
Edición I Volumen II Año 2024
Lo que se desconoce no se reconoce, y se ignora por completo, porque simplemente “no me afecta, no es conmigo”. Estas palabras que suenan tan peyorativamente, engloban la realidad de muchísimas personas que viven con una enfermedad neuromuscular y desmielinizante y que luchan diariamente por tener una vida con calidad.
Esta Herramienta Interactiva, es u homenaje a todos aquellos que parecen no tener voz ante muchas instancias, pero claman por tener un trato digno, acceso y sobre todo humanidad. Porque la enfermedad no los define, ni minimiza esa fuerza interior que sirve de aliciente para luchar y seguir adelante, por ellos, por sus familias, por sus sueños ...

interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis
Atrofia Muscular
Herramienta
y
Espinal profesionalesdelsalvador.com/asenm
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Contenido
Capítulo III
Neuromielitis Óptica
• ¿Qué es?
• ¿Por qué surge?
• Manifestaciones clínicas
• Banderas rojas, ¿cómo diferenciarlas?
• Detección y proceso diagnóstico
• Tratamiento
• Manejo y seguimiento
• Cuidados
Capítulo IV
Atrofia Muscular Espinal
• ¿Qué es?
• ¿Por qué surge?
• Manifestaciones clínicas
• Banderas rojas, ¿cómo diferenciarlas?
• Detección y proceso diagnóstico
• Tratamiento
• Manejo y seguimiento
• Cuidados
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Presentación
Porque todo sueño comienza con una simple idea, con mucha gratitud, optimismo y esperanza, les presentamos la primera edición de la “Herramienta Interactiva para el diagnóstico, Tratamiento, Manejo y Seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal producida por nuestra organización. Con información de primera mano recopilada a partir de diversas entrevistas y foros con diversos médicos especialistas de El Salvador y de otros países de América Latina, esta herramienta recopila material acorde a nuestras realidades y necesidades, pero también refleja el expertise y trayectoria de muchos especialistas que han adoptado nuestro sueño, como propio, al facilitar mecanismos y herramientas que acercan el conocimiento de las enfermedades neuromusculares y desmielinizantes a la región, visibilizando la existencia de estas con el firme propósito de alcanzar una detección pronta y un diagnóstico oportuno. Porque en la medida que logremos una mayor capacidad resolutiva de nuestros niveles de atención en salud, mejoraremos la calidad de vida de todos los pacientes y por ende de sus seres queridos.
Porque lidiar con una patología de esta índole no solo afecta a quien carga con ella sino a todo su entorno familiar a nivel social, económico y sobre todo afectivo. De ahí que nuestra organización estableció como meta no solo empoderar a los pacientes sino ir más allá, a las aulas universitarias, a los centros de salud para difundir y capacitar con evidencia y bases científicas, todo ese conocimiento que permita salvaguardar la vida de los pacientes. Sin duda, una labor titánica pero no imposible de lograr y sobrepasar.
Sabemos que el camino que nos hemos trazado no es fácil pero con el trabajo que hemos venido desarrollando estamos sembrando para el mañana.
De parte de nuestra organización, agradecemos el apoyo de los médicos especialistas: Dr. Daniel Enrique Pereira, neurólogo salvadoreño, médico internista y, presidente de la Asociación Salvadoreña de Neurología; Dra. Ericka López Torres, neuróloga salvadoreña; Dr. Hugo Gálvez, neuropediatra guatemalteco, especialista en enfermedades neuromusculares; Dr. Carlos Ortes González, neuropediatra salvadoreño, especialista en enfermedades neuromusculares; Dr. Andrés Nascimento Osorio, neuropediatra venezolano, especialista en enfermedades neuromusculares y al Dr. Alfonso Gutiérrez, neuropediatra costarricense, especialista en enfermedades neuromusculares, quienes abrieron un espacio en sus ajetreadas agendas para poder recopilar toda la información necesaria para que esta Herramienta Interactiva sea hoy una realidad.
Mil gracias y en Asenm seguimos construyendo ...
Ana Silvia Barahona Rosales
Presidente
Asenm El Salvador
+506 2236-6610
506+8814-8891
www.alteacomunicacion.com
Editora General Msc. Ma. Martha Mesén Cepeda mmesen@alteacomunicacion.com
Redacción e investigación
Licda. Claudia Pineda Herrera cpineda@alteacomunicacion.com
Nadia Aguilar Chinchilla naguilar@alteacomunicacion.com
Coordinación de Diseño Natalia Valverde Vega nvalverde@alteacomunicacion.com
Gestión de proyecto
Mtr. Ronny Garro Ureña rgarro@alteacomunicacion.com
Ana
+503 7769 6656
asenm.elsalvador19@gmail.com
profesionalesdelsalvador.com/asenm
Junta Directiva Presidente y Directora Ejecutiva
Barahona
Silvia
Rosales
Agradecimiento a los médicos especialistas






Neuropediatra
Neuropediatra
Dr. Daniel Enrique Pereira Contreras Neurólogo
Dr. Carlos Ortez González
Dra. Ericka López Torres Neuróloga
Dr. Andrés Nascimento Osorio
Dr. Hugo Gálvez Quiñones Neuropediatra
Dr. Alfonso Gutiérrez Mata Neuropediatra
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Introducción
Esta Herramienta Interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal, constituye un reto personal y profesional para nuestra organización, dado que es la primera vez que una organización de pacientes, de América Central va más allá de su labor de empoderamiento y de divulgación, para facilitar un documento que involucre a diversos especialistas que de una forma llana y didáctica abordan cada capítulo y temática de manera comprensible para todos. Porque el conocimiento empodera y debe de estar al alcance de nuestros países latinoamericanos, esta primera edición constituyó todo un desafío para nosotros. No solo por lograr los espacios con cada especialista participante durante las entrevistas o con sus participaciones durante el I Foro Centroamericano que realizamos sino en el procesamiento de la información.
Sin duda, esta primer edición, es un gran paso, en todo lo que hemos venido construyendo desde Asenm El Salvador y que nos enorgullece al convertirnos en un generador de espacios para el debate, el intercambio y la retroalimentación de conocimiento en el área neurológica de todo lo que involucra las patologías de origen neuromuscular.
De ahí que cada capítulo lleva un hilo conductor, comenzando por qué son las enfermedades neuromusculares y
desmielinizantes, hasta abordar los cuidados, manejo y tratamiento de cada una de las patologías que como organización acogemos.
Asimismo en cada uno de los ocho capítulos que complementan esta guía encontrarán audiovisuales que los llevarán al criterio del especialista en mención.
De esta manera se plasma cada una de las manifestaciones y características clínicas de estas enfermedades, que son de vital importancia poderlas detectar en la primera infancia (en muchos de los casos) con el fin de mejorar la calidad de vida de quien lo padece así como de sus familias.
En Asenm El Salvador estamos comprometidos por seguir desarrollando más espacios que abran el conocimiento. Agradecemos a cada uno de los pacientes, parientes y amigos que han confiado en nuestro trabajo y a su vez en los profesionales de la salud que nos han brindado su apoyo en todo momento. Así como a la casa farmacéutica Roche quien por medio de su programa “Juntos Mejor” hizo posible que pudiéramos desarrollar esta Herramienta Interactiva.
Gracias infinitas a todos esos valientes que son parte de nuestra organización y nuestra razón de seguir luchando.
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Capítulo III
La oscuridad que ciega: una mirada médica a la Neuromielitis Óptica
9 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Neuromielitis Óptica (NMO)
¿Qué es?
La Neuromielitis Óptica (NMO) es una enfermedad autoinmune rara que afecta principalmente el sistema nervioso central y, en particular, la médula espinal y el nervio óptico. La enfermedad se caracteriza por la presencia de anticuerpos anti-acuaporina 4 (anti-AQP4), que se unen a las células nerviosas y provocan su destrucción.
¿Por qué surge?
Esta patología se produce cuando el sistema inmunológico del cuerpo ataca y destruye la mielina, una sustancia que cubre las fibras nerviosas en la médula espinal y el cerebro, y los axones del nervio óptico.
La NMO es una enfermedad rara y se estima que su
prevalencia varía en diferentes partes del mundo, aunque se cree que es más común en Asia y América Latina que en otras partes del mundo. Según la literatura científica, la prevalencia de la NMO oscila entre 0,52 y 4,4 por cada 100.000 habitantes en todo el mundo.
La NMO se caracteriza por la presencia de anticuerpos anti-acuaporina 4 (anti-AQP4), que se unen a las células nerviosas y provocan su destrucción. La destrucción de las células nerviosas puede causar una amplia gama de síntomas, que incluyen pérdida de la visión, dolor en la espalda y extremidades, debilidad muscular, problemas de coordinación y trastornos del habla y la deglución.
La inflamación que ocurre en la NMO también puede afectar otros órganos, como los riñones y los pulmones. En algunos casos, la NMO puede ser difícil de diferenciar de otras
10 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
enfermedades autoinmunitarias, como la esclerosis múltiple (EM), pero la presencia de anticuerpos antiAQP4 es un indicador clave de la NMO.
La NMO puede tener una amplia variedad de síntomas neurológicos, que se producen cuando el sistema inmunológico del cuerpo ataca y destruye la mielina. El diagnóstico temprano y el tratamiento adecuado son importantes para prevenir las complicaciones y mejorar la calidad de vida de los pacientes con NMO.
Manifestaciones clínicas
No se conoce con exactitud qué causa la NMO, aunque se cree que puede ser una combinación de factores genéticos y ambientales. Las manifestaciones clínicas de la NMO pueden variar, pero incluyen pérdida de la visión, dolor en la espalda y extremidades, debilidad muscular, problemas de coordinación y trastornos del habla y la deglución.
11 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal

12 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Las banderas rojas de la NMO incluyen la presencia de síntomas visuales graves, como la pérdida completa de la visión, la presencia de dolor intenso en la espalda o en los brazos o piernas, y la aparición repentina de síntomas neurológicos. La NMO también puede presentarse junto con otras enfermedades autoinmunitarias, como el lupus y la esclerosis múltiple.
Es importante que los profesionales de la salud sepan
cómo detectar un posible caso de Neuromielitis Óptica (NMO) para poder referir adecuadamente y en
tiempo al paciente adonde un neurólogo o a un centro especializado.
Es importante destacar que la NMO es una enfermedad rara y que puede ser difícil de diagnosticar debido a la amplia variedad de síntomas que presenta y a la falta de conocimiento general sobre la enfermedad. Por lo tanto, es fundamental trabajar de manera conjunta con neurólogos y otros especialistas para hacer un diagnóstico preciso y brindar el tratamiento adecuado a los pacientes.
13 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Guía de diagnóstico para la Neuromielitis Óptica:
Evaluación clínica e identificación de la enfermedad
1. Reconocimiento de los síntomas: La NMO se caracteriza por una variedad de síntomas neurológicos, incluyendo dolor en la espalda y extremidades, debilidad muscular, problemas de coordinación y trastornos del habla y la deglución. Además, la pérdida de la visión en uno o ambos ojos es un síntoma dis�n�vo de la NMO que no se encuentra en otras enfermedades autoinmunitarias, como la Esclerosis Múl�ple (EM). De ahí que se deba prestar atención a la presentación de estos síntomas en sus pacientes, y si se sospecha que hay una causa neurológica, considerar la posibilidad de la NMO.
2. Antecedentes médicos: Es importante recopilar la historia médica completa de los pacientes, ya que hay ciertos factores que pueden aumentar el riesgo de desarrollar NMO, como la presencia de otras enfermedades autoinmunitarias, la presencia de ciertos an�cuerpos en la sangre, la exposición a ciertas infecciones virales, entre otros.
3. Pruebas de laboratorio: Para detectar la NMO, se pueden realizar pruebas de laboratorio para detectar la presencia de an�cuerpos an�-AQP4 en la sangre o en el líquido cefalorraquídeo (LCR). Estos an�cuerpos se encuentran en la mayoría de los pacientes con NMO y son una herramienta diagnós�ca clave.
4. Imágenes cerebrales: Las imágenes de resonancia magné�ca (IRM) del cerebro y la médula espinal son una herramienta diagnós�ca importante para la NMO. Las imágenes de la IRM pueden mostrar lesiones caracterís�cas en la médula espinal y en el cerebro, que pueden ayudar a confirmar el diagnós�co.
Fuente: National Institute of Neurological Disorders and Stroke. Neuromyelitis Optica Fact Sheet. https://www.ninds.nih.gov/Disorders/PatientCaregiver-Education/Fact-Sheets/Neuromyelitis-Optica-Fact-Sheet Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007 Sep;6(9):805-15. Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:8.
14 Capítulo 3
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Detección y proceso diagnóstico
El diagnóstico de la NMO se realiza mediante una combinación de pruebas de laboratorio y clínicas, que incluyen la detección de anticuerpos anti-AQP4 en el suero o en el líquido cefalorraquídeo y la realización de una resonancia magnética de la médula espinal y el cerebro. El tratamiento de la NMO incluye el uso de medicamentos inmunosupresores para reducir la inflamación y prevenir las recaídas, así como terapias de rehabilitación para ayudar a mejorar la función muscular y neurológica.
El proceso de detección y diagnóstico de la NMO implica una serie de pruebas clínicas y de laboratorio, incluyendo
una resonancia magnética de la médula espinal y el cerebro, así como la detección de anticuerpos anti-AQP4 en el suero o en el líquido cefalorraquídeo.
El tratamiento y manejo de la NMO incluyen la administración de medicamentos para reducir la inflamación y la supresión del sistema inmunológico, así como el uso de terapias de rehabilitación para ayudar a mejorar la función muscular y neurológica. También se deben tomar medidas para prevenir las recaídas, como evitar el estrés y el exceso de esfuerzo físico y llevar un estilo de vida saludable.
Fuentes y referencias:
Dra. Erika López Torres, médico neuróloga, del Hospital Nacional Rosales en San Salvador
Dr. Enrique Daniel Pereira Contreras, médico neurólogo salvadoreño, máster en trastornos del movimiento. Actualmente es el Presidente de la Asociación de Ciencias Neurológicas de El Salvador.
National Institute of Neurological Disorders and Stroke. Neuromyelitis Optica Fact Sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-
Education/Fact-Sheets/Neuromyelitis-Optica-Fact-Sheet
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189.
Kim SH, Kim W, Li XF, Jung IJ. Clinical spectrum of neuromyelitis optica in Korea. J Clin Neurol. 2011;7(4):161-167.
National Institute of Neurological Disorders and Stroke. Neuromyelitis Optica Fact Sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-
Education/Fact-Sheets/Neuromyelitis-Optica-Fact-Sheet
Kim SH, Kim W, Li XF, Jung IJ. Clinical spectrum of neuromyelitis optica in Korea. J Clin Neurol. 2011;7(4):161-167.
Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:8.
15 Capítulo 3

Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Capítulo IV
Cuando los músculos pierden su
VOZ: ATROFIA MUSCULAR ESPINAL
17 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Atrofia Muscular Espinal (AME)
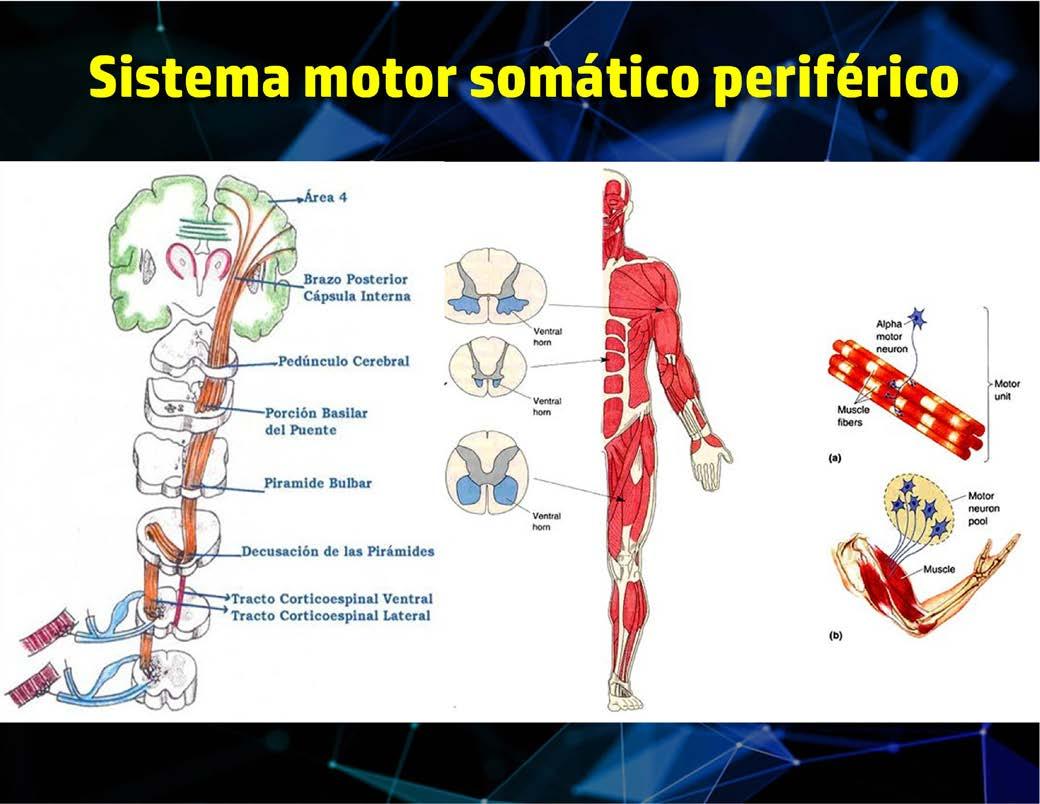
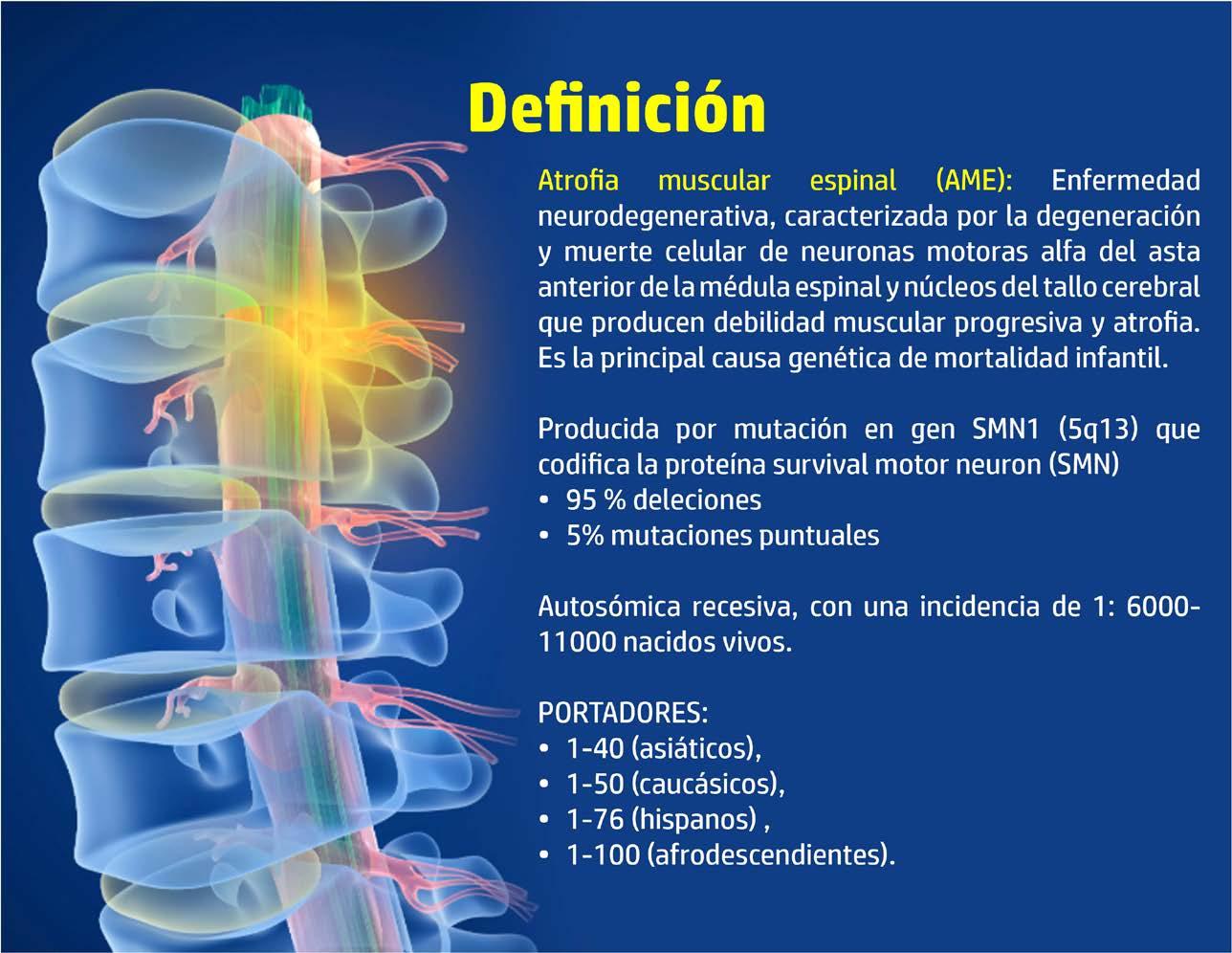
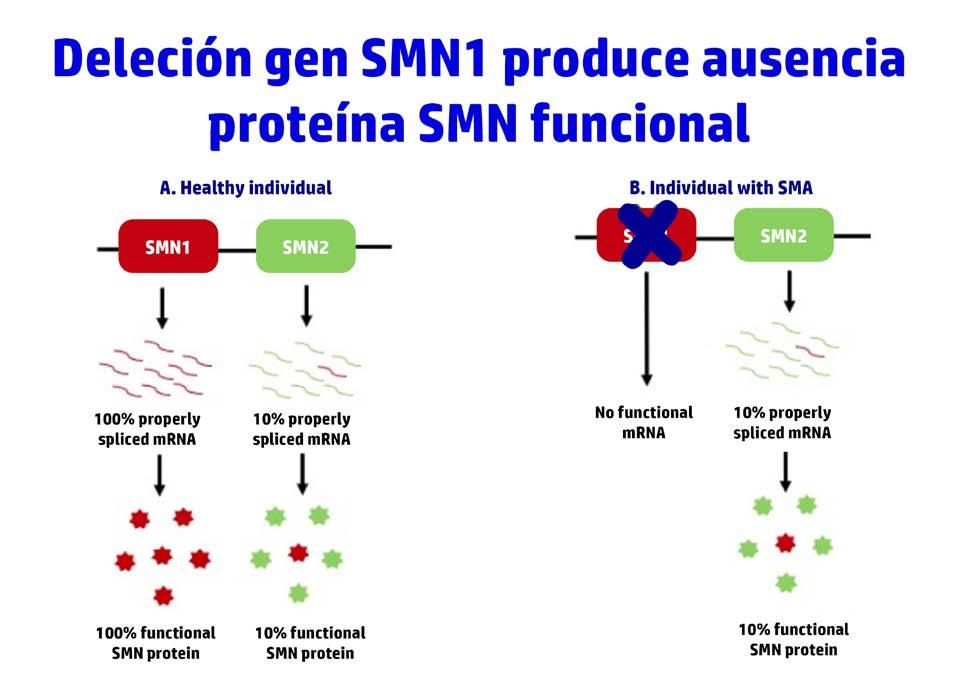
La Atrofia Muscular Espinal (AME) se presenta como una enfermedad neurodegenerativa que se caracteriza por la degeneración y fallecimiento de las neuronas motoras alfa, localizadas en el asta anterior de la médula espinal y en los núcleos del tallo cerebral. Estas neuronas son fundamentales para el control muscular y, cuando sucede su pérdida, se produce debilidad y atrofia muscular progresiva. Sin lugar a dudas, la AME se ha consolidado como la principal causa genética de mortalidad en la infancia.
Este gen se encarga de la producción de una proteína vital conocida como SMN (Survival of Motor Neuron), la cual juega un papel esencial en la supervivencia y el mantenimiento de las células nerviosas encargadas de controlar los músculos voluntarios. En la gran mayoría de los pacientes con AME, se detecta una deleción del gen SMN1, lo que implica una producción insuficiente de la proteína SMN. La cantidad de proteína SMN generada por el gen SMN2, que es una copia modificada del gen SMN1, es determinante en la gravedad de la enfermedad.
La AME presenta una incidencia que oscila entre 1 cada 6000 a 11000 nacimientos vivos. Esta cifra refleja la relevancia clínica y el impacto que esta enfermedad tiene en la salud de la población infantil.
18 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Al ser una enfermedad genética autosómica recesiva, significa que se hereda un gen defectuoso de cada progenitor.
Fuente: Dr. Alfonso Gutiérrez Mata, neuropediatra costarricense
CLICK PARA VER VIDEO 19 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
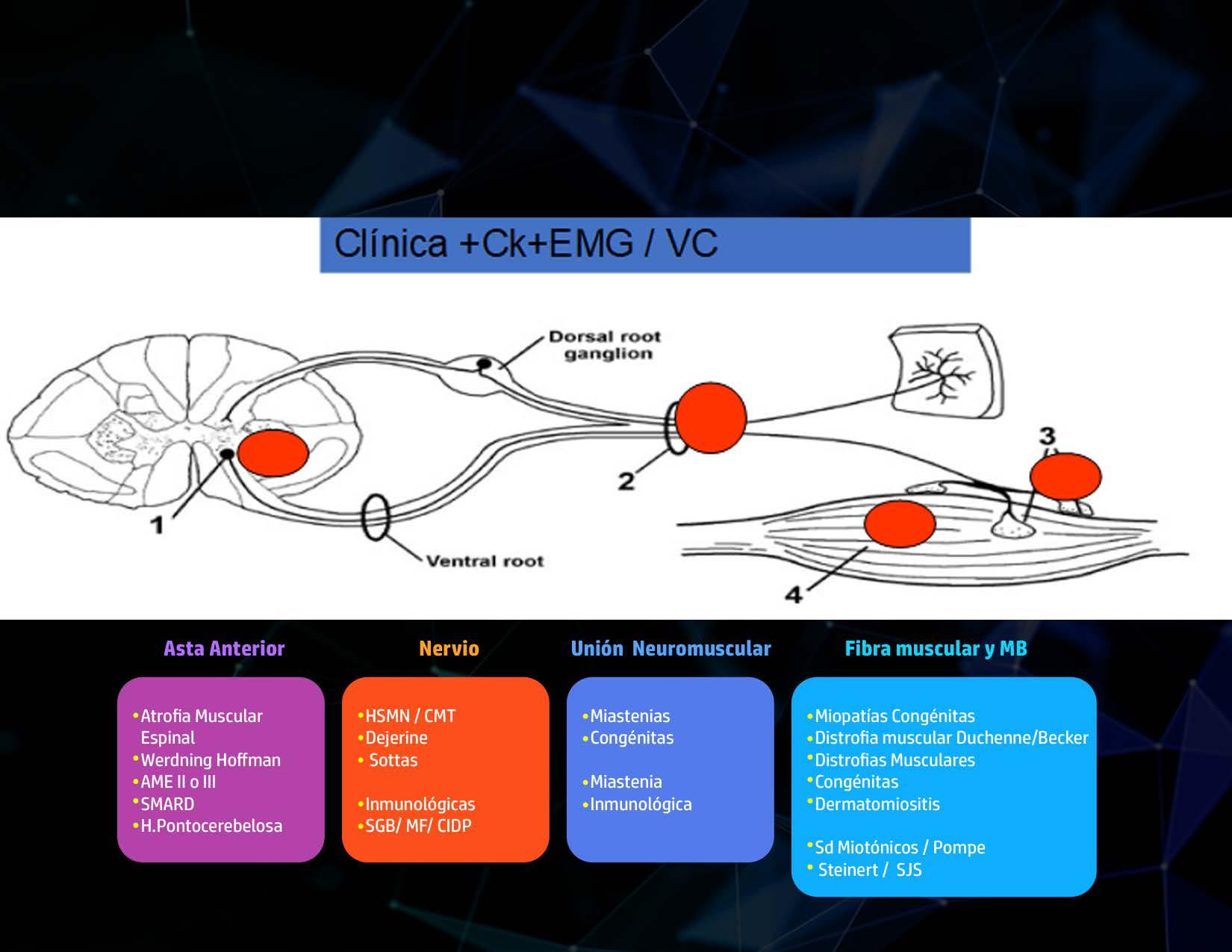
Principales desórdenes de la Unidad
Motora Debemos intentar ubicar el defecto
20 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
CLICK PARA VER VIDEO 21 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Espectro Clínico
La Atrofia Muscular Espinal se clasifica en cuatro tipos principales, según la edad de inicio y la gravedad de los síntomas:
AME tipo 1
(Werdnig-Hoffmann): es la forma más grave y comienza en los primeros meses de vida. Los pacientes afectados tienen dificultades para respirar, tragar y moverse. La mayoría no puede sentarse sin ayuda y no sobreviven más allá de los primeros años de vida.
AME tipo 2: comienza en la infancia temprana o tardía. Los pacientes logran mantener sedestación autónoma pero no logran caminar.
AME tipo 3
(KugelbergWelander): comienza en la infancia tardía o en la adolescencia. Los pacientes logran caminar aunque con dificultades e incluso pueden en la evolución perder la capacidad de caminar de manera autónoma.
AME tipo 4:
Los pacientes manifiestan debilidad distal generalmente leve.
La detección temprana es un eje clave para mejorar la historia natural de la AME.
22 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Manifestaciones clínicas
Entre las manifestaciones clínicas más comunes se encuentran la hipotonía o falta de tono muscular, la debilidad generalizada, la disminución o ausencia de los reflejos osteotendinosos, las fasciculaciones, la
dificultad para respirar y tragar, así como la dificultad para alcanzar los hitos motores del desarrollo, como sentarse o caminar.
Es importante tener en cuenta que estas manifestaciones pueden variar en su presentación y gravedad dependiendo del tipo de AME.
CLICK PARA VER VIDEO 23 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Por ejemplo en la Atrofia Muscular Espinal Tipo 1 (AME proximal infantil o enfermedad de WerdnigHoffmann) es la forma más grave y se presenta en el período neonatal o en los primeros meses de vida. Los principales rasgos clínicos pueden incluir:
Hipotonía axial y periférica (tono muscular disminuido) desde el nacimiento.
Debilidad muscular progresiva y atrofia, especialmente en los músculos proximales (más cercanos al centro del cuerpo), como los músculos del cuello, hombros, caderas y muslos.
Dificultad o incapacidad para mantener la cabeza erguida o sentarse sin soporte.
Ausencia de habilidad para caminar o moverse de forma independiente.
Problemas respiratorios significa�vos debido a la debilidad de los músculos respiratorios, lo que puede requerir el uso de soporte ven�latorio.
Pronós�co desfavorable con una esperanza de vida limitada, aunque los avances en la atención médica han mejorado la supervivencia y la calidad de vida, de ahí la importancia de la detección temprana y oportuna.
En la AME tipo 2 una de las características clínicas es la postura cifótica cuando los pacientes se sientan. La postura cifótica se refiere a una curvatura excesiva de la columna vertebral en la región torácica, lo que provoca que la espalda se arquee hacia atrás. Esta postura puede ser el resultado de la debilidad muscular en los músculos extensores del tronco y en los músculos posturales que sostienen la columna vertebral.
La debilidad muscular progresiva en el AME tipo 2 también afecta a otros grupos musculares, como los músculos del cuello, los hombros, las caderas y los muslos. A medida que la enfermedad avanza, los pacientes pueden experimentar dificultades para levantar objetos pesados, realizar movimientos finos de las manos además de presentar debilidad en los músculos respiratorios, lo que puede llevar a problemas respiratorios.
Además de la debilidad muscular, pueden presentar otros síntomas, como hipotonía (tono muscular disminuido), fasciculaciones (movimientos musculares involuntarios) y retracciones articulares (contracturas musculares que limitan la movilidad de las articulaciones).
24 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Es importante destacar que la progresión de los síntomas puede variar entre los pacientes con AME tipo 2. Algunos niños(as) pueden experimentar una progresión más lenta de los síntomas y mantener la capacidad de mantenerse sentado sin apoyo, mientras que otros pueden experimentar una progresión más rápida y una mayor discapacidad.
En el caso de la AME tipo 3 (enfermedad de KugelbergWelander) es una forma más leve que se manifiesta en la infancia o en la edad escolar.
Las características clínicas típicas son:
Debilidad y atrofia muscular, que generalmente afecta a los músculos proximales.
Habilidades motoras preservadas inicialmente, como la capacidad para caminar, pero con un deterioro gradual de la función motora con el �empo.
Dificultades para correr, saltar o subir escaleras
25 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
En el caso de la AME tipo 4 (AME adulta de inicio tardío o enfermedad de adulto) es la forma más leve de la AME y suele presentarse en la edad adulta.
A diferencia de los otros tipos de AME, el inicio de los síntomas ocurre después de los 18 años.
Las características clínicas del AME tipo 4 pueden variar significativamente de un paciente a otro.
1. Debilidad
muscular: los pacientes con AME �po 4 pueden experimentar debilidad y atrofia muscular progresiva, afectando principalmente los músculos proximales, como los músculos del muslo y la cadera. Esta debilidad muscular puede dificultar ac�vidades como levantarse de una silla o subir escaleras.
Es importante destacar que las características clínicas de la AME tipo 4 pueden variar ampliamente entre los pacientes, incluso dentro de la misma familia. Por lo tanto, el seguimiento médico especializado y una evaluación individualizada son fundamentales para establecer un diagnóstico preciso y un plan de tratamiento adecuado.
3. Problemas respiratorios: en algunos casos, los pacientes con AME �po 4 pueden experimentar debilidad en los músculos respiratorios, lo que puede manifestarse como dificultad para respirar profundamente o para mantener una función respiratoria óp�ma. Sin embargo, los problemas respiratorios en el AME �po 4 generalmente son menos severos que en los �pos más graves de AME.
2. Dificultades en la marcha: la debilidad muscular puede afectar la marcha, provocando una marcha anormal, dificultades para caminar distancias largas o problemas para subir escaleras.
4. Espas�cidad: algunos pacientes con AME �po 4 pueden presentar espas�cidad, que se caracteriza por rigidez y espasmos musculares involuntarios. Esto puede afectar la movilidad y la función de los músculos.
26 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal

Las "banderas rojas" en el diagnóstico de la AME son señales de alarma que sugieren la posibilidad de esta enfermedad. Estas señales incluyen la presencia de debilidad muscular grave y progresiva, ausencia de reflejos osteotendinosos en los miembros inferiores, fasciculaciones musculares (movimientos musculares involuntarios), escoliosis temprana y retraso significativo en el desarrollo motor.
CLICK PARA VER VIDEO 27 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Es fundamental diferenciar la AME de otras condiciones neuromusculares, como la distrofia muscular de Duchenne o las neuropatías periféricas. Para ello, es necesario realizar una evaluación clínica exhaustiva, que incluya una historia clínica detallada, un examen físico minucioso y pruebas complementarias, como análisis genéticos y estudios electrofisiológicos.
Además, reconocer las manifestaciones clínicas y las "banderas rojas" es fundamental para un diagnóstico temprano y preciso. La colaboración interdisciplinaria entre neuropediatras, genetistas y otros especialistas es esencial para brindar un enfoque integral en el manejo y tratamiento de los pacientes con AME.

CLICK PARA VER VIDEO 28 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Diagnóstico
El diagnóstico de la AME se basa en la presentación clínica, la historia familiar y los resultados de pruebas genéticas. Es importante realizar una evaluación neurológica completa, que incluye pruebas de función muscular y reflejos, y buscar signos de atrofia muscular y debilidad muscular simétrica en las extremidades
proximales y distales. También se deben realizar pruebas genéticas para detectar la mutación en el gen SMN1. En algunos casos, se pueden realizar biopsias musculares y pruebas de electromiografía (EMG) para evaluar la función neuromuscular.
CLICK PARA VER VIDEO 29 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Retraso motor
Hipo
Arreflexia
Hipotonía Debilidad
Enfoque diagnóstico
Fasciculaciones
30 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
El punto de partida es una sospecha clínica, lo cual resalta la importancia de la detección temprana en este tipo de patologías. De manera que hay cuatro signos o síntomas clave que revisten gran importancia: hipotonía o debilidad muscular, especialmente en los niños(as) más pequeños que no pueden realizar movimientos antigravitatorios; retraso en las habilidades motoras, como la incapacidad de sentarse a los seis meses y más allá; ausencia de reflejos adecuados al utilizar un martillo de forma correcta para evaluar los reflejos; y la presencia de fasciculaciones en la lengua o los músculos proximales, como los hombros o las mejillas. La presencia de estas características clínicas nos lleva a considerar directamente la posibilidad de una Atrofia Muscular Espinal (AME).
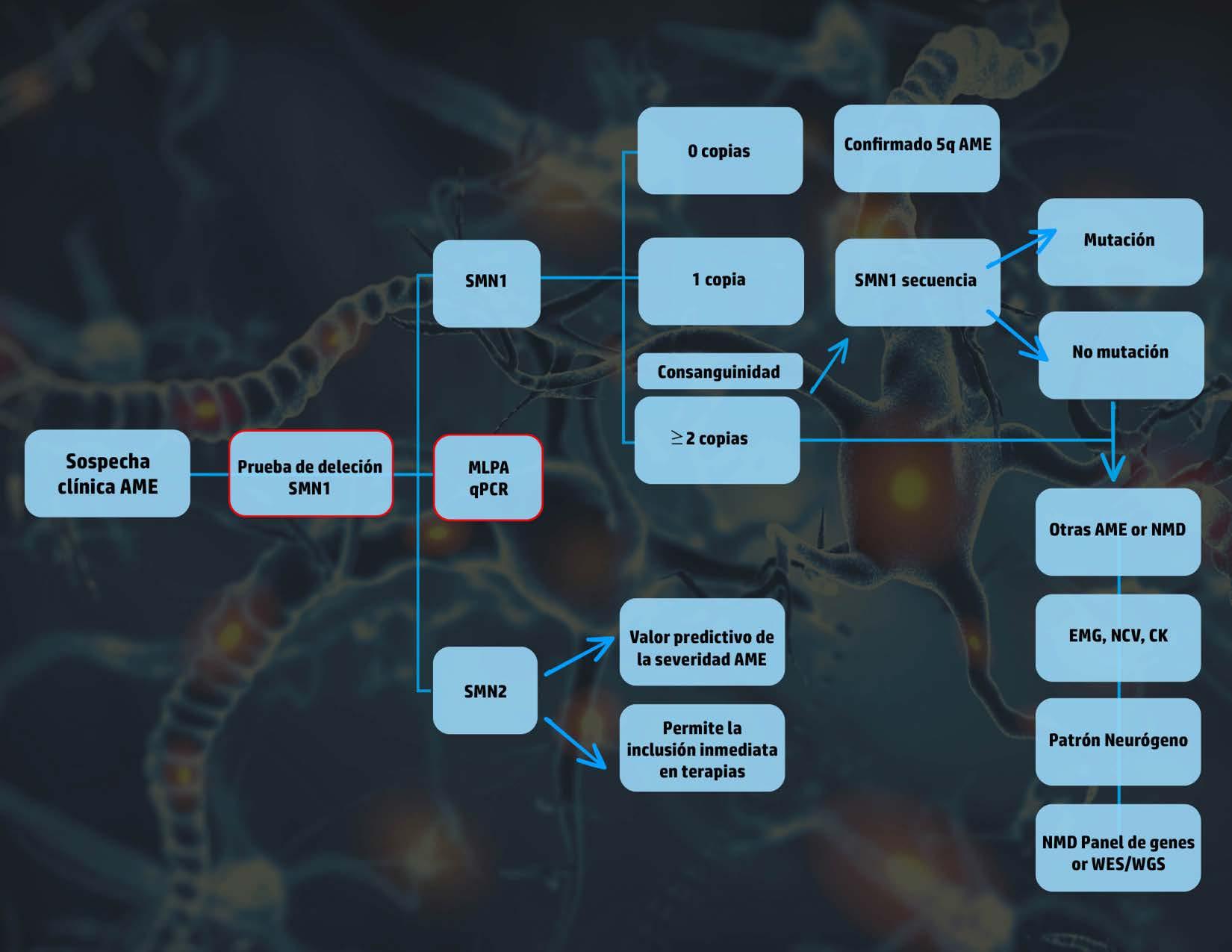
¿Cuál es el algoritmo a seguir?
1. Sospecha clínica
2. Referencia a un centro terciario con capacidad diagnós�ca
3. Confirmación gené�ca
Una vez se tenga una sospecha clínica, es crucial buscar un centro especializado con la capacidad de realizar el diagnóstico adecuado. Este algoritmo ha sido propuesto y validado a nivel internacional.
31 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
32 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Importancia de las escalas funcionales
Las escalas funcionales desempeñan un papel fundamental en el ámbito de las patologías neuromusculares. En un contexto en el que el diagnóstico temprano cobra cada vez más importancia, estas escalas nos brindan una herramienta objetiva para evaluar la evolución clínica de los pacientes. Además, nos permiten observar cómo responden no solo a los nuevos tratamientos, sino también a la gestión integral y atención que cada individuo requiere.
Estas escalas no solo resultan relevantes en el contexto de la Atrofia Muscular Espinal, sino también en otras enfermedades tratables como la enfermedad de Pompe, las miopatías congénitas, las enfermedades mitocondriales, distrofias musculares entre otras. En estos campos se están produciendo importantes avances, y contar con escalas funcionales se vuelve crucial para medir y evaluar los resultados de dichos avances.
Importancia de escalas funcionales para los clínicos
Seguimiento longitudinal de pacientes
Seguimiento de historia natural
Pronós�co
Eficacia de tratamientos
33 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal

CLICK PARA VER VIDEO 34 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Es importante saber que existen diversas escalas funcionales y que deben adaptarse según la condición clínica del paciente y que deben empezar a medirse desde que el paciente se diagnostica para poder dar seguimiento.
La evaluación funcional de los pacientes con AME es esencial para comprender el grado de afectación y el impacto de la enfermedad en su vida diaria. Permite a los médicos y profesionales de la salud diseñar planes de tratamiento personalizados y monitorear la progresión de la enfermedad a lo largo del tiempo.
En el caso de pacientes ambulantes con AME, se evalúa su capacidad para caminar y realizar actividades relacionadas con la movilidad. Se pueden utilizar escalas de evaluación como la Escala de Hammersmith (Hammersmith Functional Motor Scale) y la Escala de Evaluación Funcional para la AME (SMA-FRS) para medir la fuerza muscular, el equilibrio, la coordinación y la habilidad para realizar tareas específicas.
En cuanto a los pacientes no ambulantes con AME, la
evaluación funcional se enfoca en evaluar la capacidad para realizar actividades cotidianas básicas, como el cuidado personal, la alimentación, la comunicación y la interacción social. Se pueden utilizar escalas como la Escala de Evaluación Funcional para la AME en No Ambulantes (SMA-FRS-2) y la Escala de Evaluación Funcional para la AME en No Ambulantes Extendida (SMA-FRS-ER) para evaluar y categorizar las habilidades funcionales de estos pacientes.
Estas escalas de evaluación han sido desarrolladas y validadas por investigadores y expertos en el campo de la AME. Proporcionan una medida estandarizada y objetiva de la función motora y las habilidades cotidianas, lo que facilita la comparación entre pacientes y la monitorización de los cambios a lo largo del tiempo.
Es importante saber que las escalas funcionales deben hacerse de manera periódica y no distantes. Generalmente se realizan cada 6 meses para ver la evolución del paciente. Además, paralelo a ellos se debe realizar el tratamiento con un abordaje integral.
35 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Pulmonar
Manejo integral
Rehabilitación
Neuromuscular
Cuidados agudos
Coordinador
Paciente con AME
Ortopedia
Mercuri E, et al. Neuromuscular Disorder 2018;28(3):103-115
Citado por el Dr. Ortez, en el I Foro Centroamericano sobre AME
Nutrición gastrointes�nal y salud ósea
Familia
Medicación
Afectación de otros órganos
36 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Las guías de manejo deben enfocarse en prevenir las posibles complicaciones asociadas con la AME. Esto implica implementar medidas preventivas, como terapia física y ocupacional para mantener la función motora y prevenir la debilidad muscular, así como terapia respiratoria para prevenir problemas respiratorios y neumonías.
Además, el manejo de la AME debe basarse en el concepto de salud, que busca no solo tratar los síntomas y complicaciones, sino también promover la salud en general del paciente. Esto implica brindar atención integral que aborde aspectos físicos, emocionales y sociales.
Es importante reconocer que la AME es una enfermedad crónica y degenerativa, pero esto no significa que el paciente no pueda mantenerse saludable dentro de su condición. Se deben implementar estrategias que promuevan el bienestar físico, como la actividad física adaptada a sus capacidades, y el manejo adecuado de la alimentación y la nutrición.
Además, es esencial brindar apoyo emocional y psicológico tanto al paciente como a su familia. La AME
puede tener un impacto significativo en la calidad de vida de los pacientes y sus cuidadores, por lo que es importante contar con recursos y servicios de apoyo que ayuden a hacer frente a los desafíos emocionales y promover la salud mental.
El manejo de la AME debe ser llevado a cabo por un equipo multidisciplinario de profesionales de la salud, incluyendo médicos neuropediatras, fisioterapeutas, terapeutas ocupacionales, neumólogos, nutricionistas, psicólogos y trabajadores sociales, entre otros. Este enfoque colaborativo garantiza una atención integral y personalizada para el paciente, abordando todas las áreas relevantes para su bienestar.
El manejo de la AME deben centrarse en evitar complicaciones, promover la salud integral del paciente y garantizar una atención multidisciplinaria. Al mantener al paciente en un estado de bienestar físico, social y emocional óptimo, se mejora su calidad de vida y se le proporciona el mejor apoyo posible en su lucha contra la enfermedad.
37 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
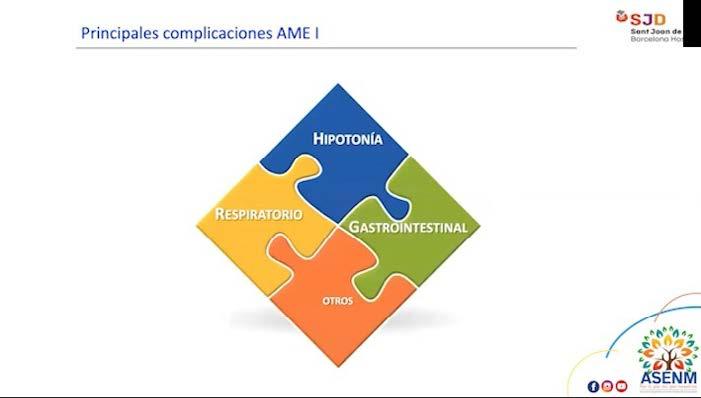
Es fundamental garantizar que el manejo respiratorio de los niños con Atrofia Muscular Espinal (AME) sea realizado por un neumólogo especialista en enfermedades neuromusculares. La participación de un neumólogo con experiencia en el manejo de estas condiciones específicas ayuda a prevenir y abordar posibles complicaciones respiratorias, mejorando así la calidad de vida del paciente.
La AME es una enfermedad neuromuscular que afecta principalmente a los músculos responsables del movimiento y la respiración. A medida que la enfermedad progresa, la debilidad muscular puede comprometer la capacidad respiratoria y aumentar el
riesgo de infecciones pulmonares y otros problemas respiratorios.
Un neumólogo especializado en enfermedades neuromusculares posee los conocimientos necesarios para evaluar y monitorear la función respiratoria del paciente con AME de manera regular. Esto implica realizar pruebas de función pulmonar, como la espirometría y la medición de los volúmenes pulmonares, para detectar posibles alteraciones en la función respiratoria y establecer un plan de cuidado personalizado.
El papel del neumólogo también es fundamental en la educación y capacitación de los cuidadores y familiares del paciente con AME. Proporcionar información sobre la importancia del manejo respiratorio adecuado, los signos de alarma de complicaciones respiratorias y las técnicas de manejo de emergencias respiratorias puede CLICK PARA VER VIDEO
Además, el neumólogo puede recomendar y supervisar estrategias de manejo respiratorio específicas, como la fisioterapia respiratoria, el uso de dispositivos de asistencia respiratoria no invasiva, como los ventiladores de presión positiva, y la monitorización continua de la oxigenación y la ventilación del paciente.
38 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
ser crucial para prevenir y actuar de manera oportuna ante cualquier situación que ponga en peligro la salud respiratoria del niño(a).
En cuanto a la alimentación, es fundamental entender que los niños(as) con debilidad muscular presentan una serie de requerimientos, especialmente según el tipo
de AME que tengan. Es importante recalcar que cada caso de AME es único, y los requerimientos dietéticos pueden variar según las necesidades individuales y la etapa de la enfermedad. Por lo tanto, es fundamental trabajar de manera interdisciplinaria.

CLICK PARA VER VIDEO 39 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Guías de atención de pacientes AME II y III
El obje�vo principal de los estándares de atención es mantener el estado clínico del paciente y evitar complicaciones posteriores
No sedestación Sedestación Marcha autónoma
Debilidad muscular
Dificultades de control postural (incluye cabeza/ cuello)
Contracturas ( extremidades, cuello, mandíbulas)
Escoliosis
Deformidades de la pared torácica
Plagiocefalia
Dolor
Fa�ga (poca resistencia muscular)
Movilidad deteriorada
Dislocación de cadera
Rotura de piel
Oblicuidad pélvia
Fracturas
Debilidad muscular
Dificultades de control postural (incluye cabeza/ cuello)
Contracturas ( extremidades, cuello, mandíbulas)
Escoliosis y oblicuidad pélvica
Movilidad deteriorada
Función pulmonar alterada
Deformación de pies
Temblores de manos
Fracturas
Debilidad muscular Asimetría
Movilidad deteriorada
Fa�ga (poca resistencia muscular)
Caídas
Contracturas e inflexibilidad
Resistencia reducida
Mercuri E, et al. Neuromuscular Disorder 2018;28: 103-15
Dr. Carlos Ortez, neuropediatra
40 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Guías de atención de pacientes AME II y III
Manejo pulmonar y nutricional
No sedestación
Manejo pulmonar:
Examen �sico, estudio del sueño
Fisioterapia torácica, aspiración oral
VNI equipado por un fisioterapeuta calificado
Manejo nutricional
Tragar con seguridad es muy importante
Monitoreo de la ingesta de calorías, líquidos y fibra
Sedestación
Manejo pulmonar:
Examen �sico, estudio del sueño
Fisioterapia torácica, aspiración
oral
VNI equipado por un fisioterapeuta calificado
Manejo nutricional
Tragar con seguridad es muy importante
Monitoreo de la ingesta de calorías, líquidos y fibra
Medicamentos, suplementos e inmunizaciones
Ambulantes
Manejo pulmonar:
Examen clínico de la eficacia de la tos y evaluación de la hipoven�lación nocturna
Manejo nutricional
Las dificultades para alimentarse son raras; la obesidad puede limitar la deambulación
Se recomiendan vacunas anuales contra la influenza y neumococos para todos los pacientes
1.Mercuri E, et al. Neuromuscular Disord. 2018;28: 103-15
2. Finfel RS, et al Neuromuscular Disord. 2018;28: 197-207 Dr. Carlos Ortez, neuropediatra
41 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
De ahí que bajo la premisa de que, al mantener al paciente en un estado de salud y bienestar óptimos, se pueden maximizar las posibilidades de respuesta a las nuevas terapias disponibles, lo que a su vez puede contribuir a mejorar su calidad de vida.
Porque, la estabilidad es fundamental para asegurar
1. Manejo nutricional: garantizar una adecuada nutrición es esencial para promover la salud general del paciente. Esto implica trabajar en colaboración con un especialista en nutrición para desarrollar un plan dietético personalizado que aborde las necesidades específicas del paciente, considerando factores como la capacidad de alimentación, la ingesta calórica y los requerimientos de nutrientes
una base sólida en el tratamiento y la atención médica. Esto implica abordar cuidadosamente las necesidades nutricionales, manejar los síntomas y complicaciones de manera eficaz, y brindar un apoyo integral tanto a nivel físico como emocional. Algunas consideraciones importantes incluyen:
2. Control de síntomas: la AME puede presentar una serie de síntomas, como debilidad muscular, dificultades respiratorias y problemas de deglución. Es importante trabajar en estrecha colaboración con un equipo multidisciplinario para manejar estos síntomas de manera eficaz. Esto puede implicar terapias de rehabilitación, intervenciones respiratorias y adaptaciones en la alimentación, según las necesidades individuales del paciente.
3. Soporte emocional y psicosocial: el impacto de la AME en la vida del paciente y su familia puede ser significativo. Es fundamental brindar un apoyo emocional y psicosocial integral para ayudar a enfrentar los desafíos que puedan surgir. Esto puede incluir la participación de psicólogos, trabajadores sociales y grupos de apoyo para proporcionar un entorno de cuidado y comprensión.
42 Capítulo 4
Herramienta
y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Al mantener al paciente con AME en un estado de estabilidad, se busca optimizar su respuesta a las terapias existentes, como el tratamiento farmacológico y las terapias génicas. Estos avances en el campo de la AME ofrecen la esperanza de mejorar la calidad de vida y ralentizar la progresión de la enfermedad. Al mantener una atención médica integral y centrada en el paciente, podemos trabajar hacia el objetivo común de brindar una calidad de vida óptima a las personas que viven con AME. Ya que las nuevas terapias están cambiando la historia natural de esta enfermedad.

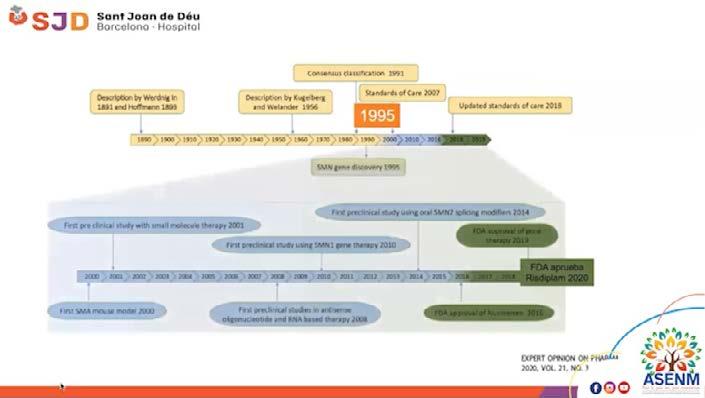
Avances terapéuticos
Cambiando la historia de la AME ...
CLICK PARA VER VIDEO CLICK PARA VER VIDEO 43 Capítulo 4
interactiva
diagnósti-
para el
co, tratamiento, manejo
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
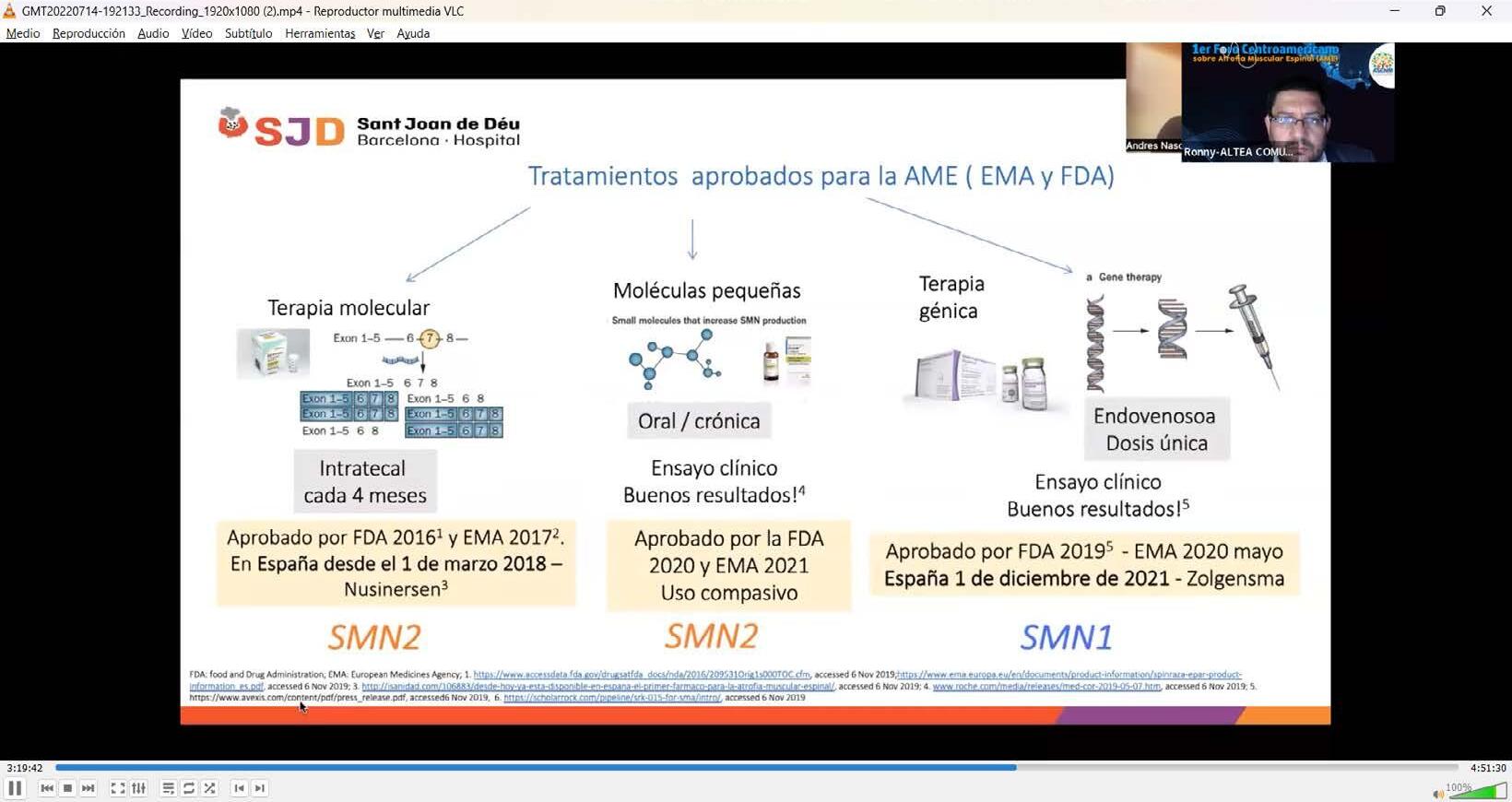
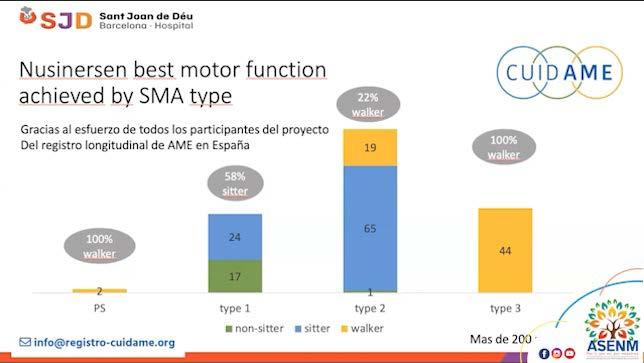
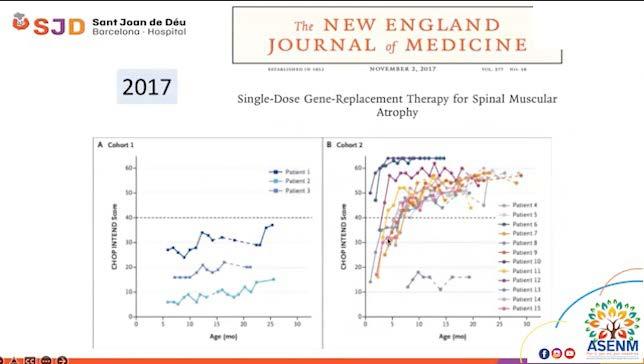
En España desde el
44 Capítulo 4
Fuente: Dr. Andrés Nascimento Osorio, neuropediatra
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
 CLICK PARA VER VIDEO
CLICK PARA VER VIDEO
45 Capítulo 4
CLICK PARA VER VIDEO
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal

CLICK PARA VER VIDEO 46 Capítulo 4
Herramienta interactiva para el diagnóstico, tratamiento, manejo y seguimiento de la Esclerosis Múltiple, Neuromielitis Óptica, Miastenia Gravis y Atrofia Muscular Espinal
Fuentes consultadas:
Finkel RS, et al. (2015). Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscular Disorders, 26(12), 925-939.
Finkel RS, et al. (2015). Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements, and immunizations; other organ systems; and ethics. Neuromuscular Disorders, 26(10), 754-766.
Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115. doi:10.1016/j.nmd.2017.11.005
Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197-207. doi:10.1016/j.nmd.2017.11.004
Oskoui M, Kaufmann P. Spinal muscular atrophy. Neurotherapeutics. 2008;5(4):499-506. doi:10.1016/j.nurt.2008.08.007
Prior TW. Spinal Muscular Atrophy. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2019.
Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-49.
Iannaccone ST. Modern management of spinal muscular atrophy. J Child Neurol. 2007;22(8):974-8.
L. Iannaccone et al., "Consensus statement for standard of care in spinal muscular atrophy." Journal of Child Neurology, vol. 22, no. 8, 2007, pp. 1027-1049.
D. Finkel et al., "Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study." Lancet, vol. 388, no. 10063, 2016, pp. 3017-3026.
Cure SMA. "Nutrition and Spinal Muscular Atrophy." www.curesma.org, www.curesma.org/nutrition-and-sma/.
I. G. Marini-Bettolo et al., "Nutritional Care for Neuromuscular Diseases." Nutrients, vol. 11, no. 3, 2019, p. 609.
R. Tizzano et al., "Management of spinal muscular atrophy from a neurologist's perspective." Neuromuscular Disorders, vol. 31, no. 3, 2021, pp. 222-229.
47 Capítulo 4

