MANUAL PARA LA PRODUCCION DE TRICHODERMA SPP

PROYECTO: ESTUDIO DE Trichoderma spp. EN VIVEROS FORESTALES DE LA PROVINCIA DE CHIMBORAZO Y SUELOS AGRICOLAS EN SANTACRUZ, ISLAS GALAPAGOS. Ing. Daniel Román Dr. Pablo Álvarez Dr. Henri Herrera Ing. Álvaro Rivera Ing. Juan Guerra Riobamba – 2022 ESPOCH
INDICE
..........................................................................................................................7
..................................................................................7
..........................................................................................................................9
..........................................................................................................10
Materiales
..............................................................13 MATERIALES....................................................................................................................13 Procedimiento 13 PROCESAMIENTO DE MUESTRAS EN LABORATORIO 14 MATERIALES....................................................................................................................14 PROCEDIMIENTO.............................................................................................................14 SIEMBRA POR DILUSIONES SERIADA....................................................................................15 MATERIALES....................................................................................................................15 PREPARACION DE SOLUCIONES Y MEDIOS DE CULTIVO. ..................................................15 MEDIO PDA: 15 PREPARACIÓN DE SOLUCIÓN MADRE DE SUELO 16 PREPARACIÓN DE CAJAS PETRI 17 DISPENSADO DE MEDIO DE CULTIVO...............................................................................17 PREPARACIÓN DE BLANCOS DE DILUCIÓN .......................................................................18 PROCEDIMIENTO DE DILUSIONES SEREADAS Y SIEMBRA 18
INTRODUCCIÓN 4 EL GENERO TRICHODERMA 5 Mecanismos de acción. 5 Micoparasitismo 6 Competencia.....................................................................................................................6 Antibiosis
Inducción de resistencia en plantas.
LABORATORIO
Normas generales 9 Normas personales 9 Normas de seguridad............................................................................................................9 Asepsia del Laboratorio.......................................................................................................10 Lavado de materiales
Desinfección de ambiente. 10 AISLAMIENTO DE TRICHODERMA 11 FASE DE RECOLECCION DE MUESTRAS DE SUELO 11
11 Procedimiento 11 FASE DE RECOLECCIÓN DE MUESTRAS VEGETALES
MATERIALES
MATERIALES....................................................................................................................21 PROCEDIMIENTO.............................................................................................................21
MATERIALES: 22 Procedimiento 22 IDENTIFICACION
MORFOLÓGICA .....................................................................23 MATERIALES....................................................................................................................23 PROCEDIMIENTO
IDENTIFICACIÓN
Extracción
PCR
Electroforesis
Secuenciación. ................................................................................................................26
MATERIALES
PREPARACION
INOCULACIÓN .................................................................................................................29 INCUBACIÓN ...................................................................................................................29 PREPARACIÓN
............................................................................................30 CALIBRACIÓN DE
PRODUCTO FINAL 31 ANEXOS 32 BIBLIOGRAFÍA .........................................................................................................................34
PURFICACION DE LOS AISLADOS SELECCIONADOS 20
20 PROCEDIMIENTO.............................................................................................................20 CULTIVO MONOSPORICO....................................................................................................21
CONSERVACION DE AISLADOS.............................................................................................22
CULTURAL Y
23
MOLECULAR 24
del ADN genómico 24
25
26
Análisis de secuencias......................................................................................................26 Análisis Filogenéticos 27 PRODUCCIÓN DE TRICHODERMA 28
28
DE SUSTRATO...........................................................................................28
DE INÓCULO
CONCENTRACIÓN.................................................................................30
INTRODUCCIÓN
En la agricultura convencional moderna, los fungicidas son la principal herramienta empleada para el control de hongos fitopatógenos. El uso continuado de estos productos químicos para el control de estos microorganismos a provocado a través del tiempo el desarrollo de resistencia a los fungicidas utilizados. Por ello, la tendencia actual ha sido racionalizar el uso de fungicidas y desarrollar nuevas alternativas de control a través del uso de agentes de control biológico. Por otra parte, se ha desarrollado a nivel Europeo y Norteamericano un importante mercado que demanda productos hortofrutícolas obtenidos en procesos limpios de producción, generados en sistemas agrícolas que no utilizan pesticidas sintéticos durante el proceso productivo. En este marco, en la actualidad, se han desarrollado biopreparados en base a microorganismos antagonistas como Trichoderma spp, que pueden reducir el impacto de los patógenos vegetales. Este hongo antagonista permite el control de patologías fungosas en árboles frutales, forestales y hortalizas, es posible aislarlo localmente y reproducirlo en forma masiva para su aplicación a nivel de campo. El objetivo de este manual es colaborar a la obtención y aplicación correcta del hongo antagonista Trichoderma spp.

EL GENERO TRICHODERMA
El género Trichoderma es uno de los más estudiados, entre numerosos agentes de control biológico, por sus características de antagonismo en condiciones naturales. Él se comporta como hiperparásito frente a diversos patógenos, atacando directamente y produciendo la ruptura del micelio de los hongos productores de enfermedades de las plantas.
Las especies de este género son hongos de vida libre, altamente interactivos en las raíces suelo y ambiente foliar, también se ha descrito en este género una gran capacidad de inactivar exudados originados en las semillas en germinación, que estimulan el desarrollo de propágulos de hongos patógenos de plantas en el suelo, (Howell, 2002; Harman 2004). Su estudio ha estado dirigido a aumentar la densidad del inóculo, mediante prácticas agrícolas o por introducción directa en el suelo (Chet, 1987), Es un hongo imperfecto perteneciente a la Subdivisión Deuteromycotina: Clase: Deuteromycetes (Aleuxopulus (1979)
El género Trichoderma se encuentra ampliamente distribuido en todo el mundo, encontrándose en prácticamente todos los suelos y otros hábitats naturales, especialmente en aquellos que contienen materia orgánica. Trichoderma es un colonizador secundario pudiendo ser aislado de materia orgánica descompuesta, superficie de raíces de plantas, madera en descomposición y en esclerocios o propágulos de otros hongos. (Papavizas, 1985; Howell 2002).

Mecanismos de acción.
Trichoderma como antagonista, puede tener diferentes mecanismos de acción entre los que se encuentran el micoparasitismo, competencia, antibiosis e inducción de resistencia en las plantas.
Micoparasitismo
Este proceso de micoparasitismo sería uno de los principales mecanismos involucrados en el antagonismo de Trichoderma como agente biocontrolador. Este proceso incluiría el crecimiento del antagonista hacia el patógeno, desarrollándose alrededor de éste o formando estructuras similares a ganchos o apresorios en la superficie del hospedero, que le permitirían, penetrar al interior (del patógeno) y por acción de enzimas líticas (quitinasa y ß-1,3-glucanasa) degradar su pared celular (Elad et al, 1993, Garza et al., 1997; Metcalf y Wilson 2001; Brunner et al 2003).


Competencia
La competencia por sustrato y nutrientes sería otro mecanismo que explicaría el efecto antagónico de Trichoderma. Este proceso ocurre cuando dos o más organismos requieren el mismo recurso y el uso de éste por uno, reduce la cantidad disponible para el otro. La competencia fundamentalmente se produce por recursos esenciales como carbono, nitrógeno, hierro y espacio físico. Un mecanismo que recientemente ha ganado adherentes es la competencia que se produce a nivel de la rizosfera cuando se aplican cepas de Trichoderma a las semillas, produciéndose un rápido crecimiento en conjunto con el desarrollo radicular de las plantas tratadas, compitiendo entonces a este nivel por nutrientes y espacio (Howell et al 2000; Harman 2000).
Antibiosis
La actividad antibiótica de Trichoderma se debería a la secreción de sustancia antibióticas o metabolitos que inhiben la actividad parasítica de los patógenos (Dubos, 1987). Estos metabolitos serían volátiles y no volátiles, del tipo antibióticos como viridín, trichodermin, glioviridin, gliotoxin y harzaniolide (Lumsden et al 1992). De todas estas micotoxinas la más representativa es Trichodermin que actuaría inhibiendo la actividad ribosomal de los patógenos, por lo tanto, su reproducción (Ghisalberti, 1991).

Inducción de resistencia en plantas.
Otro mecanismo propuesto para explicar la actividad biocontroladora de especies de Trichoderma, es la inducción de resistencia en las plantas hospederas tratadas con el agente biocontrolador. Este concepto ha sido fundamentado por Yedidia et al 1999 y Yedidia et al 2000, quien demostró que raíces de cucurbitáceas inoculadas con T. harzianum, presentan una respuesta defensiva tanto en las raíces como en las hojas de las plantas tratadas, demostraron que las hifas del hongo biocontrolador penetran la epidermis y corteza superior de la raíz de la cucurbitácea.

La planta responde con una marcada actividad de la enzima peroxidasa frecuentemente asociada con la producción de fungitoxinas, un incremento en la actividad quitinasa y la deposición de celulosa en la superficie interna de la pared celular. Se ha comparado esta relación a la que se establece entre vegetales y micorrizas.
Se han identificado tres tipos de compuesto producidos por cepas de Trichoderma que son responsables de inducir resistencia en las plantas, entre ellas se encuentran: proteínas con función enzimática, homólogos de proteínas, oligosacaridos y otros compuestos de bajo peso molecular, que son liberados desde el hongo o pared celular de la planta por la actividad de enzimas de Trichoderma. (Baker et al 1997; Harman 2004)
LABORATORIO
El laboratorio destinado para la producción de bioinsumos debe contar con políticas y normas que aseguren el correcto funcionamiento y eviten contaminaciones de material.
Normas generales
No fumar, comer o beber dentro del laboratorio.
Usar una bata y mantenerla abrochada.
Guarda tus prendas y objetos personales en un armario y no dejar objetos personales en los mesones de trabajo.
Mantener las puertas cerradas, para evitar el flujo de contaminantes dentro y fuera del laboratorio.
Restringir la entrada a personas ajenas al personal del laboratorio, en horas de trabajo.
Normas personales
Usar ropa cómoda y evitar prendas sueltas.
Evitar peinados sueltos, no usar anillos, pulseras o accesorios en las manos.
Mantener las manos limpias y secas, evitar actividades si presenta heridas en las manos.
Evitar visitar otros laboratorios si no es cuestión de trabajo.
Si tiene que salir del laboratorio dejar el mandil.
Normas de seguridad
Mantener el área de trabajo limpia y ordenada
Evitar verter solido en los fregaderos, deben emplearse recipientes para residuos que se encuentran en el laboratorio.
Contar con un botiquín y mantener al personal al tanto de su ubicación.
Usar equipos de protección (guantes, lentes) al manipular recipientes que contengan sustancias peligrosas (agua caliente, acido, etc.)
Usar los materiales de acuerdo a las instrucciones indicadas para cada equipo.
Asepsia del Laboratorio.
La limpieza dentro del laboratorio es un requisito fundamental para trabajar con organismos como hongos. Esta condición, puede garantizar que se realice un trabajo confiable y seguro. También se requiere de la limpieza frecuente para garantizar el buen funcionamiento del equipo con el que se cuenta, realizar esta desinfección cada vez que se realice una actividad.
Algunas de las actividades que se mencionan deben realizarse periódicamente, o varias veces a la semana, según se recomiende para evitar la presencia de problemas de contaminación.
El aseo del laboratorio se debe realizar diariamente en el área de incubación y de dos a tres veces por semana en el área de secado, a fin de evitar crecimiento en techos y paredes, usando la solución de hipoclorito de sodio al 0,5%. En otras áreas como la de esterilización, e incubación debe asearse diariamente.
Lavado de materiales
En el laboratorio es fundamental lavar todo material inmediatamente después de ser usado, asegurando así, que no se tenga crecimiento de organismos en los materiales.
Para hacer el lavado, es recomendable tallar los materiales con una fibra fina y jabón detergente. El enjuague debe hacerse con agua limpia dos veces, preferentemente el agua debe ser esterilizada.
La esterilización de los materiales como cristalería y medios de cultivos, nos aseguran que estas se encuentran en asepsia y permite realizar los trabajos sin dificultades. La esterilización, debe hacerse en un autoclave preferentemente, a 15 libras de presión durante 20 minutos como mínimo.
Desinfección de ambiente.
El alcohol al 70% es comúnmente utilizado para realizar esta actividad, ya que desnaturaliza las proteínas, disolviendo las capas lipídicas y como agente deshidratante. Esta solución se usa para desinfectar la cámara de flujo laminar, otras superficies y las manos antes de realizar alguna actividad que represente riesgos de contaminación.
Otro de los productos usados es el hipoclorito de sodio al 5% se usa para desinfectar paredes, techos, mesas, pisos, puertas y ventanas. Hacer una limpieza con esta solución en todo el cuarto de inoculación, siempre antes de realizar un trabajo en ella. Para así reducir al mínimo el riesgo de contaminación.
AISLAMIENTO DE TRICHODERMA


FASE DE RECOLECCION DE MUESTRAS DE SUELO
Materiales









Procedimiento
Una vez identificada los lugares a muestrear nos dirigimos hacia estos, realizamos una observación previa y determinamos el método de muestreo. El método de muestreo va estar definido por la situación geográfica del lugar, el tipo de cultivo o bosque, la accesibilidad. Pero en todo sentido se busca recolectar submuestras que nos permitan lograr obtener una muestra
GPS PALA CUADRADA BARRENO FUNDAS ZIPLOC
MARCADORES ETIQUETA COOLER GUANTES
AGUA DESTILADA CLORO
BALDE
compacta y representativa del lugar muestreado. Seleccionamos el sitio donde vamos a tomar la submuestra, identificamos el punto gps y procedemos a desinfectar la herramienta a utilizar. Procedemos a tomar las submuestras con la ayuda de la pala y en forma de V tomamos la muestra desde la parte superficial y hasta los 15 cm de profundidad y colocamos en el balde.

Repetimos esta actividad con las demás submuestras establecidas.
Finalmente mezclamos bien las submuestras obtenidas en el balde y extraemos la muestra compuesta. Colocamos en la funda ziploc aproximadamente 500g, y etiquetamos. Es aconsejable manejarse con muestras de respaldo para mayor seguridad. Colocamos las bolsas en el cooler y las trasladamos hasta el laboratorio.
Recolección de muestras (Proyecto de Investigación)
FASE DE RECOLECCIÓN DE MUESTRAS VEGETALES
MATERIALES



Procedimiento
Para la recolección de muestras vegetales, de igual manera en primer lugar se selecciona el sitio a muestrear y las plantas o árboles, se identifica el sitio georreferencial y se procede a la toma de muestras, se toma cual sea el caso (tallo, hojas o fruto) se coloca en las bolsas de papel, se etiquetan y colocan en el cooler para su traslado hasta el laboratorio
 PODON DE ALTURA FUNDAS DE PAPEL TIJERA DE PODAR GPS
MARCADORES ETIQUETA COOLER GUANTES
PODON DE ALTURA FUNDAS DE PAPEL TIJERA DE PODAR GPS
MARCADORES ETIQUETA COOLER GUANTES
PROCESAMIENTO DE MUESTRAS EN LABORATORIO
MATERIALES



ETIQUETA
PROCEDIMIENTO
En el procesamiento de las muestras lo primero que debemos hacer es colocarlas sobre un papel y dejarlas secar, el tiempo va a variar de acuerdo al tipo de muestra y la humedad que esta haya contenido. Posterior con la ayuda del mortero tratamos de triturar lo más que sea posible las partículas de suelo. Luego tamizamos obteniendo una muestra con partículas muy diminutas. Colocamos en las fundas ziploc y etiquetamos. Se recomienda mantener las muestras conservadas en refrigeración normal o en un lugar al aire libre que no modifique su estructura original.
 MORTERO TAMIZ PAPEL CRAFT FUNDAS ZIPLOC
MORTERO TAMIZ PAPEL CRAFT FUNDAS ZIPLOC
SIEMBRA POR DILUSIONES SERIADA
MATERIALES
Solución stock
Blancos de dilución
Micropipeta
Asa de drigalsky
Cajas de Petri con medio de cultivo PDA

Cajas de Petri con medio de cutivo Rosa de bengala
Parafilm
Marcador.
PREPARACION DE SOLUCIONES Y MEDIOS DE CULTIVO.
MEDIO PDA:
Para el medio PDA solo se utiliza lo siguiente:
PDA (3,9g)
Agua (100ml)
Colocar antibiótico luego de esterilizar el medio, 1ml de antibiótico x cada 100ml de medio.
Procesamiento de muestras (Proyecto de investigación)
Puntas
PREPARACIÓN DE SOLUCIÓN MADRE DE SUELO
Pesar la muestra a relación de 1g en 9ml H2O, lo que me da una cantidad de suelo a utilizar de 27,77g y agua destilado 250 ml, con 2,125 g de sal.
Colocar los 250ml de H2O en el Erlenmeyer con 27,77g de suelo ya pesado.


Después colocar en agitación a 150RPM por 12-24 horas a una temperatura de 25°C.

“Si los medios no van a ser colocados en las cajas petri es necesario someterlos a baño maría.”
Preparación solución stock (Proyecto de investigación)
PREPARACIÓN DE CAJAS PETRI
Lavar las cajas Petri con jabón y agua común, después sumergirlas en agua destilada y dejarlas secar.


Después envolverlos en papel craft, ajustarlos y sellarlos adecuadamente.
Colocarlos en el auto clave para su respectiva esterilización.
DISPENSADO DE MEDIO DE CULTIVO
1. Introducir los medios a utilizar en la cámara de flujo desinfectándolos.
2. Colocar el antibiótico.
3. Colocar una cantidad razonable de los medios en las cajas Petri.
4. Realizar la respectiva codificación del medio.
Preparación de cajas (Proyecto de Investigación)
Dispensado del medio de cultivo (Proyecto de Investigación)
PREPARACIÓN DE BLANCOS DE DILUCIÓN
Colocamos 9ml de agua en los tubos de ensayo anteriormente utilizados y se lo esteriliza.
PROCEDIMIENTO DE DILUSIONES SEREADAS Y SIEMBRA
1. Trabajamos con los tubos de ensayo con 9ml de agua esterilizados.
2. La solución madre que se encuentra en agitación la retiramos y con la micropipeta medimos 1ml de dicha solución.
3. Lo colocamos en el primer tubo de ensayo que es 10-1 .
4. Al tubo de ensayo lo sometemos al bortex (agitador) durante 5seg.
5. Cambiar la punta de la micropipeta e introducirla en el tubo de ensayo 10-1, tomar 1ml y lo colocamos en el tubo 10-2

6. El mismo procedimiento lo realizamos sucesivamente hasta 10-6
7. Utilizar el tubo de ensayo 10-3, 10-4 y 10-5 y con la micropipeta medir 0,1ml de gota del suelo sereado, para colocarlo en los medios anteriormente realizados (PDA, Rosa de Bengala).
8. Con la ayuda del mechero, alcohol y asa de drigasly expandir la gota que se encuentra en el medio.
9. Después lo colocamos en la incubadora a 25oC y evaluamos el crecimiento de hongos todos los días.
10. “Cada vez que se utiliza la espátula para expandir la gota es necesario someterla al fuego y posteriormente al alcohol para que se encuentre desinfectada.”
Blancos de dilución
t




 Siembra en cajas Petri (Proyecto de Investigación)
Siembra en cajas Petri (Proyecto de Investigación)
Siembra en cajas Petri (Proyecto de Investigación)
Siembra en cajas Petri (Proyecto de Investigación)
PURFICACION DE LOS AISLADOS SELECCIONADOS
MATERIALES
Cajas de Petri con medio PDA Palillos
Parafilm
Marcador.
PROCEDIMIENTO.

Luego de haber sembrado por dilución seriada debemos realizar un seguimiento a las cajas de Petri en la incubadora, como nuestro objetivo es aislar trichoderma debemos buscar estructuras que nos indiquen su crecimiento, los días que comienzan a aparecer estas estructuras en las cajas no son definidos, pues esto depende del tipo de aislado que sea, por tal motiva se recomienda dejar las cajas por lo menos de 15 a 20 días y luego desecharlas.
A medida que se trabaja se va adquiriendo la habilidad de acertar al momento de purificar los aislados y coincidir que estos sean Trichoderma, pero algunas claves son, el micelio que produce es blanco y muy pronunciado con rápida difusión, otros por lo general producen pigmentación verde-amarillenta al revés de la caja y otros tiene una capacidad de esporular con gran rapidez, entonces si observamos estas características debemos repicarlos en cajas por separado hasta lograr su purificación, tomar en cuenta siempre la codificación.
El proceso consiste en tomar una pequeña porción del hongo seleccionado dentro de la caja de Petri y con la ayuda del palillo de dientes previamente esterilizado, transferir a una nueva caja de Petri con medio PDA para darle las condiciones adecuadas para su desarrollo. Sellamos la caja con parafilm etiquetamos y llevamos a incubación a 25ºC de 8-10 días.
Purificación de aislados (Proyecto de Investigación)
CULTIVO MONOSPORICO
MATERIALES.
Cajas de Petri con media Agar

Cajas de Petri con medio PDA
Tubos de ensayo con agua estéril
Parafilm
Marcadores.
PROCEDIMIENTO
Este procedimiento se realiza con la finalidad de obtener cultivos desarrollados de una espora simple. Para lo anterior, se toma un tubo con 5m de agua destilad estéril y con la ayuda de una aguja de disección se pasa una pequeña proporción del crecimiento de una placa de Petri, se tapa el tubo de ensayo y se agita por 30 segundos, esta suspensión se vierte en una placa de Petri con medio Agar y enseguida se desecha el exceso en un recipiente. Luego, las cajas se incuban de forma vertical a 25°C yse observan diariamente con la ayuda del estereoscopio deprecisión hasta ver la germinación de una espora la misma que inmediatamente es transferida a una placa Petri con medio de cultivo PDA, continuando con la incubación hasta el desarrollo total de la colonia (Koneman, 2001)
Cultivo Monospórico (Proyecto de Investigación)
CONSERVACION DE AISLADOS
MATERIALES:
Tubos ependorf
Agua destilada esteril
Sorbetes esterilizados
Procedimiento
Para este procedimiento se siguió la metodología utilizada por (Mafia y Alfenas, 2016: pp.9495). En donde se sugiere el almacenamiento en agua por método de Castellani ya que es muy utilizado para almacenar aislados fúngicos. Para hongos u organismos crecidos en medio sólido, por tiempo suficiente que permitió la retirada de los discos de cultivo con micelio, los cuales fueron colocados en micro tubos estériles con aproximadamente 1 ml de agua destilada estéril, se colocaron 5 discos por cada micro tubo, se conservaron a una temperatura de 10 °C o se los llevó a una refrigeradora para que estos se mantengan hidratados.
Conservación de aislados (Proyecto de Investigación)

Palillos
Micropipeta
Puntas
IDENTIFICACION CULTURAL Y MORFOLÓGICA
MATERIALES
Cajas de Petri con medio de cultivo PDA
Cajas de Petri
Papel absorbente
Bases de vidrio
Placas porta objetos
Placas cubre objetos
Claves de identificación
PROCEDIMIENTO
Para la identificación cultural se repica los aislados en cajas de Petri con medio de cultivo PDA y se realiza una evaluación donde se describen aspectos como velocidad de crecimiento hasta llenar el plato Petri, color del anverso y reverso de la caja, estructura del micelio, produce o no pigmentos o exudados, tipo de borde y color del aislado.
Para una identificación morfológica se debe realizar microcultivo y llevarlo a microscopio para poder observar las estructuras.
Para el método de microcultivo se siguie la metodología utilizada por Mafia y Alfenas (2016: pp.216-217) colocando papel filtro en la base de la caja petri, seguido de un bastón de vidrio como base que sirvió de soporte de la placa porta objetos, se colocó un fragmento de medio de cultivo (SNA) con cerca de 1 cm2 de área y 2 mm de espesor, se transfirió un fragmento de micelio e inmediatamente se colocó la placa cubre objetos, la lámina quedó fijada y para ello se presionó ligeramente con un bisturí, transcurridos los 2-5 días dependiendo del cultivo se revisó el crecimiento, se observaron las estructuras y esporas del aislado con el lente 40X del microscopio óptico, es importante recalcar que todos los materiales deben estar previamente esterilizados.

IDENTIFICACIÓN MOLECULAR
Después de obtener los aislados puros se caracterizaron y se seleccionaron. La biomasa de los hongos filamentosos se obtuvo utilizando un medio líquido descrito a continuación, para ello, se tomó discos de micelio de 7 mm y se sembró en tubos de ensayo y cajas Petri con medio líquido Los tubos fueron incubados a 25° durante 336 h.
También se puede obtener biomasa fúngica sembrando un disco del aislado en una caja Petri en mediodecultivoAgar Avena eincubarloa 25°±2°. Elmicelioproducidoseextraeenunmortero, se añade nitrógeno líquido y se macera hasta obtener un producto polvoriento del aislado, el material obtenido se almacena en un tubo de ensayo de 2mL, y se coloca en congelación.
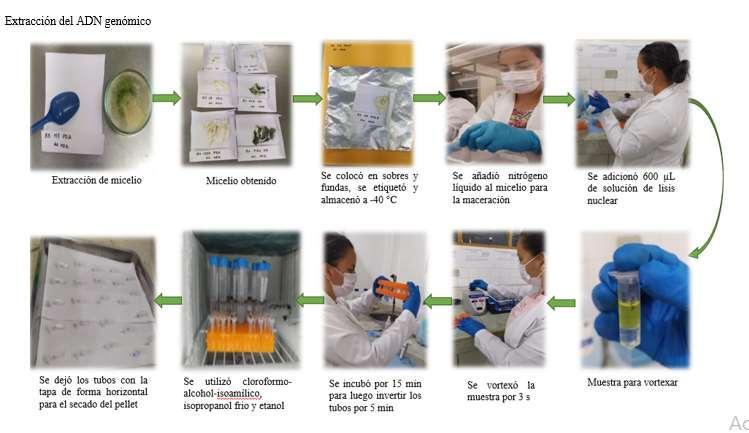
Extracción del ADN genómico
Se realizó la extracción de ADN genómico para la caracterización molecular de Trichoderma spp. con la ayuda del Kit Promega Wizard® de acuerdo a las recomendaciones del fabricante con las siguientes modificaciones:
Se tomó una pequeña cantidad de micelio macerado del tubo y se transfirió a microtubos de 1,5 mL. Posteriormente se adicionaron 600 µL de solución de lisis nuclear y se agitó en el vórtex por 3 s y después se incubó a 65 ºC por 15 min, invertiendo los tubos cada 5 min.
Posteriormente se adicionaron 200 µL solución de precipitación de proteína, colocando en el vortéx por 20 s, luego se centrifugó por 10 min a 14000 rpm. El precipitado de proteínas formó un pellet, seguidamente se transfirió el sobrenadante (600 µL) conteniendo ADN para un nuevo microtubo (1,5 mL), se adicionaron 600 µL de cloroformo-alcohol-isoamílico (24:1) se colocó en el vórtex por 20 s. Luego se centrifugó por 8 min a 14000 rpm y se transfirió el sobrenadante (± 600 µL) conteniendo el ADN para un nuevo microtubo.
Después se adicionaron 600 µL de isopropanol frio cuidadosamente mezclando la solución invirtiendo los tubos. Luego se dejó la solución en el congelador por 10 min, finalmente se centrifugaron los tubos a 14000 rpm por 10 min.
Observando que existió la formación del pellet se descartó cuidadosamente el sobrenadante. Seguidamente se procedió a realizar dos lavados con etanol al 70% frío, en cada lavado se adicionaron 600 µL de etanol y se centrifugó a 14000 rpm por 5 min.
Luego se descartó el etanol y se dejaron los tubos de forma horizontal con la tapa abierta sobre el papel toalla para el secado del pellet.
Finalmente, después de aproximadamente 15 horas, se resuspendió el pellet con 50 µL de TE y 2 µL de RNasa en cada tubo, se incubó a 37° C por 5 horas y se almacenó el ADN a -20°C hasta su posterior uso.
PCR
Para la identificación molecular de los diferentes morfotipos obtenidos, se empleó la técnica de amplificación cadena del ADN polimerasa (PCR), con los primers descritos a continuación: Nombre Gen Amplificado Secuencia
Forward 10 µM frbp25f RNA Polimerasa subunidad 2 (Rpb2)
5'-GAYGAYMGWGATCAYTTYGG-3'
Reverse 10 µM 7CR 3'-CCCATRGCTTGYTTRCCCAT-5'
Para la elaboración del mix de la reacción se utilizaron los siguientes reactivos: 6,25 µL GoTaq® Colorless Master Mix, 1 µL (pmol) primer forward (frbp25f), 1 µL (pmol) primer reverse (7CR) y 2,25 µL de agua ultra pura estéril. Se realizó la preparación de las muestras para PCR mediante el siguiente proceso: Se colocó en tubos de 0,2 mL un volumen del mix de 11,5 µL más 2 µL de ADN genómico, dando un volumen total de 12,5 µL por cada reacción de PCR, el control negativo de la PCR se lo realizó sustituyendo el ADN por agua ultra pura estéril y se colocaron los tubos en el termociclador EPPENDORF modelo vapo.protect con el siguiente programa:
1. Desnaturalización inicial a 95 oC por 5 min. 2. 38 ciclos de:
Desnaturalización 95 °C por 1 min.
Alineación 53 °C por 2 min.
Extensión 72 °C por 2 min.
3. Extensión final 72 °C por 10 min.
Finalmente, se dejó los productos de PCR en el ultracongelador a -40 oC
Electroforesis
Para comprobar la amplificación de los productos de PCR se realizó una electroforesis en gel de agarosa a una concentración de 1%, adicionando el agente intercalante de ADN UniSafe Dye Nucleic Acid Staining Solution (20,000x) (5 µL en 100 µL de agarosa) en el gel, usando Tris borato EDTA (TBE) a una concentración 1X en agua destilada estéril.
Se procedió a verter la solución en la cuba para la formación del gel, además se colocó las vinchas para que se formaran los pocillos. Una vez gelificada la solución se vertió la solución buffer TBE y luego las muestras de los productos de PCR fueron cargadas en los pocillos en un volumen de 2 µL con 3 µL de buffer de carga. Después de la electroforesis los productos de PCR fueron visualizados en un transiluminador marca Fisher Scientific 88A.
Secuenciación.
Los productos de PCR obtenidos fueron secuenciados usando la tecnología SANGER en la empresa MACROGEN KOREA.
Análisis de secuencias
Las secuencias obtenidas se analizaron con el programa Chromas 2.6.6 y BLASTn del NCBI. Primero, se verificó la calidad de las secuencias por revisión de los cromatogramas en el programa Chromas 2.6.6. Aquellas de buena calidad se depuraron eliminando los extremos y los fragmentos que presentaban ruido. Para la confirmación de la identidad de cada especie, con las secuencias depuradas se realizaron alineamientos de nucleótidos mediante el algoritmo BLASTn del NCBI, considerando los siguientes parámetros: % de identidad, % de cobertura, Evalue contra las secuencias reportadas en el Centro Nacional para la Información Biotecnológica (NCBI por sus siglas en inglés).
Análisis Filogenéticos
Los análisis filogenéticos utilizando el gen Rpb2 fueron realizadas con base en la identificación inicial del GenBank. Todas las secuencias recuperadas fueron alineadas con las secuencias de los aislados fúngicos utilizando el software Muscle®, implementado en el programa MEGA 10.0 (Molecular Evolutionary Genetics Analysis) (Kumar et al., 2016). Los análisis filogenéticos fueron basados en Maximum likelihood (ML) y fueron realizados con RAxML v. 8.2.12 (Stamatakis, 2014). Los parámetros utilizados fueron: análisis bootstrap rápido (con 1000 replicatas) y la búsqueda para el mejor puntaje del ML tree. Todas las filogenias fueron realizados a través de la interfaz online del CIPRES Science Gateway (Miller et al., 2010). Los árboles resultantes fueron visualizados en el software FigTree y editados en el programa gráfico Inkscape 0.92.
MATERIALES


PREPARACION DE SUSTRATO
El sustrato utilizado para la producción masiva de trichoderma puede estar definido de acuerdo a la accesibilidad, economía y volumen a producir. Se puede utilizar (arroz, arrocillo, maíz, trigo, sorgo, caña de azúcar o sus residuos). En nuestro caso hemos seleccionado el arrocillo o arroz partido como sustrato de producción.

En un inicio se realiza varios lavados del sustrato y se deja secar en un colador para que pierda el exceso de humedad.
Una vez se haya secado procedemos a colocar en botellas de vidrio autoclavables para proceder a realizar su esterilización en un ciclo de autoclave a 121° por el lapso de 25 minutos.


Una vez esterilizado el sustrato, el mismo está listo para pasar a la etapa de inoculación.
 Arrocillo Coladera Botellas
Tarrinas plásticas Jeringas Botellas oscuras
PRODUCCIÓN DE TRICHODERMA
Arrocillo Coladera Botellas
Tarrinas plásticas Jeringas Botellas oscuras
PRODUCCIÓN DE TRICHODERMA
INOCULACIÓN
Partimos de una caja de Petri con el aislado seleccionado (esta debe estar esporulada), con la ayuda de una cucharita plástica esteril sacamos toda la biomasa de la caja y la colocamos en un vaso de precipitación, posterior añadimos 50mL de agua destilada esteril y mezclamos cuidadosamente para obtener el inóculo de partida.


El arrocillo esteril lo vamos colocando en tarrinas (previamente esterilizadas) hasta cubrir las 3/4 partes del volumen. Con la ayuda de una jeringa vamos inoculando cada tarrina con aproximadamente 10mL de inóculo.
INCUBACIÓN
Las tarrinas con el sustrato inoculadas fueron colocadas en una cabina de esporulación diseñada para el control de luminosidad con una intensidad periódica de 12h de luz y 12h de oscuridad para promover la colonización por parte del hongo en el sustrato además esta cabina cuenta con un calefactor que permite regular la temperatura. Se incuban por alrededor de 10 días, cada 2 días se revisa el crecimiento y se revuelve el sustrato para facilitar la inoculación.
PREPARACIÓN DE INÓCULO
Luego de transcurrido el periodo de incubación procedemos a preparar el inóculo que estará listo para aplicación en cultivo. Para lo cual requerimos botellas estériles, agua destilada estéril, un colador, cucharas y una licuadora. Colocamos poco a poco el sustrato inoculado en la licuadora y colocamos agua destilada estéril, licuamos por alrededor de dos minutos. Colamos por un tamiz el más fino posible para evitar que restos de sustrato pasen y ocasionen problemas en los equipos de aplicación en campo (taponen boquillas de bombas). El mismo proceso lo realizamos con todos los aislados que hemos seleccionado para su producción masiva.

CALIBRACIÓN DE CONCENTRACIÓN
Para calibrar la concentración del producto final se realiza un conteo de esporas en una cámara de neubauer o hemacitómetro. Colocamos una gota de agua en el canal central de la cámara, fijamos la placa portaobjetos, por el canal lateral cuidadosamente colocamos una pequeña porción de la suspensión obtenida hasta que se disperse por toda la placa, llevamos al microscopio y procedemos a cuantificar las esporas, con la fórmula propuesta determinamos la concentración final.
PRODUCTO FINAL
El producto final de la producción masiva de Trichoderma se lo puede obtener tanto de forma líquida o sólida, depende de la finalidad u objetivo que se está desarrollando. Hay que tener en cuenta que los productos deben conservarse en lugares frescos con temperaturas bajas, para que no pierdan su eficiencia, además es recomendable que los envases plásticos en caso de producción líquida sean oscuros.


ANEXOS



BIBLIOGRAFÍA
CHET I. ET AL. 1987. Trichoderma, modo de acción y potencial como un agente de control biológico de hongos patógenos de la planta. pp. 137-159. Wiley, New York.
Elad Y, Chet I, Boyle P, Hennies Y. Parasitism of Trichoderma sp. on Rhizoctonia solani and Sclerotium rolfsii scanning electron microscopy and fruorencence microscopy. Phytopathol. 1983;73:78.
HARMAN, G. 2000. Myths and dogmas of biocontrol changes in perceptions derived fron research on Trichoderma Harzianum. pp. 84.
HOWELL, ET AL, 2002. Aplicación de Trichoderma, modo de acción y potencial como un agente de control biológico de hongos patógenos de la planta.
R. D. LUMSDEN, C. J. RIDOUT, M. E. VENDEMIA, D. J. HARRISON, R. M. WATERS, AND J. F. WALTER. Characterization of major secondary metabolites produced in soilless mix by a formulated strain of the biocontrol fungus Gliocladium virens Canadian Journal of Microbiology 38(12): 1274-1280. https://doi.org/10.1139/m92-210
SAMUELS, G. & HEBBAR, P. Identification and Agricultural Applications. The American Phytopathological Society. U.S.A. 2015 pp 35-160. [Consulta: 24 febrero 2022] ISBN: 9780890544846
SAMUELS, G.; Chaverri, D.; Farr, & McCray, E. Trichoderma Online: Systematic Mycology and Microbiology Laboratory, ARS, USDA. (A. U. Systematic Mycology and Microbiology Laboratory, Productor). Trichoderma Online: Systematic Mycology and Microbiology Laboratory, ARS, USDA. 2015. [Consulta: 25 febrero 2022] Disponible en: http://taxadescriptions/keys/Trichoderma Index.cfm

