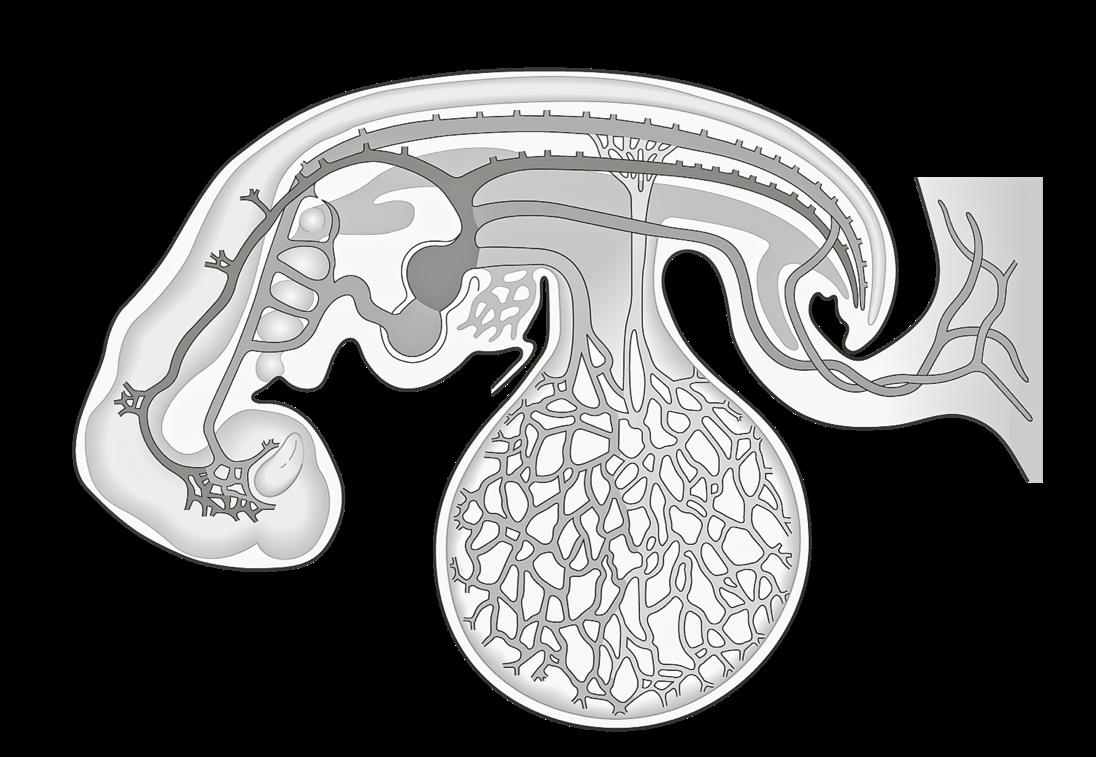
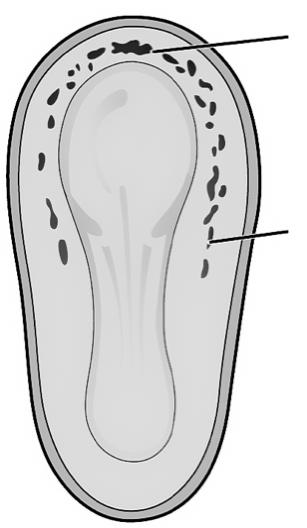
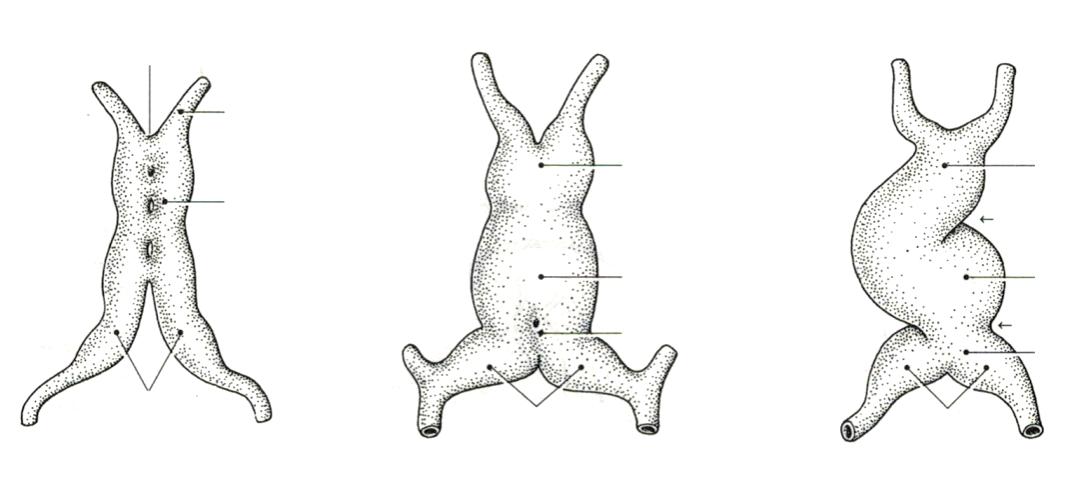
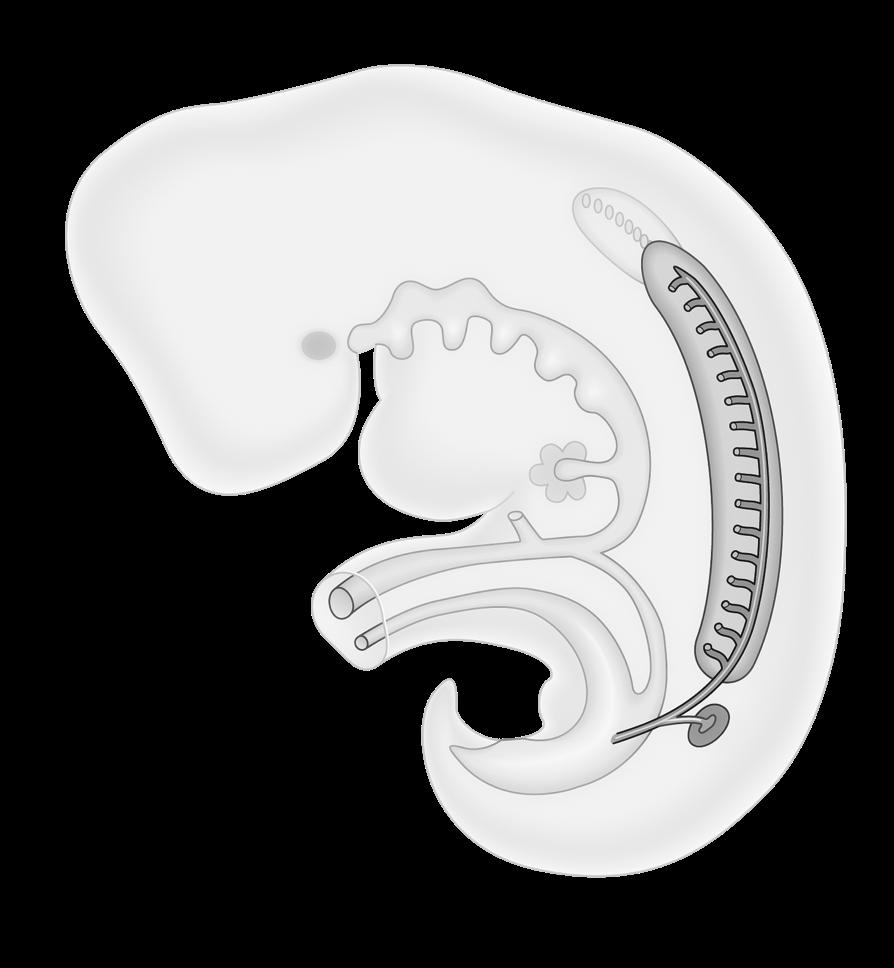
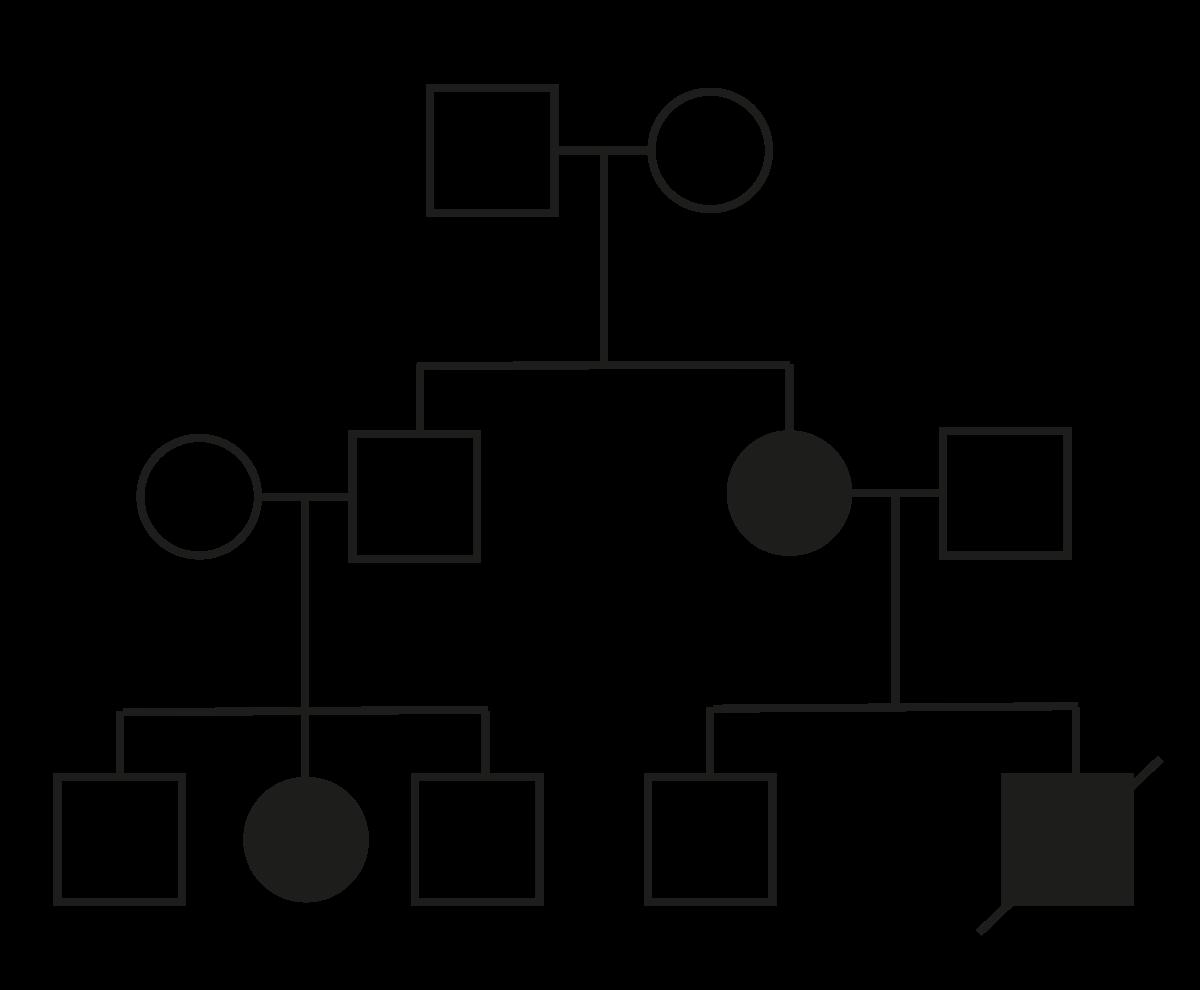
VIVES - bachelor vroedkunde - embriologie en genetica
Opleiding bachelorvroedkunde
Opleidingsfase Fase1
Voorwoord
“Sexual reproduction demands the collapse of an organism into a single cell, but then requires that single cell to expand back into an organism.”
Thomas Morgan (1920)
Embryologie bestudeert de ontwikkeling van een menselijk organisme vanuit één cel.
Genetica is de studie van erfelijke informatie en de wijze waarop deze wordt overgedragen.
De vorige editie van deze syllabus (2013-2022) werd doorheen de jaren verschillende keren aangepast, maar in 2022 was de tijd rijp voor een grondige vernieuwing. De wetenschap evolueerde immers razendsnel. Zo was er van de NIP-test in 2013 nog geen sprake. Ondertussen is deze vorm van prenatale screening goed ingeburgerd.
Op vlak van terminologie zijn in deze cursus heel wat verouderde begrippen/technieken weggelaten en nieuwe in de plaats gekomen. De meeste figuren zijn vervangen door recentere exemplaren. De bijschriften en aanduidingen op de figuren werden vertaald en aangepast om beter aan te sluiten bij de leerstof.
Ook qua opbouw is de cursus gewijzigd, om beter in te spelen op mijn eigen ervaring met de leerstof, suggesties van mededocenten en opmerkingen van studenten.
Zo vertrekt de cursus voor het eerst vanuit een gemeenschappelijk basisdeel (de eerste 5 hoofdstukken), dat de fundamenten van genetica en embryologie bevat. In deel 2 ligt de focus op embryologie, in deel 3 op genetica. Op verschillende plaatsen worden raakvlakken tussen genetica en embryologie besproken om verbanden te verduidelijken.
Per hoofdstuk zijn concrete leerdoelen opgenomen. Deze vormen de basis voor het schriftelijk examen (let wel: examenvragen zijn nooit letterlijke vertalingen van leerdoelen!).
Tot slot is ervoor gekozen om de bredere context van embryologie & genetica (zoals de geschiedenis van deze wetenschappen, de ethische vragen die nieuwe technieken oproepen, …) verspreid doorheen de cursus op te nemen (i.p.v. in afzonderlijke hoofdstukken). Zo zet dit nu op het juiste moment aan tot nadenken en zorgt het voor een betere verwerking van de leerstof.
Dit is de tweede editie van deze volledig vernieuwde syllabus (vanaf academiejaar 23-24).
Dank aan dr. M. Deschoemaeker voor het nalezen van het originele manuscript, Bijzondere dank aan de Kortrijkse studenten vroedkunde (academiejaar 22-23) voor het delen van hun ervaringen met de cursus en het signaleren van enkele resterende foutjes.
Eventuele vragen, opmerkingen of onduidelijkheden hoor ik graag van jullie.
Dr. Maarten Biesbrouck, lector, Augustus, 2023 maarten.biesbrouck@vives.be
Legende van de gebruikte iconen
Legende van de gebruikte iconen
Leerdoelen
Belangrijk om te onthouden
Figuur die zelf getekend wordt. Op het examen moet één van deze figuren getekend worden.
Studeeraanwijzing of opmerking over de opbouw van de leerstof
Historische ontdekking
Bedenking bij de leerstof
Voorbeeld, toepassing uit de praktijk
Extra verdieping
Denkvraag
Oefening
Oplossingen op Toledo.
Ethische dilemma’s
Deze zetten aan tot nadenken, het afwegen van argumenten en het vormen van een genuanceerde mening. Dilemma’s kun je niet zomaar beantwoorden met ‘juist’ of ‘fout’.
Tip: bekijk steeds de leerdoelen om te weten of iets tot de leerstof behoort of niet.
Studeeraanwijzingen
Aan het begin van elk hoofdstuk vind je telkens de leerdoelen.
Aan het begin van deze lijst met leerdoelen vind je telkens dezelfde, algemene leerdoelen.
Hieronder volgt wat extra toelichting bij deze extra leerdoelen:
Algemene leerdoelen
Na het studeren van deze cursus moet je in staat zijn om …
Figuren uit elk hoofdstuk toe te lichten = beschrijven wat op de figuur is afgebeeld, welke structuur, welk proces te zien is, …
Figuren uit dit hoofdstuk te benoemen (enkel wat aangeduid is met pijlen).
= structuren benoemen die aangeduid zijn met deze pijlen: Hierbij wordt verwacht dat je de correcte, wetenschappelijke terminologie hanteert
Belangrijke begrippen te verklaren
(Deze staan altijd minstens 1 keer vetgedrukt in de tekst)
= een bondige uitleg geven voor die term bv. aan de hand van een korte definitie of beschrijving aan de hand van een voorbeeld.
met de afbeeldingen uit dat zichtbaar als je de symbolen aanklikt).
Tip: zet de leermodules open bij het studeren. Je vindt er naast de afbeeldingen – ook de leerdoelen per hoofdstuk, oplossingen van oefeningen en extra beeldmateriaal om de leerstof te verwerken.
Nomenclatuur
¨ Enkele vaak gebruikte afkortingen
H1 Hoofdstuk 1
D1* Dag 1
W1* Week 1
Chr. Chromosoom
Li Links
Re Rechts
NB Neurale buis
*aanduidingen van embryonale of foetale leeftijd zijn altijd vanaf de conceptie (en dus niet sinds de laatste menstruatie).
¨ Definities van termen embryo en foetus.
We spreken van een embryo vanaf de bevruchting (zygote) tot en met de 2e maand (8e week) van de ontwikkeling.
Vanaf de 3e maand (9e week) spreken we van een foetus Vanaf de 9e week spreken we van de foetale periode
¨ Aanduiding van anatomische posities bij het embryo:
Let vooral op de verschillen in plaatsaanduiding tussen het embryo en het volwassen lichaam. Bij het embryo wordt ‘superior’ vervangen door ‘rostraal’ (= aan de kant van de neus) of craniaal (= aan de kant van het hoofd). ‘inferior’ wordt er vervangen door caudaal (letterlijk: aan de kant van de staart).
Tot laat in de Middeleeuwen geloofde men in het preformationisme, de idee dat het mannelijke zaad volledige mensjes bevatte die dan moesten groeien in het vrouwelijke 'bed'. Zelfs Van Leeuwenhoek (de uitvinder van de microscoop) beweerde van zo'n minimensjes te kunnen waarnemen. Pas in de negentiende eeuw kwamen theorieën naar voor die een wetenschappelijke verklaring boden voor de overerving van kenmerken.
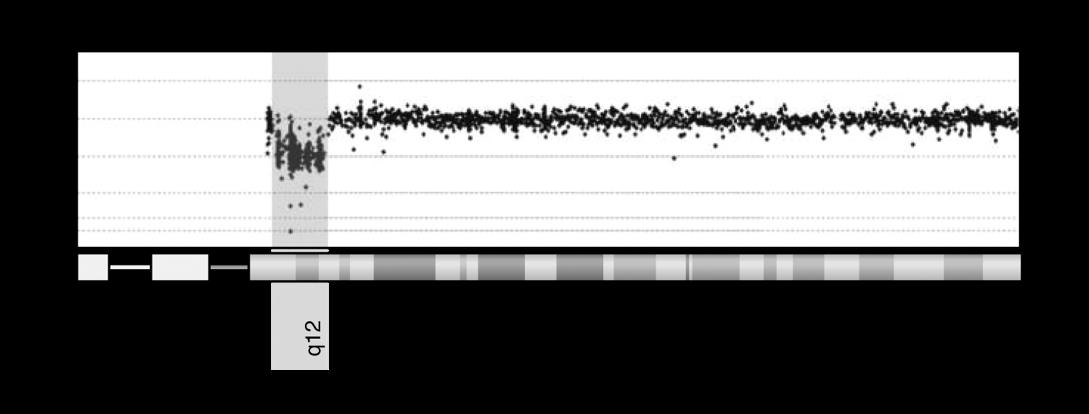
Figuur 2
1 Erfelijk materiaal
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Te beschrijven hoe genetisch materiaal in de celkern wordt opgeslagen.
De termen chromosoom, chromatine en zusterchromatide uit te leggen.
De structuur van een chromosoom te beschrijven.
Enkele functies van het chromosoom toe te lichten.
Uit te leggen wat een karyogram is, hoe dit bekomen wordt en welke informatie hierin te vinden is. Je kunt dit ook helder uitleggen aan een patiënt.
Een karyotype te noteren volgens de ISCN-notatie.
De ploïdie van de mens te kunnen omschrijven.
Te duiden vanwaar de stelling ‘mannen zijn genetisch het zwakke geslacht’ komt.
De termen haploïd, diploïd, triploïd, tetraploïd, polyploïd en aneuploïd correct te omschrijven en toe te passen.
Te duiden waarom een cel tijdens de S-fase van de celcyclus niet tetraploïd wordt.
De afkorting ‘DNA’ voluit te schrijven.
Uit te leggen hoe de bouw van DNA bijdraagt tot de functie ervan.
Kort uit te leggen wat bedoeld wordt met de ‘polariteit’ van DNA.
De belangrijkste functies van DNA te beschrijven.
Bij benadering weer te geven hoeveel genen een mens bezit.
De term ‘locus’ te kunnen uitleggen aan de hand van een zelfgekozen voorbeeld.
Bij een gegeven ‘locus’ kunnen uitleggen wat dit betekent.
Bij een gegeven ‘lengte’ van een gen (in bp) kunnen uitleggen wat dit betekent.
1.1
Chromosomen
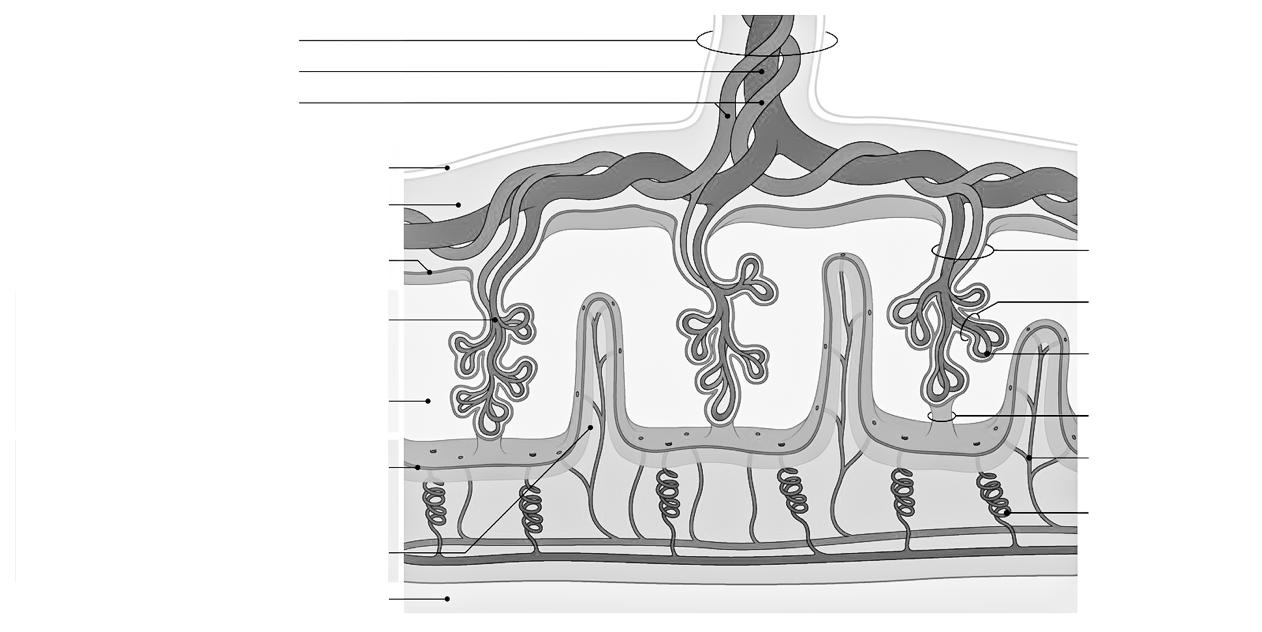
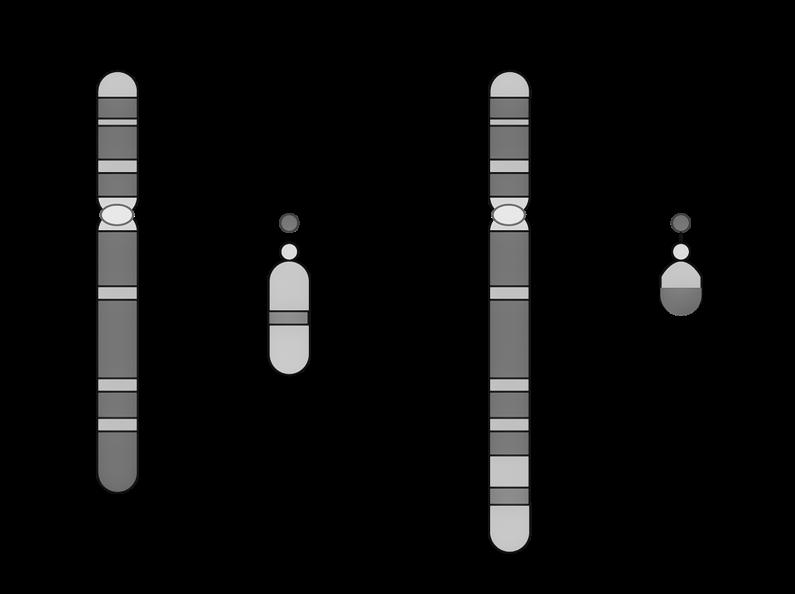
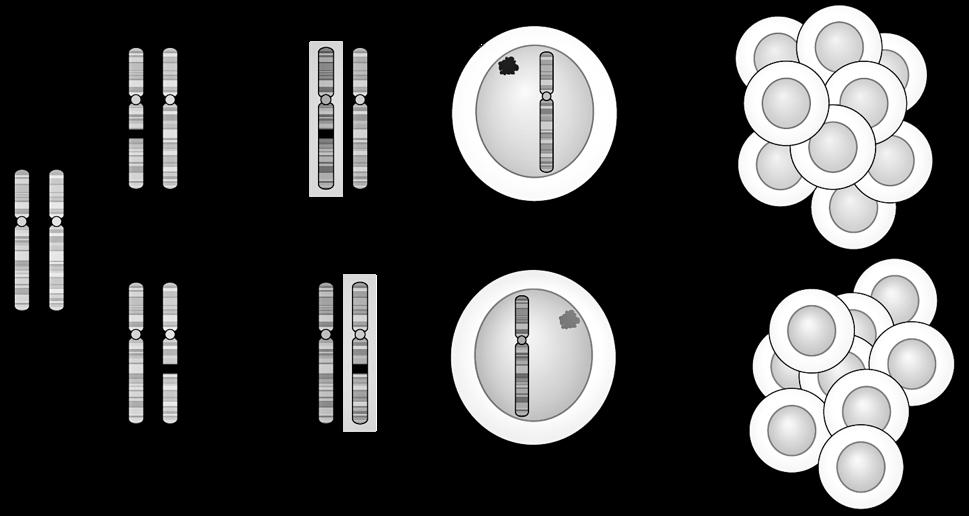
De mens is een eukaryoot organisme, wat betekent dat menselijke cellen een duidelijke, aflijnbare celkern (=nucleus) bezitten. In die celkern wordt het erfelijk materiaal opgeslagen onder de vorm van 46 chromosomen.
Chromosomen zijn draadjes kernmateriaal die enkel met een microscoop te zien zijn als de cel zich aan het delen is. Bij een niet-delende cel zijn de chromosomen ook aanwezig, alleen zijn ze dan niet van elkaar te onderscheiden.
De donkere, gevlekte kernsubstantie bij een niet-delende cel heten we chromatine.
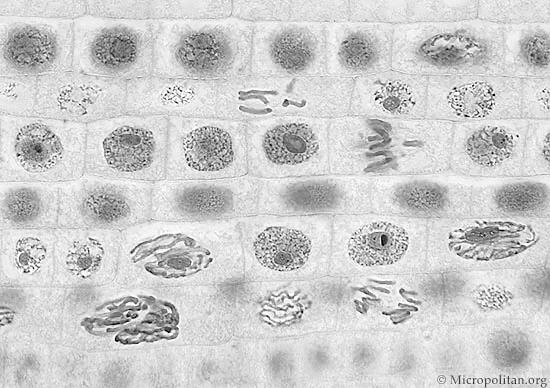
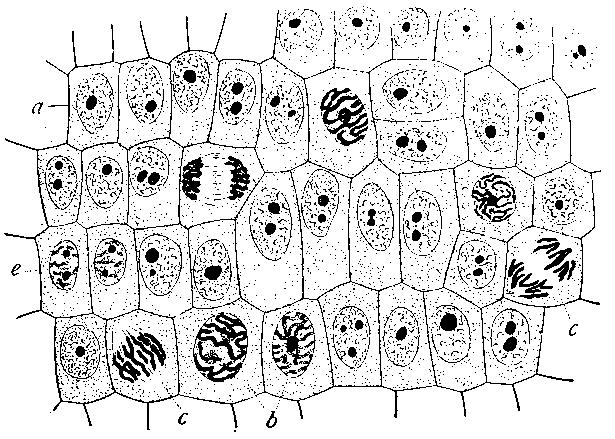
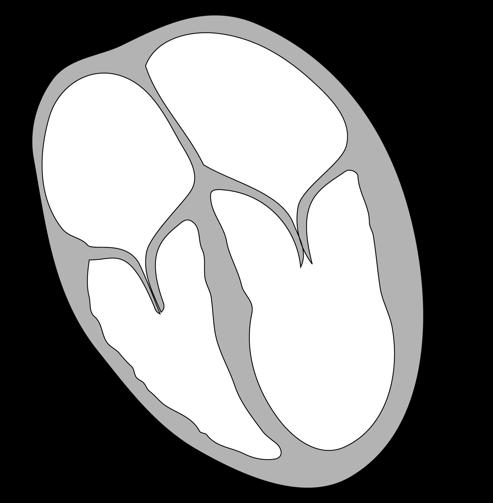
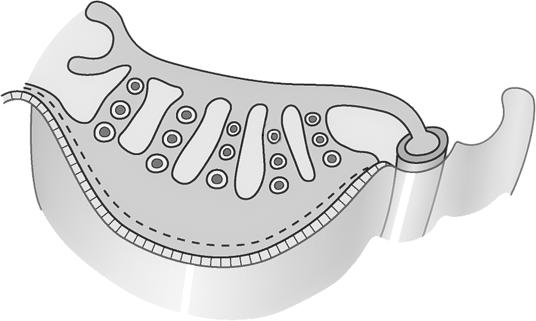
Figuur 3 Cellen in de tip van een ajuin (ook een eukaryoot organisme) vertonen verschillende stadia van celdeling (links: microscopische foto, rechts: tekening).
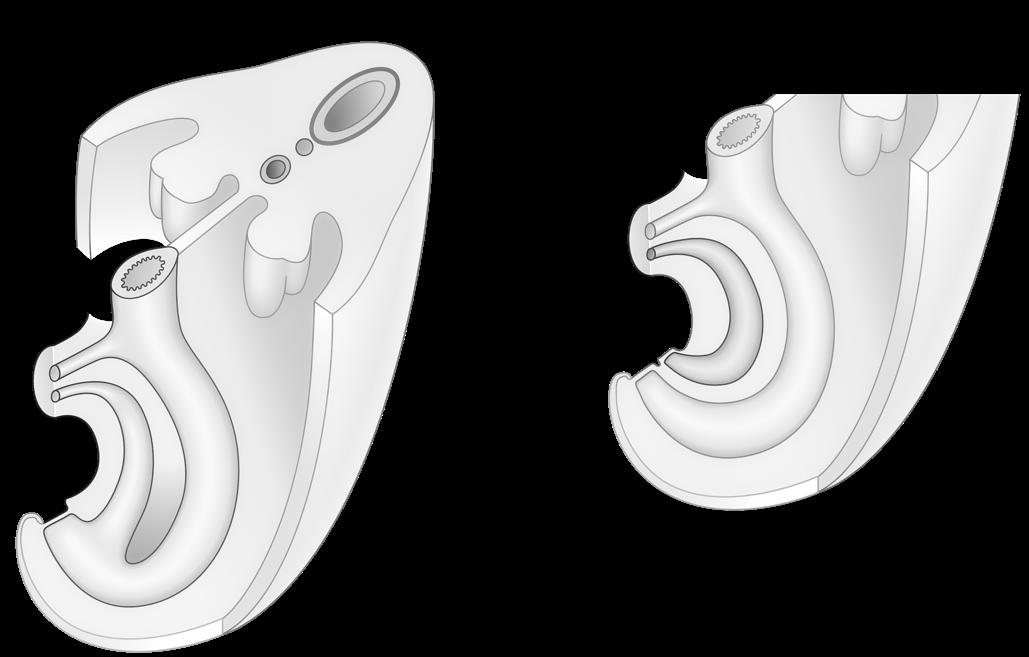
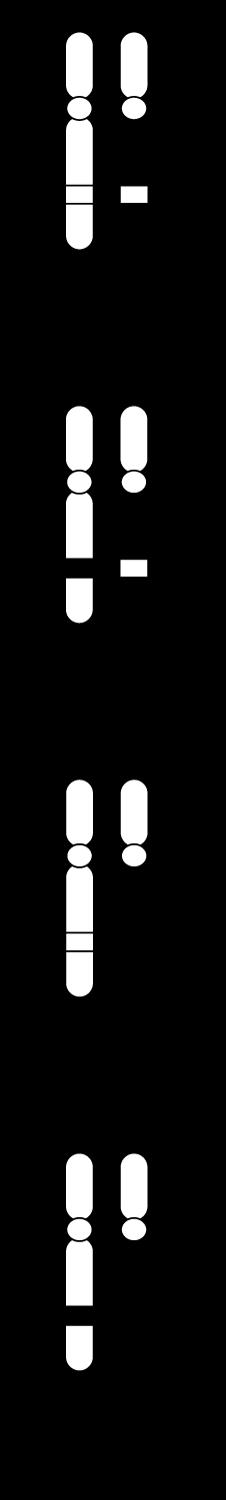
Chromosomen hebben allen een gelijkaardige opbouw:
We onderscheiden
c: een centrale insnoering (centromeer)
t: twee kapjes aan de uiteinden (telomeren)
p: een korte arm (van het Franse petit)
q: een lange arm (de letter nà p in het alfabet).
Figuur 4
Het centromeer kun je beschouwen als een ankerpunt tijdens de celdeling. Ter hoogte van dit centromeer klikken de chromosomen immers vast aan de spoeldraden.
Telomeren fungeren dan weer als beschermkapjes, die ervoor zorgen dat de uiteinden van de chromosomen niet uitrafelen en aan elkaar kleven (vgl. met de plastic eindkapjes op schoenveters). Telkens een cel deelt, worden de telomeren een klein beetje korter. Naarmate de telomeren verkorten, worden de chromosomen gevoeliger aan schade. Dit zou een belangrijke rol spelen in het verouderingsproces.
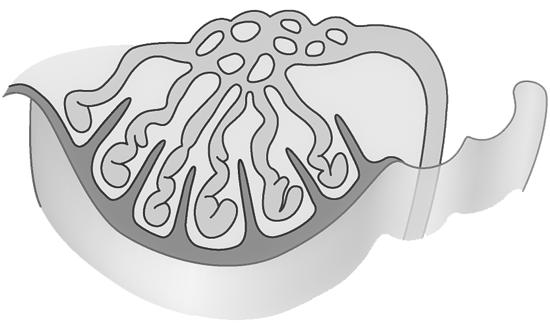
Tijdens de celdeling worden de chromosomen onder de microscoop zichtbaar. Op dat moment zijn ze al verdubbeld (zie hoofdstuk 3, celdeling).
JAAR: 1888
W. Fleming ontdekt kleurbaar materiaal in de celkern en heet dit 'chromatine', van het Griekse chromos (kleur). Tijdens de celdeling gaan deze kleurbare structuren uiteen in draden. W. Waldeyer heet deze draden later chromosomen ('gekleurde lichaampjes') (Waldeyer, 1888).
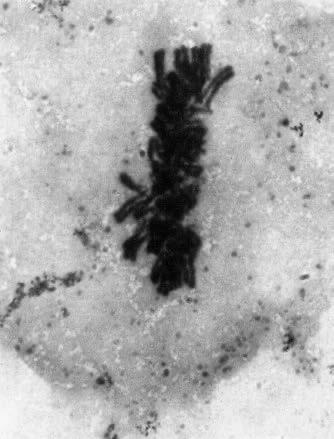
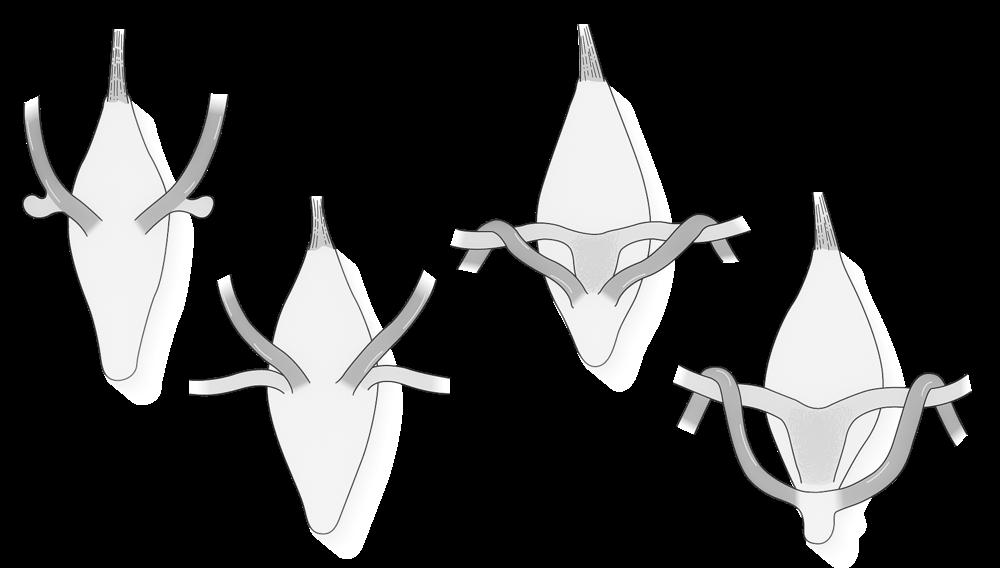
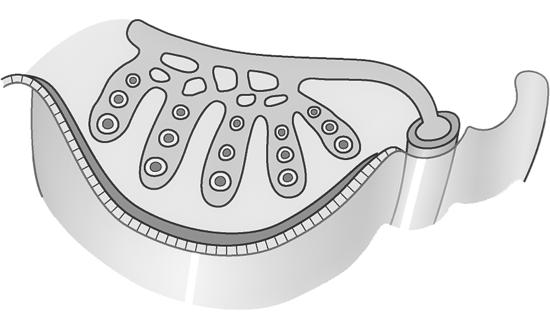
Omdat elk duplicaat aan het origineel blijft hangen ter hoogte van het centromeer krijgen de chromosomen tijdens de profase hun typische, gekruiste vorm. We spreken niet meer van een ‘chromosoom’ maar van een ‘verdubbeld chromosoom’ met 2 zusterchromatiden. Bij het begin van de celdeling bevat de cel dus 46 verdubbelde chromosomen ofwel 92 zusterchromatiden.
zusterchromatiden centromeer
telomeer
Figuur 5
Boven: tekening van verdubbeld chromosoom, bestaande uit 2 zusterchromatiden (kopieën van elkaar).
Links: verdubbelde chromosomen op de evenaar tijdens de metafase (mitose) (Turnpenny, 2017).
Tijdens de verdere celdeling (anafase) gaan deze zusterchromatiden terug uit elkaar en worden ze weer afzonderlijke chromosomen, die gelijk verdeeld worden over de dochtercellen.
JAAR: 1903
Lange tijd is niet duidelijk wat de samenstelling of functie is van chromosomen. Sutton formuleert in 1903 de hypothese dat op die chromosomen ‘partikeltjes’ aanwezig zijn die een rol spelen in de overerving (Sutton, 1903).
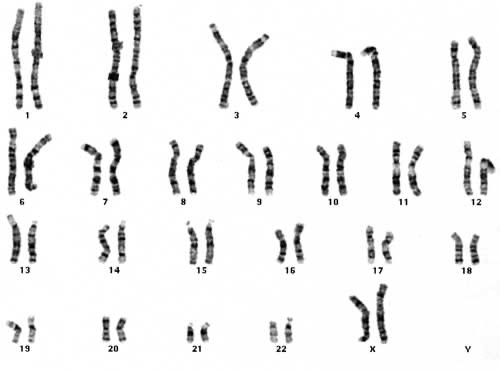
1.2 Karyogram
1.2.1 Wat leren we uit een karyogram?
JAAR: 1956
Tjio & Levan tonen aan dat de mens 46 chromosomen heeft. Dit is meer dan een fruitvlieg (die heeft er 8) of een kat (38) maar minder dan een chimpansee (48) of een hond (78). Behalve virussen bezitten alle organismen op aarde chromosomen (Tjio & Levan, 1956).
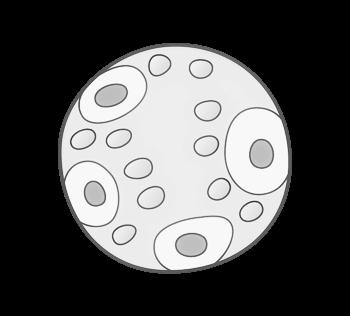
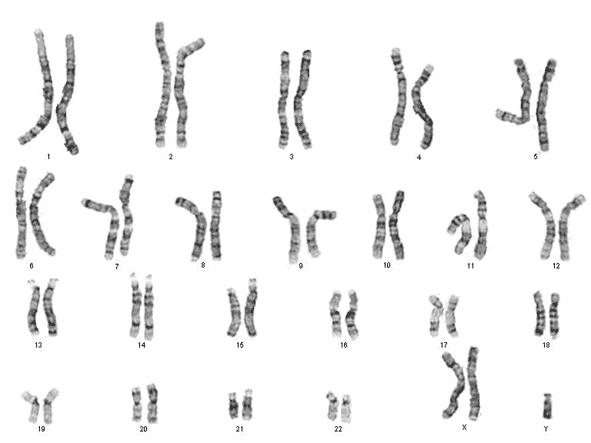
In een laboratorium kan men de chromosomen uit de kern van een witte bloedcel (verkregen via bloedafname) plukken en onder de microscoop rangschikken van groot naar klein. Zo verkrijgt men een karyogram (ook wel karyotype) genoemd.
Figuur 6 Normaal, vrouwelijk karyotype (46,XX) onder de lichtmicroscoop (vergroting x 10.000)
Uit karyotypering is ook de nummering van chromosomen ontstaan (van groot naar klein), waarbij chromosoom 1 het grootste is. Let wel: chromosoom 21 (en dus niet 22) is het kleinste chromosoom (het Y-chromosoom niet meegerekend). Het inzicht dat chromosoom 22 het kleinste is kwam er pas jaren nadat de nummering tot stand kwam.
Onder een microscoop zijn chromosomen niet enkel te herkennen aan hun lengte maar ook aan hun typische banderingspatroon. Door het toevoegen van bepaalde kleurstoffen verschijnen horizontale strepen op de chromosomen die uniek zijn voor elk chromosomenpaar.
Een normaal, humaan karyogram bevat 23 paren chromosomen:
22 paar autosomen of lichaamsbepalende chromosomen.
Dit zijn 22 paren van homologe (=gelijkwaardige, even grote) chromosomen. Deze chromosomenparen worden genummerd van groot naar klein, waarbij chromosoom 1 het grootste is en chromosoom 21 (en dus niet 22) het kleinste.
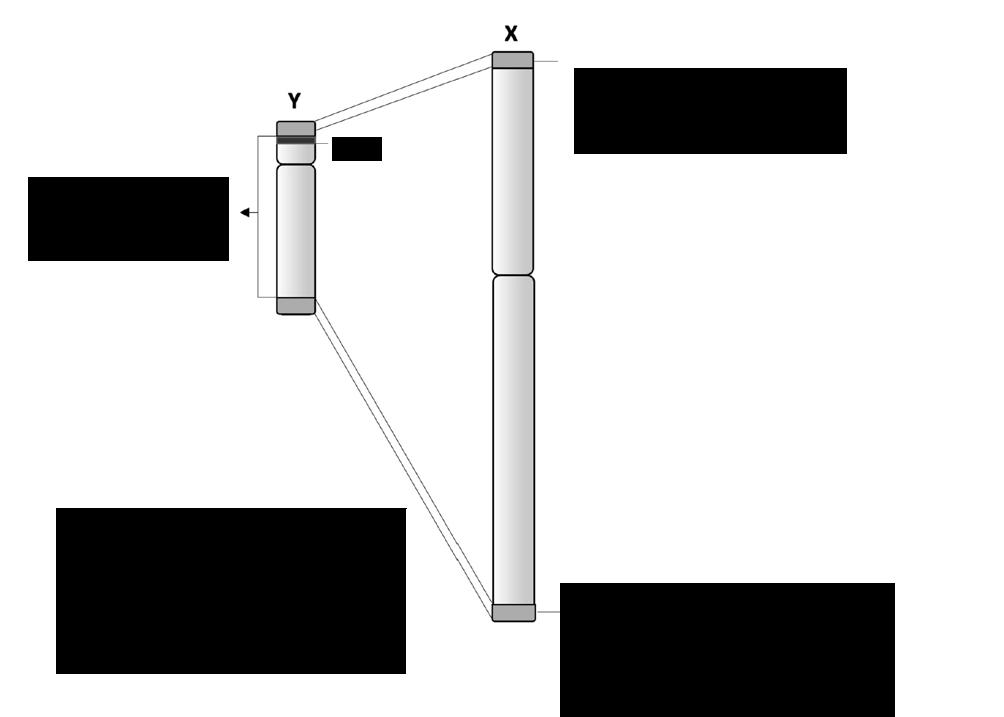
1 paar geslachtschromosomen: = chromosomen XX voor een vrouw en XY voor een man.
De geslachtschromosomen zijn niet homoloog. Het X-chromosoom is veel groter en bevat naar schatting 10 à 15 keer meer erfelijke informatie dan het Y-chromosoom. Het Y-chromosoom is het allerkleinste chromosoom. De vrijwel enige functie van dit chromosoom bestaat erin om het embryo te doen ontwikkelen als een man. Alle andere erfelijke informatie is in de loop der millennia uit het chromosoom verdwenen.
Karyotypering is tegelijk een vorm van genetisch onderzoek die toelaat om na te gaan welke chromosomen aan- of afwezig zijn, en of er eventueel grote, structurele afwijkingen zichtbaar zijn. Vanzelfsprekend is een karyogram echter een ruw onderzoek, waarbij kleine beschadigingen aan het erfelijk materiaal niet zichtbaar zijn.
1.2.2 Hoe noteren we een karyotype?
Een karyotype wordt afgekort volgens de ISCN-nomenclatuur (International System for Human Cytogenomic Nomenclature, 2020). Hierbij schrijven we eerst het totaal aantal chromosomen gevolgd door een komma (niet vergeten!) en de aanwezige geslachtschromosomen. De autosomen worden enkel vermeld als er afwijkingen zijn: ze worden bv. voorafgegaan door een + of – als het aantal autosomen afwijkt.
46, XX ± ...
totaal aantal chromosomen geslachtschromosomen overtollige, ontbrekende autosomen
Enkele voorbeelden:
ISCN-notatie
Man, normaal 46, XY
Vrouw, één extra chromosoom 21 47, XX +21
Man, één ontbrekend chromosoom 22 45, XY -22
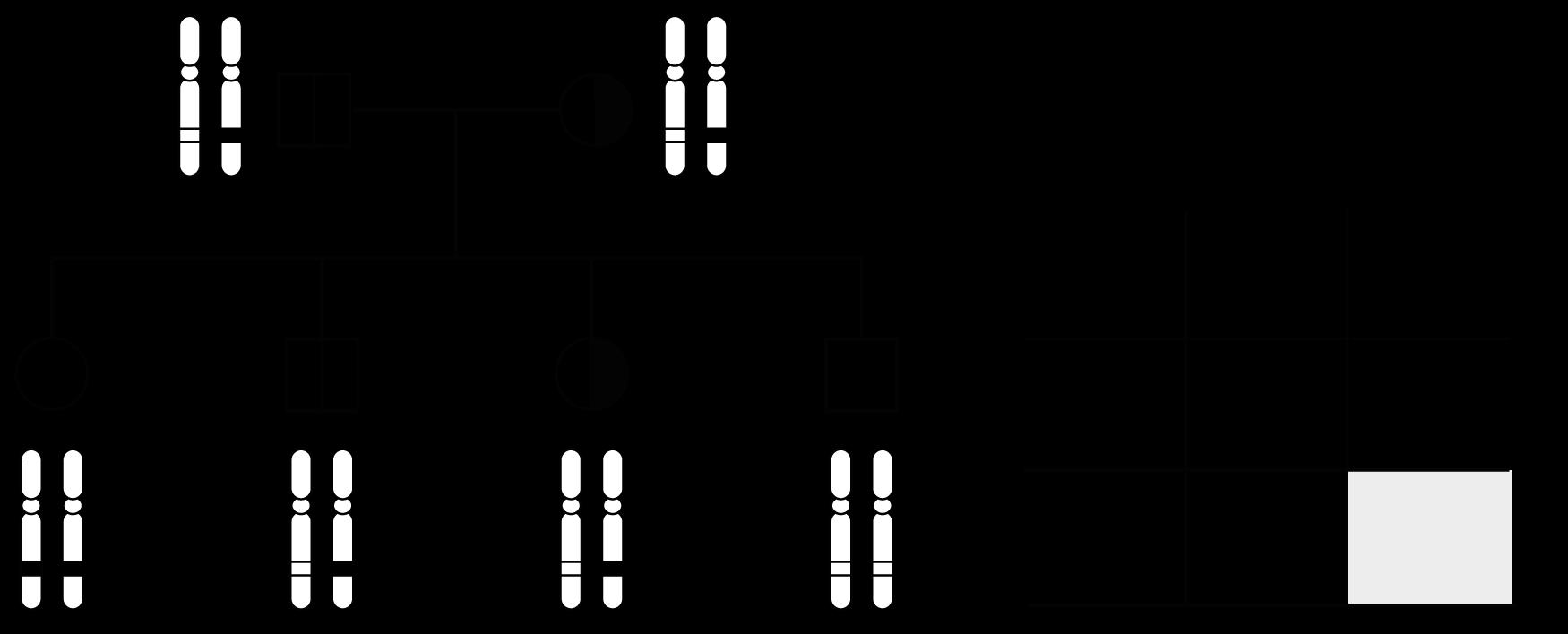
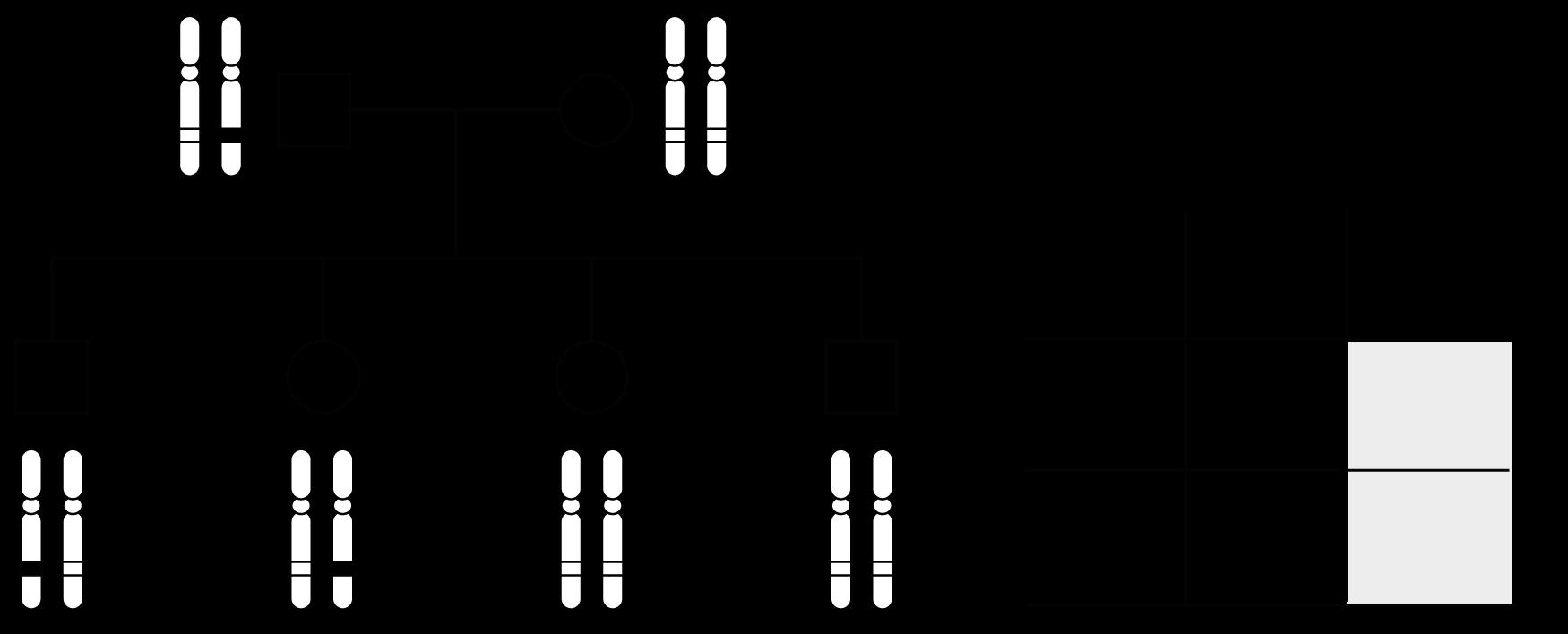
Oefening 1.
Noteer het karyotype (ISCN-notatie) voor…
Een man met een overtollig chromosoom 8
Een vrouw met een overtollig chromosoom 15 èn ontbrekend chromosoom 22
Een man met een extra X-chromosoom
1.3 Het menselijk genoom is diploïd
1.3.1
Begrippenkader
GENOOM
De complete genetische samenstelling van een organisme, cel of virus = alle erfelijke informatie in één celkern.
PLOÏDIE
Het aantal complete sets chromosomen (n) per celkern.
Het menselijk genoom, al onze erfelijke informatie, zit verpakt als 23 paar chromosomen. Je kan het wat vergelijken met een 46-delige encyclopedie, waarbij de chromosomen de boekdelen zijn. Opmerkelijk hierbij is dat we elk boekdeel dubbel hebben, zodat alle erfelijke informatie in tweevoud aanwezig is. Om die reden worden mensen diploïde wezens genoemd.
Het voordeel hiervan is evident: indien schade is opgetreden in één van beide chromosomen kan het homologe chromosoom als ‘back-up’ dienen (althans theoretisch).
Niet alle menselijke cellen zijn diploïd. In rijpe zaadcellen en eicellen is van elk chromosoom slechts één exemplaar aanwezig. Gameten (geslachtscellen = zaadcellen en eicellen) worden daarom haploïd genoemd.
Ploïdie Aantal sets Term Aantal chromosomen (mens) n 1 haploïd 23 2n 2 diploïd 46
3n 3 triploïd 69
4n 4 tetraploïd (onmogelijk bij de mens)
1.3.2 Het ‘genetisch’ zwakke geslacht
TOEPASSING: DE HONINGBIJ
Vrouwelijke honingbijen bezitten 32 chromosomen (2n), terwijl de mannelijke darren er slechts 16 (n) bezitten. De haploïde darren zijn dus ‘genetisch’ het zwakke geslacht: ze hebben zelfs geen angel en kunnen zichzelf niet voeden. Hun enige taak bestaat erin om de bijenkoningin te bevruchten, waarna ze sterven en niet mee overwinteren met de vrouwelijke werkbijen.
Bij de mens is de situatie (gelukkig) anders, hoewel we ook merken dat mannen vatbaarder zijn voor ziektes en een lagere, gemiddelde levensverwachting hebben dan vrouwen. De reden hiervoor is ook genetisch, hoewel mannen, net als vrouwen, toch diploïd (2n) zijn.
Kun je dit trachten te verklaren?
Antwoord:
Een man heeft X en Y als geslachtschromosomen. Deze chromosomen zijn niet homoloog. Dit betekent dat een man eigenlijk niet volledig diploïd is. Voor het X en Y chromosoom is er immers geen back-up. Dat kan al deels verklaren waarom een man vatbaarder is voor ziekte.
Daarnaast komt een Y-chromosoom altijd ongepaard voor (in tegenstelling tot X, die bij de vrouw wel per paar voorkomt) Het Y chromosoom heeft dus nooit een back-up vanwaaruit beschadigde informatie kan hersteld worden. Het Y-chromosoom geldt om die reden als het meest kwetsbare deel van ons erfelijk materiaal.
Van mannen zegt men dus ook wel eens dat ze genetisch het ‘zwakke geslacht’ zijn.
1.3.3 Polyploïdie
POLYPLOÏDIE
Het aantal complete sets chromosomen (n) per celkern is groter dan 2.
Een voorbeeld hiervan is triploïdie (3n), wat vaak leidt tot spontane abortus of een beperkte levensverwachting.
Tetraploïdie (4n) is bij de mens niet mogelijk.
In het plantenrijk komt polyploïdie vaak voor: bv. bananen zijn triploïd (3n) en aardbeien octoploïd (8n). Hierdoor zijn deze gewassen weliswaar onvruchtbaar en kunnen ze geen zaden of pitten aanmaken in hun vruchten. Dit vergemakkelijkt wel de consumptie (pitloos!). Heel wat groenten- en fruitsoorten worden om die reden bewust polyploïd gekweekt.
JUIST OF FOUT?
“Als een diploïde cel zich klaarmaakt om te delen, en de hoeveelheid DNA verdubbelt tijdens de S-fase van de celcyclus (zie hoofdstuk 3), dan is de cel tijdelijk tetraploïd (4n)”
Antwoord:
Deze stelling is fout. Tijdens de S-fase verdubbelt weliswaar de hoeveelheid erfelijk materiaal, maar elke kopie blijft aan het origineel hangen (zie eerder). We kunnen dus wel stellen dat de cel tijdelijk 92 zusterchromatiden bevat (men noteert dit als 4c), maar geen 92 afzonderlijke chromosomen (4n). Bij het uiteengaan van de zusterchromatiden krijgt elke cel terug 46 afzonderlijke chromosomen (2n).
1.3.4 Aneuploïdie
ANEUPLOÏDIE
Het aantal chromosomen in een celkern is geen veelvoud van n.
Aneuploïdie is de meest voorkomende, chromosomale abnormaliteit bij de mens. Dit leidt (net zoals polyploïdie) vaak tot een spontane abortus.
De frequentste vormen van aneuploïdie bij levendgeborenen zijn trisomie 13, 18 en 21. Het bekendste voorbeeld is trisomie 21, waarbij een extra chromosoom 21 in de celkern voorkomt (2n + 1). Hier vinden we dus 47 chromosomen terug in het totaal. Het karyotype noteren we als 47, XX+21 (voor een meisje) of 47, XY+21 (voor een jongen).
Het ziektebeeld dat hiermee gepaard gaat staat bekend als het syndroom van Down.
Doorheen de cursus komt aneuploïdie verschillende keren aan bod:
In 4.4 gaan we verder in op de oorzaken van aneuploïdie
In 5.4.3 bespreken we de vaakst voorkomende vormen van aneuploïdie bij de zygote
In 6.4 linken we aneuploïdie aan het optreden van spontane abortus
In 20.3 en 20.5 bespreken we enkele levensvatbare syndromen als gevolg van aneuploïdie
1.4 DNA
1.4.1 Bouw van DNA
JAAR: 1944
Na de ontdekking van de chromosomen duurde het meer dan 50 jaar vooraleer men de samenstelling ervan konden achterhalen. Wetenschappers dachten lang dat het ging om een soort eiwit, maar Avery, McCarty en MacLeod ontdekten in 1944 dat chromosomen bestaan uit een ‘zuur’. De term desoxyribo-nucleïnezuur (of 'deoxyribonucleic acid' in het Engels) werd hiervoor bedacht, internationaal afgekort als DNA (Avery et al., 1944).
Het ‘erfelijk materiaal’ in de chromosomen vinden we terug als desoxyribonucleïnezuur (DNA), een gespecialiseerde molecule die toelaat om informatie op te slaan en door te geven. DNA behoort tot de familie van de nucleïnezuren
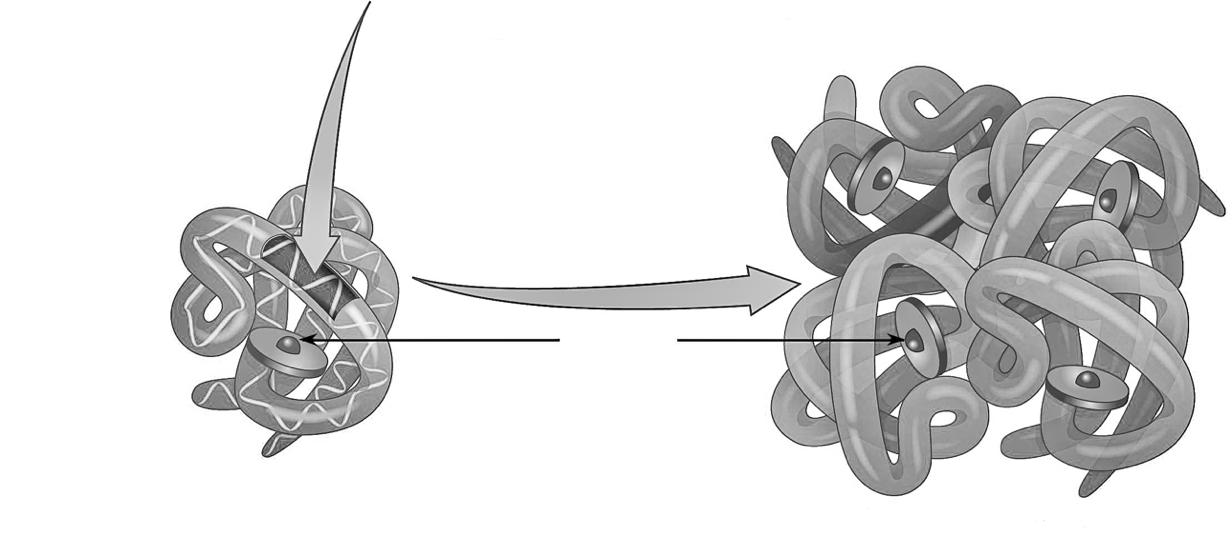
Chromosomen bestaan uit opgerolde desoxyribonucleïnezuur (DNA)-moleculen met verpakkingseiwitten (histonen) eromheen.
JAAR: 1952-53
chromosoom
Steuneiwitten (histonen)
DNA-helix
Figuur 7 Chromosomen bestaan uit opgerold DNA.
Nadat Rosalind Franklin in 1952 een röntgenfoto nam van gekristalliseerd DNA (“photo 51”) kwam de zoektocht naar de bouw van de DNA-molecule in een stroomversnelling. James Watson & Francis Crick puzzelden als eersten een (metalen) DNA-model ineen dat klopte met alle waarnemingen en berekeningen. De ontdekking van de dubbele helix leverde hun de Nobelprijs op in 1963 (Watson & Crick, 1953).
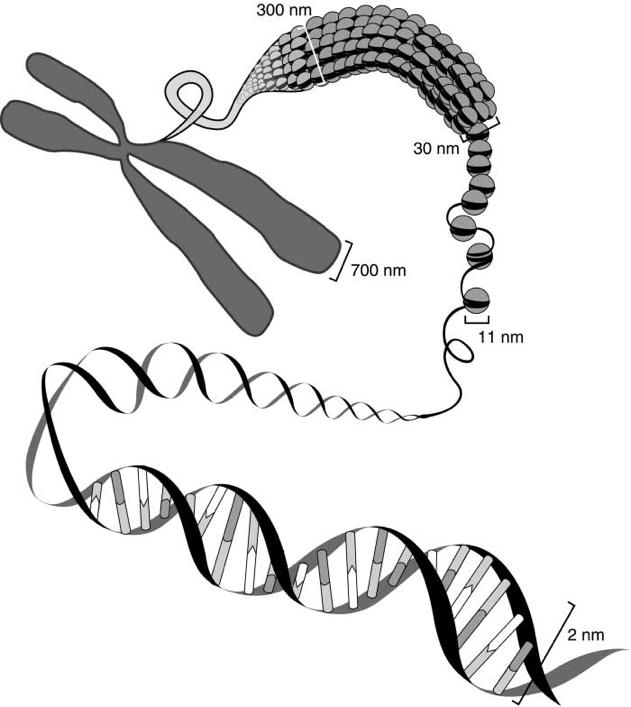
Watson & Crick ontdekten dat elk van deze DNA-moleculen opgebouwd is uit een
dubbele helix, die bestaat uit een lange, spiraalvormige ‘vlecht’ van nucleotiden
Nucleotiden zijn dus de eigenlijke bouwstenen van DNA.
Elke nucleotide bestaat uit:
Een vijfhoekige suikermolecule (deoxyribose),
o de koolstofatomen (hoeken) worden met een cijfer aangeduid (1’ t.e.m. 5’).
Een fosfaatgroep (P), die de deoxyribosen aan elkaar ‘lijmt’,
o dit gebeurt ter hoogte van hun 5’ en 3’ uiteinden
Één van 4 basen
o Adenine (A), Thymine (T), Cytosine (C) en Guanine (G).
fosfaatgroep base (cytosine) nucleotide
deoxyribose
Figuur 8 Nucleotiden vormen de bouwstenen van het DNA (Let op de polariteit van de strengen, aangegeven met pijlen)
De deoxyribosen en fosfaatgroepen vormen hierbij twee tegengesteld lopende strengen: de strengen zijn antiparallel
Elke streng heeft een duidelijke richting of polariteit
• 5'-uiteinde van het eerste deoxyribose = begin van de streng
• 3'-uiteinde van het laatste deoxyribose = einde van de streng
Een DNA-streng loopt dus steeds van 5’ naar 3’:
Figuur 9 Polariteit van de DNA-streng
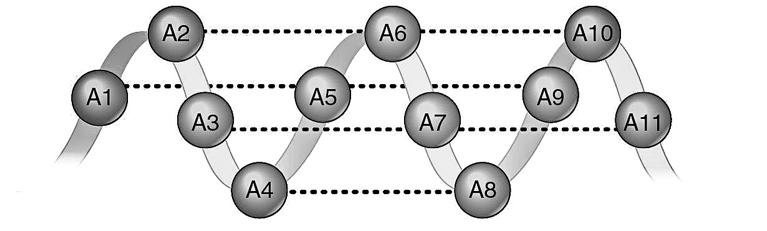
Aan de binnenkant van elke streng vinden we de basen, (A, C, G of T), die telkens vasthangen aan een deoxyribose De basen uit elke streng komen hierbij tegenover elkaar te liggen. Hierdoor trekken ze elkaar aan en vormen ze twee of drie waterstofbrugjes. Dit zijn chemische verbindingen die voldoende krachtig zijn om de strengen bij elkaar houden en te zorgen dat een dubbele helix ontstaat.
Basen die tegenover elkaar liggen heten we een basepaar (bp).
Figuur 11 DNAhelix
Figuur 10 Dubbelstrengig DNA met waterstofbrugjes (stippellijnen)
Als we kijken naar welke basen elkaar aantrekken stellen we vast:
T C G
Adenine (A) bindt enkel met Thymine (T)
Cytosine (C) bindt enkel met Guanine (G)
Er bestaan dus maar 2 soorten baseparen: AT en CG. Hierop worden geen uitzonderingen gemaakt.
Op het eerste gezicht lijkt dit misschien een banale bevinding, maar dit heeft een belangrijk gevolg: beide nucleotidestrengen moeten altijd precies complementair zijn met elkaar.
Dit betekent dat de code in de ene streng precies tegengesteld is aan de code in de andere streng (vergelijk met het ‘negatief’ van een foto). Dezelfde informatie zit erin vervat, maar dan omgekeerd.
We spreken van de coderende en de niet-coderende streng.
1.4.2
Functies van DNA
§ Opslag
§ Replicatie
1.4.2.1 Opslag
§ Regulatie
§ Recombinatie
Hieronder verstaan we:
Bewaren van alle informatie die nodig is voor de bouw en functie van een organisme.
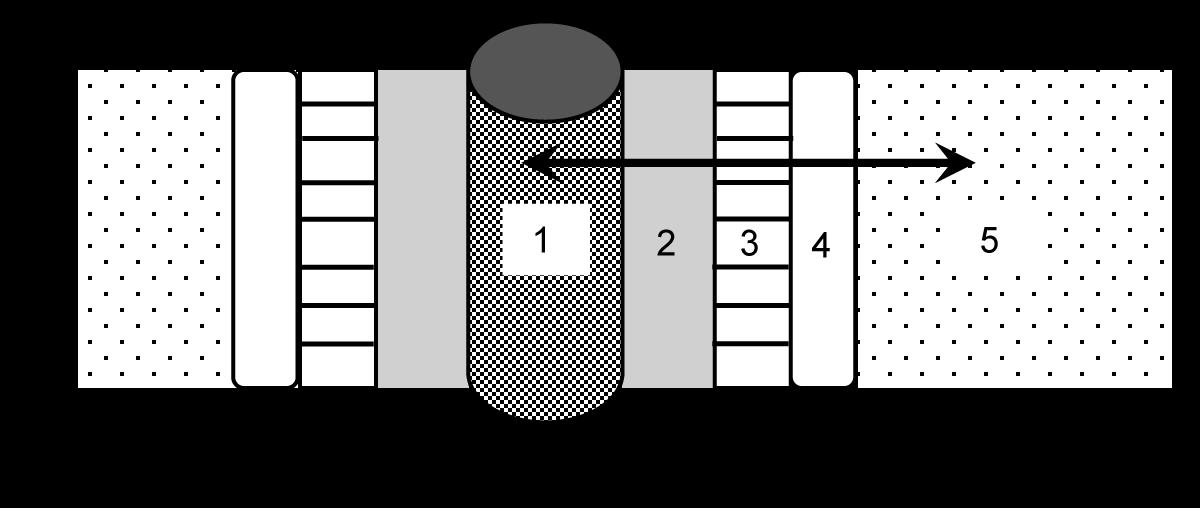
Uitgezonderd enkele virussen maken alle organismen op aarde gebruik van DNA. Bij de mens zit deze ‘handleiding’ in elke haploïde celkern als een genoom van 3,2 miljard baseparen.
Het samendrukken als chromosomen zorgt hierbij voor een enorme schaalverkleining. Elke celkern bevat immers 2 meter (!) DNA. Het 'verknippen' en oprollen in 46 afzonderlijke draadjes maakt dat het DNA compacter en ordelijker kan worden opgeborgen.
Op een stabiele manier doorgeven van erfelijke informatie, over generaties
Door de talrijke waterstofbruggen is DNA erg stevig. Enkel bij temperaturen van 70°C of meer gaan de ketens uit elkaar en gaat de helix-structuur verloren. We heten dit proces denaturatie Dit betekent trouwens niet dat het DNA afgebroken is. De ketens zèlf zijn enorm duurzaam en het duurt duizenden jaren vooraleer de strengen afbreken in de vrije natuur. De DNA-code kan dus vele malen langer overleven dan zijn gastheer.
In 2006 ontdekten Belgische wetenschappers in een Ardense grot een DNA-fragment van 123 baseparen. Het DNA bevond zich in de melktand van een Neanderthaler-kind dat ongeveer 100.000 jaar geleden leefde (Orlando et al., 2006).
1.4.2.2 Replicatie
Het vermenigvuldigen van onze erfelijke informatie is belangrijk om… weefsels te kunnen laten groeien door het aanmaken van nieuwe cellen.
deze informatie te kunnen doorgeven naar het nageslacht.
DNA-replicatie gebeurt tijdens de S-fase van de celcyclus. Vóór de celdeling gaan de beide nucleotideketens uit elkaar en wordt aan elke streng een nieuwe, complementaire streng gesynthetiseerd. Zo ontstaan nieuwe, identieke, kopieën van de oorspronkelijke helix. De oorspronkelijke, dubbelstrengige helix wordt hierbij niet gewijzigd.
1.4.2.3 Regulatie (van expressie)
Er zijn tal van systemen in de cel die bepalen welke erfelijke informatie uit ons genoom tot uiting komt. We gaan hier dieper op in, in volgende hoofdstukken (zie o.a. 2.7).
1.4.2.4 Recombinatie
Recombinatie (herordenen) van erfelijk materiaal is belangrijk voor het bevorderen van variatie. Zo kunnen nieuwe eigenschappen ontstaan die het organisme geschikter maken aan het overleven aan de (eventueel veranderende) omgeving.
Ons DNA wordt op verschillende momenten efficiënt gerecombineerd:
Tijdens de vorming van geslachtscellen, door crossing over (zie 3.3.2) en het optreden van spontane mutaties (zie 16.4.1) Elke, menselijke gamete bevat naar schatting ± 30 nieuwe mutaties!
Bij de bevruchting, door het samenstellen van een nieuwe zygote, zie hoofdstuk 5.
1.5 Gen
JAAR: pre-1900
In de 2e helft van de 19e eeuw werd volop gespeculeerd over de kleinste, ondeelbare dragers van erfelijke informatie. Darwin had het over ‘gemmulen’, Mendel over ‘vormen’ en ‘factoren’, Sutton over ‘partikels’ en De Vries over ‘pangenen’ (verwijzend naar pangenese = de oorsprong van alles). Pas in 1909 werd dit door Johannsen afgekort tot ‘gen’.
1.5.1 Definitie
Een gen is de kleinste, ondeelbare drager van een stuk erfelijke informatie. In de praktijk komt dit overeen met een stukje DNA dat codeert voor een specifiek eiwit. Dit eiwit wordt ook het genproduct genoemd.
Het aantal menselijke genen wordt geschat op 20.687. Dit is meer dan het aantal studenten in VIVES (17.000 in 2022).
Een gen wordt meestal benoemd met een afkorting (evt. met een cijfer erbij).
Enkele voorbeelden: AKT1, BRCA1, IGF1, MAX, NEMO, SMURF1, TNF, …
Vaak zijn genen genoemd naar de ziekte die ze uitlokken als het gen defect is. Vanuit biologisch perspectief is dit absurd. Bv. het huntingtine-gen (HTT, zie 20.1.6) codeert voor een eiwit dat een beschermende functie heeft in het zenuwstelsel. Toch is het gen genoemd naar de ziekte die het veroorzaakt als het gen niet goed werkt (de ziekte van Huntington).
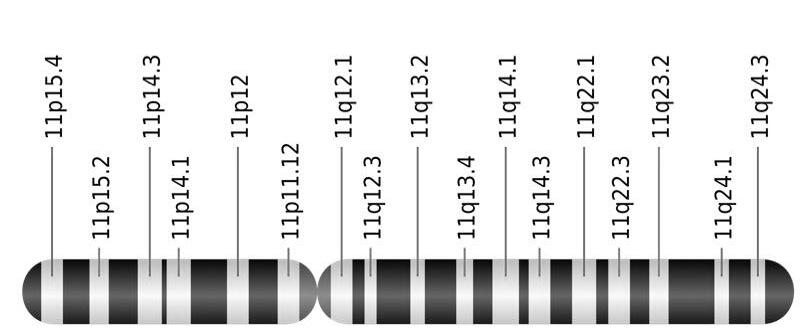
INS, het gen voor insuline is een afgelijnd stuk DNA dat we terugvinden op chromosoom 11. Het bevat de code die noodzakelijk is om insuline aan te maken.
Figuur 12 insuline als voorbeeld van een gen
Insuline is een polypeptide (een klein eiwit) dat de suikerspiegel doet dalen. Het wordt afgegeven door de b-cellen van de pancreas na de maaltijd.
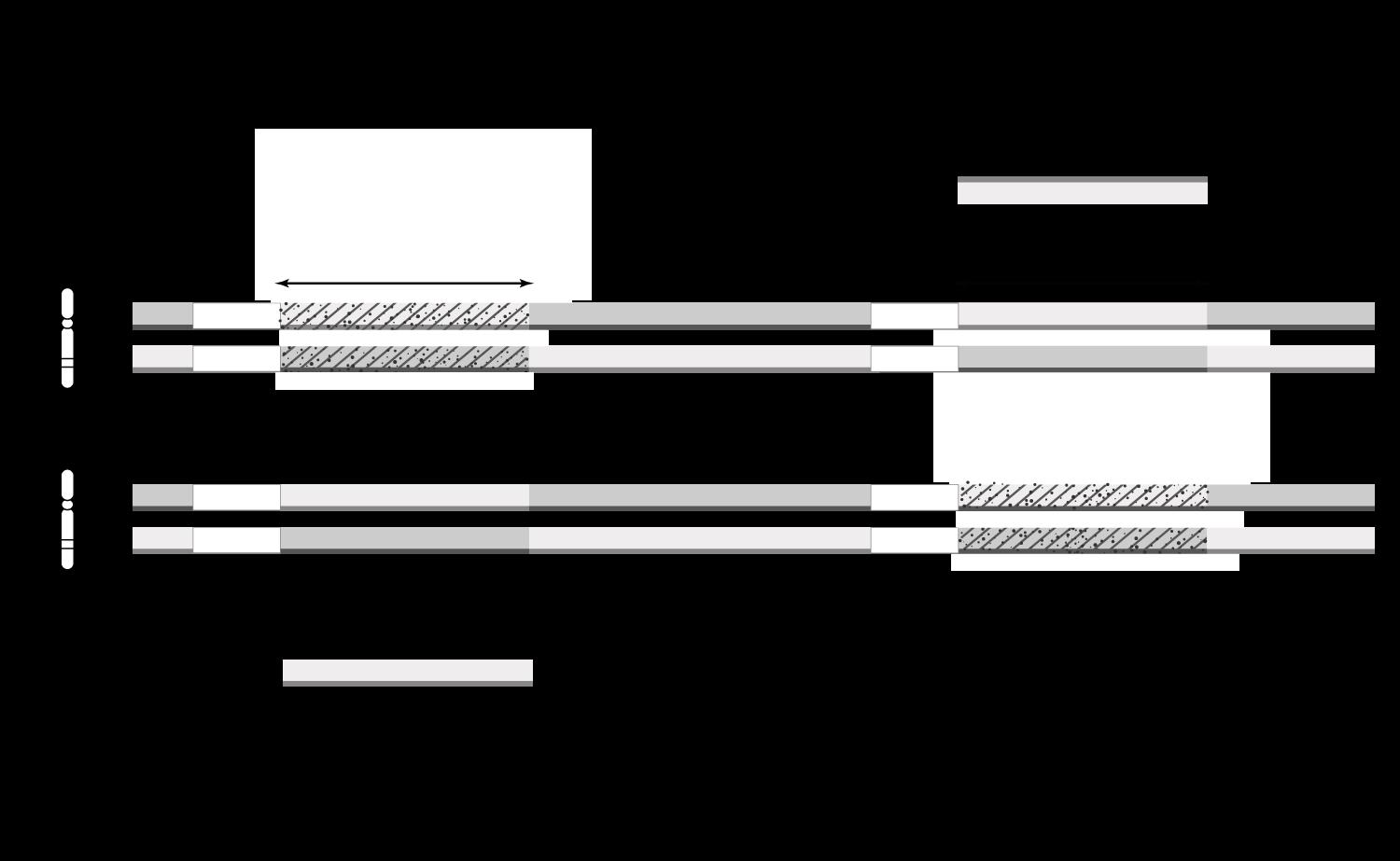
1.5.2 Locus
Een locus betekent een unieke plaats in het genoom. Vaak zal men van een gen zeggen op welke locus het zich bevindt. Een dergelijke locus bestaat uit het
Chromosoomnummer
De chromosoomarm (p of q)
Een plaatsnummer volgens het gekleurde banderingspatroon.
INS, het gen voor insuline bevindt zich op de korte arm (p) van chromosoom 11, op positie 15.5 (helemaal aan de tip). De locus is bijgevolg 11p15.5
Oefening 2.
Verklaar de volgende loci:
13 chromosoom 11
Figuur
1.5.3
Lengte
De lengte van een gen wordt uitgedrukt in eenheden van baseparen (bp).
1 1 bp
1 kilobase (kb) 1000 bp
1 Megabase (Mb) 1.000.000 bp
1 Gigabase (Gb) 1.000.000.000 bp
Het totale menselijk, haploïde genoom is 3,2 Gb lang. Genen zijn gemiddeld 24 kb lang, maar er bestaat een zeer grote onderlinge variatie:
Het SRY-gen op locus Yp11.2 is slechts 828 basen (0,828 kb) lang maar het genproduct bepaalt wel of een embryo als meisje of als jongen ontwikkelt.
® Meer over SRY (Sex Determining Region): zie 14.1.2
Het DMD-gen op locus Xp21.1 èn Xp21.2 is maar liefst 2.241.933 basen (2,241 Mb) lang. Het genproduct zorgt voor het verankeren van spierfilamenten.
® Meer over DMD (Duchenne Muscular Dystrophy) zie 20.1.10
Oefening 3.
Surf naar www.genecards.org en ga via de zoekfunctie op zoek naar de gegevenskaart van het gen voor albumine (één van de vaakst voorkomende eiwitten in ons bloed).
Hoe wordt dit gen benoemd/afgekort?
Vind je de locus terug in de lijst van gegevens?
Welke lengte heeft het? (Tip: zoek naar ‘size’ bij ‘latest assembly’)
2 Eiwitsynthese
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De rol van DNA, mRNA en tRNA in de eiwitsynthese te beschrijven
De verschillen in bouw tussen DNA en RNA kort te omschrijven.
Uit te leggen wat de transcriptie van een gen inhoudt
Uit te leggen wat de translatie van mRNA inhoudt
De juiste aminozuursequentie te noteren bij een reeks codons (de tabel met codons en overeenstemmende aminozuren krijg je hierbij gegeven).
Uit te leggen a.d.h.v. een voorbeeld wat post-translationele modificatie inhoudt.
Uit te leggen hoe een genetisch defect een ziekte als mucoviscidose kan veroorzaken. In je uitleg komt hierbij de rol van het CFTR-eiwit aan bod.
Ten minste vijf verschillende eiwitfuncties te kunnen opnoemen.
Het centrale dogma van Francis Crick toe te lichten aan de hand van een voorbeeld.
De manier waarop genexpressie gebeurt te beschrijven, incl. de rol van een promotor en transcriptiefactoren.
Een concreet voorbeeld te geven van genexpressie.
Het belang van het Human Genome Project toe te lichten aan de hand van enkele zaken die de wetenschap hieruit heeft geleerd.
Het belang van project ENCODE toe te lichten aan de hand van enkele zaken die de wetenschap hieruit heeft geleerd.
In eigen woorden uit te leggen wat bedoeld wordt met genome sequencing.
DNA is de blauwdruk voor de bouw van een levend wezen. Hiervoor dienen de genen in het DNA stapsgewijs te worden vertaald naar werkzame eiwitten
We bespreken deze stappen van de eiwitsynthese aan de hand van een concreet voorbeeld, namelijk het CFTR-eiwit, dat een rol speelt in het ontstaan van de aandoening mucoviscidose (taaislijmziekte, cystic fibrosis).
2.1 Toepassing: mucoviscidose
Mucoviscidose is een erfelijke aandoening die het gevolg is van een fout in het CFTRgen. CFTR staat voor cystic fibrosis transmembrane regulator.
Het CFTR-gen bevindt zich op locus 7q31.2, is 190 kb lang en bevat de instructies voor het bouwen van een eiwit dat bestaat uit 1480 aminozuren.
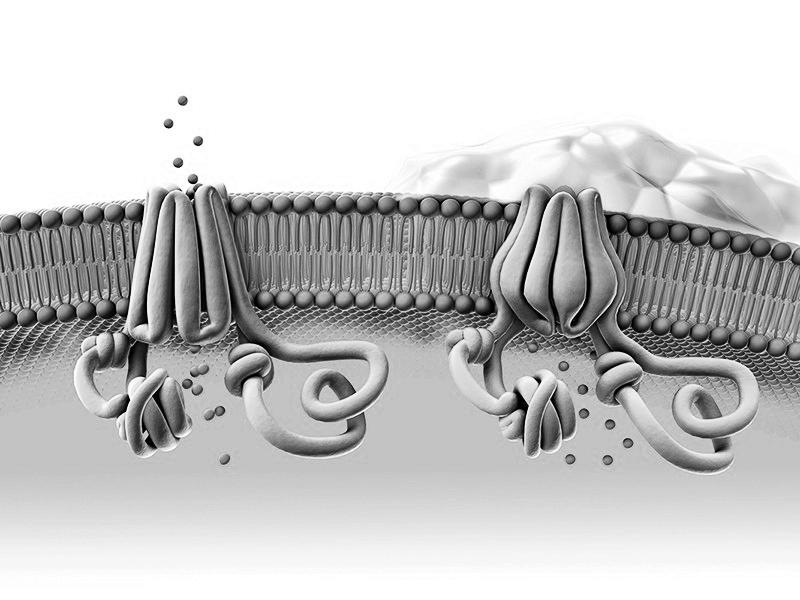
Het afgewerkte CFTR-eiwit vouwt zich samen tot een tunnel (‘kanaal’) die zich verankert in de celmembraan. Dankzij dit kanaal kunnen elektrolyten (bv. chloor, natrium) in en uit de cel worden gepompt
chloor-ionen slijmopstapeling kanaal is dicht
celmembraan
Figuur 14 Een 3D-model van het gezonde CFTR-eiwit (links) en het afwijkende CFTR eiwit (rechts) (Nature, 2020)
Bij een fout in het gen wordt een defect CFTR-eiwit geproduceerd. Zo’n defect CFTReiwit vouwt zich niet op de correcte manier, waardoor de kanaalfunctie geheel of gedeeltelijk verloren gaat. Hierdoor geraken elektrolytenconcentraties uit evenwicht en stapelen zich taaie slijmen op o.a. in de luchtwegen en het maagdarmstelsel.
De gebrekkige werking van CFTR veroorzaakt dus het ziektebeeld.
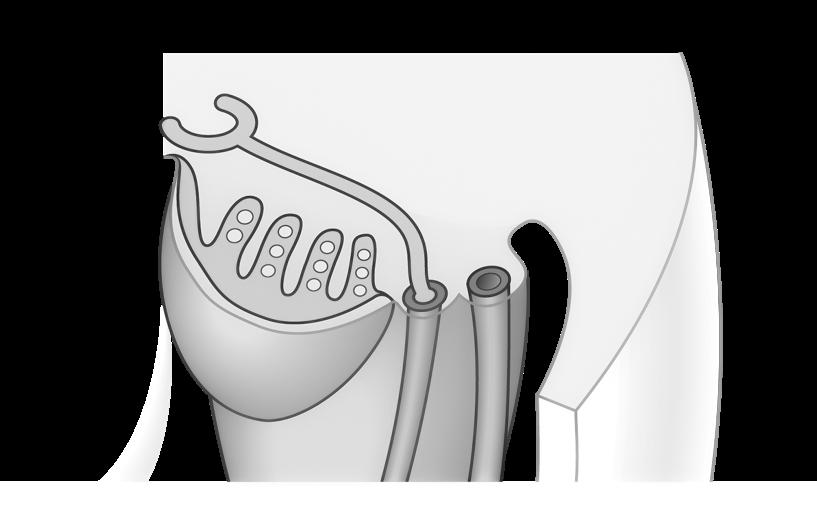
2.2 Transcriptie
De eerste stap in de eiwitsynthese is het overschrijven van de code. Tijdens deze transcriptiefase wordt het dubbelstrengig DNA gedeeltelijk uit elkaar gewikkeld zodat het gen toegankelijk wordt. Door een enzym, het RNA-polymerase, wordt vervolgens de niet-coderende streng gebruikt om een nieuwe, coderende streng aan te maken.
Deze nieuwe, enkelstrengige kopie heten we het messenger-RNA (mRNA).
RNA staat voor ribonucleïnezuur (ribonucleic acid). In tegenstelling tot DNA is er geen desoxyribose-suiker in aanwezig, maar een ribosesuiker. Ook thymine (T) is vervangen door uracil (U). Verder lijkt het mRNA heel sterk op DNA (het zijn beide nucleïnezuren) en bevat het in feite dezelfde informatie als de coderende streng.
(coderende streng) (niet-coderende streng) DNA
Figuur 15 Transcriptie van een gen dat bestaat uit 3 exonen (E1, E2 en E3).
(coderende streng) (coderende streng)
DNA codeert dus niet rechtstreeks voor eiwitten. Eerst gebeurt een tussenstap naar mRNA en de reden hiervoor is vooral praktisch. Eiwitsynthese gebeurt immers in het cytoplasma en het DNA is te groot om de nucleus te verlaten. mRNA beweegt dan weer zonder probleem door openingen in de kernmembraan naar het cytoplasma.
Vooraleer het mRNA de celkern verlaat ondergaat het nog enkele veranderingen.
Onnodige, niet-coderende fragmenten (intronen) worden eruit geknipt en afgebroken. De noodzakelijke, coderende fragmenten (exonen) worden aan elkaar gehecht tot één, nieuw geheel. Dit proces heten we splicing
De meeste humane genen bevatten veel meer intronen dan exonen. Er wordt dus heel wat onnodig DNA weggeknipt.
TOEGEPAST OP MUCOVISCIDOSE
Het CFTR-gen bestaat uit 190.000 basen met daarin 27 exonen. Na splicing vormen deze exonen aaneengeschakeld een mRNA van ± 6500 basen (96% van het gen wordt dus ‘weggeknipt’!) Alle 27 exonen zijn van belang voor het vormen van een correct functionerend CFTR-eiwit (Hamilton, 2020).
2.3 Translatie
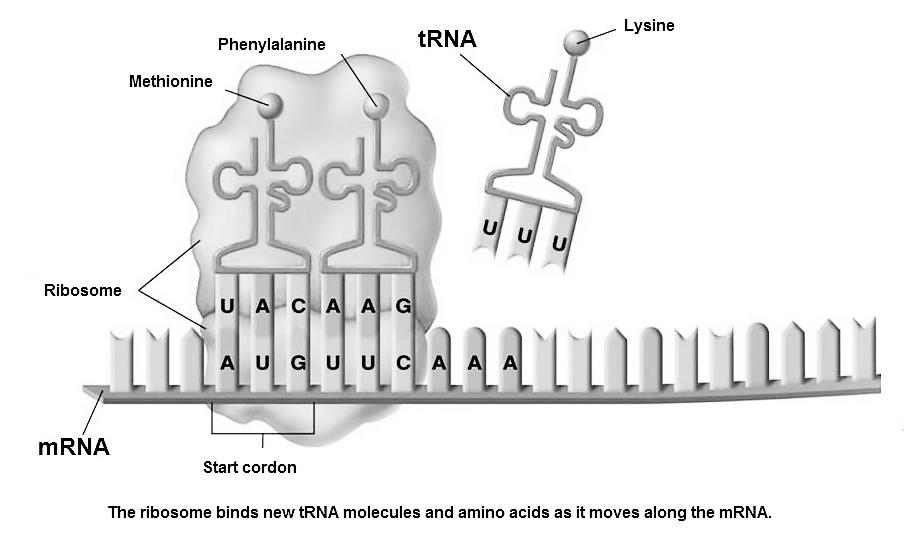
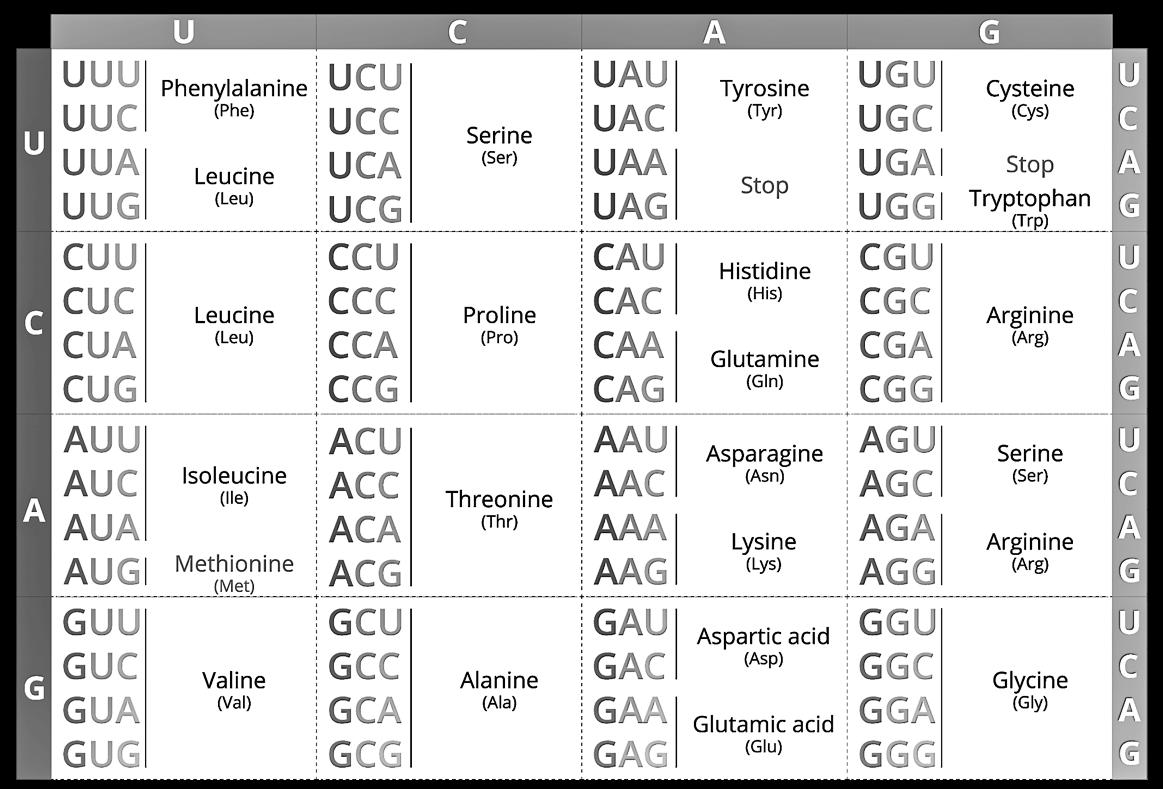
In het cytoplasma wordt het mRNA ter hoogte van het ribosoom gebruikt als instructie om een eiwit (proteïne) aan te maken. Het ribosoom is een complexe structuur die bestaat uit rRNA (ribosomaal RNA) en eiwitten. Het mRNA wordt hier afgelezen van 5’ naar 3’ en stapsgewijs gebruikt om aminozuren te selecteren.
Het aaneenrijgen van deze aminozuren (AZ) vormt een ketting (= een proteïne of eiwit).
aminozuur 1
aminozuur 2
aminozuur 3
anticodon
ribosoom
codon
Figuur Eiwitsynthese
Het aflezen van het mRNA gebeurt per triplet (groepje van 3 basen). Een triplet dat leidt tot de selectie van een aminozuur heten we een codon. Per codon wordt één AZ aan de ketting gehecht.
Aangezien elk codon plaats heeft voor 3 basen en er 4 mogelijke basen zijn, kunnen we hieruit berekenen dat er 64 verschillende codons mogelijk zijn (43 = 4 x 4 x 4 = 64)
De 22 mogelijke AZ worden aangevoerd door een transfer-RNA (tRNA) dat eveneens een groepje van 3 basen bevat, specifiek voor hun gebonden aminozuur.
Om te kunnen binden aan het mRNA moeten de 3 basen van het tRNA tegengesteld zijn aan het codon. Vandaar heten we dit een anticodon.
TOEGEPAST OP MUCOVISCIDOSE
Het CFTR-gen start – net zoals elk gen – met het codon AUG. Dit heten we het startcodon. Het startcodon bindt met het tRNA dat het anticodon UAC bevat. Op dit tRNA zit het aminozuur Methionine gebonden. Aan het eind van elk gen vinden we een stopcodon.
TWEEDE LETTER
Figuur 17 De codons en hun overeenstemmende aminozuren (in het Engels).
(Je hoeft dit schema niet uit het hoofd te kennen.)
Deze code is zo goed als universeel. Op enkele, kleine uitzonderingen na gebruiken vrijwel alle organismen precies dezelfde code voor het aanmaken van eiwitten.
Oefening 4.
a) Hoeveel startcodons en stopcodons herken je in Figuur 17?
b) Juist of fout? “Voor elk codon is er 1 aminozuur.”
c) Denkvraag: stel, je krijgt een basesequentie gegeven van 1000 basen. Hoe zou je kunnen tellen hoeveel genen er in die sequentie aanwezig zijn.
d) Kun je de aminozuursequentie bepalen voor de eerste 6 codons van het CFTRgen (mRNA):
AUG- CAG- AGG- UCG- CCU- CUG
e) Antivirale vaccins bevatten normaal een stukje virus-eiwit dat herkend wordt door ons immuunsysteem. Recente, antivirale vaccins bevatten geen eiwit, maar enkel mRNA. Hoe werkt zo’n vaccin?
2.4
Post-translationele modificatie
Nadat de aminozurenketting de ribosoom-tunnel verlaat, zijn nog verschillende aanpassingen nodig om tot een werkzaam, driedimensionaal eiwit te komen. Dit heten we post-translationele modificatie.
We bespreken hier enkel het voorbeeld van hemoglobine: Hemoglobine is een eiwit dat zorgt voor het O2 en CO2-transport in de rode bloedcellen.
2.4.1 Primaire structuur
= de aminozurenketting zelf.
globine-eiwitten bevatten 145 aminozuren (A)
2.4.2 Secundaire structuur
= delen van de ketting nemen een spiraal- of plaatvorm aan
waterstofbrugjes
2.4.3 Tertiaire structuur
= het volledige eiwit vouwt en neemt een specifieke, driedimensionale vorm aan. heem
ijzeratoom
Figuur 18 Primaire eiwitstructuur
Figuur 19 Secundaire eiwitstructuur: spiraal (helix)
“hemoglobine”
Figuur 20 (Li) Tertiaire structuur (Re) Quaternaire structuur
In het globine-eiwit zit een inkeping, waarin een heem-molecule met een ijzeratoom wordt geplaatst. Dit zal het makkelijker maken om O2 en CO2 te binden.
2.4.4 Quaternaire structuur
= meerdere tertiaire structuren werken samen om een groter geheel te vormen
Het afgewerkte hemoglobine-eiwit bestaat uit 4 subunits, (2 keer a en 2 keer b) elk met hun eigen heem-molecule. Nu is het eiwit helemaal klaar om zijn functie te vervullen.
Cruciaal voor de post-translationele modificatie is een correcte, primaire eiwitstructuur. Fouten in de DNA-code kunnen de eiwitstructuur ernstig verstoren. Zelfs in gevallen waar maar één aminozuur is gewijzigd, kan de secundaire, tertiaire en quaternaire structuur volledig falen, waardoor het eiwit niet werkt (zie 16.5)
Net zoals DNA kunnen proteïnen denatureren oiv externe factoren (bv. hitte). Hierbij gaat de secundaire, tertiaire en quaternaire structuur verloren. Dit is precies wat er gebeurt bij het bakken of kloppen van ei-wit uit een kippenei.
Enzym: vult een metabole taak uit, bv. omzetting, afbraak van een stof.
Structuureiwit: bv. collageen en keratine vormen steunvezels in de huid
Transporteiwit: bv. transferrine voor ijzer, hemoglobine voor O2 en CO2
Transcriptiefactor: regelt de expressie van genen; bepaalt de identiteit van de cel
Signaalmolecule: brengt een signaal over bv. van aan het celoppervlak naar de kern.
Kanaal: creëert een opening doorheen de celmembraan naar de buitenwereld (bv. voor Na+ , Cl, glucose, …) of een tunnel naar een andere cel (= gap junctions, bv. voor Ca2+ in spiercellen), waardoor cellen elektrisch verbonden worden.
Immunoglobulines: antistoffen, die binden met ziekteverwekkers
Hormonen: stimuleren de activiteit van weefsels door het binden op receptoren
Receptoren: binden signaalmoleculen of hormonen
Recent werd een opvallende, nieuwe eiwitfunctie ontdekt. “Hero”-eiwitten zijn hittebestendige proteïnen die andere proteïnen beschermen tegen denaturatie (bv. door te werken als een hitteschild) (Tsuboyama et al., 2020).
2.6 Het centrale dogma
Op dit punt in de cursus zou het moeten duidelijk zijn hoe het erfelijk materiaal in ons lichaam wordt opgeslagen. Alle erfelijke informatie samen heten we het genoom.
We weten ondertussen ook hoe de informatie in dat genoom gebruikt wordt om een organisme te bouwen. De informatie wordt eerst overgeschreven naar mRNA en vertaald naar eiwitten.
Deze eiwitten vervullen de chemische en structurele functies die een levende cel doen ontstaan en in stand houden. Als kleine machines vervullen duizenden eiwitten continu de gespecialiseerde taken waarvoor ze gebouwd zijn.
Dit is precies wat Francis Crick beschreef toen hij het in 1970 in Nature had over het ‘centrale dogma van de moleculaire biologie’. Met ‘dogma’ bedoelde Crick eigenlijk de centrale, verklarende ‘wet’ voor het verband tussen DNA en eiwitten (Crick, 1970).
"DNA maakt RNA maakt eiwit"
mRNA “boodschap”
DNA “gen”
Eiwit “functie”
Figuur 21 Centrale dogma (Crick, 1970 en Mukherjee, 2016)
De pijlen geven de richting van de informatieoverdracht weer.
Wie dit principe goed begrijpt zal in de volgende hoofdstukken ook begrijpen hoe fouten in het DNA tot ziektes leiden.
Enkele begrippen die hier nog bij aansluiten:
GENOTYPE
Het deel van het genoom dat de informatie bevat voor een bepaalde functie of kenmerk heten we het genotype. In de praktijk komt dit meestal overeen met een gen.
FENOTYPE
Het kenmerk of de functie die aanwezig is in het organisme heten we het fenotype.
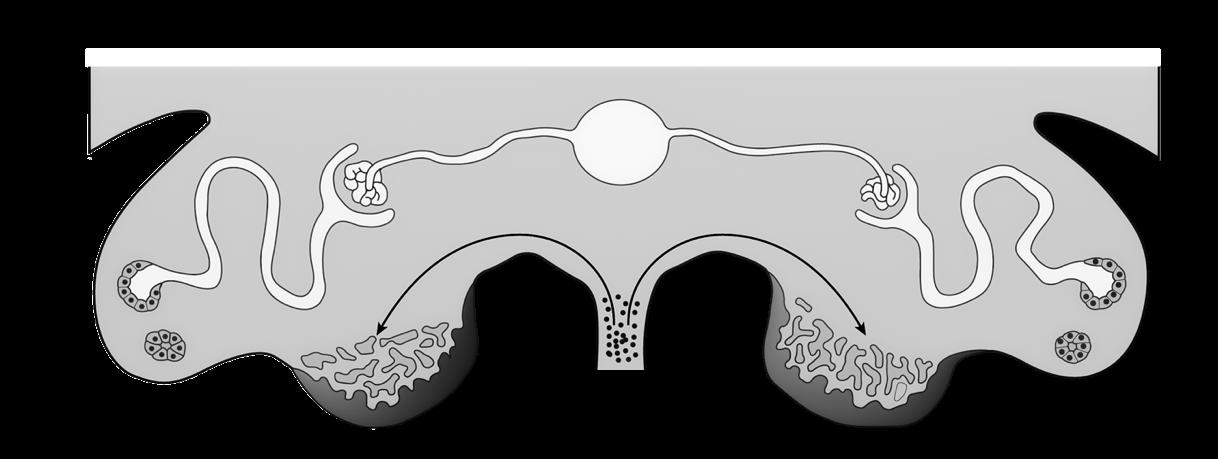
2.7 Genexpressie
Expressie = tot uiting laten komen van erfelijke informatie door eiwitproductie.
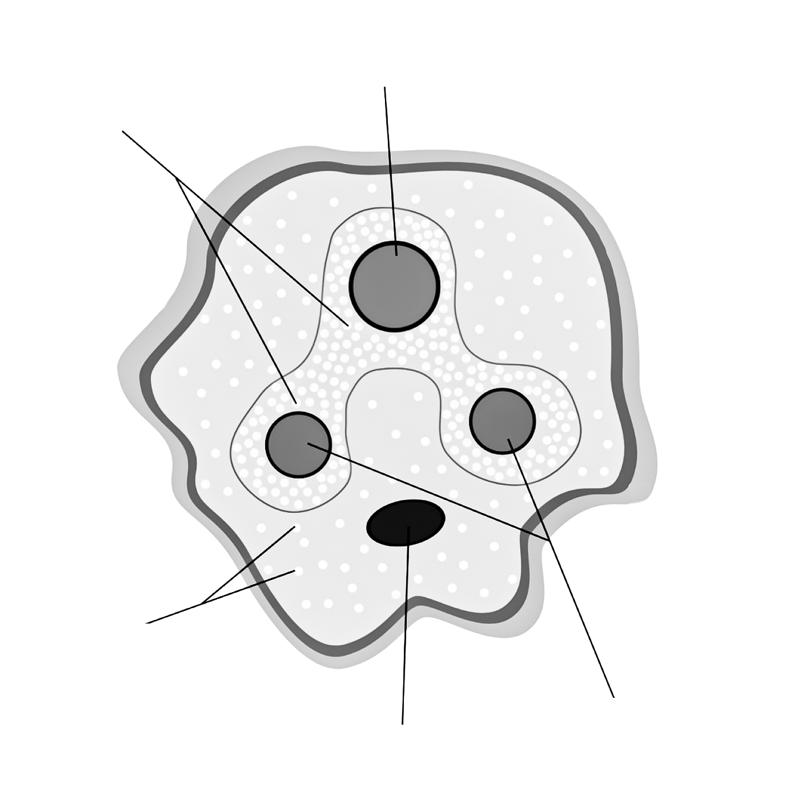
Hoewel het volledige genoom in elke celkern van ons lichaam aanwezig, komen niet alle genen in elke cel tot expressie. Cellen en weefsels produceren enkel de eiwitten die nodig zijn om hun functies te vervullen.
In elke cel zijn er hiervoor regelmechanismen die toelaten dat bepaalde genen worden aan- of uitgeschakeld.
1
eiwit 1 eiwit 4
Figuur 22 Genexpressie: enkel gen 1 en 4 komen tot expressie. GEN 1 GEN 2 GEN 3 GEN 4
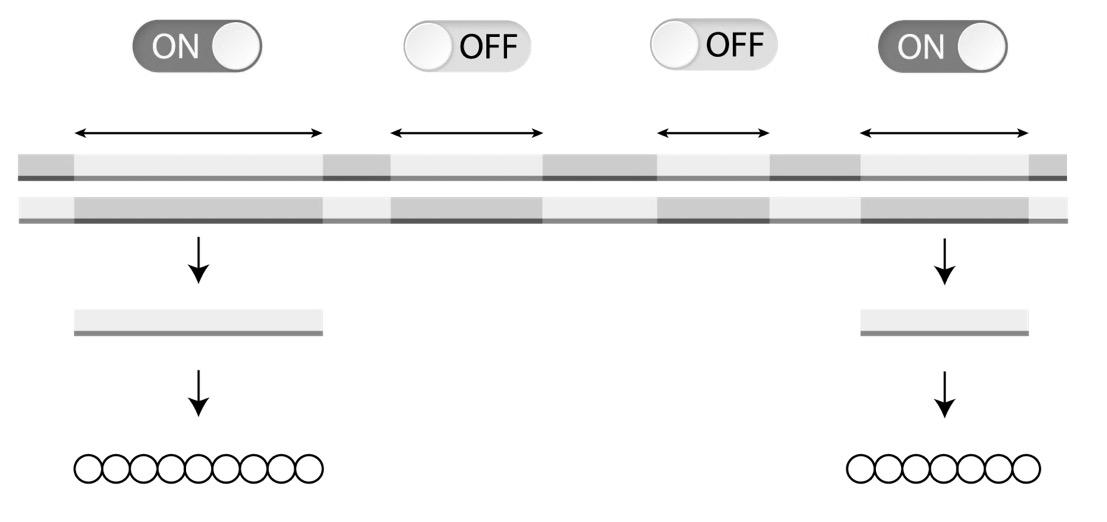
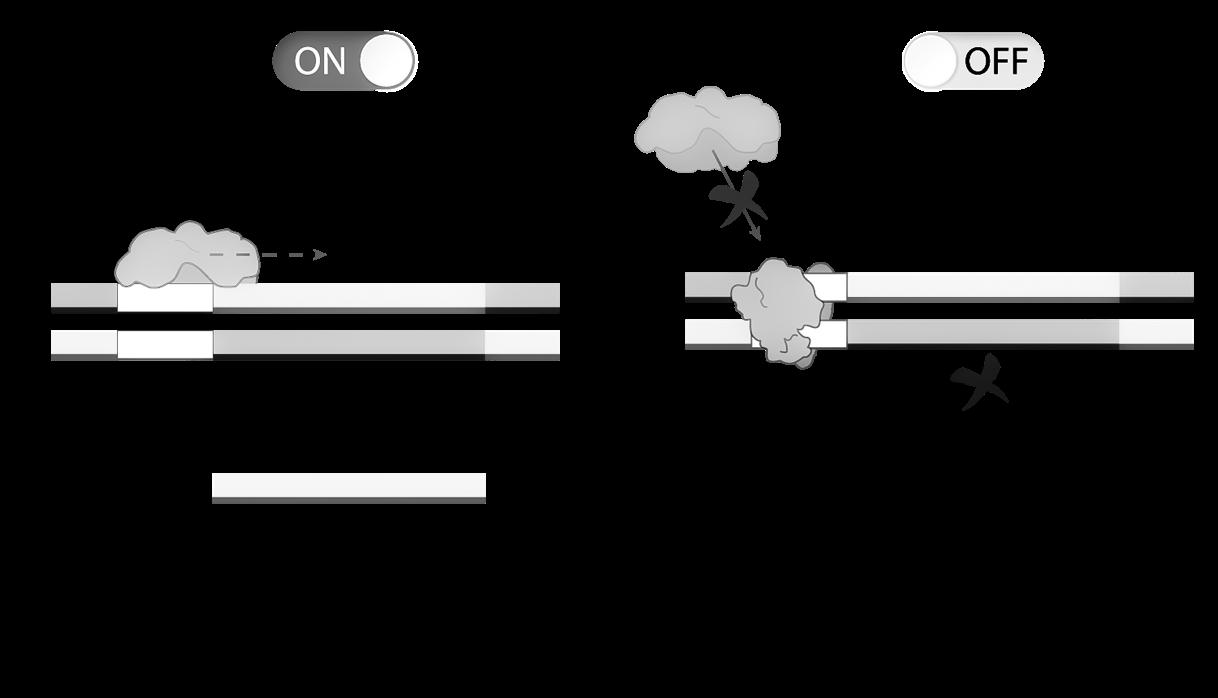
De expressie van een gen wordt geregeld door een promotor aan het 5’-uiteinde. Dit is een deel van de DNA-code die niet wordt overgeschreven tot eiwit, maar dient voor het aanhechten van het RNA-polymerase (het enzym dat de transcriptie uitvoert, zie 2.2).
Een transcriptiefactor blokkeert de promotor, waardoor RNA-polymerase niet kan binden. De transcriptie wordt hierdoor stilgelegd (=’silencing’).
Figuur 23 De rol van promotoren en transcriptiefactoren.
De promotor-regio wordt gebruikt door transcriptiefactoren die hier binden om de transcriptie stil te leggen (zie Figuur 23) of te bevorderen.
Daarnaast zorgen transcriptiefactoren ook voor het ontrollen van stukjes DNA-helix, waardoor de genen toegankelijker worden voor het RNA-polymerase.
Sommige transcriptiefactoren werken als ‘hoofdschakelaars’ en gaan op hun beurt tientallen andere transcriptiefactoren activeren.
Het aan- of uitschakelen van genen kan definitief zijn (bv. bij genen die na de embryogenese geen nut meer hebben), maar is vaak tijdelijk (bv. nadat er voldoende voorraad is geproduceerd van een bepaald eiwit).
Voorbeeld 1: embryologie:
Tijdens de embryologische ontwikkeling worden stap voor stap genen aan- en uitgeschakeld. Hun genproduct speelt een rol speelt in het aansturen van de embryogenese. Na de embryogenese worden deze genen inactief.
Voorbeeld 2: reukreceptoren:
Op chromosoom 11 liggen 155 genen naast elkaar die elk coderen voor een andere reukreceptor. Elke zenuwcel in de neus laat telkens maar één van die 155 receptoren tot expressie komen bv. enkel de receptor voor vanille, de receptor voor peper, limoen, gember etc… Deze keuze is definitief. Zo kunnen we duizenden geuren van elkaar onderscheiden.
Voorbeeld 3: het schildklier-stimulerend hormoon TSH:
Dit eiwit wordt geproduceerd door cellen van de hypofyse. Na afgifte in de bloedbaan stimuleert dit hormoon de schildklier om schildklierhormoon (T4) aan te maken. Als er voldoende T4 circuleert in de bloedbaan wordt de synthese van TSH afgezwakt. Als er veel te veel T4 circuleert in de bloedbaan (zoals bij bepaalde ziektes) wordt de synthese van TSH zelfs volledig stilgelegd.
Het bestaan van een promotor & transcriptiefactoren toont dat er voor elk gen een mechanisme bestaat om het aan- of uit te schakelen. Dit schakelsysteem wordt natuurlijk - op zijn beurt - weer gecontroleerd door andere signalen.
In het deel genetica (zie 18.2) bespreken we enkele bijkomende mechanismen die de expressie van genen aansturen
2.8 Het menselijke genoom in kaart brengen
In 1.2 maakten we al kennis met karyotypering, een ruwe manier om het genoom te onderzoeken door het te bekijken onder de microscoop.
Sinds de jaren ’80 zijn technieken ontwikkeld die een gedetailleerde studie van de DNA of RNA-code zelf mogelijk maken. Dit heten we sequencing
Sequencing (sequenering) is het bepalen van de nucleotidevolgorde van DNA of RNA.
Deze volgorde van nucleotiden heten we een sequentie.
JAAR: 1990-2003
Door het ontdekken van sequencing ontstond het ambitieuze ‘Human Genome Project’ (1990-2003), een internationale inspanning om het volledige genoom van één mens te sequencen. In 2003 waren de resultaten hiervan bekend, zie de QR-code links.
het humaan genoom
Dankzij het Human Genome Project (HGP)…
… hebben we een ‘stafkaart’ van het menselijk genoom
Telkens een nieuw genoom wordt geanalyseerd kunnen we het met deze referentie vergelijken: https://www.ncbi.nlm.nih.gov/genome/guide/human/?
… weten we dat het menselijk genoom 20 687 genen bevat.
Van 40% van de genen kennen we de eiwitfunctie nog steeds niet (we kunnen wel voorspellen hoe het eiwit eruitziet).
… weten we dat – alle genen aaneengeschakeld – slechts 2% van ons DNA uitmaken. De overige 98% van ons DNA wordt – soms wat oneerbiedig – het junk DNA geheten.
JAAR: 2003-heden
Na het Human Genome Project startte project ENCODE (Encyclopedia of DNA elements), om de betekenis van het junk-DNA na te gaan. Hierin werden oa. promotor-regio’s gevonden, maar ook stukken DNA die coderen voor RNA-moleculen die niet worden overgeschreven tot eiwitten bv. tRNA, rRNA etc… Voorlopig blijkt dat 80% van het DNA een functie heeft.
Het Human Genome Project duurde 13 jaar en kostte 3 miljard dollar. Door de sterke technologische vooruitgang duurt sequencing van een menselijk genoom tegenwoordig (in 2022) amper een halve dag en is de kostprijs gedaald tot 100 à 200 euro Dit maakt genoomanalyse stilaan toegankelijk voor het grote publiek. Diverse private bedrijven bieden deze testen al aan, al of niet met bijhorend, persoonlijk advies over het resultaat
De laatste jaren is er een enorme ‘boom’ in genomics: het onderzoek naar alle facetten van het menselijk genoom, inclusief de mogelijkheden om aanpassingen uit te voeren (zie 20.2.2).
3 Celdeling
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De rol van telomerase in het verouderingsproces toelichten.
Het doel van mitose en meiose te vergelijken.
Het verloop van de mitose en de meiose met elkaar te vergelijken.
De concepten van mitose en meiose toepassen op de rest van de leerstof.
De term ‘crossing over’ grondig toe te lichten, inclusief het tijdstip van optreden en de bedoeling hierachter.
Aan te geven welke chromosomen niet deelnemen aan crossing-over.
3.1
Celcyclus
De celcyclus is een serie gebeurtenissen die een cel doorloopt om te groeien en delen.
1 profase
2 prometafase
3 metafase
4 anafase
5 telofase
Figuur 24 De celcyclus bij mitose (Klug, 2019)
Het grootste gedeelte van de tijd zijn cellen niet aan het delen en bevinden ze zich in de interfase Chromosomen zijn op dat moment niet te onderscheiden in de celkern.
De interfase bestaat uit 3 opeenvolgende fasen: G1, S en G2
Tijdens deze 3 fasen groeit en differentieert de cel. Maar enkel tijdens de S-fase wordt het kernmateriaal verdubbeld. Door DNA-synthese kopieert de cel z'n erfelijk materiaal. Na dit verdubbelen beschikt de celkern over 92 zusterchromatiden (=4c)
Merk op dat er cellen zijn die op een bepaald moment stoppen met deze cyclus te doorlopen. Ze kunnen dus niet meer delen en bevinden zich permanent in het G0stadium, bv. de meeste zenuwcellen (neuronen) na de leeftijd van 18 maanden.
Zelfs actief delende cellen doorlopen deze cyclus maximaal 50 keer (=de Hayflick-limiet). Wellicht heeft dit te maken met het korter worden van de telomeren. Na ongeveer 50 delingen zijn de telomeren immers zodanig kort geworden dat de celcyclus stopt.
Een interessante vaststelling is dat telomeren opnieuw opgebouwd kunnen worden door het enzym telomerase. Dit proces zou in de meeste van onze cellen niet gebeuren, maar bv. wel in embryonale cellen, stamcellen, kiemcellen en kankercellen. Deze cellen worden dus niet ouder (Gomes et al., 2010; Tedone et al., 2019).
Bepaalde diersoorten zoals reuzenschildpadden en platwormen maken levenslang telomerase aan. Hierdoor kunnen ze heel erg oud worden (van platwormen wordt zelfs gezegd dat ze onsterfelijk zijn). Hun cellen blijven delen, waardoor de weefsels zich telkens vernieuwen. Jonathan, de laatste van een soort Seychellen-schildpadden, werd geboren in 1832 en verblijft sinds 1882 op het eiland Sint-Helena.
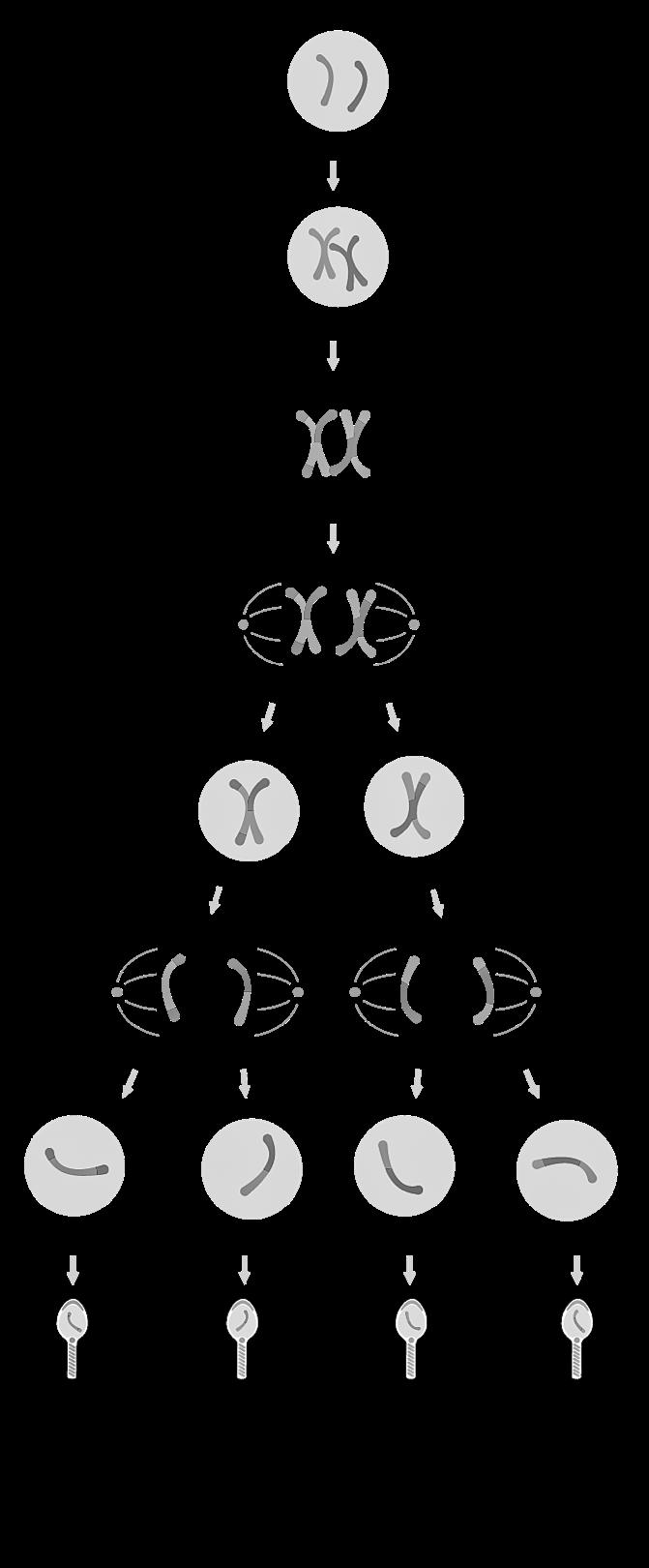
3.2 Mitose
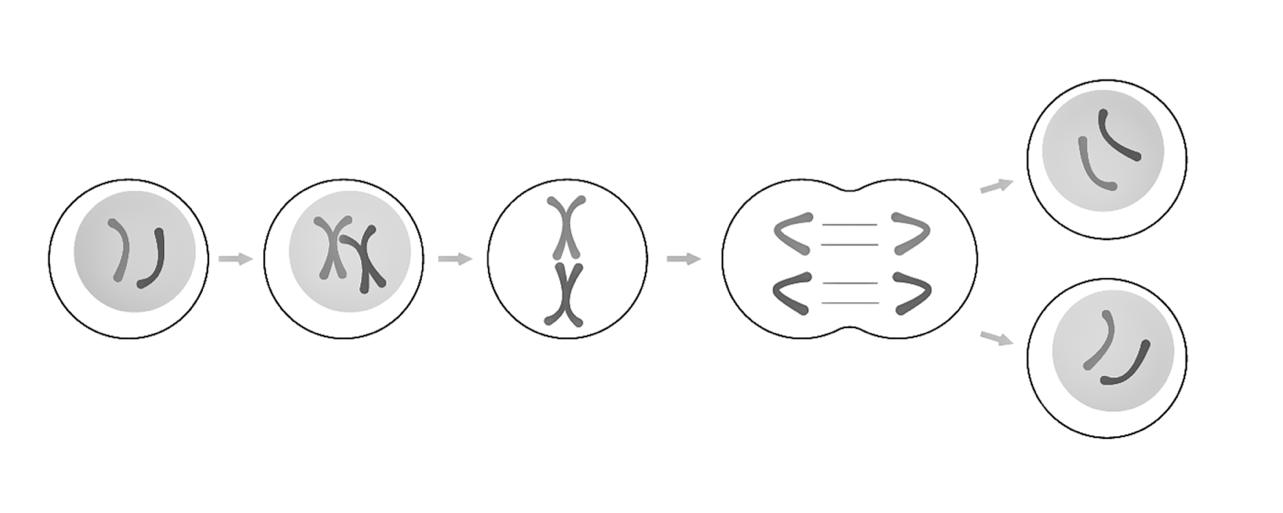
Mitose = celdeling waarbij uit één diploïde cel (2n) twee diploïde (2n) cellen worden gevormd. Doel: cellen vermeerderen, 2 identieke kopieën maken.
Mitose is de celdeling die ervoor zorgt dat weefsels groeien. Je kan dus stellen dat de belangrijkste drijvende kracht achter de embryogenese, foetale groei en groei tijdens de kinderleeftijd mitose is.
DNA-duplicatie (metafase) (anafase)
Figuur
Profase 46 verdubbelde chromosomen (4c) worden zichtbaar in de celkern.
Prometafase Spoelfiguur met trekdraden verschijnt.
Metafase Verdubbelde chromosomen aligneren zich op de evenaar van de spoel.
Anafase De zusterchromatiden gaan uiteen naar de polen.
Telofase Cytoplasmadeling tot 2 identieke dochtercellen (2 keer 2n)
3.3 Meiose
3.3.1
Verloop van de meiotische deling
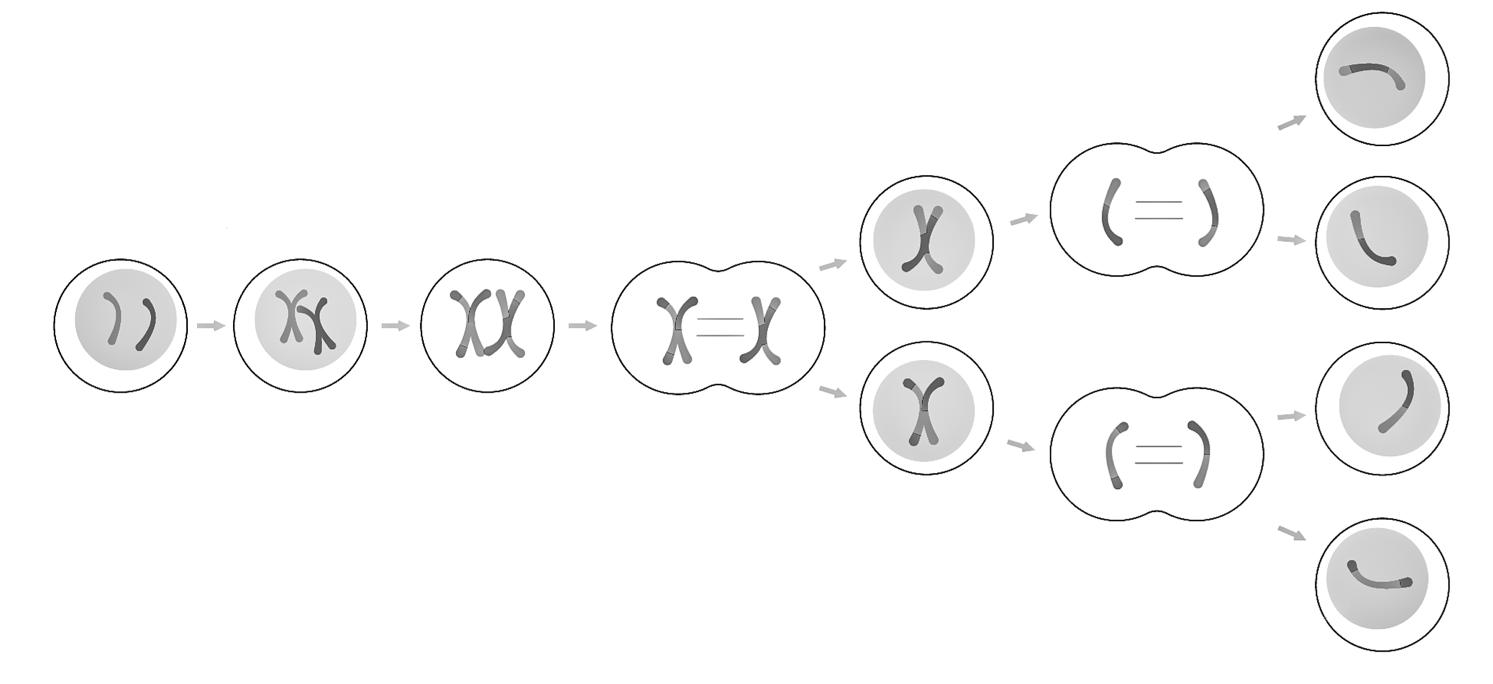
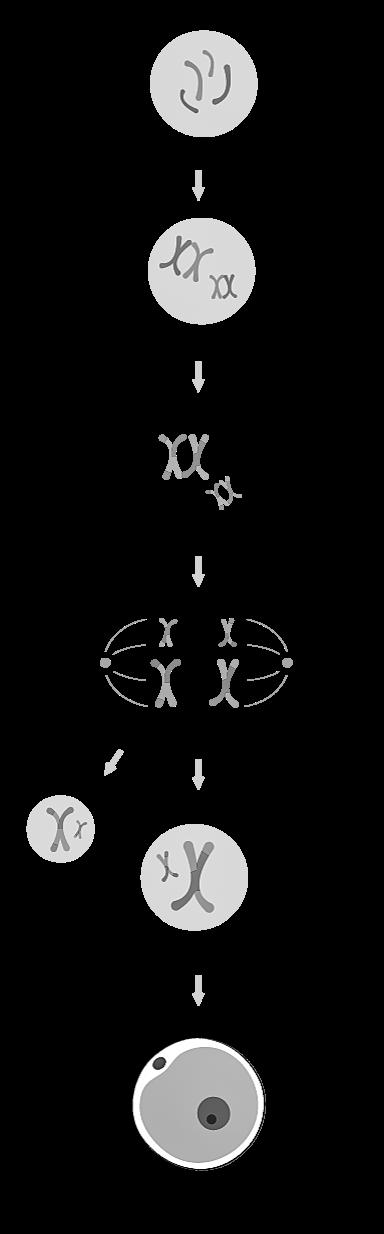
Meiose = celdeling waarbij uit één diploïde cel (2n) vier haploïde cellen (n) worden gevormd. Doel: geslachtscellen (gameten) creëren
Meiose is een speciale vorm van celdeling waarbij de hoeveelheid erfelijk materiaal gereduceerd wordt. We maken dus geen identieke kopieën zoals bij mitose. De meiotische deling wordt ook geslachtsdeling of reductiedeling genoemd.
Een meiotische deling bestaat altijd uit twee opeenvolgende celdelingen, aangeduid als meiose I en meiose II. De 2e meiotische deling lijkt hierbij heel sterk op een mitotische deling. Tussen meiose I en meiose II is er geen nieuwe interfase.
DNA duplicatie
(profase I)
I) (anafase II)
Profase I Crossing over van tetraden (4c) (zie 3.3.2)
Metafase I Tetraden aligneren zich op de evenaar van de spoelfiguur.
Anafase I Tetraden worden gesplitst in verdubbelde chromosomen die uiteengaan.
Telofase I Cytoplasmadeling tot 2 cellen met 23 verdubbelde chromosomen (2 keer 2c).
Profase II 23 verdubbelde chromosomen worden zichtbaar.
Metafase II Verdubbelde chromosomen aligneren zich op de evenaar van de spoel.
Anafase II De zusterchromatiden gaan uiteen naar de polen.
Telofase II Cytoplasmadeling tot 2 cellen met 23 chromosomen (2 keer n).
VIDEO
https://www.youtube.com/watch?v=zGVBAHAsjJM
Mooie mitose – meiose simulatie met kralen.
MEIOSE I
MEIOSE II
MEIOSE I
MEIOSE II
Figuur 26 Meiose
3.3.2
Crossing over
Crossing over (letterlijk: overkruisen) is een uniek fenomeen dat enkel plaatsvindt tijdens de 1e meiotische deling.
Tijdens profase I zoeken de verdubbelde chromosomen hun ‘partnerchromosoom’ op om een verbinding van 4 zusterchromatiden te vormen (tetraden). Elke tetrade bestaat dan uit een verdubbeld maternaal en een verdubbeld paternaal chromosoom.
zuster chromatiden zuster chromatiden
Figuur 27 Crossing over van een chromosoom (vereenvoudigde voorstelling)
Vervolgens wisselen de maternale en paternale chromosomen gelijkwaardige stukjes DNA uit. Dit gebeurt ter hoogte van een chiasma (meervoud: chiasmata), waar de nietzusterchromatiden geknipt (en weer geplakt) worden. Hierbij worden nieuwe, unieke zusterchromatiden gevormd, die na de 2e meiotische deling in de geslachtscellen (zaadcellen of eicellen) terechtkomen.
Door crossing over wordt het erfelijk materiaal sterk door elkaar geschud. Deze recombinatie van maternaal en paternaal DNA zorgt voor een grotere variabiliteit bij het nageslacht. Door deze eigenschap is de meiose een cruciaal onderdeel van de seksuele voortplanting (zie hoofdstukken 4 en 5).
Dieren die zich ook aseksueel kunnen voortplanten (zoals platwormen) splitsen zich tijdens het volwassen leven in twee en produceren twee nieuwe organismen, louter en alleen door mitose. Hoewel dit op het eerste gezicht minder omslachtig lijkt, ontstaat er door deze manier van voortplanten geen genetische variabiliteit. Dit proces van splitsing is trouwens vergelijkbaar met hoe een monozygote (eeneiige) tweeling ontstaat; ook deze zijn genetisch identiek (zie 15.2).
Crossing over gebeurt bij alle chromosomen, behalve bij het X en Y chromosoom bij de man. Het Y-chromosoom wordt dus telkens vrijwel onveranderd doorgegeven van vader op zoon.
TETRADE
CHIASMA
4 Gametogenese
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De algemene principes van spermatogenese en oögenese met elkaar te vergelijken en hierbij de verschillen te benadrukken.
Aan te tonen dat mitose en meiose een belangrijke rol spelen bij zowel spermatogenese als oögenese.
De verschillende stapjes in de spermatogenese te beschrijven.
Kort te beschrijven hoeveel procent dysmorfe spermatozoa voorkomen.
Toe te lichten welke aanpassingen gebeuren tijdens de spermiogenese.
De verschillende stapjes in de oögenese te beschrijven.
Het proces van folliculaire atresie te schetsen, met bijzondere aandacht voor het tijdsverloop ervan (=wanneer in het leven dit optreedt).
Aan te geven welke twee ‘rustmomenten’ optreden tijdens de oögenese.
Te bespreken wat een non-disjunctiefenomeen betekent en te verklaren waarom dit vaker voorkomt tijdens de oögenese.
Een schets (schema) te kunnen maken van een non-disjunctiefenomeen dat optreedt tijdens meiose I of meiose II.
4.1 Situering
Gametogenese is het proces dat ervoor zorgt dat rijpe geslachtscellen (zaadcellen of eicellen) kunnen gevormd worden vanuit primordiale kiemcellen.
Bij de man spreken we van spermatogenese, bij de vrouw van oögenese
De geslachtscellen (ook gameten genoemd) worden bij beide geslachten gevormd in de interne genitalia, vanuit voorlopercellen. Deze voorlopercellen, primordiale kiemcellen, worden ook stamcellen of kiembaancellen genoemd.
In dit hoofdstuk bespreken we enkel de vorming van de geslachtscellen zelf. De vorming van de interne genitalia komt later aan bod (zie 14.1). De fysiologie van de menstruele cyclus bij de vrouw komt uitgebreider in andere cursussen aan bod.
Gametogenese is een mooie toepassing van mitose en meiose (zie hoofdstuk 3).
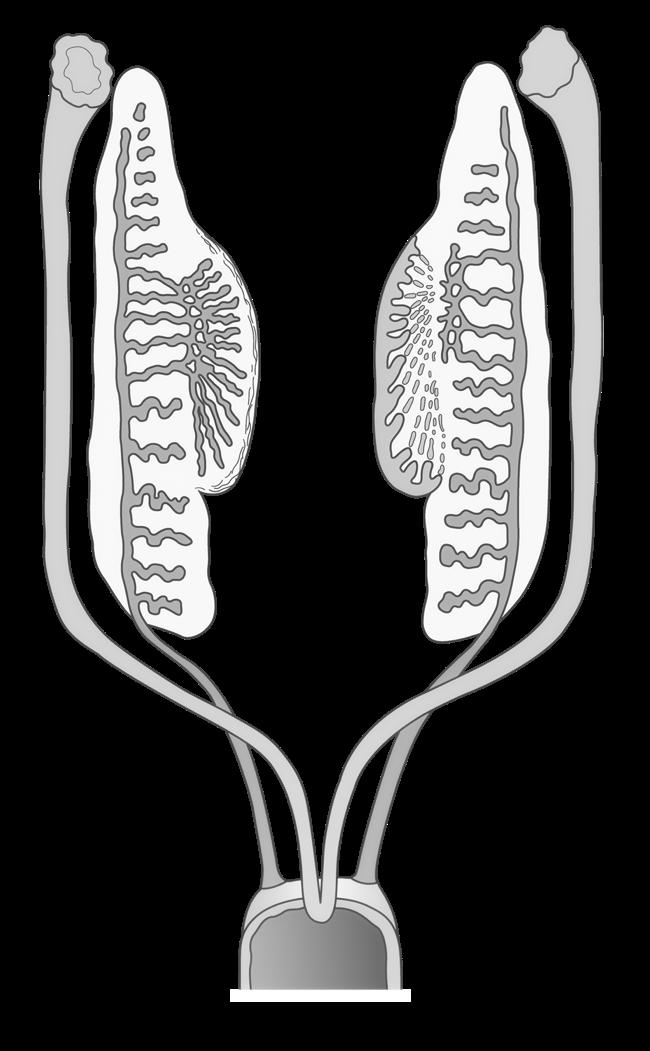
4.2 Spermatogenese
4.2.1 Algemene principes
• De spermatogenese begint vanaf de puberteit
• Het doel ervan is om een groot aantal gameten te produceren, zo’n 150 tot 275 miljoen spermatozoa per dag vanaf de vruchtbare leeftijd.
• Elke kiemcel rijpt hierbij uit tot 4 gelijkwaardige spermatozoa
• De man is continu vruchtbaar en dit levenslang
4.2.2 Van kiemcel naar spermatide
Spermatogenese begint vanaf de puberteit. Pas dan en onder invloed van hormonale veranderingen ontwaken de primordiale kiemcellen in de testiculaire zaadbuisjes (zie 14.1.3) De primordiale kiemcellen ontwikkelen tot spermatogonia en door mitose nemen ze fors toe in aantal.
Terwijl een deel van de spermatogonia blijft delen via mitose, start een ander deel met de meiose. Vanaf nu heten ze primaire spermatocyten. Na meiose I worden dit secundaire spermatocyten en na meiose II spermatiden
Uit elke primaire spermatocyt worden via de meiotische deling 4 haploïde spermatiden gevormd.
DNA-replicatie
Crossing over
spermatogonium (2n)
primaire spermatocyt
secundaire spermatocyt
spermatide (n)
spermatozoön (n)
Meiose I
Meiose II
Figuur 28 de spermatogenese
4.2.3
Van spermatide naar spermatozoön
Spermatiden delen niet meer verder, maar differentiëren via een rijpingsproces (spermiogenese) tot rijpe spermatozoa. Dit proces duurt 64 dagen.
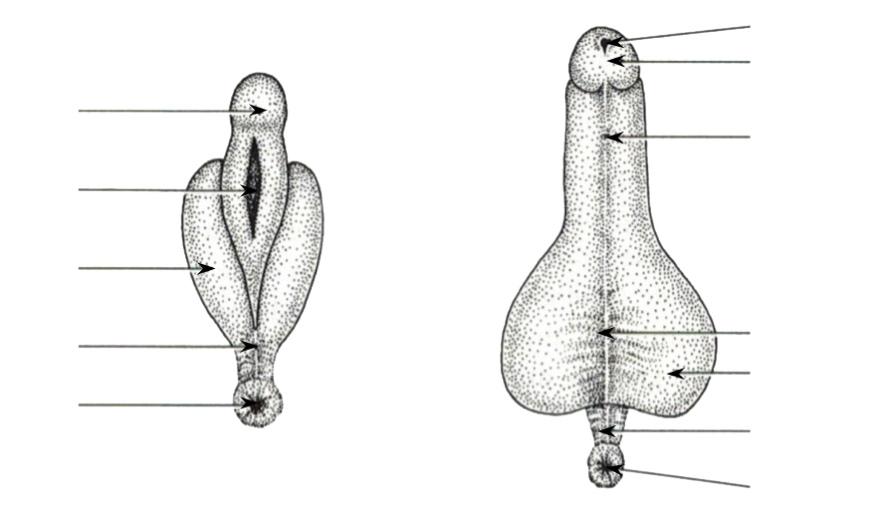
Hiermee gaan enkele grondige, morfologische veranderingen gepaard:
Kop (5 µm)
Middenstuk (7 µm)
Zweepstaart (50 µm)
acrosoom
nucleus mitochondriën
Figuur 29 de spermiogenese. (Li) Tekening (Re) electronenmicroscopie
(1) Vorming van het acrosoom: een celorganel dat talrijke enzymen bevat die assisteren in het doordringen van de beschermlaag rond de eicel. Het acrosoom vormt a.h.w. de 'kop' van de zaadcel.
(2) Condensatie (samendrukken) van het chromatine in de celkern.
(3) Verlies van het grootste gedeelte van het cytoplasma
(4) Vorming van middenstuk en zweepstaart (flagellum). In het tussenstuk komen mitochondriën voor die a.h.w. de motor vormen van de zweepstaart.
Na de spermiogenese zijn de spermatozoa uitermate gespecialiseerd voor hun functie, nl. het zelfstandig voortbewegen en een pakketje DNA afleveren aan de eicel.
De rijpe spermatozoa worden vervolgens vanuit de testiculaire zaadkanaaltjes naar de epididymis (bijbal) gestuwd voor opslag en verdere maturatie.
4.2.4 Ejaculatie
Bij ejaculatie bewegen de nu volledig mature spermatozoa razendsnel door de zaadleider (ductus deferens) en worden ze vermengd met secreties uit de zaadblaasjes en prostaat.
Het ejaculaat bedraagt doorgaans zo'n 2 à 6 ml vocht en bevat 40 à 250 miljoen spermatozoa die op zoek gaan naar een te bevruchten eicel. Naar schatting kunnen spermatozoa tot 72u na coïtus overleven binnen het vrouwelijk lichaam.
4.2.5 Dysmorfe spermatozoa
Tijdens de spermatogenese gebeuren vaak fouten. Naast fouten in het genetisch materiaal zelf (zie non-disjunctiefenomeen 4.4) zijn 10% van de spermatozoa dysmorf, wat betekent dat ze een afwijkend uitzicht hebben zoals:
Ontdubbelde kop of staart
Defect flagellum
Malformatie van het acrosoom
Dysmorfe spermatozoa hebben een gebrekkige motiliteit en een veel lagere kans op succesvolle bevruchting. Als meer dan 20% dysmorfe spermatozoa aanwezig zijn kan verminderde vruchtbaarheid optreden.
4.3 Oögenese
4.3.1 Algemene principes
• De oögenese begint tijdens de foetale ontwikkeling.
• Het doel is om één sterke eicel te produceren.
• De mitose van kiemcellen gebeurt enkel tijdens de foetale ontwikkeling.
• De aanwezige gameten organiseren zich in follikels die continu in aantal afnemen.
• De meiotische deling kent twee periodes van lange pauze.
• Elke kiemcel rijpt uit tot slechts 1 ovum (en 3 kleinere poollichaampjes).
• De vrouw is cyclisch vruchtbaar en dit van de puberteit tot aan de menopauze.
4.3.2 Van kiemcel naar secundaire oöcyt
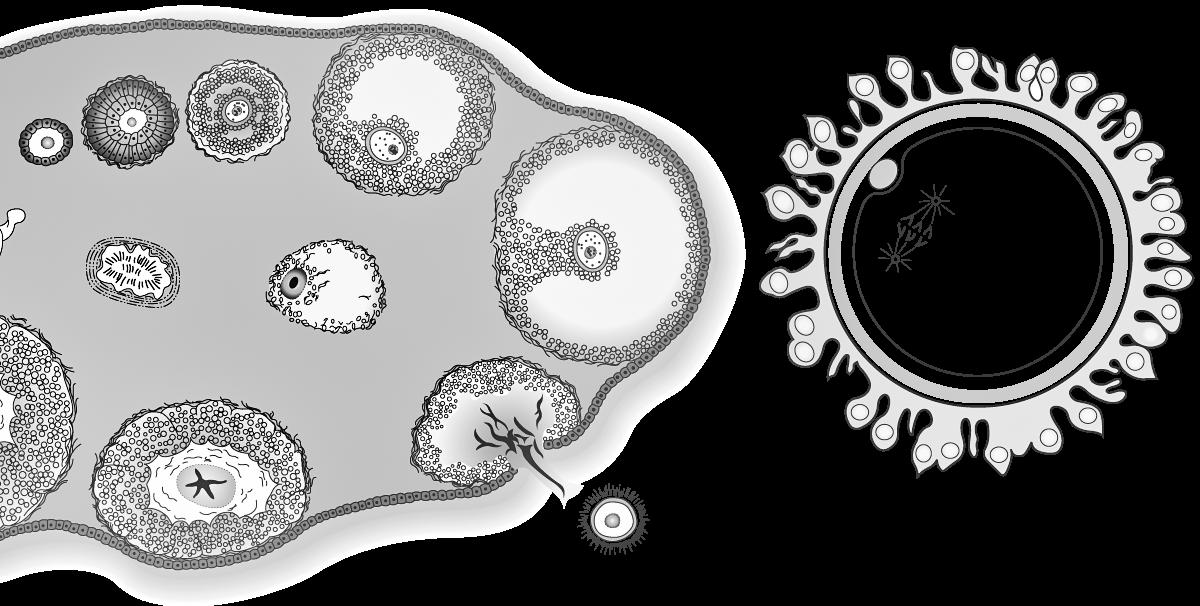
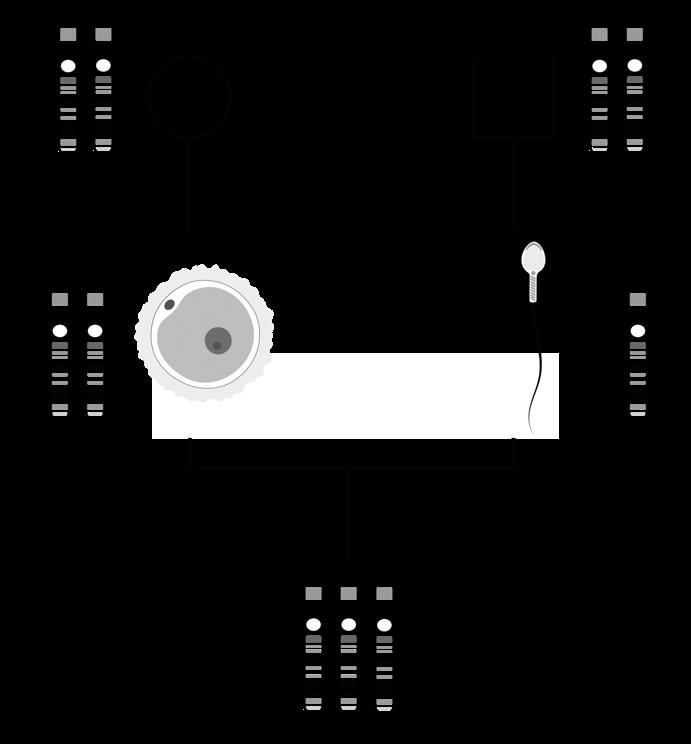
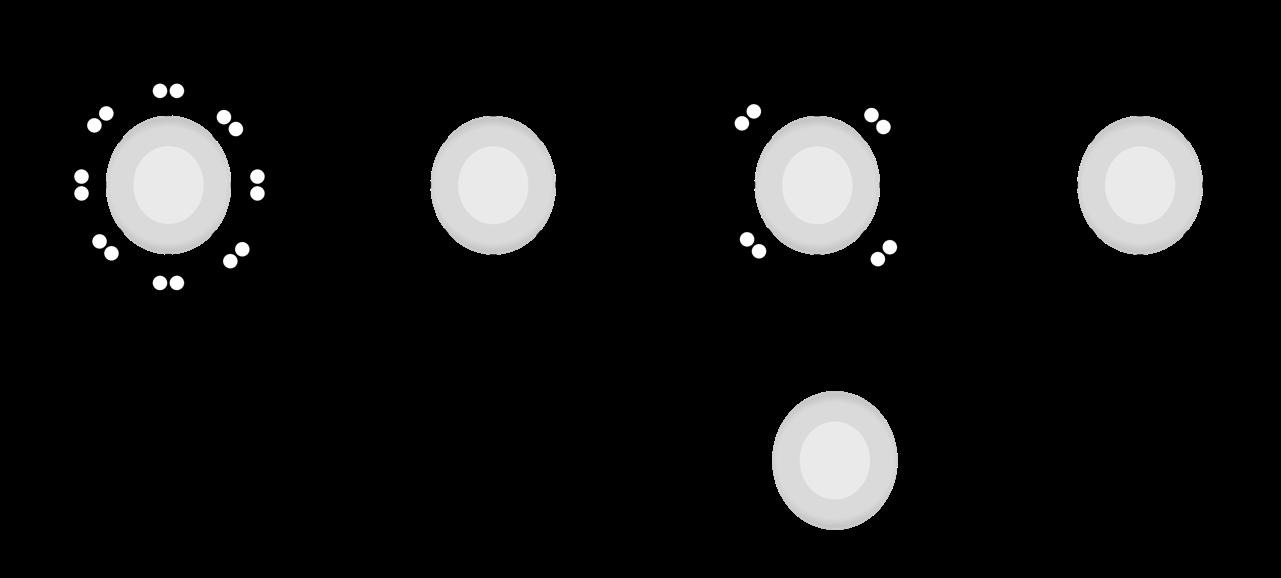
De oögenese bij de vrouw begint al vroeg tijdens het foetale leven. Vanaf een zwangerschapsleeftijd van twee maanden vormen de primordiale kiemcellen in de embryonale ovaria om tot oögonia die door mitose heel sterk in aantal toenemen Een deel van deze oögonia start de meiose. We heten ze nu primaire oöcyten. Deze cellen laten zich omringen door platte, voedende cellen (follikelcellen) en een beschermlaag (de zona pellucida) en vormen op die manier primordiale follikels. Het aantal follikels stijgt spectaculair: van enkele duizenden in de 2e maand tot een maximum van ± 7 miljoen in de 5e maand (zie: 14.1.4)
Kort nadien begint al het proces van folliculaire atresie, waarbij het grootste gedeelte van deze met follikelcellen omringde oöcyten terug verloren gaan door een vorm van geprogrammeerde celdood. Bijkomend stopt ook de mitose van oögonia tijdens het foetale leven, waardoor het totale aantal follikels voortdurend afneemt. Na de geboorte worden geen nieuwe follikels meer aangemaakt
mitose
DNA-replicatie
Crossing over
1e polair lichaampje
oögonium (2n)
primaire oöcyt
DICTYOTEEN
Meiose I wordt gestopt in de profase
secundaire oöcyt
Meiose II wordt pas afgewerkt na fertilisatie
ovum (n)
Figuur 30 Oögenese
Meiose I
Bij de geboorte van een meisje zijn nog ca ± 600.000 tot 800.000 follikels over. Door het verdergaan van folliculaire atresie zijn dit er bij het begin van de puberteit nog slechts ± 40.000 en minder dan 500 zullen tijdens het leven uiteindelijk uitrijpen en een eisprong ondergaan. Op het ogenblik van de menopauze zijn alle follikels atretisch geworden.
In de follikels die niet afsterven vinden we telkens één primaire oöcyt waarbij meiose I gestart is (tijdens het foetale leven), maar gestopt is aan het eind van de profase, net nà crossing over. Deze pauze wordt het dictyoteen geheten en kan pas worden beëindigd vanaf de puberteit, wanneer maandelijks 15 à 20 van deze follikels verder uitrijpen onder invloed van het follikelstimulerend hormoon (FSH). Enkel onder impuls van een piek in luteïniserend hormoon (LH) wordt meiose I afgewerkt. Eén oöcyt krijgt hierbij het grootste gedeelte van het cytoplasma, de ander vormt een poollichaampje. Nu kunnen we spreken van een secundaire oöcyt.
De secundaire oöcyt vangt vervolgens meiose II aan maar blijft opnieuw hangen, ditmaal in de metafase, doorgaans zo'n 3-tal uur voor de ovulatie.
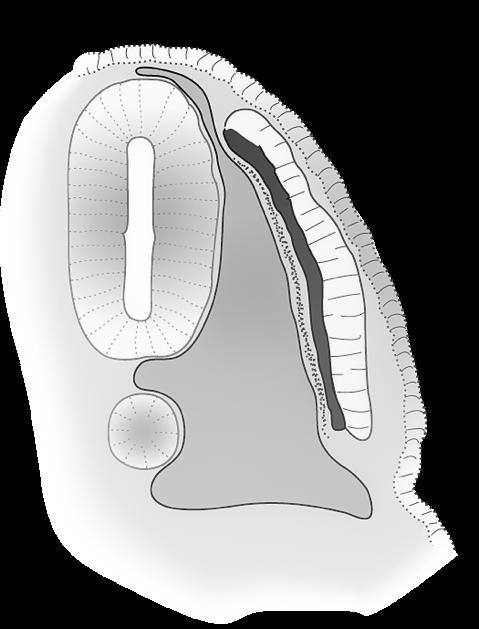
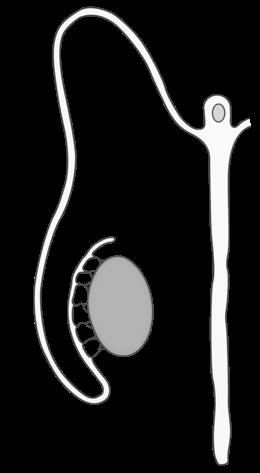
Uiteindelijk bereikt elke cyclus slechts één follikel het volwassen stadium (15-20 mm) van de tertiaire, Graafse follikel die ovulatie ondergaat. De andere follikels gaan in atresie. Bij de ovulatie barst de Graafse follikel, komt de secundaire oöcyt vrij en wordt deze opgevangen in de fimbriae van de eileider. De secundaire oöcyt is op dat moment nog steeds omgeven door de zona pellucida en een krans van granulosacellen (corona radiata).
primordiale follikel
follikelrijping
poollichaampje
tertiaire follikel
ovulatie
atretische follikels
corpus luteum (geel lichaam)
Figuur 31 (Li) follikelvorming in een eierstok (Re) secundaire oöcyt na de ovulatie (vergroot)
corona radiata
secundaire oöcyt
zona pellucida
Meiose II wordt enkel vervolledigd bij bevruchting. Als dit niet plaatsvindt ten laatste 24 uur na ovulatie sterft de eicel af. Ook bij vervolledigen van meiose II krijgt één rijpe eicel alle cytoplasma en vormt de ander een polair lichaampje.
Oefening 5. Test jezelf na het doornemen van 4.2 en 4.3
Leg de juiste verbanden: vul in de kolom onder de pijl telkens de letter van het overeenstemmende begrip in. Voor elk begrip of formulering in de linkertabel is er één bijpassende uit de rechtertabel.
Oögenese A
Primordiale kiemcel B
Acrosoom C
Spermatide D
Zona pellucida E
Tetraden F
Na de S-fase G
Spermatogenese H …
4.4 Non-disjunctiefenomeen
92 zusterchromatiden
Vanaf de puberteit …
Diploïd (2n)
Vanaf het foetale leven
Kop van de zaadcel …
Haploïd (n)
Beschermlaag rond oöcyt
Crossing over
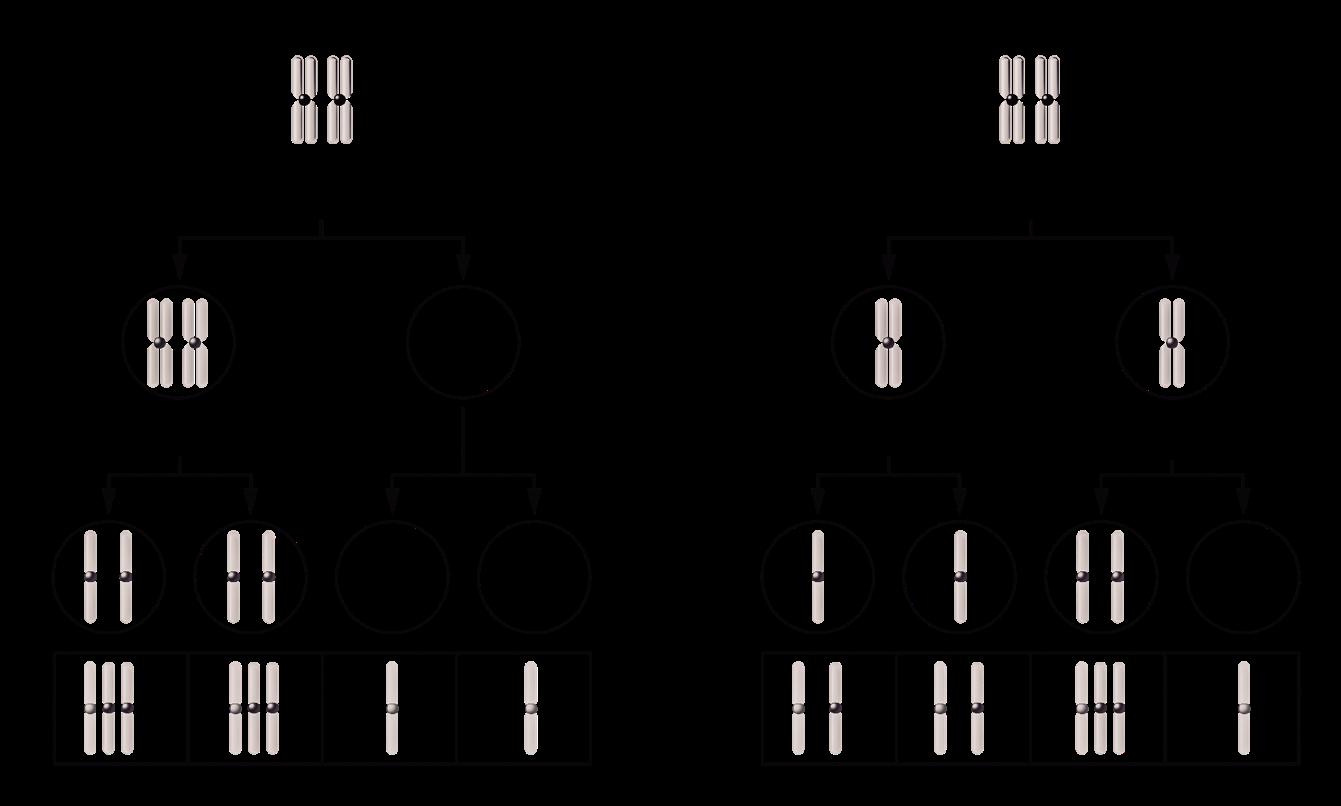
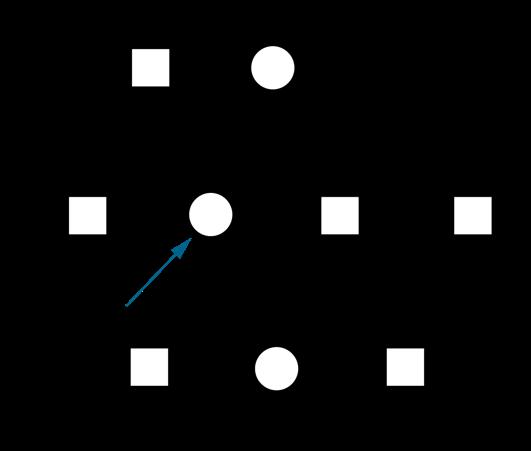
Een non-disjunctiefenomeen is een fout tijdens mitose, meiose I of meiose II waarbij de chromosomen tijdens de anafase niet op een correcte manier uiteengaan.
NON-DISJUNCTIE
DISJUNCTIE
1
DISJUNCTIE
2
DISJUNCTIE
NON-DISJUNCTIE
Figuur 32 Non-disjunctie in Meiose I (Li) of Meiose II (Re) (Klug, 2019)
Indien non-disjunctie optreedt tijdens de meiose worden gameten gevormd die niet precies haploïd zijn. Sommige gameten zullen een extra chromosoom hebben, andere gameten ontbreken een chromosoom.
Uit onderzoek blijkt dat het non-disjunctiefenomeen vaker bij de moeder gebeurt tijdens oögenese, en dan vooral tijdens meiose I (Gianaroli et al., 2005)
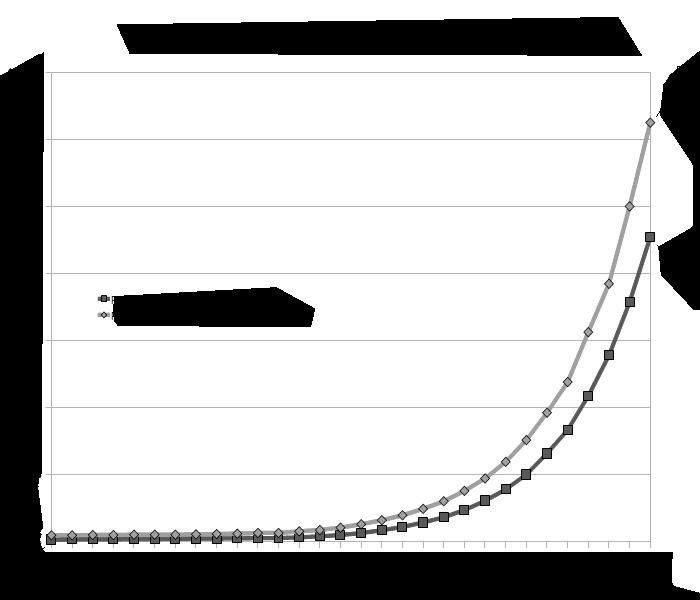
Er blijkt hierbij ook een belangrijk leeftijdseffect te bestaan: hoe ouder de moeder, hoe groter de kans op non-disjunctie Vanaf 35 jaar neemt het risico op non-disjunctie exponentieel toe Dit houdt verband met het feit dat meiose I al in het foetale leven aanvangt en dan gedurende vele jaren stilvalt (=het dictyoteen, zie 4.3).
Als het dictyoteen langer dan 35 jaar duurt, zijn de cellulaire mechanismen minder in staat om meiose I tot een goed eind te brengen. Hoe langer deze pauze, hoe meer kans dat de normale disjunctie faalt.
risico op trisomie 21 risico op alle trisomies (13,18,21,X & Y)
MATERNALE LEEFTIJD
Figuur 33 Verband tussen aneuploïdie en maternale leeftijd.
Als een aneuploïde gamete leidt tot fertilisatie zal de som van alle chromosomen in de zygote (bevruchte eicel) ook aneuploïd zijn (zie 5.4.3), wat bv. kan leiden tot trisomie 21 (syndroom van Down).
De maatschappelijke trend om later aan kinderen te beginnen, brengt dus een hoger risico op aneuploïdie met zich mee. De gemiddelde leeftijd van een moeder bij de geboorte van haar eerste kind bedraagt nu 29,3 jaar; een toename van ± 2 jaar t.o.v. het jaar 2000 en ± 5 jaar t.o.v. 1970 (Statbel, 2021).
5 Bevruchting
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De verschillende processen (reacties) te beschrijven die deel uitmaken van de bevruchting.
Uit te leggen op welke manier(en) de eicel polyspermie voorkomt.
Het verband tussen polyspermie en polyploïdie toelichten.
Precies aan te geven op welk moment de secundaire oöcyt haar 2e meiotische deling (meiose II) vervolledigt.
Te omschrijven hoe de versmelting van de pronuclei gebeurt.
De afkorting IVF voluit te schrijven en hiervan een korte definitie te geven.
Kort te omschrijven wat polaire lichaampjes zijn en wat hiermee gebeurt na de bevruchting.
Aan te geven welke ouder het geslacht bepaalt.
Een verklaring te geven voor het feit dat iets meer jongens (51,5%) worden geboren dan meisjes.
De link tussen aneuploïdie en non-disjunctie te bespreken.
Met een procent te schetsen hoe vaak aneuploïdie zygotes bij de mens worden gevormd.
Minstens 3 voorbeelden te geven van aneuploïdie door autosomen.
Minstens 3 voorbeelden te geven van aneuploïdie door geslachtschromosomen.
Uit te leggen wat uniparentale disomie betekent en hoe dit tot stand komt.
De term ‘genetisch determinisme’ uit te leggen aan de hand van een voorbeeld.
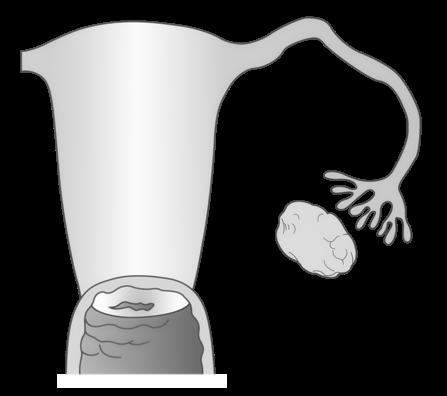
5.1 Situering
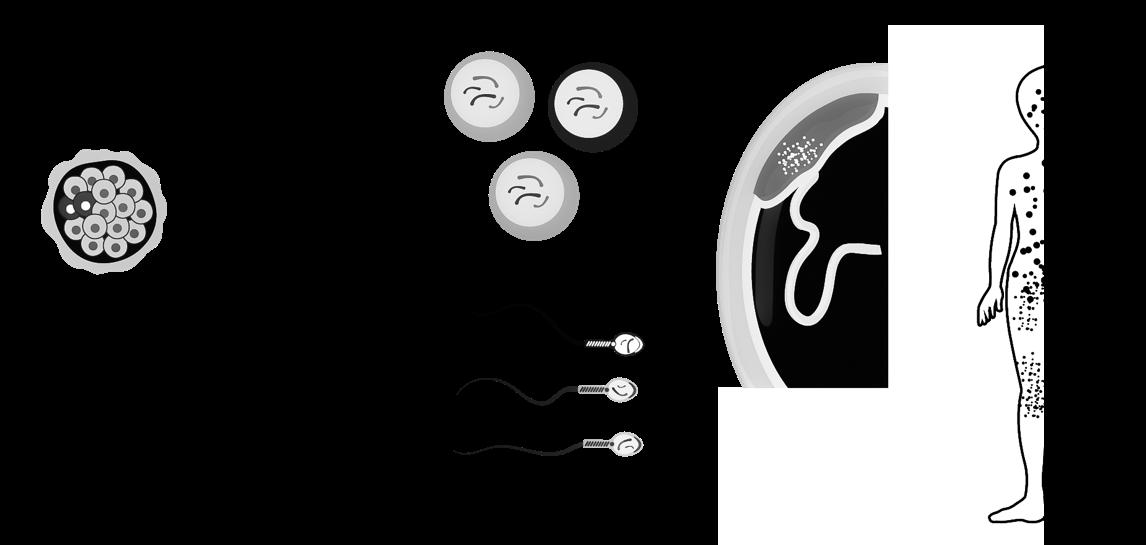
Onder bevruchting (fertilisatie) verstaan we de versmelting van een zaadcel en een eicel. Hierbij wordt het genetisch materiaal van beide, haploïde gameten gecombineerd tot een diploïde cel, de bevruchte eicel of zygote
JAAR: 1978
Op 25 juli 1978 wordt de Britse Louise Brown geboren als eerste mens na een zwangerschap die tot stand kwam door kunstmatige bevruchting (in-vitrofertilisatie, IVF). Bij deze medische procedure gebeurt de bevruchting buiten het lichaam. Verkregen embryo(s) worden nadien teruggeplaatst in de baarmoederholte. Bij koppels met vruchtbaarheidsproblemen kan op deze manier toch een zwangerschap tot stand komen (Steptoe & Edwards, 1978).
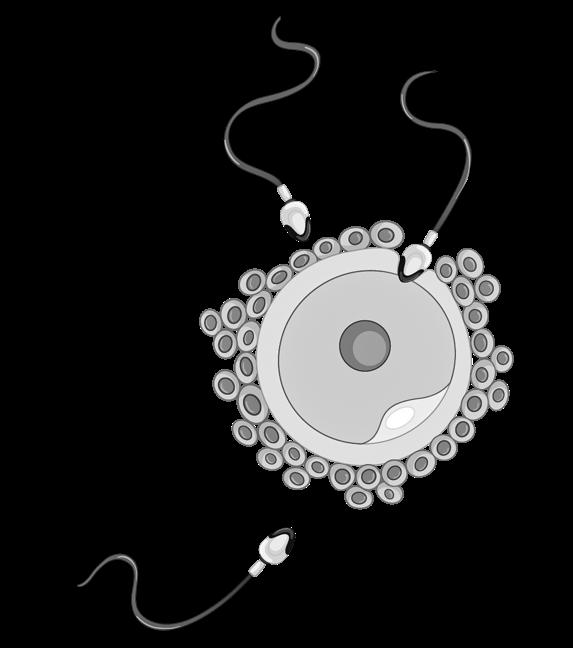
De ontmoeting tussen zaadcel en eicel gebeurt meestal in de ampulla tubae. Slechts 300 à 500 zaadcellen bereiken deze plaats, dit door hun eigen staartbeweging en contracties van de uterus.
secundaire oöcyt
corona radiata
zona pellucida spermatozoön
1e poollichaampje
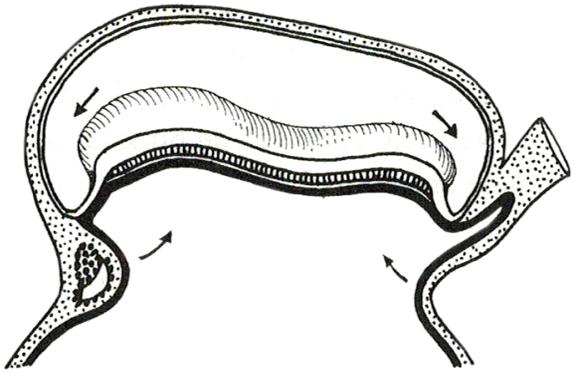
De bevruchting is geen enkelvoudige gebeurtenis maar een aaneenschakeling van processen (ook reacties genoemd) die door de zaadcel en eicel (eigenlijk nog een secundaire oöcyt) worden uitgevoerd
We bespreken ze hieronder in chronologische volgorde
Figuur 34 De bevruchting (Carlson, 2019)
5.2 De rol van de zaadcel
5.2.1 Capacitatiereactie
De bevruchting start wanneer de zaadcellen de corona radiata ontmoeten, de buitenste laag rond de eicel. In deze laag vinden we granulosacellen, ingebed in een matrix van eiwitten en koolhydraten zoals hyaluronzuur. Door afgifte van een enzym, hyaluronidase, breken de zaadcellen deze matrix af (=capacitatiereactie).
Door hun staartbewegingen kunnen de spermatozoa vervolgens deze corona radiata doorboren.
5.2.2 Acrosoomreactie
capacitatiereactie
zona pellucida
acrosoomreactie
perivitelline ruimte plasmamembraan eicel
corona radiata
membraanfusie
De zona pellucida , de binnenste laag rond de eicel, bevat receptoren voor spermatoz a. Dit laat een stevige binding toe van de zaadcellen die de corona radiata doorbreken. Bij de binding de inhoud van het acrosoom vrijgegeven (
Hierdoor komen
5.2.3 Membraanf
Eens de zaadcel doorheen de ZP is gedrongen volgt een korte doortocht doorheen de perivitelline ruimte. Nadien versmelten de celmembranen van de zaadcel en de secundaire oöcyt. Kop, middenstuk en staart van de zaadcel komen nu binnenin de secundaire oöcyt terecht.
acrosoom
5.3 De rol van de eicel
Nadat een zaadcel het erfelijk materiaal van de vader heeft binnengebracht, heeft de eicel een belangrijke taak: het voorkomen van polyspermie (meervoudige bevruchting van één eicel).
Polyspermie kan immers leiden tot een triploïde zygote:
1 EC (n) + 1 ZC (n) + 1 ZC (n) = zygote (3n)
Preventie van polyspermie gebeurt op 2 manieren:
Corticale reactie (snel)
Zonale reactie (traag)
5.3.1 Corticale reactie
Twee à drie seconden na de membraanfusie wijzigt de eicel de elektrische lading op haar plasmamembraan. Hierdoor kan geen nieuwe membraanfusie plaatsvinden.
5.3.2 Zonale reactie
Een tweede, tragere reactie bestaat in de afgifte van stoffen in de perivitelline ruimte. Deze dringen door in de zona pellucida en breken de receptoren voor spermatozoa af. Hierdoor kunnen geen spermatozoa meer binden aan de ZP
5.3.3 Vervolledigen van de 2e meiotische deling
Pas op dit moment vervolledigt de secundaire oöcyt meiose II, waardoor de definitieve haploïde oöcyt (ovum) en een tweede poollichaampje worden gevormd. Net als het eerste poollichaampje komt dit in de perivitelline ruimte terecht.
5.4 De zygote
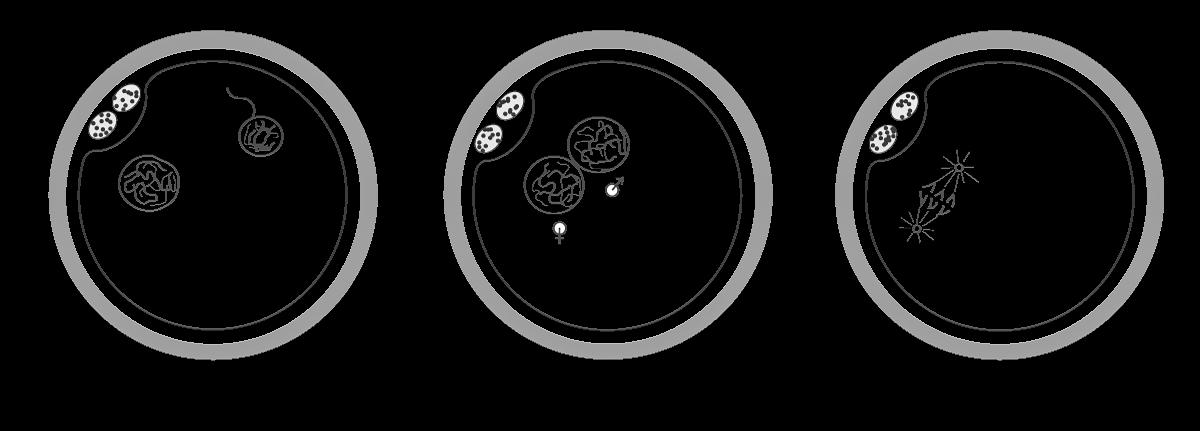
5.4.1 Versmelten van de pronuclei
Binnenin de eicel valt de zaadcel uiteen. Middenstuk, staart en de geringe hoeveelheid celorganellen (vooral mitochondriën) die zijn meegereisd worden afgebroken. Enkel de kern van de zaadcel wordt behouden, waarvan het chromatine zich breder uitsmeert om de mannelijke pronucleus te vormen.
Ook de kern van eicel ontwikkelt zich ondertussen tot een pronucleus.
De vorming van de pronuclei duurt zo’n 6 à 8 uren.
vrouwelijke pronucleus
mannelijke pronucleus
poollichaampjes
spoelfiguur van 1e mitose
36 Versmelten van de pronuclei
Terwijl de pronuclei naar elkaar toe bewegen wordt aan elk van hun 23 chromosomen een zusterchromatide gevormd. Je kunt dit vergelijken met de S-fase tijdens de celcyclus. Eens de pronuclei met elkaar in contact komen, verliezen ze hun kernmembraan, waarna de maternale en paternale chromosomen zich met elkaar kunnen vermengen. De 23 maternale en 23 paternale chromosomen vormen op die manier terug een diploïd aantal.
Vervolgens worden de (al ontdubbelde) chromosomen georganiseerd op spoeldraden om de eerste mitose te kunnen starten. (M.o.: bij de bevruchting gebeurt dus geen crossing-over tussen de maternale en paternale chromosomen).
Vanaf nu kunnen we spreken van een zygote, een bevruchte eicel die klaar is om te groeien en te ontwikkelen tot een menselijk embryo.
The Zinc Spark
Figuur 37 The Zinc Spark, zoals deze te zien is onder een fluorescentie-microscoop, 34 minuten na de bevruchting.
De eerste uren na de bevruchting geeft de zygote miljarden zink-atomen vrij, een fenomeen dat gekend staat als the ‘zinc spark’ of zink-lichtflits. Zink is nodig voor de meiose, maar vanaf de bevruchting moeten de zinkspiegels in de zygote snel dalen om de mitose te bevorderen. Onder een fluorescentie-microscoop is deze zink-uitstroom te zien als een heldere lichtflits. Deze lichtuitbarsting zou bovendien ook verband houden met de kwaliteit van de cel; een zygote die feller oplicht zou hogere slaagkansen hebben m.b.t. de verdere ontwikkeling (Duncan et al., 2016).
Figuur
5.4.2
Wat gebeurt er met de polaire lichaampjes?
De kleine, polaire lichaampjes degenereren in de eerste dag na de bevruchting. Hoewel het eerste polaire lichaampje in theorie ook nog eens kan delen (meiose II) is dit meestal niet het geval.
Geslachtsbepaling
Binnen elk chromosomenpaar wordt dus één chromosoom verkregen van de moeder en één van de vader. Voor de geslachtschromosomen kan een vrouw enkel een Xchromosoom doorgeven aan haar kinderen. Het feit of de vader een X of Y chromosoom doorgeeft zal dus het geslacht van het kind bepalen.
De vader bepaalt het geslacht.
Wereldwijd worden trouwens iets meer jongens geboren dan meisjes. Voor België zijn dit zo'n 51,5% jongens. Een mogelijke verklaring hiervoor is dat zaadcellen die een Ychromosoom dragen iets lichter zijn en bijgevolg een klein voordeel in motiliteit hebben.
Voor de eenvoud zullen we er in de rest van de cursus van uit gaan dat de kans op een jongen of meisje steeds 50% – 50 % bedraagt.
5.4.3 Aneuploïde zygotes
5.4.3.1 Vormen
Als een gamete met een afwijkend aantal chromosomen deel uitmaakt van de bevruchting, zal het totaal aantal chromosomen in de zygote ook afwijken.
Door het frequent voorkomen van aneuploïdie tijdens de gametogenese worden bij de mens opvallend vaak zygotes gevormd die aneuploïd zijn
30% (!) van de menselijke zygotes is aneuploïd.
Het gaat hierbij om:
Trisomie (2n + 1)
Er is een extra chromosoom aanwezig (totaal: 47) bv. 47,XX +21
Monosomie (2n – 1)
Er ontbreekt een chromosoom (totaal: 45) bv. 45,X
Bv. het ontstaan van trisomie 13
Figuur 38 Ontstaan van trisomie 13 door fertilisatie van een aneuploïde eicel (M = maternaal, P = paternaal).
Ingeval van trisomie beschikken de embryonale cellen dus over twee chromosomen van de ene ouder en één chromosoom van de andere ouder. Van elk gen op dit chromosoom komen dus 3 allelen tot expressie Door deze overexpressie worden te veel eiwitten geproduceerd, wat de celfunctie ontregelt.
Bij embryo’s met aneuploïdie is er hierdoor een sterk verhoogd risico dat de implantatie faalt, de embryonale ontwikkeling stilvalt en/of de zwangerschap spontaan afbreekt.
Enkel de trisomies 13,18 en 21 zijn levensvatbaar bij de mens.
§ Enkele voorbeelden van autosomale trisomie
ISCN-notatie Levensvatbaar?
Trisomie 13 (syndroom van Patau) 46, XY+13 Ja
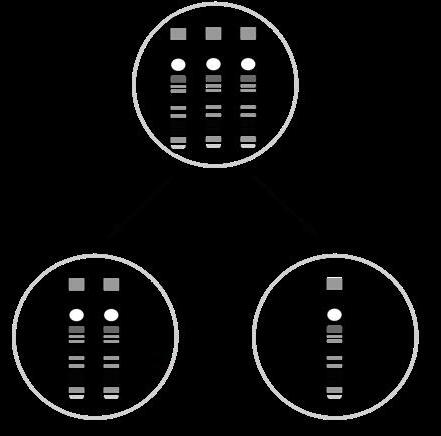
Trisomie 16
47, XX+16 Nooit
Trisomie 18 (syndroom van Edwards) 47, XY+18 Ja
Trisomie 21 (syndroom van Down) 47, XX+21 Ja
Trisomie 22
47, XY+22 Nooit
§ Trisomie van de geslachtschromosomen wordt beter verdragen dan trisomie van de autosomen. Bij de monosomie is enkel de monosomie X levensvatbaar.
ISCN-notatie Levensvatbaar?
Monosomie X (syndroom van Turner) 45, X Ja
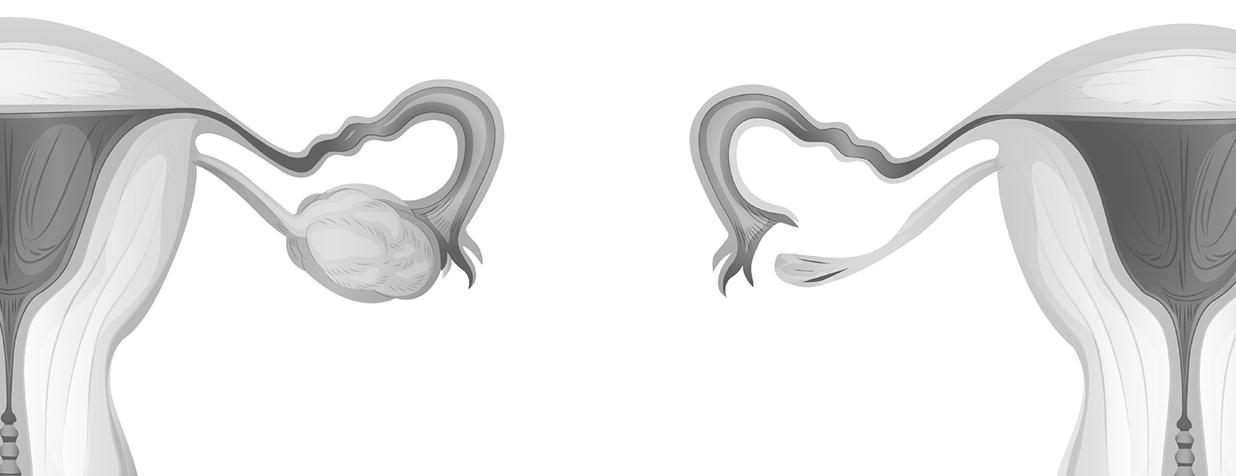
Trisomie X (Triple X-syndroom)
Syndroom van Klinefelter
Super Male Syndroom
47, XXX Ja
47, XXY Ja
47, XYY Ja
5.4.3.2
Correctie tot uniparentale disomie (UPD)
Een aneuploïde zygote kan zichzelf corrigeren. Dit gebeurt ofwel bij de versmelting van de pronuclei of tijdens de eerste, mitotische klievingsdelingen (zie 6.1)
Deze correctie kan echter leiden tot uniparentale disomie, de situatie waarbij de twee overblijvende chromosomen van dezelfde ouder afkomstig zijn.
Bv. correctie van trisomie 13
M
M P
Figuur 39 (Li) Correctie tot uniparentale disomie door het ‘uitstoten’ van het paternale chromosoom tijdens de mitose. (Re) Monosomie van het paternale chromosoom.
Bij een trisomie met twee paternale chromosomen kan het omgekeerde scenario optreden (het maternale chromosoom gaat verloren bij de correctie). Alleen komt een trisomie met twee paternale chromosomen minder vaak voor. Hoe zou dit komen? (Tip: zie 5.4.2)
UPD leidt dus tot het herstellen van het normale, diploïde aantal chromosomen. Toch kan dit problemen veroorzaken! We komen hier later op terug (zie 20.6.6).
M
M
5.4.4
Genetisch determinisme
Wanneer de zygote gevormd wordt, ligt het genoom van het organisme volledig vast. Als we op dat moment het genoom zouden analyseren, kunnen we dus bepaalde fenotypische eigenschappen afleiden: etnische afkomst, geslacht, oogkleur, het optreden van een genetische ziekte zoals mucoviscidose of het syndroom van Down.
Binnen het gerechtelijk onderzoek is het sinds kort mogelijk om het aangezicht van een dader te voorspellen, op basis van teruggevonden DNA (bv. uit een haar). Dit gebeurt door analyse van tientallen genen die een rol spelen in het vormen van bv. mond, lippen, neus (neusbrug, neuspunt, nasolabiale plooien), ogen (kleur en vorm van de ogen, afstand tussen de ogen), …. Op basis van deze informatie genereert een computer een schets van het aangezicht van de dader (Pośpiech et al., 2022; Richmond et al., 2018).
Begin 20e eeuw waren sommige wetenschappers ervan overtuigd dat alle eigenschappen van de mens volledig erfelijk bepaald waren (=genetisch determinisme), dus ook je persoonlijkheid, intelligentie, gedrag, interesses enz. Ondertussen weten we dat dat niet klopt.
De meeste fenotypische eigenschappen bij de mens zijn immers het gevolg van een complexe interactie tussen genen (nature) en omgevingsfactoren (nurture). Op basis van het genoom in de zygote alleen kun je een mens dus niet volledig gaan ‘voorspellen’.
Zo kan iemand wel degelijk erfelijke aanleg hebben voor type 2-diabetes, maar de ziekte nooit ontwikkelen omwille van bv. een gezond voedings- en bewegingspatroon.
Zeg dus niet te snel: “het zit allemaal in de genen”.
Genetisch determinisme in de verzekeringswereld.
Vind jij dat een verzekeraar een genetische test mag eisen bij een klant die een levensverzekering vraagt? De redenering is als volgt: als zou blijken dat bij de klant bepaalde genen aanwezig zijn die een verhoogd risico geven op ziektes (bv. kanker, hartinfarct) dan kan een hogere verzekeringsbijdrage worden aangerekend.
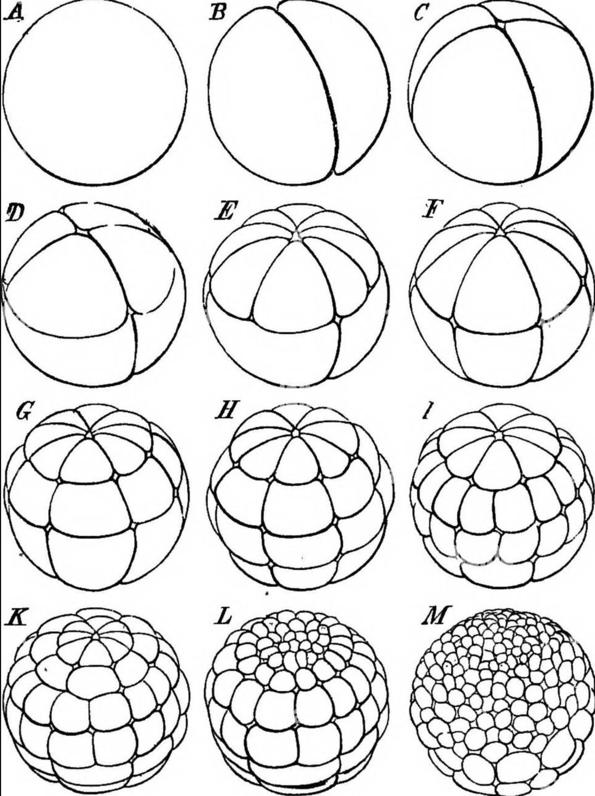
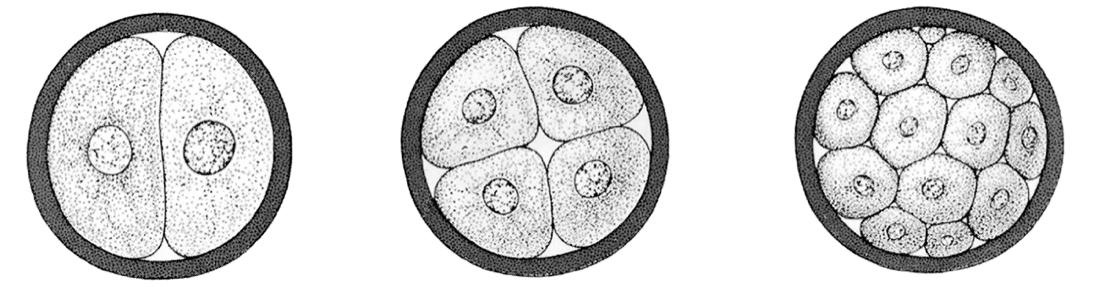
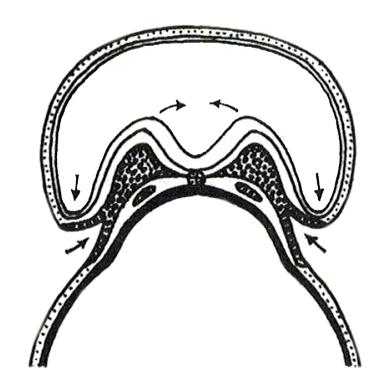
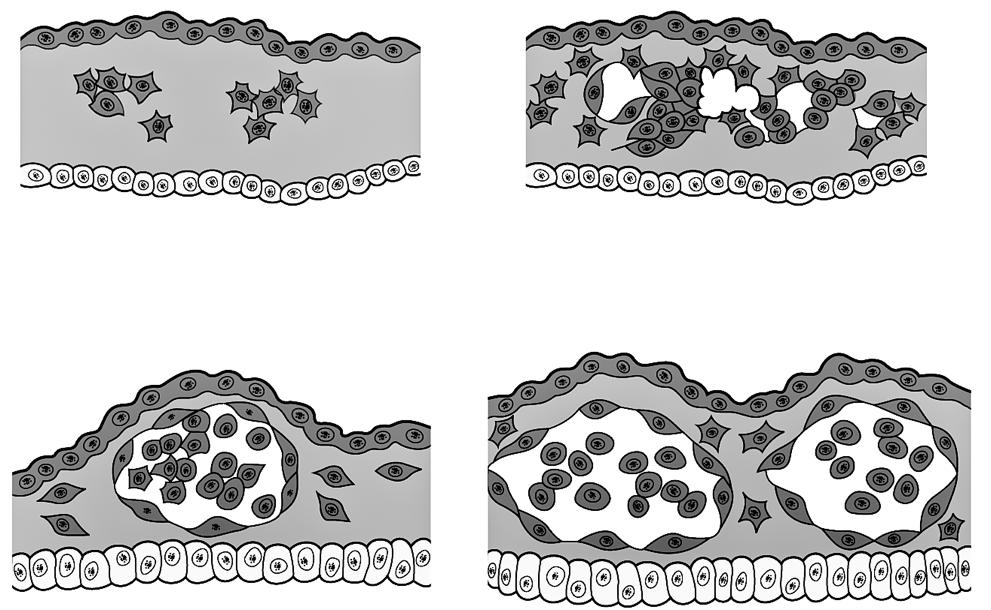
Figuur 40 De klievingsdelingen van een kikkerembryo (uit ‘The Evolution of Man’, Ernst Haeckel, 1912)
6
Eerste
week
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De klievingsdelingen te beschrijven.
Uit te leggen wat bedoeld wordt met (somatisch) mozaïcisme bij het embryo.
Te bespreken wat de mogelijke gevolgen zijn van mozaïcisme bij het embryo.
Het karyotype van een organisme met mozaïcisme correct te noteren (ISCN)
Toe te lichten in welk embryonaal stadium de embryotransfer gebeurt bij IVF.
Het stadium van de blastocyst te bespreken.
Het stadium van de innesteling te bespreken.
Kort aan te geven wat de Belgische wet toestaat m.b.t. pre-implantatie genetische testing (PGT).
De meest frequente, genetische oorzaken van spontane abortus (miskraam) te bespreken.
Een mogelijke verklaring te geven voor het feit dat mozaïcisme minder vaak leidt tot spontaan miskraam.
Aan te geven welke vormen van aneuploïdie bij de mens het vaakst voorkomen.
Aan te geven welke vormen van aneuploïdie bij de mens levensvatbaar zijn.
Te verklaren waarom chromosomenonderzoek bij een foetus in het 3e trimester van de zwangerschap minder vaak afwijkingen vertoont dan bij een embryo in het 1e trimester.
De eerste week begint bij de bevruchting en eindigt bij de implantatie
6.1 Klievingsdelingen (D1-D3)
6.1.1 Normaal verloop
Figuur 41 Klievingsdelingen van het embryo (Langman, 2018)
Na de bevruchting gaat de zygote zich verder delen via mitose. Deze delingen verlopen traag en vinden plaats binnen de rigide zona pellucida, die blijft zitten tot dag 4. Hierdoor worden de cellen bij elke deling kleiner. We heten ze blastomeren.
Drie tot vier dagen na bevruchting telt de vrucht ongeveer 16 cellen. Dit heten we het morula (moerbei-) stadium. Blastomeren zijn identiek en omnipotent. Dat wil zeggen dat ze kunnen differentiëren tot alle mogelijke weefsels (=stamcellen).
6.1.2 Mozaïcisme
Bij de vroege klievingsdelingen gebeuren heel vaak fouten. Als bv. tijdens de eerste klievingsdeling een non-disjunctiefenomeen optreedt (zie Figuur 42, links) zal de helft van alle dochtercellen aneuploïd zijn:
cellen met genoom 1 Bv. 46,XY cellen met genoom 2 Bv. 47,XY+18
mozaïcisme (50%)
Figuur 42 Het optreden van mozaïcisme tijdens de klievingsdelingen, van vroeg (Li) naar laat (Re). m.o.: in werkelijkheid blijft de ZP even groot en worden de blastomeren kleiner (naar Campbell, 2015)
mozaïcisme (20%)
Het embryo wordt op deze manier een genetisch mozaïek van cellen met een normaal genoom en cellen met een afwijkend genoom. We spreken van mozaïcisme
Wanneer verschillende genotypes voorkomen binnen hetzelfde organisme spreekt men van mozaïcisme
De verdere gevolgen hangen af van de uitgebreidheid van dat mozaïcisme. Hoe vroeger het mozaïcisme is opgetreden, hoe groter het aantal dochtercellen dat de genetische afwijking zal vertonen. Bij een hooggradig mozaïcisme (bv. meer dan 50% van de cellen) kan het embryo zelfs afsterven (zie ook spontane abortus, 6.4).
Bij een laaggradig mozaïcisme sterft het embryo niet af. Recent werd zelfs ontdekt dat het mozaïcisme weer kan verdwijnen door het uitstoten van afwijkende cellen uit de morula. De verloren cellen worden hierbij volledig vervangen door nieuwe, gezonde blastomeren (het zijn tenslotte stamcellen) en de normale ontwikkeling zet zich verder (Campbell et al., 2015; Orvieto et al., 2020; Starosti et al., 2020).
Als het mozaïcisme na het morulastadium behouden blijft, wordt het voor de rest van het leven meegenomen. We bespreken de impact op het fenotype bij 18.5
ISCN-Notatie van het karyotype bij mozaïcisme
De beide karyotypes worden na elkaar geschreven, gescheiden door een schuine streep, bv.
46, XY / 47,XY+18
m.o.: de graad van mozaïcisme kunnen we niet uit deze notatie afleiden.
Oefening 6.
Noteer het karypotype (ISCN-notatie) van een individu met …
Een normaal, mannelijk karyotype, afgewisseld met een trisomie 13.
Een normaal, vrouwelijk karyotype, afgewisseld met een trisomie van het X-chromosoom.
Een man met een trisomie 21, die in sommige van zijn cellen geen Y-chromosoom heeft.
NORMAAL TRISOMIE 18
6.2
Blastocyst (D4)
Op dag 4 bevat het embryo ongeveer 58 cellen. Dit is het tijdstip dat de vrucht aankomt in de baarmoederholte en de zona pellucida afbreekt ('hatching').
Door vochtinsijpeling tussen de blastomeren ontstaat een holte: de blastocystholte (of blastulaholte), de voorloper van de dooierzak. De vrucht zelf heet nu blastocyst (of blastula)
Voor het eerst kunnen we nu verschillen tussen de cellen waarnemen. In de blastula onderscheiden we:
De binnenste celmassa of embryoblast (=voorloper van het embryo).
Afgevlakte cellen die de blastocystholte aflijnen: de trofoblast (=voorloper van de placenta en vliezen).
Figuur 43 Blastocyst (Langman, 2018) embryoblast blastocystholte trofoblast
Bij In Vitro Fertilisatie (IVF) gebeurt de embryotransfer (het terugplaatsen van het embryo) in het blastocyststadium. Vooraf kunnen de embryo’s getest worden op diverse genetische aandoeningen Zo kan een gerichte selectie gebeuren van de embryo’s met de hoogste kans op innesteling en het tot stand komen van zwangerschap.
Pre-implantatie Genetische Test (PGT)
Bij IVF worden - voor implantatie - enkele cellen van de blastocyst verwijderd voor onderzoek Naast onderzoek naar aneuploïdie en mozaïcisme kunnen hierbij ook allerlei andere, erfelijke aandoeningen opgespoord worden (bv. de ziekte van Huntington). Daarnaast kunnen ook bepaalde uiterlijke kenmerken en zelfs het geslacht van de embryo’s gedetecteerd worden. Maar in België mag men alleen embryo’s voor transfer selecteren om ‘ernstige aandoeningen te voorkomen’ (Wet betreffende de medisch begeleide voortplanting, 2007).
Vind jij dit terecht? Of zou embryoselectie volgens jou ook moeten kunnen op basis van andere kenmerken zoals milde aandoeningen (bv. kleurenblindheid) uiterlijke kenmerken, geslacht, …?
6.3 Innesteling (D6)
uterien epitheel
uterien stroma
blastocystholte
trofoblast
embryoblast
trofoblast
Figuur 44 Innesteling (Langman, 2018)
Op deze tekening bevindt de baarmoederholte zich onderaan.
Op dag 6 begint de blastocyst z'n implantatie (innesteling of nidatie) in het baarmoederslijmvlies. Dit wordt vergemakkelijkt doordat zowel cellen van het endometrium als cellen van de trofoblast enzymen produceren die de oppervlakkige epitheelcellen afbreken. Trofoblastcellen die zich ter hoogte van de embryonale pool van de blastocyst bevinden, dringen vervolgens binnen in het endometrium en nemen door mitose sterk toe in aantal.
De implantatie is een cruciaal proces voor de verdere ontwikkeling van het embryo. De normale implantatieplaats voor de blastocyst is de achterwand van de baarmoederholte.
Wanneer de implantatieplaats zich buiten de baarmoederholte bevindt, spreekt men van een ectopische zwangerschap. Dit komt voor bij 1 à 2% van de zwangerschappen. Mogelijke implantatieplaatsen zijn dan eileider, ovarium of buikholte. Mogelijke oorzaak is een slecht doorgankelijke eileider, bv. door ontstekingen. Ectopische zwangerschappen kunnen niet voldragen worden en geven vrijwel steeds aanleiding tot levensbedreigende complicaties (peritonitis, shock). Bij iedere zwangere vrouw met klachten van abdominale pijn of vaginaal bloedverlies, moet men bedacht zijn op een ectopische zwangerschap.
De implantatie is te beschouwen als een lichte verwonding van het endometrium, waarbij een gering bloedverlies mogelijk is. Indien deze implantatiebloeding opgemerkt, wordt kan dit verkeerdelijk als menstruatie worden beschouwd en leiden tot een te korte schatting van de zwangerschapsduur.
6.4
Spontaan miskraam
Naar schatting 25% van de zwangerschappen mondt uit in een spontaan miskraam (< 20 weken). Dit gebeurt meestal nog tijdens het 1e trimester. 30 à 50% van de gevallen van spontaan miskraam wordt veroorzaakt door chromosomale abnormaliteiten, meestal een autosomale trisomie (Dugas & Slane, 2022; Gianaroli et al., 2010; Lockwood, 2022).
Aneuploïdie is de belangrijkste oorzaak van spontaan miskraam.
Het gaat hierbij vooral over trisomie 16, 22 en 21
Bij een vroeg spontaan miskraam is de vrouw zich vaak nog niet bewust van de zwangerschap en het bloedverlies dat met zo'n spontane abortus gepaard gaat, kan ook verkeerdelijk worden aanzien als een (eventueel hevigere) menstruatie.
In geval van mozaïcisme is het embryo veel vaker levensvatbaar, wellicht door de eerder besproken correctiemechanismen (zie 6.1.2).
Het roept veel vragen op dat het menselijke embryo zo vatbaar is voor chromosomale abnormaliteiten en spontaan miskraam.
Vanuit evolutionair perspectief wordt het frequent optreden van spontaan miskraam bij embryo’s met aneuploïdie soms omschreven als een ‘fail-fast’-mechanisme: een proces dat zichzelf zo snel mogelijk onderbreekt als er iets ernstig verkeerd gaat. Dit zou voorkomen dat kinderen geboren worden met erg lage overlevingskansen en/of weinig levenskwaliteit De zorg voor zo een kind vraagt bovendien ook heel veel van de ouders, wat in de prehistorie ook hun eigen overlevingskansen kon beïnvloeden.
Hoe denk jij hierover? Wat roept dit perspectief bij jou op?
7
Tweede week
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Een zorgvuldige tekening te maken van deze embryonale stadia: dag 8 (regel van 2), dag 9 (lacunaire fase), dag 11 (vorming van het extra-embryonale mesoderm).
Het stadium van de ‘regel van 2’ (D9) te bespreken.
Het stadium van de ‘lacunaire fase’ (D9) te bespreken.
Het stadium van de ‘vorming van het E.E.M.’ (D11) te bespreken.
7.1 De regel van 2 (D8)
Bij het begin van de tweede week ligt de blastocyst partieel ingebed in het endometrium.
Rond D8 gaat de trofoblast zich differentiëren in een cytotrofoblast en een syncytiotrofoblast. Deze syncytiotrofoblast is invasief groeiend weefsel met sterk fagocyterende eigenschappen en is verantwoordelijk voor de eigenlijke innesteling.
De embryoblast gaat uitgroeien in twee structuren die als twee ovaalvormige platen op elkaar liggen: de tweebladige kiemschijf, met dorsaal de epiblast (voorloper van het ectoderm) en ventraal de hypoblast (voorloper van het endoderm).
De blastocystholte wordt nu (primaire) dooierzak genoemd en grenst aan de hypoblast. In de epiblast verschijnt op dezelfde dag ook een holte, de amnionholte. Epiblastcellen die deze holte omsluiten, worden amnioblasten genoemd.
2 met vocht gevulde holtes (amnionholte en dooierzak).
Figuur 46 Dag 8
7.2
De lacunaire fase (D9)
Door uitbreiding van de syncytiotrofoblast is de vrucht omstreeks dag 9 zo goed als volledig verzonken in het endometrium. Het defect in het baarmoederslijmvlies wordt tijdelijk nog afgesloten door een fibrineprop (stollingsprop).
Andere veranderingen omstreeks dit tijdstip:
In de syncytiotrofoblast worden vacuolen (holtes) gevormd. Deze holtes versmelten met elkaar en vormen een netwerk van lacunes
Cellen van de hypoblast migreren langs de wand van de dooierzak en gaan deze holte aflijnen: de membraan van Heuser.
De stromacellen van de baarmoederwand vertonen een decidua-reactie, d.w.z. dat zij zich transformeren naar metabool actievere cellen. Later ontstaat hieruit het maternale deel van de placenta.
Figuur 47 Dag 9
7.3 Extra-embryonaal mesoderm (D11)
Rond de 11e dag wordt losmazig bindweefsel opgehoopt tussen de hypoblast en amnionepitheel aan de ene zijde en de cytotrofoblast aan de andere zijde. We spreken van het extra-embryonaal mesoderm (E.E.M.)?
Al vrij snel ontstaan in dit losmazige mesoderm intracellulaire holten. De uitbreiding van deze holten leidt tot het ontstaan van het extra-embryonaal coeloom of chorionholte.
Aan de wand van deze nieuwe holte blijft een laag mesodermcellen bestaan, we spreken van pariëtaal mesoderm (tegen de cytotrofoblast) en visceraal mesoderm (tegen de amnioblasten of membraan van Heuser). Deze mesodermlagen hebben vooral een steunfunctie.
*met trofoblast bedoelen we hier cytotrofoblast èn syncytiotrofoblast. Na verloop van tijd blijft de syncytiotrofoblast enkel over aan de placentazijde (waar de chorionvilli gevormd worden, zie hoofdstuk 10).
Op één plaats blijft een volledige verbinding bestaan tussen de tweebladige kiemschijf en de cytotrofoblast: de hechtsteel. Deze voorloper van de navelstreng ontspringt vanuit het chorion en bevindt zich steeds aan de caudale zijde van de kiemschijf. In deze mesodermale verbinding zullen bloedvaten ontstaan die tussen het embryo en de placenta lopen.
Verder omstreeks dag 11:
De vrucht is volledig verzonken. Er is geen fibrineprop meer zichtbaar.
De lacunes in de syncytiotrofoblast reiken diep in het stroma en maken contact met maternale (gedilateerde) bloedvaten, sinusoïden, genaamd. Op die manier ontstaat bloedcirculatie in de lacunes en komt de uteroplacentaire circulatie op gang.
De uitlopers van het visceraal mesoderm in het embryo heten we splanchnisch mesoderm. De uitlopers van het pariëtaal mesoderm in het embryo heten we het somatisch mesoderm (zie 8.3)
8
Derde week
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Een zorgvuldig schema te maken van het embryo, eind 3e week, zowel een overlangs aanzicht (longitudinaal), als dwarse doorsnede (transversaal).
Te omschrijven wat gastrulatie betekent (incl. het tijdstip van optreden).
Toe te lichten wanneer en hoe de oriëntatie van de kiemschijf wordt vastgelegd.
Het onderscheid te maken tussen de vorming van het extra-embryonale mesoderm en het intra-embryonale mesoderm.
Te omschrijven wat neurulatie betekent (incl. het tijdstip van optreden).
Te omschrijven hoe de neurale lijst wordt gevormd.
De verschillende soorten mesoderm te beschrijven.
De verschillende delen van het somiet te beschrijven.
Te omschrijven wat bedoeld wordt met segmentering.
Uit te leggen op welke manier een gerichte genexpressie bijdraagt aan segmentering.
De derde week begint met gastrulatie, een verbluffend (en nog niet volledig begrepen) proces waarbij het intra-embryonale mesoderm wordt gevormd. Hierdoor ontstaan de drie definitieve kiembladen en wordt het bouwplan van het organisme neergelegd.
8.1 Gastrulatie
“It is not birth, marriage, or death, but gastrulation that is truly the most important time in your life.”
Lewis Wolpert (1984)
8.1.1
Gastrulatie is de vorming van het intra-embryonale mesoderm. De gastrulatie wordt uitgevoerd door cellen van de epiblast.
Primitiefstreep en -knoop
In week 3 doorbreken de epiblastcellen hun uniformiteit. Afhankelijk van hun positie in het kiemblad brengen ze nu andere genen tot expressie. Hierdoor gaan de epiblastcellen zich meer en meer van elkaar onderscheiden.
primitiefknoop (of kuiltje)
primitiefstreep (of groeve)
OP vorming notochord
amnion (doorgesneden)
OP = orofaryngeale membraan
C = cloacale membraan
Aan de caudale zijde van de kiemschijf migreren epiblastcellen naar mediaal. Op de middellijn drukken ze tegen elkaar aan en vormen ze op die manier de primitiefstreep, een lokale verdikking van de epiblast. In die primitiefstreep duiken epiblastcellen in de diepte om zo terecht te komen tussen epiblast en hypoblast. Ze vormen er op die manier het 3e kiemblad, het mesoderm. Door deze invaginatie van cellen verschijnt een groeve aan het oppervlak van de primitiefstreep: de primitieve groeve.
Aan de tip van de primitiefstreep is het invaginatieproces het sterkst. Door ophoping van cellen ontstaat daar de primitiefkno(o)p. Sommige cellen in die primitiefknoop vervullen de taak van ‘regisseur’: ze signaleren aan andere cellen waar ze heen moeten en stuwen
Figuur 49 Gastrulatie (bovenaanzicht op de epiblast) (Langman, 2018)
ze vooruit met trilharen Vergelijkbaar met hoe de primitieve groeve ontstaat, verschijnt een kuil in de primitiefknoop: het primitiefkuiltje.
primitiefkuiltje
primitieve groeve
epiblast
DZ AH
hypoblast invaginatie
Figuur 50 Dwarse doorsnede doorheen kiemschijf (met gedeeltelijk bovenaanzicht op de epiblast) (Langman, 2018)
amnioblasten
membraan v Heuser
AH = amnionholte
DZ = dooierzak
8.1.2
Notochord
Vanuit de primitiefknop vertrekt een stroom van cellen die zich naar craniaal bewegen in het nieuwe, mesodermale kiemblad. Deze vormen een staafvormige structuur op de middellijn, het notochord (of chorda dorsalis).
Het notochord heeft een erg belangrijke steunfunctie voor het embryo en is de voorloper van ons axiale skelet (de wervelkolom zal er zich later rond vormen). Het notochord wordt ook het axiale mesoderm genoemd.
AH DZ notochord
mesoderm OP C
prechordale plaat allantoïs hechtsteel
Figuur 51 Craniocaudale snede doorheen de kiemschijf, vorming notochord (naar Carlson, 2019)
AH = amnionholte
DZ = dooierzak
OP = orofaryngeale membraan
C = cloacale membraan
Het notochord groeit door tot aan de prechordale plaat, een craniaal gelegen celmassa die o.a. een rol zal spelen in het ontstaan van het hoofd-halsgebied.
Door gastrulatie – m.b. de vorming van het notochord - krijgt de kiemschijf vanaf nu een definitieve oriëntatie: craniaal-caudaal-middellijn-links-rechts worden voorgoed vastgelegd.
Bij 1/10.000 mensen loopt de oriëntatie van de kiemschijf verkeerd omdat de primitiefknoop zijn regisserende rol niet vervult. Dit leidt tot situs inversus, een aandoening waarbij de interne organen in spiegelbeeld liggen: het hart rechts, de lever en de appendix links. Meestal wordt dit pas later in het leven – toevallig – vastgesteld tijdens een medisch onderzoek. Op zichzelf is situs inversus onschuldig, maar als de oorzaak bij een genetisch defect in de trilharen ligt, zijn er wel medische gevolgen (bv. frequent bronchitis).
8.1.3 Endoderm en allantoïs
Tijdens de gastrulatie wordt een gedeelte van de epiblastcellen naar de hypoblast gestuurd om deze cellaag te vervangen. Op die manier ontstaat het definitieve endoderm.
Tegelijk met de vorming van het endoderm ontstaat de allantoïs als een kleine uitstulping van de dooierzak. Net zoals de dooierzak is de allantoïs een evolutionaire restant zonder functie. Bij vogels en reptielen voorziet de sterker ontwikkelde dooierzak in voedingsstoffen voor het embryo en collecteert de sterker ontwikkelde allantoïs afvalstoffen. Bij de mens worden al deze taken door de placenta uitgevoerd.
8.1.4 Driebladige kiemschijf
Niet alle cellen van de epiblast nemen deel aan de gastrulatie. De epiblastcellen die niet migreren vormen het ectoderm (zie verder, neurulatie)
Na gastrulatie bestaat de kiemschijf uit 3 definitieve kiembladen:
ECTODERM De ‘buitenkant’
MESODERM De ‘tussenliggende’ steunlaag
ENDODERM De ‘binnenkant’
In vergelijking met week 2 is de kiemschijf dikker en langwerpiger geworden doordat het embryo sneller groeit aan het craniale uiteinde.
Na gastrulatie verdwijnen primitiefstreep en- knoop.
Het gastrulatieproces bij de mens is sterk vergelijkbaar met dat bij vogels en reptielen, waarbij drie kiembladen worden gevormd die 'als een stapel pannenkoeken' boven op een dooierzak liggen (de ‘gastrula’). De lichaamsvorm is tijdens week 3 nog niet te zien.
8.1.5 OP en C-membraan
Gastrulatie zorgt voor de vorming van het intra-embryonale mesoderm.
Tussen ecto- en endoderm zit nu overal mesoderm.
Op twee plaatsen blijven ecto- en endoderm echter rechtstreeks met elkaar in contact.
Craniaal heten we dit de orofaryngeale membraan (OP-membraan), van het Engelse oropharyngeal), posterieur de cloacale membraan (C-membraan).
Dit zijn de voorlopers van de lichaamsopeningen naar de buitenwereld:
• OP-membraan Þ mond- en keelholte
• C-membraan Þ urogenitale, anorectale openingen
8.2 Neurulatie
Neurulatie is de vorming van de neurale buis, de voorloper van het centrale zenuwstelsel.
De neurulatie wordt uitgevoerd door cellen van het ectoderm.
Bij het begin van de 3e week ontstaat een verdikking van het middengedeelte van het ectoderm: de neurale plaat. Vervolgens vouwen de laterale randen van de neurale plaat opwaarts en bewegen ze naar elkaar toe op de middellijn.
Zo ontstaat een diepe groeve in het ectoderm.
randen van de neurale plaat
NEURALE PLAAT
neurale lijst
Figuur 52 Neurulatie in 3 stappen
Na enkele dagen komen de randen samen en versmelten ze met elkaar. Hierdoor wordt een buisvormige structuur afgesplitst van het ectoderm: de neurale buis. Deze ligt – net als het notochord – op de middellijn van het embryo.
Het centrale kanaal in de neurale buis is een afsnoering van de amnionholte.
De volledige sluiting van de buis gebeurt pas in de 4e week en het laatst aan de uiteinden, meer bepaald thv de neuroporus anterior (craniaal) op dag 24 en de neuroporus posterior (caudaal) op dag 28.
Een regel in de embryologie: craniale structuren rijpen altijd iets sneller uit dan caudale structuren. Een ander voorbeeld is de aanleg van de ledematen. Dit gebeurt iets vroeger bij de bovenste ledematen.
neurale plaat
primitiefknop
Uit de neurale buis ontstaan de hersenen en het ruggenmerg.
Falen van de sluiting leidt tot neurale buisdefecten (NBD) zoals spina bifida of anencefalie. Uit studies blijkt dat inname van foliumzuur de kans op neurale buisdefecten doet dalen (zie 19.2.2).
Tijdens de neurulatie komen een aantal cellen van de neurale plaat lateraal van de neurale buis te liggen. Dit worden de cellen van de neurale lijst genoemd.
neuroporus anterior
neuroporus posterior somieten
Het zijn de voorlopers van o.a. melanocyten (pigmentcellen in de huid), delen van de ogen, dorsale ganglia en cellen van het bijniermerg. Ze spelen ook een rol in de aanleg van het hart en de grote bloedvaten.
Omdat cellen van de neurale lijst een belangrijke bijdrage leveren aan de vorming van tal van organen, worden ze soms als ‘vierde kiemblad’ beschouwd.
8.3 Segmentering van het mesoderm
8.3.1 Overzicht
Aan het einde van de 3e week, na gastrulatie en neurulatie, beginnen ook de meer lateraal gelegen mesodermcellen afgrensbare structuren te vormen.
We onderscheiden naast het axiale mesoderm (=notochord), nu ook het paraxiale, intermediaire en laterale plaat- of zijplaatmesoderm.
De eerste drie zijn buisvormige strengen, de laatste dankt zijn naam aan de afgeplatte vorm. Het axiale mesoderm is één structuur, op de middellijn. De andere structuren komen in tweevoud voor, aan beide zijden van de middellijn.
Figuur 54 Dwarse doorsnede doorheen de driebladige kiemschijf, De stippellijnen geven de begrenzing aan van de verschillende delen van het mesoderm (naar Larsen, 2015).
8.3.2 Het paraxiale mesoderm
8.3.2.1 Segmentering
Aan het eind van de 3e week segmenteert het paraxiale mesoderm in klompjes, mesodermale cellen die we somieten heten.
Segmentering betekent het opdelen van een structuur in serie van kleinere, functioneel gelijkwaardige delen (segmenten).
De aanwezigheid van deze aaneengeschakelde weefselblokjes is typisch voor de segmentaire anatomie van heel wat organismen.
Bij sommige dieren (bv. de regenworm) kunnen we deze segmenten zelfs met het blote oog waarnemen. Bij gewervelde dieren zien we de segmentaire anatomie bv. aan de bouw van wervelkolom (aaneenschakeling van wervellichamen).
Figuur 55 Elektronenmicroscopische opname van een kippenembryo, waarbij een deel van het oppervlakkig ectoderm is weggenomen. Op die manier is de segmentering heel duidelijk zichtbaar.
De segmentering start aan het craniale uiteinde van het paraxiale mesoderm en loopt stapsgewijs verder naar caudaal. Zo neemt het aantal somieten toe van 1 à 4 (eind 3e week) naar ± 42 (eind 5e week).
Elke somiet deelt zich vervolgens op in een
(1) Sclerotoom: ontwikkelt tot bot en kraakbeen
(2) Myotoom: ontwikkelt tot spierweefsel
(3) Dermatoom: ontwikkelt tot dermis (lederhuid)
N.B.
ectoderm ectoderm
N.B.
Figuur 56 Dwarse doorsnede die opdeling van de somieten toont, vroeg (links) en (laat).
Let op hoe het sclerotoom zich rond het notochord en neurale buis wikkelt. (naar Carlson, 2019) voorloper van ligamenten
8.3.2.3 Genetische sturing van de segmentatie
Segmentering van het paraxiale mesoderm in somieten (somitogenese) is een proces dat gestuurd wordt door tal van genen die cranio-caudaal worden geactiveerd (van kop tot teen).
Twee opmerkelijke genen die hieraan meehelpen zijn:
De notch genen (en eiwitten) werken als een timer Elke 5 uur signaleren ze dat het tijd is om een nieuw paar somieten af te splitsen. Hierdoor zijn de somieten allen even groot.
De hoxgenen zijn transcriptiefactoren. In elke somiet schakelen ze een uniek patroon aan genen aan of uit. Zo leggen ze de regionale ‘identiteit’ van elke somiet vast.
Bv. de atlas, de eerste cervicale wervel C1 heeft een unieke vorm en functie. Deze wervel wordt maar één keer gevormd, vanuit het sclerotoom van een
craniaal caudaal
Hox-genen geven instructies
Notch-genen bewaken de timing
Figuur 57 Genetische sturing van de segmentatie.
De cijfers geven het aantal somietenparen weer.
De segmentatie van het mesoderm wordt gestuurd door een indrukwekkend genetisch draaiboek met een strikt tijdspad en gedetailleerde instructies.
8.3.3 Het intermediaire mesoderm
Het intermediaire mesoderm segmenteert gedeeltelijk. Hieruit ontstaat het urogenitaal stelsel (zie hoofdstuk 13 en 14)
8.3.4 Het laterale plaat mesoderm
De laterale plaat (zijplaat) segmenteert niet. Deze structuur, bestaat uit twee lagen mesoderm, met daartussen een ruimte; het intra-embryonaal coeloom. Uit het laterale plaatmesoderm ontstaan de lichaamsholten, de bloedcirculatie en de ledematen.
coccygeale (zie 9.3, 9.4, 11)
OVERLANGS
DWARS
Figuur 59
Figuur 59
9 Vierde t.e.m. achtste week
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Te beschrijven welke krommingen het embryo ondergaat in de 4e week.
Minstens drie gevolgen van de krommingen aan te geven.
Uit te leggen waarom de 3e tot 8e week een kritische periode is voor het optreden van aangeboren afwijkingen.
Aan te geven uit welk kiemblad een orgaan of structuur ontstaat.
De rol van de embryonale kieuwbogen kort te duiden.
Het onderscheid toelichten tussen het extra-embryonaal coeloom en het intraembryonaal coeloom.
De drie lichaamsholtes te benoemen die ontstaan uit het intra-embryonaal coeloom.
Kort te omschrijven welke taak het dorsale mesenterium heeft in de buikholte.
Kort te omschrijven welke taak het septum transversum heeft.
In grote lijnen te beschrijven hoe de ledematen ontwikkelen.
Minstens 3 voorbeelden te geven van aangeboren afwijkingen aan de ledematen.
Een correcte verklaring te geven voor het feit dat embryologische stadia bij veel organismen sterke gelijkenissen vertonen.
Een correcte verklaring te geven voor het feit dat evolutionaire restanten gevormd worden tijdens de embryologische ontwikkeling.
9.1
Krommingen (W4)
Om van een platte schijf te kunnen overgaan naar een menselijke vorm, zijn nog verschillende aanpassingen noodzakelijk. De uitwendige lichaamsvorm begint vorm te krijgen, vanaf de vierde week en dit gebeurt door twee gelijktijdige krommingen:
• De transversale kromming, of dwarse kromming, waarbij de laterale uiteinden van de driebladige kiemschijf naar elkaar toe groeien.
• De cranio-caudale kromming, of voor-achterwaartse kromming, waarbij de anterieure en posterieure uiteinden van de kiemschijf naar elkaar toe groeien.
Deze krommingen ontstaan doordat de drie lagen van de kiemschijf en het amnion sneller groeien dan de dooierzak.
primitieve hart
Figuur
Deze krommingen hebben verschillende, belangrijke gevolgen:
Het ectoderm komt aan de buitenkant te liggen, het endoderm aan de binnenkant en het mesoderm tussenin
Het platte endoderm vormt zich om tot een buis, de voorloper van het maagdarmstelsel.
Er ontstaat een inwendige lichaamsholte (coeloom), later borst- en buikholte
Het embryo neemt de foetale positie aan en wordt volledig omgeven door de amnionholte. Deze 'vochtballon' rond het embryo heeft een belangrijke schokdempende functie. Daarnaast maakt het vrije bewegingen mogelijk, voorkomt het vergroeiingen en bevordert het de groei van het embryo zelf. Uiteindelijk zal het embryo zich sluiten door het ventraal fusioneren van de randen van de kiemschijf. Het menselijk lichaam heeft ventraal op die manier een (onzichtbare) middellijn waarlangs het embryo werd 'dichtgeritst'.
60 Transversale kromming (rechts) en cranio-caudale kromming (links)
Falen van deze sluiting in het gezicht leidt tot schisis (lipverhemeltespleet). Falen ter hoogte van de borst- of buikwand leidt tot zeldzamere afwijkingen waarbij organen buiten het lichaam liggen, hetzij het hart (ectopia cordis), de buikorganen (gastroschisis) of de blaas (blaasextrofie).
9.2 Organogenese (W3-W8)
De derde tot en met de achtste week is dé kritische periode voor de aanleg van alle weefsels en organen in het lichaam (=organogenese)
Vanuit de kiembladen ontstaan in deze periode op korte tijd alle orgaansystemen.
In deze periode is het embryo ook het meest gevoelig voor schadelijke factoren van buitenaf (=teratogenen, bv. alcohol, rubella, geneesmiddelen, toxoplasmose, …). Grote malformaties (misvormingen) van het embryo vinden hun oorsprong in deze periode.
VAN ORGANEN
weken >
zeer gevoelig voor teratogenen
VERDERE UITRIJPING minder gevoelig voor teratogenen
Figuur 61 Timing van organogenese (aangepast, naar Langman, 2018)
In dit hoofdstuk geven we voorlopig enkel een globaal overzicht van de organogenese per kiemblad (ectoderm, mesoderm, endoderm). In latere hoofdstukken wordt de aanleg van bepaalde orgaansystemen verder verdiept.
In het 2e jaar gaan we dieper in op de verschillende soorten teratogenen en congenitale malformaties (cursus algemene pathologie, teratologie en toxicologie).
9.2.1
Ectoderm
9.2.1.1 Centraal zenuwstelsel (CZS)
Het CZS ontstaat uit de wand van de neurale buis.
§ Het craniale deel verbreedt en plooit mee met de craniocaudale kromming. Er ontstaan 3 (en later 5) verwijdingen, de hersenblaasjes, waaruit de hersenen ontwikkelen, een enorm complex orgaan met tientallen miljarden neuronen.
§ Uit het nauwere, caudale gedeelte van de neurale buis zal het toekomstige ruggenmerg evolueren.
Het centrale kanaal van de neurale buis ontwikkelt tot de met vocht gevulde hersenkamers (hersenventrikels) en centrale ruggenmergkanaal.
Uit ectodermale placodes (lokale verdikkingen van het ectoderm) ontstaat gespecialiseerd zintuigweefsel bv. de ooglens uit de oogplacode, het binnenoor uit de oorplacode, het reukorgaan uit de olfactaire placode, ….
9.2.1.2 Huid
Enkel de opperhuid is van ectodermale oorsprong. In week 4 is dit nog een éénlagig, bedekkend epitheel, maar dit differentieert uit tot een meerlagig epitheel, waarin de oppervlakkige laag verhoornt (keratinisatie) door het neerzetten van een hard eiwit (keratine) in het cytoplasma.
Door het keratinisatieproces sterven de oppervlakkige huidcellen af, waardoor ze afschilferen en in het vruchtwater terecht komen.
Hoewel de ontwikkeling van haren (en talgklieren) al begint in de 12e week zijn de eerste haren pas zichtbaar omstreeks 20 weken (eerst op de wenkbrauwen en bovenlip). Deze fijne, eerder kleurloze, zijdeachtige haren worden lanugoharen genoemd. Op het eind van de zwangerschap worden de donsharen op het lichaam afgestoten en vervangen door definitieve haren. Prematuren bevatten bij geboorte nog veel lanugoharen.
Bij de geboorte is de huid (vooral in de huidplooien) bedekt met vernix caseosa een witte, vette substantie, bestaande uit talg, dode afgeschilferde cellen en haren.
9.2.1.3
Neurale lijst
Cellen van de neurale lijst vormen zoals eerder aangehaald de melanocyten (pigmentcellen) in de huid. De grootte van de pigmentkorrels in de cellen bepaalt hierbij de huidskleur van het kind. De klassieke ‘moedervlekjes’ (naevi) zijn goedaardige ophopingen van melanocyten. Uit de neurale lijst ontstaan ook nog delen van de ogen, dorsale ganglia, bijniermerg, hart en bloedvaten.
9.2.2
9.2.3
Mesoderm
In de hoofdstukken 11, 13 en 14.1 gaan we uitgebreid in op de structuren die zich uit het mesoderm ontwikkelen: hart, bloedvaten, nieren, blaas, genitalia.
Endoderm
Door de transversale kromming ontstaat een buisvormige structuur, de primitieve darm, waarin we een voor-, midden- en einddarm onderscheiden.
De OP-membraan scheurt door in de 4e week om het vruchtwater toegang te geven tot de voordarm. Deze voordarm stulpt uit in een reeks van 5 kieuwbogen (ook kieuwzakjes genoemd, opnieuw een soort segmentering). Elke kieuwboog bevat een eigen arterie (een vertakking van de dorsale aorta), een eigen craniale zenuw en een eigen spier- en kraakbeencomponent. De bogen stulpen uit in het bovenliggende ectoderm.
1e kieuwboog (I)
2e kieuwboog (II)
3e kieuwboog (III)
4e kieuwboog ( IV)
6e kieuwboog ( VI)
kieuwboogarterie (craniale) zenuw kraakbeen
Figuur 62 Doorsnede door het endoderm (W4), met zicht op de kieuwbogen (kieuwzakjes) (Mitchell, 2009)
De kieuwbogen worden aangeduid met een Romeins cijfer: I, II, III, IV en VI (kieuwboog V wordt bij de mens niet aangelegd). Na de embryonale periode blijven relatief frequent restanten achter van de kieuwbogen, als goedaardige cystjes in de hals op het traject van de kieuwbogen. Zo heeft 1% van de pasgeborenen een pre-auriculair putje als restant van de eerste kieuwboog.
De einddarm breekt pas in de 6e week open via de C-membraan. Het meest caudale deel van de einddarm is verwijd en wordt cloaca genoemd. Via deze cloaca worden afvalstoffen uitgescheiden naar het amnionvocht.
Tussen de voor- en einddarm vinden we de middendarm, die nog in verbinding staat met de dooierzak via de dooierzaksteel (ductus vitellinus).
Craniaal van de dooierzaksteel ontstaat de lever vanuit een verdikking van het endoderm.
9.3 Lichaamsholtes (W4)
9.3.1 Inleiding
Figuur 63 De lichaamsholtes
We bespreken de vorming van:
De buikholte (1)
De pericardholte (hartzakje) (2)
De twee pleuraholtes (3)
Deze lichaamsholtes ontstaan als compartimenten van het intra-embryonale coeloom (IEC); dat ontstaat in week 4, als gevolg van de dwarse kromming.
We kunnen dit schematisch als volgt voorstellen:
In wat volgt bespreken we de vorming van deze holtes meer in detail. We beginnen met de buikholte, want deze is het eenvoudigst te begrijpen.
9.3.2
De buikholte
Het intra-embryonale coeloom bestaat aanvankelijk uit een linker- en rechterdeel. Beide, U-vormige holtes liggen in het laterale plaatmesoderm. Door de dwarse kromming versmelten ze tot één holte, het intra-embryonaal coeloom (IEC). Ter hoogte van de buikorganen wordt dit de peritoneale holte (=buikholte).
Bij de vorming van de buikholte wordt het splanchnisch mesoderm peritoneum (tegen de organen aan). De somatische mesodermlaag wordt het pariëtale peritoneum (tegen de buikwand aan).
Ventraal is de versmelting volledig, maar dorsaal blijft een verbinding bestaan van (de meeste) organen met de achterste lichaamswand. Deze verbinding, het dorsale mesenterium, bestaat uit twee peritoneale bladen met wat mesoderm tussen. Het dorsale mesenterium zorgt ervoor dat de darm tijdens het leven minder bewegingsruimte heeft, waardoor een darmverstrengeling bij de mens zelden voorkomt.
Het dorsale mesenterium krijgt later nog een hoofdrol in de ontwikkeling van de inwendige geslachtsorganen (zie 14.1.1)
9.3.3 De pericardholte
De pericardholte ligt craniaal van de buikholte en staat er eerst nog mee in verbinding Ook hier: door de dwarse kromming versmelten de rechter- en linkerdelen van het intraembryonale coeloom (IEC) anterieur tot één geheel.
Het hart zelf ontstaat als twee hartbuizen (endocardbuizen) in het splanchnisch mesoderm (laterale plaatmesoderm). Door de krommingen groeien ze naar elkaar toe en op dag 22 versmelten ze tot één primaire buis.
Re dorsale aorta (zie 11.1.3)
neurale groeve
notochord
(fusie van) primaire hartbuizen (zie 11.2.2)
primitieve darm NB
neurale lijst pericardholte dorsale aortae
visceraal pericard
pariëtaal pericard
* = waar de longen zullen ontstaan
Figuur 66 Ontstaan van de pericardholte: dag 20 (boven Li), dag 21 (boven Re), dag 22 (onder) Reeks van 3 tekeningen, met toegenomen transversale kromming (naar Larsen 2015 & Carlson, 2019).
hartbuis
hartbuis
hartbuis
Li dorsale aorta
9.3.4
Is er geen mesenterium in de pericardholte?
Als je goed kijkt op bovenstaande figuren zul je zien dat er ook een ventrale en dorsale verbinding ontstaat tussen het hart en de lichaamswand. Zowel het ventrale als het dorsale mesocardium verdwijnen echter, waardoor het hart in feite ‘los’ komt te liggen in de pericardholte, weliswaar op zijn plaats gehouden door de grote bloedvaten.
Naast de dwarse kromming moeten we hier ook rekening houden met de overlangse kromming. Hierdoor wordt het primitieve hart naar ventraal/caudaal getrokken en komt het binnen de borstkas te liggen, vóór de voordarm, vóór de pleuraholtes en tegen de toekomstige lever.
Tussen het hart en de lever groeit op dag 22 het septum transversum, een dwarse, dikke mesodermale massa die het grootste gedeelte van het latere middenrif (diafragma)zal vormen: de scheiding tussen buikholte & de borstkas.
De pleuraholten blijven twee afzonderlijke holtes.
In de pleuraholten onderscheiden we ook een viscerale en pariëtale pleura.
De twee primitieve pleuraholten vormen de verbindingen tussen de pericardholte en de buikholte. Ze worden ook pericardio-peritoneale kanalen genoemd. Aan het eind van de vierde week ontstaan hierin aan beide zijden de longknopjes, voorlopers van de longen. Door het groeien van de longen nemen de pleuraholtes toe in omvang.
Figuur (Carlson, 2019)
viscerale pleura
pariëtale pleura
Re pleuraholte
Li long Re long
NB hart
notochord
slokdarm
extremiteitsknop
Li pleuraholte
pericardholte
Figuur 68 Dwarse doorsnede door het (nu gebogen) embryo in vooraanzicht. De beide pleuraholten (Li + Re) komen op deze doorsnede bovenop De pericardholte te liggen (Carlson, 2019).
Later worden de pleuraholtes definitief van de pericardholte en de buikholte gescheiden door (pleuro-pericardiale) plooien die ingroeien vanuit de lichaamswand. Hierdoor is het middenrif, de scheiding van thorax en abdomen, compleet.
Figuur 69 Doorsnede doorheen het toekomstige middenrif (Carlson, 2019)
In het middenrif blijven uiteraard wel openingen voor de aorta, de slokdarm en de vena cava inferior. Als er bijkomende openingen zijn (bv. door het onvoldoende ingroeien van de pleuropericardiale plooien) komt de buikinhoud prenataal in de thorax terecht. Deze levensbedreigende malformatie heet congenitale hernia diafragmatica (zie 2e jaar).
9.4
De ledematen (W4)
9.4.1 Basisaanleg (W4-8)
De ontwikkeling van de ledematen begint aan het eind van de vierde week, onder impuls van de eerdergenoemde hox-genen.
De bovenste ledematen ontwikkelen hierbij iets sneller dan de onderste.
De ledematen ontstaan als knopjes van het laterale plaatmesoderm, die bedekt worden met ectoderm. Deze extremiteitsknopjes werken onafhankelijk van de rest van het embryo en bevatten al het nodige om te ontwikkelen tot een arm of been Binnenin ontwikkelt het meegroeiende paraxiale mesoderm uit tot bot-, kraakbeen- en spierweefsels.
Later worden de lidmaten tweeledig (bovenarm/onderarm, bovenbeen/onderbeen) en in week 6-7 worden de vingers en tenen van elkaar losgemaakt door een proces van geprogrammeerde celdood (apoptose)
handplaat
Figuur 70 Geprogrammeerde celdood leidt tot de opsplitsing
Van de handplaat in vijf vingers (Webster, 2012)
Heel wat aangeboren afwijkingen zijn het gevolg van een verstoring van het ontwikkelingsproces van de ledematen.
Enkele voorbeelden:
Webbing: een web tussen de vingers – door het falen van de apoptose
Syndactylie: aan elkaar gegroeide vingers – door het falen van de apoptose
Polydactylie: extra vingertje(s) – door verstoorde signaaloverdracht in de handplaat
Aansluiteind op de ontwikkeling van de ledematen bespreken we hier kort de ontwikkeling van de nagels. Dit gebeurt pas vanaf week 10, dus buiten de periode van embryogenese.
De nagels ontstaan vanaf de 10e week als een verdikking van de opperhuid (het primaire nagelveld) op de dorsale zijde van elke vinger
primaire nagelveld nagelwortel nagelbed
nagelbed
eindkootje
nagel(plaat) )
nagelwortel
Figuur 71 Ontwikkeling van de nagels (Li) tijdens de 4e maand en (Re) vlak voor de geboorte. (Carlson, 2019)
In de diepte van de proximale nagelwal ontstaat de nagelwortel, vanwaar uit de nagel(plaat) wordt aangemaakt. Deze bestaat uit gekeratiniseerde cellen die een harde, compacte massa vormen. De nagelplaat groeit aan en schuift van proximaal tot distaal over het nagelbed (zie de dikke pijlen op Figuur 71).
De vingernagels zijn rond de 32ste week gegroeid tot aan de vingertoppen. De teennagels bereiken de teentoppen rond de 36ste week.
Het knippen van de nagel is meestal pas nodig zo’n 4 à 6 weken na de geboorte.
Keratine is een eiwit dat gebruikt wordt bij verschillende fenotypische eigenschappen bij de mens. Zowel haren, nagels als de verhoorde bovenlaag van de opperhuid bestaan uit keratine. Al deze kenmerken worden dus bepaald door één enkel gen. Dit heten we pleiotropie, nl. als meerdere fenotypische eigenschappen bepaald worden door één enkel gen.
9.5 Overzicht (eind W4)
9.5.1 Beschrijving van het embryo
Aan het eind van de 4e week is het embryo ongeveer 4 mm groot. Door de krommingen heeft het de typische, gebogen foetushouding aangenomen (als een letter “C”)
De lichaamsholten zijn gevormd en de basis is gelegd voor de verdere aanleg van alle grote, orgaansystemen.
oorplacode
aangezichts welving kieuwbogen (I, II, III, IV en VI)
lensplacode
voorloper neusholte
hartbult
navelstreng
II III IV
staart
somieten extremiteitsknopjes
Figuur 72 Zijnaanzicht embryo, eind 4e week (naar Carlson, 2019)
De OP-membraan en de kleine, zesde kieuwboog (VI) zijn niet te zien op de figuur.
Aan beide zijden van de neurale buis zijn rijen somieten zichtbaar doorheen het bovenliggende ectoderm. De aanleg van de ledematen is te zien aan de vorming van de 4 extremiteitsknopjes.
Het hoofd (en het craniale deel van de neurale buis) is sterk gegroeid maar weinig gedifferentieerd. Er zijn wel verschillende ectodermale placodes te zien.
Ter hoogte van de buik zien we een – in verhouding – grote navelstreng en een opvallende ‘hartbult’ (het hart klopt vanaf dag 22!).
Het embryonale lichaam eindigt in een prominente, gekrulde staart. Tegen week 8 is deze staart verdwenen. Dit gebeurt door geprogrammeerde celdood en fusie van de caudale wervellichamen. Wat overblijft is het os coccyx (staartbeentje).
I
9.5.2
Overzicht per kiemblad
ECTODERM
Huid (haar, nagels, zweet- en talgklieren)
Mondopening met tandglazuur
Anaal kanaal
Binnenoor
Oog: ooglens, hoornvlies (cornea)
Adenohypofyse
*Neurale buis
Hersenen
Neurohypofyse
Zintuigcellen
Ruggenmerg
Oog: retina, oogzenuw
*Neurale lijst
Melanocyten (pigmentcellen in de huid),
Dorsale (spinale) ganglia
Oog: oogspieren, choroidea (vaatvlies)
MESODERM
Dermatomen
Myotomen
Sclerotomen (kraakbeen, bot)
Hart
Bloed- en lymfecirculatie
Milt
Urogenitaal stelsel
Bijnierschors
Dentine
ENDODERM
Kieuwbogen
Maagdarmstelsel
Longen
Pancreas
Lever
Schildklier
Bijschildklieren
Speekselklieren
9.5.3
Vergelijkende embryologie
De vergelijkende embryologie bestudeert en vergelijkt de embryologische ontwikkeling van diverse organismen. We staan hier kort even bij stil.
JAAR: 1866
Ernst Haeckel, een Duits bioloog, trok voor het eerst een parallel tussen embryologie en de evolutietheorie van Charles Darwin (1959). Haeckel formuleerde de hypothese dat de embryologische ontwikkeling van een organisme een versnelde herhaling is van de evolutie die tot dat organisme heeft geleid. Deze recapitulatietheorie was volgens Haeckel de verklaring voor de gelijkenissen in vroege embryologische stadia van diverse organismen. Een ‘verder geëvolueerd’ organisme (bv. de mens) doorloopt dan eerst de primitieve organismen (bv. cel à meercellige à vis à amfibie à reptiel àzoogdier à mens).
Ondertussen weten we dat de recapitulatietheorie van Haeckel niet klopt. Doordat we allen afstammen van een gemeenschappelijke voorouder doorlopen we tijdens de embryologische ontwikkeling veel gelijkaardige stappen, maar niet precies alle stappen die tijdens de evolutie plaatsvonden.
Toch zijn de gelijkenissen die Haeckel observeerde frappant. De 4-weken-embryo’s van een mens, kip of konijn zijn amper van elkaar te onderscheiden.
Daarnaast is het ook opvallend dat we tijdens de menselijke embryogenese structuren aanleggen die voor ons niet nuttig zijn: een dooier(zak), de allantoïs, kieuwbogen, een staart. Dit heten we evolutionaire restanten.
De verklaring voor deze bevindingen is wellicht dat de natuur ‘goede oplossingen voor moeilijke problemen’ graag hergebruikt. Dit is veel efficiënter dan telkens opnieuw van nul te beginnen. De embryologie zit vol met dergelijke, complexe problemen die – eens ze opgelost zijn – in alle afstammende organismen op een gelijkaardige manier verlopen.
De segmentering van het mesoderm is hiervan een mooi voorbeeld. We zagen eerder al dat dit gebeurt door een draaiboek van genen die het notochord in stukjes knippen en aan elk stukje een identiteit toekennen. Dit is een formidabele oplossing voor een moeilijk probleem, nl. hoe bouw je een organisme op een gestructureerde manier op; waar komt wat?
Om die reden is dit hox-draaiboek bij heel veel diersoorten aanwezig, weliswaar met aanpassingen: bv. bij een fruitvlieg bepalen de hox-genen waar de poten, de vleugels en voelsprieten komen; bij de mens waar armen en benen komen.
Grote wijzigingen aan het draaiboek zijn echter niet zomaar mogelijk. Zo is de vorming van een staart een essentieel onderdeel van het segmenteringsmechanisme. Daarom is het bij de mens eenvoudiger om de staart eerst te vormen en hem dan nadien weer te laten verdwijnen.
10 Placenta, navelstreng en vliezen
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De verschillende fasen in de vorming van chorionvilli te omschrijven.
In grote lijnen te beschrijven hoe de maternale en foetale circulatie zich organiseren om zo efficiënt mogelijk stoffen uit te wisselen.
De rol van de placentabarrière toe te lichten.
Minstens 4 soorten stoffen te omschrijven die doorheen de placentabarrière kunnen worden uitgewisseld.
De structuur van de navelstreng te bespreken.
De structuur van de placenta te bespreken.
Aan te geven welke delen van de placenta hetzelfde genetisch materiaal bezitten als het embryo (/de foetus).
In grote lijnen uit te leggen waarom voorzichtigheid geboden is bij het innemen van medicatie tijdens de zwangerschap.
De afmetingen van placenta en navelstreng bij benadering kunnen aangeven.
De fysiologische navelstrengbreuk te kunnen onderscheiden van de aangeboren navelstrengbreuk.
In eenvoudige taal uit te leggen waarom het oogsten van stamcellen uit de navelstreng een veelbelovende behandeling kan zijn voor ziektes zoals een hartinfarct of hersentrombose.
H10. Placenta, navelstreng en vliezen
Vanaf dit hoofdstuk verlaten we de chronologie van de embryologische ontwikkeling. We keren eerst terug naar week 2 voor de start van de ontwikkeling van de placenta.
Net zoals vele zoogdieren ontwikkelt het menselijke embryo door middel van een placentaire zwangerschap. De placenta (of moederkoek) wordt aangemaakt vanuit het embryonale weefsel om tegemoet te komen aan de grote energiebehoefte van het groeiende embryo.
10.1 Chorionvilli
Aan het einde van de tweede week gaat de cytotrofoblast ingroeien in de omliggende syncytiotrofoblast. Hierdoor worden ter hoogte van de implantatieplaats de chorionvlokken (-villi) gevormd. Dit proces vormt de basis voor de placentavorming en is essentieel voor het verdere verloop van de zwangerschap.
Figuur 73 De verschillende stadia in de placentavorming( naar Carlson, 2019)
De vorming van chorionvilli verloopt in verschillende stadia:
Primaire villi: dit zijn louter uitstulpingen van de cytotrofoblast.
basale plaat
Secundaire villi: bevatten nu ook een kern van mesodermale cellen.
Tertiaire villi: bevatten bloedvaten (embryonale circulatie) in hun mesoderm.
Vanuit de tip van de tertiaire villi groeien de cytotrofoblastcellen door om de basale plaat te vormen, een grenslaag met de decidua basalis. Villi die zich vastzetten in deze basale plaat worden ankervilli genoemd.
tijd
H10. Placenta, navelstreng en vliezen
Het deel van de chorion waaruit de vlokken ontspringen wordt de chorionplaat genoemd. De met bloed gevulde ruimtes tussen de villi heten de intervilleuze ruimtes
JAAR: 1983
Bij een chorionvillusbiopsie (=vlokkentest) worden – onder echogeleiding – enkele chorionvlokken geaspireerd voor genetisch onderzoek. De techniek werd begin jaren ’80 op punt gesteld door Giuseppe Simone in Milaan. Aangezien de cellen in de chorionvilli foetaal van oorsprong zijn laten ze toe van het foetaal karyotype te bepalen tijdens het eerste trimester (Simoni et al., 1983).
10.2 Placentaire circulatie
10.2.1 Functie
Door het vlokkensysteem ontstaat een nauwe relatie tussen de maternale en embryonale circulatie. Dit vergemakkelijkt het uitwisselen van gassen, voedings- en afvalstoffen tussen de beide circulaties.
10.2.2
De vorming van villi leidt tot een groot contactoppervlak tussen de circulaties; van ± 5 m² op 28 weken tot ± 12 m² op 40 weken! Ter vergelijking: in de volwassen nieren bedraagt het totale contactoppervlak van de glomerulaire capillairen ‘slechts' ± 6 m².
Uteroplacentaire circulatie
In het mesoderm van hechtsteel, chorion & tertiaire villi ontstaan de embryonale bloedvaten. Ze vertakken zich in de villaire boomstructuur om het bloed zo dicht mogelijk bij de intervilleuze ruimtes te brengen.
navelstreng v. umbilicalis aa. umbilicales
amnion chorionplaat trofoblastlaag
tertiaire villus
intervilleuze ruimte trofoblastlaag
deciduaal septum myometrium
tertiaire villus (ankervillus)
maternale vene spiraalarterie decidua basalis
Figuur 74 Uteroplacentaire circulatie (Amboss, 2022)
H10. Placenta, navelstreng en vliezen
In de intervilleuze ruimtes wordt het maternale bloed uitgestort door de spiraalarteries. Door ingroeiende cytotrofoblastcellen zijn deze maternale bloedvaten sterk toegenomen in diameter Op die manier neemt de aanvoer van bloed naar de intervilleuze ruimtes toe.
Een gebrekkige trofoblastinvasie in de maternale spiraalarteries ligt aan de basis van (pre)-eclampsie of zwangerschapsvergiftiging.
10.2.3 Placentabarrière
De maternale en foetale circulatie verbinden niet rechtstreeks met elkaar. Gedurende de zwangerschap blijven ze strikt van elkaar gescheiden door enkele cellagen die samen de placentabarrière vormen. Doorheen deze barrière kunnen de volgende stoffen worden doorgegeven: O2, CO2, voedingsstoffen, afvalstoffen, hormonen, IgG-antistoffen, vitaminen, virussen (rodehond, zika-virus, …), geneesmiddelen, …
1: foetaal capillair
2: mesoderm
3: cytotrofoblast
4: syncytiotrofoblast
5: intervilleuze ruimte
De placentabarrière wordt dunner naarmate de zwangerschap vordert. Op het einde van de zwangerschap bestaat de barrière enkel nog uit de syncytiale membraan (4) en de wand van de foetale capillairen (1).
Geneesmiddelen zijn doorgaans erg kleine moleculen die makkelijk doorheen de placentabarrière bewegen (uitzonderingen zijn bv. insuline of heparine). Vooraleer medicatie toegediend wordt aan een zwangere vrouw moet dus steeds bekeken worden of het geneesmiddel schadelijke effecten kan hebben op het embryo. Voorzichtigheid is vooral geboden tijdens de periode van organogenese.
Aan het einde van de zwangerschap treden er in de placenta tal van veranderingen op die erop wijzen dat er een verminderde uitwisseling bestaat tussen de maternale en foetale circulatie. Zo neemt het bindweefsel toe in het centrum van de villus, neemt de
Figuur 75 De placentabarrière
VILLUS
H10. Placenta, navelstreng en vliezen
dikte van de wand van de foetale capillairen toe, gaan enkele van de kleine villi dicht en treden verkalkingen (calcificaties) op. Dit kan leiden (zeker in geval van postdatisme) tot placentaire insufficiëntie.
10.3
Navelstreng
De voorloper van de navelstreng is de hechtsteel, ontstaan in de chorionholte, uit het extra-embryonale mesoderm. In die brede hechtsteel ontstaan bloedvaten:
1 vena umbilicalis die zuurstofrijk bloed naar de foetus brengt.
2 arteriae umbilicales die zuurstofarm bloed naar de placenta voeren.
Rond de bloedvaten beschermende laag, de gelei van Wharton.
In de gelei van Wharton zijn krachtige stamcellen aanwezig, die in de juiste omstandigheden kunnen differentiëren tot cellen uit de drie kiembladen. Dit biedt mogelijkheden in de behandeling van ziekten waarbij weefsels onomkeerbaar beschadigd zijn bv. hartinfarct of hersentrombose (Stefánska et al., 2020).
Na de krommingen in de vierde week vervoegen de platgevallen dooierzak en allantoïs zich bij deze hechtsteel. Pas nu spreken we echt van de navelstreng.
v. umbilicales
Gelei van Wharton (perivasculair deel)
Gelei van Wharton (intervasculair deel) amnion
Aan het eind van de derde maand verdwijnt de chorionholte door het vergroten van de amnionholte zodat de navelstreng nu volledig wordt omgeven door het amnion. Omstreeks dat tijdstip zijn dooierzak, allantoïs en ductus vitellinus meestal verdwenen. de dooierzak(steel) is niet afgebeeld (Stefànska, 2020). aa. umbilicales
restant allantoïs
H10. Placenta, navelstreng en vliezen
Tussen de 6e en de 10e week wordt de buikholte (tijdelijk) te klein voor de darm van het embryo (door de snelle lengtegroei van de primaire darm en de gelijktijdige forse groei van de lever). Daarom stulpt de darm tijdelijk uit in het navelstrenggedeelte. Dit is een fysiologische navelstrengbreuk.
m.o.: dit is niet hetzelfde als een (aangeboren) navelbreuk, één van de vaakst voorkomende congenitale afwijkingen. Deze goedaardige, onderhuidse uitstulping van het peritoneum (en eventuele buikinhoud: darm en/of vetweefsel) is het gevolg van het onvoldoende verstevigen van de navelring in de buikwand. In de meeste gevallen verdwijnt zo’n navelbreuk spontaan in de eerste levensjaren.
10.4
Overzicht (eind 5e maand)
chorion laeve decidua capsularis
decidua pariëtalis
myometrium (baarmoederholte)
decidua basalis
chorion frondosum chorionplaat
restant dooierzak
baarmoederhals amnion vruchtzak
Figuur 77 Placenta aan het eind van de 5e maand (Carlson, 2019). De pijlen aan de linkerzijde van de figuur staan dicht opeen, maar zijn heel precies gezet. Deze verschillende lagen liggen dicht tegen elkaar (zie de figuur op Toledo voor een groter beeld).
Op het einde van de zwangerschap bedekt de placenta 25 à 30% van de uterus. De placenta is dan een platte schijf met een diameter van 15 à 25 cm, een dikte van 3 cm en een gewicht van 500 à 600 gram. De navelstreng is 50 à 60 cm lang en vertoont typisch meerdere windingen.
De placenta bestaat uit een foetale zijde en een maternale zijde:
De foetale zijde bestaat uit de chorionplaat met villi. Men spreekt ook van het villeuze chorion of chorion frondosum (letterlijk: loofrijk). Dit is het deel van de
H10. Placenta, navelstreng en vliezen
trofoblast dat bij de innesteling betrokken was en het sterkst is ontwikkeld. De rest wordt het chorion laeve (letterlijk: glad) genoemd.
De maternale zijde ontstaat uit gedecidualiseerd endometrium, de decidua basalis. Dit het gedeelte van het endometrium tussen myometrium en de foetus Tijdens de vierde maand van de zwangerschap dringen vanuit de decidua basalis tussenschotten (deciduale septa) binnen in de intervilleuze ruimtes. Deze tussenschotten groeien door tot aan de chorionplaat. Op die manier ontstaat een onderverdeling in cotyledonen (lobben). Op het einde van de zwangerschap telt de placenta 15 à tot 20 cotyledonen.
Verder onderscheiden we buiten de placenta nog de decidua capsularis (boven op de foetus, tussen foetus en baarmoederholte) en decidua pariëtalis (endometrium dat niet met de foetus in contact komt). Bij het verder groeien van de foetus verdwijnt de baarmoederholte en versmelten decidua capsularis en pariëtalis.
11 Hart en bloedvaten
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Kort te argumenteren waarom de bloedcirculatie al vroeg functioneel wordt.
Het proces van ‘vasculogenese’ te beschrijven en hierbij aan te geven waar en wanneer dit optreedt.
Het verschil tussen vasculogenese en angiogenese toe te lichten.
De link tussen de kieuwboogarteries en de kieuwbogen kort te beschrijven.
De aanleg van het hart zorgvuldig en stapsgewijs te kunnen beschrijven, vanaf het stadium van het primaire hartveld, tot en met de kromming van de hartbuis.
De septering van de atria te beschrijven.
De septering van de ventrikels beschrijven.
Enkele belangrijke gebeurtenissen in de aanleg van het hart op een tijdslijn te kunnen situeren (vorming van het primaire hartveld, fusie van de hartbuizen, kromming van het hart, septering van de atria/ventrikels).
Het optreden van aan aangeboren atrium- of septumdefect te kunnen verklaren vanuit de embryologie.
De weg van het bloed in de foetale circulatie te kunnen beschrijven (in grote lijnen).
Aan te geven welke shunts voorkomen in de foetale circulatie. `
Op een figuur van de foetale circulatie de shunts kunnen aanduiden.
Uit te leggen wat het nut is van elke shunt in de foetale circulatie.
11.1 De vorming van bloedvaten
Na het doornemen van vorige hoofdstukken vroeg je je misschien al af hoe bloedvaten gevormd worden (bv. in de hechtsteel). In deze paragraaf geven we hierop een antwoord.
11.1.1 Waarom is een bloedcirculatie nodig?
De eerste 3 weken is het embryo voor de aanvoer van O2 en voedingsstoffen volledig afhankelijk van passieve diffusie uit het maternale bloed in de lacunes.
Omdat diffusie slechts mogelijk is over een afstand van ± 10 cellen wordt dit mechanisme echter snel ontoereikend.
Vandaar dat de bloedcirculatie zich al in week 3 ontwikkelt en als een van de eerste orgaansystemen begint te functioneren vanaf week 4
11.1.2 Vasculogenese
11.1.2.1 In het extra-embryonale mesoderm
De vorming van bloedvaten (=vasculogenese) begint in het mesoderm van de dooierzakwand, hechtsteel en chorion (buiten het embryo dus!).
Sommige van deze mesodermale cellen gaan oiv gerichte genexpressie differentiëren tot hemangioblasten, voorlopers van bloedcellen. Deze groeperen zich in bloedeilandjes
bloedeilandjes hemangioblasten
DOOIERZAK WAND
Figuur 78 de verschillende fasen van vasculogenese in de dooierzakwand (aangepast uit Carlson, 2019)
(buitenste cellen platten af tot) endotheel
visceraal mesoderm
membraan van Heuser
Bloedeilandjes bestaan uit:
Een buitenste laag cellen die afplatten en omvormen tot endotheel Een binnenste laag cellen die zich omvormen tot grote rode, gekernde megaloblasten, de voorlopers van alle bloedcellen.
Door fusie van de zich uitbreidende bloedeilandjes ontstaan netwerken van bloedvaten. Later ontstaan ook nieuwe bloedvaten door het ontwikkelen van zijtakken uit bestaande vaten (=angiogenese)
Vasculogenese vindt alleen plaats tijdens de embryologische ontwikkeling. Angiogenese daarentegen kan ook in de rest van het leven plaatsvinden.
11.1.2.2 In het intra-embryonale mesoderm
De intra-embryonale vaten ontstaan iets later, nl. bij het begin van week 4.
In het splanchnische mesoderm (laterale plaat-mesoderm) vindt hiervoor een gelijkaardig proces van vasculogenese, angiogenese en fusie van bloedvaten plaats.
11.1.3 Overzicht van de embryonale circulatie (eind W4)
De embryonale circulatie komt op gang door het verbinden van de intra- en extraembryonale bloedvaten en het starten van de pompfunctie van het hart (dag 22).
vena cardinalis communis
ventrale aorta
kieuwboogarteries
primitieve hersencirculatie
hart
dorsale aorta navelstreng
bloedcirculatie in de dooierzak (verdwijnt)
Figuur 79 Embryonale circulatie eind 4 e week. De pijlen (stippellijnen) op de tekening geven de richting van de bloedflow aan (aangepast uit Carlson, 2019)
Maarten Biesbrouck
Enkele belangrijke bloedvaten, eind 4e week zijn:
De aorta, die ventraal vertakt in 5 kieuwboogarteries (I, II, III, IV en VI, één per kieuwboog), om dan samen te komen in één dorsale aorta Eerst zijn dit 2 dorsale aortae, maar eind 4e week zijn deze al versmolten tot 1 bloedvat. Ook de kieuwboogarteries verdwijnen en versmelten grotendeels. De definitieve aortaboog ontstaat grotendeels uit de IVe kieuwboogarterie.
Een systeem van cardinaalvenen (vv. cardinales) brengt het veneuze bloed terug naar het hart via een gemeenschappelijke v. cardinalis communis.
11.2 De vorming van het hart
11.2.1 Het primaire hartveld (D18-20)
Het hart ontstaat uit een speciaal deel van het mesoderm, het primaire hartveld
In dit craniolaterale deel van het splanchnisch mesoderm (ook het cardiogene mesoderm genoemd) is de vasculogenese zeer actief. Hierdoor ontstaat omstreeks dag 18 een hoefijzervormige cluster van bloedeilandjes.
neurale plaat
primaire hartveld
bloedeilandjes die de (toekomstige) Li hartbuis vormen
Figuur 80 Vasculogenese in het primaire hartveld. Zicht op de driebladige kiemschijf, vanuit de amnionholte (D18) (Openstax, 2022)
Aan beide zijden van de kiemschijf clusteren de bloedeilandjes samen tot grote bloedvaten; de hartbuizen Aan de binnenzijde worden deze afgelijnd door endocard, een gespecialiseerd endotheel.
11.2.2 Fusie van de hartbuizen (D22)
Door de overlangse kromming draait het hart 180° om een dwarse as en komt het caudaal van de OP-membraan te liggen. Omstreeks hetzelfde tijdstip vindt de overlangse kromming plaats, waardoor de beide hartbuizen naar elkaar toe groeien.
CRANIAAL
Op dag 22 fusioneren de beide hartbuizen tot één hartbuis (mooi weergegeven op Figuur 66) en vormt het omliggende zijplaatmesoderm een mantel van primitieve hartspiercellen. Dit zorgt ervoor dat de enkelvoudige hartbuis op dag 22 al begint te kloppen. De hartfrequentie is eerst zo'n 100 slagen per minuut. Deze stijgt naar 160/min op 8 weken, 150/min na 15 weken en daalt geleidelijk tot aan de geboorte.
De primitieve hartbuis ontwikkelt vervolgens enkele verdikkingen: een instroomkanaal (sinus venosus), een primitief atrium & ventrikel en een uitstroomkanaal (bulbus cordis).
arteriële zijde
1e aortaboog
fusie hartbuizen
bloedflow
veneuze zijde
1e aortaboog
ventrikel atrium sinus venosus
Figuur 81 Stadia in de ontwikkeling van het hart (Lohman & Ten Donkelaar, 1997)
11.2.3 Kromming van de hartbuis (D22-D28)
sinus venosus
(1) de primaire hartplooi (2) de atrioventriculaire hartplooi
Op dag 23 wordt de symmetrie van de pasgevormde hartbuis doorbroken en buigt ze ter hoogte van het ventrikel door naar ventraal en naar rechts
Het hart is het eerste, asymmetrische orgaan dat verschijnt in het embryo.
Door de kromming van de hartbuis ontstaan twee karakteristieke plooien in de buis:
(1) De primaire hartplooi, die zich bevindt op de toekomstige overgang tussen de linkerventrikel en rechterventrikel (die uit de bulbus cordis zal ontstaan).
(2) De atrioventriculaire (AV-)plooi, die zich bevindt op de overgang tussen de atria en het primitieve ventrikel. Men spreekt ook van atrioventriculair kanaal
Op D23 wordt de identiteit van de verschillende delen van de hartbuis vastgelegd. Aan de veneuze zijde (caudaal van de AV-plooi), wordt de atriale identiteit aangenomen. Aan de arteriële zijde wordt de ventriculaire identiteit aangenomen. De aanwezigheid van Vitamine A speelt hierin een cruciale rol. Een tekort aan vit A, of inname van medicatie die de werking van vit A beïnvloedt (bv. Roaccutane®) kan ernstige hartafwijkingen veroorzaken.
bulbus cordis
bulbus cordis
ventrikel
(1) (2)
Aan het eind van de 4e week (D28) is het buigproces volledig. Het nu S-vormige hart bestaat uit een dorsaal gelegen atrium en ventraal gelegen ventrikel. De binnenkant is bekleed met endocard (lijkt sterk op endotheel). De hartspiercellen zijn verder ontwikkeld en het hart vertoont al een vrij goed gecoördineerde, unidirectionele pompfunctie ondanks het feit dat er nog geen kleppen aanwezig zijn.
e aortaboog
11.2.4
toekomstige
venosus
Figuur 82 Kromming van het hart (D22-D24-D28) (A=atrium, V=ventrikel, VCS/VCI = vena cava superior/ inferior)
Septering van het hart (W4-5)
Het menselijk hart is een dubbele pomp, die twee bloedsomlopen bedient (longcirculatie en systeemcirculatie) In het hart zijn er om die reden twee atria en twee ventrikels aanwezig, die van elkaar gescheiden zijn door tussenschotten (septa). Deze ontwikkelen zich vanaf dag 28.
Septering is het proces waar bij tussenschotten (septa) worden gevormd.
rechter-atrium (RA)
tricuspidalisklep
rechter-ventrikel (RV)
linker-atrium (LA)
atriaal septum
linker-ventrikel (LV) mitraalklep
ventriculair septum
Figuur 83 Vierkamerbeeld van een aangelegd hart (Grote bloedvaten zijn weggelaten)
We bespreken hier enkel de atriale en ventriculaire septering. De vorming van de atrioventriculaire scheiding (met kleppen) laten we hier buiten beschouwing.
bulbus cordis
11.2.4.1
Septering van de atria
Dit gebeurt in volgende stappen:
Vorming van het septum primum:
Dit sikkelvormige, dunne septum groeit vanuit het dak van het primitief atrium, tot aan een verdikking van het endocard (endocardkussen) op de bodem ervan.
Er blijft een opening over aan de bodem van het septum: het ostium primum. Hierdoor stroomt tijdelijk bloed van RA naar LA.
Het ostium primum sluit vrij snel, terwijl tegelijkertijd – door apoptose – een tweede opening verschijnt aan de top van het septum: het ostium secundum. Hierdoor kan het bloed blijven stromen van RA naar LA.
Figuur 84 Vorming septum primum (aangepast uit Carlson, 2019)
Vorming van het septum secundum:
LA RA LA septum primum septum secundum foramen ovale septum primum septum secundum VCI VCS
Dit tweede, dikkere, maar ook sikkelvormige septum groeit aan de rechterzijde van het septum primum. Ook in dit septum ontstaat een (grotere), blijvende opening aan de basis: het foramen ovale (ovale venster). Ook door deze opening stroomt bloed van RA (naar ostium secundum) naar LA.
De pijl (rechtertekening) geeft de richting van de bloedflow weer. (aangepast uit Carlson, 2019) septum primum ostium secundum ostium primum endocard kussen
De vorm van het rechteratrium wordt zo gemodelleerd dat het aangevoerde bloed vanuit de vena cava inferior in de richting van het foramen ovale wordt gestuwd.
De atriale septa en hun openingen fungeren als een éénrichtings-klep. Ze laten toe dat bloed van RA naar LA stroomt, maar niet in de omgekeerde richting.
De betekenis van het foramen ovale wordt verder duidelijk (zie: 11.2.5).
11.2.4.2
Septering van de ventrikels
Aan het oppervlak van de gekromde buis is al vroeg een zichtbare scheiding aanwezig tussen de toekomstige linker- en rechterventrikel (de primaire hartplooi).
primaire hartplooi
RV LV
endocard kussen
verhoging myocard
Figuur 86 Ophoging van het myocard (aangepast uit Carlson, 2019)
Aan de binnenzijde van het hart is op die plaats ook een verhoging van het myocard (spierweefsel) te zien, het toekomstige ventriculaire septum. Vanuit de bodem van het primitieve ventrikel groeit deze ook naar het endocardkussen toe. Tijdelijk bestaat hierdoor ook een interventriculair foramen. Uiteindelijk versmelten het endocardkussen en de myocardverhoging tot een definitief (inter-)ventriculair septum.
Het definitieve, ventriculaire, septum bestaat uit een klein, membraneus deel (afkomstig van het endocardkussen) en een groot, spierig deel (uit myocardverdikking).
trabekels
membraneus deel (dun) klepvorming
spierig deel
Figuur 87 Vorming van het (inter) ventriculaire septum (aangepast uit Carlson, 2019)
Tijdens de septering van de ventrikels ondergaat het myocard een verregaande specialisatie waarin, de typische spierbalkjes (trabeculae) worden gevormd. Deze verhogen de kracht waarmee de hartspier kan samentrekken. Tegelijk ontstaan de hartkleppen, die het terugstromen van bloed voorkomen.
Aan het eind van de 5e week is de septering volledig. Na ontdubbeling van de truncus arteriosus (in aorta en truncus pulmonalis) is de parallelle circulatie gevormd.
Als er een opening in een septum blijft bestaan, spreekt men van een atrium-septumdefect (ASD) of ventrikel-septum-defect (VSD). Het VSD is de vaakst voorkomende, aangeboren hartafwijking. Kleine septumdefecten sluiten doorgaans vanzelf, terwijl grotere defecten chirurgisch moeten behandeld worden.
11.2.5
De foetale circulatie
11.2.5.1
De 3 shunts
De menselijke bloedcirculatie wordt uiteraard opgebouwd voor een leven buiten de baarmoeder. Centraal hierin is de longcirculatie, die toelaat dat O2 wordt opgenomen in het bloed. Maar prenataal wordt O2 uitsluitend aangevoerd via de placenta. De foetale longen zijn gevuld met vruchtwater en nog niet functioneel.
Hoewel deze tegenstrijdigheid op het eerste gezicht een onoverkomelijk probleem creëert, is er een elegante oplossing: de bloedcirculatie wordt opgebouwd voor een leven buiten de baarmoeder, maar door 3 tijdelijke, slimme verbindingen (‘wegomleggingen’) wordt het O2-aanbod naar de foetus gemaximaliseerd.
Shunts zijn bloedvaten die werken als een bypass Ze omzeilen delen van de circulatie die nog niet functioneren omdat ze in opbouw zijn (bv. de longen). Hierdoor wordt het O2rijke bloed van de placenta zo snel mogelijk verspreid wordt in de foetale weefsels.
Concreet gaat het om deze 3 shunts: (1) Foramen ovale (zie 11.2.4.1)
Het O2-rijke bloed van de placenta bereikt RA via de vena cava inferior. Dankzij het foramen ovale wordt dit bloed naar LA gestuurd, waarna het relatief snel in de aorta, hersen- en systeemcirculatie terechtkomt. Op die manier zijn de foetale hersenen een van de eerste ‘afnemers’ van O2-rijk bloed. Dankzij foramen ovale hoeft het meeste bloed ook niet onnodig door de (niet-functionele) longcirculatie.
Na de geboorte stijgt de druk in LA, waardoor de bloedflow doorheen het foramen ovale uiteindelijk toch omkeert. Hierdoor worden de twee septa tegen elkaar geduwd worden sluit de opening.
(2) Ductus arteriosus (DA, ductus van Botalli)
Dit bloedvat ontstaat uit de zesde kieuwboogarterie (IV) en vormt een verbinding tussen truncus pulmonalis (TP) en aorta. 90% van het O2-rijk bloed dat toch in TP is terechtgekomen stroomt via DA naar de aorta en systeemcirculatie.
Postnataal sluit de DA en blijft een bindweefselstreng over: lig. arteriosum.
(3) Ductus venosus (DV, ductus van Aranti)
Een directe verbinding tussen de vena umbilicalis en de vena cava inferior. Hierdoor stroomt een deel van het bloed dat van de placenta afkomstig is rechtstreeks naar de VCI (en dus niet onnodig door de lever).
Postnataal sluit de DV en blijft een bindweefselstreng over: lig. venosum.
rechterlongcirculatie
aorta TP
bovenste lichaamshelft & hersencirculatie
ductus arteriosus
linkerlongcirculatie
vena cava superior
foramen ovale
vena cava inferior
lever
ductus venosus
v. umbilicalis v. porta
placenta
nier
darm
navelstreng
Figuur 88 Foetale circulatie. De shunts zijn in het vet aangeduid. (Carlson, 2019).
Embryologie en Genetica – dr. Maarten Biesbrouck
aa. umbilicales
dorsale aorta (aorta descendens)
bijnier
11.2.5.2 Beschrijving van de foetale bloedcirculatie
Voor de volledigheid volgt hier nog eens een volledige beschrijving van de weg die het bloed aflegt. Leer dit niet uit het hoofd, maar tracht deze beschrijving zelf te reconstrueren aan de hand van de figuur op de vorige bladzijde.
Voor de geboorte stroomt het bloed via de v. umbilicalis van de placenta naar de foetus
Dit gebeurt soms onder verhoogde druk door uteriene contracties. Het aangevoerde bloed is voor 80% verzadigd met O2.
Wanneer het bloed de lever bereikt stroomt het grootste deel direct via de ductus venosus naar de v. cava inferior waarbij het de lever omzeilt. Slechts een klein deel komt in het vaatstelsel van de lever zelf terecht. In de ductus venosus is een sfinctermechanisme aanwezig die de toevoer van het bloed reguleert uit de v. umbilicalis zodat bij een contractie van de uterus de stroom van het veneuze bloed niet te groot wordt om zo een overbelasting van de foetale circulatie te voorkomen.
Vervolgens stroomt het bloed via de v. cava inferior het rechter atrium binnen. Daarna stroomt het via het foramen ovale voor het grootste deel direct door naar het linker atrium. Een klein deel blijft achter in het rechter atrium waar het zich vermengt met het zuurstofarme bloed dat uit het hoofd en de armen via de v. cava superior naar het hart terugstroomt.
Vanuit het linkeratrium, waar het bloed vermengt met een kleine hoeveelheid bloed uit de longen, stroomt het bloed naar het linkerventrikel en de aorta. De coronaire arteries alsook de aa. carotes takken als eerste af van de aorta descendens en voorzien de hartspier en de hersenen van voldoende O2-rijk bloed.
Het O2-arme bloed vanuit de v. cava superior stroomt via het rechter atrium het rechterventrikel binnen en gaat zo naar de a. pulmonalis (=truncus pulmonalis, TP)
Tijdens het foetale leven is de weerstand van de longvaten echter zeer groot en daarom stroomt het grootste deel van dit bloed (90%) direct door de ductus arteriosus naar de aorta descendens, waar het zich vermengt met het bloed uit de proximale aorta. Van hier loopt het bloed verder via de aa. umbilicales naar de placenta.
Wanneer het bloed van de placenta naar de verschillende organen van de foetus stroomt neemt het zuurstofgehalte langzaamaan af omdat het zich op verschillende plaatsen mengt met bloed van een lager zuurstofgehalte.
12 Ademhalingsstelsel
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De verschillende fasen van de embryonale longontwikkeling op te lijsten
De rol van het tracheo-oesofagale septum te beschrijven
De longknopjes te kunnen situeren binnen de ontwikkeling van de pleuraholtes.
Het vertakken van de bronchiaalboom als een voorbeeld van oppervlaktevergroting te benoemen, net als de vorming van chorionvilli.
Te verklaren vanuit de embryologie van de longen waarom de grens voor levensvatbaarheid bij een prematuur geboren kind omstreeks 24 weken ligt.
De rol van surfactant in het uitrijpen van de longblaasjes bespreken.
Te verklaren vanuit de embryologie waarom longrijping wordt toegediend bij dreigende vroeggeboorte.
De longen ontstaan – net als vele organen – in week 4, maar rijpen zeer traag uit, nl. tot de leeftijd van ±6 jaar. Een orgaan dat nog trager uitrijpt zijn de hersenen (tot ±25 jaar!).
12.1 Embryonale fase (W4)
De longen ontstaan uit de voordarm omstreeks week 4.
Net achter de kieuwzakjes (zie 9.5.1) vormt zich eerst een uitstulping, het laryngotracheale divertikel. Vanuit dit divertikel ontwikkelt zich later de larynx, trachea en de volledige bronchiaalboom.
Het divertikel is omgeven door mesoderm dat zal uitgroeien tot alle omliggende steunweefsels (bv. de kraakbeenringen rond de grote luchtwegen). Het epitheel van deze structuren is van endodermale afkomst.
laryngo-tracheaal divertikel voordarm voordarm
spitsing in 2 hoofdbronchiën
Figuur 89 Laryngo-tracheale divertikel (doorgesneden) (Mitchell, 2005)
Het laryngo-tracheaal divertikel groeit tijdens de 4e week naar caudaal en wordt gescheiden van de toekomstige slokdarm (die ook voortkomt uit de voordarm) door middel van een bindweefselig . De aanleg van het respiratoir gedeelte zal in open verbinding blijven met de
Figuur 90 Ingroei van het tracheo-oesofagaal septum (doorsnede) (Mitchell, 2005)
Een zeldzame aangeboren afwijking is een tracheo-oesofagale fistel. Hierbij blijft een verbinding bestaan tussen de trachea en de (vaak onvolledig doorgankelijke) slokdarm. Dit ontstaat wanneer het tracheo-oesofagale septum niet goed ontwikkelt.
12.2
Pseudo-glandulaire fase (W5-17)
Eind 4e week splitst het caudale uiteinde van het divertikel zich in twee kleinere structuren. Dit zijn de eerste vertakkingen van de bronchiaalboom, de 2 hoofdbronchiën.
Samen met het omliggende mesoderm vormen deze de longknopjes, die elk uitgroeien in hun pleuraholte (zie 9.3.4) en nog 22 keer zullen vertakken.
Er zijn dus 23 bifurcatie-niveaus in totaal. De tweede rij vertakkingen vormt de secundaire of lobaire bronchi (rechts drie, links twee; één voor elke lob dus).
longknopjes (2 hoofdbronchiën)
lobaire bronchiën (3+2)
Figuur 91 Vertakkingen van de bronchiaalboom (week 5 tot week 7) (pseudo-glandulaire fase) (Mitchell, 2005)
In week 17 zijn de meeste vertakkingen gevormd, hoewel er nog geen spoor is van de longblaasjes. Microscopisch ziet het longweefsel er bijgevolg wat uit als een klier met 'afvoerkanaaltjes'. Deze fase duidt men daarom aan als de pseudo-glandulaire fase.
In deze periode wordt het volledig buizensysteem van de longen dus aangelegd. Anders gezegd: alle grote elementen van de long aanwezig behalve diegene die betrokken zijn bij de gasuitwisseling. Ademen is bijgevolg niet mogelijk.
Foetussen die geboren worden tijdens deze periode kunnen niet overleven gezien de afwezigheid van het alveolair systeem, noodzakelijk voor de uitwisseling van O2 en CO2. Mechanische beademing is dus ook niet mogelijk.
niveau I niveau II niveau III ….
slokdarm
trachea
12.3 Canaliculaire fase (W13-25)
Pas nu begint het respiratoire deel van de bronchiaalboom te ontwikkelen. De distale bronchiolen verwijden zich, worden beter doorbloed en vertakken zich in twee of meer respiratoire bronchiolen. Dit zijn kanaaltjes (trechtertjes) die de overgang zullen maken naar de toekomstige alveolen. Er is wat overlap met de pseudo-glandulaire fase doordat craniale longsegmenten sneller matureren.
12.4 Alveolaire fase (W24-geboorte)
12.4.1 Ontwikkeling van de longblaasjes
Pas vanaf de 24e week beginnen de longblaasjes (alveoli) te ontwikkelen. Vanuit de respiratoire bronchiolen ontstaan blind eindigende zakjes van afgeplat epitheel die uitstulpen in het omliggende steunweefsel en nauw contact zoeken met de omliggende capillairen.
De latere bloed-lucht barrière is hiermee gevormd. Gasuitwisseling is nu (in theorie) mogelijk, hoewel de longen prenataal gevuld blijven met vruchtwater.
Een foetus die tijdens deze periode wordt geboren kan in leven worden gehouden mits intensieve zorgen en beademing. Vaak zal dit echter falen omwille van immaturiteit van het systeem. Hoewel gasuitwisseling mogelijk is, zijn er immers nog een aantal bijkomende aanpassingen nodig zijn om de longblaasjes open te houden en goed te laten werken.
alveolus
terminale
bronchiole
capillairen
respiratoire bronchiole
surfactant (latere) bloed-luchtbarrière
12.4.2 Productie van surfactant
Vanaf week 20 ontstaan geleidelijk aan gespecialiseerde, alveolaire epitheelcellen die surfactant produceren, een detergentachtige vloeistof bestaande uit fosfolipiden die
Figuur 92 Alveolaire fase (Carlson, 20119)
zich als een film over de binnenste wand van die alveoli uitspreidt. Surfactant zorgt ervoor dat de oppervlaktespanning van de alveolen verlaagt waardoor zij beter kunnen uitzetten en niet dichtklappen bij uitademing.
Het natuurkundig principe hierachter is precies hetzelfde als bij het blazen van zeepbellen. Zonder toevoegen van een beetje detergent (bv. afwasmiddel) lukt het nauwelijks om bellen te blazen, of ze spatten heel snel uiteen.
Vanaf 35 weken is er doorgaans voldoende surfactant aanwezig om een efficiënte gasuitwisseling te bekomen. Bij prematuren (vooral < 35 weken) bestaat het risico dat een pasgeborene ernstige ademhalingsmoeilijkheden ontwikkelt (het respiratoire distress syndroom, zie 2e jaar)
Bij een dreigende vroeggeboorte wordt daarom longrijping toegediend (=cortisone IM) aan de zwangere om de productie van surfactant te bespoedigen.
12.5
Late foetale & postnatale fase
Voor de geboorte zijn de longen gevuld met vruchtwater. Wanneer de ademhaling start bij de geboorte wordt de grootste hoeveelheid vocht in de longen snel geresorbeerd door bloed- en lymfecapillairen. Wanneer de vloeistof uit de alveoli is geresorbeerd blijft het surfactant achter als een dun fosfolipidenlaagje op de alveolaire celmembranen.
Ook na de geboorte matureren de longen nog verder, onder andere door de bijkomende vorming en rijping van longblaasjes. Stabilisatie van het morfologisch patroon van de longen wordt pas bereikt op de leeftijd van 6 jaar.
13 Urinair stelsel
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De indeling van het intermediair mesoderm (nefrogene streng) te beschrijven.
De drie soorten primitieve nieren op te noemen, incl. het ogenblik waarop ze ontwikkelen (en eventueel verdwijnen).
De ontwikkeling van de definitieve nieren te bespreken.
Het vertakken van de ureterknop als een voorbeeld van oppervlaktevergroting te benoemen, net als de vertakking van de bronchiaalboom en de vorming van chorionvilli.
De rol van de Gang van Wolff (ductus mesonefricus) in de ontwikkeling van de urinewegen te beschrijven.
De rol van het urorectaal septum in het opdelen van de cloaca te bespreken
Embryologisch en anatomisch gezien zijn de urinewegen en het voortplantingsstelsel nauw met elkaar geassocieerd. De beide ontstaan uit het intermediaire mesoderm en tijdens de vroege ontwikkeling monden ze uit in een gemeenschappelijke holte, de cloaca, het verwijde deel van de einddarm.
13.1 Hoge urinewegen: nieren & ureter
Het urinair stelsel wordt afgeleid uit het bilaterale, intermediaire mesoderm, tussen het paraxiale en het zijplaatmesoderm (zie 8.3.1).
Tijdens week 4 vormt dit intermediaire mesoderm aan beide zijden van het embryo een langwerpige nefrogene streng die van craniaal naar caudaal groeit en waarin een lumen (centrale holte) ontstaat.
In deze segmenterende streng worden de nieren 3 keer aangelegd:
nefrotomen
Figuur 93 Intermediair mesoderm (schema). De stippelijnen geven de craniocaudale segmentering weer.
Van craniaal naar caudaal:
• Pronefros (voornier)
• Mesonefros (oernier)
• Metanefros (definitieve nier)
13.1.1 Pronefros (W4)
Deze primitieve (voor)nier ontstaat aan de kop van de nefrogene streng.
DEFINITIEVE NIER
Bij het begin van week 4 segmenteert deze streng hiervoor in kleine (niet-functionele) celgroepjes, de nefrotomen.
Er wordt een afvoergang gevormd die caudaal doorgroeit tot aan de cloaca.
Nog vóór het einde van week 4 verdwijnt de pronefros volledig, alsook het kopgedeelte van de afvoergang (zie Figuur 94).
Het resterende gedeelte van de afvoergang blijft bestaan en wordt de ductus mesonefricus of gang van Wolff genoemd. PRONEFROS MESONEFROS METANEFROS CRANIAAL
1
hartbult leverknop
d. vitellinus allantoïs
urogenitale sinus
voordarm (met kieuwzakjes)
PRONEFROS
MESONEFROS
d. mesonefricus (gang van Wolff)
METANEFROS
Het pronefros is al grotendeels verdwenen. (Mitchell, 2009)
13.1.2 Mesonefros (W4-8)
Deze tweede, tijdelijke nier ontstaat in het middengedeelte van de nefrogene streng. Ook hier vindt eerst een gelijkaardig proces van segmentatie plaats.
Hieruit ontstaan primitieve nefronen waarbij een kapsel van Bowman een glomerulair capillair omgeeft. Afvoer gebeurt via de gang van Wolff. De mesonefros is functioneel en werkt als interim-nier tot de definitieve nier is ontwikkeld.
2
dorsale aorta gang van Wolff mesenterium
3
nefrogene streng primitief nefron
4
kapsel van Bowman glomerulus gang van Wolff
Figuur 95 Vorming van het primitieve nefronen in het mesonefros, doorsnede (Mitchell, 2009)
Rond week 8 verdwijnt het mesonefros volledig bij de vrouw. Bij de man ontstaan hieruit nog de bijbal en de zaadleider (zie 14.1.3).
13.1.3 Metanefros (vanaf W5)
De definitieve nier ontwikkelt caudaal in de nefrogene streng uit een samenspel van twee structuren:
De ureterknop is een uitstulping van de gang van Wolff die de voorloper zal vormen van de ureter, het nierbekken, de nierkelken en de verzamelbuisjes. De ureterknop vertakt hiervoor ongeveer 15 keer (vergelijk met de vertakkingen van de bronchiaalboom, zie 12.3).
nierkelken (nierbekken)
gang van Wolff
ureterknop MNB
nierparenchym (met nefronen)
ureter
Figuur 96 Vertakkingen van de ureterknop (Mitchell, 2009)
Het metanefrogeen blasteem (MNB) ligt als een kapje om de ureterknop en zal aanleiding geven tot de vorming van de definitieve nefronen (kapsel van Bowman, proximale tubulus, lis van Henle, distale tubulus)
Tussen week 6 en 9 bewegen de definitieve nieren vervolgens opwaarts om terecht te komen ter hoogte van de bovenste ledenwervels.
indifferente gonade (zie 14.1.1)
metanefros
mesonefros
testis penis 1 2 3 bijbal
metanefros
gang van Wolff tuberculum genitale (zie 14.2)
Figuur 97 Opwaartse beweging van het metanefrogeen blasteem (definitieve nier), week 6, 7 en 8 – mannelijk embryo. (Carlson, 2019)
13.2 Lage urinewegen: blaas en urethra
In week 6 ontwikkelt zich een urorectaal septum dat de einddarm (cloaca) verdeelt in (dorsaal) het anorectaal kanaal en (anterieur) de urogenitale sinus. Uit deze endodermale verbreding ontstaan zowel blaas als urethra Eerst staat de urogenitale sinus nog in verbinding met de allantoïs (en navelstreng) De allantoïs verdwijnt uiteindelijk en vormt een bindweefselstreng, het lig. suspensorium vesicae.
genitale plooi (zie 14.1.1
NB notochord darm
uro-genitale sinus
d. vitellinus allantoïs
cloacale membraan
Figuur 98 Septering van de einddarm (cloaca) door het urorectaal septum. Doorsnede op 6 weken (Li) en 7 weken (Re) anorectaal kanaal
uro-rectectaal septum
De blaas ontstaat uit het craniale gedeelte van de urogenitale sinus. Opmerkelijk is dat de ureterknopjes hun eigen uitmondingen krijgen in de urogenitale sinus (zie Figuur 99). Met de groei van de blaas schuiven deze openingen mee naar craniaal en lateraal. De openingen van de gangen van Wolff blijven aan de basis van de urogenitale sinus en monden uit in de urethra De urethra ontstaat uit het meest caudale gedeelte van de urogenitale sinus.
lig.susp.vesicae
gang van Wolff
ureterknop
lig.susp.vesicae
ureter
ureter
achterwand van de blaas
gang van Wolff urethra
Figuur 99 Dorsaal zicht op de zich ontwikkelende blaas. Let vooral op hoe de ureters op een bepaald moment hun eigen uitmonding in de blaas krijgen (Carlson, 2019)
14 Genitaal stelsel
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De indifferente fase in de ontwikkeling van de interne en externe genitalia te beschrijven.
De ontwikkeling van de testes vanuit de indifferente gonade te bespreken.
De ontwikkeling van de ovaria vanuit de indifferente gonade te bespreken.
De rol van SRY te bespreken in de ontwikkeling van het embryo.
De hormonen op te lijsten die een rol spelen in het differentiëren van de interne gonaden tot mannelijk of vrouwelijk.
De rollen van de gang van Wolff en de gang van Müller te bespreken in de ontwikkeling van de interne gonaden bij de man en de vrouw.
De ontwikkeling van de ovaria te linken aan de oögenese (m.b. het vormen van primordiale follikels en het proces van folliculaire atresie).
Uterus didelphys en bicornis te kunnen verklaren vanuit de embryologie.
Het bestaan van ambigue genitalia te kunnen verklaren vanuit de embryologie.
Advies te geven aan de ouders van een pasgeboren jongen m.b.t. cryptorchidie.
Een vergelijking te maken van de ontwikkeling van de interne en externe genitalia bij man & vrouw.
14.1
Interne genitalia
Hoewel het geslacht van het embryo al bij de bevruchting wordt vastgelegd, ontwikkelen de geslachtsorganen zich de eerste 6 weken op exact dezelfde manier.
We heten dit de indifferente fase.
14.1.1 Indifferente fase (W5-6)
Primordiale kiemcellen (kiembaancellen) ontstaan vanuit de epiblast, maar na de gastrulatie (zie 8.1) komen ze via de primitiefstreep terecht in de wand van dooierzak.
In week 4-5 migreren ze terug naar het lichaam. Terwijl de kiemcellen delen (mitose) en in aantal toenemen, bewegen ze langs de darm en het dorsale mesenterium naar het intermediaire mesoderm van de achterste buikwand (zie de gebogen pijlen, Figuur 100).
Hierdoor worden aan beide zijden van de achterste buikwand de genitale plooien gevormd, de voorlopers van de interne geslachtsorganen (zie ook Figuur 98).
Deze genitale plooien ontwikkelen zich ventraal en mediaal van elk mesonefros.
Figuur 100 Vorming van de genitale plooien (Carlson, 2019) migratie van kiemcellen
De kiemcellen worden goed beschermd in de achterste buikholte:
Bij aankomst van de kiemcellen, vormt het oppervlakkige epitheel van de achterste buikholte een dikke beschermlaag: het mesotheel
Vanuit dit mesotheel vertrekken celstrengen in de diepte, tussen de primordiale kiemcellen: de primaire geslachtsstrengen. Hierin differentiëren ook steuncellen, die de kiemcellen gaan omhullen.
Aan het einde van de 6e week ontstaat een bijkomende afvoergang, lateraal van de gang van Wolff, nl. de ductus paramesonefricus (gang van Müller). Deze afvoergang ontstaat aan beide zijden van de genitale plooien, door invaginatie van het epitheel van de achterste buikwand.
Op dit moment is de indifferente gonade afgewerkt.
kiemcellen
steuncellen
primaire geslachtsstrengen
verdikt mesotheel
14.1.2
mesonefros
Figuur 101 Indifferente gonade (eind 6e week)
Het SRY-gen
lichaamswand
gang van Müller
gang van Wolff
De indifferente gonade zal verder ontwikkelen tot ovaria, tenzij het embryo een Ychromosoom bevat. Op dit Y-chromosoom ligt immers het belangrijkste geslachtsgen: SRY: Sex-determining-Region Y.
Dit gen codeert voor een transcriptiefactor die het volledige embryo doet evolueren in de mannelijke richting. Het gen werkt hierbij als een ‘hoofdschakelaar’, die de expressie van tientallen andere, geslachtsbepalende genen aanstuurt bv. voor biologische kenmerken als testisdifferentiatie en skeletale groei maar wellicht ook psychologische kenmerken als genderidentiteit (Mukherjee, 2017).
JAAR: 1989
Goodfellow ontdekt de rol van het SRY-gen door het bestuderen van vrouwen met het (erg zeldzame) Swyer-syndroom. Dit zijn vrouwen met karyotype 46, XY (=genetisch mannelijk).
Door een mutatie in hun SRY-gen ontwikkelen ze echter in elk opzicht als vrouw, ook al hebben ze een Y-chromosoom (Goodfellow & Lovell-Badge, 1993).
14.1.3 Vorming van de testes (♂ vanaf W7)
De testes ontstaan uit het merg van de indifferente gonaden.
kiemcellen + steuncellen
primaire geslachtsstrengen
tunica albuginea onstaat schors gang van Wolff
gang van Müller
Figuur 102 Ontwikkeling testes (Mitchell, 2005)
¨ De primaire geslachtsstrengen verlengen zich tot diep in het merg van de gonaden. Doordat een lumen ontstaat in de strengen vormen ze om tot (zaad-)buisjes. Ze vertakken in een netwerk van nog fijnere buisjes (rete testis) om uiteindelijk aan te sluiten op de restanten van het mesonefros en de gang van Wolff: hieruit ontstaan afvoerende zaadkanaaltjes, de bijbal (epididymis), zaadleider & zaadblaasjes.
rete testis
primaire geslachtsstrengen
kiemcellen + Sertoli-cellen
schors verdwijnt
gang van Wolff
Figuur 103 Ontwikkeling testes (Mitchell, 2005)
mesoderm (met Leydig-cellen)
tunica albuginea
¨ De kiemcellen worden in de wand van de zaadbuisjes opgenomen en omgeven door steuncellen. Deze steuncellen differentiëren tot Sertoli-cellen, die de mitose van de kiemcellen stilleggen (tot in de puberteit, zie 4.2). Deze cellen produceren ook een anti-Mülleriaanse factor (AMF), waardoor de gang van Müller verdwijnt.
¨ In het stromale mesoderm tussen de kanaaltjes ontstaan Leydig-cellen, die testosteron (het mannelijk geslachtshormoon produceren). Samen met de AMF zorgt testosteron voor het verdwijnen van de gang van Müller.
¨ Rond de testes wordt de tunica albuginea gevormd, een dikke, bindweefselige mantel. Vanhieruit groeien verschillende septa die teelbal in lobjes indelen. De schors van de gonade verdwijnt.
rete testis
zaadbuisjes
kiemcel
gang van Wolff
Sertoli-cel septa
tunica albuginea
Figuur 104 Ontwikkeling van de testes, met een detail van de zaadbuisjes (tubuli seminiferi), waarin vanaf de puberteit de spermatozoa ontwikkelen Leydig-cellen zijn niet afgebeeld (Mitchell, 2005).
¨ Tijdens de verder groei worden de testes ronder en komen ze meer en meer los van de achterste buikwand.
14.1.4 Ontwikkeling
van de ovaria (♀ vanaf W7)
De ovaria ontstaan hoofdzakelijk uit de schors van de indifferente gonaden.
kiemcellen + steuncellen
primaire geslachtsstrengen
gang van Wolff
gang van Müller
schors
¨ De primaire geslachtsstrengen verdwijnen geleidelijk aan.
¨ De kiemcellen vormen om tot oögonia en primaire oöcyten. Door mitose neemt hun aantal sterk toe (zie 4.3.2).
¨ De steuncellen (uit de schors) differentiëren tot follikelcellen die loskomen van de primaire geslachtsstrengen en de primaire oöcyten omsluiten. Hierdoor komen primordiale follikels tot stand De follikelcellen leggen de eerste meiotische deling stil tot de puberteit (=het dictyoteen) (zie 4.3.2).
¨ In het mesodermaal stroma rond de follikels ontstaan thecacellen, die tijdens de menstruele cyclus oestrogenen produceren. Door de afwezigheid van testosteron en AMF verdwijnt de gang van Wolff (& blijft de gang van Müller bestaan).
primaire geslachtsstrengen verdwijnen geleidelijk aan primordiale follikels
Figuur 106 Ontwikkeling overia
gang van Wolff
gang van Müller mesoderm (met theca-cellen)
¨ Vorming van de tunica albuginea
Deze bindweefselige mantel is veel dunner dan bij de testes en dient vooral om de primordiale follikels te scheiden van het mesotheel van de achterste buikwand.
¨ Loskomen van de buikwand
De ovaria komen gedeeltelijk los van de buikwand, maar er blijft steeds een verbinding bestaan (lig. suspensorium ovarii) waarlangs ook de bloedvaten lopen.
Figuur 105 Ontwikkeling ovaria (Mitchell, 2005)
primordiale follikels
primaire oöcyt follikel-cel
lig.suspensorium ovarii
gang van Wolff gang van Müller
Figuur 107 Ontwikkeling van de ovaria (primaire geslachtsstrengen zijn verdwenen), met een detail van de primordiale follikels. Thecacellen zijn niet afgebeeld (Mitchell, 2005).
14.1.5 Ontwikkeling van de afvoergangen
Bij de vrouw wordt de gang van Müller behouden en verdwijnt de gang van Wolff.
Bij de man wordt de gang van Wolff behouden en verdwijnt de gang van Müller.
Figuur 108 Ontwikkeling van de interne genitalia bij de vrouw en de man aan het begin van week 7, in vergelijking met de definitieve anatomie (onderaan links & rechts) (Carlson, 2019)
Bij de vrouw ontwikkelen de gangen van Müller ontwikkelen craniaal verder tot de beide eileiders (die via hun ostium in open verbinding staan met de buikholte).
De caudale delen van de gangen van Müller bewegen naar de middellijn om er te fusioneren tot de baarmoeder en het bovenste deel van de vagina.
Een verstoorde fusie van de gangen van Müller leidt tot malformaties aan de baarmoeder bv. uterus didelphis (ontdubbelde baarmoeder) of uterus bicornis (tweehoornige baarmoeder).
Dit kan een invloed hebben op vruchtbaarheid of leiden tot zwangerschapscomplicaties.
Bij de man komen de testes volledig los van de buikholte en dalen ze af, doorheen het lieskanaal, naar het scrotum. Dit proces vindt vrij laat plaats (maand 7), waardoor het bij de geboorte nog niet altijd voltooid is (zeker bij prematuren).
Als de testes niet aanwezig zijn in het scrotum bij de geboorte spreken we cryptorchidie
Indien ze tijdens de eerste 6 maanden niet spontaan indalen is verder onderzoek aangewezen.
Als we de ontwikkeling van de interne genitalia vergelijken bij man & vrouw – omstreeks de 5e maand – bekomen we onderstaand overzicht.
VOORLOPERSTRUCTUUR MAN
Indifferente gonade
Primordiale kiemcel
Steuncel
Stromale cel
Gang van Wolff
Testis
Spermatogonia
Sertoli-cel
Leydig-cel
Zaadkanaaltjes
VROUW
Ovarium
Primaire oöcyt
Follikel-cel
Theca-cel
Epididymis, Zaadleider, zaadblaasjes / Gang van Müller / Eileiders
Baarmoeder
Vagina
14.2 Externe genitalia
14.2.1
Indifferente fase (W4-9)
De uitwendige geslachtsorganen ontstaan door een complexe interactie tussen ecto- en mesodermale weefsels rond de cloaca. Net zoals de interne genitalia begint deze ontwikkeling met een indifferente fase.
Al in de 4e week ontstaan rond de cloacale opening de cloacale plooien. Lateraal hiervan vinden we de genitale wallen.
De cloacale plooien komen craniaal samen in het tuberculum genitale. Deze uitstulping wordt geleidelijk aan groter en wordt vanaf de 6e week de phallus genoemd.
Omstreeks datzelfde tijdstip heeft het urorectaal septum de urogenitale opening gescheiden van de anorectale opening. De plooien die langs de urogenitale opening liggen worden nu urethrale plooien genoemd.
tuberculum genitale
genitale plooien
genitale wallen
cloaca
urogenitale opening
anus
Figuur 109 Externe genitale (indifferente fase) (Lohman & Ten Donkelaar, 1999)
14.2.2 Verdere ontwikkeling bij de man (vanaf W9)
urthrale plooien phallus
anale plooien
Enkel in aanwezigheid van testosteron (geproduceerd door de bijnier en de Leydigcellen) ontwikkelen de externe genitalia tot de mannelijke vorm. Hierdoor zal phallus uitgroeien tot de penis.
Op het eind van de 3e maand vergroeien de urethrale plooien aan de onderzijde en ontstaan in de diepte rond de urethra de zwellichamen.
De urethrale groeve wordt dus aan de onderzijde, de overblijvende naad heet raphe. Tot slot wordt een nieuwe urethrale opening (meatus) gemaakt op de glans penis. Het preputium (de voorhuid) ontstaat uit een ectodermale plooi die over de glans penis groeit.
glans penis
urethrale groeve
genitale wal
perineum
anus
meatus
glans penis
raphe raphe
scrotum
perineum
anus
14.2.3 Verdere ontwikkeling bij de vrouw (vanaf W9)
Bij meisjes vormt de phallus de clitoris. De urethrale plooien ontwikkelen zich tot de labia minora (kleine schaamlippen) en de genitale wallen worden de labia majora (grote schaamlippen).
phallus
urethrale plooi
genitale wal
perineum
anus
urethra
vagina
hymen
clitoris
labium minus labium majus
Vanaf W12 kan het geslacht echografisch worden bepaald door observatie van de externe genitalia. ≈
Soms worden kinderen geboren met onduidelijk gedifferentieerde externe genitalia. Dit wordt omschreven als ambigue genitalia (of DSD, disorder of sex development).
Karyotypering is hierbij nodig om het geslacht van het kind te bepalen. DSD kan veel mogelijke oorzaken hebben, bv. een teveel aan testosteronproductie vanuit de bijnier, waardoor de externe (en interne) genitalia van een 46,XX-meisje (geheel of gedeeltelijk) in de mannelijke richting ontwikkelen.
Figuur 110 Externe genitalia bij de man (Lohman & Ten Donkelaar, 1999)
Figuur 111 Externe genitalia bij de vrouw (Lohman & Ten Donkelaar, 1999)
15 Tweelingen
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Aan te geven welke type tweeling het vaakst voorkomt (monozygoot of dizygoot).
Te verklaren hoe monozygote tweelingen ontstaan.
Te verklaren hoe Siamese tweelingen ontstaan.
De verschillende configuraties van placenta en vliezen bij tweelingen te bespreken.
Enkele argumenten te geven voor het nut van tweelingstudies.
15.1 Voorkomen
Wereldwijd komen tweelingen steeds vaker voor, dit door de grotere kans hierop bij een medisch geassisteerde bevruchting (IVF). België volgt die trend niet meer omdat in ons land slechts één embryo per IVF-cyclus wordt teruggeplaatst.
Het aantal natuurlijk verwekte tweelingen stijgt wel. 1/3e hiervan zijn monozygote (eeneiige) tweelingen en 2/3 dizygote (twee-eiige) tweelingen.
In Vlaanderen werden in het jaar 2019 981 tweelingen en 15 drielingen geboren.
Relatief gezien betekent dat dat 3,2% van de geboren kinderen tot de categorie meerlingen behoort (Kind In Vlaanderen, 2020).
15.2 Ontstaan
Dizygote tweelingen ontstaan na fertilisatie van twee eicellen. Dit veronderstelt dus een dubbele eisprong (geen dubbele bevruchting! Zie 5.3)
Monozygote tweelingen ontstaan door het splitsen van het vroege embryo in twee genetisch identieke delen. Vermoed wordt dat deze splitsing meestal plaatsvindt in de embryoblast (blastocyst-stadium).
De splitsing is wellicht het gevolg van verstoorde communicatie tussen de cellen. Hierdoor gaat het ‘overzicht’ verloren en beginnen 2 of meer celgroepjes zich te gedragen als een afzonderlijk embryo
Er is een familiaal voorkomen van tweelingzwangerschap, vooral langs de zijde van de moeder. Een hypothese hiervoor is dat variaties in genen die de oöcyt tot expressie brengt, hiertoe kunnen bijdragen. Vaders brengen deze genen niet tot expressie in hun gameten, maar kunnen ze wel doorgeven aan hun dochters.(Hoekstra et al., 2008; Moore et al., 2004).
15.3 Tweelingstudies
Vanuit het perspectief van genetisch onderzoek, zijn monozygote tweelingen erg interessant. Hun genotype is immers identiek. Eventuele wijzigingen in het fenotype zijn dus steeds het gevolg van omgevingsfactoren.
De term concordantie (in %) wordt hierbij gebruikt om het aantal tweelingen te beschrijven dat hetzelfde fenotype (bv. ziekte als multiple sclerose) ontwikkelt. Hoe lager de concordantie, hoe groter de invloed van omgevingsfactoren.
De vingerafdrukken van een monozygote tweeling zijn nooit volledig identiek. Ze kunnen wel erg op elkaar gelijken omdat ze deels genetisch bepaald worden.
15.4
Configuraties van placenta en vliezen
Bij meerlingenzwangerschappen zijn verschillende configuraties mogelijk van de placenta en vliezen.
Een dizygote tweeling ontwikkelt volledig autonoom. Zowel amnion, chorion als placenta zijn van elkaar te onderscheiden, hoewel de placenta's soms later in de zwangerschap vergroeien.
Bij een monozygote tweeling hangt de samenstelling van placenta en vliezen af van het tijdstip waarop de splitsing is gebeurd:
splitsing embryoblast
splitsing kiemschijf
Figuur 112 Configuratie van placenten en vliezen bij tweelingen (Mitchell, 2009)
§ Bij een vroege splitsing (D1-4, in één derde van de gevallen), wanneer chorion en amnion nog niet zijn gevormd, zal de placenta van de tweeling gelijk zijn aan de placenta van twee-eiige tweelingen.
§ Bij een intermediaire splitsing, (D4-7, in twee derde van de gevallen), zal de placenta van het type zijn met één chorion en twee amnions.
§ Bij een late splitsing, (na D8 2 à 3% van de gevallen) is de placenta van het type met één chorion en één amnion. Dit is steeds het geval bij Siamese tweelingen.
De term 'Siamese tweelingen' verwijst naar Siam, het vroegere Thailand, de geboorteplaats van de (levenslang) aan het sternum verbonden tweeling 1874). Als curiosum trokken ze eerst het ganse land en later de ganse oor verdienden ze een aardige cent en konden een plantage oprichten in North . Ze trouwden met twee zussen envermits beide broers een eigen huis hadden wisselden ze om de drie dagen van woning. Chang kreeg 10 kinderen, Eng 11.
De Duitse professor Virchow berekende de te volgen scheidingslijn waarlangs de tweeling zou moeten worden gescheiden wanneer één van hen overleed. Toen Cheng vrij plots stierf aan de gevolgen van een CVA verzocht Eng een arts om deze scheiding uit te voeren. Zover is het echter nooit kunnen komen want enkele uren later stierf Eng ook, vermoedelijk door diffuse intravasculaire stolling die optrad na de dood van Chang.
Intermezzo: de foetale periode
De foetale periode begint vanaf de derde maand (week 9). Vanaf dat tijdstip is de organogenese grotendeels voorbij en breekt een periode van groei en rijping aan. Hieronder wordt enkel een beknopt overzicht gegeven. Dit komt immers uitgebreid aan bod in andere cursussen.
Een normale zwangerschapsduur bij de mens bedraagt 38 weken (266 dagen) van bevruchting tot geboorte of 40 weken (280 dagen) na de laatste menses. In de praktijk spreekt men echter al van een voldragen zwangerschap (of à terme geboorte) vanaf 37 (volledige) weken na de laatste menses.
Vroeger werd de foetale periode (vanaf week 9) nogal eens als minder belangrijk bestempeld omdat alle organen toch al min of meer waren aangelegd. Ondertussen weten we echter dat een correcte foetale groei- en gewichtstoename bijdragen tot een normale ontwikkeling van de verschillende orgaanfuncties. Kinderen die intra-uteriene groeivertraging ondergingen (=IUGR of intra-uteriene groei-restrictie), hebben om die reden een hoger risico om later een hoge bloeddruk of diabetes te ontwikkelen.
Tijdens de derde, vierde en vijfde maand treedt er voornamelijk lengtegroei op. De gewichtstoename gebeurt vooral in de laatste twee maanden van de zwangerschap.
Opvallend tijdens de foetale periode is dat eerst de lengte van het hoofd proportioneel sterk uitgesproken is en dat dit in het verder verloop van de zwangerschap relatief minder groeit. Zo is aan het begin van de derde maand de lengte van het hoofd ongeveer de helft van de CRL (= kruin-stuitlengte), aan het begin van de vijfde maand ongeveer 1/3de en bij de geboorte ongeveer 1/4de van de kruin-hiellengte.
§ Veranderingen tijdens de derde maand:
Het gezicht krijgt een menselijke aanblik. De oren krijgen hun definitieve positie.
De ogen komen aan de voorzijde van het gezicht te liggen. De extremiteiten gaan zich in proportie ontwikkelen tot de rest van het lichaam. Daarnaast kunnen op het einde van de derde maand reflexen worden opgewekt bij de foetus. Dit wijst op spieractiviteit.
§ Veranderingen tijdens de vierde maand:
In deze periode komt de lengtegroei snel tot uiting terwijl het gewicht weinig toeneemt. In de helft van de zwangerschap bedraagt het gewicht nog geen zesde van het geboortegewicht. De lengte bedraagt op dit moment al de helft. De foetus is bedekt met lanugohaartjes. Wenkbrauwen en hoofdbeharing zijn aanwezig. De foetus beweegt heftiger, de moeder voelt deze bewegingen.
§ Ontwikkeling tijdens de laatste maanden:
Het gewicht van de foetus neemt toe en de orgaanstelsels (o.a. het centraal zenuwstelsel en de longen) rijpen verder uit.
In de zesde maand vertoont de foetus nog een rimpelig uitzicht door de afwezigheid van onderhuids vetweefsel, doch naarmate de zwangerschap vordert wordt dit onderhuids vetweefsel meer gevormd en krijgt de foetus een voller uitzicht.
Naar het einde van de zwangerschap toe wordt de huid bedekt met een witachtig vet, de vernix caseosa (afgescheiden door de talgklieren).
Bij de geboorte heeft het hoofd nog steeds de grootste diameter; Het gemiddelde geboortegewicht bedraagt tussen 3 en 3,5kg. De gemiddelde lengte bedraagt 50cm.
“I think”, krabbelde Charles Darwin in 1837 boven een tekening in zijn notities. Na het bezoeken van de Galapagoseilanden kwam hij tot de vaststelling dat op elk van de eilanden een andere vinkensoort leefde, die bovendien aangepast waren aan het voedselaanbod op dat eiland. Zo kwam hij tot de hypothese dat deze vinkensoorten ontstaan waren uit een gemeenschappelijke vooroudervink, als vertakkingen van een boom. Zijn tekening geeft dit proces visueel weer. Dit idee was revolutionair, aangezien men totdantoe aannam dat de natuur onveranderlijk was en alle diersoorten in hun definitieve vorm geschapen waren door God.
16 Mutaties
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Toe te lichten waarom een mutatie niet noodzakelijk iets ‘negatief’ is.
Over mutaties te spreken met de termen ‘wild-type’ allel en ‘mutant’ allel.
De termen heterozygoot en homozygoot toe te lichten.
De verschillende soorten mutaties te bespreken.
De verschillende oorzaken van mutaties te bespreken.
Een voorbeeld te geven van een mutatie-hotspot.
Enkele maatregelen op te noemen, ter preventie van geïnduceerde mutaties
De mogelijke gevolgen van een mutatie te bespreken, zowel op vlak van eiwitfunctie, fenotype als de gevolgen binnen de populatie.
Bij een gegeven basesequentie (met mutatie) te kunnen aangeven over welke soort mutatie het gaat (deletie, insertie, inversie of duplicatie).
Bij een gegeven basesequentie (met mutatie) te kunnen aangeven over welke type mutatie het gaat (synonieme mutatie, vervangingsmutatie, stopmutatie, doorleesmutatie of frameshift-mutatie).
Het ‘dominant’ of ‘recessieve’ gedrag van een allel kunnen verklaren vanuit de functie van het eiwit waarvoor het gen codeert.
Een voorbeeld kunnen geven van hoe omgevingsfactoren die het effect van een mutatie op het fenotype kunnen beïnvloeden.
Een voorbeeld kunnen geven van hoe omgevingsfactoren die het voorkomen van bepaalde mutaties in een populatie kunnen beïnvloeden.
Bij een genetische eigenschap of aandoening kunnen argumenteren waarom dit de biological fitness van het organisme verlaagt of verhoogt.
Kort toe te lichten wat het belang is van mutaties in de evolutie der soorten (evolutieleer van Darwin)
Te beschrijven hoe forensisch DNA-onderzoek tot daderidentificatie of ouderschapsbepaling kan leiden.
16.1 Definities
16.1.1 Mutatie
Bij het woord mutatie denken de meeste mensen aan een ‘fout’ in het DNA, maar eigenlijk klopt dit niet. Een DNA-sequentie kan immers niet als ‘juist’ of ‘fout’ worden bestempeld. Er bestaan enkel variaties in de code, met een verschillende mate aan voorkomen.
Het vertonen van variatie is een belangrijke eigenschap van DNA. De term ‘mutatie’ is afgeleid van het Latijnse mutare wat verandering betekent.
Een mutatie is een variatie in de genetische code.
16.1.2 Allel
Hoewel elk mens in het bezit is van de 20.687 genen die ons ‘mens’ maken, blijken er –tussen mensen onderling – toch variaties te bestaan in de DNA-code voor heel wat van die genen. We weten dit door het genoom van duizenden mensen met elkaar te vergelijken (zie Human Genome Project, 2.8).
Elke mogelijke variant van eenzelfde gen heten we een allel. Allelen ontstaan door mutaties in de DNA-code.
Aangezien de mens diploïd is, hebben we op elke locus plaats voor 2 allelen.
16.1.3 Homo- en heterozygoot
Als een individu voor een bepaalde locus eenzelfde allel heeft dan is die persoon homozygoot voor dat gen. Als de beide allelen verschillend zijn (ook al is dat maar 1 bp) dan is die persoon heterozygoot voor dat gen.
HOMOZYGOOT HETEROZYGOOT HEMIZYGOOT Figuur 114
Een bijzonder geval zijn genen op het X en Y-chromosoom bij de man. Aangezien de man slechts één X en Y-chromosoom heeft zijn de termen homo- of heterozygoot daar niet van toepassing. We gebruiken hier de term hemizygoot.
Figuur 115 Een fictief monomorf vs polymorf gen met telkens 5 allelen en hun frequentie van voorkomen in een populatie
We onderscheiden:
Monomorfe genen: genen waarvan weinig verschillende allelen in omloop zijn. Er is één allel dat héél frequent is en andere allelen zijn zeldzaam in de populatie. Het vaak voorkomend allel heten we het 'wild-type'. De zeldzame varianten heten we 'mutanten'.
Merk op dat bestudeerde populatie en omgeving hierbij ook een rol spelen. Zo zal iemand met een allel voor blond haar in Scandinavië niet zo erg opvallen (‘allel voor blond’= vaak voorkomend = wild type). In Centraal-Afrika daarentegen is deze eigenschap niet alledaags (‘allel voor blond’ = zeldzaam = mutant).
Polymorfe genen: genen waarbij veel genetische variaties in omloop zijn (talrijke allelen komen bij >1% van de populatie voor). Hier is het moeilijker om te bepalen wat het ‘wild type’ is en wat de ‘mutanten’ zijn. We spreken hier ook van een polymorfisme.
Eén derde van de genen bij de mens zijn polymorf, bv. het globine-gen (hemoglobine), de 3 allelen (A, B, O) die de bloedgroep bepalen, …
GENOTYPE
De term genotype (zie deel I) wordt vaak gebruikt om te beschrijven over welke allelen iemand beschikt voor een bepaald gen.
16.2 Soorten mutaties
16.2.1
Met soort bedoelen we hier de aard van de wijziging in het DNA. We hebben het hier enkel over structurele wijzigingen in de DNA-code zelf. Wijzigingen in het aantal chromosomen (aneuploïdie bv. trisomie 21) komen aan bod vanaf 20.3
Puntmutatie - SN(i)P
De meest voorkomende mutatie is deze waarbij één base (één nucleotide) vervangen wordt door een andere. Dit wordt beschreven als basepaar-substitutie, puntmutatie of single nucleotide polymorphism (SNiP – spreek uit ‘snip’)
CTAGATGACT
CTAGAGGACT
Figuur 116
m.o.: bij een SNiP wordt het complementaire base (in de niet-coderende) streng natuurlijk ook gewijzigd. Bij het analyseren van mutaties wordt doorgaans enkel de coderende streng (of de mRNA-code) besproken. SNiPs zijn de vaakst voorkomende mutaties. Ze spelen een belangrijke rol in de genetische variatie tussen mensen.
16.2.2 Deletie
één of meerdere baseparen gaan verloren.
CTAGATGACT
CTAGATACT
Figuur 117
Een microdeletie is een deletie van < 5.000.000 bp (=5 Mb). Verschillende genen kunnen hierbij verloren gaan.
De term microdeletie is wat misleidend, aangezien het voorzetsel ‘micro-’ onterecht lijkt voor een verlies van miljoenen baseparen. De term verwijst echter naar het feit dat deze deleties niet zichtbaar via karyotypering met de lichtmicroscoop (zie Figuur 6).
In 20.6 bespreken we enkele syndromen die het gevolg zijn van microdeleties bij de mens, incl. de manier waarop de diagnose kan gesteld worden. "
16.2.3 Insertie
16.2.4 Duplicatie
één of meerdere baseparen worden toegevoegd.
118
16.2.5 Inversie
één of meerdere baseparen worden herhaald.
CTAGATGACT CTAGATGATGACT bvb. x 2
119
een groepje baseparen wordt omgedraaid.
120
Figuur
Figuur
Figuur
16.2.7
Translocatie
een deel van een chromosoom wordt uitgewisseld met een (niet-homoloog) chromosoom.
Een translocatie is een ingrijpende mutatie, waarbij soms wat erfelijk materiaal verloren gaat of gedupliceerd wordt. Men spreekt dan van een ongebalanceerde translocatie –dit in tegenstelling tot een gebalanceerde translocatie, waarbij de hoeveelheid erfelijk materiaal gelijk blijft.
Een bekend voorbeeld van een gebalanceerde translocatie is de zgn. Philadelphiatranslocatie waarbij een uitwisseling gebeurt tussen de lange armen (q) van de chromosomen 9 en 22. Dragers van deze translocatie hebben een hogere kans op leukemie (kanker van de witte bloedcellen).
De Philadelphia-translocatie was de eerste mutatie waarbij een verband aangetoond werd met het ontstaan van kanker.
Figuur 121
Figuur 122 Het Philadelphia-chromosoom (Snusdats & Simmons, 2011)
16.4 Oorzaken
van
mutaties
16.4.1
Spontane mutaties
De meeste mutaties gebeuren spontaan. De complexiteit van DNA en het grote aantal celdelingen dat plaatsvindt tijdens een mensenleven zorgen onvermijdelijk voor foutjes tijdens het kopiëren. De meeste mutaties treden dus willekeurig en toevallig op. 2 opmerkingen hierbij:
Deze spontane mutaties gebeuren vaker op plaatsen waar basesequenties zich herhalen; per twee (bv. CGCGCG), per drie (=triplet repeats bv. CAACAACAA) per vier etc… Deze ‘stotterfrequenties’ zijn belangrijke mutatie-hotspots.
Ondanks het frequent optreden van spontane mutaties is de DNA-replicatie toch vrij accuraat. Dit komt door DNA-repair, een systeem van herstellende enzymen die in elke cel actief zijn. Ze herkennen foute basen, knippen ze eruit en herstellen het DNA. Op deze manier geraakt 99,9% van de fouten gecorrigeerd Bekende repairgenen zijn BRCA1 en BRCA2 (de ‘breast-cancer-genes, 20.7). Naarmate we ouder worden neemt de kwaliteit van DNA-repair af.
16.4.2 Geïnduceerde mutaties
Er zijn heel wat omgevingsfactoren bekend die de kans op mutaties verhogen. Dergelijke factoren heten we mutagenen. Bekende mutagenen zijn bijvoorbeeld:
Tabaksrook
Bepaalde geneesmiddelen (vb. kankertherapie)
Straling, bv. ultraviolette (UV, zonlicht) of ioniserende straling (vb. radioactiviteit)
Sommige virussen (vb. HPV) of bacteriën (vb. Helicobacter pylori)
16.4.3 Kiembaanmutaties
§ Een somatische mutatie is een mutatie die aanwezig is in de lichaamscellen bv. een verworven mutatie o.i.v. van mutagenen (bv. longkanker door roken).
o Deze mutaties zijn niet overerfbaar.
§ Een kiembaanmutatie is een mutatie die aanwezig is in de kiembaancellen (de primordiale kiemcellen, voorlopers van zaadcellen en eicellen).
o Deze mutatie is overerfbaar en zal in alle lichaamscellen van het nageslacht aanwezig zijn. Dit betekent dat je kan geboren worden met een mutatie die niet bij jou ontstaan is, maar in een vorige generatie.
16.5 Gevolgen van mutaties
Een mutatie op zichzelf veroorzaakt geen ziekte. Zoals eerder gesteld is een mutatie louter een variatie in de genetische code.
Een mutatie kan wel leiden tot ziekte, bv. doordat de functie van een bepaald eiwit erdoor verstoord geraakt. Heel wat ziekten bij de mens worden veroorzaakt door een mutatie in één enkel gen, vaak in één enkel basepaar.
Als je de voorbije hoofdstukken goed begrijpt en het centrale dogma 'DNA maakt RNA maakt eiwit' (zie 2.6) indachtig bent, kun je al voor een groot deel zelf afleiden wat mogelijke gevolgen zijn van veranderingen in het DNA.
16.5.1 Gevolgen op eiwitfunctie
Een mutatie leidt enkel tot een gewijzigde eiwitfunctie als de mutatie …
… in het exon van een gen plaatsvindt
… niet gecorrigeerd wordt door DNA-repair
… leidt tot de selectie van een ander aminozuur tijdens de transcriptie
Gewijzigde eiwitfunctie betekent:
Loss of function: de functie van het eiwit gaat geheel of gedeeltelijk verloren
Gain of function: de functie van het eiwit neemt toe of wijzigt
LOSS OF FUNCTION
GAIN OF FUNCTION NORMALE FUNCTIE
Figuur 123 Door een mutatie kan de eiwitfunctie wijzigen. In het midden zie je hoe het normale eiwit eruitziet (5 subunits). (Li) door loss of function wordt het eiwit niet correct samengesteld. Hierdoor gaat de functie verloren (Re) door gain of function wordt het eiwit afwijkend samengesteld. Hierdoor neemt de functie toe.
16.5.1.1 TOEPASSING mutaties in een exon
Oefening 7.
We beschouwen een fictief gen, het MINI-gen (MutatIoNs are Interesting) dat codeert voor een piepklein eiwit, nl. een peptide van 6 aminozuren. Dit fictieve peptide speelt een rol in het versterken van de aandacht tijdens de les genetica ;-)
Het wild-type MINI-gen: ATGGTGCACCTGACTCCTTGA
AZ Met Val His Leu Thr Pro STOP
*niet-coderende streng
OPDRACHT:
Hieronder krijg je 5 mutante allelen van het MINI-gen voorgeschoteld.
• Vul voor elk allel de DNA/mRNA/AZ-sequentie aan in de tabel
Maak hiervoor gebruik van Figuur 17 of van de webtool (zie QR-code of https://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm)
o Elk mutant allel vertegenwoordigd één type mutatie uit onderstaande lijst. Vul onder de tabel telkens aan over welk type mutatie het gaat. Met type bedoelen we het effect op de transcriptie (itt. Het soort mutatie zie 16.2)
• Synonieme mutatie
Deze mutatie leidt tot de selectie van hetzelfde aminozuur. De mutatie heeft dus geen effect op de eiwitstructuur of -functie.
• Vervangingsmutatie
Deze mutatie leidt tot de selectie van een ander aminozuur. De mutatie heeft dus wel een effect op de eiwitstructuur of -functie.
• Stopmutatie
Deze mutatie leidt tot het vroegtijdig afbreken van de eiwitsynthese omdat een prematuur stopcodon wordt ingevoegd.
• Doorleesmutatie
Deze mutatie leidt tot het laattijdig afbreken van de eiwitsynthese omdat het oorspronkelijke stopcodon verloren gaat.
• Frameshift-mutatie
Bij deze mutatie wordt het volledige leesraam (frame) verschoven. Het eiwit zal hierdoor zeer sterk afwijken van het origineel.
Mutant allel n°1: ATGGTGCACTAGACTCCTTGA
Hierbij is een mutatie opgetreden op positie 10 en 11.
DNA ATG GTG CAC TAG ACT CCT TGA
DNA* TAC CAC GTG ATC TGA GGA GGA
mRNA AZ
*niet-coderende streng
Besluit: de mutatie in mutant allel n°1 is een ____________________________mutatie.
Mutant allel n°2: ATGGTGCACCTAACTCCTTGA
Hierbij is een puntmutatie opgetreden op positie 12.
DNA ATG GTG CAC CTA ACT CCT TGA
DNA* TAC CAC GTG GAT TGA GGA GGA
mRNA
AZ
*niet-coderende streng
Besluit: de mutatie in mutant allel n°2 is een ____________________________mutatie.
Mutant allel n°3: ATGGTGCACCTGACTGCTTGA
Hierbij is een puntmutatie opgetreden op positie 16.
DNA
DNA* mRNA AZ
*niet-coderende streng
Besluit: de mutatie in mutant allel n°3 is een ____________________________mutatie.
Mutant allel n°4: ATGGTGCACCTGACTCCTTG G
Hierbij is een puntmutatie opgetreden op positie 21.
DNA
DNA*
*niet-coderende streng
Besluit: de mutatie in mutant allel n°4 is een ____________________________mutatie.
Mutant allel n°5: ATGGCTGCACCTGACTCCTTGA
Hierbij is een insertie van één base opgetreden op positie 5.
DNA
*niet-coderende streng
Besluit: de mutatie in mutant allel n°5 is een ____________________________mutatie.
Na het maken van deze oefening komen we tot de vaststelling dat er verschillende manieren zijn waarop de bouw van het MINI-eiwit kan worden verstoord.
Als we ervanuit gaan dat deze mutaties altijd leiden tot ‘loss of function’ zou dit betekenen dat een student die een mutant allel bezit minder goed kan opletten tijdens de les genetica. Maar is dat wel altijd zo?
In de volgende paragraaf bekijken we het effect op het organisme, of – anders gezegd –het effect van een mutatie in het genotype op het fenotype (zie 2.6 voor een definitie van deze termen).
16.5.2
Op het fenotype
GENOTYPE “mutatie”
FENOTYPE “gestoorde functie?”
Of een mutatie in het genotype een effect heeft op het fenotype hangt af van:
16.5.2.1 Het weefsel waarin deze mutatie optreedt
16.5.2.2 Het aantal cellen waarin deze mutatie optreedt
16.5.2.3 De rol die het tweede allel speelt
16.5.2.4 Omgevingsfactoren
Figuur 124 Overzicht van de factoren die bepalen of het genotype een impact heeft op het fenotype
16.5.2.1 Het weefsel waarin de mutatie optreedt
Mutaties komen enkel tot uiting in het fenotype als de genen waarin ze zich bevinden tot expressie worden gebracht (zie ook 2.7)
Bv. het HBB-gen dat codeert voor het b-globine (in hemoglobine, zie 2.4) komt enkel tot uiting in cellen van het beenmerg. Alle andere cellen van het lichaam bezitten echter ook dit gen. Stel dat dit gen muteert in een huidcel, dan heeft dit geen enkel gevolg op het fenotype, want in een huidcel komt dit gen niet tot expressie.
16.5.2.2 Het aantal cellen waarin deze mutatie optreedt
Mutaties komen enkel tot uiting in het fenotype als voldoende cellen de mutatie tot expressie brengen. Overgeërfde mutaties komen in alle cellen voor.
Bv. als een mutatie optreedt in het HBB-gen van één cel in het beenmerg zal dat geen effect hebben op het fenotype. Er zijn immers voldoende, andere cellen met een nl. genotype. Wie echter geboren wordt met een mutatie in het HBB-gen (=van alle beenmergcellen) zal er niet in slagen om een ‘correct’ hemoglobine te maken.
16.5.2.3 De rol van het tweede allel
Mutaties komen enkel tot uiting in het fenotype bij afwezigheid van een dominant, wildtype allel.
Doordat ons genoom diploïd, hebben voor elk gen 2 allelen (zie 1.5). Het voordeel daaraan is dat we – zelfs bij de aanwezigheid van één mutant allel – nog een ‘back-up’ hebben. Toch slaagt dat andere, wild r niet altijd in om het fenotype te ‘redden’.
Beschouw een heterozygoot genotype:
Enkel het wild-type allel kan gebruikt worden om een eiwit te synthetiseren. Hierdoor valt de productie van dat eiwit op de helft (50%).
• Voor de meeste eiwitten zoals transportkanalen is 50% voldoende. Het lichaam heeft er immers geen grote hoeveelheden nodig. Zolang er nog één wildtype allel aanwezig is, treden er geen problemen op
Þ NORMAAL fenotype dragerschap voor het mutante allel.
Þ Het wild-type allel is want het bepaalt het fenotype.
Þ Het mutante allel is want het komt niet tot uiting.
• Voor structuureiwitten is een productie van 50% te weinig. We heten dit haploinsufficiëntie. Ons lichaam heeft bv. massieve hoeveelheden collageen en elastine nodig, bouwstenen van spieren, botten, bindweefsels etc... Wanneer één allel voor deze eiwitten defect raakt, treden er al grote problemen op.
Þ AFWIJKEND fenotype, met dragerschap voor het wild-type allel.
Þ Het mutante allel is dominant want het bepaalt het fenotype.
Þ Het wild-type allel is recessief want het komt niet tot uiting.
In het volgende hoofdstuk komen we uitgebreid terug op dominantie en recessiviteit Mendel ontdekte deze eigenschappen al en suggereerde dat er een soort ‘interactie’ tussen de allelen plaatsvond, waarbij één allel een ander kon onderdrukken. Ondertussen weten we dat die interactie niet bestaat. Het dominant of recessief zijn komt voort uit de functie van het eiwit zèlf, en of een productie van 50% voldoende is voor het organisme.
Figuur 125 Heterozygoot genotype
eiwit
mutant allel
16.5.2.4
Omgevingsfactoren
Verrassend genoeg zijn er ook omgevingsfactoren die kunnen bepalen of een mutatie een invloed heeft op het fenotype (zie ook 19.2 multifactoriële overerving).
Zo is elke mens in het bezit van 2 mutante allelen van een gen dat normaal gezien codeert voor een enzym dat helpt bij de synthese van vitamine C (ascorbinezuur). Bij de mens is dit gen echter zodanig gemuteerd (meer dan helft van de exonen ontbreken) dat het niet meer werkt. Het wordt beschouwd als een pseudo-gen.
Een pseudo-gen is een DNA-sequentie die ‘lijkt’ op een gen (meestal een evolutionaire restant is van een gen), maar niet meer functioneel is.
Doordat beide allelen defect zijn, zijn wij als mensen verplicht om vit C met de voeding in te nemen (fruit dus) om scheurbuik te vermijden, een aandoening die veroorzaakt wordt door vit C-tekort. Dit illustreert dat een omgevingsfactor (voeding) in staat is om het fenotype dat past bij een mutatie te verhinderen.
Enkel bij de mens, de mensapen, de vleermuis en de cavia is dit gen stuk. Alle andere dieren hoeven geen fruit te eten om aan hun vitamine C te komen (Drouin et al., 2011).
16.5.4
Populatie-effecten
16.5.4.1 Biological fitness
We kunnen ook de bedenking maken of de mutatie een impact heeft op de overlevingskansen van het organisme in de omgeving.
Sommige mutaties zullen immers het organisme bevoordelen en zorgen dat er meer nakomelingen kunnen geproduceerd worden. Als deze mutatie via de geslachtscellen wordt doorgegeven naar het nageslacht zal ze steeds vaker voorkomen.
Biological fitness: de geschiktheid om binnen een bepaalde omgeving nakomelingen te produceren, waaraan de eigenschappen worden doorgegeven.
Organismen met de hoogste biological fitness zullen het meeste nakomelingen produceren, waardoor hun eigenschappen steeds vaker zullen voorkomen. Darwin heette dit survival of the fittest, of natuurlijke selectie.
Andere mutaties zullen net leiden tot een lagere biological fitness en vroegtijdig overlijden bv. homozygoten voor sikkelcelanemie.
In W.-Europa lijden heel wat mensen aan hemochromatose, een milde aandoening waarbij opstapeling van ijzer plaatsvindt in het lichaam. In de Middeleeuwen bood deze ziekte een verhoogde weerstand tegen de pest, waardoor de overlevingskansen van patiënten toenamen. Op termijn heeft dit gezorgd voor het vaker voorkomen van de aandoening.
16.5.4.2 Genetische diversiteit
Het optreden van spontane mutaties is geen gebrek aan het DNA, maar een bewuste eigenschap die invloed heeft op de ganse populatie. Het feit dat elk menselijk genoom beladen is met unieke SNiPs wijst erop dat het DNA a.h.w. voortdurend ‘experimenteert’ om te zien of er voordeel kan uit gehaald worden.
Genetische diversiteit in een DNA-dragend organisme is onvermijdelijk en vormt een drijvende kracht achter de evolutie van de soorten op aarde.
De keerzijde is natuurlijk dat deze mutaties ook verantwoordelijk zijn voor het bestaan van erfelijke aandoeningen en kankers.
Let wel: hoewel elk menselijk genoom uniek is (tenzij bij monozygote tweelingen), worden de verschillen doorgaans overschat. Zelfs tussen mensen met een andere etnische afkomst, verschilt het genoom maximaal 0,01%. In dat opzicht is de menselijke soort (Homo sapiens) er een met opvallend lage diversiteit.
De Homo sapiens is de enige soort binnen het geslacht ‘Homo’ (Ter vergelijking: er zijn 350.000 soorten kevers). Ooit waren er meer mensensoorten bv. Homo neanderthalensis (Neanderthaler), een uitgestorven mensensoort waarmee wij, de H. sapiens, een gemeenschappelijke voorouder hebben. Uit het Neanderthal Genome Project – gestart in 2006 - is gebleken dat de moderne mens gemiddeld 1 à 4% Neanderthal-DNA bezit, wat erop wijst dat de soorten ooit samenleefden. De Zweed Svante Päabo kreeg in 2022 de Nobelprijs Geneeskunde voor zijn onderzoek aan het Neanderthal Genome Project.
16.5.4.3 Genetische flessenhals
De lage, genetische diversiteit van de moderne mens vindt zijn verklaring in zgn. populatie-flessenhalzen. In de Oudheid zijn er verschillende momenten geweest waarop de menselijke populatie heel sterk daalde (bv. door ziekte of natuurkrachten). Hierdoor stammen wij allemaal af van een hele kleine populatie van slechts enkele honderden individuen uit de soort H. sapiens Hun genen circuleren nog steeds in onze populatie.
Dit soort flessenhalzen treedt ook op als deel van de populatie wordt afgezonderd van de rest van de genenpoel (genetische isolatie). Dit gebeurt bv. door aardrijkskundige grenzen (bv. zeeën, bergen) waarlangs genen zich niet verder kunnen verspreiden. Genetisch isolement leidt dikwijls tot het frequenter voorkomen van bepaalde aandoeningen in die bevolkingsgroepen.
Een opvallend voorbeeld van genetische vinden we op Pingelap, een eilandje in de Stille Zuidzee waar slechts enkele honderden mensen wonen. 10% van de bevolking lijdt er aan de zeldzame, erfelijke ziekte achromatopsie (volledige kleurenblindheid). In de rest van de wereld komt deze ziekte haast niet voor. De verklaring is de volgende: in 1775 stierf het grootste deel van de eilandbevolking tijdens een tyfoon. Bij de 20 overlevenden (flessenhals) waren er enkele dragers van achromatopsie (Sundin et al., 2000)
16.6 Forensische genetica
16.6.1
DNA-fingerprint
Sinds de jaren ’80 wordt DNA gebruikt om daders van een misdrijf te identificeren.
Dit is mogelijk omwille van de vele spontane, mutaties die optreden in het junk DNA (zie 2.8), vnl. ter hoogte van herhalingsfrequenties (de zgn. mutaties hot-spots, zie 16.4.1).
Het patroon van die mutaties is bij elke mens uniek.
Concreet kijkt men naar 12 short tandem repeats (STR) in het niet coderend DNA, waar kleine, herhalingen van 2 tot 9 bp voorkomen, bv.
Figuur 126 Voorbeeld van een STR (short tandem repeat). Er zijn honderden van deze loci in het menselijk genoom. Voor DNA-onderzoek worden er slechts 12 gebruikt.
STRs worden heel vaak verkeerd overgeschreven, waardoor het aantal herhalingen per locus sterk verschilt tussen mensen onderling.
Het unieke aantal herhalingen voor elk van de 12 loci vormt de DNA-vingerafdruk (DNAfingerprint) van een individu.
Een overeenkomst tussen de fingerprint van verzameld DNA en DNA van een verdachte kan dus als bewijs van schuld dienen.
16.6.2 Methode
16.6.2.1 Staalname
Op een plaats delict gaan forensische onderzoekers altijd op zoek naar DNA van mogelijke daders. Al het biologisch materiaal dat van een mens afkomstig is, kan hiervoor als bron dienen (haar, bloed, speeksel, sperma...).
16.6.2.2 Polymerase Chain Reaction (PCR)
Om verder onderzoek te vergemakkelijken wordt het gevonden DNA duizenden keren vermenigvuldigd via polymerase kettingreactie (PCR, polymerase chain reaction), een techniek die in de jaren '80 werd ontwikkeld en een ware revolutie betekende voor zowel het DNA-onderzoek als moderne, alledaagse laboratoriumtesten (de zgn. ‘PCRtesten’).
PCR werkt als een kopiemachine voor DNA. De overvloed aan DNA die ermee aangemaakt wordt, vergemakkelijkt verder onderzoek.
16.6.2.3 Elektroforese
127 elektroforese van DNA
Vervolgens wordt het DNA door enzymen in stukjes geknipt. Zo ontstaan DNAfragmenten van verschillende grootte (hoe meer herhalingen in de STR, hoe groter het fragment.) De fragmentjes worden gelabeld met een kleurstof.
Daarna worden de DNA-fragmenten in een glazen buisje gebracht waarin een kunststofgel (polyacrylamide) aanwezig is. Hierop wordt een elektrische lading aangebracht, die de (negatief geladen) DNA-fragmenten in beweging zet.
Kleinere fragmenten (kortere STRs) bewegen hierbij trager dan grote fragmenten.
Na anderhalf uur leest een detector af waar de fragmenten zich in de gel bevinden.
Elke, unieke DNA-fingerprint creëert een uniek patroon bij DNA-elektroforese.
Door het vergelijken van DNA (bv. plaats misdaad – verdachte) kan op die manier met grote betrouwbaarheid een dader geïdentificeerd worden.
Iedereen erft de helft van z'n STRs van vader en de helft van moeder, waardoor onze DNA-vingerafdruk eigenlijk een combinatie is van die van onze ouders.
Hoewel er tijdens het overerven meestal nog kleine schrijffoutjes gebeuren in de STRs, zullen ze toch nog altijd sterk gelijken op deze van hun ouders.
Dat maakt het mogelijk om - met een grote graad van waarschijnlijkheid - een ouderkind relatie te bevestigen.
Figuur
Figuur
17 Principes van overerving
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
Een stamboom te tekenen met de correcte symbolen.
De wetten van Mendel te beschrijven en telkens kort te verklaren.
De wetten van Mendel toe te passen in een oefening.
Het herhalingsrisico te berekenen voor een kenmerk of aandoening die de klassieke Mendeliaanse overervingspatronen volgt.
De verschillende processen (reacties) te beschrijven die deel uitmaken van de bevruchting.
17.1 De stamboom
Om een stamboom te tekenen maken we gebruik van deze symbolen:
(mutant fenotype komt tot uiting) (cosanguïniteit)
kinderen, van oud naar jong
(geslacht kan verschillend zijn)
(geslacht moet gelijk zijn)
de bestudeerde persoon wordt aangeduid met een pijl recessief kenmerk) recessief kenmerk)
17.2 De wetten van Mendel
JAAR: 1865
Gregor Mendel (1822-1884) was een Hongaarse monnik met een voorliefde voor tuinieren. Hij formuleerde zijn overervingswetten ca. 1865 na het jarenlang bestuderen van erwtenplanten die hij zelf kweekte in de kloostertuin. Hij voerde hiervoor bijna tienduizend kruisingsexperimenten uit, waarbij hij nauwgezet hun karakteristieken observeerde (groen, geel, gerimpeld...) en de frequentie waarmee deze voorkwamen
We spreken van Mendeliaanse overerving als een gen (of kenmerk in het fenotype) wordt overgeërfd volgens de wetten van Mendel.
17.2.1
De 1e wet: de splitsingswet
De 1e wet van Mendel stelt dat erfelijke eigenschappen bepaald worden door 2 allelen, waarvan er slechts 1 wordt doorgegeven aan het nageslacht; m.a.w. de ouderlijke allelen worden gesplitst. Mendel gebruikte hiervoor het woord segregatie.
Verklaring: De splitsingswet is een rechtstreeks gevolg van de meiotische deling tijdens de gametogenese. Hierbij worden haploïde gameten (n) gecreëerd uit diploïde (2n) voorlopercellen.
Voorbeeld: Beschouwen we een gen A met 2 mogelijke allelen (A,a).
Figuur 129 Splitsingswet gameten vader gameten moeder
§ Een vader met het genotype Aa (links) zal in de helft van z'n gameten het allel A en in de andere helft het allel a vertonen.
§ Een moeder met het genotype AA (rechts) zal in de helft van haar gameten het allel A en in de andere helft ook het allel A vertonen.
De studie van Mendeliaanse overerving maakt vaak gebruik van kansberekening. Hierbij wordt cijfermatig uitgedrukt wat de kans is dat een bepaalde uitkomst zich voordoet.
Uit de splitsingswet volgt bv. dat de keuze voor het éne of het andere allel 50% bedraagt. 50% kunnen we ook schrijven als 1/2 of 0,5. De kans op een bepaalde uitkomst ligt altijd tussen 0% en 100% (0 en 1). Hoe dichter bij 1, hoe groter de kans.
Die uiteindelijke uitkomst is volledig willekeurig en wordt bepaald door toeval.
De keuze voor het éne of andere allel wordt in realiteit per chromosoom gemaakt.
Elke ouder creëert gameten met 23 chromosomen. Voor elk chromosomenpaar wordt dus 1 chromosoom doorgegeven aan het nageslacht. De kans hierop is – per chromosoom – 50%.
17.2.2 De 2e wet: de onafhankelijkheidswet
De tweede wet van Mendel stelt dat verschillende genen (voor verschillende kenmerken) onafhankelijk van elkaar worden overgeërfd.
Verklaring: genen op verschillende chromosomen worden onafhankelijk van elkaar overgeërfd omdat de splitsing per chromosoom gebeurt (zie hoger). Voor de genen die op hetzelfde chromosoom liggen kan crossing over (zie: 3.3.2) plaatsvinden.
Voorbeeld: Beschouwen we een kenmerk of gen A met 2 mogelijke allelen (A,a) en een kenmerk of gen B met 2 mogelijke allelen (B,b).
gameten vader gameten moeder
Figuur 130 De onafhankelijkheidswet
§ Een vader met het genotype Aa en Bb (links) zal in de helft van z'n gameten het allel A en in de andere helft het allel a vertonen èn in de helft van z'n gameten het allel B en in de andere helft het allel b vertonen.
§ De keuze voor het allel A of a heeft geen invloed op de keuze voor het allel B of b.
17.2.3 De 3e wet: de dominantiewet
De derde wet van Mendel stelt dat igv een heterozygoot genotype, het fenotype zal bepaald worden door één enkel allel
Verklaring: zie 16.5.2.3 De rol van het tweede allel
Voorbeeld: Mendel kon deze 3e wet afleiden door zijn monohybride kruisingsexperiment met erwtenplanten Hieronder passen we dit toe op de mens, waarbij we een gen T beschouwen met 2 allelen (T,t). Dit gen codeert voor een witte haarlok, een zeldzaam, fenotypisch kenmerk dat alleen tot uiting komt bij homozygoten voor het mutante allel (t).
GENERATIE 1 (F1) pure homozygoten
GENERATIE 2 (F2) enkel heterozygoten
GENERATIE 3 (F3)
Figuur 131 De monohybride kruising, toegepast op de mens
Uit deze stamboom kunnen we afleiden wat Mendel ook vaststelde bij erwtenplanten, nl. dat bij de kruising van pure homozygoten (1e generatie, F1), een generatie van dragers (carriers) ontstaat (2e generatie, F2), waarbij de mutante allelen aanwezig zijn in het genotype, maar niet tot uiting komen in het fenotype.
witte haarlok
witte haarlok
Uit deze generatie van dragers ontstaat vervolgens een 3e generatie (F3) waarbij het mutante fenotype terug ‘verschijnt’, zij het wel bij een minderheid (steeds 25%) van de nakomelingen.
Maar dit bewijst wel dat:
De erfelijke informatie van in F1 doorgegeven werd naar F3.
Het allel t enkel tot uiting komt in het fenotype als T afwezig is (zie 16.5.2.3). Het allel t (mutant) is bijgevolg recessief, het allel T (wild-type) is dominant.
Bij het maken van oefeningen op de wet van Mendel vervangen we de naam van het gen vaak door een (willekeurige) letter, zoals hierboven de letter T. Dit werkt makkelijker; bv. het gen voor de blonde haarlok heet eigenlijk WS2B.
Dominante allelen worden met een hoofdletter geschreven.
Recessieve allelen worden met een kleine letter geschreven.
m.o.: als je zelf een letter kiest; kies er dan een waar hoofdletter & kleine letter duidelijk van elkaar verschillen bv. A,a – B,b – D,d … en liever niet: P,p – C,c – X,x
BEDENKING
De meeste studenten zijn al goed vertrouwd met de begrippen dominant & recessief. Dit zit immers in heel wat leerplannen uit het secundair onderwijs. De 3e wet van Mendel lijkt hierdoor wat vanzelfsprekend. Toch mogen we niet vergeten hoe bijzonder het bestaan van ‘dragers voor recessieve allelen’ is.
Vergelijk het met kleuren mengen: als we blauwe en gele verf mengen krijg je groen. De 3e wet van Mendel vertelt ons echter iets helemaal anders, nl. dat je bij het mengen van ‘blauw’ en ‘geel’ terug blauwe verf krijgt (op voorwaarde dat blauw dominant is). De informatie voor het recessieve ‘geel’ is aanwezig in je mengsel, maar komt niet tot uiting. Dat is best merkwaardig. (In 16.5.2.3 gaven we hiervoor al een verklaring.)
Later is gebleken dat de allelen van sommige genen toch hun fenotype ‘mengen’. Dit fenomeen heten we intermediaire dominante. We komen hier later op terug (zie 18.3.4 )
17.2.4
Het kruisingsrooster
= recombinatierooster; het vierkant van Punnett
Dankzij de splitsingswet van Mendel kunnen we bij een gekend genotype van de ouders de mogelijke genotypes van het nageslacht gaan voorspellen. We kunnen daarbij zelfs gaan voorspellen hoe frequent elk genotype zal voorkomen. Als we weten of een allel dominant of recessief kunnen we zelfs de fenotypes gaan voorspellen.
Een handig, visueel hulpmiddel hierbij is het
Voorbeeld
Een
In de eerste die van de andere ouder (je mag zelf kiezen wie waar komt).
Op de kruisingen van de tweede en derde rijen kolommen breng je de allelen samen.
Dit geeft 4 mogelijke combinaties = Elk van deze 4 uitkomsten heeft een kans van 25%.
133 het recombinatierooster
Op basis van onze kennis over het allel (dominant of recessief) kunnen we nu het fenotype voorspellen. Aangezien T dominant is over t betekent dat slechts 1 op 4 kinderen (25%) een witte haarlok zal hebben. 3 op 4 kinderen (75%) zal geen witte haarlok hebben, maar 2 ervan zullen wel drager zijn (zie Figuur 131).
m.o.: dit blijft natuurlijk een statistische voorspelling, de uiteindelijke samenstelling van het gezin is aan toeval onderhevig. Het kan best zijn dat er bv. 4 kinderen geboren worden waarvan geen enkele een witte haarlok heeft. We zijn enkel zeker dat de kans op een kind met een witte haarlok bij elke zwangerschap 25% bedraagt.
Figuur
17.3 Mendeliaanse overervingspatronen
We gaven eerder al een definitie van de term ‘Mendeliaanse’ overerving (zie 17.2). In de praktijk wordt deze term vooral gebruikt voor kenmerken of eigenschappen die bepaald worden door één enkel gen, de zgn. monogene kenmerken.
Monogene kenmerken zijn kenmerken waarvan het fenotype bepaald worden door één enkel gen.
De overerving van deze kenmerken volgt enkele typische patronen, die gebaseerd zijn op de Wetten van Mendel.
17.3.1 Autosomaal recessieve overerving
AUTOSOMAAL RECESSIEVE OVERERVING
Het mutante allel ligt op chromosoom 1 tem 22
Het mutante allel is recessief, het wild-type allel is dominant
Enkel wie homozygoot is voor beide mutante allelen ontwikkelt het fenotype.
17.3.1.1 Patroon
Een typisch autosomaal recessief overervingspatroon ziet er als volgt uit (zie ook 17.2.3)
Beschouwen we een gen A met allelen A (wild-type) en a (mutant)
Figuur 134 Autosomaal recessief overervingspatroon recombinatierooster
Typisch voor autosomaal recessief overgeërfde kenmerken (of aandoeningen) is dat er meer heterozygoten (dragers) zijn dan homozygoten met het fenotype.
Enkel als beide ouders (minstens) heterozygoot zijn voor het mutante allel kan een kind geboren worden met het recessieve fenotype (aa).
Als beide ouders heterozygoot zijn is die kans 25% (zie de grijze kleur in het kruisingsrooster, Figuur 134).
Dit maakt dat autosomaal recessieve kenmerken (of aandoeningen) relatief zeldzaam zijn. Zelfs binnen een familie waarin een dergelijk kenmerk wordt overgeërfd merken we dat er weinig familieleden zijn met het recessieve fenotype. Vaak worden zelfs generaties overgeslaan.
Autosomaal recessieve kenmerken komen even vaak voor bij jongens als bij meisjes.
17.3.1.2 Cosanguïniteit
De meeste mutante allelen bij de mens hebben een recessief effect.
Als dan toch een kind geboren wordt met een autosomaal recessieve aandoening (bv. mucoviscidose, albinisme) is dit een onverwachte gebeurtenis.
Een uitzondering hierop is als ouders verwant zijn aan elkaar. Als de ouders familie zijn van elkaar (tot in welke graad) is er een significant hogere kans dat ze beiden drager zijn van dezelfde recessieve allelen.
17.3.1.3
We spreken van cosanguïniteit als beide ouders aan elkaar verwant zijn. Dit verhoogt sterk de kans op autosomaal recessieve kenmerken en aandoeningen.
In de Ashkenazi Joodse gemeenschap in Centraal-Europa komt een aantal ziektes vaker voor doordat huwelijken vaker binnen de gemeenschap plaatsvinden. Het gaat hierbij bv. om de ziekte van Tay-Sachs, een ernstige, autosomaal recessieve aandoening. Tegenwoordig wordt in deze gemeenschappen routinematig gescreend op dragerschap.
Assortative mating
We spreken van assortative mating als beide ouders een gelijkaardig fenotype bezitten. De partnerkeuze is hierbij niet volledig willekeurig gebeurd
Bij de keuze van een partner kiest de mens (net zoals veel dieren) heel vaak voor iemand met een gelijkaardig fenotype (bv. lichaamslengte, interesses, …). Het kan hierbij ook gaan over eenzelfde erfelijke aandoening (bv. erfelijke doofheid). Dit betekent dat de aandoening in kwestie ook vaker kan voorkomen bij het nageslacht (Jiang et al., 2013).
17.3.2 Autosomaal dominante overerving
AUTOSOMAAL DOMINANTE OVERERVING
Het mutante allel ligt op chromosoom 1 tem 22
Het mutante allel is dominant, het wild-type allel is recessief
Vanaf één mutant allel, komt het fenotype tot uiting.
17.3.2.1 Patroon
Een typisch autosomaal dominant overervingspatroon ziet er als volgt uit
Beschouwen we een gen A met allelen A (mutant) en a (wild-type)
Figuur 135 Autosomaal recessief overervingspatroon
Voor autosomaal dominant overgeërfde kenmerken (of aandoeningen) bestaat geen dragerschap. Vanaf de aanwezigheid van één mutant allel komt het fenotype tot uiting.
Als één van beide ouders een mutant allel bezit, is de kans 50% dat dit wordt doorgegeven aan het nageslacht (zie de splitsingswet, 17.2.1).
Statistisch gezien zullen dus 50% van de nakomelingen dus ook het mutante allel (en het bijhorende fenotype) ontwikkelen (zie de grijze kleur in het kruisingsrooster, zie Figuur 135).
Binnen een familie waarin een dergelijk kenmerk wordt overgeërfd merken we dat er geen generaties worden overgeslaan. De aandoening is vaak goed gekend in de familie.
Autosomaal dominante kenmerken komen even vaak voor bij jongens als bij meisjes.
17.3.2.2 Homozygoten zijn zeldzaam
Homozygoten voor autosomaal dominante aandoeningen zijn zeldzaam. Bij dit genotype is het fenotype steeds ernstiger, of kan vroegtijdig embryonale of foetale dood optreden (bv. bij de ziekte van Marfan, achondroplasie).
17.3.2.3 Lethale allelen
Lethale allelen zijn allelen die tot de dood van het organisme leiden. Als een vroegtijdig, lethaal effect optreedt (bv. tijdens de embryonale ontwikkeling) verdwijnen de allelen hiermee uit de populatie. In zekere zin werken lethale allelen dus hun eigen reproductie tegen. .
Van elke potentieel dodelijke, erfelijke ziekte (zowel dominant of recessief) kunnen we de allelen dus als lethaal beschouwen.
In de praktijk zullen we de term vooral gebruiken als we het hebben over mutante allelen die tot vroegtijdige, embryonale dood leiden.
Bij embryo’s die homozygoot zijn voor dominante allelen komt dit vaker voor.
17.3.3 X-gebonden recessieve overerving
X-GEBONDEN RECESSIEVE OVERERVING
Het mutante allel ligt op het X-chromosoom.
Het mutante allel is recessief, het wild-type allel is dominant
Bij de vrouw: enkel vrouwen die homozygoot zijn voor beide mutante allelen ontwikkelen het fenotype. Vrouwen die heterozygoot zijn, zijn drager.
Bij de man: bij de aanwezigheid van één mutant allel komt het fenotype tot uiting. Mannen kunnen geen drager zijn van een X-gebonden recessieve aandoening.
X-gebonden overerving heten we ook geslachtsgebonden overerving. Een man heeft slechts één X-chromosoom, dat hij overgeërfd kreeg van z'n moeder. Als er op dit Xchromosoom op een bepaalde locus een afwijkend allel voorkomt, is dit al voldoende om een bepaald kenmerk of aandoening tot uiting te laten komen (hemizygotie).
De ‘spelregels’ bij vrouwen verschillen hier dus wat van mannen.
17.3.3.1 Patroon
Als we oefeningen maken met X-gebonden overgeërfde allelen kennen we geen letter toe aan het gen. Daarentegen zullen we werken met de chromosoom-namen (X en y) zelf. Het mutante allel wordt geschreven als X*, het wild-type allel als X
Beschouwen we een X-gebonden gen met allelen X (wild-type) en X* (mutant). X is hierbij dominant over X*.
op het Y-chromosoom bestaat deze locus niet
Figuur 136 X-gebonden recessief overervingspatroon recombinatierooster
Een typische situatie is een gezin waarin de moeder drager is voor het mutante allel. Het risico op een aangedane zoon is dan 50%. (zie het recombinatierooster, Figuur 136).
Het risico voor een dochter die draagster is, is eveneens 50%.
Door gebruik te maken van X* en X kunnen we in het recombinatierooster onmiddellijk het geslacht afleiden. Bij het bespreken van de mogelijke uitkomsten verkiezen we om te spreken van “…. % van de zonen/dochters” ipv “…. % van de kinderen”
Mannen met de aandoening kunnen die niet doorgeven aan hun zonen. Maar hun dochters zullen steeds het afwijkende X-chromosoom ontvangen.
Een man kan enkel een X-gebonden mutant allel ontvangen van zijn moeder.
17.3.3.1 X-gebonden recessieve aandoeningen bij meisjes
X-gebonden ('recessieve') aandoeningen kunnen ook bij meisjes voorkomen, maar dit veronderstelt dat 1) de vader de aandoening heeft en 2) de moeder minstens drager is.
Bij meisjes die heterozygoot zijn X*X hangt het opnieuw van de mutatie zelf af, of deze zich als dominant of recessief zal gedragen.
X*X-meisjes met die drager zijn voor het mutante allel voor rood-groenkleurenblindheid hebben een perfect kleurenzicht. In dat geval gedraagt het mutant allel zich dus recessief. De meisjes in kwestie zijn louter drager.
17.3.4 X-gebonden dominante overerving
X-GEBONDEN DOMINANTE OVERERVING
Het mutante allel ligt op het X-chromosoom.
Het mutante allel is dominant, het wild-type allel is recessief
Zowel bij mannen als bij vrouwen komt het fenotype vanaf één mutant allel tot uiting.
17.3.4.1 Patroon
Beschouwen we een X-gebonden gen met allelen X* (mutant) en X (wild-type). X* is hierbij dominant over X.
Figuur 137 X-gebonden dominant overervingspatroon
Het belangrijkste verschil met de X-gebonden recessieve aandoening is dat vrouwen die heterozygoot zijn ook het kenmerk of de aandoening ontwikkelen (bv. bij vrouwen die drager zijn voor het Fragile-X syndroom zien we toch vaak lichte kenmerken in het fenotype).
50% van de kinderen (of 50% van de jongens èn meisjes) zullen het kenmerk of de aandoening ontwikkelen (zie het recombinatierooster, Figuur 137).
17.3.5 Y-gebonden overerving
Y-GEBONDEN DOMINANTE OVERERVING
Het mutante allel ligt op het Y-chromosoom.
Omwille van hemizygotie kunnen we hier niet spreken van dominant of recessief.
Bij mannen komt het fenotype tot uiting vanaf één mutant allel.
17.3.5.1 Patroon
op het X-chromosoom bestaat deze locus niet
Beschouwen we een Y-gebonden gen met allelen y* (mutant) en y (wild-type).
Figuur 138 Y-gebonden overerving
Dit overervingspatroon is zeldzaam, aangezien het Y-chromosoom slechts enkele tientallen genen bevat, die vooral een rol spelen in de differentiatie van het embryo tot een man (zie 14.1.3). Een gebrekkige mannelijke differentiatie van de genitalia kan bovendien de vruchtbaarheid en de kans op voortplanting beïnvloeden.
Mutante allelen op het Y-chromosoom worden enkel doorgegeven van vader op zoon. Aangezien elke zoon het Y-chromosoom van z’n vader ontvangt, zullen alle zonen het mutante allel ontvangen.
17.4 Oefeningen op Mendeliaanse overerving
17.4.1 Aanpakken van genetische vraagstukken
17.4.1.1 De aard van de vraag: het herhalingsrisico
Koppels die een centrum menselijke erfelijkheid consulteren zitten vaak in de volgende situatie: zijzelf of één of meerdere familieleden zijn bekend met een erfelijke aandoening & daarom wil het koppel wil graag weten hoe groot de kans is dat zij een kind krijgen met die aandoening.
Dit is een vraag naar een herhalingsrisico.
Dit soort vragen komt in de oefeningen vaak terug.
17.4.1.2 De stamboom
Het beantwoorden van een vraag naar erfelijkheid begint met het opstellen van een nauwkeurige, familiale stamboom Merk op dat je hierbij meestal geen weet hebt van eventuele dragers. Vaak kan dit wel afgeleid worden.
Gebruik de correcte symbolen bij het tekenen van een stamboom (zie 17.1)
Het is ook belangrijk om volledig te zijn bij het opmaken van een stamboom. Autosomaal recessieve overervingspatronen zijn in een kleine stamboom niet altijd zichtbaar. Bovendien zijn er tal van Niet-Mendeliaanse mechanismen (bv. verminderde penetrantie) die een overervingspatroon kunnen verstoren.
m.o.: in de oefeningen… gaan wij uit van de klassieke, Mendeliaanse overerving (tenzij anders aangegeven). is de stamboom vaak ‘gegeven’. Dragers zijn hierbij wel niet aangeduid!
17.4.1.3 Het recombinatierooster
Heel nuttig bij Mendeliaanse aandoeningen, zie 17.2.4
17.4.1.4 Kansberekening
In de oefeningen op Mendeliaanse overerving maken we vaak gebruik van een klein beetje kansberekening (zie ook 17.2.1).
Dit laat toe om de vraag naar herhalingsrisico te beantwoorden met een concreet cijfer (bv. 3/4e of 0,75). Dit cijfer staat dan voor de probabiliteit dat het bestudeerde genotype in de zygote aanwezig zal zijn (bv. in 75% van de nakomelingen).
Om tot een concreet herhalingsrisico te komen moeten we vaak meerdere, veronderstellingen doen, elk met hun eigen probabiliteit.
Zo kan een autosomaal recessief genotype enkel tot stand komen op voorwaarde dat beide ouders minstens drager zijn van het recessief allel, en dat weten we niet altijd met zekerheid.
Een voorbeeld:
Wat is de kans dat twee gezonde ouders een kind krijgen met mucoviscidose?
Aangezien mucoviscidose autosomaal recessief is, weten we dat - als beide ouders drager zijn - die kans 1/4 (of 25%) bedraagt.
Maar hoe groot is die kans dat beide ouders drager zijn?
Uit DNA-onderzoek in de gezonde populatie weten we dat 1/25 mensen heterozygoot is voor het mutante allel (Mm).
Opdat - bij gezonde ouders - een kind geboren wordt met mucoviscidose moeten we dus 3 veronderstellingen doen:
1) moeder is heterozygoot (=Mm=drager): die kans is 1/25
2) vader is heterozygoot (=Mm=drager): die kans is ook 1/25
3) bij een heterozygoot ouderpaar (Mm) is de kans 1/4 op een kind met mm
Deze 3 probabiliteiten zijn volledig onafhankelijk van elkaar. Maar ze moeten wel samen voorkomen opdat een kind met mucoviscidose zou geboren worden.
Hoe groot is dan het totale herhalingsrisico?
De productregel stelt dat we de kansen op onafhankelijke gebeurtenissen die gelijktijdig moeten plaatsvinden met elkaar mogen vermenigvuldigen
De totale kans op het gelijktijdig optreden van 3 onafhankelijke voorwaarden, A,B,C:
Totale kans = (kans op A) x (kans op B) x (kans op C)
Voor mucoviscidose betekent dit
Totale kans = (kans op Mm bij de moeder) x (kans op Mm bij de vader) x 1/4 = 1/25 x 1/25 x 1/4 = 1/2500 (=het voorkomen van mucoviscidose in onze populatie)
17.4.2
Oefeningen
Oefening 8.
Zijn deze overervingspatronen autosomaal dominant of recessief? Argumenteer.
Oefening 9.
Machteld (zie pijl) lijdt aan de ziekte van Perrault, een zeldzame autosomaal recessieve aandoening die veroorzaakt wordt door een mutatie in een gen op locus 5q23.1. Deze aandoening leidt tot gehoorverlies en - bij meisjes - tot een verstoorde ontwikkeling van de ovaria, met late puberteit en vroegtijdige menopauze tot gevolg.
1 2
3 4 5
Vragen
a. Wat wordt bedoeld met ‘locus 5q23.1’
b. Wat is het genotype van Machteld?
c. Wat zijn de mogelijke genotypes van de broer en zus van Machteld?
d. Hoe groot is de kans dat de broer van Machteld drager is?
Oefening 10.
Een vrouw met blauwe ogen krijgt een kind met bruine ogen. Stel dat de bruine oogkleur bepaald wordt een autosomaal dominant overgeërfd allel B met allelen (B,b). Het allel B is wild-type (bruin), het allel b is mutant (blauw).
Vraag. Welk(e) genotype(n) kan de vader van het kind gehad hebben?
o Bb of bb o BB of Bb o alleen Bb o alleen BB o alleen bb
Oefening 11.
Rood-groen kleurenblindheid (Daltonisme) wordt overgeërfd volgens het Mendeliaanse, X-gebonden, recessief overervingspatroon. In onderstaande familie lijdt persoon 2 aan deze vorm van rood-groen-kleurenblindheid. Uit het koppel 9 en 10 wordt een zoon geboren.
Vragen.
a. Wat is er bijzonder aan de relatie tussen persoon 9 en 10?
b. Hoe groot is de kans dit hun zoon (pijl) lijdt aan daltonisme?
(m.o.: we nemen aan dat persoon 2 de enige bron is van het allel)
Oefening 12.
In deze familie komt een erfelijke vorm van syndactylie voor. Hierbij zijn de 3e en 4e vingers van beide handen met elkaar vergroeid. Het verantwoordelijke gen ‘L’ ligt op chromosoom 7.
Vraag: welk genotype heeft persoon 4 (pijl)?
Oefening 13.
Hoe groot is de kans dat, als 4 kinderen geboren worden in een gezin, dat 2 ervan meisjes zijn èn 2 ervan jongens? Toon duidelijk aan hoe je aan dit resultaat komt.
Oefening 14.
Agathe heeft een aangeboren afwijking aan haar bovenste ooglid, nl. een ptose (afhangend ooglid). Dit kenmerk wordt voor veroorzaakt door dominant (niet-lethaal) allel R dat op chromosoom 11 is gelegen. Haar vader had ook een ptose, maar haar moeder niet. Haar grootmoeder (aan vaderszijde) had normale oogleden.
Vragen.
a. Wat is het genotype van Agatha, haar vader en haar moeder?
b. Hoe groot is de kans dat Agathe kinderen krijgt met een ptose als haar man normale oogleden heeft? (Teken minstens een recombinatierooster.)
Oefening 15.
Wie kan onmogelijk de bron zijn van de genen op het X-chromosoom van een vrouw:
o De vader van haar moeder
o De moeder van haar vader
o De moeder van haar moeder
o De vader van haar vader
o De moeder van de moeder en de vader van haar moeder
Oefening 16.
Juist of fout? Een X-gebonden dominante aandoening kun je onderscheiden van een autosomaal dominante aandoening doordat bij de X-gebonden dominante aandoening geen transmissie van vader op zoon kan plaatsvinden.
Oefening 17.
Hoe gebeurt de overerving van onderstaande aandoening? Argumenteer met verschillende elementen die je opmerkt in de stamboom.
Oefening 18.
Als een vrouw lijdt aan rood-groen kleurenblindheid en haar man niet. Wat zijn dan de mogelijkheden voor het nageslacht?
Oefening 19.
Een putje in de kin is een autosomaal, dominant overgeërfd kenmerk. Met wat je kunt afleiden uit deze stamboom, hoe groot is de kans op een kind met een putje in de kin…
a)… als persoon 7 en 9 samen een kind krijgen.
b) … als persoon 8 en 10 samen een kind krijgen.
Teken voor beide koppels een recombinatierooster.
Oefening 20.
Onderstaand kenmerk wordt X-gebonden dominant overgeërfd.
Bepaal voor elk familielid het juiste genotype.
Oefening 21.
Wanneer de dochter met bruine ogen uit deze stamboom een kindje krijgt met een man met blauwe ogen, hoe groot is dan de kans dat het kind ook blauwe ogen heeft?
18 Niet-Mendeliaanse mechanismen
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (pijlen).
Belangrijke (vetgedrukte) begrippen uit dit hoofdstuk te verklaren.
De termen ‘penetrantie’ en ‘expressiviteit’ toe te passen in een casus over een erfelijke aandoening.
Een voorbeeld te geven van een ziekte met een verminderde penetrantie.
Een voorbeeld te geven van een ziekte met een variabele expressiviteit.
Het verschil tussen de termen ‘genoom’ en ‘epigenoom’ kort te omschrijven.
De X-inactivatie te bespreken.
Uit te leggen waarom een vrouw – ondanks X-inactivatie – haar diploïd voordeel niet verliest.
Uit te leggen wat bedoeld wordt met imprinting (incl. het mechanisme).
De verschillende soorten imprinting te bespreken.
Het moment aan te duiden waarbij een parentale imprint wijzigt.
De conflicthypothese bij parentale imprinting kort toe te lichten.
Het verband tussen maternale zwaarlijvigheid en imprinting toe te lichten.
In patiëntentaal uit te leggen wat bedoeld wordt met ‘epigenetische therapie’ en wat de wetenschappelijke waarde hiervan is.
De geno- en fenotypes van het ABO-bloedgroepensysteem te bespreken.
De leerstof uit dit hoofdstuk toe te passen in een oefening.
In dit hoofdstuk bespreken we de uitzonderingen op de Mendeliaanse overerving Deze niet-Mendeliaanse mechanismen verklaren waarom overervingspatronen in de realiteit vaak afwijken van wat we verwachten volgens de wetten van Mendel.
18.1
Penetrantie & expressiviteit
Tenzij voor een recessief allel, ging Mendel ervanuit dat elk allel altijd tot uiting kwam in het fenotype. Ondertussen is gebleken dat dat niet klopt. Er zijn zelfs situaties waarbij dominante genen niet tot expressie komen (!)
Penetrantie = de probabiliteit dat een mutant genoytpe in het fenotype tot uiting komt. Dit wordt uitgedrukt als een percentage (0%-100%).
Expressiviteit = de intensiteit van het fenotype (bv. de ernst van de aandoening) bij personen met hetzelfde genotype
Penetrantie kan je ook beschouwen als het aantal personen met een mutant genotype waarbij het fenotype tot uiting komt.
Bv. stel dat er bij het gen voor de witte haarlok (zie 17.2.3) een verminderde penetrantie bestaat. In dat geval zal niet iedereen die homozygoot is voor het mutant allel (tt) het bijhorende fenotype ontwikkelen:
Figuur 139 De penetrantie van de witte haarlok is hier 70% . Bovendien is er een variable expressiviteit aangezien de witte haarlok niet altijd even uitgesproken is. t t t t t t t t t t t t t t t t t t t t
Dit toont aan dat er nog andere factoren in het spel zijn (andere genen, omgevingsfactoren) die het fenotype bepalen.
Nog enkel voorbeelden:
*Bv. de penetrantie van de ziekte van Huntington (zie 20.1.6) is volledig (=100%). Dit betekent dat het afwijkende genotype altijd leidt tot de ziekte
*Bv. de penetrantie van hemochromatose – een ijzerstapelingsziekte – is bij homozygoten slechts 1%. We spreken van verminderde penetrantie.
In Figuur 139 valt ook te zien dat de witte haarlok – indien aanwezig – niet altijd even groot is. Dit heten we expressiviteit: het feit of het fenotype variabel is qua intensiteit. Expressiviteit is dus enkel van toepassing bij individuen waar de ziekte of het kenmerk tot uiting is gekomen. De meeste erfelijke aandoeningen hebben een variabele expressiviteit en het type mutatie speelt hier een grote rol in.
*Bv. tussen mucoviscidose-patiënten onderling is er een groot verschil in longfunctie Dit ondanks hetzelfde genotype (mm, waarbij m staat voor het mutante CFTR-allel)
18.2 Epigenetica
Epigenetica is de studie van alle factoren die de genexpressie beïnvloeden, zonder dat er sprake is van een wijziging in de DNA-sequentie.
Epigenetica is a.h.w. een extra-laag instructies (het epigenoom) in het erfelijk materiaal, die losstaat van de DNA-code zelf (het genoom). Vergelijk met de notities die je vermoedelijk al maakte in de kantlijn deze cursus.
Er zijn tal van mechanismen die maken dat in elk celtype een uniek epigenoom tot stand komt. Dit zorgt voor een heel gerichte expressie van eiwitten.
We bespreken hier slechts 3 epigenetische factoren:
X-IST (X-inactivatie)
Genomische imprinting: Parentale imprints
Externe imprints
OVERERFBAAR!
18.2.1
XIST (X-inactivatie)
JAAR: 1961
Mary Lyon ontdekte in de jaren ’60 dat elke vrouwelijke cel (46, XX) maar één X-chromosoom gebruikt. Enkel van dat éne X-chromosoom komen de allelen tot expressie. Het andere Xchromosoom blijft gedurende de gehele celcyclus inactief. X-inactivatie wordt ook lyonisatie genoemd, of men spreekt van de ‘wet van Lyon’ (Lyon, 1961).
Bij een vrouw met 46, XX – karyotype wordt één X-chromosoom geïnactiveerd door het volledig te bedekken met een grote, niet-coderende RNA-molecule. Men heet dit een RNA-transcript, in dit geval het XIST (X-inactive specific transcript).
Het inactieve X-chromosoom wordt gemethyleerd: aan alle cytosine (C) – basen wordt een methylgroepje (CH3) gekleefd, waardoor transcriptie onmogelijk wordt. Beschouw het als ‘chemische plakkertjes’.
18.2.2
Door deze processen krimpt het geïnactiveerde chromosoom ineen tot een klompje chromatine, onder de microscoop zichtbaar als een Barr-lichaampje.
Barr-lichaampje
Barr-lichaampje
enkel het maternale Xchromosoom komt tot expressie enkel het paternale Xchromosoom komt tot expressie (embryo)
140 X-inactivatie (Xi) (Thompson, 2016)
De keuze voor het ene of het andere X-chromosoom wordt willekeurig gemaakt tijdens de embryonale ontwikkeling van de vrouw; in het stadium van 5000 cellen.
Gemiddeld kiest hierbij 50% van de embryonale cellen voor het éne en 50% van de cellen voor het andere X-chromosoom (vergelijk met ‘kop of munt’) Eéns zo'n embryonale cel gekozen heeft, wordt de keuze door alle afstammende dochtercellen overgenomen en behouden voor de rest van het leven (Brown & Robinson, 1997).
Bij elk vrouwelijk embryo gebeurt dit proces van X-inactivatie opnieuw.
Waarom bestaat lyonisatie? Door X-inactivatie wordt een belangrijk probleem opgelost in de menselijke, eukaryote cel, nl. dat twee X-chromosomen in één cel zouden leiden tot een overmatige genexpressie. Hierdoor zou de celfunctie ernstig verstoord geraken.
Verliest de vrouw dan haar diploïd voordeel niet? Neen, aangezien ze twee Xchromosomen bezit die in een ongeveer gelijke verdeling actief zijn in het lichaam; alleen niet in dezelfde cel. (Zie ook functioneel mozaïcisme 18.5.)
Parentale imprinting
Bij parentale imprinting (ouderlijke imprinting) wordt het aan- of uitgeschakeld zijn van een gen bepaald door de ouder van wie het allel werd overgeërfd Sommige allelen lijken zich dus te ‘herinneren’ van welke ouder ze zijn overgeërfd!
§ Paternale imprint: genen die door de vader zijn geïnactiveerd, maar door de moeder actief worden gehouden.
§ Maternale imprint: genen die door de moeder zijn geïnactiveerd, maar door de vader actief worden gehouden.
Figuur
Deze geslachtsspecifieke imprints gebeuren niet willekeurig. Genen waarbij dit soort imprinting meespeelt, hebben ofwel uitsluitend een paternale imprint, andere een maternale imprint.
Het moleculaire mechanisme achter imprinting is eveneens methylering
Aan honderden genen wordt een imprint van de ouder meegegeven.
In zaadcellen vinden we voor die genen een paternale imprint, in eicellen een maternale imprint. Deze imprints worden overgenomen door de zygote.
GEN A heeft een maternale imprint, enkel het paternale allel is actief
GEN A
maternaal chromosoom paternaal chromosoom
5’ 3’ 5’ 3’ p p p p p p p p
GEN B
Gen B heeft een paternale imprint, enkel het maternale allel is actief
Figuur 141 Maternale en paternale imprinting. De rol van de promotor (p) valt hierbij weg.
JUIST OF FOUT?
Door imprinting worden we eigenlijk 'hemizygoot' voor die erfelijke informatie.
Een imprint is definitief in alle lichaamscellen, gedurende het ganse leven. In de kiembaancellen gebeurt echter een ‘reset’, waarna een nieuw maternaal of paternaal patroon wordt geplaatst, afh. van het geslacht van de ouder. Concreet betekent dit dat een vrouw al haar eicellen een maternale imprint zal meegeven (vice versa bij de man).
Waarom bestaat imprinting? Eén hypothese is dat er in het embryo een conflict plaatsvindt tussen het belang van beide ouders. Dit valt af te leiden uit de functie van de genen zelf. Paternale genen bevorderen immers de groei van het kind en de placenta (de vader wil een ‘sterk nageslacht’), terwijl maternale gen de belangen van de vader afremmen om zich vooral te focussen op het overleven van de moeder. Deze zgn. conflicthypothese biedt echter nog geen verklaring voor de – bewezen – rol van imprinting bij oa. astma, diabetes & autisme.
18.2.3
Externe imprints
Dat de genexpressie kan gewijzigd worden onder invloed van omgevingsfactoren bleek eigenlijk ook al uit 2.7. Maar we voegen er hier een belangrijke nuance aan toe, nl. dat bepaalde omgevingsfactoren imprints nalaten in het DNA die overerfbaar zijn.
Externe imprints zijn het gevolg van omgevingsfactoren. Ze zijn reversibel, overerfbaar en niet gebonden aan het geslacht van de ouder.
Een voorbeeld:
Bij obesitas (zwaarlijvigheid) worden tal van imprints aan het DNA toegevoegd. Deze kunnen worden doorgegeven aan het nageslacht. Tijdens de embryogenese verstoren deze imprints de normale genexpressie, waardoor sommige weefsels minder goed differentiëren. Zo kunnen deze ‘programmatiefouten’ levenslang invloed hebben op de weefselfunctie van het kind en – op hun beurt - leiden tot obesitas en chronische aandoeningen (Rohde et al., 2019; Reichetzeder, 2021).
Er heerst nog veel onduidelijkheid over dit soort imprinting, aangezien het een complex samenspel is van veel factoren die een subtiel, soms tijdelijk effect hebben op geno- en fenotype.
Aangezien dit soort imprints reversibel is, biedt dit in de toekomst wel aanknopingspunten voor gerichte preventie en therapie.
Externe imprints zijn niet-Mendeliaans en zelfs niet-Darwiniaans. Volgens Darwin was er immers geen overerfbare invloed van de omgeving (dat was de theorie van Lamarck)
Leestip: “Gelijk Goed Beginnen”, Tessa Roseboom, De Tijdstroom, Utrecht (2022)
De eerste 1000 dagen van een kind (van conceptie tot peuter) vormen de basis voor de rest van het leven. Dit boek laat zien wat je kunt doen om bij te dragen aan een goede start voor elk kind. Ook de rol van epigenetica komt aan bod. De auteur vergelijkt dit met de volumeknoppen op een mengpaneel. Heel wat van deze knoppen worden vroeg in het leven afgesteld door omgevingsfactoren.
Sommige alternatieve therapeuten adverteren nu al ‘epigenetische therapie’, waarbij geclaimd wordt dat door voedingsadvies en/of manuele therapie concrete wijzigingen in genexpressie kunnen bekomen worden voor de (evt. zwangere) patiënt en/of ongeboren kind. Voor deze gerichte therapieën is er momenteel nog geen klinische evidentie.
18.3 Niet-Mendeliaanse dominantie
18.3.1 Co-dominantie
Co-dominantie: meerdere allelen zijn dominant over één recessief allel.
18.3.1.1 ABO-bloedgroepen
Een voorbeeld hiervan zijn de ABO-bloedgroepen:
De bloedgroepen worden bepaald door een polymorf gen, gelegen op locus 9q34.2
Er zijn 3 allelen, A, B en O, waarbij zowel A als B dominant zijn over O (=recessief allel).
De allelen A en B coderen voor enzymen die de oppervlakte-eiwitten (antigenen) van rode bloedcellen afwerken, dit door het toevoegen van verschillende suikermoleculen aan een voorloper eiwit, het H-antigen.
§ Allel ‘A’ codeert voor een enzym dat het H-antigen omvormt tot antigen A.
§ Allel ‘B’ codeert voor een enzym dat het H-antigen omvormt tot antigen B.
§ Allel ‘O’ is een mutant allel dat door een frameshift-mutatie codeert voor een niet-functioneel eiwit (loss of function). Hierdoor kunnen geen suikermoleculen worden toegevoegd aan het H-antigen.
Dit geeft 6 mogelijk genotypes en 4 mogelijke fenotypes (bloedgroepen):
Antigenen
Genotypes
Antigen A
Antigen B
AA, AO
Antigenen A & B (enkel H-antigen)
Figuur 142 De 4 verschillende bloedtypes
rhesus-systeem (rhesusgroep), waarvan antigeen D het bekendste is. Het gen voor antigeen D (D,d) volgt D de klassieke, Mendeliaanse overerving (autosomaal recessief).Bij aanwezigheid van het D-antigen (DD of Dd) is het fenotype een ‘positieve’ bloedgroep bv. A+. Bij afwezigheid ervan (dd) is het fenotype een ‘negatieve’ bloedgroep bv. A-.
18.3.1.2
Oefeningen
Oefening 22.
Een vrouw met bloedgroep A huwt een man met bloedgroep AB. Welke bloedgroepen kunnen hun kinderen hebben?
Oefening 23.
Als een man met bloedgroep A en een vrouw met bloedgroep B een kind hebben met bloedgroep O. Hoe groot is dan de kans dat het tweede kind eveneens bloedgroep O zal hebben?
Oefening 24.
Stel dat een vrouw de bloedgroep O-negatief (O-) heeft en haar beide ouders O-positief (O+). Betekent dit dan dat zij een buitenechtelijk kind is? Toon duidelijk hoe je aan je antwoord komt. Teken hierbij minstens een recombinatierooster.
Oefening 25.
Een vrouw met bloedgroep AB krijgt een kind met bloedgroep B. Twee mannen claimen van de vader te zijn van het kind. Man 1 heeft bloedgroep A. Man 2 heeft bloedgroep B. Kan het bloedgroepensysteem hier bepalen wie de vader is, of is bijkomend onderzoek nodig.
18.3.3 Epistase
Epistase: de expressie van een gen onderdrukt de expressie van een gen op een andere locus. Het onderdrukte gen noemen we hypostatisch.
De nuance 'op een andere locus' is hierbij erg belangrijk, want anders spreken we gewoon over dominantie & recessiviteit.
Opnieuw vinden we een mooi voorbeeld bij de bloedgroepen:
0,0004% van de bevolking bezit een mutatie in beide allelen die coderen voor het Hantigen (locus 19q33.3) Hierdoor kan het H-antigen niet worden aangemaakt. Aangezien dit een voorloper is van de A en B-antigenen, betekent dat deze ook niet kunnen worden aangemaakt, ongeacht het hiervoor aanwezige genotype. De allelen op locus 9q34.2 worden in dat geval dus buitenspel gezet. Zelfs als zo iemand genotype ‘AB’ heeft, worden er immers geen A-antigenen, noch B-antigenen aangemaakt.
Deze toestand heten we de Bombay-bloedgroep, een 5e bloedgroep naast A, B, AB en O.
De mutatie in de genen voor het H-antigen zijn hierbij epistatisch over de genen die bepalen voor de antigenen A, B en O. Deze worden dan hypostatisch genoemd.
H-antigen geen antigenen
Figuur 143 (Li) De O-bloedgroep (Re) de Bombay-bloedgroep
Functioneel komt de Bombay-bloedgroep overeen met bloedgroep O.
Het grootste probleem van de Bombay-patiënten is dat zij enkel transfusies mogen krijgen met het (uiterst zeldzame) bloed van de Bombay-bloedgroep. Het H-antigen wordt door hun immuunsysteem immers als lichaamsvreemd herkend, waardoor transfusiereacties optreden bij transfusie van de bloedgroepen A, B, AB en O.
Oefening 26.
De Bombay-bloedgroep werd ontdekt door een
Indische arts die de stamboom opstelde van een patiënte (pijl) waar uit labo-analyse bleek dat ze bloedgroep ‘O’ had. Dit bleek echter ‘onmogelijk' bij het analyseren van de bloedgroepen van haar ouders en kinderen.
Vraag: welk(e) genotype(s) heeft deze patiënte met Bombay-bloedgroep?
18.3.4 Intermediaire dominante
Bij intermediaire dominantie is het fenotype een 'mix' van de verschillende allelen.
Bv. (fictief voorbeeld).
Stel een allel B bepaalt de haarkleur met allel B (witte kleur) en allel b (zwarte kleur).
Bij intermediaire dominantie gedragen de allelen zich niet als dominant of recessief.
Het genotype Bb leidt tot een grijze haarkleur, een mengsel van zwart en wit.
18.4 Dynamische mutaties
We spreken van een dynamische of onstabiele mutatie als een mutant allel telkens verder muteert, wanneer het wordt doorgegeven aan het nageslacht. Het gaat hierbij typisch om triplet repeats in een allel die – per generatie – in aantal toenemen.
Bij een te groot aantal herhalingen (na enkele generaties dus) zal het genproduct steeds langer worden, waardoor het eiwit slecht of niet functioneert. (loss of function).
Hierdoor treedt de genetische aandoening op
Vanaf 36 repeats treedt de ziekte van Hungtingon op.
HTT-gen
25 CAG-repeats
HTT-gen
45 CAG-repeats
HTT-gen
5
90 CAG-repeats
HTT-gen
7
125 CAG-repeats
Figuur 146 (Li) de stamboom van een familie met de ziekte van Huntington (Re) Het aantal CAG-herhalingen voor de aangedane familieleden. Het aantal repeats in het HTT-gen neemt elke generatie toe. Het gen (en het eiwit) wordt dus – per generatie – langer. Vanaf 36 herhalingen treedt het fenotype op (persoon 2 is dus gezond).
Fenotypisch gaat het hierbij om aandoeningen van het zenuwstelsel, bv.
Fragile X-syndroom, een X-gebonden recessieve aandoening (zie 20.1.11)
Ziekte van Huntington, een autosomaal dominante aandoening (20.1.6)
De ziekte van Huntington treedt pas op vanaf de leeftijd van 40 jaar. Bij patiënten met een groter aantal CAG-herhalingen kan dit al vroeger zijn (zelfs op kinderleeftijd). Dit fenomeen heten we anticipatie. M.a.w. hoe meer repeats, hoe vroeger het fenotype zichtbaar wordt.
18.5 Mozaïcisme
In. 6.1.2 leerden we al dat mozaïcisme vaak voorkomt bij de mens. Bij mozaïcisme vertonen sommige cellen in het lichaam kleine wijzigingen in hun genoom. In de meeste gevallen merken we hier echter niets van.
Figuur 147 De verschillende soorten mozaïcisme. De willekeurig gekleurde of gestippelde zones hebben een ander genotype dan de rest van het organisme.
We onderscheiden verschillende types mozaïcisme:
Somatisch mozaïcisme: sommige cellen van het lichaam (of embryo) hebben een ander genotype. Hiervan zal het effect enkel merkbaar zijn als het gaat om een groot aantal lichaamscellen (zie ook 16.5.2.2).
Gonadaal mozaïcisme: sommige kiembaancellen en/of gameten hebben een ander genotype, door spontane mutaties of non-disjunctie
Het effect hiervan zal enkel merkbaar zijn op het nageslacht
Placentair mozaïcisme: sommige cellen van de placenta hebben een ander genotype Dit komt voor bij 2% van alle placenta’s en kan leiden tot placentaire insufficiëntie en groeivertraging.
SOMATISCH GONADAAL PLACENTAIR FUNCTIONEEL
morula
oögonia
spermatozoa
Functioneel mozaïcisme: dit gaat over X-inactivatie: hierbij beschikken alle cellen over hetzelfde genoom, maar wordt in elke cel maar één X-chromosoom gebruikt (zie 18.2.1). Functioneel mozaïcisme is bij elke vrouw aanwezig, in een gemiddelde verdeling van 50%-50% Indien deze verdeling sterk afwijkt van dit gemiddelde dan kan het diploïde voordeel vervallen dat vrouwen normaal hebben: als bv. in 99% van de cellen het ‘gezonde’ X-chromosoom wordt geïnactiveerd dan kunnen vrouwen toch bepaalde kenmerken in het fenotype ontwikkelen.
Bij lapjeskatten is functioneel mozaïcisme zichtbaar in het fenotype. De kleur van de vacht wordt bepaald door een autosomaal gen (witte basiskleur) en een X-gebonden gen (zwarte of rosse kleurvlekken). Door X-inactivatie zal in elke cel van de vacht maar één van beide kleuraccenten tot uiting komen (zwart of ros).
18.6 Mitochondriale overerving
18.6.1
Mitochondriaal DNA
Mitochondria zijn celorganellen die fungeren als energiecentrales. Tijdens de evolutie zijn ze ontstaan als vrij-levende bacteriën en later werden ze opgenomen in een samenwerkingsverband (endosymbiose) met eukaryote cellen Dit gaf een voordeel voor beide: voor de ene betekende het bescherming, voor de andere een meer efficiëntere energieproductie.
Mitochondriën zijn celorganellen uit het cytoplasma die hun eigen DNA bevatten (buiten de celkern dus).
Dit mitochondriële DNA (mtDNA) is circulair, haploïd en bevat 16,569 baseparen die coderen voor slechts 13 eiwitten die rol spelen in het metabolisme van de mitochondriën zelf. Daarnaast worden uit het mtDNA enkele tientallen RNA-moleculen (bv. tRNA) gesynthetiseerd.
Figuur 148 Het mitochondriale, circulaire DNA lijkt (niet toevallig) heel sterk op circulair, bacteriëel DNA (Jorde, 2016)
18.6.2
Bij de bevruchting brengen de zaadcellen amper cytoplasma mee en de weinige, paternale mitochondriën worden vernietigd, waardoor de zygote uiteindelijk enkel mitochondria van de eicel ontvangt (zie ook 5.4).
mtDNA wordt uitsluitend van de moeder overgeërfd
Mitochondriale aandoeningen
Aandoeningen die veroorzaakt worden door mutaties in het mtDNA worden enkel overgeërfd worden via de maternale lijn. Dit betekent dat een moeder de mutatie doorgeeft aan al haar kinderen, maar dat enkel haar dochters dit aan hun kinderen kunnen doorgeven.
Aandoeningen die veroorzaakt worden door defecten in het mtDNA zijn vaak spieraandoeningen (door verstoorde energieproductie in de spiercellen treedt spierzwakte op).
JAAR: 2020
Aan de Universiteit Gent verwacht het team van prof. Björn Heindryckx binnen de 5 jaar de eerste drie-ouder-baby. Dit is een kind van ouders waarbij de moeder lijdt aan een mitochondriale aandoening. De oplossing voor zo’n koppel is om een derde biologische ouder te betrekken in het proces: hierbij wordt een gedoneerde eicel gecombineerd met die van de wensmama Men verwijdert het DNA uit de celkern van de donorvrouw en vervangt dat door het DNA van de wensmoeder. De ‘nieuwe’, gecombineerde eicel bevat normale, gezonde mitochondriën èn het genetisch materiaal van de wensmama
Hierdoor wordt de mitochondriale aandoening niet doorgegeven aan het nageslacht.
Bron, De Morgen, 10/03/2020 https://www.demorgen.be/tech-wetenschap/een-baby-met-drie-biologische-ouders-ugentstaat-dichtbij-de-doorbraak~bc1739b9/
18.6.3 Mitochondriale Eva
Een interessante toepassing van mtDNA is de menselijke afstamming.
Doordat mtDNA niet deelneemt aan crossing-over wordt het niet gerecombineerd. Over meerdere generaties heen wordt het dus quasi onveranderd doorgeven.
Slechts over hele lange tijdsperiodes (duizenden jaren) worden kleine, spontane mutaties geaccumuleerd in het mtDNA. Deze kleine mutaties laten toe van de afkomst van migratiestromen geografisch uit te tekenen. Aan de hand van mutatiepatronen in mtDNA kan bv. bepaald worden wanneer een populatie zich heeft opgesplitst.
Figuur 149 de Out of Africa-migratietheorie is gebaseerd op mtDNA
Elke letter stelt een subtype mtDNA voor. L1 is het startpunt.
Door zo terug te keren in de tijd leren we hieruit dat elke mens afkomstig is van een ‘oermoeder’, die de bron heeft gevormd van alle menselijke, mitochondriale DNA.
Deze Afrikaanse vrouw, ‘Mitochondriale Eva’, leefde zo’n 200.000 jaar geleden in het Zuid-Oosten van Afrika, in een kleine homo-sapiens gemeenschap van hoogstens enkele families. We weten niet hoe ze eruitzag, maar uit studie van het mtDNA weten we dat haar dichtste, moderne verwanten de San-people zijn in het huidige Botswana & Namibië (Mukherjee, 2017).
Voor het Y-chromosoom bestaat een gelijkaardig iets, de Y-chromosomale 'Adam'.
18.7 De novo-mutaties
Soms treden genetische afwijkingen op in families waar niemand met een dergelijke aandoening bekend is. Hoewel dit niet per definitie een overerving uitsluit (denk bv. aan zeldzame, autosomaal recessieve aandoeningen), kan het wel degelijk zijn dat het oorzakelijke gendefect voor het éérst in die familie is opgetreden.
Dit heten we een de novo (letterlijk: 'een nieuwe') mutatie. Hierbij is dus een nieuw, mutant allel ontstaan, door
§ Een mutatie in de kiembaan bij een van de ouders
§ Een mutatie in een van de vroege celdelingen van het embryo.
Dominante, lethale allen ontstaan bijna altijd door een de novo mutatie. Hoe zou dit komen?
18.8 Oefeningen
Oefening 27.
Het ‘long QT-syndroom’ is een autosomaal dominante aandoening die leidt tot hartritmestoornissen. De penetrantie ervan is gereduceerd en bedraagt 80%. Hoe groot is de kans dat een koppel – waarvan enkel de vrouw één mutant allel bezit – een kind krijgt dat het fenotype zal ontwikkelen?
Oefening 28.
In deze familie komt een erfelijke spieraandoening voor die mitochondriaal wordt overgeërfd. Op welke manier kun je in deze stamboom zien dat het gaat om een mitochondriale overerving? Argumenteer met voorbeelden uit de stamboom.
Oefening 29.
In 2009 werd in de VS een arts aangeklaagd voor het stellen van een verkeerde diagnose bij een kind met een erfelijke afwijking. Twee gezonde ouders kregen er een kindje met Smith-Lemli-Opitz-syndroom, een zeldzame, genetische aandoening met afwijkingen ter hoogte van het hart, een lipverhemeltespleet en mentale beperking. Er waren geen andere gevallen bekend in de familie.
Vragen.
a) Hoe kan het dat 2 gezonde ouders een kindje krijgen met dit syndroom hoewel er verder in de familie niemand bekend was? Kan dat wel? Pleit dit de arts vrij?
b) Totaal onverwacht kregen de ouders een tweede kind met hetzelfde syndroom. Hoe kan dat? Geef minstens één andere verklaring dan in vraag a).
19 Multifactoriële overerving
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (enkel wat aangeduid is met pijlen).
Belangrijke begrippen uit dit hoofdstuk te verklaren (deze staan altijd vetgedrukt).
De term multifactoriële overerving uit te leggen door gebruik te maken van de termen ‘nature’ en ‘nurture’.
Uit te leggen waarom het fenotype van een polygeen kenmerk een normaalverdeling (normaalcurve) volgt.
Een normaalverdeling te schetsen en hierbij aan te geven waar het gemiddelde fenotype ligt en waar de zeldzame fenotypes liggen.
Het belang van intermediaire dominantie toelichten bij polygenie.
Het verschil tussen een continu en een discontinu fenotype te verduidelijken aan de hand van een voorbeeld.
Uit te leggen op welke manier foliumzuur het fenotype van neurale buisdefecten beïnvloedt.
Aan te geven hoeveel foliumzuur een zwangere dient in te nemen en tijdens welke periode van de zwangerschap.
Minstens 5 concrete voorbeelden te geven van kenmerken of aandoeningen bij de mens die multifactorieel worden bepaald
De leerstof uit dit hoofdstuk toe te passen in een oefening.
Te argumenteren of de volgende stellingen juist of fout zijn:
o “De meeste, erfelijke aandoeningen volgen de klassieke overervingspatronen van Mendel.”
o “Het opvolgen van monozygote tweelingen is interessant bij de studie van multifactoriële aandoeningen, omdat zij hetzelfde genotype en fenotype hebben.”
o “Bij multifactoriële aandoeningen kunnen we het fenotype steeds voorspellen vanuit onze kennis van het genotype.”
19.1 Polygenie
19.1.1 Definities en voorbeelden
Tot nu toe besteedden we vooral aandacht aan monogene, Mendeliaanse kenmerken.
Heel wat fenotypische kenmerken en aandoeningen worden echter bepaald door het samenspel van meerdere genen. Elk gen (en eiwit) dragen dan een klein beetje bij aan het totale fenotype. Dit heten we polygenie (of polygene overerving).
Als ook omgevingsfactoren een rol spelen in het bepalen van het fenotype, spreken we van multifactoriële overerving.
Voorbeelden van multifactorieel bepaalde kenmerken & aandoeningen:
Lengte,
Body Mass Index (BMI)
Hoofdomtrek
Huidskleur, haarkleur
Intelligentie
Muzikaliteit,
Sportiviteit, atletisch vermogen
Astma
Diabetes type 1, type 2
Neurale buisdefecten (spina bifida)
Hoge bloeddruk
De vele genen die deze kenmerken aan en aandoeningen helpen bepalen, volgen elk afzonderlijk wel de (eerste twee) wetten van Mendel. Het fenotype ontstaat door een complex samenspel van de aanwezige allelen en omgevingsfactoren. Hierdoor zijn de overervingspatronen niet duidelijk af te leiden.
JAAR: 1870
Francis Galton, neef van Charles Darwin, bestudeerde de erfelijkheid van menselijke kenmerken zoals intelligentie. In zijn boek “Hereditary Genius” trachtte hij de overerving hiervan te verklaren. Doordat intelligentie echter een multifactoriële eigenschap is, die ontstaat uit een complex samenspel van factoren – wat Galton niet wist – bleek het voor Galton te moeilijk om een concreet overervingspatroon te beschrijven. Zijn strikte, genetische determinisme (zie 5.4.4) vormde wel de basis voor de latere eugenetica (zie het Nawoord).
% van de populatie
19.1.2 Polygenie leidt tot een normaalverdeling
Beschouw een fictief gen A (met allelen A,a) dat de lichaamslengte (gestalte) bepaalt. Het allel ‘A’ bepaalt hierbij een ‘grote gestalte’, het allel ‘a’ een ‘kleine gestalte’.
% van de populatie
% van de populatie
Indien A overgeërfd wordt via klassieke, monogene, Mendeliaanse overerving èn A is dominant over a, dan geeft dit:
3 genotypes: AA, Aa en aa 2 fenotypes: groot (75%), klein (25%)
% van de populatie fenotypes fenotypes fenotypes fenotypes aaBB AaBb AAbb aaBb Aabb AaBB AABb AABB aabb
Indien A overgeërfd wordt via klassieke, monogene, Mendeliaanse overerving èn er is sprake van intermediaire dominantie, dan geeft dit
3 genotypes: AA, Aa en aa 3 fenotypes: groot (25%), gemiddeld (50%), klein (25%)
Indien we een gen B (B,b) toevoegen dat precies hetzelfde effect heeft op lichaamslengte als het gen A èn er is sprake van intermediaire dominantie,
dan geeft dit
9 genotypes: aabb, aaBb, aaBB, Aabb, AaBb, 5 fenotypes: 5 gradaties van groot naar klein
Indien we nog 2 genen toevoegen (C en D) die precies dezelfde effecten hebben als de genen A & B, èn er is sprake van intermediaire dominantie,
dan geeft dit
81 mogelijk genotypes: aabbccdd, Aabbccdd, aAbbccdd, … 17 mogelijke fenotypes: 17 gradaties van groot naar klein
Naarmate er genen worden toegevoegd neemt het aantal genotypes en fenotypes dus heel snel (exponentieel) toe.
Het precieze aantal genen dat – in werkelijkheid – de volwassen gestalte bepaald is niet gekend, maar wellicht zijn het er enkele honderden. Zelfs als we zouden aannemen dat het er maar 100 zijn, komt dit overeen met miljarden mogelijke genotypes en fenotypes.
Hierdoor zijn de afzonderlijke fenotypes niet meer van elkaar te onderscheiden en vormen ze – dankzij intermediaire dominantie – een doorlopend continuüm.
We heten dit ook een continu fenotype met normaalverdeling:
% van de populatie
zeldzame fenotypes
2/3e van de populatie
zeldzame fenotypes
gemiddelde
fenotype (bv. gestalte)
Voor polygene kenmerken is er dus een grote, doorlopende variabiliteit, waarbij de meeste mensen zich in de buurt van een bepaald gemiddelde bevinden. Aan beide uitersten van de curve vind je zeldzamere fenotypes (bv. opvallend kleine gestalte, opvallend grote gestalte), maar ook deze behoren strikt gezien tot de normale variatie.
Bij polygene kenmerken lijkt het fenotype van kinderen sterk op dat van hun ouders, maar het is zelden precies hetzelfde. Het fenotype wordt dus niet helemaal ‘intact’ doorgegeven, zoals bij veel Mendeliaanse kenmerken wel het geval is.
Er zijn vaak ook verschillen per geslacht, bv. de volwassen gestalte van een man is gemiddeld 180,5 cm, voor een vrouw 166,5 cm. Mannen en vrouwen hebben voor gestalte dus elk hun eigen normaalverdeling Ook dit verschil is genetisch bepaald.
JAAR: 1830
Nog voor het ontstaan van de genetica, werd de normaalverdeling al ontdekt door wetenschappers als Adolphe Quetelet, een Belgisch bioloog. Door biometrie (opmeten van menselijke eigenschappen) ontdekte hij dat meetwaarden steeds een klokvormige curve vormden (zie Figuur 151). Quetelet was ook de grondlegger van de Body Mass Index (eerst heette dit de Quetelet Index) als maat voor overgewicht en zwaarlijvigheid.
19.2 Multifactoriële overerving
19.2.1 Definities
en voorbeelden
Bij de meeste polygene kenmerken en aandoeningen spelen ook omgevingsfactoren mee en hun op het fenotype is onvoorspelbaar.
Hoe meer genen om omgevingsfactoren betrokken zijn, hoe moeilijker het wordt om het verband tussen genotype en fenotype uit te leggen
De meeste, menselijke eigenschappen zijn multifactorieel bepaald.
Het fenotype wordt hierbij bepaald door één of meerdere genen (nature) & omgevingsfactoren (nurture). Het verband tussen genotype en fenotype is complex en onvoorspelbaar.
Enkele voorbeelden:
§ Er zijn tal van omgevingsfactoren die de lengtegroei tijdens de kinderleeftijd negatief kunnen beïnvloeden, ongeacht het erfelijk potentieel, bv. ondervoeding, chronische ziekte, langdurige medicatiegebruik (bv. corticoïden, Rilatine®)
§ Ook huidskleur is multifactorieel bepaald. De genen die de huidskleur bepalen worden bovendien actiever onder invloed van UV-straling. Dit verklaart waarom de huid donkerder wordt tijdens een zonnige vakantie.
§ Diabetes type 1 is een ziekte waarbij > 50 genen een rol spelen. Toch ontwikkelt de ziekte pas onder impuls van omgevingsfactoren (bv. bepaalde virale infecties). Bij monozygote tweelingen is de concordantie hierdoor ‘slechts’ 50%.
Multifactoriële overerving maakt ook de interpretatie van – steeds populairder wordende – DNA-testen moeilijk. Uit zo’n test kan bv. blijken dat je drager bent van een allel dat de kans op een hartinfarct verhoogt, maar dat betekent nog niet automatisch dat dat fenotype tot uiting zal komen (zie ook verminderde penetrantie, 18.1).
In de context van multifactoriële aandoeningen spreken we daarom vaak over een ‘verhoogd risico’ op een bepaalde aandoening.
Er bestaan al private bedrijven die genoomanalyse aanbieden en op basis van je genotype persoonlijk advies geven om de teruggevonden risico’s te verlagen. Dit kan bestaan uit voedingsadvies, bewegingsadvies, nagaan welke medicatie meer of minder geschikt voor je is enzovoort. Deze vorm van persoonlijk counseling is vaak nog onvoldoende wetenschappelijk onderbouwd, vnl. door de complexiteit van de nature-nurture-interactie.
19.2.2 Foliumzuur en NBD
Een bijzonder voorbeeld van multifactoriële overerving is de impact die foliumzuur heeft op het ontstaan van neurale buisdefecten (NBD).
Dit sluit aan bij de leerstof over neurulatie, zie 8.2
Het sluiten van de neurale buis is een complex, multifactorieel proces dat wordt bepaald door honderden genen en omgevingsfactoren Toch zien we dat neurale buisdefecten niet optreden in een doorlopende variatie. Ofwel is deze aandoening aanwezig, ofwel niet. We heten dit een discontinu fenotype.
Hoewel hier onderliggend ook een normaalverdeling aanwezig is (zie Figuur 152), komt het fenotype pas tot uiting vanaf een bepaald aantal gemuteerde allelen (als een bepaald percentage van de allelen een mutatie vertoont). Boven die drempelwaarde is het sluitingsproces van de neurale buis verstoord en treedt bv. spina bifida op. Onder die drempelwaarde sluit de neurale buis op normale wijze
Neurale buisdefecten zijn multifactorieel bepaald, maar treden pas op vanaf een bepaalde drempel. Het fenotype is discontinu.
Bv. spina bifida
drempel voor spina bifida
spina bifida % van de populatie
geen spina bifida
Figuur 152 Foliumzuur en het discontinu fenotype van NBD
Van foliumzuur is het bewezen dat het de drempel verhoogt waarboven NBD kunnen optreden. (Op Figuur 152 betekent dit dat de drempel naar rechts verschuift ) Hierdoor worden heel wat neurale buisdefecten vermeden, bij mensen die hiervoor erfelijke aanleg hadden.
Alle zwangeren worden aangeraden om 400 µg foliumzuur per dag in te nemen, vanaf de zwangerschapswens tot op het einde van de 12e week.
19.2.3
Slotbeschouwing na de hoofdstukken 18, 19 en 20
Hoewel de Wetten van Mendel een elegante en eenvoudige verklaring geven voor het overerven van kenmerken, zijn er – helaas – weinig eigenschappen van de mens die deze wetten strikt volgen.
Zo zijn er de niet-Mendeliaanse mechanismen (zie hoofdstuk 18) die de klassieke overervingspatronen vertroebelen. Daarnaast zijn heel wat eigenschappen bepaald door meerdere genen (polygenie) èn omgevingsfactoren: multifactoriële overerving.
Om het belang van die multifactoriële overerving nog eens in de verf te zetten, geven we in onderstaande grafiek het verband weer tussen nature (als het aantal allelen dat het fenotype bepaalt) & nurture (aantal omgevingsfactoren dat het fenotype bepaalt):
C. MULTIFACTORIELE
Figuur 153 In de moderne genetica gaat de meeste aandacht naar multifactoriële overerving
Bij punt A vinden we de klassieke, zuivere, Mendeliaanse kenmerken en aandoeningen. Hierbij bepaalt één gen/allel het fenotype, ongeacht de aanwezige omgevingsfactoren. Het fenotype valt met 100% zekerheid af te leiden uit het genotype (=genetisch determinisme). Dergelijke kenmerken zijn zeldzaam.
Bij punt B vinden we fenotypische kenmerken die enkel bepaald worden door de omgeving bv. vitamine C-tekort. Genetische factoren hebben hier totaal geen invloed.
In het grote veld C vinden we de meeste aandoeningen. Ze worden bepaald door een complex samenspel van één of meerdere, soms honderden genen èn omgevingsfactoren. Het verband tussen genotype en fenotype is complex en onvoorspelbaar In de moderne genetica gebeurt in dit veld het meeste onderzoek.
19.2.4
Oefeningen
Oefening 30.
Welk van de volgende menselijke kenmerken heeft een continu fenotype?
o Lengte (gestalte)
o Bloedgroep
o Schisis
o Rhesusfactor
Oefening 31.
Juist of fout?
o Huidskleur
o Bloedgroep
o Rood-groen-kleurenblindheid
o Intelligentiequotiënt (IQ)
Foliumzuur verlaagt de penetrantie van neurale buisdefecten.
Neurale buisdefecten hebben een variabele expressiviteit.
Oefening 32.
Je zus laat haar volledige genoom analyseren en deelt trots het resultaat op haar sociale mediaprofiel. Uit het rapport blijkt dat ze enkele mutante allelen bezit die gelinkt zijn aan (=een verhoogd risico betekenen op) vier multifactoriële aandoeningen, nl. bijziendheid, schildklierziekte, zwangerschapsdiabetes en hoge bloeddruk
Enkel die eerste is in het fenotype merkbaar, ze draagt inderdaad lenzen.
Vind je het kunnen dat je zus deze gegevens op internet deelt?
Mag het sociale-mediabedrijf deze gegevens gebruiken om nuttige advertenties te sturen (bv. brillen/lenzen, gezonde voeding ter preventie van hoge bloeddruk)?
Vertellen deze gegevens iets over jouw genoom? Hoezo?
20 Klinische genetica
Leerdoelen
Na het studeren van dit hoofdstuk moet je in staat zijn om …
Figuren uit dit hoofdstuk toe te lichten.
Figuren uit dit hoofdstuk te benoemen (enkel wat aangeduid is met pijlen).
Belangrijke begrippen uit dit hoofdstuk te verklaren (deze staan altijd vetgedrukt).
Voor elke Mendeliaanse aandoening in dit hoofdstuk een beschrijving te geven van het genotype en fenotype (incl. het eiwit dat een sleutelrol speelt).
Te definiëren wat bedoeld wordt met een ‘zeldzame aandoening’.
De methode van de traditionele gentherapie (1990-2000) kort te vergelijken met die van de hedendaagse gentherapie (2010-heden).
Twee redenen te geven waarom het gebruik van een virus als vector voor gentherapie risicovol is.
De onderdelen en de werking van CRISPR-cas9 beknopt te beschrijven.
Te argumenteren waarom CRISPR-cas9 veelbelovend is, maar momenteel nog niet breed uitgerold kan worden als gentherapeutische behandeling.
Het genotype van trisomie 13,18 en 21 kunnen omschrijven
Aan elke trisomie (13,18,21) de juiste syndroom-naam toe te kennen.
Minstens 5 morfologische kenmerken van het syndroom van Down te omschrijven.
Minstens 3 typische, medische problemen bij het syndroom van Down te omschrijven.
Enkele fenotypische kenmerken van het syndroom van Edwards en het syndroom van Patau te omschrijven.
De prognose van trisomie 13,18 en 21 te beschrijven.
Aan te geven vanaf wanneer een NIP-test voldoende betrouwbaar is.
De methode van de NIP-test te bespreken.
Uit te leggen in patiëntentaal wat bedoeld wordt met “de NIP-test is een screeningstest, geen diagnostische test”.
Minstens 3 voordelen èn 3 nadelen van de NIP-test te omschrijven.
Uit te leggen waarom placentair mozaïcisme het resultaat van de NIP-test kan beïnvloeden.
Toe te lichten waarom het belangrijk is dat ouders voldoende geïnformeerd worden over de NIP-test, voorafgaand aan de test.
Het genotype van het syndroom van Turner & Klinefelter te kunnen omschrijven.
Enkele morfologische en fenotypische kenmerken van het syndroom van Turner en het syndroom van Klinefelter te omschrijven.
Te bespreken wat ‘gonadale dysgenese’ inhoudt en op welke manier dit de fertiliteit van meisjes met het syndroom van Turner beïnvloedt
De prognose van het syndroom van Turner en het syndroom van Klinefelter te beschrijven.
Een verklaring te geven voor het verband tussen de lichaamsgroei (gestalte) en het aantal X-chromosomen.
Het karyotype van een microdeletie te noteren volgens de ISCN-notatie.
Uit het karyotype v. een microdeletie te kunnen afleiden over welke mutatie het gaat.
Te beschrijven hoe Comparative Genomic Hybridization (CGH) een rol speelt in het diagnosticeren van een microdeletie.
Enkele voor- en nadelen van CGH kort te omschrijven.
Voor elk microdeletie-syndroom in dit hoofdstuk een korte beschrijving te geven van het genotype en fenotype.
Uit te leggen waarom imprinting een rol speelt bij Prader-Willi syndroom en Angelman syndroom.
Te argumenteren waarom kanker een genetische ziekte kan worden genoemd.
Uit te leggen wat bedoeld wordt een (proto)-oncogen a.d.h.v. een voorbeeld.
Uit te leggen wat bedoeld wordt een tumor-suppressorgen a.d.h.v. een voorbeeld.
Te verklaren waarom een tumor-suppressorgen recessief is, maar toch dominant wordt overgeërfd.
Het belang van BRCA1 en BRCA2 in de ontwikkeling van borstkanker beknopt te omschrijven (incl. een ruwe schatting van het voorkomen op dragerschap en het lifetime-risk op borstkanker).
Een korte omschrijving te geven van het begrip eugenetica.
De leerstof uit dit hoofdstuk toe te passen in een oefening.
20.1 Mendeliaanse aandoeningen
20.1.1 Mucoviscidose
20.1.1.1 Beschrijving
Voorkomen 1/2500
1/25 is drager
Overerving Autosomaal recessief
Mutant allel CFTR (zie 2.1), locus 7q31.2
Eiwit
Fenotype:
CFTR: transmembraan eiwit, chloorkanaal
Het gestoorde chloortransport over de membranen leidt tot taaiere lichaamssecreties.
In de longen leiden taaie slijmen tot verstoppingen van kleine luchtwegen, chronische bronchitis en progressieve achteruitgang van de longfunctie. In het maagdarmstelsel zorgen taaie secreties (gal, pancreas) voor een slechte vertering.
Mucoviscidose is de vaakst voorkomende, autosomaal recessieve aandoening in onze streek. Naar mucoviscidose wordt sinds 2019 gescreend d.m.v. de hielprik
20.1.1.2 Oefening
Oefening 33.
Hoe groot is de kans dat 2 gezonde, niet-verwante, ouders een kind krijgen met mucoviscidose, als je weet dat dragerschap in de populatie 1/25 bedraagt.
De Europese Commissie stelt dat een ziekte is als ze bij minder dan 1/2000 mensen voorkomt. ?
20.1.2 Fenylketonurie (PKU)
20.1.2.1 Beschrijving
Voorkomen 1/10.000
Overerving Autosomaal recessief
Mutant allel PAH, locus 12q23.2
Eiwit
Fenotype:
Fenylalaninehydroxylase, enzym dat fenylalanine (Phe) afbreekt.
Stapelingsziekte door opstapeling van Phe in de weefsels.
Patiënten zijn asymptomatisch bij de geboorte. Door aanbod van Phe (via de voeding) treedt zenuwschade op. Dit leidt tot ontwikkelingsvertraging, mentale retardatie en epilepsie.
De behandeling van PKU bestaat uit een levenslang fenylvrij dieet.
Een correct gevolgd dieet verhindert dat het PKU-fenotype tot uiting komt. Anders gezegd: de penetrantie van fenylketonurie wordt gereduceerd door de voeding.
Naar fenylketonurie wordt gescreend d.m.v. de hielprik.
20.1.2.2 Oefening
Oefening 34.
Jutta is een 28-jarige, gezonde vrouw die sinds 2 jaar samen is met Wouter, een 31jarige, gezonde man. Het koppel heeft een kinderwens. In de familie van Wouter komen geen erfelijke aandoeningen voor. Bij Jutta komt fenylketonurie voor in de familie. Frits, de vader van Jutta is immers gekend met fenylketonurie. Zijn aandoening is sinds jaren goed onder controle. Jutta maakt zich wel zorgen dat haar kinderen ook fenylketonurie zouden kunnen krijgen. Ze herinnert zich dat haar moeder, Martha, zich vroeger liet testen in Leuven en uiteindelijk geen drager bleek te zijn. Jutta’s oudere broer Carl is ook gezond. De kans op dragerschap in de algemene bevolking is 1 op 50.
Vragen.
a. Teken de stamboom van de familie van Jutta en vul aan met alle gegeven (of af te leiden) informatie uit de opgave. Duid Jutta aan met een pijl.
b. Jutta zou zich graag laten testen op dragerschap. Hoe groot is de kans dat zij drager is?
c. Hoe groot is de kans dat Jutta en Wouter een kindje krijgen met fenylketonurie?
d. Welke fenotypische kenmerken verwacht je bij Frits aan te treffen?
20.1.3
Oculo-cutaan albinisme (OCA)
20.1.3.1 Beschrijving
Voorkomen 1/17.000
Overerving Autosomaal recessief
Mutant allel TYR, locus 11q14 3
Eiwit
Fenotype:
Tyrosinase: enzym dat tyrosine omzet tot melanine
Melanine (het huidpigment) kan niet worden aangemaakt, waardoor de huid vatbaarder is voor UV-schade & ontwikkelingsstoornissen optreden van het visuele zintuig (vooral het netvlies).
Bleke, witte huidskleur
Wit haar (soms eerder blond of rood)
thv de ogen: blauwe ogen (soms eerder rood tot paars), verminderd zicht of blindheid, nystagmus (snelle, zijwaartse bewegingen van de ogen), scheelzien, …
OCA is ook een voorbeeld van epistase. Dor de mutatie in tyrosinase kan immers geen melanine (huidpigment) worden aangemaakt, ongeacht het achterliggende genotype voor huidskleur. Dit laatste wordt multifactorieel bepaald.
20.1.3.2 Oefening
Oefening 35.
Hieronder vinden we drie families waarin albinisme voorkomt. Bepaal voor elke familie het genotype van de ouders en de nakomelingen. (Soms zijn meerdere genotypes mogelijk; dragers zijn niet weergegeven!)
20.1.4 Sikkelcelanemie
20.1.4.1 Beschrijving
Voorkomen 1/17.000, maar frequenter in West-Afrika
Overerving Autosomaal recessief
Mutant allel HBB, locus 11p15 4
Eiwit b-globine, deel van Hemoglobine (zorgt voor O2-transport)
Fenotype:
Het defecte hemoglobine (HbS)’ koekt’ samen waardoor RBC een sikkelvorm krijgen.
Deze sikkelcellen…
…hebben een kortere levensduur (± 17 dagen ipv ± 120 dagen bij normale RBC), waardoor patiënten een chronische bloedarmoede (anemie) ontwikkelen.
…kunnen kleine bloedvaten verstoppen (vaso-occlusieve-crisis), waardoor orgaanschade optreedt.
… worden uitgefilterd door de milt, waardoor vroegtijdig miltfalen optreedt (± leeftijd van 12 jaar). Aangezien de milt ook ziekteverwekkers uit het bloed filtert, brengt dit een risico op ernstige infecties met zich mee.
Sikkelcelanemie heeft een grote impact op biological fitness:
Bij homozygoten is deze sterk verlaagd door een hoge mortaliteit op jonge leeftijd: 7% van de patiëntjes onder de 5 jaar.
Bij heterozygoten in West-Afrika is deze sterk verhoogd doordat ze veel minder makkelijk geïnfecteerd worden door de malaria-parasiet. Het voorkomen van sikkelcelanemie volgt om die reden – geografisch – de verspreiding van malaria.
20.1.4.2 Oefening
Oefening 36.
Hoe groot is de kans dat Diallo (zie pijl) drager is van sikkelcelanemie?
20.1.5 Achondroplasie
20.1.5.1 Beschrijving
Voorkomen 1/15.000
Overerving Autosomaal dominant
Mutant allel FGFR3, locus 4p16.3
In 80% van de gevallen zijn er geen andere gevallen in de familie gekend en betreft het dus een de novo mutatie. Dat maakt het een van de meest voorkomende ziekteverwekkende de novo mutaties.
Eiwit fibroblast-groeifactor receptor type 3:
Fenotype:
Door de defecte FGFR3-receptor wordt de lengtegroei verstoord, wat leidt tot dwerggroei. De volwassen gestalte is gemiddeld 132 cm (mannen) en 125 cm (vrouwen). Andere symptomen zijn:
Verkortingen van de ledematen (vnl. proximaal)
Korte vingers
Macrocefalie (vnl. groot voorhoofd)
Trager bereiken van de motorische mijlpalen (vnl. door hypotonie)
20.1.5.2 Oefening
Oefening 37.
Kunnen twee ouders met achondroplasie een kind krijgen zonder de aandoening? Zo ja, hoe groot is die kans?
20.1.6 De ziekte van Huntington
20.1.6.1
Beschrijving
Voorkomen 1/15.000
Overerving Autosomaal dominant
Mutant allel HTT, dynamische mutatie, locus 4p16.3 (vlakbij FGFR3)
Eiwit Huntingtine, een eiwit die zenuwcellen beschermt tegen schade.
Fenotype:
Zonder huntingtine treedt de volgende symptoomtriade op vanaf de leeftijd van 40 jaar:
1) cognitieve achteruitgang: evolueert naar dementie.
3) motorische stoornissen (meestal in een later stadium):
Stoornis van de vrijwillige bewegingen: onhandigheid, moeite met spreken, coördinatie en evenwicht
Stoornis van de onvrijwillige bewegingen: chorea ('reidans') = onregelmatige, doelloze bewegingen in verschillende delen van het lichaam.
De ziekte is progressief en leidt tot de dood na 15 à 20 jaar.
Er is geen oorzakelijke behandeling.
Hoewel deze aandoening pas laat tot uiting komt is de penetrantie volledig (100%). De meeste mensen die kans hebben om de ziekte van Huntington over te erven laat zich hiervoor niet testen. Ze willen m.a.w. niet weten of ze rond hun 40e levensjaar de symptomen van deze ernstige ziekte zullen ontwikkelen.
Hoe denk jij hierover? Zou jij je laten testen?
20.1.6.2 Oefening
Oefening 38.
Een koppel bestaat uit een 38-jarige man die homozygoot is voor de ziekte van Huntington en een 27-jarige vrouw die heterozygoot is voor de ziekte.
a. Hoeveel procent van hun nakomelingen zal de ziekte hebben?
b. Speelt niet-Mendeliaanse overerving een rol bij deze overerving?
c. Hebben mannelijke nakomelingen meer kans op de ziekte?
20.1.7 Ziekte van Marfan
20.1.7.1
Beschrijving
Voorkomen 1/4.000
Overerving Autosomaal dominant
Mutant allel FBN1, locus 4p16.3 (vlakbij FGFR3)
Eiwit
Fenotype:
Fibrilline, een elastisch eiwit
Het tekort aan fibrilline zorgt voor een verlies aan steun elasticiteit in het bewegingsstelsel. De penetrantie van deze aandoening is volledig.
De klassieke symptomen van Marfan zijn:
Overmatige groei van de lange beenderen, waardoor grote gestalte, erg wijde armspan, lange vingers (arachnodactylie)
Lakse, overstrekbare gewrichten
Aneurysma (uitzetting) van de aorta ascendens, met ruptuur en plotse dood als complicatie.
Verplaatsing van de ooglens, ernstige bijziendheid
> het duimteken bij Marfan:
De combinatie van lakse gewrichten en overgroei van de vingers zorgt ervoor dat de eindkootjes van duim en pink elkaar overlappen bij het omgeven van de contralaterale pols.
Figuur 154 Het duimteken bij Marfan
20.1.7.2 Oefening
Oefening 39.
Een man met de ziekte van Marfan heeft 4 kinderen met een vrouw zonder de aandoening. Hoe groot is de kans dat alle kinderen de ziekte van Marfan hebben? ? ? ? ?
Mutant allel COL1A1, locus 7q21.33 & COL1A2, locus 7q21.3
Eiwit
Type 1 collageen, is een belangrijk structuureiwit in bindweefsels, pezen, ligamenten, huid, kraakbeen en bot.
Fenotype:
Door een tekort aan type 1 collageen zijn de botten erg broos (botfragiliteit) waardoor breuken optreden na een klein trauma of soms zelfs spontaan.
Andere symptomen zijn:
Groeiachterstand,
Doofheid (breken van de gehoorsbeentjes)
Tandafwijkingen
Blauwe sclerae.
Er is geen oorzakelijke behandeling voor OI.
De expressiviteit van OI is sterk variabel (mild tot ernstig fenotype). Mild betekent dat fracturen pas sporadisch optreden tijdens de kinderjaren. Bij matige vormen treden talrijke (vaak honderden) fracturen op vanaf de geboorte tot de puberteit. De meest ernstige vormen zijn lethaal in utero of kort na de geboorte, dit door talrijke fracturen die optreden tijdens het intra-uteriene leven of de geboorte zelf.
20.1.8.2 Oefening
Oefening 40.
In deze familie zijn verschillende personen bekend met osteogenesis imperfecta. Stel dat we het gen COL1A1 voorstellen door de letter ‘N ‘(N,n). Kun je dan het genotype bepalen van de met pijlen aangeduide familieleden?
20.1.9 Hemofilie A en B
20.1.9.1
Beschrijving
Voorkomen 1/12.000 (frequenter bij jongens; ±1/5000)
Overerving X-gebonden recessief
Mutant allel F8, locus Xq28 of F9, locus Xq27 1 Eiwit stollingsfactoren VIII (hemofilie A) of IX (hemofilie B).
Fenotype
Het klinisch beeld bestaat uit (vnl. spontane) bloedingen:
Bloedingen in de gewrichten (vnl. knieën, enkels)
Bloedingen in de spieren.
Hersenbloedingen, meest frequent bij neonaten.
Neusbloedingen
Bloedverlies via urine of stoelgang.
Daarnaast treedt bij traumatische letsels doorgaans ook buitensporig bloedverlies op.
Hemofilie kent ook een variabele expressiviteit. Het fenotype is hierbij afhankelijk van hoeveel stollingsfactor het lichaam zelf nog aanmaakt (een factorgehalte van < 1% betekent een ernstige vorm van hemofilie); De behandeling bestaat uit het preventief, IV toedienen van de ontbrekende stollingsfactor (vb. 3 x per week).
Hemofilie wordt ook 'the royal disease' genoemd door het groot aantal gevallen bij de nazaten van de Britse Queen Victoria (1819-1901). Haar 8e kind prins Leopold 'the Bleeder Prince' leed onder vele gewrichtsbloedingen en stierf na een klein hoofdtrauma in Cannes.
20.1.9.2 Oefening
Oefening 41.
Op een dag spreekt David, je buurman, je aan over de ziekte hemofilie. Zelf lijdt hij hieraan, net als de jongste van zijn twee jongere zussen (een monozygote tweeling) Ook zijn overleden grootvader (langs moederszijde) leed aan hemofilie. De beide ouders van David, zijn vrouw en zijn enige dochter Katlijn, zijn dan weer niet ziek. David vertelt je dat Katlijn recent gehuwd is met Isaac, een gezonde niet-verwante man.
a. Teken hieronder de stamboom van de familie van David en vul aan met alle gegeven (of af te leiden) informatie uit de opgave. Duid David aan met een pijl.
b. Is er een kans dat de kinderen van Katlijn en Isaac hemofilie zullen hebben? Bespreek de mogelijke genotypes en fenotypes voor hun kinderen. Teken hierbij minstens een recombinatierooster.
20.1.10 Duchenne spierdystrofie
20.1.10.1 Beschrijving
Voorkomen 1/20.000 (frequenter bij jongens; ±1/3500)
Overerving X-gebonden recessief
Mutant allel DMD, locus Xp21.1
Eiwit
Dystrofine fungeert als een anker voor spiercellen. Verlies van dit eiwit resulteert in vroegtijdige degeneratie van spiervezels.
Fenotype Het belangrijkste fenotypische kenmerk is spierzwakte die klinisch duidelijk wordt rond de leeftijd van 2 à 3 jaar.
De spierzwakte vangt aan thv de onderste ledematen, waardoor het kind eerst moeite krijgt met stappen en lopen.
> het teken van Gower bij Duchenne:
De kinderen komen hierbij op een typische manier recht vanuit zit op de grond, waarbij ze met de handen steunen op de knieën.
Kinderen worden gemiddeld rolstoelgebonden vanaf 14 jaar. In het latere stadium van de ziekte ontstaan er problemen met het hart (verzwakte hartspier) en ademhaling (verzwakte ademhalingsspieren). Er bestaat geen oorzakelijke behandeling. Rond de leeftijd van 20 jaar is doorgaans beademingsondersteuning nodig. De gemiddelde levensverwachting ligt op dit moment rond de 35 jaar.
20.1.10.2 Oefening
Oefening 42.
Hoe groot is de kans dat Benjamin en Julia, een gezond koppel, kinderen krijgen met de ziekte van Duchenne, als je weet dat de vader van Julia lijdt aan de ziekte van Duchenne? (teken minstens een stamboom).
Figuur 155 Het teken van Gower
20.1.11
Fragile X-syndroom
20.1.11.1 Beschrijving
Voorkomen ± 1/4000 jongens en ± 1/8000 meisjes
Overerving X-gebonden dominant
Mutant allel dynamische mutatie (CGG) in het FMR1-gen op locus Xq27.3 De expressie van het FMR1-gen wordt stilgelegd vanaf 200 of meer CGG-herhalingen Deze herhalingen creëren bovendien een bijkomende insnoering aan het uiteinde van het X-chromosoom. Onder de microscoop ziet het X-chromosoom er hierdoor 'gebroken' uit ('fragiele X').
Eiwit Het FMR1-eiwit speelt een rol in het transport van mRNA in zenuwcellen.
Fenotype:
Ontwikkelingsproblemen (motoriek, taal), evoluerend naar een mentale retardatie met een IQ van gemiddeld 40. Strikt medische problemen zijn er verder niet. Gedragingen en interacties die lijken op autisme worden vaak gezien vb. fladderen, moeite met sociale interactie zoals oogcontact, hypersensitiviteit, etc...
De typische dysmorfe kenmerken zijn: lang, smal gezicht met grote, afstaande oren macro-orchidie (grote testes), na de leeftijd van 8 jaar
Bij vrouwen die heterozygoot zijn voor het mutante FMR-1 allel is de penetrantie gereduceerd (±50%). Vaak zijn er lichte kenmerken bv. een randnormale intelligentie
20.1.11.2
Oefening
Oefening 43.
Willow (pijl), 21 jaar, en Scott hebben een kinderwens. In de familie van Willow zijn verschillende mensen met het Fragile X syndroom bekend. Zelf werd zij hier nooit op getest, hoewel ze tijdens haar schoolloopbaan veel leerproblemen had, net als haar eigen moeder.
Kan Willow ook het mutante FMR-allel bezitten?
Hoe groot schat je de kans dat Willow en Scott een kind krijgen met Fragile X-syndroom? ?
20.2 Gentherapie
20.2.1
Gentherapie is het behandelen van ziektes door het genoom van de patiënt te wijzigen. Hoewel het principe achter gentherapie vrij eenvoudig is, is de uitvoering ervan heel complex. Er zijn immers heel wat hindernissen te overwinnen, bv.:
*Hoe krijg je een wild-type allel in het genoom van de patiënt?
*Hoe kun je zeker zijn dat het effect van gentherapie langdurig behouden blijft?
*Hoe voorkom je bijwerkingen of schade aan andere delen van het DNA?
*Kan de techniek ook gebruikt worden voor minder goede doeleinden?
Bovendien moeten we er ook rekening mee houden dat de rol van veel genen niet gekend is.
Of omgekeerd: dat niet alle genen gekend zijn die bijdragen aan een ziektebeeld.
Gentherapie wordt ofwel uitgevoerd
In vivo: in het lichaam van de patiënt zelf (bv. door injecties, infusie)
…Ex vivo: hierbij zijn eerst cellen van de patiënt weggenomen (bv. door bloedafname). Deze worden behandeld in het labo, waarna ze terug worden ingebracht (bv. infusie).
Monogene aandoeningen zoals de ziekte van Huntington, de ziekte van Duchenne of sikkelcelanemie zijn een dankbaar studieonderwerp voor gentherapie, omdat deze ernstige ziektes slechts door één enkel gendefect worden veroorzaakt. Voor deze aandoeningen biedt enkel gentherapie een kans op definitieve genezing.
Traditionele gentherapie (ca. 1990 – 2000)
Methode:
§ Inbrengen van een wild-type allel bij de patiënt.
§ Een virus wordt hierbij gebruikt als vector (transportmiddel). Virussen bezitten immers van nature uit de eigenschap om DNA in een gastheer in te brengen.
Toen het Human Genome Project aanving (ca. 1990) was het optimisme over gentherapie zeer groot. Talrijke studies werden opgestart en de eerste resultaten waren schitterend. Zo slaagden Amerikaanse wetenschappers erin om bij twee kinderen met SCID (een monogene immuunziekte) een wild-type allel in te brengen. Hierdoor konden die kinderen terug antistoffen en afweercellen aanmaken. Enkele jaren later werden in Italië gelijkaardige resultaten bekomen (Blaese et al., 1995; Bordignon et al., 1995).
Omstreeks het jaar 2000 keerde het succes echter. In Parijs werden 22 SCID-patiëntjes behandeld met gentherapie. Vijf kinderen ontwikkelden een ongecontroleerde deling van hun witte bloedcellen (leukemie) als gevolg van de behandeling. Eén kind overleed.
Wellicht had het virus het wild-type allel op een verkeerde plaats in het genoom van de bloedcellen ingebouwd, waardoor de bloedkanker ontstond (Sheridan, 2011).
Nog in die periode kwam uit de Verenigde Staten het bericht dat Jesse Gelsinger, een 18-jarige deelnemer aan een wetenschappelijke studie, was overleden aan de gevolgen van gentherapie Zijn lichaam reageerde onverwacht op de virale vector (adenovirus) en hij stierf aan multipel orgaanfalen. Achteraf bleek dat de onderzoekers gelijkaardige bijwerkingen bij proefdieren niet ernstig hadden genomen
De negatieve publiciteit diende een mokerslag toe aan de hype rond gentherapie. Een belangrijke les hierbij was dat het gebruik van een virus als vector risico’s met zich meebrengt, bv. het risico op een verkeerde inbouw in het genoom van de patiënt, maar ook het risico op een ernstige afweerreactie.
In de periode 2000-2010 werd het wetenschappelijk onderzoek naar gentherapie wel verdergezet, maar de focus kwam meer te liggen op patiëntveiligheid, o.m. door het nauwgezetter opvolgen van studies door de overheid. Er was ook meer aandacht voor complexe, multifactoriële aandoeningen zoals diabetes, hoge bloeddruk en kanker.
Eind 2012 gaf de Europese Commissie voor het eerst groen licht voor het ter beschikking stellen van een traditionele, gentherapeutische behandeling (Glybera®) voor de behandeling van een erfelijke stoornis van het vetmetabolisme. Een adenovirus levert hierbij het wild-type allel.
20.2.2 Het CRISPR-tijdperk (ca. 2010 – …)
Methode:
§ Aanpassen van DNA (genome editing) d.m.v. CRISPR-cas9
§ Behalve virussen worden ook non-virale vectoren gebruikt bv. (gouden) nanodeeltjes
CRISPR-cas9 (Clustered Regularly Interspaced Short Palindromic Repeats) is de naam van een systeem dat veel bacteriën (bv. S. pyogenes, E. coli) gebruiken om zich te verdedigen tegen virussen.
Telkens zo’n bacterie een aanval van een virus heeft overleefd, bewaart het een stukje van het virale DNA in zijn eigen DNA, als een soort immuun-geheugen.
De volgende stap is dat deze stukjes viraal DNA worden omgezet in een RNA-transcript dat wordt gekoppeld aan een cas9-eiwit. Cas9 fungeert als een schaar die in staat is om dubbelstrengig DNA zeer nauwkeurig te knippen. Het knippen gebeurt alleen op de plaatsen die worden aangegeven door het RNA-transcript wanneer het bindt met complementair DNA.
Cas9 knipt op een zeer specifieke manier en met een precisie van één basepaar.
In bacteriën leidt CRISPR-cas9 op die manier tot het verwijderen van viraal DNA uit het bacteriële DNA. Zo overwint de bacterie makkelijker een herinfectie met hetzelfde virus.
Wetenschappers zagen in dit mechanisme een programmeerbaar hulpmiddel om menselijk DNA te gaan knippen. Dankzij de kennis over het menselijk genoom is het immers mogelijk om stukjes RNA te creëren die cas9 naar een specifieke plek in het menselijk genoom brengen om daar een stukje weg te knippen.
stukje van virale DNA-code
RNA transcript
RNA transcript
DNA van de bacterie (CRISPR-bibliotheek)
Cas9-eiwit viraal DNA
Figuur 156 CRISPR-cas9 mechanisme (vereenvoudigd, naar Klug, 2019)
Hoewel het CRISPR-cas9-mechanisme al eind de jaren ’80 werd ontdekt focuste het onderzoek zich de eerste decennia vooral op het begrijpen van systeem. Nadat de functionaliteit van CRISPR-cas9 duidelijker werd, werd het mechanisme in 2012 op punt gezet door Jennifer Doudna (VS) en Emanuelle Charpentier (FR). Als eersten opperden zij het gebruik van CRISPR-cas9 voor het wijzigen van genetische informatie.
Het bleek een enorm succes: de techniek is snel, relatief eenvoudig aan te leren en goedkoop. In 2020 werd hun ontdekking bekroond met de Nobelprijs (Singh, 2020).
De jaren na de introductie van CRISPR-cas9 gebeurden de eerste experimenten op zoogdieren en in 2015 werd CRISPR-cas9 voor het eerst gebruikt om muizen met de ziekte van Duchenne te behandelen. Door het uitknippen van een gemuteerd DMD-allel en het herintroduceren van het wild-type allel herwonnen de muizen aan spierkracht. Gelijkaardige experimenten gebeuren al met hemofilie (Ousterout et al., 2015).
knip
Doudna en andere wetenschappers beseften al vlug dat de CRISPR-cas9-techniek iets kon wat de traditionele gentherapie niet kon: een DNA-wijziging in de menselijke kiembaan inbrengen bv. door het bewerken van het DNA van een embryo.
Terwijl nagedacht werd over de mogelijke implicaties hiervan verraste de Chinese onderzoeker He Jiankui in 2018 de wereld met de aankondiging dat de eerste CRISPRbaby’s door zijn toedoen waren geboren. Een tweeling, Lulu & Nana, werden geboren in Shenzhen (China) nadat ze als embryo door CRISPR-cas9 waren bewerkt. Bij beiden werd een gen uitgeknipt dat een rol speelt in het aanhechten van het Hiv-virus. Ondanks dat hun vader Hiv-positief was zouden Lulu & Nana dus nooit besmet kunnen worden (Singh, 2020).
Jiankui's aankondiging van het bewerken van embryo's zorgde voor een schokgolf in de wetenschappelijke wereld, ondanks het mogelijk nobele doel dat hij voor ogen had. Het grootste probleem lag in het feit dat Jiankui embryo's had gemanipuleerd zonder dat de langetermijneffecten hiervan bekend waren. Bovendien introduceerde hij een nieuwe, erfelijke DNA-verandering in de menselijke kiembaan. Deze acties werden algemeen beschouwd als illegale medische praktijken, wat leidde tot Jiankui's veroordeling en een gevangenisstraf van drie jaar. Ondertussen zijn Lulu en Nana nog steeds in leven.
Was de intentie van Jiankui volgens jou te verdedigen? Of vind je de kritiek terecht?
Hoewel de kennis over CRISPR-cas9 verder blijft groeien (ook buiten de geneeskunde, bv. in de voedingssector), zijn er momenteel nog geen medische toepassingen. In de toekomst wordt het ongetwijfeld mogelijk om ziektes te behandelen en zelfs te helpen voorkomen door ‘crisperen’ (het wordt stilaan een werkwoord)
Verder onderzoek is wel nodig om de veiligheid en de ethische grenzen van de techniek te bepalen. De interacties tussen genen en de omgeving zijn zodanig complex dat het niet verstandig is om op goed geluk te gaan ‘crisperen, omdat het kan’. De wetenschap heeft hierbij duidelijk lessen getrokken uit het tijdperk van de traditionele gentherapie.
Bv. bij sikkelcelanemie zou CRISPR-cas9 een oplossing kunnen bieden. Maar wat doen we met dragers voor sikkelcelanemie die beschermd zijn tegen malaria? Als we in West-Afrika het sikkelcel-allel uit de populatie ‘crisperen’ zal sikkelcelanemie niet meer voorkomen, maar zullen veel meer mensen malaria krijgen.
Welke keuze kan de wetenschap hier maken volgens jou?
20.3 Aneuploïdie van de autosomen
Hier bespreken we enkele frequente, genetische syndromen die het gevolg zijn van aneuploïdie (zie 1.3.4).
Een syndroom is een ziektebeeld waarbij bepaalde klinische symptomen steeds samen voorkomen. Het fenotype is herkenbaar en voorspelbaar
20.3.1 Trisomie 21 (syndroom van Down)
JAAR: 1888
Eerste beschrijving van mongolisme door J. Langdon-Down. Hij beschreef voor het eerst het syndroom dat nu naar hem is vernoemd en droeg bij aan het begrip en de classificatie van intellectuele beperkingen. Opmerkelijk: hij gebruikte – naar hedendaagse normen - opvallend beledigende en racistische taal in zijn publicaties (Down, 1888). Pas 100 jaar later werd het verband tussen het syndroom en chromosoom 21 gelegd.
20.3.1.1 Voorkomen
Het syndroom van Down (DS, Down Syndrome) komt voor bij ± 1/700 à 1/1000 levendgeborenen en is hiermee de meest voorkomende chromosomale afwijking.
Door evoluties in prenatale screening is het aantal levendgeborenen met DS de laatste jaren gedaald.
Epidemiologie (SPE, 2020)
20.3.1.2 Genotype
47,XX+21 (meisje) of 47,XY+21 (jongen)
DS wordt veroorzaakt door een trisomie van chromosoom 21. Bij ongeveer 0,45% van de menselijke bevruchtingen ontstaat zo'n trisomie 21. Embryo's die ontwikkelen met een trisomie hebben een hogere kans op spontane abortus.
Figuur 157 Cijfers van het Studiecentrum voor Perinatale
20.3.1.3 Fenotype
Het fenotype wordt veroorzaakt door overexpressie van een aantal genen op chromosoom 21. Bij DS-patiënten is er een typische, afwijkende morfologie van hoofd en aangezicht (faciale dysmorfie, zie Figuur 158)
A. DYSMORFE KENMERKEN
Microcefalie: een relatief klein hoofd (microcefalie)
Korte nek met vaak overtollige huid
Upslanting: opwaarts gerichte oogstand
Epicanthusplooi bovenste ooglid bedekt mediale zijde van de ooglidspleet
Vlakke neusbrug
Laag ingeplante oren
Macroglossie: grote tong
Kleine mond met afwijkingen aan tanden, verhemelte...
Daarnaast zijn er vaak ook afwijkingen aan de extremiteiten:
Korte, brede handen met doorlopende, dwarse handplooi
Clinodactylie: gebogen stand van de pink
Sandal gap: ruimte tussen 1e en 2e teen
microcefalie
vlak aangezicht
dwarse handplooi
epicanthus
vlak achterhoofd
vlakke neusbrug
macroglossie upslanting
Sandal gap
Figuur 158 faciale dysmorfie by DS (Rajamahendran, 2014)
B. MEDISCHE PROBLEMEN
Ontwikkelingsachterstand die over het algemeen leidt tot een matig tot ernstige mentale retardatie, achterstand in taal, spraak en motoriek.
Groeiachterstand: jongens worden ±161 cm, meisjes ±147 cm (Myrelid et al., 2002) Neiging tot overgewicht en obesitas (Rubin et al., 1998).
Vroegtijdige 'neurologische' veroudering met frequent Alzheimer-type-dementie.
m.b.t. vruchtbaarheid: mannen met DS zijn doorgaans onvruchtbaar. Vrouwen met DS zijn doorgaans normaal vruchtbaar en kunnen dus zwanger worden.
Bovicelli et. al volgden 30 zwangerschappen bij patiëntes met DS. 3 zwangerschappen resulteerden in een spontaan miskraam. 10 kinderen werden geboren met DS. 18 nietaangedane kinderen werden geboren (waaronder 1 tweeling) (Bovicelli et al., 1982).
20.3.1.4
Diagnose
De diagnose van DS wordt gesteld door pre- of postnataal chromosomenonderzoek Prenataal kan dit door de vruchtwaterpunctie.
Als de diagnose van DS prenataal niet bekend was, wordt deze in 70% van de gevallen klinisch gesteld op de eerste levensdag.
Denk er steeds aan bij een neonaat met dysmorfe kenmerken ter hoogte van het gezicht of extremiteiten; de neonaat met hypotonie (zwakke lichaamstonus) of zwakke reflexen.
20.3.3 Trisomie 18 (syndroom van Edwards)
47,XX+18 (meisje) of 47,XY+18 (jongen)
1/5000 levendgeborenen.
20.3.3.1 Fenotype
Het merendeel van de gevallen overlijdt in utero. De gemiddelde postnatale overlevingsduur bedraagt 2 weken. De meeste kinderen overlijden binnen hun eerste levensjaar.
Het fenotype is variabel
Smal aangezicht met kleine mond en kaak; groot achterhoofd
>50%: aanlegstoornissen van het hart en centraal zenuwstelsel
Aanlegstoornsissen van het maagdarmstelsel (bv. persisterende navelstrengbreuk)
Overlappende vingers, klompvoeten
20.3.4 Trisomie 13 (syndroom van Patau)
47,XX+13 (meisje) of 47,XY+13 (jongen)
1/10000 levendgeborenen.
Trisomie 13 is de meest ernstige vorm van levensvatbare aneuploïdie bij de mens.
20.3.4.1 Fenotype
Het merendeel van de gevallen overlijdt in utero. De gemiddelde postnatale overlevingsduur bedraagt 1 week. De meeste kinderen overlijden binnen hun eerste levensjaar.
Lipverhemeltespleet (schisis)
>50%: aanlegstoornissen van het hart en centrale zenuwstelsel
Gebrekkige aanleg van de ogen (microftalmie, anoftalmie)
Polydactylie
20.4 Niet Invasieve Prenatale Testing (NIPT)
20.4.1
Situering
De aanwezigheid van aneuploïdie bij de foetus kan tegenwoordig al op 12 weken gedetecteerd worden door niet-invasieve prenatale testing (NIPT). ‘Niet invasief’ betekent hier: er is geen toegang tot de vruchtzak nodig, wat wel het geval is bij een vruchtwaterpunctie of chorionvillusbiopsie (zie 10.1). NIPT gebeurt immers via een veneuze bloedafname.
JAAR: 1997-2013
Lo et al. ontdekten in 1997 dat fragmentjes foetaal DNA circuleren in het maternale bloed. Dit vormde de basis voor de ontwikkeling van niet-invasieve prenatale testing (Dennis Lo et al., 1997). Sinds 2013 wordt de test uitgevoerd in België en sinds 2017 is er terugbetaling.
20.4.2 Methode
NIPT gebeurt door analyse van cel-vrij foetaal DNA (cff-DNA). Dit zijn vrij bewegende nucleïnezuren, fragmentjes van 100 à 200 baseparen die afkomstig zijn van trofoblastcellen die contact maken met maternale spieraalarteries (zie 10.2.2).
Naast cff-DNA circuleren er ook steeds celvrije, maternale DNA-fragmenten in het bloed. Deze zijn in een grotere hoeveelheid aanwezig dan de foetale fragmenten.
Bij de NIP-test worden DNA-fragmenten uit het maternaal bloed gehaald, geïdentificeerd als foetaal of maternaal en vervolgens gerangschikt volgens het chromosoom waarvan ze afkomstig zijn. Dit rangschikken gebeurt door de fragmenten te vergelijken met het referentiegenoom (zie Human Genome Project, 2.8).
DENKVRAAG
Hoe zou men kunnen achterhalen of een DNA-fragmentje foetaal of maternaal is?
Hoewel de verdere analyse op verschillende manieren kan, is de achterliggende methodiek altijd dezelfde:
Bij NIPT wordt het aantal foetale fragmenten voor elk chromosoom vergeleken met het aantal maternale fragmenten.
Het aantal foetale fragmenten heten we de foetale fractie (FF). Rond een zwangerschapsduur van 12 bedraagt een normale foetale fractie ± 10%.
Een foetale fractie > 10% kan wijzen op trisomie 13, 18 of 21
Voorbeeld: hieronder zie je twee resultaten van een NIP-test,
RESULTAAT 1
RESULTAAT 2
elk blokje stelt een chromosoomfragment 21 voor
MATERNALE FRACTIE (90%) 10%
Beoordeling:
specifieke fragmenten teruggevonden. 10 ervan
MATERNALE FRACTIE (86%) 14%
Er worden 105 chromosoom 21-specifieke fragmenten teruggevonden. 15 ervan zijn foetaal, wat overeenstemt met een verhoogde foetale fractie (14%). Deze foetus heeft met grote waarschijnlijkheid trisomie 21.
Bovenstaand voorbeeld is een sterke vereenvoudiging. In werkelijkheid is de berekening van de foetale fractie vanuit de verzamelde DNA-fragmenten een complexe, computergestuurde berekening volgens een statistisch algoritme. Bij jongens is dit trouwens iets eenvoudiger doordat het aanwezige Y-chromosoom niet in het maternale bloed voorkomt. Dit maakt het vergelijken makkelijker.
Het resultaat van de NIPT is zeer betrouwbaar, maar geen absolute zekerheid. Een afwijkende NIPT moet om die reden nog opgevolgd worden door bijkomend onderzoek.
De NIPT is wel betrouwbaarder (en vervangt) ondertussen de vroegere eerste-trimesterscreening (met oa. alfafoetoproteïne in het maternaal bloed).
De NIPT vervangt echter niet de echografische nekplooimeting op 12 weken. Bij een verdikte nekplooi is sowieso bijkomend onderzoek nodig (van den Bogaert et al., 2021).
De NIP-test is een screeningstest is en géén diagnostische test.
Als de NIP-test een verhoogd risico op trisomie aangeeft, is bijkomend diagnostisch onderzoek nodig, bij voorkeur een vruchtwaterpunctie.
De NIP-test vervangt de echografische meting van de nekplooi niet.
20.4.3 Voor- en nadelen van de test
20.4.3.1 Voordelen
Niet-invasief; een bloedstaal volstaat.
Dankzij NIPT is er minder invasieve screening nodig.
De test wordt uitgevoerd (en in België terugbetaald) vanaf 12 weken.
De test is zeer betrouwbaar. Er zijn zeer weinig vals positieven & vals negatieven.
Het geslacht van de baby is meteen gekend (weliswaar niet het doel van de test).
20.4.3.2 Nadelen
In sommige gevallen lukt NIPT niet door een te lage hoeveelheid cff-DNA (<4%, bv. te vroeg in de zwangerschap, bij maternale obesitas). In dit geval is een 2e test nodig.
De NIPT onderzoekt enkel foetaal DNA dat van de trofoblast afkomstig is. Placentair mozaïcisme (bv. placentaire trisomie) kan dus een vertekend testresultaat geven. Er is een (zeer kleine) kans op vals positieven & vals negatieven.
Bij afwijkend resultaat is sowieso bijkomend onderzoek nodig; een vruchtwaterpunctie heeft een miskraamrisico van 0,2-0,3%:
Niet alle aandoeningen worden opgespoord bv., muco, neurale buisdefecten, …
20.4.4 Evoluties in de NIP-test
Ondertussen is het technisch mogelijk om bijkomende afwijkingen op te sporen met het cff-DNA, o.a. aneuploïdie van andere autosomen, van de geslachtschromosomen, (grote) deleties en duplicaties (zie CGH 20.6.3). De betrouwbaarheid van deze bijkomende testen in de prenatale context staat echter nog niet op punt; er zijn nog (te) veel vals positieven en negatieven.
Eerder onverwacht zijn in België ook 12 gevallen van maternale kanker ontdekt na enkele sterk afwijkende NIP-testen (=0,008% van alle NIP-testen). De NIP-test detecteerde hier tumoraal DNA dat in de bloedbaan was terecht gekomen (van den Bogaert et al., 2021).
Welke informatie moet een koppel volgens jou krijgen bij een NIPT-screeningstest? Hoe bereid je mensen voor op hun testresultaat? Informeer je over evt. onverwachte resultaten (bv. maternale kanker)? Hoe kom je tot een geïnformeerde keuze?
JUIST OF FOUT?
1. De NIP-test spoort foetale cellen op in het bloed van de moeder.
2. Als de foetale fractie lager is dan 10%, is bewezen dat het kind vrij is van erfelijke aandoeningen.
3. Met de NIP-test kan het geslacht van het kind bepaald worden
20.5 Aneuploïdie van de geslachtschromosomen
20.5.1 Monosomie X (Syndroom van Turner)
20.5.1.1 Voorkomen
Turner syndroom (TS) komt voor bij ± 1/2500 levendgeboren meisjes en is de meest voorkomende oorzaak van vrouwelijke aneuploïdie.
20.5.1.2 Genotype
TS is een monosomie van het X-chromosoom, wat betekent dat er slechts één Xchromosoom in alle cellen aanwezig is. Vaak is er wel sprake van somatisch mozaïcisme, waarbij een deel van de lichaamscellen toch het normale 46,XX genotype bevat.
45,X (Turner) of 45,X/46,XX (mozaïek Turner)
Net zoals bij de autosomale trisomieën, ligt de oorzaak van Turner Syndroom in een non-disjunctiefenomeen bij de ouderlijke meiose, hier meestal bij de vader.
20.5.1.3 Fenotype
Het fenotype is steeds vrouwelijk. Hoewel het fenotype wat variabel is zijn dit de voornaamste kenmerken: groeiachterstand en kleine gestalte, ± 140 cm hart- en nierproblemen de intelligentie is meestal bewaard. Soms zijn er leerproblemen. gonadale agenese of dysgenese: dit betekent dat al vroeg in de foetale ontwikkeling een verlies optreedt van de primordiale kiemcellen en follikels en dat de ovaria niet goed ontwikkelen (=vroegtijdige ovariële insufficiëntie, vgl. met een premature menopauze). Hierdoor blijft de puberteit uit en zijn TS-patiënten onvruchtbaar.
Slecht ontwikkeld ovariëel weefsel, zonder follikels
Typische dysmorfe kenmerken zijn:
Een brede, schildvormige thorax
Korte, brede hals (webbed neck) met lage haarlijn
Korte 4e vinger, soms clinodactylie.
Bij pasgeborenen is er typisch zwelling (lymfoedeem) ter hoogte van de hals, handen en voeten.
De belangrijkste aanknopingspunten voor de behandeling zijn: behandeling van hartafwijkingen, groeistimulatie (groeihormoon) en toedienen van oestrogenen vanaf het tijdstip van de puberteit.
Ingeval van mozaïcisme is het fenotype minder uitgesproken. Milde gevallen kunnen hierdoor langdurig ongedetecteerd blijven. Mogelijk wordt de diagnose dan gesteld i.k.v. groeivertraging, late menarche of subfertiliteit.
20.5.2 Syndroom van Klinefelter
20.5.2.1 Voorkomen
Klinefelter syndroom (KS) komt voor bij ± 1/1000 levendgeboren jongens en is de frequentste oorzaak van mannelijke aneuploïdie. Dit syndroom wordt ook wel eens de ‘mannelijke’ vorm van Turner-syndroom genoemd.
20.5.2.2 Genotype”
Het klassieke genotype is het voorkomen van één extra X-chromosoom. Genotypes met twee of meer extra X-chromosomen komen ook voor, hoewel zeldzamer, en kunnen een gelijkaardig beeld geven. Net zoals bij Down Syndroom en Turner Syndroom ligt de oorzaak van Klinefelter Syndroom in een non-disjunctiefenomeen tijdens de gametogenese bij de vader of moeder
47,XXY (meestal), of 48,XXXY of 49,XXXXY …
Figuur 159 Gonadale dysgenese bij het syndroom van Turner (Re)
20.5.2.3
Bij patiënten met Klinefelter-syndroom is de wet van Lyon (X-inactivatie) van toepassing. In elke cel wordt slechts één X-chromosoom actief gehouden. Het andere wordt omgevormd tot Barr-lichaampje.
Fenotype
Het fenotype is steeds mannelijk. Het fenotype is des te meer uitgesproken, naarmate het aantal overtollige X-chromosomen toeneemt. In tegenstelling tot DS en TS zijn er geen duidelijke dysmorfe kenmerken. Kinderen zijn bij de geboorte dus asymptomatisch.
Vergelijkbaar met het ovariële falen bij TS is het testiculaire falen bij Klinefelter, dat typisch duidelijk wordt in de loop van de puberteit. De puberteit begint dus meestal wel bij Klinefelter-jongens, maar evolueert niet vlot
Daarnaast is er een kenmerkende grote gestalte, gemiddeld 186 cm, en een neiging tot zwaarlijvigheid met typische, gynaecoïde vetverdeling. Intelligentie is normaal tot randnormaal; vaak zijn er ook lichte leer- en ontwikkelingsproblemen.
Figuur 160 Karyogram van een patiënt met Klinefelter syndroom
20.5.2.4 De pseudo-autosomale regio
Het is geen toeval dat iemand met 1 X-chromosoom (Turner) kleiner is dan het gemiddelde en dat iemand met extra X-chromosomen (Klinefelter) groter is dan het gemiddelde.
genen voor de mannelijke ontwikkeling
niet te verwarren met telomeren
Op het X-chromosoom zitten immers bepaalde genen (bv. SHOX) die een belangrijke rol spelen in de groei. Deze genen bevinden zich ook op het Y-chromosoom ter hoogte van de zgn. pseudo-autosomale regio’s (PAR). Dit zijn stukjes van het X en Y chromosoom die wel homoloog zijn en zich gedragen als autosomen.
PAR1 en 2 bevinden zich aan de uiteinden van de geslachtschromosomen, zijn onderhevig aan crossing-over en ontsnappen aan de X-inactivatie.
Er speelt een dosiseffect bij deze genen: hoe meer allelen iemand bezit, hoe groter de volwassen gestalte.
SRY
20.5.3
Oefeningen
Oefening 44.
Kan een meisje met het syndroom van Turner rood-groen kleurenblind zijn? Waarom wel/niet?
Oefening 45.
Jayden, 17 jaar, wordt gediagnosticeerd met het syndroom van Klinefelter n.a.v. een moeizaam vorderende puberteitsontwikkeling. Hij blijkt ook te lijden aan rood-groenkleurenblindheid net als zijn grootvader aan moederszijde. Zijn beide ouders zijn gezond en niet kleurenblind.
Vraag. Bij welk familielid is het non-disjunctiefenomeen opgetreden dat bij Jayden tot het syndroom van Klinefelter heeft geleid?
o Moeder
o Vader
o Grootvader
o Dit valt niet af te leiden uit deze opgave.
Oefening 46.
Rekening houdend met wet van Lyon, hoeveel Barr-lichaampjes zullen we aantreffen in een cel van iemand met...
– Klinefelter-syndroom
– Turner-syndroom
– Triple-X-syndroom
– Down-syndroom (meisje)
– Super-male-syndroom
20.6 Microdeletie-syndromen
De syndromen die we in 20.3 en 20.5 hebben besproken zijn het gevolg van aneuploïdie. Hier bespreken we nog enkele syndromen die het gevolg zijn van microdeleties.
20.6.1 Definitie en eigenschappen
20.6.2
Een microdeletie-syndroom is een syndromaal fenotype dat het gevolg is van een microdeletie (een verlies van max. 5 Mb), zie ook 16.2.2
Gemiddeld gaat het over een verlies van 1 à 3 Mb. Het fenotype is het gevolg van het resulterend verlies van verschillende genen op één van beide chromosomen.
Enkele eigenschappen van microdeleties:
Ze leiden tot haplo-insufficiëntie, en hebben hierdoor een dominant effect.
De penetrantie is gereduceerd (het fenotype is soms afwezig).
Er is een variabele expressiviteit (het fenotype wisselt sterk; afhankelijk van de grootte van de onderliggende microdeletie. Het syndromaal karakter komt vooral voort uit het feit dat enkele typische orgaansystemen betrokken zijn).
ISCN-notatie
De ISCN-notatie van microdeletie-syndromen vertrekt van het karyotype, aangevuld met del (deletie), het betrokken chromosoom (tussen haakjes) en tot slot het bandje waar de deletie is opgetreden (ook tussen haakjes).
20.6.3
Diagnose
chromosoom
deletie
totaal aantal chromosomen geslachtschromosomen bandje
46, XX del( )( )
Omdat microdeleties niet zichtbaar zijn onder een lichtmicroscoop is gespecialiseerd onderzoek nodig om de diagnose te stellen. Door het ontwikkelen van nieuwe technieken en de kennis uit het Human Genome Project zijn de laatste decennia, vele nieuwe microdeleties bij de mens bekend geraakt.
Eén van die technieken is comparative genomic hybridisation (CGH) Hierbij wordt het genoom van een testpersoon vergeleken met het referentiegenoom om na te gaan of er stukken ontbreken, wat wijst op een deletie.
testpersoon -DNA (kleur 1)
referentie- DNA (kleur 2)
blekere spot
Bij CGH wordt het DNA van de testpersoon gedenatureerd, in kleine stukjes ('probes’) geknipt en gemerkt met een fluorescerende kleurstof. Het referentie-DNA ondergaat hetzelfde proces, maar wordt gemerkt met een andere, fluorescerende kleurstof.
De probes worden vervolgens op een glasplaatje(micro-array) aangebracht, waarin zich duizenden kleine kuiltjes bevinden. In elk van die kuiltjes vindt hybridisatie plaats tussen het test-DNA en het referentie-DNA. Dit gebeurt normaal in een gelijke verhouding. Hierdoor mengen de kleurstoffen normaal gezien ook in een gelijke verhouding. Wanneer echter een deletie aanwezig is, zal de kleur in dat kuiltje afwijken van de rest
Door het aflezen van de fluorescentie in alle kuiltjes kan dus heel snel opgespoord worden of het genoom overeenkomt met de referentie.
Deze analyse gebeurt geautomatiseerd en genereert een vergelijkingsgrafiek:
duplicatie?
gelijke kleur
microdeletie
163 resultaat van een array-CGH van chromosoom 15
Een voordeel aan CGH is dat verschillende soorten mutaties tegelijk kunnen opgespoord worden. Dat maakt het een goede screeningstechniek bv. bij pasgeborenen waar een genetisch syndroom vermoed wordt. Een nadeel is dat de betekenis van de verkregen informatie niet altijd duidelijk is. Stel bv. dat wordt vastgesteld dat er een deletie is op chr. 4 en 7 en een duplicatie op chr. 1 en 16. Welke afwijking is dan verantwoordelijk voor het fenotype? Kennis over de genen op die loci is nodig en die is er niet altijd.
Figuur 162 CGH
Figuur
20.6.4 22q-deletie-syndroom
De meest frequente microdeletie bij de mens is deze op bandje 11.2 van de lange arm van chromosoom 22. Door die deletie verdwijnen tientallen genen die een belangrijke rol spelen in de embryologische ontwikkeling van het hart en hoofd- en halsgebied.
In 95% van de gevallen betreft het een de novo mutatie. In de overige gevallen zit de microdeletie in de kiembaan en wordt ze dus overgeërfd.
20.6.4.1
Fenotype
46, XX del(22)(q11.2) (meisje) of 46, XY del(22)(q11.2) (jongen)
Er zijn verschillende benamingen voor het syndroom die verwijzen naar een gelijkaardige microdeletie: het velo-cardio-faciaal-syndroom (VCFS), DiGeorgesyndroom, 22q-deletie-syndroom. Het variabele, soms zeer subtiele fenotype omvat 4 mogelijke groepen van afwijkingen die in wisselende mate aanwezig zijn:
*verhemelteproblemen (velum = verhemelte)
Bv. schisis, spraak, articulatie- & slikproblemen
*aangeboren hartafwijkingen (cardio = hart)
Bv. VSD, aorta-onderbreking
*faciale dysmorfie (faciaal =gezicht)
Bv. lange, buisvormige neus met brede neusbrug, kleine mond, laagstaande oren
1/50.000 kinderen wordt geboren met het zgn. ‘cri-du-chat’-syndroom, een genetische afwijking die voor het eerst werd beschreven door Lejeune (de ontdekker van trisomie 21 als oorzaak van het syndroom van Down) in de jaren ’60.
Zoek op.
Welke microdeletie veroorzaakt het cri-du-chat-syndroom?
Noteer het karyotype voor een meisje met dit syndroom, waarbij de microdeletie ligt op bandje 15.3
Wat vind je terug over het fenotype? Noteer minstens 3 kenmerken.
20.6.6 Prader-Willi syndroom en Angelman syndroom
PWS = Prader-Willi syndroom
AS = Angelman syndroom
20.6.6.1 Genotype
PWS en AS zijn 2 syndromen die veroorzaakt worden door een microdeletie op de lange arm van chromosoom 15 (regio 15q11-13). De loci voor PWS en AS liggen er vlak naast elkaar. Elke locus bevat meerdere genen:
microdeletie van 5 Mb
paternaal chr. 15
maternaal chr. 15
PWS-locus AS-locus
Figuur 164 (Boven) CGH toont een microdeletie in de 15q11-13 regio (Onder) Schema van de PWS & AS loci
De PWS en AS-loci zijn onderhevig aan imprinting:
PWS-locus: maternale imprinting; enkele het paternale stuk is actief
AS-locus: paternale imprinting; enkel het maternale stuk is actief
Dit betekent dat deze loci niet onderhevig zijn aan haplo-insufficiëntie. Als de genen in één locus tot expressie komen veroorzaakt dit een normaal fenotype. De PWS-AS-loci gedragen zich dus recessief.
Het PWS en AS ontstaat pas als de genen op het actieve stuk niet tot expressie komen. Dit kan het gevolg zijn van een microdeletie, door een imprintingsfout of door uniparentale disomie (zie 5.4.3.2).
Bij uniparentale disomie hoeft er zelfs geen microdeletie aanwezig te zijn!
Doordat beide chromosomen 15 van dezelfde ouder afkomstig zijn, treden imprintingsfouten op.
Als beide chr. 15 maternaal zijn ontstaat het Prader-Willi syndroom
Als beide chr. 15 paternaal zijn ontstaat het Angelman syndroom
De syndromen komen even vaak bij jongens als bij meisjes voor.
20.6.6.2 Prader-Willi syndroom
PWS wordt in de neonatale periode gekenmerkt door opvallende hypotonie en voedingsproblemen. Na het eerste levensjaar wijzigt het beeld sterk en ontstaat de typische, oncontroleerbare eetlust.
De meeste PWS-patiënten zijn obees.
Vaak is er ook gebrekkige groei, trage motorische ontwikkeling, faciale dysmorfie, onderontwikkeling van de geslachtsorganen en leer- en gedragsmoeilijkheden. Er is doorgaans een lichte tot matige mentale beperking.
20.6.6.3 Angelman syndroom
Kinderen met AS hebben ernstige ontwikkelingsproblemen (achterstand in taal en motorische ontwikkeling), faciale dysmorfie, epilepsie en ernstige leerproblemen. Er is doorgaans een lichte tot matige mentale beperking.
Kinderen met AS zijn opvallend blij en vrolijk in de sociale omgang.
20.6.6.4 Oefening
Oefening 47.
Een meisje wordt geboren met een aangeboren hartafwijking (VSD). De kinderarts stelt genetisch onderzoek voor en hieruit blijkt dat het meisje lijdt aan het velo-cardio-faciaalsyndroom. Bij de moeder van het kind wordt deze mutatie ook teruggevonden (de moeder had ook een aangeboren hartafwijking, maar bij haar gebeurde nooit genetisch onderzoek). Bij de vader worden geen genetische afwijkingen teruggevonden.
Hoe groot is de kans op een kind met VCFS bij dit koppel? (Teken een recombinatierooster.)
Op welke locus ligt deze mutatie?
Stel dat zij graag nog een tweede kindje willen. Kan pre-ïmplantatie genetische testing hier een rol spelen? Waarom wel/niet?
Oefening 48.
Een jongen wordt geboren met een opvallend groot hoofd (macrocefalie) en een hartgeruis dat veroorzaakt wordt door een ASD. Uit genetisch onderzoek blijkt dat het kind lijdt aan het Sotos-syndroom. Het karyotype is 46, XY del(5)(q35).
Vraag. Leg dit karyotype uit in - voor de ouders - begrijpbare taal.
Oefening 49.
Beschouw een familie waarin Angelman-syndroom voorkomt door een erfelijk overgedragen mutatie in de AS-locus.
Vraag. Kun je afleiden uit de stamboom dat imprinting een rol speelt? Argumenteer.
drager van de microdeletie
Angelman Syndroom (AS)
20.7 Kanker
20.7.1 Kanker is een genetische ziekte
Kanker is een ziekte waarbij lichaamscellen niet langer de belangen van het organisme nastreven. Ze geraken a.h.w. gedeconnecteerd van de omliggende weefsels en streven nog slechts twee doelen na:
Proliferatie: zich zoveel mogelijk vermenigvuldigen, wat bijdraagt tot tumorgroei.
Metastase: zich zoveel mogelijk verspreiden, waardoor nieuwe letsels ontstaan.
20.7.1.1 Proto-oncogenen
Proto-oncogenen zijn genen waarvan het wild-type allel niet actief is. Bij mutatie komen ze wel tot expressie en worden het oncogenen. Hun genproducten transformeren de cel tot een kankercel (gain-of-function).
RAS-genen zijn de bekendste proto-oncogenen. Hun genproducten zorgen normaal voor correcte informatie-uitwisseling tussen cellen. De door mutaties gewijzigde eiwitten leiden tot communicatiestoornissen en ongecontroleerde celdeling. Virussen zoals HPV kunnen baarmoederhalskanker veroorzaken door het activeren van bepaalde protooncogenen in de baarmoederhalscellen.
PROTO-ONCOGENEN
KANKER
TUMOR-SUPPRESSOR-GENEN
KANKER
Figuur 165 (Li) Proto-oncogenen en (Re) tumor-supporessorgenen. In dit schema is duidelijk dat tumorsuppressor-genen 2 x beschadigd moeten worden vooral de cel transformeert tot een kankercel.
20.7.1.2 Tumor Suppressorgenen
Tumor-suppressorgenen zijn genen waarvan de genproducten de cel actief beschermen tegen ongecontroleerde deling. Het zijn genen die bijdragen aan normale celfuncties (bv. celdeling, intercellulaire communicatie, apoptose, …). Bij uitschakeling van beide allelen (loss-of-function-mutatie) transformeert de cel tot kankercel.
In meer dan de helft van alle kankers zit de mutatie in het TP53-gen op chromosoom 17. Dit gen codeert voor het beruchte p53-eiwit, een eiwit dat een belangrijke, dirigerende functie heeft in de normale celcyclus (Freed-Pastor & Prives, 2012).
20.7.2
Erfelijkheid van kanker
Zowel proto-oncogenen als tumor-suppressorgenen worden doorgegeven van generatie op generatie. Maar zoals blijkt uit Figuur 165 zijn bijkomende, schadelijke invloeden nodig (zie mutagenen 16.4.2) om de cel te kunnen transformeren tot kankercel. Bij heel wat mensen zal dat niet het geval zijn.
De kans dat deze genen muteren wordt wel groter naarmate de leeftijd toeneemt. Dit omdat de kwaliteit van DNA-repair afneemt en het effect van geaccumuleerde schade (bv. aantal jaren gerookt, aantal jaren UV-blootstelling, …) toeneemt.
20.7.2.1 (Proto-)oncogenen hebben een dominant effect
Oncogenen gedragen zich als dominante allelen, aangezien het fenotype (kanker) al tot uiting komt vanaf één mutant allel (=haplo-insufficiëntie).
Merk wel op dat deze mutaties doorgaans optreden in lichaamscellen en dus niet kunnen doorgegeven worden aan het nageslacht. Je kunt het beschouwen als een vorm van somatisch mozaïcisme.
20.7.2.2 Tumor-suppressorgenen hebben een recessief effect
Tumor-suppresorgenen (TS) gedragen zich als recessieve allelen, aangezien het fenotype (kanker) pas tot uiting komt vanaf twee mutante allelen.
Beide allelen moeten dus defect moeten zijn om kanker te veroorzaken. In de literatuur zegt men dat deze genen recht hebben op 2 'hits' (2 treffers). Hier speelt ons diploïde voordeel dus mee. Ook dit verklaart waarom heel wat kankers pas op oudere leeftijd optreden.
Een bijzondere situatie ontstaat echter wanneer zo’n mutatie in één van deze allelen (1 ‘hit)’ in de kiembaan terechtkomt:
NIET-ERFELIJKE KANKER
Van elke ouder wordt één wild-
ERFELIJKE KANKER
Van elke ouder wordt één mutant TS-allel overgeërfd. Dit geeft slechts recht op 1 ‘hit’ tijdens het leven.
Figuur 166 (Re) Overerving van een mutant TS-allel
KANKER
KANKER
M P
Op zichzelf veroorzaakt een overgeërfd TS-allel niet meteen kanker bij het nageslacht. Alleen vervalt het diploïde voordeel wel, waardoor de cel al na 1 bijkomende ‘hit’, tijdens het leven, kan transformeren tot kankercel. Dit verklaart waarom familiale vormen van kanker typisch op jongere leeftijd optreden.
In families waarin een erfelijke vorm van kanker voorkomt, wordt een mutant allel voor een tumor-suppressorgen overgedragen.
JAAR: 1872
In de 19e eeuw al beschreef Hilario De Gouvia, een Braziliaanse oogarts een familie waarin verschillende leden een zeldzame oogkanker ontwikkelden (retinoblastoma). Volgens de Gouvia lag de oorzaak bij een overgedragen, erfelijke factor. Later werd aangetoond dat het ging over een mutant allel van een tumor-suppressor-genen.
In geval van borstkanker en ovariumkanker werden 2 belangrijke TS-genen geïdentificeerd: BRCA1 op chromosoom 17 en BRCA2 op chromosoom 13 (BRCA = breast cancer). De BRCA-eiwitten spelen een belangrijke rol in DNA-repair.
Dragerschap voor mutante BRCA-allelen verschilt sterk van populatie tot populatie en ligt in onze regio wellicht rond de 1/800 à 1/1000. Hogere prevalenties zijn beschreven bij Ashkenazi Joden en mensen van Ijslandse afkomst.
Kun je voor deze twee bevolkingsgroepen een mogelijke verklaring bedenken voor het frequenter voorkomen van mutante allelen?
Wie drager is van een BRCA1 en 2 mutatie heeft een hoger risico op o.a. borst- en ovariumkanker. Ook borstkanker bij mannen is mogelijk.
Het lifetime risk (kans dat dit tijdens het leven optreedt) bedraagt:
Borstkanker Ovariumkanker
BRCA1 72 % (95% CI 65 -79 %) 69 % (95% CI 61 - 77 %)
BRCA2 44 % (95% CI 36 - 53 %) 17 % (95% CI 11 - 25 %) (Kuchenbaecker et al., 2017)
Bij mensen uit deze families is het zinvol om aan vroegtijdige kankeropsporing (vroeger dan in de algemene populatie). Bij bevestiging van dragerschap wordt soms gekozen voor een preventieve ingreep zoals bv. een bilaterale mastectomie (wegname borstklierweefsel) verkiezen. Dit verlaagt het kankerrisico immers met 90%.
De actrice Angelina Jolie onderging in 2013 een preventieve, bilaterale mastectomie toen bij haar dragerschap voor BRCA1 werd vastgesteld. Ze schreef ervaring neer in de New York Times: ‘My Medical Choice’ (zie QR-code).
Risicofamilies voor BRCA-dragerschap zijn families met minstens 3 individuen met borstkanker (waarvan min.1 jonger dan 50), families met borst- èn ovariumkanker, families met borstkanker bij mannen en families met borstkanker onder de 30 jaar.
Hou wel in het achterhoofd dat er bij 95% van de kankers geen sprake is van een familiale voorbeschiktheid en dat tijdens het leven wel degelijk '2 hits' optreden
Tot slot staan we hier nog even stil bij het overervingspatroon van deze kankers.
Het overervingspatroon is (paradoxaal genoeg) dominant.
Een voorbeeld van een familie met BRCA1-overerving:
ovarium, 49j
borst, 42j ovarium, 53j
borst, 38j
Figuur 167 BRCA-overerving in een familie met vermelding van leeftijd van diagnose en type kanker
Merk op dat we hier ook kunnen spreken van een verminderde penetrantie. Niet elk individu met een BRCA-mutatie zal immers kanker ontwikkelen. Dit wijst op een multifactoriële oorsprong, waarbij ook andere genen en omgevingsfactoren een rol spelen. Deze zijn nog niet allemaal bekend.
Nawoord
Wanneer we kijken naar de toekomst van de genetica is het ook belangrijk om de fouten uit het verleden niet te vergeten. Na het doornemen van deze cursus is het ongetwijfeld duidelijk geworden dat het onderzoeksdomein van de genetica vaak geconfronteerd wordt met ethische vragen. Denk bijvoorbeeld aan de recente CRISPR-cas-9 revolutie.
In het verleden werden deze vragen op cruciale momenten dikwijls niet gesteld of (achteraf gezien) verkeerd beantwoord.
Een voorbeeld hiervan is de geschiedenis van de eugenetica. Dit gedachtengoed gaat ervanuit dat het menselijk ras er door selectieve reproductie zou moeten naar streven om zichzelf te ‘verbeteren'.
De Brit Francis Galton, bedenker van de term eugenetica in 1883, werd één van de grondleggers van het gedachtengoed. Galton paste de theorieën van zijn neef Darwin toe op de mens en concludeerde dat onze maatschappij er niet goed aan deed om de 'zwakken' te beschermen. Dit zou tegen de principes van 'natuurlijke selectie' zijn en de kwaliteit van de mens en zijn erfelijk materiaal niet ten goede komen.
Figuur 168 Aankondiging van het 3e, Internationale Eugenetica-congres in de Verenigde Staten (1932)
In het begin van de 20e eeuw groeide wereldwijd het maatschappelijk draagvlak voor deze nieuwe wetenschap. Eugenetica werd beschouwd als een volwaardige onderzoekstak en aan diverse
universiteiten werden er faculteiten voor opgericht. De eugenetische beweging leidde vervolgens tot de gedwongen sterilisatie van duizenden mentaal geretardeerden, doven, blinden en epilepsiepatiënten oa. in Europa, maar ook in de VS (waar de beweging overigens ook heel sterk was).
In het Duitsland van de jaren '30 voedde het eugenisme het nationaal-socialisme van Hitler, dat naast de zwakken en zieken ook volkerengroepen zoals joden als 'genetisch inferieur' beschouwde. Tijdens
WO II werden er onder het nazi-regime naar schatting honderdduizenden mensen gesteriliseerd en miljoenen vermoord omwille van ziekte of afkomst. Daarnaast werd in het berucht 'project Lebensborn' gepoogd om een 'genetisch zuiver' Arisch ras te scheppen.
Na het bekendmaken van de wrede praktijken van de nazi's tijdens WO II werd het eugenisme wereldwijd verworpen. Faculteiten werden opgedoekt en veel voorstanders van het eugenisme nuanceerden of wijzigden hun vroegere standpunten. Nieuwe instanties werden opgericht om de medische ethiek te bewaken, waardoor het ook niet meer mogelijk werd om zomaar op iedereen onderzoek te doen of behandelingen uit te voeren.
Ook dankzij de 'Universele Verklaring van de Rechten van de Mens' werd de integriteit van elk mens gevrijwaard. Toch bleven de gedwongen sterilisaties in heel wat landen plaatsvinden tot ruim in de jaren '70.
De hedendaagse verwezenlijkingen van de genetica (gentherapie, PIGD, klonen, human genome project, CRISPR-cas9 etc..) doen velen huiveren voor een mogelijke terugkeer van de eugenetica.
Initiatieven zoals een spermabank waar enkel Nobelprijswinnaars mogen doneren (actief in California van 1980 tot 1999), designer-baby's met voorafbepaalde geslachtskeuze of de CRISPR-baby’s van He Jiankiu - worden dan ook steevast fel bekritiseerd.
In de meeste Westerse landen zijn op vandaag medisch-ethische commissies actief die de wetgever adviseren over mogelijke wanpraktijken. Het blijft voor deze commissies echter wel een grote uitdaging om de snel ontwikkelende genetica bij te benen en op tijd en stond adviezen te formuleren
Meer info: http://www.eugenicsarchive.org/
Bronnen
BOEKEN
CARLSON, B. Human Embryology & Developmental Biology 6th ed. 2019
HARTL, D. Essential Genetics & Genomics, 7th ed. 2020
JORDE, L. Medical genetics, 5th ed. 2016
IWASA, J. Karp’s Cell & Molecular Biology, 8th ed. 2016
KLUG, W. Concepts of Genetics, 12th ed. 2019
MITCHELL B. Embryology, an Illustrated Colour Text. 2nd ed. 2009
MUKHERJEE, S. The Gene, an Intimate History, Scribner, 2016
MULINSKY, A. Genetic Disorders and the Fetus: Diagnosis, Prevention and Treatment, 7th ed. 2015
NUSSBAUM R. Thompson & Thompson, Genetics in Medicine, 8th ed. 2016
POLLARD, T. Cell Biology, 3rd ed. 2017
SADLER, T.W. Langman’s Medical Embryology, 13th ed. 2014
SCHOENWOLF, G. Larsen’s Human Embryology, 5th ed. 2015
SNUSTAD, P. Principles of Genetics, 6th ed. 2011
TURNPENNY, P. Emery’s Elements of Medical Genetics, 15th ed. 2017
WETENSCHAPPELIJKE LITERATUUR
Avery, O. T., MacLeod, C. M., & McCarty, M. (1944). STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES. Journal of Experimental Medicine, 79(2). https://doi.org/10.1084/jem.79.2.137
Blaese, R. M., Culver, K. W., Miller, A. D., Carter, C. S., Fleisher, T., Clerici, M., Shearer, G., Chang, L., Chiang, Y., Tolstoshev, P., Greenblatt, J. J., Rosenberg, S. A., Klein, H., Berger, M., Mullen, C. A., Ramsey, W. J., Muul, L., Morgan, R. A., & Anderson, W. F. (1995). T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science, 270(5235). https://doi.org/10.1126/science.270.5235.475
Bordignon, C., Notarangelo, L. D., Nobili, N., Ferrari, G., Casorati, G., Panina, P., Mazzolari, E., Maggioni, D., Rossi, C., Servida, P., Ugazio, A. G., & Mavilio, F. (1995). Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science, 270(5235). https://doi.org/10.1126/science.270.5235.470
Bovicelli, L., Orsini, L. F., Rizzo, N., Montacuti, V., & Bacchetta, M. (1982). Reproduction in down syndrome. Obstetrics and Gynecology, 59(6).
Brown, C. J., & Robinson, W. P. (1997). XIST expression and X-chromosome inactivation in human preimplantation embryos. In American Journal of Human Genetics (Vol. 61, Issue 1).
https://doi.org/10.1086/513914
Campbell, I. M., Shaw, C. A., Stankiewicz, P., & Lupski, J. R. (2015). Somatic mosaicism: Implications for disease and transmission genetics. In Trends in Genetics (Vol. 31, Issue 7).
https://doi.org/10.1016/j.tig.2015.03.013
Crick, F. (1970). Central dogma of molecular biology. Nature, 227(5258). https://doi.org/10.1038/227561a0
Dennis Lo, Y. M., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W. G., & Wainscoat, J. S. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet, 350(9076). https://doi.org/10.1016/S0140-6736(97)02174-0
Drouin, G., Godin, J.-R., & Page, B. (2011). The Genetics of Vitamin C Loss in Vertebrates. Current Genomics, 12(5). https://doi.org/10.2174/138920211796429736
Dugas, C., & Slane, V. H. (2022). Miscarriage. StatPearls https://www.ncbi.nlm.nih.gov/books/NBK532992/
Duncan, F. E., Que, E. L., Zhang, N., Feinberg, E. C., O’halloran, T. v, & Woodruff, T. K. (2016). The zinc spark is an inorganic signature of human egg activation OPEN. https://doi.org/10.1038/srep24737
Freed-Pastor, W. A., & Prives, C. (2012). Mutant p53: One name, many proteins. Genes and Development, 26(12). https://doi.org/10.1101/gad.190678.112
Gianaroli, L., Magli, M. C., Cavallini, G., Crippa, A., Capoti, A., Resta, S., Robles, F., & Ferraretti, A. P. (2010). Predicting aneuploidy in human oocytes: key factors which affect the meiotic process. https://doi.org/10.1093/humrep/deq123
Gomes, N. M. V., Shay, J. W., & Wright, W. E. (2010). Telomere biology in Metazoa. In FEBS Letters (Vol. 584, Issue 17). https://doi.org/10.1016/j.febslet.2010.07.031
Goodfellow, P. N., & Lovell-Badge, R. (2003). SRY AND SEX DETERMINATION IN MAMMALS. Https://Doi.Org/10.1146/Annurev.Ge.27.120193.000443, 27, 71–92. https://doi.org/10.1146/ANNUREV.GE.27.120193.000443
Hoekstra, C., Zhao, Z. Z., Lambalk, C. B., Willemsen, G., Martin, N. G., Boomsma, D. I., & Montgomery, G. W. (2008). Dizygotic twinning. Human Reproduction Update, 14(1), 37–47. https://doi.org/10.1093/HUMUPD/DMM036
Irving, C. A., & Chaudhari, M. P. (2012). Cardiovascular abnormalities in Down’s syndrome: Spectrum, management and survival over 22 years. Archives of Disease in Childhood, 97(4). https://doi.org/10.1136/adc.2010.210534
Jiang, Y., Bolnick, D. I., & Kirkpatrick, M. (2013). Assortative mating in animals. American Naturalist, 181(6). https://doi.org/10.1086/670160
KB, K., JL, H., DR, B., KA, P., TM, M., MJ, R.-B., S, J., FE, van L., RL, M., N, A., DE, G., MB, T., MA, R., DF, E., AC, A., L, M., DG, E., D, B., D, F., … H, O. (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA, 317(23), 2402–2416.
https://doi.org/10.1001/JAMA.2017.7112
Langdon, J., & Down, H. (1966). Observations on an ethnic classification of idiots. Heredity, 21(4). https://doi.org/10.1038/hdy.1966.69
Lockwood, C. (2022). Congenital cytogenetic abnormalities. UpToDate
Lyon, M. F. (1961). Gene action in the X-chromosome of the mouse (mus musculus L.). Nature, 190(4773). https://doi.org/10.1038/190372a0
Moore, R. K., Erickson, G. F., & Shimasaki, S. (2004). Are BMP-15 and GDF-9 primary determinants of ovulation quota in mammals? Trends in Endocrinology and Metabolism: TEM, 15(8), 356–361. https://doi.org/10.1016/J.TEM.2004.08.008
Mukherjee, S. (2017). The Gene: An Intimate History
Myrelid, Å., Gustafsson, J., Ollars, B., & Annerén, G. (2002). Growth charts for Down’s syndrome from birth to 18 years of age. Archives of Disease in Childhood, 87(2).
https://doi.org/10.1136/adc.87.2.97
Orlando, L., Darlu, P., Toussaint, M., Bonjean, D., Otte, M., & Hänni, C. (2006). Revisiting Neandertal diversity with a 100,000 year old mtDNA sequence. In Current Biology (Vol. 16, Issue 11).
https://doi.org/10.1016/j.cub.2006.05.019
Orvieto, R., Shimon, C., Rienstein, S., Jonish-Grossman, A., Shani, H., & Aizer, A. (2020). Do human embryos have the ability of self-correction. Reproductive Biology and Endocrinology, 18(1).
https://doi.org/10.1186/s12958-020-00650-8
Ousterout, D., Kabadi, A., … P. T.-N., & 2015, undefined. (n.d.). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nature.Com. Retrieved October 2, 2022, from https://www.nature.com/articles/ncomms7244
Pośpiech, E., Teisseyre, P., Mielniczuk, J., & Branicki, W. (2022). Predicting Physical Appearance from DNA Data Towards Genomic Solutions. In Genes (Vol. 13, Issue 1).
https://doi.org/10.3390/genes13010121
Reichetzeder, C. (2021). Overweight and obesity in pregnancy: their impact on epigenetics. In European Journal of Clinical Nutrition (Vol. 75, Issue 12). https://doi.org/10.1038/s41430-02100905-6
Richmond, S., Howe, L. J., Lewis, S., Stergiakouli, E., & Zhurov, A. (2018). Facial genetics: A brief overview. Frontiers in Genetics, 9(OCT), 462.
https://doi.org/10.3389/FGENE.2018.00462/BIBTEX
Rohde, K., Keller, M., la Cour Poulsen, L., Blüher, M., Kovacs, P., & Böttcher, Y. (2019). Genetics and epigenetics in obesity. In Metabolism: Clinical and Experimental (Vol. 92).
https://doi.org/10.1016/j.metabol.2018.10.007
Rubin, S. S., Rimmer, J. H., Chicoine, B., Braddock, D., & McGuire, D. E. (1998). Overweight prevalence in persons with Down syndrome. Mental Retardation, 36(3). https://doi.org/10.1352/00476765(1998)036<0175:OPIPWD>2.0.CO;2
Sheridan, C. (2011). Gene therapy finds its niche. Nature Biotechnology, 29(2). https://doi.org/10.1038/nbt.1769
Simoni, G., Brambati, B., Danesino, C., Rossella, F., Terzoli, G. L., Ferrari, M., & Fraccaro, M. (1983). Efficient direct chromosome analyses and enzyme determinations from chorionic villi samples in the first trimester of pregnancy. Human Genetics 1983 63:4, 63(4), 349–357. https://doi.org/10.1007/BF00274761
Singh, V. (2020). An introduction to genome editing CRISPR-Cas systems. Genome Engineering via CRISPR-Cas9 System, 1–13. https://doi.org/10.1016/B978-0-12-818140-9.00001-5
Starosti, M. R., Sosin, O. A., & McCo, R. C. (2020). Single-cell analysis of human embryos reveals diverse patterns of aneuploidy and mosaicism. Genome Research, 30(6). https://doi.org/10.1101/gr.262774.120
Stefá nska, K., Hutchings, G., Popis, M., Moncrieff, L., Dompe, C., Janowicz, K., Pié nkowski, W., Gutaj, P., Shibli, J. A., Mathias Prado, W., Piotrowska-Kempisty, H., Mozdziak, P., Bruska, M., Zabel, M., Kempisty, B., & Nowicki, M. (1102). Clinical Medicine Human Wharton’s Jelly-Cellular Specificity, Stemness Potency, Animal Models, and Current Application in Human Clinical Trials. J. Clin. Med, 2020, 1102. https://doi.org/10.3390/jcm9041102
Steptoe, P. C., & Edwards, R. G. (1978). Birth after the reimplantation of a human embryo. Lancet (London, England), 2(8085), 366. https://doi.org/10.1016/S0140-6736(78)92957-4
Sundin, O. H., Yang, J. M., Li, Y., Zhu, D., Hurd, J. N., Mitchell, T. N., Silva, E. D., & Maumenee, I. H. (2000). Genetic basis of total colourblindness among the Pingelapese islanders. Nature Genetics, 25(3), 289–293. https://doi.org/10.1038/77162
Sutton, W. S. (1903). The chromosomes in heredity. Biological Bulletin, 4, 231–251.
Tedone, E., Huang, E., O’Hara, R., Batten, K., Ludlow, A. T., Lai, T. P., Arosio, B., Mari, D., Wright, W. E., & Shay, J. W. (2019). Telomere length and telomerase activity in T cells are biomarkers of high-performing centenarians. Aging Cell, 18(1). https://doi.org/10.1111/acel.12859
TJIO, J. H., & LEVAN, A. (1956). THE CHROMOSOME NUMBER OF MAN. Hereditas, 42(1–2), 1–6. https://doi.org/10.1111/J.1601-5223.1956.TB03010.X
Tsuboyama, K., Osaki, T., Matsuura-Suzuki, E., Kozuka-Hata, H., Okada, Y., Oyama, M., Ikeuchi, Y., Iwasaki, S., & Tomari, Y. (2020). A widespread family of heat-resistant obscure (Hero) proteins protect against protein instability and aggregation. PLoS Biology, 18(3). https://doi.org/10.1371/journal.pbio.3000632
van den Bogaert, K., Lannoo, L., Brison, N., Gatinois, V., Baetens, M., Blaumeiser, B., Boemer, F., Bourlard, L., Bours, V., de Leener, A., de Rademaeker, M., Désir, J., Dheedene, A., Duquenne, A., Fieremans, N., Fieuw, A., Gatot, J. S., Grisart, B., Janssens, K., … Vermeesch, J. R. (2021).
Outcome of publicly funded nationwide first-tier noninvasive prenatal screening. Genetics in Medicine, 23(6). https://doi.org/10.1038/s41436-021-01101-4
Waldeyer, W. (1888). Ueber Karyokinese und ihre Beziehungen zu den Befruchtungsvorgängen. Archiv Für Mikroskopische Anatomie 1888 32:1, 32(1), 1–122. https://doi.org/10.1007/BF02956988
Watson, J. D., & Crick, F. H. C. (1969). Molecular structure of Nucleic acids: A structure for Deoxyribose Nucleic acid (Reprinted from Nature, April 25, 1953). Nature, 224(5218). https://doi.org/10.1038/224470a0