PHARMACOLOGY


For 1st year



1. Pharmacokinetics……………..…………………………………………………………………………………….1 2. Pharmacodynamics……………...…………………………………………………………………….……….35 3. Parasympathetic nervous system……………….……………………………………………………..57 4. Sympathetic nervous system……………………………………………………….……………………..86 5. Sympatholytics …………………………………..……………………………………………………………...107 6. Autacoids …………………………………..………………………………………………………………………..119 7. Anti-bacterial drugs……………..…………………………………………………………………………...147 8. Anti-viral & anti-helminthics………………..……………………………………………………………155 9. Anti-cancer drugs……………….……………………………………………………………………………..168 10.Immuno-suppresive drugs…………..………………………………………………….………………..177 Content
Outlines
Mechanisms of drugs transport across cell membranes
Drug Absorption and drug distribution
routes of drug administration


Defenition: is the study of actions of the body on the drug including absorption, distribution and elimination (biotransformation and excretion) (ADME).
These processes involve passage of drug across cell membranes.
Pharmacokinetics 1 transport across cell membranes
Passage of drugs across cell membranes involves the following processes:
1-passive diffusion:
Most drugs cross cell membranes by passive diffusion, from an area of high concentration to an area of lower concentration

No expenditure of energy is required by this process
It includes

o passive diffusion of water-soluble drugs (aqueous diffusion)
o passive diffusion of lipid soluble drugs (lipid diffusion)
Filtration or aqueous diffusion: that involves the the passage of some watersoluble substances through aqueous channels between cells. Such passage is assisted by hydrostatic or osmotic pressure difference across the membrane. This is an important way of passage of drugs across glomerular membrane in the kidney. Lipid solubility and pH do not influence the passage of drugs into glomerular filtrate.
1
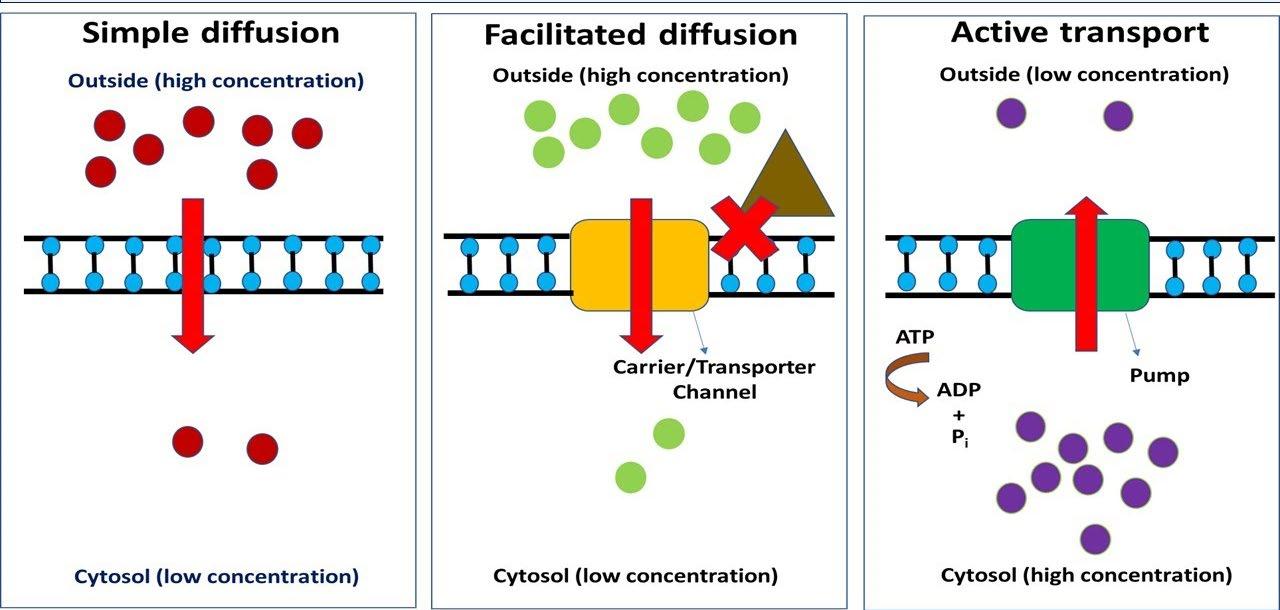
2-Carrier-mediated membrane transport: It includes: -
A. active transport: It is carrier-mediated transport system with the following characteristics:

the drug moves against concentration gradient
The process needs energy
the carrier may be selected for certain drugs
the carrier may be saturated at high concentration
the process may be competitive as drugs with similar structures may be compete for the same carrier
e.g., transport of penicillin by renal tubules.
B. facilitated diffusion: It is a carrier- mediated process in which
diffusion occurs with a concentration gradient


not require energy
e.g., permeation of glucose across a muscle cell membrane. (Not important for drugs).
3-Pinocytosis:
drugs of high molecular weight or which exist as molecular aggregates may be transported by being engulfed within membrane vesicles
It is important for some polypeptide drugs e.g., transport of insulin across blood brain barrier.
2
Influence of pH on weak electrolytes
Most drugs are weak acids or weak bases that present in solution as both non-ionized and ionized form.
The non-ionized molecules usually have high lipid solubility and can diffuse readily across the cell membrane because of high lipid solubility.
The transmembrane distribution of a weak electrolyte is determined by pH gradient across the membrane and pKa of electrolyte.

The pKa is the pH at which 50%of the drug is in ionized form and 50% is in unionized form.

The extent of ionization can be calculated using the general HendersonHasselbalch equation:
pH=pKa+log base/acid
Where, for a
o weak acid it is the acid moiety that is non ionized as the degree of ionization increases as pH increases
o weak base it is the base moiety that is non ionized as the degree of ionization decreases as pH increases.
Many drugs are weak electrolytes and tend to ionize according to environmental pH:
o weak acidic drugs (salicylate, barbiturate) become less ionized in an acidic environment (citric acid, NH4Cl)
o weak basic drugs (amphetamine, qunidine) become less ionized in an alkaline environment (NaHCO3).
Unionized and lipid soluble drug can pass readily across cell membrane, while water soluble and ionized drug have less probability to be absorbed
3
Drug Absorption
Absorption: is the movement of a drug from its site of adminstration into the systemic circulation.
Factors Affecting Absorption:
I. surface area of absorption
II. Blood flow to the site of absorption
III. concentration of the drug at site of absorption
IV. physical state of the drug (solution, suspension, solid dosage form, its water solubility)
The drug absorption occurs from different sites depending on the route of administration.
Absorption from GIT:

Intestine is the main site of absorption, due to
o the extremely large surface area of the villi of upper intestine
o the large blood supply by mesenteric vessels
Any factor that accelerates gastric emptying will accelerate the arrival of the drug in the duodenum for systemic absorption i.e., increase drug absorption, while any factor that delays gastric emptying will decrease drug absorption
Although absorption is favored when the drug is in the non-ionized form, the rate of absorption of the drug from intestine is larger than from stomach even if the drug is extremely ionized in the intestine and largely non-ionized in the stomach. For example, phenobarbital is absorbed from intestine 18 times more rapid than from stomach.




Enteric coated preparation:
Drugs that are destroyed by gastric secretion or that cause gastric irritation are sometimes given in a dosage form with enteric coating (e.g., enteric coated tablet) that prevents dissolution in gastric content.
Sustained Release Preparation:
The rate of absorption of a drug administered as tablet or other solid dosage forms is partly dependent on its rate of dissolution in GI fluids.
This is the basis of controlled release pharmaceutical preparations which produce slow uniform absorption of the drug for 8 hours or longer.
4
Drug distribution
Following absorption or systemic administration into blood stream, a drug distributed into interstitial and intracellular fluid, and various tissues.

Factor affecting distribution: the pattern of distribution of a drug from the plasma to the interstitium depends on:
I. Regional blood flow:
o Tissues that are well perfused e.g., heart, liver, kidney, brain and other well perfused organs receive most of the drug during the first few minutes after absorption.
o Delivery of the drug to the larger fraction of body mass as muscles, skin and fat is slower (poorly perfused tissues)
II. lipid solubility:
Drugs which are highly lipid soluble can distribute more readily to tissues than polar compound (water soluble).
III. Capillary permiability:
o Capillary structure varies widely in the presence of opening between endothelial cells through which the drugs move readily by passive diffusion
o Many drugs do not easily distribute into the brain in comparison with other tissues e.g., liver and spleen due to the presence of blood brain barrier
IV. Membrane transporters:
Membrane transporters may be located in certain body places to control drug distribution to specific organs e.g. P-glycoproteins(P-gp) transporters which are located specifically in blood brain barrier system of the brain to protect this special organ from different compound through its efflux mechanism .e.g. CNS side effect of digoxin (drug used in heart failure)may not be observed in individuals with over expression of P-gp transporter at BBB. The transporter then reduces passage of the drug into the CNS and increases its efflux out the cell.
Diffusion of the drug to the interstitial and cellular compartments depends on the various factors that affect membrane permeation
o lipid solubility
o extent of binding to plasma proteins
o degree of ionization.
A drug that has low lipid solubility, strong binding to plasma proteins and/or high degree of ionization has limited access to cellular sites of action .
5
Some drugs may show selective accumulation in particular tissues e.g
o iodides are concentrated in thyroid gland
o chloroquine in liver and eye
o tetracyclines in bones
o chlorinated insecticides in fats.
Some areas of the body (e.g., brain) are not readily accessible to drugs due to the presence of anatomic barriers.
Redistribution
Although termination of drug effect is usually accomplished by biotransformation and excretion
it may also result from redistribution of the drug from its site of action into other tissues.
It is, however stored there in an active form and its ultimate elimination from the body is still dependent on biotransformation and excretion
A notable example of this phenomenon of redistribution is the ultrashort acting IV anesthetic thiopental
Thiopental






Note that highly lipid soluble
single I.V. injection given repeatedly given successive doses
o immediately taken up by the brain due to its high blood flow
o it reaches maximum concentration there within a minute
o then it leaves rapidly the brain (causing rapid recovery of the patient from anesthesia).
o This is a redistribution and thiopental leave the brain unchanged i.e., in an active form and diffuses into other tissues as muscles and slowly into fat depots where it is stored there.
o As the plasma level falls, the concentration in the brain similarly falls and consciousness returns in 20-30 minutes

o initial doses saturate the storage sites
o further doses of the drug may produce a prolonged effect
o Due to the accumulation of the drug in storage sites (i.e., muscles and fats) reaches saturation; plasma concentration rises and likewise the brain concentration.
o The duration of action is prolonged and recovery may require several hours.
6
Thus, a drug that is short acting because of rapid redistribution to other sites can become long-acting when these storage sites are filled and termination of the drug action will become dependent on biotransformation and excretion
Distribution across special membranes
Blood -brain barrier:

the concept of blood brain barrier arose from the observation that many aniline dyes when given intravenously to animals stained all tissues except CSF and brain
The barrier is formed by the tight junction of capillary endothelial cells with the absence of intercellular pores. Also, there is special arrangement of pericapillary glial cell.
Drug molecule must traverse both the endothelial and pericapillary cell membrane to reach the cerebral neurons.
Penetration of the barrier by a drug depends on:
o the degree of ionization
o lipid solubility

o degree of binding to plasma proteins
Compounds that are unionized, those that are highly lipid soluble and those that are poorly bound to plasma proteins are better able to diffuse across the blood brain barrier.
Cerebral blood flow is the only limitation to permeation of CNS by highly lipid soluble drugs.
Some drugs like e.g., penicillin, penetrates the normal barrier slightly but in the presence of inflammation of meninges, the local permeability increases and the drug passes through more readily.
Placental transfer of drugs:
At one time it was postulated that the placenta acts as a barrier to drug transport, but this view is inaccurate.
Drugs cross the placenta primarily by simple diffusion and the extent of ionization, lipid solubility, plasma protein binding and the placental blood flow are the main determinants to diffusion.
7
The potential transfer of drugs across the placenta is important as drugs may cause congenital anomalies especially in the first trimester (period of organogegenesis). As the fetus is, to some extent, exposed to essentially all drugs taken by the mother
the consensus is that pregnant women should never receive drugs unless highly indicated
route Intramuscular Intrathecal Intradermal
I. Oral Route:
Subcutaneous Intravenous Intraperitoneal Intra-articular
Alimentary route (Enteral)
It is the most common method of administration
Transdermal


















The drug is swallowed and is systemically absorbed from gastrointestinal tract to the mesenteric circulation to the hepatic portal vein into the liver then to the systemic circulation
Advantages This is:
o the safest
o most convenient
8

o most economic of administration Enteral Inhalation Topical Parenteral Oral Sublingual Rectal Intra-arterial
Disadvantages:
o Not feasible in unconscious, uncooperative or severely ill patients
o Not suitable in the presence of vomiting as a result of gastrointestinal irritation and highly ionized drugs as aminoglycoside
o Some drugs are destroyed by gastric acidity (penicillin G) or digestive enzymes(insulin)
o Irregularities of absorption in presence of food or other drugs
o Drugs in gastrointestinal tract (GIT) may be metabolized by enzymes in the mucosa, intestinal flora or liver before gaining access to the general circulation (first-pass metabolism)
o Onset of action is relatively slow and so unsuitable for emergency conditions
Some drugs are given by mouth to act locally on GIT rather than by absorption as antacids, adsorbents, some purgatives and some antimicrobial drugs to treat infections of GIT as neomycin, streptomycin and some sulfonamides.
II. Sublingual and Buccal Administration:
A tablet or lozenge is placed under the tongue (sublingual) or in the cheek(buccal) to allow non-polar lipid soluble drugs to be absorbed through the epithelial lining of the mouth
Drugs used by this route (e.g., nitroglycerin) are absorbed from the buccal mucosa rapidly into the systemic circulation (through venous drainage of the mouth to the superior vena cava) i.e. bypass the liver and protected from hepatic metabolism.
III. Rectal Administration:
The drug in solution (enema) or suppository form is inserted into rectum
Advantages:
o This route is useful when the drug cannot be given by mouth as in the presence of vomiting or in an unconscious patient
o About 50% of the drug absorbed from the rectal mucosa will bypass the liver
Disadvantage:
o Rectal absorption is often irregular and incomplete
o Unsutable for drugs that cause irritation of rectal mucosa.
9
Non-alimentary route (Parenteral)( injection)
Advantages
effect is more rapid and more predictable
effective dose can be more accurately selected
suitable in unconscious, uncooperative patient and in the presence of vomiting
usually used for drugs that are poorly absorbed from GIT, for agent unstable in GIT e.g., insulin.

I. Intravenous Injection:
Disadvantages
requires aseptic conditions
relatively expensive
Pain may accompany the injection
self-medication is sometimes difficult
In this method the drug is given in aqueous Solution

Advantages:
o The effect produced is very rapid
o Very irritating solutions (as anticancer drugs) are only given by this route since the blood vessel walls are relatively insensitive
Disadvantages:
o unfavorable reactions are likely to occur since high concentrations of the drug are reached rapidly in both plasma and tissues
o once the drug is injected there is no retreat, so if there is any fault it may not be corrected or can be corrected with great difficulty
o Drugs in an oily vehicle, those that percipitate blood constituent or hemolyze erythrocytes must not be given by this rout
Intravenous bolus injections:
small volume given rapidly. The drug is directly injected into blood stream, so it rapidly distributed through the body and generally act very rapid e.g., diazoxide and adenosine
Intravenous infusion:
The drug is given Intravenously at constant input rate to maintain a relatively constant plasma level
10
II. Subcutaneous Administration:
Solutions (aqueous or fine suspension) injected subcutaneously should nonirritating. otherwise, severe pain, necrosis and sloughing may occur
The rate of absorption is often constant and slow to provide a sustained effect
The incorporation of a vasoconstrictor substance in the injected solution retards absorption
Solid pellets may be implanted subcutaneously, and absorption of the drug occurs slowly over a period of weeks or months; some hormones are effectively administered in this manner as the contraceptive levonorgestrel (Norplant).
III. Intramuscular Injection:
Absorption by this route is usually faster than by subcutaneous injection especially when the drug is water soluble
Absorption can be slowed if the drug is given in an oily solution or emulsified
Irritating substances that cannot be given subcutaneously may sometimes be given intramuscularly
This rout unsuitable for injection of anticoagulant as heparins
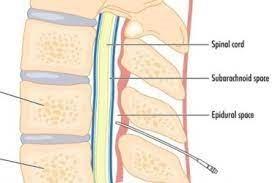
IV. Intrathecal Injection:
The drug is injected directly into the spinal subarachnoid space to provide local or rapid effects on cerebrospinal axis as in spinal anesthesia or acute CNS infections. This because the blood brain barrier and the blood-cerebrospinal fluid barrier often prevent or slow the entrance of drugs into CNS
Direct intraventricular injection may be used for treatment of brain tumors
V. Intra -arterial injection:
used to localize the drug effect in a particular tissue e.g., Treatment of liver tumors and neck and head cancers
VI. Intra -articular Injection:
The drug is injected directly into the joint
11
In rheumatoid arthritis and osteoarthritis, intraarticular injection of corticosteroids is used for treatment of episodic manifestations of acute inflammation
This should be done infrequently as repeated injections may lead to painless destruction of the joint
VII. Intradermal injection:
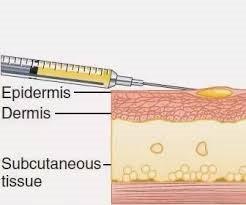
is made into skin
This method is usually used in sensitivity tests and BCG
VIII. Intraperitoneal injection:
is commonly used in experimental animals

It is rarly used clinically because the risk of infection, intestinal or vascular injury and adhesion
it was used for treatment of rabies
Topical Administration
This includes application of the drug (in lotion, ointment, cream and patch) for local effect to the skin and mucous membranes of conjunctiva, ear, nose, throat, rectum and vagina

However, significant drug absorption into the systemic circulation may occur from these sites as in:
o Intranasal administration e.g., synthetic antidiuretic hormone (desmopressin) and oxytocin are applied on to the nasal mucosa as nasal sprays
o Transdermal administration:
This route utilizes application of lipid soluble drugs to the skin for systemic effects
Absorption is dependent on the surface area over which drugs are applied and on their lipid solubility
Prolonged blood levels of some drugs can be achieved by this method because they are slowly absorbed
Nitroglycerin patches that provide sustained delivery of the drug are used for the treatment of angina pectoris
Also patches containing scopolamine placed behind the ear release sufficient drug to the systemic circulation to protect against motion sickness
Clonidine patch in hypertension
Nicotine patches for smokers
Estrogen patches for replacement therapy after menopause.
12
Inhalation
Gaseous and volatile liquids may be inhaled and absorbed through the pulmonary epithelium to produce systemic effects
Anesthetic gases and vapors are the most important drugs given by inhalation

In addition, solutions of drugs can be atomized and the fine droplets in air (aerosol) inhaled.
Advantage:
o Access to the circulation is rapid because the surface area of pulmonary alveoli is large and the blood flow through the lungs is high
o It avoids first-pass metabolism
o Aerosols of β-adrenergic agonists and corticosteroids are commonly given by inhalation to relieve bronchospasm.This actually constitutes a type of topical administration.

13
Outlines
Plasma protein binding
Tissue binding


Drug biotransformation




A variable proportion of a drug present in blood is bound to plasma proteins. chiefly albumin for acidic drugs and α1 acid glycoprotein for basic drugs. The binding is usually reversible although covalent binding of reactive drugs such as alkylating agents occasionally occurs
The binding of a drug to plasma proteins is governed by the same physicochemical factors involved in the binding of drugs with receptors i.e. drug concentration, affinity for binding sites and the number of binding sites .

The fraction of a drug that is bound to plasma proteins cannot diffuse from the vascular space and is pharmacologically inactive; only the unbound portion of a drug is pharmacologically active. Thus plasma protein binding limits the concentration of the drug in tissues and also limits its glomerular filtration although active carrier mediated transport across cell membranes is not restricted to free drug .
Free and unbound fractions are in equilibrium and free drug removed from the plasma by metabolism or excretion is replaced by drug released from the bound fraction i.e, the bound fraction is considered as drug reservoir.
1-Plasma protein binding
Pharmacokinetics P2 14
2-Tissue binding

Other structures besides plasma may bind drugs. Tissue binding of drugs usually occurs to proteins, phospholipids or nucleoproteins and is generally reversible. Less is known about tissue binding than about plasma protein binding because tissue samples can be obtained only by invasive biopsy .
Displacement of drugs from tissue binding sites may be a mechanism for drug interaction. When quinidine is given to patients who are receiving digoxin, the plasma concentration of free digoxin may double because quinidine displaces digoxin from tissue binding sites it also displaces digoxin from plasma protein binding sites as well as impairing the renal excretion of digoxin.
DRUG BIOTRANSFORMATION (Drug Metabolism)






o Biotransformation (metabolism) is a general term serves to convert hydrophopic chemical or xenobiotics into hydrophilic derivatives that easily eliminated through urine or bile. Thus affect the drug in two major ways :
1-Reducing lipid solubility
Drugs that are already water soluble are generally excreted unchanged by the kidney. Lipid soluble drugs are not easily excreted by the kidney because following glomerular filtration they are reabsorbed from the proximal tubule. Biotransformation converts these lipid soluble drugs to more polar and water soluble compounds that are more readily excreted by the kidney .
15
2-Alteration of biological activity
The end result of biotransformation is the abolition of biological activity. Various steps in between may have the following consequences :
a) conversion of a pharmacologically active to an inactive compound; this is the case with most drugs .
b) conversion of a pharmacologically active to another active compound e.g., codeine can be changed to morphine and diazepam to oxazepam. Both the drug and its metabolite are active .
c) conversion of a pharmacologically inactive to an active compound. When the metabolite itself is the active drug, the parent compound is said to be a prodrug e.g., enalapril (inactive) is converted to enalaprilat(active) and minoxidil minoxidil sulfate (active).


Sites of biotransformation

o The liver is the major site for drug biotransformation although a number of tissues including the kidney e.g. insulin and vitamin D, gut mucosa, lung and skin also contribute .
1)The majority of drugs are metabolized by the hepatic microsomal enzymes. By electron microscopy the cells of the liver and other tissues can be shown to contain a network of lipoprotein tubules distributed throughout the cytoplasm. The membranes of this endoplasmic reticulum when isolated by homogenization and fractionation of the cell, they reform into vesicles called microsomes .
These microsomes retain most of the morphologic and functional characteristics of the intact membranes.
16
One surface of these microsomes is covered with globules of RNA ribosomes) and is called rough endoplasmic reticulum; it is responsible for protein synthesis. The other surface is smooth and is called smooth endoplasmic reticulum; it contains the enzymes responsible for oxidative drug biotransformation particularly the mixed function oxygenases which comprises a large family of isozymes called cytochromes P450 .
2) Some drugs are metabolized by a variety of enzymes involved in intermediary metabolism (non-microsomal enzymes) located intracellularly (e.g. in the cytosol or mitochondria) or extracellularly e.g. in plasma.
Ethanol is inactivated by alcohol dehydrogenase



succinylcholine by plasma pseudocholinesterase,
6-mercaptopurine by xanthine oxidase and epinephrine by monoamine oxidase.
Pathways of drug biotransformation
Lipid solubility is an important requirement for a drug to be metabolized by hepatic microsomes since this property favors the penetration of the drug into the endoplasmic reticulum and the binding with the cytochrome P450 .
Phases of drug biotransformation
Phase l or Non Synthetic Reactions
Phase II or Synthetic Reactions (Conjugation)

17
1-Phase l or Non Synthetic Reactions
This phase consists of reactions that convert the parent drug to a more polar metabolite by introducing a polar functional group such as –OH, orNH2 by oxidation, reduction or hydrolysis.
o The phase I reactions, most frequently involved in drug metabolism, are catalyzed by the cytochrome P-450 system of heme-containing isozymes that are located in most cells but found mainly in the liver and intestinal tract. The CYP system is important for metabolism of many endogenous compound (steroid, prostaglandins, vitamins and free fatty acids,etc.),and for the detoxification of exogenous substances.
o There are multiple cytochromes P450 families which differ in amino acid composition, in substrate specificity and in sensitivity to inhibitors and to inducing agents .
o A number of drugs and environmental factors can induce (increase) or inhibit the microsomal enzyme system .
1.Cytochrome P-450(CYP) is composed of many families
2.CYPs have various forms (isoenzymes) such as CYP 3A4 and CYP 2D6.
3.CYP3A4 (family 3 subfamily A and gene number 4) is most abundant CYP involved in drug metabolism.
These reactions usually represent only the first stage of biotransformation. Metabolic products formed may subsequently undergo phase II or synthetic reactions prior to elimination.
Unlike the conjugated products or synthetic reactions. The metabolites of non-synthetic reactions may be pharmacologically active.

18
1-Oxidation reaction
-Oxidation reaction are carried out by CYPs, Flavin monooxygenases, (FMO) and epoxide hydrolase (EH)
-The key step in oxidation reactions is the insertion of one atom of molecular oxygen into the substrate, often producing an unstable intermediate that breaks down to yield the final product
a-microsomal
oxidation
Most oxidative reactions are carried out by cytochromes P450 isozymes which are located mainly in the liver and principally in the smooth endoplasmic reticulum (microsomal fraction). These enzymes have an absolute requirement for molecular oxygen and NADPH .
o Some types of microsomal oxidation reactions are
1. O-Dealkylation e.g., codeine.
2. N-dealkylation e.g. desipramine and diazepam.
3. Aromatic hydroxylation e.g., phenytoin, phenobarbital.
4. N-oxidation e.g. guanethidine.
5. S-Oxidation e.g., cimetidine.
6. Sulfoxide formation e.g. chlorpromazine .
7. Deamination of amines e.g. amphetamine


8. Desulfuration e.g. thiopental
19
b-non-microsomal oxidation:
Enzymes found in the cytosol or mitochondria of cells are responsible for the biotransformation of relatively few compounds. They are, however important and few examples include :
i-alcohol dehydrogenase and aldehyde dehydrogenase which oxidize ethanol to acetaldehyde and then to acetate respectively .



ii-xanthine oxidase which converts hypoxanthine to xanthine and xanthine to uric acid .
iii-monoamine oxidase which is important for the metabolism of catecholamines and serotonin .
iv-Flavin monooxygenases (FMO) which catalyze oxidation of drugs e.g., desipramine.
2-Reduction reactions:
Reduction occurs in both microsomal and non-microsomal enzyme systems; it is less common than oxidation. A notable example is the reduction of the nitro group in case of chloramphenicol and clonazepam.
3-Hydrolysis reactions:
Non microsomal hydrolases exist in a wide variety of body systems including the plasma. Examples of non-microsomal hydrolases are :
i-Estrases catalyze hydrolysis of esters as in procaine and aspirin.
ii-Amidases catalyze hydrolysis of amides as in lidocaine and indomethacin .
20
(B)Phase II or Synthetic Reactions (Conjugation)
The parent drugs or intermediates formed by phase reactions undergo coupling with an endogenous substrate such as glucuronic acid, glycine, glutathione, inorganic sulfate, methyl groups or acetyl groups to yield conjugation products. It is generally assumed that conjugation reactions lead to inactive conjugates.
Usually a conjugate is more polar and more readily water soluble that is rapidly excreted in urine and bile. In the intestine, part of the conjugated product that is excreted via the bile may also by hydrolyzed by βglucuronidases of intestinal flora with subsequent reabsorption of the active molecule (enterohepatic circulation). This can greatly extend the action of the drug .
The most important conjugation reactions are :
1 glucuronic acid conjugation: this is the most common synthetic reaction. Unlike most phase II reactions which occur in the cytosol glucuronic acid conjugation occurs in the liver microsomes. Examples are: morphine. paracetamol and chloramphenicol .
2.acetylation e.g., isoniazid, sulphonamide


3.glycine conjugation e.g., salicylic acid.
4.conjugation with sulfate e.g., steroids.
5.methylation e.g., epinephrine and captopril.
Not all drugs undergo phase I and then phase II reactions in that order. for example, isoniazid is first acetylated (phase II reactions) and then hydrolyzed to isonicotinic acid (phase I reaction). This called reversal of order of phases.
21
Factors influencing drug biotransformation:
1.Genetic Polymorphism

Genetic variation in enzymes that catalyze drug biotransformation reactions. This results in enzyme subpopulations with different drug metabolizing abilities i.e. genetic polymorphism. Accordingly, in some individuals drug biotransformation may occur so rapidly (fast metabolizers) that therapeutically effective blood and tissue levels are not achieved. In others biotransformation may be so slow (slow metabolizers) that toxic effects result with usual doses. Such individual variations might be explained by genetically determined differences in the amount of key enzyme cytochrome P450 available in the liver and differences in the affinity of the enzyme for substrates.


For example:
1.phenytoin blood levels vary from 2.5 to> 40 ug/ml in different persons given the same dose.
2. • Acetylation polymorphism is considered one of the first known examples of genetic defect in drug biotransformation. The incidence of slow acetylators has been found to vary remarkably in different ethnic groups; it is about 8085% in Egyptians, 30% in Caucasians and 10% in Japanese. The importance of acetylator status to therapy is illustrated by the example of isoniazid which may cause peripheral neuropathy in slow acetylators (isoniazid promotes excretion of pyridoxine and so pyridoxine is added to the antituberculous regimen to prevent this.
3. • Succinylcholine is less metabolized in individuals who have defects in pseudocholinestrase.
22
2-Age
-Newborn infants (premature or full term) metabolize drugs at a much slower rate than that observed in adults. This is due to impaired maturation of the various drug metabolizing enzyme system and is the basis for the increased toxicity in the neonate of drugs such as: -
• Impairment of chloramphenicol glucuronidation in newborn (gray baby syndrome) and some opioids as morphine glucuronidation (respiratory distress syndrome).
• Impairment of bilirubin glucuronidation contributes to hyperbilirubinemia of newborns (Gilbert’s syndrome).
-On the other hand, administration of vasodilator drug nifedipine to patients with old age (>65years) leads to delay in its hepatic metabolism with tendency of the drug to accumulate and cause hypotension.
-The capacity for biotransformation increases during the early months of postnatal life although the pattern for different enzymes is variable .
-The enzymes for phase I reactions are usually near adult activities after few months whereas phase II enzymes develop more slowly (2-3years) .
3-Liver microsomal enzyme Inducers or Inhibitors
-The activities of hepatic microsomal drug metabolizing enzymes are influenced by dietary and nutritional factors. hormonal changes in the body, ingestion of various chemical agents as well as environmental factors.
- An increase in the activity appears to represent an increased concentration of enzyme protein and is referred to as enzyme induction. Agents capable or inducing effects are very numerous and there is no apparent relationship between either their action or structure and their ability to induce enzymes. Among enzyme inducers are phenobarbital, polycyclic hydrocarbons, DDT, phenytoin ,tolbutamide, rifampicin. etc. It is worth mentioning that some inducers stimulate their own metabolism.




23
Some drugs can also produce reduction in the activities of drug metabolizing enzymes i.e. metabolic inhibition. Examples of enzyme inhibitors are cimetidine, ethanol (chronic ingestion), aspirin, paracetamol, valproic acid .
4-Pathological Conditions
Liver disease, cardiac insufficiency, obstructive lung disease, hypoxemia can reduce the various metabolic activities of the hepatic microsomal enzyme systems.
5-Nutritional status



Malnutrition decreases activity of microsomal enzyme systems.
24
Outlines
DRUG EXCRETION
THERAPEUTIC DRUG MONITORING
PHARMACOKINETIC VARIABLES
DRUG EXCRETION
The kidney and gastrointestinal tract are the major routes of drug excretion of most non-volatile water soluble substances.
Inhalational anesthetics are, however, excreted mainly by the lungs and few drugs are secreted by the salivary glands.
Drugs excreted in milk may have an effect on a breast-fed child e.g.
o Atenolol
o Barbiturates
o Tetracyclines






o isoniazid.
Drugs may be excreted either in an unchanged form or as metabolites. Generally the more polar compounds are excreted unchanged. The less polar lipid soluble drugs are not readily excreted until they undergo biotransformation to more polar, less lipid soluble compounds.
Renal Excretion
The most important route of excretion of most drugs is the kidney.
Excretion of drugs in urine involves three processes:
I. glomerular filtration
II. passive tubular reabsorption
III. active tubular secretion
1
Pharmacokinetics Pharmacokinetics P3 25
Glomerular filtration:The amount of drug entering the tubular lumen by filtration is dependent on the filtration rate and the degree of plasma protein binding. Protein binding reduces the amount of the drug available for glomerular filtration
Passive tubular reabsorption: The pH of urine exerts an influence on the excretion of certain weak acids and bases.
In the proximal and distal tubules:
o the non-ionized forms (which are lipid soluble) of weak acids and bases undergo net passive reabsorption



o the ionized form (which is water soluble and non-diffusible) is excreted in urine.
In alkaline urine a large portion of weak acids (e.g., salicylates and barbiturates) is ionized; thus, its tubular reabsorption is minimal and is quickly excreted .
Similarly weak- bases (as amphetamine and meperidine) are more rapidly excreted in acid urine.
The action of these substances in the body can, however, be prolonged if the urinary pH is not favorable for their excretion.
Active tubular secretion: Transport mechanisms for active tubular secretion exist in the proximal tubule and they transfer certain organic acids and bases from the plasma to the tubular fluid. There are two separate transport mechanisms:
o one for acids (e.g., penicillin, furosemide)
o one for bases (amphetamine, amiloride).
The various organic acids compete with each other for the same site of secretion e.g., probenecid competes with penicillin preventing its proximal tubular secretion, thus prolonging its duration of action. Organic bases also compete with each other but not with organic acids.
26
The biliary system acts as a specialized transport mechanism for the intestinal excretion of drugs especially those conjugated with glucuronic acid in the liver.
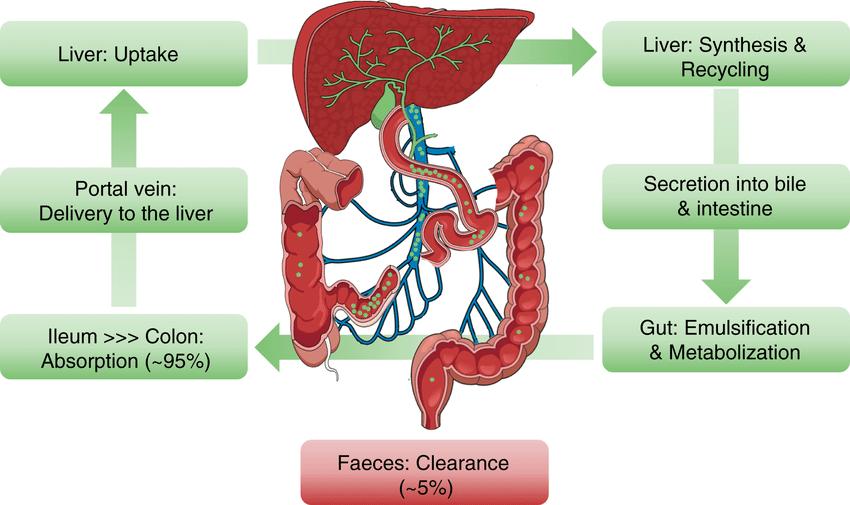
Specialized mechanisms also exist for the active transport of acids and bases from blood to bile and they closely resemble those of the renal proximal tubules .


Drugs and their metabolites that are secreted into bile are carried by the biliary ducts to the duodenum. Glucuronides that are eliminated in bile can be hydrolyzed by glucuronidases in the intestine and the free drug can be reabsorbed (enterohepatic circulation).This responsible for prolongation of half-life of some drugs e.g. estrogens.


EXCRETION
Glomerular filtration Passive tubular reabsorption Active tubular secretion
Renal
Biliary
drug monitoring (TDM) is the measurement of drug plasma level
Therapeutic
27
Biliary Excretion enterohepatic circulation THERAPEUTIC DRUG MONITORING
Indication
I. drugs with low therapeutic index where the therapeutic dose is close to the toxic dose as e.g.,
o cardiac glycosides
o antidysrhythmic drugs

o antiepileptic drugs
o Lithium
o aminoglycoside antibiotics.
II. failure of therapeutic response at a dose that is expected to be effective as in patient noncompliance and incomplete drug absorption.
III. in the event of multiple drug therapy with the possible occurrence of drug interactions.
IV. in presence of organ disease as hepatic or renal disease that adversely affect the pharmacokinetic processes.
V. to diagnose drug toxicity.
not indicated in the following conditions
I. For some drugs whose dose can be correlated with an easily measured effect as with
o antihypertensives (blood pressure measurements)
o oral anticoagulants (prothrombin time)
o hypoglycemics (blood sugar) plasma drug concentration is not worth measuring.
II. For other drugs that act irreversibly as MAO inhibitors, some anticholinesterases and cytotoxic drugs. their effect persists after the drug has left the plasma. They are named "hit and run drugs" and in such cases plasma concentration may have no correlation with effect.
III. For drugs that have more than one action depending on the dose used e.g.,
o nortriptyline in small dose inhibits the amine pump
o in large dose produces αadrenergic blockade
IV. Drugs that have active metabolites as
o some benzodiazepines plasma concentration poorly correlates with effect
28
PHARMACOKINETIC VARIABLES
Drug disposition is a term used to describe the movement of a drug throughout the body; it involves

o Absorption
o Distribution



o Biotransformation
o excretion
These pharmacokinetic parameters may differ from individual to individual according to several physiological (e.g. age) and pathological (e.g. liver, kidney or heart disease) processes.
These pharmacokinetic differences require dose adjustment in individual patients. The pharmacokinetic parameters that are of importance in this respect are
o Bioavailability
o Clearance
o volume of distribution
o elimination half-life
Bioavailability
Bioavailability (F) is the fraction (%) of the dose given that reaches the systemic circulation in unchanged form.
Also drug metabolities if active when enter the systemic circulation considered bioavailable.
It has a value between 0 and 1(0%-100%).
In case of I.V. administration it is equal to 1(100%).
For drugs administered orally, bioavailability may be less than 100% for two main reasons :
o poor absorption
o enhanced first pass metabolism.
First pass or presystemic metabolism refers to the metabolism of a drug that occurs in route from the gut lumen to the systemic circulation.
When a drug is absorbed across the GIT, it enters the portal circulation. Some drugs are metabolized in the gut wall but in most cases first pass metabolism occurs in the liver e.g., with lidocaine first pass hepatic metabolism is nearly complete and its bioavailability following oral administration is zero.
29
Clearance (CL) represents the volume (V) of biological fluid such as blood or plasma that would be completely free of the drug.

Clearance of a drug is the rate of elimination by all routes normalized to the concentration (C) of the drug in the biological fluid.
So clearance is a measure of the ability of the body to eliminate the drug and is expressed in units of volume per time (ml/min).
Plasma clearance is thus defined as the theoretical volume of plasma from which the drug is completely removed in a given period of time.
Two major sites of drug elimination are
o Kidney
o Liver
renal clearance represents clearance of unchanged drug in the urine.
In liver,drug elimination occurs via:
o biotransformation of parent drug to one or more metabolites OR


o excretion of unchanged drug into bile OR
o both.
Other routes of elimination could include loss of drug in saliva or sweat, secretion in GIT, volatile elimination from the lung and metabolism at other sites such as skin.
Total clearance can be calculated using the following equation:
Clearance of most drugs is usually constant over the range of concentrations encountered clinically. This means that elimination is not saturated and the rate of elimination of a drug is directly proportional to its concentration in plasma.
Elimination of drugs from the body follows one of two general patterns of elimination kinetics:
I. First-order kinetics
II. Zero-order kinetics.
CLsystemic=CL renal+CL hepatic+CL others
Clearance
kinetics 30
CL=Rate of elimination/Plasma concentration
First-order kinetics:
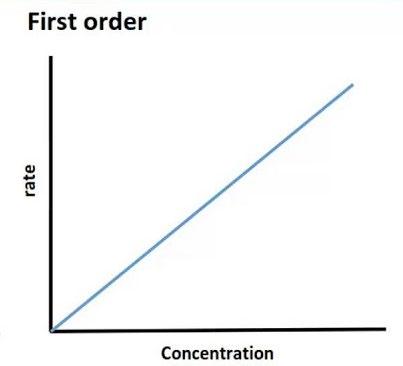
o Elimination of most drugs follows first order kinetics where the rate of elimination is directly proportional to the concentration of drug in plasma
o i.e.fixed % of drug is eliminated/time(linear relationship) that make clearance of the drug is a constant value
o Clearance (CL)=rate of elimination/plasma drug concentration (PDC)
o For example, If PDC increases from 0.01 to 0.05mg/L, and the rate of elimination increases from 0.25 to 1.25mg/h
At PDC of 0.01mg/L: CL=0.25(mg/h)/0.01(mg/L) =25L/h
At PDC of 0.05mg/L: CL=1.25(mg/h)/0.05(mg/L) =25L/h
Zero-order kinetics
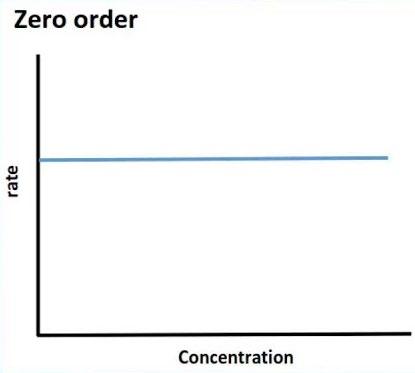
o The rate of elimination of a drug does not change in proportion to change in plasma drug concentration (nonlinear relationship). A fixed amount of the drug is eliminated per unit time but not fixed%.
o The drug is eliminated irrespective the drug concentration level.
o The drug tends to accumulate in the body when the dose is increased so the toxicity is high.
o If mechanisms of elimination of a given drug become saturated, the kinetics become zero order.
o Saturable metabolism of phenytoin is example of zero order kinetics where the rate of elimination is limited by the number of metabolizing enzymes available and once drug concentration exceeds the enzymatic capacity, the rate of metabolism of the drug will not increase even if the concentration of the drug is increased. Also, ethanol and salicylic acid in high dose are examples of drugs with zero order- kinetics


Uses of clearance:
I. Calculation of the maintenance dose:-
Maintenance dose=Steady state concentration x CL x dose interval
II. It gives an idea about the healthy state of the eliminating organ for that drug.
31
Volume of distribution
Volume of distribution (Vd) It is the apparent volume that would be required to contain all of the drug in the body at the same concentration as in plasma assuming the body acts as a single homogenous compartment for the drug.
It does not refer to real physiologic or anatomic volumes, so name” apparent”. It relates the amount of the drug in the body (A) to the concentration of the drug (C) in the blood or plasma.
The volume of distribution is determined by:
I. extent of ionization
II. degree of lipid solubility
III. degree of binding to plasma proteins
IV. degree of binding to tissue proteins.

The volume of distribution is a hypothetical volume and has no physiological or physical basis. It reflects the extent to which the drug present in extravascular tissues and not in plasma.
It can be seen from the above equation that Vd is increased when plasma concentration is decreased and vice versa. It therefore follows that high exists when the drug is greatly unionized. has high lipid solubility, has reduced binding to plasma proteins and/or has high tissue binding.



Clinical Relevance(importance) of Vd
1. When a drug is extensively distributed outside blood i.e. has high Vd , it is not easily removed from the body by hemodialysis e.g. 95% of digoxin in the body is distributed in muscle and adipose tissue leaving very small amount in plasma (Vd 500 L/70kg). In case of digoxin poisoning there is no advantage to speed elimination by hemodialysis but specific binding antibodies will be of considerable value.
2. Drugs that are completely retained within vascular compartment have small volume of distribution e.g. aspirin (11 L/70kg) and tolbutamide (7 L/70kg). These drugs are easily removed from the body by hemodialysis.
amount of the drug in the body (mg/kg) Vd(L/kg) = plasma concentration (mg/l)
Note
32
3. When a drug is extensively distributed outside blood i.e., high Vd, it provides a reservoir from which the blood is continuously replenished between doses and so fluctuations in blood concentrations are minimal
4. Uses of Vd:
to calculate the loading dose (LD): In case of some drugs e.g., digoxin it is necessary to achieve therapeutic levels within hours instead of days to get a rapid response and so we have to give a loading dose (LD)
LD =Vd x Css…………………... (1)
where: LD= loading dose
Vd =volume of distribution
Css = plasma steady state concentrations where the rate of administration equals the rate of elimination
to estimate plasma clearance (CL)
CL =Vd x Kel ……………………………... (2)
Where: Kel = elimination rate constant (Fraction of drug eliminated per time) = 0.693/t1/2
So, equation (2) can be rewritten:
CL =Vd x 0.693/tI/2…………………... (3)
to predict the steady state concentration in plasma which occurs with repetitive dosing from the kinetic behavior of a single dose.
Css = F.D/ Vd. Kel.t …………………………. (4)
where: Css =steady state concentration
F= bioavailability
D= dose
t = dose interval
From equations (2) and (4) we can suhstitute Vd Kel by CL and equation (4) can be rewritten as follows:
Css =F.D / CL .t………………................ (5)
Biological Half Life
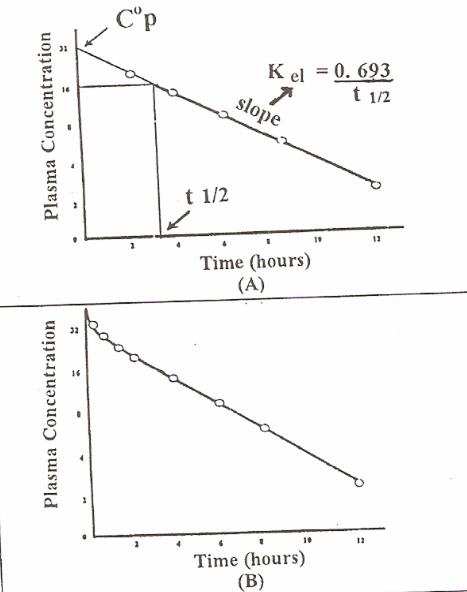
Biological Half Life (t1/2) It is the time required for the plasma concentration to fall to one half its original plasma concentration.
It can be determined either:
I. Graphically from the plot of log concentration versus time. It is the time period required for drug concentration to fall half its concentration


33
II. Mathematically T1/2=0693/Kel since Kel=CL/Vd T1/2= 0.693xVd/CL
Equation (3) clearly demonstrates that t ½ depends on both
o clearance
o volume of distribution
Value of calculating the half-life
As a rule steady state concentration is attained in 4-5 half-lives (of contious drug administration). Once the drug is stopped elimination is usually complete after 4-5 half- lives (i.e 95% the drug disapears from the body after 4-5 half- lives) .
The half-life of a drug has an important on the selection or dosage interval for maintenance therapy
Two situations illustrate this:
I. If the dosage interval is much longer than t½ - the drug is effectively eliminated before the next dose and each dose represents a new single dose.
II. 2-If the dosage interval is much shorter than t½ a true steady state is achieved with very minimal fluctuations in plasma levels.
34
PHARMACODYNAMICS
Outlines
✓ Drug receptor interaction
✓ Drug potency
✓ Drug receptors
Definition
It is the study of the biochemical and physiological
• effects of drugs on the body
• the mechanisms of drug action
• the relationship between drug concentration and effect
Drug Receptor Interactions
• Binding of a drug with its receptor formation of drug receptor complex (DR) which is responsible for triggering the biological response.
D + R = (DR) → Response

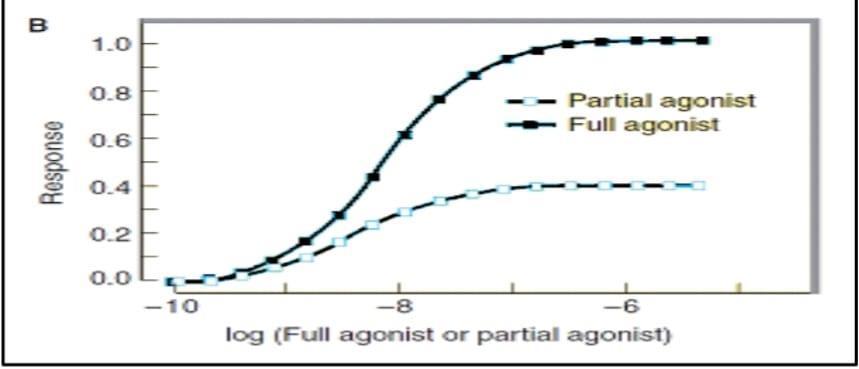
Fig 2 .When each of the two drugs is used alone and response is measured, occupancy of all the receptors by the partial agonist produces a lower maximal response than does similar occupancy by the full agonist
2 35
Drug Potency depend on Intrinsic activity or efficacy Affinity
the ability of the drug-receptor complex to produce a pharmacologic response.
-The intrinsic activity of a full agonist can be set equal to 1
-that of pure antagonist 0

-that of partial agonist to a value between 0 and 1
tendency of the drug to bind to receptors
Agent which activates a receptor to produce an effect similar to that of the physiologic signal molecule such as neurotransmitters, hormones and autacoids
Have both affinity as well as high intrinsic activity, therefore can trigger the maximal biological response.
Agent which prevents the action of an agonist on a receptor but doesn’t have any effect of its own
Agent which activates a receptor to produce a sub maximal effect. OR drugs bind to receptors and activate them but do not evoke a great response as full agonists
Have only affinity but no intrinsic activity. These drugs bind to the receptor and block the binding of an endogenous agonist.
Have affinity but with low intrinsic activity and hence are only partly as effective as agonists.
Agent which activates a receptor to produce an effect in the opposite direction to that of the agonist
Have affinity but intrinsic activity ranges between 0 to -1.
Agonist Antagonists Partial agonists Inverse agonists (Negative)
36
Example of Inverse agonists






Benzodiazepines act on specific binding sites in the CNS producing antianxiety and anticonvulsant effects. Other substances called “β-carbolines” also bind to the same binding sites blocking the effects of benzodiazepines but at the same time produce anxiety and seizures).
Note:-
Drugs will never create a new function to the cell but rather will modulate an already existing function e.g., there is no drug whatsoever that can let the thyroid gland to secrete insulin but what a drug does is to increase or decrease the secretion of thyroid hormones
Drug-Receptors
Definition
component of the cell that interacts with a drug and initiates the chain of events leading to the drug’s observed effects.
Functions
1) Ligand binding: Ligands are molecules which attach selectively to a particular receptor.
2)Message propagation of the regulatory signal in the target cell
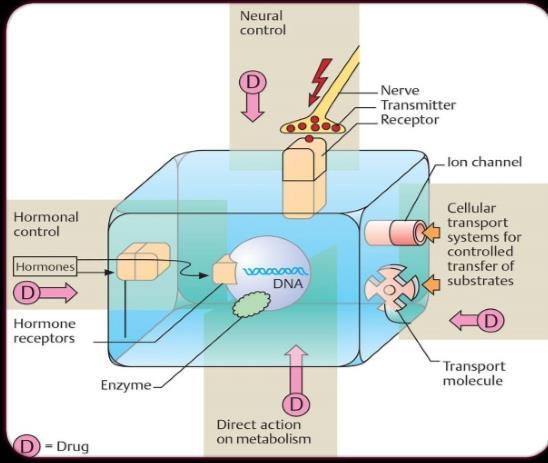
Fig 3. Transmembrane signaling mechanisms: 1. Lipid soluble chemical signal cross the plasma membrane and act on intracellular receptor. 2. The signal binds to the extracellular domain of a transmembrane receptor bound to a protein tyrosine kinase. 3. The signal binds to and directly regulates the opening of an ion channel. 4. The signal binds to a cell surface receptor linked to an effector enzyme by a G protein. R, receptor; G, G protein; Y, tyrosin

37
Types of receptors
1 Receptors linked to tyrosine kinase
These are transmembrane receptors that have two domains:
➢Extracellular domain for drug binding

➢Cytoplasmic domain with tyrosine kinase activity. Binding of the drug to the extracellular domain simulates the activity of tyrosine kinase of its cytoplasmic domain. This produces phosphorylation of target proteins on tyrosine residues.
➢Example: insulin.
Intracellular Receptors for Lipid-Soluble Agents
Several biologic ligands are sufficiently lipid-soluble to cross the plasma membrane and act on intracellular receptors
➢Example: steroids and thyroid hormone.
Do not bind neurotransmitters directly but are controlled by membrane potential; such channels are also important drug targets.
➢For example, verapamil inhibits voltage-gated calcium channels that are present in the heart and in vascular smooth muscle, producing antiarrhythmic effects and reducing blood pressure without mimicking or antagonizing any known endogenous transmitter.
4 Ligand Gated Channels
Many of the most useful drugs in clinical medicine act by mimicking or blocking the actions of endogenous ligands that regulate the flow of ions through plasma membrane channels.The natural ligands are acetylcholine, serotonin, GABA, and glutamate. All of these agents are synaptic transmitters.
➢Example: Nicotinic cholinergic receptors
2 Voltage-gated ion channels 3
38
5 G-Protein-Coupled Receptors
The drug binds with a cell surface receptor which triggers the activation of a Gprotein (Guanine nucleotide binding protein) located on the cytoplasmic face of the cell membrane: This, in turn, regulates the activity of
•Certain enzymes (adenylcyclase,phospholipases)
•Ion channels that change the concentration of an intracellular second messenger such as cAMP, inositol triphosphate, diacylglycerol or Ca++.
The family of G-proteins is quite diverse. In cases of adenylcyclase there is Gs, that stimulates and Gj that inhibits the enzyme. Examples: muscarinic cholinergic receptors, adrenergic receptors, dopaminergic receptors, 5HT receptors, opiate receptors and others.
MECHANISMS OF DRUG ACTION
Binding to specific receptors
➔Most drugs act by binding to specific receptors located on
1- Cell membrane as
-adrenoceptors
-cholinoceptors
-histamine receptors
2- Inside the cells
-steroid receptors
➔This is the most important mechanism of drug action.
Physical or physicochemical
➔Drugs may act through their physical or physicochemical properties (the purgative magnesium sulfate and the diuretic mannitol owe their action to osmotic effects.
Enzymes
➔Enzymes may act as targets for drug actions.
➔Drugs can produce effects on enzyme reactions by - substrate competition - reversibly or irreversibly modifying the enzyme.
Example
-aspirin inhibits cyclooxygenase
-allopurinol inhibits xanthine oxidase
-trimethoprim inhibits dihydrofolate reductase.
Direct chemical interaction
➔Some drugs may act by direct chemical interaction as the antacids which neutralize gastric acidity and the chelating agents which bind a variety of metal cations.
39
Drug antagonism
➢ This occurs when an antagonist prevents an agonist from interacting with its receptors to produce an effect.
➢ A receptor "antagonist" binds to the appropriate receptor but without activating it because it does not have "intrinsic activity".
➢ Receptor antagonists are classified into two main types
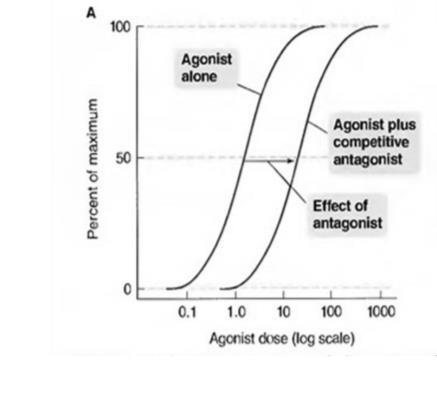
Competitive Antagonists
➢In the presence of a fixed concentration of an agonist, increasing the concentration of a "competitive antagonist" progressively inhibits the agonist response and high antagonist concentration completely prevents the response.
➢Conversely, sufficiently high concentrations of the agonist can completely prevent the effect of a given concentration of the antagonist.
➢Since the antagonism is "competitive", the presence of the antagonist increases the agonist concentration required to produce a certain degree of response. This will lead to a "parallel shift" of the agonist concentration-effect curve to the right.
➢ Examples: prazosin, propranolol.
Non-Competitive Antagonists
➢These antagonists bind to receptors irreversibly either because:
1)The antagonist affinity is so high that the receptor is unavailable for binding to the agonist.
2)the antagonist forms stronger covalent bonds with the receptor.
➢After occupying a substantial proportion of receptors by the irreversible antagonist, the number of remaining unoccupied receptors becomes so low that high concentrations of the agonist cannot overcome the antagonism and a maximal agonist response cannot be attained.
➢In the presence of an irreversible antagonist, a downward shift of the concentration-effect curve of the agonist is produced.
➢Example: phenoxybenzamine







 Receptor antagonism
Physiological antagonism
Chemical antagonism Pharmacokinetic antagonism
Competitive Non- competitive
Receptor or Pharmacological antagonism Antagonism
Receptor antagonism
Physiological antagonism
Chemical antagonism Pharmacokinetic antagonism
Competitive Non- competitive
Receptor or Pharmacological antagonism Antagonism
40
Fig 4. Agonist dose response curve in the presence of competitive and Non-Competitive Antagonists.
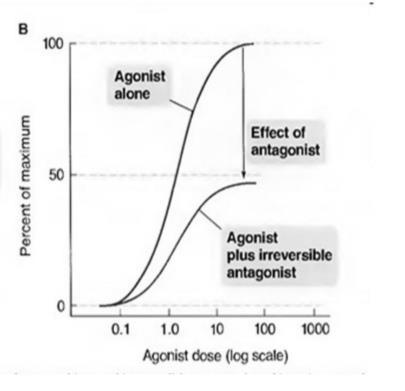
A. Competitive Antagonists has an effect illustrated by the shift of the agonist curve to the right.
B. Non-Competitive Antagonists has an effect illustrated by the shift of the agonist curve downward
Physiological or Functional Antagonism
➢Occurs when two compounds produce opposite effects on the same physiological function but act independently on two different receptors.
➢Example: epinephrine is used to physiologically antagonize the action of histamine in anaphylactic shock (it opposes the bronchoconstriction, vasodilatation and hypotension caused by histamine release through physiological antagonism on different receptors).
Chemical Antagonism
➢This occurs when two substances chemically interact to neutralize the action of each other.
➢Example: Protamine is positively charged at physiologic pH and is used clinically to counteract the effects of heparin which is a negatively charged anticoagulant.

Pharmacokinetic or Dispositional Antagonism
➔This is the alteration of the disposition of a substance (absorption, distribution, biotransformation or excretion) by another substance so that less of the agent reaches the target organ.
41
Pharmacodynamics P2
Outlines
Receptorregulation
Variationindrugresponse
Drugconcentrationandresponse
Therapeuticindex
Undesirabledrugeffects
Druginteractions
Drugdosage
Receptordown-regulation


Continuedstimulationofcellswithagonistleadstoastateofdesensitization,also referredas“downregulation”.
Thiswillcauseadecreaseintheresponsetothedrug.
Example:Repeateduseofβ-adrenergicstimulantdrugsfortreatmentofbronchial asthmaleadstoadiminishedresponseduetodecreaseinthenumberofβadrenergicreceptors.
Differentmechanismsaresuggestedforthisphenomenon:

1.Attenuation(decreasedsensitivity)ofthesignalfromthestimulatedreceptor.
2.Feed-backregulationofthesynthesisofthereceptor.
2 42
Receptorup-regulation

Long-termexposureofcellstoantagonists frequentlyleadstohyperreactivityor supersensitivitytoreceptors.
Example:abruptwithdrawalofa(βadrenergic antagonist)leadstoworseningofangina pectorisorcardiacventriculardysrhythmiasin somepatients.
Thisismainlybecauseprolongedcontactofthe adrenergicreceptorswiththeantagonistmay resultinsynthesisofnewreceptors.
Remember
Agoniststendto desensitize receptors
Antagoniststend toupregulate receptors.
Changeinthenumberofreceptorsbyothersubstances
Thenumberofphysiologicalorregulatoryreceptorscanbechangedby anotherhormone.
Example:Thyroidhormonesareknowntoincreasethenumberofβ-adrenergic receptorsinthecardiacmuscleandtoincreasethesensitivityto catecholamines,
Thisexplainsthetachycardiainpatientswiththyrotoxicosisandtheusefulness ofβ-adrenergicreceptorantagonistsintreatingthesymptomsofthisdisease.
43
Changeinthenumberofreceptorsbyadiseasestate
Certaindiseasescanarisefrom,orcausecertaindisordersin receptorsorreceptor-effectorsystems.
Examples:
Myastheniagravis,someformsofinsulin-resistantdiabetesmellitus, testicularfeminizingsyndrome
VariationinDrugResponsiveness
Individualsmayvaryconsiderablyintheir responsetoadrug.
Asingleindividualmayresponddifferentlytothe samedrugatdifferenttimesduringthecourseof treatment.
Individuals,occasionallyexhibitanunusualor idiosyncraticdrugresponse.
Quantitativevariationsindrugresponsearein generalmorecommonandmoreclinically important.
Theterm hypersensitivity usuallyrefersto allergicorother immunologic responsestodrugs.
Anindividualpatientishyporeactiveor hyperreactivetoadruginthattheintensityof effectofagivendoseofdrugisdiminishedor increasedcomparedwiththeeffectseeninmost individuals.

.
Note 44
Fivegeneralmechanismsmaycontributetovariationindrug responsivenessamongpatientsorwithinanindividualpatientatdifferenttimes:
DevelopmentofDrugTolerance
Followingtherepeatedadministrationofsomedrugs,theintensityof responsemaydecreaseduringthecourseoftreatment;thisiscalled "tolerance".
Whenthisoccursrapidlyafterdrugadministration,thefirstdoseorthefirst fewdoses,thisiscalled"tachyphylaxis",e.g.,amphetamine.
pharmacodynamic
adaptationorcellulartolerance
pharmacokinetic
drugdispositiontolerance:
✓Occursduetotheadaptationof thebodycellstothepresenceofthe drugsothatresponsetoagiven concentrationisreduced.
✓Occursduetoalterationofthe pharmacokineticsofdrugsfollowing theirrepeatedadministration.
✓Example:morphine,barbiturates.
✓Inductionoflivermicrosomal enzymesresponsibleforthe biotransformationofdrugsisoneof themajorfactorsimplicatedinthe developmentofpharmacokinetic tolerance.
✓Example:alcoholand barbiturates.
45
AlterationinConcentrationofDrugThatReachestheReceptor
Insomecases,thechangeinreceptornumberiscausedbyotherhormones;
forexample,thyroidhormonesincreaseboththenumberofβreceptorsinrat heartmuscleandcardiacsensitivitytocatecholamines.
Similarchangesprobablycontributetothetachycardiaofthyrotoxicosisin patientsandmayaccountfortheusefulnessofpropranololinameliorating symptomsofthisdisease.
Inothercases,theagonistinducesdown-regulationordesensitizationofits receptors.
ChangesinComponentsofResponseDistaltotheReceptor
Includetheageandgeneralhealthofthepatientandmostimportantlythe severityandpathophysiologicmechanismofthedisease.
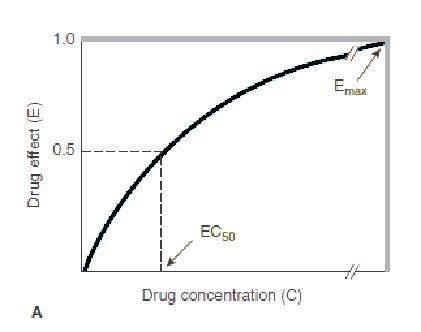
RELATIONBETWEENDRUG CONCENTRATION&RESPONSE
Generally,drugresponseincreasesindirect proportiontotheadministereddose.
Asthedoseincreases,theincrementalresponse diminishes.
Finally,dosesmaybereachedatwhichnofurther increaseinresponseisachieved.
The"hyperbolic"curvesshownincorresponding figureindicatethatanagonist
actsbybindingto,oroccupyingadistinctclassof biologicmoleculesorreceptorsduetoa characteristicaffinity.
Whenalltheavailablereceptorsorbindingsites areoccupied,nofurtherincreaseinresponseis attained
46

CHECK OUR PLATFORMS
