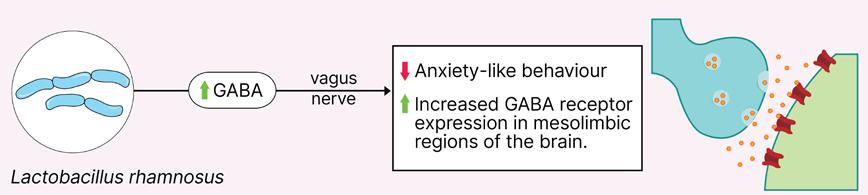
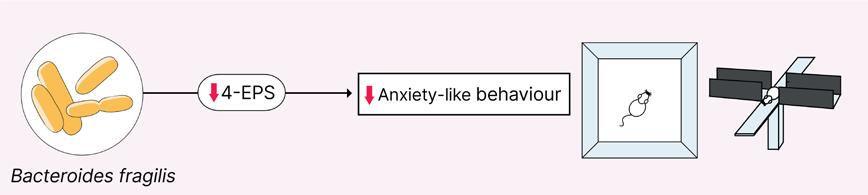
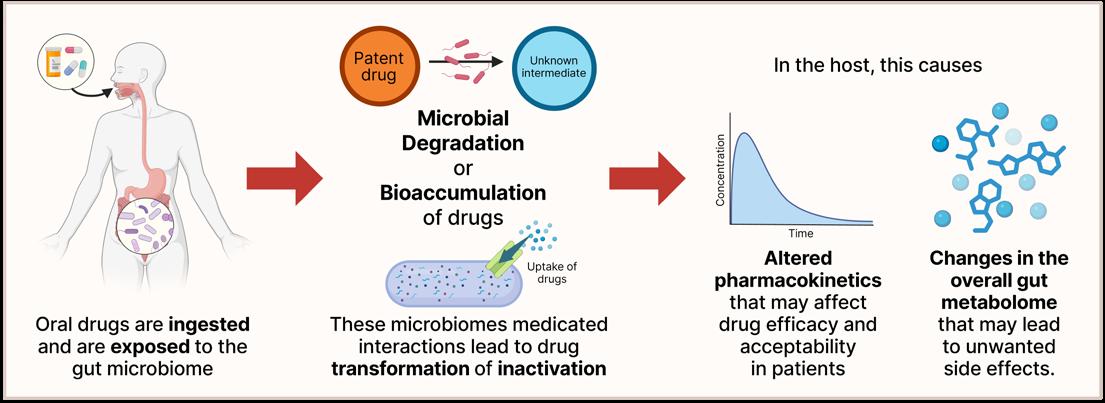
Does Gut Microbiome Composition Influence the Efficacy of Psychiatric Drugs?
Suryawinata and
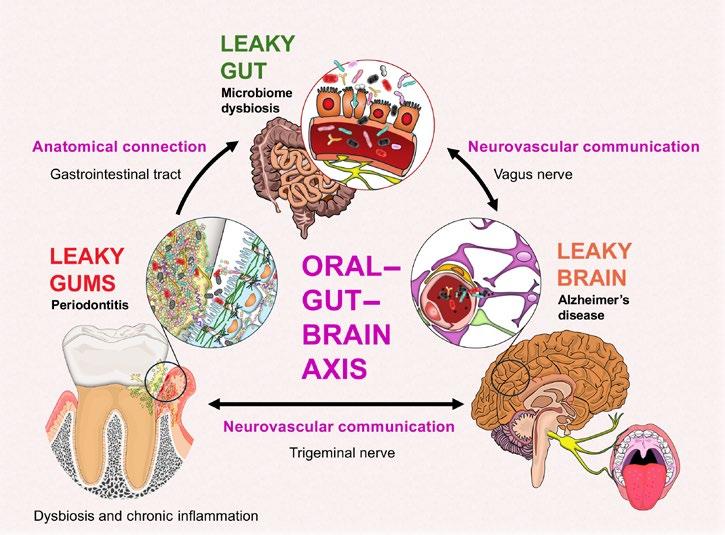
Systemic Health Implications of the Leaky Barriers within the Oral–Gut–Brain Axis and its Pathways of Communication
Kim et al.
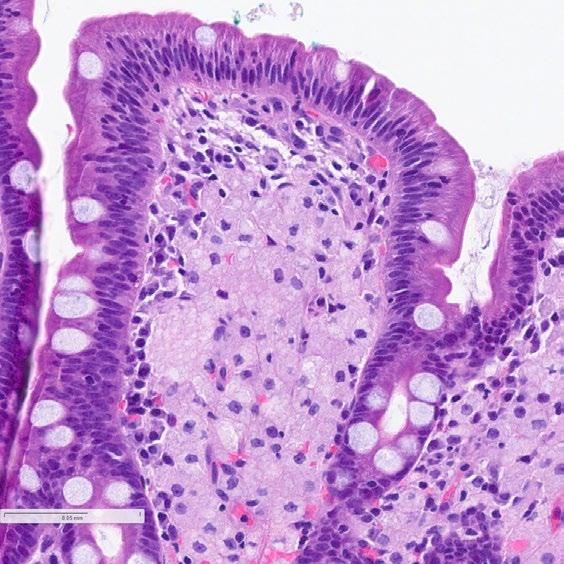
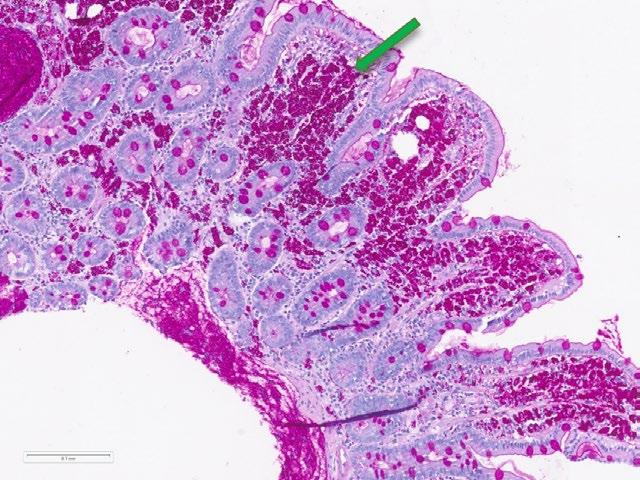
51 The Intersection of the Upper Gastrointestinal Microbiome and Oesophageal Cancer: A Review of Pathways and Therapeutic Insights
Bui et al.
Editor's Pick
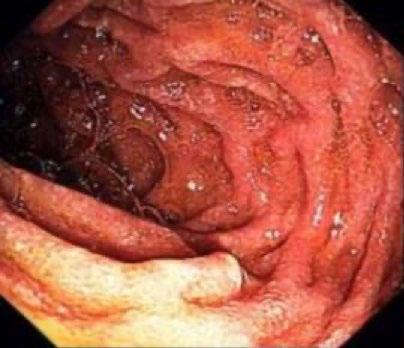
57 Whipple’s Disease, One of Medicine’s Great Imitators: A Case Report
Heard et al.
Articles
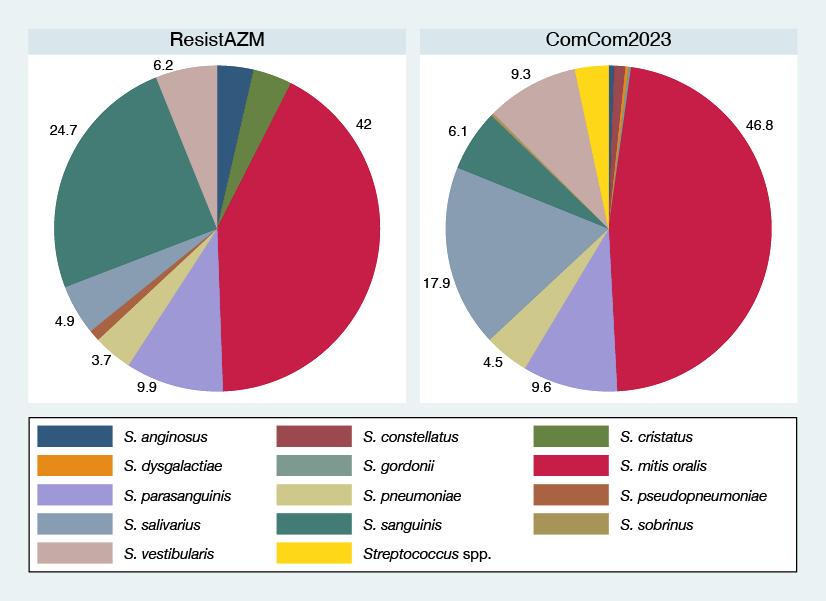
62 Azithromycin Susceptibility of Oral Streptococci in Belgian Men Who Have Sex with Men and the General Population: A Comparison of Two Cross-Sectional Surveys
Abdellati et al.
71 Infliximab-Induced Multifocal Motor Neuropathy in a Patient with Ankylosing Spondylarthritis and Crohn’s Disease: A Case Report with Anti-GM2 Antibodies
Eid and Matta
77 Rare Anti-HMGCR-Induced Immune-Mediated Necrotising Myopathy: A Case Report and Literature Review
Thayumanavan et al.
86 Unmasking the Imitator: Peritoneal Tuberculosis in the Guise of Ovarian Carcinoma: A Case Report
Moinuddin et al.
93 Prolonged Premature Preterm Rupture of Membranes:
"A lot of people have called the microbiome an additional organ"
Editorial Board
Editor-in-Chief
Prof Markus Peck-Radosavljevic
Klinikum Klagenfurt am Wörthersee, Austria
Current Chairman and Head of the Department of Gastroenterology and Hepatology, Endocrinology, Rheumatology, and Nephrology at Klinikum Klagenfurt am Wörthersee, with expertise in portal hypertension, hepatocellular carcinoma, and HIV-HCV coinfection.
Prof Ahmad Awada
Jules Bordet Institute, Belgium
Dr Abdullah Erdem Canda
Yildirim Beyazit University, Türkiye
Prof Sorin T. Barbu
“Iuliu Hațieganu” University of Medicine and Pharmacy, Romania
Prof Ian Chikanza
Harley Street Clinic, UK
Prof Lászlo Vécsei
University of Szeged, Hungary
Dr Pierfrancesco Agostoni
St. Antonius Hospital, the Netherlands
Dr Fernando Alfonso
Hospital Universitario de La Princesa, Spain
Dr Emanuele Angelucci
IRCCS Ospedale Policlinico San Martino, Italy
Dr George Anifandis
University of Thessaly, Greece
Dr Riccardo Autorino
Virginia Commonwealth University, USA
Dr Mátyás Benyó
University of Debrecen, Hungary
Prof Andrew Bush
Imperial College London, UK
Dr Hassan Galadari
United Arab Emirates University, United Arab Emirates
Dr Amir Hamzah Abdul Latiff
Pantai Hospital, Malaysia
Dr Lorenz Räber
Bern University Hospital, Switzerland
Aims and Scope
EMJ, the flagship journal of the EMJ portfolio, is an openaccess, peer-reviewed eJournal, committed to elevating the quality of healthcare globally by publishing high-quality medical content across the 18 clinical areas covered in our portfolio. The journal is published quarterly and showcases the latest developments across these clinical areas.
EMJ publishes peer-reviewed research papers, review articles, and case reports across all therapy areas of the EMJ portfolio. In addition, the journal publishes features and opinion pieces create a discussion around key topics in the field and broaden readers’ professional interests. The journal also features interviews with leading experts in various clinical disciplines.
The journal covers advances within the pharmaceutical arena by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ endeavours to increase knowledge, stimulate discussion, and contribute to the delivery of world-class updates in the clinical realm. We do not publish veterinary science papers or laboratory studies that are not linked to patient outcomes. Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
• A team of internal and independent medical writers.
Peer Review
Every review article, case report, feature, and research article published in EMJ undergoes peer review by at least two independent experts.
On submission, all manuscripts are assessed and undergo a technical check by the EMJ Editorial staff to determine their suitability for the journal and appropriateness for peer review. Editorial staff identify appropriate reviewers who are selected based on their specialist knowledge in the relevant area. All peer review is double-blind.
Following review, manuscripts are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected. Editorial staff are responsible for ensuring that necessary amendments to the manuscript have been made, with input from our Editorial Board or the original reviewers where necessary. The Editor of EMJ has final discretion over any proposed amendments. Manuscripts authored by members of the Editorial Board are subjected to the same double-blind process. Short opinion pieces are published following internal review and publication is at the discretion of the Editor. Congress-associated content authored by the EMJ Editorial staff undergoes internal quality control checks. Congress-related content sponsored or funded by our industry partners undergoes quality control checks independently. Industry-supported content that falls into any of
the categories that are eligible for peer review, undergoes the same peer review process.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields globally.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®.
EMJ is available through the websites of our leading partners and collaborating societies.
EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. EMJ is completely independent of any event reviews in this issue and the use of the organisations does not constitute endorsement or media partnership in any form whatsoever. The cover photo is of Nottingham, the location of work for the primary author of Editor's Pick.
Katie Wright, Katrina Thornber, Aleksandra Zurowska
Creative Director
Tim Uden
Design Manager
Stacey White
Creative Artworker
Dillon Benn Grove
Designers
Owen Silcox, Fabio van Paris, Shanjok Gurung
Senior Performance & Insight Lead
Darren Brace
Marketing Director
Kristina Mestsaninova
Chief Content Officer
Justin Levett
Chief Commercial Officer
Dan Healy
Founder and Chief Executive Officer
Spencer Gore
Welcome
Dear Readers,
Welcome to the first issue of the EMJ Flagship Journal for 2025. This issue brings a plethora of content and spotlights the microbiome, a key emerging player in our understanding of health and disease mechanisms.
It is becoming clear that the role of the microbiome extends beyond gut health, potentially impacting brain function, lung health, and the immune system. The human microbiome is not only a potential target for therapies but also a promising therapeutic tool itself. For instance, studies are currently exploring the use of nasal microbiota transplantation as a tool for limiting the spread of antimicrobial-resistant microbes, which would be a game-changer in combating the burden of antimicrobial resistance.1
Given that the microbiome is paving new avenues in therapeutic approaches, in this issue we have chosen to give you a glimpse of where key translational science is heading, and provide prospects for the future. Our interviewees share their own key insights and vision, ranging from the role of the microbiome in the response to cancer immunotherapy to the value of breastmilk in healthy microbiome development.
I extend my thanks to the Editorial Board, contributors, and peer reviewers for their great work on this journal. Our next issue will explore preventative healthcare, amongst other topics. Until then, I hope you find this an enjoyable, thought-provoking issue.
Reference
1. Shekhar S et al. Nasal microbiota transplantation: a gateway to novel treatments. Trends Microbiol. 2025;DOI:10.1016/j.tim.2024.12.010.
Editorial enquiries: editor@emjreviews.com
Sales opportunities: salesadmin@emjreviews.com
Permissions and copyright: accountsreceivable@emjreviews.com
Evgenia Koutsouki Editor
Reprints: info@emjreviews.com Media enquiries: marketing@emjreviews.com
Foreword
Dear Colleagues,
Welcome to the latest issue of the EMJ Flagship Journal, where we spotlight one of the most rapidly evolving areas of medical research: the microbiome. This issue explores the intricate relationship between microbial communities and human health, bringing together expert insights from a multidisciplinary perspective.
Our carefully curated content includes a selection of peer-reviewed articles, thought-provoking features, and exclusive interviews with leading experts in the field. Among this issue’s highlights is an indepth review of the upper gastrointestinal microbiome’s role in oesophageal cancer, shedding light on key pathogenic pathways and emerging therapeutic strategies. In addition to this, you will find features on the emerging role of gut bacteria in determining psychiatric drug efficacy, and the systemic health effects of microbial disruptions in the oral–gut–brain axis.
We are also proud to present interviews with two distinguished microbiome researchers: Dennis Lee Kasper, whose pioneering work has advanced our understanding of gut bacteria and immune system interactions, and Meghan Azad, a leader in neonatal and early-life microbiome research, discussing its profound implications for long-term health.
Finally, this issue’s Editor’s Pick is an insightful case report on Whipple’s disease, one of medicine’s ‘great imitators’. With this rare but often misdiagnosed condition, we highlight the importance of clinical awareness and the key role of microbiological diagnostics in patient care.
This issue explores the intricate relationship between microbial communities and human health
As always, I extend my gratitude to our authors, reviewers, interviewees, and Editorial Board members for their contributions to EMJ. I hope this issue offers valuable insights and meaningful discussions for healthcare professionals.
Prof Markus Peck-Radosavljevic
Professor of Medicine and Chairman, Department of Gastroenterology and Hepatology, Endocrinology, Rheumatology and Nephrology, Klinikum Klagenfurt am Wörthersee, Klagenfurt, Austria
New Horizons for Metachromatic Leukodystrophy with the Advent of Newborn Screening
This symposium took place in September 2024, as part of the Society for the Study of Inborn Errors of Metabolism (SSIEM) 2024 congress held in Porto, Portugal.
Speakers: Amy Gaviglio,1 Laura Adang,2 Lucia Laugwitz,3 Hanka Dekker4
1. Connetics Consulting, Minneapolis, Minnesota, USA
2. Children's Hospital of Philadelphia, Pennsylvania, USA
3. University of Tübingen, Germany
4. VKS, Zwolle, the Netherlands
Disclosure: Gaviglio has previously served on a speaker's bureau for Orchard Therapeutics Ltd. and Takeda Pharmaceuticals U.S.A., Inc. No further known disclosures on file. Some of the speakers involved received honorarium for their involvement in the symposium but no honoraria was received for involvement and/or mention in this article.
Disclaimer: This is a non-promotional article funded by Orchard Therapeutics representing a summary of an Orchard Therapeutics non-promotional sponsored symposium at the SSIEM 2024 conference.
Support: The publication of this article was sponsored by Orchard Therapeutics.
Meeting Summary
This article summarises an Orchard Therapeutic-sponsored symposium titled ‘New horizons for metachromatic leukodystrophy – with the advent of newborn screening’, which was delivered on 5th September 2024 as part of the Society for the Study of Inborn Error of Metabolism (SSIEM) annual congress in Porto, Portugal.
During the symposium, the panellists discussed the applicability of the Wilson and Jungner criteria to metachromatic leukodystrophy (MLD), which they considered a strong candidate for newborn screening (NBS) thanks to the existing supporting evidence. This includes the availability of a screening test, the agreement on how to confirm diagnosis after positive screening, the presence of a prospective population-based newborn screening project that identified at least one infant with the condition, and the evidence that an early identification through NBS leads to better health outcomes.
In the symposium, the speakers also reminded the audience of the existence of a validated three-tier screening algorithm of recent publication and the availability of two consensus guidelines that have been published in both the EU and the USA, and which unanimously support the implementation of NBS for MLD.
PHARMA
PARTNERSHIP
Introduction
The decision of whether or not to screen for a disease still relies heavily on a framework developed almost 40 years ago, when the WHO commissioned a report on screening from James Maxwell Glover Wilson, then Principal Medical Officer at the Ministry of Health in London, UK, and Gunner Jungner, then Chief of the Clinical Chemistry Department of Sahlgren’s Hospital in Gothenburg, Sweden. The report, published in 1968, was titled ‘Principles and practice of screening for disease and it has since become a public health classic’.1
Despite the admirable method of combating disease, the practice of screening comes with some challenges, and the authors were preoccupied with the notion that: “The central idea of early disease detection and treatment is essentially simple. However, the path to its successful achievement (on the one hand, bringing to treatment those with previously undetected disease, and, on the other, avoiding harm to those persons not in need of treatment) is far from simple, though sometimes it may appear deceptively easy.”1
The Wilson and Jungner Criteria and Newborn Screening
For this reason, Wilson and Jungner attempted to define screening criteria to guide the selection of conditions that would be suitable for screening, based, among other factors, on the capacity to detect the condition at an early stage and the availability of an acceptable treatment:
1. The condition sought should be an important health problem.
2. There should be an accepted treatment for patients with recognised disease.
3. Facilities for diagnosis and treatment should be available.
4. There should be a recognisable latent or early symptomatic stage.
5. There should be a suitable test or examination.
6. The test should be acceptable to the population.
7. The natural history of the condition, including development from latent to declared disease, should be adequately understood.
8. There should be an agreed policy on whom to treat as patients.
9. The cost of case-finding (including diagnosis and treatment of patients diagnosed) should be economically balanced in relation to possible expenditure on medical care as a whole.
10. Case-finding should be a continuing process and not a ‘once and for all’ project.
These criteria are particularly challenging when applied to rare diseases, conditions affecting less than 1 in 2,000 people.2 It is currently estimated that there are over 7,000 rare diseases, and while 80% of rare diseases have an identified genetic origin, they can also be caused by disordered immunity, infections, allergies, deterioration of body tissues and organs, or disruption to development while in the womb.
MLD is a rare inherited lysosomal storage disease affecting 1 in 100,000 newborns and caused by deficiency of arylsulfatase A (ARSA), due to mutations in the ARSA gene.3 Reduced ARSA activity results in the accumulation of sulfatides in the CNS and peripheral nervous system, leading to progressive demyelination, neuroinflammation, and neurodegeneration.4-6 These events result in progressive motor and cognitive deterioration, with loss of motor and neurocognitive functions, and ultimately death.4,5,7 Three clinical forms are commonly described on the basis of age at first symptom onset: late-infantile (LI; ≤30 months), juvenile (subdivided into early juvenile [30 months–6 years] and late
juvenile [7–16 years]), and adult MLD (≥17 years), with earlier age at onset or presence of motor symptoms at onset associated with a more severe and rapid disease course.4-6,8,9 Regardless of the clinical variant, the underlying disease pathophysiology is similar for all phenotypic forms of MLD.4,7,10
The fast progression of the LI subtype of the disease (accounting for the majority of the MLD cases, or about 60% of the affected population), clearly highlights the importance of identifying children with urgency. Affected children initially show a normal development, but once symptoms start to become evident, patients may no longer be eligible for treatment, entering a disease progression phase that leads to irreversible and progressive neurological damage and ultimately death, usually in the first decade of life.11
The early detection of the disease during the asymptomatic phase is therefore paramount to increase the chances of a timely intervention and better clinical outcomes. Should MLD be considered a good candidate for newborn screening, then? As explained by Amy Gaviglio, a Genetic Counsellor, Public Health Genetics and Rare Disease Consultant from Minneapolis, Minnesota, USA, with a wealth of experience in the expansion of newborn screening programmes, to answer this question, we must first consider several criteria: 1) Is there a screening test available for use at a population level in the newborn period? 2) Is there an agreed-upon way for a clinical specialist to confirm the diagnosis after a positive screening? 3) Is there a prospective, population-based newborn screening project that has identified at least one infant with the condition? 4) Does early identification through newborn screening lead to better health outcomes compared to usual clinical identification?
The Principles and Practice of Newborn Screening
As for any disease, the inclusion of MLD on the newborn screening panel requires the knowledge and the generation of evidence
to confirm its readiness for population newborn screening. To achieve this goal, an international group of scientists, patient advocates, and MLD experts started three key retrospective studies, using dried blood spots to validate the screening algorithm.12-14 The promising results, data collected, and knowledge acquired led to the initiation of 11 additional prospective investigatorinitiated newborn screening studies for MLD, which are active throughout the USA, Europe, and the Middle East, with more than 300,000 newborns screened to date (as of September 2024). Most importantly, through these prospective studies, five newborns with no known previous MLD family history were screened positive and subsequently diagnosed with the disease. The evidence generated is being used to support the nomination to add MLD to the newborn screening panel in Europe (including the UK, Ireland, France, and Germany) and in the USA, and Norway is the first country in the world to add MLD to the national newborn screening programme, officially starting on 6th January 2025.
Lucia Laugwitz, a clinician scientist and child neurologist in training in the Department of Neuropediatrics and the Institute for Medical Genetics and Applied Genomics at the University of Tuebingen, Germany, explains that when starting a newborn screening, the first and most important aspect is to establish the existence of a screening test available for use at a population level in the newborn period. In 2024, Laugwitz et al.15 validated and published the results of a threetier screening algorithm for MLD; the protocol includes two biochemical tests (sulfatides and enzyme activity) followed by a third genetic test not only for the ARSA gene, but also the SUMF1 and PSAP genes associated with multiple sulfatides deficiency and prosaposin B deficiency respectively.
Due to the subtleness of MLD, it is important that all three genes are tested, because, at a biochemical level, both SUMF1 and PSAP can mimic MLD but have currently no approved treatments. The combination of two sulfatides (C16:0 and C16:1-OH) is used to reduce the number
First tier: Sulfatides screening
0.17 µmol/L for C160 or ≥0.050 µmol/L for C161 OH
of false positives in the first tier, and the subsequent need for second-tier enzymatic biochemical testing, which in turn improves the feasibility for implementation at a national level (Figure 1).
Selecting Conditions Suitable for Newborn Screening
In 2024, two complementary consensus guidelines on MLD have been published: one from the MLD initiative (MLDi)16,17 stressing the importance, among other things, of implementing newborn screening to promptly identify children and ensure proper management and care; and one in the USA with a prominent focus on patient care and specific monitoring requirements.18
As highlighted by Laura Adang, Assistant Professor of Neurology at the Children’s Hospital of Philadelphia, Pennsylvania, USA, the most relevant aspect of consistency between the two documents is that all the authoring experts unanimously support
Second tier: ARSA enzyme
µmol/L/h
Third tier: Genetic sequencing
the implementation of newborn screening for MLD, whose subacute nature makes it difficult to be detected until children become symptomatic, affecting their ability to potentially receive appropriate treatment. The same experts also agree and strongly recommend initiating treatment in identified individuals before symptom onset, the best option to help the patients and their families.
As clearly shown in Figure 1, the screening algorithm includes a biochemical confirmation of ARSA enzyme activity in leukocytes and urinary sulfatides (performed in MLD expert centres) followed by genetic confirmation, which is required to help predict MLD subtypes (family history, genotype, and ARSA enzyme activity). In general, 80% of the time experts anticipate what MLD subtype the child will have; therefore, the consensus guidelines are particularly relevant to support the management and monitoring of the 20% of the patients with uncertain subtypes, with the goal of early intervention.
Figure 1: Validated screening algorithm for metachromatic leukodystrophy.15
Adapted from Laugwitz et al.15
There are several known and recognised challenges, in particular, genetic variants of unknown significance and pseudo deficiencies; their existence is not dismissed by the expert’s community, who recognise the need to establish essential and wider collaboration. With time, the guidelines will be refined and updated to reflect new discoveries; gradually, new variants of unknown significance will emerge once national newborn screening programmes are implemented, and in parallel, the understanding of the entire MLD community of clinicians, scientists, and experts will improve, leading to better patient care and treatment outcome, so long as the information is shared as openly as possible.
Laugwitz et al.17 were not only able to implement a newborn screening algorithm but also a comprehensive care pathway going from the confirmation of MLD diagnosis to the clinical assessment and subtype prediction and treatment decision. Screening is only the first step of this process, but how should the identified MLD children be managed? Is early identification through newborn screening the necessary step leading to better health outcomes compared to clinical identification?
In a paper published by Claire Horgan, Senior Clinical Research Fellow in paediatric bone marrow transplant, CAR-T, and stem cell gene therapy at Royal Manchester Children’s Hospital in Manchester, UK, it was reported that in the year following NHS approval of an active treatment for MLD, 17 UK patients with MLD were referred for treatment.19 Four patients met eligibility criteria and were treated, whereas 11 patients failed screening; 10 due to symptomatic disease (LI subtype) and one with early juvenile disease and cognitive decline. Two further patients with later onset subtypes did not meet the approval criteria, and three out of four treated patients were diagnosed by screening after MLD was diagnosed in a symptomatic older sibling. This result is a clear testament to the challenges of diagnosing MLD in a timely manner before symptom onset, which further supports the need for newborn screening.
The key to progress in this field is cooperation within the expert’s community. In Europe, the establishment of a network of MLD experts under the umbrella of the MLDi is an example of this cooperation. The MLDi is an international patient registry for MLD and an academic collaborative network, and data in the MLDi registry can be used for academic research, regulatory decision-making, and drug development. In the USA, the establishment of the Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN), a consortium of scientists, industry stakeholders, and patient advocacy leaders, promotes advances in the diagnosis and treatment of leukodystrophies, and specifically, it seeks to create a robust research infrastructure that will allow for collection and analysis of longitudinal natural history data, development of novel clinical outcome assessments, and identification of surrogate biomarkers, ultimately paving the way for transformative therapeutic trials across the leukodystrophies.
Conclusion
MLD strongly fulfils the Wilson and Jungner criteria for newborn screening due to the availability of a screening test for use at a population level in the newborn period, the availability of an agreed way for a clinical specialist to confirm the diagnosis after a positive screen, the presence of a prospective, populationbased newborn screening project that has identified at least one infant with the condition, and the potential of better health outcomes of an early identification through newborn screening compared to usual clinical identification.
When thinking about diseases and what makes them good candidates for newborn screening, it is paramount they meet certain criteria, including the feasibility of testing for that disease in the newborn period. Typically, this is done for the purpose of newborn screening using a dried blood spot matrix on dried blood that is collected from the newborn around 24–72 hours after birth, while ensuring that that test has low false positive rates and low false
negative rates, as well. A second important criteria to consider is whether the natural history of the disease and the type targeted with newborn screening are sufficiently understood. MLD is a spectrum; therefore, it is fundamental to recognise what that spectrum looks like and how to target the various subtypes that may have effective treatments. The third condition considers the feasibility of the test, to make sure the process works. For this reason, prospective pilot studies that look at the feasibility of this testing all the way through the diagnosis and administration of early pre-symptomatic interventions are exceptionally useful. Lastly, the existence of an effective treatment, which is needed asymptomatically to improve outcomes, completes the picture.
For a family, hearing that their child has a fatal disorder is extremely difficult, and they require appropriate social and psychological support, explained Hanka Dekker, Director of VKS, Zwolle, the
References
1. Wilson JMG, Jungner G. Principles and practice of screening for disease Geneva: WHO; 1968. Available at: http://www.who. int/bulletin/volumes/86/4/07-050112BP. pdf Last accessed: 10 February 2025.
2. Department of Health and Social Care. The UK Rare Disease Framework. 2021. Available at: https://www.gov. uk/government/publications/uk-rarediseases-framework/the-uk-rarediseases-framework Last accessed: 3 January 2025.
3. Fumagalli et al. Lentiviral haematopoietic stem-cell gene therapy for earlyonset metachromatic leukodystrophy: longterm results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399:372-83.
4. van Rappard DF et al. Metachromatic leukodystrophy: disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29:261-73.
5. Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy– an update. Neuropediatrics. 2010;41:1-6.
6. Von Figura K et al., “Metachromatic leukodystrophy,” Scriver CR, BA, Sly WS (eds.), The Metabolic and Molecular Bases of Inherited Diseases (2001), New York: McGraw-Hill, pp. 3695.
7. Elgun S et al. Phenotypic variation between siblings with metachromatic leukodystrophy. Orphanet J Rare Dis. 2019;14:136.
Netherlands. In certain situations, and until now, parents had to care for both an index patient and their younger sibling, both diagnosed with the disease, witnessing their children losing all their abilities within years. In many cases, these challenges led to the family break-up. Newborn screening can change all this by identifying the index patient early enough to be treated, and the resulting psychological follow-up for these families will be much better.
As explained by Gaviglio, when considering these criteria and applying them to MLD, it is appropriate to say that the disease meets them all and should be included in newborn screening panels. This will help the early identification of patients with MLD while still in the asymptomatic phase, allowing for early access to potential disease-modifying treatments and improved health outcomes that would otherwise be negated, leading to irreversible neurodegeneration and early death of affected children.
8. Kehrer C et al, on behalf of the German Leukonet. The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol. 2011;53:850-85.
9. Kehrer C et al. Association of age at onset and first symptoms with disease progression in patients with metachromatic leukodystrophy. Neurology. 2021;96:e255-66.
10. Biffi A et al. Metachromatic leukodystrophy – mutation analysis provides further evidence of genotype–phenotype correlation. Clin Genet. 2008;74:349-57.
11. Von Figura K, Gieselmann V, Jaeken J, “Metachromatic leukodystrophy,” Scriver CR, BA, Sly WS (eds.), The Metabolic and Molecular Bases of Inherited Diseases (2001) 8th edition, Vol. 3, New York, NY: McGraw-Hill, pp. 3695-372.
12. Hong X et al. Toward newborn screening of metachromatic leukodystrophy: results from analysis of over 27,000 newborn dried blood spots. Genetics in Medicine. 2020;23(3):555-61.
13. Pettazzoni M et al. LC-MS/MS quantification of three C16 sulfatide species in dried blood spots for the diagnosis and treatment monitoring of metachromatic leukodystrophy. Mol Genet Metab Rep. 2023;138(2):107265.
14. Wu TH. Improving newborn screening test performance for metachromatic leukodystrophy: Recommendation from
a pre-pilot study that identified a lateinfantile case for treatment. Mol Genet Metab Rep. 2024;142(1):108349.
15. Laugwitz et al. Newborn screening and presymptomatic treatment of metachromatic leukodystrophy. N Engl J Med. 2024;391:1256-8.
16. The MLD initiative. To improve disease management of metachromatic leukodystrophy through an international disease registry and multistakeholder collaboration. 2024. Available at: https:// www.mldinitiative.com/. Last accessed: 18 February 2025.
17. Laugwitz L et al. Newborn screening in metachromatic leukodystrophy – European consensus-based recommendations on clinical management. Eur J Paediatr Neurol. 2024;49:141-54.
18. Adang LA et al. Consensus guidelines for the monitoring and management of metachromatic leukodystrophy in the United States. Cytotherapy. 2024;26(7):739-48.
19. Horgan C et al. A retrospective cohort study of Libmeldy (atidarsagene autotemcel) for MLD: What we have accomplished and what opportunities lie ahead. JIMD Reports. 2023;64(5):1-7.
March 2025
Prescribing Information and Adverse Event reporting can be found in the Disclaimers section below.
Retracted: Ozanimod▼ in Ulcerative Colitis: Key New Data
A promotional summary of selected data presented at the United European Gastroenterology Week (UEGW) held in Vienna, Austria, from 12th–15th October 2024 and the American College of Gastroenterology’s (ACG) Annual Scientific Meeting held in Philadelphia, Pennsylvania, USA, from 25th–30th October 2024.
Authors: James O. Lindsay,1 David T. Rubin,2 Nicholas Scalzo,3 Joana Torres4
1. Barts and the London School of Medicine and Dentistry, UK
2. Inflammatory Bowel Disease Center, University of Chicago Medicine, Illinois, USA
3. Icahn School of Medicine at Mount Sinai, New York, USA
4. Division of Gastroenterology, Hospital da Luz, Lisbon, Portugal
Disclosure: Lindsay has received consulting and/or speaker fees from AbbVie, Bristol Myers Squibb, Celgene, Celltrion, Eli Lilly, Engitix, Ferring, Galapagos, Gilead Sciences, GlaxoSmithKline, Janssen, MSD, Napp, Orchard Therapeutics, Pfizer, Shire, and Takeda; and investigator-led research grants from AbbVie, Gilead Sciences, and Takeda. Rubin has received consulting and/or speaker fees from AbbVie, AltruBio, Aslan Pharmaceuticals, Athos Therapeutics, Bellatrix Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Chronicles, ClostraBio, Connect BioPharma, EcoR1, Eli Lilly, Genentech/Roche, Gilead Sciences, Iterative Health, Janssen, Kaleido Biosciences, Pfizer, Prometheus Biosciences, Reistone, Seres Therapeutics, Syneos, Takeda, Target RWE, and Trellus Health; and grant/research support from Takeda. Nicholas Scalzo reported no conflicts of interest. Torres has received consulting and/ or speaker fees from AbbVie, Bristol Myers Squibb, Janssen, Pfizer, and Sandoz; and grants from Abbvie and Janssen.
Acknowledgements: Writing assistance was provided by Nicola Humphry, Nottingham, UK.
Disclaimer: The opinions expressed in this article belong solely to the authors of the posters.
Ozanimod▼ is indicated for the treatment of adult patients with relapsing remitting multiple sclerosis (RRMS) with active disease as defined by clinical or imaging features, and for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent.
Ozanimod® is subject to additional monitoring. This will allow quick identification of new safety information.
Prescribing information for HCPs in the UK can be found here. Prescribing information for HCPs in Ireland can be found here.
Adverse events should be reported. Reporting forms and information can be found via: Great Britain & Northern Ireland – The Yellow Card Scheme at: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App store; Ireland –HPRA Pharmacovigilance at www.hpra.ie
Adverse events should also be reported to Bristol-Myers Squibb via medical.information@bms.com or 08007311736 (Great Britain & Northern Ireland); 1 800 749 749 (Ireland).
Support: The publication of this article was funded by Bristol Myers Squibb.
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
This article has now been retracted and a retraction statement has been published.
Retraction : Ozanimod▼ in Ulcerative Colitis: Key New Data
Interviews
EMJ had the pleasure of interviewing Dennis Lee Kasper and Meghan Azad, two pioneers in the emerging field of the microbiome. Kasper discusses the fascinating interaction between the gut microbiota and immune system, highlighting the complexity of this newly discovered ‘organ’. Azad explores how infant nutrition and breastfeeding shape the early microbiome, with significant implications for long-term health and development of chronic diseases.
Featuring: Dennis Lee Kasper and Meghan Azad
Dennis Lee Kasper Professor of Medicine and Immunology, Harvard
Medical School, Boston, Massachusetts, USA
Our gut microbiome profoundly shapes our immune system; it both regulates our immune status and can throw it off kilter
Q1Your major research focus lies in the interaction between the gut microbiota and the immune system. How have you seen the field evolve since the start of your career?
Early in my career, there was a small group of investigators interested in commensal microbes, and these were basic scienceoriented microbiologists. There was a flurry of papers in the 60s and 70s about the role of commensal microbes (a.k.a. anaerobic bacteria) in disease, bringing about a more general awareness of commensals. Along with this awakening, antibiotics specifically effective against anaerobic bacteria were developed. Of interest were infections arising from commensal colonised sites where there was leakage or spread of commensal bacteria to normally sterile sites, including the peritoneum, lung, brain, and liver. These infections often manifested themselves as abscesses.
Groups in Germany and the USA who were studying microbes in the gut were able to raise germ-free mice, and they quickly learnt that these mice were very susceptible to infection. When their isolators got contaminated, these mice often died. It then became more or less known that commensal microbes had something to do with fortifying the immune system, but nothing specific was understood.
For the prior 30 years, I had been studying infection and abscess formation, and we had focused on a gut anaerobic organism called Bacteroides fragilis. When I looked at clinical studies that were enumerating anaerobic bacteria associated with diseases like peritonitis or lung abscesses, B. fragilis kept coming up as an important contributor. It did become clear very early that multiple commensal organisms were often isolated from infectious sites, unlike classic infections like pneumonia, meningitis, or sepsis, where typically one organism is responsible.
We made a number of observations about B. fragilis that clearly differentiated it from the typical gram-negative pathogen. Pathogens, such as pneumococci, meningococci, streptococci or Escherichia coli have one capsular polysaccharide in a given strain. When we were trying to isolate the capsule of B. fragilis, we were getting different chemical results from grow up to grow up, which was very confusing. Then, around the late 80s, we started to understand that each organism made several polysaccharides and could express them as capsules, which was very unusual. When B. fragilis was sequenced by the Sanger Centre, we learnt that it has loci to produce at least eight polysaccharides. Now, it's known that some other Bacteroides in the gut can make more than one polysaccharide.
A typical polysaccharide may have 3–15 genes responsible for its synthesis, and those genes occur in loci or operons. These are groups of genes that are flanked together and regulated by a single promoter. Work done with a former postdoc in my lab, Laurie Comstock, University of Chicago, Illinois, USA, showed that B. fragilis had at least eight loci for the production of polysaccharides, and that there was an unusual genetic mechanism that regulated it. We named these polysaccharides as A, B, C, D, E, F, G, and H. It turned out polysaccharide A (PSA) was the most important, and also the most abundant.
When we began studying PSA, we observed some unanticipated immunologic responses to this molecule. For context, all the childhood vaccines against pneumococcus, influenza, and meningococcus are conjugate vaccines: polysaccharides
chemically coupled to proteins. The reason behind this is that these polysaccharides themselves, particularly in young children, were not immunogenic, so that is why they were coupled to proteins. It was a dogma in immunology that polysaccharides don't activate T cells; they were T cell-independent, and by coupling the polysaccharide to a protein, T cell help was activated.
However, former postdoc Brian Cobb, Case Western Reserve University, Cleveland, Ohio, USA, found that PSA was activating T cells in the absence of a protein, and that became a major focus of our work. PSA is processed and presented by antigen presenting cells (APCs). Former postdoc Arthur Tzianabos, Lifordi Immunotherapeutics, Inc, Boston, Massachusetts, USA, showed that these APCs induce CD4+ T cells to make a cytokine called IL-10, which actually turns off immune responses and inflammation. I thought, “This organism’s induction of T cells to make IL-10 must have something to do with it living in the gut.” In the gut, you have 100 trillion organisms, and you and I are sitting here talking to each other, and we're fine. But, how is it that if you had 100 trillion organisms in your blood, you would not be alive? There’s an immunologic phenomenon called tolerance, which is very important to health and to preventing autoimmune disease. When you break tolerance, you start making immune responses to yourself.
In the late 90s, Jeff Gordon, Washington University, St. Louis, USA, focused on identification and enumeration of specific microbiota, and understanding microbiota populations in health and disease. He focused on genomic and metabolic relationships of the host and their commensal microbes. I
think the word ‘microbiome’ was actually coined by Nobel Laureate Joshua Lederberg, from Stanford University, California, USA, and Gordon capitalised on the term ‘microbiome’. This was a turn of the century, and a rumination was getting louder that said, “Gee, these organisms may be important.”
About 25 years ago was when I decided it was time to turn our attention from pathogenesis to anaerobic organisms living in the gut. Work by my former postdoc Sarkis Mazmanian, California Institute of Technology, USA, found that our gut microbiome profoundly shapes our immune system; it both regulates our immune status and can throw it off kilter, making you more susceptible to certain diseases. Without the gut microbiome, you don't have normal development of T cell populations systemically, and immune tissues such as the spleen and lymph nodes are deficient in T cells and have abnormal histology.
Q2
You have elucidated the role of B. fragilis, an important intestinal commensal, in immune system modulation. Can you explain how PSA on the surface of this organism stimulates the immune system?
I often use B. fragilis as a model to try to understand at a mechanistic level how molecules of gut bacteria stimulate the immune system, but B. fragilis and PSA are just models for the interaction of microbial molecules in the gut with the immune system, they're not the whole story.
PSA stimulates both the innate and adaptive immune system. PSA binds to innate Toll-like receptors (TLR4) and C-lectin receptors (dectin-1) on APCs and is transported to the endosome,
where it gets depolymerised by nitric oxide into smaller subunits, usually about eight or 10 sugars long. The structure of PSA has one characteristic that differs it from most bacterial polysaccharides: it is zwitterionic, meaning it has positive and negative charges on each repeating unit. Most polysaccharides have no charge groups or only negative charge groups. Because of its zwitterionic nature, PSA actually binds to the major histocompatibility complex class II (MHC II) cleft on APCs. This cleft is loaded with positive and negatively charged amino acids. It's an ionic binding between the amino acids in MHCII and the zwitterionic charge of the depolymerised PSA. Most other polysaccharides get digested by nitric oxide or reactive oxygen species, but they don't bind, so they never get presented because they lack the positive and negative charge groups. PSA activates T cells because it gets presented by MHC II to the T cell receptor, and this induces IL-10 production.
Q3You also discovered a role for PSA in shaping mammalian immune development and mediating Th1/Th2 balance. Can you elaborate on this mechanism, and its potential implications for autoimmune or inflammatory disorders?
There are several types of CD4 T cells; two of the major types are called Th1 and Th2. Th1 cells are primarily involved with cellular immunity, while Th2 cells are is primarily humoral immunity stimulating. In the immune system of a germ-free mouse, T cells are heavily Th2-skewed. If you colonise germ free mice with B. fragilis or give PSA orally to a sterile mouse, you actually balance Th1 and Th2 cells in the systemic immune system, bringing it back to normal. One of the other interesting things we found at that time was that, with no bacteria, the spleen and lymph nodes have abnormal histology, but when you give them PSA early in life, they end up with normal tissue in the spleen and lymph nodes. The
interactions of molecules of gut bacteria with the immune system has been a primary focus of ours in the last 20 years.
There’s another subset of T cells that live in the gut, called regulatory T cells, which shut off inflammation mostly using IL10. It is thought that if you give patients with Crohn’s disease a healthy dose of regulatory T cells, that would make the disease quiescent. PSA induces these regulatory T cells to make IL10. There is another subset of T cells in the gut lamina propria called natural killer T cells (NKT) cells, and we showed that Bacteroides make a molecule which is a glycosphingolipid, not a polysaccharide, that actually turns off the pro-inflammatory response of NKT cells. So, those are both examples of specific T cell subsets regulated by gut bacteria.
The literature now contains papers about associations of gut bacteria with neurologic diseases like autism, Parkinson's
A lot of people have called the microbiome an additional organ and I believe that we haven't really come to grips with how it works or the full scope of its impact
disease, and even schizophrenia. Nearly all human systems have in one fashion or another been associated with the microbiome. Each of us has a few hundred microbial species in our gut, with a total organism load of around 100 trillion microbes. Interestingly, the effect of these organisms on the immune system is strain-specific. You could have a Clostridium that affects regulatory T cells, and I could have a different bacterial species impacting on the same cell type, making understanding this so complex. It's going to take science a long time to really understand the full scope of the microbiome. A lot of people have called the microbiome an additional organ and I believe that we haven't really come to grips with how it works or the full scope of its impact.
Q4
Group B Streptococcus (GBS) is the main cause of serious neonatal bacterial infections. Can you tell us about your contributions to the development of vaccines against GBS?
We basically conceived of the possibility of a vaccine for this devastating neonatal disease in one of my first papers. In 1975–76, we showed that women whose babies got GBS infection lacked antibody to the polysaccharide, but if the mother had antibodies to the capsular polysaccharide, the baby was protected through transplacental passage of the antibodies. So that led me on a 30-year intense effort to try to get companies interested in immunising pregnant women.
We identified the polysaccharides that represent each of different serotypes of GBS, solved the structure of all the polysaccharides, and made conjugate vaccines by
covalently linking them to carrier proteins. These vaccines were immunogenic in adult humans and the plan was to immunise women and protect their neonates by transplacental passage of antibodies. Unfortunately, at the time, industry was not interested in further development because of the fear of liability associated with immunising pregnant women. Currently, however, there are some companies moving ahead with these conjugate vaccines using the capsular polysaccharides and a similar chemical approach to that we discovered.
Q5 How can your work in GBS inform the design of vaccines for other age-specific or immunecompromised populations?
We designed the vaccine for GBS and made many basic science contributions to the understanding of it, but the basic idea of taking polysaccharides and coupling them to proteins for a vaccine was developed in the 1940s with pneumococcal capsular polysaccharides. So, the idea of enhancing T cell help to make antibodies to polysaccharides was known. In fact, there was already a vaccine on the market that used that basic approach with Haemophilus influenzae type B capsular polysaccharides. Much of the pneumococcal vaccine development that was done by some pharma companies followed the work we did with GBS, although our vaccine never made it to the market.
One basic concept that came from our work on both B. fragilis PSA and GBS conjugate vaccines was that T cell receptors are able to recognise carbohydrates if they are presented in the presence of the MHCII molecule.
Q6
You have also led fascinating research on Francisella tularensis, a potential agent of bioterrorism. Can you tell us more about the approach you used to develop a vaccine against this pathogen?
There was a huge scare in the world about anthrax as a biologic weapon in the early 2000s, and that fear spread to all of the organisms with potential for use in warfare. Tularaemia is certainly one of those organisms
For F. tularensis, we took the lipopolysaccharide (LPS) of the organism, called endotoxin of the organism. We chemically cut off the toxic lipid A end of the LPS, and we coupled the polysaccharide to carrier proteins to make a conjugate vaccine. We also did studies on what optimises the polysaccharide immunogenicity in these conjugates, and found that longer polysaccharides made better conjugate vaccines than smaller polysaccharides.
Q7
Moving beyond microbiome-wide associations to identify causative microbial agents has been a significant challenge in the field. How did your team overcome this barrier?
Our work has primarily taken a reductionist approach where we have tried to solve mechanistic relationships of host and microbe. Microbiomewide associations have not fully solved the questions that arise when studying the microbiome. For example, just because we have an important species like B. fragilis, it does not mean all the organisms belonging to the species B. fragilis will act the same. There is enough genetic variation between strains of a given species to result in different
functions. So, you really have to talk about individual strains.
For example, it became known about 10 years ago that the microbiome has an effect on the efficacy of checkpoint blocking antibodies used for cancer immunotherapy. The main targets of immunotherapy currently are PD-1, PD-L1, and CTLA4. It became clear that gut colonisation with certain organisms, as well as some patient microbiomes, had a negative effect on checkpoint blockade efficacy in mouse models. This has now blossomed into an important field. When we began work in this field, it was difficult to find an organism in a given patient’s microbiome that inhibited checkpoint blockade.
So, we developed a model, for which I published a paper in 2017 with my former postdoc and colleague Neeraj K Surana, Duke University, Durham, North Carolina, USA. We applied that model to this question of checkpoint inhibition working with Arlene Sharpe and Gordon Freeman, from Harvard Medical School, Massachusetts, USA. First, we found microbiomes that inhibited checkpoint blocking in a mouse model, and then, using specific antibiotics and various other manipulations, our postdocs Francesca Gazzaniga, Harvard Medical School, and Joon Seok Park, University of Chicago, sorted through microbes and came out with a small group of organisms from which we eventually could isolate one organism that had a pretty significant effect on checkpoint blocking. I hadn't even heard of this species when we found it. It’s called Coprobacillus catenaformis.
Using models and studying the response of cells in tumours and in tumour-draining lymph nodes,
we learnt that, when mice were colonised with C. catenaformis, another checkpoint-blocking molecule called PD-L2 was actually lowered in the tumourdraining lymph nodes on dendritic cells. It was known from the literature that PD-L2 had two different binding receptors. One is PD-1, which is a main target of checkpoint blocking, and the other was called repulsive guidance molecule b (RGMb). It turned out that it was the impact on RGMb that affected the efficacy of PD-(L)1 therapy. If you took a monoclonal antibody to RGMB or to PD-L2 and used it in combination with the antibody to PD-(L)1, you actually could overcome tumour resistance. It was a pretty nifty mechanistic approach to figuring out a complex biological problem.
To return to your question, how do we overcome this barrier? One of the reasons we've been successful with this is that, even though I'm in an immunology department, I also use a lot of microbiology, chemistry, and genetics. It takes an interdisciplinary approach to solve complex problems in the microbiome because you couldn't ask for a more complex system. People who only consider
the whole microbiome, to me, are missing part of the story. It's really the molecules on, or made by, the microbes in the microbiome that are having an immunological effect. The complete picture requires figuring out the mechanisms by which microbial molecules interact with the immune system. So, taking an interdisciplinary approach has been very helpful in working through these host/microbe interactions. The unfortunate thing is that it's really slow work and requires microbiology, chemistry, immunology, and cell biology tools. I'm hoping that someday someone will figure out a better way to do it.
Q8
Finally, having mentored over 100 trainees, what advice would you give to young scientists entering the fields of microbiome and immunology, especially regarding interdisciplinary collaboration?
You shouldn't be inhibited by dogma if your data tell you something different. I use PSA as the example. It was quite a bit of work to prove to colleagues that polysaccharides can activate T cells. What we did with PSA is that we've defined what it is about
the structure of the molecule that allows it to activate T cells, and why most polysaccharides don't. I heard many times in the late 90s and early 2000s that PSA must be contaminated with peptides or proteins because it's activating T cells, because the dogma taught that polysaccharides don't activate T cells. So, my main advice would be to not be constrained and to keep an open mind. You have to believe in your data if you are sure it is accurate.
Another thing that I try and teach my students and postdocs by example, is that I've never considered what I do as ‘work’. I go to my lab every day because what I do there is have fun solving challenging questions. I have a good time, and it's always been that way. It's something I look forward to. I get energised from our data and thinking about new experiments. So, one, enjoy your work, and do something that's exciting, and two, don't be constrained by dogma in the field.
Finally, in my experience, interdisciplinary collaboration works best when each side offers unique expertise in solving a problem.
Meghan Azad Professor of Pediatrics and Child Health, University of Manitoba, Canada
One of our major discoveries was how breastfeeding was crucial to microbiome development
What led you to specialise in the field of infant nutrition and the microbiome?
I had done my PhD in cancer cell biology and was looking for a change. My original motivation to pursue a career in science was to influence human health, and I realised through my PhD work that very basic science, while important, was quite far removed from clinical impact. I audited a course in epidemiology and realised this was more aligned with my goals, so I looked for a postdoc position in this area and was fortunate to connect with Anita Kozkyrskyj, an epidemiologist who had just received a grant to study the microbiome in babies from the CHILD Cohort Study. One of our major discoveries was how breastfeeding was crucial to microbiome development. So, when I started my own lab, I decided to dive deeper into this topic and find out how breast milk shapes microbiomes. The CHILD study had collected breast milk, so I had a fantastic opportunity to expand my postdoc research and explore this question.
Q2
You are Director of the THRiVE Discovery Lab, which studies how early life nutrition shapes the infant microbiome and child health, and lead the CHILD Cohort Study, which follows 3,500 children from mid-pregnancy into adolescence. Can you tell us more about these two initiatives? What have your key discoveries been to date?
CHILD is a birth cohort study that started around 2010 and
is following 3,500 pregnant women from four centres across Canada. It was established to investigate the recent epidemic of childhood allergies and asthma and understand how genes and the environment (very broadly speaking) influence the development of these conditions. It’s a massive initiative led by a fantastic multidisciplinary team of researchers and staff across the country. As a postdoc, I was lucky to join the CHILD team to help with some of the first microbiome analyses in this cohort, and when I started my own lab a few years later, I took the lead on breast milk research within CHILD. It has been a wonderful place to ‘grow up’ as a scientist! There have been tons of discoveries from CHILD, including many related to the microbiome and breastfeeding and how they relate to various child health outcomes, ranging from allergies and asthma to obesity and behaviour. The CHILD babies are now entering their teenage years and we are still following them!
THRiVE is my research program, focused on understanding how early nutrition shapes the microbiome and lifelong health. Building on my research with the CHILD study, we now have additional projects focused on breast milk and child development in other global contexts, donor human milk for premature infants, colostrum (the very first milk produced, which has unique immunological properties), and education about breastfeeding for professional and public audiences. Our team is very multidisciplinary, with members specialising not only in nutrition and microbiology, but also neonatology,
anthropology, midwifery, data science, knowledge translation, and more.
Our key discoveries range from epidemiologic evidence on breastfeeding and chronic disease prevention to mechanistic research on breast milk compounds and the infant microbiome. Our clinical research shows that breastfeeding shapes the early immune system, and is linked to lower rates of childhood asthma and obesity.
Our clinical research shows that breastfeeding shapes the early immune system
We dive deeper into these questions than many other studies, paying attention to nuances in the duration, exclusivity, and method of feeding. We’ve shown that pumped milk, while still beneficial, is not equivalent to feeding at the breast, and we are doing more research to understand why. Our breast milk research shows that milk composition is highly variable and personalised, with some components (like omega-3 fats) affected by maternal diet, others
(like prebiotic sugars) controlled by genetics, and still others (like microbes) more influenced by the physical environment. We have linked specific milk components to infant gut and nasal microbiome development and shown that these relationships are important for supporting respiratory health and preventing asthma.
Q3
Focusing on your work with the International Milk Composition (IMiC) Consortium, what insights have emerged about the variability in human milk composition and its impact on the microbiome?
IMiC is still underway, but our results so far demonstrate enormous variability in milk composition among women, across time from early to later lactation, and between settings (Canada, Pakistan, Tanzania, and Burkina Faso). Some milk components (like microbes) are highly variable, while others (like macronutrients) are less variable. Some are affected by maternal diet while others are not. Some compounds are yet-to-be identified; we have analysed over 50,000 metabolites and many of these chemical structures are still unknown. Our initial focus is to understand how milk composition relates to infant growth, but we are excited to explore relationships with infant
microbiomes as a next step. The four studies participating in IMiC all have microbiome data, so this is a feasible and exciting prospect!
Q4 How can donor human milk be optimised to better support preterm neonates’ microbiome development and overall health?
Donor human milk is very important for preterm neonates and their fragile microbiomes. Unfortunately, some bioactive compounds in human milk are lost or diminished through the pasteurisation process. Researchers are working on improving this process to maintain safety and preserve as much bioactivity as possible. It might also be possible to ‘match’ milk donors to infant recipients in ways that optimise their microbiomes.
Q5 What role does the microbiome play in resilience against chronic conditions, such as asthma, allergies, and diabetes? How could clinical interventions be designed to harness this potential?
Microbes help train the immune system during early life. When this process is disrupted, conditions like allergies and asthma can result. By understanding how microbes support immune development during the critical
first months of life, we can develop microbiome-targeted preventive approaches to avoid these conditions. For example, we are finding that some microbes are missing from babies who go on to develop asthma later in childhood. Some of these particular microbes are found in breast milk and/or rely on breast milk sugars to survive, so it might be possible to design asthma prevention strategies that replenish these microbes through probiotic supplements given during breastfeeding, or alongside breast milk compounds.
Q6
What are the most significant challenges in translating microbiome research into public health policies or clinical guidelines for early nutrition?
Microbiomes are dynamic (always changing) and personalised (unique to each individual). We are still learning what a ‘healthy’ microbiome actually is, and how this might differ among individuals of different ages, geographies and circumstances.
We are still learning what a ‘healthy’ microbiome actually is, and how this might differ among individuals of different ages, geographies, and circumstances
The ideal microbiome is probably different for a baby or child in Canada versus Tanzania or Pakistan. So, it’s a challenge to identify the specific microbes or microbial products necessary for optimal health, and to translate this knowledge into effective
products. However, is abundantly clear that human milk, which is also dynamic and personalised, supports human microbiome development in early life, so we should do everything possible to support breastfeeding through public policies and clinical guidelines. For scenarios where exclusive breastfeeding isn’t possible, we can draw inspiration from breast milk and its impact on the microbiome to inform alternative feeding strategies.
Q7
Looking ahead, what do you envision as the next major breakthrough in infant microbiome research, and what role do you see your work playing in that evolution?
Our new research shows that timing is critical in early microbiome development: it is not only about having the ‘right microbes’ (or microbial functions), they need to arrive in the right sequence, at the right time. This is true not only in the gut, but also in the nose, and likely other body sites, too. Breastmilk is key to this progression because it gradually changes over time and guides this delicate maturation of the microbiome. I think the next major breakthroughs in infant microbiome research will centre on understanding this progression at a deeper resolution, enabling us to pinpoint specific milk components and microbes that temporally orchestrate microbiome development during infancy. Understanding these processes will facilitate the development of microbiome-targeted ‘tools’ for optimising health during infancy and across the lifespan.
The 32nd United European Gastroenterology Week, held from 12th–15th October 2024 in Vienna, Austria, brought together over 11,500 participants from more than 115 countries to discuss groundbreaking developments in the diagnosis and treatment of digestive system diseases. This year’s interdisciplinary sessions focused on innovative, non-invasive techniques for managing gastrointestinal conditions, alongside cutting-edge translational and basic research.
INTRODUCTION
The gut microbiome plays a critical role in maintaining health and mediating disease, as discussed by three leading researchers during a session entitled ‘Future Microbiome Therapeutics’ on microbiotabased therapies. Harry Sokol, Saint Antoine Hospital, Paris, France, explored bacterial consortia as a targeted approach to managing inflammatory bowel disease (IBD); while Rafael Valdes, University of Navarre, Pamplona, Spain, presented innovative bacteriophage therapies aimed at pathogenic gut bacteria. To conclude the presentations, Benjamin H. Mullish, Imperial College London, UK, shed light on the importance of gut microbial metabolites and their therapeutic potential.
BACTERIAL CONSORTIA: A TARGETED APPROACH
In his presentation, ‘Bacterial Consortia’, Sokol outlined innovative strategies for harnessing the gut microbiota to treat diseases such as IBD. He emphasised that the gut microbiota is a crucial player in the pathogenesis of IBD, making it a compelling therapeutic target. While traditional approaches like faecal microbiota transplantation (FMT) have demonstrated utility in acute conditions, their limitations
have prompted researchers to develop more refined methods, including bacterial consortia.
Moving Beyond Faecal Microbiota Transplantation
Although FMT has proven effective in treating recurrent Clostridioides difficile (C. diff) infections, Sokol noted several challenges to its broader application, particularly in chronic diseases like IBD. A significant issue is the lack of standardisation as up to 50% of sequences identified in human stool cannot be mapped, and the functions of many identified genes remain unknown.1 Additionally, a large proportion of gut metabolites, critical players in microbiome–host interactions, remain unidentified. This complexity, coupled with the variability in FMT efficacy, emphasises the need for well-defined, scalable microbiome-based therapies.
The Concept of Bacterial Consortia
Bacterial consortia represent a promising alternative to FMT as these carefully designed groups of bacteria are selected for their complementary functions and synergistic interactions. By assembling bacteria that collaborate metabolically or immunologically, consortia can achieve specific therapeutic effects. Sokol
The ultimate goal is to develop a "super consortium" that recapitulates the functions of a healthy microbiota
of sequences identified in human stool cannot be mapped
highlighted several examples, including a pioneering study that identified 17 bacterial strains capable of inducing regulatory T cells in mice, which are crucial for reducing inflammation in colitis models.2 This research laid the foundation for clinical developments, including an ongoing Phase II trial that is testing a bacterial consortium for mild-to-moderate IBD.3
Another promising application of bacterial consortia is decolonising harmful bacteria, such as antibiotic-resistant Enterobacteriaceae, from the gut. Recent research demonstrated that an 18-strain consortium could successfully eliminate Klebsiella pneumoniae from the gut microbiota of mice by outcompeting it for resources, such as gluconate.4 These findings suggest that bacterial consortia could provide an ecological solution to combat multidrug-resistant organisms, a pressing public health concern.
Defining the Next Generation of Microbiome-Based Therapies
Beyond small consortia, researchers are exploring the potential of highly defined, large-scale consortia that mimic the complexity of a healthy gut microbiota. In a groundbreaking study, scientists identified 119 bacterial strains that collectively represented a functional human microbiome.5 When introduced into germfree mice, this artificial microbiome restored key immune, metabolic, and bile acid functions, demonstrating its potential to replicate the benefits of a natural gut microbiota.5
At the other end of the spectrum, singlestrain approaches also hold promise. For instance, Faecalibacterium prausnitzii, a bacterium known for its anti-inflammatory properties, is reduced in patients with IBD, and early-stage clinical trials have shown that administering this strain is safe and could lead to new treatments targeting specific microbial deficits.
A pioneering study identified 17 bacterial strains capable of inducing regulatory T cells in mice
Challenges and Future Directions
While bacterial consortia offer exciting possibilities, Sokol described several hurdles that must be overcome, emphasising that ensuring safety is paramount, particularly in chronic disease settings as researchers must design bacterial formulations to avoid unintended interactions or persistence in unintended environments. Although regulatory frameworks for live biotherapeutics are evolving, they remain complex and challenging for developers. Additionally, understanding the nuances of microbiome–host interactions is crucial for optimising the efficacy of these therapies.
Looking ahead, Sokol describes how the ultimate goal is to develop a “super consortium” that recapitulates the functions of a healthy microbiota and can be tailored to treat diverse conditions, including IBD, cancer, and liver diseases.
BACTERIOPHAGES IN INFLAMMATORY BOWEL DISEASE
In his talk titled ‘Bacteriophages’, Valdes detailed an innovative approach to treating IBD by targeting specific pathogenic bacteria with bacteriophage therapy. He outlined the challenges of identifying shared IBD-associated microbiome signatures and the limitations of current treatments, which primarily target downstream adaptive immune responses rather than addressing the underlying bacterial contributors to the disease.
The Role of Klebsiella pneumoniae in Inflammatory Bowel Disease Pathogenesis
Using data from four diverse IBD cohorts totalling 537 samples, Valdes and his team
identified a bacterial signature associated with Crohn’s disease and ulcerative colitis.6 The study’s findings observed Klebsiella pneumoniae (KP2) as a key pathobiont enriched during IBD flare-ups and linked to a distinct profile of antibiotic resistance genes.
Germ-free mouse experiments were used to test the causal relationship between KP2 strains and IBD, showing that colonisation with KP2 strains resulted in reduced IL-10 production and elevated interferon-γ levels in splenic cells. These results confirmed the pro-inflammatory potential of these bacteria, thus providing a strong rationale for targeting KP2 strains as a therapeutic strategy.
Bacteriophages: Advancing to Human Trials
Valdes turned to bacteriophages as a precision tool to target IBD-associated KP2 strains. Bacteriophages have two unique advantages, their high host specificity, and their safety, as they do not infect human cells. However, they also present challenges, such as the potential for bacterial resistance and their interaction with the immune system, which can trigger strong immune responses.
To address these challenges, Valdes developed an iterative approach to isolate and combine bacteriophages that target KP2 strains while maintaining efficacy. Testing 18 different phage combinations, each containing threeto-five bacteriophages, demonstrated varying levels of efficiency. In preclinical mouse models, phage therapy successfully reduced intestinal inflammation by suppressing KP2 strains, as evidenced by improved histopathological scores and decreased pro-inflammatory cytokines.6
GUT MICROBIAL METABOLITES
In his talk, ‘Microbial Metabolites in Engineered Probiotics’, Mullish explored the profound significance of gut microbial metabolites and their therapeutic applications. Mullish’s own research has
provided valuable insights into the role of microbial metabolites in combating infections like C. diff and multidrug-resistant organisms (MDRO). In a 2023 study, mice exposed to antibiotics or stool from healthy donors exhibited increased nutrient levels and decreased metabolites.7 This reduction in metabolites following antibiotic treatment contributed to the rise of MDROs; however, supplementing mice with a mixture of metabolites led to a significant reduction in the colonisation of carbapenem-resistant E. coli, a key MDRO.
Similarly, FMT has shown promise in restoring metabolite balance as, after FMT, valerate levels were restored to those of healthy donors. Valerate was shown to inhibit the growth of C. diff in vitro in a dose-dependent manner.8
Bile Acids: A Double-Edged Sword
Mullish also discussed the impact of bile acids on gut health. In antibiotic-treated guts, there is a marked drop in bile salt hydrolase activity and an increase in stool taurocholic acid, a primary conjugated bile acid that promotes C. diff infection.9 Whilst secondary bile acids like deoxycholic acid could potentially reduce C. diff colonisation, they pose risks such as increased colonic cancer rates, highlighting the complexity of using bile-metabolising enzymes or small molecules therapeutically.
Challenges in Therapeutic Applications
While the therapeutic potential of microbial metabolites is immense, Mullish outlined several challenges. One significant issue is the palatability of these compounds, as many metabolites, particularly shortchain fatty acids, have unpleasant smells and tastes that make them difficult to administer. Stability and volatility further complicate their use, as these compounds are often unstable and degrade rapidly. Another major obstacle lies in targeting and absorption; it is challenging to deliver metabolites precisely to their intended sites of action within the body. Ensuring proper dosing is equally problematic, as incorrect dosing can lead to toxicity or adverse effects. Responses to metabolites can also vary widely between individuals, adding another layer of complexity and, finally, the regulatory and intellectual property landscapes pose additional hurdles.
Engineered Bacteria: A Promising Solution
To overcome these obstacles, engineered bacteria offer a promising approach to restoring gut microbial metabolites. Probiotic strains such as E. coli Nissle and various Bacteroides species, which have established safety profiles, are particularly attractive for this purpose. These bacteria can be engineered using a range of techniques, from classical plasmid
One significant issue is the palatability of these compounds, as many metabolites have unpleasant smells and tastes
transfection to advanced methods like CRISPR-Cas systems, allowing precise modifications to the bacterial chromosome.
Despite promising results in animal studies, translating these findings to human practice is fraught with challenges. For example, SYNB1020, an engineered E. coli Nissle 1917 designed to convert ammonia into L-arginine, showed success in reducing systemic hyperammonaemia and improving survival in mouse models of urea cycle disorder and liver injury.10 Phase I human trials indicated the treatment was welltolerated and quickly reached a steady state; however, a subsequent Phase IB/II study was halted due to a lack of efficacy.
Future Perspectives
Mullish discussed potential future directions for engineered probiotics. One approach involves using native gut bacteria instead of traditional probiotic strains, as the former may colonise more effectively. Another notable development involves engineering
References
1. Pasolli E et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176(3):649-62.e20.
2. Atarashi K et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500(7461):232-6.
3. Vedanta Biosciences. VE202. Available at: https://www.vedantabio.com/ pipeline-programs/ve202/. Last accessed: 21 January 2025.
4. Furuichi M et al. Commensal consortia decolonize Enterobacteriaceae via ecological control. Nature. 2024;633(8031):878-86.
bacteria not only to produce metabolites but also to sense them. In one study, E. coli was designed to detect gut sulphates, a marker of inflammation. Upon sensing these sulphates, the bacteria activated a base-editing system that produced a colourimetric signal, recorded the inflammatory episode in DNA, and released an immunomodulatory molecule to reduce inflammation.11 This multifunctional system demonstrated the potential for bacteria to serve as both sensors and therapeutics.
CONCLUSION
The session provided a compelling overview of the cutting-edge developments in microbiota-based therapies, showcasing the convergence of scientific innovation and clinical application. From refining bacterial consortia for chronic disease to targeting specific pathogens with phages and leveraging engineered probiotics, these approaches offer promising avenues to restore gut health and combat disease.
5. Cheng S et al. The intrinsic and extrinsic effects of TET proteins during gastrulation. Cell. 2022;185(17):316985.e20.
6. Federici S et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell. 2022;185(16):287998.e24.
7. Yip AYG et al. Antibiotics promote intestinal growth of carbapenemresistant Enterobacteriaceae by enriching nutrients and depleting microbial metabolites. Nat Commun. 2023;14(1):5094.
8. McDonald JAK et al. Inhibiting growth of Clostridioides difficile by restoring valerate, produced by the intestinal
9. Mullish BH et al. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut. 2019;68(10):1791-800.
10. Kurtz CB et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci Transl Med. 2019;11(475):eaau7975.
11. Zou ZP et al. Biomarker-responsive engineered probiotic diagnoses, records, and ameliorates inflammatory bowel disease in mice. Cell Host Microbe. 2023;31(2):199-212.e5.
Does Gut Microbiome Composition Influence the Efficacy of Psychiatric Drugs?
Authors: *Nadia Suryawinata,1 Sarkis K. Mazmanian1
1. California Institute of Technology, Pasadena, California, USA
*Correspondence to nsuryawi@caltech.edu
Disclosure: Mazmanian has received consulting fees from Axial Therapeutics and Nuanced Health for work unrelated to the current manuscript. The authors have declared no conflicts of interest.
Received: 03.02.25
Accepted: 24.02.25
Keywords: Anxiety, biomarkers, depression, gut microbiome, microbiome composition.
Altered gut microbiome profiles correlate with anxiety and depression in humans, and work in animal models has identified specific bacterial taxa and/or microbiome-derived metabolites that influence complex emotional behaviours. Intriguingly, many pharmaceuticals, including widely used oral treatments for anxiety and depression, can be chemically modified by microbes in the gastrointestinal tract, which may lead to drug inactivation. The authors highlight the importance of integrating research across microbial culture systems, animal models, and multi-omics analyses of clinical cohorts to gain mechanistic insights into whether microbiome composition determines efficacy, bioavailability, and tolerability of neuropsychiatric medications. This hypothesis, if validated, may have profound implications for personalised drug treatment plans and microbiomebased biomarker development.
THE RECIPROCAL RELATIONSHIP BETWEEN THE GUT MICROBIOME AND MEDICATIONS
The gut microbiome, comprising a staggering 3.8x1013 bacteria along with
microscopic fungi, archaea, and viruses in humans,1 plays crucial roles in shaping and maintaining host health. Gut microbes support a wide range of physiological functions including digestion, immune modulation, metabolism, and neuronal signaling. Disruptions in host-microbe interactions are associated with a range of human diseases, such as inflammatory bowel disease (IBD),2 cancer,3 Type 2 diabetes,4 and neurological disorders.5
The gut microbiome is highly dynamic, with community composition influenced by intrinsic factors such as host genetics,6 but also strongly determined by extrinsic/ environmental contributors,7 including diet and medication.8 Because diet and drugs are modifiable, understanding the interactions between environmental factors and the gut microbiome offers an exciting and tractable opportunity for development of personalised medicines.
Most pharmaceuticals are administered orally. These substances are either absorbed in the small intestine, where the microbiome is sparse, or pass to the colon, where the densest and most diverse microbial communities reside. Additionally, drugs absorbed in the small intestine may be modified (or not) and secreted back into
the intestine, creating new opportunities for exposure to the gut microbiome.9 Consumption of antibiotics, unsurprisingly, has profound effects on the gut microbiome. Acute exposure to a single course of antibiotics can result in the transient reduction or loss of microbial taxa that are important for basic metabolic functions such as carbohydrate fermentation,10 energy production, bile acid transformation,11 and lipid absorption. While most individuals treated with antibiotics experience a rapid recovery of microbiome composition, for some it may take up to 6 months to fully recover their original (pre-drug) microbiome.12 Loss of community stability and, consequently, compromise of normal metabolic functions of the microbiome may lead to opportunistic infections,12,13 deficits in gut barrier integrity,14 weakening of the immune system,15,16 and other unintended consequences. While antibiotics likely have the most profound impact on microbiome function, emerging evidence suggests that other medications may also compromise the microbiome, albeit to a subtler degree.
The vast majority of pharmaceutical drugs were developed against human targets (e.g., proteins, molecules, metabolic pathways), are diverse in structure, and are often consumed for extended periods of time, making it challenging to predict their direct or indirect effects on the microbiome. However, some drug–microbiome interactions have been uncovered. The common Type 2 diabetes medication metformin alters gut microbiome composition in patients, increasing microbial taxa that promote glucose metabolism and thereby increasing its therapeutic effect.17 Methotrexate, a first-line treatment for rheumatoid arthritis, alters microbiome composition in patients and in human microbiome colonised mice, with transplantation of a drug-modified microbiome into drug-naïve mice being sufficient to reduce immune activation.18 The benzisoxazole ring structure in risperidone, an atypical antipsychotic used for schizophrenia and bipolar disorder, is chemically modified by gut microbes, leading to its rapid excretion and thus potentially reducing efficacy and altering dosing regimens in ways that may vary between patients.19
Informed by these findings, there is growing interest in understanding how the gut microbiome may be influenced by, and may influence the efficacy of, various drug classes. Emerging evidence has identified novel microbial transformations of drugs that may alter the intended outcomes of medications.9,20 Given the emerging and likely intricate relationship between gut bacteria and brain function, drug–microbiome interactions in the context of neuropsychiatric disorders represent a particularly interesting area of study. This perspective will first examine how the gut microbiome influences drug metabolism in vivo, drawing from studies in mice and humans with anxiety and major depressive disorder (MDD). The authors will then review known drug–microbiome interactions, primarily through examples beyond neuropsychiatric medications, as these well-characterised cases provide insights into the methodologies needed for future study of microbial metabolism of psychiatric drugs. Finally, the authors will discuss how integrating these approaches can provide an actionable framework for understanding the role of microbial influences on the efficacy and other features of psychiatric drugs.
THE GUT MICROBIOME AND BRAIN HEALTH: INSIGHTS FROM ANXIETY AND DEPRESSION
Anxiety disorders represent the most common class of neuropsychiatric conditions, and are characterised by a persistent avoidance response even in the absence of imminent danger.21 Often co-occurring with depression,22 which is marked by a prolonged loss of interest in activities, anxiety disorders significantly impact quality of life in up to a quarter of the USA and European populations.23,24
In recent years, studies conducted in both mouse models and human cohorts have described a functional role for the gut microbiome in the development of anxiety and depression (Figure 1A). The gut microbiome is stereotypically altered in individuals with anxiety or MDD,25,26 and is speculated to affect symptoms via altered
neurotransmitter production,27 inflammation/ cytokines,28 the hypothalamic–pituitary–adrenal (HPA) axis,29 vagus nerve,27 and other potential mechanisms. These associations are supported by animal models. Germ-free mice, which are raised without any exposure to microbes, exhibit reduced anxiety-like behaviour, and the reintroduction of a normal microbiome early in life is sufficient to restore anxiety-like traits of standard laboratory mice.30 Transplantation of microbiomes from mice that have experienced chronic stress into naïve recipient mice induces behaviours consistent with depression, and supplementation with Lactobacillus alleviates this effect.31
The gut and brain communicate through various pathways (neuronal, endocrine, immunological) and these interactions involve factors that can be influenced by a diverse array of microbes and their products. For instance, treatment with the bacterium Lactobacillus rhamnosus (JB-1) has been shown to alleviate anxiety and depression-like behaviours in mice.27 This effect occurs through the differential regulation of GABA receptors in the brain and is dependent on the vagus nerve, as vagotomised mice do not exhibit the same behavioural improvements when treated with L. rhamnosus (Figure 1B). Gut bacteria can also produce small molecule
Figure 1: The microbiome and microbially-derived metabolites modulate host nervous system function.
Figure 1A is generated on Biorender.com.
Created on Biorender.com.
metabolites that then travel to the brain and alter cell function: the gut microbial metabolite 4-ethylphenyl sulfate (4-EPS) impairs oligodendrocyte differentiation in mice and increases anxiety-like behaviour.32 In contrast, treating mice with the human commensal Bacteroides fragilis is able to alleviate anxiety-like and autism-associated features (Figure 1C).33
In humans, large cohort studies surveying the microbiomes of depressed patients have revealed stereotypical alterations. Notably, MDD patients often show depletion of genera such as Subdoligranulum and Coprococcus, and an increase in Eggerthella, alongside changes in their metabolomes, particularly increased lipid metabolism.26,34 A recent study integrated microbiome sequencing data from faecal samples of individuals with anxiety and depressive disorders, including those taking medications, to train machine learning algorithms that could successfully predict both the presence of these disorders and medication use based on microbiome profiles alone.35 While effect sizes in human studies remain modest and may necessitate further replication, research to date on the potential pathogenic or protective effects of the gut microbiome in neuropsychiatric conditions represents an exciting frontier of research at the intersection of microbiology, neuroscience, and human health.
Microbiome Modulation of Neuropsychiatric Drugs
Selective serotonin reuptake inhibitors (SSRIs) and selective norepinephrine reuptake inhibitors (SNRIs) are first-line treatments for both anxiety and MDD.36 While these drug classes have been shown to be more effective than placebo for generalised anxiety disorder, their benefits are often accompanied by a therapeutic lag and significant variability in response rates, particularly in terms of long-term acceptability and sustained efficacy.37 Given that SSRIs and SNRIs are orally administered, an important question is whether their interaction with the gut microbiome contributes to the substantial differences in therapeutic acceptability observed across patient populations. Interindividual variations in gut microbiome composition may influence drug metabolism and bioavailability, potentially explaining why some patients respond better to treatment than others.
Recent studies with high-throughput, in vitro culture-based screening systems have revealed extensive drug–microbiome interactions (Figure 2). In one study, researchers exposed 76 individual strains of diverse human gut bacterial taxa to over one hundred commonly prescribed drugs, including medications for anxiety.38
Figure 2: The microbiome modulates drugs, potentially affecting their therapeutic function in the host.
This work found that metabolic reactions were taxon-specific; i.e., Bacteroidetes primarily hydrolysed drugs with ester or amide groups, while most other strains metabolised drugs containing a nitro or azide group. Of note, 10% of the strains chemically transformed anxiolytics, significantly reducing active drug levels in culture, with the SSRI fluoxetine emerging as the most widely metabolised anxiolytic across isolated bacterial strains. Another study incubated different complex communities derived from human faecal samples with drugs used to treat anxiety, again finding that microbiome composition can broadly influence drug metabolism.39 Some communities had the metabolic capacity to degrade specific drugs, while others did not, highlighting interindividual variability in microbiomedriven drug metabolism.
Researchers have also leveraged publicly available repositories to develop models to predict drug–microbiome interactions, such as SIMMER (Similarity algorithms that Identify MicrobioMe Enzymatic Reactions)40 and AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2).41 SIMMER combines metagenomeassembled genomes, protein homology, and enzyme databases to predict bacterial drug metabolism. This tool identified candidate gut bacterial enzymes, primarily carboxypeptidase G2-like enzymes, with sequence similarity to an environmental enzyme known to hydrolyse methotrexate.40 Experimental testing of strains containing these enzymes confirmed methotrexate degradation. AGORA2 provides a resource for reconstructing metabolic pathways from metagenomic datasets, and incorporates clinical parameters such as BMI and age to facilitate rapid prediction of drug metabolism in epidemiological cohorts.41 Both SIMMER and AGORA2 provide interactive frameworks, allowing researchers to prioritise microbial species, gene products, and pathways of particular relevance for a given disorder and drug class.
While high-throughput screening and large-scale dataset analyses have provided valuable insights, efforts to fully characterise drug–microbiome interactions remain ongoing, with only a few examples
to date that have identified products of drug metabolism and even fewer cases tested functionally. For instance, Levodopa (L-DOPA), the first-line treatment for Parkinson’s disease, is degraded by Eggerthella lenta and Enterococcus faecalis 42 These bacterial species were shown to contain enzymes for conversion of L-DOPA into m-tyramine through decarboxylation and dihydroxylation, which may reduce L-DOPA bioavailability and impact treatment efficacy. Another well-known example of a drug–microbiome interaction is 5-aminosalicylic acid (5ASA), used to treat IBD, whose efficacy is reduced by microbial metabolism.43 By longitudinally monitoring IBD patients on 5-ASA treatment using metagenomics, metatranscriptomics, and metabolomics, researchers identified twelve previously uncharacterised microbial acetyltransferases that were upregulated in non-responders. In vitro assays confirmed that these enzymes acetylate 5-ASA into an inactive form, providing a mechanistic link between microbial metabolism and drug response.
In addition to metabolising drugs, some gut bacteria have been shown to actively transport and bioaccumulate drugs in vitro without modifying their chemical structure (Figure 2).44 Duloxetine, an SNRI, bioaccumulates in diverse gut species, including many from the Firmicutes phylum (Streptococcus salivarius, Clostridium bolteae, Clostridium saccharolyticum, Ruminococcus gnavus, Lactobacillus plantarum, and Lacticaseibacillus paracasei), resulting in altered endogenous metabolism and secretion profiles. Duloxetine modulates Caenorhabditis elegans movement in a dose-dependent manner, and colonisation with the Escherichia coli IAI1 strain that is capable of bioaccumulating duloxetine attenuates this behaviour, highlighting that drug–microbiome interactions can impact behavioural outcomes.44
Finally, the gut microbiome can regulate host drug transporters, thus influencing pharmacokinetics. Differences in microbiome composition, such as between conventionally-raised and germ-free animals, alter the expression of the efflux
transporter P-glycoprotein (P-gp/ABCB1),45 which may contribute to pharmacokinetic variability for P-gp substrate drugs, including the SSRI sertraline and the antipsychotic risperidone. However, whether degradation, modification, bioaccumulation and/or altered transport of SSRIs or SNRIs impact anxiety or depression-like behaviours in mammalian model systems remains unexplored to date, defining a frontier of future research.
TOWARDS A HOLISTIC UNDERSTANDING OF DRUG–MICROBIOME INTERACTIONS
While microbial cell culture-based experiments offer rigorous insights into drug–microbiome interactions, these systems are unable to capture the physiology of an organism and its associated microbiome, with studies in freely behaving animals required to advance this research toward understanding effects on emotional behaviours. Recent in vitro findings have also revealed that reductions in drug levels do not necessarily indicate microbial metabolism.20 Abiotic factors, including spontaneous degradation, ion suppression, surface adsorption, and bioaccumulation, can have strong effects on drug activity.
To ensure reproducible and clinically relevant results, it is important to test drug–microbiome interactions within their native host context, minimising artefacts introduced by culture conditions. Moreover, it is possible that long-term medication use can reshape the gut environment and microbiome composition, which then secondarily influences symptoms or treatment outcomes, though this concept remains hypothetical in the absence of empiric evidence.
References
1. Sender R et al. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533.
2. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427-34.
Disentangling these factors requires an integrated approach, combining multiomics analyses of diverse human cohorts with rodent models or non-human primate models that are amenable to experimental approaches to define functional outcomes. Given that microbial bio-transformations largely fall within a defined set of reaction types, such as reduction, hydrolysis, decarboxylation, and dealkylation, identifying overarching principles governing these transformations may be feasible. Leveraging large-scale machine learning models trained on high-resolution microbiome and metabolomics datasets could offer a powerful strategy to predict drug modifications and their downstream effects, ultimately guiding the design of more precise and effective therapeutic interventions.
As our understanding of drug–microbiome interactions becomes more refined, the development of predictive frameworks for drug efficacy and tolerability based on an individual’s symptoms, lifestyle, medication history, and microbiome status will be increasingly feasible. Such tools could one day help tailor pharmacological treatments to maximise therapeutic benefit, ultimately advancing precision medicine. It is conceivable that gut microbiome variations explain inter-individual responses to numerous classes of oral drugs, beyond those for neuropsychiatric conditions, and potentially even injectables via microbiome modulation of immune profiles (e.g., immune checkpoint inhibitors)46-48 and metabolic states (e.g., weight loss drugs).49,50 Identifying microbiome-based markers that quantitatively predict variance in drug response in defined patient populations may streamline drug discovery and development, improve efficacy rates and response times, and reduce side effects.
3. Louis P et al. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12(10):661-72.
4. Qin J et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55-60.
5. Morais LH et al. The gut microbiotabrain axis in behaviour and brain
disorders. Nat Rev Microbiol. 2021;19(4):241-55.
6. Vujkovic-Cvijin I et al. Host variables confound gut microbiota studies of human disease. Nature. 2020;587(7834):448-54.
7. Rothschild, D et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555(7695):210-5.
8. Culp EJ et al. Microbial transformation of dietary xenobiotics shapes gut microbiome composition. Cell. 2024;187(22):6327-45.e20.
9. Zimmermann M et al. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science. 2019;363(6427):eaat9931.
10. Flint HJ et al. The role of the gut microbiota in nutrition and health. Nat. Rev. Nat Rev Gastroenterol Hepatol. 2012;9(10):577-89.
11. Funabashi M et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature. 2020;582(7813):566-70.
12. Palleja A et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol. 2018;3(11):1255-65.
13. Dethlefsen L et al. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280.
14. Sawaed J et al. Antibiotics damage the colonic mucus barrier in a microbiotaindependent manner. Sci Adv. 2024;10(37):eadp4119.
15. Yang JH et al. Antibiotic-induced changes to the host metabolic environment inhibit drug efficacy and alter immune function. Cell Host Microbe. 2017;22(6):757-65.e3.
16. Watanabe K et al. Microbiomemediated neutrophil recruitment via CXCR2 and protection from amebic colitis. PLoS Pathog. 2017;13(8):e1006513.
17. Wu H et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23(7):850-8.
18. Nayak RR et al. Methotrexate impacts conserved pathways in diverse human gut bacteria leading to decreased host immune activation. Cell Host Microbe. 2021;29(3):362-77.e11.
19. Meuldermans W et al. The metabolism and excretion of risperidone after oral administration in rats and dogs. Drug Metab Dispos. 1994;22(1):129-38.
20. Garcia-Santamarina S et al. Emergence of community behaviors in the gut microbiota upon drug treatment. Cell. 2024;187(22):634657.e20.
21. Bandelow B, Michaelis S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin Neurosci. 2015;17(3):327-35.
22. Kessler RC et al. Prevalence, severity, and comorbidity of 12-Month DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62(6):617-27.
Corrected and republished from: Arch Gen Psychiatry. 2005 Jul;62(7):709.
23. Terlizzi E et al. Symptoms of anxiety and depression among adults: United States, 2019 and 2022. Natl Health Stat Report. 2024;(213):CS353885.
24. Hajek A et al. Depression and anxiety in later COVID-19 waves across Europe: new evidence from the European COvid Survey (ECOS). Psychiatry Res. 2022;317:114902.
25. Valles-Colomer M et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat Microbiol. 2019;4(4):623-32.
26. Radjabzadeh D et al. Gut microbiomewide association study of depressive symptoms. Nat Commun. 2022;13(1):7128.
27. Bravo JA et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A. 2011;108(38):16050-5.
28. Berk, M. et al. So depression is an inflammatory disease, but where does the inflammation come from?. BMC Med. 2013;11:200.
29. Wu WL et al. Microbiota regulate social behaviour via stress response neurons in the brain. Nature. 2021;595(7867):409-14.
30. Sudo N et al. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J Physiol. 2004;558(Pt 1):263-75.
31. Chevalier G et al. Effect of gut microbiota on depressive-like behaviors in mice is mediated by the endocannabinoid system. Nat Commun. 2020;11(1):6363.
32. Needham BD et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature. 2022;602(7898):647-653.
33. Hsiao EY et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451-63.
34. Amin N et al. Interplay of metabolome and gut microbiome in individuals with major depressive disorder vs control individuals. JAMA Psychiatry. 2023;80(6):597-609.
35. Dilmore AH et al. Medication use is associated with distinct microbial features in anxiety and depression. Mol Psychiatry. 2025;DOI:10.1038/ s41380-024-02857-2.
36. Cassano GB et al. Psychopharmacology of anxiety disorders. Dialogues Clin Neurosci. 2002;4(3):271-85.
37. Cipriani A et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357-66.
38. Zimmermann M et al. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462-7.
39. Javdan B et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell. 2020;181(7):1661-79.e22.
40. Bustion AE et al. SIMMER employs similarity algorithms to accurately identify human gut microbiome species and enzymes capable of known chemical transformations. eLife. 2023;12:e82401.
41. Heinken A et al. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat Biotechnol. 2023;41(11):1320-31.
42. Maini Rekdal V et al. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science. 2019;364(6438):eaau6323.
43. Mehta RS et al. Gut microbial metabolism of 5-ASA diminishes its clinical efficacy in inflammatory bowel disease. Nat Med. 2023;29(6):700-9.
44. Klünemann M et al. Bioaccumulation of therapeutic drugs by human gut bacteria. Nature. 2021;597(7875):533-8.
45. Degraeve AL et al. Gut microbiome modulates tacrolimus pharmacokinetics through the transcriptional regulation of ABCB1. Microbiome. 2023;11(1):138.
46. Routy B et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91-7.
47. Gopalakrishnan V et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97-103.
48. Matson V et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104-8.
49. Forslund K et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262-6.
50. Wang L et al. Structural modulation of the gut microbiota and the relationship with body weight: compared evaluation of liraglutide and saxagliptin treatment. Sci Rep. 2016;6:33251.
Systemic Health Implications of the Leaky Barriers within the Oral–Gut–Brain Axis and its Pathways of Communication
Authors: Rachel Y. Kim,1 Ryutaro Kuraji,2 *Yvonne L. Kapila1
1. Sections of Biosystems and Function and Periodontics, School of Dentistry, University of California, Los Angeles, USA
2. Department of Periodontology, The Nippon Dental University School of Life Dentistry at Tokyo, Japan
*Correspondence to ykapila@dentistry.ucla.edu
Disclosure: The authors have declared no conflicts of interest.
The human body is a holistic system, interconnected by the circulatory, nervous, lymphatic, endocrine, and immune systems that facilitate communication throughout the body. While these physiological highways allow distant organs to work together to perform biological functions, such as through the gut–brain and the hypothalamic-pituitary-adrenal (HPA) axes, our intricately connected network is inherently vulnerable to obstructions or perturbations, and pathogens can exploit it as a transportation system to invade or impact different parts of the body.1 Thus, maintaining these pathways free from obstructions and ensuring proper insulation of communication channels by physical and chemical barriers is essential for preserving health and preventing pathogens and their components from travelling alongside the body’s normal signals.1,2 In conjunction with these built-in crosstalks, increasing evidence suggests that the human microbiota plays an integral role in promoting human health by actively interacting with the host immune system and contributing to the development and
regulation of physiological communication pathways.3,4 In this brief review, the authors discuss the interplay between the two largest human microbiome reservoirs, the oral cavity and gut, within the context of the oral–gut–brain axis and the physiological pathways these microbes could utilise to induce neuroinflammation, infiltrate the brain, and potentially contribute to the onset of Alzheimer’s disease (AD).
DISCUSSION
Living in an unsterile world, we constantly interact with bacteria, fungi, archaea, protozoa, and viruses that populate our body surfaces, these are collectively termed the microbiota.1,4,5 While the previously estimated 10-to-1 ratio of bacteria to human cells is outdated, the human body is estimated to host about one bacterium for every human cell.6 Not only are these human–microbe interactions unavoidable, they are essential, as our body actively depends on their contributions during development and throughout life. For instance, the complete absence of microbiota during a critical developmental window irreversibly impaired the HPA axis
function in postnatal mice and decreased neurogenesis in adult mice.3,4 At the same time, microbial dysbiosis (i.e., alterations in microbial abundance and diversity) leads to systemic inflammation and increases the risks of diseases such as diabetes, metabolic diseases, cardiovascular diseases, colorectal cancer, and AD.1,4,5,7,8 Interestingly, a few studies reveal that taking antibiotics can reduce certain pathologies, such as amyloid-beta plaque deposition and microglial activation in AD mouse models, suggesting that transient suppression of dysbiosis or overall microbially induced neuroinflammation can improve symptoms in the brain.3-5 However, the impact of antibiotics against dysbiosis remains speculative as others have found adverse effects, such as reduced overall cognitive abilities.3-5
In line with the microbiota’s direct health impacts, faecal transplantation from healthy to diseased individuals improved cognitive health in clinical and in vivo animal studies, while the opposite outcome occurred when diseased faecal matter was transplanted into healthy mice.3,4 Thus, these studies illustrate that not all microorganisms are equal and, instead, act as a double-edged sword. Different microorganisms participate in distinct functions and produce specific molecules that lead to unique responses. Therefore, the composition and delicate balance of bacteria and other members of the microbiota (e.g., viruses and fungi) hold immense power to either induce or reverse pathology, making the study of microbiota highly complex. For this article, the authors have focused on the bacterial influence, where most of these essential players reside in the gut, which receives a continuous influx of bacteria from the oral cavity, the anatomical starting point of the gastrointestinal (GI) tract.1,9
Because microbiota are not an intrinsic part of our body but constituents of the unsterile environment, it is not surprising that the oral cavity and the gut are the two most densely populated areas with microbes, given their direct exposure to the external environment. At the intersection of these constant microbial–host interactions is the mucosal immune system, which lines
the oral cavity and GI tract, providing both physical and molecular protection against pathogenic bacteria while tolerating the commensal microbes. Under conditions of health, commensal bacteria keep the pathogenic bacterial load in check and secrete metabolites such as short-chain fatty acids (SCFA) that aid the mucosal barrier integrity.1
Moreover, IgA neutralises foreign antigens and tolerates bacterial colonisation of the mucosa while providing non-inflammatory or non-destructive protection against pathogenic invasion.10 However, microbial dysbiosis overwhelms the local immune system of the oral cavity or gut, impairing mucosal barrier integrity by causing inflammation and downregulating junctional proteins.1,9,10 Consequently, a leaky barrier develops that enables microbes to leave their residence and access the body’s communication pathways, ultimately migrating to distant organs like the liver, spleen, and brain.1,7,8 Such phenomena have been observed following periodontitis, an infection-driven, bone-resorbing inflammatory disease of the mouth that puts the body under a low-grade inflammatory state and is recognised as a risk factor for AD.1,9 Additionally, periodontal pathogens can directly induce gut dysbiosis, which is also associated with the onset and progression of AD.1,9 Hence, the impact of oral pathogens spans these separate organs, suggesting the existence of the oral–gut–brain communication axis (Figure 1).
Since the largest and most diverse microbiota in the body reside in the gut, the gut microbiome has been extensively studied.2-4,9 Most of the body’s immune cells are localised here, managing hundreds of trillions of microbes, and these microbe–host interactions are monitored by 100 million enteric neurons, with the vagus nerve acting as the main bidirectional communication pathway between the gut and the brain.1,3,4 This intricate communication between the microbiota, immune, and nervous systems has been characterised as the microbiota–gut–brain axis, where these microorganisms are involved in a wide array of physiological functions, including digestion.4 Emerging
studies are recognising that the oral cavity is the gateway to the gut and hosts the second-largest human microbiome, with over a trillion oral bacteria reaching the gut daily via their anatomical connection.1,2,9 A comprehensive investigation of the microbial influence across the entire oral–gut–brain axis found that saliva from periodontal disease patients directly induced gut dysbiosis and impaired the intestinal barrier integrity in an AD mouse model while concurrently leading to neuroinflammation, memory deficits, and amyloid-beta plaque accumulation.9 Another study found similar results where a topically applied polymicrobial periodontal inoculum significantly increased molecular hallmarks of AD (e.g., neuroinflammation, amyloidbeta accumulation, and phosphorylated tau formation) and mice brain microbiome dysbiosis. Moreover, the same infection regimen was found to downregulate tight junctions and cause inflammation in the murine gut epithelium, as well as elevate lipid depositions in the liver.7,8 Lastly, elevated levels of periodontal pathogens have been detected in postmortem brains in patients with AD and in experimental studies, which suggest that oral pathogens
can physically traverse the oral–gut–brain axis to exert inflammatory responses in both the gut and the brain (as well as other organs, such as the liver), contributing to AD pathologies.1,7
Notably, oral pathogens such as Porphyromonas gingivalis (Pg) exist in healthy individuals, albeit at low levels, and often enter the bloodstream after normal activities such as eating and brushing teeth. These pathogens can induce an inflammatory response that can influence brain functions and, in some cases, lead to atheroma formation that can obstruct the circulatory highway.1 Interestingly, the aforementioned study using the polymicrobial infection regimen detected Pg in the brains of two-thirds of the control mice, although at significantly lower DNA copy numbers than the infected group.7 Since Pg is not a natural member of the mouse microbiome, this finding raises the possibility of experimental contamination. Yet, only Pg was found in control brains and not the other three pathogens in the polymicrobial inoculum administered at equal doses. The presence of Pg in the brains of uninfected mice, despite the antimicrobial
Figure 1: Pathways of communication within the oral–gut–brain axis.
wash treatments that inhibited natural oral microbiota, raises the question of whether these pathogens in the oral cavity of healthy individuals could also exist at low levels in healthy brains, or if the presence of these oral pathogens in the brain is part of an impending disease progression.7 More studies are needed to characterise how low levels of these oral pathogens in healthy individuals influence the brain via the oral–brain axis, which is connected through the neurovascular bundles that include the trigeminal nerves, as well as the circulatory and lymphatic systems.
CONCLUSION
Nevertheless, the chronological events of oral or gut microbial dysbiosis, local inflammatory response, and barrier impairment seem to be the common thread that primes the conditions for pathogens to access our interconnected system, infecting distant organs, and leading to pathologies.9 The elderly population is particularly susceptible to this disease progression due to age-associated alteration in their microbial profile, gut-dysbiosis-induced
References
1. Sansores-España LD et al. oral-gutbrain axis in experimental models of periodontitis: associating gut dysbiosis with neurodegenerative diseases. Frontiers in aging. 2021;2781582.
2. Kerstens R et al. balancing the oralgut-brain axis with diet. Nutrients. 2024;16(18):3206.
3. Capocchi JK. Symposium: what does the microbiome tell us about prevention and treatment of AD/ADRD? J Neurosci. 2024;44(41):e1295242024.
4. Luczynski P. Growing up in a bubble: using germ-free animals
barrier dysfunction, which leads to systemic inflammation and infection, and immunosenescence that limits their ability to combat infections.4,5,10 Remarkably, this association between ageing and increased likelihood of infections is not observed in germ-free mice, which, by design, lack the opportunity for infections and tend to live longer.4 Recognising that microbial–host interactions are an inevitable part of normal life and direct determinants of health, reshaping the dysbiotic microbiota by using prebiotics, probiotics, synbiotics, or postbiotics (commensal bacterial metabolites, such as nisin) may hold the key to achieving therapeutic efficacy.3,5,7,8 Interestingly, despite their advanced age, centenarian populations from blue zones carry distinct microbiota compositions across their bodily compartments and have superior oral health conditions compared to their corresponding controls.2 In light of this, another productive strategy may be to keep oral health in check and thereby remove one of the key contributors to gut dysbiosis and systemic inflammation, ultimately protecting both the brain and overall health along the oral–gut–brain axis.1,2,5,9
to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol. 2016;19(8):pyw020.
5. Kesika P et al. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer's disease. Life Sci. 2021;264:118627.
6. Sender R et al. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337-40.
7. Zhao C et al. Nisin a probiotic bacteriocin mitigates brain microbiome dysbiosis and Alzheimer's disease-like neuroinflammation
triggered by periodontal disease. J Neuroinflammation. 2023;20(1):228.
8. Kuraji R et al. Nisin lantibiotic prevents NAFLD liver steatosis and mitochondrial oxidative stress following periodontal disease by abrogating oral, gut and liver dysbiosis. NPJ Biofilms and Microbiomes. 2024;10(1):3.
9. Lu J et al. Periodontitis-related salivary microbiota aggravates Alzheimer's disease via gut-brain axis crosstalk. Gut Microbes. 2022;14(1):2126272.
10. Feller L et al. Oral mucosal immunity. JOOO. 2013;116(5):576-83.
The Intersection of the Upper Gastrointestinal Microbiome and Oesophageal Cancer: A Review of Pathways and Therapeutic Insights
1. Department of Surgery, Henry Ford Health, Detroit, Michigan, USA
2. Department of Surgery, Corewell Health East William Beaumont University Hospital, Royal Oak, Michigan, USA
3. Section of Thoracic Surgery, Department of Surgery, University of Michigan, Ann Arbor, USA
*Correspondence to reddyrm@med.umich.edu
Disclosure:
Rishindra serves as a teaching site consultant for Intuitive Surgical, is on the advisory board and receives grants from Atricure and On Target Laboratories, serves on the advisory board for Medtronic and Genentech, is a speaker for BMS, and works as a consultant for Trinity Health. The other authors have declared no conflict of interest.
The upper gastrointestinal microbiome, a complex ecosystem of microorganisms that have historically been difficult to identify, may play a pivotal role in the development of oesophageal cancer and postoperative outcomes. Dysbiosis, characterised by imbalances in microbial composition, is believed to drive tumorigenesis in various gastrointestinal cancers through mechanisms such as chronic inflammation, immune suppression, and epithelial barrier dysfunction. Additionally, dysbiosis may contribute to postoperative complications, including anastomotic leaks and infections following surgery. Most research to date has focused on colorectal cancer, demonstrating these complex relationships. Pathogenic bacteria exacerbate the dysregulation processes through mechanisms including pro-inflammatory cytokine release, immune evasion, and biofilm formation. Therapeutic strategies targeting microbiome hold promise for restoring microbial balance, reducing systemic inflammation, and improving surgical outcomes. This review synthesises current evidence on the microbiome's role in oesophageal cancer pathogenesis and postoperative outcomes, highlighting opportunities for therapeutic interventions and the potential for integrating microbiome strategies into oesophageal cancer management protocols.
Key Points
1. Dysbiosis promotes chronic inflammation, immune suppression, and epithelial barrier dysfunction, which can lead to tumorigenesis.
2. Microbial imbalances can exacerbate postoperative outcomes like anastomotic leaks and infections, as well as possible resistance to chemo- and radiation therapy.
3. Strategies include prebiotics and probiotics to restore microbial balance, targeted antimicrobial therapy to eliminate specific pathogens, and personalised microbiome-based diagnostics for tailored interventions.
INTRODUCTION
The gastrointestinal microbiome, a complex ecosystem of microorganisms residing in the oral cavity to the lower gut, may play a significant role in maintaining overall health. In recent years, the upper gastrointestinal (UGI) microbiome involvement in the pathogenesis of oesophageal cancer (EC) has garnered considerable attention. Emerging evidence suggests that dysbiosis, or the imbalance of the UGI microbiome, contributes to chronic inflammation, immune dysregulation, and carcinogenic processes, potentially increasing the risk of developing EC.1,2
This review aims to explore the multifaceted role of the UGI microbiome in EC, focusing on its contribution to cancer development and its influence on treatment outcomes. It will examine the mechanisms by which microbial dysbiosis promotes tumorigenesis, including its effects on inflammation, immune modulation, and epithelial barrier function. Additionally, the authors highlight the potential impact of the UGI microbiome on complications and recovery after EC surgery, such as infections, anastomotic healing, and resistance to adjuvant therapy. By synthesising current evidence, the authors aim to identify opportunities for therapeutic interventions, such as microbiome modulation, and to provide insights into the potential for integrating microbiome-targeted strategies into the management of EC.
UPPER GASTROINTESTINAL MICROBIOME IN OESOPHAGEAL CANCER
While the direct role of oesophageal microbiota in the pathogenesis of EC remains incompletely understood, emerging
evidence highlights significant interactions between these microbes and tumour biology. Among these findings, microbial dysbiosis has gained recognition as a critical factor contributing to EC development.3 Recent studies highlight the microbiome’s role in promoting tumorigenesis through mechanisms involving chronic inflammation, immune modulation, and disruption of epithelial barrier function.1,2,4 Pathogenic bacteria such as Porphyromonas gingivalis (P. gingivalis) and Fusobacterium nucleatum (F. nucleatum), commonly associated with periodontal disease or gingivitis, are found in higher abundance in the oesophageal tissues of patients with EC.5,6 These bacteria can induce persistent inflammation by activating toll-like receptors and the NF-κB signalling pathway, triggering the production of proinflammatory cytokines.5,6 This inflammatory cascade fosters a microenvironment that supports cellular proliferation, angiogenesis, and the survival of transformed cells.7 Furthermore, chronic inflammation associated with microbial dysbiosis creates conditions favourable to DNA damage, oxidative stress, and impaired DNA repair mechanisms, all of which contribute to tumour initiation and progression.8
In addition to promoting inflammation, these pathogens can modulate the immune response, facilitating immune evasion by cancer cells. For instance, F. nucleatum has been shown to bind to immune receptors like TIGIT on T cells, suppressing their cytotoxic function.9 Simultaneously, the dysbiotic microbiota may recruit immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells to the tumour microenvironment, further dampening antitumor immune responses.10 This immunosuppressive milieu not only allows tumour cells to evade immune surveillance but can also facilitate their growth and metastasis.
Furthermore, microbial dysbiosis can compromise the integrity of the oesophageal epithelial barrier. Pathogenic bacteria associated with dysbiosis degrade tight junction proteins, such as occludin and claudins, weakening the epithelial barrier.11 This breach enables bacterial translocation and systemic dissemination of microbial metabolites, such as lipopolysaccharides. Lipopolysaccharides act as a potent endotoxin, binding to Toll-like receptors on epithelial and immune cells, triggering chronic inflammation and promoting epithelial-mesenchymal transition, a key process in cancer metastasis.4
UPPER GASTROINTESTINAL MICROBIOME IMPACT ON OESOPHAGEAL CANCER TREATMENT OUTCOMES
Beyond its role in cancer development, the UGI microbiome may also significantly impact outcomes following EC surgery. Postoperative complications, such as anastomotic leaks and infections, have been linked to microbial imbalances in the gastrointestinal tract, including the oral cavity.12,13 Bacterial translocation during surgery, compounded by preexisting dysbiosis, may exacerbate these complications.14 Furthermore, the UGI microbiome’s influence on systemic inflammation and immune modulation could affect wound healing and recovery, emphasising the importance of understanding its role in surgical outcomes.15
Prior work investigated the relationship between the diversity of oral and gastric microbiomes and the incidence of anastomotic leaks following esophagectomy.12 They found that patients who developed post-operative anastomotic leaks exhibited greater variability in their oral and gastric microbiomes at the time of surgery compared to those who did not experience leaks.12 This increased variance suggests a potential link between microbiome diversity and the risk of postoperative complications. The impact of the microbiome on postoperative colorectal anastomotic leaks has been demonstrated multiple times, supporting this
paradigm.13,16-19 The microbiome may also play a role in influencing cancer treatment outcomes. In gastrointestinal cancers, the microbiome can modulate drug resistance, with studies showing that gut bacteria impact the metabolism of chemotherapy agents, alter immune responses, and reshape the tumour microenvironment.20 For example, the gut microbiota can exacerbate chemoresistance through mechanisms such as enhancing senescence-associated secretory phenotypes, as seen in F. nucleatum, which promotes oesophageal squamous cell carcinoma resistance to platinum-based chemotherapy by activating DNA damage response pathways.21 Similarly, microbiota dysbiosis can affect the efficacy of immunotherapies and radiotherapies, with pathogenic bacteria fostering an environment that undermines treatment.20,21
F. nucleatum and P. gingivalis have been increasingly recognised as key players in systemic inflammation, impaired wound healing, and heightened susceptibility to postoperative infections.5,6 These bacteria are known to produce virulence factors that can degrade host tissues and evade immune responses. F. nucleatum is capable of adhering to and invading host cells, disrupting tight junction proteins, and compromising the integrity of epithelial barriers.6,22 This not only facilitates bacterial translocation from the oral cavity to distant surgical sites but also allows for the systemic dissemination of bacterial toxins and metabolites, further fuelling inflammation.
Once these pathogenic microbes translocate to surgical sites, they contribute to local tissue damage and immune dysfunction, significantly increasing the risk of complications such as anastomotic leaks and surgical site infections. The presence of bacteria such as P. gingivalis at the surgical site has been shown to stimulate the release of pro-inflammatory cytokines like IL-6 and TNF-α, which exacerbate tissue damage and delay the healing of anastomotic junctions.23 Furthermore, bacterial biofilm formation on sutures or wound surfaces can act as a physical barrier to immune cells and antibiotics, further complicating infection management.24
OPPORTUNITIES FOR THERAPEUTIC INTERVENTIONS
Therapeutic interventions targeting the microbiome present a promising frontier in the management and treatment of EC. The microbiome plays a critical role in modulating inflammation, immune responses, and tissue repair, all of which are central to cancer progression and surgical outcomes. By addressing dysbiosis, clinicians may be able to improve patient outcomes by reducing inflammation, enhancing immune function, and promoting epithelial barrier integrity. Maintaining oral health appears to be crucial for improving dysbiosis, which may influence the development of UGI tumours and impact treatment outcomes.1,25 Poor oral health and its associated microbiome changes have been linked to various disease processes.26-28 Tobacco use has been shown to disrupt the microbiome, while tobacco cessation could improve oral health and potentially mitigate microbial dysbiosis.29,30
One potential intervention is the use of probiotics and prebiotics to restore microbial balance. Probiotics, which introduce beneficial bacteria, can help suppress the growth of pathogenic species such as F. nucleatum and P. gingivalis, both of which have been implicated in EC progression and postoperative complications.31 Prebiotics, which serve as nutrients for beneficial bacteria, can further encourage a healthy microbiome composition.32 This combination can reduce systemic inflammation, improve immune regulation, and potentially mitigate adverse outcomes like anastomotic leaks and infections following esophagectomy.
Another promising strategy involves targeted antimicrobial therapies, such as the use of narrow-spectrum antibiotics or bacteriophages to selectively eliminate pathogenic bacteria while preserving beneficial species.33 Unlike broad-spectrum antibiotics, which disrupt the entire microbial community, targeted approaches can address specific pathogens without causing collateral damage to the microbiome. Such therapies could be particularly useful in preoperative settings, reducing microbial burden and the risk of translocation of harmful bacteria during surgery.
In addition, recent research has underscored the significant influence of the microbiome on the efficacy of immunotherapy in EC. Studies have demonstrated that specific microbial profiles are associated with improved responses to immune checkpoint inhibitors, while dysbiosis correlates with treatment resistance.34,35 For instance, certain bacterial species within the gut microbiome can enhance antitumour immunity by promoting the infiltration and activation of cytotoxic T cells within the tumour microenvironment.34 Conversely, the presence of pathogenic bacteria may foster an immunosuppressive milieu, hindering the effectiveness of immunotherapeutic agents.34
Interventions aimed at modulating the microbiome are being explored to improve immunotherapy outcomes in patients with EC. Approaches such as the administration of probiotics, prebiotics, and dietary modifications seek to enrich beneficial microbial populations, thereby fostering a more favourable immune environment.31,32 Additionally, faecal microbiota transplantation has been investigated as a means to restore a healthy microbiome composition, with some studies showing enhanced responses to immune checkpoint inhibitors following faecal microbiota transplantation.36 These therapeutic strategies aim to shift the microbiome towards a state that supports robust antitumour immunity, potentially overcoming resistance to immunotherapy.
Finally, the emerging field of microbiomebased diagnostics and personalised medicine offers exciting opportunities for tailoring treatments. By analysing the microbiome composition of individual patients, clinicians could identify high-risk microbial profiles associated with poor outcomes.37 This information could guide interventions, such as preoperative microbiome modulation or postoperative monitoring for complications. Additionally, combining microbiome therapies with conventional treatments like chemotherapy or immunotherapy could enhance their efficacy by mitigating treatment-associated dysbiosis and inflammation. Continued research into these therapeutic avenues holds the potential to transform the management of EC.
CONCLUSION
The UGI microbiome may potentially play a critical role in the development of EC and in influencing postoperative outcomes. Dysbiosis can drive chronic inflammation, immune suppression, and epithelial barrier dysfunction, which contribute to tumorigenesis. Additionally, microbial imbalances exacerbate postoperative complications, including anastomotic leaks and infections, by promoting systemic inflammation, delaying tissue healing, and enabling bacterial translocation to surgical sites. These imbalances can also contribute to poor treatment outcomes by
References
1. Liu X et al. From microbes to medicine: harnessing the power of the microbiome in esopha-geal cancer. Front Immunol. 2024;15:1450927.
2. Talapko J et al. The impact of oral microbiome dysbiosis on the aetiology, pathogenesis, and development of oral cancer. Cancers. 2024;16(17):2997.
3. Baima G et al. The gum–gut axis: periodontitis and the risk of gastrointestinal cancers. Can-cers. 2023;15(18):4594.
4. Sharma T et al. Cross-talk between the microbiome and chronic inflammation in esophageal cancer: potential driver of oncogenesis. Cancer Metastasis Rev. 2022;41(2):281-99.
5. Gao S et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect Agent Cancer. 2016;11(1):3.
6. Lei J et al. Fusobacterium nucleatum promotes the early occurrence of esophageal cancer through upregulation of IL ‐32/ PRTN3 expression. Cancer Sci. 2023;114(6):2414-28.
7. Cao Y et al. NF-κB signaling pathway in tumor microenvironment. Front Immunol. 2024;15:1476030.
8. Ivleva EA, Grivennikov SI. Microbiotadriven mechanisms at different stages of cancer de-velopment. Neoplasia. 2022;32:100829.
9. Gur C et al. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory re-ceptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344-55.
fostering resistance to chemotherapy and other therapeutic interventions.
Therapeutic strategies targeting the microbiome offer significant potential to improve EC management. Approaches such as prebiotics, probiotics, and targeted antimicrobial therapies could restore microbial balance, reduce inflammation, and enhance immune responses. While these strategies are promising, further research is needed to better understand the microbiome’s complex role in EC and to validate the clinical benefits of microbiome-based therapies.
10. Mohseni AH et al. Potential links between the microbiota and T cell immunity determine the tumor cell fate. Cell Death Dis. 2023;14(2):154.
11. Quante M et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21(1):36-51.
12. Reddy RM et al. Increased variance in oral and gastric microbiome correlates with esoph-agectomy anastomotic leak. Ann Thorac Surg. 2018;105(3):865-70.
13. Russ A, Casillas M. Gut microbiota and colorectal surgery: impact on postoperative compli-cations. Clin Colon Rectal Surg. 2016;29(03):253-7.
14. Doudakmanis C et al. Bacterial translocation in patients undergoing major gastrointestinal surgery and its role in postoperative sepsis. World J Gastrointest Pathophysiol. 2021;12(6):106-14.
15. Kleinstein SE et al. Inflammatory networks linking oral microbiome with systemic health and disease. J Dent Res. 2020;99(10):1131-9.
16. Boatman S et al. The influence of the microbiome on anastomotic leak. Clin Colon Rectal Surg. 2023;36(2):127-32.
17. Hajjar R et al. Modulating gut microbiota prevents anastomotic leak to reduce local Implan-tation and dissemination of colorectal cancer cells after surgery. Clin Cancer Res. 2024;30(3):616-28.
18. Hajjar R et al. Basal levels of microbiota-driven subclinical inflammation are associated with anastomotic leak in patients with colorectal cancer. Gut. 2024;73(6):1031-3.
19. Hajjar R et al. Gut microbiota influence
anastomotic healing in colorectal cancer surgery through modulation of mucosal proinflammatory cytokines. Gut. 2023;72(6):1143-54.
20. Garajová I et al. The role of the microbiome in drug resistance in gastrointestinal cancers. Expert Rev Anticancer Ther. 2021;21(2):165-76.
21. Zhang JW et al. Fusobacterium nucleatum promotes esophageal squamous cell carcinoma progression and chemoresistance by enhancing the secretion of chemotherapyinduced senescence-associated secretory phenotype via activation of DNA damage response pathway. Gut Microbes. 2023;15(1):2197836.
22. Fan Z et al. Fusobacterium nucleatum and its associated systemic diseases: epidemiologic studies and possible mechanisms. J Oral Microbiol. 2023;15(1):2145729.
23. Pollreisz A et al. Enhanced monocyte migration and proinflammatory cytokine production by Porphyromonas gingivalis infection. J Periodontal Res. 2010;45(2):239-45.
24. Zhao A et al. Understanding bacterial biofilms: from definition to treatment strategies. Front Cell Infect Microbiol. 2023;13:1137947.
25. Sadeghi F et al. Oral microbiome dysbiosis is associated with precancerous lesions and dis-orders of upper gastrointestinal tract: A population-based study. Am J Gastroenterol. 2024;doi:10.14309/ ajg.0000000000003279.
26. Pathak JL et al. The role of oral microbiome in respiratory health and diseases. Respir Med. 2021;185:106475.
27. Liccardo D et al. Periodontal disease: a risk factor for diabetes and
cardiovascular disease. Int J Mol Sci. 2019;20(6):1414.
28. Herrera D, Sanz M, Shapira L, et al. Association between periodontal diseases and cardio-vascular diseases, diabetes and respiratory diseases: Consensus report of the Joint Work-shop by the European Federation of Periodontology ( EFP ) and the European arm of the World Organization of Family Doctors ( WONCA Europe). J Clin Periodontol. 2023;50(6):819-41.
29. Kumar PS et al. Tobacco smoking affects bacterial acquisition and colonization in oral bio-films. Morrison RP, ed. Infect Immun. 2011;79(11):4730-8.
30. Kadam S et al. Looking beyond the
smokescreen: can the oral microbiome be a tool or tar-get in the management of tobacco-associated oral cancer? Ecancermedicalscience. 2021;15:1179.
31. Zhu Y et al. Competition between yogurt probiotics and periodontal pathogens in vitro. Acta Odontol Scand. 2010;68(5):261-8.
32. Zhou P et al. Unveiling the therapeutic symphony of probiotics, prebiotics, and postbiotics in gut-immune harmony. Front Nutr. 2024;11:1355542.
33. Stone VN, Xu P. Targeted antimicrobial therapy in the microbiome era. Mol Oral Microbiol. 2017;32(6):446-54.
34. Dadgar N et al. The influence of the microbiome on immunotherapy for gastroesophageal cancer. Cancers.
2023;15(18):4426.
35. Zhang W et al. Immune checkpoint inhibitors for esophageal squamous cell carcinoma: a narrative review. Ann Transl Med. 2020;8(18):1193.
36. Kim MS et al. Lung cancer surgery for patients with reduced left ventricular ejection frac-tion: clinical outcomes and long-term survival. Eur J Cardiothorac Surg. 2023;63(5):ezad152.
37. Shukla V et al. Targeting the microbiome to improve human health with the approach of personalized medicine: Latest aspects and current updates. Clin Nutr ESPEN. 2024;63:813-20.
Whipple’s Disease, One of Medicine’s Great Imitators: A Case Report
Editor's Pick
The editor’s pick for this issue is a rare case of Whipple’s disease, often referred to as a 'great imitator' due to its non-specific symptoms. The patient presented with a history of chronic wasting, lymphadenopathy, and pulmonary hypertension. Due to the absence of typical symptoms, they underwent an extensive diagnostic journey, ultimately revealing Whipple’s disease. Following appropriate treatment, the patient made a full recovery, including resolution of pulmonary hypertension. This case highlights the importance of considering Whipple’s disease even when classical symptoms are absent, especially in patients with unexplained multisystem disease.
Markus Peck-Radosavljevic Professor of Medicine and Chairman, Department of Gastroenterology and Hepatology, Endocrinology and Nephrology, Klinikum Klagenfurt am Wörthersee, Austria
Authors: *Francesca Heard,1 Elizabeth Hart,1 Vicki M Fleming,2 Claudia Santos,3 Ross Thomson4
1. Department of Infectious Diseases, Nottingham City Hospital, Nottingham University Hospitals Trust, UK
2. Department of Microbiology, Nottingham University Hospitals Trust, UK
3. Department of Histopathology, Nottingham University Hospitals Trust, UK
4. Faculty of Medicine and Health Sciences, University of Nottingham, UK
*Correspondence to francesca.heard@nhs.net
Disclosure: The authors have declared no conflicts of interest.
The authors describe a rare case of a 46-year-old woman, presenting with a 1-year history of a wasting disease and widespread mediastinal lymphadenopathy on imaging. The patient had a history of pulmonary hypertension diagnosed 7 years prior and had been treated with macitentan (an endothelin receptor antagonist) and sildenafil. Following an admission to the authors’ centre with severe anaemia and workup for suspected haematological malignancy, duodenal biopsy confirmed the diagnosis of Whipple’s disease. Interestingly, the patient did not report the cardinal symptoms of arthralgia, fevers, or gastrointestinal upset, emphasising the importance of investigating for Whipple’s disease even in the absence of these symptoms, particularly in the context of malabsorption or chronic wasting disease. Whipple’s disease can be regarded as a ‘great imitator’, often with non-specific signs and symptoms, which can present a diagnostic challenge. Following diagnosis, appropriate antibiotic therapy was commenced, and the patient made a complete clinical recovery, including resolution of pulmonary arterial hypertension, which likely was the presenting feature of this case of Whipple’s Disease, a rare phenomenon associated with this infection.
Key Points
1. The prevalence of Whipple’s disease is likely to be vastly underestimated due to the symptoms presenting a diagnostic challenge, resulting in underdiagnosis.
2. In this case report, the authors report a case of Whipple’s disease where the initial presenting symptoms occurred 7 years prior to diagnosis and imitated various other multi-system diseases.
3. Clinicians should consider investigating for Whipple’s disease even if the ‘classical’ symptoms do not appear to be present, and should maintain awareness of complications such as pulmonary arterial hypertension.
INTRODUCTION
The authors present a rare case of Whipple’s disease (WD) in a patient with a background of pulmonary hypertension and on treatment that presented with chronic wasting disease and severe anaemia. The diagnosis was unexpected as there was a strong suspicion of malignancy given the nature of the presentation and investigation findings of mediastinal lymphadenopathy. WD can be seen as a ‘great imitator’ given its multisystem presentation and is often challenging to diagnose.1 Interestingly, this case was likely associated with pulmonary arterial hypertension, which had developed years prior to diagnosis, a rare but reported complication of WD.2
CASE
A 46-year-old Caucasian woman was admitted to the authors’ centre with severe symptomatic anaemia, which had required iron replacement therapy and blood transfusions over the preceding 6 months. In addition, they reported significant weight loss (20 kg), nausea, and poor appetite for 1 year prior to presentation. The patient denied any other symptoms, including diarrhoea, skin lesions, arthralgia, fevers, or neurological disturbance.
The patient had a background of learning disability and idiopathic primary pulmonary arterial hypertension (PAH). This had been diagnosed following an episode of hospitalisation with pericardial effusion and pulmonary oedema 7 years prior to presentation and was under longterm follow-up at a regional pulmonary hypertension centre after being commenced on macitentan and sildenafil.
No other causes for the PAH were found at that time
Outpatient investigations preceding admission included a CT scan that revealed mediastinal lymphadenopathy and a possible lymphoproliferative disorder. However, a bone marrow biopsy did not confirm this diagnosis, nor did endobronchial ultrasoundguided lymph needle biopsy. Their condition progressed, and they developed ascites requiring paracentesis for symptomatic relief.
Following hospital admission, a full blood count revealed haemoglobin levels of 55 g/L (reference range: 130–175 g/L). Further blood tests showed evidence of malabsorption with red cell folate 2.5 µg/L (reference range: 3.1-20 µg/L), serum iron 2.9 µmol/L (reference range 9–30.4 µmol/L), transferrin saturation 8%, serum albumin 15 g/L (reference range: 35–52 g/L), and 25-hydroxyvitamin D 22 nmol/L (reference range: <30 nmol/L; vitamin D deficiency).
Blood pressure, respiratory rate, oxygen saturation, and temperature were normal. BMI at admission was 13.2 with a weight of 37.5 kg. Further investigations were performed to identify the cause of anaemia and look for evidence of lymphoproliferative malignancy. CT imaging of the abdomen confirmed anterior peritoneal thickening, raising concerns for peritoneal malignancy. Further diagnostic procedures performed included a gastroscopy with duodenal biopsies (Figure 1), a colonoscopy, peritoneal biopsy, and ascitic drainage. Ascitic fluid cytology revealed a population of clonal B cells by immunophenotyping. Significant clinical events during hospital admission included the development of a left lower limb deep vein thrombosis and a subdural haematoma.
Duodenal mucosa showing a lamina propria markedly expanded by numerous foamy macrophages.
Figure 1: Endoscopic image of duodenum.
Figure 2: Haematoxylin and eosin stain of duodenal biopsy (x100).
Histological examination of the duodenal biopsies revealed foamy macrophages consistent with a diagnosis of Whipple’s disease (WD; Figure 2), and periodic acid-Schiff stain was positive (Figure 3). Tropheryma whippelei DNA was subsequently detected via PCR of the duodenal tissue, whole blood, and ascitic fluid samples. No features of malignancy were detected on histological examination of tissue biopsies and ascitic fluid.
The patient was transferred to the care of the infectious diseases team and received four weeks of intravenous ceftriaxone 2 g once a day. Cerebrospinal fluid examination was not possible due to the potential complications of concurrent subdural haematoma and anticoagulation associated with lumbar puncture; therefore, the initial phase of treatment was selected to provide cover for probable central nervous system involvement. Echocardiogram confirmed no features of infective endocarditis were present.
Inflammatory markers and haemoglobin were normalised during the 8-week admission. The patient was discharged with a follow-on regimen of oral co-trimoxazole
960 mg twice a day for 12 months. At the time of discharge, the patient’s BMI was 16.2 with a weight of 38.9 kg (with no ascites). At a 6-month follow-up appointment, the patient’s weight had increased by a further 14 kg, with no side effects of the antibiotic course and a complete clinical recovery.
DISCUSSION
WD is caused by the gram-positive bacteria T.whippelei and is characterised by progressive malabsorption, usually accompanied with gastrointestinal symptoms such as diarrhoea, abdominal pain, and weight loss, often for many years before diagnosis. It is a multisystem disorder with fever and arthralgia being cardinal symptoms in many cases.1 Anaemia and hypoalbuminaemia are often present as a manifestation of malabsorption syndrome. Lymphadenopathy can also be seen.3 Cardiac involvement is thought to be present in one-third of patients, with endocarditis an acknowledged manifestation. Pericarditis has also been reported at autopsy in up to two-thirds of cases.4
Figure 3: Periodic acid-Schiff stain of duodenal biopsy (x10).
Green arrow identifies T.whipplei bacteria.
Host factors are thought to play an important role in the pathogenesis of WD. Patients with WD are thought to have impaired immune function of monocytes and macrophages, resulting in a muted inflammatory response to the organism that is carried in the gastrointestinal tract.2 Reduced expression of CD11b, the α-chain of the phagocytic receptor CR3, has been implicated in particular.2 The organism has a predilection for small-bowel macrophages; hence, duodenal biopsy with periodic acidSchiff staining is the gold-standard method for diagnosis.1An important consideration in this case is the onset of PAH 7 years prior to the diagnosis of WD. This was treated with macitentan and sildenafil. Macitentan is an endothelin receptor antagonist approved for use for PAH. It works by occupying a pocket in the endothelin A receptor, blocking the action of endothelin-1, a potent vasoconstrictor which is found in increased levels in the plasma and pulmonary vascular endothelium of patients with PAH.5 PAH has been reported, although thought to be exceedingly rare in WD. At the completion of antibiotic treatment, clinical resolution of PAH symptoms was noted, and repeat echocardiogram revealed no evidence of PAH. It is likely that this was the initial presentation of WD given the apparent reversal of PAH with targeted antibiotic treatment. The pathophysiology of WD-PAH is poorly understood; however, evidence of bacterial invasion of the tunica media has been reported on histological examination, which may play a role in vascular resistance.6,7 Following the resolution of PAH with WD treatment, sildenafil and macitentan were gradually stopped with careful monitoring for recurrence.
References
1. Antunus C, Singhal M, Whipple Disease [Internet] (2023). Treasure Island: StatPearls. Available at: https://www. ncbi.nlm.nih.gov/books/NBK441937/. Last accessed: 15 December 2024.
2. Lyle PL et al. Reversible pulmonary hypertension in Whipple disease: a case report with clinicopathological implications, and literature review. BMJ Case Rep. 2009;2009:bcr0620080095.
The time to diagnosis from symptom onset in this case was approximately 18 months. WD is regarded as a rare diagnosis; however, the exact prevalence is unknown. One European epidemiological study estimated a prevalence of three in one million.8 The diagnosis can be challenging owing to the variety of clinical signs and symptoms that can mimic other systemic disorders, in particular malignancy, as in this case. This can result in a considerable delay between presentation and diagnosis. This case demonstrates the importance of considering investigating for WD in patients who present with evidence of severe systemic inflammation, anaemia, or malabsorption of chronic duration, even if the cardinal symptoms of arthralgia, fever, or diarrhoea are not reported.
The authors believe that this is the first reported case of WD with initial presentation of pulmonary arterial hypertension years prior to WD diagnosis.
Patient Perspective
I have found taking antibiotics for a long time very hard. I feel very happy the treatment has worked, and I feel normal again. My family said I am much better and have put on weight. I am glad my life is back to normal again.
Evidence of Patient-Informed Consent
The authors obtained written, informed consent from the patient to publish the case report and any accompanying images prior to publication.
3. Obst W et al. Whipple's Disease. Viszeralmedizin. 2014;30(3):167-72.
4. Iqbal T et al. Whipple's disease with constrictive pericarditis: a rare disease with a rare presentation. Can J Cardiol. 2009;25(3):e89-91.
5. Belge C, Delcroix M. Treatment of pulmonary arterial hypertension with the dual endothelin receptor antagonist macitentan: clinical evidence and experience. Ther Adv Respir Dis. 2019;13:1753466618823440.
6. Camboulice A et al. Reversible pulmonary hypertension associated with multivisceral Whipple’s Disease. Eur Respir J. 2021;57(2):2003132.
7. James TN, Bulkley BH. Whipple Bacilli within the tunica media of pulmonary arteries. Chest. 1984;86(3):454-8.
8. Biagi F et al. Prevalence of Whipple’s disease in north-western Italy. Eur J Clin Microbiol Infect Dis. 2015;34(7):1347-8.
Azithromycin Susceptibility of Oral Streptococci in Belgian Men Who Have Sex with Men and the General Population: A Comparison of Two Cross-Sectional Surveys
1. Department of Clinical Sciences, Institute of Tropical Medicine Antwerp, Belgium
2. University of Cape Town, South Africa *Correspondence to ckenyon@itg.be
Disclosure: The authors have declared no conflicts of interest. Abdellati, Vanhout, Manoharan-Basil, and Kenyon contributed equally to this work.
Acknowledgements: Kenyon, Abdellati, Vanbaelen, and Manoharan-Basil conceptualised the study. Kenyon, Vanbaelen, Gestels, and Abdellati collected the samples. Abdellati and Gestels generated the laboratory results. Abdellati, Gestels, Manoharan-Basil, and Kenyon verified and analysed the data. Kenyon wrote the first draft of the manuscript. All authors reviewed and approved the final manuscript. The authors thank all the study participants for their participation in the two studies.
Background: Excessive antimicrobial consumption may saturate the relationship between subsequent antimicrobial exposure and resistance. The ResistAZM randomised controlled trial in Belgium found that 2 g of azithromycin had no effect on the prevalence of macrolide resistance in oral streptococci. At baseline, macrolide resistance was, however, pervasive in this population of men who have sex with men with high exposure to antimicrobials.
Methods: The authors used the same sampling and laboratory protocol to assess if the streptococcal azithromycin susceptibilities in the ResistAZM study were higher than those from isolates obtained from the Belgian general population with lower consumption of antimicrobials (ComCom2023 study).
Results: Streptococcal azithromycin minimum inhibitory concentrations (MIC) were higher in the ResistAZM (median: 32 mg/L; interquartile range: 12–96 mg/L) than in the ComCom2023 study (median: 1 mg/L; (interquartile range: 0.5–12 mg/L; P<0.00001). The azithromycin
MICs of S. mitis/oralis, S. parasanguinis, and S. sanguinis were all significantly higher in the ResistAZM study.
Interpretation: The authors found that the streptococcal macrolide MIC distribution of the ResistAZM participants was significantly higher than that of the Belgian general population in 2023. These findings are compatible with the saturation hypothesis and strengthen the argument that more discriminatory methods are needed to evaluate the effects of antimicrobials on antimicrobial resistance in populations exposed to high levels of antimicrobials. The authors’ findings add support to stewardship initiatives that aim to reduce antimicrobial consumption in this population.
Key Points
1. Streptococcal macrolide minimum inhibitory concentrations were high in both the general population and men who had sex with men.
2. Streptococcal macrolide minimum inhibitory concentrations were significantly higher in men who have sex with men than those in the general population.
3. More discriminatory methods are needed to evaluate the effects of antimicrobials on antimicrobial resistance in populations exposed to high levels of antimicrobials.
INTRODUCTION
A recent randomised controlled trial came to the surprising conclusion that dual gonococcal treatment with azithromycin/ ceftriaxone did not increase the proportion of commensal Neisseria spp. or streptococci with azithromycin resistance compared to ceftriaxone monotherapy.1 Two reasons for this finding were advanced in the ResistAZM trial. First, measuring the minimum inhibitory concentrations (MIC) distribution of target bacteria is a more sensitive method of detecting reduced susceptibility than the proportion of colonies that are resistant.2-4 Second, the extent of antimicrobial resistance (AMR) seen in this study may have saturated the relationship between antimicrobial consumption and resistance.1 Almost half the participants reported using antimicrobials in the year prior to the study, and hence, their streptococci and Neisseria spp. from their baseline visit displayed high levels of AMR. For example, all the individuals had streptococcal isolates with azithromycin MICs greater than 2 mg/L.
To explore the first explanation, the authors repeated the analyses using single colony MICs and confirmed that the samples from
14 days post-treatment with azithromycin did have higher azithromycin MICs for both streptococci and commensal Neisseria spp. compared to baseline samples.2 In the current manuscript, the authors explore the second hypothesis by comparing the streptococcal azithromycin MIC distributions of the baseline samples of the ResistAZM study with samples from the Belgian general population that were collected using a very similar methodology to that used in the ResistAZM trial.
A second objective was to compare the streptococcal azithromycin MICs from the general population with those from previous surveys. This comparison aimed to determine if there has been a shift in azithromycin MIC over time. The data from previously published surveys on streptococcal macrolide resistance in Belgium are not comparable with the authors’ study. One study in 2001 found that the prevalence of macrolide resistance in viridans group streptococci detected from pharyngeal swabs of 154 Belgian individuals aged 17–25 years was 71%.5
A randomised controlled trial of medical students in Antwerp, Belgium, between 2002–2003 found that approximately 25%
of participants had at least one isolate of oral streptococci with erythromycin resistance (erythromycin MIC >2 mg/L) in their baseline samples.6 Individual colony MICs and species identities were not reported in either of these studies. The only comparable data the authors found was from the Antimicrobial Testing Leadership and Surveillance (ATLAS) collection of Streptococcus pneumoniae isolates from Belgian sputum isolates. For this second objective, the authors, therefore, limited their analysis to comparing the azithromycin MICs of S. pneumoniae in the ATLAS collection with those from their general population sample.
METHODS
ComCom2023 Survey Population
The antimicrobial susceptibility of Commensals in the Community 2023 (ComCom2023) study was conducted over the course of two weekends in October 2023. Thirty-five randomly selected families attending children’s sports events at a municipal sporting facility in Antwerp, Belgium, were recruited. The full study methodology has been published elsewhere.7 Briefly, the inclusion criteria required being a member of a family where at least one child (aged 5–13 years) and one adult who was either the parent or a first-degree relative living with the child were willing to participate. The participating child needed to be present with at least one of the parents.
ResistAZM Study Population
The ResistAZM study methodology has been published elsewhere.8 Briefly, this was an randomised controlled trial comparing the effect on the resistome of ceftriaxone 1 g intramuscular injection plus azithromycin (AZM) 2 g orally versus ceftriaxone 1 g intramuscular injection alone for the treatment of N. gonorrhoeae. Twenty men who have sex with men (MSM) with genital, anorectal, or pharyngeal N. gonorrhoeae infection and no consumption of a macrolide in the preceding 6 months were randomised into the ceftriaxone/azithromycin arm,
and 22 to the ceftriaxone arm. Secondary outcomes included the proportion of oral streptococci and commensal Neisseria spp. that were resistant to azithromycin taken from oral rinse samples.
Sampling Procedure
The same sampling method and sample processing were used in both studies. For both studies, participants were instructed to rinse/gargle their mouths with 15 mL sterile water for 30 seconds, after which this was collected in a sterile container.9 Immediately upon arrival at the laboratory (within 6 h after sample collection), 750 µL of each sample was added to 750 µL of skim milk with 30% glycerol and stored at -80 °C until further processing in batch.
Sample Processing
Culture, MIC determination, and identification of streptococcal species
One aliquot of each sample in skim milk was thawed at room temperature, vortexed, and 100 µL was spread plated onto Columbia CNA agar plates with 5% sheep blood (Beckton-Dickinson, Belgium). Plates were incubated for up to 48 h at 37 °C in 5–7% CO2 incubator. After 24–48 hours of incubation, a minimum of four colonies with streptococcal morphology were randomly selected. Species identities were confirmed by matrix-assisted laser desorption ionisation-time of flight mass spectrometry (MALDI-TOF MS) as previously described.10 S. mitis and S. oralis were identified as S. mitis/oralis 11 MICs were ascertained via Etests (BioMérieux, France) on Mueller Hinton agar with 5% horse blood and 20 mg/l ß-NAD (BioMérieux, France).
Historical S. pneumoniae azithromycin susceptibility: ATLAS collection
The authors used the ATLAS database to provide a historical comparison of azithromycin MICs in the Belgian general population. ATLAS holds 6 million anonymised MICs for 3,919 pathogen–antibiotic pairs isolated from 630,000 individuals in 88 countries between 2004–2017.12 Comparisons of ATLAS antimicrobial susceptibility results with those from other datasets, such as the European Centre for Disease Prevention and
Control (ECDC) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST), have revealed that the isolates in ATLAS have a similar but slightly higher MIC distribution per species-antibiotic pair than the other datasets.12
The ATLAS dataset contains azithromycin MICs for 706 S. pneumoniae isolates from Belgium. Of these, the authors used the 468 that were isolated from sputum isolates.12 These isolates were obtained between 2005–2017 (median 2013; interquartile range [IQR]: 2009–2015).
Statistics
When MICs were reported as less or more than the lowest or highest concentration
Time since last antibiotic use, n (%) <7 days 7–30 days 1–3 months 4–6 months >6 months
antibiotics in the past 12 months (ResistAZM), n (%)
Used antibiotics in the past 6 months (ComCom2023), n (%)
*Willcoxon rank sum test (age) or Fisher’s exact test (sex) IQR: interquartile range; N/A: not applicable.
tested, these were replaced with the lowest or highest concentrations tested, e.g., azithromycin MIC >256 mg/L was recorded as 256 mg/L.
Differences in MICs were compared between groups using Wilcoxon rank sum tests. The Bonferroni correction was applied to account for multiple comparisons. Statistical analyses were conducted using Stata v16.1 (StataCorp, LLC College Station, Texas).
Ethics
Ethics approval was obtained from ITM’s Institutional Review Board (1574/22) and the Ethics Committee of the University of Antwerp (3831).
Table 1: Population characteristics.
RESULTS
Survey Population Data
The participants of the ResistAZM were older than those in the Comcom2023 study but similar in age to the adults in the ComCom2023 study (Table 1).
While all the participants in the ResistAZM study were men, only 51% (51/88) of the participants were men in the ComCom2023 study. Eighteen of 42 (42.9%) participants in the ResistAZM study reported using antimicrobials in the 12 months prior
to the study, whereas 8/88 (9.1%) of the ComCom2023 study reported antimicrobial use in the prior 6 months. None of the ResistAZM participants had taken macrolides in the prior 6 months. In the 12 months before recruitment, 14 had consumed beta-lactams, seven tetracyclines, one a fluoroquinolone, and one a macrolide.
Streptococcal Species Composition
S. mitits/oralis was the most abundant streptococcal species in the ResistAZMbaseline-visit (34/81 isolates, 42%) and the
The pie charts depict the percent of each streptococcal species isolated out of all streptococcal isolates from that study (only the baseline results are shown for the ResistAZM study).
Figure 1: Streptococcal species composition in the ResistAZM and ComCom2023 studies.
ComCom2023 (146/312, 46.8%) studies (Figure 1; Supplementary Table 1). S. sanguinis was detected more frequently in the ResistAZM (24.7%) than in the ComCom2023 study (6.1%; P<0.001). In contrast, S. salivarius was detected more frequently in the ComCom2023 (17.9%) than in the ResistAZM study (4.9%; P<0.001).
Antimicrobial Susceptibility of Streptococci
Streptococcal azithromycin MICs were higher in the ResistAZM (median: 32 mg/L; IQR: 12–96 mg/L) than the ComCom2023 study (median: 1 mg/L; IQR: 0.5–12mg/L; P<0.00001; Table 2). The azithromycin MICs of S. mitis/oralis, S. parasanguinis, and S.
Table 2: Azithromycin MIC (mg/L) in the ComCom2023 and the baseline samples of the ResistAZM study.
MIC: minimum inhibitory concentration; N: number.
sanguinis were all significantly higher in the ResistAZM study (P<0.0001; Table 2). There was no significant difference between the streptococcal species composition or azithromycin MICs by species between the children and the adults in the ComCom2023 study (Supplementary Table 2).
Using the EUCAST azithromycin breakpoint of 0.25 mg/L for S. pneumoniae and Group A, B, C, and G streptococci, only one of 81 (1.2%) isolates from the ResistAZM study was susceptible to azithromycin compared to 29/331 (9.3%; P<0.001) in the ComCom2023 study (Supplementary Table 3).13 If a breakpoint of 2 mg/L is used, then 79.1% of ResistAZM isolates and 46.0% of ComCom23 isolates would be classified as resistant (Supplementary Table 3 and Supplementary Table 4).
Switching to considering the prevalence of AMR per person, 78/88 (88.6%) of individuals in the ComCom2023 study had streptococci with MICs >2 mg/L (Supplementary Table 5). The same was true for 100% of the ResistAZM cohort.
ATLAS versus ComCom2023
The median azithromycin MIC of the sputum S. pneumoniae isolates in the ATLAS collection was 0.06mg/L, IQR 0.06-0.25mg/ L (Supplementary Table 6). This was significantly lower than the S. pneumoniae MICs from the ComCom2023 study (median 8mg/L; IQR: 0.38-16; P=0.0003).
DISCUSSION
The authors found that the streptococcal azithromycin MICs of the participants in the ResistAZM study were markedly higher than those from the ComCom2023 study. This was driven by higher azithromycin MICs in three of the most prevalent streptococcal species (S. mitis/oralis, S. parasanguinis, and S. sanguinis). Only minor differences in the composition of streptococcal species were found between the studies.
These findings are compatible with the AMR saturation hypothesis. Close to 100% of isolates from the ResistAZM would be classified as resistant to azithromycin according to the EUCAST breakpoint. The median azithromycin MIC was 32 mg/L in this study, with only 20.9% of isolates having a MIC below 2 mg/L. These high MICs would be expected to decrease the chance of detecting an azithromycin-induced increase in the proportion of colonies with azithromycin resistance (defined as >2 mg/L in the ResistAZM study).
It is likely that high consumption of antimicrobials is responsible for the high azithromycin MICs in the ResistAZM population. Almost half of the study population reported consuming antimicrobials in the prior year. The association between antimicrobial consumption and resistance is, however, not straightforward. Consumption of macrolides in the prior 6 months was an exclusion criterion for study entry. Only one individual had thus consumed a macrolide, and this was between 6–12 months prior. The effect of macrolide consumption on oral streptococcal macrolide resistance has been shown to last for 6 months, but the possibility that this effect could last longer cannot be excluded.6 It is also possible that the other antimicrobials could select for resistance to macrolides. However, a multivariate linear regression analysis found no association between consumption of antimicrobials and median streptococcal azithromycin MIC per individual.1,14
Various lines of evidence suggest that population-level consumption of
antimicrobials plays a key role. There are at least five different populationlevel pathways between antimicrobial consumption and resistance.15,16 The authors have established that macrolide consumption in their MSM preexposure prophylaxis cohort was until recently up to seven-fold greater than macrolide consumption thresholds associated with macrolide resistance in bacteria such as M. genitalium and S. pneumoniae 17,18 From detailed analyses of this cohort, the authors have confirmed that oral Neisseria species do have higher macrolide and ciprofloxacin MICs than those from the Belgian general population.10 Surprisingly, these elevated MICs were found in both the MSM who had and who had not consumed antimicrobials in the prior 6 months.10 There was no difference in the MICs between these two groups of MSM.10 Likewise, individual-level analyses found no association between recent antimicrobial consumption and ceftriaxone, azithromycin, or ciprofloxacin MICs.14 These findings, plus the extensive evidence of transmission of resistant bacteria between individuals, have led the authors and others to conclude that population-level consumption of antimicrobials is a key determinant of high MICs in this population.10,19,20
The azithromycin MICs of the general population (ComCom2023) were also concerningly high. The authors found that the pneumococcal azithromycin MICs were higher than those in ATLAS, which were collected between 2005–2017. The authors have very little metadata for the ATLAS collection, and therefore, these differences may simply stem from selection biases. It is likely that ATLAS contains more invasive isolates of pneumococcus, which have been found to have lower beta-lactam MICs than non-invasive isolates.21 Previous comparisons of ATLAS with other datasets have, however, found that ATLAS tends to be slightly biased toward less-susceptible isolates.12
There are a number of other limitations to the authors’ analysis. The authors did not assess the MICs in duplicate. The sample sizes of the studies are small. ResistAZM and ComCom2023 were not conducted in the same season, which may mean that
season variations in MIC may contribute to the differences in MIC the authors found.22 Species identification relied on MALDI-TOF MS, which lacks sufficient discrimination to distinguish species such as S. mitis from S. oralis. Finally, the authors did not identify the molecular basis of macrolide resistance. Future studies could circumvent these limitations by conducting similar but simultaneous surveys in general populations and key populations with high antimicrobial consumption using larger sample sizes. Antimicrobial susceptibilities to a broader range of antimicrobials could be assessed. Whole genome sequencing of isolates would enable optimised species identification and comparisons of the prevalence of macrolide-resistanceassociated mutations.
References
1. Vanbaelen T et al. Effect on the resistome of dual vs monotherapy for the treatment of Neisseria gonorrhoeae: results from a randomised controlled trial (ResistAZM Trial). Open Forum Infect Dis. 2023;10(10):ofad462.
2. Vanbaelen T et al. Four recent insights suggest the need for more refined methods to assess the resistogenicity of doxycycline post exposure prophylaxis. Curr Res Microb Sci. 2024;6:100234.
3. Vanbaelen T et al. 45 years of tetracycline post exposure prophylaxis for STIs and the risk of tetracycline resistance: a systematic review and meta-analysis. BMC Infect Dis. 2024;24(1):376.
4. Zawack K et al. Monitoring antimicrobial resistance in the food supply chain and its implications for FDA policy initiatives. AAC. 2016;60(9):5302-11.
5. Malhotra-Kumar S et al. Oropharyngeal carriage of macrolide-resistant viridans group streptococci: a prevalence study among healthy adults in Belgium. J Antimicrob Chemother. 2004;53(2):271-6.
6. Malhotra-Kumar S et al. Effect of azithromycin and clarithromycin therapy on pharyngeal carriage of macrolide-resistant streptococci in healthy volunteers: a randomised, double-blind, placebo-controlled study. The Lancet. 2007;369(9560):482-90.
7. Abdellati S et al. Antimicrobial susceptibility of commensal Neisseria spp. in parents and their children in belgium: a cross-sectional survey. FEMS Microbi-
CONCLUSION
The authors found that the streptococcal macrolide MIC distribution of the ResistAZM participants was alarmingly high and significantly higher than that of the general population in 2023. These findings are compatible with the saturation hypothesis and add extra weight to the argument that more discriminatory methods than the proportion resistant are needed to evaluate the effects of antimicrobials on AMR in this population.3 Crucially, the authors’ findings add support to stewardship initiatives that aim to reduce antimicrobial consumption in this population. As an example, a single intervention in the authors’ HIV preexposure prophylaxis clinic was able to reduce macrolide consumption by fourfold.23
ol Lett. 2024;371:fnae069.
8. Vanbaelen T et al. Effect on the resistome of dual- vs monotherapy for the treatment of Neisseria gonorrhoeae: results from a randomised controlled trial (ResistAZM Trial). Open Forum Infect Dis. 2023;10(10):ofad462.
9. Laumen JGE et al. A novel method to assess antimicrobial susceptibility in commensal oropharyngeal Neisseria—a pilot study. Antibiotics. 2022;11(1):100.
10. Laumen JGE et al. Antimicrobial susceptibility of commensal Neisseria in a general population and men who have sex with men in Belgium. Sci Rep. 2022;12(1):1-10.
11. Marín M et al. Accurate differentiation of Streptococcus pneumoniae from other species within the Streptococcus mitis group by peak analysis using MALDI-TOF MS. Front microbiol. 2017;8:698.
12. Catalán P et al. Seeking patterns of antibiotic resistance in ATLAS, an open, raw MIC database with patient metadata. Nat Commun. 2022;13(1):2917.
13. European Committee on Antimicrobial Susceptibility Testing (EUSCAT). EUCAST Clinical Breakpoint Tables. 2024. Available at: https://www.eucast.org/ eucast_news/news_singleview?tx_ ttnews%5Btt_news%5D=566&cHash=db55f3a8829726044512a1fe74cce41b. Lat accessed: 12 November 2024.
14. Vanbaelen T et al. Lack of association between antimicrobial consumption and antimicrobial resistance in a hiv preexposure prophylaxis population: a cross-sectional study. Antibiotics. 2024;13(2):188.
15. Lipsitch M, Samore MH. Antimicrobial use and antimicrobial resistance: a population perspective. Emerg Infect Dis. 2002;8(4):347-54.
16. Kenyon CR, Schwartz IS. Effects of sexual network connectivity and antimicrobial drug use on antimicrobial resistance in Neisseria gonorrhoeae. Emerging Infectious Diseases. 2018;24(7):1195-203.
17. Kenyon C. Dual azithromycin/ceftriaxone therapy for gonorrhea in PrEP cohorts results in levels of macrolide consumption that exceed resistance thresholds by up to 7-fold. J Infect Dis. 2021;224(9):1623-4.
18. 1Kenyon C et al. Is there a resistance threshold for macrolide consumption? Positive evidence from an ecological analysis of resistance data from Streptococcus pneumoniae, Treponema pallidum, and Mycoplasma genitalium. MDR. 2021;27(8):1079-86.
19. khimiukor OO et al. A bottom-up view of antimicrobial resistance transmission in developing countries. Nat Microbiol. 2022;7(6):757-65.
20. Kenyon C. Positive association between the use of macrolides in food-producing animals and pneumococcal macrolide resistance: a global ecological analysis. Int J Infect Dis. 2022;116:344-7.
21. Higgs C et al. Comparison of contemporary invasive and non-invasive
Streptococcus pneumoniae isolates reveals new insights into circulating anti-microbial resistance determinants. Antimicrob Agents Chemother. 2023;67(11):e0078523.
22. Martinez EP et al. Seasonality of antimicrobial resistance rates in respiratory bacteria: a systematic review and meta-analysis. PLoS One. 2019;14(8):e0221133.
23. Vanbaelen T et al. Screening for STIs is one of the main drivers of macrolide consumption in PrEP users. International Journal of STD & AIDS. 2021:32(12):1183-4. FOR REPRINT QUERIES PLEASE CONTACT: INFO@EMJREVIEWS.COM
Infliximab-Induced Multifocal Motor Neuropathy in a Patient with Ankylosing Spondylarthritis
and Crohn’s Disease:
A
Case Report with Anti-GM2 Antibodies
Authors: *Dollen Eid,1 Christian Matta1
1. Department of Neurology, Hôtel-Dieu de France Hospital, Beirut, Lebanon
*Correspondence to dolleneid@gmail.com
Disclosure: The authors have declared no conflicts of interest.
Received: 23.12.24
Accepted: 06.02.25
Keywords: Anti-GM2 antibodies, immune-mediated neuropathy, infliximab-induced multifocal motor neuropathy, intravenous immunoglobulin (IVIg), multifocal motor neuropathy (MMN).
Background: Multifocal motor neuropathy (MMN) is a rare immune-mediated disorder characterised by progressive, asymmetric muscle weakness primarily affecting distal limb nerves without sensory involvement. While MMN is estimated to affect only one-to-two individuals per 100,000, the presence of anti-GM2 antibodies in MMN is exceedingly rare, with fewer than five reported cases in the medical literature.
Case presentation: This case report describes a 51-year-old female with a history of ankylosing spondylarthritis and Crohn’s disease who developed left wrist weakness and sensory disturbances following infliximab infusion and a SARS-CoV-2 vaccination. Electromyography revealed neurogenic changes and motor conduction block in the left radial nerve, which is consistent with the MMN diagnosis. Further investigation showed elevated anti-GM2 antibodies in serum, a rare finding associated with MMN. Treatment with intravenous immunoglobulin (IVIg) resulted in significant symptom improvement.
Conclusion: This case highlights the diagnostic challenges in MMN associated with anti-GM2 antibodies, emphasising the importance of early recognition and targeted management in similar clinical presentations. Although the patient’s symptoms improved after IVIg treatment, the aetiology of MMN and the role of anti-GM2 antibodies remained unclear.
Key Points
1. Multifocal motor neuropathy (MMN) is a rare immune-mediated disorder affecting one-to-two per 100,000 individuals, with anti-GM2 antibodies being exceedingly rare, making early recognition and management critical for preventing long-term disability.
2. The authors present the case of a 51-year-old female with MMN associated with anti-GM2 antibodies following infliximab infusion and COVID-19 vaccination, highlighting diagnostic challenges and treatment response to intravenous immunoglobulin.
3. Clinicians should consider MMN in patients with progressive asymmetric weakness, even with atypical antibody profiles, and explore potential links between vaccines, immunomodulatory therapies, and autoimmune neuropathies.
INTRODUCTION
Multifocal motor neuropathy (MMN) is a rare immune-mediated condition recently documented in the medical literature. MMN typically manifests with gradual, progressive, asymmetric muscle weakness in the distal limbs, predominantly affecting the ulnar, median, radial, and tibial nerves.1-4 MMN has been found to affect predominantly men, with a maleto-female ratio of approximately 3:1, and typically manifests around the age of 40. Prevalence rates vary globally, with estimates ranging from 0.6 to 2 cases per 100,000 individuals, with no significant differences reported across ethnicities.1-3 A defining electrophysiological feature of MMN is multifocal chronic conduction block (CB) affecting motor neurons only.5-7 MMN is thought to result from an autoimmune attack on peripheral motor nerves, mediated by antibodies targeting gangliosides such as GM1, GD1a, and, rarely, GM2. Approximately 30–80% of patients with MMN exhibit elevated levels of IgM antibodies against GM1, and less commonly GM2 and GD1a, detectable in their blood.8 These antibodies are thought to disrupt nerve conduction by targeting gangliosides in the myelin sheath of motor nerves, and their disruption leads to conduction block and muscle weakness. Intravenous immunoglobulin (IVIg) therapy is highly effective in treating MMN. IVIg works by modulating the immune system, neutralising autoantibodies, and suppressing inflammatory pathways. Multiple studies have demonstrated its efficacy, with significant improvement in muscle strength observed in up to 70–80% of patients.9,10 However, up to 20% of patients with MMN experience moderate-to-severe impairment, primarily in the upper limbs.1
PATIENT INFORMATION
A 51-year-old female with a medical history of ankylosing spondylarthritis and Crohn’s disease presented with left wrist weakness and sensory disturbances following infliximab (originator) infusion and SARSCoV-2 vaccination. The patient had been previously treated with three doses of infliximab biosimilar, at the standard dose of 5 mg/kg every 8 weeks without adverse effects. Due to a drug shortage in Lebanon, she was switched to infliximab originator at the same dose. However, during the first 30 minutes of infliximab infusion, she developed generalised pruritus and a rash on her hands, chest, and back, a reaction that occurs in approximately 3–10% of patients receiving infliximab.11
Treatment with cortisone and desloratadine resulted in the disappearance of itching and rash after approximately one hour, except for persistent itching on the soles of her feet; therefore, the infusion was allowed to continue after this time. Three weeks after the infusion, she presented with left wrist weakness, tingling sensations in her feet, hypoesthesia resembling walking on cotton, and calf contractures. Notably, 20 days prior to infliximab infusion, the patient had completed a course of treatment with cefixime 400 mg (0-1-0) and metronidazole 500 mg (1-0-1) for severe gastroenteritis exacerbated by consumption of raw meat. Additionally, the patient had received two doses of the Pfizer-BioNTech COVID-19 vaccine, with the second dose administered 3 days prior to her last infliximab infusion.
CLINICAL FINDINGS
On physical examination, motor strength was intact in bilateral upper and lower limbs except for left wrist extension weakness
(3+/5). Reflex testing revealed diminished responses in the left triceps (0/4) compared to the right triceps, biceps, brachioradialis, patellar, and Achilles tendon (2/4). Sensory examination indicated preserved vibration sense over toes bilaterally and intact light touch at the knees and fingertips bilaterally. Upper extremity sensory testing showed diminished light touch sensation in the left hand, particularly over the dorsum.
TIMELINE
Day 0: Received second dose of Pfizer-BioNTech COVID-19 vaccine.
Day 3: Infusion of infliximab originator after a drug shortage of infliximab biosimilar.
Day 23: Onset of left wrist weakness and sensory disturbances.
Day 25: Initial electromyography (EMG) showing conduction block in the left radial nerve (Figure 1).
Day 32: Second EMG confirming motor conduction block in both radial nerves (Figure 2).
Day 60: Third EMG showing resolution of conduction blocks but persistent fibrillations (Figure 3).
DIAGNOSTIC ASSESSMENT
The patient was evaluated with EMG, which showed a normal sensory study but reduced recruitment patterns and neurogenic motor unit changes in the left extensor indicis propius, with a suspected conduction block in the left radial nerve (Figure 1). Two days later, the patient presented to the ER for further degradation of her left wrist extension. Motor exam showed left wrist extension of 3-/5 with an absent left triceps reflex, which was also present in the initial physical exam.
Lumbar puncture was performed, and cerebrospinal fluid (CSF) analysis showed that the protein level was 0.35 g/L (normal values: 0.12–0.6 g/L), while the white blood cell count was 9x106 /L (normal
values: 0–5x106 /L). Lab workup, including CBC, biochemistry panel, C3, C4, antiHelicobacter pylori antibody, parathyroid hormone, brucellosis antibodies, rheumatoid factor, folic acid, vitamin B12, vitamin D, TSH, Widal test, antinuclear antibody (ANA), anti-Sjögren’s syndrome-related antigen A (anti-SSA; also called antiRo), anti-Sjögren’s syndrome-related antigen B (anti-SSB; also called anti-La), anti-Smith antibody (anti-Sm), antiribonucleoprotein/Smith antibody (antiRNP/Sm), anti-scleroderma 70 antibodies or anti-topoisomerase I antibody (anti-Scl 70; associated with systemic sclerosis), anti-histidyl-tRNA synthetase antibody (anti-Jo 1; associated with polymyositis and dermatomyositis), anti-double-stranded DNA immunoglobulin G (anti-dsDNA IgG; highly specific for systemic lupus erythematosus), and anti Campylobacter jejuni and fetus, were within normal range.
Anti-ganglioside antibody analysis of the serum revealed high levels of anti-GM2 with 102 (reference range: 0–30). The blots for other anti-gangliosides (antiGM1, anti-GD1a, anti-GQ1b, anti-GD1b, and anti-MAG) were negative.
THERAPEUTIC INTERVENTION
The patient was treated 1 month after the initial symptoms with IVIg at 0.4 g/ kg daily for 5 days. She completed the course of IVIg and was discharged with marked improvement in left wrist extension strength (from 3-/5 to 4+/5) and resolution of tingling sensations in her feet.
The patient was scheduled to receive 1 g/ Kg IVIg daily for 5 days, 28 days following discharge. A third EMG was performed 4 weeks after the initial IVIg treatment and showed the disappearance of conduction block in both radial nerves (Figure 2) but with persistent fibrillations of muscles in the left extensor indicis propius, the left brachioradialis, and the left extensor carpi radialis longus. The patient then received two courses of IVIg at 0.5 g/kg each on two consecutive days, 28 days apart. A fourth EMG revealed a neurogenic motor unit change in the left extensor indicis propius with no conduction block (Figure 3).
Figure 1: The initial electromyography findings obtained 2 weeks after symptom onset.
A) Motor nerve conduction study of the left radial nerve shows there is a conduction block at the lateral arm stimulation point.
B) Motor nerve conduction study of the right radial nerve shows there is a conduction block at the lateral arm stimulation point.
Figure 2: The electromyography findings obtained 4 weeks after IVIg treatment.
A) Motor nerve conduction study of the left radial nerve shows there is a disappearance of the conduction block at the lateral arm stimulation point.
B) Motor nerve conduction study of the right radial nerve shows there is a disappearance of the conduction block at the lateral arm stimulation point.
FOLLOW-UP AND OUTCOMES
Treatment with IVIg resulted in significant improvement of symptoms in the author’s patient, who exhibited high levels of antiGM2 IgM antibodies, a rare association with MMN. The patient reported marked improvement in left wrist extension strength and resolution of sensory disturbances.
DISCUSSION
MMN is a rare inflammatory neuropathy characterised by progressive, asymmetric weaknessin the distal limbs without sensory loss.4 Diagnosis typically requires the presence ofconduction blocks in at least two peripheral nerves.4 In a substantial percentage of cases (30–80%), patients exhibit elevated levels of IgM anti-GM1 ganglioside antibodies, though a negative result does not exclude MMN.8
The main differential diagnoses for MMN include Lewis-Sumner syndrome (LS) and Guillain-Barré syndrome (GBS).8 LS is distinguished from MMN by the presence of significant sensory symptoms and neuropathic pain, often accompanied by
diminished sensory nerve potentials.8 GBS and Fisher syndrome, while similar in some respects, can be differentiated through comprehensive laboratory and electrophysiological evaluations, as demonstrated in the authors’ case. GBS and Fisher syndrome were excluded based on normal cerebrospinal fluid protein levels without cytoalbuminologic dissociation, absence of ophthalmoplegia or ataxia, and EMG findings consistent with motor conduction blocks rather than demyelination or axonal damage. The pathogenesis of MMN suggests an autoimmune basis because of the association with anti-GM1 antibodies and robust response to immunomodulatory treatments, particularly IVIg. IVIg remains the first-line treatment for MMN based on multiple studies and randomised trials.9
While anti-GM2 antibodies have been implicated in other neuropathic conditions such as GBS and Fisher syndrome, their implication in MMN remains unclear due to limited research.10,12 Anti-GM2 antibodies are exceedingly rare in MMN, with fewer than five reported cases in the literature. This rarity underscores the uniqueness of the author’s case and highlights the
Figure 3: Illustrating the third electromyography findings 8 weeks post-treatment.
Motor nerve conduction study of the left radial shows no conduction block.
need for further research into the role of anti-GM2 antibodies in immune-mediated neuropathies. Anti-GM2 antibodies have been described in patients with GBS subsequent to cytomegalovirus hepatitis,10 and in mycoplasma pneumoniae-associated acute disseminated encephalomyelitis,10 but nothing is known about their occurrence in MMN or other chronic immune-mediated neuropathies.8 The author’s case excluded GBS, LS, and Fisher syndrome by way of laboratory and electrophysiological examination. The diagnosis of MMN was supported by the characteristic clinical features, including CBs in both radial nerves, as well as the positive response to IVIg therapy. Treatment with IVIg resulted in significant improvement of symptoms in the authors’ patient, who exhibited high levels of anti-GM2 IgM antibodies, a rare association with MMN.
It is plausible that the Pfizer-BioNTech COVID-19 vaccine primed the immune system, predisposing the patient to an adverse reaction to infliximab originator. This hypothesis is supported by the temporal association between vaccination, infliximab infusion, and symptom onset. Further research is needed to explore potential interactions between vaccines and immunomodulatory therapies.
A similar case was described by Eren et al.13 involving a 52-year-old man who presented with progressive weakness in the lower extremities and gait disturbance
References
1. Cats EA et al. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology. 2010;75(9):818-25.
2. Nobile-Orazio E. Multifocal motor neuropathy. J Neuroimmunol. 200;115(1-2):4-18.
3. Van Asseldonk JTH et al. Multifocal motor neuropathy. Lancet Neurol. 2005;4(5):309-19.
4. Vlam L et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol. 2011;8(1):48-58.
5. Chad DA et al. Slow resolution of multifocal weakness and fasciculation: a reversible motor neuron syndrome.
over the course of several weeks. There was no disease or functional disability in his previous medical history. He had no history of trauma, fever, night sweats, bowel and bladder dysfunction, or infection. His symptoms began 25 days after receiving the second dose of the Pfizer-BioNTech COVID-19 vaccine.
CONCLUSION
MMN associated with anti-GM2 antibodies is a rare entity, which should be a differential in patients presenting with rapidly progressive neurological symptoms. A thorough neurological examination and testing with EMG could aid in prompt diagnosis and management with anti-GM2 antibodies where appropriate. The authors’ patient had reported improvement in symptoms after receiving treatment with IVIg. Further research is warranted to investigate the potential association between COVID-19 vaccination and the development of MMN with anti-GM2 antibodies. Additionally, studies should explore the mechanisms underlying infliximab-induced MMN to guide clinicians in managing patients with autoimmune conditions.
ETHICAL CONSIDERATIONS
Written informed consent was obtained from the patient involved in this study for the publication of their data and images.
Neurology. 1986;36(9):1260-3.
6. Roth G et al. Motor neuropathy with proximal multifocal persistent conduction block, fasciculations and myokymia. Evolution to tetraplegia. Eur Neurol. 1986;25(6):416-23.
7. Parry GJ, Clarke S. Multifocal acquired demyelinating neuropathy masquerading as motor neuron disease. Muscle Nerve. 1988;11(2):103-7.
8. Jusufović E et al. Multifocal motor neuropathy: case reports. Acta Clin Croat. 2018;57(3):581-7.
9. Léger JM et al. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther Adv Neurol Disord. 2015;8(3):109-22.
10. Tsukaguchi M et al. IgM anti-GM2 antibody in a patient with GuillainBarré syndrome subsequent to cytomegalovirus hepatitis cross reacts with N-acetylgalactosaminyl GD1a. J Neurol Neurosurg Psychiatry. 1998;65(3):407-8.
11. Lichtenstein L et al. Infliximab-Related Infusion Reactions: Systematic Review. J Crohns Colitis. 2015 Sep;9(9):806-15.
12. Xia C, Chen HS. Anti-GM2 antibodies in mycoplasma pneumoniaeassociated acute encephalomyelitis. Can J Neurol Sci. 2020;47(2):258-60.
13. Eren F et al. Multifocal motor neuropathy after SARS-CoV-2 vaccination: a causal or coincidental association? J Int Med Res. 2022;50(7):3000605221110709.
Rare Anti-HMGCR-Induced Immune-Mediated Necrotising Myopathy: A Case Report and Literature Review
An acute necrotising myopathy is a distinct form of uncommon muscle disease characterised by the rapid advancement of weakness affecting the limbs, neck, pharyngeal, respiratory, and occasionally cardiac muscles. It frequently arises as part of idiopathic inflammatory myopathies, which include conditions like polymyositis, dermatomyositis, and inclusion body myositis. Anti-hydroxy-3-methylglutaryl-coenzyme A reductase represents an infrequent immune-mediated necrotising myopathy. This case study involves a 55-year-old African American woman experiencing muscle weakness and recurrent falls over 2 months with elevated creatine kinase levels, indicating an inflammatory disease process. The patient received symptomatic management after excluding any critical illness. After initial treatment, she underwent outpatient follow-up along with necessary investigations that led to the definitive diagnosis.
Key Points
1. Statins, the most commonly prescribed medication, are linked to anti-hydroxy-3-methylglutaryl-coenzyme A reductase-induced myopathy. Early recognition is crucial due to its significant impact on muscle function.
2. This case report describes the diagnostic challenges and immunotherapy in a 55-year-old woman with antihydroxy-3-methylglutaryl-coenzyme A reductase immune-mediated necrotising myopathy.
3. Early statin discontinuation and combination immunotherapy improve outcomes in necrotising autoimmune myopathy, with multi-agent therapy being more effective in resistant or complex cases.
INTRODUCTION
Inflammatory myopathies constitute a wide range of diseases characterised by muscle inflammation and weakness. Dermatomyositis and polymyositis are the most widely recognised and extensively researched among these conditions.1 However, recent progress in medical research has revealed new distinct inflammatory myopathies,2 emphasising the complexity and diversity of these disorders. One such ailment is immune-mediated necrotising myopathy, also known as necrotising autoimmune myopathy (NAM), a form of autoimmune myopathy characterised by the acute or sub-acute onset of severe, symmetrical weakness primarily affecting the proximal musculature. It includes a rare subset associated with antibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR).3
Idiopathic inflammatory myopathies affect individuals across different ages and ethnicities, although they are generally uncommon. These myopathies feature persistent muscle inflammation, leading to progressive muscle weakness and disability. Identifying myositis-specific autoantibodies has improved diagnostic accuracy, enabling better categorisation and comprehension of these illnesses. Despite their overall rarity, their impact on patients’ lives is profound, demanding precise diagnosis and effective treatment strategies.
Anti-HMGCR myopathy poses unique challenges in clinical practice due to its infrequency as well as its potential association with statin use, commonly prescribed for hyperlipidaemia and cardiovascular risk reduction, the aspect known as statin-induced necrotising autoimmune myopathy.4 This condition represents an intersection between pharmacology and autoimmunity, where the therapeutic benefits of a medication need to be weighed cautiously against a severely debilitating muscular disease.
The presented case report discusses a 55-year-old woman with hypertension, dyslipidaemia, Type 2 diabetes, and obesity who developed muscle weakness and recurrent falls. The subsequent diagnostic workup revealed significantly elevated creatine kinase levels, indicating an inflammatory disorder. Following extensive investigations, she was diagnosed with anti-HMGCR-induced immunemediated necrotising myopathy. This case emphasises the importance of considering rare neuromuscular consequences and highlights the need for comprehensive diagnostic approaches to distinguish various causes of myopathy.
CASE PRESENTATION
Patient Information and History
A 55-year-old African American woman, a clerical worker with a past medical history of hypertension, dyslipidaemia, Type 2 diabetes, and obesity, presented to the emergency department. She complained of gradually worsening weakness over the past 2 months that affected her ability to perform daily activities such as combing her hair, rising from a seated position, and climbing stairs. She initially considered her symptoms to be the result of vitamin deficiency but later sought medical attention when the symptoms worsened. Her primary care physician ordered some blood tests that showed elevated liver enzymes and creatine kinase levels (11,000 IU/L), suspected to be due to adverse effects of statins. She also reported intermittent sharp chest pains not associated with physical exertion or other symptoms such as nausea or diaphoresis. She then was referred to the emergency department for further evaluation.
Clinical Findings
On examination, she had a significantly elevated blood pressure of 223/111 mmHg and proximal muscle weakness in her lower extremities.
Timeline
As detailed in Table 1, the timeline of her case presentation highlights key events and interventions.
Diagnostic Assessment
Further evaluation of complete blood count, comprehensive metabolic panel, EKG, troponin, D-dimer, creatine kinase, B-12, and aldolase was scheduled, along with discontinuation of ezetimibe and atorvastatin till investigation results were pending (Table 2).
The authors initially excluded the known and reversible causes of muscle weakness. The patient was admitted later to the hospital for further evaluation over the next month. Various infectious and autoimmune tests were performed, all yielding largely negative results.
A provisional diagnosis of myopathy was made, and further investigations such as nerve conduction studies, electromyography, muscle biopsy, and MRI were considered. The MRI report revealed oedema (Figure 1) in the thigh muscles, while the nerve conduction study results appeared normal. Electromyography showed irritable myopathy affecting the proximal more than the distal muscles of the upper and lower extremities.
A consistent muscle biopsy result diagnosed inflammatory necrotising myopathy. Striated muscle fibres with prominent myofibre size variation, including rounded myofibres, focal endomysial chronic inflammation (lymphocyte predominant), presence of necrotic myofibers and regenerative features, as well as patchy endomysial fibrosis and fatty replacement (perimysial>endomysial), were identified on haematoxylin and eosin (H&E) stained sections of the formalin-fixed, paraffinembedded ‘left deltoid’ muscle biopsy material. There was no evidence of vasculitis. The collective H&E features were suggestive of inflammatory myopathy. Further antibody testing through a myopathy panel revealed positive HMGCR antibody and PM SCL 75 (polymyositis/scleroderma 75) antibody presence (Table 3).
Therapeutic Interventions and Management
The management of the authors’ patient followed a multi-dimensional and comprehensive course starting with the withdrawal of statin therapy at presentation. The treatment was initiated by aggressive immunosuppression, which included the intravenous administration of methylprednisolone at 1,000 mg/day for 3 days. This was then followed by oral prednisone at a dose of 1 mg/kg/day. Close attention was paid towards monitoring her blood glucose levels, owing to her concurrent diabetes, and her medications were adjusted accordingly, thus mandating a close collaboration with the department of endocrinology. Simultaneously, supportive care was instituted, which included calcium supplementation at 1,200 mg/day with vitamin D supplementation. Blood pressure and glycaemic control were monitored closely, and medication was adjusted appropriately throughout this period.
The patient’s creatine kinase levels gradually decreased to 9,299 U/L, 9,671 U/L, and 8,466 U/L over the course of several days following treatment. However, the levels plateaued and did not normalise. As the disease course continued to progress, treatment was escalated to include secondline agents. Intravenous immunoglobulin (IVIG) therapy was started with a loading dose of 2 g/kg, given over a period of 5 days, and thereafter dosed monthly at 1 g/kg. Due to the necessity of long-term immunosuppressive therapy, steroid-sparing agent- mycophenolate mofetil was initiated at 500 mg twice-daily dosing and titrated stepwise based on clinical response and tolerability. It included regular monitoring of complete blood count and liver function throughout this period. Due to partial response to mycophenolate mofetil, another immunosuppressive agent was considered.
Biological therapy with rituximab was instituted after proper screening for hepatitis B. Before initiating rituximab, a thorough premedication regimen was instituted with the aim of minimising infusion reactions. Induction doses of 1,000 mg were administered at Weeks 0 and 2 intravenously. The maintenance schedule
Date
Day 0
Day 60
Day 98
Day 168
Onset of muscle weakness, difficulty standing, and ground-level falls.
Initial ER Visit: the patient presented with generalised muscle weakness, recurrent falls, and elevated CK (11,000 IU/L). Extensive investigations were done. The plan included high-dose IV methylprednisolone, fluid administration, EKG, and other workups.
Follow-up: imaging showed muscle oedema, and EMG indicated irritable myopathy, leading to a muscle biopsy that confirmed necrotising myopathy. Continued weakness persisted, prompting consideration for IVIG treatment.
Improved status: occasional night cramps, no subjective weakness. Objective strength still diminished. Prednisone taper started.
Day 350 Medication update: prednisolone was tapered further. MMF was started at 500 mg BID and subsequently increased to 1,000 mg BID. Rituximab was also added to the treatment regimen.
was based on clinical response and B cell monitoring.
Rehabilitation played an important role in this patient’s recovery. Physical therapy intervention started with a baseline functional assessment. This was then followed by the implementation of a graduated exercise programme, progressive resistive exercises, range of motion exercises, gait training, and balance exercises with muscle strengthening. Occupational therapy centred on assessing and modifying the person’s home, teaching energy conservation techniques, and training the individual in adaptive equipment use as required.
Long-term management was based on an individualised tapering schedule of immunosuppression, keeping watch for relapse of disease. It would demand a multi-disciplinary team care involving specialists from rheumatology, neurology, endocrinology, and physical medicine and rehabilitation. Education of the patient was detailed with information regarding the understanding of the disease, warning signs of disease relapse, adherence to medications, and activity modifications
appropriately. Lifestyle modification was provided in the form of exercise prescription, dietary counselling, stress management, and sleep hygiene.
Significant attention was also given to the in-place support systems. This included family education and involvement, contacting a patient support group if necessary, regular psychological assessment, and social work consultation where necessary. This holistic management approach ensured that all aspects of the condition, including both the physical and psychosocial features of the patient’s condition, were taken into account.
DISCUSSION
Statin-induced NAM is an uncommon yet significant adverse effect of statin treatment. Due to its infrequency, specific figures for its occurrence and frequency are not well established. It affects a small proportion of individuals who use statins, a frequently prescribed medication for controlling high cholesterol levels and reducing cardiovascular risk.3,5
3: Myositis panel results. (continued.)
Despite being rare, statin-induced NAM is a severe illness. It results in considerable morbidity due to severe muscle weakness and functional limitations. While the mortality directly caused by NAM is not well-defined, it significantly affects quality of life and can lead to disability. The longlasting muscle weakness and functional limitations in patients even getting proper care and treatment emphasise the persistent nature of the illness.
The condition, predominantly but not always triggered by statins, is identified by muscle weakness in the torso area, elevated serum creatine kinase levels, and myopathic patterns detected through electromyography.4,6,7 Some studies suggest a direct relation between creatine kinase levels and muscle strength.8 Clinical symptoms, ranging from myalgia to rhabdomyolysis,9 often improve alongside decreased levels of anti-HMGCR antibodies. For instance, after treatment, undetectable antibody levels were observed in four out of seven patients.10
The pathogenesis of statin-induced NAM includes an immune-related assault on muscle fibres, marked by present antibodies against HMGCR. Statins increase the production of HMGCR in damaged muscle cells, potentially initiating an autoimmune reaction in vulnerable individuals.3,5 Statininduced NAM is treated by stopping statins and starting immunosuppressive therapy. The initial treatment usually involves corticosteroids, sometimes combined with other immunosuppressants like methotrexate or azathioprine.6,7,11
The outlook for individuals with statininduced necrotising autoimmune myopathy can differ. While some patients experience a positive response to treatment, as shown by the disappearance of anti-HMGCR antibodies and improved muscle strength, others may continue to experience lasting disability, as evidenced by persistent functional limitations and reduced Karnofsky Performance Status (KPS) scores.10 On average, KPS scores decreased from 89/100 before diagnosis to 68/100 at the most recent follow-up assessment.
Additional research is critical for improving the management of statin-induced NAM. Prospective clinical trials are necessary to establish standardised treatment protocols and explore the long-term outcomes of different therapeutic approaches. Investigating the mechanisms underlying persistent functional impairments despite clinical remission could provide valuable insights into improved supportive care and rehabilitation methods. Furthermore, researchers can identify genetic or other biomarkers predicting susceptibility to statin-induced NAM and can make personalised prevention strategies for patients who are on statins.
The management of anti-HMGCR immunemediated necrotising myopathy is challenging, and standardised treatment protocols have not been established yet. Recent literature varies with the response rate to different therapeutic approaches. In a multicentre study on 100 patients with anti-HMGCR myopathy, 90% required multiple immunosuppressive agents for adequate disease control. Complete response to initial treatment with highdose corticosteroids at 1 mg/kg/day was achieved in only 25% of cases.7,8 Second-line agents, particularly IVIG, have been promising, with one retrospective study showing improvement in 85% of patients receiving combination therapy with IVIG and corticosteroids.11 Rituximab has become an effective treatment for refractory cases, demonstrating complete or partial response in 75% of patients who failed first-line therapy.11-13 Notably, early aggressive therapy does appear to correlate with improved results, as reported by Christopher-Stine et al.,6 in that patients treated with combination immunotherapy within the first 3 months of presentation had significantly more complete remissions (68% versus 30%) than those treated later.6 Long-term follow-up data suggested by Turrin et al.10 appear to suggest that about 40% of these patients remain well at 24 months, and 20% relapse into disease upon attempts to taper their immunosuppression.4,10 Studies have also focused on the role of exercise in reducing steroid dosage and improving the quality of life in patients with multiple subtypes of
idiopathic inflammatory myopathies.14 More research needs to be done specifically for statin-induced myopathies.
Patient Perspective
From the patient’s perspective, they expressed high satisfaction with the treatment and demonstrated consistent compliance, attributing their positive experience to an accurate diagnosis.
Informed consent
Informed consent was obtained from the patient prior to the commencement of the case report.
CONCLUSION
Statin-induced NAM is a rare but significant adverse effect of statin therapy, characterised by severe muscle weakness, elevated serum creatine kinase levels, and myopathic changes in electromyography. Despite its infrequency, the impact on affected individuals is substantial, often leading to considerable morbidity and functional limitations. Due to its autoimmune nature, marked by anti-HMGCR antibodies, there is a need for careful monitoring and management of patients on statins.
The standard treatment involves discontinuing statin use and initiating immunosuppressive therapy, primarily
References
1. Raychaudhuri S, Mitra A. Polymyositis and dermatomyositis: disease spectrum and classification. Indian Journal of Dermatology. 2012;57(5):366.
2. Pinal-Fernandez I et al. Immunemediated necrotizing myopathy. Current Rheumatology Reports. 2018;20(4):21.
3. Fink NS et al. Anti-HMGCR (hydroxy3-methylglutaryl-CoA reductase) myopathy: a rare cause of proximal muscle weakness. Cureus. 2024;16(5):e61094.
with corticosteroids, often supplemented with additional immunosuppressants like methotrexate or azathioprine. While some patients respond well to treatment, achieving remission and improvement in muscle strength, others continue to experience persistent disability and reduced quality of life, as reflected in decreased KPS scores.
THE WAY FORWARD
Further research is essential to enhance the management and understanding of statin-induced NAM. Prospective clinical trials are crucial for developing standardised treatment protocols and for evaluating the long-term efficacy and safety of different therapeutic regimens. Additionally, investigating the underlying mechanisms of apparent clinical remission with persistent functional impairments could lead to better supportive care and rehabilitation strategies.
Moreover, identifying genetic or other biomarkers that predict susceptibility to statin-induced NAM is critical. Such advancements could enable personalised prevention strategies, ensuring that patients at higher risk can be monitored more closely or offered alternative treatments. Ultimately, improving the management of statin-induced NAM will mitigate the adverse effects on patients’ quality of life and enhance the overall safety profile of statin therapy.
4. Mammen A L et al. Autoantibodies against 3-hydroxy-3-methylglutarylcoenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis & Rheumatism. 2011;63(3):713-21.
5. Kennedy N et al. HMGCR-associated myositis: a New Zealand case series and estimate of incidence. Internal Medicine Journal. 2016;46(5):622-5.
6. Christopher‐Stine L et al. A novel autoantibody recognizing 200‐kd and 100‐kd proteins is associated with an immune‐mediated necrotising myopathy. Arthritis & Rheumatism. 2010;62(9):2757-66.
7. Watanabe Y et al. (2016). Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. Journal of Neurology, Neurosurgery & Psychiatry. 2016;87(10):1038-44.
8. Allenbach Y et al. Immune-mediated necrotising myopathy: clinical features and pathogenesis. Nature Reviews Rheumatology. 2020;16(12):689-701.
9. Stanley M et al. Rhabdomyolysis [Internet] (2024). Treasure Island: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/ NBK448168/. Last accessed: 28 January 2025.
10. Turrin M. Statins and immunemediated necrotising myopathy. Clinical Management Issues. 2019;12(1).
11. Ramanathan S et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotising autoimmune myopathy.
Peritoneal tuberculosis (TB) is a form of extrapulmonary TB that primarily affects areas such as the omentum, liver, intestines, spleen, and female reproductive organs. Diagnosing peritoneal TB can be challenging, as its presentation often resembles that of advanced ovarian conditions. Among the estimated 10 million TB cases worldwide, India has the highest number, with 2.8 million cases. In this case, a 14-year-old female presented with abdominal pain, distention, weight loss, and amenorrhoea. She had elevated CA-125 levels and left-sided pleural effusion observed on X-ray. An ultrasound showed significant ascites, and a contrastenhanced CT scan of the abdomen and pelvis revealed widespread peritoneal thickening, marked ascites, and necrotic lymph nodes in the mesentery. A diagnosis of peritoneal TB was considered, and a biopsy of peritoneal deposits confirmed TB without malignancy. The patient was treated with anti-tubercular drugs, leading to substantial clinical improvement on follow-up. In cases presenting with massive ascites and high CA-125 levels without ovarian enlargement, clinicians should consider peritoneal TB as a potential diagnosis, alongside peritoneal carcinomatosis and advanced ovarian cancer, especially in resource-limited settings. Imaging is crucial in guiding diagnosis and narrowing differential options, while ultrasound-guided biopsy with histological analysis provides definitive confirmation.
Key Points
1. Peritoneal tuberculosis (TB) is a form of extrapulmonary TB that occurs in 1–2% of patients, with a higher incidence in developing countries.
2. This case report highlights the diagnostic importance, as well as the challenge, of distinguishing peritoneal TB from ovarian carcinoma to avoid misdiagnosis and ensure proper treatment. This distinction directly impacts the patient’s quality of life and long-term outcomes.
3. Peritoneal TB can cause remarkable elevation in serum CA-125 levels. Timely and accurate diagnosis is essential to ensure proper treatment, such as antitubercular therapy in peritoneal TB rather than surgical or oncological interventions.
INTRODUCTION
Peritoneal tuberculosis (TB) is a significant public health issue in regions where the disease is endemic, ranking as the sixth most prevalent form of extrapulmonary TB, constituting ~3% of all extrapulmonary TB globally and 11–13% of cases in India.1 Abdominal TB typically impacts the omentum, intestines, liver, spleen, female reproductive organs, and the parietal and visceral layers of the peritoneum.2
Diagnosing peritoneal TB is challenging due to its vague symptoms, which can sometimes be mistaken for gynaecological cancers, such as advanced ovarian carcinoma.3,4 The disease frequently presents with nonspecific abdominal or pelvic symptoms, including masses, ascites, and elevated cancer antigen (CA) 125 levels, which are also associated with advanced ovarian cancer, and overlapping of these symptoms can lead to diagnostic confusion.5 In 2009, the WHO estimated there were 9.4 million TB cases globally, with the majority occurring in developing regions.6
Abdominal TB generally manifests in four forms: visceral TB affecting solid organs, lymph nodal TB, gastrointestinal TB, and peritoneal TB.7,8 Peritoneal TB is often characterised by ascites, mesenteric adhesions, lymphadenopathy, septations within ascitic fluid, and involvement of the omentum, all of which closely resemble the manifestations of primary peritoneal carcinoma or advanced ovarian carcinoma.9 Serum CA 125 levels can be raised in both peritoneal TB and ovarian cancer cases.7
Diagnostic imaging, including ultrasonography and CT, is crucial for evaluating such cases.10 For differential diagnosis, abdominal TB with ascites should be considered when ovarian cancer is suspected due to its potential to mimic widespread ovarian carcinoma.11
CASE REPORT
A 14-year-old female presented with abdominal pain and distension, low-grade fever, reduced appetite, weight loss, and irregular menstrual cycles over 2 months. She was previously in good health with no known history of chronic illness or TB. There was no family history of cancer. Due to her worsening symptoms, she sought evaluation at a tertiary care hospital.
Physical examination revealed abdominal dullness and a doughy abdomen without tenderness or a palpable mass. Abdominal assessment revealed a slighty distended abdomen with normal bowel sounds. On palpation, a positive impression of muscular defence was obtained. Tenderness in the umbilical region was found, but there was no chessboard phenomenon. The liver and spleen were within normal limits.
Laboratory tests showed the patient was anaemic, with a haemoglobin level of 9.0 g/dL, reduced albumin levels at 2.8 g/dL, and thrombocytosis with a platelet count of 499,000/L. Her CA 125 level was elevated to 136.4 U/mL (normal range: <35 U/mL).
Imaging studies included an initial chest radiograph, which showed clear lung fields and left pleural effusion (Figure 1A). Abdominal and pelvic ultrasonography detected gross ascites, mild right hydronephrosis, left pleural effusion, omental thickening, necrotic mesenteric lymph nodes, and mild free fluid in the pouch of Douglas. The uterus, ovaries, and endometrium appeared normal on ultrasound (Figure 1B).
A contrast-enhanced CT scan of the abdomen and pelvis showed gross ascites, diffuse peritoneal thickening, clumping of bowel loops, mild right hydronephrosis, left pleural effusion, necrotic mesenteric lymphadenopathy, and mild free fluid in
1: Chest radiograph and ultrasound of abdomen and pelvis.
B
A) Chest radiograph showed minimal left pleural effusion (red arrows). B) Ultrasound of abdomen and pelvis showed gross ascites (yellow arrows), omental thickening (orange arrow), necrotic mesenteric lymph node (green arrow), and fluid in the pouch of Douglas. Uterus (yellow arrow), ovaries (blue arrow), and endometrium (violet arrow) appear normal.
2: Contrast-enhanced CT of the abdomen and pelvis.
Contrast-enhanced CT of the abdomen and pelvis showed gross ascites (red arrows), diffuse peritoneal thickening in bilateral iliac fossa (yellow arrows) and hypogastrium (orange arrows), clumping of bowel loops (blue arrows), mild right hydronephrosis (yellow arrow), moderate left pleural effusion (green arrow), necrotic mesenteric lymphadenopathy (violet arrow), and fluid in the pouch of Douglas (red arrow). Both ovaries (violet arrows) and uterus (yellow arrows) appeared normal.
Figure
Figure
the pouch of Douglas. Both ovaries and uterus appeared normal. Peritoneal thickening was observed in the bilateral iliac fossa and hypogastrium (Figure 2).
Differential diagnoses included peritoneal TB, peritoneal carcinomatosis, and advanced ovarian cancer. Pertaining to imaging findings and elevated CA 125 levels, peritoneal TB was considered likely. An ultrasound-guided biopsy was performed from the peritoneal thickening (Figure 3A). Histopathological analysis showed a granulomatous reaction typical of TB, including epithelioid granulomas with caseating necrosis, giant cells, and chronic inflammatory infiltrates, with no evidence of malignancy (Figure 3B).
Mycobacterium TB was isolated on AcidFast Bacilli smear. Thus, a diagnosis of peritoneal TB was confirmed.
The patient was enrolled in the Directly Observed Treatment Short-course (DOTS) programme and began receiving anti-tubercular medications. A follow-
up ultrasound after 1 month showed reduced ascites, resolution of the previous right hydronephrosis, and clearance of the left pleural effusion. The patient exhibited marked clinical improvement and completed 6 months of anti-TB treatment.
DISCUSSION
Abdominal involvement is the most common extrapulmonary manifestation of TB, accounting for about 5% of all TB cases worldwide.4 The gastrointestinal tract, peritoneum (e.g., ascites), lymph nodes, and solid organs (e.g., the liver, spleen, and pancreas) are common sites of involvement in abdominal TB.12 The diagnosis of abdominal TB is challenging, as it typically presents with nonspecific symptoms, making it difficult and timeconsuming to identify. However, with the advancement of imaging techniques like CT scans, it is now possible to detect lesions caused by chronic inflammation and distinguish them from malignancies.13
A) An ultrasound-guided biopsy (yellow arrow) was performed on the peritoneal thickening (blue arrow).
B) Histopathological analysis showed a granulomatous reaction typical of tuberculosis (dark blue arrows), epitheloid granulomas with caseating necrosis (yellow arrow), giant cells (red arrows), and chronic inflammatory infiltrates, with no evidence of malignancy.
Figure 3: Ultrasound-guided biopsy and histopathological analysis.
One rare manifestation of abdominal TB is tuberculous peritonitis, which occurs in less than 4% of patients with TB.14 Despite its rarity, it remains a significant cause of ascites in endemic regions. High-risk groups for developing TB include individuals with alcoholism, cirrhosis, renal failure, diabetes, malignancy, and immunodeficiencies such as AIDS.3,15 Symptoms of tuberculous peritonitis, such as abdominal distension, ascites, pelvic or adnexal masses, and elevated CA 125 levels are often similar to those seen in advanced ovarian carcinoma, and both conditions can present with fever and weight loss.12 In regions with high TB incidence, tuberculous peritonitis is an important cause of ascites.15 While tuberculous peritonitis and ovarian cancer are distinct conditions, they can be difficult to differentiate without proper diagnostic evaluation. Tuberculous peritonitis generally has a favourable prognosis with appropriate treatment, except in older or otherwise frail patients. Therefore, clinicians must be vigilant in considering TB as a possible diagnosis.16
Differentiating peritoneal TB from malignant abdominal tumours can be challenging, as they share the same symptoms.12,17
The spread of Mycobacterium TB from a pulmonary infection to the abdominal cavity through haematogenous dissemination is thought to contribute to peritoneal TB. The primary pulmonary focus typically heals without leaving clinical or radiological signs.8,10 Diffuse peritoneal diseases such as carcinomatosis, peritoneal lymphomatosis, malignant peritoneal mesothelioma, diffuse peritoneal leiomyomatosis, peritoneal TB, or IgG4-related disease present with similar imaging features, including nodular or diffuse peritoneal thickening, omental caking, and mesenteric invasion.1,18,19 Accurate early diagnosis is critical because the prognosis and treatment approach vary significantly between these conditions.20
Peritoneal carcinomatosis, for example, requires aggressive locoregional therapy, while tuberculous peritonitis may be managed effectively with anti-TB drugs.21 On imaging, peritoneal TB can resemble advanced ovarian cancer or other nontuberculous granulomatous diseases.19,22 Tuberculous peritonitis
can occur due to reactivation of latent TB foci in the peritoneum, haematogenous spread from a pulmonary infection, or ingestion of bacilli that spread via the intestinal mucosa and mesenteric lymph nodes.23 Another mechanism involves the contiguous spread of TB from the intestines or fallopian tubes.23 Peritoneal TB is categorised into three types based on the characteristics of the ascitic fluid: wet (free or loculated ascites), fixed fibrotic (omental and mesenteric masses with matted bowel loops), and dry plastic (thickened peritoneum and necrotic lymph nodes).22,24,25 Given the overlap in symptoms, a high index of suspicion is necessary to avoid unnecessary surgeries and ensure timely treatment.26
CA 125, a non-specific marker for peritoneal inflammation and a coelomic epithelial glycoprotein,28,30 is often elevated in conditions like ovarian carcinoma, endometriosis, and pelvic inflammatory disease, and it can also be elevated in peritoneal TB.29,30,32 Though CA 125 is used to monitor ovarian cancer treatment, its elevated levels are not definitive for malignancy.34 In some cases, CA 125 levels above 1,000 U/mL have been associated with malignancy, but there have also been instances of high CA 125 levels in peritoneal TB, making it unreliable as a sole diagnostic marker.31,32,33
A contrast-enhanced abdominal CT scan findings of peritoneal TB may include diffuse thickening of the peritoneum, nodules, and a ‘cake-like’ appearance of the omentum.36 Ascites are present in 70–90% of cases,27 and the mesenteric root fat planes may show densification with necrotic lymphadenopathy.35,36 Imaging techniques such as ultrasound, CT, and MRI can reinforce diagnosis. Ultrasound-guided tru-cut biopsy5 is a precious and minimally invasive method to obtain tissue for microbiological and histopathological analysis. A final diagnosis is typically made with microbiological and histopathological examination of biopsy samples, which reveal granulomas composed of epithelioid cells, lymphocytes, Langhans giant cells, and central caseous necrosis.37 Other than that, GeneXpert (Cepheid, Sunnyvale, California, USA) sputum and stool examinations, positive smear or culture from
peritoneal fluid, histological appearance of granuloma, acid resistant bacillus test, and adenosine deaminase levels of ascitic fluid. and it has high sensitivity and specificity in non-TB endemic areas.35-37 Treatment for extra-pulmonary TB, including abdominal TB, typically involves a 6-month course of anti-TB drugs such as isoniazid, rifampicin, pyrazinamide, and ethambutol.38
Correct diagnosis is crucial, as treatment for peritoneal TB differs from that for ovarian cancer. While peritoneal TB can be treated with anti-TB medications, ovarian cancer requires surgical intervention and, in some cases, chemotherapy.39 Accurate diagnosis can thus help avoid unnecessary surgeries and enable appropriate treatment, particularly in high-incidence TB regions such as India.5
Informed Consent
Written informed consent was obtained from the patient.
CONCLUSION
Peritoneal TB is a significant diagnostic challenge due to its ability to closely mimic other conditions, particularly ovarian malignancy. This case report highlights the masquerades of peritoneal TB, with elevated serum CA 125 levels further complicating the diagnosis. Clinicians must be vigilant and consider TB as a differential diagnosis, especially in endemic areas and among immigrant populations, as it can closely
References
1. Diop AD et al. CT imaging of peritoneal carcinomatosis and its mimics. Diagn Interv Imaging. 2014;95(9):861-72.
2. PC Hopewell, “Overview of clinical tuberculosis,” Bloom BR (eds.), Tuberculosis: Pathogenesis, Protection, and Control (1994), ASM Press, Washington, DC, USA.
3. Koc S et al. Peritoneal tuberculosis mimicking advanced ovarian cancer: a retrospective review of 22 cases. Gynecol Oncol. 2006;103(2):565-9.
4. Sharma JB et al. Abdomino-peritoneal tuberculosis masquerading as ovarian cancer: a retrospective study of 26 cases. Arch Gynecol Obstet.
resemble ovarian carcinoma. With appropriate treatment, peritoneal TB generally has a favourable prognosis. Early and accurate diagnosis can help avoid unnecessary surgeries, as pre-operative minimally invasive methods like ultrasound-guided biopsy can be utilised. Timely treatment and followup care are crucial for improving patient outcomes. Greater awareness and the use of radiological investigations are key to enabling early detection and effective management of this condition.
SIGNIFICANCE
Peritoneal TB is one of the most challenging forms of extrapulmonary TB to diagnose and requires more awareness because the clinical symptoms are nonspecific. Biological tests such as adenosine deaminase may be useful but are not always available in developing countries. Ultrasound and contrast-enhanced CT have the advantage of being cheaper, widely available, and easy to perform. In addition, it has high diagnostic cost-effectiveness and may be used in certain situations to guide peritoneal biopsy. No single test can effectively diagnose peritoneal TB, but a combination of history and radiological, immunologic, molecular, and cytologic tests are important. Clinicians need to consider peritoneal TB as a differential diagnosis in patients presenting with abdominal pain and distension, particularly in high TB-incidence countries such as India. A delay in the diagnosis and treatment of peritoneal TB may lead to worse clinical outcomes.
2010;282(6):643-8.
5. Oge T et al. Peritoneal tuberculosis mimicking ovarian cancer. Eur J Obstet Gynecol Reprod Biol. 2012;162(1):105-8.
6. Lönnroth K et al. Tuberculosis control and elimination 2010-50: cure, care, and social development. Lancet. 2010;375(9728):1814-29.
7. Thomas A et al. Abdominal tuberculosis mimicking ovarian cancer: a diagnostic dilemma. J Obstet Gynaecol India. 2020;70(4):304-309.
8. Debi U et al. Abdominal tuberculosis of the gastrointestinal tract: revisited. World J Gastroenterol. 2014;20(40):14831-40.
9. Maheshwari A et al. Clinical and laboratory characteristics of patients with peritoneal tuberculosis mimicking advanced ovarian cancer. South Asian J Cancer. 2021;10(2):102-6.
10. Purbadi S et al. Peritoneal tuberculosis mimicking advanced ovarian cancer case report: Laparoscopy as diagnostic modality. Int J Surg Case Rep. 2021;88:106495.
11. Gosein MA et al. Peritoneal tuberculosis mimicking advanced ovarian carcinoma: an important differential diagnosis to consider. BMC Res Notes. 2013;6:88.
12. Bilgin T et al. Peritoneal tuberculosis with pelvic abdominal
mass, ascites and elevated CA 125 mimicking advanced ovarian carcinoma: a series of 10 cases. Int J Gynecol Cancer. 2001;11(4):290-4.
13. Lingenfelser T et al. Abdominal tuberculosis: still a potentially lethal disease. Am J Gastroenterol. 1993;88(5):744-50.
15. Na-ChiangMai W et al. CT findings of tuberculous peritonitis. Singapore Med J. 2008;49(6):488-91.
16. Chow KM et al. Tuberculous peritonitis-associated mortality is high among patients waiting for the results of mycobacterial cultures of ascitic fluid samples. Clin Infect Dis. 2002;35(4):409-13.
17. Bulut Gökten D et al. A case report of peritoneal tuberculosis: a challenging diagnosis. Case Rep Infect Dis. 2018;2018:4970836.
18. Vadi SK et al. IgG4-Related disease simulating carcinoma colon with diffuse peritoneal carcinomatosis on 18F-FDG PET/CT. Clin Nucl Med. 2018;43(7):e247-49.
19. Hedgire SS et al. The spectrum of IgG4-related disease in the abdomen and pelvis. AJR Am J Roentgenol. 2013;201(1):14-22.
20. Sia DS et al. Peritoneal lymphomatosis mimicking peritoneal carcinomatosis: important imaging clues for correct diagnosis. Singapore Med J. 2013;54(4):e93-6.
21. Pickhardt PJ, Bhalla S. Primary neoplasms of peritoneal and sub-peritoneal origin: CT findings. Radiographics. 2005;25(4):983-95.
22. Balachandran A, Silverman PM, “Mesenteric and Omental lesions,” Gore RM, Levine MS (eds.) Textbook of Gastrointestinal radiology (2007) 3rd ed. Saunders vol. 2, pp.2139-43.
23. Vaid U, Kane GC. Tuberculous peritonitis. Microbiol Spectr. 2017;5(1):10.1128/microbiolspec. tnmi7-0006-2016.
24. Van Crevel R, Hill PC. Tuberculosis. Infect Dis. 2017;2:271-84.e1.
25. Akhan O, Pringot J. Imaging of abdominal tuberculosis. Eur Radiol. 2002;12(2):312-23.
26. Mahdavi A et al. Peritoneal tuberculosis disguised as ovarian cancer: an emerging clinical challenge. Gynecol Oncol. 2002;84(1):167-70.
27. Kiu MC et al. Elevated serum CA-125 in tuberculous peritonitis: report of a case. J Formos Med Assoc. 1994;93(9):816-8.
28. Spitzer M et al. Maternal CA-125 levels in pregnancy and the puerperium. J Reprod Med. 1998;43(4):387-92.
29. Zakrzewska I . Antygen CA125 [CA-125 antigen]. PostepyHig Med Dosw 2002;56(1):29-38.
30. Moss EL, Hollingworth J, Reynolds TM. The role of CA125 in clinical practice. J Clin Pathol. 2005;58(3):308-12.
31. Markman M. The Role of CA-125 in the Management of Ovarian Cancer.
Oncologist. 1997;2(1):6-9.
32. Le Thi Huong Du et al. Spécificité du marqueur tumoral CA 125. Etude de 328 observations de médecine interne [Specificity of CA 125 tumor marker. A study of 328 cases of internal medicine]. Presse Med. 1988;17(43):2287-91.
33. Piura B et al. Peritoneal tuberculosis mimicking ovarian carcinoma with ascites and elevated serum CA-125: case report and review of literature. Eur J Gynaecol Oncol. 2002;23(2):120-2.
34. da Rocha EL et al. Abdominal tuberculosis: a radiological review with emphasis on computed tomography and magnetic resonance imaging findings. Radiol Bras. 2015;48(3):181-91.
35. Lee DH et al. Sonographic findings in tuberculous peritonitis of wet-ascitic type. Clin Radiol. 1991;44(5):306-10.
36. Koff A, Azar MM. Diagnosing peritoneal tuberculosis. BMJ Case Rep. 2020;13(2):e233131.
37. Dasgupta A et al. Abdominal tuberculosis: a histopathological study with special reference to intestinal perforation and mesenteric vasculopathy. J Lab Physicians. 2009;1(2):56-61.
38. Jullien S et al. Six-month therapy for abdominal tuberculosis. Cochrane Database Syst Rev. 2016;11(11):CD012163.
39. Agha RA et al; SCARE Group. The SCARE 2020 Guideline: Updating Consensus Surgical CAse REport (SCARE) Guidelines. Int J Surg. 2020;84:226-30.
Prolonged Premature Preterm Rupture of Membranes: A Successful Case From the Dominican Republic
Authors: Megan Dorame,1 *Rahina Sheikh2
1. Departments of Anthropology and Public Health, University of Minnesota Twin-Cities, USA
2. Department of Public Health, University of Nevada Las Vegas, Nevada, USA
*Correspondence to Rahina.sheikh123@gmail.com
Disclosure: The authors have declared no conflicts of interest.
Acknowledgements: The authors would like to acknowledge the Global Health Leaders program, especially the research advisor, for providing guidance, tools, and resources to write the case study. Thank you to the hospital and medical team for releasing information about this significant case. The medical staff obtained informed, written consent from the patient for the publication of this case report and any accompanying images.
Preterm premature rupture of the membranes (PPROM) impacts 5–7% of all pregnancies. A pregnancy is considered previable, the fetus having a low chance of surviving before 22 weeks of gestation. Preterm birth is the leading cause of neonatal mortality. PPROM is associated with health complications for the neonates and mothers. Some of the neonatal morbidities include respiratory distress syndrome, infection, pulmonary hypoplasia, and sepsis. Some of the maternal complications are chorioamnionitis, endometritis, and placental abruption. This case study describes a clinical case from the Dominican Republic where there was a PPROM that occurred at 16 weeks of gestation. Furthermore, the latency period from the initial rupture to preterm birth was approximately 18 weeks, which is longer than the average latency period. Despite these complications, the patient successfully gave birth at 33.6 weeks to a relatively healthy baby.
Key Points
1. Prolonged preterm premature rupture of membranes (PPROM) affects 5–7% of pregnancies and is a major contributor to both neonatal and maternal health complications.
2. A case-control study was conducted to examine a clinical case from the Dominican Republic where PPROM occurred at 16 weeks gestation, with a prolonged latency of 18 weeks.
3. Despite complications, effective PPROM management enabled the patient to deliver a healthy baby. Further research is needed to understand PPROM prevalence in the Dominican Republic and evaluate current treatment and management strategies.
INTRODUCTION
This case study describes a clinical case from the Dominican Republic where there was a preterm premature rupture of membranes (PPROM) that occurred at 16 weeks of gestation. It is important to note that there is a lack of specific PPROM statistics and information from the Dominican Republic.1 A pregnancy is determined to be previable when the fetus is at a stage of maturity where it has a low chance of surviving outside of the uterus. Generally, anything before 22 weeks of gestation is thought to be nonviable.2 PPROM occurs in approximately 5–7% of all births,3 but PPROM occurring “near the limit of fetal viability”4 only complicates around 0.04% of pregnancies and has a higher morbidity.4 The cause for the premature rupture is unknown, but there are several risk factors, such as history of prematurity, maternal age (adolescence or being over the age of 35 years), decreased BMI, multiple pregnancies, and genital infections.5 There are many complications associated with PPROM for both the neonate and the mother. In neonates, some of the common morbidities include respiratory distress syndrome (RDS), infection, pulmonary hypoplasia, arterial hypotension, bronchopulmonary dysplasia (BPD), and sepsis.4 Some of the maternal complications are chorioamnionitis, endometritis, and placental abruption.4 There are two management options for mothers with PPROM, expectant management and the termination of pregnancy (TOP). Mothers who chose the expectant management option have a 60.2% chance of developing maternal morbidity, which is 3.47 times more likely than those who chose TOP.6 Globally, complications from preterm birth before 37 weeks are the leading cause of neonatal mortality.7 The literature on PPROM shows varying survival rates for infants from approximately 20% to 56%. However, in instances where PPROM
occurred before 20 weeks of gestation, the survival rate is around 18%.8 Moreover, only approximately 15.7% of cases where the neonate survives to discharge the mother does not experience any kind of morbidity.6
Although there has been a steady decline in preterm births, preterm neonates have a higher mortality rate in developing nations.9 Sociodemographic factors that increase the risk of PPROM include low socioeconomic status, lack of education, and race/ethnicity.5 Lower socioeconomic status and poverty severely restrict access to health services, including prenatal and newborn care, nutritious foods, and other vital amenities. Moreover, those with lower incomes are more likely to have difficult working conditions, such as strenuous and labour-intensive jobs with long working hours, which is associated with PPROM.5
This clinical case is significant because both the mother and infant were determined to be relatively healthy after birth despite PPROM occurring at 16 weeks before the fetus was deemed to be viable and the presence of anhydramnios. Furthermore, the latency period between the initial PPROM and birth was approximately 18 weeks, which is much longer than the average latency period. There is a lack of information and statistics on PPROM from the Dominican Republic, which makes this clinical case that much more important.
CASE
The patient is a 32-year-old woman from the Dominican Republic. This is the patient’s first pregnancy and has no reported history of toxic habits, surgical, or medical history. However, it is noted that the patient has A RH-positive blood. The patient had their first menstruation at 12 years old and had a regular cycle that typically lasted 4 days following the first occurrence.
The patient went to the emergency room after experiencing transvaginal fluid leakage for 3 hours. During the initial physical examination, 12 weeks sonography was extrapolated with a caudal cephalic length of 5.4 cm, dating a gestational age of 16 weeks. The results of the initial physical examination showed that the patient’s head, neck, chest, heart, genitalia, and extremities had no pathologies. An examination of the lungs revealed the presence of a vesicular murmur and no pathologic rales. The examination of the patient’s abdomen documented that it was globose at the expense of adipose tissue and pregnant uterus, had adequate peristalsis, indeterminate at Leopold’s maneuvers, uterine height 17 cm, no uterine dilation, fetal heart rate 149, depressible, manageable, and not painful on palpation. The speculoscopy showed that the patient had a cervix of normal appearance and configuration, and outflow of fluid in slight amounts with the performance of the Valsalva maneuver, obtaining sample for arborization test (Figure 1A). The vaginal examination determined the patient had a normothermic vagina; posterior cervix, 2 cm long, permeable to the external cervical os; the bony pelvis had a sacral promontory that was not palpable, divergent spines, and a broad subpubic arch. The patient had weekly
follow-up appointments to check on the patient’s and the fetus’ progress following the positive diagnosis with PPROM.
During the diagnostic assessment, an arborization test, sonography, and physical examination were performed to determine the prognosis. Expectant management was considered on an outpatient basis, and frequent laboratory tests were required. These laboratory tests were performed during the assessment period and continued to be performed during the monitoring period until birth: blood count, glycaemia, urine test, C-reactive protein, culture of vaginal secretion, and urine culture (Table 1 and 2).
Upon admission at 16 weeks, the patient was given 875 mg of amoxicillin and 125 mg of clavulanic acid to take every 8 hours for a period of 48 hours. From 16–24 weeks, the patient was given 300 mg of ferrous sulfate, 5 mg of folic acid, and 600 mg of calcium to take every 24 hours. At the 24-week consultation, the patient was admitted to the hospital and the medical team administered intravenous ampicillin and erythromycin every 6 hours for 48 hours. This was followed by oral amoxicillin and erythromycin every 8 hours for 5 days. Additionally, the medical team administered
A) Arborization test results. The crystallisation present in the arborization test indicates a positive test result. This confirms the occurrence of a rupture of membranes. B) Fetal Doppler Test. Index of Pulsality for the Pulmonary Artery in 3.15 seconds.
Figure 1: Arborization test results and a fetal Doppler test.
Pulmonary artery
Table 1: First, second, and third trimester results.
HIV negative, VDRL negative, HBSAG negative, HVC negative
HIV negative, VDRL negative, HBSAG negative, HVC negative
Fosfomycin-sensitive klebsiella SPP
6 mg of dexamethasone intramuscularly every 12 hours for 48 hours to induce fetal lung maturity. Following this admission, weekly follow-ups were scheduled where blood count was measured, and urine reactive test and obstetrical sonography were conducted. At 30 weeks, a vaginal discharge culture tested positive for Klebsiella, the patient was treated with fosfomycin for 7 days and repeated the control. A biophysical profile was measured every week from 28 weeks onwards, and a fetal doppler with pulmonary artery pulsatility index was done at 33 weeks (Figure 1B).
At 33.6 weeks, the patient was admitted to the hospital centre and presented a pattern of three moderate contractions in 10 minutes. A vaginal examination revealed a normothermic vagina; a posterior cervix, 1.5 cm long; the bony pelvis spines were not prominent, had a wide subpubic arch, and was non-palpable. A diagnosis of threatened preterm labour and incorrect pelvic presentation was given. The patient gave birth at 33.6 weeks via a caesarean section and was found to have anhydramnios. The preterm newborn was developmentally appropriate for a gestational age of 34 weeks. The newborn was a male, with a head circumference of 34 cm, thoracic perimeter of 32 cm, was 45 cm long, had an Apgar score of 8/9, and weighed 2,760 g at birth. The newborn
HIV negative, VDRL negative, HBSAG negative, HVC negative
spent 2 weeks inpatient in the perinatology admission area and was discharged in stable condition. A follow-up for both the mother and baby was conducted 2 months later by the obstetrics department.
DISCUSSION
Premature rupture of membranes in pregnancy complicates 0.04% of pregnancies.4 It may be spontaneous or follow an invasive procedure such as amniocentesis or fetal surgery. Significant maternal complications related to premature rupture of membrane in previable pregnancy include chorioamnionitis, genital infections, sepsis, placental abruption, and retained placenta. With complications, such as bleeding, cramping, or fever, abortion is considered inevitable, and the uterus is evacuated. However, in cases without these complications, expectant management is an option. Neonatal morbidity is related to prematurity. In the clinical case, this patient experienced premature rupture of the membranes at 16 weeks, with a latency period of 18 weeks and anhydramnios.
In the Dominican Republic, medical facilities generally follow WHO guidelines for the treatment and management of PPROM. Larger hospitals in metropolitan areas are fully equipped, so they are
Table 2: Infection test results.
HBSAG: hepatitis B surface antigen; HVC: hepatitis C virus; VDRL: venereal disease research laboratory.
more likely to follow general guidelines. However, practices may differ from those in rural communities with fewer resources. Notably, there is a lack of research on the management of previable premature rupture in these settings. In this case, the hospital staff were thorough in providing care and routinely conducting follow-up care. The medical staff followed general clinical guidelines for PPROM.10 Due to prolonged rupture coupled with a pelvic presentation that threatened preterm labour, the patient gave birth before reaching full term. At 24 weeks, the medical team initiated the Mercer Protocol, which involves administering intravenous ampicillin and erythromycin followed by oral amoxicillin and erythromycin. The staff also administered dexamethasone, an antenatal corticosteroid therapy for fetal lung maturation that is widely prescribed and recommended by WHO PPROM guidelines.11 Corticosteroids have been shown to reduce the probability of severe neonatal outcomes and not increase the risk of infection for the mother or fetus.11 Generally, the administration of antenatal corticosteroids is recommended and should be offered to PPROM patients who have no signs of infection from 24 weeks gestation onwards.10,11 It should also be noted that the WHO PPROM guidelines caution against the use of antenatal corticosteroids in prolonged PPROM cases and when characteristics of sepsis are present.11 At 16 weeks, the patient was given amoxicillin plus clavulanic acid 875 and 125 mg every 8 hours for 48 hours. The administration of both amoxicillin and clavulanic acid is not widely agreed upon. In some PPROM cases, if uterine dilation
References
1. Pan American Health Organization. Perinatal information system. Available at: https://www.sipplus.org/. Last accessed: 23 June 2024.
2. Gkiougki E et al. Periviable birth: a review of ethical considerations. Hippokratia. 2021;25(1):1-7.
3. Herzlich J et al. Neonatal outcomes in women with preterm premature rupture of membranes at periviable gestational age. Sci Rep. 2022;12(1):11999.
has not started, administering both drugs is considered beneficial because the combination is able to treat a wider range of microorganisms.12 However, in other cases, it is not recommended because of an increased risk of developing necrotising enterocolitis (NEC), a severe intestinal disease in neonates, and instead the use of erythromycin is advised.11,13 An additional recommendation to reduce neonatal morbidity is to consider other options for fetal lung maturation therapies instead of prescribing dexamethasone. This is recommended because of the 18-week latency period between the rupture of membranes and birth in this case.
Given the lack of current statistics on the prevalence and management of PPROM in the Dominican Republic, the level of significance of this case is yet unknown. When looking at the current statistics available worldwide, this case does show clinical significance due to its success. In order to gain a better understanding of the level of its significance in the Dominican Republic, it is necessary to conduct further research. This case study is noteworthy not only for its success but also for its unique nature, with the potential to enhance neonatal and maternal health outcomes.
ETHICAL CONSIDERATIONS
There were no ethical issues when conducting the case study. Informed, written consent was given by the medical staff and there was no personal, identifiable information linking the case to the patient.
4. Kraft K et al. Pre-viable preterm premature rupture of membranes under 20 weeks of pregnancy: a retrospective cohort analysis for potential outcome predictors. Eur J Obstet Gynecol Reprod Biol. 2022;278:177-82.
5. Bouvier D et al. Risk factors and outcomes of preterm premature rupture of membranes in a cohort of 6968 pregnant women prospectively recruited. J Clin Med. 2019;8(11):1987.
6. Sklar A et al. Maternal morbidity
after preterm premature rupture of membranes at <24 weeks’ gestation. Am J Obstet Gynecol. 2022;226(4):558.e1-11.
7. World Health Organization (WHO). Preterm birth. 2023. Available at: https://www.who.int/news-room/ fact-sheets/detail/pretermbirth#:~:text=Key%20facts, deaths%20in%202019%20(2). Last accessed: 16 June 2024.
8. Gauthier‐Moulinier H et al. Outcomes of pregnancies with preterm premature rupture of membranes
occurring before 24 weeks of gestation: an 11‐year observational study. Int J Gynaecol Obstet. 2023;162(2):590-5.
9. Díaz-Rodríguez A et al. Risk factors associated with preterm birth in the Dominican Republic: a case-control study. BMJ. 2021;11(12):e045399.
10. Thompson AJ.; Royal College of Obstetricians and Gynaecologists. Care of women presenting with
suspected preterm prelabour rupture of membranes from 24+0 weeks of gestation. BJOG. 2019;126(9):e15266.
11. World Health Organization (WHO). WHO recommendations on interventions to improve preterm birth outcomes. 2015. Available at: https:// iris.who.int/bitstream/handle/10665/ 183037/9789241508988_eng.pdf? sequence=1. Last accessed: 14 July 2024.
12. Fetal Medicine Barcelona. Rotura Prematura De Membranas A Término Y Pretérmino. Available at: https:// fetalmedicinebarcelona.org/wpcontent/uploads/2024/02/roturaprematura-de-membranas-hcp-hsjd. pdf Last accessed: 14 July 2024.
13. Bohlițea RE et al. Expectant management of PPROM improves neonatal outcome—a retrospective study of 562 patients. J Clin Med. 2021;11(1):214.