan (ref. MN908947.3 GenBank), amb trenta substitucions no sinònimes, tres delecions (del) curtes i una inserció (ins): A67V, DEL69-70, T95I, G142D, del143-145, del211-212, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493K, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K,
54
Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple
P681H, N764K, D796Y, N856K, Q954H, N969K, L981F. Algunes d’aquestes mutacions estan relacionades amb l’increment de la transmissibilitat per l’augment d’unió del virus al receptor ACE2 i/o per l’escapament a la resposta immunitària; moltes d’aquestes mutacions, a més, han estat observades de manera individual en altres variants (Zahradník et al. , 2021). Per exemple, aquesta variant és portadora de les mutacions S477N, E484A, Q498R i N501Y (aquesta última comuna entre les variants alfa, beta i gamma), mutacions clau del domini d’unió al receptor (RBD, de l’anglès receptor binding domain) per a unirse a ACE2. També conté mutacions de l’espícula observades en altres variants com la del69-70 (escapament a resposta immunitària detectada en alfa), del143-145 (delta), K417N (beta), T478K (delta), H655Y (gamma), N679K (gamma) o P681H (alfa en el lloc de tall de la Furina). A més, aquesta variant presenta altres canvis en diferents regions genòmiques (Kim et al. , 2020): K38R, V1069I, del1265, L1266I i A1892T en la proteïna no estructural 3 (nsp3); T492I en nsp4; P132H en nsp5; del105-107 i A189V en nsp6; P323L en nps12; I42V en nsp14; així com T9I en la proteïna estructural (E); D3G, Q19E i A63T en la proteïna M; P13L, del31-33, R203K i G204R en la proteïna N.
El que hom espera, per experiència d’altres pandèmies, és que el virus evolucioni cap a variants més transmissibles però més silencioses i esdevingui un virus estacional com els coronavirus estacionals (OC43, NL63, HKU1 o 229E). I és per aquesta evasió immunològica que, de fet, la majoria de les hospitalitzacions i morts s’estan donant en pacients no vacunats, i s’ha observat un increment de les infeccions en persones amb vacunació completa (infeccions irruptives o breakthough infections), encara que sense evolucionar a malaltia greu. És ben sabut que les vacunes contra els virus respiratoris, com en el cas del virus de la grip, no protegeixen de la infecció, però sí de la malaltia greu i l’hospitalització per complicacions clíniques. Cal esmentar que, degut al gran nombre de mutacions a la proteïna de l’espícula, la majoria dels anticossos monoclonals per al tractament de la malaltia en els primers estadis de la infecció no serveixen ja per a òmicron perquè aquests virus han demostrat in vitro ser-hi resistents (Chen et al., 2022).
L’aparició sobtada d’òmicron ha obert les portes a especular sobre el seu origen, i s’han proposat diverses opcions del que hauria pogut passar:
1. D’una manera similar a com es va suggerir per a la variant alfa (B.1.1.7) (Haseltine, 2021), aquestes variants amb gran nombre de mutacions podrien tenir l’origen en pacients amb una resposta immunitària compromesa (immunosuprimits). En aquests pacients, la infecció pot passar a ser una infecció de llarga durada, ja que són pacients que pateixen algun tipus d’immunosupressió, de manera que la seva resposta immunitària no és prou efectiva per a eliminar el virus. Quan això passa, el virus pot continuar infectant i replicant-se, produint noves partícules virals i acumulant mutacions (indels, mutacions puntuals…) durant el procés de replicació viral, normalment sota la pressió selectiva d’un tractament amb activitat antiviral que pot, a més a més, condicionar l’emergència de variants amb cert grau de resistència. Per exemple, si apareix una mutació que li permeti escapar a un tractament en curs amb anticossos monoclonals o d’un antiviral, especialment en monoteràpia, aquest mutant pot ser seleccionat i el virus pot adquirir resistència.
Per què hi ha sospites que òmicron hauria pogut sorgir al sud de l’Àfrica? A Sud-àfrica trobem un ambient propici a la selecció de noves variants. Per una banda, tenim una alta taxa d’incidència de noves infeccions, i per l’altra, una població amb taxes d’immunosupressió molt altes degudes a la prevalença elevada de la infecció per VIH (KFF, 2021; UNAIDS, 2022); són pacients amb una resposta immunitària feble, i això fa factible la selecció de noves variants en aquest escenari, tal com hem esmentat anteriorment.
2. Una altra hipòtesi és que òmicron hagi aparegut en un país on no es faci un seguiment de les variants, i des d’allà s’hagi transmès de manera silenciosa i hagi arribat a Sud-àfrica i d’aquí a la resta del món. En aquest sentit, la seqüenciació a tot el continent africà és molt inferior a la que es duu a terme a països desenvolupats i les variants intermèdies d’òmicron haurien pogut passar desapercebudes. De totes maneres, no hi ha encara una explicació per a saber per què no s’han detectat variants intermèdies prèvies a òmicron. Caldria, a més, que en aquesta regió on podria haver sorgit l’òmicron el virus estigués sotmès a una pressió selectiva i a infeccions persistents.
3. També hauria pogut ser per zoonosi reversa (transmissió d’humà a animal i tornada a humà). Hi ha consens en la comunitat científica de l’origen zoonòtic del SARS-CoV-2 (Andersen et al., 2020; Calisher et al., 2021; World Health Organization, 2021; Zhou et al., 2020;
Treballs de la Societat Catalana de Biologia, 72: 51-59
Wacharapluesadee et al. , 2021). Es proposa que l’origen devia ser en un virus de ratpenat (el més proper fins ara és el del RaTG13), des del qual va passar a un hoste intermediari, on va evolucionar a partir de múltiples salts des de l’animal salvatge fins a l’humà (zoonosi) i viceversa (zooantroponosi), fins que va aparèixer el SARS-CoV-2 (Sikkema i Koopmans, 2021; Jia et al., 2021). La seqüenciació massiva va ser essencial per a demostrar que els brots del SARS-CoV-2 a les granges de visons de Dinamarca i dels Països Baixos van ser causats per humans, que el virus va poder evolucionar per a adaptar-se a infectar de manera eficient els visons, i de nou va reinfectar els humans ja lleugerament evolucionat. Tot i que s’ha demostrat transmissió d’home a animals salvatges com lleons en els zològics, i també en l’àmbit domèstic, com ara en gossos i gats, aquestes infeccions han dut a punts i final, és a dir, no s’ha pogut traçar que des d’aquests animals infectats el virus infectés altres animals o tornés als humans, i per tant, fins ara, no hi ha proves d’una transmissió eficient en aquests animals. Per tant, ara per ara, amb les variants actuals, es descarta que aquests animals tinguin un paper destacat en la pandèmia. De totes maneres, cal ser prudents i evitar la infecció massiva d’animals per a no facilitar l’adaptació viral a aquests nous hostes, cosa que afavoriria l’emergència de variants amb efectes impredictibles (Jia et al ., 2021). Aquesta hipòtesi, que explicaria l’origen de la variant òmicron, té algunes consideracions que la posen en dubte i calen més dades per a demostrar-la. Els nostres estudis (Andrés et al., 2020) demostren que el virus ha patit un procés d’adaptació a l’humà a mesura que ha infectat la població. Les variants de preocupació alfa, beta i delta presenten freqüències de delecions que generen virus defectius més baixes que les variants a l’inici de la pandèmia B.1.5, B.1.1 o B.1.177, la qual cosa pot anar acompanyada de càrregues virals més elevades (observat en la variant delta) i, en conseqüència, suposar un dany tissular més important en el tracte respiratori (Tegally et al., 2020 i 2021; Campbell et al., 2021). Un virus que s’està adaptant a l’humà augmenta la seva capacitat de replicació i transmissió (també anomenada fitness), que va unida a la seva eficiència de transmissió. El salt a una altra espècie animal suposa un canvi d’ambient, i el virus ha de readaptar-se a les noves condicions, la qual cosa té un cost de fitness (Domingo, 2016) Perquè el virus s’adapti, necessita un cert temps per a adquirir mutacions que li permetran ser millor en el nou hostatger a costa de perdre fit-
55
ness en l’hostatger original (l’humà). Si la hipòtesi d’origen animal d’òmicron fos certa, caldria temps per a explicar-la, la qual cosa implicaria que el virus s’hauria adaptat temporalment a infectar un hoste no humà i que en el moment de tornar a infectar l’humà aquesta nova variant hauria necessitat temps per a recuperar fitness per a arribar a tenir l’avantatge selectiu respecte de la variant delta i la resta de variants. De fet, els estudis epidemiològics i filogenètics mostren més semblança d’òmicron amb variants de mitjan 2020 que respecte d’alfa, beta o delta.
Ara per ara, la discussió sobre l’origen d’òmicron està oberta i no es pot descartar cap de les tres possibilitats, ni tampoc cap que resulti de la seva combinació.
3. Què és l’NGS i què hem après de la pandèmia del SARS-CoV-2?
La tècnica de seqüenciació de nova generació o NGS consisteix en la seqüenciació de milions de fragments de DNA en una mateixa carrera de seqüenciació. Altres maneres d’anomenar aquesta metodologia són seqüenciació massiva, seqüenciació profunda, seqüenciació massiva en paral·lel i seqüenciació d’alt rendiment, entre d’altres, totes igualment vàlides. La sigla NGS (next generation sequencing) es va emprar per primer cop l’any 2000 i encara és acceptada com a oposició a la metodologia de seqüenciació clàssica coneguda com a seqüenciació de Sanger, basada en una reacció química de síntesi i l’ús de dideoxinucleòtids trifosfat (ddNTP) com a terminadors de la reacció de còpia d’un DNA i lectura per electroforesi capil·lar. Amb la metodologia de seqüenciació de Sanger, obtenim una sola seqüència per capil·lar i, per tant, si tenim una barreja de genomes virals en la mostra que hem d’estudiar, la seqüència que obtindrem serà la seqüència consens, és a dir, que a cada posició de nucleòtids veurem el nucleòtid o nucleòtids més representats, però que no és fidel a tota la diversitat genètica present en la mostra. Per tant, no es podran observar totes les variants al·lèliques que hi són presents, especialment les minoritàries. De fet, llegint els pics de l’electroferograma d’una carrera ( run) de seqüenciació de Sanger, sí que es poden intuir mutacions, però només les presents per sobre d’un 20 %. En canvi, a més de permetre incrementar el rendiment dels processos de seqüenciació, per a estudiar genomes complets i per a un gran nombre de mostres, l’NGS permet identificar i quantificar genomes virals individuals en una barreja complexa, ja sigui
estudiant el fragment d’un genoma en profunditat o inclús el genoma sencer per assemblatge de les seqüències que resulten d’aquest procés.
Així, l’NGS s’ha convertit en una eina clau per a la identificació i caracterització ràpides de nous agents virals. La detecció ràpida d’un agent infecciós permet implementar mesures de control en el moment que hi ha pocs casos, de manera que augmenta l’efectivitat de les accions quan es produeixen brots o l’emergència de nous virus. Qualsevol endarreriment en la identificació o la no identificació d’un patogen suposa una reducció de l’eficàcia de la resposta. En el cas de la pandèmia del SARSCoV-2, l’NGS va permetre seqüenciar en poques setmanes el genoma complet d’aquest nou virus dels primers casos diagnosticats a finals de desembre de 2019; la seqüència genètica va estar disponible públicament al banc de dades del Centre Nacional per a la Informació Biotecnològica (NCBI) el 17 de gener de 2020 (MN908947.3). Disposar d’aquesta seqüència va permetre un gran avenç en la lluita contra la pandèmia.
La seqüenciació del SARS-CoV-2 ha ajudat a caracteritzar les regions clau del genoma viral per a:
a) Desenvolupar vacunes altament efectives basades en plataformes d’mRNA/DNA en un temps rècord de sis mesos (World Health Organization, 2021). Cal destacar que es van trigar trenta-quatre anys (1954-1988) per a des envolupar una vacuna per a la varicel·la, quinze per al virus del papil·loma humà (19912006), nou per al xarampió (1954-1963), set per a la primera vacuna de la poliomielitis (1948-1955) i quatre per a les galteres (19631967).
b) Desenvolupar solucions diagnòstiques qualitatives i quantitatives basades en PCR per a confirmar la presència o absència del virus en mostres respiratòries i no respiratòries (Chan et al., 2020; Corman et al., 2020).
c) Detectar mutacions rellevants associades a un escapament a la resposta immunitària adquirida per la vacunació o infecció natural, resistència als antivirals o als tractaments basats en anticossos monoclonals o plasma convalescent (Ruiz-Rodriguez et al., 2021).
d) Estudiar la composició de les quasiespècies presents en els teixits d’un pacient (Piñana et al., 2021).
e) Fer el seguiment de l’evolució natural del virus per a identificar la circulació de noves variants i associar-ne la prevalença a la situació epidemiològica del moment. A més, ha
Treballs de la Societat Catalana de Biologia, 72: 51-59
permès seqüenciar milions de genomes i compartir-los a la plataforma GISAID (Global Initiative on Sharing All Influenza Data) (Elbe i Buckland-Merrett, 2017; Shu i McCauley, 2017; John Hopkins University [JHU], 2022), que ha servit com a font de dades per a altres webs que permetien visualitzar-ne l’evolució i la distribució: Nextstrain: https://nextstrain.org/ncov/ gisaid/global; Outbrak.info: https://virological.org/ t/outbreak-info-sars-cov-2-mutation-situation -reports/629; CoV-lineages: https://cov-lineages.org/ lineage_list.html
Tot el monitoratge de variants, llinatges i subllinatges en circulació es duu a terme principalment mitjançant la seqüenciació de genoma complet, tasca clau per al seguiment de l’evolució de la pandèmia i la presa de decisions per les institucions de salut pública.
f ) Estudiar com canvia genèticament el virus al llarg del temps, quines mutacions poden anar associades a una transmissibilitat i/o gravetat més elevades, l’origen de les noves variants, la seva ruta i/o eficàcia de transmissió, i dur a terme estudis de les reinfeccions, dels escapaments vacunals i de la infecció persistent (Hodcroft et al., 2021; Ruiz-Rodriguez et al., 2021; Andrés et al., 2022).
4. Ús de l’NGS per a identificar i caracteritzar virus/patògens emergents, reemergents o nous Es poden seguir diferents estratègies metodològiques per a la seqüenciació de genomes virals, depenent de si en som o no coneixedors del genoma (encebadors a l’atzar o random primers en anglès), o bé si disposem d’un mínim d’informació (enriquiment per amplicons o panells de captura). Aquestes estratègies estan esquematitzades en la figura 2.
4.1. Seqüenciació de genoma complet emprant encebadors a l’atzar Pel que fa a la seqüenciació de genoma complet (WGS, de l’anglès whole-genome sequencing), els encebadors a l’atzar (o random primers) més emprats són els que tenen sis nucleòtids de llargària (hexàmers), de cadena simple i inclouen totes les possibles combinacions de les quatre bases nucleotídiques (A, C, T i G), de manera que s’obté una barreja de 4.096 (46) hexàmers diferents. Com que tots els hexàmers possibles estan presents, es poden unir a qualsevol fragment d’RNA o DNA i actuar com a iniciadors de còpia del material genò-
56
Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
mic i permetre’n l’amplificació i seqüenciació posterior.
Avantatges:
— No cal tenir informació del genoma.
Permet identificar tots els patògens de qualsevol mostra (plasma, sèrum, femtes, aigües residuals, etc.), cosa que permet fer estudis metagenòmics.
El cost dels encebadors per còpia i amplificació és molt baix.
Desavantatges:
— Els genomes minoritaris sempre queden infrarepresentats i moltes vegades no es detecten; per tant, és una tècnica que té una baixa sensibilitat.
Cal tenir una quantitat elevada del material genòmic de l’agent que volem detectar, ja que la proporció de seqüències de l’agent viral infecciós que volem detectar sempre és molt inferior.
El material genòmic de l’hostatger o de microorganismes presents és, de fet, un contaminant, i la seva presència, a més de reduir l’eficiència de la seqüenciació, suposa també un cost computacional. Una possible solució és dur a terme mètodes d’enriquiment, que en-
careixen el cost de la tècnica (Fernandez-Cassi et al., 2018; Chrzastek et al., 2017).
4.2. Captura de seqüències, recuperació de fragments i NGS Un mètode d’enriquiment del patogen que hom vol seqüenciar es basa en l’ús de panells de captura i consisteix a emprar petites sondes complementàries al genoma de l’agent patogen.
Avantatges:
— Podem enriquir el genoma de l’agent patogen en una barreja complexa de genomes.
— La seqüenciació del genoma complet té un cost baix perquè no cal una gran profunditat de seqüenciació per a obtenir les seqüències del virus que podrem assemblar per a la seva detecció i caracterització.
— Les variants minoritàries es preserven i permeten obtenir una imatge més fidel de la variabilitat de la mostra.
Desavantatges:
— Per a dissenyar les sondes de captura cal conèixer la seqüència del microorganisme.
— No permet seqüenciar nous patògens, però sí patògens molt relacionats genètica-
Treballs de la Societat Catalana de Biologia, 72: 51-59
ment en compartir regions altament conservades.
— Requereix gran expertesa en la preparació de la mostra com a pas previ a la seqüenciació.
— És molt dependent de la profunditat de seqüenciació.
4.3.
Ús d’amplicons
Consisteix en el disseny d’encebadors específics que permetin amplificar un segment del genoma (targeted sequencing), o tot el genoma complet (WGS) emprant encebadors de fragments encavalcats. Aquesta estratègia ha estat la més emprada en el nostre laboratori (Gregori et al., 2014; Quer et al., 2015; RodríguezFrías et al., 2017; Chen et al., 2020) i el mètode escollit per a la seqüenciació del SARS-CoV-2 amb la metodologia ARTIC (Quick, 2020).
Avantatges:
— És el mètode més sensible i el que ofereix més confiança per a estudis de variabilitat.
— Permet amplificar específicament un agent infecciós enmig d’una barreja complexa de genomes minimitzant l’efecte del fons (background).
— Permet obtenir una bona cobertura del genoma (coverage), inclús a baixes concentracions del patogen en la barreja.
— Permet detectar variants presents a molt baixa freqüència.
Desavantatges:
— Cal conèixer la seqüència del microorganisme que volem detectar i dissenyar-ne els encebadors específics.
— És un mètode laboriós.
— És altament dependent de la qualitat dels encebadors, de la seva especificitat. Hi pot haver canvis en la regió de l’encebador, per exemple, per deriva genètica natural del virus, que poden crear un biaix en l’amplificació i seqüenciació, o inclús fer que no sigui possible.
— Cal emprar un nombre gran de cicles d’amplificació i, per tant, cal tenir en compte que poden aparèixer mutacions artefactuals generades en aquest procés de laboratori.
La seqüenciació massiva és, doncs, una de les eines més poderoses que tenim per a la identificació i caracterització genètica de virus emergents, reemergents o nous. El risc de noves pandèmies existeix, i la història ens demostra que l’amenaça és real. En aquesta cursa, els virus, amb la seva enorme capacitat de generar variabilitat, ens porten un gran avantatge. Enfront d’això, la tecnologia actual no permet preveure ni quan ni com apareixerà la
57
Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple
MOSTRA Amplicons Panells de captura Randomprimers Reduir contaminants (àcids nucleics, bacteris, RNA, DNA…) Plataforma d’NGS (Illumina-Nanopore) Solucions bioinformà�ques. Pipelines d’anàlisi: 1. Estudis de variabilitat 2. Filogènia molecular 3. Iden�ficar mutacions i regions altament conservades 4. Desenvolupar tests diagnòs�cs Generarun INFORME final Extracció RNA/DNA
propera
Figura 2. Esquema general de les metodologies per a la seqüenciació de genomes virals. Elaboració pròpia.
pandèmia. El que sí que podem fer és estar cada cop més ben preparats, i una de les eines clau que tenim és l’NGS. Cal crear xarxes de vigilància interconnectades, reforçar els centres de seqüenciació actualment en actiu creant nous nodes i millorar les metodologies de seqüenciació on encara hi ha recorregut per fer. L’eficàcia en la resposta a una pandèmia és
Bibliografia
Alm, E. [et al.] (2020). «Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020». Eurosurveillance, 25 (32). DOI: 10.2807/1560-7917.ES.2020.25.32.2001410.
Amanat, F.; Krammer, F. (2020). «SARS-CoV-2 vaccines: Status report». Immunity, 52(4): 583-589. DOI: 10.1016/j.immuni.2020.03.007.
Andersen, K. G. [et al.] (2020). «The proximal origin of SARS-CoV-2». Nature Medicine [Estats Units], 26: 450-452. DOI: 10.1038/s41591-020-0820-9.
Andrés, C. [et al.] (2020). «Naturally occurring SARSCoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients». Emerging Microbes & Infections [Estats Units], 9 (1): 1900-1911. DOI: 10.1080/22221751.2020. 1806735.
(2022). «A year living with SARS-CoV-2: An epidemiological overview of viral lineage circulation by whole-genome sequencing in Barcelona city (Catalonia, Spain)». Emerging Microbes & Infections [Estats Units], 11 (1): 172-181. DOI: 10.1080/22221751.2021. 2011617.
Calisher, C. H. [et al.] (2021). «Science, not speculation, is essential to determine how SARS-CoV-2 reached humans». Lancet [Londres, Anglaterra], 398 (10296): 209-211. DOI: 10.1016/S0140-6736(21)01419-7.
Campbell, F. [et al.] (2021). «Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021». Eurosurveillance, 26 (24): 1-6. DOI: 10.2807/1560-7917.es.2021.26.24.2100509.
Carcereny, A. [et al.] (2021). «Monitoring emergence of the SARS-CoV-2 B.1.1.7 variant through the Spanish national SARS-CoV-2 wastewater surveillance system (VATar COVID-19)». Environmental Science & Technology, 55 (17): 11756-11766. DOI: 10.1021/acs. est.1c03589.
Chan, J. F.-W. [et al.] (2020). «Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens». Journal of Clinical Microbiology, 58 (5). DOI: 10.1128/JCM.00310-20.
Chen, J. [et al.] (2022). «Omicron variant (B.1.1.529): Infectivity, vaccine breakthrough, and antibody resistance». Journal of Chemical Information and Modeling, 62 (2): 412-422. DOI: 10.1021/acs.jcim.1c01451.
Chen, Q. [et al.] (2020). «Deep-sequencing reveals broad subtype-specific HCV resistance mutations associated with treatment failure». Antiviral Research [Elsevier B. V.], 174: 104694. DOI: 10.1016/j.antiviral. 2019.104694.
Chrzastek , K. [et al.] (2017). «Use of sequence-independent, single-primer-amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses». Virology, 509: 159-166. DOI: 10.1016/j.virol.2017.06.019.
Corman, V. M. [et al.] (2020). «Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR». Eurosurveillance , 25 (3). DOI: 10.2807/1560-7917. ES.2020.25.3.2000045.
Domingo, E. (2016). Virus as populations: Composition, complexity, dynamics, and biological implications. 1a ed. Amsterdam: Academic Press. 412 p.
— (2020). «Molecular basis of genetic variation of viruses: error-prone replication». A: Virus as populations. 2a ed. Londres: Academic Press, 35-71. DOI: 10.1016/ B978-0-12-816331-3.00002-7.
més alta com més ràpida sigui la detecció, identificació i caracterització genètiques de l’agent infecciós.
Agraïments
Volem donar les gràcies als finançadors dels nostres projectes: Pla Estratègic de Recerca i Innovació en Salut (PERIS) de la Direcció Ge-
Eckerle, L. D. [et al.] (2010). «Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing». PLoS Pathogens [Estats Units], 6 (5): e1000896. DOI: 10.1371/ journal.ppat.1000896.
Elbe, S.; Buckland-Merrett, G. (2017). «Data, disease and diplomacy: GISAID’s innovative contribution to global health». Global Challenges [Hoboken, NJ], 1 (1): 33-46. DOI: 10.1002/gch2.1018.
European Centre for Disease Prevention and Control (ECDC) (2022). Assessment of the further spread and potential impact of the SARS-CoV-2 Omicron variant of concern in the EU/EEA, 19th update. Estocolm: ECDC.
Fernandez-Cassi, X. [et al.] (2018). «Viral concentration and amplification from human serum samples prior to application of next-generation sequencing analysis». Methods in Molecular Biology [Clifton, N. J., Estats Units], 1838: 173-188. DOI: 10.1007/978-1-49398682-8_13.
Gibb, R. [et al.] (2020). «Zoonotic host diversity increases in human-dominated ecosystems». Nature [Anglaterra], 584 (7821): 398-402. DOI: 10.1038/s41586-020-2562-8.
Gregori, J. [et al.] (2014). «Inference with viral quasispecies diversity indices: Clonal and NGS approaches». Bioinformatics, 30 (8): 1104-1111. DOI: 10.1093/bioinformatics/btt768.
Gribble, J. [et al.] (2020). «The coronavirus proofreading exoribonuclease mediates extensive viral recombination». BioRxiv. Cold Spring Harbor Laboratory. DOI: 10.1101/2020.04.23.057786.
Haseltine, W. A. (2021). «Persistently infected Covid-19 patients: A potential source for new variants». Forbes [en línia] <https://www.forbes.com/sites/williamha seltine/2021/02/16/persistently-infected-covid-19 -patients-a-potential-source-for-new-variants/ ?sh=771761de208a> [Consulta: 14 març 2022].
Hodcroft, E. B. [et al.] (2021). «Spread of a SARS-CoV-2 variant through Europe in the summer of 2020». Nature [Anglaterra], 595 (7869): 707-712. DOI: 10.1038/ s41586-021-03677-y.
Jia, P. [et al.] (2021). «New approaches to anticipate the risk of reverse zoonosis». Trends in Ecology & Evolution, 36 (7): 580-590. DOI: 10.1016/j.tree.2021.03.012.
John Hopkins University (JHU) (2022). COVID-19 dashboard by the Center for Systems Science and Engineering (CSSE) at John Hopkins University (JHU) [en línia]. Baltimore, Estats Units. <https://coronavirus. jhu.edu/map.html> [Consulta: 14 març 2022].
KFF (2021). «The global HIV/AIDS epidemic» [en línia]. < https://www.kff.org/global-health-policy/fact -sheet/the-global-hivaids-epidemic/ > [Consulta: 14 març 2022].
Kim, D. [et al.] (2020). «The architecture of SARS-CoV-2 transcriptome». Cell , 181 (4): 914-921.e10. DOI: 10.1016/j.cell.2020.04.011.
Korber, B. [et al.] (2020). «Tracking changes in SARSCoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus». Cell, 182 (4): 812-827. e19. DOI: 10.1016/j.cell.2020.06.043.
López, M. G. [et al.] (2021). «The first wave of the COVID19 epidemic in Spain was associated with early introductions and fast spread of a dominating genetic variant». Nature Genetics, 53 (10): 1405-1414. DOI: 10.1038/s41588-021-00936-6.
Mallapaty, S. (2022). «The hunt for the origins of Omicron». Nature, 602 (3 febrer): 26-28.
Treballs de la Societat Catalana de Biologia, 72: 51-59
neral de Recerca i Innovació en Salut (DGRIS), Generalitat de Catalunya; Centro para el Desarrollo Tecnológico Industrial (CDTI) del Ministeri d’Economia i Competitivitat, núm. IDI-20200297; Beca de Gilead GLD21_00006, i Beca PI19/00301 Instituto de Salud Carlos III, cofinançada per European Regional Development Fund (ERDF).
Nextstrain (2021). Genomic epidemiology of SARSCoV-2 [en línia]. <https://nextstrain.org/ncov/global> [Consulta: 14 març 2022]
NICD (2022). Wastewater-based epidemiology for SARSCOV-2 in South Africa [en línia]. <https://www.nicd. ac.za/diseases-a-z-index/disease-index-covid-19/ surveillance-reports/weekly-reports/wastewater -based-epidemiology-for-sars-cov-2-in-south-africa/> [Consulta: 4 desembre 2022].
O’Toole, Á. [ et al. ] ( 2022). SARS-CoV-2 lineages [en línia]. <https://cov-lineages.org/lineage_list.html> [Consulta: 14 març 2022].
Piñana, M. [et al.] (2021). «Viral populations of SARSCoV-2 in upper respiratory tract, placenta, amniotic fluid and umbilical cord blood support viral replication in placenta». Clin. Microbiol. Infect. , 27 (10): 1542-1544. DOI: 10.1016/j.cmi.2021.07.008.
Quer, J. [et al.] (2015). «High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods». Journal of Clinical Microbiology, 53 (1): 219-226. DOI: 10.1128/ JCM.02093-14.
Quick, J. (2020). nCoV-2019 sequencing protocol [en línia]. <https://www.protocols.io/view/ncov-2019-sequenc ing-protocol-v3-locost-bp2l6n26rgqe/v3> [Consulta: 19 setembre 2022].
Rambaut, A. [et al.] (2020). «A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology». Nature Microbiology [Anglaterra], 5 (11): 1403-1407. DOI: 10.1038/s41564-020-0770-5.
Rodríguez-Frías, F. [et al.] (2017). «High HCV subtype heterogeneity in a chronically infected general population revealed by high-resolution hepatitis C virus subtyping». Clinical Microbiology and Infection , 23 (10): 775.e1-775.e6. DOI: 10.1016/j.cmi.2017.02. 007.
(2021a). «Microorganisms as shapers of human civilization, from pandemics to even our genomes: Villains or friends? A historical approach». Microorganisms, 9 (12). DOI: 10.3390/microorganisms9122518.
(2021b). «Evolutionary and phenotypic characterization of two spike mutations in European lineage 20E of SARS-CoV-2». MBio , 12 (6): e0231521. DOI: 10.1128/mBio.02315-21.
Roger, F. [et al.] (2021). Origin of the Covid-19 virus: The trail of mink farming [en línia]. <https://theconversat ion.com/origin-of-the-covid-19-virus-the-trail-of -mink-farming-155989> [Consulta: 14 març 2022].
Ruiz-Rodriguez, P. [et al.] (2021). «Evolutionary and phenotypic characterization of two spike mutations in European Lineage 20E of SARS-CoV-2». MBio, 12 (6): e02315-21. DOI: 10.1128/mBio.02315-21.
Shu, Y.; McCauley, J. (2017). «GISAID: Global initiative on sharing all influenza data - from vision to reality». Eurosurveillance: 30494. DOI: 10.2807/1560-7917. ES.2017.22.13.30494.
Sikkema, R. S.; Koopmans, M. P. G. (2021). «Preparing for emerging zoonotic viruses». A: Encyclopedia of virology [en línia]. Elsevier, 256-266. DOI: 10.1016/ B978-0-12-814515-9.00150-8.
Smith, E. C.; Denison, M. R. (2013). «Coronaviruses as DNA wannabes: A new model for the regulation of RNA virus replication fidelity». PLoS Pathogens , 9 (12): e1003760. DOI: 10.1371/journal.ppat.1003760.
Tegally, H. [et al.] (2020). «Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple
58
Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
spike mutations in South Africa». MedRxiv, 2020.12. 21.20248640.
Tegally, H. [et al.] (2021). «Detection of a SARS-CoV-2 variant of concern in South Africa». Nature , 592 (7854): 438-443. DOI: 10.1038/s41586-021-03402-9.
Tollefson , J. (2020). «Why deforestation and extinctions make pandemics more likely». Nature [Anglaterra ], 584: 175-176. DOI: 10.1038/d41586-02002341-1.
UNAIDS (2022). Global data on HIV epidemiology and response [en línia]. < https://aidsinfo.unaids.org/ > [Consulta: 14 març 2022].
Wacharapluesadee, S. [et al.] (2021). «Evidence for SARS-CoV-2 related coronaviruses circulating in bats
Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple
and pangolins in Southeast Asia». Nature Communications, 12 (1): 972. DOI: 10.1038/s41467-021-21240-1.
Wearn, O. R. [et al.] (2012). «Extinction debt and windows of conservation opportunity in the Brazilian Amazon». Science [Nova York, Estats Units], 337 (6091): 228-232. DOI: 10.1126/science.1219013.
World Health Organization (2021). WHO-convened global study of origins of SARS-CoV-2: China Part [en línia]. Ginebra, Suïssa: WHO. <https://www.who.int/ publications/i/item/who-convened-global-study-of -origins-of-sars-cov-2-china-part> [Consulta: 14 març 2022].
Wouters, O. J. [et al.] (2021). «Challenges in ensuring global access to COVID-19 vaccines: production, af-
fordability, allocation, and deployment». Lancet [Londres, Anglaterra], 397 (10278): 1023-1034. DOI: 10.1016/S0140-6736(21)00306-8.
Zahradník, J. [et al.] (2021). «SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution». Nature Microbiology [Anglaterra], 6 (9): 1188-1198. DOI: 10.1038/s41564-02100954-4.
Zhou, H. [et al.] (2020). «A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein». Current Biology , 30 (11): 2196-2203.e3. DOI: 10.1016/j. cub.2020.05.023.
Treballs de la Societat Catalana de Biologia, 72: 51-59
59
NORMES DE PRESENTACIÓ D’ARTICLES PER ALS AUTORS DE TREBALLS DE LA SOCIETAT CATALANA DE BIOLOGIA
Abast
La revista treballs de la societat catalana de biologia (abreujat, Treb. Soc. Cat. Biol.), editada per la Societat Catalana de Biologia (SCB), filial de l’Institut d’Estudis Catalans (IEC), publica articles de l’àmbit de les ciències de la vida en llengua catalana (i ocasionalment en altres llengües).
La revista inclou un conjunt d’articles de recerca o de revisió sobre un tema monogràfic que tracta d’alguna qüestió científica concreta. Un coordinador, expert en el tema, encarrega els articles a un equip d’autors i en supervisa la redacció. Si voleu fer de coordinador d’un tema del vostre interès, poseu-vos en contacte amb la secretaria de la SCB (scb@iec.cat).
Presentació d’articles
Els articles han de tenir una extensió entre 20.000 i 40.000 caràcters (comptant els espais) i han d’incloure els apartats següents: títol; noms i cognoms dels autors; filiació de tots els autors; autor per a la correspondència (cal indicar-ne les adreces postal i electrònica, i, preferiblement, el telèfon); resum en català (d’una extensió màxima de 1.000 caràcters); paraules clau en català (com a màxim cinc); títol, resum i paraules clau en anglès (que han de ser fidels, en extensió i contingut, als corresponents en català); text de l’article (amb un màxim de dos nivells d’apartats); bibliografia; taules, figures i peus de figura. En cas que la llargària no s’adeqüi als criteris especificats o que el nombre de taules o figures es consideri excessiu, la SCB pot proposar canvis pel que fa a aquests aspectes abans d’acceptar l’article.
Els articles s’han d’enviar en un arxiu en format Microsoft Word. Aquest arxiu ha de contenir només text (article, bibliografia, taules, peus, etc.). Les figures s’han d’enviar en arxius a part i, si contenen text (a part dels peus), han d’estar en un format que permeti editar-lo. Si la taula o la figura té copyright, cal indicar-ho.
La bibliografia s’ha de compondre tal com s’exemplifica tot seguit: Codina, C. et al. (1989). «Potencial biotecnològic del cultiu de cèl·lules vegetals per a l’obtenció de productes farmacèutics». Treb. Soc. Cat. Biol., 40: 47-70.
Mellado, R. P. (1987). «Vectores utilizados para la manipulación y expresión de genes».
A: Vicente, M.; Renart, J. (ed.). Ingeniería genética. Madrid: CSIC, 21-30.
Wolffe, A. (1995). Chromatin structure and function. Londres: Academic Press.
Les referències bibliogràfiques completes han d’aparèixer ordenades alfabèticament al final dels articles i, si contenen més de dos autors, cal escriure només el primer, seguit de la indicació et al. entre claudàtors. Les remissions a la bibliografia dins el text han de seguir el sistema de cognom i any, no pas un sistema numèric, i, si contenen més de dos autors, cal escriure només el primer, seguit de la indicació et al., en aquest cas sense claudàtors.
En el moment de lliurar els articles per a sol·licitar-ne la publicació els autors accepten els termes següents:
— Els autors cedeixen a la SCB els drets de reproducció, comunicació pública i distribució dels articles presentats per a ser publicats a treballs de la societat catalana de biologia
— Els autors responen davant la SCB de l’autoria i l’originalitat dels articles presentats.
— És responsabilitat dels autors l’obtenció dels permisos per a la reproducció de tot el material gràfic inclòs en els articles.
Procés editorial
Els articles, un cop rebuts, se sotmeten a un procés de revisió externa de forma i de contingut. En acabat, poden ser: a) acceptats sense canvis, b) rebutjats o c) acceptats amb esmenes proposades als autors. En aquest darrer cas, és un requisit indispensable per a publicar-los que els autors acceptin introduir les esmenes proposades.
Els autors rebran unes galerades perquè les revisin. En aquest procés només poden introduir-hi esmenes de caràcter lingüístic i tècnic, però no de contingut. En casos especials, la SCB pot demanar als autors revisions addicionals.
Els autors rebran sense càrrec un exemplar de la revista un cop publicada.
Protecció de dades personals
L’Institut d’Estudis Catalans (IEC) compleix el que estableix el Reglament general de protecció de dades de la Unió Europea (Reglament 2016/679, del 27 d’abril de 2016). De conformitat amb aquesta norma, s’informa que, amb l’acceptació de les normes de publicació, els autors autoritzen que les seves dades personals (nom i cognoms, dades de contacte i dades de filiació) puguin ser publicades en el volum corresponent de treballs de la societat catalana de biologia
Aquestes dades seran incorporades a un tractament que és responsabilitat de l’IEC amb la finalitat de gestionar aquesta publicació. Únicament s’utilitzaran les dades dels autors per a gestionar la publicació de treballs de la societat catalana de biologia i no seran cedides a tercers, ni es produiran transferències a tercers països o organitzacions internacionals. Un cop publicada la revista, aquestes dades es conservaran com a part del registre històric d’autors. Els autors poden exercir els drets d’accés, rectificació, supressió, oposició, limitació en el tractament i portabilitat, adreçant-se per escrit a l’Institut d’Estudis Catalans (carrer del Carme, 47, 08001 Barcelona), o bé enviant un correu electrònic a l’adreça dades.personals@iec.cat, en què s’especifiqui de quina publicació es tracta.
2
Editorial. Jaume Pellicer
El racó de la SCB. Lisa Pokorny

Articles
3 La iniciativa catalana per a l’Earth BioGenome Project
Elisabet Tintó-Font, Helga Simon-Molas, Oriane Hidalgo, Roderic Guigó i Montserrat Corominas
10 Sobre la necessitat d’estudiar trets genètics que influeixen en l’organització i l’estructura del genoma en projectes de seqüenciació de plantes
Jaume Pellicer, Oriane Hidalgo, Joan Vallès i Teresa Garnatje
16 Del genoma als gens
Ferriol Calvet i Roderic Guigó
21 Avenços en les tecnologies de seqüenciació del DNA
Berta Fusté, Elena Vila i Mònica Bayés
28 Assemblatge de genomes a escala cromosòmica per redescobrir i conservar la biodiversitat catalana Jèssica Gómez-Garrido, Fernando Cruz, Marc PalmadaFlores i Tyler Alioto
34 Genòmica de quelicerats: la desconstrucció dels aràcnids i la base genòmica de la seda, els verins i altres trets de rellevància biològica
Miquel A. Arnedo i Julio Rozas
43 Protists, la principal font de diversitat genòmica en eucariotes
Ramon Massana, Ramiro Logares, David López-Escardó i Javier del Campo
51 Variabilitat i seqüenciació massiva de virus. El SARS-CoV-2 com a exemple
Cristina Andrés, David Tabernero, Tomás Pumarola, Andrés Antón i Josep Quer
Volum 72, 2022
CREU DE SANT JORDI 2012 PLACA NARCÍS MONTURIOL 2003 1912 2012 Si t’interessa la biologia i vols… ...conèixer els darrers avenços ...participar en l’organització de seminaris ...rebre la revista ...gaudir de descomptes en llibres, cursos i jornades... ...per què no t’hi associes? http://scb.iec.cat
