
Review of the
ESMO Congress 2025
Editor’s Pick:
Integrated Multi-omics Approaches in Non-small Cell Lung Cancer for Biomarker and Pathway Discovery

Interviews:
Komal Jhaveri and ESMO President, Fabrice André, discuss the current and future landscape of breast cancer

10 Review of the European Society for Medical Oncology (ESMO) Congress 2025, 17th-21st October 2025
Congress Features
23 Expanding the First-Line Option in Triple-Negative Breast Cancer: Pivotal Trials of Datopotamab Deruxtecan and Sacituzumab Govitecan at the European Society for Medical Oncology (ESMO) Congress 2025
François Cherifi
27 AI and Cancer Care
Helena Bradbury
31 The New Frontiers of Personalised Cancer Prevention
Katie Wright Symposium Reviews
36 Emerging Evidence-Based Treatment Strategies in Metastatic Castration-Resistant Prostate Cancer
47 Navigating a Dynamic Treatment Landscape: Established Therapies and Emerging Modalities for Patients with Lung Cancer
58 Optimising Patient Care: Cutting-Edge Nutritional Strategies in Oncology
71 Exploring New Horizons and Emerging Topics in Squamous Anal Cancer and Colorectal Cancer
82 Abstract Highlights Congress Interview
89 Fabrice André
95 Komal Jhaveri
Infographic
100 The Evolving Biomarker Landscape in GI Cancers
Articles
102 Editor's Pick: Integrated Multi-omics Approaches in Non-small Cell Lung Cancer for Biomarker and Pathway Discovery
Sahli M
116 Immune Checkpoint Inhibitor-Associated Hydropneumothorax: A Rare Case Report with Histopathologic Insights
Fernandez J et al.
124 Recurrent and Aggressive Solitary Plexiform Neurofibroma with KRAS and AKT1 Alterations: A Case Report
Espinoza IRG et al.
133 Favourable Response of Unresectable Giant Pinealoblastoma After Induction Chemotherapy and Craniospinal Radiotherapy: A Case Report
Laraichi R et al.
140 Immunotherapeutic Strategies Based on CAR-T Cells in Hepatocellular Carcinoma
Hemmati N
154 Aggressive Angiomyxoma of Vulva with Term Pregnancy: A Case Report
Khan FH et al.

"Year on year, the society continues to grow and lead innovations in oncology, prioritising education, scientific dissemination, and supporting members in their journey"
Editorial Board








Editor-in-Chief
Prof Ahmad Awada
Chirec Cancer Institute, Brussels, Belgium
Head of the Oncology Department and Director of the Chirec Cancer Institute in Brussels, Belgium. With over 35 years of experience in the field, he is a renowned and trusted expert.
Dr Divyanshu Dua
Canberra Hospital, Australia
Dr Caroline Michie
Edinburgh Cancer Centre & University of Edinburgh, UK
Dr Aniket Mohite
Novo Solitaire Care and Jhangir Hospital, India
Dr Mohammad Akheel
Greater Kailash Hospitals, India
Dr Jyoti Dabholkar
King Edward Memorial Hospital and Seth Gordhandas Sunderdas Medical College, India
Dr Jad Degheili
Ibn Sina Hospital, Kuwait
Prof Dr Yves Chalandon
Geneva University Hospitals, Switzerland
Dr Javier Cortés
International Breast Cancer Center, Spain
Prof Paul Dent
Virginia Commonwealth University, USA
Dr Abdulmajeed Hammadi
Alyermook Teaching Hospital, Iraq
Prof Antoine Italiano
Institut Bergonié, France
Dr Katarazyna Rygiel
Medical University of Silesia (SUM), Poland
Dr Francesco Sclafani
Institut Jules Bordet, Belgium
Dr Klaus Seiersen
Aarhus University Hospital, Denmark
Prof Yong Teng
Georgia Cancer Center, USA
Aims and Scope
EMJ Oncology is an open access, peer-reviewed ejournal committed to helping elevate the quality of practices in interventional cardiology globally by informing healthcare professionals on the latest research in the field.
The journal is published annually, six weeks after the European Society for Medical Oncology (ESMO) Congress, and features highlights from this event, alongside interviews with experts in the field, reviews of abstracts presented at the congress, as well as in-depth features on sessions from this event. The journal also covers advances within the clinical and pharmaceutical arenas by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ Oncology also publishes peer-reviewed research papers, review articles, and case reports in the field. In addition, the journal welcomes the submission of features and opinion pieces intended to create a discussion around key topics in the field and broaden readers’ professional interests. EMJ Oncology is managed by a dedicated editorial team that adheres to a rigorous double-blind peer-review process, maintains high standards of copy editing, and ensures timely publication.
EMJ Oncology endeavours to increase knowledge, stimulate discussion, and contribute to a better understanding of practices in the field. Our focus is on research that is relevant to all healthcare professionals in this area. We do not publish veterinary science papers or laboratory studies not linked to patient outcomes. We have a particular interest in topical studies that advance knowledge and inform of coming trends affecting clinical practice in oncology.
Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
Peer Review
On submission, all articles are assessed by the editorial team to determine their suitability for the journal and appropriateness for peer review.
Editorial staff, following consultation with either a member of the Editorial Board or the author(s) if necessary, identify three appropriate reviewers, who are selected based on their specialist knowledge in the relevant area.
All peer review is double blind. Following review, papers are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected.
Editorial staff have final discretion over any proposed amendments.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields across Europe.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®; selected articles are indexed in PubMed Central®.
EMJ is available through the websites of our leading partners and collaborating societies. EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
This Publication
Launch Date: 2013
Frequency: yearly Online ISSN: 2054-619X
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. EMJ is completely independent of the review event (ESMO 2025) and the use of the organisations does not constitute endorsement or media partnership in any form whatsoever. The cover photo is of Berlin, Germany, the location of ESMO 2025.
Front cover and contents photograph: Brandenburg, Germany © Daisha / stock.adobe.com

Editorial Director
Andrea Charles
Managing Editor
Darcy Richards
Copy Editors
Noémie Fouarge, Sarah Jahncke
Editorial Leads
Helena Bradbury, Ada Enesco
Editorial Co-ordinators
Bertie Pearcey, Alena Sofieva
Editorial Assistants
Niamh Holmes, Katrina Thornber, Katie Wright, Aleksandra Zurowska
Creative Director
Tim Uden
Design Manager
Stacey White
Senior Designers
Tamara Kondolomo, Owen Silcox
Creative Artworker
Dillon Benn Grove
Designers
Shanjok Gurung, Fabio van Paris
Junior Designer
Helena Spicer
Head of Marketing
Stephanie Corbett
Business Unit Lead
Nabihah Durrani
Project Manager
Aghattha Villaflor
Chief Executive Officer
Justin Levett
Chief Commercial Officer
Dan Healy
Founder and Chairman
Spencer Gore
Welcome
Dear Readers,
We are delighted to welcome you to the 2025 issue of EMJ Oncology, bringing you up to date with the key highlights from this year’s European Society for Medical Oncology (ESMO) Congress, which took place in Berlin, Germany. With a record number of attendees and a programme covering the breadth of the field, from new diagnostics and emerging treatments to early diagnosis and prevention, the event was a hub of excitement and insights.
Within this issue, you can find our event coverage, alongside a timely feature exploring the pivotal trial data updates for expanding first-line treatment in triple-negative breast cancer that were presented. Plus, don’t miss our exclusive interview with the ESMO Congress President, Fabrice André, who discusses the Society’s main goals, current challenges in research and patient care, targeted therapies, and much more!
Among our peer-reviewed content, you will find a comprehensive review article summarising emerging T cellbased immunotherapies for hepatocellular carcinoma, as well as several interesting case reports, and an infographic focusing on the evolving diagnostic, prognostic, and predictive biomarker landscape of gastrointestinal cancers.
We would like to take this opportunity to thank our Editorial Board, authors, peer reviewers, and interviewees who have contributed to the creation of this issue. We hope you enjoy reading and find useful takeaways for your day-to-day clinical practice and beyond!
Contact us
Editorial enquiries: editor@emjreviews.com
Sales opportunities: salesadmin@emjreviews.com
Permissions and copyright: accountsreceivable@emjreviews.com
Reprints: info@emjreviews.com Media enquiries: marketing@emjreviews.com


Foreword
Welcome to the latest issue of EMJ Oncology, where we present a focused selection of the most compelling developments shaping cancer research and clinical practice in 2025. This edition offers a concise blend of peer-reviewed research, expert insights, and congress highlights that collectively reflect the rapid evolution of oncology.
We are pleased to feature interviews with two leading voices in oncology: Fabrice André, President of the European Society for Medical Oncology (ESMO), and Komal Jhaveri, an internationally recognised expert in breast cancer and drug development.
This issue includes six diverse, peerreviewed articles spanning rare case reports and forward-looking reviews. They include a rare presentation of immune checkpoint inhibitor-associated hydropneumothorax; a recurrent, aggressive, solitary plexiform neurofibroma with KRAS and AKT1 alterations; and a compelling case of unresectable giant pinealoblastoma responding favourably to induction chemotherapy followed by craniospinal radiotherapy. We also highlight a review of CAR-T cell-based immunotherapeutic strategies in hepatocellular carcinoma, a clinically significant case of aggressive angiomyxoma in term pregnancy, and a multi-omics exploration in non-small cell lung cancer aimed at advancing biomarker and pathway discovery.


While our infographic offers a concise and accessible overview of one of the fastest-moving areas in cancer diagnostics and treatment, this issue also includes a dedicated review of the 2025 ESMO Congress, with features detailing the role of ChatGPT (OpenAI, San Francisco, California, USA) and AI in cancer care, emerging advances in personalised cancer prevention, and pivotal data expanding first-line treatment options in triple-negative breast cancer.
This edition offers a concise blend of peer-reviewed research, expert insights, and congress highlights that collectively reflect the rapid evolution of oncology
My sincere thanks to the EMJ team, the Editorial Board, our authors, interviewees, and peer reviewers for their invaluable contributions. I hope you enjoy reading!
Prof Ahmad Awada Head of the Oncology Department and Director of Chirec Cancer Institute, Brussels, Belgium
ESMO 2025

Year on year, the society continues to grow and lead innovations in oncology, prioritising education, scientific dissemination, and supporting members in their journey
Review of the European Society for Medical Oncology (ESMO) Congress 2025 Congress Review
Location: Berlin, Germany
Date: 17th–21st October 2025
Citation: EMJ Oncol. 2025;13[1]:10-22.
https://doi.org/10.33590/emjoncol/GBXP9968
THE EUROPEAN Society for Medical Oncology (ESMO) Congress
2025 took place in Berlin, Germany, a dynamic epicentre known for its blend of innovation, culture, and historical significance. Home to approximately 3.7 million people, Berlin is Germany’s largest and most diverse city, offering a vibrant backdrop for an international gathering of oncologists, researchers, and healthcare professionals.
Fabrice André, the ESMO President, opened the Congress, highlighting the society’s 50th anniversary. Year on year, the society continues to grow and lead innovations in oncology, prioritising education, scientific dissemination, and supporting members in their journey, now with over 45,000 members. André highlighted the five integral pillars of ESMO: new drugs, strategies of treatment and care, delivery of care, toxicity management, and prevention. Looking to future initiatives, he spotlighted the society’s desire to simplify clinical research, revitalise academic research, and develop individuals’ careers. He stressed the need to invest in personalised prevention and post-cancer care, as well as fostering responsible integration of AI and digital tools, and developing a health economics approach.
So, what are the highlights of this year’s programme? Scientific Co-Chairs Myriam Chalabi, Netherlands Cancer Institute, Amsterdam, the Netherlands; and Toni Choueiri, Dana-Farber Cancer Institute, Boston, Massachusetts, USA, took the stage to outline them. The overarching theme of this Congress was one of broken records. A record 5,677 abstracts
were submitted, over 600 more than last year’s 5,030. Attendees also reached an all-time high at 35,676, a steep increase from 33,830 the previous year. Of the 5,677 submitted abstracts, 2,926 were accepted for presentation across presidential symposia (12), proffered papers (158), mini orals (213), posters (1,894), and ePosters (649). In total, the programme featured 2,181 sessions, including three major presidential symposia showcasing practice-changing, practice-informing, and forward-looking research. Read on for an in-depth coverage of the presentations in the presidential symposia.
During the opening ceremony, several prestigious awards recognised individuals for their leadership and contributions to oncology. Thierry Conroy, University of Lorraine, Nancy, France, recipient of the ESMO Award, spoke on the progress made in pancreatic cancer research, improving pancreatic cancer’s 5-year survival from under 3% in 2000 to 13% in 2025. Christina Curtis, Stanford University, Palo Alto, California, USA, recipient of the ESMO Award for Translational Research, presented
on harnessing AI to enhance the prediction of cancer progression and personalise treatment. Rolf Stahel, President of the European Thoracic Oncology Platform –International Breast Cancer Study Group (ETOP IBCSG) Partners Foundation, who received the ESMO Lifetime Achievement Award, reflected on his longstanding commitment to mentorship, teamwork, and the development of initiatives such as the ESMO Clinical Practice Guidelines and the ETOP IBCSG Partners Foundation. The final awardees were Natasha Leighl, University of Toronto, Canada, who received the ESMO Women for Oncology Award, and Glenda Ramos Martinez, Sociedad de Lucha Contra el Cancer, Guayaquil, Ecuador, who received the ESMO Oncologist of the Year Award.
The ESMO Congress 2025 was a landmark event, standing at the forefront of major scientific advances and reaffirming the collective commitment to improving outcomes for patients around the world. Read on for the major takeaways from ESMO 2025, and make sure to join us next year for ESMO 2026 in Madrid, Spain, from 23rd–27th October 2026.
A record 5,677 abstracts were submitted, over 600 more than last year’s 5,030. Attendees also reached an all-time high at 35,676, a steep increase from 33,830 the previous year

Anthracycline-Free Regimen Improves Outcomes in Early HER2+ Breast Cancer
INTERIM
results from the Phase III DESTINY Breast11 trial (291O), presented at the ESMO 2025 Congress, indicate that trastuzumab deruxtecan (T DXd) followed by paclitaxel, trastuzumab, and pertuzumab (T DXd THP) significantly increased pathological complete response (pCR) rates compared with the standard anthracycline-based regimen in patients with high-risk HER2-positive early breast cancer (eBC).1
The trial evaluated two T DXd regimens, T DXd alone or T DXd THP, against dose dense doxorubicin and cyclophosphamide followed by THP (ddAC THP). The openlabel, randomised, multicentre study enrolled adults with untreated high-risk disease (≥T3, node positive [N1–3], or inflammatory HER2-positive eBC).
Treatments were administered in eight neoadjuvant cycles: T DXd (5.4 mg/kg every 3 weeks), T DXd THP (four cycles of T DXd followed by weekly paclitaxel plus 3-weekly trastuzumab and pertuzumab), or ddAC THP (A+C every 2 weeks for four cycles followed by THP). The primary endpoint was pCR (ypT0/is ypN0); secondary endpoints included event-free survival and safety.
By March 2025, 321 patients were randomised to T DXd THP and 320 to ddAC THP. The T DXd THP regimen achieved a pCR rate of 67.3% compared with 56.3% for ddAC THP, an absolute difference of 11.2% (95% CI: 4.0–18.3; p=0.003). Improvements were seen across hormone receptor subgroups: 61.4% versus 52.3% in hormone
receptor-positive and 83.1% versus 67.1% in hormone receptor-negative disease. An early favourable trend for event-free survival was reported (hazard ratio: 0.56; 95% CI: 0.26–1.17).
Safety data showed lower rates of highgrade toxicity with T DXd THP. Grade ≥3 adverse events (AE) occurred in 37.5% (T DXd THP) versus 55.8% (ddAC THP) of patients, and serious AEs in 10.6% (T DXd THP) versus 20.2% (ddAC THP).
Adjudicated interstitial lung disease or pneumonitis occurred in 4.4% (T DXd THP) versus 5.1% (ddAC THP), and left ventricular dysfunction occurred in 1.9% (T DXd THP) versus 9.0% (ddAC THP). No AEs prevented surgery in any treatment arm.
The results suggest that T DXd THP provides superior efficacy and improved tolerability compared with ddAC THP, supporting its potential as an anthracyclinefree neoadjuvant option for high-risk HER2positive eBC. The T DXd-only arm, which halted enrolment following Independent Data Monitoring Committee advice in 2024, will be reported separately.
The trial evaluated two T DXd regimens, T DXd alone or T DXd THP, against dose dense doxorubicin and cyclophosphamide followed by THP

Trastuzumab Deruxtecan Outperforms Trastuzumab Emtansine in High-Risk HER2+ Early Breast Cancer
IN ONE of the most closely watched trials in early breast cancer, trastuzumab deruxtecan (T-DXd) has delivered a major step forward for patients with HER2-positive disease and residual invasive cancer after neoadjuvant therapy. Interim findings from the DESTINY-Breast05 Phase III trial, presented at the ESMO 2025 Congress, show that T-DXd cut the risk of invasive disease recurrence or death by more than half compared with the long-standing standard of care, trastuzumab emtansine (T-DM1).2
The open-label trial enrolled 1,635 patients with residual HER2-positive invasive breast cancer following neoadjuvant taxane-based chemotherapy and HER2-targeted therapy. Participants were randomised to receive either T-DXd (5.4 mg/kg) or T-DM1 (3.6 mg/kg) every 3 weeks for 14 cycles. After a median follow-up of nearly 30 months, T-DXd demonstrated a 53% reduction in the risk of invasive disease-free survival events (hazard ratio [HR]: 0.47; 95% CI: 0.34–0.66; p<0.0001) compared with T-DM1. The benefit was mirrored in disease-free survival results (HR: 0.47; 95% CI: 0.34–0.66; p<0.0001).
A trend towards improved brain metastasisfree interval was also observed with T-DXd (HR: 0.64; 95% CI: 0.35–1.17), highlighting its potential to delay or prevent central nervous system involvement, which is a growing concern in HER2-positive disease.
Safety outcomes were largely consistent with prior T-DXd studies. Grade ≥3 adverse events occurred in about half of patients in both groups (50.6% with T-DXd versus 51.9% with T-DM1). Interstitial lung disease was more frequent with T-DXd (9.6% versus 1.6%), though most cases were Grade 1–2; two Grade 5 events were reported.
Taken together, these interim data establish T-DXd as a new benchmark for post-neoadjuvant treatment in patients with high-risk HER2-positive early breast cancer, offering not just incremental but transformative improvement in outcomes for this challenging population.
T-DXd demonstrated a 53% reduction in the risk of invasive disease-free survival events compared with T-DM1
Perioperative Enfortumab Vedotin–Pembrolizumab Sets New Standard for Muscle-Invasive Bladder Cancer
A STUDY presented at the ESMO 2025 Congress reported that adding perioperative enfortumab vedotin (EV) plus pembrolizumab to surgery significantly and meaningfully improves outcomes in patients with muscle-invasive bladder cancer (MIBC) who are cisplatin-ineligible.3
Radical cystectomy combined with pelvic lymph node dissection is the standard treatment for patients with MIBC who are cisplatin-ineligible. Perioperative therapy may improve outcomes in these patients. Therefore, researchers investigated the addition of perioperative EV plus pembrolizumab to standard treatment for MIBC.
The Phase III KEYNOTE-905/EV-303 study evaluated the efficacy and safety of perioperative EV plus pembrolizumab with radical cystectomy and pelvic lymph node dissection compared to radical cystectomy and pelvic lymph node dissection alone in adult patients with MIBC (T2-T4aN0M0 or T1T4aN1M0) who were cisplatin-ineligible or declined cisplatin. Patients were randomised 1:1 to EV plus pembrolizumab (three cycles of EV 1.25 mg/kg on Day 1 and Day 8 plus pembrolizumab 200 mg on Day 1 every 3 weeks, followed by radical cystectomy and pelvic lymph node dissection, then six cycles of EV plus 14 cycles of pembrolizumab) versus control (radical cystectomy and pelvic lymph node dissection only). Therapy continued until disease progression, unacceptable adverse events, withdrawal of consent, or completion of planned treatment.
In total, 170 patients were randomised to EV plus pembrolizumab and 174 patients to control. Over 80% of patients were cisplatin-ineligible, as determined by the Galsky criteria. As of 6th June 2025, the median follow-up time was 25.6 months (range: 11.8–53.7). In this study, 149 patients (87.6%) underwent surgery in the EV plus pembrolizumab group and 156 (89.7%) in the control group.
The results revealed that EV plus pembrolizumab significantly improved event-free survival (hazard ratio: 0.40; 95% CI: 0.28–0.57; p<.001), overall survival (hazard ratio: 0.50; 95% CI: 0.33–0.74; p<.001), and pathological complete response rate (57.1% versus 8.6%; estimated difference: 48.3%; 95% CI: 39.5–56.5; p<.001) versus control. Treatmentemergent adverse events occurred in 100% of patients in the EV plus pembrolizumab arm (of which
EV plus pembrolizumab significantly improved event-free survival, overall survival, and pathological complete response rate
71.3% were at least Grade 3), and in 64.8% of patients in the control group (of which 45.9% were at least Grade 3). The most frequent Grade ≥3 adverse events of special interest were severe skin reactions (11.4%) for pembrolizumab and skin reactions (10.8%) for EV.
In conclusion, the addition of perioperative EV plus pembrolizumab significantly improved event-free survival, overall survival, and pathological complete response rate in patients with MIBC who were predominantly cisplatin-ineligible. Additionally, the safety profile of EV plus pembrolizumab was manageable and consistent with prior reports. This is the first perioperative regimen to improve outcomes compared to standard treatment, and may become a new standard of care.
New Study Supports Ivonescimab for Squamous Lung Cancer
A RECENT Phase III study, presented at the ESMO 2025 Congress, has shown that ivonescimab combined with chemotherapy significantly improves progression-free survival (PFS) compared with tislelizumab plus chemotherapy in patients with untreated advanced squamous non-small cell lung cancer, regardless of programmed death-ligand 1 (PD-L1) expression.4
This trial included 532 patients with Stage IIIB–IV disease, randomised equally to receive ivonescimab or tislelizumab alongside paclitaxel and carboplatin for four cycles, followed by maintenance monotherapy. Randomisation was stratified by disease stage and PDL1 tumour proportion score (TPS). The primary endpoint was PFS, assessed by an independent radiographic review committee in line with RECIST v1.1, with overall survival as a key secondary endpoint.
Baseline characteristics were balanced between the two groups, with 63.2% of patients presenting central tumours, 8.8% tumour cavitation, and 17.5% major blood vessel encasement. Ivonescimab plus chemotherapy demonstrated a statistically significant PFS improvement versus tislelizumab plus chemotherapy, with a hazard ratio (HR) of 0.60 (95% CI: 0.46–0.78; p<0.0001). Median PFS was 11.1 months in the ivonescimab arm versus 6.9 months in the tislelizumab arm. The benefit was consistent across key subgroups:
patients with PD-L1 TPS <1% had a median PFS of 9.9 versus 5.7 months (HR: 0.55), while those with PD-L1 TPS ≥1% showed 12.6 versus 8.6 months (HR: 0.66). Safety profiles were comparable, with treatmentrelated serious adverse events reported in 32.3% and 30.2% of patients, and Grade ≥3 haemorrhagic events occurring in 1.9% and 0.8%, for the ivonescimab and tislelizumab groups, respectively.
These findings suggest that ivonescimab in combination with chemotherapy may represent a new first-line standard of care for patients with advanced or metastatic squamous non-small cell lung cancer, offering a meaningful extension in PFS while maintaining a manageable safety profile.
These findings suggest that ivonescimab in combination with chemotherapy may represent a new first-line standard of care
Phase III OptiTROP-Lung04 Trial Shows Sacituzumab Tirumotecan Extends Survival in EGFR-Mutated Non-small Cell Lung Cancer
A PHASE III trial presented at the ESMO 2025 Congress has shown that sacituzumab tirumotecan (sac-TMT), a novel TROP2-directed antibody–drug conjugate, delivers a significant survival advantage over standard platinumbased chemotherapy in patients with EGFR-mutated non-small cell lung cancer (NSCLC) who have progressed on EGFR tyrosine kinase inhibitors (TKI).5
The findings position sac-TMT as a promising new treatment option for a population with limited therapeutic alternatives.
The randomised, multicentre study enrolled 376 patients with advanced EGFR-mutated NSCLC who received prior third-generation EGFR TKI therapy, with or without platinum-based chemotherapy. Participants were randomised 1:1 to receive either sac-TMT monotherapy or a platinum doublet (pemetrexed plus carboplatin or cisplatin). The study’s primary endpoint was progression-free survival (PFS), assessed by a blinded independent review committee, with overall survival (OS) as a key secondary endpoint.
At a median follow-up of 18.9 months, sac-TMT achieved a median PFS of 8.3 months compared with 4.3 months for chemotherapy (hazard ratio [HR]: 0.49; 95% CI: 0.39–0.62; p<0.0001). The 12-month PFS rate was 32.3% with sac-TMT versus 7.9% with chemotherapy. OS data also favoured sac-TMT, with a median OS not yet reached versus 17.4 months for chemotherapy (HR: 0.60; 95% CI: 0.44–0.82; p=0.0006). Adjusted OS analysis confirmed these findings (HR: 0.56; p=0.0002). The objective response rate was 60.6% for sac-TMT compared with 43.1% for chemotherapy, with a median response duration of 8.3 versus 4.2 months, respectively.
Treatment-related adverse events of Grade 3 or higher occurred in 49.5% of patients receiving sac-TMT and 52.2% receiving chemotherapy. Serious treatment-related adverse events were less frequent with sac-TMT (7.4% versus 17.0%), and no
drug-related interstitial lung disease or pneumonitis was reported in either group.
According to the study chair, the results represent a major advancement in the post-TKI treatment landscape, as sacTMT becomes the first TROP2-targeted antibody–drug conjugate to show both PFS and OS superiority over chemotherapy in this setting.

The findings position sac-TMT as a promising new treatment option for a population with limited therapeutic alternatives
Radioligand Therapy Improves Survival in Metastatic Prostate Cancer
AT THE ESMO 2025 Congress, the Phase III PSMAddition trial (LBA6) demonstrated that combining the radioligand [177Lu]Lu-PSMA-617 with standard androgen deprivation therapy (ADT) and an androgen receptor pathway inhibitor (ARPI) significantly improved radiographic progression-free survival (rPFS) in patients with prostate-specific membrane antigen (PSMA)positive metastatic hormone-sensitive prostate cancer (mHSPC).6
The trial involved 1,144 adults with treatment-naïve or minimally treated (≤45 days) mHSPC and at least one PSMApositive metastatic lesion detected by [68Ga]Ga-PSMA-11 PET/CT. Participants were randomised (1:1) to receive [177Lu] Lu-PSMA-617 (7.4 GBq every 6 weeks, six cycles) plus ADT and ARPI, or standard ADT and ARPI alone. Randomisation was stratified by disease volume (high or low), age (≥70 or <70 years), and primary tumour treatment (yes or no). Patients in the control arm with centrally confirmed radiographic progression could cross over to [177Lu]LuPSMA-617 if eligible.
At the second interim analysis (data cutoff: 13th January 2025; median follow-up: 23.6 months), the primary endpoint was met. rPFS was significantly improved in the [177Lu]Lu-PSMA-617 arm, with 139 events (24.3%) compared with 172 (30.1%) in the control arm, yielding a hazard ratio of 0.72 (95% CI: 0.58–0.90; p=0.002). Median rPFS was not estimable in either arm. A positive trend in overall survival was observed (hazard ratio: 0.84, 95% CI: 0.63–1.13; p=0.125), with 85 events (14.9%) in the [177Lu]Lu-PSMA-617 arm versus 99 (17.3%) in the control arm. The objective response rate also favoured the [177Lu]Lu-PSMA-617 arm at 85.3% (95% CI: 79.9–89.6; n=224) versus 80.8% (95% CI: 74.8–85.8; n=213).
The findings establish that [177Lu] Lu-PSMA-617 added to ADT and ARPI significantly improves rPFS in PSMApositive mHSPC without notable impairment in safety or quality of life
Safety findings were consistent with prior experience. Any adverse event (AE) occurred in 98.4% of patients receiving [177Lu]Lu-PSMA-617 and 96.6% in controls.
Grade ≥3 AEs were reported in 50.7% versus 43.0% in the control arm, and serious AEs in 31.9% versus 28.7% in the control arm. Dry mouth, mainly Grade 1–2, was the most common AE (41.0% Grade 1 and 4.8% Grade 2 versus 3.4% and 0.4% in controls).
Grade ≥3 cytopenias occurred in 14.4% of patients receiving [177Lu]Lu-PSMA-617 versus 5.0% in the control arm. Time to deterioration in quality of life, measured by Functional Assessment of Cancer TherapyProstate (FACT-P) and EuroQol-5 Dimension (EQ-5D), did not differ meaningfully between groups.
The findings establish that [177Lu]LuPSMA-617 added to ADT and ARPI significantly improves rPFS in PSMApositive mHSPC without notable impairment in safety or quality of life. Long-term followup for overall survival and durability of benefit is ongoing.

Circulating Tumour DNA-Guided Atezolizumab Extends Survival in High-Risk Bladder Cancer
A NOVEL circulating tumour DNA (ctDNA)-guided approach to postoperative therapy has delivered a major advance for patients with muscle-invasive bladder cancer (MIBC). Interim results from the Phase III IMvigor011 trial (NCT04660344), presented at the ESMO 2025 Congress, show that adjuvant atezolizumab significantly improves both disease-free and overall survival compared with placebo in patients who test positive for ctDNA following radical cystectomy.7
The global, randomised, double-blind trial enrolled 761 patients with high-risk MIBC and no radiographic evidence of disease. Participants were entered into a year-long ctDNA surveillance programme beginning 6–24 weeks after surgery. Only those who tested ctDNA-positive, indicating the presence of minimal residual disease, were randomised 2:1 to receive either atezolizumab (1,680 mg every 4 weeks) or placebo for up to 12 cycles. Patients who remained ctDNA-negative throughout surveillance received no adjuvant treatment.
After a median follow-up of 16.1 months, atezolizumab reduced the risk of recurrence or death by 36% among patients who were ctDNA-positive compared with placebo (disease-free survival hazard ratio: 0.64; 95% CI: 0.47–0.87; p=0.0047). The overall survival benefit was also statistically significant, with a 41% reduction in mortality risk (hazard ratio: 0.59; 95% CI: 0.39–0.90; p=0.0131).
Median disease-free survival was 9.9 months with atezolizumab versus 4.8 months with placebo, while the 12-month survival rate
reached 85.1% in the atezolizumab arm compared with 70.0% for placebo.
Toxicities were manageable and in line with previous reports for atezolizumab. Grade 3–4 adverse events occurred in 28.5% of atezolizumab-treated patients versus 21.7% with placebo; treatment-related events were reported in 7.3% and 3.6%, respectively.
Fatal treatment-related events were rare (1.8% versus 0%).
Importantly, for the 357 patients who remained persistently ctDNA-negative, the outcomes were excellent: 95.4% were disease-free at 1 year and 88.4% at 2 years, suggesting that these patients may safely avoid adjuvant therapy altogether.
By integrating molecular monitoring with precision immunotherapy, IMvigor011 marks a pivotal step towards personalising postoperative treatment in MIBC, offering therapy only when it’s truly needed, while sparing others from unnecessary toxicity.

By integrating molecular monitoring with precision immunotherapy, IMvigor011 marks a pivotal step towards personalising postoperative treatment in MIBC
Circulating Tumour DNA-Guided De-escalation Reduces Toxicity in Stage III Colon Cancer
PRIMARY analysis of the circulating tumour DNA (ctDNA)-negative cohort from the randomised AGITG DYNAMIC-III trial, presented at the ESMO 2025 Congress, has demonstrated that ctDNA-guided de-escalation is feasible and substantially reduces oxaliplatin exposure and adverse events, with outcomes approaching standard management, especially for clinical low-risk tumours.8
The individual benefit of adjuvant chemotherapy is not well understood. Therefore, researchers sought to investigate whether post-surgery ctDNA testing could support risk-adjusted treatment selection and guide the de-escalation of adjuvant chemotherapy. The DYNAMIC-III study explored adjuvant chemotherapy deescalation or escalation, informed by postsurgery ctDNA results.
In this multicentre, randomised, Phase II/III trial, patients with Stage III colon cancer who underwent tumour-informed ctDNA testing 5–6 weeks post-surgery were randomised to receive either ctDNA-guided or standard management. For patients who received ctDNAguided management, ctDNA-negative results prompted adjuvant chemotherapy de-escalation: from 6 to 3 months of fluoropyrimidine or observation, from 3 months of doublet to singleagent fluoropyrimidine, or from 6 months of doublet to 3 months of doublet or single-agent fluoropyrimidine. In this study, the primary endpoint was 3-year recurrence-free survival.
ctDNA-guided de-escalation is feasible and reduces oxaliplatin exposure and adverse events, with outcomes approaching standard management
In total, 968 patients were enrolled, of whom 702 (72.5%) were ctDNA-negative. Within this group, 353 patients were assigned to ctDNA-guided treatment management, and 349 to standard management. At a median follow-up of 45 months, 319 (90.4%) patients received ctDNAguided per-protocol de-escalation.
The analysis revealed that ctDNA-guided treatment de-escalation reduced oxaliplatinbased chemotherapy use to 34.8%, compared to 88.6% with standard management (p<0.001). Additionally, ctDNA-guided treatment lowered Grade 3+ adverse events of special interest (6.2% versus 10.6%; p=0.037) and treatmentrelated hospitalisation (8.5% versus 13.2%; p=0.048). However, the analysis did not confirm non-inferiority of ctDNA-guided treatment de-escalation, and the 3-year recurrence-free survival was 85.3% versus 88.1% (97.5% lower CI: –8.0%). The pre-planned subgroup analysis suggested de-escalation may be non-inferior in clinical low-risk tumours (T1-3N1), with a 3-year recurrence-free survival of 91.0% versus 93.2% (97.5% lower CI: –7.2%).
In summary, the results of the DYNAMIC-III study suggest that ctDNA-guided de-escalation is feasible and reduces oxaliplatin exposure and adverse events, with outcomes approaching standard management. Benefit may be seen, particularly for patients with low-risk tumours, but further research is needed to confirm this.
Bemarituzumab Significantly Improves Survival in Gastric Cancer
A RECENT Phase III trial, presented at the ESMO 2025 Congress, has demonstrated that bemarituzumab (BEMA), a first-in-class anti-FGFR2b antibody, significantly improves overall survival (OS) in patients with FGFR2b-overexpressing, non-HER2-positive, unresectable, locally advanced or metastatic gastric or gastro-oesophageal junction cancer (G/GEJC).9
BEMA functions by blocking oncogenic FGFR2b signalling and engaging antibodydependent cell-mediated cytotoxicity. In this study, 547 patients were randomised to receive BEMA plus mFOLFOX6 or a matched placebo plus mFOLFOX6, with primary analysis focused on those with FGFR2b ≥10% tumour cell staining. Key secondary endpoints included progression-free survival, objective response rate, and safety.
At the primary analysis, with a median follow-up of 11.8 months, BEMA significantly improved OS compared with placebo, with a median OS of 17.9 months versus 12.5 months and a hazard ratio of 0.61 (95% CI: 0.43–0.86; p=0.005). In the descriptive follow-up analysis, at a median follow-up of 19.4 months, the median OS remained numerically higher with BEMA (14.5 versus 13.2 months), although the treatment effect was attenuated (hazard ratio: 0.82; 95% CI: 0.62–1.08). The safety profile was consistent with expectations, with higher rates of Grade ≥3 treatment-emergent adverse events observed in the BEMA arm, primarily driven by corneal events.
These results suggest that BEMA offers a meaningful survival benefit in patients with FGFR2b-overexpressing G/GEJC, particularly in those with ≥10% tumour cell staining. While the effect was somewhat reduced with longer follow-up, the initial findings support the potential of BEMA as a new targeted therapy in this setting. Further studies, including the upcoming FORTITUDE-102 trial, will help to better define the long-term efficacy and safety of BEMA, offering hope for improved outcomes in this difficult-to-treat patient population.
These results suggest that BEMA offers a meaningful survival benefit in patients with FGFR2b-overexpressing G/GEJC
References
1. Harbeck N et al. DESTINY-Breast11: neoadjuvant trastuzumab deruxtecan alone (T-DXd) or followed by paclitaxel + trastuzumab + pertuzumab (T-DXdTHP) vs SOC for high-risk HER2+ early breast cancer (eBC). Abstract 291O. ESMO Congress, 17-21 October, 2025.
2. Geyer CE et al. Trastuzumab deruxtecan (T-DXd) vs trastuzumab emtansine (T-DM1) in patients (pts) with high-risk human epidermal growth factor receptor 2–positive (HER2+) primary breast cancer (BC) with residual invasive disease after neoadjuvant therapy (tx): interim analysis of DESTINY-Breast05. LBA1. ESMO Congress, 17-21 October, 2025.
3. Vulsteke C et al. Perioperative (periop) enfortumab vedotin (EV) plus pembrolizumab (pembro) in participants (pts) with muscleinvasive bladder cancer (MIBC) who are cisplatin-ineligible: the phase III
KEYNOTE-905 study. Abstract LBA2. ESMO Congress, 17-21 October, 2025.
4. Lu S et al. Phase III study of ivonescimab plus chemotherapy versus tislelizumab plus chemotherapy as first-line treatment for advanced squamous non-small cell lung cancer (HARMONi-6). Abstract LBA4. ESMO Congress, 17-21 October, 2025.
5. Zhang L et al. Sacituzumab tirumotecan (sac-TMT) vs platinumbased chemotherapy in EGFR-mutated (EGFRm) non-small cell lung cancer (NSCLC) following progression on EGFR-TKIs: results from the randomized, multi-center phase III OptiTROP-Lung04 study. Abstract LBA5. ESMO Congress, 17-21 October, 2025.
6. Tagawa ST et al. Phase III trial of [177Lu]Lu-PSMA-617 combined with ADT + ARPI in patients with PSMApositive metastatic hormone-sensitive prostate cancer (PSMAddition).
Abstract LBA6. ESMO Congress, 17-21 October, 2025.
7. Powles TB et al. IMvigor011: a phase III trial of circulating tumour (ct) DNA-guided adjuvant atezolizumab vs placebo in muscle-invasive bladder cancer. LBA8. ESMO Congress, 17-21 October, 2025.
8. Tie J et al. ctDNA-guided adjuvant chemotherapy de-escalation in stage III colon cancer: primary analysis of the ctDNA-negative cohort from the randomized AGITG DYNAMIC-III trial (Intergroup study of AGITG and CCTG). Abstract LBA9. ESMO Congress, 17-21 October, 2025.
9. Rha SY et al. Bemarituzumab (BEMA) plus chemotherapy for advanced or metastatic FGFR2b-overexpressing gastric or gastroesophageal junction cancer (G/GEJC): FORTITUDE-101 phase III study results. Abstract LBA10. ESMO Congress, 17-21 October, 2025.
Expanding the First-Line Option in TripleNegative Breast Cancer: Pivotal Trials of Datopotamab Deruxtecan and Sacituzumab Govitecan at the European
Society for Medical Oncology (ESMO) Congress
2025
Author: *François Cherifi1
1. Medical Oncology Department, Cancer Centre François Baclesse, Caen, France
*Correspondence to francois.cherifi@gmail.com
Disclosure: Cherifi has received grants from Novartis and Gilead, with payment to the institution; consulting fees from Gilead; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Pharmaand; and support for attending meetings and/or travel from Roche, Gilead, and Chugai.
Citation: EMJ Oncol. 2025;13[1]:23-26.
https://doi.org/10.33590/emjoncol/SEBR8422
THIS YEAR, the field of triple-negative breast cancer (TNBC) has witnessed significant advancements in the first line. In June, during the American Society of Clinical Oncology (ASCO) Annual Meeting 2025, the ASCENT-04 trial proposed sacituzumab govitecan (SG) in combination with immunotherapy at first line for patients who are programmed death-ligand 1 (PD-L1) positive. Then, at the European Society for Medical Oncology (ESMO) Congress 2025, two highly anticipated studies for patients who are PD-L1 negative were unveiled: the ASCENT-03 trial and the TROPION-Breast02 trial. These studies mark groundbreaking progress in the treatment landscape for TNBC with the advancement of antibody-drug conjugates (ADC) and ‘chemo free’ firstline treatments.
INTRODUCTION
TNBC remains the most aggressive subtype of breast cancer, and is characterised by the absence of oestrogen receptor, progesterone receptor, and HER2 expression, as well as by a high likelihood of early relapse and visceral spread.1 Despite progress with immune checkpoint inhibitors, response remains variable and unpredictable. Moreover, a substantial proportion of patients with metastatic TNBC are ineligible for PD-L1-based therapy due to PD-L1-negative status or comorbidities.2 For these individuals, chemotherapy has
long represented the default first-line option, offering modest benefit with a median progression-free survival (PFS) of less than 6 months and a median overall survival (OS) rarely exceeding 18 months.1,3
The emergence of ADCs has reshaped therapeutic strategies in pretreated TNBC, with SG and datopotamab deruxtecan (Dato-DXd) both demonstrating robust antitumour activity in later-line settings. The 2025 congress season marked a major inflexion point for the field: the ASCENT-04/ KEYNOTE-D19 study presented at ASCO 2025 established SG plus pembrolizumab
as a new standard of care for PD-L1positive, previously untreated metastatic TNBC.4 Simultaneously, two Phase III trials presented at ESMO 2025, TROPIONBreast02 (Dato-DXd versus chemotherapy) and ASCENT-03 (SG versus chemotherapy), reported practice-changing results in patients who are PD-L1 negative or immunotherapy ineligible.5,6
The emergence of ADCs has reshaped therapeutic strategies in pretreated TNBC
DATOPOTAMAB DERUXTECAN IN TROPION-BREAST02
The TROPION-Breast02 trial5 was a randomised, open-label, Phase III study evaluating Dato-DXd, a trophoblast cell surface antigen 2 (TROP2)-directed ADC with a DXd payload, versus the investigator’s choice of chemotherapy in patients with locally recurrent inoperable or metastatic TNBC for whom immunotherapy was not an option. Eligible patients had received no prior systemic therapy for advanced disease and were randomised 1:1 to Dato-DXd 6 mg/kg intravenously every 3 weeks or to standard single-agent chemotherapy. The dual primary endpoints were PFS by blinded independent central review and OS.
At the August 2025 data cutoff, DatoDXd significantly improved PFS compared with chemotherapy, reducing the risk of progression or death by 43% (hazard ratio [HR]: 0.57; 95% CI: 0.47–0.69; p<0.0001).
Median PFS was 10.8 months with DatoDXd versus 5.6 months with chemotherapy. The trial also demonstrated a significant OS improvement (HR: 0.79; median OS: 23.7 versus 18.7 months). The objective response rate was 62.5% with Dato-DXd versus 29.3% with chemotherapy, with complete responses in 9.0% versus 2.5% of patients. Median duration of response exceeded 1 year. Safety findings were consistent with prior reports, with stomatitis, nausea, and dry eye as the most common
adverse events. Despite a median treatment duration twice that of chemotherapy, Grade ≥3 toxicities and treatment discontinuations were comparable.
SACITUZUMAB GOVITECAN IN ASCENT-03
The ASCENT-03 trial6 evaluated SG, a TROP2-directed ADC conjugated to SN-38, versus physician’s choice chemotherapy in previously untreated, locally advanced inoperable or metastatic TNBC not eligible for PD-L1 therapy. A total of 558 patients were randomised 1:1 to SG or to chemotherapy (paclitaxel, nab-paclitaxel, or gemcitabine/carboplatin). SG achieved a statistically significant and clinically meaningful PFS improvement, with a 38% reduction in risk of progression or death (HR: 0.62; 95% CI: 0.50–0.77; p<0.0001). Median PFS was 9.7 months with SG versus 6.9 months with chemotherapy. Twelve-month PFS rates were 41% and 24%, respectively.
Although OS data were immature, a numerical improvement was observed (median OS: 21.5 versus 20.2 months). Median duration of response was longer with SG (12.2 versus 7.2 months). Safety analysis showed manageable toxicity consistent with prior studies, including neutropenia, diarrhoea, and alopecia. Treatment-related discontinuations were lower with SG (4%) than with chemotherapy (12%).
EVOLVING THERAPEUTIC LANDSCAPE AND CLINICAL IMPLICATIONS
The 2025 congress data mark the advent of a new era in TNBC therapy. The parallel success of ASCENT-03 and TROPIONBreast02 represents a decisive shift toward ADCs as front-line standards in TNBC, displacing traditional taxanebased chemotherapy.
ASCENT-04 (SG plus pembrolizumab) and likely TROPION-Breast057 (Dato-DXd in combination with durvalumab) serve PD-L1-positive disease, while SG or DatoDXd monotherapy serve PD-L1-negative
or immunotherapy-ineligible populations, heralding an ADC-dominant paradigm for first-line TNBC management.
EXPERT PERSPECTIVE
The ESMO 2025 data consolidate a unified ADC paradigm in metastatic TNBC. For clinicians, the challenge now lies not in whether to use an ADC, but which ADC to use first. SG and Dato-DXd both target
TROP2, but differ in linker chemistry and payload composition, influencing their pharmacologic profiles and toxicity spectra.8,9 Dato-DXd’s improved tolerability and prolonged PFS make it an attractive option for patients prioritising quality of life, while SG, alone or with pembrolizumab, remains the most clinically validated agent across disease settings. Future research will likely explore long-term outcomes, sequencing and cross-resistance between ADCs, and biomarker-driven personalisation.

References
1. Gennari A et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann Oncol. 2021;32(12):1475-95.
2. Buisseret L et al. The long and winding road to biomarkers for immunotherapy: a retrospective analysis of samples from patients with triple-negative breast cancer treated with pembrolizumab. ESMO Open. 2024;9(5):102964.
3. Punie K et al. Unmet need for previously untreated metastatic triplenegative breast cancer: a real-world study of patients diagnosed from 2011 to 2022 in the United States. Oncologist. 2025;30(3):oyaf034.
4. Tolaney SM et al. Sacituzumab govitecan plus prembrolizumab vs chemotherapy plus pembrolizumab in patients with previously untreated, PD-
L1 positive, advanced or metastatic triple-negative breast cancer: primary results from the randomized, phase 3 ASCENT-04/KEYNOTE-D19 study. ASCO Annual Meeting, 30 May-3 June, 2025.
5. Dent RA et al. First-line datopotamab deruxtecan (Dato-DXd) vs chemotherapy in patients with locally recurrent inoperable or metastatic triple-negative breast cancer (TNBS) for whom immunotherapy was not an option: primary results from the randomised, phase 3 TROPIONBreast02 trial. ESMO Congress, 17-21 October, 2025.
6. Cortés J et al. Primary results from ASCENT-03: a randomised phase 3 study of sacituzumab govitecan vs chemotherapy in patients with previously untreated metastatic triplenegative breast cancer who are unable to receive PD-(L)1 inhibitors. ESMO Congress, 17-21 October, 2025.
7. Schmid P et al. TROPION-Breast05: a randomized phase III study of Dato-DXd with or without durvalumab versus chemotherapy plus pembrolizumab in patients with PDL1-high locally recurrent inoperable or metastatic triple-negative breast cancer. Ther Adv Med Oncol. 2025:17:17588359251327992.
8. Cherifi F et al. Pharmacokinetics and pharmacodynamics of antibodydrug conjugates for the treatment of patients with breast cancer. Expert Opin Drug Metab Toxicol. 2024;20(12):45-59.
9. Hong Y et al. Population pharmacokinetic analysis of datopotamab deruxtecan (Dato-DXd), a TROP2-directed antibody-drug conjugate, in patients with advanced solid tumors. CPT Pharmacometrics Syst Pharmacol. 2025;DOI:10.1002/ psp4.70118.
AI and Cancer Care
Author: Helena Bradbury, EMJ, London, UK
Citation: EMJ Oncol. 2025;13[1]:27-30. https://doi.org/10.33590/emjoncol/RYOB7684
AI WAS a highlight of the 2025 European Society for Medical Oncology (ESMO) Congress, held in Berlin, Germany, with sessions focused on how it works and its potential impact on clinical oncology. One such session, entitled ‘ChatGPT and Cancer Care’ provided a timely exploration of AI’s capabilities in cancer care, as well as the practical and ethical considerations that come with integrating these tools into clinical practice.
LARGE LANGUAGE MODELS AND AI AGENTS IN ONCOLOGY
Jakob Kather, TU Dresden, University of Heidelberg, Germany, opened the session by exploring the world of large language models (LLM) and AI agents in oncology. Stating that AI is already an active part of clinical practice, Kather noted that several AI-driven medical devices have received regulatory approval and are now certified for patient use. Most of these applications focus on image analysis and interpreting X-rays, pathology slides, or endoscopy videos. However, Kather emphasised that each of these systems is built to perform a single, highly specific task on a single data type. This task-specific design limits scalability in clinical settings, where hundreds of different analytic processes occur routinely, each potentially requiring its own dedicated AI system.
First launched in 2022, ChatGPT (OpenAI, San Francisco, California, USA) is now a household application, and has transformed the way we receive and consume information. Kather noted the improvements seen in speed, accuracy, and utility of these LLMs over time as they have gathered more information. In terms of use, there are typically three types of interaction with LLMs. The first, and most basic interaction, is called ‘zero-shot’, which comprises a simple question and response, and uses
the embedded knowledge the model was trained on. The second approach, which is more common with recent models, is where LLMs scan the internet for relevant sources to incorporate into the answer. Issues with this, as flagged by Kather, are that it can often pull out-of-date or unrelated information. This can negatively influence the output. Finally, the interaction recommended for enhanced accuracy involves the user asking a direct question with an attached file for the LLM to draw additional information from in its answer.
The medical oncology community must approach emerging technologies with optimism, while upholding the principles of evidence-based medicine
In closing, Kather emphasised that the medical oncology community must approach emerging technologies with optimism, while upholding the principles of evidence-based medicine. He stated that, whenever claims are made about a new technology enhancing our understanding of cancer or improving patient empowerment, it is essential that rigorous clinical trials are conducted to thoroughly test and validate those claims.
LARGE LANGUAGE MODELS FOR DRUG DEVELOPMENT AND CANCER THERAPY
Opening his talk, Loic Verlingue, Centre Léon Bérard, Centre de Recherche en Cancérologie de Lyon, France, shared the benefits that clinical trials offer, namely increasing access to innovative treatments for patients, providing physicians with expertise on new treatments, and improving the economic benefit for societies.
According to Verlingue, there are approximately 9,000 FDA-approved small molecule drugs, but there are thought to be 1,062 potential pharmaceutical compounds. There are also believed to be 20,244 human proteins, but the 3D structure is only known for approximately 6,200 of them, and there are only approximately 2,700 that could act as potential drug targets. As stressed by Verlingue, the use of AI is therefore imperative to predict the structure of proteins and also screen for novel pharmaceuticals.1
As an initial example, Verlingue discussed Cell2Sentence (C2S; Google, Mountain View, California, USA; Yale University, New Haven, Connecticut, USA), a novel computational method that converts singlecell gene expression data into textual representations known as ‘cell sentences’. Using this framework, researchers trained a family of LLMs, named C2S-Scale, on
approximately 50 million single-cell profiles and their associated texts.2
To evaluate the platform’s ability to support novel biological discovery, the authors conducted a virtual drug screen using C2SScale. They simulated the effects of roughly 4,000 drug candidates on single-cell data under a specific condition: identifying a drug that could enhance immune signalling in cancer cells that exhibit low interferon levels, which are insufficient on their own to induce antigen presentation. The model identified silmitasertib, a casein kinase 2 inhibitor, predicting that it would enhance major histocompatibility complex Class I-mediated antigen presentation (HLA-A, HLA-B, HLA-C), but only in the context of an already activated immune response. This distinction is important, as it indicates that silmitasertib does not initiate immune activation itself, but rather amplifies it once interferon has already triggered the response.2
Verlingue then raised an important question: while these AI models may be effective at generating new therapeutic drugs, how well do these approaches translate in the clinical setting? Citing a 2024 study,3 and the first analysis of the clinical pipeline of AI-native biotech companies, it was reported that, in Phase 1, AI-discovered molecules had an 80–90% (21/24) success rate of drugs meeting their clinical endpoint, which is higher than the classical success


rate of 40–60%. This is a promising statistic suggesting that AI-generated drug candidates may progress through early clinical stages more efficiently and with greater precision than traditionally developed therapies.
Looking more broadly, Verlingue then discussed the global landscape for oncology clinical trials, and how LLMs can assist in their design and recruitment. Analysing data from 87,748 clinical trials conducted between 2000–2021 across high-income, upper-middle income, lowermiddle income, and low-income countries, a 2024 study reported that, despite an absolute mean annual rise of 266.6 trials, there had been no new trials initiated by 2024.4 These delays may be attributed to several factors, such as patient availability, industry funding, or clinical infrastructure. To address this challenge, he suggested that AI could assist in matching patients to potential clinical trials, helping to increase attrition rates from 8% to over 20%.
Finally, Verlingue spoke on Evo 2 (Arc Institute, Palo Alto, California, USA; Nvidia, Santa Clara, California, USA),5 a new, large-scale, generative AI model that analyses and generates DNA, RNA, and protein sequences to predict protein function, identify pathogenic mutations, and even design new genomes. Trained on
9.3 trillion DNA base pairs from genomes spanning all domains of life, it represents one of the largest biological models ever built, assessing the functional impact of mutations, including in the non-coding regions, splice sites, and clinically relevant genes, without the need for task-specific fine-tuning.
AGENTIC AI IN ONCOLOGY
Closing this timely session, Daniel Truhn, University Hospital Aachen, Germany, summarised the strengths and limitations of AI implementation in oncology. Highlighting findings from a 2023 study on the use of AI chatbots for cancer treatment recommendations,6 Truhn noted a staggeringly high number of inaccuracies. The study reported that 13 of 104 (12.5%)
With the development of more advanced systems, there is now greater assurance and consistency in their answers
responses contained hallucinations, meaning recommendations that did not align with any formal clinical guidelines. Furthermore, while the chatbot provided at least one recommendation for 102 of
the 104 prompts (98%), 35 of those 102 responses (34.3%) included one or more treatments that were not concordant with clinical guidelines.6
Since 2023, have we seen improvements in the use of AI in clinical decision-making in oncology? In a study published this year,7 researchers set out to evaluate an AI agent tailored to interact with and draw conclusions from multiple patient data. It was evaluated on 20 realistic multimodal patient cases and demonstrated an 87.5% accuracy, drawing clinical conclusions in 91.0% of cases and accurately citing relevant guidelines in 75.5% of the time.7
The study reported that 13 of 104 (12.5%) responses contained hallucinations
References
1. Rifaioglu AS et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform. 2019;20(5):1878-912.
2. Rizvi et al. Scaling large language models for next-generation single-cell analysis. bioRxiv. 2025;DOI:10.1101/20 25.04.14.648850.
Speaking more broadly, Truhn commented on the tone of AI models and their increasing confidence in generated outputs. He noted that, historically, these models were designed to satisfy the user and could easily alter their responses if challenged. However, with the development of more advanced systems, there is now greater assurance and consistency in their answers.
In closing, Truhn remarked: “You now know about AI, and you have the tools available, or if you don’t, you will soon. What matters is how you use these tools, that you use them responsibly, and that you help us continue to develop them.”
CONCLUSION
At a time where AI is increasingly utilised in all aspects of people’s lives, this session served as a useful reference point. The presentations underscored that, while AI offers faster, more accurate insights, human oversight remains essential to ensure reliability, ethical use, and patient safety. The future of oncology will likely be shaped by a hybrid model, combining the speed and scalability of AI with the expertise and judgment of clinicians.
3. Jayatunga MK et al. How successful are AI-discovered drugs in clinical trials? A first analysis and emerging lessons. Drug Discov Today. 2024;29(6):104009.
4. Izarn F et al. Globalization of clinical trials in oncology: a worldwide quantitative analysis. ESMO Open. 2025;10(1):104086.
5. Brixi GB et al. Genome modeling and design across all domains of life with
Evo 2. bioRvix. 2025;DOI:10.1101/2025. 02.18.638918.
6. Chen S et al. Use of artificial intelligence chatbots for cancer treatment information. JAMA Oncol. 2023;9;(10):1459-62.
7. Ferber D et al. Development and validation of an autonomous artificial intelligence agent for clinical decisionmaking in oncology. Nat Cancer. 2025;6:1337-49.
The New Frontiers of Personalised Cancer Prevention
Author: Katie Wright, EMJ, London, UK
Citation: EMJ Oncol. 2025;13[1]:31-35. https://doi.org/10.33590/emjoncol/DZRT1764
THIS YEAR, the highly anticipated European Society for Medical Oncology (ESMO) Congress 2025 was held in Berlin, Germany, from 17th –21st October 2025. The Sunday afternoon session, entitled ‘The state-of-the-art of personalised prevention’, brought together leading international experts to explore emerging strategies for reducing cancer risk and improving early detection across multiple cancer types.1 Chaired by Harry de Koning, Erasmus MC University Medical Centre, Rotterdam, the Netherlands; and Suzette Delaloge, Gustave Roussy, Villejuif, France, the session featured a series of presentations on cutting-edge approaches in cancer prevention. Together, the speakers outlined the current landscape of personalisedcancer prevention and the steps needed to translate research into equitable, effective clinical practice.
LUNG CANCER
Translating Screening Evidence into Public Health
To begin the session, de Koning took the stage to explore the transformative role of low-dose CT in shifting lung cancer diagnosis towards earlier, more treatable stages. Drawing on comparative registry data from the Netherlands, he demonstrated the dramatic redistribution of stage at diagnosis that accompanies the implementation of structured CT screening. In the general population, approximately 50% of lung cancers are detected at Stage IV and only 7% at Stage IA. Under CT screening conditions, this profile reverses, with 50% detected at Stage IA and only 10% at Stage IV. This “stage shift,” de Koning explained, represents the fundamental advantage of CT-based detection over symptom-driven diagnosis or older imaging methods such as chest radiography.
He highlighted key clinical evidence establishing this principle: the National Lung Screening Trial (NLST)2 in the USA and the European NELSON trial.3 Both large-scale
RCTs demonstrated a significant reduction in lung cancer-specific mortality with lowdose CT screening compared to chest X-ray or no screening. Importantly, the NELSON trial achieved even greater mortality reduction, with hazard ratios of 0.76 in males and 0.41 in females after 8 years.
Refining Eligibility and Outcomes
de Koning then presented recently published analyses exploring why NELSON achieved superior outcomes compared to NLST.4 The findings indicate that histologyspecific mortality reductions were a key differentiator: in the NELSON cohort, CT screening significantly reduced mortality from squamous cell carcinoma, a benefit not observed in the NLST trial. This histological insight may explain the European advantage, though de Koning cautioned that further validation is needed.
The same study also examined risk stratification by smoking intensity and cessation status. Contrary to traditional assumptions, individuals with lower cumulative tobacco exposure (<30 packyears) and former smokers (those who quit
≥5 years prior) appeared to benefit equally or even more from screening compared with heavy current smokers. This suggests a broader potential eligibility range for national screening programmes beyond only high-intensity smokers.
Focusing on national projections, de Koning discussed modelling from the Netherlands, estimating the impact of biannual CT screening in high-risk groups.5 Simulations indicate that introducing screening in 2022 could yield an 18% reduction in national lung cancer mortality, representing thousands of prevented deaths over time.
Further analyses demonstrated that early detection directly translates to a survival advantage. In a stage-specific comparison of mortality prevention probabilities,6 earlystage detection (IA–IB) corresponded to an 80% reduction in disease-specific mortality. However, de Koning acknowledged the inevitable trade-off between lives saved and overdiagnosis: based on earlier modelling,7 approximately 500 deaths are prevented per 100,000 screened individuals, alongside 200 cases of overdiagnosis and overtreatment, a balance considered acceptable given the magnitude of benefit.
From Evidence to Implementation
Turning to implementation, de Koning reviewed the European Council’s 2022 recommendations expanding cancer screening programmes to include lung, prostate, and gastric cancers. Several countries, including Croatia, Czechia, Poland, Italy, Hungary, and the Netherlands, have now initiated pilots or national
programmes. The UK, he noted, is a frontrunner with its large-scale Targeted Lung Health Check initiative, having issued 1.8 million invitations and conducted 360,000 scans to date, yielding a 1.3% cancer detection rate with 62% of cases found at Stage I. Encouragingly, preliminary analyses show a 22% reduction in latestage disease, with evidence that screening may narrow socioeconomic disparities, as early-stage detection rates are now higher among the most deprived populations.
Finally, de Koning introduced ongoing and future studies designed to refine screening intervals and population selection, including the 4-IN-THE-LUNG-RUN trial, the SOLACE project (focusing on women and underserved groups), and LAPIN, which aims to evaluate both tobacco and non-tobacco risk factors such as radon exposure. He concluded by emphasising the synergistic role of improved treatment and screening in enhancing survival, referencing recent Dutch data showing marked mortality improvements linked to modern therapies.8
BREAST CANCER
Limitations of Standard Screening
Delaloge began her presentation by outlining the rationale and emerging direction for personalised prevention in breast cancer, emphasising that rising incidence, widening health inequalities, and the limitations of standard screening programmes necessitate a shift in strategy. She noted that global breast cancer incidence has doubled over the
last 30 years, with French epidemiological analyses showing that, while demographic changes account for part of this increase, approximately 50% is attributable to modifiable risk factors linked to lifestyle and environmental exposures. As a result, prevention and screening approaches developed in the 1990s are no longer sufficient, especially given the substantial financial and treatment burden associated with later-stage diagnoses.
Delaloge illustrated the treatment implications of stage at diagnosis, showing that women diagnosed at Stage I require significantly less systemic therapy compared with those at Stages II or III, where extended endocrine therapy, immunotherapies, and targeted agents are increasingly used. The clinical and economic burden of treating later-stage disease, therefore, reinforces the importance of early detection. She highlighted data from France demonstrating that participation in organised screening programmes is associated with lower excess mortality, and importantly, that organised screening reduces the impact of social deprivation on outcomes.9 However, participation in standard mammography screening programmes is declining, particularly among younger women and socioeconomically disadvantaged groups, presenting a pressing public health challenge.
The
Multi-Factorial
Framework of Risk Assessment
To address these limitations, Delaloge presented the emerging framework of personalised or ‘interception’ prevention, which combines risk assessment, risk reduction, and tailored early detection.
Germline genetics remains a cornerstone of identifying high-risk groups. Evidence-based strategies for carriers of BRCA1/2 and other high-penetrance genes include MRI from the age of 30 years and, where appropriate, riskreducing interventions.10,11 However, Delaloge emphasised growing attention to polygenic risk scores (PRS), where the cumulative effects of single-nucleotide polymorphisms can markedly refine risk stratification.
Two validated approaches for risk assessment in the general population were highlighted: the integration of PRS with clinical and hormonal risk factors, and AIbased image-derived risk modelling, the latter now being evaluated prospectively in the SMART trial.12,13
Tailored Strategies: Trials and Interventions
The implementation of risk-based screening is currently being tested in major randomised trials. Delaloge highlighted the MyPeBS trial in Europe, which she leads, and the WISDOM trial in the USA.14,15 These studies compare standard age-based screening to risk-adjusted intervals informed by PRS, breast density, and clinical factors. Results, expected in 2027, will determine whether personalised screening reduces rates of Stage II+ disease, while maintaining safety, feasibility, and acceptability.
Delaloge then reviewed risk-reduction strategies, including risk-reducing mastectomy, which may be cost-effective for women aged 30–55 years with a ≥35% lifetime risk;16 endocrine prevention using tamoxifen or aromatase inhibitors; and lifestyle interventions, noting evidence that lifestyle modification can produce mortality reductions comparable to some pharmacological approaches.17
She concluded by emphasising the need to integrate prevention and screening, address social inequities, and establish sustainable care pathways to support long-term implementation. Personalised prevention, Delaloge argued, represents not only a scientific evolution but a necessary public health transition.
COLORECTAL CANCER
Aspirin Chemoprevention
Andrew Chen, Massachusetts General Hospital, Boston, USA, presented on the emerging strategies for personalised prevention of gastrointestinal and colorectal cancer. He began by emphasising the critical role of colorectal cancer (CRC)
Personalised prevention represents not only a scientific evolution but a necessary public health transition

screening while advocating for more individualised approaches to primary prevention, particularly through the use of aspirin.
Chen outlined two main areas: precision prevention using age-based screening and molecularly guided aspirin therapy for localised CRC. Multiple case-control and cohort studies demonstrate consistent reductions in CRC incidence among aspirin users across diverse populations, supported by five RCTs showing lower recurrence of adenomas or CRC in high-risk individuals. Furthermore, over 50 cardiovascular prevention trials with linked CRC outcomes consistently showed reduced CRC incidence and mortality in aspirin users.18
The CAPP2 trial in patients with Lynch syndrome demonstrated a long-term reduction in CRC risk with aspirin.19 These findings informed the 2016 US Preventive Services Task Force (USPSTF) recommendation supporting low-dose aspirin in adults aged 50–59 years with ≥10% 10-year cardiovascular risk, marking a milestone in cancer prevention via medication.20 However, in 2022, the USPSTF reversed this recommendation, citing insufficient evidence for CRC prevention, largely due to the ASPREE trial, which randomised 19,114 adults aged ≥70 years (or ≥65 for USA minorities) to 100 mg aspirin versus placebo over 4.7 years. ASPREE found increased cancer mortality (hazard ratio: 1.31) in the aspirin arm, without differences in overall cancer incidence.21 Further analysis revealed that
the excess mortality was driven by higher incidence of Stage IV cancers.22
These findings contrast with prior trials, highlighting the importance of age and duration in aspirin prevention. Epidemiologic studies, including the Nurses’ Health Study and Health Professionals Follow-Up Study, indicate that initiating aspirin before the age of 70 years, particularly between 15–69 years, reduces CRC incidence by approximately 25%, whereas starting after 70 years of age offers no benefit.23 Similarly, the Japan Prevention of Atherosclerosis in Diabetes trial confirmed that aspirin’s protective effect is limited in older adults.24 Lifestyle factors also influence benefit: a CRC risk score based on five lifestyle factors predicts that individuals with poorer lifestyles experience the greatest absolute benefit from aspirin.25,26
Molecular-Guided Therapy
Molecularly-guided aspirin therapy is an exciting precision prevention approach. Chen’s group showed that adjuvant aspirin reduced CRC-specific mortality in patients with activating PIK3CA mutations, whereas wild-type tumours did not benefit.27 These findings were validated in the ALASCCA trial26 and supported by the SAC study in Switzerland, which, despite limited enrolment, suggested similar trends in PI3K pathway-altered cancers.28,29 Mechanistically, aspirin may enhance antitumour immunity by blocking thromboxane A2 and prostaglandin
Molecularly-guided aspirin therapy is an exciting precision prevention approach
signalling, rejuvenating exhausted T cells to eradicate PI3K-mutant tumour cells.30
While aspirin use represents a model for personalised CRC prevention, age of initiation, lifestyle, and tumour molecular profile (especially PIK3CA mutations) are critical determinants of possible benefit. Routine aspirin use is justified
References
1. de Koning et al. The state-of-the-art of personalised prevention. ESMO Congress, 17-21 October, 2025.
2. National Lung Screening Trial Research Team et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395-409.
3. de Koning HJ et al. Reduced lungcancer mortality with volume CT screening in a randomized trial. N Engl J Med. 2020;382(6):503-13.
4. Welz M et al. A comparative analysis of heterogeneity in lung cancer screening effectiveness in two randomised controlled trials. Nat Commun. 2025;16(1):8060.
5. de Nijs K et al. Projected effectiveness of lung cancer screening and concurrent smoking cessation support in the Netherlands. EClinicalMedicine. 2024;71:102570.
6. de Nijs K et al. Stage- and histologyspecific sensitivity for the detection of lung cancer of the NELSON screening protocol—a modeling study. Int J Cancer. 2025;157(11):2248-58.
7. de Koning HJ et al. Benefits and harms of computed tomography lung cancer screening strategies: a comparative modeling study for the U.S. Preventive Services Task Force. Ann Intern Med. 2014;160(5):311-20.
8. de Nijs K et al. Influence of changing patterns in lung cancer treatment and survival on the cost-effectiveness of CT screening: a modeling study. eClinicalMedicine. 2023;88:103446
9. Poiseuil M et al. Impact of organized and opportunistic screening on excess mortality and on social inequalities in breast cancer survival. Int J Cancer. 2025;156(3):518-28.
10. Kotsopoulos J et al. Bilateral oophorectomy and all-cause mortality in women with BRCA1 and BRCA2 sequence variations. JAMA Oncol. 2024;10(4):484-92.
for patients with Lynch syndrome, with emerging evidence supporting its use in adjuvant therapy for localised CRC with PI3K alterations. These findings highlight a paradigm shift toward inexpensive, lowcost, and personalised strategies to prevent and treat one of the most significant global cancers.
11. Lubinski J et al. MRI surveillance and breast cancer mortality in women with BRCA1 and BRCA2 sequence variations. JAMA Oncol. 2024;10(4):493-9.
12. Lee A et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med. 2019;21(8):1708-18.
13. Lehman CD et al. Deep learning vs traditional breast cancer risk models to support risk-based mammography screening. J Natl Cancer Inst. 2022;114(10):1355-63.
14. Ipina MA et al. Risk-based breast cancer screening: an expert Delphi consensus assessment of evidence under the EUCanScreen initiative. Abstract 67. The Early Detection of Cancer Conference, 21-23 October, 2025.
15. Ipina MA et al. Risk-based breast cancer screening: mapping three decades of evidence under the EUCanScreen initiative. Abstract 68. The Early Detection of Cancer Conference, 21-23 October, 2025.
16. Wei X et al. Defining lifetime risk thresholds for breast cancer surgical prevention. JAMA Oncol. 2025;11(9):1072-82.
17. Chlebowski RT et al. Dietary modification and breast cancer mortality: long-term follow-up of the women's health initiative randomized trial. J Clin Oncol. 2020;38(13): 1419-28.
18. Burn J et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year followup and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855-63.
19. Burn J et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081-7.
20. US Preventive Services Task Force. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;315(23):2564-75.
21. McNeil JJ et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med. 2018;379(16):1509-18.
22. McNeil JJ et al. Effect of aspirin on cancer incidence and mortality in older adults. J Natl Cancer Inst. 2021;113(3):258-65.
23. Guo CG et al. Aspirin use and risk of colorectal cancer among older adults. JAMA Oncol. 2021;7(3):428-35.
24. Okada S et al. Effect of aspirin on cancer chemoprevention in Japanese patients with type 2 diabetes: 10Year observational follow-up of a randomized controlled trial. Diabetes Care. 2018;41(8):1757-64.
25. Wang K et al. Healthy lifestyle, endoscopic screening, and colorectal cancer incidence and mortality in the United States: a nationwide cohort study. PLoS Med. 2021;18(2):e1003522.
26. Sikavi DR et al. Aspirin use and incidence of colorectal cancer according to lifestyle risk. JAMA Oncol. 2024;10(10):1354-61.
27. Liao X et al. Aspirin use, tumor PIK3CA mutation, and colorectalcancer survival. N Engl J Med. 2012;367(17):1596-606.
28. Martling A et al. Low-dose aspirin for PI3K-altered localized colorectal cancer. N Engl J Med. 2025;393(11):1051-64.
29. Güller U et al. Adjuvant aspirin treatment in PIK3CA-mutated colon cancer patients: the SAKK 41/13 prospective randomized placebocontrolled double-blind trial. Clin Cancer Res. 2025;31(15):3142-9.
30. Yang J et al. Aspirin prevents metastasis by limiting platelet TXA2 suppression of T cell immunity. Nature. 2025;640:1052-61.
Emerging Evidence-Based Treatment Strategies in Metastatic CastrationResistant Prostate Cancer
This satellite symposium took place on 19th October 2025, as part of the European Society for Medical Oncology (ESMO) Congress held in Berlin, Germany
Support: This content was funded by Bayer.
Chairperson: Rana McKay1
Speakers: Bertrand Tombal,2 Pedro Barata,3 Kambiz Rahbar4
1. University of California, San Diego, USA
2. Cliniques Universitaires Saint-Luc, Université Catholique de Louvain, Brussels, Belgium
3. University Hospitals Seidman Cancer Center, Cleveland, Ohio, USA
4. University Hospital Münster, Germany
Disclosure:
McKay has served as a consultant/advisor for Ambrx, Arcus, AstraZeneca, Aveo, Bayer, Blue Earth Diagnostics, Bristol Myers Squibb, Calithera, Caris, Daiichi-Sankyo, Dendreon, Exelixis, Johnson & Johnson, Lilly, Merck, Myovant, Neomorph, Novartis, Pfizer, Sanofi, SeaGen, Sorrento Therapeutics, Telix, and Tempus; and received institutional research funding from Artera AI, AstraZeneca, Bayer, Bristol Myers Squibb, Exelixis, Oncternal, and Tempus. Tombal has served as an investigator and paid advisor for Amgen, Astellas, Bayer, Ferring, Janssen, Myovant, Pfizer, and Sanofi. Barata has received honoraria from Astellas, AstraZeneca, Bayer, BMS, Caris Life Sciences, Dendreon, Eisai, EMD Serono, Exelixis, Guardant Health, Ipsen, Janssen, Myovant, OncLive, Pfizer, Seattle Genetics, Targeted Oncology, and UroToday; and undertaken contracted research for AstraZeneca, Caris Life Sciences, Exelixis, ESSA Pharma, Merck, Merus, and Myovant. Rahbar declares potential conflicts of interest with AAA/Novartis, ABX GmbH, ABX-CRO, Amgen, AstraZeneca, Bayer, Janssen-Cilag, Pharmtrace, SIRTEX, Siemens, and Urotrials.
Acknowledgements: Writing assistance was provided by Samantha Stanbury, PhD, Stockport, UK.
Disclaimer: The content of this article is based on a non-promotional symposium organised and funded by Bayer, for healthcare professionals only. The opinions expressed in this article belong solely to the named speakers.
Keywords: Androgen receptor pathway inhibitor (ARPI), combination therapy, lutetium-177, lutetium-177 prostate-specific membrane antigen-617 (177Lu-PSMA-617), metastatic castration-resistant prostate cancer (mCRPC), metastatic hormone-sensitive prostate cancer (mHSPC), poly(ADP-ribose) polymerase inhibitor (PARPI), prostate cancer, radiopharmaceutical, radium-223, treatment sequencing.
Citation: EMJ Oncol. 2025;13[1]:36-46. https://doi.org/10.33590/emjoncol/MIVW2802
Meeting Summary
This satellite symposium at the European Society for Medical Oncology (ESMO) Congress 2025, chaired by Rana McKay, Professor of Medicine and Urology at the University of California, San Diego, USA, explored evidence-based treatment strategies for metastatic castration-resistant prostate cancer (mCRPC) in the evolving treatment landscape. Bernard Tombal, Professor of Medicine at Cliniques Universitaires Saint-Luc, Université Catholique de Louvain, Brussels, Belgium, discussed combination treatment strategies with androgen receptor pathway inhibitors (ARPI) and radiopharmaceuticals. Pedro Barata, Medical Oncologist at the University Hospitals Seidman Cancer Center, Cleveland, Ohio, USA, addressed considerations for patients with homologous recombination repair (HRR) gene mutations, and the role of poly(ADPribose) polymerase inhibitors (PARPI) in this patient group. Kambiz Rahbar, Professor of Nuclear Medicine at University Hospital Münster, Germany, looked at emerging data on treatment sequencing in mCRPC following intensified treatment in the hormonesensitive phase. The faculty illustrated how the data they presented inform treatment strategies by applying it to treatment decisions, for example, clinical cases.
Patients with Metastatic CastrationResistant Prostate Cancer Today: Unmet Needs and Treatment Goals
Rana McKay
McKay opened this symposium by describing the natural history of mCRPC. Among patients experiencing disease recurrence after definitive treatment of localised prostate cancer, some may progress to metastatic disease while the cancer remains hormone sensitive (mHSPC) and later become castration resistant, while others may have rising prostate-specific antigen (PSA) while on androgen deprivation therapy (ADT), indicating castration resistance, before metastases develop. Both disease courses culminate in mCRPC, which McKay described as a ‘universally lethal’ disease state. Real-world median survival is only slightly longer than 2 years from the onset of mCRPC,1 although longer overall survival (OS) has been achieved in Phase III trials,2,3 suggesting a need to optimise use of the available treatment options to prolong the survival of patients with mCRPC. Recent data suggest that over 20% of patients developing mCRPC do not receive lifeprolonging therapy, and of those receiving first-line treatment for mCRPC, only about half go on to receive second-line therapy, further diminishing in subsequent lines.1
There is therefore an unmet need to improve treatment and outcomes for patients with mCRPC. McKay summarised the goals of mCRPC treatment as prolonging survival, optimising safety, and preserving and improving quality of life, emphasising the importance of considering patients’ concerns and the goals that are most important to them, within a shared decision-making framework.
Evolving Landscape and New Options in Metastatic CastrationResistant Prostate Cancer Treatment
Rana McKay,
Bertrand Tombal
McKay gave an overview of the treatment landscape across clinical phases of prostate cancer. ADT is the backbone of prostate cancer treatment. As the treatment landscape evolves, ADT is increasingly integrated with a variety of other treatment options, including ARPIs, PARPIs, and radiopharmaceuticals. ARPIs have a prominent role across the disease continuum, and may be used in high-risk localised disease, as well as mHSPC and mCRPC.4 McKay highlighted treatment advances in the mHSPC setting in the last decade, with increasing recognition
that intensified therapy with ARPIs improves progression-free survival when added to ADT, with or without taxane chemotherapy.5-10 She noted that this shapes the treatment history of patients entering the mCRPC setting, as increasing numbers of patients receive doublet or triplet therapy (ADT+ARPI±docetaxel) for mHSPC. However, recent data suggest that as many as 40% of patients with mHSPC progress to mCRPC without having received ARPI treatment.11,12
Tombal’s presentation focused on this patient group. For ARPI-naïve patients progressing on ADT, with or without docetaxel, the standard of care is an ARPI, with abiraterone or enzalutamide recommended as first-line options.13,14 Tombal posed the question of whether ARPI monotherapy is enough, and went on to discuss combination treatment approaches. PARPIs are indicated only for patients with HRR gene mutations; ARPI+PARPI combinations in this population were the subject of Barata’s presentation (below). Tombal discussed the role of radiopharmaceuticals in combination treatment with ARPIs, presenting recent clinical trial data that support this approach.
Radiopharmaceutical Plus
Androgen Receptor Pathway Inhibitor
Combination Therapy for Patients with Metastatic Castration-Resistant Prostate Cancer
Radium-223 is an α-emitting radionuclide which selectively targets metastases in the bone.15 Osteoblastic activity in bone metastases leads to high bone turnover; radium-223 is a calcium-mimetic and becomes incorporated into the bone matrix, where emission of α particles destroys both cancer cells and osteoblasts and osteoclasts. This disrupts a vicious cycle of positive feedback between osteoblasts and cancer cells, and makes the bone matrix ‘infertile soil’ for metastatic growth, thereby reducing the risk of bone complications and their negative impact on survival in prostate cancer.15
A significant survival benefit with radium-223 monotherapy was demonstrated in the ALSYMPCA trial,16 and supported by real-world OS findings in the REASSURE prospective observational study,17 which also confirmed a favourable long-term safety profile during 7 years’ follow-up.18
The Phase III EORTC-1333/PEACE-3 trial is an investigator-led academic trial, sponsored by the European Organisation for Research and Treatment of Cancer (EORTC), which assessed the effect of adding radium-223 to the ARPI enzalutamide in patients with mCRPC.2 Patients had ≥4 bone metastases but no known visceral metastases, and were asymptomatic or mildly symptomatic, with a World Health Organization performance status (WHO PS) of 0 or 1. All patients were receiving ADT; prior treatment with abiraterone and/or chemotherapy for mHSPC was permitted. In practice, very few enrolled patients had received abiraterone, while approximately 30% had received docetaxel. Patients were randomised to receive open-label treatment with enzalutamide 160 mg once daily, alone or with radium-223 55 kBq/kg every 4 weeks for six cycles. The primary endpoint was radiographic progression-free survival (rPFS); OS was a key secondary endpoint. A total of 446 patients were enrolled in PEACE-3, with a median age of 70 years in each treatment arm. The majority (88%) of patients randomised to radium-223 completed six cycles.2 Commenting on the high completion rate, Tombal recommended early use of radium-223 to allow completion of the treatment course to optimise efficacy, as completion of five or six cycles is associated with improved survival outcomes compared with fewer cycles.19,20
Addition of radium-223 to enzalutamide significantly increased median rPFS (19.4 versus 16.4 months; hazard ratio [HR]: 0.69; 95% CI: 0.54–0.87; p=0.0009; Figure 1A).2 At data cut-off for the final rPFS analysis, 80% of expected OS events had occurred. Interim OS analysis showed a significant OS benefit: median OS was extended by over 7 months, as of the data cut-off, with combination therapy versus enzalutamide alone (42.3 versus 35.0 months; HR: 0.69; 95% CI: 0.52–0.90; p=0.0031; Figure 1B).2 An Independent Data Monitoring
Figure 1: Efficacy of enzalutamide+tadium-223 versus enzalutamide monotherapy in the PEACE-III trial.2
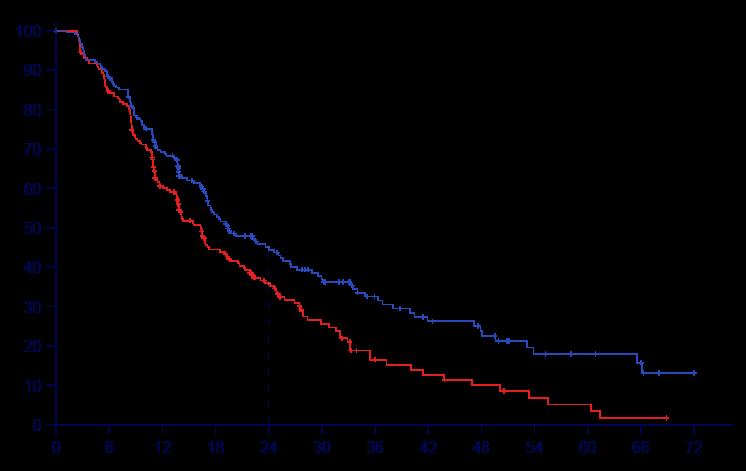
ENZA ENZA+Ra-223
ENZA ENZA+Ra-223
HR (95% CI) 0.69 (0.54–0.87)
Log-rank p-value 0.0009
Assumption of proportional hazard achieved
Patients at risk
Number of cumulative events
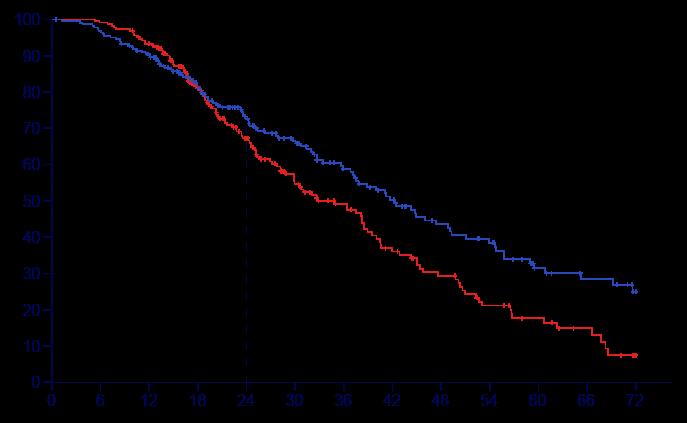
ENZA ENZA+Ra-223 ENZA ENZA+Ra-223
Patients
Number of cumulative events
HR (95% CI) 0.69 (0.52–0.90)
Log-rank p-value 0.0031*
*Pre-set level of significance for interim analysis was ≤0.0034.
A) rPFS (primary endpoint); and B) OS (interim analysis).
rPFS and OS were analysed using a Cox proportional hazards survival model stratified by baseline pain score, prior docetaxel, and BPA use at randomisation. The assumption of proportional hazards was not met for OS, and restricted mean survival time sensitivity analyses did not give unequivocal significance. Therefore, the study has continued to final OS analysis.
BPA: bone-protecting agent; ENZA: enzalutamide; HR: hazard ratio; OS: overall survival; Ra-223: radium-223; rPFS: radiographic progression-free survival.
Committee recommended continuation of the study to final OS analysis, with 100% of OS events to power the final analysis according to the statistical analysis plan, to confirm and further characterise results.
Tombal updated the audience with news that the trial has reached its final OS endpoint, and EORTC has revealed that the final analysis reinforces the findings of the interim analysis, confirming that the addition of radium-223 to enzalutamide significantly prolonged OS (unpublished data). Time to next systemic treatment was also significantly delayed by addition of radium-223 to enzalutamide: at 24 months’ follow-up, over 50% of patients randomised to enzalutamide alone had started their next line of therapy, while only 30% of patients on enzalutamide plus radium-223 had started further therapy, approximately 18 months after completing six cycles of radium-223.2 PSA data further supported a synergistic effect of radium-223 combined with enzalutamide. A significantly higher proportion of patients achieved a PSA response (≥90% decline) in the combination arm compared with the enzalutamide monotherapy arm (51% versus 34% at 6 months; 55% versus 38% at 12 months), with significantly shorter time to achieving confirmed PSA response.21 Safety data showed limited additional toxicity associated with adding radium-223 to enzalutamide. A slight increase in Grade 3/4 drug-related adverse events was observed (28% versus 19% of patients), but no individual adverse event increased in incidence by more than 5%.2
Tombal also presented efficacy data from a Phase II trial of the radioligand lutetium-177 prostate-specific membrane antigen-617 (177Lu-PSMA-617), in combination with enzalutamide. rPFS and OS were prolonged in patients with mCRPC receiving enzalutamide plus 177Lu-PSMA-617 (n=83) compared with enzalutamide alone (n=79),22 warranting validation of this regimen in a Phase III trial.
Protecting Bone Health in Patients with Metastatic Castration-Resistant Prostate Cancer
Tombal emphasised the importance of administering a bone-protecting agent (BPA) to patients undergoing treatment for mCRPC. Patients with bone metastases are at particularly high risk of skeletal-related events, including fractures, and guidelines recommend preventive administration of BPAs such as zoledronic acid or denosumab,4,13,14 although Tombal noted that adherence to this recommendation is suboptimal in real-world practice. During the PEACE-3 trial, a protocol amendment mandated the use of BPAs. Approaches to BPA use varied between study centres before it was mandated for all patients. Among approximately 120 patients enrolled before the protocol amendment, approximately half received preventive BPA before/during study treatment, while the other half received no BPA, or started BPA only after a fracture. An exploratory analysis in these patient subgroups showed that, as well as decreasing fracture rate,23 BPAs appeared to enhance the efficacy of the main study treatments, with considerably longer rPFS and OS in patients who were taking BPAs than those who were not.24 This hypothesis-generating posthoc analysis suggests a synergistic effect beyond bone protection.
Clinical case challenge #1: Androgen receptor pathway inhibitor- and docetaxel-naïve patient
Throughout the symposium, speakers contextualised the data they presented by applying it to treatment decisions for an example patient, with clinical characteristics reflecting the different settings they discussed. McKay introduced their model patient as a 65-year-old male, a retired teacher with a history of hypertension.
In the first scenario, the patient was treatment naïve, having presented to his primary care physician in January 2016 for routine health maintenance, where screening revealed PSA of 20 ng/ mL. MRI of the prostate showed locally extensive T3a disease, and CT of the chest, abdomen, and pelvis showed pelvic and retroperitoneal nodes, and an
isolated bone metastasis in the iliac bone, confirmed on bone scan. Prostate biopsy confirmed prostatic adenocarcinoma with Gleason score 4+3=7. He started ADT in February 2016, and received an external beam radiotherapy to the primary cancer and nodes. He achieved a PSA nadir <0.01 ng/mL after 3 months and remained on ADT for 3 years, but discontinued in February 2019 due to toxicity. The patient was monitored, and by July 2022, his PSA level had risen to 10 ng/mL, and the disease had progressed to T3b (locally invasive into the seminal vesicle) on MRI. No metastases were present on CT or bone scan. ADT monotherapy was restarted in August 2022; PSA returned to <0.01 ng/ mL after 2 months, and was controlled for approximately 2 years. However, in November 2024, PSA was rising, and by July 2025, PSA was 14 ng/mL, with a doubling time of 2 months. CT revealed pelvic and retroperitoneal nodes and metastases in the pelvis, lumbar spine, and ribs, confirmed on bone scan. Tumour somatic gene profiling showed no HRR mutations. He remained asymptomatic, with an Eastern Cooperative Oncology Group performance score (ECOG PS) of 0.
After presenting her overview of the mCRPC treatment landscape, McKay asked the audience how they would treat this patient on emergence of metastatic disease. The majority (53%) selected ARPI monotherapy.
Tombal repeated the question, considering the same patient case, at the end of his presentation. After seeing the PEACE-3 data, the majority of respondents (84%) selected enzalutamide+radium-223.
Treating Metastatic
Castration-Resistant Prostate Cancer in Patients with Homologous Recombination Repair Gene Mutations
Pedro Barata
HRR-related gene mutations are prevalent in patients with mCRPC (approximately
25% of patients)25 and are associated with worse prognosis than HRR mutation (HRRm)-negative status.25-27 Genetic testing is underused, with less than 40% of patients in Europe undergoing testing.28
The efficacy and safety of various ARPI+PARPI combinations have been investigated in clinical trials,29-35 including the Phase III TALAPRO-2 trial, which enrolled over 1,000 patients, including a cohort with various HRR mutations.35 Significant improvements in rPFS and OS were seen in HRRm-positive (HRRm+) patients receiving talazoparib plus enzalutamide compared with those on enzalutamide alone; median OS was extended by 14.0 months (45.1 versus 31.1 months; HR: 0.62; 95% CI: 0.48–0.81; p<0.0005).3 Barata remarked that this represents meaningful benefit to patients, highlighting the value of genetic testing to identify HRRm+ patients and give them the chance to benefit from ARPI+PARPI treatment. However, toxicities must be managed to keep patients on treatment. The combination regimen was associated with increased incidence of haematological adverse events, most commonly anaemia, which typically occurred in the first 3–4 months of treatment.3 Barata discussed how trial investigators had learned to manage anaemia, with strategies including dose adjustments and blood transfusions, and suggested that there would be a learning curve for urologists who lack experience with PARPIs.
ARPI+PARPI regimens that have demonstrated efficacy in mCRPC are now also being investigated in the hormonesensitive setting. The AMPLITUDE trial investigated nariparib plus abiraterone in patients with mHSPC and ≥1 HRR mutation. Barata showed results for a subgroup of patients with BRCA1/2 alterations, in whom addition of nariparib to standardof-care ARPI significantly improved rPFS.36 However, benefits of combination treatment must be balanced against increased toxicity. Barata commented that it will be important to see more safety data, with longer treatment exposure and follow-up, as PARPIs move into earlier disease settings.
Radiopharmaceutical+PARPI
Combination Therapy for Patients with HRR Mutation-Positive Metastatic Castration-Resistant Prostate Cancer
The Phase II COMRADE trial investigated the efficacy of radium-223 in combination with olaparib in patients with mCRPC. Although the HRRm+ subgroup was small (n=23), clinical benefit was observed in this cohort, with median rPFS extended from 4.7 to 5.5 months (HR: 0.47; 90% CI: 0.22–1.01) when radium-223 was added to olaparib.37
The combination of olaparib with 177LuPSMA-617 has also shown promising results in a Phase I study.38 Efficacy signals in these studies provide proof-of-concept for combining radiopharmaceuticals and PARPIs, warranting further investigation to explore the balance of efficacy and safety.
Clinical case challenge #2: Homologous recombination repair mutation-positive patient
Barata revisited the patient case described earlier by McKay, describing a slightly different scenario. In this case, the patient followed the same clinical course, with progression on ADT, but genetic testing on progression revealed a BRCA2 mutation (the most common HRR gene alteration found in mCRPC).35
Based on the clinical trial data Barata presented, ARPI+PARPI combination therapy would be the preferred treatment option for this patient. Discussing sequencing of ARPIs and PARPIs in ARPI-naïve patients, he advocated early use of combination therapy as a rational approach to optimising suppression of androgen signalling pathways. A treatment sequence that does not use the most effective treatment option first, Barata commented, risks losing the patient to follow-up before they move on to second-line therapy.
The combination of a PARPI with a radiopharmaceutical agent remains an investigational approach in the HRR+ mCRPC setting at present, but Phase I/ II data suggest this could be a promising approach in the future.
Sequencing of Treatments for Metastatic Castration-Resistant Prostate Cancer
Rana McKay, Kambiz Rahbar
The data on ARPI combinations presented by Tombal and Barata were from trials that included predominantly ARPI-naïve patients. However, McKay reiterated, ARPIs are indicated for mHSPC and non-metastatic CRPC, and increasing numbers of patients developing mCRPC will have received prior ARPI treatment. For patients progressing to mCRPC on an ARPI, several studies suggest that switching to a different ARPI provides limited benefit.29,30,39-42 Adding docetaxel can be beneficial for taxanenaïve patients,43,44 but for those who have received prior docetaxel, rechallenge is minimally effective.45,46 Alternative taxane chemotherapy with cabazitaxel can be considered post-docetaxel and ARPI.42 However, other treatment options also have a place in the management of pre-treated patients who are not candidates for PARPIs.
Role of Radiopharmaceuticals in Pre-treated Patients with Metastatic Castration-Resistant Prostate Cancer
Radiopharmaceuticals are indicated for second/third-line treatment following ARPIs and docetaxel for mCRPC,4,13,14 but may also have a role in earlier lines for patients with mCRPC who have already received ARPIs and/or docetaxel in the mHSPC setting. Two radiopharmaceutical products are approved for mCRPC: radium-223 for patients with bone metastases and no visceral metastases, and 177Lu-PSMA-617 for those with ≥1 PSMA-positive metastasis at any site. These products differ in terms of both type of radiation and targeted delivery of that radiation; Rahbar outlined their respective properties. Radium-223, as Tombal explained earlier, is an α-emitting radionuclide.15 α radiation has high energy but short range; radium-223 induces double-stranded DNA breaks that are difficult for tumour cells to repair,15 but has limited penetration. Cytotoxic effects are therefore localised to sites of uptake in bone metastases, limiting damage to healthy tissues.15 177Lu-PSMA-617 is a
radioligand that delivers β-particle radiation to PSMA-expressing tissues,47 including PSMA-positive metastases in bone and other organs. β radiation has lower energy, inducing single-stranded DNA breaks, but greater penetration than α radiation. Due to the penetrative nature of the radiation delivered by 177Lu-PSMA-617, patients are advised to avoid close contact with other people, including sleeping separately from partners, for up to 15 days after each administration (specific recommendations vary between countries). Both are administered over six cycles, radium-223 with a body weight-adapted dosage of 55 kBq/kg every 4 weeks, and 177Lu-PSMA-617 at a fixed dose of 7,400 MBq every 6 weeks. Antiemetics are recommended prior to the administration of 177Lu-PSMA-617.
Given their differing and complementary mechanisms of action, sequencing of radiopharmaceuticals is a rational approach. RaLu was a retrospective, multicentre study evaluating outcomes with 177LuPSMA-617 in approximately 200 patients who had received ≥1 cycle of radium-223 (Figure 2).48,49 Median OS from the start of 177Lu-PSMA-617 was 12 months (Figure 2),49 which is in line with published values for OS with 177Lu-PSMA-617, including in the VISION trial.47 Median OS from the start of radium-223 treatment was 33 months (Figure 2).49 No increase in toxicity was observed when 177Lu-PSMA-617 was given after radium-223, compared with its established safety profile, supporting the feasibility of this sequencing approach.48,49 The reverse sequence has been investigated in a small retrospective cohort study, LuRa (n=19), which suggested that radium-223 can be given after 177LuPSMA-617.50
Rahbar went on to describe the RADIANT trial (NCT04597125),51 an ongoing Phase IV study investigating the efficacy and safety of radium-223 in early lines of therapy in mCRPC. The study population comprises patients with mCRPC, with ≥2 bone metastases and no visceral metastases, who have progressed on or after one line of ARPI for an approved prostate cancer indication (mHSPC or mCRPC) plus taxane chemotherapy unless contraindicated or refused. A range of treatment histories are
represented in the trial population (Figure 3) to provide data on radium-223 sequencing in the contemporary mCRPC landscape. Over 600 patients have been randomised to receive either six cycles of radium-223 or a second ARPI (different to their firstline ARPI). The primary endpoint is OS, and results are expected in 2026.51
Clinical case challenge #3:
Pre-treated patient
The third clinical case challenge featured a patient with a history of prior ADT+ARPI+docetaxel triplet therapy for mHSPC. McKay reframed the example patient’s history, a 65-year-old male with no other significant medical history, but with a more substantial disease on diagnosis of prostate cancer than previous examples.
The patient had a high PSA level of 99 ng/ mL on initial presentation, and Stage T3a prostate cancer was detected on MRI. CT revealed pelvic and retroperitoneal nodes. Lung metastasis and multiple bone metastases were also detected and confirmed by bone scan and lung biopsy. In this scenario, it is appropriate to intensify therapy in the mHSPC setting; first-line treatment was darolutamide and docetaxel in addition to ADT, initiated in August 2022.
The patient did well on triplet therapy and achieved a PSA nadir <0.01 ng/mL after 2 months. Two years later (late 2024), PSA was rising, but lung metastases had resolved, and bone metastases were stable. However, new and increased bone metastases were found in the spine, pelvis, and ribs in July 2025. Lymph node metastases had also developed, but no visceral metastases were detected. PET-CT was performed and showed that lesions were PMSA-negative.
Rahbar considered treatment options for this patient, asking the audience to vote on their preferred treatment choice for firstline treatment of mCRPC following prior triplet therapy for mHSPC. Half indicated that they would select a second taxane chemotherapy regimen, and half opted for radiopharmaceuticals (predominantly radium-223, given this patient’s metastatic profile of PMSA-negative bone metastases and absence of visceral metastases).
Figure 2: Overall survival and safety in patients receiving sequential treatment with radium-223 and 177Lu-PMSA in the RaLu study.49
mCRPC diagnosis
Median time from diagnosis to first dose of 177Lu-PSMA: 37.8 months
Pre-baseline period
Life-prolonging therapies:
≥1 injection of radium -223: 100%
≥5 injections of radium -223: 69%
Taxanes: 75%
Abiraterone and enzalutamide: 60%
≥4 life-prolonging therapies: 58%
Baseline
Median duration of 177Lu-PSMA: 3.5 months
177Lu-PSMA treatment
Grade 3–4 TEAEs: 25%
Grade 3–4 laboratory abnormalities:
• 3 32% anaemia
• 1 17% thrombocytopenia
• 4 4% neutropenia
Follow-up period
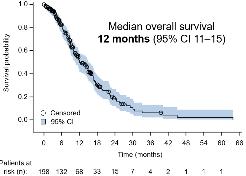
mOS from the start of 177Lu-PSMA 12 months (95% CI: 11 –15)
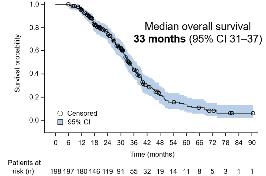
mOS from the start of radium-223 33 months (95% CI: 31–37)
mCRPC: metastatic castrate-resistant prostate cancer; mOS: median overall survival; PMSA: prostate membrane specific androgen; TEAE: treatment-emergent adverse event.
Figure 3: Prior treatments for metastatic hormone-sensitive prostate cancer and/or metastatic castrate-resistant prostate cancer in patients enrolling in the RADIANT trial.
ADT+ARPI (mHSPC)

ADT+ARPI (mCRPC)

ADT+ARPI (mCRPC)
ADT+chemotherapy (mCRPC)



ADT+ARPI+chemotherapy (mHSPC)
ADT+chemotherapy (mCRPC) RADIANT patients
ADT+ARPI (mCRPC)

ADT+ARPI (mCRPC)

ADT+chemotherapy (mCRPC)
RADIANT ClinicalTrials.gov ID: NCT04597125. ADT, androgen deprivation therapy; ARPI, androgen receptor pathway inhibitor; mCRPC, metastatic castration-resistant prostate cancer; mHSPC, metastatic hormone-sensitive prostate cancer. ClinicalTrials.gov. NCT04597125: https://clinicaltrials.gov/ct2/show/NCT04597125. Accessed October 2025.
ADT+chemotherapy (mHSPC)
ADT+ARPI (mHSPC)
ADT: androgen deprivation therapy; ARPI: androgen receptor pathway inhibitor; mCRPC: metastatic castrationresistant prostate cancer; mHSPC: metastatic hormone-sensitive prostate cancer.
Conclusions and Future Perspectives
The symposium concluded with a panel discussion. The speakers concurred that ADT+ARPI+chemotherapy triple therapy sets a high bar for efficacy and is likely to be standard-of-care for patients requiring escalation of therapy for mHSPC for the foreseeable future. In the rapidly evolving mCRPC landscape, research to understand where different treatment options sit in the treatment paradigm is important. The evidence presented in this symposium supports the use of combination therapy with ARPIs+radiopharmaceuticals early in the disease course in mCRPC, including asymptomatic disease. An audience poll indicated that, after seeing the PEACE-3 data presented in the symposium, most (>60%) saw a future role for radium-223 plus enzalutamide as first-
References
1. Freedland SJ et al. Real-world treatment patterns and overall survival among men with metastatic castrationresistant prostate cancer (mCRPC) in the US Medicare population. Prostate Cancer Prostatic Dis. 2024;27(2):32733.
2. Tombal B et al. Enzalutamide plus radium-223 in metastatic castrationresistant prostate cancer: results of the EORTC 1333/PEACE-3 trial. Ann Oncol. 2025;36(9):1058-67.
3. Fizazi K et al. Talazoparib plus enzalutamide in men with HRRdeficient metastatic castrationresistant prostate cancer: final overall survival results from the randomised, placebo-controlled, phase 3 TALAPRO-2 trial. Lancet. 2025;406(10502):461-74.
4. Cornford P et al. EAU - EANM - ESTRO - ESUR - ISUP - SIOG Guidelines on Prostate Cancer. 2025. Available at: https://uroweb.org/guidelines/prostatecancer. Last accessed: 22 October 2025.
5. Fizazi K et al. Abiraterone plus prednisone in metastatic, castrationsensitive prostate cancer. N Engl J Med. 2017;377(4):352-60.
6. Chi KN et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N Engl J Med. 2019;381(1):13-24.
7. Armstrong AJ et al. ARCHES: a randomized, phase III study of androgen
line therapy for mCRPC. ARPI+PARPI, and potentially radiopharmaceutical+PARPI, combinations have a role in the HRRm+ portion of the mCRPC population. Alongside anti-cancer agents, BPAs are a critical element of the management of mCRPC, to mitigate risk of fractures.
Data were also shown that support the approach of sequencing radiopharmaceuticals. Tombal noted that it is not a case of choosing between radium-223 and 177Lu-PMSA-617, but rather determining the position of each in the treatment sequence. This will depend on patient characteristics including metastatic sites and biomarkers, but Tombal stated that, in his view, many patients could benefit from early treatment with radium-223, while metastases are confined to the bones, followed by 177Lu-PMSA-617.
deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J Clin Oncol. 2019;37(32):2974-86.
8. Fizazi K et al. Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castrationsensitive prostate cancer (PEACE-1): a multicentre, open-label, randomised, phase 3 study with a 2 × 2 factorial design. Lancet. 2022;399(10336):1695707.
9. Smith MR et al. Darolutamide and survival in metastatic, hormonesensitive prostate cancer. N Engl J Med. 2022;386(12):1132-42.
10. Saad F et al. Darolutamide in combination with androgen-deprivation therapy in patients with metastatic hormone-sensitive prostate cancer from the phase III ARANOTE trial. J Clin Oncol. 2024;42(36):4271-81.
11. Castro E et al. Real-world treatment patterns and treatment sequences in the metastatic castration-resistant prostate cancer settings across Europe. Value Health. 2024. 27(6):S227.
12. Raval AD et al. Real-world evidence of combination therapy use in metastatic hormone-sensitive prostate cancer in the United States from 2017 to 2023. JCO Oncol Pract. 2025;21(8):1174-84.
13. Parker C et al. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(9):1119-34.
14. Fizazi K, Gillessen S. Updated treatment recommendations for prostate cancer from the ESMO Clinical Practice Guideline considering treatment intensification and use of novel systemic agents. Ann Oncol. 2023;34(6):557-63.
15. Farinea G et al. A new era for radium-223? Optimizing treatment by balancing efficacy and toxicity through combination therapies. Crit Rev Oncol Hematol. 2025;214:104819.
16. Parker C et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-23.
17. Higano CS et al. Clinical outcomes and treatment patterns in REASSURE: planned interim analysis of a real-world observational study of radium-223 in metastatic castration-resistant prostate cancer. EClinicalMedicine. 2023;60:101993.
18. Sartor AO et al. Long-term safety of radium-223 (Ra-223) in metastatic castration-resistant prostate cancer (mCRPC): 7-year follow-up from the largest global prospective study. J Clin Oncol. 2025;43(16_suppl):5048.
19. Buscombe J et al. Quantifying the survival benefit of completing all the six cycles of radium-223 therapy in patients with castrate-resistant prostate cancer with predominant bone metastases. World J Nucl Med. 2020;20(2):139-44.
20. Lunan-Taylor M et al. Radium-223 in men with metastatic castrationresistant prostate cancer: a systematic literature review of real-world outcomes in observational studies. Eur Urol Oncol. 2025;8(4):1150-64.
21. Choudhury A et al. PSA and alkaline phosphatase changes in the EORTC-1333 PEACE-3 study evaluating the addition of six cycles of radium 223 in metastatic castrationresistant prostate cancer (mCRPC) starting enzalutamide. J Clin Oncol. 2025;43(16_suppl):5062.
22. Emmett L et al. [177Lu]Lu-PSMA-617 plus enzalutamide in patients with metastatic castration-resistant prostate cancer (ENZA-p): an open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2024;25(5):563-71.
23. Gillessen S et al. Decrease in fracture rate with mandatory bone-protecting agents in the EORTC 1333/PEACE-3 trial comparing radium-223 combined with enzalutamide versus enzalutamide alone: a safety analysis. Eur Urol. 2025;87(3):285-8.
24. Saad F et al. Impact of bone protecting agents (BPA) on the efficacy and safety of enzalutamide vs. combination of Radium-223 (Ra223) and enzalutamide in asymptomatic or mildly symptomatic patients with bone metastatic castration-resistant prostate cancer (mCRPC): Sub-group analysis from EORTC-GUCG 1333/PEACE-3, an EORTC/CTI/CUOG/LACOG/UNICANCER randomized phase III study. Abstract 0518. EAU Congress, 21-24 March, 2025.
25. Lukashchuk N et al. Impact of DNA damage repair alterations on prostate cancer progression and metastasis. Front Oncol. 2023;13:1162644.
26. Uemura H et al. The prevalence of gene mutations in homologous recombination repair pathways in Japanese patients with metastatic castration-resistant prostate cancer in real-world clinical practice: The multiinstitutional observational ZENSHIN study. Cancer Med. 2023;12(5):526574.
27. Olmos D et al. Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Ann Oncol. 2024;35(5):458-72.
28. Castro E et al. Real-world treatment patterns and genetic testing in a metastatic castration-resistant prostate cancer setting in Europe. Future Oncol. 2025;21(9):1085-99.
29. de Bono J et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-102.
30. Hussain M et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383(24):2345-57.
31. Fizazi K et al. Rucaparib or physician's choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719-32.
32. Chi KN et al. Niraparib and abiraterone acetate for metastatic castrationresistant prostate cancer. J Clin Oncol. 2023;41(18):3339-51.
33. Chi KN et al. Niraparib and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: final overall survival analysis for the phase 3 MAGNITUDE trial. Eur Urol Oncol. 2025;8(4):986-98.
34. Saad F et al. Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(10):1094-108.
35. Fizazi K et al. First-line talazoparib with enzalutamide in HRR-deficient metastatic castration-resistant prostate cancer: the phase 3 TALAPRO-2 trial. Nat Med. 2024;30(1):257-64.
36. Attard G et al. Phase 3 AMPLITUDE trial: niraparib (NIRA) and abiraterone acetate plus prednisone (AAP) for metastatic castration-sensitive prostate cancer (mCSPC) patients (pts) with alterations in homologous recombination repair (HRR) genes. Abstract LBA5006. ASCO Annual Meeting, 30 May-3 June, 2025.
37. McKay R et al. A multicenter, randomized, phase 2, investigatorinitiated ETCTN trial of olaparib + radium-223 vs. radium-223 in men with castration-resistant prostate cancer (CRPC) with bone metastases (BM) (COMRADE): initial efficacy and biomarker analysis. Abstract 5007. ASCO Annual Meeting, 30 May-3 June, 2025.
38. Sandhu S et al. LuPARP: Phase I trial of [177Lu]Lu-PSMA-617 (LuPSMA) and olaparib in patients (pts) with metastatic castration resistant prostate cancer (mCRPC). Poster 2391P. ESMO Congress, 17-21 October, 2025.
39. Morris MJ et al. 177Lu-PSMA-617 versus a change of androgen receptor pathway inhibitor therapy for taxanenaive patients with progressive metastatic castration-resistant prostate cancer (PSMAfore): a phase 3, randomised, controlled trial. Lancet. 2024;404(10459):1227-39.
40. Fizazi K et al. Final overall survival and safety analyses of the phase III PSMAfore trial of [177Lu]Lu-PSMA-617 versus change of androgen receptor pathway inhibitor in taxane-naive patients with metastatic castrationresistant prostate cancer. Ann Oncol. 2025;36(11):1319-30.
41. Agarwal N et al. Cabozantinib plus atezolizumab in metastatic prostate cancer (CONTACT-02): final analyses from a phase 3, open-label, randomised trial. Lancet Oncol. 2025;26(7):860-76.
42. de Wit R et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med. 2019;381(26):2506-18.
43. de Bono JS et al. Subsequent chemotherapy and treatment patterns after abiraterone acetate in patients with metastatic castration-resistant prostate cancer: post hoc analysis of COU-AA-302. Eur Urol. 2017;71(4):65664.
44. Merseburger AS et al. Continuous enzalutamide after progression of metastatic castration-resistant prostate cancer treated with docetaxel (PRESIDE): an international, randomised, phase 3b study. Lancet Oncol. 2022;23(11):1398-408.
45. Lavaud P et al. Anticancer activity and tolerance of treatments received beyond progression in men treated upfront with androgen deprivation therapy with or without docetaxel for metastatic castration-naïve prostate cancer in the GETUG-AFU 15 phase 3 trial. Eur Urol. 2018;73(5):696-703.
46. Mahler M et al. UnCHAARTED territory: the role of docetaxel rechallenge following chemohormonal therapy for metastatic castration-sensitive prostate cancer. Urol Oncol. 2022;40(12):539. e17-539.e22.
47. Sartor O et al. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. 2021;385(12):1091-103.
48. Rahbar K et al. 177Lu-prostate-specific membrane antigen therapy in patients with metastatic castration-resistant prostate cancer and prior 223Ra (RALU Study). J Nucl Med. 2023;64(12):192531.
49. Rahbar K et al. Lutetium-177–prostatespecific membrane antigen (177LuPSMA) therapy in patients (pts) with prior radium-223 (223Ra). Ann Oncol. 2024;35(S2):S983-4.
50. Zarka J et al. LuRa: efficacy and tolerability of radium-223 following [177Lu]Lu-PSMA-617 in patients with metastatic castration-resistant prostate cancer. Poster 2403P. ESMO Congress, 17-21 October, 2025.
51. Bayer. A phase 4, randomized, openlabel, multicenter efficacy and safety study of standard dose of radium-223 dichloride vs. standard doses of novel anti-hormonal therapy (NAH) in patients with bone dominant metastatic castration resistant prostate cancer (mCRPC) progressing on/after one line of NAH (RADIANT). NCT04597125. https://clinicaltrials.gov/ct2/show/ NCT04597125.
Support:
Navigating a Dynamic Treatment Landscape: Established Therapies and Emerging Modalities for Patients with Lung Cancer
This symposium took place on 18th October 2025 as part of the European Society for Medical Oncology (ESMO) Congress held in Berlin, Germany
This industry-sponsored symposium is a medical education activity that is organised and funded by the medical department of Bristol Myers Squibb.
Chairperson: Rosalyn Juergens1
Speakers: Pasi A. Jänne,2 John V. Heymach3
1. Medical Oncology, Juravinski Cancer Centre, Hamilton, Ontario, Canada
2. Dana-Farber Cancer Institute, Boston, Massachusetts, USA
3. MD Anderson Cancer Center, Houston, Texas, USA
Disclosure:
Juergens has received advisory board/lecture fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, EMD Serono, GlaxoSmithKlein, Janssen, Eli Lilly, Merck Sharp & Dohme, Novartis, Pfizer, Roche, Sanofi, and Takeda; and research funding from Alkermes, Amgen, Astellas, AstraZeneca, Bold Therapeutics, Bristol Myers Squibb, Conjupro, Janssen, Merck Sharp & Dohme, Pfizer, and SignalChem.
Jänne has served as a consultant/advisor for AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Chugai Pharmaceuticals, Eli Lilly, SFJ Pharmaceuticals, Voronoi, Daiichi Sankyo, Biocartis, Novartis, Sanofi, Takeda Oncology, Mirati Therapeutics, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Eisai, Allorion Therapeutics, Accutar Biotech, Abbvie, Monte Rosa Therapeutics, Scorpion Therapeutics, Merus, Frontier Medicines, Hongyun Biotechnology, Duality Biologics, Blueprint Medicines, Dizal Pharma, GlaxoSmithKline, Tolremo, Myris Therapeutics, and Bristol Myers Squibb; received research funding from AstraZenenca, Daiichi Sankyo, PUMA, Eli Lilly, Boehringer Ingelheim, Revolution Medicines, Takeda Oncology, and Troper Wojcicki Foundation, with payments to institution; and post-marketing royalties from Dana Farber Cancer Institute owned intellectual property on EGFR mutations licensed to Lab Corp.
Heymach has received advisory board/lecture fees from AbbVie, Abdera Therapeutics, Amgen, AnHeart Therapeutics, Arrivent, AstraZeneca, BioNTech AG, Boehringer-Ingelheim, Bristol Myers Squibb, Curio Science, DAVA Oncology, Eli Lily & Co, EMD Serono, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Mirati Therapeutics, Moffitt Cancer Center, ModeX, Novartis Pharmaceuticals, OncoCyte, Pfizer, Regeneron, Sanofi, Spectrum Pharmaceuticals, and Takeda; research funding from AstraZeneca, Boehringer-Ingelheim, Mirati, Bristol Myers Squibb, and Takeda; and licensing/royalties from Spectrum Pharmaceuticals.
Acknowledgements: Medical writing assistance was provided by BGB Group, New York, USA.
Disclaimer: This summary is intended for educational use. The content within may refer to treatment, indications, or uses not approved by your local regulatory agency in your home country. Please always refer to your local prescribing information.
Bristol Myers Squibb does not endorse the promotion of unapproved products or indications.
The opinions expressed in this article belong solely to the named speakers.
Keywords: Antibody–drug conjugates (ADC), bispecific antibodies, epigenetic modifiers, immuno-oncology (I-O), KRAS, metastatic NSCLC (mNSCLC), non-small cell lung cancer (NSCLC), programmed death ligand 1 (PD-L1), small cell lung cancer (SCLC).
Citation: EMJ Oncol. 2025;13[1]:47-57. https://doi.org/10.33590/emjoncol/UWVT4034
Meeting Summary
Over the past two decades, thoracic oncology has undergone a profound transformation, moving from limited chemotherapy regimens to highly personalised strategies utilising targeted therapies and immuno-oncology (I-O). This symposium, featuring three leading experts, reviewed the current management of metastatic nonsmall cell lung cancer (mNSCLC) by focusing on three critical areas: optimising I-Obased regimens, particularly for programmed death-ligand 1 (PD-L1) tumour expression <1%; the evolving landscape of KRASG12C inhibition and resistance; and the promise of novel drug modalities, including bispecific antibodies, select epigenetic modifiers, and antibody–drug conjugates (ADC). Data presented reinforced the durable, long-term survival achieved with dual I-O and chemotherapy regimens in patients with PD-L1 negative disease and other difficult-to-treat subgroups, the clinical efficacy of approved KRASG12C inhibitors and those under investigation, and the exciting potential of novel targets and treatments for the treatment of both NSCLC and small-cell lung cancer (SCLC). Key discussions highlighted that, while outcomes for NSCLC have dramatically improved, challenges remain in achieving curative outcomes, managing resistance mechanisms, and developing effective biomarkers beyond PD-L1 status.
Introduction
The symposium presentation explored the established I-O landscape for first-line (1L) mNSCLC without actionable genomic alterations (AGA), the emerging landscape for KRASG12C-mutated NSCLC, and the development of various novel targets and agents for both NSCLC and SCLC. A major focus of the symposium was the dynamic nature of the treatment landscape for lung cancer, highlighting the need for physicians to be well-informed about both established therapies and emerging modalities in order to make informed treatment decisions for their patients.
Informing Optimal Treatment
Decisions with Immuno-oncologyBased Regimens for Patients with Programmed Death-Ligand 1 <1% Metastatic Non-small Cell Lung Cancer Rosalyn Juergens, Medical Oncology, Juravinski Cancer Centre, Hamilton, Ontario, Canada, began the symposium with an overview of the increasing number of 1L treatment options available for patients with mNSCLC without AGAs based on tumour PD-L1 expression levels. NSCLC accounts for nearly 90% of lung cancer cases, with most patients presenting with metastatic disease at the time of diagnosis. Select genomic alterations in NSCLC allow
PHARMA
for targeted therapy use where available. However, in patients with no known AGAs and those with AGAs for which there is no approved treatment in 1L (e.g., KRAS), tumour PD-L1 expression is key to informing treatment decisions in 1L mNSCLC.1,2 For patients with PD-L1 Tumor Proportion Score (TPS) ≥50%, various I-O monotherapies and I-O+chemotherapy combinations are commonly used. For patients with PD-L1 1–49% or PD-L1 <1%, combination regimens involving I-O+chemotherapy or dual I-O+chemotherapy, summarised in Figure 1, represent key standards of care.3-6
Outcomes for patients with tumour programmed death-ligand 1 <1% Juergens presented data for the KEYNOTE-189 trial, noting that the median overall survival (OS) of 22 months with pembrolizumab+chemotherapy in the intention-to-treat population was more than double that observed with placebo+chemotherapy in the same population. However, the median OS was numerically lower in patients with TPS PD-L1 <1%. The 5-year OS rate was 19% with pembrolizumab+chemotherapy across all levels of PD-L1 expression and 10% in the PD-L1 <1% subgroup.7
Figure 1: Landscape overview of key first-line programmed death-ligand 1-based immuno-oncology agents approved for treatment of metastatic non-small cell lung cancer.
Landscape overview of key 1L PD-(L)1-based I-O*
I-O mono
I-O + chemo



Atezolizumab
Pembrolizumab
Atezolizumab
Cemiplimab
Nivolumab + chemo
Pembrolizumab + chemo
Atezolizumab + chemo ± bevacizumab (NSQ only)
Cemiplimab + chemo
Cemiplimab + chemo
Sugemalimab + chemo
Tislelizumab + chemo (SQ only)

Tislelizumab + chemo (NSQ only)
Nivolumab + ipilimumab
I-O + I-O
I-O + I-O + chemo
Nivolumab + ipilimumab
Nivolumab + ipilimumab + two cycles of chemo
Durvalumab + tremelimumab + four cycles of chemo




*This diagram is intended for illustrative purposes only, and the treatment algorithm may vary by region.
†Refer to local materials such as prescribing information and/or Summary of Product Characteristics for each agent.
1L: first line; chemo: chemotherapy; I-O: immuno-oncology; mono: monotherapy; NSQ: non-squamous; PD-1: programmed death receptor-1; PD-L1: programmed death ligand 1; SQ: squamous.
Pembrolizumab
This was followed by 5-year data from a pooled analysis of KEYNOTE-189 and KEYNOTE-407. At Year 5, OS rates were 13% and 9%, for pembrolizumab+chemotherapy and placebo+chemotherapy, respectively, demonstrating limited clinical benefit for 1L pembrolizumab+chemotherapy treatment for patients with PD-L1 tumour expression <1%.8 Similarly, real-world data demonstrated that patients with tumour PD-L1 expression <1% had poorer longterm outcomes compared with those with tumour PD-L1 expression ≥50% receiving 1L I-O therapy+chemotherapy. At Year 5, OS rates were 11% and 25%, respectively, in patients with mNSCLC.9 These data demonstrate a remaining unmet need for treatments with long-lasting efficacy in patients with tumour PD-L1 expression <1%.
Dual immuno-oncology-based regimens for patients with PD-L1 <1% Anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and anti-PD-(L)1 treatments have distinct but complementary mechanisms of action to reactivate the immune system. Anti-CTLA-4 treatment induces de novo anti-tumour responses and promotes the emergence of memory T cells, while anti-PD-(L1) treatment restores anti-tumour T cell function and enhances pre-existing T cell response.10-12 The combination of dual I-O and chemotherapy has demonstrated long-term benefit in the 1L setting in NSCLC.13,14
Juergens discussed two key trials that evaluated dual I-O–based regimens: CheckMate 9LA and POSEIDON. In CheckMate 9LA, the 5-year OS rate in patients with tumour PD-L1 <1% was 22% for nivolumab+ipilimumab+chemotherapy versus 8% for chemotherapy alone, and the 6-year OS rates were 20% and 7%, respectively.13 In the POSEIDON trial, the 5-year OS rate was 6% in patients treated with durvalumab+tremelimumab+chemotherapy versus 4% for chemotherapy alone.14
While the median duration of response (DOR) with single I-O+chemotherapy in KEYNOTE-189 was 10.8 months, the
median DOR for dual I-O+chemotherapy in CheckMate 9LA was almost 18 months.7,13 Real-world data from Germany in the FINN study are consistent with outcomes from CheckMate 9LA, indicating that these durable outcomes are achievable in routine practice.15
Data show that the rate of high-grade (Grade 3/4) treatment-related adverse events (TRAE) for dual I-O+chemotherapy (CheckMate 9LA/POSEIDON) is similar to that of single agent I-O+chemotherapy (KEYNOTE 189), approximately 50% of patients. Crucially, the rate of treatment discontinuation due to TRAEs is consistent across these regimens (20–26% of patients), and treatment-related deaths remain rare.7,13,16 In an exploratory analysis, discontinuation due to TRAEs did not appear to negatively impact survival rates for patients in CheckMate 9LA.13 Analysis of real-world safety data found that the safety experiences of patients treated with 1L nivolumab+ipilimumab±chemotherapy were numerically similar to those treated with other approved I-O+chemotherapy combination therapies, supporting the use of these regimens in routine practice.17
Building Momentum in Research of Targeted Therapies for Patients with KRASG12C-Mutated Non-small Cell Lung Cancer
Pasi Jänne, Dana-Farber Cancer Institute, Boston, Massachusetts, USA, opened with an overview of the prevalence of KRAS mutations, which account for 25% of mutations in lung adenocarcinoma, with KRASG12 being the most common subtype.1,18 KRAS acts as a molecular ‘ON/OFF’ switch. In its ‘ON’ state, GTP binding to KRAS results in the activation of several cellsignalling pathways promoting cell growth and survival. The G12C mutation causes a persistent ‘ON’ state, and until recently, KRAS was considered undruggable.19 Discovery of the switch II binding pocket led to the development of KRASG12C-selective inhibitors, which irreversibly lock KRASG12C in the ‘OFF’ state.19,20
KRASG12C Inhibitors
Two KRASG12C inhibitors, adagrasib and sotorasib, received accelerated/conditional approvals in the USA and EU based on Phase II trials (KRYSTAL-1 and CodeBreak 100, respectively).21,22 Confirmatory Phase III trials KRYSTAL-12 and CodeBreak 200 compared these inhibitors to docetaxel in patients with locally advanced or mNSCLC who had prior treatment with platinum-based chemotherapy and antiPD-(L)1 therapy.22,23 In the KRYSTAL-12 trial, progression-free survival (PFS) improved from 3.8 to 5.5 months versus docetaxel, with an objective response rate (ORR) of 32% and a median DOR of 8.3 months with adagrasib.23 In KRYSTAL-1, an intracranial ORR of 42% was observed in patients with untreated central nervous system (CNS) metastases; this data set resulted in the only National Comprehensive Cancer Network (NCCN) Category 2A recommendation for patients with KRASG12C mNSCLC with CNS metastases.24,25 CodeBreak 200 demonstrated improved PFS with sotorasib versus docetaxel (5.6 versus 4.5 months). There was an ORR of 28% and a median DOR of 8.6 months with sotorasib.22 Sotorasib has an NCCN Category 2B recommendation for patients with KRASG12C mNSCLC with CNS metastases.25
KRASG12C
Inhibitors in Combination with Other Therapies
Improving efficacy with KRASG12C inhibitors remains crucial. KRASG12C inhibitor monotherapy can lead to resistance via multiple mechanisms.26 Combination therapies are being explored to circumvent these resistance pathways, including KRASG12C inhibitors combined with I-O±chemotherapy or anti-epidermal growth factor receptor (EGFR) therapies.27-35
Early data for I-O and KRASG12C inhibitors in the 1L setting are promising, particularly in patients with PD-L1 TPS ≥50%. For adagrasib+pembrolizumab (KRYSTAL-7, Phase II), the response rate was 61%, with a median PFS of 27.7 months in patients with PD-L1 TPS ≥50%.27 Separately, in a pooled analysis of Phase I LOXO-RAS 20001 and Phase III SUNRAY-01, there was a 78% response rate with olomorasib
and pembrolizumab in patients with PD-L1 TPS ≥50%.28 Calderasib+pembrolizumab demonstrated a response rate of 77% in patients with PD-L1 TPS ≥1% in the Phase I trial KANDLELIT-001.29 KRASG12C inhibitors+pembrolizumab±chemotherapy are being explored in numerous Phase III trials, e.g., KRYSTAL-7, KRYSTAL-4, SUNRAY-01, KRAScendo-2, and KANDLELIT-004.27-31
However, initial combinations of sotorasib with I-O (atezolizumab and pembrolizumab) as part of the CodeBreak 101 trial exhibited high rates of Grade 3 and 4 hepatic toxicities, despite attempts to reduce toxicity via a ‘lead-in’ phase.32 Jänne highlighted that he believes that combination strategies integrating I-O and KRAS inhibitors will ultimately be fruitful, noting that toxicity issues may be specific to certain drugs rather than the target itself.
Though combinations of sotorasib with I-O in CodeBreaK 101 exhibited high rates of adverse events, combining sotorasib with chemotherapy in a separate arm of the same trial demonstrated a 65% response rate and a PFS of 10.8 months.33 The ongoing Phase III Code Break 202 trial is investigating sotorasib and chemotherapy in patients with PD-L1 TPS <1%.34 Combinations of KRASG12C inhibitor+EGFR inhibitors are also being investigated. The KROCUS study, which evaluated fulzerasib with cetuximab, achieved a response rate of 80% and a modified PFS (mPFS) of 12.5 months.35
Selected ongoing Phase III trials of KRASG12C inhibitors in both 1L and 2L+ NSCLC and the potential future directions of KRAS-directed therapy are summarised in Figure 2.
Relentless Research for Novel Treatment Options: Meeting the Diverse Needs of Patients with Lung Cancer
John Heymach, MD Anderson Cancer Center, Houston, Texas, USA, launched this segment by emphasising that, while
Figure 2: Summary of select ongoing Phase 3 trials of KRASG12C inhibitors for KRASG12C non-small cell lung cancer and potential future directions for KRAS-directed therapy.
1L setting
I-O Combination Therapies
KRYSTAL-727
Tumour PD-L1 expression ≥50%
Adagrasib + pembrolizumab vs pembrolizumab
SUNRAY-01 Part A28
Tumour PD-L1 expression ≥50%
Olomorasib + pembrolizumab vs PBO + pembrolizumab
KRAScendo-231
PD-L1 all-comers
Divarasib + pembrolizumab vs pembrolizumab + chemo
KANDLELIT-00429
Tumour PD-L1 expression ≥50%
Calderasib + pembrolizumab vs PBO + pembrolizumab
Chemotherapy combinations
CodeBreaK 20234
Tumour PD-L1 expression ≤1%
Sotorasib + chemo vs pembrolizumab + chemo
I-O + Chemotherapy combinations
KRYSTAL-430
PD-L1 all-comers
Adagrasib + pembrolizumab + chemo vs PBO + pembrolizumab + chemo
SUNRAY-01 Part B28
PD-L1 all-comers
Olomorasib + pembrolizumab + chemo vs PBO + pembrolizumab + chemo
*Being investigated in solid tumours.
2L+ setting
Monotherapies
KRYSTAL-1223
PD-L1 all-comers
Adagrasib vs docetaxel
CodeBreaK 20022
PD-L1 all-comers
Sotorasib vs docetaxel
Potential future directions36-38
KRAS 'ON' inhibitors
KRASG12C: RMC-6291
KRASG12X: Daraxonrasib (RMC-6236)
Pan-KRAS inhibitors
Proteolysis-targeting chimeras
Pan-RAS inhibitors
Daraxonrasib (RMC-6236)*
Mutant selective inhibitors beyond KRASG12C
Novel I-O approaches
Tumor-infiltrating lymphocyte therapy
T cell receptor therapy
SHP2 inhibitor: TNO155, RMC-4630, JAB-3312
SOS1 inhibitor: BI1701963
MEK inhibitor: trametinib
MEK + FAK inhibitor: avutometinib and defactinib
mTOR inhibitor: everolimus*
AURKA inhibitor: LY329566*
1L: first line; 2L: second line; AURKA: aurora kinase A; chem: chemotherapy; FAK: focal adhesion kinase; I-O: immuno-oncology; MEK: mitogen-activated protein kinase; mTOR: mammalian target of rapamycin; PD-L1: programmed death-ligand 1; PBO: placebo; PROTAC: proteolysis targeting chimera; SHP2: SH2 domain-containing tyrosine phosphatase 2; SOS1: son of sevenless homologue 1; vs: versus.
significant progress has been achieved, the lack of curative outcomes highlights the need for a diverse range of new therapeutic modalities.
Novel Treatment Combinations and Modalities Shaping the Future of Non-small Cell Lung Cancer Treatment
Beyond PD-L1 status, co-occurring genomic alterations can play a role in outcomes for patients with NSCLC and thus help guide treatment decisions. For example, co-mutations in STK11 or KEAP1 are known to drive resistance to PD-(L)1 regimens in NSCLC.
Patients with KEAP1 wild-type tumours have a median OS of 16.6 months when treated with anti-programmed cell death protein 1 (PD-1)+chemotherapy, versus 7.6 months for those with KEAP1 mutations.39 These co-mutations also impact KRASG12C inhibitor efficacy.26 Intriguingly, while STK11/KEAP1 mutations are associated with resistance to anti-PD-1, they appear associated with greater sensitivity to anti-CTLA-4 therapy. Data from POSEIDON showed that, in the STK11/KEAP1 mutant subgroup, there was a greater relative benefit from adding tremelimumab to durvalumab+chemotherapy (hazard ratio: 0.64), suggesting that genomics can help providers select the most appropriate I-O regimen.39
Novel therapeutic approaches and emerging drug classes are being explored to improve outcomes in mNSCLC, with select classes, targets, and agents summarised in Figure 3. The first among these to be discussed by Heymach was lymphocyte-activation gene 3 (LAG3) immunotherapy. LAG-3 negatively regulates T cell proliferation and function. Combining LAG-3 and PD-1 inhibition has the potential to enhance antitumour activity through a synergistic effect.42,52 Relatlimab (approved for melanoma in combination with nivolumab) and fianlimab are anti-LAG-3 agents currently being investigated, while eftilagimod alpha is a soluble LAG-3 agent under investigation.42 In the RELATIVITY-104 study, relatlimab+nivolumab+chemotherapy showed improved median PFS (6.7 versus 6.0 months) and ORR (51.3% versus 43.7%) compared to nivolumab+chemotherapy alone, with no dramatic increase in adverse events. Survival benefit was observed in patient subgroups, with an mPFS of 9.8 versus 6.1 months in patients with tumour PD-L1 ≥1% and an mPFS of 8.3 versus 6.0 months in patients with non-squamous histology.40 Phase III trials of relatlimab (RELATIVITY-1093) and eftilagimod alpha (TACTI-004) are currently ongoing.41,43
Next, Heymach shifted focus to protein arginine methyltransferase 5 (PRMT5) and methionine adenosyltransferase 2A (MAT2A) inhibitors, which are examples of epigenetic modifiers. PRMT5 inhibitors are an exciting new class of drug for tumours
with MTAP deletions. The MTAP gene, commonly deleted alongside CDKN2A, is associated with poor prognosis and immunotherapy resistance. The deletion creates a vulnerability: inhibiting PRMT5 leads to a build-up of methylthioadenosine (MTA), which forms a complex with PRMT5 that is highly enriched in tumour cells. This complex is a synthetic lethal target that can be inhibited by MTA-cooperative PRMT5 inhibitors.53 Several second-generation PRMT5 inhibitors are showing substantial activity in Phase I/II trials across MTAPdeleted tumours. In a Phase I trial, BMS agent BMS-986504 (Bristol Myers Squibb, Princeton, New Jersey, USA) demonstrated 29% ORR, 10.5 months median DOR, and 80% disease control rate in patients with pretreated mNSCLC.44 Amgen’s agent AMG193 (Amgen, Thousand Oaks, California, USA) demonstrated 11.7% ORR and 8.3 months median DOR in the Phase I trial MTAPESTRY 101.45 Ideaya Biosciences’ MAT2A inhibitor IDE397 (Ideaya Biosciences, South San Francisco, California, USA) demonstrated an ORR of 38% (squamous) and 22% (non-squamous), and high disease control rate in the Phase I trial IDE397-001.46 These drugs are generally well-tolerated, with Grade ≥3 adverse event rates ranging from 14–18%.44-46 PRMT5 inhibitors are rapidly moving into Phase II/III trials as monotherapies and in combination with pembrolizumab and/or abemaciclib (cyclin-dependent kinase [CDK]4/6 inhibition), including Phase II/III trial of BMS-986504+pembrolizumab+ chemotherapy in 1L MTAP-del NSCLC (MountainTAP-29).54
Bispecific antibodies targeting both PD-(L)1 and VEGF were the next emerging drug class discussed by Heymach. This dual targeting of PD-(L)1 and vascular endothelial growth factor (VEGF) is hypothesised to both enhance immune response and antiangiogenesis and may be more effective than targeting either pathway alone.55 In 1L NSCLC, median PFS was 13.6 months, and ORR was 47.1% with pumitamig, an anti-PD-L1 x VEGF bispecific antibody in a Phase I/IIb study.47 A randomised study in China (HARMONi-2) demonstrated a striking difference in median PFS with ivonescimab, an anti-PD-1 x VEGF bispecific antibody
Figure 3: Emerging drug classes and key targets being investigated in non-small cell lung cancer.
Emerging drug class
I-O
Epigenetic modifiers
Bispecific antibodies
Antibody–drug conjugates
Example target(s)
Key agents
Relatlimab40,41 Eftilagimod alpha42,43 Fianlimab42 LAG-3
PRMT5 MAT2A
x VEGF
BMS-98650444 AMG 19345 IDE39746
Pumitamig47 Ivonescimab48
DB-131049 SHR-A200950 Iza-bren51 HER3
EGFR: epidermal growth factor receptor; HER3: human epidermal growth factor receptor 3; LAG-3: lymphocyte-activation gene 3; MAT2A: methionine adenosyltransferase 2A; PD-1: programmed death receptor-1; PD-L1: programmed death ligand 1; PRMT5: protein arginine N-methyltransferase 5; VEGF: vascular endothelial growth factor.
(11.1 months with ivonescimab versus 5.8 months for pembrolizumab alone).48 These drugs have tolerable safety profiles as single agents, with rates of Grade ≥3 TRAEs between 20–29%.47,48 The Phase II/III global study ROSETTA-LUNG-02 is investigating pumitamig combinations with chemotherapy and/or pembrolizumab, and multiple global Phase III studies (HARMONi-3 and HARMONi-7) are currently investigating ivonescimab combinations with chemotherapy and/or pembrolizumab.56-58
ADCs utilise an antibody to target specific cells, which are internalised, leading to the release of a cytotoxic payload to kill tumour cells, often with a bystander effect on nearby cells.59 Monospecific anti-human epidermal growth factor receptor (HER)3 ADCs under investigation for treatment of EGFR-mutant NSCLC include DB1310 and SHR 82009, which have shown ORRs of 28% and 36%, respectively, in early phase trials.49,50 The drug iza-bren (BLB 01D1), a potential first-in-class EGFR x HER3-targeting bispecific ADC with a topoisomerase I inhibitor (Ed-04) payload, demonstrated a high 54% response rate in patients with EGFR-mutant NSCLC who have progressed on third-generation
tyrosine kinase inhibitors but are chemo naive in a Phase I/II trial in China (NCT05194982/NCT05880706).51 Toxicities across this class of drugs are largely haematologic.49-51
Novel Strategies to Address Unmet Need in Small Cell Lung Cancer
There remains an unmet need for novel therapies with long-term outcomes and manageable safety in SCLC. Adding antiPD-(L)1 regimens to platinum-based chemotherapy has only shown modest OS improvement in patients with 1L extensive-stage SCLC, though recently, lurbinectedin+atezolizumab maintenance further improved survival.60-62 The bispecific T cell engager tarlatamab has recently demonstrated improved survival over chemotherapy in the relapsed/refractory setting and is under investigation in the 1L setting.63 Several innovative approaches are currently under rigorous investigation for SCLC, including innate immune inducers, bispecific antibodies, ADCs, and radiopharmaceuticals.
Atigotatug (anti-fucosylmonosialoganglioside-1 [Fuc-GM1]) is a
PD-(L)1
x EGFR
specific innate immune inducer currently being investigated for the treatment of SCLC. Targeting the ganglioside Fuc-GM1 has the potential to trigger various antitumour mechanisms, including complementdependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and antibodydependent cellular phagocytosis.64
As in NSCLC, combining checkpoint blockade (PD-1/PD-L1) with antiangiogenesis (VEGF inhibition) is currently under investigation in SCLC for treatment with the use of bispecific PD-(L)1 x VEGF antibodies pumitamig (BNT327) and ivonescimab.55
Numerous ADCs against promising new targets are in development for the treatment of SCLC, including ifinatamab deruxtecan (I-DXd), ZL-1310, and sacituzumab govitecan.65
Radiopharmaceuticals, antibodies linked to a radionucleotide or radioactive compound, combine the precise targeting of a monoclonal antibody with the potent cytotoxic effect of radiation. Among those discussed was RYZ101 (225Ac-DOTATATE), a first-in-class, highly potent alphaemitting radiopharmaceutical therapy being developed for somatostatin receptor Type 2 positive solid tumours, including SCLC.66
These novel treatment options and modalities represent relentless research efforts aimed at meeting the diverse needs of patients with lung cancer and overcoming the unmet need for improved long-term outcomes in SCLC.
Patient Cases and Panel Discussion
Two patient cases were discussed by faculty during the symposium, highlighting treatment options and the importance of making informed decisions about patient treatment.
The first case, presented by Juergens, focused on I-O treatment options, and the audience was asked to select which I-O-based therapy regimen they would choose for the patient with TTF1+
metastatic adenocarcinoma. The second case presented by Jänne focused on 1L treatment options for a hypothetical patient with KRASG12C mutations and tumour PD-L1 expression <1%. Questions for the audience included whether they would choose standard-of-care I-O+chemotherapy or enrolment in a clinical trial of the treatment options discussed earlier in Jänne’s presentation.
The interactive discussion with all faculty members focused on several critical challenges in thoracic oncology. Audience questions spurred discussions about the need for biomarkers beyond PD-L1, the exciting future for KRAS therapy, the desire for the presence of co-mutations STK11 and KEAP1 within routine next-generation sequencing panels, and brain penetrance of large molecules and inclusion of patients with baseline brain metastases in clinical studies.
The symposium concluded by addressing the most significant hurdles remaining in lung cancer treatment. Despite advances with next-generation drugs, a residual drug-tolerant population exists, and antitumour immunity remains largely unutilised in oncogenic driver settings (e.g., PD-1 inhibitors add limited benefit). Unlike the chemo-I-O setting, which produces a plateau on the survival curve, targeted therapies historically do not cure patients with advanced oncogene-addicted lung cancer. Better biomarkers are needed to select patients for various combination strategies, determine who requires treatment escalation in locally advanced disease, and understand complex drug modalities like ADCs. For ADCs, optimising treatment requires understanding not just of the cell surface target, but also payload sensitivity (e.g., sensitivity to topoisomerase inhibitors versus tubulin inhibitors). Finally, as longterm survival rates increase due to durable I-O and combination regimens, the challenge of managing survivorship and determining optimal duration of follow-up for long-term survivors is becoming increasingly relevant.
References
1. Bubendorf L et al. Nonsmall cell lung carcinoma: diagnostic difficulties in small biopsies and cytological specimens. Eur Respir Rev. 2017;26(144).
2. Goldschmidt JH et al. Treatment patterns and clinical outcomes among patients with metastatic non–small cell lung cancer without actionable genomic alterations previously treated with platinum-based chemotherapy and immunotherapy. Drugs Real World Outcomes. 2024;11(4):425-39.
3. National Cancer Institute (NIH). Drugs approved for lung cancer. 2025. Available at: https://www.cancer.gov/ about-cancer/treatment/drugs/lung. Last accessed: 5 November 2025.
4. European Medicines Agency. Medicines authorised for non-small-cell lung cancer. 2025. Available at: https:// www.ema.europa.eu/en/medicines/ download-medicine-data. Last accessed: 5 November 2025.
5. Government of Canada. Notice of Compliance with conditions (NOC/c). 2025. Available at: https://www.canada. ca/en/health-canada/services/drugshealth-products/drug-products/noticecompliance/conditions.html. Last accessed: 5 November 2025.
6. Pharmaceuticals and Medical Devices Agency. List of approved new drugs. Available at: https://www.pmda.go.jp/ review-services/drug-reviews/reviewinformation/p-drugs/0010.html. Last accessed: 5 November 2025.
7. Garassino MC et al. Pembrolizumab plus pemetrexed and platinum in nonsquamous non–small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 study. J Clin Oncol. 2023;41(11):1992-8.
8. Gadgeel SM et al. Pembrolizumab plus chemotherapy for metastatic NSCLC with programmed cell death ligand 1 tumor proportion score less than 1%: pooled analysis of outcomes after five years of follow-up. J Thorac Oncol. 2024;19(8):1228-41.
9. Waterhouse D et al. Five-year real-world survival outcomes of patients with metastatic non-small cell lung cancer receiving first-line immunotherapy-based regimens. Abstract 5946. AACR Annual Meeting, 25-30 April, 2025.
10. Das R et al. Combination therapy with anti-CTLA-4 and anti-PD1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194(3):950-9.
11. Wang C et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846-56.
12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64.
13. Ciuleanu TE et al. Nivolumab plus ipilimumab with chemotherapy as first-line treatment of patients with metastatic non-small-cell lung cancer: final, 6-year outcomes from CheckMate 9LA. ESMO Open. 2025;DOI:10.1016/j. esmoop.2025.105123.
14. Peters S et al. Durvalumab with or without tremelimumab in combination with chemotherapy in first-line metastatic NSCLC: five-year overall survival outcomes from the phase 3 POSEIDON trial. J Thorac Oncol. 2024;20(1):76-93.
15. Schumann C et al. First-line nivolumab plus ipilimumab with two cycles of platinum-based chemotherapy in patients with metastatic non-small cell lung cancer: interim data from the German non-interventional study FINN. Abstract 42P. ELCC, 26-29 March, 2025.
16. Peters S et al. Durvalumab ± tremelimumab + chemotherapy in firstline metastatic NSCLC: 5-year overall survival update from the POSEIDON study. Abstract LBA3. ESMO I-O Annual Congress, 6-8 December, 2023.
17. Betts KA et al. Real-world safety of first-line immuno-oncology combination therapies for advanced non-small-cell lung cancer. Future Oncol. 2024;20(13):851-62.
18. Yang H et al. New horizons in KRASmutant lung cancer: dawn after darkness. Front Oncol. 2019;9:953.
19. Liu P et al. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B. 2019;9(5):871-9.
20. Christensen JG et al. Targeting Krasg12c-mutant cancer with a mutation-specific inhibitor. J Intern Med. 2020;288(2):183-91.
21. Mok TSK et al. KRYSTAL-12: phase 3 study of adagrasib versus docetaxel in patients with previously treated locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Abstract LBA8509. ASCO Annual Meeting, 31 May-4 June, 2024.
22. de Langen AJ et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRASG12C mutation: a randomised, open-label, phase 3 trial. Lancet. 2023;401(10378):733-46.
23. Barlesi F et al. Adagrasib versus docetaxel in KRASG12C-mutated nonsmall-cell lung cancer (KRYSTAL-12): a randomised, open-label, phase 3 trial. Lancet. 2025;406:615-26.
24. Negrao MV et al. Intracranial efficacy of adagrasib in patients from the
KRYSTAL-1 trial with KRASG12Cmutated non–small-cell lung cancer who have untreated CNS metastases. J Clin Oncol. 2023;41(28):4472-7.
25. Referenced without permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for non-small cell lung cancer V.8.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. Available at: https://www.nccn.org/professionals/ physician_gls/pdf/cns.pdf. Last accessed: 17 Septemeber 2025.
26. Negrao MV et al. Co-mutations and KRAS G12C inhibitor efficacy in advanced NSCLC. Cancer Discov. 2023;13(7):1556-71.
27. Jänne PA et al. First-line adagrasib (ADA) with pembrolizumab (PEMBRO) in patients with advanced/metastatic KRASG12C-mutated non-small cell lung cancer (NSCLC) from the phase 2 portion of the KRYSTAL-7 study. Abstract 8500. ASCO Annual Meeting, 30 May-3 June, 2025.
28. Johnson ML et al. Efficacy and safety of 1L olomorasib plus pembrolizumab in KRAS G12C-mutant NSCLC: results from LOXO-RAS-20001 and SUNRAY-01. Abstract MA02.06. IASCLC WCLC, 6-9 September, 2025.
29. Sacher A et al. MK-1084 for KRAS G12C-mutated metastatic non–small-cell lung cancer: results from KANDELIT-001. Abstract 8605. ASCO Annual Meeting, 30 May-3 June, 2025.
30. Mirati Therapeutics Inc. A study of adagrasib plus pembrolizumab plus chemotherapy vs. placebo plus pembrolizumab plus chemotherapy in participants with previously untreated non-squamous non-small cell lung cancer with KRAS G12C mutation (KRYSTAL-4). NCT06875310. https:// clinicaltrials.gov/study/NCT06875310.
31. Hoffmann-La Roche. A study evaluating the efficacy and safety of divarasib and pembrolizumab versus pembrolizumab and pemetrexed and carboplatin or cisplatin in participants with previously untreated, KRAS G12C-mutated, advanced or metastatic non-squamous non-small cell lung cancer (Krascendo 2). NCT06793215. https://clinicaltrials. gov/study/NCT06793215.
32. Li BT et al. CodeBreaK 100/101: First report of safety and efficacy of sotorasib in combination with pembrolizumab or atezolizumab in advanced KRAS p.G12C NSCLC. Abstract OA03.06. WCLC, 6-9 August, 2022.
33. Li BT et al. Sotorasib plus carboplatin and pemetrexed in KRAS G12C advanced NSCLC: updated analysis from the international CodeBreaK 101 trial. Abstract 8512. ASCO Annual Meeting, 31 May-4 June, 2024.
34. Barlesi F et al. Trial in progress: sotorasib versus pembrolizumab in combination with platinum doublet chemotherapy as first-line treatment for metastatic or locally advanced, PD-L1 negative, KRAS G12C-mutated NSCLC (CodeBreaK 202). Abstract 103TiP. ELCC, 20-23 March, 2024.
35. Majem M et al. First-line (1L) fulzerasib + cetuximab in KRAS G12Cm advanced NSCLC: updated efficacy and safety from KROCUS study. Abstract LBA1. ELCC, 26-29 March, 2025.
36. Miyashita H et al. KRAS G12C inhibitor combination therapies: current evidence and challenge. Front Oncol. 2024;14:1380584.
37. Skoulidis F et al. Molecular determinants of sotorasib clinical efficacy in KRASG12C-mutated non-small-cell lung cancer. Nat Med. 2025;31:2755-67.
38. Cordani N et al. Proteolysis targeting chimera agents (PROTACs): new hope for overcoming the resistance mechanisms in oncogene-addicted non-small cell lung cancer. Int J Mol Sci. 2024;25(20):11214.
39. Skoulidis et al. CTLA4 blockade abrogates KEAP1/STK11-related resistance to PD-(L)1 inhibitors. Nature. 2024;635:462-71.
40. Girard N et al. Nivolumab plus relatlimab with platinum-doublet chemotherapy vs nivolumab plus platinum-doublet chemotherapy as first line treatment for stage IV or recurrent NSCLC: results from the randomized phase 2 RELATIVITY-104 study. Abstract LBA53. ESMO, 13-17 September, 2024.
41. Bristol-Myers Squibb. A study to compare the efficacy of nivolumab and relatlimab plus chemotherapy vs pembrolizumab plus chemotherapy for stage IV/recurrent non-squamous non-small cell lung cancer with PD-L1 expression ≥ 1% (RELATIVITY1093). NCT06561386. https://www. clinicaltrials.gov/study/NCT06561386.
42. Maruhashi T et al. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. 2020;8(2):e001014.
43. Immutep S.A.S. Study of eftilagimod alfa (efti) in combination with pembrolizumab and chemotherapy versus placebo in combination with pembrolizumab and chemotherapy in participants with metastatic non-small cell lung cancer (NSCLC) (TACTI-004). NCT06726265. https://www. clinicaltrials.gov/study/NCT06726265.
44. Jänne PA et al. BMS-986504 in patients with homozygous MTAP-deletion: clinical results in patients with NSCLC enrolled in CA240-0007. Abstract OA08.01. IASLC WCLC, 6-9 September, 2025.
45. Sacher A et al. Phase 1 dose escalation and initial dose expansion results of AMG 193, an MTA-cooperative PRMT5 inhibitor, in patients (pts) with MTAPdeleted solid tumors. Abstract 604O. ESMO, 13-17 September, 2024.
46. Herzberg B et al. Phase 1 expansion results of IDE397, a first-in-class, oral, MAT2A inhibitor in MTAP deleted nonsmall cell lung cancer and urothelial cancer. Abstract LBA 501. EORTC-NCIAACR, 23-25 October, 2024.
47. Wu C et al. A phase lb/ila trial to evaluate the safety and efficacy of PM8002/BNT327, a bispecific antibody targeting PD-L1 and VEGF-A, as a monotherapy in patients with advanced NSCLC. Abstract 8533. ASCO Annual Meeting, 31 May-4 June, 2024.
48. Xiong A et al. Ivonescimab versus pembrolizumab for PD-L1-positive nonsmall cell lung cancer (HARMONi-2): a randomised, double-blind, phase 3 study in China. Lancet. 2025;405:83949.
49. Lisberg AE et al. DB-1310, a HER3targeted ADC, in patients with advanced solid tumors: preliminary results from the Phase 1/2a trial. Abstract 3000. ASCO Annual Meeting, 30 May-3 June, 2025.
50. Zhou Q et al. Phase 1 study of SHR-A2009, a HER3-targeted ADC, in pretreated EGFR-mutated NSCLC. Abstract 642P. ESMO, 13-17 September, 2024.
51. Fang W et al. Phase I/II study of iza-bren(BL-B01D1) as monotherapy in patients with locally advanced or metastatic EGFR mutated NSCLC. Abstract OA10.03. IASLC WCLC, 6-9 September, 2025.
52. Ruffo E et al. Lymphocyte-activation gene 3 (LAG3): the next immune checkpoint receptor. Semin Immunol. 2019;42:101305.
53. Fan N et al. Methylthioadenosine phosphorylase deficiency in tumors: a compelling therapeutic target. Front Cell Dev Biol. 2023;11:1173356.
54. Bristol-Myers Squibb. A study to compare the combination of BMS986504 with pembrolizumab and chemotherapy versus placebo plus pembrolizumab and chemotherapy in first-line metastatic non-small cell lung cancer participants with homozygous mtap deletion (MountainTAP-29). NCT07063745. https://www. clinicaltrials.gov/study/NCT07063745.
55. Chen W et al. Bispecific antibody for lung cancer: mechanisms and clinical insights. Front Immunol.
2025;16:1572802.
56. Peters S et al. A global phase 2/3, randomized, open-label trial of BNT327/PM8002 in combination with chemotherapy (chemo) in first-line (1L) non-small cell lung cancer (NSCLC). Abstract TPS8670. ASCO Annual Meeting, 30 May-3 June, 2025.
57. Summit Therapeutics. Clinical study of ivonescimab for first-line treatment of metastatic NSCLC patients. NCT05899608. https://www. clinicaltrials.gov/study/NCT05899608.
58. Summit Therapeutics. Clinical study of ivonescimab for first-line treatment of metastatic NSCLC patients with high PD-L1 (HARMONi-7). NCT06767514. https://www.clinicaltrials.gov/study/ NCT06767514.
59. Coleman N et al. Antibody-drug conjugates in lung cancer: dawn of a new era? NPJ Precis Oncol. 2023;7(5):1-12.
60. Paz-Ares L et al. Durvalumab, with or without tremelimumab, plus platinumetoposide in first-line treatment of extensive-stage small-cell lung cancer: 3-year overall survival update from CASPIAN. ESMO Open. 2022;7(2):100408.
61. Horn L et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220-9.
62. Paz-Ares L et al. Efficacy and safety of first-line maintenance therapy with lurbinectedin plus atezolizumab in extensive-stage small-cell lung cancer (IMforte): a randomised, multicentre, open-label, phase 3 trial. Lancet. 2025;405(10495):2129-43.
63. Rudin CM et al. Tarlatamab versus chemotherapy as second-line treatment for small cell lung cancer (SCLC): primary analysis of the phase 3 DeLLphi-304 study. Abstract LBA8008. ASCO Annual Meeting, 30 May-3 June, 2025.
64. Paz-Ares L et al. The TIGOS trial: a randomized double-blind, phase 3 trial of atigotatug + nivolumab fixeddose combination with chemotherapy vs atezolizumab with chemotherapy in patients with first-line extensivestage small cell lung cancer. Abstract TPS8127. ASCO Annual Meeting, 30 May-3 June, 2025.
65. Wang H et al. The evolving landscape of antibody-drug conjugates in small cell lung cancer: from research progress to clinical application. Biochim Biophys Acta Rev Cancer. 2025;1880(6):189445.
66. Puri S et al. RYZ101 (225Ac-DOTATATE) + carboplatin + etoposide + atezolizumab in somatostatin receptor expressing extensive-stage small-cell lung cancer. Abstract P1.13A.09. IASLC WCLC, 7-10 September, 2024.
Optimising Patient Care: Cutting-Edge Nutritional Strategies in Oncology
This satellite symposium took place on the 20th October 2025 as part of the European Society of Medical Oncology (ESMO) Congress held in Berlin, Germany
Support: The symposium and publication of this article was funded by Nestlé Health Science.
Chairperson: Florian Scotté1,2
Speakers: Florian Scotté,1,2 Jann Arends,3 Alessandro Laviano,4 Paula Ravasco,5 Riccardo Caccialanza6,7
1. Institut Gustave Roussy, Villejuif, France
2. Université Paris Saclay, France
3. University of Freiburg, Freiburg im Breisgau, Germany
4. Sapienza University Hospital Sant'Andrea, Rome, Italy
5. Universidade Católica Portuguesa, Lisbon, Portugal
6. University of Milan, Italy
7. Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
Disclosure: Scotté has acted as a consultant/advisory board member/speaker for Helsinn, Sanofi, MSD, Prostrakan (now acquired by Kyowa Kirin), LEO Pharma, Janssen, Hospira (now acquired by Pfizer), Boehringer, Amgen, Pierre Fabre Oncologie, Vifor Pharma, Pfizer, BMS, GSK, BeiGene (now known as BeOne Medicines), Gilead, Daiichi Sankyo, Arrow, Viatris, Pharmanovia, Chugai, Fresenius, Nutricia, and Nestlé. Arends has received honoraria from Nestlé, Danone, Fresenius Kabi, and Pfizer. Laviano has received honoraria for independent lectures at industry-sponsored events; is a member of the Nutricia Oncology advisory board; is a consultant for EO3; and sits on the Board of Directors for Danone Nutricia Campus. Ravasco has received honoraria for independent lectures at industry-sponsored events and grants/ research support. Caccialanza has received grants/research support and/or acted as a paid consultant/speaker bureau member for Astellas, Akern, Baxter, B. Braun, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, Fresenius Kabi, Lionhealth, Mediam, MSD, Nestlé Health Science, Novartis, Nutrica, Nutrisens, Pfizer, Roche, Servier, Takeda, and Viatris.
Acknowledgements: Writing assistance was provided by Helen Boreham, HB Medical (UK) Ltd, Wetherby, UK.
Keywords: Anabolism, cachexia, cancer, immunonutrition, malnutrition, muscle wasting, nutrition, oral nutritional supplements (ONS).
Citation: EMJ Oncol. 2025;13[1]:58-70. https://doi.org/10.33590/emjoncol/PWPE6967
Meeting Summary
During this symposium at the European Society of Medical Oncology (ESMO) Congress 2025, leading experts in oncology and nutrition discussed state-of-theart nutritional strategies aimed at optimising clinical outcomes and quality of life (QoL) for patients with cancer. Chairperson Florian Scotté, Gustave Roussy Institute and Université Paris Saclay, France, opened the session by emphasising that, as cancer survival improves, QoL has become a key outcome, making supportive care (including nutrition) an essential component of comprehensive oncology care. The pathophysiological mechanisms underlying malnutrition and cancer-associated wasting were then explored by Jann Arends, University of Freiburg, Germany, highlighting their impact on treatment response, survival, and QoL. Alessandro Laviano, Sapienza University, Rome, Italy, reviewed current and emerging therapeutic approaches for malnutrition and cachexia management, focusing on the latest guideline recommendations and stressing the need for early multimodal interventions combining nutrition, physical activity, and pharmacological support. Strategies to overcome anabolic resistance and enhance nutrient balance were then presented by Paula Ravasco, Catholic University in Lisbon, Portugal, who emphasised the importance of adequate dietary intake and tailored counselling. Finally, Riccardo Caccialanza, University of Milan; and Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, summarised the robust clinical evidence supporting immunonutrition in surgical oncology and its emerging applications in systemic treatment.
Introduction: The Foundation of Nutritional Oncology
Due to major advances in anticancer therapies, many patients now live longer with cancer as a chronic disease, making QoL a key care priority, Scotté explained. The success of oncology care increasingly depends on the quality of supportive care, which includes pain, psychological, and nutritional management, as defined by the Multinational Association of Supportive Care in Cancer (MASCC).
Both prehabilitation and rehabilitation are crucial to optimise patients’ functional status and QoL during and after treatment. Prehabilitation, in particular, can help patients better tolerate the toxicity of anticancer therapies and mitigate the adverse effects of malnutrition, he emphasised.1,2 Global publication trends show that both sarcopenia and malnutrition have become major research focuses in modern oncology.3 There is also increasing evidence supporting the importance of
adopting a multidisciplinary approach to sarcopenia management, using specialist tools and involving experts in nutrition, exercise, and pharmacological care.4
Evidence consistently demonstrates that low muscle mass is linked to increased dose-limiting toxicity from anticancer therapy and a higher likelihood of treatment discontinuation.5-7 Weight loss and malnutrition also negatively impact overall survival (OS), stressed Scotté, as shown in several recent studies. In a longitudinal analysis of 1,406 patients with incurable cancer, the severity of malnutrition, graded by a combination of weight loss and BMI, was associated with reduced OS.8 This link between sarcopenia and increased rates of both all-cause and cancer-specific mortality was further confirmed in a recent study involving over 1,000 patients with cancer.9 Similarly, a meta-analysis of 100 studies demonstrated an association between lean mass/sarcopenia and mortality across a range of cancer types.10
Scotté highlighted a recent analysis presented at this year’s ESMO Congress that analysed five different cachexia indicators: skeletal muscle index, weight loss, modified Glasgow prognostic score, loss of appetite, and level of growth differentiation factor (GDF)-15 (cut-off of 2,320 pg/mL). An increased number of these cachexia-related factors was associated with reduced OS in patients with solid cancer.11 Data from Nutrition Day 2024 (unpublished) confirmed the significantly greater impact of malnutrition on cohorts of patients with cancer versus patients without cancer, with an associated increase in 30-day mortality and hospital readmission rates.
Collectively, this evidence underscores the importance of early screening for malnutrition at diagnosis and throughout treatment, Scotté stressed. Cancer-related malnutrition affects not only individual patients but also the healthcare system, leading to more postoperative complications and infections, longer hospital stays, greater readmissions, and increased hospital costs.12-20 Results from a survey of over 700 individuals with a broad range of tumours have also highlighted the importance of nutrition from a patient perspective. Overall, 83% of patients with cancer considered nutrition as important during their treatment and recovery, and 59% mentioned that the topic of nutrition should be addressed earlier.21 This highlights the need for early nutritional assessment and improved patient education.
Scotté reviewed ESMO Clinical Practice Guidelines on Cancer Cachexia, which provide recommendations for evaluation and management across different clinical settings.22 He also presented the Gustave Roussy model for early assessment, integrating nutrition with global symptom management.
Scotté concluded by calling for the development of “Nutritional Oncology,” involving both cancer specialists and supportive-care professionals, and invited the clinicians to join MASCC to advance multidisciplinary practice and education in this area.
Decoding Malnutrition and CancerAssociated Wasting: Understanding the Clinical Landscape
Arends began by highlighting the high prevalence of nutrition-impact symptoms in patients with cancer, which include anorexia, nausea, dysphagia, and diarrhoea.23 These problems may be associated with the tumour itself, anticancer treatments, or metabolic derangements. Overall, approximately 30% of patients with cancer have signs of malnutrition, with prevalence exceeding 50% in upper gastrointestinal (GI) cancers, and this has a significant impact on clinical outcomes.24 Large-scale studies, each involving more than 3,000 patients, have confirmed that malnutrition, whether defined by weight loss or Global Leadership Initiative for Malnutrition (GLIM) criteria, is consistently associated with reduced OS, regardless of cancer stage.25-29 Malnutrition is also associated with reduced tumour responsiveness to treatment and decreased QoL, Arends explained, making it “of high relevance to clinical oncology.”
Arends stressed the need to adapt nutritional care to patients’ disease stage and prognosis.22 In advanced cancer, cachexia cannot be reversed in the last weeks of life. At the end of life, care should instead focus on alleviating symptoms, avoiding invasive interventions like tube feeding or parenteral nutrition. However, for patients with a survival probability of more than a few months or weeks, regular screening and nutritional intervention are warranted.22 Arends confirmed that most guidelines on nutritional care advocate for repeated screening of patients for the risk or presence of malnutrition. This should be followed by an in-depth diagnostic assessment, including food intake, nutrition impact symptoms, weight loss, BMI, and metabolic derangements.
On the subject of diagnosis, Arends clarified that cachexia is often mistakenly equated with complete muscle wasting, which represents a very late stage of the condition. In reality, cachexia can appear much earlier and is now defined as ≥5% involuntary weight loss combined with
metabolic changes such as systemic inflammation.30 He explained that there are two basic subtypes of malnutrition: starvation-type malnutrition, with normal metabolism or ketosis in response to inadequate food intake; and diseaseassociated malnutrition, characterised by metabolic changes, systemic inflammation, and cachexia.30 Treatment for starvationtype malnutrition involves basic support for food intake or feeding to meet energy and protein requirements. In contrast, Arends stressed that management of diseaseassociated malnutrition remains “a major unsolved problem.” These patients typically present with insulin resistance, glucose intolerance, and anabolic resistance, meaning that protein provision does not elicit the expected anabolic response seen in healthy individuals. Additional challenges include anorexia, fatigue, and activated catabolism affecting multiple organs (including the heart, skeletal muscle, fat, kidney, gut, and brain), underscoring the systemic nature of this condition.
The systemic inflammation associated with disease-associated malnutrition is driven by the interaction between the immune system and malignant cells. The tumour microenvironment and surrounding stroma produce proinflammatory mediators, such as TNF-α, IL-6, and IL-1, which spill into the circulation and act systemically. These mediators promote fat depletion and muscle wasting, signal anorexia and fatigue in the central nervous system, and trigger metabolic changes in the liver, including altered protein synthesis.31,32 Unsurprisingly, this systemic inflammation is linked to adverse clinical outcomes, with studies showing increased infection rates and reduced survival. Multicentre studies confirm that patients with malnutrition or systemic inflammation have significantly higher hazard ratios for death (up to threefold compared to those without inflammation) and poorer survival even in early-stage or palliative settings.33-35
Arends added that other issues in patients with advanced cancer can also interfere with food intake, such as chronic pain, depression, psychological distress, and social barriers. To address these,
a multidisciplinary approach is crucial, involving nurses, psychologists, social workers, oncologists, palliative care/ rehabilitation specialists, and dietitians/nutritionists.22
In conclusion, Arends emphasised the importance of early screening for malnutrition and supporting with food intake/feeding when metabolism is normal. For patients with systemic inflammation or complex issues interfering with intake, multiprofessional care should be initiated. At the end of life, the focus should remain on symptomatic care only.
Breaking the Cycle: Therapeutic Strategies for Malnutrition and Muscle Wasting
“We have learned a lot about cachexia since the original consensus definition was published in 2011,” noted Laviano.36 Cachexia is now recognised as a systemic disease, rather than merely a nutritional syndrome, associated with immune suppression and complex changes across multiple organ systems, including the brain, liver, and gut microbiota.37
Cachexia is highly prevalent, although the true incidence depends on how it is defined. In the TRACERx lung-cancer study, 29% of patients met muscle loss criteria for cachexia, while over half (51%) showed changes in body composition. Notably, these changes in body composition were associated with worse cancer-specific survival outcomes.38
Beyond survival, QoL remains a critical but often overlooked dimension in cancer treatment. According to a recent analysis, only 10% of studies supporting the approval of new oncology drugs considered QoL as an outcome.39 Global QoL is closely linked to cachexia, and evidence shows that patients with poor QoL due to nutritional impairment at the start of their clinical journey rarely improve.40,41 This highlights a major gap in care: extending survival without preserving QoL is not enough. If patients live 6 more months, but spend 3 or 4 months bedridden
and dependent, something is missing, Laviano commented. Oncology care must therefore aim not only to maximise efficacy and minimise toxicity but also to maintain patients’ functional independence and wellbeing.
Encouragingly, results from a recent study in colorectal cancer indicate that patients with low muscle mass at the outset of their
clinical journey, who are able to improve muscle mass, can achieve a survival curve similar to those without adverse body composition changes.42 To achieve these improvements, Laviano highlighted the importance of adopting a parallel approach in which the oncological pathway is closely aligned with the metabolic nutritional pathway throughout the clinical journey (Figure 1).
Oncological PathwayMetabolic Nutritional Pathway
Disease staging Nutritional screening and assessment (max within 4 weeks from cancer diagnosis) Nutritional metaboloic interventions (tailored to patients’ specific needs, drugs) First-line therapy
Second-line treatment“Upper level” nutritional/ metaboloic strategies (tailored ar tificial nutrition, specific nutrition)
Figure 1: Integrating nutrition and oncology.6
A recent study described a potential genetic predisposition to cachexia development or resistance in skeletal human muscle, characterised by different molecular subtypes.43 Although genetic predisposition may play a role, current strategies must focus on preventable and treatable factors.44 Nutritional intervention remains vital and should address energy and protein requirements alongside key nutrients such as amino acids and derivatives, omega-3 fatty acids, and vitamin D. For patients with poor calorie and protein intake, tools such as dietary counselling, nutritional supplements, and enteral or parenteral nutrition are recommended by the ESMO guidelines.22 As highlighted by other speakers, multimodal intervention encompassing nutritional, exercise, pharmaceutical, and psychosocial aspects is also key.
Timing of treatment is another critical factor influencing cachexia development and clinical outcomes. A recent study from Japan showed a substantial increase in cachexia prevalence from 34% to 50% in the 1-month period between suspicion of cancer and final diagnosis, underscoring the need for early intervention.45 In the EFFORT prospective randomised trial, early screening for malnutrition and provision of nutritional support reduced 30-day mortality risk by 43% across different types of cancers.46 Exercise and physical activity have also been linked to improved diseasefree survival in patients with colorectal cancer.47 However, Laviano explained that anabolic resistance can counteract these positive interventions. For example, secondary analysis of the EFFORT trial revealed that higher baseline inflammation, as measured by C-reactive protein levels, was associated with lower clinical benefit from nutritional support.48
For the specific treatment of anorexia, olanzapine has shown clinical benefits.49 Pharmacological approaches to cachexia are also under development, including anti-GDF-15 antibody (ponsegromab), anti-IL monoclonal antibody (tocilizumab), and anti-senescence-associated secretory phenotype combination therapies (quercetin and dasatinib).37
In summary, malnutrition in patients with cancer is a complex condition contributing to poor outcomes. The key message is that cachexia and malnutrition are both preventable and treatable, provided that intervention begins early. Early diagnosis is essential for meaningful results, and optimal prevention and treatment should address all contributing factors, which may evolve throughout the clinical journey.
Optimising Nutrient Balance: Strategies to Enhance Anabolism in Oncology
Low muscle mass has an adverse impact on cancer outcomes, Ravasco reiterated, including increased postoperative complications, higher chemotherapyinduced toxicity, and reduced survival.50 In the recently published LEANOX trial, chemotherapy dosing based on lean body mass was associated with a 47% lower risk of developing significant neurotoxicity and showed a trend towards improved Grade ≥2 neurotoxicity-free survival compared to standard body surface area dosing. Importantly, there was no compromise in relapse-free or OS with this muscle massbased dosing approach.51
Muscle protein synthesis and degradation can become unbalanced in patients with cancer due to anabolic resistance. Anabolic resistance describes a decline in muscle responsiveness to normally robust anabolic stimuli such as protein intake and resistance exercise. It can be exacerbated by long periods of muscle disuse and is more common in older adults.52,53 “But we have a way of overcoming this anabolic resistance if we maintain an adequate stimulus with protein intake during the whole course of the journey,” she confirmed.
Individualised nutritional counselling has been shown to prevent the deterioration of nutritional status and reduce the incidence of malnutrition in patients with head and neck squamous cell carcinoma (HNSCC) undergoing chemo/radiotherapy.54 More recently, the PRIMe trial demonstrated the positive impact of dietary counselling on
protein intake. Over half of patients in the 2.0 g/kg/day group maintained or gained muscle mass after 12 weeks of targeted nutritional intervention.55
However, maintaining optimal levels of protein intake through diet alone is challenging, making oral nutritional supplements (ONS) a critical component of care. In a study comparing dietary counselling alone to counselling plus ONS, use of ONS significantly reduced interruptions and the need for changes in scheduled anticancer treatments.56 Adequate dosing of ONS is key to achieving these clinical benefits, with evidence showing that higher energy and protein intake from ONS leads to better outcomes than lower amounts.57 In the EFFORT trial,
the integration of nutritional support was associated with a 43% reduction in 30-day mortality, decreased functional decline, and improved QoL (Figure 2).46
In addition to protein, Ravasco highlighted omega-3 fatty acids as important nutrients that can mitigate inflammatory and catabolic responses in patients with cancer. A systematic review showed that supplementation of omega-3s favoured better recovery from weight loss and may reduce acute chemotherapy toxicity, including mucosal toxicity, peripheral neuropathy, and GI toxicity.58
Several specific protocols have been developed to aid in the implementation of nutritional therapy in routine clinical
OR: odds ratio; vs: versus.
Figure 2: EFFORT trial: individualised nutrition support reduced 30-day mortality.46
Kaplan–Meier survival estimates
oncology practice. One example is PRONTO, which integrates European Society for Clinical Nutrition and Metabolism (ESPEN) and ESMO guidelines and defines three checkpoints for assessing patients’ nutrition risk when starting or continuing anticancer therapy.59,60 The ESMO guideline is very explicit: every patient with cancer should undergo systematic screening and individualised nutritional assessment, considering nutritional status, symptoms, clinical history, and metabolic dysfunctions. These elements must be integrated to design a tailored intervention adapted to each patient’s needs.22
Patient compliance to nutritional intervention is critical to achieving effective results. ONSs are highly effective, especially when combined with individualised counselling. “We need to work with the patient to find common ground for greater acceptance of ONSs,” Ravasco confirmed. For example, it is important to consider loss/alterations in taste that can occur as a consequence of cancer treatment. Evidence indicates that compliance is improved when patients are offered a wide variety of flavours.60 Higher compliance has also been observed with high-energy-dense ONS (≥2 kcal/mL versus ≤1.5 kcal/mL), low volumes, and clear formulas.59-61 When oral intake is insufficient or not feasible, enteral and parenteral nutrition must be considered and integrated into the care plan to ensure adequate nutritional support.
In summary, Ravasco reiterated the importance of integrating nutritional intervention into standard oncology pathways to deliver state-of-the-art cancer care for patients.59 This approach helps maintain or restore nutritional status, improving physical function, metabolic health, and QoL. Adequate nutrition corrects macro and micronutrient deficits, reduces the frequency and duration of treatment interruptions, and lowers rehospitalisation rates, Ravasco confirmed. Stimulation of physical activity is also essential to reverse frailty and reduce disability. Ultimately, these interventions will help to achieve the ultimate goal, which is increasing patient survival, she concluded.
Navigating Immunonutrition: Established Foundations and New Frontiers
As in the wider oncology setting, Caccialanza emphasised that nutritional status impacts postoperative outcomes in cancer surgery, including survival and QoL. Malnutrition also imposes a significant economic burden: every 1 USD invested in nutrition therapy for hospitalised patients can save over 50 USD in hospital costs.62
However, nutrition is not just calories and protein. A new concept, immunonutrition, has emerged, defined as the modulation of immune system activity or its consequences through nutrients or specific food components provided in amounts above those normally consumed in the diet.63 The effectiveness of immunonutrition in oncological surgery is supported by a robust evidence base of over 100 RCTs and 62 meta-analyses across multiple cancer types. As a result, perioperative immunonutrition is now included in prehabilitation programmes and protocols, including the Enhanced Recovery After Surgery (ERAS) protocol, as metabolic preparation for surgical stress. The recently updated ERAS guidelines recommend pre- and postoperative immunonutrition, including arginine, omega-3 fatty acids, and nucleotides, for all patients undergoing colorectal surgery, not just those who are malnourished.64 Similarly, ESPEN guidelines recommend immunonutrition for patients undergoing major tumour surgery and those with GI cancers.65 In a step towards implementing this evidence in the realworld setting, Caccialanza and colleagues in Lombardy, Italy, created a Clinical Nutrition Network. This initiative aims to overcome inequalities in nutritional care management by making nutritional screening mandatory in all hospitals. One of the key targets is to provide immunonutrition to at least 70% of patients with cancer.
Caccialanza reviewed extensive evidence demonstrating that immunonutrition reduces the length of hospital stay for patients with cancer.66 Recent data indicate reductions over 2 days for colorectal cancer, 3 days for oral cancer, and nearly 2 days
3: The IMPATOX trial.82
A) PFS and B) OS according to study treatment in compliant patients (compliance ≥75%). mo: months; OS: overall survival; PFS: progression-free survival.
Figure
for gastric cancer.67-73 These reductions translate into millions of EUR in potential cost-savings, he stressed. Few studies have been undertaken in bladder cancer, but preliminary case-series data on the use of immunonutrition in radical cystectomy are promising, and randomised trials are ongoing.74,75
Beyond the surgical setting, Caccialanza described immunonutrition in systemic treatment as “the new frontier.”
Immunonutrition modulates the tumour microenvironment towards a cytotoxic profile, reducing inflammation (a key driver of cachexia) and enhancing immune system activation to counteract neoplastic growth.76-78 Bibliometric analysis has shown that immunonutrition during hospitalisation can reduce mortality and improve QoL in patients with cancer.79
Immunonutrition may also act as a potential enhancer of systemic therapies through immune-inflammatory modulation. Small studies have shown improvements in inflammatory markers and immune responses in patients with HNSCC undergoing radiochemotherapy.80,81 The larger Phase 3 double-blind IMPATOX trial evaluated the impact of immunomodulating nutritional formula in patients with HNSCC receiving adjuvant chemoradiotherapy. Although immunonutrition did not reduce severe mucositis (primary endpoint),
References
1. Silver JK. Cancer prehabilitation and its role in improving health outcomes and reducing health care costs. Semin Oncol Nurs. 2015;31(1):13-30.
2. Gillis C et al. Prehabilitation versus rehabilitation: a randomized control trial in patients undergoing colorectal resection for cancer. Anesthesiology. 2014;121(5):937-47.
3. Liu R et al. Global trends in sarcopenia and cancer over the past 10 years: a bibliometric analysis. Discov Oncol. 2025;16(1):1358.
4. Park WT et al. Multidisciplinary approach to sarcopenia: a narrative review. J Yeungnam Med Sci. 2023;40(4):352-63.
5. Daly L et al. A window beneath the skin: how computed tomography assessment of body composition can
compliant patients showed improved longterm survival (Figure 3).82
However, patients in the IMPATOX study did not receive nutritional counselling, which is a key limitation, Caccialanza noted.83 To address this, two new studies are ongoing. The first is an RCT comparing nutritional counselling plus immunonutrition ONS versus counselling plus isocaloric/ isonitrogenous ONS in patients with HNSCC undergoing chemoradiotherapy. The primary endpoint is toxicity. The second trial is a multicentre, randomised, open-label, Phase 2 study evaluating immunonutrition in improving immunotherapy efficacy in patients with metastatic non-small cell lung cancer. Patients will receive counselling plus immunonutrition ONS or counselling alone.84,85 Preliminary trials have shown positive results in survival and chemotherapy completion rates in patients receiving immunonutrition plus systemic anticancer therapy.86-88
Future directions may include combining immunotherapy with immunonutrition and exploring interactions with the gut microbiome, Caccialanza suggested.89,90 Hopefully, in the next few years, there will be a clear idea of the real effectiveness of immunonutrition in patients with cancer during systemic treatment, from both a clinical and economic point of view, he concluded.
assist in the identification of hidden wasting conditions in oncology that profoundly impact outcomes. Proc Nutr Soc. 2018;77(2):135-51.
6. Muscaritoli M et al. From guidelines to clinical practice: a roadmap for oncologists for nutrition therapy for cancer patients. Ther Adv Med Oncol. 2019;11:1758835919880084.
7. Hsueh SW et al. A comparison of the MNA-SF, MUST, and NRS2002 nutritional tools in predicting treatment incompletion of concurrent chemoradiotherapy in patients with head and neck cancer. Support Care Cancer. 2021;29(9):5455-62.
8. Vagnildhaug OM et al. The applicability of a weight loss grading system in cancer cachexia: a longitudinal analysis. J Cachexia Sarcopenia Muscle. 2017;8(5):789-97.
9. Cui F et al. Association of sarcopenia with all-cause and cause-specific mortality in cancer patients: development and validation of a 3-year and 5-year survival prediction model. BMC Cancer. 2025;25:919.
10. Au PCM et al. Sarcopenia and mortality in cancer: a meta-analysis. Osteoporos Sarcopenia. 2021;7(Suppl 1):S28-33.
11. Gottmann L et al. Cancer-associated cachexia indicators in patients with solid cancer. Oral presentation 2804MO. ESMO Congress, 17-21 October, 2025.
12. Zhao B et al. The impact of preoperative underweight status on postoperative complication and survival outcome of gastric cancer patients: a systematic review and meta-analysis. Nutr Cancer. 2018;70(8):1254-63.
13. Zheng HL et al. Effects of preoperative malnutrition on short- and long-term outcomes of patients with gastric cancer: can we do better? Ann Surg Oncol. 2017;24(11):3376-85.
14. Pressoir M et al. Prevalence, risk factors and clinical implications of malnutrition in French Comprehensive Cancer Centres. Br J Cancer. 2010;102(6):966-71.
15. Bossi P et al. Malnutrition management in oncology: An expert view on controversial issues and future perspectives. Front Oncol. 2022;12:910770.
16. D’Almeida CA et al. Prevalence of malnutrition in older hospitalized cancer patients: a multicenter and multiregional study. J Nutr Health Aging. 2020;24(2):166-71.
17. Na BG et al. Nutritional status of patients with cancer: a prospective cohort study of 1,588 hospitalized patients. Nutr Cancer. 2018;70(8):1228-36.
18. Kwaan MR et al. Readmission after colorectal surgery is related to preoperative clinical conditions and major complications. Dis Colon Rectum. 2013;56(9):1087-92.
19. van Vugt JLA et al. Low skeletal muscle mass is associated with increased hospital expenditure in patients undergoing cancer surgery of the alimentary tract. PLoS One. 2017;12(10):e0186547.
20. Planas M et al. Prevalence of hospital malnutrition in cancer patients: a sub-analysis of the PREDyCES® study. Support Cancer Care. 2016;24(1): 429-35.
21. Blanchard H et al. Perspective and experience of patients on nutritional care and education across the cancer journey. Clinical Nutrition ESPEN. 2024;63:1012.
22. Arends J et al. Cancer cachexia in adult patients: ESMO Clinical Practice Guidelines. ESMO Open. 2021;6(3):100092.
23. Walsh D et al. Symptoms and prognosis in advanced cancer. Support Care Cancer. 2022;10(5): 385-8.
24. Marshall K et al. Prevalence of malnutrition and impact on clinical outcomes in cancer services: a comparison of two time points. Clin Nutr. 2019;38(2):644-51.
25. Dewys WD et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med. 1980;69(4):491-7.
26. Martin L et al. Diagnostic criteria for the classification of cancer-associated weight loss. J Clin Oncol. 2015;33(1):90-9.
27. Yin L et al. Association of malnutrition, as defined by the PG-SGA, ESPEN 2015, and GLIM criteria, with complications in esophageal cancer patients after esophagectomy. Front Nutr. 2021;8:632546.
28. GlobalSurg Collaborative and NIHR Global Health Unit on Global Surgery. Impact of malnutrition on early outcomes after cancer surgery: an international, multicentre, prospective cohort study. Lancet Glob Health. 2023;11(3):e341-9.
29. Shachar SS et al. Prognostic value of sarcopenia in adults with solid tumours: a meta-analysis and systematic review. Eur J Cancer. 2016;57:58-67.
30. Cederholm T et al. GLIM criteria for the diagnosis of malnutrition - a consensus report from the global clinical nutrition community. Clin Nutr. 2019;38(1):1-9.
31. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860-7.
32. Arends J et al. ESPEN expert group recommendations for action against cancer-related malnutrition. Clin Nutr. 2017;36(5):1187-96.
33. Liu Y et al. Prognostic role of Glasgow prognostic score in patients with colorectal cancer: evidence from population studies. Sci Rep. 2017;7(1):6144.
34. Laird BJ et al. Prognostic factors in patients with advanced cancer: a comparison of clinicopathological factors and the development of an inflammation-based prognostic system. Clin Cancer Res. 2013;19(19):5456-64.
35. Li W et al. Nutritional management interventions and multi-dimensional outcomes in frail and pre-frail older adults: a systematic review and meta-analysis. Arch Gerontol Geriatr. 2024;125:105480.
36. Fearon K et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12(5):489-95.
37. Thakir TM et al. Cancer therapy and cachexia. J Clin Invest. 2025;135(15):e191934.
38. Al-Sawaf O et al. Body composition and lung cancer-associated cachexia in TRACERx. Nat Med. 2023;29(4):846-58.
39. Tibau A et al. Predictors of withdrawal of anticancer drug indications granted accelerated approval: a retrospective cohort study. EClinicalMedicine. 2025;84:103088.
40. Zhang Y et al. Sarcopenia is a prognostic factor of adverse effects and mortality in patients with tumour:
a systematic review and metaanalysis. J Cachexia Sarcopenia Muscle. 2024;15(6):2295-310.
41. Singhal S et al. Nutritional impairment and quality of life trajectories among older adults with advanced cancer. J Am Geriatr Soc. 2025;73(9):2789-97.
42. Li Z et al. Association of perioperative skeletal muscle index change with outcome in colorectal cancer patients. J Cachexia Sarcopenia Muscle. 2024;15(6):2519-35.
43. Bhatt BJ et al. Molecular subtypes of human skeletal muscle in cancer cachexia. Nature. 2025;646:973-82.
44. Prado CM et al. Nutrition interventions to treat low muscle mass in cancer. J Cachexia Sarcopenia Muscle. 2020;11(2):366-80.
45. Katsushima U et al. Impact of time to treatment initiation on the development of cachexia and clinical outcomes in lung cancer. Jpn J Clin Oncol. 2025;55(5):505-13.
46. Bargetzi L et al. Nutritional support during the hospital stay reduces mortality in patients with different types of cancers: secondary analysis of a prospective randomized trial. Ann Oncol. 2021;32(8):1025-33.
47. Courneya K et al. Structured exercise after adjuvant chemotherapy for colon cancer. N Engl J Med. 2025;393(1): 13-25.
48. Merker M et al. Association of baseline inflammation with effectiveness of nutritional support among patients with disease-related malnutrition. JAMA Netw Open. 2020;3(3):e200663.
49. Othman N et al. Olanzapine for anorexia in patients with incurable cancer and cachexia (OlAnCa): a double-blind, placebo-controlled, randomized clinical trial. J Natl Compr Canc Netw. 2025;23(9):385-92.
50. Pamoukdjian F et al. Prevalence and predictive value of pre-therapeutic sarcopenia in cancer patients: a systematic review. Clin Nutr. 2018;37(4):1101-13.
51. Assenat E et al. Impact of lean body mass-based oxaliplatin dose calculation on neurotoxicity in adjuvant treatment of stage III colon cancer: results of the phase II randomized LEANOX trial. J Clin Oncol. 2025;43(23):2616-27.
52. McKendry J, et al. Resistance exercise, aging, disuse, and muscle protein metabolism. Compr Physiol. 2021;11(3):2249-78.
53. Breen L, Phillips SM. Skeletal muscle protein metabolism in the elderly: interventions to counteract the “anabolic resistance” of ageing. Nutrition & Metabolism. 2011;8(1):68.
54. Orell H et al. Nutritional counseling for head and neck cancer patients undergoing (chemo) radiotherapy-a prospective randomized trial. Front Nutr. 2019;6:22.
55. Ford KL et al. Feasibility of two levels of protein intake in patients with colorectal cancer: findings from the Protein Recommendation to Increase Muscle (PRIMe) randomized controlled pilot trial. ESMO Open. 2024;9(7):103604.
56. Cereda E et al. Nutritional counseling with or without systematic use of oral nutritional supplements in head and neck cancer patients undergoing radiotherapy. Radiother Oncol. 2018;126(1):81-8.
57. Seguy D et al. Compliance to oral nutritional supplementation decreases the risk of hospitalisation in malnourished older adults without extra health care cost: prospective observational cohort study. Clin Nutr. 2020;39(6):1900-7.
58. Mateus C et al. N-3 fatty acids supplementation and chemotherapy induced toxicity: scoping review. Clinical Nutrition ESPEN. 2023;54: 576-7.
59. Muscaritoli M et al. Oncology-led early identification of nutritional risk: a pragmatic, evidence-based protocol (PRONTO). Cancers (Basel). 2023;15(2):380.
60. Ravasco P. Aspects of taste and compliance in patients with cancer. Eur J Oncol Nurs. 2005;9(Suppl 2):S84-91.
61. Hubbard GP et al. A systematic review of compliance to oral nutritional supplements. Clin Nutr. 2012;31(3):293-312.
62. Wischmeyer PE et al. American society for enhanced recovery and perioperative quality initiative joint consensus statement on nutrition screening and therapy within a surgical enhanced recovery pathway. Anesth Analg. 2018;126(6):1883-95.
63. Grimble R. Basics in clinical nutrition: immunonutrition – nutrients which influence immunity: effect and mechanism of action. e-SPEN the European e-Journal of Clinical Nutrition and Metabolism. 2009;4(1):e10-3.
64. Gustafsson UO et al. Guidelines for perioperative care in elective colorectal surgery: Enhanced Recovery After Surgery (ERAS) Society recommendations 2025. Surgery. 2025;184:109397.
65. Weimann A et al. ESPEN guideline on clinical nutrition in surgery - update 2025. Clin Nutr. 2025;53:222-61.
66. García-Malpartida K et al. Effects of immunonutrition on cancer patients
undergoing surgery: a scoping review. Nutrients. 2023;15(7):1776.
67. Matsui R et al. Impact of perioperative immunonutrition on postoperative outcomes for patients undergoing head and neck or gastrointestinal cancer surgeries: a systematic review and meta-analysis of randomized controlled trials. Ann Surg. 2024;279(3):419-28.
68. Matsui R et al. Impact of perioperative immunonutrition on postoperative outcomes in patients with upper gastrointestinal cancer: a systematic review and meta-analysis of randomized controlled trials. Nutrients. 2024;16(5):577.
69. Li J et al. Comparison of enteral immunonutrition and enteral nutrition in patients undergoing gastric cancer surgery: a systematic review and meta-analysis of randomized, controlled trials. J Int Med Res. 2024;52(1):3000605231220870.
70. Wong CS et al. Effects of enteral immunonutrition in laparoscopic versus open resections in colorectal cancer surgery: a meta-analysis of randomised controlled trials. Eur J Surg Oncol. 2025;51(2):109488.
71. Hiraoka SI et al. Beneficial outcomes of immunoenhancing nutritional interventions in perioperative care for oral cancer: a systematic review and meta-analysis. Cancers (Basel). 2025;17(11):1855.
72. Xin C et al. Effect of perioperative immunonutrition on outcomes in gastric cancer surgery patients: a systematic review and evidence map. Clin Nutr ESPEN. 2025;67:90-104.
73. Goyal A et al. Perioperative immunonutrition in gastrointestinal oncology: a comprehensive umbrella review and meta-analysis on behalf of TROGSS-The Robotic Global Surgical Society. Nutrients. 2025;17(14):2304.
74. Cianflone F et al. Effect of perioperative immunonutrition on early-postoperative complications in patients undergoing radical cystectomy for bladder cancer: a case series. J Clin Med. 2025;14(6):1992.
75. Da Prat V et al. Effectiveness of preoperative immunonutrition in improving surgical outcomes after radical cystectomy for bladder cancer: study protocol for a multicentre, open-label, randomised trial (INu-RC). Healthcare (Basel). 2024;12(6):696.
76. D’Ignazo A et al. Preoperative oral immunonutrition in gastrointestinal surgical patients: how the tumour microenvironment can be modified. Clin Nutr ESPEN. 2020;38:153-9.
77. Molfino A et al. Effects of oral immunonutrition on histological changes of inflammatory infiltration
of the tumor microenvironment among patients with a new diagnosis of gastric cancer. Nutrition. 2023;105:111855.
78. Ambrosio MR et al. Paving the path for immune enhancing nutrition in colon cancer: modulation of tumor microenvironment and optimization of outcomes and costs. Cancers (Basel). 2023;15(2):437.
79. De Felice F et al. Mapping the landscape of immunonutrition and cancer research: a comprehensive bibliometric analysis on behalf of NutriOnc Research Group. Int J Surg. 2024;110(1):395-405.
80. Machon C et al. Immunonutrition before and during radiochemotherapy: improvement of inflammatory parameters in head and neck cancer patients. Support Care Cancer. 2012;20(12):3129-35.
81. Talvas J et al. Immunonutrition stimulates immune functions and antioxidant defense capacities of leukocytes in radiochemotherapytreated head & neck and esophageal cancer patients: A double-blind randomized clinical trial. Clin Nutr. 2015;34(5):810-7.
82. Boisselier P et at. A double-blind phase III trial of immunomodulating nutritional formula during adjuvant chemoradiotherapy in head and neck cancer patients: IMPATOX. Am J Clin Nutr. 2020;112(6):1523-31.
83. Caccialanza R et al. Immunonutrition in head and neck cancer patients undergoing chemoradiotherapy: an alternative approach for overcoming potential bias. Am J Clin Nutr. 2021;113(4):1053-4.
84. Caccialanza R et al. The efficacy of immunonutrition in improving tolerance to chemoradiotherapy in patients with head and neck cancer, receiving nutritional counseling: study protocol of a randomized, open-label, parallel group, bicentric pilot study. Ther Adv Med Oncol. 2021;13:17588359211025872.
85. Caccialanza R et al. Multicentre, randomised, open-label, parallelgroup, clinical phase II study to evaluate immunonutrition in improving efficacy of immunotherapy in patients with metastatic non-small cell lung cancer, undergoing systematic nutritional counseling. BMC Cancer. 2022;22(1):1212.
86. Dechaphunkul T et al. Benefits of immunonutrition in patients with head and neck cancer receiving chemoradiation: a phase II randomized, double-blind study. Clin Nutr. 2022;41(2):433-40.
87. Araki S et al. The usefulness of immunonutrition in chemotherapy and
chemoradiotherapy for esophageal cancer. Oncol Lett. 2025;30(3):428.
88. Muangwong P et al. Effect of immunonutrition during concurrent chemoradiotherapy on acute oral mucositis in head and neck cancer patients: a prospective
randomized study. PLoS One. 2025;20(3):e0320145.
89. Pilotto S et al. Nutritional support in lung cancer: time to combine immunonutrition with immunotherapy? Nutrition. 2022;98:111637.
FOR REPRINT QUERIES PLEASE CONTACT:
90. Mattavelli E et al. Nutritional status, immunonutrition, and gut microbiome: a coming of age for immunotherapy? Front Immunol. 2025;16:1612567.
Exploring New Horizons and Emerging Topics in Squamous Anal Cancer and Colorectal Cancer
This symposium took place on 20th October 2025, as part of the European Society for Medical Oncology (ESMO) Congress 2025 held in Berlin, Germany, between 17th–21st October 2025
Support: This content was developed independently by EMJ with support from an unrestricted educational grant from Incyte Biosciences International Sàrl.
Chairperson: Sheela Rao1
Speakers: Christelle de la Fouchardière,2 Gunnar Folprecht,3 Dominik Modest,4 Sheela Rao1
1. Gastrointestinal Unit, Royal Marsden Hospital, London, UK
2. Paoli-Calmettes Institute, Marseille, France
3. University Hospital Carl Gustav Carus, Dresden, Germany
4. Charité Universitätsmedizin, Berlin, Germany
Disclosure: de la Fouchardière has served on advisory boards for AbbVie, Amgen, Astellas, AstraZeneca, Bristol Myers Squibb, Daiichi-Sankyo, MSD, Pierre Fabre Oncologie, Roche, and Servier; has received invitations to attend meetings from AstraZeneca, Amgen, Incyte, MSD, Roche, and Servier; has received clinical trial sponsorship from Amgen, MSD, Pierre Fabre Oncologie, Servier, and Summit Therapeutics; and is affiliated with the Unicancer Gastrointestinal Group (UCGI), where she previously served as President, as well as with the Prodige Group and European Organisation for Research and Treatment of Cancer (EORTC), where she leads the Gastrointestinal Tract Cancer Group (GITCG)-Gastric and Oesophageal Task Force. Folprecht has served on advisory boards or received lecture honoraria from Roche, Merck, Bristol Myers Squibb, Servier, MSD, AstraZeneca, Incyte, GlaxoSmithKline, Daiichi-Sankyo, Takeda, BeiGene, Amgen, and AbbVie; has received invitations to attend meetings from Roche, Merck, Pierre Fabre Oncologie, and AstraZeneca; and is affiliated with the University Hospital Carl Gustav Carus in Dresden. Modest has received honoraria for lectures from 21up, Amgen, Aptitude Health, AstraZeneca, Bristol Myers Squibb, COR2ED, Cureteq, G1, GlaxoSmithKline, IKF GmbH, Incyte, Medscape, Merck, MSD, Onkowissen, Pierre Fabre Oncologie, Regeneron, Sanofi, Seagen, Servier, Taiho, and Takeda; has received institutional research funding from Amgen and Servier; and has received travel support from Amgen and Servier. Rao has served in consulting or advisory roles for AstraZeneca, Bayer, BeiGene, Hookipa, Merck Serono, Seagen, Servier, and Incyte; has acted as a speaker for Bayer, Merck Serono, Servier, and Incyte; has received travel grants from Servier and Incyte; and has provided expert testimony for Boehringer Ingelheim.
Acknowledgements: Writing assistance was provided by Rachel Danks, RSD Medical Communications Ltd, Gloucestershire, UK.
Disclaimer: The opinions expressed are those of the speakers and do not necessarily reflect those of the sponsor or the publisher. The company had no role in content development or editorial review.
Keywords: Colorectal cancer (CRC), immunotherapy, squamous cell anal cancer (SCAC).
Citaion: EMJ Oncol. 2025;13[1]:71-81. https://doi.org/10.33590/emjoncol/PUYU5693
Meeting Summary
PARTNERSHIP
Anal and colorectal cancers are complex gastrointestinal malignancies that continue to pose therapeutic and diagnostic challenges worldwide. This article is based on the Industry Satellite Symposium, ‘Exploring New Horizons and Emerging Topics in Squamous Anal Cancer and CRC’, delivered by four world-renowned experts during the European Society for Medical Oncology (ESMO) 2025 Annual Congress through a series of informative talks, case studies, and an interactive panel discussion. Christelle de la Fouchardière, Medical Oncologist at Paoli-Calmettes Institute, Marseille, France, opened the meeting with an introduction to squamous cell anal cancer (SCAC), describing its epidemiology, aetiology, and diagnosis, as well as strategies for prevention. Gunnar Folprecht, Professor and Medical Oncologist at the University Hospital Carl Gustav Carus, Dresden, Germany, then described multidisciplinary approaches to early-stage SCAC. This was followed by a presentation from Dominik Modest, Professor and Medical Oncologist at the Charité Universitätsmedizin, Berlin, Germany, who discussed current treatment strategies for advanced SCAC. The final two presentations were given by Sheela Rao, Consultant Medical Oncologist at the Royal Marsden Hospital, London, UK, who was also chairing the meeting. Rao first described the evolving landscape in advanced anal cancer and concluded by discussing present and future targets in colorectal cancer (CRC), with a focus on KRAS inhibition and transforming growth factor beta (TGF-β).
Squamous
Anal Cancer: A Rare Human PapillomavirusDriven Tumour where Early Diagnosis is Key
Introduction to Squamous Anal Cancer de la Fouchardière began her presentation by explaining that, although SCAC accounts for the majority of anal cancer cases, it remains an uncommon malignancy, with an estimated 55,000 new diagnoses and 22,000 deaths worldwide in 2020.1,2 SCAC occurs more frequently in women, particularly those over 50 years of age, but an increasing incidence among younger men has been observed.3 Data from population-based registries in the UK and the USA indicate a 2–3% annual rise in incidence,4,5 with increases in incidence and mortality predicted worldwide to 2045.6
Human papillomavirus (HPV) infection is the principal risk factor for SCAC, being identified in approximately 90% of cases, most commonly with genotypes 16 and 18.7 Other risk factors include immunosuppression, particularly in individuals with HIV infection or recipients of solid organ transplants, as well as a history of long-term steroid use.8,9
HPV is a small, non-enveloped DNA virus comprising an 8-kilobase circular genome, with early (E) and late (L) genes encoding the viral proteins.10 Among over 180 HPV genotypes, 12 are classified as oncogenic, of which types 16 and 18 are most frequently implicated in SCAC.10,11 The virus targets proliferating basal cells in the squamous epithelium,10,11 then stable integration of the HPV genomic element into the host genome and subsequent expression induces premalignant lesions, which may progress to cancer.8
HPV infection is widespread, with a peak incidence around the age of 25 years.12 Most infections are asymptomatic and clear spontaneously within 2 years; however, persistent infection with oncogenic subtypes may progress to high-grade lesions and invasive cancer.12,13 de la Fouchardière explained that HPV positivity is also an important prognostic factor in SCAC. HPV-positive tumours typically exhibit improved responses to chemoradiotherapy, with lower recurrence rates and superior overall survival compared with HPV-negative tumours.14 Conversely, adverse prognostic features include ulceration, lymph node involvement, and male sex.15
Clinical Presentation and Diagnostic Workup
According to the ESMO 2021 clinical practice guidelines, diagnosis of SCAC should begin with digital rectal examination followed by biopsy for histopathological confirmation and p16 immunohistochemistry, with additional HPV testing when indicated.16 Pelvic MRI and PET-CT with [18F]2-fluoro-2-deoxyD-glucose are recommended for staging, providing superior accuracy for local and nodal assessment.16
Most patients with anal cancer present with localised or locoregional disease,17 with diagnosis at an earlier clinical stage strongly correlating with improved overall survival (OS).18 This highlights the importance of early detection and provides a rationale for targeted screening. High-risk populations include men who have sex with men, individuals with HIV, women with prior HPV-related gynaecological cancer, and immunosuppressed individuals.19
HPV vaccination represents a cornerstone of primary prevention, substantially reducing the incidence of HPV infection and associated malignancies.20 Current immunisation programmes target both girls and boys aged 9–14 years, with catch-up schedules for young adults,21 although global vaccination coverage remains suboptimal at only 40–55%.20
de la Fouchardière concluded that SCAC is a rare but increasingly prevalent malignancy in which HPV infection acts as the primary tumour-initiating event.6,7 Anal cancer should be diagnosed in line with local guidelines and should include histological confirmation of SCAC,16 with a timely diagnosis essential to improve OS.18 de la Fouchardière noted that although HPV vaccination programmes have the potential to significantly reduce the burden of HPV-related malignancies such as SCAC,20 vaccination rates among high-risk groups remain suboptimal, even in high-income countries, underscoring the need for continued public health efforts.20
Navigating Early-Stage Squamous Anal Cancer: Multidisciplinary Treatment Approaches
In his presentation, Folprecht described current treatment approaches for patients with early-stage SCAC, drawing on the case of a female in her late 60s presenting with well-differentiated, p16-positive squamous cell carcinoma as an illustrative example.
The patient underwent a comprehensive diagnostic workup, including pelvic MRI and fluorodeoxyglucose PET, which clearly delineated the primary lesion, confirmed the absence of metabolic activity in regional lymph nodes, and excluded adrenal or distant metastases. Imaging demonstrated a tumour of approximately 1 cm in diameter with a cranio-caudal length of 4 cm, without nodal involvement. The patient’s medical history included smoking-related emphysema, HIV serology was negative, and dihydropyrimidine dehydrogenase function was normal.
Based on the tumour, node, and metastasis classification, the lesion was categorised as Stage IIA (T2N0M0), representing a localised, yet not early microinvasive disease.18,22 Early-stage/localised anal cancer is considered to fall within the range of Stage I to IIIa.18,22 Folprecht explained that the only surgical option for the patient would be abdominal perineal resection with a permanent stoma, which was ruled out in preference to combined radiotherapy and chemotherapy with mitomycin C (MMC)
or cisplatin, as recommended by both the American Society of Clinical Oncology (ASCO) and ESMO guidelines.16,23
Evidence from the UK Collaborative for Cancer Clinical Research (UKCCCR) ACT I trial showed that the addition of 5-fluorouracil (5-FU) and MMC to radiotherapy among 577 patients with squamous, basaloid, or cloacogenic cell carcinoma of the anal canal/margin resulted in a substantially reduced risk of death from anal cancer.24
Questions regarding the role of induction or maintenance chemotherapy have been addressed in subsequent investigations. Induction therapy prior to chemoradiation did not improve disease-free survival in the RTOG 9811 trial, and maintenance therapy after definitive treatment failed to demonstrate progression-free benefit in ACT II.25,26 Therefore, concurrent fluoropyrimidine plus MMC with radiotherapy remains the standard of care for early-stage/localised anal cancer.16 In current chemotherapy protocols, 5-FU is administered either by continuous infusion during Weeks 1 and 5, or alternatively, oral capecitabine, combined with MMC, is given once or twice throughout treatment. Radiotherapy dosing typically ranges from 50 Gy or more for advanced lesions, with lower doses occasionally used for smaller T1–T2 tumours. In the present case, the patient received chemoradiation with MMC, including nodal basins within the radiation field.
Prognosis after definitive chemoradiation depends primarily on tumour size and nodal status. Patients with T2N0 tumours can expect a 5-year progression-free survival (PFS) of approximately 70% and a high probability of cure.27 However, some patients do not achieve complete response and require salvage surgery. Post-treatment analyses have shown that early surgical assessment, within 11 weeks after chemoradiation, may lead to overtreatment, as some tumours continue to regress beyond this period. Re-evaluation at approximately 26 weeks allows more accurate distinction between residual disease and delayed response (Figure 1).28
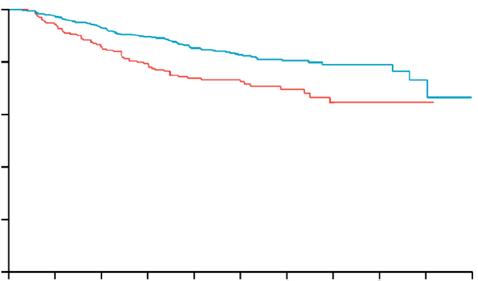
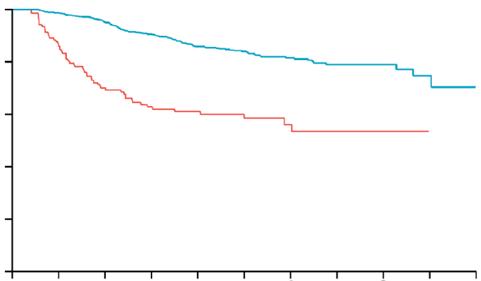
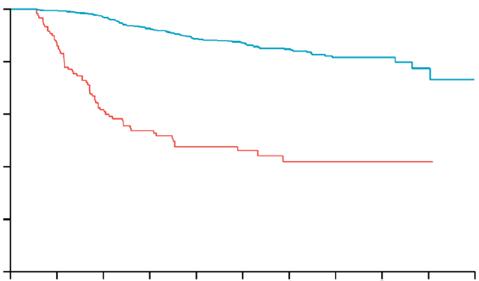
Adapted from Glynne-Jones R et al.28 and licensed under CC BY 4.0.
cCR: complete clinical response; HR: hazard ratio.
Figure 1: ACT-2 post-hoc analysis.28
Surgery remains an important option for selected patients. For tumours of the anal margin confined outside the canal, local excision preserving the sphincter may be feasible, provided a histologic margin of at least 1 mm is achieved. MRI assists in assessing suitability for conservative surgery.16,29 Nevertheless, for most anal canal lesions, sphincter-preserving surgery is rarely possible.16,29
Salvage abdominoperineal resection is indicated for persistent or recurrent disease, but is associated with substantial morbidity and the need for permanent stoma.16 Despite these limitations, a proportion of patients undergoing salvage surgery achieve durable remission, underscoring the importance of multidisciplinary management and careful patient selection.29
In conclusion, Folprecht noted that only a small subset of anal cancers are responsive to primary surgery, and that most of these are early anal margin cancers (cT1N0M0).16 Based on current guidelines, early-stage/localised disease should be treated with chemoradiotherapy (fluoropyrimidine+MMC+radiotherapy) in the first-line setting,16 while induction or maintenance therapies do not appear to result in survival benefits for patients.25,26 In addition, salvage surgery should be considered in patients with residual or locally recurrent disease, although this is a significant procedure, requiring lifelong management.16 Folprecht also remarked that a multidisciplinary approach is mandatory, involving radiation oncologists, medical oncologists, surgeons, radiologists, and pathologists.16
Advanced Squamous Anal Cancer: Current Treatment Strategies
Systematic Assessment of Chemotherapy Regimens
Modest began by noting that management of advanced or metastatic SCAC has evolved significantly over recent years, guided by both pragmatic clinical experience and emerging evidence from
collaborative trials. Two major areas of development have shaped current practice: the optimisation of chemotherapy backbones for unresectable disease and the integration of newer, less toxic regimens for improved survival and tolerability.
Historically, treatment of metastatic SCAC relied on small observational studies and institutional experiences to direct therapeutic choices, where combinations of platinum compounds with fluoropyrimidines were commonly employed.30 MMC, long used in concurrent chemoradiation, was also included in early metastatic regimens, though its use has since declined. Secondline approaches were inconsistent, often involving taxanes such as paclitaxel or irinotecan after progression on platinumbased therapy.30 Reported PFS ranged from 6–9 months, with OS rarely exceeding 12–15 months, underscoring the urgent need for more standardised treatment strategies.30
The pivotal InterAAct trial marked a major step forward by establishing carboplatin and paclitaxel as a modern first-line standard of care. This international randomised Phase II study compared carboplatin–paclitaxel with cisplatin–5-fluorouracil and demonstrated comparable response rates, but a more favourable safety profile for the carboplatin–paclitaxel regimen.31 Although PFS was similar between arms, OS was favoured by carboplatin–paclitaxel, reaching a median of approximately 20 months, which is considered a meaningful improvement for this rare malignancy.31 The regimen’s tolerability and feasibility in a global multicentre setting solidified its adoption as the preferred firstline backbone in current guidelines.
The choice of carboplatin–paclitaxel has since become the foundation for ongoing research and combination studies. Its balanced efficacy and safety make it suitable for a broad range of patients, including those with comorbidities or reduced performance status.16 For subsequent therapy after platinum failure, treatment options remain heterogeneous. Irinotecan-based regimens are commonly used, although no second-line standard has been firmly established. Immunotherapy with programmed cell death protein 1
(PD-1) and programmed death-ligand 1 (PD-L1) inhibitors has emerged as a promising approach for this group, offering durable responses in selected patients with previously treated metastatic disease.16
Parallel efforts to intensify systemic therapy have produced encouraging results. The French Epitopes-HPV02 trial investigated a triplet regimen combining docetaxel, cisplatin, and 5-FU, administered either every 3 weeks or in a modified biweekly schedule.32 The study achieved high response rates and prolonged PFS compared with historical doublet regimens. Notably, the biweekly modification substantially reduced the incidence of severe haematological and gastrointestinal toxicities, making it more manageable for clinical practice.32 Although triplet therapy may not be suitable for frail or comorbid patients, it represents a valuable option for those requiring rapid tumour control, particularly when disease burden is high.
Across studies, the consistency of clinical outcomes supports a growing optimism in this challenging field.31-34 Median OS for patients with advanced or metastatic SCAC now exceeds 20 months, with some regimens achieving response rates above 70%. These gains reflect both improved systemic therapies and the benefits of structured multidisciplinary collaboration in managing a rare and complex cancer type.
From a molecular perspective, therapeutic innovation remains limited by the relatively simple genomic landscape of SCAC.35 Actionable mutations are uncommon, and current molecular profiling has yet to identify consistent targets amenable to precision therapies.35 While exploratory work is ongoing into PI3K inhibitors and poly(ADP-ribose) polymerase-targeted agents, the heavy prior exposure to platinum chemotherapy complicates their potential benefit.35 As such, future progress is expected to come primarily from immunotherapy and rationally designed combination approaches rather than from highly specific molecular targeting.
Modest concluded by emphasising that carboplatin–paclitaxel remains the established standard of care for patients with locally advanced, unresectable, or metastatic SCAC based on current evidence.31 The modified docetaxel, cisplatin, and 5-FU regimen has shown promising efficacy and may represent a viable alternative chemotherapy backbone.32 Ongoing advances in combined checkpoint inhibition further reinforce the role of carboplatin–paclitaxel as the foundation of emerging standardof-care strategies in this setting.34
Immunotherapy and Beyond: The Evolving Treatment Landscape in Advanced Anal Cancer
Rao opened her presentation by noting that, as an HPV-driven malignancy, SCAC displays viral antigens that elicit chronic immune activation, leading to T cell exhaustion within the tumour microenvironment.36 This biological profile provides a strong rationale for PD-1 and PD-L1 blockade, which can restore antitumour immune activity and modify disease trajectory.
For patients with advanced or metastatic disease, a combination of carboplatin and paclitaxel remains the standard first-line therapy.16 Historically, second-line regimens relied on repurposed chemotherapy combinations with limited benefit, highlighting the need for novel approaches.16 The recent FDA approval of the PD-1 inhibitor, retifanlimab, represents a pivotal development, with retifanlimab becoming the first immunotherapy licensed in the USA specifically for advanced anal cancer.37
Early single-agent immunotherapy studies established proof of concept. Nivolumab, pembrolizumab, and retifanlimab each demonstrated activity in previously treated patients, producing response rates between 11–24%, with median OS around 10–12 months.38-40 Rao noted that although response rates were modest, these studies help to establish immunotherapy as a clinically meaningful option and pave the way for combination regimens.
Immuno-oncology in Combination with Chemotherapy in First-Line Advanced Anal Cancer
The Phase II SCARCE-PRODIGE 60 trial evaluated atezolizumab with the modified docetaxel, cisplatin, and 5-FU regimen in untreated advanced SCAC.33 The study did not improve PFS compared with chemotherapy alone, but Rao observed that exploratory analysis hinted that patients with higher PD-L1 expression might derive greater benefit. These findings reinforce the rationale for combining immune checkpoint inhibitors with chemotherapy.
More conclusive evidence came from the Phase III POD1UM-303/InterAAct 2 trial, which investigated carboplatin–paclitaxel with or without retifanlimab in the first-line setting in 308 patients with locally recurrent or metastatic SCAC (Figure 2).34,41 The study met its primary endpoint, demonstrating improved PFS (9.3 months with retifanlimab
Figure 2: POD1UM-303/InterAACT 2: study design.34,41,42
versus 7.4 months with chemotherapy alone; p=0.0006).34 Early separation of the survival curves suggested a sustained treatment effect, with median OS extending to 29 months in the immunotherapy arm (Figure 3).34,41 The addition of retifanlimab nearly doubled the median duration of response from approximately 7 to 14 months and increased the overall response rate to approximately 56%. Toxicities were consistent with prior experience, largely immune-related but manageable, without compromising chemotherapy delivery.34,41
Rao commented that these findings suggest that carboplatin–paclitaxel plus retifanlimab should be considered as a new global standard of care for first-line advanced SCAC. The regimen provides durable benefit, high disease control, and acceptable tolerability in a patient population that historically had limited effective treatment options.
Primary PFS by BICR (HR: 0.67 at >80% power, alpha=0.025 [1-sided])
Secondary OS (key secondary, alpha=0.025 [1-sided], if PFS is statistically significant), ORR, DOR, DCR, safety, PK
Exploratory PROs, HIV control, immunogenicity
*Standard-dose carboplatin–paclitaxel. Carboplatin: area under the curve 5 mg/mL on Day 1; Paclitaxel: 80 mg/m2 IV on days 1, 8, and 15. Each cycle=28 days. 6 months/24 weeks (6 cycles).
AUC: area under the curve; BICR: blinded independent central review; DCR: disease control rate; DOE: duration of response; HR: hazard ratio; IV: intravenous; mo: months; NA: North America; PD: progressive disease; PK: pharmacokinetics; PRO: patient-reported outcome; Q4W: every 4 weeks; ROW: rest of the world; SCAC: squamous cell anal cancer; vs: versus.
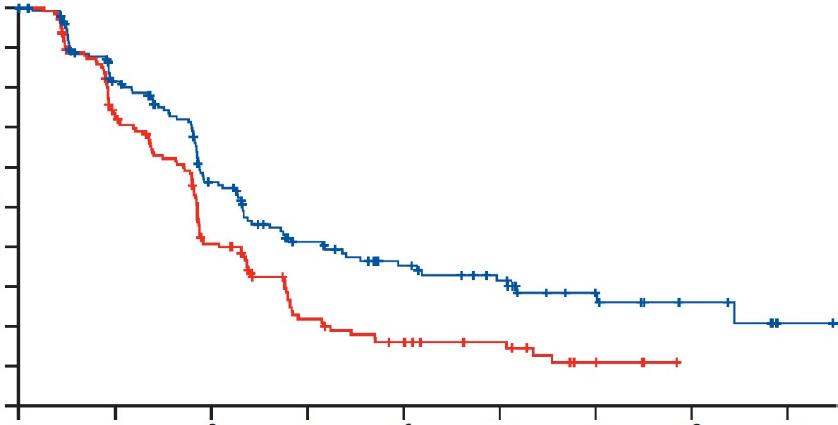
*Stratified log-rank test with a one-sided significance level of 2.5%. Stratification factors: region of the world, extent of disease, and PD-L1 expression status.
Adapted from Rao S et al.34 and licensed under CC BY 4.0.
BICR: blinded independent central review; HR: hazard ratio; PD-L1: programmed death-ligand 1; PFS: progressionfree survival.
Future
Outlook: Immunotherapy and Beyond
Looking ahead, the therapeutic landscape for anal cancer continues to expand rapidly, with a number of trials exploring multiple immunotherapy strategies across disease stages.43-45 Rao commented that the ongoing trials of vaccines in combination with immunotherapy look particularly interesting, but suggested that more work is needed to investigate a possible role for targeted treatments in this disease area.
Present and Future Targets in Colorectal Cancer: Focus on KRAS and TGF-β
In the final presentation, Rao turned the attention of the audience towards CRC, which remains one of the most prevalent malignancies worldwide.46 Although advances in screening, surgery, chemotherapy, and targeted therapies have improved outcomes, metastatic disease continues to carry a poor prognosis.47 Five-year survival falls sharply once distant metastases are present, emphasising the need for more precise molecularly targeted approaches.48
Figure 3: POD1UM-303/InterAACT 2: PFS by BICR (primary endpoint).34
The therapeutic landscape of CRC has become increasingly complex, with multiple signalling pathways under investigation. The approval of bevacizumab and cetuximab in 2004 signalled the arrival of targeted therapies in CRC,47 and since then, targeted therapies have transformed the treatment paradigm in CRC.47,49 As a result, multiple novel targeted therapies, directed against a diverse array of cellular pathways in CRC, are being explored in CRC.47,50 Among these, KRAS mutations represent a particularly important challenge and opportunity for innovation.
Within the KRAS-mutant population, the most common alterations include G12D, G12V, and G13D, while G12C accounts for only a small proportion.51 These mutations are associated with inferior prognosis and reduced sensitivity to anti-EGFR therapy.51 Despite decades of effort, direct targeting of RAS proteins was long considered impossible; however, Rao explained that the development of allele-specific inhibitors and degraders has changed this paradigm.
Several KRAS inhibition strategies are now being explored.52,53 Mutation-specific inhibitors selectively bind either the active, GTP-bound (‘on’) or inactive, GDP-bound (‘off’) conformations of KRAS, locking the protein in its inactive form.53,54 RMC-6236 is an oral RASMULTI(ON) tri-complex inhibitor that binds to cyclophilin A, resulting in a binary complex that potently binds to RAS(ON) to form a tri-complex, blocking downstream signalling.55 In another approach, proteolysis-targeting chimaeras direct the degradation of RAS proteins through the ubiquitin–proteasome system.56
The KRASG12D-selective degrader ASP3082 is currently under evaluation in a Phase I, open-label, multicentre study in adults with KRASG12D-mutant advanced solid tumours (NCT05382559).57,58 Meanwhile, the selective G12D inhibitor, INCB161734, has shown encouraging early-phase results, with manageable safety profiles and preliminary evidence of antitumour activity in patients with solid tumours.59 Collectively, these agents represent the first generation of rationally designed RAS-targeted therapies and are expected to reshape management strategies once mature efficacy data emerge.
Beyond KRAS, attention has turned to the TGF-β pathway, which plays a pivotal role in epithelial–mesenchymal transition and tumour progression.60 Overexpression of TGF-β is a key pathway in the development of CRC, promoting epithelial–mesenchymal transition, angiogenesis, and immunosuppression.61,62 Due to its role in tumour progression, TGF-β expression has been studied as a potential prognostic biomarker in CRC.61,63
Several drugs targeting TGF-β signalling are in development in CRC, although numerous drugs targeting TGF-β signalling have stopped development due to toxicity concerns/pleiotropic effects.62-66 Examples of promising candidates in this area include the bifunctional fusion protein targeting TGF-β and PD-L1, bintrafusp alfa, which demonstrated modest antitumour activity and a manageable safety profile in patients with heavily pretreated, advanced CRC.67 In addition, the first-in-class bispecific antibody, INCA33890, targets PD-1-positive cells only, thereby sparing normal tissues and reducing off-target effects.68-70 In a Phase I, open-label, multicentre study in participants with advanced or metastatic solid tumours, INCA33890 demonstrated encouraging activity, including responses in patients with liver metastases.71 Toxicities were largely immune-related and manageable, with fatigue, rash, pruritus, and diarrhoea as the most frequent adverse events. Importantly, severe treatment discontinuations remain uncommon, suggesting a favourable therapeutic window.71
Rao concluded her presentation, commenting that CRC remains a biologically diverse disease with substantial unmet need. The rapid expansion of KRAS inhibitors and tumour-specific TGF-β modulators represents one of the most dynamic areas of current oncology research. Together, these innovations signal a shift toward increasingly personalised, tumour-specific targeted strategies capable of addressing molecular subtypes that have long resisted conventional therapy.
References
1. Gondal TA et al. Anal cancer: the past, present and future. Curr Oncol. 2023;30(3):3232-50.
2. Global Cancer Observatory. Anus. Available at: https://gco.iarc.who. int/media/globocan/factsheets/ cancers/10-anus-fact-sheet.pdf. Last accessed: 26 October 2025.
3. Deshmukh AA et al. Recent trends in squamous cell carcinoma of the anus incidence and mortality in the United States, 2001-2015. J Natl Cancer Inst. 2020;112(8):829-38.
4. National Cancer Institute. Cancer stat facts: anal cancer. Available at: https:// seer.cancer.gov/statfacts/html/anus. html. Last accessed: 26 October 2025.
5. Smittenaar CR et al. Cancer incidence and mortality projections in the UK until 2035. Br J Cancer. 2016;115(9):1147-55.
6. Cancer tomorrow. Dataviz. Available at: https://gco.iarc.fr/tomorrow/en/ dataviz/tables. Last accessed: 26 October 2025.
7. Silva Dalla Libera L et al. Human papillomavirus and anal cancer: prevalence, genotype distribution, and prognosis aspects from Midwestern region of Brazil. J Oncol. 2019;2019:6018269.
8. Wang CJ et al. Human immunodeficiency virus/AIDS, human papillomavirus, and anal cancer. Surg Oncol Clin N Am. 2017;26(1):17-31.
9. Shridhar R et al. Anal cancer: current standards in care and recent changes in practice. CA Cancer J Clin. 2015;65(2):139-62.
10. Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev. 2012;25(2):215-22.
11. Nyitray AG et al. The association between body mass index and anal canal human papillomavirus prevalence and persistence: the HIM study. Hum Vaccin Immunother. 2019;15(7-8):1911-9.
12. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Biological Agents (2012) 110B. Lyon IARC Publications.
13. Molina MA et al. HPV integration and cervical cancer: a failed evolutionary viral trait. Trends Mol Med. 2024;30(9):890-902.
14. Parwaiz I et al. A systematic review and meta-analysis of prognostic biomarkers in anal squamous cell carcinoma treated with primary chemoradiotherapy. Clin Oncol (R Coll Radiol). 2019;31(12):e1-13.
15. Bartelink H et al. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J Clin Oncol. 1997;15(5):2040-9.
16. Rao S et al. Anal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(9):1087-100.
17. Feng M et al. Radiation therapy for anal squamous cell carcinoma: an ASTRO clinical practice guideline. Pract Radiat Oncol. 2025;15(4):367-86.
18. Janczewski LM et al. Updates on the Version 9 American Joint Committee on cancer staging system for anal cancer. Ann Surg Oncol. 2024;31(7):4155-8.
19. Clifford GM et al. A meta-analysis of anal cancer incidence by risk group: toward a unified anal cancer risk scale. Int J Cancer. 2021;148(1):38-47.
20. Han J et al. Global HPV vaccination programs and coverage rates: a systematic review. E Clin Med. 2025;84:103290.
21. European Center for Disease Prevention and Control (ECDC). Vaccine Scheduler. Available at: https://vaccine-schedule.ecdc.europa. eu/Scheduler/. Last accessed: 26 October 2025.
22. Janczewski LM et al. Survival outcomes used to generate version 9 American Joint Committee on Cancer staging system for anal cancer. CA Cancer J Clin. 2023;73(5):516-23.
23. Morris VK et al. Systemic therapy for stage I-III anal squamous cell carcinoma: ASCO Guideline. J Clin Oncol. 2025;43(5):605-15.
24. Northover J et al. Chemoradiation for the treatment of epidermoid anal cancer: 13-year follow-up of the first randomised UKCCCR Anal Cancer Trial (ACT I). Br J Cancer. 2010;102(7):1123-8.
25. Ajani JA et al. Fluorouracil, mitomycin, and radiotherapy vs fluorouracil, cisplatin, and radiotherapy for carcinoma of the anal canal: a randomized controlled trial. J Am Med Assoc. 2008;299(16):1914-21.
26. James RD et al. Mitomycin or cisplatin chemoradiation with or without maintenance chemotherapy for treatment of squamous-cell carcinoma of the anus (ACT II): a randomised, phase 3, open-label, 2 × 2 factorial trial. Lancet Oncol. 2013;14(6):516-24.
27. Ajani JA et al. Prognostic factors derived from a prospective database dictate clinical biology of anal cancer: the intergroup trial (RTOG 98-11). Cancer. 2010;116(17):4007-13.
28. Glynne-Jones R et al. Best time to assess complete clinical response after chemoradiotherapy in squamous cell carcinoma of the anus (ACT II): a post-hoc analysis of randomised controlled phase 3 trial. Lancet Oncol. 2017;18(3):347-56.
29. Congedo A et al. An updated review on imaging and staging of anal cancer – not just rectal cancer. Tomography. 2023;9(5):1694-710.
30. Sclafani F et al. Platinumfluoropyrimidine and paclitaxel-based chemotherapy in the treatment of advanced anal cancer patients. Oncologist. 2017;22(4):402-8.
31. Rao S et al. International rare cancers initiative multicenter randomized phase II trial of cisplatin and fluorouracil versus carboplatin and paclitaxel in advanced anal cancer: InterAAct. J Clin Oncol. 2020;38(22):2510-8.
32. Kim S et al. Docetaxel, cisplatin, and fluorouracil chemotherapy for metastatic or unresectable locally recurrent anal squamous cell carcinoma (Epitopes-HPV02): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2018;19(8):1094-106.
33. Kim S et al. Atezolizumab plus modified docetaxel, cisplatin, and fluorouracil as first-line treatment for advanced anal cancer (SCARCE C1702 PRODIGE 60): a randomised, non-comparative, phase 2 study. Lancet Oncol. 2024;25(4):518-28.
34. Rao S et al. Retifanlimab with carboplatin and paclitaxel for locally recurrent or metastatic squamous cell carcinoma of the anal canal (POD1UM-303/InterAACT-2): a global, phase 3 randomised controlled trial. Lancet. 2025;405(10495):2144-52.
35. Morris V et al. Comprehensive genomic profiling of metastatic squamous cell carcinoma of the anal canal. Mol Cancer Res. 2017;15(11):1542-50.
36. Incyte. ESMO 2024 Incyte Investors presentation. 2024. Available at: https://investor.incyte.com/staticfiles/b479ebfe-f65d-481c-a341574f257ff6a6. Last accessed: 26 October 2025.
37. Incyte. Incyte announces FDA approval of Zynyz® (retifanlimab-dlwr) making it the first and only approved first-line treatment for advanced anal cancer patients in the United States. Available at: https://investor.incyte.com/ news-releases/news-release-details/
incyte-announces-fda-approvalzynyzr-retifanlimab-dlwr-makingit?utm_source=chatgpt.com. Last accessed: 26 October 2025.
38. Morris VK et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18(4):446-53.
39. Marabelle A et al. Pembrolizumab for previously treated advanced anal squamous cell carcinoma: results from the non-randomised, multicohort, multicentre, phase 2 KEYNOTE-158 study. Lancet Gastroenterol Hepatol. 2022;7(5):446-54.
40. Rao S et al. A phase II study of retifanlimab (INCMGA00012) in patients with squamous carcinoma of the anal canal who have progressed following platinum-based chemotherapy (POD1UM-202). ESMO Open. 2022;7(4):100529.
41. Rao S et al. POD1UM-303/InterAACT 2: phase III study of retifanlimab with carboplatin-paclitaxel (c-p) in patients (Pts) with inoperable locally recurrent or metastatic squamous cell carcinoma of the anal canal (SCAC) not previously treated with systemic chemotherapy (Chemo). Ann Oncol. 2024;35(Suppl 2):LBA2.
42. Rao S et al. POD1UM-303/InterAACT 2: phase III study of retifanlimab with carboplatin-paclitaxel (c-p) in patients (Pts) with inoperable locally recurrent or metastatic squamous cell carcinoma of the anal canal (SCAC) not previously treated with systemic chemotherapy (Chemo). Oral presentation LBA2. ESMO Congress, 13-17 September, 2024.
43. Spehner L et al. Present and future research on anal squamous cell carcinoma. Cancers (Basel). 2021;13(15):3895.
44. Bicara Therapeutics. Study of safety and tolerability of BCA101 monotherapy and in combination therapy in patients with EGFRdriven advanced solid tumors. NCT04429542. https://www. clinicaltrials.gov/study/NCT04429542.
45. National Cancer Institute. Combination immunotherapy in subjects with advanced HPV associated malignancies. NCT04287868. https:// clinicaltrials.gov/study/NCT04287868.
46. Bray F et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229-63.
47. Xie YH et al. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020;5(1):22.
48. National Cancer Institute. Cancer stat facts: colorectal cancer. Available at: https://seer.cancer.gov/statfacts/html/ colorect.html. Last accessed: 26 October 2025.
49. Van Cutsem E et al. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):1-9.
50. Salva de Torres C et al. Current and emerging treatment paradigms in colorectal cancer: integrating hallmarks of cancer. Int J Mol Sci. 2024;25(13):6967.
51. Bteich F et al. Targeting KRAS in colorectal cancer: a bench to bedside review. Int J Mol Sci. 2023;24(15):12030.
52. Lee JK et al. Comprehensive pancancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis Oncol. 2022;6(1):91.
53. Ash LJ et al. KRAS: biology, inhibition, and mechanisms of inhibitor resistance. Curr Oncol. 2024;31(4):2024-46.
54. Takeda M et al. The role of KRAS mutations in colorectal cancer: biological insights, clinical implications, and future therapeutic perspectives. Cancers (Basel). 2025;17(3):428.
55. Spira AI et al. Preliminary safety and pharmacokinetic profiles of RMC6236, a first-in-class, RAS-selective, tri-complex RASMULTI(ON) inhibitor in patients with KRAS mutant solid tumors on the Phase 1 trial RMC6236-00. Abstract PR010. AACR, 11-15 October, 2023.
56. Kos T, Saur D. Breaking down KRAS: small-molecule degraders for cancer therapy. Signal Transduct Target Ther. 2025;10(1):86.
57. Yoshinari T et al. Discovery of KRAS(G12D) selective degrader ASP3082. Commun Chem. 2025;8(1):254.
58. Astellas Pharma Inc. A study of ASP3082 in adults with advanced solid tumors. NCT05382559. https:// clinicaltrials.gov/study/NCT05382559.
59. Desai J et al. Preliminary phase I results of INCB161734, a novel oral KRAS G12D inhibitor, in patients with advanced or metastatic solid tumors. Abstract 9160. ESMO Congress, 17-21 October, 2025.
60. Villalba M et al. Role of TGF-β in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370(1):29-39.
61. Bakrim S et al. Recent advances and molecular mechanisms of TGF-β signaling in colorectal cancer, with focus on bioactive compounds targeting. Biomed Pharmacother. 2024;177:116886.
62. Fasano M et al. TGF-β modulated pathways in colorectal cancer: new potential therapeutic opportunities. Int J Mol Sci. 2024;25(13):7400.
63. Li X et al. TGF-β Signaling in metastatic colorectal cancer (mCRC): from underlying mechanism to potential applications in clinical development. Int J Mol Sci. 2022;23(22):14436.
64. Jing H et al. Recent advances in therapeutic use of transforming growth factor-beta inhibitors in cancer and fibrosis. Front Oncol. 2025;15:1489701.
65. Tolcher AW et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(4):673-80.
66. Herbertz S et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479-99.
67. Spira A et al. Bintrafusp alfa: a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with pretreated colorectal cancer: results from a phase i trial. Oncologist. 2023;28(2):e124-7.
68. Wang LCS et al. INCA33890, a novel PD-1×TGFβR2 bispecificantibody conditionally antagonizes TGFβ signaling inprimary immune cells co-expressing PD-1. Cancer Res. 2023;83(7 Suppl):2936.
69. Guan J et al. PD-1xTGFβR2 bispecific Biclonics® antibody INCA33890 augments human T-cell effector functions invitro and ex vivo. Cancer Res. 2025;85(8 Suppl 1):6071.
70. Kinder M et al. INCA33890 increases CD8+ T-cell effector function compared with pembrolizumab asassessed by single-cell RNA sequencing in human PD-1xTGFβR2 knock-in mouse model. Cancer Res. 2025;85(8 Suppl 1):4861.
71. Garralda E. A Phase 1 study of INCA33890, a TGFβR2×PD-1 bispecific antibody, for advanced solid tumours. Abstract 3297. ESMO, 17-21 October, 2025.
Abstract Highlights
Citation: EMJ Oncol. 2025;13[1]:82-88. https://doi.org/10.33590/emjoncol/WYFP9862
The following highlights showcase key research presented at the European Society for Medical Oncology (ESMO) 2025 Congress. Together, these studies span major advances in breast, oesophageal, urothelial, oropharyngeal, and ovarian cancers, illuminating emerging strategies in treatment, personalisation, and long-term survivorship. From the safety of pausing endocrine therapy to pursue pregnancy, to the nuanced role of immunotherapy across tumour types, and the survival impact of intensified chemotherapy, these findings reflect the evolving intersection of biology, patient priorities, and therapeutic innovation.

POSITIVE: Temporary Endocrine Therapy Interruption for Pregnancy in Early Breast Cancer
UPDATED results from the POSITIVE trial, presented at the ESMO Congress 2025, further support the safety and feasibility of temporarily interrupting adjuvant endocrine therapy (ET) for pregnancy in women with hormone receptor-positive early breast cancer. Previous early follow-up findings (median: 41 months) demonstrated that this approach was viable. New data provide longer-term insight into breast cancer outcomes, pregnancy success, and ET resumption.1
The POSITIVE trial was a single-arm prospective study enrolling 518 eligible women between December 2014–December 2019. Participants paused ET after 18–30 months, for up to 2 years, to attempt pregnancy. Outcomes were compared with a matched external control group from SOFT/ TEXT. At median follow-up of 71 months in POSITIVE and 80 months in SOFT/TEXT, 5-year cumulative incidence of breast cancer-free interval (BCFI) events was 12.3% in POSITIVE versus 13.2% in SOFT/ TEXT (difference: -0.9%; 95% CI: -4.2–2.6%). Similarly, 5-year distant recurrence-free interval events were 6.2% in POSITIVE and 8.3% in SOFT/TEXT (difference: -2.1%; 95% CI: -4.5–0.4%), confirming no increased risk associated with treatment interruption.
Among the 497 women followed for pregnancy outcomes, 377 (75.8%) achieved at least one pregnancy, and 343 (69%) had at least one live birth, resulting in 440
newborns, including 75 women with more than one live birth. An 18-month landmark analysis showed comparable BCFI outcomes between patients who became pregnant and those who did not.
Of the 180 patients (36%) who underwent embryo/oocyte cryopreservation before enrolment, the 5-year cumulative incidence of BCFI events was 14.0% (95% CI: 9.6–20.2%), versus 11.5% (95% CI: 8.4–15.7%) in those who did not cryopreserve. Among 429 women followed for at least 2 years and remaining disease-free, 352 (82%) resumed ET per protocol.
Although long-term follow-up is still required, these findings suggest that temporary ET interruption to pursue pregnancy does not increase breast cancer recurrence risk at 5 years, with high rates of successful pregnancies and ET resumption.

SKYSCRAPER-07: New Direction for Unresectable Oesophageal Cancer?
DATA from the Phase III SKYSCRAPER-07 trial, presented at the ESMO Congress 2025, offer new insights into the potential role of immunotherapy following definitive chemoradiotherapy (dCRT) in patients with unresectable, locally advanced oesophageal squamous cell carcinoma (ESCC). Although dCRT remains the established standard of care, more than half of patients experience disease recurrence, highlighting the need for more effective post-treatment options.2
SKYSCRAPER-07 evaluated the efficacy and safety of tiragolumab combined with atezolizumab, as well as atezolizumab combined with placebo, in adults ≥18 years with Eastern Cooperative Oncology Group (ECOG) performance status 0–1 and Stage II–IVA, and select Stage IVB, locally advanced ESCC. All participants were deemed unsuitable for curative surgery and had completed dCRT before enrolment. A total of 760 patients were randomised in a 1:1:1 ratio to receive tiragolumab plus atezolizumab, atezolizumab plus placebo, or placebo plus placebo. Treatment was administered intravenously every 3 weeks for up to 17 cycles, or until disease progression or unacceptable toxicity. The primary endpoints were assessed using a hierarchical testing strategy, beginning with investigator-assessed progressionfree survival (PFS) for tiragolumab plus atezolizumab compared with placebo, followed by overall survival (OS) comparisons.
With a median follow-up of 25.0 months, baseline characteristics were consistent across treatment arms. Median PFS was 20.8 months for patients receiving tiragolumab plus atezolizumab and 16.6 months for those receiving placebo, a difference that did not reach statistical significance. Median OS was 38.6 months in the tiragolumab plus atezolizumab arm and 36.4 months in the placebo arm, while median OS was not reached in the atezolizumab plus placebo group.
Adverse events were reported in the majority of patients across all arms, with treatment-related events more frequent in the tiragolumab combination arm. Importantly, no new safety concerns emerged.
Although the trial did not meet its primary endpoint and no survival benefit was demonstrated for the tiragolumab and atezolizumab combination, the findings indicate that atezolizumab plus placebo delivered clinically meaningful improvements in both PFS and OS compared with placebo alone. These results support further exploration of atezolizumab-based treatment strategies for patients with unresectable locally advanced ESCC.
A total of 760 patients were randomised in a 1:1:1 ratio to receive tiragolumab plus atezolizumab, atezolizumab plus placebo, or placebo plus placebo
Adjuvant Nivolumab: 5-Year Outcomes from the CheckMate 274 Trial
THE EXTENDED 5-year follow-up results of the CheckMate 274 trial were presented at the ESMO Congress 2025, demonstrating that adjuvant nivolumab (NIVO) continues to provide meaningful clinical benefit for patients with high-risk muscleinvasive urothelial carcinoma (MIUC) following radical surgery, improving diseasefree survival and extending long-term outcomes compared with placebo.3
The research team found that ctDNA was detected in 18.8% of patients, and 40.6% of patients were ctDNA positive
The Phase III CheckMate 274 trial has previously shown that NIVO in patients with MIUC after radical surgery significantly improves disease-free survival in intent-to-treat patients and patients with tumour programmed death ligand 1 (PD-L1) expression ≥1%. Next, the researchers analysed the 5-year follow-up data, as well as the exploratory analysis of circulating tumour DNA (ctDNA) data with the Natera Signatera (Natera, Inc., Austin, Texas, USA) assay. In this study, 709 patients who had radical surgery, neoadjuvant chemotherapy, and were at high risk of recurrence were randomised 1:1 to either NIVO 240 mg IV (n=353) or placebo (n=356) every 2 weeks for 1 year.
With a median follow-up of 43.4 months, the results revealed an improvement in disease-free survival with NIVO, and the overall survival and disease-specific survival were longer with NIVO compared to placebo. This was true for both intentto-treat and PD-L1 ≥1% patients.
With the exploratory ctDNA analysis, the research team found that ctDNA was detected in 18.8% of patients, and 40.6% of patients were ctDNA positive. Additionally, the results demonstrated that improvement in disease-free survival was seen in patients who were ctDNA positive at Cycle 1 Day 1, but not in those who were ctDNA negative at the same time point.
In summary, the 5-year results of the Phase 3 CheckMate 274 trial show continued improvement in disease-free survival with NIVO, consistent with prior observations. These long-term results support the continued use of adjuvant NIVO as a standard of care for patients with high-risk MIUC. Furthermore, ctDNA-based risk assessment may help refine future treatment pathways, enabling more individualised decisionmaking and potentially guiding immunotherapy selection in clinical practice.
CompARE: Neoadjuvant and Adjuvant Durvalumab in Locally Advanced Oropharyngeal Cancer
DATA from the Phase III CompARE trial, presented at the ESMO Congress 2025, provide new insight into the role of immunotherapy (IO) combined with chemoradiotherapy (CRT) in locally advanced oropharyngeal cancer (OPC). Previous trials of IO in the radical CRT setting have failed to demonstrate its benefit. This has been attributed to concurrent administration with CRT or delayed initiation of adjuvant IO. CompARE evaluated the impact of neoadjuvant IO followed by adjuvant IO started shortly after CRT completion.4
CompARE is a multicentre Phase III RCT (ISRCTN41478539) using an adaptive, multi-arm, multi-stage design. Patients aged 18–70 years with an Eastern Cooperative Oncology Group (ECOG) performance status 0–1 and either intermediate-risk OPC (HPV-positive TNM7 N2b+ with more than 10 pack-year smoking history, N3 or T4) or high-risk OPC (HPV-negative) were randomised to standard therapy (70 Gy in 35 fractions with concurrent cisplatin; Arm 1) or neoadjuvant durvalumab 1,500 mg followed by standard therapy, and then durvalumab 1,500 mg every 4 weeks for 6 months, initiated 2–6 weeks after CRT (Arm 5). The primary endpoint was overall survival (OS), with interim assessment of event-free survival. Secondary outcomes included toxicity, quality of life, swallowing function, and gastrostomy dependence.
A total of 594 patients were recruited across 34 centres (306 in Arm 1 and 288 in Arm 5). In total, 85% had intermediate-risk disease and 15% had high-risk disease. In Arm 5, 98% received induction durvalumab and 81% received adjuvant treatment. At median follow-up of 37 months (95% CI: 28–37), 3-year OS was 84% (95% CI: 79–88%) in Arm 1 and 82% (95% CI: 76–86%) in Arm 5
(stratified log-rank p=0.99). Cox regression analysis showed a hazard ratio (HR) for Arm 5:Arm 1 of 0.97 (95% CI: 0.65–1.46).
In the intermediate-risk group, 3-year OS was 90% (95% CI: 84–93) in Arm 1 and 84% (95% CI: 78–89) in Arm 5, HR: 1.24 (95% CI: 0.75–2.03; p=0.40). In the high-risk group, 3-year OS was 52% (95% CI: 35–67) in Arm 1 and 65% (95% CI: 45–80) in Arm 5; HR: 0.60 (95% CI: 0.30–1.24; p=0.17).
PD-L1 biomarker analysis is ongoing, and secondary outcomes and updated follow-up will be presented.
Although the addition of neoadjuvant and adjuvant durvalumab to standard care did not improve overall survival in the overall OPC population, a potential signal of benefit in high-risk HPV-negative patients was observed. These findings support further investigation of PD-1 and PD-L1 inhibition strategies in this subgroup.
The primary endpoint was overall survival (OS), with interim assessment of event-free survival
Dose-Dense Regimen Extends Survival in Ovarian Cancer
ICON8B, a Phase III randomised clinical trial, presented at the ESMO Congress 2025, evaluated whether adding dose-dense weekly paclitaxel (ddwT) to standard three-weekly carboplatin (q3w C) and bevacizumab (BEV) improves outcomes for patients with high-risk Stage III–IV epithelial ovarian cancer (EOC). Earlier analysis demonstrated that ddwT increased median progression-free survival by 5.5 months compared with standard q3w paclitaxel (q3w T) when both were combined with carboplatin and BEV. The current report presents the final overall survival (OS) outcomes following long-term follow-up.5
Eligible patients included those with Stage III disease with >1 cm residual tumour after primary surgery or those requiring primary chemotherapy, as well as all Stage IV cases. Participants were randomised to standard therapy (Arm B1: q3w C + q3w T + BEV) or the experimental regimen (Arm B3: q3w C + ddwT + BEV). A third arm without BEV(B2) was closed early due to the lack of progression-free survival benefit. Treatment consisted of up to six cycles of chemotherapy and 18 cycles of BEV. OS was a key secondary endpoint, and followup continued until December 2024.
Between 2015–2020, 579 patients were randomised to Arms B1 or B3. The population reflected typical high-risk EOC: median age of 64 years, 91% with high-grade serous carcinoma, and 93% with Stage IIIC–IV disease. Most (84%) commenced treatment with primary chemotherapy.
After a median follow-up of 72 months, 411 deaths occurred. Median OS was significantly improved in the ddwT arm at 49.8 months versus 39.6 months with standard paclitaxel (hazard ratio: 0.79; p=0.010). Among patients receiving primary chemotherapy, OS remained superior in the ddwT group (47.3 versus 37.1 months).
The findings demonstrate a clinically meaningful 10-month OS improvement with ddwT when combined with carboplatin and BEV. The authors conclude that this regimen should be considered a new first-line
standard of care for high-risk Stage III–IV EOC, while future research should explore the impact of homologous recombination deficiency and tumour chemosensitivity on treatment benefit.

References
1. Peccatori FA et al. POSITIVE trial investigators. Temporary interruption of adjuvant endocrine therapy for pregnancy in women with hormone receptor–positive early breast cancer: Updated outcomes. Abstract LBA12. ESMO Congress, 17-21 October, 2025.
2. Chau I et al. SKYSCRAPER-07: A phase III, randomised study of atezolizumab (atezo) with or without tiragolumab (tira) in patients (pts) with unresectable esophageal squamous cell carcinoma (ESCC) that has not progressed following definitive concurrent chemoradiotherapy (dCRT). Abstract 2094O. ESMO Congress, 17-21 October, 2025.
3. Galsky MD et al. Adjuvant nivolumab vs placebo for high-risk muscleinvasive urothelial carcinoma: 5-year efficacy and ctDNA results from CheckMate 274. Abstract 3068O. ESMO Congress, 17-21 October, 2025.
4. Mehanna HM et al. CompARE: A phase III randomised evaluation of neoadjuvant and adjuvant durvalumab combined with chemoradiotherapy in oropharyngeal cancer (OPC). Abstract 13170. ESMO Congress, 17-21 October, 2025.
5. Clamp A et al. ICON8B: GCIG phase III randomised trial comparing first-line weekly dose-dense chemotherapy + bevacizumab to three-weekly chemotherapy + bevacizumab in
high-risk stage III-IV epithelial ovarian cancer (EOC): Final overall survival (OS) analysis. Abstract 10640. ESMO Congress, 17-21 October, 2025.
Congress Interview
This year, we had the pleasure of speaking with Fabrice André, Director of Research and Professor in the Department of Medical Oncology at Institut Gustave Roussy, Villejuif, France, and current President of the European Society for Medical Oncology (ESMO). As a global leader in precision oncology, André reflects on his career journey, the evolving molecular landscape of breast cancer, and the transformative role of biomarkers, AI, and emerging technologies in patient care. He also shares insights into the vision shaping ESMO Congress 2025 and the future of oncology practice worldwide.
Featuring: Fabrice André


Fabrice André
Director of Research and Professor, Department of Medical Oncology, Institut Gustave Roussy, Villejuif, France; President, European Society for Medical Oncology (ESMO)
Citation: EMJ Oncol. 2025;13[1]:89-94. https://doi.org/10.33590/emjoncol/COAN3250
Q1
Could you begin by sharing some insights into your professional journey and what led to your current role as Director of Research at Gustave Roussy, Villejuif, France?
I went into cancer care and cancer research because, when I was a fellow, it was the medical specialty with the largest number of challenges; survival was very poor. At that time, it was also the beginning of molecular oncology, meaning that people had identified the first oncogene. It was the beginning of the work in immunology. There was this conjunction where a lot of patients were dying, but we could see the first signal that something was going to happen. Because of this, I went into oncology. Then, I had the opportunity to get a permanent position at Gustave Roussy and work on breast cancer.
I sub-specialised and worked on gaining a deeper understanding of breast cancer and developing
new therapies for patients. After I came back from a stay at the MD Anderson Cancer Center, Houston, Texas, USA, I had the opportunity to start my own research team focused on how to predict treatment efficacy thanks to biomarkers. Then, because Gustave Roussy began developing a lot of translational research, they needed someone involved in this type of research to lead the research division.
That’s the journey, starting from challenges and identifying opportunities, and then step by step, growing the field of translational research.
Q2
As the director of UMR 981, could you share some of the most exciting molecular predictors and new targets your team is currently investigating, particularly in relation to breast cancer?
My team performs molecular analysis at a large scale in situations that have not been well investigated, and where there are some important challenges. For example, we could report the first genomic landscape of metastatic breast cancer using whole exome sequencing, and identify some new genomic alteration that could be involved in resistance. That was an important report. We could identify, subsequently, that some molecular activation, like programmed death-ligand 1 (PDL1) amplification, could predict sensitivity to immunotherapeutics. We could describe how antibodydrug conjugates, for example, work, and identify the protein that could be involved in resistance (SLS4). This is what has been done in the last few years: describing the molecular landscape of a very specific situation, like metastatic breast cancer, and from this, focusing on a few biomarkers. What we are doing more often now is investigating the mechanism of action of new complex anticancer therapies.
Q3How would you describe the current genomic landscape of metastatic breast cancer, and what emerging developments or advancements are on the horizon for its treatment and understanding?
So, when you analyse the genomic landscape of metasticmetastatic breast cancer, and I will be focusing more precisely on hormone receptor positive breast cancer, there are several key findings: firstly, most of the molecular alterations that are enriched in metastatic breast cancer are connected to therapy resistance. For example, ESA1, AKT1, ERBB2, KMT2C, and RB1 mutations. This is a very important point, and the general concept of this has also been found by other teams. What it tells us is that there are not many genomic alterations, in terms of mutation, that could explain the metastatic process. The molecular clues of the metastatic process could probably fall more on the side of epigenetics, for example.
Secondly, there is an evolution of the genome. It seems that, when we look at the mutational signature acquired during the evolution of the resistance and metastatic process, it could be in relation to (apolipoprotein B mRNA editing enzyme catalytic polypeptide) APOBEC. So, is it APOBEC that mediates this evolution, or is it something else to do with the APOBEC signature? We don't know, but it's a mutational scar and signature that we can see.
Something else that I think is important is that the more these mutational signatures are active, the worse the outcome is. When there is an activation of the mutational process, and we can see it in the cancer, then suddenly the outcome of the patient becomes very poor. This is also something used to identify the patients who have a high risk of death very quickly.
Q4
As a key contributor to the SAFIR02BREAST IMMUNO trial exploring durvalumab in triple-negative breast cancer (TNBC), could you share the key findings, particularly regarding the correlation between CD274 gene amplification and increased PD-L1 expression in TNBC?

The aim was really to identify which subtype of breast cancer could be more sensitive to maintenance therapy with an anti-PD-L1 antibody. The trial tested durvalumab compared to maintenance chemotherapy in patients with metastatic breast cancer without genomic alteration identified. A key finding is that durvalumab improved outcomes in patients with TNBC, but not in the other group of patients. Interestingly, we could find copy number alterations because the patients were pre-screened and all received genomic testing. Because we had the data on copy number alteration at a

We tested 1,400 patients and trained the AI, and interestingly, the AI came with a model that could identify a group of patients with high risk of relapse

large scale, we could observe in an exploratory retrospective analysis that the amplification of CD274 was associated with higher sensitivity to anti-PD-L1 antibody. CD274 is the gene that encodes for PD-L1, so it is an interesting finding; we can see that the more the gene copies code for PD-L1, the more sensitivity there is to anti-PD-L1 antibodies (in breast cancer specifically). Interestingly, some other colleagues have reported similar analyses. There is one paper on lung cancer, for example, with the same findings.
Q5 Your research on using bioinformatics and AI to predict relapse in patients with breast cancer is fascinating. How close are we to developing a clinical tool that can reliably identify patients at risk of recurrence based on these advanced techniques?
One major step regarding patients with early-stage breast cancer is identifying which patients present a high risk of relapse as soon as
possible. Why? Because then the patients with a very low risk of relapse can be treated locally with potential de-escalation. Meanwhile, for patients at a high risk of relapsing, we must develop new drugs. From this need and this gap, we developed a project, supported by the French government, to re-identify patients with a very high risk of relapse as soon as possible. The strategy was to use AI-assisted pathology. We tested 1,400 patients and trained the AI, and interestingly, the AI came with a model that could identify a group of patients with high risk of relapse. When we looked at the features that were identified, we could see that the AI identified some features that are already known to be associated with a poor outcome. This means that the AI, without prior knowledge, could identify the features that we already know but that took us several decades to discover. This shows us that AI can rediscover things that are relevant for the patient.
The other part of this very large programme is about the genomic side of the disease and identifying which genomic alterations predict outcomes at the DNA and RNA level. Though it has not yet been reported, we could indeed identify that some DNA mutations or gene amplification could predict the outcome. What I want to highlight is the need to do this, because otherwise we are going to develop new drugs that are very expensive and cause some toxicity in a population that does not need new drugs and will, in fact, never relapse.
Q6 With the European Society for Medical Oncology (ESMO) Congress 2025 set in Berlin, Germany, are there any sessions or speakers you’re especially excited about, and why?
The ESMO Congress is a big event. There are around 35,000 participants; most attend physically, but some attend virtually as well. The Congress is organised in two different parts.
We have some sessions that are mostly for educational purposes. For these, the programme is available. We also have a large part of the Congress dedicated to new data. This is based on abstract submission, and we usually have very interesting presidential sessions about practice-changing trials in cancer care.
In terms of the educational part, the aim is really to train all the oncologists on innovation and equip them with knowledge. A lot of innovations are on the way, and it's our job as a scientific society to train doctors to be able to understand these innovations and use them in an optimal way for the patient. For example, we have a new track on AI, which is becoming very important in cancer care. Interestingly, we also developed a new track on molecular oncology, which means that we are going to classify metastatic cancer based on biology rather than the organ of the (primary) tumour. This really is a new way of classifying cancer and practising oncology based on molecular alteration. Digital oncology is becoming more and more prevalent, and we need to implement and use it in an optimal way.
We also have a track dedicated to prevention. Oncologists are becoming more and more involved in prevention, so it's important for them to learn more about it, such as how to prevent secondary cancer.
Then we have the usual tracks. I will call them the historical tracks: breast cancer, gastrointestinal cancer, lung cancer, etc. Inside these tracks, what we are really trying to do is once again provide oncologists with training on the latest advances. So, most of the sessions are focused on new
drugs, and what the optimal use of these new drugs looks like in terms of target populations, but also in terms of safety and toxicity management.
Q7
As the ESMO President, what are the main goals and areas of focus for this year’s Congress, and how do you plan to address the current challenges in oncology research and patient care?
ESMO is a scientific society with 45,000 members globally, so which challenges are we facing now in the field of oncology, and how can ESMO address them? Firstly (not necessarily by order of priority), it’s training doctors on innovation. For this, we are developing new conferences and new journals on AI and digital oncology, but also prevention and all these emerging elements that have never been on oncologists’ radars before.
The ESMO Congress is a big event. There are around 35,000 participants; most attend physically, but some attend virtually as well
Secondly, it’s moving forward to develop new cancer classifications based on molecular analysis. This is a new vision for oncology, especially in the metastatic setting. We want oncologists to understand the disease in order to better treat the patient. Our vision is for the way that we practise oncology in the future to be based on an understanding of the disease, in addition to the current evidence. This is the major plan for oncology.
Besides the educational and scientific dissemination, we want ESMO to become an organisation that develops tools that will shape oncology. My first example of this is that ESMO developed the Magnitude of Clinical Benefit Scale (MCBS), which allows oncologists to rank the efficacy and utility of drugs. It's a tool that we can use as oncologists to shape our practice. My second example is the ESMO Scale for Clinical Actionability of molecular


Targets (ESCAT). It's used to classify genomic activation. It’s a framework that is going to shape and optimise the way we do oncology. We want ESMO to not only be an educational platform or a platform for scientific dissemination, but also an organisation that develops new tools and frameworks that will change the way we practice oncology and make it more optimal. We want to reach out and help more members globally. We want to be sure that any member, wherever he or she is located, can access our services, and that our services are aligned with local needs.
We want to develop solutions for areas of the world that do not have access to the same innovations that we do. This is a big problem, and it's an ethical
We want ESMO to not only be an educational platform or a platform for scientific dissemination, but also an organisation that develops new tools and frameworks that will change the way we practice oncology and make it more optimal
issue. Collectively, ESMO and others need to find concrete solutions instead of just having discussions. In order to address this, ESMO has developed the International Cancer Foundation (ICF) where we aim to help colleagues working in areas where there is a lack of access to innovation.
We also have a very good working group on AI and how to integrate the use of generative AI in
educational activities, as well as in the decision-making process in the field of oncology. Again, this is going to be an important topic.
To summarise: we are 1) developing activities to train oncologists about innovation, 2) developing a new vision around molecular oncology and new classifications, 3) trying to reach members globally and provide solutions for access to innovation, and 4) trying to integrate AI and
the way we make decisions.
Q8
Given your extensive experience in precision medicine, how do you envision the integration of genomics, biomarkers, and targeted therapies evolving in oncology over the next decade?
What are the challenges in the field of precision oncology that we have to overcome in order to deliver the right treatment? The first is how to move beyond genomics. Most of the drugs that are being developed now are complex therapies. A lot of them are coming from the biotechnology sector. More and more, we need to integrate a new dimension of biology with biomarkers, meaning we need to integrate protein profiling, assessment of the tissue microenvironment, and spatial assessment. This means that we are moving towards a situation where we will have multiple dimensions of the biology that we can use to predict drug efficacy. This is one part of the vision. The second part is how to make the first generation of testing available everywhere, and for these, there are two solutions.
The first solution is developing tools based on the pathology, assisted by AI, to screen at a very large scale and identify patients with a high likelihood of presenting a gene alteration. The aim here is very simple: can we, based on very cheap, simple technology, and assisted by AI, identify groups of patients on a large scale that we will then test with genomics or more complex technology?
The other way to increase the number of patients who receive genomic testing is to move to circulating tumour DNA (ctDNA), because when you use ctDNA,
there is no need to perform a biopsy, which saves money (the patient doesn’t stay at the hospital and there’s no need for complex infrastructure). Therefore, the use of ctDNA is likely going to increase the number of patients who get access to a test. This is very important.
Lastly, we have the development of new drugs. It is about understanding the mechanism of action of new drugs in patients in order to derive new biomarkers. In the past, we used to derive biomarkers based on what was happening in pre-clinical studies or what we thought was the mechanism of action. Now, there are more efforts worldwide focused on performing all treatment biopsies to re-understand how these complex drugs are working, and this will facilitate the development of biomarkers.
Q9What advice would you give to young researchers aspiring to specialise in oncology?
I would really invite my young colleagues to join this community
Cancer is a very aggressive disease. Through our connection with the patient, we really get to the core of what humanity is
of doctors working on cancer care. Why? First, because we are facing a major challenge. It's always very important to start your professional life with a challenge, because during several decades of effort, you will see a lot of these challenges being solved, which is very rewarding. It's a very important consideration when you start your career: where is the field going to be in the next 3 or 4 decades? Clearly oncology is going to grow. We will have new drugs and new solutions for patient. It's a field with optimism.
The second reason why younger colleagues should become cancer care doctors is because the relationship with the patients is very special. Cancer is a very aggressive disease. Through our connection with the patient, we really get to the core of what humanity is. When you begin this relation, helping the patient is very important, but you also learn a lot about yourself, and you become another person. I'm sure that with the development of AI and new technologies, the most important people in the future will be the ones who have more humanity. I think humanity is going to come back because of these technologies. This part of the relation between patients and doctors is at the core of the practice of oncology; it gives us a lot of humanity and humility as well.
My last point is that, because this is a medical field where you can rediscover things and the challenges are so big, you can really, as a doctor, make new observations, undertake research, and contribute to transforming the field and making a difference.
Interview
This year we had the pleasure of speaking with Komal Jhaveri, Breast Medical Oncologist and Early Drug Development Specialist at Memorial Sloan Kettering Cancer Center, New York, USA. Jhaveri reflected on the evolution of targeted therapies in breast cancer, the transformative impact of antibody–drug conjugates, and the importance of tackling resistance mechanisms through precision medicine. She also discusses the vital roles of survivorship care, patient advocacy, and health equity, while looking ahead to the exciting potential of theranostics and precision-guided treatment strategies to shape the future of breast cancer care.


Breast Medical Oncologist and Early Drug Development Specialist, Memorial Sloan Kettering Cancer Center, New York, USA
When I think about breast cancer, I think about how it challenges us to pair cutting-edge science and innovation with humanity and genuine compassion
Citation: EMJ Oncol. 2025;13[1]:95-99. https://doi.org/10.33590/emjoncol/MTNO4310
Q1
What initially inspired you to specialise in breast cancer?
When I think about breast cancer, I think about how it challenges us to pair cutting-edge science and innovation with humanity and genuine compassion. I feel truly inspired by the hope patients carry, the strength, the resilience, and the courage they show, and the opportunity to then turn that hope into progress through science. More importantly, I would say that it's a real privilege to build a long-term, longitudinal relationship with patients who have breast cancer and their families, because you get to walk alongside them through their diagnosis, treatment, and survivorship. That ongoing connection really gives incredible meaning to the science and the care that we deliver.
Q2
What do you see as the most transformative recent breakthroughs in targeted therapies for metastatic breast cancer, particularly in oestrogen receptor (ER)-positive, human epidermal growth factor (HER)2positive, HER2-low, and triplenegative subtypes?
We have come a very long way in the management of breast cancer and breast cancer subtypes. As you pointed out, the ER-positive, HER2-positive, and triple-negative breast cancer subtypes are examples where I think we're now matching treatments to tumour biology. We have targeted therapies against specific mutations. In the ER-positive realm alone, in the last 2 years, we've had four approvals by matching these precision-based oncology therapies. More importantly, antibody–drug conjugates (ADC) have also shaped our subtyping and blurred the subtype boundaries. We now have ADCs, not just for HER2-positive or triple-negative breast cancer, but for categories such as HER2-low
Komal Jhaveri
I think that ADCs are here to stay, and I envision them taking over chemotherapy in the future

and HER2-ultra-low, subtypes that didn't exist a few years ago.
Last but not least, we're also now seeing therapies against central nervous system metastases or brain metastases, with agents and combination strategies in that space as well. This really speaks to how we’ve been able to overcome some of the previous unmet needs. Again, there is a lot more work to do, but I think that we have definitely come a long way from where we started.
Q3How are novel ADCs shaping the treatment landscape and improving patient outcomes?
I think that ADCs are here to stay, and I envision them taking over chemotherapy in the future. They provide a targeted way of delivering a very potent and otherwise toxic payload directly to the tumour, with the idea of having slightly less toxicity than what we normally would have seen with systemic chemotherapy. These highly potent payloads, which otherwise could not be
given on their own, have also improved efficacy. This has really transformed our landscape and treatment paradigms.
With HER2-positive breast cancer, we had the very first prototype ADC, trastuzumab emtansine, in 2013, based on the EMILIA trial. Since then, we've had a newer generation of ADCs with improved linker technologies and more potent payloads, such as trastuzumab deruxtecan, sacituzumab govitecan, and datopotamab deruxtecan. We know that trastuzumab deruxtecan has been approved in HER2-positive disease. It has shown activity in second-line settings and beyond, and now we also have data for patients with brain metastases and in the firstline metastatic setting.
At this year’s European Society for Medical Oncology (ESMO) Congress, we also saw practice-changing data in the early-stage setting with trastuzumab deruxtecan. This is truly transformative in terms of what we have been able to
achieve. Sacituzumab govitecan was initially approved for triplenegative breast cancer and was later approved for heavily pre-treated hormone receptorpositive breast cancer. We also saw promising new data at ESMO in the first-line triple-negative metastatic setting from the ASCENT-03 trial.
Lastly, the third agent to join the treatment paradigm is datopotamab deruxtecan. This is a trophoblast cell surface antigen 2 (TROP2)-directed ADC, already approved based on the TROPIONBreast01 study in hormone receptor-positive breast cancer. We now have practice-changing overall survival benefits, first reported as a press release, and later presented at the annual ESMO Congress from the TROPIONBreast02 study in triple-negative breast cancer. These are very exciting times across all breast cancer subtypes, as we are now seeing how these agents can be used earlier in metastatic and even early-stage settings to further improve outcomes for our patients.
Q4
What strategies are being explored to overcome resistance mechanisms in breast cancer therapies?
When we think about resistance, the ER-positive subtype is a great example. There, we know that the most important target is the oestrogen receptor itself. While we have endocrine therapies that are very effective, we have also learned that, unfortunately, resistance is inevitable in the majority of our patients. That is why our research efforts have focused on identifying the mechanisms of endocrine resistance and developing therapies to target those resistance pathways.
This has led to real success stories. We now have four agents targeting the PI3K–AKT–mTOR pathway. We started with everolimus, which targets the most downstream node, mTOR, and achieved approval for that in 2012. Then, in 2019, we had approval for alpelisib, a PI3K inhibitor used with endocrine therapy, based on the SOLAR-1 trial. In 2023, we saw approval for the protein kinase B (AKT) inhibitor capivasertib, in combination with fulvestrant, based on the CAPItello-291 study for PI3K/ AKT/PTEN-altered tumours. Most recently, we had a triplet strategy using another alpha-specific PI3K inhibitor, inavolisib, in combination with fulvestrant and palbociclib
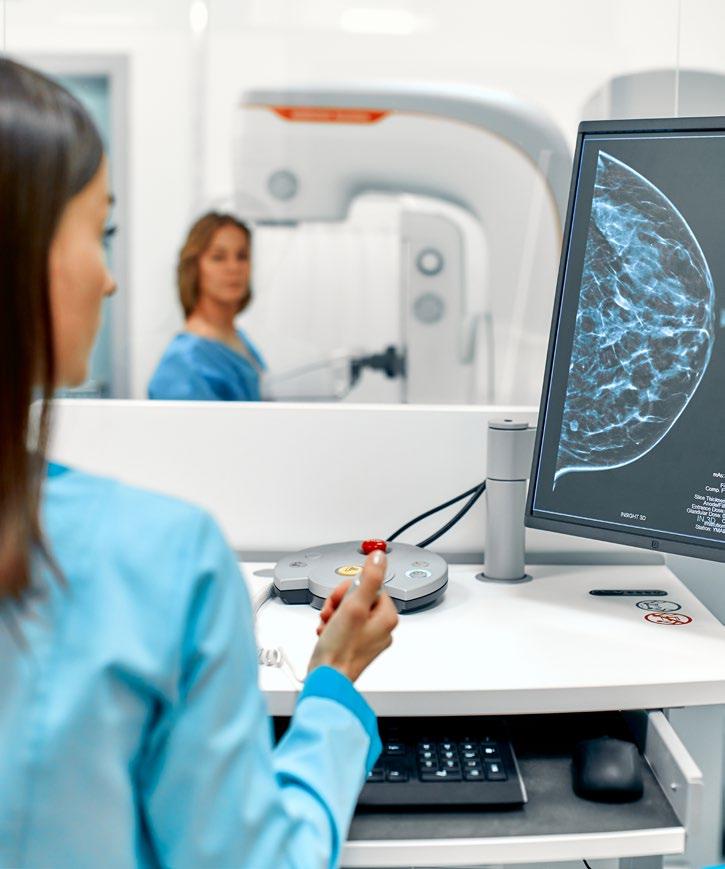
in the first-line PIK3CA-mutated, endocrine-resistant setting. That triplet strategy, for the first time with this pathway, showed not only a progression-free survival benefit but also an overall survival benefit, which has been very meaningful and practice-changing for us.
Another good example of overcoming resistance in ERpositive breast cancer is the development of ESR1 mutations, which arise under the selective pressure of aromatase inhibitors in the metastatic setting. Here, we are not necessarily seeing other stem-cell-like mechanisms driving resistance; rather, we see the main target itself mutating, yet remaining relevant, because the pathway continues to be oestrogen-receptor-dependent, even though it becomes ligandindependent. This justifies the use of novel endocrine agents such as oral selective oestrogen receptor degraders (SERD), which have now been approved.
We had approval in January 2023 for elacestrant, the first oral SERD, and most recently, this past month, we had approval for imlunestrant, another oral SERD, based on the EMBER-3 trial. In the future, we may also see approval for proteolysis-targeting chimeras such as vepdegestrant. These are great examples of how we have achieved significant progress by targeting mutations and addressing endocrine resistance in ER-positive breast cancer.
A further example, which applies even to triple-negative breast cancer, is the use of poly(ADPribose) polymerase (PARP) inhibitors for germline carriers. PARP inhibitors are already approved for germline BRCA1 and BRCA2 mutations, and we have also seen data for somatic
BRCA1/2 and germline PALB2 mutations. We look forward to seeing label expansions for these PARP inhibitors in that setting as well. This illustrates not only how we tackle resistance, but also how we target germline and somatic mutations that may be driving resistance.
Finally, for very rare yet important mechanisms such as NTRK and RET fusions, we now have neurotrophic receptor tyrosine kinase (NTRK) inhibitors and RET proto-oncogene receptor tyrosine kinase (RET) inhibitors approved. For tumours with high mutational burden, we have pembrolizumab, a checkpoint inhibitor, approved across subtypes, including ERpositive disease. Altogether, I think this captures how far we have come in addressing resistance in breast cancer.
Q5 October is Breast Cancer Awareness Month. From your perspective, why are these awareness months important?
October really is a reminder, in my mind, that behind every pink ribbon, there is a person, a family, and a story. It creates a powerful moment of collective focus, which unites patients, clinicians, researchers, and the public.
To me, I think it's a first step towards prevention, early detection, and ultimately progress. But it is not just that, it is also a call to action for all of us to close the gaps of inequities in cancer care. I think that awareness brings change, but action is what saves lives. That's how I think October brings us together and makes us act together to ultimately save lives.

October really is a reminder, in my mind, that behind every pink ribbon, there is a person, a family, and a story
Q6
Beyond awareness campaigns, what additional steps can the medical community, policymakers, and the public take to further amplify awareness and drive meaningful change in breast cancer prevention and care?
Beyond awareness, I think there are a few things that we could do together. We can strengthen prevention and early detection strategies by expanding access to genetic counselling, promoting evidence-based screening programmes, and investing in lifestyle-based prevention as well.
We can also bridge gaps in equity and access, as we were talking about, by reducing disparities in diagnosis and treatment and ensuring timely access. Certainly, we can continue to drive innovation with science and data, investing not just in translational research but also leveraging AI and big data, and encouraging cross-disciplinary collaborations.
We may also need to shape policy and infrastructure by expanding coverage beyond treatment to include prevention and screening. And last but not least, I think we really need to empower the public and our survivors, to promote not just health literacy but also to include the voices of survivors and foster strong community partnerships.
Q7 Can you discuss the importance of survivorship care in long-term breast cancer management?
I think that beating cancer is not the finish line; it’s really the start of a lifelong journey. Survivorship must therefore be seen as an essential part of treatment, not just an afterthought. It’s about how we turn survival into lasting health, dignity, and empowerment, because true progress means ensuring that every survivor has access to care that addresses not just the disease, but the whole person: their physical, emotional, and social wellbeing.
We really need to focus on survivorship by addressing not only the early side effects but also the late ones, supporting mental and emotional health, and coordinating continuity of care beyond treatment. We must also promote lifestyle changes and secondary prevention so that each survivor’s journey is meaningful, with the best possible quality of life throughout. And we have to do all of this by ensuring equity.
So I believe that embedding survivorship into every stage of treatment, and making it equitable and accessible to everybody should be our goal. And for that, we need to plan right at the time of therapy and never lose sight of it.
Q8
How do you view the role of patient advocacy in guiding research priorities and improving equity in clinical care?
I think that patient advocates are critical. We want to encourage them, support them, hear them, and implement their voices. Their voices and insights really help us to design trials that actually matter. They help us improve access to therapies and ensure that scientific progress translates into meaningful outcomes for diverse communities.
They ensure that the clinical priorities they highlight reflect real-world needs. They highlight the gap in equity and hold the system accountable, so that there is benefit for all patients alike, and not just a subset of patients that enrol in clinical trials.
I think they are the reason that we push ourselves to focus on quality of life, on access, and on equity. They make research and care more human-centred and more inclusive, which is exactly why we do what we do.
I’m in breast cancer because it's at the intersection of science and compassion. And I think listening to our patients and actually hearing them gives us that human-centred and inclusive approach that we want to bring into our day-to-day care.
Q9 Looking ahead, what excites you most about the potential of precision medicine in reshaping the future of breast cancer care?
I think we’re on the path we set out on, one that focuses on personalising and individualising care. That path, to me, is important, and will remain so in the future, because I truly believe the future of breast cancer treatment is personal.
By that, I mean not just precisely matching the right drug to the right biology, but also developing novel, precision-based strategies and delivering those therapies more effectively, as we’ve seen with ADCs. The goal is to combine precise matching and precise delivery to achieve smarter, safer, and more effective therapies.
One particular new wave of treatments that I’m excited about for the future of breast cancer is what we’ve learned from prostate cancer with theranostics, or radiopharmaceutical therapies. The theranostic trials that showed improvements in progressionfree survival, overall survival, and quality of life in prostate cancer could be equally meaningful for breast cancer patients, especially because they fit the same personalised, precision-based approach I was referring to.
Theranostics uses imaging to find a target, and once that target is identified, it’s treated with a radioligand therapy, a true ‘see and treat’ approach that allows us to precisely target the disease while minimising side effects and improving quality of life. So really, a precision-guided evolution is what I'm focusing on for the future of breast cancer care.

I think that beating cancer is not the finish line; it’s really the start of a lifelong journey
The Evolving Biomarker Landscape in GI
Citation: EMJ Oncol. 2025;13[1]:xx-xx. https://doi.org/10.33590/emjoncol/GNSE9148.
2025;13[1]:100-101. https://doi.org/10.33590/emjoncol/GNSE9148.
Types and Roles of Biomarkers
There are various ways biomarkers can be used:
1. Diagnostic - aid in disease detection
2. Prognostic - help determine clinical outcome, regardless of treatment received
3. Predictive - determine the potential benefit or lack of response to a treatment
4. Monitoring - assess the effect of a certain treatment2
Biomarker testing is usually performed by taking a blood sample or biopsy of the tumour and sending it to a pathology laboratory, where tests are done to determine abnormalities in DNA, RNA, hormones, or proteins.3
Prevalence of Gastrointestinal Cancers
• The main types of GI cancers are cancers of the oesophagus, stomach, colon and rectum, liver, and pancreas.1
Hepatocellular carcinoma:12
• The most common serum protein marker is AFP.
• The cut-off for AFP in diagnosing HCC is generally 20 ng/mL. However, the sensitivity is around 60%.
Abbreviations
Stomach cancer:10-11
• Predictive biomarkers include PDL-1, CLDN18.2 and MET
• In a 2024 US study of gastric and gastroesophageal junction adenocarcinomas, 44.4% of tumours were CLDN18.2positive overall, with 51.4% positivity specifically in gastric adenocarwcinomas.10
• A PD-L1 CPS of 1% or higher is seen in over 80%; however, this can vary with different populations.11
References AFP: alpha-fetoprotein; CA19-9: carbohydrate antigen 19-9; CEA: carcinoembryonic antigen; CLDN18.2: claudin 18.2; ctDNA: circulating tumour; CPS: Combined Positive Score; DNA; dMMR: mismatch repair deficiency; DPD: dihydropyridine dehydrogenase enzyme; EAC: oesophageal adenocarcinoma; ERBB2: Erb-B2 receptor tyrosine kinase 2; GC-MS: gas chromatography mass spectrometry; GI: gastrointestinal; HCC: hepatocellular carcinoma; HDI: high development
HER2: human epidermal growth factor receptor 2; MSI-H:
instability;
and localiser of BRCA2; PD-L1: programmed death ligand 1; VEGFR2: vascular endothelial growth factor receptor 2; VOC: volatile organic compounds.

• In 2022, there were over 4,000,000 new cases and 3,000,000 deaths from the five GI cancers, accounting for 23.9%, and 33.2% of all new cancer cases and deaths worldwide, respectively.1 1. Li M et al. Cancer Communications. 2025;45(7):774-88. 2. Chai X et al. Journal of Gastroenterology and Hepatology. 3. Colorectal Cancer Alliance. Available at: https://colorectalcancer.org/treatment/ types-treatment/why-biomarkers-matter#:~:text=Anyone%20who%20is%20 diagnosed%20with,does%20not%20respond%20to%20treatment. accessed: 23 October 2025.
4. Rai V et al. Int J Mol Sci. 2023;24(4):3316.
5. Dhakras P et al. Transl Gastroenterol Hepatol. 2022:5:55.
index;
microsatellite
PALB2: partner
GI Cancers
Oesophageal cancer:4
• Predictive biomarkers to determine the potential treatment response for EAC are HER2, MSI-H, and PD-L1.
• Diagnostic and prognostic biomarkers include HER2, and PD-L1 5
Innovations in Detection Technologies
Circulating Nucleic Acids:
• ctDNA is emerging as a promising non-invasive approach for cancer detection.
• A 2024 study showed ctDNA profiling improved therapy matching, survival, and assessment of tumor heterogeneity 14
Colorectal cancer:6
• Elevated levels of CEA post-surgery could indicate recurrence of the cancer.
• Approximately 40-50% of colorectal cancer patients have a KRAS mutation.7
• The most common KRAS mutations are G12V, G12D, G13D, and G12A
• A BRAF mutation is found in 10% of colorectal cancer patients, the most common mutation being V600E.
• Other common biomarkers include MSI-H and HER2/ERBB2, which both act as prognostic and predictive biomarkers.8
Microbiome-Based Biomarkers:
• A 2023 study showed that gut microbiome and metabolite changes, including Flavonifractor plautii enrichment and altered bile acid, choline, and tryptophan metabolism, distinguished early-onset colorectal cancer from controls.15
Pancreatic cancer:13
• KRAS codon 12 mutations are the most common genetic alterations in PDAC, occurring in over 90% of cases.13
• Other common genetic alterations include TP53, CDKN2A, and SMAD4 13
2025;40(5):1059-69. https://colorectalcancer.org/treatment/ types-treatment/why-biomarkers-matter#:~:text=Anyone%20who%20is%20
6. Colorectal Cancer Alliance. Available at: https://colorectalcancer.org/treatment/ types-treatment/why-biomarkers-matter/types-biomarkers. Last accessed: 23 October 2025.
7. Martianov AS et al. Int J Mol Sci. 2023;24(5):4868.
8. Battaglin F et al. Clin Adv Hematol Oncol. 2018;16(11):735-45.
9. Gastric Cancer Foundation. Available at: https://gastriccancer.org/biomarkers/. Last accessed: 23 October 2025.
10. Waters R et al. JCO Precis Oncol. 2024;8:e2300543.
11.
Breath Analysis:
• A 2022 study showed that analysing VOCs in exhaled breath can noninvasively detect advanced adenomas and colorectal cancer in screening populations, achieving about 77–80% sensitivity and 70% specificity, thus potentially improving colorectal cancer screening accuracy in the future.16
TM et al. Int J Gynecol Pathol. 2021;40(6):563-74.
YT et al. Molecular Cancer. 2024;23(1):189.
M et al. Clinica Chimica Acta. 2025;569:120176.
Jenkins
12. Chan
13. Chen
14. Nakamura Y et al. Nat Med. 2025;31(1):165-75.
15. Kong C et al. Gut. 2023;72(6):1129-42.
16. Cheng HR et al. Clin Transl Gastroenterol. 2022;13(11):e00518.
Integrated Multi-omics Approaches in Non-small Cell Lung Cancer for Biomarker and Pathway Discovery
Editor's Pick
I have selected this review as my Editor’s Pick for this issue of EMJ Oncology because multi-omics integration is rapidly emerging as a cornerstone of precision cancer medicine. Non-small cell lung cancer remains one of the most challenging malignancies to diagnose and treat, and advancing biomarker and pathway discovery is essential to improving outcomes. This article provides a timely and comprehensive exploration of how integrated multi-omics approaches are reshaping our understanding of non-small cell lung cancer and opening new avenues for personalised therapy.
Ahmad Awada Head of the Oncology Department and Director of the Chirec Cancer Institute, Brussels, Belgium
Author: *Malika Salhi1


1. Université Paris Cité, Hématologie, Oncogenèse, Biothérapies et Biotechnologies, France *Correspondence to mmsali@outlook.fr
Disclosure: The author has declared no conflicts of interest.
Received: 26.05.25
Accepted: 13.11.25
Keywords: Biomarker discovery, multi-omics, non-small cell lung cancer (NSCLC), personalised therapy, therapeutic resistance.
Citation: EMJ Oncol. 2025;13[1]:102-115. https://doi.org/10.33590/emjoncol/OEHS9513
Abstract
Lung cancer is one of the most common malignant tumours worldwide, with non-small cell lung cancer (NSCLC) accounting for the largest number of cases among both men and women. Poor patient prognosis due to therapeutic resistance remains a current issue, underscoring the need for a more comprehensive understanding of the underlying biology of the pathogenesis and progression mechanisms of NSCLC. Integrating multi-omics approaches, such as genomics, transcriptomics, proteomics, and metabolomics, has become crucial for studying the underlying biology of complex diseases like lung cancer. Applying these methods not only enhances knowledge of the mechanisms of lung cancer but also plays a pivotal role in identifying biomarkers and therapeutic targets for implementing personalised treatment plans. This review quantitatively analyses the predictive capability of integrated multi-omics models by synthesising findings from studies utilising clinical data (including survival outcomes and treatment response) with multi-omics technologies to pinpoint essential biomarkers and pathways associated with NSCLC. The author focused on comparing the reported predictive accuracy metrics of these models and the consistency
of identified key biomarkers across different studies. The author highlights the importance of integrating multi-omics analyses in the development of targeted therapies, and offers a roadmap for future clinical applications, emphasising challenges in data integration and biomarker validation, alongside opportunities for novel clinical trial designs. This review aims to provide a comprehensive quantitative assessment of the current state of integrated multi-omics in NSCLC, ultimately informing the design of more effective personalised therapeutic strategies and future research directions.
Key Points
1. Non-small lung cancer (NSCLC) accounts for approximately 85% of lung malignancies and remains the leading cause of cancer mortality worldwide, underscoring the urgent need for biomarker-driven precision strategies.
2. This systematic review synthesised evidence from 50 original and peer reviewed research articles published during the last 5 years, integrating genomics, transcriptomics, proteomics, metabolomics, and epigenomics to evaluate biomarker discovery, patient stratification, and therapy optimisation in NSCLC.
3. Multi-omics integration enables clinically actionable biomarker identification, enhances prediction of immunotherapy response, and informs personalised treatment frameworks. Collaborative validation and translational pipelines are essential to embed these insights into routine NSCLC care.
INTRODUCTION
Lung cancer remains the most frequently diagnosed malignancy and the leading cause of cancer tumour-related mortality worldwide.1 Non-small cell lung cancer (NSCLC) is the primary subtype of lung cancer, accounting for 85% of lung cancer cases.2 Despite advances in targeted therapies and immunotherapies, therapeutic resistance remains the principal barrier to durable responses and long term survival.3,4 Resistance mechanisms span the genome, epigenome, transcriptome, proteome, metabolome, and tumour microenvironment (TME), and evolve dynamically during treatment. Understanding how these mechanisms interact across molecular layers is essential for improving treatment efficacy and patient outcomes.4
NSCLC arises from a multifactorial aetiology involving both environmental and intrinsic factors. Environmental exposures such as tobacco smoke, air pollution, and occupational carcinogen exposure induce chronic epithelial damage and mutagenesis, while internal factors, including genetic predisposition, immune dysregulation, and chronic inflammation, contribute to tumour initiation, progression, and immune escape. These aetiologic drivers vary in impact
across histological subtypes and patient populations, underscoring the need for stratified prevention and therapy strategies.
Histologically, NSCLC is classified into three major subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (Supplementary Table 1). Each subtype exhibits distinct pathological characteristics, clinical behaviours, and molecular signatures. Biomarkers such as EGFR, ALK, and programmed death-ligand 1 (PD-L1) are routinely used to guide diagnosis and therapeutic decisions, particularly in the context of targeted therapy and immune checkpoint inhibition.5-7 However, their predictive power is limited by the emergence of acquired resistance, which often involves bypass signalling, epigenetic reprogramming, and TME-mediated immune evasion.
While blood-based biomarkers such as circulating proteins and cytokines offer a minimally invasive route for early detection and longitudinal monitoring, their clinical translation remains limited by challenges in specificity and validation across diverse populations. Expression levels of IL-6, IL-8, and colony stimulating factor 1 (CSF-1), for example, are influenced by systemic inflammation, comorbid conditions, and sampling variability, complicating their
interpretive value in NSCLC.8,9 Similarly, epigenomic markers, particularly DNA methylation signatures, show promise for subtype classification and risk stratification. However, methylation patterns are highly context dependent, varying with tumour subtype, anatomical sites, environmental exposures, and immune status.10,11 These limitations underscore the need for multilayered integration and robust validation frameworks, reinforcing the rationale for multi-omics approaches that triangulate signals across platforms to improve biomarker fidelity and clinical relevance.12-16
Recent concepts in tumour biology highlight how cancers remodel their microenvironment and hijack systemic homeostasis, including neuroendocrine signalling pathways that influence immune function, metabolism, and stress response.12 These interactions shape the natural history of NSCLC and complicate therapeutic design, reinforcing the need for narrative frameworks that capture tumourintrinsic and host-mediated dynamics.
Multi-omics technologies offer a comprehensive approach in interrogating NSCLC biology that predicts therapeutic efficacy and for discovering personalised targets.13,14 The term ‘omics’ refers to a suite of technologies: genomics (DNA sequencing to identify actionable mutations), transcriptomics (RNA profiling reveals regulation and treatment response), proteomics (protein level analysis revealing tumour progression and resistance mechanisms), metabolomics (small metabolites profiling for therapeutic targeting and disease monitoring), and epigenomics (genome wide assessment of epigenetic regulation). The integration of these platforms enables the discovery of biomarkers that predict therapeutic efficacy, inform personalised treatment strategies, and support the development of precision oncology studies.13,15
The TME, a complex ecosystem of cancer cells, stromal components, immune infiltrates, and extracellular matrix, plays a pivotal role in NSCLC resistance progression. Single-cell multi-omics technologies have revealed profound TME heterogeneity and cell-type-specific drivers of resistance and progression, while
systems biology approaches integrate multilayer data to establish a causal link between molecular alterations and phenotypic outcomes.15-17 Proteomics studies, for example, connect genomic alterations with protein-level consequences, exposing therapeutic vulnerabilities that would be missed by single-platform analyses. Emerging evidence also implicates the microbiome as a modulator of host immunity and therapeutic response, particularly in immunotherapy.17,18 Metagenomic and whole-genome sequencing of microbial communities, including bacteria, fungi, viruses, and archaea, have revealed associations between microbial signatures and treatment outcomes.
Pharmacokinetic-pharmacodynamic modelling represents another frontier in NSCLC research. These mathematical frameworks simulate drug–tumour interactions, enabling the optimisation of radiotherapy and chemotherapy schedules and the prediction of resistant subclones, and guiding the design of regimens that minimise resistance and enhance efficacy.19,20
In summary, NSCLC exemplifies the complexity of cancer biology, where genetic, epigenetic, metabolic, and microenvironmental factors converge to drive resistance. Multi-omics integration provides the most promising avenue to unravel this complexity, enabling the development of biomarker-guided strategies that anticipate resistance, personalise therapy, and ultimately improve patient outcomes.
MULTI-OMICS INTEGRATION IN NON-SMALL CELL LUNG CANCER RESISTANCE
Investigating the intricate molecular changes that drive resistance to targeted therapies is crucial for understanding tumour cell survival and clinical progression during treatment. In recent years, therapies targeting EGFR, BRAF, and KRAS mutations and ALK, ROS-1, RET, NTRK fusions/ rearrangements have improved survival in subsets of patients. Nevertheless, treatment responses remain incomplete, with acquired resistance emerging as the rule rather than
the exception.21 Resistance mechanisms, including genetic mutations, epigenetic reprogramming, metabolic adaptation, and TME-mediated immune evasion, complicate the durability and the effectiveness of targeted and immune-based therapies (Supplementary Table 2). Therefore, multiomics approaches, by interrogating tumour biology both at baseline and longitudinally during therapy, present promising tools to address these challenges.21,22
The interplay between genetic mutations and the TME is central to NSCLC complexity. Understanding these factors is vital for developing effective therapies and overcoming resistance in patients with NSCLC.
Emerging concepts further highlight how tumours regulate their environment and hijack systemic homeostasis through neuroendocrine signalling, influencing immune function, metabolism, and stress responses. These interactions shape the natural history of NSCLC and complicate therapeutic design, reinforcing the need for frameworks that capture both tumourintrinsic and host-mediated dynamics.12
While blood-based biomarkers such as circulating proteins and cytokines offer a minimally invasive route for early detection and longitudinal monitoring, their clinical translations remain limited by challenges in specificity, reproducibility, and validation across diverse populations.
Expression levels of IL-6, IL-8, and CSF-1, for example, are influenced by systemic inflammation, comorbid conditions, and sampling variability, complicating their interpretive value in NSCLC.8,17 Similarly, epigenomic markers, particularly DNA methylation signatures, show promise for subtype classification and risk stratification. However, methylation patterns are highly context-dependent, varying with tumour subtype, anatomical site, environmental exposures, and immune status.23,11 These limitations underscore the need for multilayered integration and robust validation frameworks, reinforcing the rationale for multi-omics approaches that triangulate signals across platforms to improve biomarker fidelity and clinical relevance.12,16
From DNA to RNA and ultimately to proteins, the amount and complexity of information progressively increases. Genomics has revealed key mutations, supporting the development of targeted therapies in personalised medicine.17 In NSCLC, genomic methods have identified aberrant activation of the PI3K/protein kinase B (AKT)/mTOR pathway as a driver of resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI). The discovery led to drugs targeting mTOR, EGFR, and ALK, which have shown clinical effectiveness in lung cancer treatment.24 However, the intricate nature of cancer mechanisms makes it difficult to establish clear links between tumours and specific genetic variants, necessitating a more nuanced approach to tackle its challenges.
Transcriptomics captures dynamic RNA profiles reflecting cellular states,17,25 while proteomics reveal functional protein changes, including post-translational modifications (phosphorylation, acetylation, and glycosylation) that change proteins’ structure and functionality.25
Next-generation sequencing and mass spectrometry have transformed the understanding of cancer biology by enabling comprehensive genomic, transcriptomic, and proteomic profiling. Next-generation sequencing allows for both genomic DNA and RNA sequencing.25 RNA sequencing explores transcripts, isoforms, splice variants, single-nucleotide polymorphisms, and chimeric gene fusions with high sensitivity and accuracy.17,22 Massspectrometry-based proteomics quantifies proteins in cells, tissues, and fluids.25 Large-scale initiatives such as The Cancer Genome Atlas (TCGA) programme integrate publicly available multi-omics databases across thousands of patients, providing a reference framework for NSCLC.21,26
Karaman et al.,15 conducted a network-based integrative analysis of RNA sequencing and DNA methylation data across lung, breast, colorectal, and kidney cancers identifying common prognostic biomarkers. This approach led to the identification of several significant biomarkers, including the SEC61G and the PTDSS1 genes associated with poor survival outcomes.15
Metabolomics has emerged as a powerful tool for identifying metabolic alterations.13 Shestakova et al.27 used targeted metabolomic profiling to distinguish patients with NSCLC from healthy individuals, identifying changes in tryptophan metabolism, the tricarboxylic acid cycle, and lipid metabolism, and developed a machine learning model with high diagnostic accuracy (area under the curve: 0.96), highlighting the potential of metabolomics in NSCLC diagnostics.27 Plasma and serum metabolomics offer minimally invasive diagnostics, but disease-specific plasma metabolites remain difficult to validate.13
Despite these advances, integration remains challenging. Multi-omics datasets are complex, heterogeneous, and high dimensional, requiring sophisticated computational methods.28-30
Machine learning and deep learning approaches are increasingly applied,31,32 but reproducibility and interpretability remain obstacles. Addressing these challenges demands advanced computational methods and substantial resources, but overcoming them could result in significant advancements in precision oncology and personalised medicine for NSCLC.33
This review quantitatively synthesises the predictive performance of integrated multiomics models and evaluates the impact of open-source tools on translational research. By examining studies that leverage clinical data and multi-omics technologies to identify key biomarkers and relevant pathways in NSCLC, the author provides a roadmap for future clinical implementation, highlighting both critical challenges (specificity, reproducibility, context-dependence) and emerging opportunities in the field.
METHODS
For this systematic review, the author conducted a structured literature search using PubMed, covering publications from January 2020–March 2025. The objective was to identify original research articles reporting multi-omics data with clinically relevant biomarker findings in NSCLC. Three keywords-based strategies were applied
using the ‘[Title] AND [Title/Abstract]’ filed tags to capture studies addressing omics modality, disease intersection, biomarker relevance, subtype stratification, and TME.
The search strings were:
• lung cancer and omics ([Title/Abstract]) AND (lung cancer [Title])) AND (omics [Title/Abstract];
• NSCLC and omics([Title/Abstract]) AND (NSCLC[Title])) AND (omics [Title/ Abstract]; and
• NSCLC, omics, and resistance([Title/ Abstract]) AND (omics [Title])) AND (resistance [Title/Abstract]).
Filters included “LUAD,” “LUSC,” and “tumour microenvironment” to refine results towards subtype-specific and clinically actionable findings. Additional keywords such as “biomarker,” “diagnostic,” “prognostic,” “therapeutic,” “survival,” and clinical outcome” were used to identify studies with translational potential.
Studies included were original research articles reporting multi-omics data on NSCLC biomarkers that focused on human subjects, provided subtype-specific insights (lung adenocarcinoma [LUAD], lung squamous cell carcinoma [LUSC]), and were published in peer-reviewed journals. The studies that were excluded were reviews, editorials, or conference abstracts, as well as studies lacking omics integration or clinical relevance.
The PubMed searches yielded 196, 30, and three records, corresponding to search one, two, and three, respectively. After deduplication and relevance screening, 30 articles were shortlisted, of which six were selected for detailed synthesis based on methodological rigour and translational value. To ensure currency and alignment with the review’s objectives, two additional studies published in March 2025 were manually incorporated into the final analysis (Figure 1).
All included studies were appraised for methodological clarity, completeness
of omics layers, subtype stratification, and relevance to clinical endpoints. Bias was mitigated through predefined eligibility criteria, transparent screening, and standardised data extraction, with emphasis on studies demonstrating reproducibility, multi-layer integration, and external validation.
RESULTS
Multi-omics analyses identified a broad spectrum of biomarkers with diagnostic, prognostic, therapeutic, and mechanistic relevance in NSCLC. To enhance clarity and clinical utility, findings were synthesised into functional domains and molecular pathways, highlighting convergent drivers, resistance mechanisms, and survival determinants.
Oncogenic Drivers and Tumour Suppressors
Mutations in TP53 and EGFR were the most consistent signals across studies. TP53, the most frequently mutated gene in NSCLC,31 was associated with recurrence, immune evasion, and resistance to conventional therapies,22,31,33 positioning it as both a prognostic and predictive biomarker. Its mutation status also influenced sensitivity to immune checkpoint inhibitors.18,33,32 In LUAD compared with LUSC, TP53 mutations were linked to altered tumour microbiota, immune infiltration, and histological architecture, and could be predicted using multimodal deep learning models integrating histopathology, microbiome, and transcriptomic features.31
EGFR mutations, present in over half of early-stage NSCLC cases, predicted shorter disease-free survival and recurrence.32
EGFR-TKIs remain a cornerstone of precision medicine,34 and integrative multiomics analyses confirmed favourable overall survival with afatinib in patients who were EGFR-positive.35 Resistance to EGFR-TKIs frequently emerged through secreted phosphoprotein 1 (SPP1) overexpression and activation of the yes-associated protein (YAP)/ transcriptional coactivator with PDZ-binding motif (TAZ) driven epithelialmesenchymal transition (EMT), conferring stem-like and invasive properties.34,36,37 Other genomic signals included TTN
mutations, correlating with chemotherapy and immunotherapy response,22,32 and ZNF71, which stratified patients into prognostic groups.38,39
Interpretation
TP53 and EGFR exemplify convergent drivers that shape both prognosis and therapy, while EMT-related pathways highlight recurrent mechanisms of therapeutic escape with the need for adaptive monitoring.
Immune Modulation and Systemic Inflammation
Immune-related biomarkers were prominent across omics layers, ZFHX3 mutations correlated with enhanced immune responses and immunotherapy sensitivity.36 Circulating cytokines such as IL6, IL8, CSF1, and C-X-C motif chemokine ligand 13 (CXCL13) predicted poor survival.8 Integrated microbiomic, metabolomic, and proteomic analyses identified C-reactive protein (CRP), lipopolysaccharide binding protein (LBP), and cluster of differentiation (CD)14 as systemic inflammatory markers reflecting systemic immune activation.40
Interpretation
Immune biomarkers provide stratification for immunotherapy, but also signal systemic inflammation, reinforcing the need for integrated host–tumour profiling.
Epigenetic and Metabolic Signatures
DNA methylation alterations (MGMT, CDKN2A, PCDH17, IRX1, TBX5, and HSPB6) were recurrently implicated in carcinogenesis.10,11 Large-scale, genomewide association studies linked methylation biomarkers to NSCLC risk,11 while epigenomic–transcriptomic integration nominated novel methylation biomarkers with diagnostic and therapeutic relevance.10
Metabolomic profiling differentiated LUAD from LUSC,41 while integrated metabolomic–proteomic analyses identified biomarkers predictive of immunotherapy sensitivity.33
Interpretation
Epigenetic and metabolic biomarkers highlight contextdependent signals that enrich stratification but require integration with genomic and transcriptomic data for reliable application.
Survival-Associated Biomarkers
Survival outcomes were linked to both tumour intrinsic signals and systemic mediators. Transcriptomic signals such as NKX21, CAV1, YBX1, FN1, and CDH3 were associated with LUAD survival.42
Circulating proteins (IL6, IL8, CSF1, matrix metallopeptidase 12 [MMP12], CXCL13) predicted poor outcomes.8 Immunerelated genes (TNS3, SEPT7, PUS1, IRF9, COMP, KLRB1, CD45, CD244) further correlated with survival.17,9,43,44
Interpretation
Survival reflects the dual importance of tumour biology and host immune response, reinforcing the need for integrated biomarker panels in prognostic modelling.
Longitudinal and Clinical Insights
Several studies emphasised the value of tracking biomarker patterns over time. Serial sampling and timeseries omics data enabled the monitoring of tumour evolution, the emergence of resistance, and the adaptive responses to therapy.31,34,45 Dynamic cytokine profiling revealed immune shifts,41 while comparative analyses distinguished NSCLC from sarcoma.42 Importantly, leptomeningeal metastasis was identified as a severe complication in EGFR-mutant NSCLC. Cerebrospinal fluidbased biomarkers enabled early detection/ identified central nervous system (CNS) dissemination in EGFR-mutant LUAD and CNS-specific monitoring.8
Interpretation
Longitudinal profiling demonstrates how biomarkers evolve under therapeutic pressure, offering realtime insights into resistance and progression, and reinforcing the need for adaptive, multi-omics-guided clinical strategies.
Leptomeningeal metastasis highlights the importance of extending biomarker discovery to CNS involvement.
Integrated Insights
Synthesising across pathways reveals three dominant themes:
• Convergent drivers (TP53, EGFR) underpin both prognosis and therapeutic response, making them central to biomarker-guided strategies.
• Resistance mechanisms (SPP1, YAP/ TAZ, EMT, inflammatory cytokines) recur across omics layers, highlighting the importance of longitudinal monitoring and adaptive therapy.
• Context dependent signals (methylation, metabolic fingerprints, immune mediators) enrich stratification but require multilayer integration for reproducible application.
This pathway-based synthesis distils actionable conclusions from a data rich literature base, bridging molecular complexity with clinical translation. It highlights the need for biomarker frameworks that integrate tumour drivers, resistance pathways, and systemic host responses to guide precision oncology in NSCLC.
DISCUSSION
Recent years have witnessed a marked increase in studies applying multi-omics approaches to biomarker discovery in NSCLC. In the context of NSCLC, transcriptomics and genomics remain the dominant platforms, while proteomics and metabolomics provide complementary insights into treatment response and patient stratification.
Epigenomics, immunogenomics, and microbiomics play supporting roles in specific biomarker identification, enabling a more holistic molecular characterisation of NSCLC.
The advancements in biomarker discovery through multi-omics approaches hold great promise for clinical applications in NSCLC. In summary, the clinical outcomes associated with biomarker identification in the selected research studies can be applied to the following.
Prognosis
Multi-omics approaches have advanced prognostic assessment by extending beyond canonical oncogenes such as TP53 and EGFR to include immunerelated factors (CXCL13, MMP12, and CSF-1) and metabolites as candidate biomarkers to predict disease progression and patients’ response to targeted treatments and immunotherapy.22,31-34 For example, TP53 mutations not only correlate with poor outcomes but also impair antigen presentation and modulate cytokine signalling, contributing to immune evasion and reduced immunotherapy efficacy.35 These findings highlight how integrated biomarker panels can refine risk stratification, although reproducibility across cohorts remains a critical challenge. Collectively, they reflect a broader shift towards understanding the tumour’s molecular complexity and its interactions with both the immune system and the TME, with the ultimate goal of advancing targeted therapy and patients’ stratification.35,46
Early-Stage Detection
Blood-based biomarkers offer a minimally invasive route for early lung cancer detection, but their translation is limited by specificity and reproducibility issues. Genomic signatures and network analyses have identified a 12-gene signature that can detect lung cancer in biological fluids at early stages of the disease and is associated with a poor disease outcome, enabling routine screening and early intervention to improve patient outcomes.38 Several studies also used multi-omics techniques (transcriptomics, genomics, and metabolomics) to identify biomarkers that differentiate NSCLC from normal tissue.39,47 Integrating metabolomics and lipidomics emphasises the importance of characterising the metabolome and
lipidome in the plasma of patients with lung cancer to create a comprehensive metabolic fingerprint and identify potential clinical diagnostic markers for lung cancer.39,23
Therapy Response Prediction
Multi-omics platforms have identified biomarkers predicting therapy response, confirming the importance of oncogenes such as CCND1, TP53, MYC, and EGFR in identifying the susceptibility of NSCLC to therapies such as TKIs and immunocheckpoint inhibitors.33,10 Furthermore, metabolomic and proteomic analyses refine immunotherapy response prediction,33 while SPP1 overexpression exemplifies how tumour-intrinsic changes remodel the TME to drive EGFR-TKI resistance, promoting tumour-associated macrophage infiltration and immunosuppressive signalling and ultimately leading to poor survival outcomes.40 Other candidates, such as ZFHX3 mutations, correlate with an increased immune response, suggesting predictive utility for immunotherapy in patients with lung cancer.11 Multi-omics platforms have identified predictors of therapy response, confirming that oncogenes (such as HMGB3 overexpression) are linked to worse survival in small cell lung cancer, while CASP10 is associated with better prognosis, indicating their potential as prognostic biomarkers. These examples underscore both the promise and the need for robust validation frameworks to ensure reproducibility.11
Chemotherapy and Immunotherapy Guidance
Combined metabolomic and transcriptomic analyses distinguish LUAD and LUSC subtypes, identify prognostic markers, and predict immunotherapy sensitivity.33,48 A proposed 14-gene signature serves as a biomarker panel to guide immunotherapy and chemotherapy, supporting personalised treatment for patients with NSCLC.45 Importantly, the integration of omics data into treatment algorithms must account for tumour heterogeneity and dynamic changes in therapy, reinforcing the need for longitudinal sampling.
Figure 1: Preferred Reporting Items for Systematic reviews and Meta-Analyses flow diagram of study selection for non-small cell lung cancer multi-omics biomarker review.
IDENTIFICATION
#1
Pubmed Search #2
Records Screened
Multi-omics techniques for NSCLC biomarkers
Records Excluded
Not relevant to multi-omics techniques for NSCLC biomarkers 198
KEY FINDINGS Studies Included in Analysis
Recurrent Biomarkers Identified
TP53
• Appears across multiple studies
• Key biomarker for prognosis
• Associated with treatment response
• Relevant across various omics contexts
• Linked to treatment response
• Specifically associated with resistance to targeted therapies
• Recurrent across clinical contexts
EGFR Clinical Contexts: Prognosis, Treatment Response, Carcinogenesis
Note: This PRISMA diagram illustrates the systematic review process for identifying multi-omics biomarkers in Non-Small Cell Lung Cancer (NSCLC). The review identified TP53 and EGFR as key recurrent biomarkers with significance across multiple omics platforms and clinical contexts.
NSCLC: non-small cell lung cancer; PRISMA: Preferred Reporting Items for Systematic reviews and Meta-Analyses.
Survival Prediction
Multi-omics approaches have revealed molecular markers associated with survival outcomes.37-41 Easily measurable serum proteins (e.g., IL-6, IL-8) offer non-invasive biomarkers for predicting poor outcomes,34 though their interpretive value is confounded by systemic inflammation and comorbidities. These findings highlight the potential of multi-omics to stratify patients by survival risk, while also underscoring the importance of context-dependent validation.
Carcinogenesis
Epigenomic studies highlight methylationbased biomarkers as potential diagnostic and therapeutic targets.8,42 Genome-wide association studies confirm that genetic and environmental factors shape methylation patterns, reinforcing their relevance in NSCLC development.8 However, methylation signatures remain highly variable and context dependent, influenced by tumour subtype, anatomical site, and immune status. This variability underscores the importance of multi-layered integration to capture the carcinogenic process more faithfully.
Translational Framework and Future Directions
The integration of multi-omics into clinically actionable endpoints remains challenging due to high dimensionality, heterogeneity, and computational complexity.28,30 Advanced machine learning and deep approaches31,32 are increasingly applied, but reproducibility and interpretability remain obstacles. The author’s proposed framework (Figure 2) maps five omics domains (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) onto diagnostic, prognostic, therapeutic, mechanistic, and longitudinal categories. By stratifying outputs across NSCLC subtypes, this framework supports biomarker discovery, resistance profiling, and personalised therapy guidance, reinforcing the trajectory from molecular insight to patient-centred outcomes.
CONCLUSION
Multi-omics approaches are reshaping NSCLC research, transforming biomarker discovery, diagnosis, prognosis, and treatment optimisation. By integrating genomics, transcriptomics, proteomics, metabolomics, and epigenomics, these strategies move beyond single-layer analyses to capture the molecular complexity of tumours and their interactions with the microenvironment,22,31-34 enabling more precise patient stratification and accelerating the transition towards personalised therapeutic strategies.35-43
Recent advances highlight three major implications: multi-omics expands the repertoire of clinically actionable biomarkers,33,10 refines patient stratification to predict therapy responses,11,45 and decodes tumour complexity and resistance pathways to inform targeted interventions.40,41-47
Multi-omics approaches provide valuable insights into the molecular complexity of tumours and their interactions with the TME, but several challenges remain. The absence of standardised protocols, incomplete datasets, and the biological heterogeneity of NSCLC limit reproducibility and clinical translation.36,42 High-dimensional data demand advanced computational pipelines, rigorous statistical frameworks, and collaborative validation across diverse cohorts. Without these, the promise of multi-omics risks remaining confined to research settings.17,42
Incomplete Datasets
Missing data are common due to limitations in sample availability, costs, and experimental issues. These missing values can hinder data integration and compromise the validity of downstream analyses. To address this, researchers have developed sophisticated imputation methods that leverage the correlations among different omics layers to estimate missing data more accurately.9
Figure 2: Multi-omics integration framework for non-small cell lung cancer biomarker discovery: mapping molecular inputs to functional clinical endpoints.
Multi-omics Intergration Framework for NSCLC Biomarker Discovery
From Molecular Data to Functional Clinical Categories
Biomarker Landscape
50+ validated biomarkers
5 functional categories
Multi-omics integration
Genomics EGFR TP53 TTN
Key Features
• Cross-omics validation
• Network-basedis covery
• Functional categorisation
• Clinical relevance
• Dynamic monitoring
• Therapeutic targets
Epigenomics
Multi-Omics Integration and Analysis
Network analysis • Machine learning • Pathway enrichment Cross-omics correlation • Systems biology • Integrative modelling
Diagnostic Biomarkers
Cyfra21, CEA
• 12/14-gene signatures
• Plasma metabolites IncRNA/circRNA networks
• Keratin (KRT6A/14/17) Methylation markers
Early detection Disease differentiation
Prognostic Biomarkers
TP53 (poor prognosis)
• TTN (therapy response)
ZNF71 (stratification)
Therapeutic Response Mechanic Pathways
• CXCL13, MMP12, IL-6/8 DNA methylation patterns EGFR (TKI therapy)
• SPP1 (TKi resistance)
• CCND1, MYC, CASP10 ZFHX3 (immunotherapy) • LM predictors (CNS)
Clinical Outcomes and Patient Management
Early diagnosis • Risk stratification • Personalised therapy
Treatment monitoring
Improved survival
Quality of life PIMs CXCL13 MMP12
Longitudinal & Clinical Insights
Serial CCND1/TP53/MYC
• EGFR dynamics Immune modulation
• NSCLC vs sarcoma
• CSF-based tracking
Plasma lipids Metabolites Metabolic fingerprints
• Screening programmes
• Precision medicine
• Drug selection
• Resistance monitoring
• Clinical trials
• Biomarker panels Applications
This layout reflects the progression from molecular input to patient-centred outcomes, supporting biomarker classification and clinical application.
AKT: protein kinase B; CEA: carcinoembryonic antigen; circRNA: circular RNA; CNS: central nervous system; CSF: cerebrospinal fluid; CXCL13: C-X-C motif chemokine ligand 13; Cyfra21: cytokeratin fragment 21-1; EGFR: epidermal growth factor receptor; IRX1: iroquois homeobox 1; LM: leptomeningeal metastasis; lncRNA: long non-coding RNA; miRNA: micro RNA; MMP12: matrix metallopeptidase 12; NSCLC: non-small cell lung cancer; PCDH17: protocadherin 17; PTM: posttranslational modifications; SPP1: secreted phosphoprotein 1; TAZ: transcriptional co-activator with PDZbinding motif; TBX5: T-box transcription factor 5; TKI: tyrosine kinase inhibitor; vs: versus; YAP: yes-associated protein.
Biological Complexity
Integrating these techniques remains challenging due to the heterogeneity among different cancer types and within tumours of the same type. An accurate interpretation of complex genetic and molecular data still faces barriers due to difficulties in integrating genomics, transcriptomics, proteomics, and metabolomics data from diverse sources.17,43 Handling multiple high-dimensional datasets and interpreting complex biological systems also represents a barrier, requiring advanced computational utilities and rigorous statistical methods to guarantee accurate data
interpretation.17 Therefore, the identification of reliable biomarkers still poses a hurdle and requires the collaboration and combined efforts of different sources.17,43
Looking forward, open-source tools and collaborative research networks will be pivotal in bridging the gap between discovery and clinical implementation.9,17 By offering accessible, scalable, and reproducible analytical frameworks, opensource platforms empower researchers to integrate and analyse complex multi-omics datasets more effectively.
Advancing NSCLC care requires coordinated, multidisciplinary efforts to bridge the gap between research and clinical implementation, thereby accelerating progress in diagnostics, prognostics, and therapeutic strategies. The integration of multi-omics not only enriches understanding of tumour biology but also establishes a foundation for precision medicine. Sustained collaboration and innovation are essential to translate these insights into practice, ultimately improving patient outcomes and shaping the future of cancer care.
To operationalise multi-omics insights into patient-centric strategies, the author proposes a decision-making framework (Figure 3) that unifies five omics layers (genomics, transcriptomics, proteomics, metabolomics, and epigenomics) and maps them to actionable biomarkers, subtype-specific therapies, and resistance mechanisms. This model supports clinical decision-making by linking molecular profiles to targeted treatments, the prediction of immunotherapy sensitivity, and longitudinal monitoring, thereby reinforcing the translational trajectory from molecular discovery to personalised oncology.
Figure 3: Multi-omics decision-making framework for non-small cell lung cancer: integrating molecular profiles to guide subtype-specific treatment and monitoring.
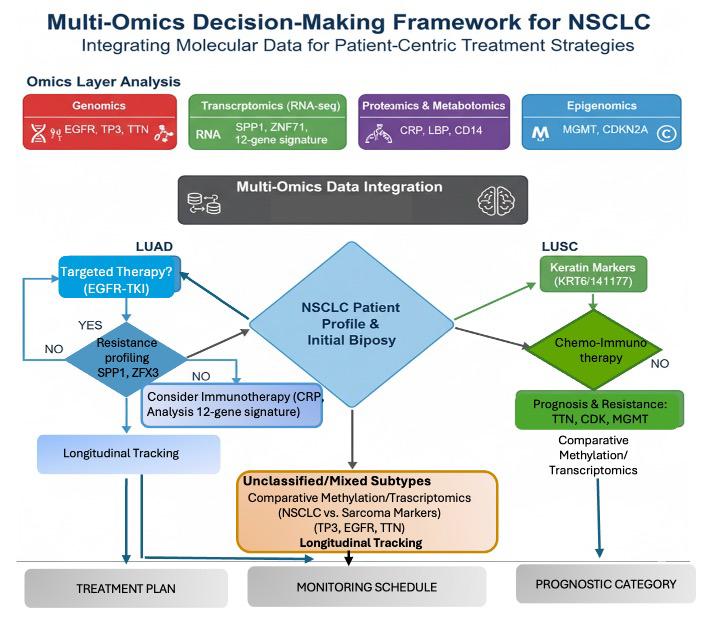
Genomics EGFR, TP53, TTN
Transcrptomics (RNA-seg) SPP1, ZNF71, 12-gene signature RNA
Targeted Therapy? (EGFR-TKI)
Proteomics and Metabolomics CRP, LBP, CD14 Epigenomics MGMT, CDKN2A Resistance profiling SPP1, ZFX3
Unclassified/Mixed Subtypes
Comparative Methylation/Transcriptomics (NSCLC vs. Sarcoma Markers) (TP3, EGFR, TTN) Longitudinal Tracking
This patient-centric, decision-making framework for NSCLC integrates multi-omics data to inform subtype-specific therapeutic strategies. Molecular inputs, including genomics (EGFR, TP53, TTN), transcriptomics (RNA sequencing, gene expression, splicing), proteomics/metabolomics (protein expression, post-translational modifications, metabolites), and epigenomics (MGMT, CDKN2A), are consolidated through a centralised integration pipeline. The framework anchors on the initial biopsy and NSCLC patient profile, branching into tailored pathways for LUAD, LUSC, and unclassified/mixed subtypes. Each pathway incorporates molecular markers to guide treatment selection (e.g., EGFR-TKI, immunotherapy, chemo-immunotherapy), resistance profiling, and longitudinal tracking. Outcome categories include treatment planning, monitoring schedules, and prognostic stratification, reinforcing the translational trajectory from molecular insight to personalised care.
CD: cluster of differentiation; CRP: C-reactive protein; EGFR: epidermal growth factor receptor; LBP: lipopolysaccharide binding protein; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; NSCLC: non-small cell lung cancer; SPP1: secreted phosphoprotein 1; TKI: tyrosine kinase inhibitor; vs: versus; ZFX3: zinc finger protein x linked; ZNF71: zinc finger protein 71.
Keratin Markers (KRT6/141177)
Prognosis & Resistance: TTN, CDK, MGMT
Multi-Omics Data Integration
References
1. Bray F et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229-63.
2. Deshpand R et al. Evolving trends in lung cancer: epidemiology, diagnosis, and management. Indian J Cancer. 2022;59(Suppl 1):S90-105.
3. Wang M et al. Toward personalized treatment approaches for nonsmall-cell lung cancer. Nat Med. 2021;27(8):1345-56.
4. Meador CB, Hata AN. Acquired resistance to targeted therapies in NSCLC: updates and evolving insights. Pharmacol Ther. 2020;210:107522.
5. Zou K et al. Etiology of lung cancer: evidence from epidemiologic studies. J Natl Cancer Cent. 2022;2(4):216-25.
6. Alduais Y et al. Non-small cell lung cancer (NSCLC): a review of risk factors, diagnosis, and treatment. Medicine (Baltimore). 2023;102(8):e32899.
7. Yang IA et al. Genetic susceptibility to lung cancer and co-morbidities. J Thorac Dis. 2013;5(Suppl 5):S454-62.
8. Zhao X et al. Identification of genetically predicted DNA methylation markers associated with non–small cell lung cancer risk among 34,964 cases and 448,579 controls. Cancer. 2024;130(6):913-26.
9. Mohr AE et al. Navigating challenges and opportunities in multi-omics integration for personalized healthcare. Biomedicines. 2024;12(7):1496.
10. Takata S et al. Prospective exosomefocused translational research for afatinib (EXTRA) study of patients with nonsmall cell lung cancer harboring EGFR mutation: an observational clinical study. Ther Adv Med Oncol. 2023;15:17588359231177021.
11. Liu Q et al. Proteogenomic characterization of small cell lung cancer identifies biological insights and subtype-specific therapeutic strategies. Cell. 2024;187(1):184-203. e28.
12. Slominski RM et al. How cancer hijacks the body’s homeostasis through the neuroendocrine system. Trends Neurosci. 2023;46(4):263-75.
13. Olivier M et al. The need for multiomics biomarker signatures in precision medicine. Int J Mol Sci. 2019;20(19):4781.
14. Song X et al. Spatial multi-omics revealed the impact of tumor ecosystem heterogeneity on immunotherapy efficacy in patients with advanced non-small cell lung cancer treated with bispecific
antibody. J Immunother Cancer. 2023;11(2):e006234.
15. Karaman ED, Işık Z. Multi-omics data analysis identifies prognostic biomarkers across cancers. Med Sci (Basel). 2023;11(3):44.
16. Sabit H et al. Leveraging singlecell multi-omics to decode tumor microenvironment diversity and therapeutic resistance. Pharmaceuticals (Basel). 2025;18(1):75.
17. Menyhárt O, Győrffy B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput Struct Biotechnol J. 2021;19:949-60.
18. McQuade JL et al. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019;20(2):e77-91.
19. Ghita M et al. Optimizing radiotherapy with chemotherapy using PKPD modeling for lung cancer. 2022 IEEE 20th Jubilee World Symposium on Applied Machine Intelligence and Informatics (SAMI) [Internet]. 2022 [cited 2025 Apr 1]. p. 000299–304.
20. Ghita M et al. Model calibration of pharmacokinetic-pharmacodynamic lung tumour dynamics for anticancer therapies. J Clin Med. 2022;11(4):1006.
21. Park M-K et al. Deep-learning algorithm and concomitant biomarker identification for NSCLC prediction using multi-omics data integration. Biomolecules. 2022;12(12):1839.
22. Chen Z et al. Integrative multiomics analysis for identifying novel therapeutic targets and predicting immunotherapy efficacy in lung adenocarcinoma. Cancer Drug Resist. 2025;8:3.
23. Yan F et al. Integration of clinical phenoms and metabolomics facilitates precision medicine for lung cancer. Cell Biol Toxicol. 2024;40(1):25.
24. Yan Y et al. Cross-omics strategies and personalised options for lung cancer immunotherapy. Front Immunol. 2024;15:1471409.
25. Chakraborty S et al. Onco-multiomics approach: a new frontier in cancer research. BioMed Res Int. 2018;2018(1):9836256.
26. Chang JTH et al. The impact of the cancer genome atlas on lung cancer. Transl Res. 2015;166(6):568-85.
27. Shestakova KM et al. Targeted metabolomic profiling as a tool for diagnostics of patients with nonsmall-cell lung cancer. Sci Rep. 2023;13(1):11072.
28. Nicora G et al. Integrated multi-omics analyses in oncology: a review of machine learning methods and tools. Front Oncol. 2020;10:1030.
29. Acharya D, Mukhopadhyay A. A comprehensive review of machine learning techniques for multi-omics data integration: challenges and applications in precision oncology. Brief Funct Genomics. 2024;23(5):549-60.
30. Subramanian I et al. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights. 2020;14:1177932219899051.
31. Tong S et al. Unveiling the distinctive variations in multi-omics triggered by TP53 mutation in lung cancer subtypes: an insight from interaction among intratumoral microbiota, tumor microenvironment, and pathology. Comput Biol Chem. 2024;113:108274.
32. Peng H et al. Molecular and immune characterization of Chinese earlystage non-squamous non-small cell lung cancer: a multi-omics cohort study. Transl Lung Cancer Res. 2024;13(4):763-84.
33. Wu R et al. Multi-omics analysis reveals the sensitivity of immunotherapy for unresectable non-small cell lung cancer. Front Immunol. 2025;16:1479550.
34. Parra ER et al. Multi-omics analysis reveals immune features associated with immunotherapy benefit in patients with squamous cell lung cancer from phase III lung-MAP S1400I trial. Clin Cancer Res. 2024;30(8):1655-68.
35. Qian X et al. Integrated microbiome, metabolome, and proteome analysis identifies a novel interplay among commensal bacteria, metabolites and candidate targets in non‐small cell lung cancer. Clin Transl Med. 2022;12(6):e947.
36. Shen S et al. A multi-omics study links TNS3 and SEPT7 to long-term former smoking NSCLC survival. NPJ Precis Onc. 2021;5(1):39.
37. Lo Russo G et al. PEOPLE (NTC03447678), a phase II trial to test pembrolizumab as first-line treatment in patients with advanced NSCLC with PD-L1 <50%: a multiomics analysis. J Immunother Cancer. 2023;11(6):e006833.
38. Kaya IH et al. Integrated analysis of transcriptomic and genomic data reveals blood biomarkers with diagnostic and prognostic potential in non-small cell lung cancer. Front Mol Biosci. 2022;9:774738.
39. Liu B et al. Characterizing microbiota and metabolomics analysis to identify candidate biomarkers in lung cancer. Front Oncol. 2022;12:1058436.
40. Wang Z et al. The multi-omics analysis of key genes regulating EGFR-TKI resistance, immune infiltration, SCLC transformation in EGFR-mutant NSCLC. J Inflamm Res. 2022;15:649-67.
41. Takahashi S et al. Predicting deep learning based multi-omics parallel integration survival subtypes in lung cancer using reverse phase protein array data. Biomolecules. 2020;10(10):1460.
42. Sun X et al. An integrated epigenomictranscriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance. Theranostics. 2021;11(11):5346-64.
43. Song M et al. A review of integrative imputation for multi-omics datasets. Front Genet. 2020;11:570255.
44. Tan Y et al. Multi-omics analysis reveals PUS1 triggered malignancy and correlated with immune infiltrates in NSCLC. Aging (Albany NY). 2023;15(21):12136-54.
45. Zhu J et al. Identification of molecular subtypes, risk signature, and immune landscape mediated by necroptosisrelated genes in non-small cell lung cancer. Front Oncol. 2022;12:955186.
46. Chakraborty S et al. Multi-OMICS approaches in cancer biology: new era in cancer therapy. Biochim Biophys Acta Mol Basis Dis. 2024;1870(5):167120.
47. Shi S et al. Integrative omics analysis reveals metabolic features of groundglass opacity-associated lung cancer. J Cancer. 2024;15(7):1848-62.
48. Thaiparambil J et al. Integrative metabolomics and transcriptomics analysis reveals novel therapeutic vulnerabilities in lung cancer. Cancer Med. 2023;12(1):584-96.
Immune Checkpoint Inhibitor-Associated Hydropneumothorax: A Rare Case Report with Histopathologic Insights
Authors: *Javier Fernandez,1 Lynn Feun,1 Jaylou Velez Torres,2 Dao Nguyen3
1. Department of Medicine, University of Miami, Miller School of Medicine, Florida, USA
2. Department of Pathology and Laboratory Medicine, University of Miami, Miller School of Medicine, Florida, USA
3. Department of Surgery, University of Miami, Miller School of Medicine, Florida, USA
*Correspondence to jdf108@med.miami.edu
Disclosure: The authors have declared no conflicts of interest. Written informed consent for publication of this case report, including clinical details and histopathological findings, was obtained from the patient. The consent process followed established ethical and legal standards, ensuring that the patient was provided with clear, understandable information regarding the purpose, risks, benefits, and alternatives to participation. Documentation of consent is maintained in according with institutional policy and the CARE Checklist.
Received: 25.03.25
Accepted: 13.10.25
Keywords: Checkpoint inhibitors, hydropneumothorax, pneumonitis, pneumothorax.
Citation: EMJ Oncol. 2025;13[1]:116-123. https://doi.org/10.33590/emjoncol/QJSI5311
Abstract
Immune checkpoint inhibitors (ICI) targeting programmed death-ligand 1 have transformed the management of advanced malignancies, yet are associated with a spectrum of immunerelated pulmonary toxicities, most notably checkpoint inhibitor pneumonitis (CIP). While CIP is a well-documented immune-related adverse event, the occurrence of hydropneumothorax as a manifestation of programmed death-ligand 1 inhibitor-induced pulmonary immunerelated adverse event is exceedingly rare, with very limited prior reports. The authors present a case of hydropneumothorax in a 75-year-old Hispanic male with hepatocellular carcinoma who developed respiratory symptoms 3 weeks after initiating atezolizumab and bevacizumab, following extensive prior exposure to ICIs. Hydropneumothorax occurred after initiation of atezolizumab and bevacizumab, and CIP was favoured clinically after alternative causes, including bevacizumab, prior thoracic radiation, and talc pleurodesis were evaluated. This case contributes to the growing literature by presenting one of the earliest reports of ICI-associated hydropneumothorax with histopathological characterisation, while also highlighting potential contributing factors to this complication. Clinicians should be aware of hydropneumothorax as a potential manifestation of CIP as it may require acute surgical intervention and inform future diagnostic and therapeutic strategies.
Key Points
1. The authors demonstrate one of the earliest documented cases of immune checkpoint inhibitor-associated hydropneumothorax in a 75-year-old man with hepatocellular carcinoma treated with atezolizumab and bevacizumab, expanding the spectrum of immune checkpoint inhibitor-related pulmonary toxicity.
2. Pleural biopsy revealed chronic pleuritic with focal non-necrotising granulomatous inflammation, providing novel tissue-level evidence that supports an immune-mediated mechanism of injury.
3. The authors’ case highlights the need for clinicians to recognise hydropneumothorax as a potential manifestation of checkpoint inhibitor pneumonitis, which may require prompt surgical intervention and multidisciplinary management.
INTRODUCTION
Immune checkpoint inhibitors (ICI) are monoclonal antibodies capable of reversing cancer-induced immune evasion and promoting tumour death. Through blockade of critical regulatory proteins on T cells, including cytotoxic T lymphocyte antigen 4 and the programmed cell death 1/programmed cell death 1 ligand axes, ICIs have revolutionised cancer therapy and sustained remission across multiple tumour types.1,2 Toxicities stemming from ICI therapy, commonly referred to as immune-related adverse effects (irAE), are not uncommon. While most reactions are mild, some patients may experience fatal outcomes stemming from irAEs, including pneumonitis, hepatitis, colitis, neurotoxicity, and myocarditis.3 Checkpoint inhibitor pneumonitis (CIP) is defined as the new onset of dyspnoea, cough, fever, chest pain, or fatigue, alongside pulmonary exudates and evidence of interval changes in imaging not due to infection or underlying disease progression.4 Rarely, the inflammatory changes associated with CIP may be complicated by pneumothorax, with only a handful of documented case reports in the literature.5-8 Hydropneumothorax, defined by the abnormal presence of both air and serous fluid in the pleural space, is even more uncommon, with only one documented case in a patient with metastatic melanoma to the lungs.9
In this paper, the authors present the case of a patient with an extensive history of ICI therapy for treatment of hepatocellular carcinoma (HCC), with presentation of
hydropneumothorax and respiratory symptoms 3 weeks after initiation of atezolizumab and bevacizumab combination therapy. To the authors’ knowledge, this is among the earliest reported cases of hydropneumothorax occurring in this clinical context with histopathological findings. Unlike prior cases, this patient had an extensive history of ICI exposure, with the complication occurring shortly after the initiation of combination atezolizumab and bevacizumab therapy. This report adds to the growing body of evidence on the pulmonary complications of ICIs by highlighting hydropneumothorax as a rare but significant and potentially lifethreatening presentation of CIP.
CASE PRESENTATION
A 75-year-old Hispanic male with a history of HCC recently started on atezolizumab and bevacizumab (3 weeks prior) presented to the outpatient oncology clinic with exertional dyspnoea for 4 days. The patient was a never-smoker with no prior history of chronic lung disease including asthma, COPD, or interstitial lung disease. He was initially diagnosed with HCC after an incidental hepatic mass was detected during routine abdominal imaging. The aetiology of his HCC was attributed to non-alcoholic steatohepatitis given his risk factors, which included a past medical history of coronary artery disease, hypertension, and prediabetes. He subsequently underwent surgical resection of the mass, which was located in segment 7. Over the course of 4 years, the patient
underwent multidisciplinary surveillance and management with several therapeutic interventions to treat cancer recurrence and progression. In terms of systemic therapy, he completed 6 weeks of daily lenvatinib, nine cycles of tremelimumab and durvalumab, and four cycles of nivolumab/ ipilimumab combination immunotherapy followed by eight cycles of nivolumab alone. During this time, he also received stereotactic body radiation therapy (SBRT) and microwave ablation (MWA) 2 years after diagnosis, for treatment of new focal hepatic lesions, along with transhepatic arterial chemoembolisation the following year, and 18 fractions of proton therapy the subsequent year. He initiated atezolizumab and bevacizumab, and developed exertional dyspnoea approximately 7 weeks later, leading to his presentation to the oncology clinic (Figure 1).
The symptoms began 4 days prior to presentation and were not accompanied by cough, fever, or chest pain. The patient, who permanently resides in a foreign country, stated that he noted feeling mildly short of breath while ambulating in his home. On physical examination, the patient was in no acute respiratory distress and his vital signs were within normal limits with an O2 saturation of 96% on room air. Auscultation was notable for decreased breath sounds at the right lung base, with no evidence of jugular venous distension or lower extremity oedema. A chest X-ray (CXR; Figure 2A) and CT scan of the chest (Figure 2B) showed right-sided hydropneumothorax with mediastinal shift to the left and possibility of right heart strain. Laboratory tests including white blood cell count and comprehensive metabolic panel were within normal limits and at the patient’s baseline. In the emergency department, the patient was haemodynamically stable and underwent tube thoracostomy, which was placed to -20 cmH2O suction without evidence of a persistent air leak. The chest tube remained in place for 10 days, during which interval improvements in pleural effusion and resolution of pneumothorax were observed. Pleural fluid analysis showed a lactate dehydrogenase of 305 U/L and serum lactate dehydrogenase of 162 U/L, suggesting an exudative process (Table 1). Pleural fluid
and blood cultures were unremarkable for an infectious process. Cytologic evaluation was negative for malignancy with reactive mesothelial cells. Due to persistent chest output totalling 1 L and fluid re-accumulation, thoracic surgery was consulted, and the patient underwent talc pleurodesis with pleural biopsy to induce adhesion and prevent recurrent pneumothorax. Pleural biopsy specimens were obtained immediately before talc insufflation and demonstrated chronic pleuritis with focal non-necrotising granulomatous inflammation (Figure 3). No birefringent talc particles were identified, and special stains including acid-fast bacillus, Grocott methenamine silver, and periodic acid–Schiff were negative for infectious organisms. Two pleural fluid samples were also sent for cytology and not concerning for malignancy. The procedure was well tolerated, and the patient was started on systemic corticosteroids after ruling out an infectious aetiology, with subsequent symptomatic improvement. The event was classified as a Grade 3 adverse event per Common Terminology Criteria for Adverse Events (CTCAE) v5.0, as the hydropneumothorax required chest tube placement and hospitalisation.
FOLLOW-UP AND OUTCOMES
Ten days after discharge, he presented to the oncology clinic endorsing improvement in shortness of breath. On physical examination, persistent decreased breath sounds in the right lung base were appreciated and a repeat CXR showed bilateral effusions increased in size compared to prior study, but no evidence of pneumothorax. During followup appointment 1 week later, he reported worsening shortness of breath and bilateral lower extremity swelling. A repeat CXR demonstrated stable trace bilateral pleural effusions, though there was no evidence of recurrent pneumothorax. He was started on an oral diuretic regimen for symptom control and subsequently underwent a repeat thoracentesis. Pleural fluid analysis showed a pleural protein of 1.6 g/dL, serum protein of 6.5 g/dL, and a pleural/serum protein ratio <0.5 consistent with a transudative process. The apparent shift in pleural fluid
Figure 1: Clinical course of the patient.
C: cycle; HCC: hepatocellular carcinoma; MWA: microwave ablation; R-CHOP: rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin), vincristine sulfate, and prednisone; SBRT: stereotactic body radiation therapy; w/: with.
profile compared to his admission was most likely attributable to recent diuretic use, resulting in a pseudo-exudate. The patient reported improved shortness of breath during follow-up visits thereafter and has discontinued atezolizumab/bevacizumab with anticipated initiation of oral regorafenib following microwave ablation of two new focal hepatic lesions.
of tremelimumab /durvalumab
SBRT
6Cs of R-CHOP Hepatic segmentectomy
Figure 2: Radiologic findings obtained on admission.
A and B) Right-sided hydropneumothorax (red arrows); B) Near complete collapse of the right lower lobe aside and partial collapse of the right middle lobe with mediastinal shift to the left. A B
1: Pleural fluid analyses from the patient’s two thoracenteses.
Microbiology
Diuretic
*ULN at the authors’ institution for serum LDH was 225 g/dL.
Light’s criteria define an exudative effusion if one or more of the following are met: (1) pleural fluid protein/serum protein ratio >0.5; (2) pleural fluid LDH/serum LDH ratio >0.6; or (3) pleural fluid LDH greater than two-thirds the ULN for serum LDH. The first thoracentesis met exudative criteria, whereas the second demonstrated transudative profile.
LDH: lactate dehydrogenase; ULN: upper limit of normal; WBC: white blood cells.
Table
Figure 3: Pleural biopsy specimen demonstrating non-necrotising granulomatous inflammation (red arrow).
DISCUSSION
Hydropneumothorax is an exceedingly rare complication of ICI-related pneumonitis, with only one previously documented case in the literature.9 The authors performed a PubMed/Embase search from January 2011–May 2025 using the terms “immune checkpoint inhibitor,” “pneumothorax,” “hydropneumothorax,” and “pleural effusion,” identifying only one previously reported case of ICI-associated hydropneumothorax and several cases of pneumothorax.8-11 CIP and pulmonary toxicity associated with ICIs are well-documented in the scientific literature, with an incidence of approximately 3–5% based on systematic reviews and meta-analyses of RCTs.12,13 However, some studies suggest that the incidence may be as high as 19% in patients with lung cancer.14 While CIP typically manifests with mild pulmonary symptoms such as dyspnoea, cough, and low-grade fever, some patients progress to respiratory failure, significantly increasing their risk of mortality.3,15 Unlike typical presentations, the authors’ patient exhibited a relatively mild clinical course despite severe imaging findings. Pneumothorax remains a rare but recognised complication of CIP, with only a handful of cases reported. Given the reported CIP-associated mortality rate of 10–17%, the progression to pneumothorax or hydropneumothorax may further worsen prognosis and increase the risk of recurrence.8,16 The authors’ patient underwent surgical intervention to prevent future recurrence, placing him at increased risk for mortality both from the procedure and the underlying pathology. To the authors’ knowledge, this is among the first cases documenting histopathologic findings in a patient with ICI-related hydropneumothorax, adding novel insight into the underlying pathophysiology of this complication.
In evaluating the aetiology of the patient’s hydropneumothorax, several potential contributors were considered. CIP was favoured given the close temporal association with atezolizumab initiation, the presence of inflammatory and nonnecrotising granulomas on biopsy, symptomatic improvement with steroids,
and the absence of infection or tumour involvement on microbiologic and cytologic studies. However, alternative causes also merit discussion. Bevacizumab, an antivascular endothelial growth factor agent that is administered concurrently, may impair wound healing and predispose patients to bronchopleural fistula formation, which could increase pneumothorax risk.17 Furthermore, the patient’s history of prior thoracic interventions, including SBRT and MWA, are established cases of delayed pulmonary and pleural complications, including fibrosis and spontaneous pneumothorax months to years after exposure.18,19 Talc pleurodesis, performed at the time of biopsy, can also include granulomatous inflammation that may mimic immune-mediated pathology.20 However, this is less likely given that specimens were obtained prior to talc insufflation, and no birefringent talc particles were identified histologically. Finally, infectious aetiologies were excluded through negative bacterial, fungal, and mycobacterial cultures. Altogether, the combination of negative microbiologic work-up, lack of tumour cavitation, supportive histopathology, and temporal relationship with ICI exposure favours CIP as the most likely driver, though a multifactorial process cannot be excluded, especially in the setting of prior SBRT and MWA. The authors suspect the mechanism involved ICI-induced subpleural inflammation with subsequent air leak, compounded by impaired healing and fistula risk from concurrent bevacizumab therapy along with underlying parenchymal fragility from prior thoracic radiation.
Another novel aspect of the authors’ case was the capturing of histopathologic results from the patient’s pleural biopsy. The most common histopathologic finding in patients with CIP is organising pneumonia; however, other patterns of lung injury including acute fibrinous inflammatory changes and diffuse alveolar damage may also be seen.4,21 Nonetheless, there are currently no specific histologic findings specific for CIP. The authors’ patient’s biopsy results revealed focal non-necrotising granulomatous inflammation, which is less common in patients with ICI-related pneumonitis.21 These biopsy findings are consistent with chronic
pleuritis and sarcoid-like granulomatous reactions that are well-described with ICIs, with a prior history of SBRT and MWA as plausible contributing factors. Infection and talc pleurodesis may also precipitate granulomatous reaction; however, this is less likely given the negative microbiological cultures and sampling of the pleura prior to insufflation during talc pleurodesis. Future research with a larger cohort of patients may help clarify the significance of these histopathological findings and their relationship with long-term ICI use, ultimately guiding clinicians in the identification of highrisk patients and the development of targeted therapeutic strategies to mitigate the risk of severe complications.
The precise mechanisms underlying irAEs, including pneumonitis, remain incompletely understood. It has been suggested that patients with autoimmune conditions and specific HLA mutations may be more susceptible to these toxicities and experience worse outcomes.22,23 Wang et al.24 identified inflammatory cytokines, such as serum IL-17A and IL-35, as potential biomarkers for predicting the severity of ICI-induced pneumonitis. Additionally, the association between HLA-DR4 mutations and increased risk of ICI-induced diabetes suggests a genetic predisposition to irAEs.25 Given the authors’ patient’s remote history of hypothyroidism secondary to Hashimoto’s thyroiditis, it is plausible that he had a heightened susceptibility to immune-mediated pulmonary toxicity. Current evidence suggests that irAEs may arise from increased T cell diversity, cross-reactivity between tumour and self-antigens, and imbalances between effector and regulatory T cells.26 These findings emphasise the potential benefit of pre-treatment genetic screening to identify high-risk individuals and implement personalised strategies for mitigating toxicity. In this case, the development of hydropneumothorax may have resulted from chronic pleural and parenchymal inflammation, predisposing the patient to spontaneous pneumothorax. However, due to the rarity of this presentation, further research is needed to elucidate the precise pathophysiologic mechanisms linking ICI therapy to hydropneumothorax.
One of the key considerations in this case is the relationship between the duration of ICI therapy and its prognostic implications. Most cases of CIP present early, with a median time to onset of 2.8 months from ICI initiation.27 Huang et al.28 reported that patients with early-onset CIP (within six weeks of treatment initiation) experience higher mortality rates compared to those with late-onset CIP. The authors’ patient had received multiple rounds of ICI therapy, including durvalumab, nivolumab, and atezolizumab, over 4 years before presenting with hydropneumothorax. This prolonged exposure to ICIs may have influenced his relatively stable in-hospital and perioperative course. While this case suggests that delayed onset of ICI-induced pulmonary complications may be associated with more favourable outcomes, further studies are needed to establish the clinical significance of this observation.
CONCLUSION
This case report expands the current understanding of ICI-related pulmonary toxicity by documenting hydropneumothorax as a potentially rare and severe manifestation of CIP. Histopathological characterisation of such rare complications has remained largely unexplored. The authors’ report is, to their knowledge, among the first to provide detailed histopathological evidence of chronic pleuritis with focal non-necrotising granulomatous inflammation in the setting of ICI-associated hydropneumothorax, thereby offering new insights into the tissue-level inflammatory processes underlying this complication. Clinicians should maintain a high index of suspicion for hydropneumothorax in patients with prolonged ICI exposure who develop new respiratory symptoms, as patients may require surgical intervention if there is no improvement with medical management, as in this case. Histopathological evaluation may be considered when the diagnosis is uncertain or when management decisions may be impacted. This case highlights the evolving spectrum of ICI-induced pulmonary adverse events and the importance of integrating clinical, radiographic, and histopathologic data to optimise patient care.
PATIENT PERSPECTIVE
The patient appreciated the multidisciplinary approach and felt that his concerns were addressed throughout his care. The patient noted that understanding of the potential risks and benefits of ongoing immunotherapy was important to him and his family, and he valued being involved in decisions regarding his treatment plan.
References
1. Martins F et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563-80.
2. Borgeaud M et al. Novel targets for immune-checkpoint inhibition in cancer. Cancer Treat Rev. 2023;120:102614.
3. Wang DY et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018;4(12):1721-8.
4. Naidoo J et al. Chronic immune checkpoint inhibitor pneumonitis. J Immunother Cancer. 2020;8(1):e000840.
5. Kucukarda A et al. Secondary pneumothorax during immunotherapy in two patients with metastatic solid tumors; a new entity. Immunotherapy. 2021;13(7):565-70.
6. Leow L et al. Lung erosion following adjuvant immunotherapy with pembrolizumab: a case report. J Med Case Rep. 2023;17(1):420.
7. Sardeli C et al. Acute pneumothorax due to immunotherapy administration in non-small cell lung cancer. Respir Med Case Rep. 2020;31:101258.
8. Shin YE et al. A potential pneumothorax induced by immune checkpoint inhibitors: a case report and literature review. Medicina (Kaunas). 2024;60(10):1634.
9. Sanchez A et al. Unilateral pneumonitis and hydropneumothorax following pembrolizumab. Brown J Hosp Med. 2022;2(1):39747.
10. Suresh K et al. Immune checkpoint immunotherapy for non-small cell lung cancer: benefits and pulmonary toxicities. Chest. 2018;154(6):1416-23.
11. Reese SW et al. Lessons from pharmacovigilance: pulmonary
immune-related adverse events after immune checkpoint inhibitor therapy. Lung. 2021;199(2):199-211.
12. Su Q et al. Risk of pneumonitis and pneumonia associated with immune checkpoint inhibitors for solid tumors: a systematic review and metaanalysis. Front Immunol. 2019;10:108.
13. Wang Y et al. Treatment-related adverse events of pd-1 and pd-l1 inhibitors in clinical trials: a systematic review and meta-analysis. JAMA Oncol. 2019;5(7):1008-19.
14. Lin MX et al. Immune checkpoint inhibitor-related pneumonitis: research advances in prediction and management. Front Immunol. 2024:15:1266850.
15. Rapoport BL et al. Pulmonary toxicities associated with the use of immune checkpoint inhibitors: an update from the immuno-oncology subgroup of the neutropenia, infection & myelosuppression study group of the multinational association for supportive care in cancer. Front Pharmacol. 2021:12:743582.
16. Zhai X et al. The mechanism and risk factors for immune checkpoint inhibitor pneumonitis in non-small cell lung cancer patients. Cancer Biol Med. 2020;17(3):599-611.
17. Zhang H et al. Bevacizumab and wound-healing complications: a systematic review and meta-analysis of randomized controlled trials. Oncotarget. 2016;7(50):82473-81.
18. Yoshimatsu R et al. Delayed and recurrent pneumothorax after radiofrequency ablation of lung tumors. Chest. 2009;135(4):1002-9.
19. Zhao J et al. Simple factors associated with radiation-induced lung toxicity after stereotactic body radiation therapy of the thorax: a pooled analysis of 88 studies. Int J Radiat Oncol Biol Phys. 2016;95(5):1357-66.
20. van den Heuvel MM et al. Talc-induced inflammation in the pleural cavity. Eur Respir J. 1998;12(6):1419-23.
21. Larsen BT et al. Clinical and histopathologic features of immune checkpoint inhibitor-related pneumonitis. Am J Surg Pathol. 2019;43(10):1331-40.
22. Buendía-Roldán I et al. A major genetic determinant of autoimmune diseases is associated with the presence of autoantibodies in hypersensitivity pneumonitis. Eur Respir J. 2020;56(2):1901380.
23. Adegunsoye A et al. Autoimmune hypothyroidism as a predictor of mortality in chronic hypersensitivity pneumonitis. Front Med (Lausanne). 2017:4:170.
24. Wang YN et al. Elevated levels of IL-17A and IL-35 in plasma and bronchoalveolar lavage fluid are associated with checkpoint inhibitor pneumonitis in patients with nonsmall cell lung cancer. Oncol Lett. 2020;20(1):611-22.
25. Stamatouli AM et al. Collateral damage: insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes. 2018;67(8):1471-80.
26. König D, Läubli H. Mechanisms of immune-related complications in cancer patients treated with immune checkpoint inhibitors. Pharmacology. 2021;106(3-4):123-36.
27. Wu Y et al. Late-onset immune checkpoint inhibitor-related pneumonitis after cessation of sintilimab: a case report and literature review. Exp Ther Med. 2023;25(2):83.
28. Huang A et al. Radiographic features and prognosis of early- and lateonset non-small cell lung cancer immune checkpoint inhibitorrelated pneumonitis. BMC Cancer. 2021;21(1):634.
Recurrent and Aggressive Solitary Plexiform Neurofibroma with KRAS and AKT1 Alterations: A Case Report
Authors: *Iván Romarico González Espinoza,¹ Abraham Castro Ponce,¹ Salma Andrea Ávila Lozano,² José Antonio García Villaseñor,³ Gabriela Juárez Salazar¹
1. Comprehensive Oncology Center, Hospital Ángeles Puebla, Mexico
2. Universidad Anáhuac Puebla, Mexico
3. Benemérita Universidad Autónoma de Puebla, Mexico
*Correspondence to vanvanmed@hotmail.com
Disclosure:
Espinoza has received fees from Roche as a paid speaker. The other authors have declared no conflicts of interest. Written informed consent for publication of clinical details and images was obtained from the patient; the signed form is held by the authors. The data supporting the findings of this case report are available at the Comprehensive Cancer Centre of Hospital Ángeles Puebla. However, restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are available from the authors upon reasonable request and with permission of the Hospital Ángeles Puebla ethical board.
Acknowledgements: The authors would like to express their deepest gratitude to the entire team at the Centro Oncológico Integral and the Hospital Angeles Puebla, including Sergio Sánchez Sosa, Eric Acosta Ponce de Leon, Maria de Jesús González Blanco, and all those who supported the authors with their expertise and knowledge during the management of this patient. Espinoza conceived the idea for the case report and provided the essential information and data regarding the patient. Ponce reviewed and edited the final manuscript. Lozano and Villaseñor prepared the first draft of the manuscript. Salazar supervised and coordinated the team. All authors read and approved the final manuscript.
Received: 16.04.25
Accepted: 13.10.25
Keywords: AKT1 mutation, genomic profiling, KRAS mutation, plexiform neurofibroma (PNF), precision oncology, targeted therapy.
Citation: EMJ Oncol. 2025;13[1]:124-132. https://doi.org/10.33590/emjoncol/BAPM1469
Abstract
Plexiform neurofibromas (PNF) are benign peripheral nerve sheath tumours classically associated with neurofibromatosis Type 1 (NF1). Isolated PNFs in patients without clinical or genetic evidence of NF1 are exceptionally rare and may pose diagnostic and therapeutic challenges. This case report describes a female who first presented in childhood with a congenital solitary PNF of the left hemiface and, as an adult, demonstrated rapid regrowth following multiple excisions. Comprehensive genomic profiling identified KRAS p.K117N and an AKT1 in-frame indel (W80_T81>CRQRTSS) with no germline or somatic NF1 alteration, suggesting alternative oncogenic activation of rat sarcoma (RAS)–MAPK and PI3K–protein kinase B (AKT) pathways. Multimodal management included numerous surgeries and targeted therapy (trametinib plus pazopanib), which achieved partial reduction but was limited by toxicity and access constraints. Subsequent chemoradiotherapy conferred
minimal additional benefit. The patient was referred to the Undiagnosed Diseases Network (UDN) of the National Institutes of Health (NIH) for further evaluation. This report highlights the importance of precise clinicopathological characterisation and broad molecular testing in atypical PNFs, and underscores gaps in consensus guidance for solitary, non-NF1 PNFs where surgery is not feasible. Precision oncology may offer rational options, although durable control remains challenging in highly aggressive and infiltrative lesions.
Key Points
1. Solitary plexiform neurofibromas (PNF) without clinical or genetic neurofibromatosis Type 1 are rare and challenging. This case showed rapid regrowth after multiple excisions and required multidisciplinary assessment beyond standard surgery. It is, to the authors’ knowledge, the first reported case from Mexico.
2. Broad sequencing revealed KRAS p.K117N and an AKT1 in-frame indel, supporting alternative activation of rat sarcoma (RAS)–MAPK and PI3K–protein kinase B (AKT) pathways in non-neurofibromatosis Type 1 PNF.
3. Targeted therapy produced partial regression but was constrained by toxicity and access. Chemoradiotherapy offered limited additional benefit. The case underscores the lack of specific guidance for solitary PNF and the need for personalised decisions when complete resection is not feasible.
INTRODUCTION
Plexiform neurofibromas (PNF) are benign, often infiltrative tumours of the peripheral nerve sheath classically linked to neurofibromatosis Type 1 (NF1), appearing in about 30% of patients with this disease.1 However, a small subset, known as ‘isolated’ or ‘solitary’ PNFs, occurs in patients who do not meet the clinical diagnostic criteria or harbour the germline mutations associated with NF1.2
To date, the best epidemiologic study of solitary PNFs is the systematic review performed by Ho et al.,3 which identified 35 studies comprising 39 subjects with a total of 41 documented isolated mucocutaneous PNF cases.3
These present with a similar histopathologic architecture as NF1-associated PNFs but tend to be solitary, well-circumscribed, and benign, so diagnosis rests on clinical and genetic exclusion of NF1 and careful radiological–pathological correlation.4 Because their molecular and biological profiles differ from NF1-related PNFs, direct treatment extrapolation is uncertain. Surgery remains the mainstay, although local recurrence may occur.5
This report presents, to the authors’ knowledge, the first reported case in Mexico of a 17-year-old female with a recurrent, congenital, hemifacial, solitary PNF that harboured KRAS p.K117N and AKT1 in-frame indel (W80_T81>CRQRTSS), detailing its diagnostic work-up and multidimensional management.
CASE
A 17-year-old Mexican female presented to the authors’ centre with a congenital left hemifacial lesion and malformed left auricle previously treated in both Mexico and the USA. Family history for neurocutaneous disorders was negative. Personal history included multiple craniofacial procedures performed in childhood (auricular reconstructions 15–18 years prior to initial presentation; mastoidectomy with temporal lesion resection 13 years prior; excisions of anterior neck, tongue, and tonsillar masses 12 years prior; and removal of a temporal osteoma/chondroma 11 years prior). Nine years prior to initial presentation, a left cavernous internal carotid artery fusiform aneurysm was treated endovascularly after a successful balloon test occlusion, achieving complete occlusion with platinum coils and Onyx-34 (Medtronic, Dublin, Ireland), and no complications (Table 1).
Table 1: Summary of patient’s procedures and findings.
Timeline after initial presentation (years, months)
Initial presentation at the age of 2
1 y, 8m
1 y, 10m
Procedure
Paediatric discharge note
MRI face/brain/neck
Maxillofacial CT (±contrast)
1y, 11m PICC placement
2y, 2m
2y, 4m
2y, 5m
3y, 5m
Temporal bone CT (bilateral)
Temporal bone CT (bilateral)
Temporal bone CT with contrast
Brain MRI
4y, 4m CT brain/neck
4y, 4m
4y, 11m
5y, 4m
MRI brain/neck
Brain MRI
Maxillofacial CT
5y, 11m CT angiography head
6y
6y
6y, 4m
MRI/MRA brain
Endovascular embolisation
Paediatric haematologyoncology consult
9y Neurosurgery operation note (MRI)
9y, 10m Psychology letter
Key findings/notes
Location
Retro-auricular mass; salivary gland abscess; ‘neoplasm under study’ Mexico
Large left facial mass with temporal bone involvement; fatty atrophy of left tongue; minimal inferior temporal fossa invasion USA
Benign cartilaginous bone tumour (osteochondroma vs giant cel tumour vs fibrous dysplasia) in left petrous temporal bone; postoperative collections USA
Long-term IV access for mastoiditis; uneventful USA
Interval mastoidectomy changes; extensive bony sclerosis/thickening USA
No significant interval change; partial resection described USA
Stable post-resection changes; no abscess or osteomyelitis USA
Interval reduction of left mass post-surgeries; residual heterogeneous disease USA
Persistent temporal bone overgrowth; fatty hemi-glossal atrophy; no enhancing soft-tissue mass USA
Matches CT; enlarged temporal bone with exostosis; abnormal enhancing soft tissue in pinna USA
Stable residual exostoses; no intracranial mass; mild meningeal enhancement focus USA
Marked deformity of temporal–maxillary–sphenoid bones; auricular ossification; ICA canal narrowing USA
Left cavernous ICA fusiform aneurysm approximately 9–10 mm; rest of circulation normal USA
Stable 10 mm ICA aneurysm; no parenchymal lesion USA
Balloon test occlusion then coil plus Onyx-34 (Medtronic, Dublin, Ireland) occlusion of left ICA (cavernous and proximal petrous); complete occlusion; no complications USA
Mosaic epidermal naevus syndrome phenotype considered; radiotherapy not advised USA
Left craniofacial chondromatous disease stable; dysphagia/snoring under evaluation; no neurosurgical indication USA
Adjustment disorder with depressed mood; ongoing therapy USA
Table 1: Summary of patient’s procedures and findings. (Continued)
Timeline after initial presentation (years, months)
Procedure
12y, 5m Non-contrast head CT
14y, 7m
Chest X-ray and cervical spine radiograph
15y Retro-auricular biopsy plus IHC
15y
FoundationOne CDx, Foundation Medicine, Inc, (acquired by Roche) Boston, Massachusetts, USA (tumour)
15y Chest CT
15y for 6 m
Targeted therapy
Key findings/notes
Persistent left craniofacial osseous distortion and soft-tissue nodules; lytic mandibular ramus focus; suggests polyostotic fibrous dysplasia
No acute cardiopulmonary disease; minimal cervical changes
Plexiform neurofibroma; S-100 focally weak positive; benign morphology; prior surgery likely R1
Location
Mexico
Mexico
Mexico
MSS; TMB 1 mut/Mb; KRAS K117N and AKT1 W80_T81>CRQRTSS; no NF1 alteration Mexico
Tiny calcified upper-lobe nodules (healed granulomas); otherwise clear
Mexico
Trametinib 2 mg od plus pazopanib 800 mg od (rationale: no selumetinib available; empirical MPNST-style targeting). CTCAE Grade 3 AEs (nausea, headache) → discontinuation Mexico
15y, 6m Neck CT (with contrast) 31×22 mm left palatine tonsil lesion; left SCM atrophy
15y, 6m Head CT (±contrast)
16y Concurrent chemoradiotherapy
16y, 4m Post-CRT MRI
18y NIH/UDN multidisciplinary review
20-21y Follow-up
Multiple craniofacial exostoses; 24×14× 16 mm nodule at left inferior turbinate; coarse auricular cartilage calcifications
Cisplatin every 3 weeks plus 60 Gy of 3D-RT in 25 fractions; limited clinical benefit
New intracranial extension toward left parieto-occipital region; enhancing lentiform component 63×53×23 mm
Comprehensive clinical and genomic evaluation; NF1 excluded clinically and genetically; confirms KRAS/AKT1; recommends delayed debulking, nutrition and psychosocial support; surveillance
Symptom-guided surveillance; quality-of-life-centred management; no evidence of MPNST transformation
Mexico
Mexico
Mexico
Mexico
USA
Mexico
AE: adverse event; CRT: chemoradiotherapy; CTCAE: Common Terminology Criteria for Adverse Events; ICA: internal carotid artery; IHC: immunohistochemistry; IV: intravenous; m: months; Mb: megabase; MPNST: malignant peripheral nerve sheath tumour; MRA: magnetic resonance angiography; MSS: microsatellite stable; mut: mutation; NF1: neurofibromatosis Type 1; NIH: National Institutes of Health; od: once daily; PICC: peripherally inserted central catheter; RT: radiotherapy; SCM: sternocleidomastoid muscle; TMB: tumour mutational burden; UDN: Undiagnosed Diseases Network; vs: versus; y: years.
Cross-sectional imaging from 2006–2014 consistently demonstrated a large, heterogeneously enhancing left facial mass with temporal bone overgrowth/exostoses
extending into infratemporal, masticator, and parapharyngeal spaces; ossification of the left pinna; narrowing of the left carotid canal; and fatty atrophy of the posterior left
tongue. Intracranial parenchyma remained unremarkable. Radiological differentials across reports favoured benign cartilaginous bone tumour/osteochondroma and polyostotic fibrous dysplasia; a paediatric haemato-oncology assessment in 9 years prior to initial presentation considered epidermal naevus syndrome (mosaicism).
After a period without specialist follow-up, a head CT without contrast performed 3 years prior in Mexico showed extensive left craniofacial osseous remodelling with soft-tissue nodules, including a dominant periorbital–malar component measuring approximately 61×60 mm, a lytic left mandibular ramus focus (24×18 mm), and hyperdense material along the left internal carotid artery, compatible with prior embolisation.
Retroauricular biopsy 1 month prior to initial presentation confirmed plexiform neurofibroma, showing plexiform proliferations of wavy spindle cells in a collagenous stroma without atypia or mitoses, and weak, focal S-100 immunoreactivity. The patient was thus referred to the authors’ centre.
After clinical assessment, tumour nextgeneration sequencing was indicated (1 month after initial presentation), demonstrating microsatellite stability, tumour mutational burden (one mutation per megabase), and co-occurring KRAS p.K117N and AKT1 W80_T81>CRQRTSS alterations, with no NF1 variant detected. A chest CT in the same month showed only tiny calcified upper-lobe nodules consistent with prior granulomatous disease. Head and neck CT 3 months later again documented left temporal bone enlargement with exostoses, a 31×22 mm left palatine tonsillar lesion, and a 24×14×16 mm nodule at the left inferior turbinate; long-standing fatty hemiglossal atrophy persisted.
Because the lesion was unresectable owing to vascularity and skull-base infiltration, an empirical, biology-based regimen was employed, given the aggressive behaviour, which initiated 1 month after initial presentation with trametinib 2 mg once daily plus pazopanib 800 mg
once daily (selumetinib was unavailable locally). After approximately 6 months, imaging documented a partial radiological response (index component reduced from approximately 61×60 mm to approximately 31×22 mm) with symptomatic improvement. However, therapy was discontinued because of Common Terminology Criteria for Adverse Events (CTCAE) Grade 3 adverse events (notably nausea and headache) and financial constraints.
Alternative treatment with concurrent chemoradiotherapy (for 6 months the following year) was initiated, consisting of cisplatin every 3 weeks and external-beam radiotherapy to a total dose of 60 Gy in 25 fractions using 3D conformal radiation therapy. However, it yielded limited benefit, as post-treatment MRI nearly 1.5 years after initial assessment at the authors' centre demonstrated intracranial extension towards the left parieto-occipital region with an enhancing lentiform component measuring 63×53×23 mm, together with persistent soft-tissue and osseous disease (Figure 1).
Because of the atypical phenotype (solitary PNF without clinical or genomic NF1) and suboptimal local control, a referral was made to the Undiagnosed Diseases Network (UDN) of the National Institutes of Health (NIH). Multidisciplinary NIH reassessment 3 years after the first assessment at the authors' centre confirmed the prior KRAS and AKT1 somatic alterations without additional drivers, excluding NF1 syndrome clinically and genomically again. This concluded that, despite the extension and infiltrative behaviour of the lesion, resection would be the most appropriate disease-modifying option. Given malnutrition, anxiety, and functional impairment at that time, active surveillance with symptom-directed care was recommended as they continued to assess resection options. At the latest review, the clinical picture comprises persistent left hemifacial deformity with auricular ossification, reduced oral aperture, dysphagia, left facial palsy, ocular surface exposure symptoms due to incomplete eyelid closure, and reduced ipsilateral hearing. There is no clinical or histological evidence of malignant peripheral nerve
A B
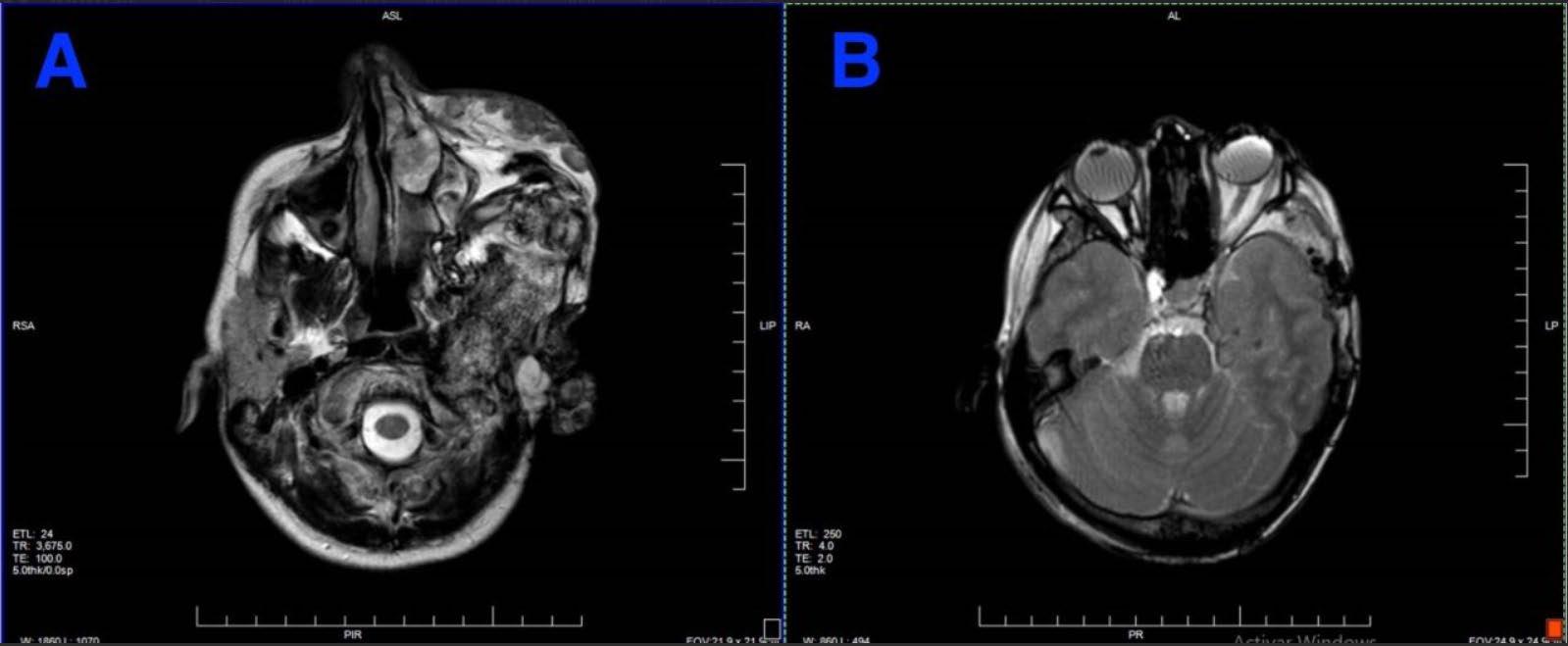
A) Heterogeneous, infiltrative soft-tissue and osseous involvement of temporal, maxillary, and zygomatic regions.
B) T2-weighted image showing mass effect with posterior displacement of the left orbital contents.
sheath tumour transformation. Management focuses on surveillance, nutritional optimisation, ocular protection, and psychological support, with re-evaluation of surgical options as functional status allows.
DISCUSSION
Solitary PNFs are exceedingly rare entities that challenge the dogma that the ‘plexiform’ growth pattern is exclusively associated with NF1.6 Their clinical features, genetic landscape, pathogenesis, and therapeutic management remain unclear.7
Mean age of presentation was 19.6 years, with nearly 64% of the reported cases being paediatric patients and 49% within the first decade of life, and there was a slight male predominance (53.8% versus 46.2%) in the systematic review by Ho et al.3 The most common site was head and neck, followed by the trunk, hands and, less commonly, the lower limbs in cutaneous lesions, while 90% of mucosal lesions occurred in the oral cavity.3 In this case, the lesion originated at the left ear and was resected multiple times, but with limited
efficacy, as the infiltrative behaviour of the tumour allowed regrowth multiple times.
The diagnosis of a solitary PNF is complex, as clinicians usually relate this entity to von Recklinghausen’s disease.8 Unfortunately, pathological assessment of the lesions resected during childhood was not available; however, it was probable that PNF was excluded from the differential diagnosis due to its lack of clinical criteria, as little was known about solitary PNFs at that time.9 However, after 2 decades, it has been recognised as a different disease with unique molecular and biological behaviour.10
Unlike PNFs in the NF1 setting, where biallelic NF1 loss and a characteristic low mutational burden dominate the molecular signature,1 the genomic landscape in isolated cases remains incompletely defined. Previous cases have discussed a possible NF1 mosaicism or segmental forms of NF1 that are clinically unapparent outside of the tumour tissue,11 demonstrating NF1 inactivation through an insertion of chromosomal bands (1p36-35 at 17q11.2) in one allele and a deletion
Figure 1: Axial MRI demonstrating extensive left hemifacial plexiform neurofibroma.
in the other, leading to an isolated plexiform neurofibroma in a 13-year-old boy.12 However, more recent cases have proposed alternative NF1-independent pathways, like the case presented by Stallworth et al.,13 in which an activating KRAS mutation and an inactivating mutation in PHF6 were observed.
The detection in this tumour of an activating KRAS variant (p.K117N) together with an AKT1 in-frame indel provides a plausible mechanism for sustained pathway activation that phenocopies the biological consequences of NF1 loss.14 Although there is no approved, direct targeted therapy for KRAS p.K117N in this disease context, such alterations offer a mechanistic rationale for pathway-directed interventions (Figure 2).
Specifically, mitogen-activated protein kinase kinase (MEK) inhibition can dampen
MAPK signalling downstream of KRAS, while anti-angiogenic blockade may modulate the hypervascular, stroma-dependent microenvironment that frequently characterises large, infiltrative PNFs.
In this context, over the past years, clinical activity of MEK inhibitors in NF1associated PNFs has been demonstrated, with meaningful reductions in tumour volume and symptom burden (particularly in children), whereas earlier attempts with imatinib, cabozantinib, miR farnesyltransferase inhibitors, or mTOR inhibition achieved only modest disease stabilisation.15 Extrapolating from this biology, trametinib was used to counteract KRAS-driven MAPK activation as selumetinib was unavailable in the authors’ country. Pazopanib, which targets vascular endothelial growth factor receptor/platelet-derived growth factor receptor/fibroblast growth factor receptor,
Figure 2: Rat sarcoma–MAPK and PI3K–protein kinase B–mTOR signalling with candidate therapeutic targets.
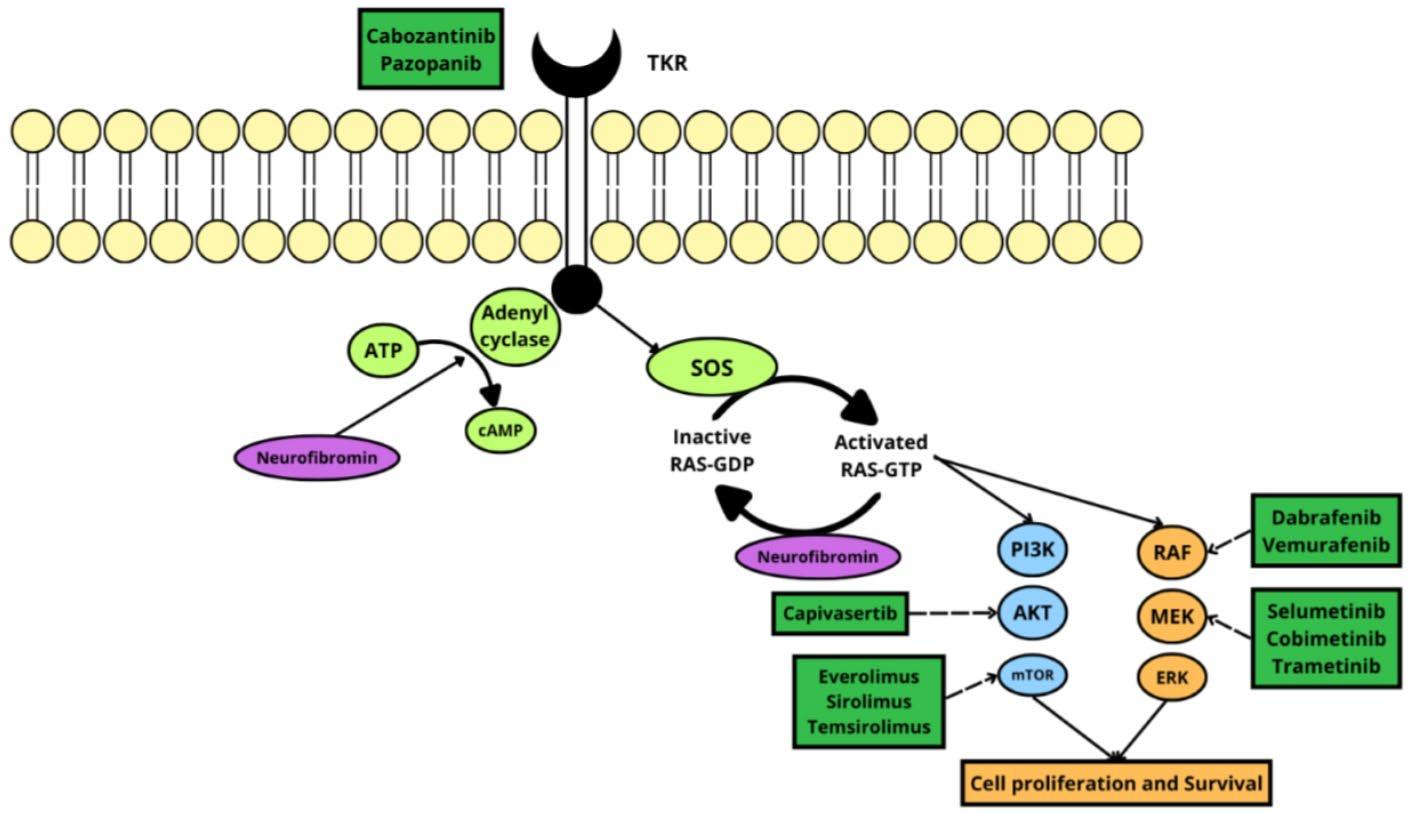
Dysregulation of RAS–MAPK and PI3K–AKT–mTOR signalling can drive proliferation; candidate drug targets are shown. AKT: protein kinase B; cAMP: cyclic adenosine monophosphate; ERK: extracellular signal-regulated kinase; GDP: guanosine diphosphate; GTP: guanosine triphosphate; MEK: mitogen-activated protein kinase kinase; RAS: rat sarcoma; RAF: rapidly accelerated fibrosarcoma serine/threonine-protein kinase; SOS: son of sevenless protein; TKR: tyrosine kinase receptor.
was combined, given the pronounced vascularity and the expectation that reducing angiogenic signalling and stromal support could enhance disease control, which has been demonstrated in soft-tissue sarcomas.16 The combination achieved a partial radiological response consistent with pathway plausibility; however, durability was limited by toxicity and access constraints, underscoring real-world barriers even when a coherent biological strategy is available.
Given these considerations and the lesion’s unresectability, an alternative treatment with cisplatin-based chemoradiotherapy was considered despite not being recommended for NF1-related PNFs due to the risk of radiation-induced malignancy in susceptible tissues.15 However, as this was an unresectable isolated PNF with substantial symptoms, it was considered the best option after a multidisciplinary review and explicit consent from the patient, attending to organat-risk constraints and malignancy risk.17
Given the limited benefit this regimen provided, and without any options left in the authors’ country, referral to the NIH UDN was considered; however, the COVID-19 pandemic delayed the patient’s travel until 3 years after her initial presentation. After a comprehensive review, it was determined that it was indeed a solitary PNF, for which surgery remained the most promising option for durable local control. However, active surveillance was favoured given the current performance status and competing risks.18
This report has several limitations. Pathology material from childhood procedures performed abroad was unavailable, precluding central histopathological review and comparison across time. Access to MEK inhibitors formally approved for NF1PNF (e.g., selumetinib, mirdametinib)19 was constrained in this setting, which influenced
References
1. Friedman JM, "Neurofibromatosis 1," Adam MP et al (eds.), GeneReviews® [Internet] (1993) Seattle: University of Washington. Available at: http://www. ncbi.nlm.nih.gov/books/NBK1109/. Last accessed: 22 September 2025.
therapeutic choices. Although trametinib combined with pazopanib achieved a partial response, the experience reflects off-label use and should be interpreted cautiously; in similar cases, MEK inhibitors or multikinase inhibitors (e.g., cabozantinib) may be considered according to the tumour’s mutational profile, access, and risk–benefit assessment. However, from a patient-centred perspective, and acknowledging the scarcity of consensus documents specific to isolated PNF, it was considered that supportive care was not ancillary but central, providing structured nutritional optimisation, pain management, functional rehabilitation (including speech and swallowing), and embedded psychological support in the care plan until resection is feasible.
CONCLUSION
This case demonstrates that the diagnosis of solitary PNFs is a challenge, as it requires not only clinical assessment but also genomic testing. Moreover, it expands the clinical and genomic spectrum of isolated PNFs by documenting concomitant KRAS p.K117N and an AKT1 in-frame indel in a congenital, hemifacial lesion with aggressive regrowth and is, to the authors’ knowledge, the first solitary PNF reported from Mexico. It highlights how comprehensive molecular profiling can uncover non-NF1 drivers that justify pathway-directed therapy, while also revealing the current limitations in durability and access. In the absence of dedicated guidelines, management should be personalised, multidisciplinary, and explicitly quality-of-life-centred, reserving systemic or locoregional treatments for unresectable, progressive, or highly symptomatic disease and revisiting surgical options as patient factors evolve.
2. Lin V et al. Is a plexiform neurofibroma pathognomonic of neurofibromatosis type I? Laryngoscope. 2004;114(8):1410-4.
3. Ho JD et al. Isolated, nonsyndromic mucocutaneous plexiform neurofibromas: a systematic review of the clinicopathologic features. Am J Dermatopathol. 2022;44(12):904-12.
4. Bechtold D et al. Plexiform neurofibroma of the eye region occurring in patients without neurofibromatosis type 1. Ophthal Plast Reconstr Surg. 2012;28(6):413-5.
5. Lange F et al. [Management of plexiform neurofibroma isolated in childhood: four patients]. Ann Chir Plast Esthet. 2013;58(6):694-9. (In French)
6. Fisher DA et al. Solitary plexiform neurofibroma is not pathognomonic of von Recklinghausen’s neurofibromatosis: a report of a case. Int J Dermatol. 1997;36(6):439-42.
7. Ou XL et al. Rare recurrent solitary plexiform neurofibroma of the dorsum of the finger: a case report. J Clin Images Med Case Rep. 2022;3(6):1882.
8. Peltonen S, Pöyhönen M, “Clinical diagnosis and atypical forms of NF1,” Upadhyaya M, Cooper DN (eds.), Neurofibromatosis Type 1: Molecular and Cellular Biology (2012), Heidelberg: Springer, pp.17–30.
9. Zwane NP et al. Solitary oral plexiform neurofibroma: review of literature and report of a case. Oral Oncol. 2011;47(6):449-51.
10. Jungmann J et al. Genetic basis of a solitary familial plexiform neurofibroma without verified associated
neurofibromatosis. J Dtsch Dermatol Ges. 2016;14(5):525-7.
11. Singh GB et al. A rare case of paediatric solitary plexiform neurofibroma of the lip. J Oral Maxillofac Surg Med Pathol. 2020;32(2):164-6.
12. Beert E et al. Biallelic inactivation of NF1 in a sporadic plexiform neurofibroma. Genes Chromosomes Cancer. 2012;51(9):852-7.
13. Stallworth JY et al. Plexiform neurofibroma with activating KRAS mutation and segmental presentation involving the unilateral eyelid. Ophthalmic Plast Reconstr Surg. 2022;38(4):e104-6.
14. Longo JF et al. Recent advances in the diagnosis and pathogenesis of neurofibromatosis type 1 (NF1)associated peripheral nervous system neoplasms. Adv Anat Pathol. 2018;25(5):353.
15. Pellerino A et al. Diagnosis and treatment of peripheral and cranial nerve tumors with expert recommendations: an EUropean
Network for RAre CANcers (EURACAN) initiative. Cancers. 2023;15(7):1930.
16. Antonia F et al. Trametinib combined with pazopanib for treating malignant peripheral nerve sheath tumor in NF-1 patients: a case report and a review of the literature. Ann Case Rep. 2025;10:2156.
17. Robertson TC et al. Isolated plexiform neurofibroma: treatment with threedimensional conformal radiotherapy. Laryngoscope. 2004;114(7):1139-42.
18. Nebiki H et al. A rare case of plexiform neurofibroma of the liver in a patient without neurofibromatosis type 1. Clin J Gastroenterol. 2020;13(6):1297-302.
19. Moertel CL et al. ReNeu: a pivotal, phase IIb trial of mirdametinib in adults and children with symptomatic neurofibromatosis type 1-associated plexiform neurofibroma. J Clin Oncol. 2025;43(6):716-29.
Favourable Response of Unresectable Giant Pinealoblastoma After Induction Chemotherapy
and
Craniospinal Radiotherapy: A Case Report
Authors: *R. Laraichi,¹,³ O. Lamsyah,²,³ A. Lachgar,¹,³ K. Nouni,¹,³ H. El Kacemi,¹,³ S. Boutayeb,²,³ I. El Ghissassi,²,³ H. M’rabtI,²,³ H. Errihani,²,³ T. Kebdani,¹,³ K. Hassouni¹,³
1. Department of Radiotherapy, National Institute of Oncology, Rabat, Morocco
2. Department of Medical Oncology, National Institute of Oncology, Rabat, Morocco
3. Faculty of Medicine and Pharmacy, Mohammed V University, Rabat, Morocco
*Correspondence to rachidalaraichi2@gmail.com
Disclosure: The authors have declared no conflicts of interest. Informed consent of the patient was obtained verbally and in writing.
Acknowledgements: The authors would like to express their sincere gratitude to the entire medical and research staff who contributed to the diagnosis, treatment, and follow-up of this case. Special thanks are extended to the patient and her family for their trust and cooperation throughout the course of treatment.
Laraichi and Lamssayah had full access to all the data and take responsibility for the integrity and accuracy of the case report.
Received: 23.07.25
Accepted: 09.10.25
Keywords: Case report, chemotherapy, craniospinal radiotherapy, pinealoblastoma.
Citation: EMJ Oncol. 2025;13[1]:133-139. https://doi.org/10.33590/emjoncol/AZCV4269
Abstract
Introduction: Pinealoblastomas are rare, aggressive, Grade 4 tumours of the pineal gland, predominantly affecting children. Their occurrence in adults is exceedingly rare, posing significant diagnostic and therapeutic challenges due to the lack of standardised management protocols.
Case Presentation: The authors present the case of a 23-year-old woman with a 3-month history of hearing loss. Brain MRI revealed a large (6.5×5 cm), unresectable pineal region tumour causing obstructive hydrocephalus. Biopsy confirmed the diagnosis of pinealoblastoma. Initial attempts at radiotherapy were precluded by severe agitation. A multidisciplinary team decision led to treatment with induction chemotherapy (cisplatin and etoposide) followed by craniospinal radiotherapy (54 Gy total dose) using the volumetric modulated arc therapy technique.
Outcomes: The patient tolerated the treatment well, with significant improvement in her neuropsychiatric status. A post-therapeutic MRI at 3 months showed an 80% tumour
regression (near-complete remission) and resolution of hydrocephalus. The patient made a full neurological recovery and successfully resumed her university studies.
Conclusion: This case demonstrates that a sequential approach of induction chemotherapy followed by high-dose radiotherapy can be a highly effective strategy for managing unresectable pinealoblastoma in adults, leading to excellent oncological and functional outcomes. It underscores the need for adaptive, multidisciplinary management and highlights the potential of non-surgical modalities.
Key Points
1. Pinealoblastoma is a rare, aggressive, Grade 4 tumour of the pineal gland that is exceptionally uncommon in adults and poses significant diagnostic and therapeutic challenges due to a lack of standardised management protocols.
2. The authors describe an adult case of unresectable pinealoblastoma managed successfully with induction chemotherapy followed by high-dose craniospinal radiotherapy, achieving near-complete remission and full neurological recovery.
3. A sequential strategy combining induction chemotherapy and craniospinal radiotherapy can be effective for unresectable adult pinealoblastoma, emphasising the importance of multidisciplinary, adaptive management when surgery is not feasible.
INTRODUCTION
The pineal gland is a small endocrine gland that regulates the circadian rhythm by secreting melatonin. Tumours of the pineal region represent less than 1% of all intracranial neoplasms, among which pinealoblastoma is distinguished as a primitive neuroectodermal tumour.1
Pinealoblastomas represent between 24–50% of pineal parenchymal tumours and occur mainly in infants and young children and occasionally in adults.1 Considered the most aggressive among pineal parenchymal tumours, they are classified as Grade 4 tumours according to the WHO classification.2 Due to compression of the cerebral aqueduct, pinealoblastoma is almost systematically associated with obstructive hydrocephalus. Furthermore, this tumour presents a high risk of leptomeningeal and extracranial dissemination.
Pinealoblastoma cases occurring in adults are extremely rare and represent less than 10% of cases reported in the literature.3 Due to this rarity, the data available to establish standardised management of the
disease remain limited, and no consensus currently exists on the optimal combination of multimodal treatment involving surgery, radiotherapy, and/or chemotherapy.
OBJECTIVE
In this work, the authors describe the case of an unresectable giant pinealoblastoma in a 23-year-old patient. The treatment consisted of induction chemotherapy followed by craniospinal radiotherapy. The post-treatment outcome was favourable, with almost complete remission after the end of treatment.
This case illustrates a particularly rare presentation of pinealoblastoma in an adult patient, which is unusual given that the majority of cases occur in children. Furthermore, the tumour was deemed unresectable, and the favourable outcome following a sequential strategy of induction chemotherapy followed by craniospinal radiotherapy highlights an alternative, nonsurgical approach with excellent results.
Table 1: Timeline of patient’s care.
Date/Period
Initial presentation (3 months before diagnosis)
At diagnosis
At diagnosis
At diagnosis
Immediately after diagnosis
Shortly after diagnosis
Following 3 months
Subsequent month
3 months after RT
At follow-up
Event
Onset of hearing loss leading to medical consultation
Brain MRI: large pineal mass (6.5×5 cm) with hydrocephalus
Ventriculo-cysternostomy + biopsy → diagnosis of pinealoblastoma
Spinal MRI: no abnormalities, no dissemination
Tumour deemed unresectable by neurosurgeons
Attempted RT, not tolerated due to severe agitation → referral to neuropsychiatry, initiation of anxiolytics
Three cycles of induction chemotherapy (cisplatin + etoposide) → partial tumour response + clinical improvement
Curative craniospinal radiotherapy with VMAT: 36 Gy/20 fractions + boost of 18 Gy (total 54 Gy)
Evaluation MRI: approximately 80% tumour regression, near-complete remission
Clinical recovery: neurological and physical function improved
RT: radiotherapy; VMAT: volumetric modulated arc therapy.
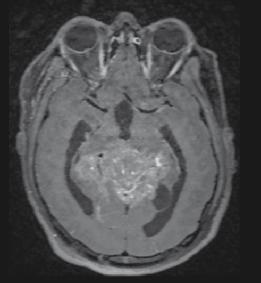
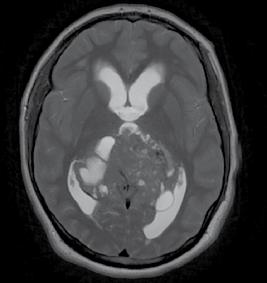
A B C
A) T1: heterogeneous expansive process centred on the pineal gland with moderate enhancement after gadolinium injection.
B) T2: heterogeneous cystic tissue structure with hypo calcification signals.
C) T2: compression of the aqueduct of the brain responsible for triventricular hydrocephalus.
Figure 1: Tumour iconography on initial brain MRI.
Figure 2: Microscopic image of pinealoblastoma made up of round monomorphic cells with reduced cytoplasm equipped with anisokaryotic and hyperchromatic nuclei, arranged in a sheet, clump, and rosette.
CASE PRESENTATION
A 23-year-old, right-handed female university student with no significant past medical history presented with a primary complaint of bilateral progressive hearing loss over 3 months. Neurological examination showed no cranial nerve deficits, papilledema, or focal motor signs. Baseline blood tests were within normal limits. There were no reported headaches, visual disturbances, nausea, or vomiting at initial presentation. A detailed timeline of the patient’s care is presented in Table 1
Brain MRI revealed a large (6.5×5 cm), heterogeneous, cystic mass in the pineal region with calcifications. The mass compressed the cerebral aqueduct, causing triventricular obstructive hydrocephalus (Figure 1). The patient underwent a ventriculocisternostomy to relieve hydrocephalus and a concurrent biopsy of the pineal lesion.
Histopathological examination showed a highly cellular tumour composed of sheets of small, round, monomorphic cells with hyperchromatic nuclei and scant cytoplasm, with evidence of rosette formation (Figure 2). Immunohistochemical analysis showed tumour cell positivity for synaptophysin and neuron-specific enolase, with negative glial fibrillary acidic protein staining. The Ki-67 proliferative
index was estimated at 40–50%, consistent with the highly proliferative nature of pinealoblastoma. These findings helped distinguish pinealoblastoma from other pineal region tumours such as pineocytoma, germ cell tumours, and ependymoma. Although no molecular or cytogenetic analyses were performed in this case, such investigations are increasingly recognised as valuable for identifying prognostic and therapeutic biomarkers.
A spinal MRI showed no evidence of leptomeningeal dissemination. Given the tumour’s large size and proximity to the brainstem, it was deemed unresectable by neurosurgery. An initial attempt to administer radiotherapy failed due to severe treatment-related agitation, requiring neuropsychiatric intervention and anxiolytics. A multidisciplinary tumour board decided on a novel sequence for this context:
• Induction Chemotherapy (3 months following diagnosis): Three cycles of cisplatin and etoposide, resulting in partial tumour response.
• Radiotherapy (1 month later): Following a good partial response and clinical improvement, the patient received craniospinal irradiation to 36 Gy in 20 fractions, with a simultaneous integrated boost to the tumour bed (18 Gy), to a
Figure 3: Curative craniospinal radiotherapy using volumetric modulated arc therapy technique at a total dose of 36 Gy with dose overprinting on the tumour at a dose of 18Gy in normal fractionation of 1.8GY/fr.
A), B), and C) Target craniospinal volumes + boost and 95% isodoses.
D) Statistics of doses delivered on target volumes and constraints at the level of organs at risk
total dose of 54 Gy. This was delivered using the volumetric modulated arc therapy technique (Figure 3).
The primary outcome was radiological response. A follow-up MRI 3 months post-radiotherapy showed approximately 80% regression of the tumour mass. The secondary outcome was clinical status; the patient’s hearing loss and agitation resolved completely. She recovered full neurological function and resumed her academic studies without limitation. No acute or long-term toxicities from treatment were reported at the 3-month follow-up.
DISCUSSION
Pinealoblastomas represent a major therapeutic challenge in neuro-oncology, where a lack of consensus on optimal management persists.4 The authors’ observation of an unresectable giant pinealoblastoma in a 23-year-old patient illustrates the adaptive strategies required in this rare pathology.
The initial tumour extent and neuropsychiatric status compromising radiotherapy guided the authors’ therapeutic sequence. This choice is
consistent with the data of Barlas et al.,4 where a non-surgical approach (stereotactic biopsy, cerebrospinal fluid diversion, and radiotherapy) resulted in an overall survival of 80% at 28 months in six patients with unresectable tumours. While several studies confirm that complete surgical resection followed by radiotherapy remains the ideal option,3,5 the authors’ case highlights the effectiveness of alternatives when surgery is impossible.
The dose of radiotherapy delivered to the primary tumour has a significant impact on overall survival. The recommended total dose should exceed 50 Gy.6 Conventional fractionation is recommended, preferably using recent techniques such as intensitymodulated radiotherapy. Regarding target volume delineation, although the most adopted strategy is based on craniospinal irradiation followed by local boost, some authors have reported treatment focused only on the tumour site. However, a cranial dose of ≥40 Gy was associated with improved survival.7
The use of neoadjuvant chemotherapy has proven crucial. As Lombardi et al.8 pointed out, adult protocols remain extrapolated from paediatrics, with radio-chemotherapy combinations (alkylating agents/platinum/ vincristine) offering 60–70% progression-free survival at 5 years for non-metastatic forms.8 The authors’ choice of cisplatin–etoposide is consistent with the observations of Biswas et al.,9 where this regimen was the most frequently used in adult pinealoblastoma, while the results of Lee et al.7 are a reminder that the impact of chemotherapy remains variable according to subpopulations.
This success should not mask the molecular heterogeneity of pinealoblastoma. Recent work has identified distinct subgroups, including tumours with MYC proto-oncogene amplification sensitive to mTOR inhibitors, and those with RB1 (RB transcriptional corepressor 1) loss vulnerable to WEE1
inhibitors. In this context, clinical trials evaluating targeted therapies (such as NCT02574728 and NCT02095132) or immunotherapies could potentiate conventional regimens. Systematic molecular characterisation becomes essential to explain exceptional responses and guide personalised strategies.10
In addition to pinealoblastomas, other tumours of the sellar and suprasellar regions, such as craniopharyngiomas,11 may also present with similar clinical features and therapeutic challenges. Recent advances in radiomics have further expanded diagnostic possibilities, providing imagingbased biomarkers that may complement histopathological evaluation in rare central nervous system tumours.12
CONCLUSION
This observation validates the efficacy of an induction chemotherapy sequence followed by high-dose craniospinal radiotherapy for unresectable pinealoblastoma. The cisplatin–etoposide regimen merits evaluation in prospective trials dedicated to adults, combined with molecular analysis to identify candidates for innovative therapies. The integration of these targeted approaches represents the future of treatment for these rare tumours.
PATIENT PERSPECTIVE
Being diagnosed with a pinealoblastoma was a difficult experience, especially since surgery was not an option. Thanks to chemotherapy followed by radiotherapy, I experienced significant tumour regression and neurological recovery. This treatment allowed me to resume my studies and regain hope for the future.
References
1. Gener MA et al. Clinical, pathological, and surgical outcomes for adult pineoblastomas. World Neurosurg. 2015;84(6):1816-24.
2. Louis DN et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231-51.
3. Ai P et al. Complete regression of adult pineoblastoma following radiotherapy: a case report and review of the literature. Oncol Lett. 2015;10(4):2329-32.
4. Barlas O et al. Non-resective management of pineoblastoma. Minim Invasive Neurosurg. 2000;43(3):163-70.
5. Clark AJ et al. Tumor control after surgery and radiotherapy for pineocytoma. J Neurosurg. 2010;113(2):319-24.
6. Kumar N et al. Role of radiotherapy in residual pineal parenchymal tumors. Clin Neurol Neurosurg. 2018;166:91-8.
7. Lee JY et al. Management and survival of pineoblastoma: an analysis of 34 adults from the brain tumor registry of Japan. Neurol Med Chir (Tokyo). 2005;45(3):132-41.
8. Lombardi G et al. Diagnosis and treatment of pineal region tumors in adults: a EURACAN overview. Cancers (Basel). 2022;14(15):3646.
9. Biswas A et al. Treatment outcome and patterns of failure in patients of
pinealoblastoma: review of literature and clinical experience from a regional cancer center in northern India. Childs Nerv Syst. 2015;31(8):1291-304.
10. Jiang Z et al. Recent advances in pineoblastoma research: molecular classification, modeling and targetable vulnerabilities. Cancers (Basel). 2025;17(5):720.
11. Diaz MJ et al. Current approaches to craniopharyngioma management. Front Biosci (Landmark Ed). 2022;27(12):328.
12. Rai P et al. Radiomics in pediatric brain tumors: from images to insights. Discov Oncol. 2025;16(1):1563.
Author:
Immunotherapeutic Strategies Based on CAR-T Cells in Hepatocellular Carcinoma
*Negar Hemmati1
1. Department of Microbiology and Immunology, University of Tehran, Iran
2. Shiraz University of Medical Sciences, Iran *Correspondence to Negar.hemmati@ut.ac.ir
Disclosure: The author has declared no conflicts of interest.
Received: 19.05.25
Accepted: 22.10.25
Keywords: Chimeric antigen receptors (CAR), hepatocellular carcinoma (HCC), immunotherapy, T lymphocytes, tumour microenvironment (TME).
Citation: EMJ Oncol. 2025;13[1]:140-153. https://doi.org/10.33590/emjoncol/TABT3057
Abstract
Hepatocellular carcinoma (HCC) is the sixth leading cause of cancer-related mortality worldwide. Despite the availability of therapeutic options such as surgical resection, radiofrequency ablation, molecular-targeted agents, and liver transplantation, HCC shows a poor prognosis and limited responsiveness to conventional treatments. The tumour immune microenvironment (TME) influences key processes in HCC, including selection pressure on tumour cells, immune evasion, tumour evolution, treatment resistance, and recurrence. Among immune components within the TME, T cells, dominant among tumour-infiltrating lymphocytes (TIL), exert both suppressive and promotive effects on tumour growth. Thus, T cell-mediated immune responses are fundamental to cancer surveillance and elimination. Research highlights the crucial role of TILs in HCC prognosis, pathogenesis, and immunotherapy. Subpopulations such as Foxp3+ regulatory T cells, CD8+ cytotoxic T cells, and CD3+/CD4+ helper T cells show complex and often contrasting roles. However, the TME often induces T cell exhaustion or dysfunction, facilitating tumour progression and immune evasion. Understanding immune dysregulation is vital for improving anti-tumour immunity and refining T cell function. This review examines TIL subpopulation roles in HCC, emphasising their plasticity and therapeutic relevance. It also covers emerging T cell-based immunotherapies, especially TIL-based adoptive transfer and CAR-T cell therapy, both showing promise in preclinical and early clinical trials. These novel approaches offer new hope for enhancing immune-driven tumour eradication and improving HCC outcomes.
Key Points
1. Hepatocellular carcinoma remains a leading cause of cancer related mortality worldwide, and its immunosuppressive tumour microenvironment limits the efficacy of conventional treatments, highlighting the need for advanced immune based therapeutic strategies.
2. This narrative review explores recent advances in T cell-based immunotherapies, including tumourinfiltrating lymphocyte and chimeric antigen receptor-T cell approaches, and their emerging role in treating hepatocellular carcinoma.
3. Integrating chimeric antigen receptor-T or tumour-infiltrating lymphocyte therapies with immune checkpoint blockade and microenvironment modulation may enhance antitumour efficacy, offering new personalised and combinatorial treatment avenues for patients with advanced hepatocellular carcinoma.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth leading cause of cancer-related mortality worldwide.1 Despite therapeutic modalities such as surgical resection, radiofrequency ablation, molecular-targeted agents, and liver transplantation being available, HCC is frequently associated with a poor prognosis and limited responsiveness to conventional interventions.2 Therapeutic modalities vary based on the tumour characteristics, hepatic activity, and physiological condition of the patients.3 To prevent liver parenchyma damage, various mechanisms function to inherently avert undesirable immune responses elicited by contact with microbial antigens and conserved molecular motifs termed danger or pathogen-associated molecular patterns, rendering the liver a predominantly immunesuppressive microenvironment.4 The liver immune microenvironment exhibits operational heterogeneity, characterised by the diverse roles of stromal cells such as liver sinusoidal endothelial cells, hepatic stellate cells, liver resident macrophages (Kupffer cells), and various components of the adaptive immune response, such as CD4+ and CD8+ T lymphocytes and natural killer (NK) cells (Figure 1).5
Immunotherapy treatment methods developed in recent years, such as monoclonal antibodies that simultaneously suppress the programmed cell death protein 1 (PD-1) axis and cytotoxic T lymphocyte-associated protein 4 (CTLA4), could stimulate anti-tumour immunity.6,7 Furthermore, locoregional treatments such as trans-arterial chemoembolisation and ablation produce cell death locally and promote CD8+ cell infiltration into the tumour microenvironment, which supports the use of combination PD-1 blockers.8 In
order to circumvent immune escape and trigger anti-tumour responses, autologous T cell transplantation also entails ex vivo activation of mixed T cell/NK cells through cytokine-induced stimulation and reinfusion into the patient. Additionally, ex vivo stimulation of dendritic cells can be used with anti-tumour vaccines against immunodominant peptides of oncofetal proteins to enhance efficient antigen presentation (Figure 2).
T cells are an important part of the immune system, and cancer-specific T lymphocytes can experience a gradual decrease in functional activity once subjected to continuous antigenic stimulus. This decrease exhibits several characteristics, such as persistent generation of restrictive receptors, reduced cytokine production, modified metabolic characteristics, inadequate memory re-establishment, and unique transcriptional and epigenomic characteristics, a dynamic phenomenon commonly referred to as T cell exhaustion.9 The present emphasis of immunotherapy is on reversing T cell depletion, seeking to mitigate the detrimental impacts of continuous antigenic induction and offering a pathway for the therapy of HCC. T cells undergo terminal differentiation during cancer progression due to sustained antigenic induction, resulting in T cell exhaustion.10 Naive CD8+ T cells can differentiate into various degrees of exhaustion, characterised by diminishing working and proliferative abilities, which can eventually suffer from overstimulationinduced cell death.11,12 A thorough understanding of the procedure and its fundamental processes is essential to successfully preventing T cell exhaustion. It is necessary to relocate T lymphocytes from a defective developmental pathway before they reach terminal exhaustion.13 In recent
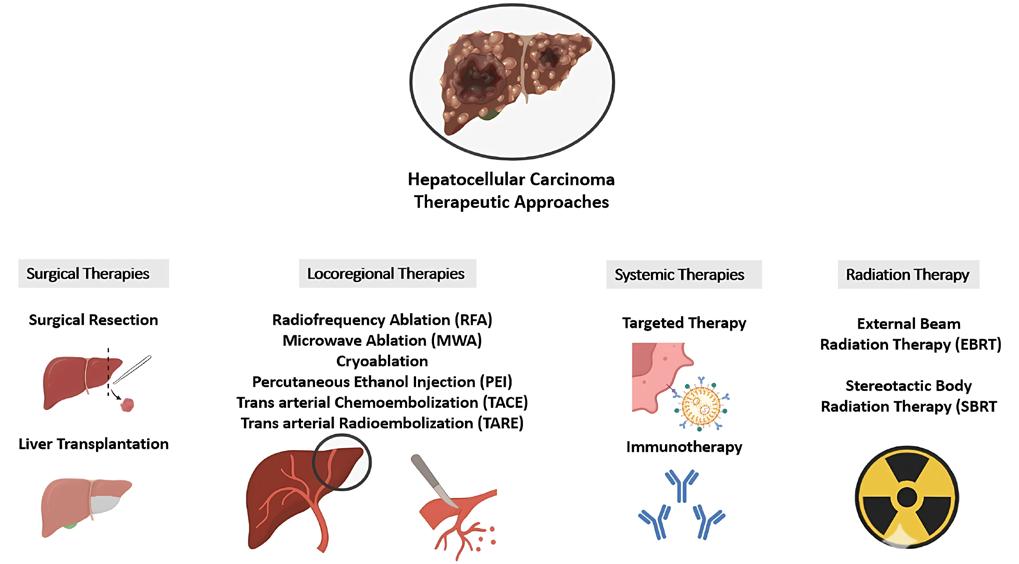
years, the efficacy of PD-1/L1 targeted therapy using monoclonal antibodies has led to promising outcomes in solid tumours, such as HCC, and prompted growing efforts to focus on immunotherapy to reactivate T cell function for tumour treatment.14
In this light, adoptive cell transfer, like tumour-infiltrating lymphocytes (TIL), and chimeric antigen receptor (CAR)-T cell therapy, not only modulates immune equilibrium within the tumour microenvironment but has also demonstrated significant efficacy in treating various malignancies, thereby heightening clinical researchers’ interest in this area.15 Nonetheless, the use of both adoptive cell treatment and immune-targeted therapies will bring about the challenge of T cell exhaustion.16-18 This study will outline the significant T cell subsets found in HCC and examine existing T cell-based immunotherapies in HCC, focusing on TILs and CAR-T cell-based therapies.
SUBTYPES OF TUMOURINFILTRATING T CELLS IN HEPATOCELLULAR CARCINOMA
CD8+ Cytotoxic T Lymphocytes
CD8+ cytotoxic T lymphocytes are crucial for mediating antigen-specific cytotoxicity through tumour-associated antigens presented by antigen-presenting cells, including dendritic cells. The cytotoxic T cell receptor (TCR) binds to the tumour antigen presented by major histocompatibility complex class I, and becomes mature. Then CD8+ mature T lymphocytes induce cytotoxicity by immediate destruction of neoplastic cells and the release of enzymes along with destructive mediators, including perforin/granzyme B, interferon gamma (IFNγ), and TNFα 19 Perforin disrupts the integrity of the tumour cell membrane, while granzyme B triggers apoptosis through caspase activation, via interaction with Fas ligand expressed by CD8+ T lymphocytes with Fas receptors on cancerous cells, and initiates the caspase signalling axis. TNF induces inflammatory responses and
Figure 1: Current therapeutic modalities for hepatocellular carcinoma.
Figure 2: Integrated immunotherapeutic strategies combining checkpoint blockade and locoregional therapies.
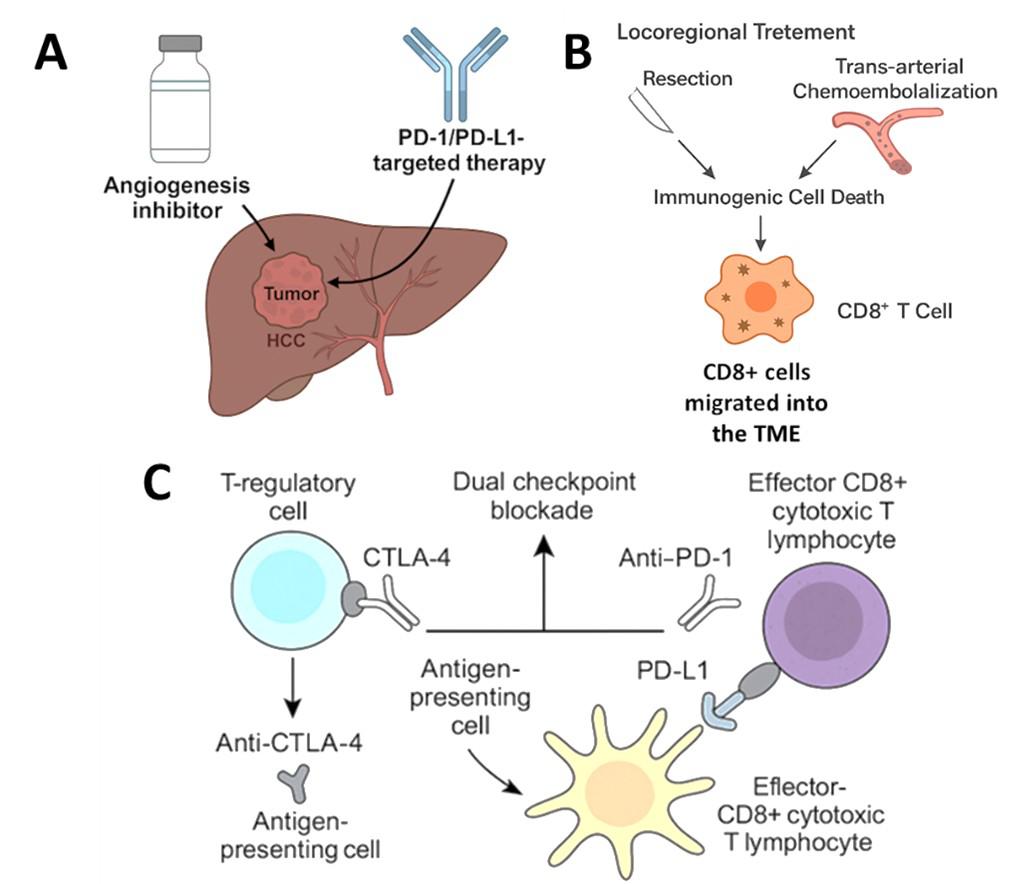
A) Schematic depiction of the combination of angiogenesis inhibitors and PD-1/PD-L1-targeted therapy.
B) Locoregional treatments, including resection and trans-arterial chemoembolisation, serve as locoregional stimulants of immunogenic cell death and promote CD8+ cells to migrate into the tumour microenvironment, hence justifying the use of a combination PD-1 blocker. C) Concurrent blocking of CTLA-4 and the PD-1 pathway with monoclonal antibodies. The impact of dual checkpoint on T-cell immune reconstitution is shown, with CTL-4 primarily influencing T-regulatory cells and antigen-presenting cells, while PD-1 affects effector CD8+ cytotoxic lymphocytes.
CTLA-4: cytotoxic T-lymphocyte-associated protein 4; PD-1: programmed death-1; PD-L1: programmed death-ligand 1; TME: tumour microenvironment.
apoptosis by activating caspase-mediated signalling pathways in tumour cells.20-22
Clinically, considerably higher levels of CD8+ T cells are found in HCC, particularly within or around neoplastic tissue, identifying them as key components of tumour-infiltrating lymphocytes for monitoring HCC development.23 Cytotoxic lymphocytes, along with an elevated CD4+/CD8+ ratio, are correlated with a reduced risk of relapse following hepatic transplantation.24 The effectiveness of antitumour immunological response requires not only antigen presentation and T cell priming, but also efficient infiltration and persistence
of effector T cells within the tumour parenchyma. Immune-desert phenotype, immune-excluded phenotype, and inflamed phenotype are three tumour penetration types based on pathology investigations. The amount and quality of invading T cells both enhance anti-cancer effectiveness.25,26
Previous research has proved that the proportion of infiltrating CD8+ T cells is considerably lower in HCC tissues compared to non-cancerous liver tissue.27 Furthermore, the immunosuppressive function of regulatory T cells (Tregs) and CD8+ T cells within the tumour microenvironment often exhibits functional
defects, which are evident through exhaustion and immune tolerance. This pathologic state entails reduced cytotoxic activity and elevated expression of inhibitory immune checkpoint receptors, such as PD-1, LAG-3, TIM-3, and CTLA-4. The loss of CD8+ T cells is also promoted by the immunosuppressive activities of Tregs, which together compromise the effectiveness of the anti-tumour immune response. Inside the tumour microenvironment, CTLs may undergo activation-induced cell death, a process which is induced by chronic antigen stimulation and inflammatory stress.12 Empirical evidence based on murine models of HCC has demonstrated increased rates of apoptosis in intratumoral CD8+ T cells, thus substantiating the belief that the tumour microenvironment is actively engaged in facilitating T cell dysfunction and consequent loss of cells. Collectively, these findings demonstrate that CD8+ T cells are crucial for the mediation of antitumour immunity; their quantity, quality, and longevity in the TME ultimately determine their therapeutic potential in HCC.28-30
Regulatory T Cells
Regulatory T cells are a distinct subset of CD4+ T lymphocytes that play a critical role in the induction of immune tolerance and are critical in maintaining immune homeostasis. In the tumour microenvironment of HCC, the existence of Tregs facilitates immune evasion by cancer cells by suppressing effective anti-tumour immune responses.31 In tumour tissues, Tregs are generally divided into two major subsets: natural Tregs and inducible Tregs. Natural Tregs with the CD4+CD25+FOXP3+ phenotype are the prominent subset and play a critical role in the maintenance of immunological homeostasis through the regulation of peripheral inflammation.32,33 These cells possess the capacity to exert immune suppressive function in the tumour via the Fas-FasL pathway, and importantly, induce apoptosis in natural killer cells and CD8+ cytotoxic T lymphocytes in a granzyme B/ perforin-dependent process.34,35 Inducible Tregs, that may or may not express FOXP3, perform immunosuppressive activities by mainly secreting anti-inflammatory
cytokines IL-10 and transforming growth factor-TGF-β (TGF-β), and by producing adenosine. All these events serve to inhibit the activation and functionality of effector B and T lymphocytes following contact with an antigen. While Tregs are essential under normal circumstances to prevent autoimmunity and regulate inflammatory responses, their inappropriate expansion and activity in the TME of HCC facilitate tumour immune evasion.36 The immunosuppressive role of Tregs operates via multiple mechanisms, including the release of inhibitory cytokines, direct elimination of effector T cells through granzyme and perforin pathways, and the alteration of dendritic cell differentiation and their antigen-presenting ability.37 The mechanism above entails the regulatory signalling mediated by inhibitory receptors like CTLA-4 and LAG-3.38 Surprisingly, the concurrent activation of both the TCR and IL-2 receptor is central to the survival of Tregs and their immunosuppressive functions.39 In HCC, Tregs predominantly undermine anti-tumour immunity through the inhibition of CD8+ T cell activities. Mechanistic studies have also shown that Tregs derived from HCC can downregulate the co-stimulatory molecules CD80 and CD86 on DCs in a CTLA-4-dependent manner, dampening antigen presentation and inhibiting effector T cell responses further.40,41
Clinical observations have revealed significantly increased percentages of CD4+CD25+ Tregs in peripheral blood as well as liver tissues of patients with HCC compared to healthy controls. Such an increase is linked with disease progression and poor prognosis. Additionally, a high intratumoural Treg-to-CTL ratio has been identified as a prognostic factor, where higher ratios are correlated with poorer overall survival and disease-free survival.42 In a study of 19 patients with advanced HCC, lower frequencies of PD1+ and FOXP3+ Tregs were correlated with increased survival after sorafenib treatment, suggesting their utility as predictive biomarkers. Additionally, evidence exists that Treg accumulation can be causally responsible for HCC recurrence following liver transplantation through a CXCL10–
CXCR3 chemokine axis. In preclinical models, depletion of Tregs using monoclonal antibodies against CD25 has been effective at preventing HCC progression, highlighting a potential therapeutic avenue that involves reprogramming the immunosuppressive TME.43,44
Th1 and Th2 Helper T Cells
T helper cells are a heterogeneous population of CD4+ T lymphocytes that orchestrate immune responses by secreting various cytokine profiles.45,46 Th1 and Th2 cells are two principal subsets among them, which have different cytokine secretion patterns and immunological roles. Th1 cells are mainly responsible for producing IFNγ, IL-2, IL-22, and TNFβ, whereas Th2 cells are characterised by the secretion of IL-4, IL-5, IL-9, IL-10, and IL-13.47,48 These polarised subsets play complementary but often opposite functions: Th1 cells play a central role in inducing cell-mediated immunity and delayed-type hypersensitivity, whereas Th2 cells induce humoral immune responses and are involved in regulating the production of antibodies.49-51 In HCC, the functional distinction between Th1 and Th2 cells is crucial for determining the nature of the TME and the course of the disease. Th1 cells, on recognising peptide-major histocompatibility complex class II complexes and interacting with co-stimulatory molecules, produce pro-inflammatory cytokines like IFNγ, which are accountable for the recruitment and activation of CD8+ CTLs in tumour tissue.52,53 This Th1-induced immune activation enables productive anti-tumour immunity. The shift from Th1-dominant to Th2-dominant cytokine milieu is frequently observed in HCC and signifies the setup of an immunosuppressive TME.54,55 While Th1-associated cytokines IL-1α, IL-1β, IL-2, and IFNγ are typically associated with good clinical outcomes and improved prognosis, overexpression of Th2 cytokines IL-4, IL-5, and IL-10 is correlated with more invasive tumour behaviour and poor prognosis. This Th1-to-Th2 transition has been regarded as a hallmark of immune evasion in HCC.56,57 Moreover, increased circulating Th2 cells have been associated with progressed HCC and decreased efficacy of treatment options available. Research demonstrated
that individuals with lower Treg levels at baseline exhibit an improved Th1/Th2 ratio prior to trans-arterial chemoembolisation, a widely applied locoregional therapy for HCC. Importantly, a higher Th1/Th2 ratio was linked to more intense anti-tumour immunity and prolonged survival rates. Although cancer vaccines targeting helper T cells have reported promising activity in a number of malignancies, the specific contribution of Th1 responses to promoting effector T cell activation in HCC remains ill-defined. Further research is required to establish the therapeutic utility of adjusting the Th1/Th2 balance as a strategy to reverse immunosuppression and enhance antitumour immunity in HCC.58
CRITICAL SIGNALLING PATHWAYS MODULATING TUMOURINFILTRATING LYMPHOCYTES IN HEPATOCELLULAR CARCINOMA
TGFβ Signalling
The TGFβ family includes TGFβs, activins, inhibins, bone morphogenetic proteins, and growth and differentiation factors.59,60 The initiation of this axis begins with the attachment of ligands to the extracellular domains of TGFβ Type I and Type II receptors (TβRI and TβRII), stimulating both suppressor of mothers against decapentaplegic homolog (SMAD)-reliant and SMAD-non-reliant axis to elicit the downstream pathway. The TGFβ axis is implicated in almost every phase of tumorigenesis in HCC.61 The TME exhibited elevated levels of TGFβ in both cancer cells and several immunological cells. In the first phase of HCC, TGFβ inhibited the growth of pre-malignant hepatocytes. However, in the developed phase of HCC, it facilitated cancer growth by modulating immunological cells, including Tregs, CTLs, TAMs, and NKs.62 TGFβ may up-regulate FOXP3 in CD4+CD25naïve T cells by promoting the SMAD-reliant axis, facilitating the development of Tregs. Tregs from peripheral blood penetrated HCC tumour tissues, thereby inhibiting the therapeutic efficacy of CTLs. Moreover, HCC cells demonstrate elevated levels of TGFβ, thus underlying the upregulation of PD-1 levels on CD8+ CTLs. CTLs subsequently attach to PD-L1 on malignant cells and
antigen-presenting cells, resulting in CTL exhaustion.63,64 TGFβ activation may regulate the innate immune system by inhibiting NK cells directly and facilitating M2 macrophage polarisation to promote immune evasion. TGFβ signalling pathways were extensively implicated in the control of TILs, ultimately facilitating HCC progression.61,62
JAK/STAT3 Signalling
The STAT protein family has seven members, including STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. STAT3 has been the subject of several studies concerning HCC management. It was believed to be triggered swiftly and temporarily in normal cells by various mediators, including IL-6/ IL-10, IFNs, and growth factors like vascular endothelial growth factor. Nonetheless, it was consistently and aberrantly activated in neoplastic cells.65-68 Recent research indicated that phosphorylated STAT3 was present in approximately 60% of HCC samples, correlating with a worse prognosis. The STAT3 axis primarily promotes tumorigenesis by modulating dendritic cells and developing tumour-associated macrophages, NK cells, and Tregs.68,69 In DCs, IL-6 released through HCC cells and hepatic carcinomaassociated fibroblasts can attach to the IL-6 receptor on DCs, activating JAK and initiating the downstream phosphorylation STAT3 axis, which inhibits T cell growth and enhances Tregs expansion.70 Furthermore, the stimulation of STAT3 has been shown to suppress the activating receptor NKG2D on NK cells and its associated ligands, major histocompatibility complex class I-related protein A/B on malignant cells, hence obstructing NK cell stimulation and leading to impaired immune surveillance in HCC.71 Inhibition of STAT3 signalling in HCC may reactivate NK cells to perform anti-tumour functions via modifying cytokine levels in the TME, specifically by decreasing IL-10 levels. Additionally, the IL-6-STAT3 axis promoted tumorigenesis by diminishing M1 macrophage polarisation and enhancing M2 differentiation of TAM. STAT3 activation affected many immunological cells, contributing to the progression of HCC.72,73
Wnt/β-Catenin Signalling
The Wnt signalling pathway becomes active when Wnt proteins engage with their specific membrane-bound receptors, initiating a cascade of intracellular events. One key player in this pathway is β-catenin, a protein usually associated with cell-to-cell adhesion or found in the cytoplasm. Upon pathway activation, β-catenin accumulates and translocates to the nucleus, where it influences gene expression. In HCC, persistent activation of the Wnt/βcatenin axis has been frequently observed and is linked to immune evasion. This activation impairs the infiltration and function of tumour-infiltrating lymphocytes, especially by reducing dendritic cell presence, which plays a crucial role in antigen presentation and T cell priming. Consequently, this weakens the recruitment and cytotoxic function of CD8+ T cells. Chronic stimulation of this axis also contributes to cytotoxic T lymphocyte dysfunction, ultimately fostering an immunosuppressive environment favourable to tumour progression. Overall, the aberrant Wnt/β-catenin signalling in HCC is a critical factor in disrupting immune surveillance and supporting tumour immune escape mechanisms (Table 1).44,74-78
TUMOUR-INFILTRATING LYMPHOCYTE THERAPY IN HEPATOCELLULAR CARCINOMA
TIL therapy is a promising immunotherapeutic approach for HCC, leveraging the patient’s own tumour-reactive T cells. These lymphocytes are isolated from the tumour, expanded in vivo, often with IL-2, and reintroduced after lymphodepletion to enhance their persistence and efficacy.79-81 This strategy boosts the body’s immune response by restoring and amplifying tumour-specific T cell activity. However, challenges remain, including difficulty in isolating sufficient TILs from the liver’s immunosuppressive environment and ensuring effective tumour antigen recognition. Current research aims to optimise expansion techniques, tailor conditioning regimens, and overcome immune resistance. Continued refinement is essential to establish TIL therapy as a reliable treatment option for HCC.82-86
Table 1: Critical immunoregulatory signalling pathways affecting tumour-infiltrating lymphocytes in hepatocellular carcinoma.
Signalling Pathway
Ligand Source Receptor
TGFβ HCC cells, immune cells in TME
TβRI/TβRII SMAD-dependent and independent in T cells, NKs, and macrophages
IL-6/STAT3 HCC cells, hCAFs IL-6R JAK → STAT3 in DCs, macrophages, NKs
Wnt/β-Catenin HCC cells Frizzled/ LRP5/6 β-catenin nuclear translocation in tumour cells
Immune Cells Affected Immunological Outcome
Tregs, CTLs, NKs, TAMs ↑ Tregs via FOXP3 upregulation, ↓ NK cell cytotoxicity, ↑ PD-1 expression on CTLs, ↑ M2 macrophage polarisation
DCs, Tregs, NKs, TAMs ↑ Tregs through DC modulation, ↓ CD8+ T cell priming, ↓ NKD2G receptor on NK cells, ↑ M2 macrophages
DCs, CD8+ T cells (indirectly) ↓ DC recruitment and antigen presentation, ↓ CD8+ T cell infiltration, ↑ and immune evasion
References
(63-66)
(67-74)
(46, 76-80)
CTL: cytotoxic T lymphocyte; DC: dendritic cell; hCAF: hepatic cancer-associated fibroblast; HCC: hepatocellular carcinoma; NK: natural killer; PD-1: programmed death-1; TAM: tumour-associated macrophage; TGFβ: transforming growth factor-β; TME: tumour microenvironment; Tregs: regulatory T cells.
RECENT ADVANCES AND CLINICAL APPLICATIONS
Recent advances in tumour-infiltrating lymphocyte therapy have positioned it as a promising immunotherapeutic option for HCC. Early-phase clinical trials, such as the Phase I trial of BST02, a TIL-derived product, have demonstrated encouraging outcomes in patients with advanced HCC. Notably, combining TILs with immune checkpoint inhibitors, particularly PD-1/ PD-L1 blockade, appears to overcome the immunosuppressive liver microenvironment, leading to more durable antitumor responses compared to TIL monotherapy.87-90 To further improve efficacy, researchers have employed adjunct strategies such as IL-2 to enhance TIL proliferation and function. Pretreatment lymphodepletion with agents like cyclophosphamide has also shown benefit by facilitating TIL engraftment and expansion. However, the tumour microenvironment, characterised by high PD-L1 expression and suppressive cytokines, continues to limit therapeutic success. To address this,
novel approaches, including oncolytic virotherapy and lymphodepleting regimens, are being evaluated for their synergistic potential to reprogram the TME.91,92 In parallel, TIL engineering strategies such as CAR expression have shown promise in preclinical models, offering increased tumour specificity. Phase II trials report that combination regimens remain safe and tolerable, even in patients with chronic hepatitis B, although tumour regression is modest in this subgroup. Overall, the future of TIL therapy in HCC lies in personalised approaches informed by immune profiling and the strategic integration of combination therapies. These advancements may ultimately establish TILs as a foundational component of immunotherapy in liver cancer (Table 2).25,93,94
BIOMARKERS AND PREDICTIVE INDICATORS
Biomarker identification is pivotal for optimising patient selection and improving
Table 2: Recent studies on tumour-infiltrating lymphocytes in hepatocellular carcinoma.
Study/Strategy
TIL+Checkpoint Inhibitors (Atezolizumab+Bevacizumab)
Combination of TIL therapy with PD-1/PD-L1 inhibitors to reverse T cell exhaustion in the tumour microenvironment
BST02 Trial TIL-derived product (BST02) for patients with HCC
TIL+IL-2 Cytokine Support Using IL-2 to improve the survival and function of TILs
CAR-T in HCC
Oncolytic Virus+ Lymphodepletion+TIL
TIL+ICIs in Patients with Chronic HBV
Engineering T cells to express chimeric antigen receptors targeting tumour antigens
Modifying the tumour microenvironment using oncolytic viruses and chemotherapy to enhance TIL efficacy
Assessing the safety and effectiveness of TIL+checkpoint inhibitors in patients with chronic hepatitis B
Phase II (89-92, 95)
Phase I (85, 88, 96)
Preclinical and Observational (81, 82, 84, 85)
Preclinical/Early Clinical (93, 94, 97-99)
Under Investigation (83, 84, 94, 100)
Phase II (90, 91, 101)
Personalised TIL Therapy Based on Immune Profile Tailored TIL treatments based on individual patient immune signatures In Development (101-104)
HBV: hepatitis B virus; HCC: hepatocellular carcinoma; PD-1: programmed death-1; PD-L1: programmed death-ligand 1; TIL: tumour-infiltrating lymphocytes.
the therapeutic efficacy of TIL therapy in HCC. Accumulating evidence suggests that a high frequency of CD8+ TILs within tumour tissues is associated with superior clinical outcomes, particularly when TIL therapy is combined with immune checkpoint inhibitors. Tumours enriched in CD8+ T cells may demonstrate enhanced immune responsiveness, making them more amenable to TIL-based interventions. Nonetheless, the predictive roles of additional immune biomarkers, such as PD-L1 expression and TCR diversity, remain under active investigation. Furthermore, both liver functional status and HCC staging are important considerations in determining patient eligibility. Individuals with preserved liver function and early-stage disease are more likely to benefit from adoptive TIL therapy.95, 101 Recent clinical studies have yielded variable results regarding its therapeutic impact. A Phase I trial published in 2022 demonstrated the technical
feasibility of TIL isolation, expansion, and reinfusion in patients with HCC; however, clinical responses were limited, particularly in heavily pre-treated individuals.102 More recent investigations have evaluated the combination of TIL therapy with PD-1 inhibitors, showing promising early efficacy, especially in tumours with high PD-L1 expression.96,103 These findings underscore the need for improved patient stratification strategies and potentially the incorporation of additional combination therapies to enhance response. Moreover, TMEinduced immunosuppression continues to be a major barrier to TIL functionality in HCC. Observational studies confirm that overcoming the TME’s suppressive effects is critical for maximising TIL efficacy, aligning with previous research emphasising the central role of the TME in modulating immune responses in liver cancer.105,106
ADVANCES IN CAR-T CELL THERAPY FOR HEPATOCELLULAR CARCINOMA
CAR-T cell therapy is being actively explored for HCC, with recent innovations addressing key challenges, such as immune evasion, tumour heterogeneity, and a suppressive microenvironment. Glypican-3 (GPC3), highly expressed in HCC and rarely in normal tissues, is a leading CAR-T target. Early clinical results show anti-tumour activity, though efficacy is limited by immunosuppression (Figure 3). To counter this, GPC3-CAR-T cells are engineered to release PD-1 or TGFβ inhibitors, enhancing persistence and function. Co-administering checkpoint inhibitors (e.g., anti-PD-1) also helps reduce T cell exhaustion.97,104,107 Tandem CARs (TanCARs), recognising GPC3 and AFP, boost tumour detection and reduce immune escape. SynNotch CARs use
a two-step activation model, first sensing one antigen, then triggering CAR expression via another, ensuring tumour specificity and minimising off-target effects. Both methods improve tumour targeting in HCC models.44,98,108 HCC’s immunosuppressive environment hinders CAR-T activity. New CAR-Ts are modified to secrete cytokines like IL-15, supporting T cell survival. Targeting vascular endothelial growth factor improves infiltration, while combining with TGFβ inhibitors reprogrammes the microenvironment to be more immunepermissive.108 Additionally, Universal CARs employ a modular 'lock-and-key' system, allowing redirection to multiple tumour antigens. This approach addresses tumour heterogeneity and lowers production costs, improving feasibility for personalised treatments.99,109 CAR-T efficacy is improved by blocking inhibitory receptors such as PD-1, LAG-3, and TIM-3. Inhibiting CD39,
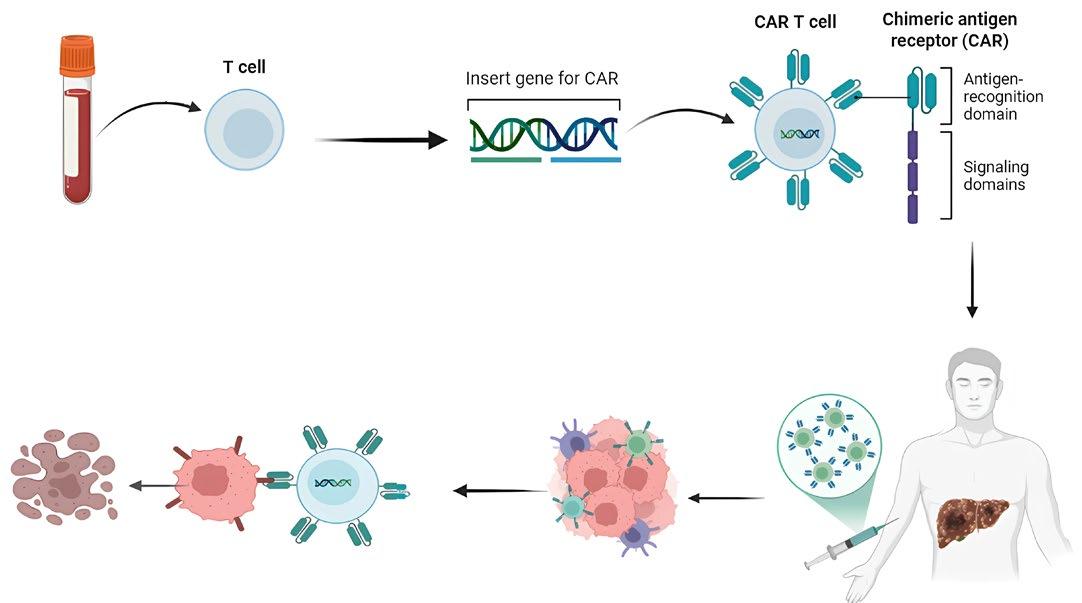
Peripheral blood T cells are isolated and genetically engineered to express CAR. The modified CAR-T cells are expanded and re-infused into the patient, where they recognise and kill tumour cells in the liver through antigenspecific cytotoxicity.
CAR: chimeric antigen receptor.
Figure 3: Schematic representation of CAR-T cell therapy in liver cancer.
a driver of adenosine-mediated suppression, restores function in HCC. Combining CAR-T with anti-LAG-3 antibodies further revives exhausted T cells.110-112 Engineered cytokine receptors like 4/7 ICR convert suppressive IL-4 signals into stimulatory IL-7-like effects. Advanced 4/21 ICR CAR-T cells adopt a Th17-like phenotype under IL-4, enhancing anti-tumour activity and persistence in HCC.113,114 To prevent recurrence, CAR-T cells are engineered to produce IL-7 or IL-21, promoting memory cell development. Smallmolecule agents like T-lymphokine-activated killer cell-originated protein kinase inhibitors and CAR signalling modifications improve survival. Combining CAR-T with chemo, radiation, or oncolytic viruses further boosts immune response and tumour antigen visibility.115,116
CONCLUSION
Recent evidence has revealed that cancer progression is not only driven by local immune and molecular alterations but also by systemic neuroendocrine regulation. Tumours can hijack neuroendocrine signalling pathways to manipulate host homeostasis, thereby creating conditions favourable for their growth and immune evasion. Through the release of stressrelated hormones and neuropeptides, the tumour can modulate immune cell activity, suppress cytotoxic CD8+ T lymphocytes, and promote Treg expansion within the tumour microenvironment. This neuroendocrine–immune interaction leads to impaired anti-tumour immunity and contributes to T cell exhaustion, a key obstacle to effective immunotherapy. Understanding how HCC exploits
References
1. Samant H et al. Addressing the worldwide hepatocellular carcinoma: epidemiology, prevention and management. J Gastrointest Oncol. 2021;12(Suppl 2):S361-S373.
2. Sachdeva M, Arora SK. Prognostic role of immune cells in hepatocellular carcinoma. EXCLI J. 2020;19:718-33.
neuroendocrine mechanisms to suppress immune surveillance may open new avenues for combination therapies that integrate immunomodulation with neuroendocrine targeting to restore both immune balance and systemic homeostasis.100 Recent advances in immunotherapy have rekindled hope for HCC treatment, with T cell exhaustion receiving more attention. This pathological condition is one of the driving forces for HCC development, but its causative mechanisms are only half understood and require more clinical investigations. Clarification of the molecular mechanisms of T cell exhaustion is essential for unravelling the immune microenvironment of HCC and developing new therapeutic interventions. Both TILs and CAR-T cells possess double-edged roles, both fighting cancer and potentially supporting tumour growth, via a complex network of signalling interactions within the tumour microenvironment. Further research is necessary to unveil the occult functions and mechanisms of TILs, particularly how they mould and are moulded by the evolving HCC environment. Longitudinal studies of the functions of TILs and CAR-T cells in HCC development will provide additional information on immune-tumour interactions and guide the development of next-generation immunotherapeutic approaches. As a narrative review, this study is limited by the lack of a systematic search strategy, which may introduce selection bias. Additionally, due to the rapidly evolving nature of immunotherapy research, some recent data may not have been captured at the time of writing. Finally, many referenced findings originate from early-phase or preclinical studies, which limits the strength and generalisability of the conclusions.
3. Jelic; ESMO Guidelines Working Group. Hepatocellular carcinoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annal Oncol. 2009;DOI: 10.1093/ annonc/mdp124.
4. Zhou G et al. Immune suppressive checkpoint interactions in the tumour microenvironment of primary liver cancers. Br J Cancer. 2022;126(1):10-23.
5. Fujita T, Narumiya S. Roles of hepatic stellate cells in liver inflammation: a new perspective. Inflamm Regen. 2016;36(1):1.
6. Song Q et al. A study on the efficacy and Safety Evaluation of a novel PD-1/CTLA-4 bispecific antibody. Immunobiology. 2024;229(6):152844.
7. Wang K et al. Combination anti-PD-1 and anti-CTLA-4 therapy generates waves of clonal responses that include progenitor-exhausted CD8+ T cells. Cancer Cell. 2024;42(9):1582-97.
8. Zhu H-D et al. Transarterial chemoembolization with PD-(L) 1 inhibitors plus molecular targeted therapies for hepatocellular carcinoma
(CHANCE001). Signal Transduct Target Ther. 2023;8(1):58.
9. Catakovic K et al. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal. 2017;15(1):1.
10. Zhu Y et al. Molecular insight into T cell exhaustion in hepatocellular carcinoma. Pharmacol Res. 2024;203:107161.
11. Muthiah KA et al. Prospective evaluation of procalcitonin in sepsis in the Illawarra area of Australia: PEPSIA study. Crit Care Resusc. 2007;9(2):13742.
12. Dolina JS et al. CD8+ T cell exhaustion in cancer. Front Immunol. 2021;12:715234.
13. Hu Y et al. Reversal of T-cell exhaustion: mechanisms and synergistic approaches. Int Immunopharmacol. 2024;138:112571.
14. Sharafi F et al. A comprehensive review about the utilization of immune checkpoint inhibitors and combination therapy in hepatocellular carcinoma: an updated review. Cancer Cell Int. 2022;22(1):269.
15. Aggeletopoulou I et al. Chimeric antigen receptor T cell therapy for hepatocellular carcinoma: where do we stand? Int J Mol Sci. 2024;25(5):2631.
16. Gumber D, Wang LD. Improving CAR-T immunotherapy: overcoming the challenges of T cell exhaustion. EBioMedicine. 2022;77:103941.
17. Kang K et al. T cell exhaustion in human cancers. Biochim Biophys Acta Rev Cancer. 2024;1879(5):189162.
18. Li R et al. Cellular kinetics and biodistribution of adoptive t cell therapies: from biological principles to effects on patient outcomes. AAPS J. 2025;27(2):55.
19. Abdou AG et al. Role of CD8 cytotoxic T lymphocytes in hepatocellular carcinoma: an immunohistochemical study. Medical Journal of Cairo University. 2019;87:4061-9.
20. Annibaldi A, Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med. 2018;24(1):49-65.
21. Volpedo G et al. Chapter 4 - The Fas/ FasL pathway as a target for enhancing anticancer adoptive cell therapy. Immunotherapy in Resistant Cancer: From the Lab Bench Work to Its Clinical Perspectives. Elsevier. 2021:2;47-68.
22. Metkar SS et al. Cytotoxic cell granulemediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity. 2002;16(3):417-28.
23. Chen Z et al. AI-based tumor-infiltrating lymphocyte scoring system for assessing HCC prognosis in patients undergoing liver resection. JHEP Rep. 2024;7(2):101270.
24. Xin H et al. The CD68+ macrophages to CD8+ T-cell ratio is associated with clinical outcomes in hepatitis B virus (HBV)-related hepatocellular carcinoma. HPB (Oxford). 2021;23(7):1061-71.
25. Zhang L et al. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys Acta Rev Cancer. 2020;1874(2):188441.
26. Mellman I et al. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. 2023;56(10):2188-205.
27. Ikeguchi M et al. CD8+ lymphocyte infiltration and apoptosis in hepatocellular carcinoma. Eur J Surg Oncol. 2004;30(1):53-7.
28. Alim LF et al. Molecular mechanisms of tumour necrosis factor signalling via TNF receptor 1 and TNF receptor 2 in the tumour microenvironment. Curr Opin Immunol. 2024;86:102409.
29. Huan L et al. Targeting tumor metabolism to augment CD8+ T cell anti-tumor immunity. J Pharm Anal. 2025;15(5):101150.
30. Fu C et al. Manganese improves CD8+ T cell recruitment via cGASSTING in hepatocellular carcinoma. International Immunopharmacology. 2024;143:113591.
31. González-Navajas JM et al. The impact of Tregs on the anticancer immunity and the efficacy of immune checkpoint inhibitor therapies. Front Immunol. 2021;12:625783.
32. Peterson RA. Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol Pathol. 2012;40(2):186-204.
33. Valencia X, Lipsky PE. CD4+ CD25+ FoxP3+ regulatory T cells in autoimmune diseases. Nat Clin Pract Rheumatol. 2007;3(11):619-26.
34. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109-18.
35. Vignali DA et al. How regulatory T cells work. Nat Rev Immunol. 2008;8(7): 523-32.
36. Sharma MD et al. Indoleamine 2, 3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood, The Journal of the American Society of Hematology. 2009;113(24):6102-11.
37. Schmidt A et al. Molecular mechanisms of treg-mediated T cell suppression. Frontiers in immunology. 2012;3:51.
38. Zong Y et al. Regulation of Treg cells by cytokine signaling and costimulatory molecules. Front Immunol. 2024;15:1387975.
39. Shouse AN et al. Interleukin-2 receptor signaling acts as a checkpoint that influences the distribution of regulatory T cell subsets. iScience. 2024;27(12):111248.
40. Chen X et al. Tumor-derived CD4+ CD25+ regulatory T cells inhibit dendritic cells function by CTLA-4. Pathology-Research and Practice. 2017;213(3):245-9.
41. Tekguc M et al. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proceedings of the National Academy of Sciences. 2021;118(30):e2023739118.
42. Yan W et al. CXCL10 mediates CD8+ T cells to facilitate vessel normalization and improve the efficacy of cetuximab combined with PD-1 checkpoint inhibitors in colorectal cancer. Cancer Letters. 2023;567:216263.
43. Zhu Y, Qin L-X. Strategies for improving the efficacy of immunotherapy in hepatocellular carcinoma. Hepatobiliary & Pancreatic Diseases International. 2022;21(5):420-9.
44. Muhammed TM al. T lymphocytebased immune response and therapy in hepatocellular carcinoma: focus on TILs and CAR-T cells. NaunynSchmiedeberg's Archives of Pharmacology. 2025:1-18.
45. Romagnani S. Th1 and Th2 in human diseases. Clinical immunology and immunopathology. 1996;80(3):225-35.
46. Eagar TN, Miller SD. Helper T-cell subsets and control of the inflammatory response. Clinical immunology: Elsevier; 2019. p. 235-45. e1.
47. Pesce B et al. TNF-α affects signature cytokines of Th1 and Th17 T cell subsets through differential actions on TNFR1 and TNFR2. Int J Mol Sci. 2022;23(16):9306.
48. Kaiko GE et al. Immunological decision‐making: how does the immune system decide to mount a helper T‐cell response? Immunology. 2008;123(3):326-38.
49. Aleebrahim-Dehkordi E et al. T helper type (Th1/Th2) responses to SARSCoV-2 and influenza A (H1N1) virus: from cytokines produced to immune responses. Transplant immunology. 2022;70:101495.
50. Zhang Y et al. Th1/Th2 cell’s function in immune system. T helper cell differentiation and their function. 2014:45-65.
51. Biedermann T et al. TH1 and TH2 Lymphocyte development and
regulation of TH cell–mediated immune responses of the skin. Journal of Investigative Dermatology Symposium Proceedings; 2004: Elsevier.
52. Ryba-Stanisławowska M. Unraveling Th subsets: insights into their role in immune checkpoint inhibitor therapy. Cellular Oncology. 2024:1-18.
53. Lin Y et al. New insights on anti-tumor immunity of CD8+ T cells: cancer stem cells, tumor immune microenvironment and immunotherapy. Journal of Translational Medicine. 2025;23(1):341.
54. Maggi E et al. T cell landscape in the microenvironment of human solid tumors. Immunology Letters. 2024:106942.
55. Lin J et al. LncRNA MEG3 suppresses hepatocellular carcinoma by stimulating macrophage M1 polarization and modulating immune system via inhibiting CSF-1 in vivo/vitro studies. International Journal of Biological Macromolecules. 2024;281:136459.
56. Qin L-X. Inflammatory immune responses in tumor microenvironment and metastasis of hepatocellular carcinoma. Cancer Microenvironment. 2012;5(3):203-9.
57. Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. Journal of leukocyte biology. 2006;80(6):1197-213.
58. Song Y, Yang JM. Role of interleukin (IL)-17 and T-helper (Th) 17 cells in cancer. Biochemical and biophysical research communications. 2017;493(1):1-8.
59. Wu MY, Hill CS. TGF-β superfamily signaling in embryonic development and homeostasis. Developmental cell. 2009;16(3):329-43.
60. Liu S et al. Molecular evolution of transforming growth factor-β (TGF-β) gene family and the functional characterization of lamprey TGF-β2. Frontiers in Immunology. 2022;13:836226.
61. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends in molecular medicine. 2019;25(11):101023.
62. Thangaraj JL et al. Disruption of TGF-β signaling pathway is required to mediate effective killing of hepatocellular carcinoma by human iPSC-derived NK cells. Cell Stem Cell. 2024;31(9):1327-43. e5.
63. Qin D et al. Targeting tumor-infiltrating tregs for improved antitumor responses. Frontiers in Immunology. 2024;15:1325946.
64. Fu S et al. TGF-β induces Foxp3+ T-regulatory cells from CD4+ CD25−
precursors. American Journal of Transplantation. 2004;4(10):1614-27.
65. Babaei G et al. STAT protein family and cardiovascular diseases: overview of pathological mechanisms and therapeutic implications. Molecular Biology Reports. 2024;51(1):440.
66. Shi D et al. Structure, function, signaling pathways and clinical therapeutics: The translational potential of STAT3 as a target for cancer therapy. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2024:189207.
67. Thuya WL et al. Insights into IL-6/ JAK/STAT3 Signaling in the Tumor Microenvironment: Implications for Cancer Therapy. Cytokine Growth Factor Rev. 2025;85:26-42.
68. Songbai H et al. STAT3 Orchestrates Immune Dynamics in Hepatocellular Carcinoma: A Pivotal Nexus in Tumor Progression. Critical Reviews in Oncology/Hematology. 2025:104620.
69. Makino Y et al. STAT3 is activated by CTGF-mediated tumor-stroma cross talk to promote HCC progression. Cellular and molecular gastroenterology and hepatology. 2023;15(1):99-119.
70. Xu J et al. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Frontiers in oncology. 2021;11:760971.
71. Xing S, Ferrari de Andrade L. NKG2D and MICA/B shedding: a ‘tag game’between NK cells and malignant cells. Clinical & translational immunology. 2020;9(12):e1230.
72. Tian Z et al. Macrophages and hepatocellular carcinoma. Cell & bioscience. 2019;9:1-10.
73. Hu R et al. STAT3: A key signaling molecule for converting cold to hot tumors. Cancer letters. 2020;489:2940.
74. Zhang Z et al. Wnt/β-catenin signaling in the development and therapeutic resistance of non-small cell lung cancer. Journal of Translational Medicine. 2024;22(1):565.
75. Duchartre Y et al. The Wnt signaling pathway in cancer. Critical reviews in oncology/hematology. 2016;99:141-9.
76. Morita M et al. Role of β-catenin activation in the tumor immune microenvironment and immunotherapy of hepatocellular carcinoma. Cancers. 2023;15(8):2311.
77. Huang Y et al. Activation of Wnt/βcatenin signaling promotes immune evasion via the β-catenin/IKZF1/CCL5 axis in hepatocellular carcinoma. International Immunopharmacology. 2024;138:112534.
78. Zhao Z et al. Wnt/β-catenin signaling pathway in hepatocellular carcinoma:
pathogenic role and therapeutic target. Frontiers in Oncology. 2024;14:1367364.
79. König D et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in combination with nivolumab in patients with advanced melanoma. Immuno-Oncology and Technology. 2024;24:100728.
80. Lickefett B et al. Lymphodepletion–an essential but undervalued part of the chimeric antigen receptor T-cell therapy cycle. Frontiers in immunology. 2023;14:1303935.
81. Tennant MD et al. Efficient T cell adoptive transfer in lymphoreplete hosts mediated by transient activation of Stat5 signaling. Molecular Therapy. 2023;31(9):2591-9.
82. Shen W et al. Immunotherapeutic approaches for treating hepatocellular carcinoma. Cancers. 2022;14(20):5013.
83. Matsueda S et al. Recent clinical researches and technological development in TIL therapy. Cancer Immunology, Immunotherapy. 2024;73(11):232.
84. Amaria RN et al. Entering a new era of tumor-infiltrating lymphocyte cell therapy innovation. Cytotherapy. 2025;27(7):864-73.
85. Poschke IC et al. The outcome of ex vivo TIL expansion is highly influenced by spatial heterogeneity of the tumor T-cell repertoire and differences in intrinsic in vitro growth capacity between T-cell clones. Clinical cancer research. 2020;26(16):4289-301.
86. Dougé A et al. Adoptive T cell therapy in solid tumors: state-of-the art, current challenges, and upcoming improvements. Molecular Cancer Therapeutics. 2024;23(3):272-84.
87. Dong Y et al. Recent advances and future prospects in immune checkpoint (ICI)-based combination therapy for advanced HCC. Cancers. 2021;13(8):1949.
88. Chen X et al. Unveiling the role of tumor-infiltrating T cells and immunotherapy in hepatocellular carcinoma: a comprehensive review. Cancers. 2023;15(20):5046.
89. Li Q et al. PD-1/PD-L1 checkpoint inhibitors in advanced hepatocellular carcinoma immunotherapy. Frontiers in immunology. 2022;13:1070961.
90. Castet F et al. Atezolizumab plus bevacizumab: a novel breakthrough in hepatocellular carcinoma. Clinical Cancer Research. 2021;27(7):1827-9.
91. Shi D et al. Chimeric antigen receptorglypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clinical Cancer Research. 2020;26(15):3979-89.
92. Zhou Y et al. CAR-T cell therapy for hepatocellular carcinoma: current trends and challenges. Frontiers in Immunology. 2024;15:1489649.
93. Lin T-Y, Su T-H. Progression of portal hypertension after atezolizumab plus bevacizumab for hepatocellular carcinoma-report a case and literature review. Journal of the Formosan Medical Association. 2024;123(8):916-9.
94. Ramadan A et al. Personalized treatment approaches in hepatocellular carcinoma. Arab Journal of Gastroenterology. 2025;26(1):122-8.
95. Pelizzaro F, Farinati F, Trevisani F. Immune checkpoint inhibitors in hepatocellular carcinoma: current strategies and biomarkers predicting response and/or resistance. Biomedicines. 2023;11(4):1020.
96. Ji JH et al. Predictive biomarkers for immune-checkpoint inhibitor treatment response in patients with hepatocellular carcinoma. International Journal of Molecular Sciences. 2023;24(8):7640.
97. Ozer M et al. Adoptive cell therapy in hepatocellular carcinoma: a review of clinical trials. Cancers. 2023;15(6):1808.
98. Yang Y et al. The predictive value of PD‐L1 expression in patients with advanced hepatocellular carcinoma treated with PD‐1/PD‐L1 inhibitors: A systematic review and meta‐analysis. Cancer Medicine. 2023;12(8):9282-92.
99. Chen Y et al. The current advances and future directions of PD-1/ PD-L1 blockade in head and neck squamous cell carcinoma (HNSCC) in the era of immunotherapy.
International Immunopharmacology. 2023;120:110329.
100. Liu X et al. The immunosuppressive role of MDSCs in HCC: mechanisms and therapeutic opportunities. Cell Communication and Signaling. 2025;23(1):155.
101. Arvanitakis K, Germanidis G. Immunity revealed: Stratifying hepatocellular carcinoma for precision immunotherapy. Molecular Therapy Oncology. 2025;33(1).
102. Devan AR et al. The role of glypican-3 in hepatocellular carcinoma: Insights into diagnosis and therapeutic potential. European Journal of Medical Research. 2024;29(1):490.
103. Eghbali S, Heumann TR. NextGeneration Immunotherapy for Hepatocellular Carcinoma: Mechanisms of Resistance and Novel Treatment Approaches. Cancers. 2025;17(2):236.
104. Tehrani HA et al. GPC-3 in Hepatocellular Carcinoma; A Novel Biomarker and Molecular Target. Experimental Cell Research. 2024:114391.
105. Batra SA et al. Glypican-3–specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. 2020;8(3):309-20.
106. Li X, Hu D. Ligand-restricted synNotch switches enable precision cell therapy. Trends Immunol. 2025;46(2):91-3.
107. Wu Z et al. Universal CAR cell therapy: challenges and expanding applications. Translational Oncology. 2025;51:102147.
108. Chen T et al. The construction of modular universal chimeric antigen receptor T (MU-CAR-T) cells by covalent linkage of allogeneic T cells and various antibody fragments. Molecular Cancer. 2024;23(1):53.
109. Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumormediated inhibition. J Clin Invest. 2016;126(8):3130-44.
110. Cai L et al. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. 2023;16(1):101.
111. Liu Y et al. Review immune response of targeting CD39 in cancer. Biomarker Research. 2023;11(1):63.
112. Wu Y et al. Strategies for the enhancement of IL-21 mediated antitumor activity in solid tumors. Cytokine. 2024;184:156787.
113. Wang Y et al. An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solidtumor microenvironment. Frontiers Immunol. 2019;10:1691.
114. Ponterio E et al. Oncolytic virus and CAR-T cell therapy in solid tumors. Frontiers Immunol. 2024;15:1455163.
115. Štach M et al. Interleukin 21 enhances survival and expansion of CAR T cells Via inhibition of their terminal differentiation during interaction with tumor target cells. Blood. 2018;132(Supplement 1):4545.
116. Slominski RM et al. How cancer hijacks the body’s homeostasis through the neuroendocrine system. Trends in neurosciences. 2023;46(4):263-75.
Aggressive Angiomyxoma of Vulva with Term Pregnancy: A Case Report
Authors: Farah Hassan Khan,1 Aisha Shahid,1 Memoona Kashaf,1 Komal Khan,2 *Saba Ambreen Aftab,2 Zarafshan Rehan1
1. Jinnah Postgraduate Medical Center (JPMC), Jinnah Sindh Medical University, Karachi, Pakistan
2. Ziauddin Medical College, Ziauddin University, Karachi, Pakistan *Correspondence to sabaambreen24@gmail.com
Disclosure: Written informed consent was obtained from the patient for the publication of this case report and accompanying images.
Received: 27.08.25
Accepted: 02.11.25
Keywords: Angiomyxoma, pregnancy, surgical excision, vulval mass.
Citation: EMJ Oncol. 2025;13[1]:154-157. https://doi.org/10.33590/emjoncol/HCXR8243
Abstract
Aggressive angiomyxoma (AA) is a rare, locally invasive, benign tumour that mostly affects women of reproductive age. Its occurrence during pregnancy is extremely rare and may delay diagnosis due to its painless, gradual growth and resemblance to common vulvovaginal lesions. The authors report a 23-year-old primigravida who presented in active labour with a large vulval mass. The mass had progressively enlarged during pregnancy, causing discomfort in daily activities. An emergency lower segment caesarean section was performed after excision of the 30×21×10 cm mass. Histology confirmed AA. Postoperative recovery was uneventful, and the patient was discharged with plans for long-term follow-up. This case emphasises the clinical importance of AA during pregnancy, its potential to mimic other vulval lesions, and the necessity for surgical management and histopathological diagnosis, along with close follow-up due to its high risk of local recurrence.
Key Points
1. Aggressive angiomyxoma is a rare, locally infiltrative tumour that can enlarge significantly during pregnancy and mimic benign vulval lesions, delaying diagnosis and increasing the risk of obstructed labour and complex surgical intervention.
2. This case report describes a 23-year-old primigravida who presented in active labour with a giant vulval aggressive angiomyxoma requiring emergency caesarean section and multidisciplinary surgical excision of a 30×21×10 cm mass.
3. Clinicians should maintain a high index of suspicion for aggressive angiomyxoma in enlarging vulval masses during pregnancy, as optimal management requires complete excision, histopathological confirmation, and long-term follow-up due to high recurrence rates.
INTRODUCTION
Aggressive angiomyxoma (AA) is a rare, slow-growing, but locally infiltrative mesenchymal tumour primarily affecting women of reproductive age and most commonly affecting the vulva, perineum, and pelvis.1 Its incidence in pregnancy is exceptionally uncommon, with only a few occurrences reported in literature. These tumours can develop to significant proportions and are usually distinguished by their painless, soft, gelatinous nature. They frequently resemble benign lesions such as Bartholin gland cysts, lipomas, or fibromas.1 Malignant transformation and distant metastasis are exceedingly rare during pregnancy, but must be investigated.2 In this context, the authors report a rare case of a giant vulval AA discovered during pregnancy, which resulted in obstructed labour and necessitated an emergency caesarean section followed by surgical excision of the mass.
CASE PRESENTATION
A 23-year-old primigravida at 37+3 weeks of gestation was brought to a gynaecological emergency room with complaints of labour pains and the presence of a large obstructing vulval mass. She noticed a pea-sized mass in the vulva during her first trimester and, over the duration of her pregnancy, the mass progressively enlarged in size, beginning to interfere with her daily activities. Notably, she had a similar vulval swelling 2 years prior, which resolved with oral medication. This recurrence was temporally associated with hot-wax hair removal, after which she opted not to seek treatment due to concerns regarding fetal harm. As the patient presented directly to the emergency department in active labour, there was no opportunity to perform any advanced imaging such as CT or MRI. Only a routine obstetric ultrasound was undertaken to assess fetal well-being. Given the obstructed labour and need for urgent intervention, surgery could not be delayed for further imaging. Additionally, the patient was unbooked, limiting the availability of prior antenatal investigations. The patient had been prescribed progesterone support
by her local general practitioner due to suspected preterm labour, as she reported experiencing intermittent lower abdominal pain during pregnancy. She was also given multiple courses of antibiotics by the same practitioner, prescribed prophylactically in anticipation of a possible underlying infection, although no confirmed diagnosis was documented.
She was haemodynamically stable at the time of admission. Abdominal examination revealed a term-sized uterus consistent with the symphysio-fundal height, with normal fetal heart sounds and movements. Vaginal examination revealed a 5 cm dilated cervix with a soft, anterior os, but significant obstruction at the introitus due to the vulval mass. In view of the obstructed labour and patient preference for mass removal, a decision was made to proceed with emergency lower segment caesarean section and concurrent excision of the mass under general anaesthesia. Intraoperatively, the uterus showed signs of obstructed labour (Bandl’s ring was observed). A healthy 3 kg male infant was delivered with good Appearance, Pulse, Grimace, Activity, and Respiration (Apgar) scores.
The mass, measuring 30×21×10 cm (Figure 1), was excised by a combined gynaecology and plastic surgery team using sharp and blunt dissection. It was noted to have both cystic and solid components. Excess vulval skin was excised, and the defect was closed primarily with adjacent skin flaps using sutures. A closed-suction drain was placed, and an aseptic dressing was applied. The total surgery time spanned approximately 4–5 hours.
Postoperatively, the patient was monitored in the ICU on ventilatory support due to the prolonged duration of anaesthesia, but was successfully extubated and transferred to the general ward within 24 hours. She started on broad-spectrum antibiotics, iron supplementation, and was advised on wound care with sitz baths using warm water, hydrogen peroxide, and povidoneiodine. Iron supplementation was continued throughout the patient’s pregnancy, as empirical iron therapy is routinely practised in the authors' region due
to the high prevalence of iron deficiency among pregnant women. Although commonly administered, the patient herself was not anaemic. She was discharged on the fifth postoperative day with instructions for follow-up on Day 7 for suture and drain removal.
Histopathological examination of the excised mass confirmed the diagnosis of AA, a rare, locally infiltrative mesenchymal tumour known for its recurrence rate in women of reproductive age. The patient was counselled extensively about the benign, yet locally aggressive, nature of the tumour; the potential for recurrence; and the need for long-term surveillance, including routine imaging and clinical evaluation.
DISCUSSION
AA is a rare, benign, hormonally sensitive mesenchymal tumour that primarily affects women of reproductive age.3 While women of reproductive age account for the majority of cases, there are occasional occurrences in premenstrual women and children. This
can be explained by the tumour hormone’s responsiveness, as its growth is promoted by oestrogen and progesterone.4 In the present case, the patient had received progesterone support during pregnancy, which may have contributed to the tumour’s growth due to its hormonal sensitivity. The tumour is primarily found in the pelvic and perineal area. Uncommon locations include the orbit, larynx, liver, and lung.5 The tumour may occur in male patients, in sites such as the inguinal canal and scrotum.6 The female to male ratio is 6:1.6 Although its pathophysiology is unclear, the high mobility protein isoform C, which is associated with DNA transcription, has recently been shown to have atypical manifestations as a result of a translocation at the level of chromosome 12.7
Typically, AA manifests as a soft, compressible, progressive vulvar or perineal tumour. It displaces pelvic organs and is encapsulated, extending into the paravaginal, pararectal, or retroperitoneal regions. It looks grey, glossy, and uniform, with potential bleeding and necrosis, on gross examination. Under a microscope, it has a loose myxoid stroma with mesenchymal
Figure 1: Vulval mass measuring 30x21x10 cm at 37+3 weeks gestation.
cells that are stellate and spindle shaped, distinctive, irregularly distributed blood vessels and peripheral finger-like projections. Mitoses rarely occur. Smooth muscle actin, desmin, and vimentin show positive immunohistochemistry, which suggests fibroblastic or myofibroblastic origin.8
It is frequently misdiagnosed as a vulvar abscess, Bartholin’s cyst, vaginal prolapse, vaginal mass or polyp, pelvic floor hernia, and more on clinical inspection.9 In this case, multiple empirical courses of antibiotics were prescribed by local practitioners under the assumption of an infectious aetiology, delaying accurate diagnosis and definitive management. To determine the tumour’s extent and schedule surgical removal appropriately, preoperative imaging is crucial. It appears as a cystic or hypoechoic mass on ultrasound.10 On MRI and CT, the bulk exhibits a clear characteristic of layered and swirling tissue with alternating low and high signal strands, which represent the tumour’s myxoid and fibrous septa.11 These imaging results make it easier to accurately estimate the depth and margins of the tumour, which is essential when planning a wide local excision to decrease the likelihood of it reoccurring.12 The infiltrative nature of the tumour and its proximity to important tissues make its total excision difficult.12
References
1. Bagga R et al. Aggressive angiomyxoma of the vulva in pregnancy: a case report and review of management options. MedGenMed. 2007;9(1):16.
2. Geng J et al. Aggressive angiomyxoma: an unusual presentation. Korean J Radiol. 2012;13(1):90-3.
3. Bigby SM et al. Aggressive angiomyxoma [corrected] of the female genital tract and pelvis— clinicopathologic features with immunohistochemical analysis. Int J Gynecol Pathol. 2011;30(5):505-13.
4. Orfanelli T et al. A case report of aggressive angiomyxoma in pregnancy: do hormones play a role? Case Rep Obstet Gynecol. 2016;2016:6810368.
5. Gaurav A et al. Aggressive angiomyxoma of the vulva - a rare entity: case report and review of
Gonadotropin-releasing hormone agonists (e.g., triptorelin, leuprolide) and antagonists (relugolix) target oestrogen and progesterone receptors, and have demonstrated potential in preoperative tumour volume reduction, residual disease treatment, and recurrence management.13 In cases of resistance, aromatase inhibitors may also be helpful when used with gonadotropin-releasing hormone agonists.14
CONCLUSION
AA is a rare, hormonally sensitive, and locally infiltrative mesenchymal tumour that presents significant diagnostic and management challenges, especially during pregnancy. Delays in detection are frequently caused by its benign histology, slow growth, and clinical resemblance to common vulvovaginal lesions. This case highlights the importance of maintaining a high index of suspicion for AA in women presenting with enlarged vulvar masses, especially during pregnancy. Hormonal therapy can help reduce the size of the tumour and manage its recurrence. For optimal management, timely imaging, multidisciplinary surgical planning, and histological confirmation are crucial.
literature. Int J Reprod Contracept Obstet Gynecol. 2020;9(6):2605-9.
6. Das BP et al. An aggressive angiomyxoma of vulva - a rare entity – a case report. J Midlife Health. 2016;7(3):140-3.
7. Rezai S et al. Aggressive angiomyxoma of the vulva in a teenager, a case report and review of literature. Obstet Gynecol Int J. 2016;4(6):192-6.
8. McCluggage WG. Aggressive angiomyxoma of pelvic parts exhibits oestrogen and progesterone receptor positivity. J Clin Pathol. 2000;53(8):603-5.
9. Chen H et al. Clinicopathological features and differential diagnosis of aggressive angiomyxoma of the female pelvis. Medicine. 2017;96(20):e6820.
10. Patel T et al. Unveiling a rare case of a giant aggressive angiomyxoma of the perineum: diagnostic insights from T2-weighted MRI. Cureus. 2025;DOI: 10.7759/cureus.78971.
11. Cingoz M et al. Radiologic imaging findings of pelvic aggressive angiomyxoma correlated with surgical and pathological features. Egypt J Radiol Nucl Med. 2021;52(1):259.
12. Fucà G et al. Treatment outcomes and sensitivity to hormone therapy of aggressive angiomyxoma: a multicenter, international, retrospective study. Oncologist. 2018;24(7):e536-41.
13. Saito A et al. Recurrent aggressive angiomyxoma that responded to the gonadotropin-releasing hormone (GNRH) antagonist relugolix. Cureus. 2025;17(3):e81270.
14. Aguilar-Frasco J et al. Aggressive angiomyxoma: giant recurrence successfully treated with wide excision and adjuvant therapy with GnRH analogue. BMJ Case Rep. 2018;11(1):e226973.
If you like it,why not share it...

Share with a colleague today