DIABETES MELLITUS www.medicinaysaludpublica.com @RevistaMSP MANEJO ACTUALIZADO DE LA PANCREATITIS AGUDA EL ROL DE LA RADIOLOGÍA EN CÁNCER DE SENO VASCULITIS LEUCOCITOCLÁSTICA DESPUÉS DE LA VACUNA COVID 19

SPIRIT-P1 ACR20 AT WEEK 2: TALTZ 39% VS PLACEBO=13% SPIRIT-P2 ACR20 AT WEEK 2: TALTZ 38% VS PLACEBO=12%

Primary endpoint=ACR20 response at week 24. SPIRIT-P1 and -P2 Trial Design3-6
SPIRIT-P1 (N=417) and -P2 (N=363) were phase 3, randomized, doubleblind, placebo-controlled trials to evaluate the efficacy and safety of Taltz compared with placebo in patients with active psoriatic arthritis. Patients in SPIRIT-P1 were biologic-naive. Patients in SPIRIT-P2 were tumor necrosis factor inhibitor (TNFi)- experienced, having had an inadequate response and/or intolerance to 1 or 2 prior TNFis. In both trials, the primary efficacy endpoint was the proportion of patients achieving ACR20 response at week 24. All patients were ≥18 years of age and had ≥3 swollen and ≥3 tender joints. Patients were randomized to placebo or Taltz 80 mg every 2 or 4 weeks following a 160 mg starting dose. In SPIRIT-P1, an active reference arm of adalimumab 40 mg every 2 weeks was included. Patients in all study
arms were allowed to continue taking stable background medications during the trial. Inadequate responders (as defined by blinded criteria of <20% improvement in tender and in swollen joint counts) at week 16 received rescue therapy and were analyzed as nonresponders after week 16 until the primary endpoint. After receiving rescue therapy, inadequate responders in the placebo and adalimumab arms were re-randomized to Taltz 80 mg every 2 or 4 weeks. NRI methods were used for categorical efficacy analyses during the double-blind treatment period.
ACR=American College of Radiology; TNFi=tumor necrosis factor inhibitor; NRI=nonresponder imputation.
VAS Injection Site Pain Score Immediately Following Injection7
Taltz is indicated for adult patients with active ankylosing spondylitis, for adult patients with active psoriatic arthritis (PsA), and for adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation. Taltz is also indicated for adult patients and pediatric patients aged 6 years or older with moderate-to-severe plaque psoriasis (PsO) who are candidates for systemic therapy or phototherapy.
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients.
Taltz may increase the risk of infection. In clinical trials of adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). A similar increase in risk of infection was seen in placebocontrolled trials of adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis. Serious infections have occurred. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a serious infection develops, discontinue Taltz until the infection resolves. Pre-Treatment Evaluation for Tuberculosis Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Closely monitor patients receiving Taltz for signs and symptoms of active TB during and after treatment.
¶P<.0001; based on VAS 0-100
‡Same active ingredient §Vs original formulation; immediately after injection; based on VAS 0-100
No new National Drug Codes (NDCs)
No new Rx needed for existing Taltz patients
No new PAs to transition existing Taltz patients
No gaps in therapy
Once inventory of Taltz original formulation is depleted, Only citrate-free formulation will be available
The Citrate-Free Bioequivalence Study (N=245) was a 2-arm, subjectblind, parallel-design study in healthy subjects age 18-75 years to evaluate bioequivalence of Taltz citrate-free (CF) formulation compared to the original formulation of Taltz. Subjects were stratified into 1 of 3 weight categories (low: <70.0 kg; medium: 70.0-80.0 kg; high: >80.0 kg). Participants were then randomized within the 3 weight categories 1:1 to a single subcutaneous dose of either 80 mg Taltz original formulation (n=126) or 80 mg Taltz CF formulation (n=119). Subjects in each group were sub-randomized 1:1:1 to injection site (arm, thigh, or abdomen). Injections were administered by a medical professional using an autoinjector. The primary endpoint was bioequivalence as measured by maximum concentration (Cmax) of serum ixekizumab and area under the concentration versus time curve (AUC) of ixekizumab from time of injection through day 85 and time of injection through infinity.
Citrate-Free Injection Pain Study (N=70) was a subject-blind, randomized, crossover study in healthy subjects age 18-75 years to determine injection site pain differences between Taltz citrate-free formulation compared to the original formulation of Taltz. The primary endpoint was pain intensity on injection, as measured by VAS Pain 0-100. Subjects were randomized 1:1:1 to receive a single 1 mL subcutaneous injection of 80 mg Taltz original formulation, 80 mg Taltz citrate-free formulation 1 (CF1), or 80 mg Taltz citrate-free formulation 2 (CF2) in 1 of 3 possible treatment sequences on Days 1, 8, and 15 in a 3-period cross-over design. Injections were administered in the abdomen by a medical professional using a prefilled syringe. CF2 is not an approved formulation. Only data on the commercially available CF1 will be presented.
Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz. If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than the placebo group. During Taltz treatment, monitor patients for onset or exacerbations of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz.
Most common adverse reactions (≥1%) associated with Taltz treatment are injection site reactions, upper respiratory tract infections, nausea, and tinea infections. Overall, the safety profiles observed in adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis were consistent with the safety profile in adult patients with plaque psoriasis, with the exception of influenza and conjunctivitis in psoriatic arthritis and conjunctivitis, influenza, and urticaria in pediatric psoriasis.
Please see Brief Summary of Prescribing Information on the following pages. Please see Instructions for Use included with the device.
IX HCP ISI 07MAY2020
References: 1. Data on file. Lilly USA, LLC. DOF-IX-US-0304. 2. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79-87. 3. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327. 4. Taltz. Prescribing information. Lilly, USA. LLC. 5. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebocontrolled and active (adalimumab)-controlled period of the phase 3 trial SPIRIT-P1. Ann Rheum Dis. 2017;76(suppl):1-30. 6. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327. Supplementary appendix. 7. Chabra S, Gill BJ, Gallo G, et al. Ixekizumab citrate-free formulation: results from two clinical trials. Adv Ther. 2022;Epub (Incl Suppl Inf):1-11, 1-4. https://doi.org/10.1007/s12325-022-02126-0.
Taltz® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-IX-US-5471 05/2022 ©Lilly USA, LLC 2022. ALL RIGHTS RESERVED.
Taltz is FDA approved in a citrate-free formulation 4
Same Taltz,‡ less injection site pain§
Taltz® (ixekizumab) injection
Brief Summary: Consult the package insert for complete prescribing information.
Plaque Psoriasis—Taltz is indicated for the treatment of patients aged 6 years and older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis—Taltz is indicated for the treatment of adult patients with active psoriatic arthritis. Ankylosing Spondylitis—Taltz is indicated for the treatment of adult patients with active ankylosing spondylitis.
Non-radiographic Axial Spondyloarthritis—Taltz is indicated for the treatment of adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients (Warnings and Precautions)
Infections—Taltz may increase the risk of infection. In clinical trials in adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). Upper respiratory tract infections, oral candidiasis, conjunctivitis and tinea infections occurred more frequently in the Taltz group than in the placebo group. A similar increase in risk of infection was seen in placebo-controlled trials in patients with pediatric psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis (Adverse Reactions)
Instruct patients treated with Taltz to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a serious infection or is not responding to standard therapy, monitor the patient closely and discontinue Taltz until the infection resolves. Pre-treatment Evaluation for Tuberculosis—Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Consider anti-TB therapy prior to initiating Taltz in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Patients receiving Taltz should be monitored closely for signs and symptoms of active TB during and after treatment.
Hypersensitivity—Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz (Adverse Reactions). If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy. Inflammatory Bowel Disease—Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than in the control group (Adverse Reactions). During Taltz treatment, monitor for onset or exacerbation of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations—Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz. No data are available on the response to live vaccines.
The following adverse drug reactions are discussed in greater detail in other sections of the label:
• Infections (Warnings and Precautions)
• Hypersensitivity Reactions (Contraindications and Warnings and Precautions)
• Inflammatory Bowel Disease (Warnings and Precautions)
Clinical Trials Experience—Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Weeks 0 to 12: Three placebo-controlled trials in subjects with plaque psoriasis were integrated to evaluate the safety of Taltz compared to placebo for up to 12 weeks. A total of 1167 subjects (mean age 45 years; 66% men; 94% White) with plaque psoriasis received Taltz (160 mg at Week 0, 80 mg every 2 weeks [Q2W] for 12 weeks) subcutaneously. In two of the trials, the safety of Taltz (use up to 12 weeks) was also compared with an active comparator, U.S. approved etanercept.
In the 12-week, placebo-controlled period, adverse events occurred in 58% of the Taltz Q2W group (2.5 per subject-year of follow-up) compared with 47% of the placebo group (2.1 per subject-year of follow-up). Serious adverse events occurred in 2% of the Taltz group (0.07 per subject-year of follow-up), and in 2% of the placebo group (0.07 per subject-year of follow-up).
Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the Taltz group than the placebo group during the 12-week placebo-controlled period of the pooled clinical trials.
Table 1: Adverse Reactions Occurring in ≥1% of the Taltz Group and More Frequently than in the Placebo Group in the Plaque Psoriasis Clinical Trials through Week 12 Adverse Reactions
Taltz 80 mg Q2W (N=1167) (n%) Etanerceptb (N=287) (n%) Placebo (N=791) (n%)
Injection site reactions 196 (17) 32 (11) 26 (3)
Upper respiratory tract infectionsa
163 (14) 23 (8) 101 (13)
Nausea 23 (2) 1 (<1) 5 (1)
Tinea infections 17 (2) 0 1 (<1)
a Upper respiratory tract infections cluster includes nasopharyngitis and rhinovirus infection.
b U.S. approved etanercept.
Taltz® (ixekizumab) injection
Adverse reactions that occurred at rates less than 1% in the Taltz group and more frequently than in the placebo group during the 12-week induction period included rhinitis, oral candidiasis, urticaria, influenza, conjunctivitis, inflammatory bowel disease, and angioedema.
Weeks 13 to 60: A total of 332 subjects received the recommended maintenance regimen of Taltz 80 mg dosed every 4 weeks. During the maintenance period (Weeks 13 to 60), adverse events occurred in 80% of subjects treated with Taltz (1.0 per subject-year of follow-up) compared to 58% of subjects treated with placebo (1.1 per subject-year of follow-up). Serious adverse events were reported in 4% of subjects treated with Taltz (0.05 per subject-year of follow-up) and none in the subjects treated with placebo.
Weeks 0 to 60: Over the entire treatment period (Weeks 0 to 60), adverse events were reported in 67% of subjects treated with Taltz (1.4 per subject-year of follow-up) compared to 48% of subjects treated with placebo (2.0 per subject-year of follow-up). Serious adverse events were reported in 3% of subjects treated with Taltz (0.06 per subject-year of follow-up), and in 2% of subjects treated with placebo (0.06 per subject-year of follow-up).
Specific Adverse Drug Reactions: Injection Site Reactions: The most frequent injection site reactions were erythema and pain. Most injection site reactions were mild-to-moderate in severity and did not lead to discontinuation of Taltz.
Infections: In the 12-week, placebo-controlled period of the clinical trials in plaque psoriasis, infections occurred in 27% of subjects treated with Taltz (1.2 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.4% of subjects treated with Taltz (0.02 per subject-year of follow-up) and in 0.4% of subjects treated with placebo (0.02 per subject-year of follow-up) (Warnings and Precautions)
During the maintenance treatment period (Weeks 13 to 60), infections occurred in 57% of subjects treated with Taltz (0.70 per subject-year of follow-up) compared to 32% of subjects treated with placebo (0.61 per subject-year of follow-up). Serious infections occurred in 0.9% of subjects treated with Taltz (0.01 per subject-year of follow-up) and none in the subjects treated with placebo.
Over the entire treatment period (Weeks 0 to 60), infections were reported in 38% of subjects treated with Taltz (0.83 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.7% of subjects treated with Taltz (0.02 per subject-year of follow-up), and in 0.4% of subject treated with placebo (0.02 per subject-year of follow-up).
Inflammatory Bowel Disease: In adult subjects with plaque psoriasis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ 80 mg Q2W group (Crohn’s disease 0.1%, ulcerative colitis 0.2%) than the placebo group (0%) during the 12-week, placebo-controlled period in clinical trials (Warnings and Precautions).
Laboratory Assessment of Cytopenia: Neutropenia—Over the entire treatment period (Weeks 0 to 60), neutropenia occurred in 11% of subjects treated with Taltz (0.24 per subject-year of follow-up) compared to 3% of subjects treated with placebo (0.14 per subject-year of follow-up). In subjects treated with Taltz, the incidence rate of neutropenia during Weeks 13 to 60 was lower than the incidence rate during Weeks 0 to 12.
In the 12-week, placebo-controlled period, neutropenia ≥ Grade 3 (<1,000 cells/mm3) occurred in 0.2% of the Taltz group (0.007 per subject-year of follow-up) compared to 0.1% of the placebo group (0.006 per subject-year of follow-up). The majority of cases of neutropenia were either Grade 2 (2% for Taltz 80 mg Q2W versus 0.3% for placebo; ≥1,000 to <1,500 cells/mm3) or Grade 1 (7% for Taltz 80 mg Q2W versus 3% for placebo; ≥1,500 cells/mm3 to <2,000 cells/mm3). Neutropenia in the Taltz group was not associated with an increased rate of infection compared to the placebo group.
Thrombocytopenia—Ninety eight percent of cases of thrombocytopenia were Grade 1 (3% for Taltz 80 mg Q2W versus 1% for placebo; ≥75,000 cells/mm3 to <150,000 cells/mm3). Thrombocytopenia in subjects treated with Taltz was not associated with an increased rate of bleeding compared to subjects treated with placebo.
Active Comparator Trials: In the two clinical trials that included an active comparator, the rate of serious adverse events during weeks zero to twelve was 0.7% for U.S.-approved etanercept and 2% for Taltz 80 mg Q2W, and the rate of discontinuation from adverse events was 0.7% for U.S. approved etanercept and 2% for Taltz 80 mg Q2W. The incidence of infections was 18% for U.S. approved etanercept and 26% for Taltz 80 mg Q2W. The rate of serious infections was 0.3% for both Taltz 80 mg Q2W and U.S. approved etanercept.
Taltz was evaluated in a placebo-controlled trial in pediatric subjects with moderate-to-severe psoriasis 6 to less than 18 years of age. A total of 171 subjects were studied (115 subjects on Taltz and 56 subjects on placebo). Overall, the safety profile observed in pediatric subjects with plaque psoriasis treated with Taltz every 4 weeks is consistent with the safety profile in adult subjects with plaque psoriasis with the exception of the frequencies of conjunctivitis (2.6%), influenza (1.7%), and urticaria (1.7%).
In this clinical trial, Crohn’s disease occurred at a greater frequency in the Taltz group (0.9%) than the placebo group (0%) during the 12-week, placebo-controlled period. Crohn’s disease occurred in a total of 4 Taltz treated subjects (2.0%) in the clinical trial (Warnings and Precautions).
Taltz was studied in two placebo-controlled trials in patients with psoriatic arthritis. A total of 678 patients were studied (454 patients on Taltz and 224 on placebo). A total of 229 patients in these trials received Taltz 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with psoriatic arthritis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis with the exception of the frequencies of influenza (1.3%) and conjunctivitis (1.3%).
IX HCP BS 06JAN2022 Taltz® (ixekizumab) injection IX HCP BS 06JAN2022
Taltz was studied in two placebo-controlled trials in patients with ankylosing spondylitis. A total of 566 patients were studied (376 patients on Taltz and 190 on placebo). A total of 195 patients in these trials received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with ankylosing spondylitis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis.
In adult patients with ankylosing spondylitis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred in 2 patients (1.0%) and 1 patient (0.5%), respectively, in the Taltz 80 mg Q4W group and 1 patient (0.5%) and 0%, respectively, in the placebo group during the 16-week, placebo-controlled period in clinical trials. Of these patients, serious events occurred in 1 patient in the Taltz 80 mg Q4W group and 1 patient in the placebo group (Warnings and Precautions).
Taltz was studied in a placebo-controlled trial in patients with non-radiographic axial spondyloarthritis. A total of 303 patients were studied (198 patients on Taltz and 105 on placebo). A total of 96 patients in this trial received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with non-radiographic axial spondyloarthritis treated with Taltz 80 mg Q4W up to Week 16 is consistent with the previous experience of Taltz in other indications. Immunogenicity—As with all therapeutic proteins, there is the potential for immunogenicity with Taltz. The assay to test for neutralizing antibodies has limitations detecting neutralizing antibodies in the presence of ixekizumab; therefore, the incidence of neutralizing antibodies development could be underestimated.
By Week 12, approximately 9% of adult subjects treated with Taltz every 2 weeks developed antibodies to ixekizumab. Approximately 22% of subjects treated with Taltz at the recommended dosing regimen developed antibodies to ixekizumab during the 60-week treatment period. The clinical effects of antibodies to ixekizumab are dependent on the antibody titer; higher antibody titers were associated with decreasing drug concentration and clinical response.
Of the adult subjects who developed antibodies to ixekizumab during the 60-week treatment period, approximately 10%, which equates to 2% of subjects treated with Taltz at the recommended dosing regimen, had antibodies that were classified as neutralizing. Neutralizing antibodies were associated with reduced drug concentrations and loss of efficacy.
In pediatric psoriasis subjects treated with ixekizumab at the recommended dosing regimen up to 12 weeks, 21 subjects (18%) developed anti-drug antibodies, 5 subjects (4%) had confirmed neutralizing antibodies associated with low drug concentrations. No conclusive evidence could be obtained on the potential association of neutralizing antibodies and clinical response and/or adverse events due to small number of pediatric subjects in the study.
Psoriatic Arthritis Population
For subjects treated with Taltz 80 mg every 4 weeks for up to 52 weeks (PsA1), 11% developed anti-drug antibodies, and 8% had confirmed neutralizing antibodies.
Ankylosing Spondylitis Population
For patients treated with Taltz 80 mg every 4 weeks for up to 16 weeks (AS1, AS2), 5.2% developed anti-drug antibodies, and 1.5% had neutralizing antibodies.
Non-radiographic Axial Spondyloarthritis Population
Of patients treated with Taltz 80 mg every 4 weeks for up to 52 weeks (nr-axSpA1), 8.9% developed anti-drug antibodies, all of which were low titer. No patient had neutralizing antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to Taltz across indications or with the incidences of antibodies to other products may be misleading.
Postmarketing Experience—The following adverse reactions have been identified during postapproval use of Taltz. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Taltz exposure.
Immune system disorders: anaphylaxis (Contraindications and Warnings and Precautions)
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Pregnant women should be encouraged to enroll themselves in the registry by calling 1-800-284-1695.
Risk Summary—There are no available data on Taltz use in pregnant women to inform any drug associated risks. Human IgG is known to cross the placental barrier; therefore, Taltz may be transmitted from the mother to the developing fetus. An embryofetal development study conducted in pregnant monkeys at doses up to 19 times the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus. When dosing was continued until parturition, neonatal deaths were observed at 1.9 times the MRHD [see Data]. The clinical significance of these nonclinical findings is unknown.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data—An embryofetal development study was conducted in cynomolgus monkeys administered ixekizumab. No malformations or embryofetal toxicity were observed in fetuses
from pregnant monkeys administered ixekizumab weekly by subcutaneous injection during organogenesis to near parturition at doses up to 19 times the MRHD (on a mg/kg basis of 50 mg/ kg/week). Ixekizumab crossed the placenta in monkeys.
In a pre- and post-natal development toxicity study, pregnant cynomolgus monkeys were administered weekly subcutaneous doses of ixekizumab up to 19 times the MRHD from the beginning of organogenesis to parturition. Neonatal deaths occurred in the offspring of two monkeys administered ixekizumab at 1.9 times the MRHD (on a mg/kg basis of 5 mg/kg/week) and two monkeys administered ixekizumab at 19 times the MRHD (on a mg/kg basis of 50 mg/kg/ week). These neonatal deaths were attributed to early delivery, trauma, or congenital defect. The clinical significance of these findings is unknown. No ixekizumab-related effects on functional or immunological development were observed in the infants from birth through 6 months of age.
Risk Summary—There are no data on the presence of ixekizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Ixekizumab was detected in the milk of lactating cynomolgus monkeys. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Taltz and any potential adverse effects on the breastfed infant from Taltz or from the underlying maternal condition.
Pediatric Use—The safety and effectiveness of Taltz have been established in pediatric subjects aged 6 years to less than 18 years with moderate-to-severe plaque psoriasis. The safety and effectiveness of Taltz in other pediatric indications and for pediatric subjects less than 6 years of age have not been established.
Geriatric Use—Of the 4204 psoriasis subjects exposed to Taltz, a total of 301 were 65 years or older, and 36 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects.
PATIENT COUNSELING INFORMATION—Advise the patient and/or caregiver to read the FDAapproved patient labeling (Medication Guide and Instructions for Use) before the patient starts using Taltz and each time the prescription is renewed, as there may be new information they need to know.
Instructions on Self-Administration: Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the autoinjector or prefilled syringe correctly (Instructions for Use)
Infection: Inform patients that Taltz may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider, and contacting their healthcare provider if they develop any symptoms of infection (Warnings and Precautions).
Allergic Reactions: Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions (Warnings and Precautions).
Pregnancy: Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Advise patients to contact the registry at 1-800-284-1695 to enroll (Use in Specific Populations)
Additional information can be found at www.Taltz.com.
See Instructions for Use accompanying the product device.
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2016, 2017, 2019, 2020, 2021, 2022 Eli Lilly and Company. All rights reserved.
IX HCP BS 06JAN2022 PP-IX-US-5359

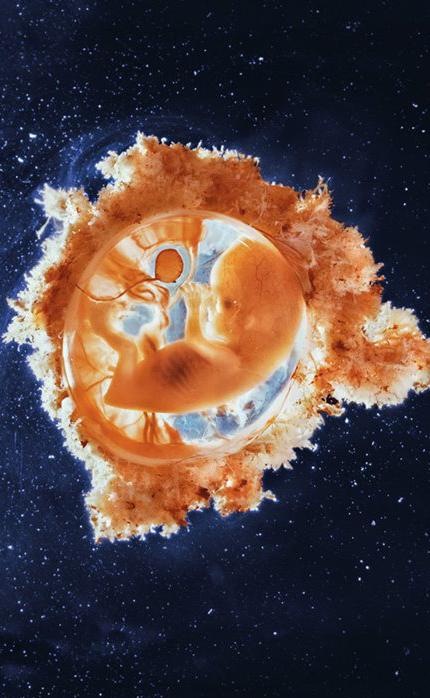
Los virus han determinado la evolución desde el principio y pese a que comprendemos su capacidad patogénica, estos organismos desempeñan funciones portentosas en el desarrollo de la especie humana. Aproximadamente el 8 % de nuestro ADN se ha originado de los virus que infectaron a nuestros antepasados cuando se introdujeron estos genes en su genoma. Hoy algunos de los genes víricos cumplen un rol importante en el desarrollo del embrión en la placenta, como la que rodea a este feto de 13 semanas de gestación.
Las pandemias por virus han asolado la humanidad desde hace siglos. Una de las primeras de las que se tiene noticia es la plaga de Galeno (165-180 d.C), bajo el emperador romano Antonino. Se produjo tras volver las legiones de una campaña en Mesopotamia. Produjo 5 millones de muertes en Italia, Asia Menor, Grecia y Egipto. La form a hemorrágica del sarampión fue la causa más probable (Muñoz-Sanz A. EIMC 2012).
El aumento de la población mundial, la invasión de ecosistemas de otros animales y los viajes frecuentes, entre otras razones, han favorecido en las últimas décadas una expansión sin precedentes de zoonosis, esto es, infecciones en humanos por virus de otras especies. Es el caso del SIDA, el COVID y la reciente viruela del mono.
Para dar el salto a los humanos, los virus de animales necesitan mutaciones de adaptación. A continuación, para transmitirse entre humanos, deben sumar más cambios genéticos. Solo tras esos dos pasos, una zoonosis puede desencadenar una pandemia. Esto ocurrió con el VIH hace 50 años, con el coronavirus hace 3 años y con el virus de la viruela del mono hace apenas 6 meses.
EDITOR FUNDADOR Juan Carlos Orengo Valverde, MD, MPH, PhD EDITOR Alberto Santiago Cornier, MD, PhD CONSEJO ASESOR Oscar Soto Raíces, MD, Ahmed Morales, MD, FACP, FACG, FASGE, AGAF, Lcda. Wanda González PRINCIPAL OFICIAL EJECUTIVO Pedro Carlos Lugo Hernández III, P.A.C. PRESIDENTA Y FUNDADORA Glorybelle Hernández Figueroa, MBA VICEPRESIDENTA Y FUNDADORA Laila Paloma Lugo, MBA CONTABILIDAD Julio Soto ADMINISTRACIÓN Marta Ivelisse Vélez Ramos, MBA, MARKETING Y SERVICIOS 360 Alexelena Cayere, Yasmin Morell PERIODISTAS Belinda Burgos, Grenda Rivera, Mayra Acevedo, Luis Penchi, Limarys Suárez DIRECCIÓN GRÁFICA Natalia Zoé Rivera Torres ARTISTAS GRÁFICOS Jhorman González, Isabel Hernandez DIRECTORA AUDIOVISUAL Fabiola Plaza REALIZADORA AUDIOVISIAL Salomé Mateus FOTOS Revista Medicina y Salud Pública DIRECCIÓN GENERAL / FUNDADOR Carlos Alexis Lugo Marrero DISTRIBUCIÓN OFICINAS Y TORRES MÉDICAS Editorial Mundo ENVÍO DE REVISTAS Y DISTRIBUCIÓN A GRUPOS MÉDICOS Servicio de correo postal/Comunicación Inteligente Para ventas y otros servicios pueden comunicarse al 787.848.3333, msp@editorialmundo.com o www.medicinaysaludpublica.com Revista Puertorriqueña de Medicina y Salud Pública ISSN 1937-8521
COMITÉ EDITORIAL Olga Rodríguez, MD - Decana Escuela de Medicina de Ponce (Puerto Rico), Vivian Green, LND, MS, PhD, Sub editora y fundadora (Puerto Rico), José Cordero, MD, MPH - Exdecano Escuela Graduada Salud Pública Recinto de Ciencias Médicas UPR (Puerto Rico), Ángeles Rodríguez, MD, MPH (Puerto Rico), Simón Carlo, MD (Puerto Rico), Bárbara Rosado, MD (Puerto Rico), Idhaliz Flores PhD (Puerto Rico), Jesús Cruz-Correa, MD, FACOG (Puerto Rico), Rafael Bredy, MD, LicMTo, MBE, MS (Puerto Rico), David Caseida, MD, FACOG, (Puerto Rico), José Capriles, MD, MHSA (Puerto Rico) Joaquín Laboy, MD, FACOG (Puerto Rico), Luis Adrian Rivera Pomales, MD, PEMBA, MPH, CMQ (Puerto Rico), Juan Fernández, MS, PhD (Puerto Rico), Nuria Sebate, MD (Puerto Rico), Pedro Amador, MD, MPH (Puerto Rico), Nydia Cappas, PsyD (Puerto Rico), Luis Franco, MD (Puerto Rico), Federico Montealegre, DVM, PhD, Msc (Puerto Rico), Nydia Ortiz, PsyD (Puerto Rico), José Pons, PhD, FPPR (Puerto Rico), Esdrás Vélez, JD, MPH (Puerto Rico), Diego Zavala, MSc, PhD, (Puerto Rico), Ana Torres-Martín, MD (Puerto Rico), Julio Cádiz, MD, MPH (Puerto Rico), Rafael Gómez-Cuevas (Colombia), José Javier Orengo, PhD(c) (España), Cesar A. Del Rey, MD (Panamá), Pedro Serrano, MD, PhD (España), Luis Serra-Majem, MD, PhD (España), José Ramón Calvo, MD, PhD (España).
Síguenos en www.medicinaysaludpublica.com, www.facebook.com/revistamsp, en Twitter @revistamsp, en LinkedIn como Revista Puertorriqueña de Medicina y Salud Pública. Las normas editoriales de la Revista Puertorriqueña de Medicina y Salud Pública para la publicación de artículos originales y cartas al editor pueden ser accesadas en la página web: www.medicinaysaludpublica.com, y solicitadas a través de msp@editorialmundo.com.
Medicina y Salud Pública es propiedad de publicaciones mundo. Medicina es una publicación de la REVISTA PUERTORRIQUEÑA DE MEDICINA Y SALUD PÚBLICA. Medicina y Salud Pública tiene como política corregir y aclarar cualquier información incorrecta que pueda ser publicada en su revista. Medicina y Salud Pública no asume responsabilidad alguna por los anuncios, artículos y otros servicios anunciados en nuestra
Puertorriqueña de Medicina y Salud Pública
Fuga de médicos.
El sistema de salud de Puerto Rico sufre una hemorragia de pediatras, generalistas, cardiólogos, nefrólogos y profesionales de la salud que obliga a todos los sectores a revisar y resolver las causas de este éxodo sin precedente en nuestra historia.
Una tormenta perfecta se está formando sobre los servicios de salud que recibe la población puertorriqueña. Cada año son más los especialistas y profesionales de la salud que escogen, obligados por las difíciles circunstancias laborales y la relación con los seguros de salud, irse de su Isla principalmente hacia los Estados Unidos. Políticas públicas difíciles de revertir y decisiones de las aseguradoras son cada día más devastadoras para la estabilidad de los profesionales de la salud. Para decenas de pacientes esto es una situación que compromete su vida debido a la aglomeración de citas con especialistas y subespecialistas debido a la escasez de especialistas.
Esta tendencia viene observándose desde que comenzó la crisis económica pero se ha hecho más evidente desde el paso del huracán María, terremotos, pandemias y el nuevo fenómeno atmosférico llamado Fiona.
Las especialidades con mayor éxodo de profesionales son pediatría, cardiología, cirugía de cabeza y cuello, medicina deportiva, y médicos generalistas.

En la isla trabajan tres nefrólogos pediátricos y un endocrinólogo pediátrico de 10 que habían en el 2015.
Los costos cada día son mayores y las tarifas que pagan las aseguradoras ni se acercan a lo que deberían pagar.
A esto hay que añadir el déficit creciente de médicos por la jubilación masiva de profesionales.
Teniendo en cuenta que la formación de un médico, requiere cerca de 12 años de estudio, aumentar la oferta no ofrece resultados a corto plazo y tampoco es suficiente para detener la corriente de fondo que desangra al sistema. De poco sirve que se aumenten las plazas si, cuando terminan la carrera, cada vez más médicos emigran a otros países huyendo de la precariedad laboral y de unas condiciones de trabajo cada vez más estresantes. Encuestas realizadas sobre la Situación de la Profesión Médica en Puerto Rico, revela que uno de cada tres médicos está insatisfecho, y los principales motivos de este descontento lo son la carga de trabajo, el nivel de exigencia y el cansancio emocional. En otros países no solo encuentran mejores condiciones profesionales, sino un salario que en Estados Unidos, el Reino Unido o Alemania se duplica, y en algunos países incluso triplica, que el que pueden llegar a cobrar en Puerto Rico
De todos estos datos se infiere que si no se interviene sobre las condiciones salariales y de trabajo de los médicos especialistas y enfermeras, difícilmente se cortará la hemorragia. Si las cosas se mantienen, Puerto Rico tendrá que importar médicos extranjeros que en muchos casos habrán recibido una formación más precaria pero que también son necesarios en sus países. El sistema de salud deber ser uno de los grandes pilares del Estado de bienestar en Puerto Rico. Dejar que se descapitalice de su principal activo, es un acto deshumanizante y temerario que debemos evitar.
Dr. Oscar Soto Raíces, reumatólogo puertorriqueño.
“Datos del Colegio de Médicos Cirujanos revelan que la clase médica del país se ha reducido a poco menos de la mitad en los últimos 13 años.”
a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo + MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.] 1,2


RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.

Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity.


Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis (TB), which may present with pulmonary or extrapulmonary disease. Test patients for latent TB before RINVOQ use and during therapy. Consider treatment for latent TB infection prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
Carefully consider the risks and benefits of treatment with RINVOQ prior to initiating therapy in patients with chronic or recurrent infection. Monitor patients closely for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy.
In a large, randomized, postmarketing safety study comparing another Janus kinase (JAK) inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years old with at least one cardiovascular (CV) risk factor, a higher rate of all-cause mortality, including sudden CV death, was observed with the JAK inhibitor. Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Lymphoma and other malignancies have been observed in patients treated with RINVOQ.
In a large, randomized, postmarketing safety study comparing another JAK inhibitor with TNF blockers in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk.
With RINVOQ, consider the benefits and risks for the individual patient prior to initiating or continuing therapy, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers. NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer. Advise patients to limit sunlight exposure by wearing protective clothing and using sunscreen.
In a large, randomized, postmarketing study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other CV risk factors. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing study comparing another JAK inhibitor to TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of thrombosis was observed with the JAK inhibitor. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated.
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients. Serious hypersensitivity reactions, such as anaphylaxis and angioedema, were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy.
Gastrointestinal (GI) perforations have been reported in clinical trials with RINVOQ. Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Promptly evaluate patients presenting with new onset abdominal pain for early identification of GI perforation.
Neutropenia
Treatment with RINVOQ was associated with an increased incidence of neutropenia (absolute neutrophil count [ANC] <1000 cells/mm3). Treatment with RINVOQ is not recommended in patients with an ANC <1000 cells/mm3. Evaluate neutrophil counts at baseline and thereafter according to routine patient management.
Absolute lymphocyte counts (ALC) <500 cells/mm3 were reported in RINVOQtreated patients. Treatment with RINVOQ is not recommended in patients with an ALC <500 cells/mm3. Evaluate at baseline and thereafter according to routine patient management.
Decreases in hemoglobin levels to <8 g/dL were reported in RINVOQ-treated patients. Treatment should not be initiated or should be interrupted in patients with hemoglobin levels <8 g/dL. Evaluate at baseline and thereafter according to routine patient management.
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Manage patients according to clinical guidelines for the management of hyperlipidemia. Evaluate patients 12 weeks after initiation of treatment and thereafter according to the clinical guidelines for hyperlipidemia.
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevation compared to placebo. Evaluate at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. If increases in aspartate aminotransferase (AST) or alanine aminotransferase (ALT) are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose. Verify pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ.
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, patients should be brought up to date on all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
LACTATION
There are no data on the presence of RINVOQ in human milk, the effects on the breastfed infant, or the effects on milk production. Available data in animals have shown the excretion of RINVOQ in milk. Advise patients that breastfeeding is not recommended during treatment with RINVOQ and for 6 days after the last dose.
RINVOQ is not recommended for use in patients with severe hepatic impairment.
The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenzalike illness, elevated liver enzymes, and rash.
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.
Dosage Forms and Strengths: RINVOQ is available in 15 mg, 30 mg, and 45 mg extended-release tablets.
in: Lancet. 2019;393(10191):2590.
RINVOQ® and its design are registered trademarks of AbbVie Biotechnology Ltd. ©2022 AbbVie Inc. North Chicago, IL 60064 US-RNQR-220149 April 2022 Printed in Puerto Rico
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions, Adverse Reactions]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with RINVOQ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions]
In a large, randomized, postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another Janus kinase (JAK) inhibitor to tumor necrosis factor (TNF) blockers, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed with the JAK inhibitor [see Warnings and Precautions]
Lymphoma and other malignancies have been observed in patients treated with RINVOQ. In RA patients treated with another JAK inhibitor, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk [see Warnings and Precautions]
In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke [see Warnings and Precautions].
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of thrombosis was observed when compared with TNF blockers. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated [see Warnings and Precautions]
Rheumatoid Arthritis
RINVOQ® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs (DMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is indicated for the treatment of adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
RINVOQ is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for ulcerative colitis, or with potent immunosuppressants such as azathioprine and cyclosporine.
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients [see Warnings and Precautions].
Serious Infections
Serious and sometimes fatal infections have been reported in patients receiving RINVOQ. The most frequent serious infections reported with RINVOQ included pneumonia and cellulitis [see Adverse Reactions]. Among opportunistic infections, tuberculosis, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis, were reported with RINVOQ.
Avoid use of RINVOQ in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating RINVOQ in patients:
• with chronic or recurrent infection
• who have been exposed to tuberculosis
• with a history of a serious or an opportunistic infection
• who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
• with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ. Interrupt RINVOQ if a patient develops a serious or opportunistic infection.
A patient who develops a new infection during treatment with RINVOQ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and RINVOQ should be interrupted if the patient is not responding to antimicrobial therapy. RINVOQ may be resumed once the infection is controlled.
Evaluate and test patients for latent and active tuberculosis (TB) infection prior to administration of RINVOQ. Patients with latent TB should be treated with standard antimycobacterial therapy before initiating RINVOQ. RINVOQ should not be given to patients with active TB. Consider anti-TB therapy prior to initiation of RINVOQ in patients with previously untreated latent TB or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but who have risk factors for TB infection.
Consultation with a physician with expertise in the treatment of TB is recommended to aid in the decision about whether initiating anti-TB therapy is appropriate for an individual patient.
During RINVOQ use, monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster) and hepatitis B virus reactivation, were reported in clinical trials with RINVOQ [see Adverse Reactions]. The risk of herpes zoster appears to be higher in patients treated with RINVOQ in Japan. If a patient develops herpes zoster, consider temporarily interrupting RINVOQ until the episode resolves. Screening for viral hepatitis and monitoring for reactivation should be performed in accordance with clinical guidelines before starting and during therapy with RINVOQ. Patients who were positive for hepatitis C antibody and hepatitis C virus RNA, were excluded from clinical trials. Patients who were positive for hepatitis B surface antigen or hepatitis B virus DNA were excluded from clinical trials. However, cases of hepatitis B reactivation were still reported in patients enrolled in the Phase 3 trials of RINVOQ. If hepatitis B virus DNA is detected while receiving RINVOQ, a liver specialist should be consulted.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed in patients treated with the JAK inhibitor compared with TNF blockers.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Malignancy and Lymphoproliferative Disorders
Malignancies, including lymphomas, were observed in clinical trials of RINVOQ [see Adverse Reactions]
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. In this study, current or past smokers had an additional increased risk of overall malignancies.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Non-Melanoma Skin Cancer
NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of major adverse cardiovascular events (MACE) defined as cardiovascular death, non-fatal myocardial infarction (MI), and non-fatal stroke was observed with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients
should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Thrombosis, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers.
If symptoms of thrombosis occur, patients should discontinue RINVOQ and be evaluated promptly and treated appropriately. Avoid RINVOQ in patients that may be at increased risk of thrombosis.
Serious hypersensitivity reactions such as anaphylaxis and angioedema were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy [see Adverse Reactions].
Gastrointestinal perforations have been reported in clinical trials with RINVOQ.
Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Evaluate promptly patients presenting with new onset abdominal pain for early identification of gastrointestinal perforation.
Treatment with RINVOQ was associated with an increased incidence of neutropenia (ANC less than 1000 cells/mm3).
Evaluate neutrophil counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation and interrupt RINVOQ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3).
Lymphopenia
ALC less than 500 cells/mm3 were reported in RINVOQ-treated patients in clinical trials.
Evaluate lymphocyte counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3).
Anemia
Decreases in hemoglobin levels to less than 8 g/dL were reported in RINVOQ-treated patients in clinical trials.
Evaluate hemoglobin at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low hemoglobin level (i.e., less than 8 g/dL).
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol [see Adverse Reactions] Elevations in LDL cholesterol decreased to pre-treatment levels in response to statin therapy. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assess lipid parameters approximately 12 weeks after initiation of treatment, and thereafter according to the clinical guidelines for hyperlipidemia. Manage patients according to clinical guidelines for the management of hyperlipidemia.
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevations compared to treatment with placebo.
Evaluate liver enzymes at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Administration of upadacitinib to rats and rabbits during organogenesis caused increases in fetal malformations. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with RINVOQ and for 4 weeks following completion of therapy [see Use in Specific Populations]
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, it is recommended that patients be brought up to date with all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
The following clinically significant adverse reactions are described elsewhere in the labeling:
• Serious Infections [see Warnings and Precautions]
• Mortality [see Warnings and Precautions]
• Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions]
• Major Adverse Cardiovascular Events [see Warnings and Precautions]
• Thrombosis [see Warnings and Precautions]
• Hypersensitivity Reactions [see Warnings and Precautions]
• Gastrointestinal Perforations [see Warnings and Precautions]
• Laboratory Abnormalities [see Warnings and Precautions]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Patients with Rheumatoid Arthritis
A total of 3833 patients with rheumatoid arthritis were treated with upadacitinib in the Phase 3 clinical trials of whom 2806 were exposed for at least one year.
Patients could advance or switch to RINVOQ 15 mg from placebo, or be rescued to RINVOQ from active comparator or placebo from as early as Week 12 depending on the trial design.
A total of 2630 patients received at least 1 dose of RINVOQ 15 mg, of whom 1860 were exposed for at least one year. In trials RA-I, RA-II, RA-III and RA-V, 1213 patients received at least 1 dose of RINVOQ 15 mg, of which 986 patients were exposed for at least one year, and 1203 patients received at least 1 dose of upadacitinib 30 mg, of which 946 were exposed for at least one year.
Table 1: Adverse Reactions Reported in ≥ 1% of Rheumatoid Arthritis Patients Treated with RINVOQ 15 mg in Placebo-controlled Trials
Adverse Reaction Placebo RINVOQ 15 mg n=1042 (%) n=1035 (%)
Upper respiratory tract infection (URTI)* 9.5 13.5
Nausea 2.2 3.5 Cough 1.0 2.2 Pyrexia 0 1.2
*URTI includes: acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
Four integrated datasets are presented in the Specific Adverse Reaction section:
Placebo-controlled Trials: Trials RA-III, RA-IV, and RA-V were integrated to represent safety through 12/14 weeks for placebo (n=1042) and RINVOQ 15 mg (n=1035). Trials RA-III and RA-V were integrated to represent safety through 12 weeks for placebo (n=390), RINVOQ 15 mg (n=385), and upadacitinib 30 mg (n=384). Trial RA-IV did not include the 30 mg dose and, therefore, safety data for upadacitinib 30 mg can only be compared with placebo and RINVOQ 15 mg rates from pooling trials RA-III and RA-V.
MTX-controlled Trials: Trials RA-I and RA-II were integrated to represent safety through 12/14 weeks for MTX (n=530), RINVOQ 15 mg (n=534), and upadacitinib 30 mg (n=529).
12-Month Exposure Dataset: Trials RA-I, II, III, and V were integrated to represent the long-term safety of RINVOQ 15 mg (n=1213) and upadacitinib 30 mg (n=1203).
Exposure adjusted incidence rates were adjusted by trial for all the adverse events reported in this section.
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, infections were reported in 218 patients (95.7 per 100 patient-years) treated with placebo and 284 patients (127.8 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, infections were reported in 99 patients (136.5 per 100 patient-years) treated with placebo, 118 patients (164.5 per 100 patient-years) treated with RINVOQ 15 mg, and 126 patients (180.3 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Infections were reported in 127 patients (119.5 per 100 patient-years) treated with MTX monotherapy, 104 patients (91.8 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 128 patients (115.1 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Infections were reported in 615 patients (83.8 per 100 patient-years) treated with RINVOQ 15 mg and 674 patients (99.7 per 100 patient-years) treated with upadacitinib 30 mg.
Serious Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, serious infections were reported in 6 patients (2.3 per 100 patient-years) treated with placebo, and 12 patients (4.6 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, serious infections were reported in 1 patient (1.2 per 100 patientyears) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 7 patients (8.2 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Serious infections were reported in 2 patients (1.6 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 8 patients (6.4 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Serious infections were reported in 38 patients (3.5 per 100 patient-years) treated with RINVOQ 15 mg and 59 patients (5.6 per 100 patient-years) treated with upadacitinib 30 mg. The most frequently reported serious infections were pneumonia and cellulitis.
Placebo-controlled Trials and MTX-controlled Trials: In the placebo-controlled period, there were no active cases of tuberculosis reported in the placebo, RINVOQ 15 mg, and upadacitinib 30 mg groups. In the MTX-controlled period, there were no active cases of tuberculosis reported in the MTX monotherapy, RINVOQ 15 mg monotherapy, and upadacitinib 30 mg monotherapy groups.
12-Month Exposure Dataset: Active tuberculosis was reported for 2 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg. Cases of extra-pulmonary tuberculosis were reported.
Opportunistic Infections (excluding tuberculosis)
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, opportunistic infections were reported in 3 patients (1.2 per 100 patient-years) treated with placebo, and 5 patients (1.9 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, opportunistic infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (7.1 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Opportunistic infections were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 0 patients
treated with RINVOQ 15 mg monotherapy, and 4 patients (3.2 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Opportunistic infections were reported in 7 patients (0.6 per 100 patient-years) treated with RINVOQ 15 mg and 15 patients (1.4 per 100 patient-years) treated with upadacitinib 30 mg.
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, malignancies excluding NMSC were reported in 1 patient (0.4 per 100 patient-years) treated with placebo, and 1 patient (0.4 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, malignancies excluding NMSC were reported in 0 patients treated with placebo, 1 patient (1.1 per 100 patient-years) treated with RINVOQ 15 mg, and 3 patients (3.5 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Malignancies excluding NMSC were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 0 patients treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Malignancies excluding NMSC were reported in 13 patients (1.2 per 100 patient-years) treated with RINVOQ 15 mg and 14 patients (1.3 per 100 patient-years) treated with upadacitinib 30 mg.
Placebo-controlled Trials: There were no gastrointestinal perforations (based on medical review) reported in patients treated with placebo, RINVOQ 15 mg, and upadacitinib 30 mg.
MTX-controlled Trials: There were no cases of gastrointestinal perforations reported in the MTX and RINVOQ 15 mg group through 12/14 weeks. Two cases of gastrointestinal perforations were observed in the upadacitinib 30 mg group.
12-Month Exposure Dataset: Gastrointestinal perforations were reported in 1 patient treated with RINVOQ 15 mg and 4 patients treated with upadacitinib 30 mg.
Placebo-controlled Trials: In RA-IV, venous thrombosis (pulmonary embolism or deep vein thrombosis) was observed in 1 patient treated with placebo and 1 patient treated with RINVOQ 15 mg. In RA-V, venous thrombosis was observed in 1 patient treated with RINVOQ 15 mg. There were no observed cases of venous thrombosis reported in RA-III. No cases of arterial thrombosis were observed through 12/14 weeks.
MTX-controlled Trials: In RA-II, venous thrombosis was observed in 0 patients treated with MTX monotherapy, 1 patient treated with RINVOQ 15 mg monotherapy and 0 patients treated with upadacitinib 30 mg monotherapy through Week 14. In RA-II, no cases of arterial thrombosis were observed through 12/14 weeks. In RA-I, venous thrombosis was observed in 1 patient treated with MTX, 0 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg through Week 24. In RA-I, arterial thrombosis was observed in 1 patient treated with upadacitinib 30 mg through Week 24.
12-Month Exposure Dataset: Venous thrombosis events were reported in 5 patients (0.5 per 100 patient-years) treated with RINVOQ 15 mg and 4 patients (0.4 per 100 patient-years) treated with upadacitinib 30 mg.
Arterial thrombosis events were reported in 0 patients treated with RINVOQ 15 mg and 2 patients (0.2 per 100 patient-years) treated with upadacitinib 30 mg.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, alanine transaminase (ALT) and aspartate transaminase (AST) elevations ≥ 3 x upper limit of normal (ULN) in at least one measurement were observed in 2.1% and 1.5% of patients treated with RINVOQ 15 mg, and in 1.5% and 0.7% of patients treated with placebo, respectively. In RA-III and RA-V, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 1.0% of patients treated with RINVOQ 15 mg, 1.0% and 0% of patients treated with upadacitinib 30 mg and in 1.3% and 1.0% of patients treated with placebo, respectively.
In MTX-controlled trials, for up to 12/14 weeks, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 0.4% of patients treated with RINVOQ 15 mg, 1.7% and 1.3% of patients treated with upadacitinib 30 mg and in 1.9% and 0.9% of patients treated with MTX, respectively.
Upadacitinib treatment was associated with dose-related increases in total cholesterol, triglycerides and LDL cholesterol. Upadacitinib was also associated with increases in HDL cholesterol. Elevations in LDL and HDL cholesterol peaked by Week 8 and remained stable thereafter. In controlled trials, for up to 12/14 weeks, changes from baseline in lipid parameters in patients treated with RINVOQ 15 mg and upadacitinib 30 mg, respectively, are summarized below:
• Mean LDL cholesterol increased by 14.81 mg/dL and 17.17 mg/dL.
• Mean HDL cholesterol increased by 8.16 mg/dL and 9.01 mg/dL.
• The mean LDL/HDL ratio remained stable.
• Mean triglycerides increased by 13.55 mg/dL and 14.44 mg/dL.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related increases in creatine phosphokinase (CPK) values were observed. CPK elevations > 5 x ULN were reported in 1.0%, and 0.3% of patients over 12/14 weeks in the RINVOQ 15 mg and placebo groups, respectively. Most elevations >5 x ULN were transient and did not require treatment discontinuation. In RA-III and RA-V, CPK elevations > 5 x ULN were observed in 0.3% of patients treated with placebo, 1.6% of patients treated with RINVOQ 15 mg, and none in patients treated with upadacitinib 30 mg.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in neutrophil counts, below 1000 cells/mm3 in at least one measurement occurred in 1.1% and <0.1% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in neutrophil counts below 1000 cells/mm3 in at least one measurement occurred in 0.3% of patients treated with placebo, 1.3% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg. In clinical trials, treatment was interrupted in response to ANC less than 1000 cells/mm3
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.9%
and 0.7% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.5% of patients treated with placebo, 0.5% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, hemoglobin decreases below 8 g/dL in at least one measurement occurred in <0.1% of patients in both the RINVOQ 15 mg and placebo groups. In RA-III and RA-V, hemoglobin decreases below 8 g/dL in at least one measurement were observed in 0.3% of patients treated with placebo, and none in patients treated with RINVOQ 15 mg and upadacitinib 30 mg.
Adverse Reactions in Patients with Psoriatic Arthritis
A total of 1827 patients with psoriatic arthritis were treated with upadacitinib in clinical trials representing 1639.2 patient-years of exposure, of whom 722 were exposed to upadacitinib for at least one year. In the two Phase 3 trials, 907 patients received at least 1 dose of RINVOQ 15 mg, of whom 359 were exposed for at least one year.
Two placebo-controlled trials were integrated (640 patients on RINVOQ 15 mg once daily and 635 patients on placebo) to evaluate the safety of RINVOQ 15 mg in comparison to placebo for up to 24 weeks after treatment initiation.
Overall, the safety profile observed in patients with active psoriatic arthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis. During the 24-week placebo-controlled period, the frequencies of herpes zoster and herpes simplex were ≥1% (1.1% and 1.4%, respectively) with RINVOQ 15 mg and 0.8% and 1.3%, respectively with placebo. A higher incidence of acne and bronchitis was also observed in patients treated with RINVOQ 15 mg (1.3% and 3.9%, respectively) compared to placebo (0.3% and 2.7%, respectively).
Adverse Reactions in Patients with Atopic Dermatitis
Three Phase 3 (AD-1, AD-2, and AD-3) and one Phase 2b (AD-4) randomized, double-blind, placebo-controlled, multicenter trials evaluated the safety of RINVOQ in patients with moderate-to-severe atopic dermatitis. The majority of patients were White (68%) and male (57%). The mean age was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. In these 4 trials, 2612 patients were treated with RINVOQ 15 mg or 30 mg orally once daily, with or without concomitant topical corticosteroids (TCS).
In the Phase 3 clinical trials (AD-1, AD-2, and AD-3), a total of 1239 patients received RINVOQ 15 mg, of whom 791 were exposed for at least one year and 1246 patients received RINVOQ 30 mg, of whom 826 were exposed for at least one year.
Trials AD-1, AD-2, and AD-4 compared the safety of RINVOQ monotherapy to placebo through Week 16. Trial AD-3 compared the safety of RINVOQ + TCS to placebo + TCS through Week 16.
Weeks 0 to 16 (Trials AD-1 to AD-4)
In RINVOQ trials with and without TCS (Trials AD-1, 2, 3 and 4) through Week 16, the proportion of patients who discontinued treatment because of adverse reactions in the RINVOQ 15 mg, 30 mg and placebo groups were 2.3%, 2.9% and 3.8%, respectively. Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% in the RINVOQ 15 mg or 30 mg groups during the first 16 weeks of treatment.
Table 2: Adverse Reactions Reported in ≥ 1% of Patients with Atopic Dermatitis Treated with RINVOQ 15 mg or 30 mg
Adverse Reaction
Placebo RINVOQ 15 mg RINVOQ 30 mg n=902 (%) n=899 (%) n=906 (%)
Upper respiratory tract infection (URTI)* 17 23 25
Acne** 2 10 16
Herpes simplex*** 2 4 8
Headache 4 6 6
Increased blood creatine phosphokinase 2 5 6 Cough 1 3 3
Hypersensitivity**** 2 2 3
Folliculitis 1 2 3 Nausea 1 3 3
Abdominal pain***** 1 3 2 Pyrexia 1 2 2
Increased Weight 1 2 2 Herpes zoster****** 1 2 2
Influenza <1 2 2 Fatigue 1 1 2
Neutropenia <1 1 2
Myalgia 1 1 2
Influenza like illness 1 1 2
* Includes: laryngitis, laryngitis viral, nasopharyngitis, oropharyngeal pain, pharyngeal abscess, pharyngitis, pharyngitis streptococcal, pharyngotonsillitis, respiratory tract infection, respiratory tract infection viral, rhinitis, rhinolaryngitis, sinusitis, tonsillitis, tonsillitis bacterial, upper respiratory tract infection, viral pharyngitis, viral upper respiratory tract infection
** Includes: acne and dermatitis acneiform
*** Includes: genital herpes, genital herpes simplex, herpes dermatitis, herpes ophthalmic, herpes simplex, nasal herpes, ophthalmic herpes simplex, herpes virus infection, oral herpes
**** Includes anaphylactic reaction, anaphylactic shock, angioedema, dermatitis exfoliative generalized, drug hypersensitivity, eyelid oedema, face oedema, hypersensitivity, periorbital swelling, pharyngeal swelling, swelling face, toxic skin eruption, type I hypersensitivity, urticaria
***** Includes abdominal pain and abdominal pain upper
****** Includes herpes zoster and varicella
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg and/or 30 mg group and at a higher rate than in the placebo group through Week 16 included anemia, oral candidiasis, pneumonia, and the adverse event of retinal detachment.
The safety profile of RINVOQ through Week 52 was generally consistent with the safety profile observed at Week 16.
Overall, the safety profile observed in patients with AD treated with RINVOQ was similar to the safety profile in patients with RA. Other specific adverse reactions that were reported in patients with AD included eczema herpeticum/Kaposi’s varicelliform eruption.
Eczema Herpeticum/Kaposi’s Varicelliform Eruption
Placebo-controlled Period (16 weeks): Eczema herpeticum was reported in 4 patients (1.6 per 100 patient-years) treated with placebo, 6 patients (2.2 per 100 patient-years) treated with RINVOQ 15 mg and 7 patients (2.6 per 100 patient-years) treated with RINVOQ 30 mg.
12-Month Exposure (Weeks 0 to 52): Eczema herpeticum was reported in 18 patients (1.6 per 100 patient-years) treated with RINVOQ 15 mg and 17 patients (1.5 per 100 patient-years) treated with RINVOQ 30 mg.
Adverse Reactions in Patients with Ulcerative Colitis
RINVOQ was studied up to 8 weeks in patients with moderately to severely active ulcerative colitis in two randomized, double-blind, placebo-controlled induction studies (UC-1, UC-2) and a randomized, double-blind, placebo controlled, dose-finding study (UC-4; NCT02819635). Long term safety up to 52-weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (UC-3) and a long-term extension study.
In the two induction studies (UC-1, UC-2) and a dose finding study (UC-4), 1097 patients were enrolled of whom 719 patients received RINVOQ 45 mg once daily.
In the maintenance study (UC-3), 746 patients were enrolled of whom 250 patients received RINVOQ 15 mg once daily and 251 patients received RINVOQ 30 mg once daily.
Adverse reactions reported in ≥2% of patients in any treatment arm in the induction and maintenance studies are shown in Tables 3 and 4, respectively.
Table 3. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (UC-1, UC-2 and UC-4)
Adverse Reaction
Placebo RINVOQ 45 mg Once Daily N= 378 (%) N = 719 (%)
Upper respiratory tract infection* 7 9
Acne* 1 6
Increased blood creatine phosphokinase 1 5
Neutropenia* <1 5 Rash* 1 4
Elevated liver enzymes** 2 3 Lymphopenia* 1 3 Folliculitis 1 2
Herpes simplex* <1 2
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury and cholestasis.
Other adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 8 included herpes zoster and pneumonia.
Table 4. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (UC-3)1
Adverse Reaction Placebo RINVOQ 15 mg Once Daily RINVOQ 30 mg Once Daily n = 245 (%) n = 250 (%) n = 251 (%)
Upper respiratory tract infection* 18 16 20
Increased blood creatine phosphokinase 2 6 8
Neutropenia* 2 3 6
Elevated liver enzymes** 1 6 4 Rash* 4 5 5
Herpes zoster 0 4 4
Folliculitis 2 2 4
Hypercholesterolemia* 1 2 4 Influenza 1 3 3
Herpes simplex* 1 2 3
Lymphopenia* 2 3 2
Hyperlipidemia* 0 2 2
1 Patients who were responders to 8 weeks induction therapy with RINVOQ 45 mg once daily
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury, and cholestasis.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Overall, the safety profile observed in patients with ulcerative colitis treated with RINVOQ was generally similar to the safety profile in patients with RA and AD.
Induction Studies: In UC-1, UC-2, and UC-4, serious infections were reported in 5 patients (8.4 per 100 patient-years) treated with placebo and 9 patients (8.4 per 100 patient-years) treated with RINVOQ 45 mg through 8 weeks.
Placebo-controlled Maintenance Study: In UC-3, serious infections were reported in 8 patients (6.3 per 100 patient-years) treated with placebo, 8 patients (4.5 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (3.1 per 100 patient-years) treated with RINVOQ 30 mg through 52 weeks.
In studies UC-1, UC-2, and UC-4, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 1.5% of patients treated with RINVOQ 45 mg, and 0% of patients treated with placebo for 8 weeks. AST elevations to ≥ 3 x ULN occurred in 1.5% of patients treated with RINVOQ 45 mg, and 0.3% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN occurred in 0.4% of patients treated with RINVOQ 45 mg and 0% of patients treated with placebo.
In UC-3, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 4% of patients treated with RINVOQ 30 mg, 2% of patients treated with RINVOQ 15 mg, and 0.8% of patients treated with placebo for 52 weeks. Elevations of AST to ≥ 3 x ULN in at least one measurement were observed in 2% of patients treated with RINVOQ 30 mg, 1.6% of patients treated with RINVOQ 15 mg and 0.4% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN were observed in 0.8% of patients treated with 30 mg, 0.4% of patients treated with 15 mg, and 0.4% of patients treated with placebo.
Overall, laboratory abnormalities observed in patients with ulcerative colitis treated with RINVOQ were similar to those described in patients with RA.
Adverse Reactions in Patients with Ankylosing Spondylitis
A total of 596 patients with ankylosing spondylitis were treated with RINVOQ 15 mg in the two clinical trials representing 577.3 patient-years of exposure, of whom 228 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active ankylosing spondylitis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis and psoriatic arthritis.
During the 14-week placebo-controlled period in Trial AS-I, the frequency of headache was 5.4% with RINVOQ 15 mg and 2.1% with placebo. During the 14-week placebo-controlled period in Trial AS-II, the frequency of headache was 3.3% with RINVOQ 15 mg and 1.4% with placebo.
Strong CYP3A4 Inhibitors
Upadacitinib exposure is increased when RINVOQ is co-administered with a strong CYP3A4 inhibitor (such as ketoconazole and clarithromycin), which may increase the risk of RINVOQ adverse reactions. Monitor patients closely for adverse reactions when co-administering RINVOQ 15 mg once daily with strong CYP3A4 inhibitors.
For patients with atopic dermatitis, coadministration of RINVOQ 30 mg once daily with strong CYP3A4 inhibitors is not recommended.
For patients with ulcerative colitis taking strong CYP3A4 inhibitors, reduce the RINVOQ induction dosage to 30 mg once daily. The recommended maintenance dosage is 15 mg once daily.
Strong CYP3A4 Inducers
Upadacitinib exposure is decreased when RINVOQ is co-administered with strong CYP3A4 inducers (such as rifampin), which may lead to reduced therapeutic effect of RINVOQ. Coadministration of RINVOQ with strong CYP3A4 inducers is not recommended.
Pregnancy
Risk Summary
Available data from the pharmacovigilance safety database and postmarketing case reports on use of RINVOQ in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. Based on animal studies, RINVOQ has the potential to adversely affect a developing fetus. Advise patients of reproductive potential and pregnant patients of the potential risk to the fetus.
In animal embryo-fetal development studies, oral upadacitinib administration to pregnant rats and rabbits at exposures equal to or greater than approximately 1.6 and 15 times the 15 mg dose, 0.8 and 7.6 times the 30 mg dose, and 0.6 and 5.6 times the maximum recommended human dose (MRHD) of 45 mg (on an AUC basis) resulted in dose-related increases in skeletal malformations (rats only), an increased incidence of cardiovascular malformations (rabbits only), increased post-implantation loss (rabbits only), and decreased fetal body weights in both rats and rabbits. No developmental toxicity was observed in pregnant rats and rabbits treated with oral upadacitinib during organogenesis at exposures approximately 0.29 and 2.2 times the 15 mg dose, 0.15 times and 1.1 times the 30 mg dose, and at 0.11 and 0.82 times the MHRD (on an AUC basis).
In a pre- and post-natal development study in pregnant female rats, oral upadacitinib administration at exposures approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and the same as the MRHD (on an AUC basis) resulted in no maternal or developmental toxicity (see Data)
The background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages are 2-4% and 15-20%, respectively.
Report pregnancies to the AbbVie Inc.’s Adverse Event reporting line at 1-888-633-9110, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
In an oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 5, 25, and 75 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that consisted of misshapen humerus and bent scapula) at exposures equal to or greater than approximately 1.7 times the
15 mg dose, 0.9 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 5 mg/kg/day and higher). Additional skeletal malformations (bent forelimbs/hindlimbs and rib/vertebral defects) and decreased fetal body weights were observed in the absence of maternal toxicity at an exposure approximately 84 times the 15 mg dose, 43 times the 30 mg dose, and 31 times the MRHD (on an AUC basis at a maternal oral dose of 75 mg/kg/day).
In a second oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 1.5 and 4 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that included bent humerus and scapula) at exposures approximately 1.6 times the 15 mg dose, 0.8 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 4 mg/kg/day). No developmental toxicity was observed in rats at an exposure approximately 0.29 times the 15 mg dose, 0.15 times the 30 mg dose, and 0.11 times the MRHD (on an AUC basis at a maternal oral dose of 1.5 mg/kg/day).
In an oral embryo-fetal developmental study, pregnant rabbits received upadacitinib at doses of 2.5, 10, and 25 mg/kg/day during the period of organogenesis from gestation day 7 to 19. Embryolethality, decreased fetal body weights, and cardiovascular malformations were observed in the presence of maternal toxicity at an exposure approximately 15 times the 15 mg dose, 7.6 times the 30 mg dose, and 5.6 times the MRHD (on an AUC basis at a maternal oral dose of 25 mg/kg/day). Embryolethality consisted of increased post-implantation loss that was due to elevated incidences of both total and early resorptions. No developmental toxicity was observed in rabbits at an exposure approximately 2.2 times the 15 mg dose, 1.1 times the 30 mg dose, and 0.82 times the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
In an oral pre- and post-natal development study, pregnant female rats received upadacitinib at doses of 2.5, 5, and 10 mg/kg/day from gestation day 6 through lactation day 20. No maternal or developmental toxicity was observed in either mothers or offspring, respectively, at an exposure approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and at approximately the same exposure as the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
There are no data on the presence of upadacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Available pharmacodynamic/toxicological data in animals have shown excretion of upadacitinib in milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with RINVOQ, and for 6 days (approximately 10 half-lives) after the last dose.
A single oral dose of 10 mg/kg radiolabeled upadacitinib was administered to lactating female Sprague-Dawley rats on post-partum days 7-8. Drug exposure was approximately 30-fold greater in milk than in maternal plasma based on AUC0-t values. Approximately 97% of drug-related material in milk was parent drug.
Females and Males of Reproductive Potential Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ [see Use in Specific Populations]
Based on animal studies, upadacitinib may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations]. Advise female patients of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose.
Juvenile Idiopathic Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The safety and effectiveness of RINVOQ in pediatric patients with juvenile idiopathic arthritis, psoriatic arthritis, and ankylosing spondylitis have not been established.
The safety and effectiveness of RINVOQ in pediatric patients 12 years of age and older weighing at least 40 kg with atopic dermatitis have been established. A total of 344 pediatric patients aged 12 to 17 years with moderate to severe atopic dermatitis were randomized across three trials (AD-1, AD-2 and AD-3) to receive either RINVOQ 15 mg (N=114) or 30 mg (N=114) or matching placebo (N=116) in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the pediatric patients and adults. The adverse reaction profile in the pediatric patients was similar to the adults [see Adverse Reactions].
The safety and effectiveness of RINVOQ in pediatric patients less than 12 years of age with atopic dermatitis have not been established.
Ulcerative Colitis
The safety and effectiveness of RINVOQ in pediatric patients with ulcerative colitis have not been established.
Rheumatoid Arthritis and Psoriatic Arthritis
Of the 4381 patients treated in the five clinical trials, a total of 906 rheumatoid arthritis patients were 65 years of age or older, including 146 patients 75 years and older. Of the 1827 patients treated in the two psoriatic arthritis Phase 3 clinical trials, a total of 274 patients were 65 years of age or older, including 34 patients 75 years and older. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of overall adverse events, including serious infections, in patients 65 years of age and older.
Of the 2583 patients treated in the three Phase 3 clinical trials, a total of 120 patients with atopic dermatitis were 65 years of age or older, including 6 patients 75 years of age. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of serious infections and malignancies in those patients 65 years of age or older in the 30 mg dosing group in the long-term trials.
Of the 1097 patients treated in the controlled clinical trials, a total of 95 patients with ulcerative colitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ulcerative colitis to determine whether they respond differently from younger adult patients.
For patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), or severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2).
For patients with atopic dermatitis, the maximum recommended dosage is 15 mg once daily for patients with severe renal impairment. No dosage adjustment is needed in patients with mild or moderate renal impairment.
For patients with ulcerative colitis, the recommended dosage for severe renal impairment is 30 mg once daily for induction and 15 mg once daily for maintenance. No dosage adjustment is needed in patients with mild or moderate renal impairment.
RINVOQ has not been studied in patients with end stage renal disease (eGFR <15 mL/min/1.73m2). Use in patients with atopic dermatitis or ulcerative colitis with end stage renal disease is not recommended.
The use of RINVOQ has not been studied in patients with severe hepatic impairment (Child Pugh C), and therefore not recommended for use in patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ulcerative colitis, or ankylosing spondylitis.
For patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment.
For patients with ulcerative colitis, the recommended dosage for mild to moderate hepatic impairment is 30 mg once daily for induction and 15 mg once daily for maintenance
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients that they may be more likely to develop infections when taking RINVOQ. Instruct patients to contact their healthcare provider immediately during treatment if they develop any signs or symptoms of an infection [see Warnings and Precautions]
Advise patients that the risk of herpes zoster is increased in patients taking RINVOQ and in some cases can be serious [see Warnings and Precautions]
Inform patients that RINVOQ may increase their risk of certain cancers and that periodic skin examinations should be performed while using RINVOQ.
Advise patients that exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen [see Warnings and Precautions]
Major Adverse Cardiovascular Events
Inform patients that RINVOQ may increase their risk of major adverse cardiovascular events (MACE) including myocardial infarction, stroke,
and cardiovascular death. Instruct all patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see Warnings and Precautions]
Inform patients that events of deep venous thrombosis and pulmonary embolism have been reported in clinical trials with RINVOQ. Instruct patients to seek immediate medical attention if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions]
Advise patients to discontinue RINVOQ and seek immediate medical attention if they develop any signs and symptoms of allergic reactions [see Warnings and Precautions].
Inform patients that gastrointestinal perforations have been reported in clinical trials with RINVOQ and that risk factors include the use of NSAIDS or history of diverticulitis. Instruct patients to seek medical care immediately if they experience new onset of abdominal pain, fever, chills, nausea, or vomiting [see Warnings and Precautions].
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ [see Adverse Reactions]
Inform patients that RINVOQ may affect certain lab tests, and that blood tests are required before and during RINVOQ treatment [see Warnings and Precautions]
Vaccinations
Advise patients to avoid use of live vaccines with RINVOQ. Instruct patients to inform their healthcare practitioner that they are taking RINVOQ prior to a potential vaccination [see Warnings and Precautions]
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to RINVOQ during pregnancy may result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions and Use in Specific Populations]
Advise females of reproductive potential that effective contraception should be used during treatment and for 4 weeks following the final dose of upadacitinib [see Use in Specific Populations]
Advise females patients who are exposed to RINVOQ during pregnancy to contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Advise women not to breastfeed during treatment with RINVOQ and for 6 days after the last dose [see Use in Specific Populations]
Administration
Advise patients not to chew, crush, or split RINVOQ tablets. Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd. ©2019-2022 AbbVie Inc.
Ref: 20071734 Revised: April 2022
LAB-7082 MASTER
El cáncer de seno es el cáncer más comúnmente diagnosticado, y la primera causa de muerte, en mujeres. El Colegio Americano de Radiología recomienda el cernimiento con mamografía o tomosíntesis a partir de los 40 años para aquellas pacientes con un riesgo promedio para cáncer. El radiólogo especialista juega un rol importante en la detección temprana de esta condición utilizando varias modalidades diagnósticas. El cernimiento se hace mediante mamografía o su modalidad asociada conocida como tomosíntesis. La tomosíntesis tiene mayor sensibilidad y especificidad que la mamografía regular y delimita con más detalle los bordes en lesiones identificadas.
El ultrasonido o sonomamografia juega un rol importante en la evaluación de lesiones sólidas y quísticas, y contribuye en la evaluación de lesiones vistas en otras modalidades. El MRI se utiliza para evaluar extensión de enfermedad, envolvimiento de nódulos linfáticos y para evaluación de algunos casos especiales. La biopsia de seno es un procedimiento mínimamente invasivo en el cual se extrae una pequeña muestra de tejido mamario utilizando agujas guiadas por imágenes.
Cancer de seno, mamografia, tomosintesis, sonomamografia, magnetic resonance imaging (MRI) de seno, biopsia de seno, BI-RADS.
El cáncer de seno sigue siendo el cáncer más común diagnosticado, y la primera causa de muerte, en mujeres. Sin embargo, gracias a la mamografía, la mortalidad por cáncer de seno ha bajado hasta un 41%.1 La radiología juega un rol crucial en la detección temprana de cáncer de seno. Dentro del campo de la radiología existe la subespecialidad en imágenes de seno. Este radiólogo especialista interpreta las imágenes por las distintas modalidades y también realiza las biopsias de seno.

El Colegio Americano de Radiología recomienda el cernimiento con mamografía o tomosíntesis a partir de los 40 años para aquellas pacientes con un riesgo promedio para cáncer.
Sin embargo, aquellas pacientes con historial familiar de primera línea (madre o hermana) para cáncer de seno se debe comenzar 10 años antes de la edad del diagnóstico del familiar, pero no menos de 25 años. También existen pacientes con mutaciones genéticas qué deben empezar su cernimiento antes. Es importante que las pacientes discutan con sus médicos cuáles son sus riesgos para saber cuándo deben comenzar a examinarse.
El Colegio Americano de Radiología recomienda que las evaluaciones de seno sean clasificadas en base a un léxico estandarizado conocido como la clasificación BI-RADS (Breast Imaging and Reporting Data System). El léxico se utiliza para poder utilizar un lenguaje universal estandarizado referente al diagnóstico y clasificación de cáncer de seno y poder ofrecer a los referidos recomendaciones claras y consistentes. Además, el mismo contribuye a estandarizar estadísticas de diagnóstico.2
Gracias a la mamografía, podemos hacer el cernimiento de cáncer de seno y detectar lesiones pequeñas cuando aún no son palpables o sin síntomas.
La mamografía es una modalidad de rayos x. La técnica consiste en obtener dos imágenes comprimiendo cada seno, se obtienen vistas craneocaudal y mediolateral oblicua. El seno se compone de grasa y tejido mamario glandular. Mientras más tejido glandular mamario, más denso (o blanco) se ve el seno en la mamografía. La sensibilidad de la mamografía para detectar cáncer de seno varía entre un 70% a un 85% dependiendo de la densidad. 3, 4
La tecnología de la mamografía ha seguido evolucionando y la Food and Drug Administration (FDA) en el 2011 aprobó una modalidad de mamografía más sofisticada llamada tomosíntesis. Con ésta se obtiene la mamografía 2D tradicional y además se realiza una evaluación tridimensional (3D). La tecnología utiliza una angulación en arco que toma múltiples imágenes en dos proyecciones (frente y de perfil) del seno. Las imágenes obtenidas son procesadas brindando al radiólogo la posibilidad del análisis en tres dimensiones del seno.
La tomosíntesis tiene mayor sensibilidad y especificidad en la detección de imágenes en las diferentes variedades de densidad mamaria; sobre todo en el tejido mamario fibroglandular y heterogéneamente denso.3 Esta modalidad disminuye hasta un 17% la llamada al paciente para vistas adicionales.5,6 Y puede aumentar la detección de cáncer hasta un 53%.7, 8 También delimita mucho mejor los bordes de las lesiones que la mamografía tradicional.
En la mamografía o tomosíntesis podemos detectar microcalcificaciones, masas, asimetrías o distorsiones. Algunas microcalcificaciones basadas en su morfología irregular pueden estar asociadas a la presentación más temprana de cáncer de seno, carcinoma ductal in situ (DCIS), y la mamografía o tomosíntesis son la mejor modalidad para detectarlas.5
Es un estudio que utiliza ondas de sonidos para generar las imágenes. Es dependiente del operador, técnica y experiencia. El sonografista obtiene las imágenes y se someten para interpretación por el radiólogo. Las indicaciones incluyen evaluar hallazgos que se ven en la mamografía, tomosíntesis, MRI o masas palpables.9 El ultrasonido se utiliza para discernir si una masa es sólida o quística. Es importante recalcar que el ultrasonido o sonomamografía no sustituye la mamografía o tomosíntesis como cernimiento de cáncer de seno.
Es una modalidad mucho más sofisticada o especializada que utiliza campos magnéticos y ondas de radio para crear imágenes generadas por computadora del tejido mamario. Se inyecta contraste intravenoso de gadolinium para destacar y caracterizar las lesiones sólidas en el seno. Las indicaciones incluyen pacientes recién diagnosticados con cáncer de seno para evaluar extensión o enfermedad oculta y envolvimiento de los nódulos linfáticos. Ayuda en evaluar la respuesta en pacientes con terapia neoadyuvante antes y después del tratamiento.10 En pacientes de alto riesgo, tales como mutaciones genéticas, historial familiar de primer grado o pacientes con biopsias previas que muestran atipia, el MRI es otra herramienta que contribuye en el cernimiento. Otra indicación es evaluar la integridad de los implantes de silicona.
Revista Puertorriqueña de Medicina y Salud Pública
Es un procedimiento mínimamente invasivo en el cual se extrae una pequeña muestra de tejido mamario utilizando agujas guiadas por imágenes.
Luego las muestras son enviadas a analizar por el patólogo. Es un procedimiento ambulatorio, la paciente no tiene que estar en ayuna y se utiliza anestesia local.
Las biopsias se pueden hacer dirigidas por mamografía, también conocida como estereotaxia, por sonografía o por MRI. El procedimiento se debe hacer en la modalidad que se vea mejor la lesión.6 En la medida que se pueda tratamos de hacerlo por sonografía ya que es mucho más cómodo y fácil para el paciente.
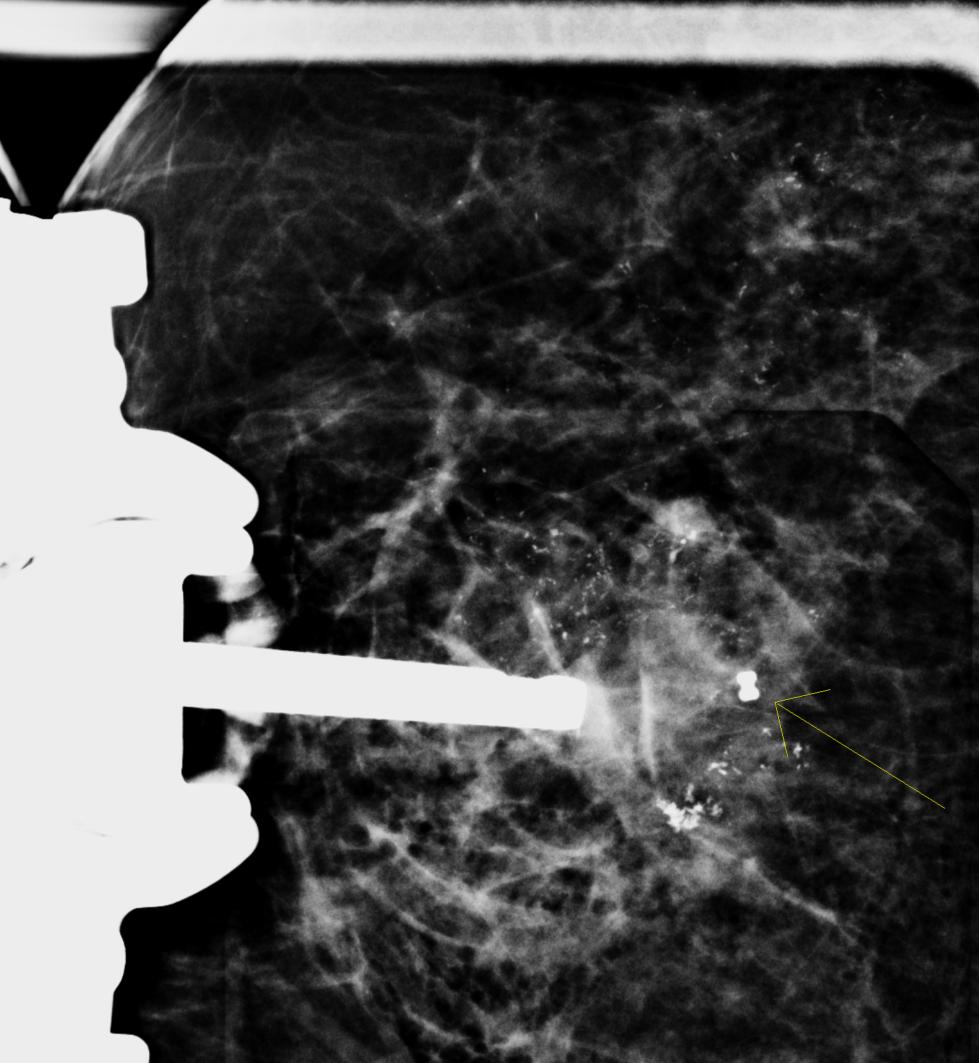
Luego de hacer la biopsia se coloca un marcador con el propósito de identificar el área por si requiere cirugía o tratamiento, o como referencia en futuros exámenes si el tejido sale benigno. Este marcador no interfiere en otros estudios como MRI o detectores de metales.
El resultado usualmente demora menos de una semana. El radiólogo tiene la responsabilidad de hacer un reporte por escrito del procedimiento que incluye la impresión y recomendación basada en la correlación de cómo se veía la lesión en las imágenes y si concuerda con el resultado de la patología. El resultado se le informa al paciente y también se envía reporte al médico referidor.
Gracias a la radiología podemos detectar cáncer de seno de una manera más temprana. Es importante conocer todas las modalidades radiológicas con sus indicaciones para así poder ayudar a nuestras pacientes. Sin embargo, la modalidad número uno para cernimiento y detección temprana sigue siendo la mamografía o tomosíntesis.
La mamografía o tomosíntesis no previenen el cáncer de seno, pero pueden salvar vidas si se detecta el cáncer lo más temprano posible.
Evaluación de ultrasonido en área palpable que demuestra un quiste sencillo. Cáncer avanzado invasivo ductal del seno izquierdo con invasión al músculo pectoral.
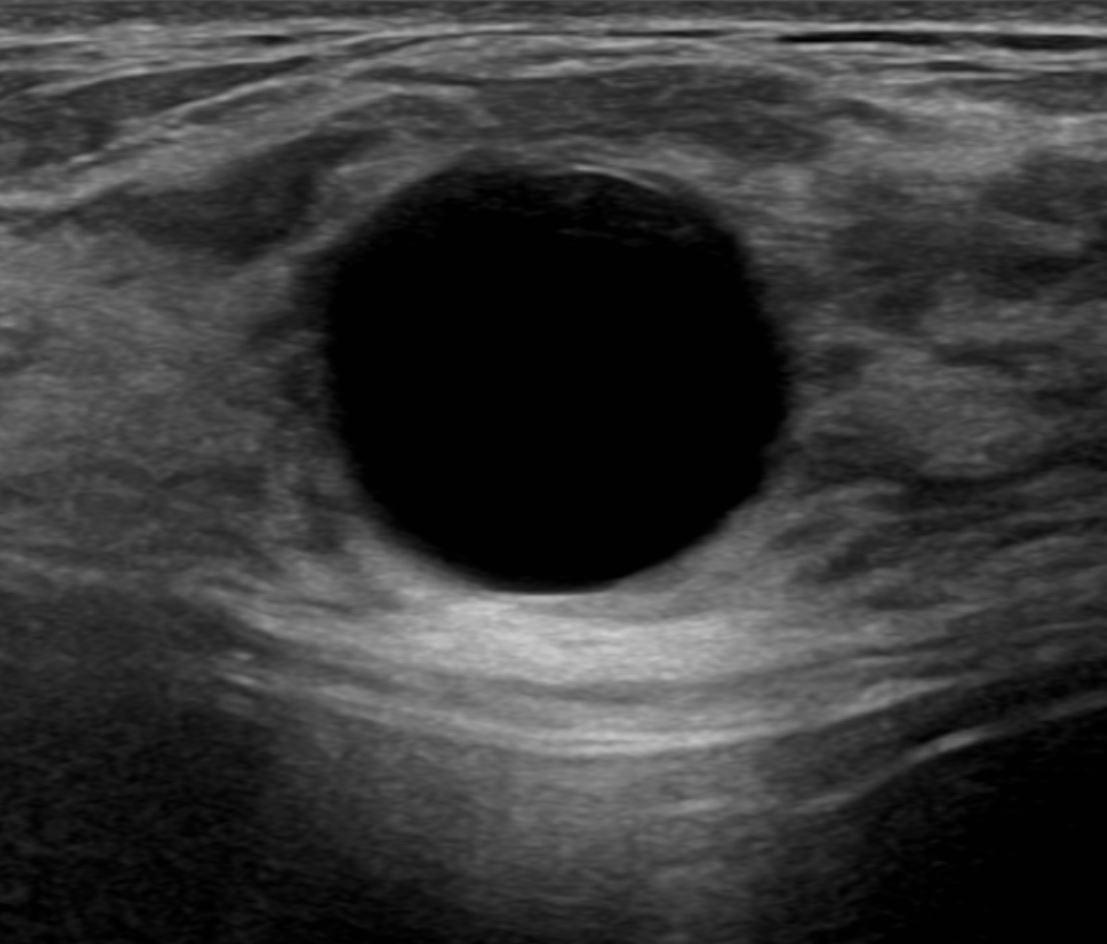
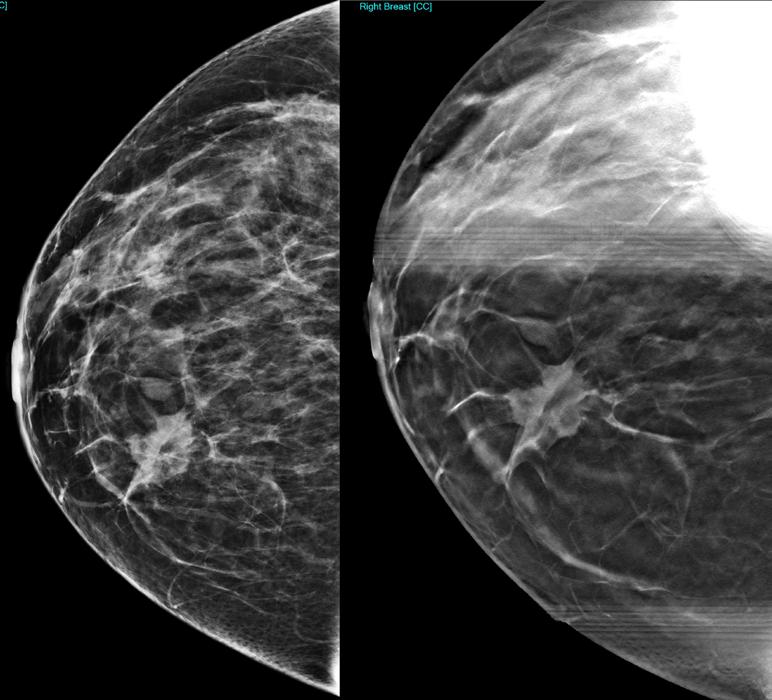
A A
Imagenes de cáncer invasivo ductal. Se puede comparar
B
el detalle de los bordes espiculados de la masa entre la mamografía 2D
(A) versus mamografía 3D o tomosíntesis (B). Mamografía de cernimiento con vistas mediolateral oblicua (A) y craneocaudal (B).
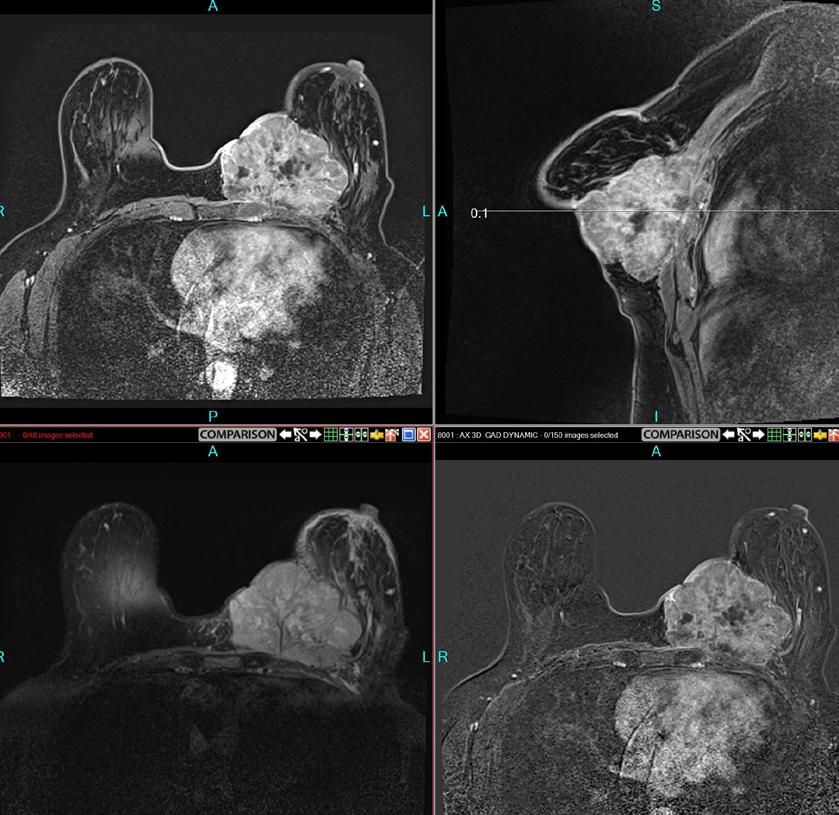
B
A B
Biopsia de seno dirigida por ultrasonido y colocación de marcador dentro de la masa. La densidad del seno graso (A) versus seno denso (B) , el tejido mamario denso se ve más blanco en la imagen.
She needs a treatment shown to reduce risk of recurrence in high-risk early breast cancer (EBC) 1

The first FDA-approved addition to adjuvant ET in nearly 2 decades 1-9
ET=endocrine therapy; HER2−=human epidermal growth factor receptor 2–negative; HR+=hormone receptor–positive.
VERZENIO® (abemaciclib) is indicated in combination with endocrine therapy (tamoxifen or an aromatase inhibitor) for the adjuvant treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, node-positive, early breast cancer at high risk of recurrence and a Ki-67 score ≥20% as determined by an FDA-approved test.1


Severe diarrhea associated with dehydration and infection occurred in patients treated with Verzenio. Across four clinical trials in 3691 patients, diarrhea occurred in 81 to 90% of patients who received Verzenio. Grade 3 diarrhea occurred in 8 to 20% of patients receiving Verzenio. Most patients experienced diarrhea during the first month of Verzenio treatment. The median time to onset of the first diarrhea event ranged from 6 to 8 days; and the median duration of Grade 2 and Grade 3 diarrhea ranged from 6 to 11 days and 5 to 8 days, respectively. Across trials, 19 to 26% of patients with diarrhea required a Verzenio dose interruption and 13 to 23% required a dose reduction.
Instruct patients to start antidiarrheal therapy, such as loperamide, at the first sign of loose stools, increase oral fluids, and notify their healthcare provider for further instructions and appropriate follow-up. For Grade 3 or 4 diarrhea, or diarrhea that requires hospitalization, discontinue Verzenio until toxicity resolves to ≤Grade 1, and then resume Verzenio at the next lower dose.
Neutropenia, including febrile neutropenia and fatal neutropenic sepsis, occurred in patients treated with Verzenio. Across four clinical trials in 3691 patients, neutropenia occurred in 37 to 46% of patients receiving Verzenio. A Grade ≥3 decrease in neutrophil count (based on laboratory findings) occurred in 19 to 32% of patients receiving Verzenio. Across trials, the median time to first episode of Grade ≥3 neutropenia ranged from 29 to 33 days, and the median duration of Grade ≥3 neutropenia ranged from 11 to 16 days. Febrile neutropenia has been reported in <1% of patients exposed to Verzenio across trials. Two deaths due to neutropenic sepsis were observed in MONARCH 2. Inform patients to promptly report any episodes of fever to their healthcare provider.
Monitor complete blood counts prior to the start of Verzenio therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, or delay in starting treatment cycles is recommended for patients who develop Grade 3 or 4 neutropenia. Severe, life-threatening, or fatal interstitial lung disease (ILD) or pneumonitis can occur in patients treated with Verzenio and other CDK4/6 inhibitors. In Verzenio-treated patients in EBC (monarchE), 3% of patients experienced ILD or pneumonitis of any grade: 0.4% were Grade 3 or 4 and there was one fatality (0.1%). In Verzenio-treated patients in MBC (MONARCH 1, MONARCH 2, MONARCH 3), 3.3% of Verzenio-treated patients had ILD or pneumonitis of any grade: 0.6% had Grade 3 or 4, and 0.4% had fatal outcomes. Additional cases of ILD or pneumonitis have been observed in the postmarketing setting, with fatalities reported.
Monitor patients for pulmonary symptoms indicative of ILD or pneumonitis. Symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams. Infectious, neoplastic, and other causes for such symptoms should be excluded by means of appropriate investigations. Dose interruption or dose reduction is recommended in patients who develop persistent or recurrent Grade 2 ILD or pneumonitis. Permanently discontinue Verzenio in all patients with Grade 3 or 4 ILD or pneumonitis.
monarchE was a phase III clinical trial that enrolled 5,637 peri- and postmenopausal adult women and men with HR+, HER2−, node-positive EBC at high risk of recurrence. High risk was defined as 4+ positive nodes, or 1-3 positive nodes with Grade 3 disease or tumor size ≥5 cm (central Ki-67 testing was conducted retrospectively for patients with untreated breast tissue samples), or 1-3 positive nodes with Ki-67 ≥20%. All patients completed primary treatment prior to 1:1 randomization to receive either 150-mg, twice-daily Verzenio plus SoC ET or SoC ET alone for 2 years. ET continued through 5-10 years as clinically indicated. The primary endpoint was IDFS.1,2


Grade ≥3 increases in alanine aminotransferase (ALT) (2 to 6%) and aspartate aminotransferase (AST) (2 to 3%) were reported in patients receiving Verzenio. Across three clinical trials in 3559 patients (monarchE, MONARCH 2, MONARCH 3), the median time to onset of Grade ≥3 ALT increases ranged from 57 to 87 days and the median time to resolution to Grade <3 was 13 to 14 days. The median time to onset of Grade ≥3 AST increases ranged from 71 to 185 days and the median time to resolution to Grade <3 ranged from 11 to 15 days. Monitor liver function tests (LFTs) prior to the start of Verzenio therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, dose discontinuation, or delay in starting treatment cycles is recommended for patients who develop persistent or recurrent Grade 2, or any Grade 3 or 4 hepatic transaminase elevation.
Please see Select Important Safety Information throughout and Brief Summary of full Prescribing Information for Verzenio on the following pages.

HR=hazard ratio; OS=overall survival.
See the breakthrough results at VerzenioData.com/EBC

At 3 years, Verzenio reduced the risk of recurrence by more than a third1


86.1% of patients remained recurrence-free with Verzenio plus ET vs 79.0% with ET alone.1


The number of events at the time of analysis was 104 with Verzenio plus ET vs 158 with ET alone.1

OS was immature. A total of 95 (4.7%) patients had died. Long-term follow-up is planned.1,2
This post hoc efficacy analysis was performed at a median follow-up of 27.1 months. Additional exploratory analyses were performed at this time; efficacy results for the subpopulation with high-risk clinicopathological features and Ki-67 ≥20% are provided.3*
Statistical significance was achieved for this subpopulation earlier at the final IDFS analysis. The result in this post hoc analysis cannot be interpreted as statistically significant.1
Venous thromboembolic events (VTE) were reported in 2 to 5% of patients across three clinical trials in 3559 patients treated with Verzenio (monarchE, MONARCH 2, MONARCH 3). VTE included deep vein thrombosis, pulmonary embolism, pelvic venous thrombosis, cerebral venous sinus thrombosis, subclavian and axillary vein thrombosis, and inferior vena cava thrombosis. In clinical trials, deaths due to VTE have been reported in patients treated with Verzenio.
Verzenio has not been studied in patients with early breast cancer who had a history of VTE. Monitor patients for signs and symptoms of venous thrombosis and pulmonary embolism and treat as medically appropriate. Dose interruption is recommended for EBC patients with any grade VTE and for MBC patients with a Grade 3 or 4 VTE.

Verzenio can cause fetal harm when administered to a pregnant woman, based on findings from animal studies and the mechanism of action. In animal reproduction studies, administration of abemaciclib to pregnant rats during the period of organogenesis caused teratogenicity and decreased fetal weight at maternal exposures that were similar to the human clinical exposure based on area under the curve (AUC) at the maximum recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Verzenio and for 3 weeks after the last dose. Based on findings in animals, Verzenio may impair fertility in males of reproductive potential. There are no data on the presence of Verzenio in human milk or its effects on the breastfed child or on milk production. Advise lactating women not to breastfeed during Verzenio treatment and for at least 3 weeks after the last dose because of the potential for serious adverse reactions in breastfed infants.
The most common adverse reactions (all grades, ≥10%) observed in monarchE for Verzenio plus tamoxifen or an aromatase inhibitor vs tamoxifen or an aromatase inhibitor, with a difference between arms of ≥2%, were diarrhea (84% vs 9%), infections (51% vs 39%), neutropenia (46% vs 6%), fatigue (41% vs 18%), leukopenia (38% vs 7%), nausea (30% vs 9%), anemia (24% vs 4%), headache (20% vs 15%), vomiting (18% vs 4.6%), stomatitis (14% vs 5%), lymphopenia (14% vs 3%), thrombocytopenia (13% vs 2%), decreased appetite (12% vs 2.4%), ALT increased (12% vs 6%), AST increased (12% vs 5%), dizziness (11% vs 7%), rash (11% vs 4.5%), and alopecia (11% vs 2.7 %).
The most frequently reported ≥5% Grade 3 or 4 adverse reaction that occurred in the Verzenio arm vs the tamoxifen or an aromatase inhibitor arm of monarchE were neutropenia (19.6% vs 1%), leukopenia (11% vs <1%), diarrhea (8% vs 0.2%), and lymphopenia (5% vs <1%).
Lab abnormalities (all grades; Grade 3 or 4) for monarchE in ≥10% for Verzenio plus tamoxifen or an aromatase inhibitor with a difference between arms of ≥2% were increased serum creatinine (99% vs 91%; .5% vs <.1%), decreased white blood cells (89% vs 28%; 19.1% vs 1.1%), decreased neutrophil count (84% vs 23%; 18.7% vs 1.9%), anemia (68% vs 17%; 1% vs .1%), decreased lymphocyte count (59% vs 24%; 13.2 % vs 2.5%), decreased platelet count (37% vs 10%; .9% vs .2%), increased ALT (37% vs 24%; 2.6% vs 1.2%), increased AST (31% vs 18%; 1.6% vs .9%), and hypokalemia (11% vs 3.8%; 1.3% vs 0.2%). Strong and moderate CYP3A inhibitors increased the exposure of abemaciclib plus its active metabolites to a clinically meaningful extent and may lead to increased toxicity. Avoid concomitant use of ketoconazole. Ketoconazole is predicted to increase the AUC of abemaciclib by up to 16-fold. In patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the Verzenio dose to 100 mg twice daily with concomitant use of strong CYP3A inhibitors other than ketoconazole. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the Verzenio dose to 50 mg twice daily with concomitant use of strong CYP3A inhibitors. If a patient taking Verzenio discontinues a strong CYP3A inhibitor, increase the Verzenio dose (after 3 to 5 half-lives of the inhibitor) to the dose that was used before starting the inhibitor. With concomitant use of moderate CYP3A inhibitors, monitor for adverse reactions and consider reducing the Verzenio dose in 50 mg decrements. Patients should avoid grapefruit products. Avoid concomitant use of strong or moderate CYP3A inducers and consider alternative agents. Coadministration of strong or moderate CYP3A inducers decreased the plasma concentrations of abemaciclib plus its active metabolites and may lead to reduced activity.
With severe hepatic impairment (Child-Pugh C), reduce the Verzenio dosing frequency to once daily. The pharmacokinetics of Verzenio in patients with severe renal impairment (CLcr <30 mL/min), end stage renal disease, or in patients on dialysis is unknown. No dosage adjustments are necessary in patients with mild or moderate hepatic (Child-Pugh A or B) and/or renal impairment (CLcr ≥30-89 mL/min).
Please see Select Important Safety Information throughout and Brief Summary of full Prescribing Information for Verzenio on the following pages.
AL HCP ISI_mE 12OCT2021
REFERENCES: 1. Verzenio [package insert]. Indianapolis, IN: Eli Lilly and Company. 2. Johnston SRD, Harbeck N, Hegg R, et al; monarchE Committee Members and Investigators. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, nodepositive high-risk, early breast cancer (monarchE). J Clin Oncol. 2020;38(34):3987-3998. doi:10.1200/JCO.20.02514. 3. Harbeck N, Rastogi P, Martin M, et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: updated efficacy and Ki-67 analysis from the monarchE study. Ann Oncol. 2021. doi:10.1016/j.annonc.2021.09.015. 4. Breast International Group (BIG) 1-98 Collaborative Group, Thürlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353(26):2747-2757. doi:10.1056/NEJMMoa052258. 5. Baum M, Buzdar A, Cuzick J, et al; ATAC (Arimidex, Tamoxifen Alone or in Combination) Trialists’ Group. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer. 2003;98(9):1802-1810. doi:10.1002/cncr.11745. 6. Howell A, Cuzick J, Baum M, et al; ATAC Trialists’ Group. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365(9453):60-62. doi:10.1016/SO140-6736(04)17666-6. 7. Mayer EL, Dueck AC, Martin M, et al. Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2021;22(2):212222. doi:10.10160/S1470-2045(20)30757-9. 8. A trial to evaluate efficacy and safety of ribociclib with endocrine therapy as adjuvant treatment in patients with HR+/HER2− early breast cancer (NATALEE). https://clinicaltrials. gov/ct2/show/ NCT03701334. Accessed February 12, 2021. 9. Loibl S, Marmé F, Martin M, et al. Palbociclib for residual high-risk invasive HR-positive and HER2-negative early breast cancer—the Penelope-B trial. J Clin Oncol. 2021;39(14):1518-1530. doi:10.1200/JCO.20.03639.
PP-AL-US-3237 03/2022 ©Lilly USA, LLC 2022. All rights reserved. Verzenio® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries or affiliates.

therapy (tamoxifen or an aromatase inhibitor) alone. Patients were randomly assigned to receive 150 mg of VERZENIO orally, twice daily, plus tamoxifen or an aromatase inhibitor, or tamoxifen or an aromatase inhibitor, for two years or until discontinuation criteria were met. The median duration of VERZENIO treatment was 24 months.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, leukopenia, diarrhea, and lymphopenia.
Fatal adverse reactions occurred in 0.8% of patients who received VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor), including: cardiac failure (0.1%), cardiac arrest, myocardial infarction, ventricular fibrillation, cerebral hemorrhage, cerebrovascular accident, pneumonitis, hypoxia, diarrhea and mesenteric artery thrombosis (0.03% each).
Permanent VERZENIO treatment discontinuation due to an adverse reaction was reported in 19% of patients receiving VERZENIO, plus tamoxifen or an aromatase inhibitor. Of the patients receiving tamoxifen or an aromatase inhibitor, 1% permanently discontinued due to an adverse reaction. The most common adverse reactions leading to VERZENIO discontinuations were diarrhea (5%), fatigue (2%), and neutropenia (0.9%).
Dose interruption of VERZENIO due to an adverse reaction occurred in 62% of patients receiving VERZENIO plus tamoxifen or aromatase inhibitors. Adverse reactions leading to VERZENIO dose interruptions in ≥5% of patients were diarrhea (20%), neutropenia (16%), leukopenia (7%), and fatigue (5%).
Dose reductions of VERZENIO due to an adverse reaction occurred in 44% of patients receiving VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor). Adverse reactions leading to VERZENIO dose reductions in ≥5% were diarrhea (17%), neutropenia (8%), and fatigue (5%).
The most common adverse reactions reported (≥20%) in the VERZENIO, plus tamoxifen or an aromatase inhibitor, arm and ≥2% higher than the tamoxifen or an aromatase inhibitor arm were: diarrhea, infections, neutropenia, fatigue, leukopenia, nausea, anemia, and headache. Adverse reactions are shown in Table 1 and laboratory abnormalities are shown in Table 2.
Table 1: Adverse Reactions (≥10%) of Patients Receiving VERZENIO Plus Tamoxifen or an Aromatase Inhibitor [with a Difference between Arms of ≥2%] in monarchE VERZENIO Plus Tamoxifen or an Aromatase Inhibitor N=2791
Tamoxifen or an Aromatase Inhibitor N=2800
All Gradesa % Grade 3 % Grade 4 % All Gradesb % Grade 3 % Grade 4 %
Gastrointestinal Disorders
Diarrhea 84 8 0 9 0.2 0 Nausea 30 0.5 0 9 <0.1 0 Vomiting 18 0.5 0 4.6 0.1 0 Stomatitisc 14 0.1 0 5 0 0
Infections and Infestations
Infectionsd 51 4.9 0.6 39 2.7 0.1
General Disorders and Administration Site Conditions
Fatiguee 41 2.9 0 18 0.1 0
Nervous System Disorders
Headache 20 0.3 0 15 0.2 0
Dizziness 11 0.1 0 7 <0.1 0
Metabolism and Nutrition Disorders
Decreased appetite 12 0.6 0 2.4 <0.1 0
Skin and Subcutaneous Tissue Disorders
Rashf 11 0.4 0 4.5 0 0
Alopecia 11 0 0 2.7 0 0
a Includes the following fatal adverse reactions: diarrhea (n=1), and infections (n=4)
b Includes the following fatal adverse reactions: infections (n=5)
c Includes mouth ulceration, mucosal inflammation, oropharyngeal pain, stomatitis.
d Includes all reported preferred terms that are part of the Infections and Infestations system organ class. Most common infections (>5%) include upper respiratory tract infection, urinary tract infection, and nasopharyngitis.
e Includes asthenia, fatigue.
f Includes exfoliative rash, mucocutaneous rash, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculo-papular, rash maculovesicular, rash morbilliform, rash papular, rash papulosquamous, rash pruritic, rash vesicular, vulvovaginal rash.
Clinically relevant adverse reactions in <10% of patients who received VERZENIO in combination with tamoxifen or an aromatase inhibitor in monarchE include:
• Pruritus-9%
• Dyspepsia-8%
• Nail disorder-6% (includes nail bed disorder, nail bed inflammation, nail discoloration, nail disorder, nail dystrophy, nail pigmentation, nail ridging, nail toxicity, onychalgia, onychoclasis, onycholysis, onychomadesis)
• Lacrimation increased-6%
• Dysgeusia-5%
• Interstitial lung disease (ILD)/pneumonitis-3% (includes pneumonitis, radiation pneumonitis, interstitial lung disease, pulmonary fibrosis, organizing pneumonia, radiation fibrosis – lung, lung opacity, sarcoidosis)
• Venous thromboembolic events (VTEs)-3% (includes catheter site thrombosis, cerebral venous thrombosis, deep vein thrombosis, device related thrombosis, embolism, hepatic vein thrombosis, jugular vein occlusion, jugular vein thrombosis, ovarian vein thrombosis, portal vein thrombosis, pulmonary embolism, subclavian vein thrombosis, venous thrombosis limb)
Table 2: Laboratory Abnormalities (≥10%) in Patients
Receiving VERZENIO Plus Tamoxifen or an Aromatase Inhibitor [with a Difference between Arms of ≥2%] in monarchE
VERZENIO Plus Tamoxifen or an Aromatase Inhibitor N=2791
Tamoxifen or an Aromatase Inhibitor N=2800
All Grades % Grade 3 % Grade 4 % All Grades % Grade 3 % Grade 4 %
Creatinine increased 99 0.5 0 91 <0.1 0 White blood cell decreased 89 19 <0.1 28 1.1 0
Neutrophil count decreased 84 18 0.7 23 1.6 0.3 Anemia 68 1.0 0 17 0.1 0 Lymphocyte count decreased 59 13 0.2 24 2.4 0.1
Platelet count decreased 37 0.7 0.2 10 0.1 0.1 Alanine aminotransferase increased 37 2.5 <0.1 24 1.2 0 Aspartate aminotransferase increased
31 1.5 <0.1 18 0.9 0 Hypokalemia 11 1.2 0.1 3.8 0.1 0.1
Effect of
CYP3A Inhibitors
Strong and moderate CYP3A4 inhibitors increased the exposure of abemaciclib plus its active metabolites to a clinically meaningful extent and may lead to increased toxicity.
Avoid concomitant use of ketoconazole. Ketoconazole is predicted to increase the AUC of abemaciclib by up to 16-fold.
In patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the VERZENIO dose to 100 mg twice daily with concomitant use of strong CYP3A inhibitors other than ketoconazole. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the VERZENIO dose to 50 mg twice daily with concomitant use of strong CYP3A inhibitors. If a patient taking VERZENIO
VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021 VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021
Verzenio, AL HCP BS_MonE 12OCT2021 - 7 x 10
Revista Puertorriqueña de Medicina y Salud Pública
discontinues a strong CYP3A inhibitor, increase the VERZENIO dose (after 3-5 half-lives of the inhibitor) to the dose that was used before starting the inhibitor. Patients should avoid grapefruit products.
With concomitant use of moderate CYP3A inhibitors, monitor for adverse reactions and consider reducing the VERZENIO dose in 50 mg decrements, if necessary.
Coadministration of strong or moderate CYP3A inducers decreased the plasma concentrations of abemaciclib plus its active metabolites and may lead to reduced activity. Avoid concomitant use of strong or moderate CYP3A inducers and consider alternative agents.
Based on findings in animals and its mechanism of action, VERZENIO can cause fetal harm when administered to a pregnant woman. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus. In animal reproduction studies, administration of abemaciclib during organogenesis was teratogenic and caused decreased fetal weight at maternal exposures that were similar to human clinical exposure based on AUC at the maximum recommended human dose (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
In an embryo-fetal development study, pregnant rats received oral doses of abemaciclib up to 15 mg/kg/day during the period of organogenesis. Doses ≥4 mg/kg/day caused decreased fetal body weights and increased incidence of cardiovascular and skeletal malformations and variations. These findings included absent innominate artery and aortic arch, malpositioned subclavian artery, unossified sternebra, bipartite ossification of thoracic centrum, and rudimentary or nodulated ribs. At 4 mg/kg/day in rats, the maternal systemic exposures were approximately equal to the human exposure (AUC) at the recommended dose.
There are no data on the presence of abemaciclib in human milk, or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed infants from VERZENIO, advise lactating women not to breastfeed during VERZENIO treatment and for 3 weeks after the last dose.
Based on animal studies, VERZENIO can cause fetal harm when administered to a pregnant woman.
Verify pregnancy status in females of reproductive potential prior to initiating treatment with VERZENIO.
Advise females of reproductive potential to use effective contraception during VERZENIO treatment and for 3 weeks after the last dose.
Based on findings in animals, VERZENIO may impair fertility in males of reproductive potential.
The safety and effectiveness of VERZENIO have not been established in pediatric patients.
Of the 2791 VERZENIO-treated patients in monarchE, 15% were 65 years of age or older and 2.7% were 75 years of age or older.
Of the 900 patients who received VERZENIO in MONARCH 1, MONARCH 2, and MONARCH 3, 38% were 65 years of age or older and 10% were 75 years of age or older. The most common adverse reactions (≥5%) Grade 3 or 4 in patients ≥65 years of age across MONARCH 1, 2, and 3 were: neutropenia, diarrhea, fatigue, nausea, dehydration, leukopenia, anemia, infections, and ALT increased.
No overall differences in safety or effectiveness of VERZENIO were observed between these patients and younger patients.
No dosage adjustment is required for patients with mild or moderate renal impairment (CLcr ≥30-89 mL/min, estimated by Cockcroft-Gault [C-G]). The pharmacokinetics of abemaciclib in patients with severe renal impairment (CLcr <30 mL/min, C-G), end stage renal disease, or in patients on dialysis is unknown.
No dosage adjustments are necessary in patients with mild or moderate hepatic impairment (Child-Pugh A or B).
Reduce the dosing frequency when administering VERZENIO to patients with severe hepatic impairment (Child-Pugh C).
Additional information can be found at www.verzenio.com
VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021 VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021
Verzenio,
Revista Puertorriqueña de Medicina y Salud Pública

AUTORES:
Vivian S. Green, PhD, MS, LND Public Health Program, Ponce Health Sciences University vgreen@psm.edu (Vivian S. Green es el autor para correspondencia)
Soreli Santana Public Health Program, Ponce Health Sciences University Ssantana13@stu.psm.edu
Mayra Roubert Public Health Program, Ponce Health Sciences University mroubert@psm.edu
Yemile Ron-Suarez, MD Merck & Co., Inc., Carolina, PR, USA yemile_ron@merck.com
Luis Carlos Mejia-Rivera, PhD, MD Merck & Co., Inc., Carolina, PR, USA luis.mejia-rivera@merck.com
Felipe Arbelaez, MD, MBA Merck & Co., Inc., Kenilworth, NJ, USA felipe_arbelaez@merck.com
Homero Monsanto, PhD Merck & Co., Inc., Carolina, PR, USA homero_monsanto@merck.com
Edgar I. Miranda, PhD Merck & Co., Inc., Carolina, PR, USA edgar.i.miranda.avalo@merck.com
Cecile Marqués-Goyco, MD Merck & Co., Inc., Carolina, PR, USA cecile_marques-goyco@merck.com
Juan C. Orengo, MD, MPH, PhD Public Health Program, Ponce Health Sciences University jorengo@psm.edu
CONFLICTO DE INTERÉS: YR, HM, LCM-R, EIM y CM-G son empleados de Merck & Co., Inc., Carolina, PR, EEUU.; FA es Merck & Co., Inc., Kenilworth, NJ, Estados Unidos; JCO era un empleado de Merck & Co., Inc., Carolina, PR, EEUU cuando se diseñó el estudio.
FINANCIACIÓN: MSD (IA) LLC, Carolina, Puerto Rico
CONTRIBUCIONES DE LOS AUTORES: VSG, SS, MR y JCO trabajaron en la implementación del estudio, análisis de datos, interpretación de resultados y redacción del manuscrito; YR, HM, FA, LCM-R, EIM, CM-G y JCO colaboraron en el diseño de la investigación. El manuscrito fue revisado y aprobado por todos los autores.
AGRADECIMIENTOS: Queremos agradecer a los pacientes que participaron voluntariamente en el estudio y a los médicos que colaboraron en la implementación del proyecto.
PALABRAS CLAVE: Hipoglucemia, diabetes, hispano, Puerto Rico, calidad de vida
KEYWORDS: hypoglycemic, diabetes, Hispanic, Puerto Rico, quality of life
En Puerto Rico la prevalencia de la diabetes mellitus tipo 2 (DMT2) es del 15,5% (2016). Los objetivos de nuestro estudio fueron: 1) evaluar la prevalencia de hipoglucemia en pacientes con DMT2 que toman sulfonilureas; 2) evaluar la prevalencia de pacientes con DMT2 que no están en la meta de HbA1c de <7%; 3) evaluar la evaluación del autocuidado, la calidad de vida y la adherencia general al tratamiento. Se realizó un estudio exploratorio transversal en consultorios médicos privados en todo Puerto Rico (2016-2017). Los pacientes con diagnóstico de DMT2 tratados con sulfonilureas fueron invitados a participar en el estudio. Los datos sobre el cumplimiento, la calidad de vida, el estilo de vida y los factores conductuales fueron informados por el participante y se recopilaron los datos clínicos de la historia clínica. Se utilizaron estadísticas descriptivas e inferenciales para resumir los datos generales de los pacientes. (260 participantes fueron inscritos en el estudio). Durante los últimos seis meses la hipoglucemia autoinformada fue del 41,2%, siendo el 58,9% la prevalencia de pacientes con la última HbA1c <7%. El 85,3% de los participantes siempre utilizaron sus medicamentos según lo prescrito por su médico. El 60,3% de los participantes no sigue una dieta especial para diabéticos y el 67,2% no sigue un programa de ejercicios de rutina. La puntuación del Índice EQ-5D para los pacientes hipoglucémicos fue de 0,77 (DE=0,21) y para los pacientes no hipoglucémicos fue de 0,85 (DE=0,14) (p <0,05). La prevalencia de hipoglucemia autoinformada se encuentra en el rango de otros estudios realizados en Asia y Europa; el objetivo de HbA1c<7 no se alcanzó en el 41,1% de la muestra. Estos hallazgos resaltan la necesidad médica insatisfecha de tratamiento que puede ayudar a los pacientes a alcanzar los objetivos del tratamiento con DMT2.
In Puerto Rico the prevalence of Type 2 Diabetes Mellitus (T2DM) is 15,5% (2016). The objectives of our study were: 1) to assess the prevalence of hypoglycemia in T2DM patients taking sulfonylureas; 2) to assess the prevalence of T2DM patients who are not at the HbA1c goal of <7%; 3) to assess self-care evaluation, quality of life and the general adherence to treatment. An exploratory cross-sectional study was conducted in private medical offices across Puerto Rico (2016-2017). Patients with a diagnosis of T2DM treated with sulfonylureas were invited to participate in the study. Data on adherence, quality of life, lifestyle/behavioral factors were reported by the participant and clinical data from the medical record were collected. Descriptive and inferential statistics were used to summarize general data of the patients. (260 participants were enrolled in the study).
During the last six months the self-reported hypoglycemia was 41,2%, being 58,9% the prevalence of patients with the last HbA1c <7%. 85,3% of the participants always used their medicines as prescribed by their doctor. The 60,3% of the participants do not follow a special diabetic diet and 67,2% do not follow a routine exercise program. The EQ-5D Index score for hypoglycemic patients was 0.77 (SD=0.21) and for non-hypoglycemic patients was 0.85 (SD=0.14) (p <0.05). The prevalence of self-reported hypoglycemia is in the range of other studies conducted in Asia and Europa; the target of HbA1c < 7 was not achieved by the 41,1% of the sample. These findings highlight the unmet medical need for treatment that can help patients achieve T2DM treatment goals.
La prevalencia de diabetes mellitus (DM) en Puerto Rico se encuentra entre las más altas de América Latina y el Caribe 1 . Se estima que casi 400 000 personas entre 20 a 79 años tienen diabetes en Puerto Rico. Según el Sistema de Vigilancia de los Factores de Riesgo del Comportamiento (BRFSS, por sus siglas en inglés) de los Centros para el Control y la Prevención de Enfermedades (CDC) en 2010, al 8,7% de la población de los Estados Unidos se le había informado que tenía DM y en 2018 la prevalencia era del 11%, siendo para Puerto Rico de un 15,5% 2
La DM fue la tercera causa de muerte en Puerto Rico en el 2016 y así desde el 2004 3,4 . La tasa de mortalidad por DM más alta en Puerto Rico fue durante el 2011 (75,5 por cada 100 000 habitantes), pero datos de 2016 mostraron que la tasa de mortalidad fue de 66,3 por cada 100 000 habitantes 3,4 . En Puerto Rico, en el 2013, el presupuesto invertido a DM ascendió a $138 416 722 y el grupo de edad entre 65 a 69 años invirtió aun más con aproximadamente $25,4 millones.
La diabetes mellitus tipo 2 es la forma más común de diabetes mellitus (DM2) y representa el 90% de todos los casos diagnosticados en todo el mundo
A su vez es la causa principal de morbilidad y mortalidad5 , particularmente con enfermedad cardiovascular, neuropatía y enfermedad renal crónica6,7. Además, dado que las enfermedades cardiovasculares y la DM son la primera y la tercera causa de muerte en Puerto Rico (2016) respectivamente, la prevención y el tratamiento de la diabetes mellitus son de máxima prioridad4 . Se estima que el 80-90% de los pacientes con DM2 requieren tratamientos farmacológicos de un tipo u otro.
El objetivo del tratamiento es controlar los parámetros glucémicos (HbA1c Objetivo <7%,
American Diabetes Association, 2014) para minimizar el riesgo de complicaciones a largo plazo y aliviar cualquier síntoma8 Sin embargo, se ha documentado que el control glucémico intensivo puede conducir a eventos hipoglucémicos en pacientes diabéticos, por lo que actúa como un obstáculo significativo en los esfuerzos de control glucémico en el tratamiento del paciente9 . La hipoglucemia ocurre cuando los niveles de glucosa en sangre se encuentran por debajo de la línea de base, generalmente a 70 mg /dL8 o menos. En algunos pacientes diabéticos la respuesta al glucagón se ve afectada y su respuesta a otras hormonas (epinefrina y adrenalina) puede elevar los niveles de glucosa en sangre. El problema de estos pacientes surge debido a su tratamiento: la insulina y los agentes hipoglucemiantes orales (OHA), como las sulfonilureas (SU), aumentan la producción de insulina, por lo que los niveles de glucosa no pueden volver a su rango normal 10 . Según los hallazgos de tres grandes ensayos aleatorios controlados (ACCORD, ADVANCE, VADT) 11,13 , la
Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública
CARACTERÍSTICAS
25.2% 21
relevancia clínica de los eventos hipoglucémicos se ha vuelto cada vez más importante en las guías internacionales para el tratamiento de DM2. La Declaración de Consenso 2013 del Algoritmo de Manejo Integral de la Diabetes de AACE destacó que “minimizar el riesgo de hipoglucemia es una prioridad. Es una cuestión de seguridad, adherencia y costo”.14 En los estudios de Real-Life Effectiveness and Care Patterns of Diabetes Management (RECAP-DM) Asia Pacífico y RECAP DM de la Unión Europea, el 36% de los pacientes tratados en la región de Asia Pacífico y el 38% de los pacientes tratados en la región de la Unión Europea informaron síntomas de hipoglucemia.15,16 En Puerto Rico, donde la diabetes es altamente predominante, la prevalencia de hipoglucemia en pacientes tratados con SU en un entorno real aún no se ha evaluado. Por lo tanto, los objetivos generales de este estudio fueron: 1) evaluar la prevalencia de hipoglucemia (autoinformada o documentada en la historia clínica del participante) en pacientes diabéticos tipo 2 tratados con SU (como monoterapia o en combinación con metformina) en Puerto Rico y 2) estimar la prevalencia de pacientes que alcanzan la meta de HbA1c de <7%.
Se realizó un estudio exploratorio de corte transversal en consultorios médicos privados en las regiones norte y sur de Puerto Rico durante los años 2016-2017. Los pacientes con un diagnóstico de diabetes mellitus tipo 2 tratados con sulfonilureas como monoterapia o en combinación con metformina y otros criterios de inclusión fueron invitados a participar en el estudio. Los objetivos del estudio fueron, primero, evaluar la prevalencia de hipoglucemia en pacientes con diabetes mellitus tipo 2 que toman sulfonilureas en Puerto Rico; segundo, evaluar la prevalencia de pacientes con diabetes mellitus tipo 2 en Puerto Rico que no están en el objetivo de HbA1c de <7 %; y tercero, evaluar el autocuidado, la calidad de vida y la adherencia general al tratamiento. La población de estudio eran pacientes adultos diagnosticados con diabetes mellitus tipo 2 de acuerdo con los criterios de la ADA a) Síntomas de DM más concentración de glucosa en plasma casual ≥200 mg/dl (11.1 mmol/l); b) FPG ≥ 126 mg/ dl (7,0 mmol/l); y c) PG≥200 mg /dl de 2 h (11.1 mmol/l) durante un OGTT) de 30 años de edad o más que han estado tomando SU (terapia mono o en combinación con metformina) durante al menos 6 meses.
14%
11.2% 34.6%
25.2% 42.1% 13.1% 19.6%
25.2% 42.2% 14% 18.6%
13.7% 25% 19.6%
34 59.7(14.6)
29 42.4% 14.6% 17.9%
17.8% 9.2%
19
74 14 15
82 21
53 48 3 4 7 4.8 , 12.0* -4.3 , 0.7
19.6% 28.5% 31.4% 2% 3.7% 2.7%
Medio tiempo Desempleado (Buscando trabajo) Retirado Ama de casa Discapacitado
22.8% 52% 43.9%
126 59 21
72 79 47
48.6% 21.7% 1.3% 0.8%
36.4% 27.8%
39
31.5% 31.8% 15.4% 40 38 33 2 2 0
El 36,7% de la muestra eran hombres y el 63,3% mujeres. La edad media fue de 63,4 años (DE=14) y la mediana fue de 65 años. La edad promedio de las mujeres fue de 63,7 años (DE=14) y la de los hombres de 63,1 años (DE=14); las diferencias no fueron estadísticamente significativas.
La edad media al diagnóstico de diabetes mellitus (DM) fue de 53,2 años (DE=14) y la mediana de 55 años; en las mujeres, la edad promedio al diagnóstico fue de 53,7 años (DE=14) y en los hombres fue de 52,5 años (DE=15); la duración promedio de la enfermedad desde el diagnóstico fue de 10,4 años (DE=10) y la mediana de 8 años, en mujeres fue de 10,1 (DE=9,7) y en hombres de 10,9 (DE=10,4) la mediana fue de 8,5 años y 7 años para mujeres y hombres respectivamente, sin diferencias estadísticamente significativas.
En los últimos seis meses, el 41,2% de los participantes en el estudio informaron un episodio de hipoglucemia como se define en el estudio. En cuanto a la relación entre hombres y mujeres, 40% (38) y 43,1% (69) respectivamente informaron algún episodio de hipoglucemia.
En la tabla 1 podemos ver la edad media en el momento del diagnóstico y la duración promedio de la DM según el paciente que informó haber sufrido un episodio de hipoglucemia o no. Encontramos diferencias estadísticamente significativas en la edad del paciente y en la edad de diagnóstico. Los pacientes con hipoglucemia tenían una edad menor y eran más jóvenes cuando fueron diagnosticados. También podemos observar los resultados relacionados con las características sociodemográficas de la muestra, tanto para los pacientes que notificaron hipoglucemia como los que no la notificaron.
El 66,4% (168) de la muestra informó que sus padres también habían sido diagnosticados con DM, para los participantes no hipoglucémicos fue del 58,5% (89) y para los hipoglucémicos del 75,2% (79), por lo que se encontro una diferencia estadísticamente significativa (p <0.05) para estos dos grupos, lo que representa para los pacientes con hipoglucemia 2,01 veces más posibilidades (OR) de tener padres diabéticos (IC 95%: 1,16; 3,49).
El 65% (169) de la muestra nunca había fumado; aquellos que habían dejado de fumar o fumaban actualmente representaban el 25,8% (67) y 8,8% (23) respectivamente. En relación con los participantes no hipoglucémicos, el 63,4% (97) nunca había fumado, el 28,1% (43) había dejado de fumar y el 8,5% (13) continuó fumando; para los pacientes con hipoglucemia, el 67% (72) nunca había fumado, el 22,4% (24) había dejado de fumar y el 9,3% (10) fumaba actualmente.
El 49,8% (129) de los participantes informaron que realizaban algún tipo de actividad física, siendo el 53,9% (82) de los no hipoglucémicos y el 43,9% (47) de los hipoglucémicos. El 69,8% (180) de la muestra notificó que ingiere una dieta baja en azúcar, lo que corresponde al 73,5% (111) de los pacientes no hipoglucémicos y al 64,5% (69) de los hipoglucémicos. Una dieta baja en calorías es seguida por el 46,5% (111) de los participantes, esta misma dieta es seguida por el 48,7% (73) de los no hipoglucémicos y el 43,4% (46) de los hipoglucémicos.
El índice de masa corporal (IMC) medio de la muestra fue de 30 (DE=6,8) y la mediana de 28,6 en hipoglucémicos; el promedio fue de 31,3 (DE=6,7) y la mediana fue de 30,2, en no hipoglucémicos, la media fue de 29 (DE=6,8) y la mediana de 27,3; al comparar las medias encontramos una diferencia estadísticamente significativa (p <0.05) con una diferencia entre
las medias de -2,3 (95% Ci -4,0, -0,6). Los pacientes no hipoglucémicos tienen 1,9 (IC 95% 1.1.3.2) más posibilidades de tener un peso saludable que los pacientes que presentaron hipoglucemia en los últimos seis meses.
En la tabla 2 podemos observar la clasificación de los participantes según el índice de masa corporal.
En el 67,5% (172) de la muestra no se informaron cambios en el peso en los últimos seis meses ni en el 62,6% (67) de los pacientes con hipoglucemia y el 70,9% (105) de los participantes no hipoglucémicos. Por otro lado, el 9,4% (24) de los participantes informaron pérdida de peso, así como el 13,1% (14) de los pacientes con hipoglucemia y el 6,8% (10) de los pacientes sin hipoglucemia (Figura 1). No encontramos diferencias estadísticamente significativas en la ganancia o pérdida de peso entre pacientes con hipoglucemia y pacientes no hipoglucémicos. Sin embargo, se observa una mayor pérdida de peso en pacientes con hipoglucemia.
Los pacientes que informaron haber tenido un episodio de hipoglucemia en los últimos seis meses informaron haber sufrido “leve” (síntomas de bajo nivel de azúcar en la sangre definidos como poca o ninguna interrupción de las actividades, y no sienten la ayuda necesaria para controlar los síntomas), “moderado” (síntomas de bajo nivel de azúcar en la sangre definidos como cierta interrupción de actividades, pero no siente la asistencia necesaria para controlar los síntomas), “grave” (síntomas de bajo nivel de azúcar en la sangre definidos como que sentían que necesitaba la ayuda de otros para controlar los síntomas (por ejemplo, para llevar comida o bebida) o “muy grave”
(síntomas de bajo nivel de azúcar en la sangre definidos como atención médica necesaria (por ejemplo, llamar a una ambulancia, visitar una sala de emergencias u hospital, o ser visto por un médico o enfermera) síntomas en el 68,9% (73) , 29,9% (32), 6,5% (7) y 2,8% (3) respectivamente. 16,9% (25) informaron que no sentían “ninguna preocupación” por sus síntomas, 18,7% (20) “sin molestias”, 34,6 % (37) una “pequeña molestia”, 15% (16) “algo de incomodidad” y 8,4% (9) “mucha incomodidad “.
En relación con la información que el médico discutió en el momento del diagnóstico de DM encontramos que la dieta, los niveles de glucosa en la sangre, el control de la presión arterial, la salud emocional, el manejo del nivel alto de azúcar en la sangre y la cobertura del
No hypoglycemic patients
Hypoglycemic patients
0.00%
Figura 1. Prevalencia de pérdida de peso notificada por el participante
6.80% 13.10% Prevalence 2.00% 4.00% 6.00% 8.00% 10.00% 12.00% 14.00%
seguro de salud se discutieron menos con los hipoglucémicos, siendo esta diferencia estadísticamente significativo (p <0.05). En la información discutida por el médico con el paciente en una revisión de seguimiento de la patología encontramos que a los pacientes hipoglucémicos se les habla menos sobre la dieta, la salud emocional y el manejo del nivel
Tabla 3. Información discutida por el médico con el paciente al momento del diagnóstico
alto de azúcar en la sangre, siendo para estas características la diferencia estadísticamente significativa (p <0.05).
Las tablas 3 y tabla 4 presentan la información que fue discutida por el médico con el paciente en el momento del diagnóstico de DM y se dialoga en una revisión de la enfermedad, respectivamente.
107(69.9%) 57(53.8%)* 164(63.3%)
Dieta 139(90.8%) 81(76.4%)* 220(84.9%) Factores culturales 42(27.5%) 24(22.6%) 66(25.5%) Actividad f ísica 132(86.3%) 88(83.0%) 220(84.9%) Monitoreo de la glucosa 124(81 0%) 82(77 4%) 206(79 5%) Opciones de tratamiento 89(58.2%) 58(54.7%) 147(56.8%) Niveles de glucosa en sangre 113(73.9%) 61(57.5%)* 174(67.2%) Presión sanguínea 133(86 9%) 82(77 4%)* 215(83 0%) Colesterol 124(81 0%) 78(73 6%) 202(78 0%) Empleo 44(28 9%) 26(24 5%) 70(27 1%) Hábito tabáquico 51(33.6%) 37(34.9%) 88(34.1%) Ingesta de alcohol 50(32 7%) 32(30 2%) 82(31 7%) Salud emocional 94(61 4%) 41(38 7%)* 135(52 1%) Manejo de niveles altos de glucosa en sangre
Cobertura de seguro médico 51(33.3%) 22(20.8%)* 73(28.2%)
No discutió nada 2(5.6%) 2(5.9%) 4(5.7%) No recuerda 7(19 4%) 7(20 6%) 14(20 0%)
* Diferencia estadísticamente significativa (p<0.05)
Tabla
visita de seguimiento
Infor mación No Hipoglucémico Hipog lucémico
T odos
Dieta 139(90 8%) 79(74 5%)* 41(15 8%)
Factores culturales 39(25 5%) 20(18 9%) 59(22 8%)
Actividad f ísica 124(81 0%) 77(72 6%) 201(77 6%) Monitoreo de la glucosa 121(79.1%) 77(72.6%) 198(76.4%)
Opciones de tratamiento 80(52 3%) 58(54 7%) 138(53 3%) Niveles de glucosa en sangre 116(75.8%) 74(69.8%) 190(73.4%)
Presión sanguínea 129(84 3%) 80(83 4%) 2010(81 1%) Colesterol 113(73 9%) 78(73 6%) 191(73 7%) Empleo 35(23 0%) 31(29 2%) 66(25 6%) Hábito tabáquico 45(29 6%) 24(22 6%) 69(26 7%) Ingesta de alcohol 52(34 4%) 30(28 3%) 82(31 9%) Salud emocional 78(51.3%) 37(34.9%)* 115(44.6%)
Manejo de niveles bajos de glucosa en sangre
Manejo de niveles altos de glucosa en sangre
97(63.4%) 58(54.7%) 155(59.8%)
105(68.6%) 55(51.9%)* 160(61.8%)
Cobertura de seguro médico 53(34.9%) 28(26.4%) 81(31.4%) No discutió nada 1(3 1%) 3(7 5%) 4(5 6%) No recuerda 6(18.8%) 7(17.5%) 13(18.1%)
* Diferencia estadísticamente significativa (p<0.05)
El 26,3% (40) de los no hipoglucémicos y el 21% (22) de los hipoglucémicos notificaron que nunca omiten alimentos cuando trabajan, mientras que el 0,7% (1) de los no hipoglucémicos y el 3,8% (4) de ellos lo hacen. El 46,7% (121) de los participantes han sido referidos a un educador en diabetes o a un dietista o nutricionista, el 43% (46) de los pacientes con hipoglucemia informaron que fueron referidos a uno de los especialistas anteriores mientras que los no hipoglucémicos el 49,3% (75) informó haber recibido referidos. De los que recibieron un referido, el 50% (60) fue en los primeros meses, el 5% (6) en los primeros tres meses, el 10% (12) en los primeros seis meses, el 11,7% (14) en el primer año, el 12,5 % (15) después del primer año y 10,8% (13) nunca asistieron a la cita. En relación con los pacientes con hipoglucemia, el 32,6% (15) fue en los primeros meses, el 6,5% (3) en los primeros tres meses, el 4,3%
(2) en los primeros seis meses, el 21,7% (10) en el primer año, el 17,4% (8) después del primer año y el 17,4% (4) nunca asistieron a la cita en comparación con el 61.6% (45), 2,7% (2), 13,7% (10), 5,5% (4), 9,6% (7) y 6,8% (5) de pacientes no hipoglucémicos. Los pacientes que presentaron hipoglucemia visitaron al educador o al dietista en menor frecuencia durante los primeros tres meses y lo visitaron más en el primer año, siendo estas diferencias estadísticamente significativas (p <0.05).
El 58,6% (150) de la muestra informó que no desembolsó dinero para pagar los medicamentos recetados para la diabetes, el 28,5% (73) pagó un copago, el 3,9% (10) pagó por algunos de ellos y el 9% (23) pagó por todos. Para el 59,8% (153) es muy importante en el diálogo con el médico, el costo de los medicamentos en la elección de su tratamiento médico, para el 8,2% (21) no es importante.
La tabla 5 presenta los valores clínicos de la
muestra total para pacientes con episodios de hipoglucemia en los últimos seis meses y, para aquellos participantes que no los notificaron, encontramos diferencias estadísticamente significativas (p <0.05) en los valores de HDL, triglicéridos y cintura. La prevalencia de participantes con el último valor de HbA1c <7% fue del 58,9%.
En relación con la calidad de vida, se modificaron las respuestas al cuestionario EQ-5D-3L. Las tres posibles respuestas a cada dimensión se redujeron a dos, si el participante no presentaba ningún problema se consideraba “sin problemas” y si el participante respondía “algún problema” (el paciente presenta limitaciones para realizar alguna actividad) o se sentía incapacitado en la dimensión estudiada se consideró “con problemas”. En la tabla 6 podemos ver los resultados para cada
dimensión en el caso de que el participante respondió “sin problemas”, según el participante informó si había sufrido hipoglucemia o no. Para la dimensión de movilidad, dolor y ansiedad/depresión encontramos una diferencia estadísticamente significativa (p<0.05), presentando los pacientes con hipoglucemia 1.8 (IC 95%: 1.1, 3.0) más posibilidad de presentar problemas de movilidad, 1.9 (IC 95%): 1.2.3.2) más posibilidades de presentar problemas de dolor y 2,3 (IC 95%: 1.4.3.9) más posibilidades de presentar ansiedad/ depresión.
El puntaje del índice EQ-5D (las cinco preguntas del EQ-5D-3L se usaron para calcular este puntaje, el 0 se refiere al peor estado de salud (muerte) y el 1 al mejor estado de salud) fue de 0.82 (DE=0.17) para toda la muestra del estudio, siendo para los pacientes hipoglucémicos de 0,77 (DE=0,21) y para los pacientes no hipoglucémicos de 0,85 (DE=0.14), encontrando esta diferencia estadísticamente significativa (p <0.05), para una diferencia de las medias de 0,08 (IC del 95% de diferencia: - 1,26; - 0,35 )
En nuestro estudio la prevalencia de hipoglucemia informada por los pacientes con diabetes tipo 2 tratados con sulfonilureas fue del 41,2%, lo que corresponde a la prevalencia encontrada en otros estudios. En el estudio RECAP-DM de Asia-Pacífico realizado por Chan et al (2010) en una muestra de 2257 pacientes con diabetes tipo 2, la prevalencia de hipoglucemia fue del 35,8%, siendo el 41,7% la prevalencia en China (muestra de 484 pacientes), similar a la encontrada en Puerto Rico. Por otro lado, la prevalencia en Taiwán fue del 29,4% en una muestra de 609 pacientes. En general, en el estudio de Chan et al., el 11,6% informó síntomas “graves” de hipoglucemia y el 8,2% síntomas “muy graves”. En nuestro estudio encontramos 6,1% y 2,6% respectivamente, que es similar al encontrado en China que fue de 6,9% y 4,0% respectivamente16. En el estudio realizado por Alvarez-Guisasola et al (2008) en varios países europeos (España, Francia, Alemania, Reino Unido, Polonia, Noruega y Finlandia), en una muestra de 1709 pacientes
Tabla 5. Valores clínicos
Valor clínico No hipoglucémico Hipoglucémico Todos
HbA1c Media DE Mediana
7 1 1 4 6 8
7 7 7 3 6 7
7 3 4 7 6 8 Glucosa en ayunas Media DE Mediana
130 6 45 3 123 0
167 7 44 7 166 0
132 3 50 4 127 5
172 1 39 1 171 0
169 6 42 6 167 0
131 1 47 4 124 0 Colesterol Media DE Mediana
HDL * Media DE Mediana
LDL Media DE Mediana
Triglicéridos * Media DE Mediana
Presión sistólica Media DE Mediana
52 0 25 5 48 1
90 0 34 9 87 8
133 9 63 9 119 0
46 4 13 6 44 0
95 1 34 5 93 0
171 6 109 9 145
49 8 21 9 46 7
92 2 34 7 89 0
147 6 84 9 128 0
125 6 17 5 125 0
128 3 14 5 129 0
126 5 16 4 128 0 Presión diastólica Media DE Mediana
74 1 13 3 78 0
77 1 9 6 80 0
75 4 12 0 80 0 Creatinina Media DE Mediana
2 3 11 5 0 8
88 7 13 2 86 4
* Diferencia estadísticamente significativa (p<0.05)
2 6 10 7 0 8
93 1 14 3 91 4
90 4 13 8 87 0
2 4 11 1 0 8 Cintura * Media DE Mediana
diagnosticados con diabetes tipo 2 con una edad promedio de 62.9 ± 10,6 años, la prevalencia general de hipoglucemia encontrada fue de un 38,4%, la prevalencia más baja se encontró en Alemania con un 24,2% y la más alta en el Reino Unido con un 53,8%, mientras que la más similar a Puerto Rico se encontró en Francia, que fue de un 38,7 %. Como en nuestro estudio no se encontraron diferencias estadísticamente significativas relacionadas con el valor objetivo de una HbA1c en los límites recomendados entre los pacientes
que informaron hipoglucemia y los que no15. Majanovic et al (2017) en el estudio RECAP-DM en la región de los Balcanes con una muestra de 573 pacientes con diabetes tipo 2 y una edad media de 67,2 años encontraron una prevalencia de hipoglucemia del 36,6%; de estos, 62,8% tenían síntomas leves, 31,2% moderados, 4% severos y 2 % muy severos, resultados similares a los nuestros 17 González y Cols. en el estudio RECAP-DM realizado en Argentina en el 2015 con una muestra de 397 participantes y una edad
promedio de 62,5 años encontraron una prevalencia de hipoglucemia autoinformada de 45,5% y diabetes controlada del 36,4%; para Puerto Rico la prevalencia fue de 41,2% y 58,9% respectivamente. Similar a nuestro estudio, el tiempo promedio con el diagnóstico de diabetes en Argentina fue de 10 años con una edad promedio de 52,7 años al tiempo del diagnóstico, siendo Puerto Rico de 10,4 años y 53,2 años 18
En el estudio Exhype realizado en Suecia en 2010 por Petterson et al. con una muestra de 430 pacientes, la prevalencia informada de hipoglucemia fue del 34%19 . También en Suecia, Lundkvist et al (2005) encontraron una prevalencia de hipoglucemia del 37% en una muestra de 399 pacientes diabéticos tipo 2 con una edad media de 65 ± 11 años20 . Rombopoulos et al. (2013) en el estudio HYPO implementado en Grecia, en una muestra de 6631 pacientes con diabetes tipo 2, evidenciaron que la prevalencia estimada de hipoglucemia informada por los pacientes fue del 41,9%, presentando el 41% del total de la muestra los valores de la HbA1c en los límites recomendados21 . En España en 2013, Orozco-Beltrán et al. encontraron una prevalencia de hipoglucemia (eventos de salud no graves) informada por pacientes con diabetes tipo 2 del 39% 22 . Edidge et al. (2015) en un metanálisis encontraron una prevalencia del 33% (IC 95% 24% - 42%) de hipoglucemia en los paciente tratados con terapia con sulfonilureas23
En relación con el IMC, en nuestro estudio encontramos una mayor prevalencia de obesidad (51,4%) en los pacientes que informaron al menos un episodio de hipoglucemia frente a los que no notificaron hipoglucemia. Sin embargo, no encontramos diferencias estadísticamente significativas al agrupar las categorías en “sobrepeso + obesidad” y “peso saludable”. En el estudio de Álvarez-Guisasola (2008) no encontraron diferencias en el peso entre los pacientes que informaron un episodio de hipoglucemia y los que no lo informaron15 . Petterson et al. (2011) encontraron que los pacientes que no presentaban hipoglucemia o hipoglucemia leve tenían un IMC de 28.8 ± 4.4 frente a 28.5 ± 4.1 en pacientes con hipoglucemia moderada o peor 19 . Nunes et al. (2017) en un estudio realizado en 143 635 pacientes con diabetes tipo 2 que usaron sulfonilureas, el IMC promedio de la muestra total fue de 32.3 kg/m2. En pacientes con algún evento de
LOS OBJETIVOS DEL ESTUDIO FUERON, PRIMERO, EVALUAR LA PREVALENCIA DE HIPOGLUCEMIA EN PACIENTES CON DIABETES MELLITUS TIPO 2 QUE TOMAN SULFONILUREAS EN PUERTO RICO; SEGUNDO, EVALUAR LA PREVALENCIA DE PACIENTES CON DIABETES MELLITUS TIPO 2 EN PUERTO RICO QUE NO ESTÁN EN EL OBJETIVO DE HBA1C DE <7 %; Y TERCERO, EVALUAR EL AUTOCUIDADO, LA CALIDAD DE VIDA Y LA ADHERENCIA GENERAL AL TRATAMIENTO.
hipoglucemia, el IMC fue de 31,7 kg/m2 y en aquellos que no presentaron hipoglucemia, de 32,4 kg/m2. En nuestra muestra fue de 30kg/m2, 31.3 kg/m2 y 29 kg/m2 para el total de participantes, pacientes que informaron hipoglucemia y los que no respectivamente24 . Majanovic et al. (2017) encontraron que un 28,2% informó un aumento de peso en el último año, en nuestro estudio el 23,1% notificó lo mismo 17 Petterson et al. (2011) encontraron una puntuación del índice EQ-5D no ponderada para toda la muestra de 0.81 ± 0.22 similar a la encontrada por nosotros que fue de 0.82 ± 0.17. En ese estudio agruparon a los pacientes que no habían notificado hipoglucemia con los que informaron hipoglucemia leve. Ambos grupos de pacientes fueron comparados con aquellos que habían notificado una hipoglucemia moderada o peor. La puntuación del índice EQ-5D fue de 0.83 ± 0.21 y 0.75 ± 0.26 respectivamente 19 . Rombopoulos et al. encontraron que los pacientes que informaron un episodio de hipoglucemia tenían una peor calidad de vida que aquellos que no presentaron ningún evento de hipoglucemia21 . Zhang et al. (2010) en su revisión sistemática de la carga de hipoglucemia en pacientes con diabetes tipo 2 encontraron que los pacientes que presentaron algún evento de hipoglu-
cemia tenían valores de calidad de vida (EQ-5D) más bajos que aquellos que no presentaron ningún evento. También encontraron valores más bajos al evaluar el puntaje de la Escala Visual Analógica25 . Marrett et al. (2009) en un estudio basado en Internet y con una muestra de 1984 pacientes con DM2 en tratamiento con agentes antihiperglucemiantes orales encontraron que los pacientes con hipoglucemia informaron una peor puntuación del índice EQ-5D que aquellos que no la presentaron (0,78 frente a 0,86) 26 . En nuestro estudio, los valores del índice EQ-5D fueron de 0.85 ± 0.14 y 0.77 ± 0.21 para los pacientes que no informaron hipoglucemia y para aquellos que la notificaron. Respectivamente, otros autores encontraron valores similares a estos; a su vez, las puntuaciones de la Escala Visual Analógica fueron más bajas para los pacientes con hipoglucemia que para los que no la informaron20
Aproximadamente el 25% de las hospitalizaciones en pacientes de edad avanzada se deben a hipoglucemia 27. Zhang et al. (2010) encontraron un mayor costo médico y una mayor pérdida de días de trabajo. López et al. (2015) encontraron que en 367 602 pacientes con diabetes tipo 2, beneficiarios de Medicare, el 2,9% de los blancos solicitaron servicios de salud relacionados con eventos de hipoglucemia en comparación con el 3,6% de los hispanos y el 4,7% de los negros, lo que podría generar inequidad en la salud 25,28 . Estos datos refuerzan la importancia de realizar estudios como el que hemos presentado.
Las generalizaciones del presente estudio pueden estar limitadas por las características del tipo de diseño, incluso cuando se trata de un estudio del mundo real. El estudio fue exploratorio con un diseño transversal. La falta de información en Puerto Rico relacionada con los eventos de hipoglucemia en pacientes con diabetes tipo 2 justifica este tipo de estudio con el fin de encontrar respuestas y desarrollar nuevas preguntas que puedan guiar nuevos estudios. Al ser un estudio transversal, no nos permite temporalizar la información obtenida. No podemos establecer la relación causa-efecto, por ejemplo, en nuestro
estudio, los pacientes que presentaron hipoglucemia tenían un IMC más alto que aquellos que no, mientras que en la literatura encontramos resultados opuestos. Por otro lado, una mayor proporción de pacientes con hipoglucemia informaron haber perdido peso, podríamos plantearnos la siguiente pregunta: ¿los pacientes con hipoglucemia tienen un IMC similar a los que no tienen hipoglucemia y antes de un episodio de hipoglucemia presentan una disminución de peso? Otro tipo de sesgo que podemos encontrar es el de recuerdo. Incluso teniendo en cuenta estas limitaciones, el estudio es de gran importancia porque es el primero de este tipo que se lleva a cabo en Puerto Rico en un entorno del mundo real y proporciona información muy útil para pacientes y médicos. Por ejemplo, encontramos valores altos de triglicéridos y niveles bajos de HDL en pacientes con hipoglucemia que fueron estadísticamente significativos y valores más altos en colesterol, LDL, presión sistólica y diastólica, creatinina y HbA1c también en estos pacientes, con diferencias no estadísticamente significativas.
Como resumen final, podemos decir que la prevalencia de hipoglucemia en pacientes con diabetes mellitus tipo 2 tratados con sulfonilureas en Puerto Rico es similar a la encontrada en la literatura. Los pacientes que informaron un episodio de hipoglucemia tuvieron más limitaciones en la movilidad, más dolor y ansiedad/depresión, lo que evidencia una puntuación más baja de índice calidad de vida. Por último, un gran número de pacientes no tenía la HbA1c controlada.
1. IDF Diabetes Atlas 6th Edition. (n.d.). Country Estimates Table 2013. Retrieved from htpp://www. idf.org/atlasmap/atlasmap
2. Centers for Disease Control and Prevention, CDC. (2019). BRFSS Prevalence & Trends Data [online]. Retrieved Sept. 23, 2019, from https://www.cdc.gov/ brfss/brfssprevalence/
3. Sánchez-Hernández, E., Morales-González, J. J., Machín-Rivera, S., & Torres-Concepción, K. (2015, Septiembre). Informe de Salud de Puerto Rico 2015. Resumen General de la Salud en Puerto Rico. San Juan, Puerto Rico: Departamento de Salud de Puerto Rico
4. Departamento de Salud (2019). Informe Annual de Estadísticas Vitales: Defunciones, años 2015 y 2016.
5. Groenveld, Y., Petri, H., & Hermans, J. (1999). Relationship between blood glucose level and mortality in type 2 diabetes mellitus: a systematic review. Diabetes Med, 16, 2-13.
6. Gilet, H., Gruenberg, J., Bader, G., & Viala-Danten, M. (2012). Demonstrating the burden of hypoglycemia on Patients’ Quality of life in Diabetes Clinical Trials: Measurement Considerations for Hypoglycemia. Value in Health , 15, 1036-1041. doi:http:// dx.doi.org/10.1016/j.jval.2012.06.002
7. Real, J., & Carmena, R. (2004). La diabetes tipo 2 o la enfermedad incomprendida. Revista Clínica Española(1), 1-2.
8. American Diabetes Association. (2015). Hypoglycemia (Low Blood Sugar). Retrieved from http://www.diabetes.org/living-with-diabetes/ treatment-and-care/blood-glucose-control/ hypoglycemia-low-blood.html
9. Morales, J., & Schneider, D. (2014). Hypoglycemia. The American Journal of Medicine, 127(S17-S24). doi:http://dx.doi.org/10.1016/j.amjmed.2014.07.004
10. US Department of Health and Human Services, N. N. (2008, October). Hypoglycemia. Retrieved from www.diabetes.niddk.nih.gov
11. The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of Intensive Glucose Lowering in Type 2 Diabetes. Vol 358; 2008:2545-2559.
12. Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. Jan 8 2009;360(2):129-139.
13. Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. Jun 12 2008;358(24):2560-2572.
1 4. Garber AJ, Abrahamson MJ, Barzilay JI, et al. AACE Comprehensive Diabetes Management, Endocr Pract. 2013;19(Suppl 2):1-38
15. Alvarez Guisasola F, Tofe Povedano S, Krishnarajah G, Lyu R, Mavros P, Yin D. Hypoglycaemic symptoms, treatment satisfaction, adherence and their associations with glycaemic goal in patients with type 2 diabetes mellitus: findings from the
Real-Life Effectiveness and Care Patterns of Diabetes Management (RECAP-DM) Study. Diabetes Obes Metab. Jun 2008;10 Suppl 1:25-32.
16.Chan, S. P., Ji, L. N., Nitiyanant, W., Baik, S. H., & Sheu, W. H. H. (2010). Hypoglycemic symptoms in patients with type 2 diabetes in Asia-Pacific-Real-life effectiveness and care patterns of diabetes management: The RECAP-DM study. Diabetes Research and Clinical Practice,89(2). DOI:10.1016/j.diabres.2010.05.008
17.Majanovic SK . Janez A . Lefterov I . Tasic S . Cikac T. The Real-Life Effectiveness and Care Patterns of Diabetes Management Study for Balkan Region (Slovenia, Croatia, Serbia, Bulgaria): A Multicenter, Observational, Cross-Sectional Study. Diabetes Ther (2017) 8:929–940. DOI 10.1007/s13300-017-0288-x
18.Gonzalez C, et al. Prevalence of hypoglycemia among a sample of sulfonylurea-treated patients with Type 2 diabetes mellitus in Argentina: The real-life effectiveness and care patterns of diabetes management (RECAP-DM) study. Endocrinol Diabetes Nutr. 2018. https:// doi.org/10.1016/j.endinu.2018.05.014
19.Pettersson B, Rosenqvist U, Deleskog A, Journath G, Wändell P.Self-reported experience of hypoglycemia among adults with type 2 diabetes mellitus (Exhype). Diabetes research and clinical practice, 92 (2011): 19-25.
20.Lundkvist J, Berne C, ·Bolinder B, ·Jönsson L. The economic and quality of life impact of hypoglycemia. Eur J Health Econom 2005 · 50:197–202 DOI 10.1007/s10198-005-0276-3
21.Rombopoulos G, Hatzikou M, Latsou D, Yfantopoulos J.The prevalence of hypoglycemia and its impact on the quality of life (QoL) of type 2 diabetes mellitus patients (The HYPO Study). HORMONES 2013, 12(4):550-558
22.Orozco-Beltrán D,Mezquita-Raya P, Ramírez de Arellano A, Galán M. Self-Reported Frequency and Impact of Hypoglycemic Events in Spain. Diabetes Ther (2014) 5:155–168. DOI 10.1007/ s13300-014-0057-z
23.Edridge CL, Dunkley AJ, Bodicoat DH, Rose TC, Gray LJ, Davies MJ, et al. (2015) Prevalence and Incidence of Hypoglycaemia in 532,542 People with Type 2 Diabetes on Oral Therapies and Insulin: A Systematic Review and Meta-Analysis of Population Based Studies. PLoS ONE 10(6): e0126427. doi:10.1371/journal. pone.0126427
24.Nunes AP, Iglay K, Radican L, Engel SS, Yang J, Doherty MC, Dore DD. Hypoglycaemia seriousness and weight gain as determinants of cardiovascular disease outcomes among sulfonylurea users. Diabetes Obes Metab. 2017;1–11
25.Zhang Y, Wieffer H, Modha R, Balar B, Pollack M, Krishnarajah G.The Burden of Hypoglycemia in Type 2 Diabetes: A Systematic Review of Patient and Economic Perspectives. JCOM (2010); 17 (12):547-557
Movilidad limitada 83 (49 4%) 41 (26 8%) 42 (50 6%) < 0 05 1 793 (1 057, 3 042)
Autocuidado limitado 25 (9 7%) 14 (9 3%) 11 (10 3%) 0 787 ns Actividades habituales limitadas 66 (25 0%) 33 (21 7%) 33 (30 8%) 0 097 1 608 (0 916,2 824) Más dolor/incomodidad 145 (56 0%) 75 (49 3%) 70 (65 4%) < 0 05 1 942 (1 167,3 234) Más ansiedad/depresión 88 (34%) 40 (26 1%) 48 (45 3%) <0 01 2 338 (1 382,3 995)
* Diferencia estadísticamente significativa (p<0.05)
26.Marrett E1, Stargardt T, Mavros P, Alexander CM. Patient-reported outcomes in a survey of patients treated with oral antihyperglycaemic medications: associations with hypoglycaemia and weight gain. Diabetes Obes Metab. 2009 Dec;11(12):1138-44. doi: 10.1111/j.14631326.2009.01123.x. Epub 2009 Sep 16.
27. Ahrén B. Avoiding hypoglycemia: a key to success for glucose-lowering therapy in type 2 diabetes. Vascular Health and Risk Management 2013:9 155–163
28.Lopez JM1, Bailey RA1, Rupnow MF. Demographic Disparities Among Medicare Beneficiaries with Type 2 Diabetes Mellitus in 2011: Diabetes Prevalence, Comorbidities, and Hypoglycemia Events. Popul Health Manag. 2015 Aug;18(4):283-9. doi: 10.1089/pop.2014.0115. Epub 2015 Feb 3.
MCS te brinda beneficios EXTRA para que tengas salud completa. Con más de 13,000 proveedores de salud alrededor de toda la isla y varios programas de bienestar, porque tú mereces estar extra querido.


Mounjaro—the first and only approved single molecule that activates GIP and GLP-1 receptors in the body.1
Mounjaro is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use
• Mounjaro has not been studied in patients with a history of pancreatitis.
• Mounjaro is not indicated for use in patients with type 1 diabetes mellitus.
Important Safety Information
In both male and female rats, tirzepatide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2, and in patients with known serious hypersensitivity to tirzepatide or any of the excipients in Mounjaro. Please see Important Safety Information here and on the following page, including Boxed Warning about possible thyroid tumors, including thyroid cancer, and Brief Summary on the following pages.
Risk of Thyroid C-cell Tumors: Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. Pancreatitis has been reported in Mounjaro clinical trials. Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. Observe patients for signs and symptoms, including persistent severe abdominal pain sometimes radiating to the back, which may or may not be accompanied by vomiting. If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management. Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Concomitant use with an insulin secretagogue (e.g., sulfonylurea) or insulin may increase the risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by reducing the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions, sometimes severe, have been reported with Mounjaro in clinical trials. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity to Mounjaro. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown if such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury. In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, sometimes requiring hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of reported events occurred in patients who had experienced nausea, vomiting,
diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: In clinical trials, acute gallbladder disease was reported by 0.6% of Mounjarotreated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated. The most common adverse reactions reported in ≥5% of Mounjaro-treated patients in placebo-controlled trials were nausea, diarrhea, decreased appetite, vomiting, constipation, dyspepsia, and abdominal pain.
Drug Interactions: When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (such as sulfonylureas) or insulin to reduce the risk of hypoglycemia. Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications, so caution should be exercised.
Pregnancy: Limited data on Mounjaro use in pregnant women are available to inform on drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide. Use only if potential benefit justifies the potential risk to the fetus.
Lactation: There are no data on the presence of tirzepatide in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females of Reproductive Potential: Advise females using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established and use is not recommended in patients less than 18 years of age. Please see the Brief Summary on the following page, including the Boxed Warning about possible thyroid tumors, including thyroid cancer, and Medication Guide. Please see Instructions for Use included with the pen. TR HCP ISI MAY2022
Reference: 1. Mounjaro. Prescribing Information. Lilly USA, LLC.
Mounjaro™ and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-TR-US-0193 05/2022 ©Lilly USA, LLC 2022. All rights reserved.
Revista Puertorriqueña de Medicina y Salud
Mounjaro™ (tirzepatide) Brief Summary: Consult the package insert for complete prescribing information.
INDICATIONS AND USAGE Mounjaro™ is a glucose-dependent insulinotropic polypeptide (GIP) receptor and glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use: Mounjaro has not been studied in patients with a history of pancreatitis. Mounjaro is not indicated for use in patients with type 1 diabetes mellitus.
In both male and female rats, tirzepatide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro.
CONTRAINDICATIONS: Mounjaro is contraindicated in patients with: A personal or family history of medullary thyroid carcinoma (MTC), in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) and in patients with a known serious hypersensitivity to tirzepatide or any of the excipients in Mounjaro.
WARNINGS AND PRECAUTIONS:
Risk of Thyroid C-Cell Tumors: In both sexes of rats, tirzepatide caused a dose-dependent and treatment-duration-dependent increase in the incidence of thyroid C-cell tumors (adenomas and carcinomas) in a 2-year study at clinically relevant plasma exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined. Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. In clinical studies, 14 events of acute pancreatitis were confirmed by adjudication in 13 Mounjarotreated patients (0.23 patients per 100 years of exposure) versus 3 events in 3 comparator-treated patients (0.11 patients per 100 years of exposure). Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. After initiation of Mounjaro, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management. Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Patients receiving Mounjaro in combination with an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by a reduction in the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in clinical trials (e.g., urticaria and eczema) and were sometimes severe. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity reaction to tirzepatide or any of the excipients in Mounjaro.
Anaphylaxis and angioedema have been reported with GLP-1 receptor agonists. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury.
In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, which may sometimes require hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of the reported events occurred in patients who had experienced nausea, vomiting, diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe gastrointestinal adverse reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: Acute events of gallbladder disease such as cholelithiasis or cholecystitis have been reported in GLP-1 receptor agonist trials and post marketing. In Mounjaro placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic, and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Pool of Two Placebo-Controlled Clinical Trials: The data in Table 1 are derived from 2 placebo-controlled trials (1 monotherapy trial [SURPASS-1] and 1 trial in combination with basal insulin with or without metformin [SURPASS-5]) in adult patients with type 2 diabetes mellitus. These data reflect exposure of 718 patients to Mounjaro and a mean duration of exposure to Mounjaro of 36.6 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 54% were male. The population was 57% White, 27% Asian, 13% American Indian or Alaska Native, and 3% Black or African American; 25% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.1%. As assessed by baseline fundoscopic examination, 13% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 53%, 60 to 90 mL/min/1.73 m2 in 39%, 45 to 60 mL/min/1.73 m2 in 7%, and 30 to 45 mL/min/1.73 m2 in 1% of patients.
Pool of Seven Controlled Clinical Trials: Adverse reactions were also evaluated in a larger pool of adult patients with type 2 diabetes mellitus participating in seven controlled clinical trials which included two placebo-controlled trials (SURPASS-1 and -5), three trials of Mounjaro in combination with metformin, sulfonylureas, and/or SGLT2 inhibitors (SURPASS-2, -3, -4), and two additional trials conducted in Japan. In this pool, a total of 5119 adult patients with type 2 diabetes mellitus were treated with Mounjaro for a mean duration of 48.1 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 58% were male. The population was 65% White, 24% Asian, 7% American Indian or Alaska Native, and 3% Black or African American; 38% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.3%. As assessed by baseline fundoscopic examination, 15% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 52%, 60 to 90 mL/min/1.73 m2 in 40%, 45 to 60 mL/min/1.73 m2 in 6%, and 30 to 45 mL/min/ 1.73 m2 in 1% of patients.
Common Adverse Reactions: In the pool of placebo-controlled trials, the following adverse reactions were reported in ≥5% of Mounjaro-treated adult patients with type 2 diabetes mellitus (excluding hypoglycemia): (frequencies listed, respectively, as: placebo [N=235]; 5 mg [N=273]; 10 mg [N=240]; 15 mg [N=241]): nausea (4%, 12%, 15%, 18%), diarrhea (9%, 12%, 13%, 17%), decreased appetite (1%, 5%, 10%, 11%), vomiting (2%, 5%, 5%, 9%), constipation (1%, 6%, 6%, 7%), dyspepsia (3%, 8%, 8%, 5%), and abdominal pain (4%, 6%, 5%, 5%). Percentages reflect the number of patients who reported at least 1 occurrence of the adverse reaction. In the pool of seven clinical trials, the types and frequency of common adverse reactions, not including hypoglycemia, were similar.
Gastrointestinal Adverse Reactions: In the pool of placebo-controlled trials, gastrointestinal adverse reactions occurred more frequently among patients receiving Mounjaro than placebo (placebo 20.4%, Mounjaro 5 mg 37.1%, Mounjaro 10 mg 39.6%, Mounjaro 15 mg 43.6%). More patients receiving Mounjaro 5 mg (3.0%), Mounjaro 10 mg (5.4%), and Mounjaro 15 mg (6.6%) discontinued treatment due to gastrointestinal adverse reactions than patients receiving placebo (0.4%). The majority of reports of nausea, vomiting, and/or diarrhea occurred during dose escalation and decreased over time.
The following gastrointestinal adverse reactions were reported more frequently in Mounjaro-treated patients than placebo-treated patients (frequencies listed, respectively, as: placebo; 5 mg; 10 mg; 15 mg): eructation (0.4%, 3.0%, 2.5%, 3.3%), flatulence (0%, 1.3%, 2.5%, 2.9%), gastroesophageal reflux disease (0.4%, 1.7%, 2.5%, 1.7%), abdominal distension (0.4%, 0.4%, 2.9%, 0.8%).
Other Adverse Reactions: Hypoglycemia: Hypoglycemia adverse reactions in placebo-controlled trials in adults with type 2 diabetes mellitus were as follows. For the 40-week monotherapy study (frequencies listed, respectively, as: placebo [N=115]; 5 mg [N=121]; 10 mg [N=119]; 15 mg [N=120]): blood glucose <54 mg/dL (1%, 0%, 0%, 0%), severe hypoglycemia (0%, 0%, 0%, 0%). For the 40week add-on to basal insulin with or without metformin study (placebo [N=120], 5 mg [N=116], 10 mg [N=119], 15 mg [N-120]): blood glucose <54 mg/dL (13%, 16%, 19%, 14%), severe hypoglycemia (0%, 0%, 2%, 1%). Data include events occurring during 4 weeks of treatment-free safety follow up. Events after introduction of a new glucose-lowering treatment are excluded. Severe hypoglycemia was defined as episodes requiring the assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions. Hypoglycemia was more frequent when Mounjaro was used in combination with a sulfonylurea. In a clinical trial up to 104 weeks of treatment, when administered with a sulfonylurea, hypoglycemia (glucose level <54 mg/dL) occurred in 13.8%, 9.9%, and 12.8%, and severe hypoglycemia occurred in 0.5%, 0%, and 0.6% of patients treated with Mounjaro 5 mg, 10 mg, and 15 mg, respectively.
Heart Rate Increase: In the pool of placebo-controlled trials, treatment with Mounjaro resulted in a mean increase in heart rate of 2 to 4 beats per minute compared to a mean increase of 1 beat per minute in placebo-treated patients. Episodes of sinus tachycardia, associated with a concomitant increase from baseline in heart rate of ≥15 beats per minute, also were reported in 4.3%, 4.6%, 5.9% and 10% of subjects treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. For patients enrolled in Japan, these episodes were reported in 7% (3/43), 7.1% (3/42), 9.3% (4/43), and 23% (10/43) of patients treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. The clinical relevance of heart rate increases is uncertain.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in the pool of placebo-controlled trials, sometimes severe (e.g., urticaria and eczema); hypersensitivity reactions were reported in 3.2% of Mounjaro-treated patients compared to 1.7% of placebo-treated patients.
In the pool of seven clinical trials, hypersensitivity reactions occurred in 106/2,570 (4.1%) of Mounjaro-treated patients with antitirzepatide antibodies and in 73/2,455 (3.0%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Injection Site Reaction: In the pool of placebo-controlled trials, injection site reactions were reported in 3.2% of Mounjarotreated patients compared to 0.4% of placebo-treated patients.
In the pool of seven clinical trials, injection site reactions occurred in 119/2,570 (4.6%) of Mounjaro-treated patients with antitirzepatide antibodies and in 18/2,455 (0.7%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Acute Gallbladder Disease: In the pool of placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients.
Laboratory Abnormalities: Amylase and Lipase Increase: In the pool of placebo-controlled clinical trials, treatment with Mounjaro resulted in mean increases from baseline in serum pancreatic amylase concentrations of 33% to 38% and serum lipase concentrations of 31% to 42%. Placebo-treated patients had a mean increase from baseline in pancreatic amylase of 4% and no changes were observed in lipase. The clinical significance of elevations in lipase or amylase with Mounjaro is unknown in the absence of other signs and symptoms of pancreatitis.
Concomitant Use with an Insulin Secretagogue (e.g., Sulfonylurea) or with Insulin When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (e.g., sulfonylureas) or insulin to reduce the risk of hypoglycemia.
Oral Medications: Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications. Caution should be exercised when oral medications are concomitantly administered with Mounjaro. Monitor patients on oral medications dependent on threshold concentrations for efficacy and those with a narrow therapeutic index (e.g., warfarin) when concomitantly administered with Mounjaro. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro. Hormonal contraceptives that are not administered orally should not be affected.
Pregnancy: Risk Summary: Available data with Mounjaro use in pregnant women are insufficient to evaluate for a drug-related risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide during pregnancy. Mounjaro should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In pregnant rats administered tirzepatide during organogenesis, fetal growth reductions and fetal abnormalities occurred at clinical exposure in maternal rats based on AUC. In rabbits administered tirzepatide during organogenesis, fetal growth reductions were observed at clinically relevant exposures based on AUC. These adverse embryo/fetal effects in animals coincided with pharmacological effects on maternal weight and food consumption. The estimated background risk of major birth defects is 6–10% in women with pre-gestational diabetes with an HbA1c >7% and has been reported to be as high as 20–25% in women with an HbA1c >10%. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively. Clinical Considerations: Disease-Associated Maternal and/or
MOUNJAROTM (tirzepatide) Injection, for subcutaneous use TR HCP BS MAY2022 MOUNJAROTM (tirzepatide) Injection, for subcutaneous use TR HCP BS MAY2022
42 Revista Puertorriqueña de Medicina y Salud Pública MOUNJARO, TR HCP BS MAY2022 - 10.5 x 13 (9.75x11.25 Live)
Embryo/Fetal Risk: Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia-related morbidity. Data: Animal Data: In pregnant rats given twice weekly subcutaneous doses of 0.02, 0.1, and 0.5 mg/kg tirzepatide (0.03-, 0.07-, and 0.5-fold the MRHD of 15 mg once weekly based on AUC) during organogenesis, increased incidences of external, visceral, and skeletal malformations, increased incidences of visceral and skeletal developmental variations, and decreased fetal weights coincided with pharmacologically-mediated reductions in maternal body weights and food consumption at 0.5 mg/kg. In pregnant rabbits given once weekly subcutaneous doses of 0.01, 0.03, or 0.1 mg/kg tirzepatide (0.01-, 0.06, and 0.2-fold the MRHD) during organogenesis, pharmacologically-mediated effects on the gastrointestinal system resulting in maternal mortality or abortion in a few rabbits occurred at all dose levels.
Reduced fetal weights associated with decreased maternal food consumption and body weights were observed at 0.1 mg/kg. In a pre- and post-natal study in rats administered subcutaneous doses of 0.02, 0.10, or 0.25 mg/kg tirzepatide twice weekly from implantation through lactation, F1 pups from F0 maternal rats given 0.25 mg/kg tirzepatide had statistically significant lower mean body weight when compared to controls from post-natal day 7 through post-natal day 126 for males and post-natal day 56 for females.
Lactation: Risk Summary: There are no data on the presence of tirzepatide in animal or human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females and Males of Reproductive Potential: Contraception: Use of Mounjaro may reduce the efficacy of oral hormonal contraceptives due to delayed gastric emptying. This delay is largest after the first dose and diminishes over time. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established in pediatric patients (younger than 18 years of age).
Geriatric Use: In the pool of seven clinical trials, 1539 (30.1%) Mounjaro-treated patients were 65 years of age or older, and 212 (4.1%) Mounjaro-treated patients were 75 years of age or older at baseline. No overall differences in safety or efficacy were detected between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Renal Impairment: No dosage adjustment of Mounjaro is recommended for patients with renal impairment. In subjects with renal impairment including end-stage renal disease (ESRD), no change in tirzepatide pharmacokinetics (PK) was observed. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Hepatic Impairment: No dosage adjustment of Mounjaro is recommended for patients with hepatic impairment. In a clinical pharmacology study in subjects with varying degrees of hepatic impairment, no change in tirzepatide PK was observed.
OVERDOSE: In the event of an overdosage, contact Poison Control for latest recommendations. Appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms. A period of observation and treatment for these symptoms may be necessary, taking into account the half-life of tirzepatide of approximately 5 days.
PATIENT COUNSELING INFORMATION: Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use)
Missed Doses: Inform patients if a dose is missed, it should be administered as soon as possible within 4 days after the missed dose. If more than 4 days have passed, the missed dose should be skipped and the next dose should be administered on the regularly scheduled day. In each case, inform patients to resume their regular once weekly dosing schedule.
Additional information can be found <at www.Mounjaro.com/at lillymedical.com/by calling The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979)/ on Twitter @MounjaroHCP/at Click to Chat/etc.>.
See Brief Summary of Prescribing Information on adjacent page. See User Guide accompanying the product device.
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA Copyright © 2022, Eli Lilly and Company. All rights reserved.
MOUNJAROTM (tirzepatide) Injection, for subcutaneous use TR HCP BS MAY2022
MOUNJARO, TR HCP BS MAY2022 - 10.5 x 13 (9.75x11.25 Live)
Revista Puertorriqueña de Medicina y Salud Pública
La hepatitis C sigue siendo un problema de salud pública a nivel global en adición a ser la causa número uno de enfermedad hepática terminal y de trasplante de hígado en muchos países. Si embargo, hace ya más de cuatro años contamos con opciones simples y noveles de tratamiento oral que han demostrado una eficacia cercana al 100% de cura en la mayoría de los pacientes tratados irrespectivo del genotipo viral y estadio clínico en que se encuentren. ¿Entonces cuál es la problemática?

Teóricamente, tanto científica como matemáticamente, la erradicación de la enfermedad es factible, pero requerirá grandes esfuerzos para en primer
lugar, diagnosticar a los pacientes que no están conscientes de su infección, así como de un compromiso real de la arena sociopolítica y cultural para lograr este objetivo asignando los recursos necesarios.
Estamos pues, en una coyuntura histórica en la medicina moderna donde se contempla la posibilidad de erradicar o al menos convertirla en una enfermedad rara de transmisión limitada a nivel global, ya que podría ser posible con herramientas simples de identificación diagnostica, prevención manejo y tratamiento oral curativo o escoger seguir drenando el sistema de salud pública con los costos

Unidad de Investigaciones Clínicas para el SIDA (ACTU, por sus siglas en inglés) Recinto de Ciencias Médicas Universidad de Puerto Rico.
asociados a el manejo de pacientes de enfermedad avanzada con cirrosis y pacientes en necesidad de trasplante de hígado. Desafortunadamente, la hepatitis C no cuenta con una vacuna preventiva y todavía no se considera en muchos países como una prioridad de salud pública a pesar de los grandes esfuerzos de muchos organizaciones científicas y reguladores de la salud, incluida la Organización Mundial de la Salud (OMS). En adición a esto al menos a nivel doméstico, no se ha identificado ninguna figura o celebridad icónica que pueda elevar la pasión emocional de la conciencia política y comunitaria colectiva como hemos visto con otras enfermedades crónicas y terminales como el VIH y el cáncer y que estimule a las partes interesadas en la palestra público-política a asignar los fondos necesarios para aplacar esta epidemia silente.
No empecé a esto la OMS al igual que el Centro para Control Enfermedades en Atlanta (CDC) y el Departamento de Salud y Recursos Humanos (DHHS) por sus siglas en ingles han juntado esfuerzos encaminados para que a nivel global y doméstico en los Estados Unidos y Puerto Rico se establezcan unas metas de detección, enlace a cuidado y tratamiento llamada la estrategia 90-90-90 para el año 2030 donde se pretende estimular que las estructuras de salud pública y gubernamentales dirijan esfuerzos para lograr dicho cometido. Todas las personas dentro de la sociedad civil tenemos el deber de aunar esfuerzos y prosperar con determinación, asociaciones, colaboración y defensa para construir y mantener el apoyo a esta encomienda. Es necesario entablar negociaciones justas y reales con el apalancamiento farmacéutico y del gobierno para aumentar la accesibilidad y reducir
aún más el precio de estos medicamentos altamente efectivos.
Podemos y debemos, como individuos dentro de la sociedad civil, estimular la conciencia colectiva médica y comunitaria con el fin de seleccionar, identificar y diagnosticar a los individuos en riesgo para que podamos sensibilizar a los políticos, las partes interesadas clave y los líderes de opinión sobre la realidad de la respuesta de que una inversión inicial y sustancial segura y sostenida, ayudará a salvar muchas vidas y generará beneficios a largo plazo para la sociedad en conjunto. En la actualidad, Puerto Rico cuenta con las herramientas terapéuticas para poder lograr ese cometido. El sistema de Salud Vital (ASES) al igual que muchos de los planes médicos comerciales y Advantage autorizan el manejo y tratamiento de los casos identificados con estos nuevos fármacos de acción directa. Solo falta el deseo de buscar e identificar esas personas a riesgo.
Se necesita esa defensa comunitaria y política robusta, recurrente y sostenida junto con las personas que viven con la enfermedad para desarrollar e impulsar medidas efectivas, que trasciendan la necesidad existente. Si esto va a suceder en nuestras vidas, el tiempo de acción no debe esperar más. La historia siempre nos juzgara por las acciones realizadas u omitidas en el presente.
La Organización Mundial de la Salud (OMS) ha fijado el objetivo de eliminar la hepatitis C como amenaza de salud para el 2030 La nueva definición de eliminación de la OMS establece que una reducción en un 65% de la mortalidad y nuevas infecciones en un 90% 1
Debido a la eficacia limitada de las pruebas de hepatitis C basadas en el riesgo por sí solas, el Centro para el Control y la Prevención de Enfermedades (CDC) consideró estrategias para aumentar la proporción de personas infectadas que conocen su estado y están vinculadas a la atención.2
Por lo tanto, en abril de 2020, los CDC aumentaron la orientación anterior con dos nuevas recomendaciones:2
1. Hacer prueba de Hepatitis C una vez en la vida a individuos con 18 años o más
2. Realizar pruebas de la hepatitis C para todas las mujeres embarazadas durante cada embarazo, excepto en los entornos en los que la prevalencia de la infección por el VHC es <0 1%
La recomendación para las pruebas del VHC que se mantiene sin cambios es que, independientemente de la edad o de la prevalencia del entorno, todas las personas con factores de riesgo deben someterse a las pruebas de la hepatitis C, con pruebas periódicas mientras persistan los factores de riesgo
Cuando se aborda específicamente la barrera de las visitas adicionales para confirmar un diagnóstico, las directrices de la Asociación Americana para el Estudio de las Enfermedades del Hígado y la Sociedad de Enfermedades Infecciosas de América (AASLD-IDSA) recomiendan el uso de la prueba de reflejo de la hepatitis C para la prueba inicial del VHC Una prueba de anticuerpos de la hepatitis C positiva indica una infección actual, una infección previa resuelta o un falso positivo 3
La detección de la presencia del virus en la sangre se realiza con una prueba de PCR del ARN del virus de la hepatitis C para confirmar una infección actual 3
Una prueba de reflejo inmediato del VHC debería ser el foco de detección en la mayoría de los entornos clínicos, ya que sólo requiere una única muestra de sangre sin que el paciente necesite una segunda visita para una prueba de confirmación, considerada hoy en día como una barrera principal en la vinculación con el tratamiento y la curación de la hepatitis C La orden del médico debería incluir el tipo de prueba que debe utilizar el técnico 3
Tras la confirmación del paciente, un profesional sanitario debe evaluarlo, incluyendo la valoración de la enfermedad hepática y/o las comorbilidades para una selección adecuada del tratamiento del paciente También debe ofrecerse asesoramiento para reducir la progresión de la enfermedad hepática y prevenir la transmisión del virus de la hepatitis C (VHC) 3
1. WHO guidelines on hepatitis B and C testing. Geneva: World Health Organization; 2017
2. Schillie S, Wester C, Osborne M, Wesolowski L, Ryerson AB. CDC recommendations for hepatitis C screening among adults - United States, 2020. MMWR Recomm Rep 2020;69(2):1-17
3. American Association for the Study of Liver Diseases-Infectious Disease Society of America. Recommendations for testing, managing, and treating hepatitis C. https://www.hcvguidelines org/. Updated October 5, 2021. Accessed February 3, 2021..
La pancreatitis aguda es una de las causas más comunes de hospitalización dentro del campo de las enfermedades gastrointestinales. Se estima que la incidencia anual es de 4.9 a 35 pacientes por cada 100,000 habitantes. Pancreatitis es una condición inflamatoria causada por la activación intracelular de enzimas en las células acinares que se liberan hacia el tejido intersticial causando edema y daño al órgano. En un 20% de ocasiones pueden surgir complicaciones serias como necrosis la cual está asociada a complicaciones sistémicas, fallo orgánico y aumento en la mortalidad. La pancreatitis aguda se caracteriza por dolor abdominal súbito, mayormente epigástrico, y en
el 50% de los casos este dolor se irradia hacia la espalda. El mismo se asocia a náuseas, vómitos, elevación de las enzimas pancreáticas, y hallazgos radiológicos característicos de inflamación. Las dos causas más comunes son cálculos biliares y uso excesivo de alcohol. Otras causas menos comunes son: metabólicas, como la hipercalcemia e hipertrigliceridemia, medicamentos, pancreatitis hereditaria, quistes neoplásicos y tumores. A continuación estaremos discutiendo las causas, diagnóstico, complicaciones y manejo según las guías más recientes.

El páncreas es un órgano localizado en la parte superior del abdomen, detrás del estómago, dentro del espacio retroperitoneal (cabeza y cuerpo), excepto la cola, que es intraperitoneal y cercana al bazo. Éste órgano tiene funciones tanto endocrinas como exocrinas. La porción endocrina se encarga de la regulación de los niveles de glucosa mediante la síntesis de insulina o glucagón. La función exocrina contribuye a la digestión y absorción de alimentos mediante la producción de enzimas digestivas. Éstas son amilasa, tripsina y lipasa, para digerir carbohidratos, grasas y proteínas, respectivamente. El funcionamiento de este órgano Es primordial para la digestión de alimentos y
absorción adecuada de vitaminas liposolubles. Cuando ocurre algún daño que altera el drenaje de estas células o activan las enzimas prematuramente, se inicia la inflamación y autodegradación de los tejidos y vasos sanguíneos.
La causa más común de pancreatitis aguda es cálculos biliares, al que se le atribuyen 40 a 70% de los casos. Hábitos tóxicos como el consumo de alcohol o cigarrillo son la segunda causa más común de pancreatitis contribuyendo de un 25 a 35% de los casos en los Estados Unidos. Hipertrigliceridemia e hipercalcemia son otras causas. Procedimientos como la ColangioPancreatografía Retrógrada Endoscópica (mejor conocido como ERCP por sus siglas en inglés) pueden resultar en pancreatitis. También, existe la pancreatitis inducida por medicamentos, pero sucede en raras ocasiones y muchas veces asociada a otras posibles causas, el cual dificulta poder definir el origen. Algunos medicamentos ya están claramente reconocidos al demostrar que la pancreatitis recurre con su uso continuo. Otros sólo tienen reportado una posible asociación en la literatura, ya que no se volvió a comenzar el medicamento al paciente luego de su recuperación y así ver si la pancreatitis recurre. La pancreatitis hereditaria causada por mutaciones en los genes de CFTR, PRSS1 o SPINK-1 usualmente se presenta en infantes o adolescentes, y puede resultar en daños irreversibles al páncreas y hasta cáncer. No existe un tratamiento específico para prevenir o reducir los ataques de pancreatitis en esta población. En casos de daño severo o síntomas severos, se recomienda una pancreatectomía total con autotransplante de células de islote.
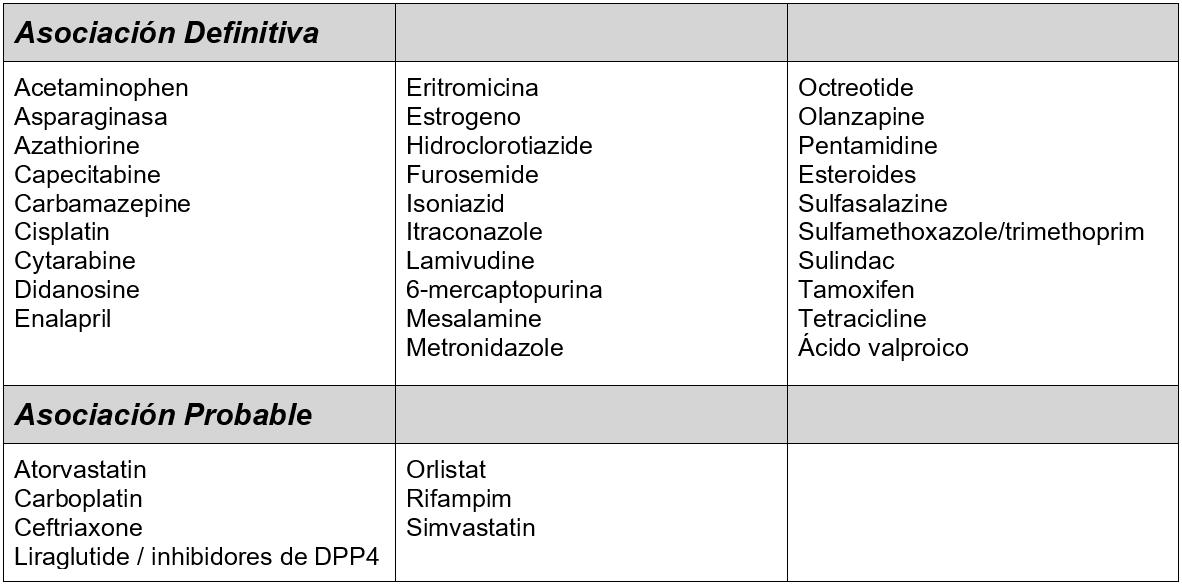
Dentro de las causas infecciosas existen algunos virus como el Coxsackie, Citomegalovirus, Varicela-Zoster, Herpes Simplex, VIH y los de hepatitis A, B y E, que pueden ser causantes de pancreatitis Ademas, algunas bacterias como Mycoplasma, Legionella, Leptospirosis, Salmonella, han sido asociadas, también como ciertos hongos (Aspergillus) y parásitos (Toxoplasma, Cryptosporidium, Ascaris lumbricoides, Fasciola hepatica).
Anomalías anatómicas del ducto biliar como quistes congénitos, unión anómala del ducto pancreáticobiliar, divertículos en el área de la ámpula de Vater, páncreas anular y páncreas divisum. Traumas abdominales que pudieran comprimir el cuerpo del páncreas contra la columna vertebral resultan en laceración o ruptura del ducto pancreático, liberación de enzimas e inflamación. Esta condición se asocia a ascitis pancreática, trombosis de los vasos sanguíneos adyacentes y/o sangrado.
Pacientes con pancreatitis aguda se presentan con un dolor epigástrico agudo que en el 50% de las ocasiones se irradia hacia la espalda. En la mayoría de los casos el dolor se alivia al sentarse o inclinar el torso hacia delante. Éste se puede acompañar de náuseas y vómitos. En casos de pancreatitis aguda severa los pacientes pudieran presentar con dificultad respiratoria asociada a irritación del diafragma o efusiones pleurales. Además del síndrome agudo de distrés respiratorio causado por la liberación de fosfolipasa, la cual degrada el surfactante intra alveolar causando fallo respiratorio. Seudoquistes
y necrosis pancreática son complicaciones locales severas.
Es necesario cumplir con al menos dos de los siguientes tres criterios para hacer el diagnóstico: presencia de dolor abdominal, elevación de la amilasa o lipasa sérica por encima de tres veces el límite superior del rango normal, o hallazgos sugestivos de inflamación en imágenes radiográficas (CT o MRI, por sus siglas en inglés). Algunos hallazgos en CT o MRI son: agrandamiento del páncreas, disminución de la captación del contraste representando áreas de necrosis, grasa peripancreática estriada y líquido peripancreático.
Estudios recientes han demostrado que el inicio de la alimentación dentro de las primeras 24 horas según sea tolerado es beneficioso para los pacientes y disminuye complicaciones infecciosas
-Pancreatitis leve, la cual se caracteriza por la ausencia de falla orgánica y complicaciones locales o sistémicas.
-Moderadamente severa, la cual se caracteriza por una falla orgánica transitoria de menos de 48 horas y/o complicaciones locales o sistémicas
-Pancreatitis aguda severa, la cual se caracteriza por falla orgánica persistente de uno o más órganos.
Es preferible la alimentación oral y en casos que el paciente no la tolere, a través de una sonda nasogástrica o nasoentérica. La alimentación parenteral sólo es recomendable cuando el paciente no tolere alimentación oral o por sonda, o cuando el tracto digestivo no se pueda utillizar por sangrado gastrointestinal o íleo paralítico. Éstos pacientes se benefician de un monitoreo constante en las primeras 24 a 48 horas de sus signos vitales, producción de orina, cambios en electrolitos, niveles de glucosa, elevación de hematocrito y niveles de BUN comparados con los niveles iniciales.
No existe un tratamiento específico dirigido a atender la inflamación del páncreas, sino que los esfuerzos son para darle apoyo al paciente mediante el reemplazo de fluídos y la eliminación de agentes ofensores. La hidratación agresiva con líquidos intravenosos, preferiblemente el lactato de Ringer, en las primeras 24 a 48 horas es sumamente importante para disminuir la morbilidad y mortalidad. Sin embargo recientemente, el estudio WATERFALL (de Madaria, N Engl J Med 2022; 387: 989) demostrò que la hidratacion moderada usando lactato de Ringer mediante un bolo 10 mL por kilogramo, sólo si el paciente estaba hipovolémico, y 1.5 mL por kilogramo de infusión continua era igualmente efectivo, pero con menos riesgo de causar edema pulmonar y fallo cardiaco que cuando se hidrataba agresivamente (bolo 20 mL/kg + 3 mL/kg infusión). Los opioides son apropiados para el manejo del dolor siempre teniendo en cuenta efectos adversos como depresión respiratoria, disminución de motilidad gastrointestinal y acumulación de metabolitos, como en el caso de Meperidine. al reducir la translocación de bacterias entéricas.
La pancreatitis aguda es una condición muy frecuente que pudiera estar asociada a complicaciones serias incluyendo fallo multiorgánico. Es de suma importancia la eliminación de factores desencadenantes. Manejo dirigido de reemplazo de fluidos acompañado de monitoreo continuo es crucial para prevenir y manejar posibles complicaciones de una manera multidisciplinaria.
Según la última revisión de la clasificación de Atlanta la pancreatitis aguda se pudiera subdividir en:
by Gastroenterologists, Primary Care Physicians, Nurse Practitioners, and Physician Assistants5
Based on the FDA-approved labels, CREON is the only PERT specifically indicated for patients with EPI due to chronic pancreatitis and pancreatectomy, in addition to cystic fibrosis and other conditions.
(CREON) for exocrine pancreatic insuf
• Porcine-derived pancreatic enzyme products contain purines. Caution should be exercised when prescribing CREON to patients with gout, renal impairment, or hyperuricemia.
• There is theoretical risk of viral transmission with all pancreatic enzyme products including CREON.
• Exercise caution when administering pancrelipase to a patient with a known allergy to proteins of porcine origin.
• were vomiting, dizziness, and cough.
• Adverse reactions that occurred in at least 1 chronic pancreatitis or pancreatectomy patient (greater than or equal bowel movements, and nasopharyngitis.
• CREON is not interchangeable with any other pancrelipase product.
Please see the following pages for Brief Summary of Full Prescribing Information.
CREON® conditions.
DOSAGE AND ADMINISTRATION
CREON is not interchangeable with other pancrelipase products.
CREON is orally administered. Therapy should be initiated at the lowest recommended dose and gradually increased. The dosage of CREON should be individualized based on clinical symptoms, the degree of steatorrhea present, and the fat content of the diet as described in the Limitations on Dosing below [see Dosage and Administration and Warnings and Precautions]
Administration
Infants (up to 12 months)
CREON should be administered to infants immediately prior to each feeding, using a dosage of 3,000 lipase units per 120 mL of formula or prior to breastfeeding. Contents of the capsule may be administered directly to the mouth or with a small amount of applesauce. Administration should be followed by breast milk or formula. Contents of the capsule should not be should be taken to ensure that CREON is not crushed or chewed or retained in the mouth, to avoid irritation of the oral mucosa.
Children and Adults
CREON capsules and capsule contents should not be crushed or chewed. Capsules should be swallowed whole.
For patients who are unable to swallow intact capsules, the capsules may be carefully opened and the contents added to a small amount of acidic soft food with a pH of 4.5 or less, such as applesauce, at room temperature. The CREON-soft food mixture should be swallowed immediately without crushing or chewing, and followed with water or juice to ensure complete ingestion. Care should be taken to ensure that no drug is retained in the mouth.
Dosage recommendations for pancreatic enzyme replacement therapy were published following the Cystic Fibrosis Foundation Consensus Conferences. CREON should be administered in a manner consistent with the recommendations of the Cystic Fibrosis Foundation Consensus Conferences (also known as Conferences) provided in the following paragraphs, except for infants. Although the Conferences recommend doses of 2,000 to 4,000 lipase units in infants up to 12 months, CREON is available in a 3,000 lipase unit capsule. Therefore, the recommended dose of CREON in infants up to 12 months is 3,000 lipase units per 120 mL of formula or per breastfeeding. Patients may be dosed on a fat ingestion-based or actual body weight-based dosing scheme.
Additional recommendations for pancreatic enzyme therapy in patients pancreatectomy are based on a clinical trial conducted in these populations.
Infants (up to 12 months)
CREON is available in the strength of 3,000 USP units of lipase thus infants may be given 3,000 lipase units (one capsule) per 120 mL of formula or per breastfeeding. Do not mix CREON capsule contents directly into formula or breast milk prior to administration [see Administration]
Children Older than 12 Months and Younger than 4 Years
Enzyme dosing should begin with 1,000 lipase units/kg of body weight units/kg of body weight per meal (or less than or equal to 10,000 lipase units/kg of body weight per day), or less than 4,000 lipase units/g fat ingested per day.
Children 4 Years and Older and Adults
Enzyme dosing should begin with 500 lipase units/kg of body weight per meal body weight per meal (or less than or equal to 10,000 lipase units/kg of body weight per day), or less than 4,000 lipase units/g fat ingested per day.
Usually, half of the prescribed CREON dose for an individualized full meal approximately three meals plus two or three snacks per day.
Enzyme doses expressed as lipase units/kg of body weight per meal should be decreased in older patients because they weigh more but tend to ingest less fat per kilogram of body weight.
The initial starting dose and increases in the dose per meal should be individualized based on clinical symptoms, the degree of steatorrhea present, and the fat content of the diet.
In one clinical trial, patients received CREON at a dose of 72,000 lipase units per meal while consuming at least 100 g of fat per day]. Lower starting doses recommended in the literature are consistent with the 500 lipase units/kg of body weight per meal lowest starting dose recommended for adults in the Cystic Fibrosis Foundation Consensus Conferences Guidelines. Usually, half of the prescribed CREON dose for an individualized full meal should be given with each snack.
Dosing should not exceed the recommended maximum dosage set forth by the Cystic Fibrosis Foundation Consensus Conferences Guidelines. If symptoms and signs of steatorrhea persist, the dosage may be increased by the healthcare professional. Patients should be instructed not to increase the dosage on their own. There is great inter-individual variation in response to enzymes; thus, a range of doses is recommended. Changes in dosage may require an adjustment period of several days. If doses are to exceed 2,500 lipase units/kg of body weight per meal, further investigation is warranted. Doses greater than 2,500 lipase units/kg of body weight per meal (or greater than 10,000 lipase units/kg of body weight per day) should be used with caution and only if they are documented to be effective by of fat absorption. Doses greater than 6,000 lipase units/kg of body weight colonopathy, in children less than 12 years of age [see Warnings and Precautions]. Patients currently receiving higher doses than 6,000 lipase units/kg of body weight per meal should be examined and the dosage either immediately decreased or titrated downward to a lower range.
None.
Fibrosing colonopathy has been reported following treatment with different pancreatic enzyme products. Fibrosing colonopathy is a rare, serious adverse reaction initially described in association with high-dose pancreatic enzyme use, usually over a prolonged period of time and most commonly
products exceeding 6,000 lipase units/kg of body weight per meal have been associated with colonic stricture in children less than 12 years of age. some patients may be at risk of progressing to stricture formation. It is recommended, unless clinically indicated, that enzyme doses should be less than 2,500 lipase units/kg of body weight per meal (or less than 10,000 lipase units/kg of body weight per day) or less than 4,000 lipase units/g fat ingested per day [see Dosage and Administration].
Doses greater than 2,500 lipase units/kg of body weight per meal (or greater than 10,000 lipase units/kg of body weight per day) should be used with caution and only if they are documented to be effective by 3-day fecal fat
Patients receiving higher doses than 6,000 lipase units/kg of body weight per meal should be examined and the dosage either immediately decreased or titrated downward to a lower range.
Potential for Irritation to Oral Mucosa
Care should be taken to ensure that no drug is retained in the mouth. CREON should not be crushed or chewed or mixed in foods having a pH greater than 4.5. These actions can disrupt the protective enteric coating resulting in early release of enzymes, irritation of oral mucosa, and/or loss of enzyme activity [see Dosage and Administration and Patient Counseling Information]
For patients who are unable to swallow intact capsules, the capsules may be carefully opened and the contents added to a small amount of acidic soft food with a pH of 4.5 or less, such as applesauce, at room temperature. The CREON-soft food mixture should be swallowed immediately and followed with water or juice to ensure complete ingestion.
Potential for Risk of Hyperuricemia
Caution should be exercised when prescribing CREON to patients with gout, renal impairment, or hyperuricemia. Porcine-derived pancreatic enzyme products contain purines that may increase blood uric acid levels.
Potential Viral Exposure from the Product Source
CREON is sourced from pancreatic tissue from swine used for food consumption. Although the risk that CREON will transmit an infectious agent to humans has been reduced by testing for certain viruses during manufacturing and by inactivating certain viruses during manufacturing, there is a theoretical risk for transmission of viral disease, including diseases
of transmission of an infectious illness associated with the use of porcine pancreatic extracts have been reported.
Caution should be exercised when administering pancrelipase to a patient with a known allergy to proteins of porcine origin. Rarely, severe allergic reactions including anaphylaxis, asthma, hives, and pruritus, have been reported with other pancreatic enzyme products with different formulations of
CREON treatment in patients with severe allergy should be taken into consideration with the overall clinical needs of the patient.
The most serious adverse reactions reported with different pancreatic enzyme products of the same active ingredient (pancrelipase) that are described allergic reactions [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly the rates observed in practice.
The short-term safety of CREON was assessed in clinical trials conducted in or pancreatectomy were treated with CREON.
Cystic Fibrosis
Studies 1 and 2 were randomized, double-blind, placebo-controlled, crossover studies of 49 patients, ages 7 to 43 years, with EPI due to CF. Study 1 included 32 patients ages 12 to 43 years and Study 2 included 17 patients ages 7 to 11 years. In these studies, patients were randomized to receive CREON at a dose of 4,000 lipase units/g fat ingested per day or matching placebo for 5 to 6 days of treatment, followed by crossover to the alternate treatment for an additional 5 to 6 days. The mean exposure to CREON during these studies was 5 days.
In Study 1, one patient experienced duodenitis and gastritis of moderate severity 16 days after completing treatment with CREON. Transient neutropenia without clinical sequelae was observed as an abnormal
In Study 2, adverse reactions that occurred in at least 2 patients (greater than or equal to 12%) treated with CREON were vomiting and headache. Vomiting occurred in 2 patients treated with CREON and did not occur in patients treated with placebo; headache occurred in 2 patients treated with CREON and did not occur in patients treated with placebo.
The most common adverse reactions (greater than or equal to 4%) in Studies 1 and 2 were vomiting, dizziness, and cough. Table 1 enumerates adverse reactions that occurred in at least 2 patients (greater than or equal to 4%) treated with CREON at a higher rate than with placebo in Studies 1 and 2. than or equal to 4%) in Cystic Fibrosis (Studies 1 and 2)
Adverse Reaction CREON Capsules n = 49 (%) Placebo n = 47 (%) 3 (6) 1 (2)
2 (4) 1 (2) 2 (4) 0
An additional open-label, single-arm study assessed the short-term safety and tolerability of CREON in 18 infants and children, ages 4 months to pancreatic enzyme replacement therapy (mean dose of 7,000 lipase units/kg/day for a mean duration of 18.2 days) followed by CREON (mean dose of 7,500 lipase units/kg/day for a mean duration of 12.6 days).
There were no serious adverse reactions. Adverse reactions that occurred in patients during treatment with CREON were vomiting, irritability, and decreased appetite, each occurring in 6% of patients.
Chronic Pancreatitis or Pancreatectomy
A randomized, double-blind, placebo-controlled, parallel group study was conducted in 54 adult patients, ages 32 to 75 years, with EPI due to chronic pancreatitis or pancreatectomy. Patients received single-blind placebo treatment during a 5-day run-in period followed by an intervening period of up to 16 days of investigator-directed treatment with no restrictions on pancreatic enzyme replacement therapy. Patients were then randomized to receive CREON or matching placebo for 7 days. The CREON dose was 72,000 lipase units per main meal (3 main meals) and 36,000 lipase units per snack (2 snacks). The mean exposure to CREON during this study was 6.8 days in the 25 patients that received CREON.
The most common adverse reactions reported during the study were related to glycemic control and were reported more commonly during CREON treatment than during placebo treatment.
Table 2 enumerates adverse reactions that occurred in at least 1 patient (greater than or equal to 4%) treated with CREON at a higher rate than with placebo.
Table 2: Adverse Reactions in at Least 1 Patient (greater than or equal to 4%) in the Chronic Pancreatitis or Pancreatectomy Trial
Adverse Reaction CREON Capsules n = 25 (%) Placebo n = 29 (%)
2 (8) 2 (7)
1 (4) 1 (3)
1 (4) 1 (3)
1 (4) 0 1 (4) 0 1 (4) 0 1 (4) 0
Postmarketing Experience
Postmarketing data from this formulation of CREON have been available post approval use of this formulation of CREON. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
constipation and nausea), skin disorders (including pruritus, urticaria and rash), blurred vision, myalgia, muscle spasm, and asymptomatic elevations of liver enzymes have been reported with this formulation of CREON.
Delayed- and immediate-release pancreatic enzyme products with different formulations of the same active ingredient (pancrelipase) have been used for
intestinal obstruction syndrome (DIOS), recurrence of pre-existing carcinoma, and severe allergic reactions including anaphylaxis, asthma, hives, and pruritus.
DRUG INTERACTIONS
been conducted.
Pregnancy
Published data from case reports with pancrelipase use in pregnant women or other adverse maternal or fetal outcomes. Pancrelipase is minimally absorbed systematically; therefore, maternal use is not expected to result in fetal exposure to the drug. Animal reproduction studies have not been conducted with pancrelipase.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
There are no data on the presence of pancrelipase in either human or animal milk, the effects on the breastfed infant or the effects on milk production. Pancrelipase is minimally absorbed systemically following oral administration; therefore, maternal use is not expected to result in clinically relevant exposure
breastfeeding should be considered along with the mother’s clinical need for CREON and any potential adverse effects on the breastfed infant from CREON or from the underlying maternal condition.
The short-term safety and effectiveness of CREON were assessed in two randomized, double-blind, placebo-controlled, crossover studies of 49 Study 1 included 8 adolescents between 12 and 17 years of age. Study 2 in pediatric patients in these studies were similar to adult patients [see Adverse Reactions and Clinical Studies]
An open-label, single-arm, short-term study of CREON was conducted in 18 infants and children, ages 4 months to six years of age, with EPI due to therapy (mean dose of 7,000 lipase units/kg/day for a mean duration of 18.2 days) followed by CREON (mean dose of 7,500 lipase units/kg/day for a mean duration of 12.6 days). The mean daily fat intake was 48 grams during treatment with usual pancreatic enzyme replacement therapy and 47 grams
during treatment with CREON. When patients were switched from their usual pancreatic enzyme replacement therapy to CREON, they demonstrated similar spot fecal fat testing results; the clinical relevance of spot fecal fat testing has not been demonstrated. Adverse reactions that occurred in patients during treatment with CREON were vomiting, irritability, and decreased appetite [see Adverse Reactions]
formulations of pancrelipase consisting of the same active ingredient (lipases, proteases, and amylases) for treatment of children with exocrine pancreatic literature and through clinical experience.
Dosing of pediatric patients should be in accordance with recommended guidance from the Cystic Fibrosis Foundation Consensus Conferences [see Dosage and Administration]. Doses of other pancreatic enzyme products exceeding 6,000 lipase units/kg of body weight per meal have been than 12 years of age [see Warnings and Precautions] Geriatric Use
65 and over to determine whether they respond differently from younger responses between the elderly and younger patients.
There have been no reports of overdose in clinical trials or postmarketing surveillance with this formulation of CREON. Chronic high doses of pancreatic colonic strictures [see Dosage and Administration and Warnings and Precautions]. High doses of pancreatic enzyme products have been associated with hyperuricosuria and hyperuricemia, and should be used with caution in patients with a history of hyperuricemia, gout, or renal impairment [see Warnings and Precautions]
Carcinogenesis, Mutagenesis, Impairment of Fertility Carcinogenicity, genetic toxicology, and animal fertility studies have not been performed with pancrelipase.
PATIENT COUNSELING INFORMATION Dosing and Administration
• Instruct patients and caregivers that CREON should only be taken as directed by their healthcare professional. Patients should be advised that the total daily dose should not exceed 10,000 lipase units/kg body weight/ day unless clinically indicated. This needs to be especially emphasized for patients eating multiple snacks and meals per day. Patients should be informed that if a dose is missed, the next dose should be taken with the next meal or snack as directed. Doses should not be doubled [see Dosage and Administration (2)]
• Instruct patients and caregivers that CREON should always be taken with food. Patients should be advised that CREON delayed-release capsules and the capsule contents must not be crushed or chewed as doing so could cause early release of enzymes and/or loss of enzymatic activity. Patients should swallow the intact capsules with adequate amounts of liquid at mealtimes. If necessary, the capsule contents can also be sprinkled on soft acidic foods [see Dosage and Administration]
Advise patients and caregivers to follow dosing instructions carefully, as doses of pancreatic enzyme products exceeding 6,000 lipase units/kg of body weight per meal have been associated with colonic strictures in children below the age of 12 years [see Dosage and Administration]
Advise patients and caregivers to contact their healthcare professional immediately if allergic reactions to CREON develop [see Warnings and Precautions]
Manufactured by: Abbott Laboratories GmbH Hannover, Germany
Marketed by: AbbVie Inc. North Chicago, IL 60064, U.S.A. © 2009-2020 AbbVie Inc.
Ref: 03-C054-R4 Revised March, 2020 LAB-3604 MASTER
Leukocytoclastic vasculitis affect small vessels causing immune complexes deposition and activation of the complement system. Dermatologic manifestations like IgA vasculitis, Kawasaki-like inflammatory syndrome and leukocytoclastic vasculitis has been reported after Covid infection and Covid vaccines exposure. Our case, a 57-year-old female without previous history of autoimmune disease reported a pinpoint rash in both legs from below the knees after first Covid vaccine. After second dose of the same vaccine the rash spread up to her trunk accompanied with systemic manifestations. Skin biopsy confirmed the diagnosis and was treated with oral steroids reaching full recovery.

SARS-CoV-2 or COVID19 as is known by the majority have been affecting millions of people around the world since 2019 manifesting with respiratory problems and can develop severe pneumonia and other systemic complications like septic shock, acute respiratory distress syndrome (Altun & Kuzucular, 2022), antiphospholipid syndrome, immune-mediated myositis and myocarditis (Cohen et al., 2021) . Dermatologic manifestations have been reported after COVID-19 infection including IgA vasculitis, kawasakilike multisystem inflammatory syndrome and leukocytoclastic vasculitis (Sandhu et al., 2021). These skin manifestations have also been observed, although rare, after vaccination against COVID-19 with Astra-Zeneca, Bharat, Jansen, Moderna, Pfizer and/or recombinant (ChAdOx1 nCoV-19) vaccine (Grossman et al., 2022; Sandhu et al., 2021).
Dermatologic manifestation of vaccines such as areas of erythema, swelling and pain on site of injection are very common. Other dermatologic manifestations less commonly seen include immune mediated reactions such as vasculitis. We present the case of a patient who presented leukocytoclastic vasculitis as an adverse reaction to the first dose of COVID-19 vaccine. The reaction after the first dose was self limited and after the second dose the reaction presented with systemic manifestations requiring the use of oral steroids.
A 57 years old female patient with history of diabetes mellitus 2 since 2015, status post thyroidectomy in 2017 and depressive disorder since 2018 came for evaluation due to history of leg rash after COVID vaccine. Patient denied toxic habits and no drug or food allergies. Chronic medications at the moment janumet 100/500mg, synthroid and sertraline.
Patient presented a rash on both legs from below the knees 5 day after her first COVID vaccine (Pfizer). Rash was described as red and pinpoint. Additional symptoms included mild pain on legs. At that moment was diagnosed with dermatitis and was treated with leg elevation, acetaminophen and antihistamine drugs, providing relief and total resolution within 2 weeks.
Three days after she received her second dose of COVID vaccine the rash reappeared but up to her thighs and trunk. Systemic symptoms included joint pain, fatigue, generalized malaise, rash on palms, with associated erythema and mild desquamation. Denied fever.
On a physical examination she had palpable painful purpuras with areas of coalescence on both lower extremities from thigh down but more prominent below the knee. Pain on joints of hands and knee but no evidence of arthritis. Patient was treated with oral steroids (30 mgs per day taper over 14 days) with improvement of symptoms and full recovery.
Results showed normal ESR of 15 mm/hr, and C3 / C4. ANA test of 1;80 homogeneous.
Site: Rt leg Gross description: punch 3mm
Microscopic description: There is fibrin in the walls of venules with a perivascular and interstitial infiltrate of neutrophils with nuclear dust.
Microscopic diagnosis: Leukocytoclastic vasculitis (early changes)
Leukocytoclastic vasculitis (LCV) or hypersensitivity vasculitis affect small vessels leading to extravasation of erythrocytes and immune complexes deposition causing activation of the complement system (Altun & Kuzucular, 2022).
Leukocytoclastic vasculitis is one of the manifestations which has been associated not only with COVID vaccine but also with COVID disease. This is a cutaneous small vessel vasculitis of the dermal capillaries and venules. LCV can be idiopathic, associated with other diseases, medications, infections and had been previously
described as associated with influenza vaccine. The mechanism of this vasculitis could be hypersensitivity or abnormal immunological activation. (Sandhu et al., 2021). The exact mechanism of COVID vaccine vasculitis is not clearly established yet; molecular mimicry, hyperactivation of immune mechanisms, loss of immune tolerance are all possible etiologies(Alojzija Hočevar et al., 2022).
In this case the patient presented with vasculitis manifestations after her first vaccine but diagnosis was not done the first time around, exposing this
patient to a second dose of the vaccine which caused her more severe manifestations and earlier than on her first episode. The patient was treated with oral steroids with good response but literature had reported more catastrophic cases which resulted in the death of the patient. The exact etiology of these immune mediated diseases is still not clear. The interplay of these possible mechanisms defers further research and we as clinicians should be compromised on reporting any type of adverse reactions to these relatively new vaccines.It is important to recognize the possible adverse events associated to the
administration of this vaccine in order to avoid possible complications associated to re-exposure to this vaccines in predispose individuals. Nevertheless, the benefits of them in the case of a pandemic such as the one we experienced with COVID19, outweighs the risks of its use. LCV and other vasculitis should be considered when patients present with the above-mentioned symptoms not only after COVID-19 vaccines but also after COVID-19 infection (Carubbi et al., 2022). We should be aware of the possible immune mediated inflammatory diseases associated to COVID 19 vaccines even in previously healthy individuals. Carubbi et al., 2022
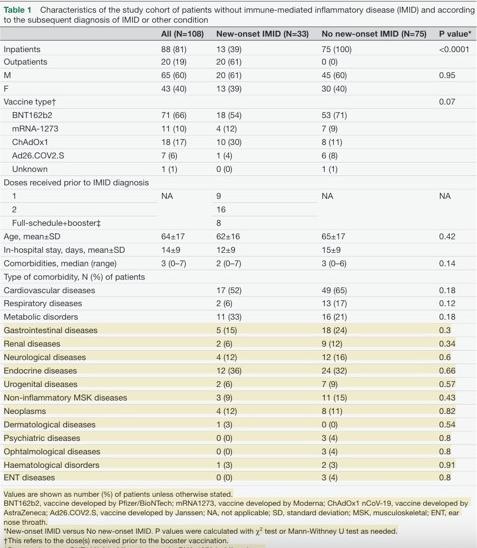
Carubbi et al., 202 Alojzija Hočevar, Zoran Simonović, Žiga Rotar, & Matija Tomšič. (2022). Vasculitis as Temporally Associated With COVID-19 Infection or Vaccination: A Single-center Experience. The Journal of Rheumatology , 49, 232–233. https://doi. org/10.3899/jrheum.210788
Grossman, M., Appel, G., Little, A., & Ko, C. (2022). Post-COVID-19 vaccination IgA vasculitis in an adult. Journal of Cutaneous Pathology, 49(4), 385–387. https://doi.org/10.1111/ cup.14168
Altun, E., & Kuzucular, E. (2022). Leukocytoclastic vasculitis after COVID-19 vaccination. Dermatologic Therapy, 35(3). https:// doi.org/10.1111/dth.15279
Carubbi, F., Alunno, A., Santilli, J., Natali, L., Mancini, B., Gregorio, N. di, Pinto, R. del, Viscido, A., Grassi, D., & Ferri, C. (2022). Immune-¬ mediated¬inflammatory¬ diseases¬after¬anti-¬ SARS-¬ CoV-¬ 2¬ vaccines:¬new¬diagnoses¬and¬ disease¬flares. Open, 8, 2460. https://doi.org/10.1136/ rmdopen-2022-002460
Cohen, S. R., Prussick, L., Kahn, J. S., Gao, D. X., Radfar, A., & Rosmarin, D. (2021). Leukocytoclastic vasculitis flare following the COVID-19vaccine. International Journal of Dermatology, 60, 1032–1033. https://doi. org/10.1111/ijd.15623
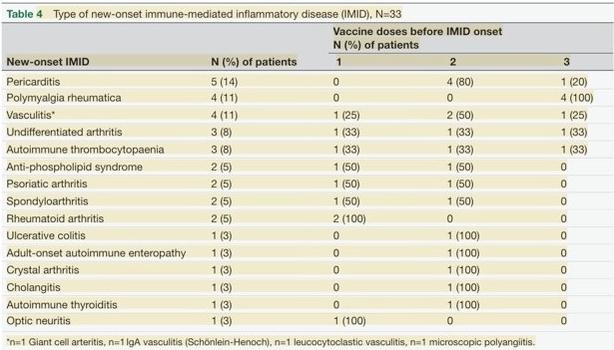
Sandhu, S., Bhatnagar, A., Kumar, H., Kumar Dixit, P., Paliwal, G., Kumar Suhag, D., Patil, C., & Mitra, D. (2021). Leukocytoclastic vasculitis as a cutaneous manifestation of ChAdOx1 nCoV-19 corona virus vaccine (recombinant). Dermatologic Therapy, 34(6). https://doi.org/10.1111/dth.15141







Ge Yang a,b,e, Yueh-Ying Han a, Erick Forno a, Edna Acosta-Perez c, Angel Colon-Semidey c,d, María Alvarez c,d, Glorisa Canino c,d, Wei Chen a, Juan C. Celedon a,*
a Division of Pediatric Pulmonary Medicine, UPMC Children’s Hospital of Pittsburgh, University of Pittsburgh, Pittsburgh, PA, USA b Department of Neonatology, Xiangya Hospital, Central South University, Changsha, Hunan, 410008, China c Behavioral Sciences Research Institute, University of Puerto Rico, San Juan, Puerto Rico d Department of Pediatrics, University of Puerto Rico, San Juan, Puerto Rico
e Third Xiangya Hospital, Central South University, Changsha, Hunan, 410083, China
Antecedentes: poco se sabe sobre la dermatitis atópica (DA) entre los niños de Puerto Rico.
Objetivo: Examinar los factores de riesgo e identificar enfoques para diagnosticar mejor la EA en niños puertorriqueños. Métodos: Estudio de casos y controles de EA entre 540 niños de 6 a 14 años en San Juan, Puerto Rico. La EA se definió como: 1) EA diagnosticada por un médico, 2) RAST-AD: síntomas de EA más IgE positiva para β1 a alérgenos, y 3) STR-AD: síntomas de EA y reactividad de la prueba cutánea al alérgeno β1. Se utilizó la regresión logística para el análisis multivariable. También evaluamos el rendimiento diagnóstico de varios enfoques al comparar su sensibilidad, especificidad, valor predictivo positivo [VPP], valor predictivo negativo [VPN] y área bajo la curva [AUC]).
Resultados: De los 70 niños con STR-AD, solo 5 (7,1%) tenían PD-AD. En niños sin asma, una IgE
positiva para Dermatophagoides (D.) pteronys sinus y signos de moho/hongos en el hogar se asociaron significativamente con probabilidades 3,3 y 5 veces mayores de STR-AD, respectivamente. Entre los niños con asma, el seguro de salud privado/del empleador y una IgE positiva para D. pteronyssinus se asociaron significativamente con aproximadamente el doble de probabilidades de STR-AD. Una combinación de síntomas de eccema actuales y una IgE positiva para D. pteronyssinus produjo una sensibilidad del 70 %, especificidad y VPN del 95 %, VPP del 88 % y un AUC de 0,85 para STR-AD. Reemplazar una IgE positiva para D. pteronyssinus con una IgE positiva para el alérgeno ??1 aumentó ligeramente la sensibilidad sin afectar otros parámetros.
Conclusiones: la EA está marcadamente subdiagnosticada por los médicos en Puerto Rico. Esto podría mejorarse evaluando los síntomas del
Background: Little is known about atopic dermatitis (AD) among children in Puerto Rico.
Objective: To examine risk factors and identify approaches to better diagnose AD in Puerto Rican children. Methods: Case-control study of AD among 540 children aged 6–14 years in San Juan, Puerto Rico. AD was defined as: 1) physician-diagnosed AD, 2) RAST-AD: AD symptoms plus 1 positive IgE to allergens, and 3) STR-AD: AD-symptoms and skin test reactivity to 1 allergen. Logistic regression was used for the multivariable analysis. We also evaluated the diagnostic performance of various approaches by comparing their sensitivity, specificity, positive predicted value [PPV], negative predictive value [NPV], and area under curve [AUC]).
Results: Of the 70 children with STR-AD, only 5 (7.1%) had PD-AD. In children without asthma, a positive IgE to Dermatophagoides (D.) pteronyssinus and signs of mold/ mildew at home were significantly associated with 3.3 and 5 times increased odds of STR-AD, respectively. Among children with asthma, private/employer-based health insurance and a positive IgE to D. pteronyssinus were each significantly associated with approximately twofold increased odds of STR-AD. A combination of current eczema symptoms and a positive IgE to D. pteronyssinus yielded a sensitivity 70%, specificity and NPV 95%, PPV 88%, and an AUC 0.85 for STR-AD. Replacing a positive IgE to D. pteronyssinus with a positive IgE to 1 allergen slightly increased sensitivity without affecting other parameters.
Conclusions: AD is markedly under-diagnosed by physicians in Puerto Rico. This could be improved by assessing eczema symptoms and measuring IgEs to common allergens.
Atopic dermatitis (AD) is a common allergic disease worldwide, affecting over 20% of children in high income countries and rising in prevalence in low to middle income countries.1,2 Among participants in a U.S.-based study, ~17.1% had eczematous symptoms but only 6% were diagnosed with AD, suggesting marked disease under-diagnosis and under-treatment.3 In Puerto Rico, where atopic asthma is a major public health problem, 24.8% of second-grade children attending two schools had parental report of symptoms of atopic dermatitis.4
The “gold standard” for a diagnosis of AD consists of a thorough history and physical exam, combined with allergy skin testing. Such diagnostic approach, however, may not be feasible in epidemiologic studies or in underserved areas with limited access to an allergist, such as Puerto Rico. Some epidemiologic studies rely on questionnaire-reported symptoms or a physician’s diagnosis to identify AD,5 while others use a G. Yang et al. combination of questionnaire-based data and objectively measured allergic sensitization.6 Since self-reported diagnosis or symptoms are subject to
recall and reporting bias, the ideal criteria to detect or diagnose AD in population-based studies remains to be established.7
AD frequently occurs during infancy, often serving as a harbinger of other atopic diseases during childhood, including allergic rhinitis and asthma. Indeed, up to 80% of children with AD may develop or have concurrent allergic rhinitis or asthma.8 In addition to such atopic dis-eases, common risk factors or co-morbidities of AD include low income,2 obesity,9 dust mite allergen exposure,10 and an elevated total serum IgE.11,12
We hypothesized that AD would be markedly under-diagnosed among Puerto Rican children living in the island of Puerto Rico, where fewer than fifteen allergists served a population of 3.7 million people before Hurricane Maria. We further hypothesized that such under-diagnosis of AD could be reduced by obtaining a history of symptoms suggestive of eczema and measuring levels of IgE to common allergens.
In this report, we estimated and compared the prevalence of AD identified through self-reported symptoms and objectively measured allergic sensitization against that of physician-diagnosed-AD among 540 Puer-
Subject recruitment
From March of 2009 to June of 2010, children were recruited for a case-control study of asthma from randomly selected households in San Juan (Puerto Rico). As previously described,15 households in the metropolitan area of San Juan were selected by a multistage probability sampling design. Primary sampling units were randomly selected neighborhood clusters based on the 2000 U.S. census. Secondary sam-pling units were randomly selected households within each primary sampling unit. A household was included if 1 resident was a child aged 6–14 years whose four grandparents were all Puerto Rican. In households with >1 eligible child, only one child was randomly selected for screening. On the basis of the sampling design, 7073 households were selected and 6401 (90.5%) were contacted. Of these 6401 households, 1111 had 1 child who met inclusion criteria. In an effort to reach a target sample size of approximately 700 children, we attempted to enroll a random sample (n ¼ 783) of these 1111 eligible children. We were able to obtain parental consent for 678 of these 783 children. There were no significant differences in age, gender, or area of residence between eligible children who did (n ¼ 678 [86.6%]) and did not (n ¼ 105 [13.4%]) agree to participate. Of the 678 participating children, 540 (79.7%) had complete data on allergy skin testing, levels of allergen specific-IgEs, and parental report of AD symptoms, and were thus included in the current analysis.
Cases had asthma, defined as parental report of physician-diagnosed asthma and at least one episode of wheeze in the previous year. Control subjects had neither parental report of physician-diagnosed asthma nor wheeze in the prior year.
Study participants completed a protocol that included questionnaires, allergy skin testing, and collection of blood (for measurement of total and allergen-specific IgEs). One of the child’s parents (usually [>93%] the mother) completed questionnaires about the child’s general and respi-ratory health, socio-demographic and household characteristics, and family
history of asthma and allergic diseases. Height and weight were measured to the nearest centimeter and pound, respectively.
Plasma levels of total IgE and IgEs to five common allergens (dust mite [Der p 1], cockroach [Bla g 2], cat dander [Fel d 1], dog dander [Can f 1], and mouse urinary protein [Mus m 1]) were determined using the UniCAP 100 system (Pharmacia & Upjohn, Kalamazoo, MI). For each allergen, an IgE 0.35 IU/ml was considered positive. Skin test reactivity (STR) to aeroallergens was assessed using a Multi Test device (Lincoln Diagnostics, Decatur, IL).
In addition to histamine (positive control) and saline solution (negative control), allergen extracts from dust mites (Dermatophagoides (D.) pteronyssinus, D. farinae and Blomia tropicalis), house dust, German cockroach (Blatella germanica), cat dander, dog dander, mixed grass pollen, mugwort sage, ragweed, mixed tree pollen, mold mix, Alternaria tenuis and mouse urinary protein were applied to the skin of the forearm in a site free of eczema (Alk-Abello, Round Rock, Texas). Skin test reactivity (STR) was defined as a maximum wheal diameter exceeding the saline diluent wheal diameter by at least 3 mm.
Written parental consent was obtained for participating children, from whom written assent was also obtained. The study was approved by the Institutional Review Boards of the University of Puerto Rico (San Juan, PR), Brigham and Women’s Hospital (Boston, MA), and the Uni-versity of Pittsburgh (Pittsburgh, PA).
We analyzed and compared several de finitions of AD, as follows: 1) physician-diagnosed AD (PD-AD), 2) current symptoms suggestive of AD (AD symptoms), defined as a positive response to the following two questions (a) “Has your child ever had a prolonged, itchy, scaly or weepy skin rash?” and (b) “Has your child had this rash in the last 12 months?”, 3) RAST-AD, de fined as AD-symptoms plus at least one positive IgE to allergens, and 4) STR-AD, defined as AD-symptoms and STR to at least one allergen.
Bivariate analyses were conducted using two-sample t-tests (for continuous variables) and chi-square tests or Fisher’s exact tests (for categorical variables). Lo -
gistic regression was used for the multivariable analysis of STR-AD. All multivariable models included age, gender, body mass index as a z-score (based on 2000 CDC growth charts13), and asthma. The following covariates were also included in the initial multivariable models, if associated with STR-AD at P 0.25 in bivariate analyses: parental education (either parent completed high school vs. none), household income (<vs. $15,000/year [near the median in-come for households in Puerto Rico in 2008–200914]),15 type of health insurance (public vs. private or employer-based), parental history of eczema, current exposure to second-hand smoke (SHS), day care attendance in the first year of life, plasma total IgE, a positive IgE to each allergen, a positive IgE to at least one allergen, and parental report of each of the following: signs of mold or mildew in the house, sighting cockroaches, and sighting mice. Because of collinearity, the initial multivariable models included only one of the significant total or allergen-specific IgE measures (i.e. we did not include total IgE and a positive IgE to dust mite in the same model). These additional covariates remained in the final models if they were associated with AD at P < 0.05 or if they changed the estimate of effect ( )by 10%. For test parameters, we calculated sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) for STR-AD, as follows: 1) Sensitivity ¼ True positives 100, 2)
True positives þ False negatives
Specificity ¼ True negatives 100, 3) Positive predictive value was plotted as sensitivity against (1 specificity). The area under the curve (AUC) of ROC was used to assess diagnostic performance. R program (Version 3.4.2) was used for all analyses.
eTable 1 shows the comparison of the main characteristics of study participants who were (n ¼ 540) and were not (n ¼ 138) included in the current analysis. Compared with children who were included in the analysis, those excluded were significantly more likely to have a house-hold income >$15,000 per year and a lower BMI z-score, but signifi-cantly less likely to be currently exposed to
parental history of eczema, day care attendance in the first year of life, signs of mold or mildew in the home, or sighting pests (cockroaches or mice) between children who were and were not included in the current analysis.
The characteristics of study participants, according to whether they did or did not have STR-AD, as well as asthma, are shown in Table 1.Of the 540 participants, 70 (13.0%) had STR-AD.
Among children with asthma, those with STR-AD were significantly more likely to have private or employer-based health insurance than those without STR-AD. Among children without asthma, those with STR-AD were significantly more likely to have a parent report signs of mold or mildew in the home, but less likely to sighting mice, than those without STR-AD.
Among children with and without asthma, those with STR-AD were significantly more likely to have current allergic rhinitis, an increased total IgE, and a positive IgE to D. pteronyssinus than children without STR-AD. There was no significant difference in current SHS or any other characteristic between children with and without STR-AD, regardless of asthma status.
We next calculated the proportion of subjects with STR-AD who were also diagnosed by a physician (i.e. who had PD-AD). Of the 70 subjects with STR-AD, only 5 (7.1%) also had PD-AD (Fig. 1, Panel A). This marked under-diagnosis of STR-AD was similar in children with and without asthma (Fig. 1, panels B and C).
Table 2 shows the results of the multivariable analysis of STR-AD, separately in children with and without asthma. In the analysis among children without asthma (which was adjusted for age, gender, and BMI z-score), having a positive IgE to D.pteronyssinus and parental report of mold or mildew at home were significantly associated with 3.3 times and 5 times increased odds of STR-AD, respectively (Model 1). Similar results were obtained when a positive IgE to D. pteronyssinus was replaced with having 1 positive IgE to allergens (Model 2) or with total IgE (Model 3). In all three multivariable models, there was a trend for an inverse asso-ciation between parental report of sighting mice and STR-AD, but such trend was non-statistically significant (models
1 and 3) or of borderline statistical significance (model 2). In the multivariable analysis of STR-AD among children with asthma (also adjusted for age, gender, and BMI z-score), having private or employer-based health insurance was signifi -cantly associated with approximately twofold increased odds of STR-AD (Model 1). Similar results were obtained when a positive IgE to D. pteronyssinus was replaced with either 1 positive IgE to allergens (Model 2) or total serum IgE (Model 3).
Table 3 shows the sensitivity, specificity, PPV, NPV and AUC of various approaches to the diagnosis of STR-AD in Puerto Rican children, which were partly based on the results of our multivariable analysis (see Table 2). PD-AD had high specificity but very low sensitivity and a low AUC for STR-AD, both in children with and without asthma. A combination of AD symptoms and a positive IgE to D. pteronyssinus yielded sensitivity 70% and specificity 95%, NPV 95% and PPV 88%, and an AUC 0.85 for STR-AD, both in children with and without asthma. Replacing a positive IgE to D. pteronyssinus with a positive IgE to 1 allergen slightly increased sensitivity (i.e. from 75.7% to 80% in all children) but had minimal impact on all other parameters (specificity, NPV, PPV or the AUC).
We found that ~93% of cases of STR-AD among children in Puerto Rico had not been diagnosed by a physician, possibly due to limited access to healthcare (particularly allergists) in Puerto Rico.15,16
A previous study of AD among children ages 6–7 years who attended two schools in Puerto Rico found that the prevalence of parental report of current eczema symptoms was 24.8%,4 and that ~70% of children with such symptoms had not been diagnosed with AD by a physician. In contrast to that study, we assessed not only eczema symptoms but also skin test reactivity to allergens and levels of total/allergen-specific IgEs in a population-based sample of school-aged children aged 6–14 years. Moreover, the response rate in the previous study (53%) was substan-tially lower than that in the current study.
Consistent with findings in other popula -
tions, we show that a positive IgE to common allergens such as house dust mite or an elevated total serum IgE is significantly associated with STR-AD.6 Indeed, dust mite allergen exposure has been linked to eczematous symptoms,2,8 and high dust mite allergen levels have been reported to be both common and associated with asthma and other allergic diseases in Latin America.17 In contrast to our findings for total IgE and STR-AD, Perkins et al reported that total IgE plays little role in prediction of visible eczema at 5 years, despite a significant association between total IgE and eczema severity.12 On the other hand, Ville et al showed that reductions in total IgE are associated with good treatment response and complete remission of AD11
Among children without asthma, we found that signs of mold or mildew in the house are associated with STR-AD. Such exposure could alter the skin barrier, a first step in AD pathogenesis.18 However, the impact of reducing mold or mildew on AD, if any, is unknown.19 Among children with asthma, we show that private or employer-based health insurance is significantly associated with STR-AD, likely due to improved access to healthcare.
We show that a combination of current eczematous symptoms and 1 positive IgE to common allergens could markedly improve the diagnostic rate of STR-AD among physicians in Puerto Rico. Indeed, such approach yielded a sensitivity of 80%, plus high: specificity, PPV, NPV, and AUC for STR-AD in all children. Of interest, replacing 1 positive IgE to al-lergens with a positive IgE to dust mite yielded a comparably high sensitivity (i.e. 76%) for STR-AD in all children, thus supporting a relatively simple and cost-effective approach to AD diagnosis and care in Puerto Rico.
Our study has substantial strengths, including a population-based sample of school-aged children and data on objective measures of allergic sensitization.4 We also recognize several study limitations. First, we cannot assess temporal relationships due to the cross-sectional study G. Yang et al. design. Second, we did not conduct direct skin examinations. However, we used a combination of STR to allergens
and questionnaire-based data for our “reference diagnosis of AD”. Third, selection bias and recall bias are possible in any observational study such as ours. However, selection bias is an unlikely explanation for our results, since there was no sig-nificant difference in most relevant characteristics (i.e. type of health insurance, parental history of eczema, signs of mold or mildew in the home) between children who were and were not included in our analysis. Similarly, poor parental understanding or recall of a physician’s diagnosis of eczema is not probable as the sole explanation for the marked under-diagnosis of STR-AD in study subjects. Finally, we had no data on some potential confounders, such as diet.
In summary, only 7.1% of cases of STRAD among school-aged Puerto Rican children were diagnosed by a physician, as reported by the child’s parents. Physicians in Puerto Rico could improve their diagnostic accu-racy for AD by inquiring about current eczematous symptoms, conducting a clinical examination of the skin, and measuring IgEs to a panel of common allergens (or, if that were non-feasible, IgE to house dust mite).
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards of the University of Puerto Rico (San Juan, PR), Brigham and Women’s Hospital (Boston, MA), and the University of Pittsburgh (Pittsburgh, PA).
Written parental consent was obtained for participating children, from whom written assent was also obtained.
Consent for publication Not applicable.
Availability of data and material Not applicable. Competing interests
Dr. Celedon has received research materials from Merck and GSK (inhaled steroids), and Pharmavite (vitamin D and placebo capsules), to provide medications free of cost to participants in NIH-funded studies, unrelated to the current work. The other authors report no competing interests.
GY and JCC conceived of the study and participated in its design, and drafted the manuscript. GY, YYH, EF, and WC performed statistical analysis. EAP, ACS, MA, and GC participated in the design and
coordi-nation of the study. All authors read and approved the final manuscript.
This work was supported by the U.S. National Institutes of Health [grants HL079966, HL117191, and HD052892], the Heinz Endowments, the China Scholarship Council, and the Third Xiangya Hospital, Central South University.
We thank children and their families for their participation in this study. Appendix A. Supplementary data Supplementary data to this article can be found online at https://doi. org/10.1016/j.waojou.2018.11.003.
1. Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124(6), 1251-1258.e23. 2009/12/01/.
2. Flohr C, Mann J. New insights into the epidemiology of childhood atopic dermatitis. Allergy. 2014;69(1):3–16.
3. Hanifin JM, Reed ML. A population-based survey of eczema prevalence in the United States. Dermatitis : contact, atopic, occupational. Drug. 2007 Jun;18(2):82–91. PubMed PMID: 17498413. Epub 2007/05/15. eng.
4. Maymi MA, Somolinos AL, Nazario CM, Sanchez JL. The prevalence of atopic dermatitis in Puerto Rican school children. Puert Rico Health Sci J. 2007 Jun;26(2): 127–133. PubMed PMID: 17722425. Epub 2007/08/29. eng.
5. Haileamlak A, Lewis SA, Britton J, et al. Validation of the International Study of Asthma and Allergies in Children (ISAAC) and U.K. Criteria for atopic eczema in Ethiopian children. Br J Dermatol. 2005;152(4):735–741.
6. Bos JD, Van Leent EJ, Sillevis Smitt JH. The millennium criteria for the diagnosis of atopic dermatitis. Exp Dermatol. 1998 Aug;7(4):132–138. PubMed PMID: 9758407. Epub 1998/10/03. eng.
7. Brenninkmeijer EEA, Schram ME, Leeflang MMG, Bos JD, Spuls PI. Diagnostic criteria for atopic dermatitis: a systematic review. Br J Dermatol. 2008;158(4): 754–765.
8. Leung DYM, Bieber T. Atopic dermatitis. Lancet. 2003;361(9352):151–160, 2003/ 01/11/.
9. Koutroulis I, Magnelli L, Gaughan J, Weiner E, Kratimenos P. Atopic dermatitis is more severe in children over the age of two who have an increased body mass index. Acta Paediatr. 2015;104(7):713–717.
10. Bremmer SF, Simpson EL. Dust mite avoidance for the primary prevention of atopic dermatitis: a systematic review and meta-analysis. Pediatr Allergy Immunol. 2015; 26(7):646–654.
11. Kiiski V, Karlsson O, Remitz A, Reitamo S. High serum total IgE predicts poor longterm outcome in atopic dermatitis. Acta Derm Venereol. 2015 Nov;95(8):943–947. PubMed PMID: 25916555. Epub 2015/04/29. eng.
12. Perkin MR, Strachan DP, Williams HC, Kennedy CTC, Golding J. Natural history of atopic dermatitis and its relationship to serum total immunoglobulin E in a population-based birth cohort study. Pediatr Allergy Immunol. 2004;15(3):221–229.
13. Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, et al. CDC growth charts: United States. Adv Data. 2000 Jun 8;(314):1–27. PubMed PMID: 11183293. Epub 2001/02/ 24. eng.
14. U.S. Census Bureau. Household Income for States: 2008 and 2009 2010 [cited 2013 September 9]. Available from: http://www. census.gov/prod/2010pubs/acsbr09-2. pdf.
15. Forno E, Celedon JC. Asthma and ethnic minorities: socioeconomic status and beyond. Curr Opin Allergy Clin Immunol. 2009 Apr;9(2):154–160. PubMed PMID: 19326508. Epub 2009/03/28. eng.
16. Nazario S, Acantilado C, Alvarez M, et al. Allergist role in asthma care in Puerto Rico. Bol Asoc Med P R. 2011 Jan-Mar;103(1):18–21. PubMed PMID: 21696098. Epub 2011/06/24. eng.
17. Hunninghake GM, Weiss ST, Celedon JC. Asthma in hispanics. Am J Respir Crit Care Med. 2006;173(2):143–163, 10/06 08/09/received 10/05/accepted. PubMed PMID: PMC2662985.
18. Kim BE, Leung DYM. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol Res. 2018 May;10(3):207–215. PubMed PMID: 29676067. Pubmed Central PMCID: PMC5911439. Epub 2018/04/21. eng.
19. Leung DYM, Guttman-Yassky E. Assessing the current treatment of atopic dermatitis: unmet needs. J Allergy Clin Immunol. 2017;139(4, Supplement):S47–S48, 2017/04/ 01/.
parental history of eczema, day care attendance in the first year of life, signs of mold or mildew in the home, or sighting pests (cockroaches or mice) between children who were and were not included in the current analysis.
The characteristics of study participants, according to whether they did or did not have STR-AD, as well as asthma, are shown in Table 1.Of the 540 participants, 70 (13.0%) had STR-AD.
Among children with asthma, those with STR-AD were significantly more likely to have private or employer-based health insurance than those without STR-AD. Among children without asthma, those with STR-AD were significantly more likely to have a parent report signs of mold or mildew in the home, but less likely to sighting mice, than those without STR-AD.
Among children with and without asthma, those with STR-AD were significantly more likely to have current allergic rhinitis, an increased total IgE, and a positive IgE to D. pteronyssinus than children without STR-AD. There was no significant difference in current SHS or any other characteristic between children with and without STR-AD, regardless of asthma status.
We next calculated the proportion of subjects with STR-AD who were also diagnosed by a physician (i.e. who had PD-AD). Of the 70 subjects with STR-AD, only 5 (7.1%) also had PD-AD (Fig. 1, Panel A). This marked under-diagnosis of STR-AD was similar in children with and without asthma (Fig. 1, panels B and C).
Table 2 shows the results of the multivariable analysis of STR-AD, separately in children with and without asthma. In the analysis among children without asthma (which was adjusted for age, gender, and BMI z-score), having a positive IgE to D.pteronyssinus and parental report of mold or mildew at home were significantly associated with 3.3 times and 5 times increased odds of STR-AD, respectively (Model 1). Similar results were obtained when a positive IgE to D. pteronyssinus was replaced with having 1 positive IgE to allergens (Model 2) or with total IgE (Model 3). In all three multivariable models, there was a trend for an inverse asso-ciation between parental report of sighting mice and STR-AD, but such trend was non-statistically significant (models
1 and 3) or of borderline statistical significance (model 2). In the multivariable analysis of STR-AD among children with asthma (also adjusted for age, gender, and BMI z-score), having private or employer-based health insurance was signifi -cantly associated with approximately twofold increased odds of STR-AD (Model 1). Similar results were obtained when a positive IgE to D. pteronyssinus was replaced with either 1 positive IgE to allergens (Model 2) or total serum IgE (Model 3).
Table 3 shows the sensitivity, specificity, PPV, NPV and AUC of various approaches to the diagnosis of STR-AD in Puerto Rican children, which were partly based on the results of our multivariable analysis (see Table 2). PD-AD had high specificity but very low sensitivity and a low AUC for STR-AD, both in children with and without asthma. A combi-nation of AD symptoms and a positive IgE to D. pteronyssinus yielded sensitivity 70% and specificity 95%, NPV 95% and PPV 88%, and an AUC 0.85 for STR-AD, both in children with and without asthma. Replacing a positive IgE to D. pteronyssinus with a positive IgE to 1 allergen slightly increased sensitivity (i.e. from 75.7% to 80% in all children) but had minimal impact on all other parameters (specificity, NPV, PPV or the AUC).
We found that ~93% of cases of STR-AD among children in Puerto Rico had not been diagnosed by a physician, possibly due to limited access to healthcare (particularly allergists) in Puerto Rico.15,16
A previous study of AD among children ages 6–7 years who attended two schools in Puerto Rico found that the prevalence of parental report of current eczema symptoms was 24.8%,4 and that ~70% of children with such symptoms had not been diagnosed with AD by a physician. In contrast to that study, we assessed not only eczema symptoms but also skin test reactivity to allergens and levels of total/allergen-specific IgEs in a population-based sample of school-aged children aged 6–14 years. Moreover, the response rate in the previous study (53%) was substan-tially lower than that in the current study.
Consistent with findings in other popula -
tions, we show that a positive IgE to common allergens such as house dust mite or an elevated total serum IgE is significantly associated with STR-AD.6 Indeed, dust mite allergen exposure has been linked to eczematous symptoms,2,8 and high dust mite allergen levels have been reported to be both common and associated with asthma and other allergic diseases in Latin America.17 In contrast to our findings for total IgE and STR-AD, Perkins et al reported that total IgE plays little role in prediction of visible eczema at 5 years, despite a significant association between total IgE and eczema severity.12 On the other hand, Ville et al showed that reductions in total IgE are associated with good treatment response and complete remission of AD11
Among children without asthma, we found that signs of mold or mildew in the house are associated with STR-AD. Such exposure could alter the skin barrier, a first step in AD pathogenesis.18 However, the impact of reducing mold or mildew on AD, if any, is unknown.19 Among children with asthma, we show that private or employer-based health insurance is significantly associated with STR-AD, likely due to improved access to healthcare.
We show that a combination of current eczematous symptoms and 1 positive IgE to common allergens could markedly improve the diagnostic rate of STR-AD among physicians in Puerto Rico. Indeed, such approach yielded a sensitivity of 80%, plus high: specificity, PPV, NPV, and AUC for STR-AD in all children. Of interest, replacing 1 positive IgE to al-lergens with a positive IgE to dust mite yielded a comparably high sensitivity (i.e. 76%) for STR-AD in all children, thus supporting a relatively simple and cost-effective approach to AD diagnosis and care in Puerto Rico.
Our study has substantial strengths, including a population-based sample of school-aged children and data on objective measures of allergic sensitization.4 We also recognize several study limitations. First, we cannot assess temporal relationships due to the cross-sectional study G. Yang et al. design. Second, we did not conduct direct skin examinations. However, we used a combination of STR to allergens
DUPIXENT es un medic amento de vent a con recet a que se utiliza par a tr at ar a adultos y niños de 6 meses o m á s co n de r m ati tis ató pi c a (e c ze m a) m o de r a d a a g r a v e , q u e n o pu e d e c o n t r o l a r s e c o r r e c t a me nt e c o n t e r a p i a s r e ce t a d a s q u e s e a p l i c a n s o b r e l a p i e l (tópic as) o en c asos en los que no se puedan utilizar terapias tópicas DUPIXENT puede utilizarse con o sin cor ticosteroides tópicos Se desconoce si DUPIXENT es seguro y efic az en niños menores de 6 meses que tengan dermatitis atópic a INFORMACIÓN IMPORTANTE DE SEGURIDAD No uses este medicamento si eres alérgico al dupilumab o a alguno de los ingredientes en DUPIXENT® Antes de usar DUPIXENT, infórmale a tu proveedor de atención médica sobre todas tus afecciones médicas, incluyendo: Si tienes problemas oculares Si tienes una infección parasitaria (helmintos). Si tienes programado recibir alguna vacuna No debes recibir una “ vacuna viva” antes o dur ante el tr at amiento con DUPIXENT Si estás embarazada o planeas quedar embarazada Se desconoce si DUPIXENT dañará a tu bebé en gestación Un registro de embarazo para mujeres que usan DUPIXENT durante el embarazo recopila información sobre tu salud y la d e t u b e b é P ar a ins cr ibir te o p ar a obte n e r más información, llama al 1-877-311-8972 o visit a https:// mother tobaby org /ongoing-study/dupixent / Si est ás amamant ando o planeas amamant ar Se desconoce si DUPIX ENT se tr ansmite a tr avés de la leche materna. Infór male a tu p rove e dor de ate n ción mé dic a sob re t o d o s l o s m e d i c a me n t o s q u e t o m a s , i n c l ui d o s l o s medic amentos de vent a libre y de vent a con recet a , las vit aminas y los suplementos a base de hierbas Especialmente, infórmale a tu proveedor de atención médica si estás tomando medicamentos corticosteroides orales, tópicos o inhalados, o si tienes dermatitis atópica y asma y usas un medicamento para el asma No cambies
ni suspendas tu medic amento cor ticos teroide u otro medic amento par a el asma sin hablar primero con tu proveedor de atención médic a Esto puede hacer que reaparezcan otros síntomas que fueron controlados por el medic amento cor ticos teroide u otro medic amento par a el asma
DUPIXENT puede c ausar efec tos secundarios graves, que incluyen:
R e a c c i o n e s a l ér g i c a s D U P I X E N T pu e d e c au s a r r e a c c i on e s a l ér g i c a s qu e a lg un a s v e c e s p u e d e n s e r g r ave s D e j a d e u s a r DU P IX E N T e i n f ó r m a l e a t u p r ove e d o r d e ate n ció n m é di c a o b u s c a ate n ció n d e em e r ge n cia d e in m e diato si tien e s a lg u n o d e l os siguientes signos o síntomas: Problemas respiratorios o sibilancias, inflamación del rostro, los labios, la boca, la lengua o la gargant a, desmayos, mareos, sens ación d e mare o, p uls o a ce le r a d o, fie b re, ur tic ar ia , d olor e n las ar ticulaciones, malestar general, picazón, erupción c u t á n e a , g a ng l io s l i n f á ti c o s i n fl a m a d o s , n á u s e a s o vómitos , o c alamb re s e n e l áre a d e l e s tómago P r o b l e ma s o c u l a r e s . I n f ó r ma l e a t u p r o v ee d o r d e atención médic a si tienes problemas oculares nuevos o si n ot as un e m pe o r a mie nto d e l os p ro b l e mas q u e te nías , i n cl ui d o d olo r o cul a r o c am b ios e n l a v isió n , como visión borrosa. Tu proveedor de atención médica puede referir te a un oft almólogo par a que te realicen un examen de la vis t a , si es neces ario Dolores en las ar ticulaciones A lgunas per sonas que usan DUPIXENT han tenido dificultades para caminar o moverse debido a los síntomas en sus ar ticulaciones y, en algunos casos, debieron ser hospitalizados Infórmale a t u p rove e d o r d e ate n ció n m é dic a s o b re cu a l q uie r síntoma nuevo en las ar ticulaciones o si empeoran los síntomas que tenías Tu proveedor de atención médica pu e d e inte r r um pir e l t r at amie nto con DU PIXENT si des arrollas sínto mas en las ar ticulaciones Los efectos secundarios más comunes en pacientes con dermatitis atópica son reacciones en el lugar de la
inyección, inflamación de los ojos y los párpados, que incluyen enrojecimiento, hinchazón y picazón, a veces con visión borrosa, herpes en la boca o en los labios, y un alto recuento de ciertos glóbulos blancos (eosinofilia).

Infórmale a tu proveedor de atención médic a si sufres algún efecto secundario que te cause malestar o que no desaparezca Estos no son todos los efectos secundarios posibles de DUPIXENT Llama a tu médico para obtener consejos mé dicos sob re los efe c tos se cun dar ios Te recomendamos informar sobre los efectos secundarios negativos de los medic amentos de vent a con recet a a la Adminis t r ación de A limentos y Me dic amentos de los E s t a dos Unidos (Fo o d an d D r ug Adminis t r ation , F D A ) V is i t a ww w f d a g o v/m e d w a t c h o l l a m a a l 1-8 0 0 -FDA-10 8 8
Us a DUPIXENT exac t amente según lo prescrito por tu proveedor de atención médic a DUPIXENT es una inyección que se administra debajo de la piel (inyección subcutánea) Tu proveedor de atención médica decidirá si t ú o t u cuid a d o r pu e de n inye c t a r DU PIX ENT N o intentes preparar e inyectar DUPIXENT hasta que tú o tu cuidador hayan sido c apacit ados por tu proveedor d e ate n ci ó n m é di c a . En ni ñ o s d e 12 a ñ o s o m á s , s e recomienda que DUPIXENT sea adminis tr ado por un a d ul to o b a j o l a s u p e r v is i ó n d e u n a d ul to En ni ñ o s menores de 12 años , un cuidador deb
Resumen breve de información importante para pacientes sobre DUPIXENT® (dupilumab) Inyección para uso subcutáneo
¿Qué es DUPIXENT?
• DUPIXENT es un medicamento con receta utilizado para lo siguiente: –moderada a grave, que no puede controlarse correctamente con terapias recetadas que se aplican sobre la piel (tópicas) o en casos en los que no se puedan utilizar terapias tópicas DUPIXENT puede utilizarse con o sin corticosteroides tópicos
• DUPIXENT actúa bloqueando dos proteínas que contribuyen a un tipo de
• que tengan dermatitis atópica ¿Quiénes no deberían usar DUPIXENT? No utilices DUPIXENT si eres alérgico al dupilumab o a cualquiera de los
para conocer la lista completa de ingredientes de DUPIXENT
¿Qué debo informarle a mi pro veedor de atención médica antes de usar DUPIXENT? Antes de usar DUPIXENT, infórmale a tu proveedor de atención médica sobre todas tus afecciones médicas, lo que incluye:
• Si tienes problemas oculares
• Si tienes una infección parasitaria (helmintos)
• Si tienes programado recibir algún tipo de vacuna No debes recibir ninguna “vacuna viva” justo antes o mientras recibes tratamiento con DUPIXENT
• Si estás embarazada o tienes planes de quedar embarazada Se desconoce si DUPIXENT puede dañar a tu bebé en gestación – Registro de exposición durante el embarazo. Hay un registro de exposición para mujeres que toman DUPIXENT durante el embarazo El objetivo de este registro es reunir información sobre tu salud y la de tu bebé Tu proveedor de atención médica puede inscribirte en este registro También puedes inscribirte por tu cuenta u obtener más información sobre el registro llamando al 1-877-311-8972 o ingresando en https://mothertobaby.org/ongoing-study/dupixent/.
• Si estás amamantando o tienes planes de hacerlo Se desconoce si DUPIXENT se transmite a través de la leche materna
Informa a tu proveedor de atención médica todos los medicamentos que tomas, incluidos los medicamentos de venta libre y de venta con receta, las vitaminas
Informa a tu proveedor de atención médica en especial en los
• Si estás tomando corticosteroides orales, tópicos o inhalados
• Si tienes dermatitis atópica y asma y usas un medicamento para el asma
No cambies ni interrumpas la administración del medicamento corticosteroide
médica Esto puede hacer que reaparezcan otros síntomas que fueron controlados con el medicamento corticosteroide u otro medicamento para el asma.
¿Cómo debo utilizar DUPIXENT?
• Consulta las “Instrucciones de uso” detalladas que se proporcionan con DUPIXENT para obtener información sobre cómo preparar
• Utiliza DUPIXENT exactamente como te lo recetó tu proveedor de atención médica
• Tu proveedor de atención médica te dirá cuánto DUPIXENT debes inyectar
• DUPIXENT viene en una jeringa precargada de dosis única con protector
– La pluma precargada de DUPIXENT es solo para uso en adultos y niños
– La jeringa precargada de DUPIXENT es solo para uso en adultos y niños
• • Si tu proveedor de atención médica decide que tú o un cuidador pueden administrar las inyecciones DUPIXENT, tú o tu cuidador deben recibir capacitación sobre la manera correcta de preparar e inyectar DUPIXENT No intentes inyectar DUPIXENT hasta que tu proveedor de atención médica te haya mostrado la adulto coloque o super vise la administración de DUPIXENT A niños menores de
• Si tu cronograma de dosis es semana de por medio y omites una dosis de DUPIXENT: a la dosis omitida y, luego, continúa con el cronograma original Si la dosis omitida para administrar la inyección de DUPIXENT
Puertorriqueña de Medicina y Salud Pública
• Si tu cronograma de dosis es cada cuatro semanas y omites una dosis de DUPIXENT: posteriores a la dosis omitida y, luego, continúa con el cronograma original
administrarte la inyección de DUPIXENT
• Si inyectas más DUPIXENT de lo que se recetó (sobredosis), busca ayuda médica o comunícate inmediatamente con un experto del Centro de Toxicología llamando al 1-800-222-1222
• Es posible que tu proveedor de atención médica te recete otros medicamentos para utilizar con DUPIXENT Utiliza los otros medicamentos recetados exactamente como te lo indique tu proveedor de atención médica ¿Cuáles son los efectos secundarios posibles de DUPIXENT? DUPIXENT puede provocar efectos secundarios graves, incluidos
• Reacciones alérgicas DUPIXENT puede causar reacciones alérgicas que algunas veces pueden ser graves. Deja de usar DUPIXENT emergencia de inmediato si tienes alguno de los siguientes síntomas:
la boca, la lengua o la garganta, urticaria, picazón, náuseas o vómitos, desmayos, mareos, sensación de mareo, dolor en las articulaciones, erupción
• Problemas oculares. Informa a tu proveedor de atención médica si tienes problemas oculares nuevos o si notas un empeoramiento de los problemas que tenías, incluido el dolor ocular o cambios en la visión, como visión borrosa Tu proveedor de atención médica puede referirte a un oftalmólogo para que te realicen un examen de la vista, si es necesario • Dolores en las articulaciones. Las personas que utilizan DUPIXENT pueden experimentar dolores en las articulaciones Algunas personas han articulaciones y, en algunos casos, debieron ser hospitalizados Informa a tu proveedor de atención médica sobre cualquier síntoma nuevo en las articulaciones o si empeoran los síntomas que tenías Tu proveedor de atención médica puede interrumpir el tratamiento con DUPIXENT si desarrollas síntomas en las articulaciones Los efectos secundarios más comunes de DUPIXENT en pacientes con dermatitis atópica son: los ojos y los párpados (que incluye enrojecimiento, hinchazón y picazón) a veces con visión borrosa, herpes en la boca o en los labios y un alto recuento de ciertos
Se han informado los siguientes efectos secundarios adicionales con el uso de DUPIXENT: Erupción facial o enrojecimiento Informa a tu proveedor de atención médica si sufres algún efecto secundario que te cause malestar o que no desaparezca Estos no son todos los efectos secundarios posibles de DUPIXENT Llama a tu médico para obtener consejos médicos sobre los efectos secundarios Puedes informar los efectos secundarios a la FDA Visita www.fda.gov/medwatch o llama al 1-800-FDA-1088.
en un prospecto de Información para el paciente No uses DUPIXENT para una afección para la que no se recetó No administres DUPIXENT a otras personas, incluso si tienen los mismos síntomas que tú Es posible que les haga daño Este es un resumen breve de la información más importante sobre DUPIXENT para este uso Si deseas recibir más información, habla con tu proveedor de atención médica Puedes pedirle a tu farmacéutico o proveedor de atención médica más información sobre DUPIXENT dirigida a profesionales de atención médica. Para obtener más información sobre DUPIXENT, visita www.DUPIXENT.com o llama al 1-844-DUPIXENT (1-844-387-4936)
¿Cuáles son los ingredientes de DUPIXENT?
Ingrediente activo: dupilumab.
Ingredientes inactivos: acetato de sodio, sacarosa y agua para inyección
Fecha de publicación: Junio de 2022 DUP.22.06.0030

Los síntomas asociados a la menopausia pueden comenzar a presentarse desde los 40 años de edad. Estos síntomas tienen usualmente su punto máximo, un año luego de la menopausia. Este proceso previo a la menopausia, se le llama perimenopausia. Usualmente está marcado por disminución en la función ovárica, y en baja de niveles de estrógeno y progesterona.
Menopausia es el tiempo en la vida de una mujer cuando deja de tener su menstruación. Esto marca el fin de la etapa reproductiva de la mujer. La edad promedio en que se experimenta la menopausia es de 51 años. No obstante, las mujeres alcanzan la menopausia cuando cesa su período durante 12 meses consecutivos, incluidos episodios de manchado. Es una transición clave en la vida de una mujer, pero siempre recordando que la experiencia de la menopausia es diferente para cada persona.

En el periodo perimenopausico se comienza a tener cambios en la cantidad del sangrado menstrual y/o tener ciclos, más o menos cortos. También es cuando las mujeres comienzan a manifestar varios de los síntomas asociados a la menopausia, entre ellos los sofocos o calentones, sudoración, rubor en la cara y/o cuello, sequedad vaginal, dispareunia, insomnio y cambios de humor.
Los sofocos llamados calentones pueden presentarse hasta en un 75 por ciento de las mujeres. En promedio duran de 5-7 años. Están caracterizados por periodos de 1-5 minutos, en los cuales puede presentarse palpitaciones, ansiedad, transpiraciones, escalofríos y sudoración. El único tratamiento que se ha visto que significativamente reduce los síntomas vasomotores son los estrógenos, ya sea solos o en combinación.
Con la menopausia, también aumenta el riesgo de osteoporosis, que pone en riesgo a la mujer de desarrollar fracturas de cadera y/o espina dorsal.
Dr. Carlos A. Fonseca Salgado, FACOG, FPMRS
Estas fracturas no solo ponen en riesgo la vida de las mujeres, si no que complica su calidad de vida, limitando su movilidad.
Además, pueden experimentar el Síndrome Genitourinario en la que la vagina comienza a ponerse, fina, seca, sensible, pierde elasticidad y cambia de acidez. Esto puede asociarse con picor vaginal, irritación y/o ardor vaginal, dolor con las relaciones sexuales, sensibilidad, disuria, frecuencia urinaria, infecciones vaginales y/o orina. El tratamiento preferido para el síndrome Genitourinario son los estrógenos vaginales. Estos no solo han probado ser efectivos en aliviar efectos locales, sino que ha probado su seguridad de tratamiento. En un estudio en Finlandia con 18,000 pacientes, el estrógeno vaginal no pudo asociarse con el riesgo a desarrollar cáncer de senos. Cuando se compara con placebo, tampoco se ha visto que incremente el riesgo a cáncer de endometrio y/o hiperplasia, por lo cual no se recomienda el uso de progesterona para protección del endometrio.
Se ha visto que el uso de estrógenos ayuda a aumentar los receptores alfa adrenérgicos en la uretra. Esto a su vez, aumentando la perfusión y vascularidad en la uretra y, por lo tanto, mejorando la coaptación y aumentando la presión uretral.
La intensidad de los síntomas asociados a la menopausia puede variar entre las mujeres. Este proceso puede transcurrir, sin presentar síntomas, con síntomas leves y/o severos. Los síntomas pueden ser tan severos, que podrían afectar su calidad vida, llevando a estas a desarrollar síntomas depresión, problemas interpersonales, de trabajo y/o matrimoniales.
El tratamiento de la menopausia ha variado a través de los años. Como los síntomas pueden variar entre persona a persona, así también varían los tratamientos. No se debe esperar a que afecte el día
a día, para buscar ayuda. Muchas veces la paciente no busca ayuda por desconocimiento. Una paciente debidamente orientada, puede reconocer estos síntomas y buscar ayuda con un profesional de la salud. Mientras más información y conocimiento, más fácil puede entender y mejorarlos síntomas en la transición hacia la menopausia.
Hoy día, para la menopausia contamos con tratamientos hormonales, no hormonales, cremas vaginales, parchos transdermales, anillos vaginales, humectantes, lubricantes, medicamentos para mejorar la absorción de calcio, suplementos, vitaminas, remedios naturales, productos de soya y ejercicios.
La menopausia es un proceso individual de cada mujer, igualmente los tratamientos varían de paciente a paciente. Sin embargo, la educación siempre puede ayudar a las mujeres a conocer sus signos, síntomas que experimenta y las opciones de cuidado. En fin, poder llevar una vida activa y plena. Para conocer más sobre la menopausia puedes acceder a www.conocetumenopausia.com. Visite a su ginecólogo, discuta sus opciones y conozca sus recomendaciones.
Cody JD, Jacobs ML, Richardson K, et al.: Oestrogen therapy for urinary incontinence in post-menopausal women. Cochrane Database Syst Rev. 10:CD001405 2012 23076892


Rahn DD, Carberry C, Sanses TV, et al.: Vaginal estrogen for genitourinary syndrome of menopause: a systematic review. Obstet Gynecol. 124:1147 2014 25415166
Ewies AA, Alfhaily F: Topical vaginal estrogen therapy in managing postmenopausal urinary symptoms: a reality or a gimmick? Climacteric. 13:405 2010 20670198


Katie K. Crean-Tate, MD; Stephanie S. Faubion, MD; Holly J. Pederson, MD; Jennifer A. Vencill, PhD, LP; Pelin Batur, MD, NCMP, CCD: Management of genitourinary syndrome of menopause in female cancer patients: a focus on vaginal hormonal therapy. American Journal of Obstetrics & Gynecology 103-112 FEBRUARY 2020
WALTERS AND KARRAM. 5th edition. UROGYNECOLOGY AND RECONSTRUCTIVE PELVIC SURGERY: Elsevier; 2022 Management of Menopausal Symptoms. Practice Bulletin Number 141. january 2014
Revista Puertorriqueña de Medicina y Salud Pública


Fotos de izquierda a derecha: Luis K. Hernández Muñiz, (Estudiante Doctorado en Psicología Clínica) junto al Presidente y CEO David Lenihan de Ponce Health Sciences University. Premio Presidencial a la Diversidad Reconocimiento por Departamento del Programa Doctoral en Psicología ReconocimientoClínica de Honor Académico (Promedio 3.90 a 4.0)
Karen L. Ocasio Rivera (Maestría en Salud Pública) junto al Presidente y CEO David Lenihan de Ponce Health Sciences ReconocimientoUniversity. de Honor Académico (Promedio 3.90 a 4.0)
Reconocimiento Academia Médica del Sur Premio Presidencial a la Diversidad

Daniela N. Martir (Graduanda del Doctorado en Medicina) a su lado Izquierdo Dr. David Lenihan Presidente y CEO de Ponce Health Sciences University. Recibe Premio Presidencial a la Diversidad













Estudiante







Estudiante
Estudiante
La
Estudiantes











La Universidad Ana G. Méndez (UAGM) confirió un total de 2,015 grados académicos en la segunda ceremonia de graduación realizada hoy en el Centro de Convenciones de Puerto Rico.
“Apuesto cien por ciento a su talento y a sus ganas de crecer. A su determinación de hacer las cosas de manera distinta. Estoy convencido de que se destacarán en sus respectivas profesiones y que seremos testigos de sus grandes aportaciones al país”, expresó José F. Méndez Méndez, presidente de UAGM, en su mensaje a los graduandos.
Durante los actos, la universidad le otorgó un doctorado honoris causa en Salud a Lilliam Rodríguez Capó, principal oficial ejecutivo y fundadora de VOCES, Coalición de Inmunización y Promoción de la Salud de Puerto Rico, por su excelente liderato para articular de forma exitosa los esfuerzos de vacunación y orientación a la población en tiempos de pandemia.



En su mensaje, Rodríguez destacó, entre otras cosas, la importancia de ser salubristas. “Todos debemos ser salubristas, no importa la carrera en que nos desempeñemos o estemos a punto de emprender. Esta pandemia, que nos ha tocado vivir tan duramente estos dos años, nos debe hacer reflexionar que no podemos vivir una vida de manera individual, sino pensando en el colectivo, en el bien común”, expresó.
Es la primera vez que la institución celebra una graduación integrada con sus tres recintos de Carolina, Cupey y Gurabo y sus ocho centros universitarios. Los recién graduados que obtuvieron sus grados en la ceremonia de hoy jueves, 16 de junio pertenecen a las divisiones académicas de: Ciencias y Tecnología, Negocios, Turismo y Emprendimiento, Ingeniería, Diseño y Arquitectura, Medicina Naturopática, Medicina Veterinaria y Dental. Se otorgaron 59 certificados técnicos,
297 grados asociados, 1,071 bachilleratos, 567 maestrías y 21 doctorados.
El 2021 trajo para ella y su organización distintos retos que ha sabido proyectar y llevar adelante junto con un equipo de personas que solo trabaja para ser útil a la sociedad en la que viven.
Conoce la trayectoria de esta ilustre dama que se convirtió en la heroína contra el Covid-19.
VOCES es una coalición multisectorial, centrada en la comunidad, dedicada a la promoción de la salud y prevención de enfermedades mediante la inmunización con estrategias diversas basadas en evidencia. Fue fundada en el 2013 por Lilliam Rodríguez Capó.
Cuenta con la colaboración de sobre 100 entidades e individuos en el campo de la salud en P.R. Actualmente, es reconocida como una organización con certificación 501c3 y en cumplimiento con las regulaciones del Estado libre Asociado de Puerto Rico.
Desde su creación, VOCES ha tenido un impacto significativo en la educación y promoción de la inmunización a través de iniciativas diversas, campañas educativas y eventos nacionales en colaboración con asociaciones profesionales, el Departamento de Salud de Puerto Rico y entidades no gubernamentales.
Estos esfuerzos de concienciación integral han dado lugar a darle un tema de la actualidad, a la inmunización, a la aprobación de política pública, mejor cobertura y regulaciones innovadoras que han acercado a Puerto Rico a garantizar un acceso adecuado a las vacunas para un segmento amplio de la población.
Con el objetivo de educar y concientizar acerca de la Hidradenitis Supurativa, desde el año 2020 la Asamblea Legislativa de Puerto Rico, en vista de que esta condición crónica de la piel, dolorosa, no contagiosa, necesitaba más visibilización por el impacto en la vida de los pacientes, declaró la primera semana de junio como la “Semana de la Concienciación de la Hidradenitis Supurativa”.




Motivados por el objetivo de educar y dar voz a los pacientes, la Revista de Medicina y Salud Pública, junto al grupo Hidradenitis Supurativa Golondrinos, serán parte del primer encuentro presencial de esta comunidad de pacientes y especialistas para conocer los avances en tratamientos y diagnóstico en el País.
Vale la pena mencionar que la condición no incapacita, pero sí genera incomodidad, porque la persona afectada comúnmente puede sentir vergüenza y rehusarse a salir en público, debido a la ubicación de estos abscesos, a la supuración y al mal olor, lo cual puede provocar tristeza o incluso, depresión. Aunque no existe cura para esta condición, el diagnóstico y tratamiento temprano pueden controlar el dolor, promover la
cicatrización de las heridas y prevenir complicaciones.
Al respecto, la Lic. Janice Marrero Irizarry, presidenta de Vocational and Transition Services, y Consejera de Rehabilitación, indicó que es muy común que se presenten situaciones de empresas que no garantizan los derechos de los pacientes con esos limitantes. “Muchas veces las personas tienen miedo y se quedan callados porque pueden que los despidan si piden un acomodo razonable. A veces sufren atropellos y no hacen valer sus derechos”.

La Licenciada, quien también cita las leyes federales, American with disability (ADA), Acta de Rehabilitación, Sección 504 y la Individual with Disability Education Act (IDEA), que protegen a las personas con discapacidades en el área laboral y educativa, es enfática en precisar que un paciente con Hidradenitis no necesariamente es una persona con discapacidad, ya que no depende de la condición misma, sino las limitaciones que trae consigo esa condición.
Por esa razón, esta Ley contempla la exhortación a todos los organismos públicos y entidades privadas, así como
la ciudadanía en general, a unirse a ser parte de la educación de esta afección para concienciar sobre esta enfermedad autoinmune.
La misma Ley 29, indica que el Departamento de Salud, así como los municipios y cualesquiera otras entidades sin fines de lucro interesadas, deberán adoptar las medidas necesarias para la consecución de los objetivos de esta Ley, mediante la organización y realización de actividades que promuevan la educación a la ciudadanía puertorriqueña sobre esta condición.
1Dermatóloga y fundadora del Programa de Ciencias Clínicas Externas de hidradenitis supurativa del Recinto de Ciencias Médicas de la Universidad de Puerto Rico. 2Residente del Departamento de Dermatología del Recinto de Ciencias Médicas 3Residente del Departamento de Dermatología del Recinto de Ciencias Médicas 4Veterana actriz puertorriqueña 5Fundadora de HS Golondrinos Puerto Rico 6Residente del Departamento de Psiquiatría del RCM 7MS RDN LND 8Ginecóloga obstetra.



A propósito del Día Internacional de Acción por la Salud de las Mujeres, Pedro Lugo, vicepresidente del Grupo Editorial Mundo y de la Revista Medicina y Salud Pública, destacó la labor de los médicos y profesionales quienes se esfuerzan por brindar su apoyo a favor de la salud de las féminas puertorriqueñas, agradeciendo a las mujeres que han encaminado su vida, como su madre, y otras que lideran en el campo de la ciencia, salud y la política.
A propósito del Día Internacional de Acción por la Salud de las Mujeres, Pedro Lugo, vicepresidente del Grupo Editorial Mundo y de la Revista Medicina y Salud Pública, destacó la labor de los médicos y profesionales quienes se esfuerzan por brindar su apoyo a favor de la salud de las féminas puertorriqueñas, agradeciendo a las mujeres que han encaminado su vida, como su madre, y otras que lideran en el campo de la ciencia, salud y la política. El doctor Oscar Soto Raíces, reumatólogo y asesor editorial de la Revista Medicina y Salud Pública, resaltó que el camino a transitar ha sido más difícil para la mujer que para el hombre, por lo que se reconoce la contribución histórica que han hecho al pueblo a través del apoyo que han dado a la salud de la mujer en Puerto Rico. Las mujeres han roto patrones y han superado obstáculos, por lo que merecen ser celebradas.
El doctor Oscar Soto Raíces, reumatólogo y asesor editorial de la Revista Medicina y Salud Pública, resaltó que el camino a transitar ha sido más difícil para la mujer que para el hombre, por lo que se reconoce la contribución histórica que han hecho al pueblo a través del apoyo que han dado a la salud de la mujer en Puerto Rico. Las mujeres han roto patrones y han superado obstáculos, por lo que merecen ser celebradas.
Por su parte, el médico y editor de la Revista Medicina y Salud Pública, Dr. Alberto Santiago Cornier, felicitó a quienes se han dedicado a la investigación y el manejo de las enfermedades en la mujer puertorriqueña
La senadora Ana Irma Rivera Lassén, enfatizó que hablar de la salud de la mujer,
del cuerpo femenino, es un tabú: “parecería que todo el mundo tiene derecho de hablar del cuerpo de las mujeres, menos las mujeres mismas. Todos se creen con derecho de controlar el cuerpo de las mujeres, hasta el Estado. Hablar de persona gestante parece darles pánico a algunas personas, porque ello significa hablar de diversidad, reconocer que hay mujeres que en sus cuerpos tienen capacidad de gestación, aunque su identidad de género sea otra”, afirmó, añadiendo que hablar de los derechos de las mujeres y de sus embarazos atañe a las mujeres y a las personas gestantes, así que se deben normalizar las conversaciones entre las mujeres, para las mujeres y por las mujeres.
Isbelia Farías Periodistaen la importancia que tiene la lactancia materna, calificándolo como un acto de amor y de ternura.
La senadora Ana Irma Rivera Lassén, enfatizó que hablar de la salud de la mujer, del cuerpo femenino, es un tabú: “parecería que todo el mundo tiene derecho de hablar del cuerpo de las mujeres, menos las mujeres mismas. Todos se creen con derecho de controlar el cuerpo de las mujeres, hasta el Estado. Hablar de persona gestante parece darles pánico a algunas personas, porque ello significa hablar de diversidad, reconocer que hay mujeres que en sus cuerpos tienen capacidad de gestación, aunque su identidad de género sea otra”, afirmó, añadiendo que hablar de los derechos de las mujeres y de sus embarazos atañe a las mujeres y a las personas gestantes, así que se deben normalizar las conversaciones entre las mujeres, para las mujeres y por las mujeres.
La senadora Migdalia González Arroyo, presidenta de la comisión de asuntos de la mujer del Senado de Puerto Rico, felicitó a las profesionales como salubristas y por sus aportes en el ámbito de la salud de las mujeres. Libia Méndez, la vicepresidenta de la Cámara de Representantes, recordó que su madre era ama de casa y su papá agricultor, por lo que los pequeños iban a la escuela para aprender y luego enseñar a la madre. Desde el 97 ocupa el cargo como representante, pero su camino no fue fácil, pues, venía de la pobreza. Hace énfasis

La senadora Migdalia González Arroyo, presidenta de la comisión de asuntos de la mujer del Senado de Puerto Rico, felicitó a las profesionales como salubristas y por sus aportes en el ámbito de la salud de las mujeres. Lidia Méndez, la vicepresidenta de la Cámara de Representantes, recordó que su madre era ama de casa y su papá agricultor, por lo que los pequeños iban a la escuela para aprender y luego enseñar a la madre. Desde el 97 ocupa el cargo como representante, pero su camino no fue fácil, pues, venía de la pobreza. Hace énfasis en la importancia que tiene la lactancia materna, calificándolo como un acto de amor y de ternura.
La mujer es la fuerza unificadora de la familia y la sociedad, por lo que se debe promover su bienestar, el cual es, más que una necesidad, es un deber que implica retos, ya que, por ser mujer, los números no siempre están a favor, basándose en el hecho de que la depresión es más alta en la mujer, las muertes por enfermedades cardiovasculares son más elevadas en la mujer, las discapacidades son más frecuentes en las mujeres, las infecciones por VIH son más altas en la mujer, quienes, además, padecen de cáncer uterino y seno, endometriosis, síndrome de ovario poliquístico, lupus, artritis, entre otras condiciones que arriesgan su salud, en Puerto Rico y en el mundo.
En este sentido, se destaca la labor de: Lilliam Rodríguez, principal oficial y fundadora de VOCES; Dra. María Ramos, cardióloga y presidenta de la sociedad de cardiología; Dra. Leticia Hernánez, endocrinóloga y presidenta de la sociedad puertorriqueña de endocrinología y diabetología; Dra. Idhaliz Flores, Investigadora en endometriosis; Dra. Esther Torres, gastroenteróloga, directora del centro de
La mujer es la fuerza unificadora de la familia y la sociedad, por lo que se debe promover su bienestar, el cual es, más que una necesidad, es un deber que implica retos, ya que, por ser mujer, los números no siempre están a favor, basándose en el hecho de que la depresión es más alta en la mujer, las muertes por enfermedades cardiovasculares son más elevadas en la mujer, las discapacidades son más frecuentes en las mujeres, las infecciones por VIH son más
Por su parte, el médico y editor de la Revista Medicina y Salud Pública, Dr. Alberto Santiago Cornier, felicitó a quienes se han dedicado a la investigación y el manejo de las enfermedades en la mujer puertorriqueña
enfermedades inflamatorias del intestino de la UPR, presidenta de la fundación Esther A. Torres; Dra. Carmen Zorrilla, catedrática del recinto de ciencias médicas y miembro de la coalición científica de Puerto Rico; Dr. Nabal Bracero, ginecólogo, presidente de Progyn, especialista en infertilidad y endocrinología reproductiva; Dra. Damaris Torres Paoli, presidenta de la sociedad dermatológica de Puerto Rico; Dra. Eva Cruz, radióloga, especialista en imágenes de la mujer; Dr. Bolívar Arboleda, director médico y director del Instituto de Seno, del Hospital HIMA, San Pablo, Caguas; Dra. Franchesca Fiorito, neuróloga especialista en cefaleas; Dra. Ángeles Rodríguez, infectóloga y exepidemióloga del Estado; Dra. Bárbara Rosado, gastroenteróloga hepatóloga; Dra. Elivette Zambrana, reumatóloga pediátrica y presidenta de la Asociación de Reumatólogos de Puerto Rico; Lcda. Linette Sánchez, directora del programa de vacunación del colegio de médicos cirujanos de Puerto Rico; Adria García, enfermera oncóloga.
altas en la mujer, quienes, además, padecen de cáncer uterino y seno, endometriosis, síndrome de ovario poliquístico, lupus, artritis, entre otras condiciones que arriesgan su salud, en Puerto Rico y en el mundo.
enfermedades inflamatorias del intestino de la UPR, presidenta de la fundación Esther A. Torres; Dra. Carmen Zorrilla, catedrática del recinto de ciencias médicas y miembro de la coalición científica de Puerto Rico; Dr. Nabal Bracero, ginecólogo, presidente de Progyn, especialista en infertilidad y endocrinología reproductiva; Dra. Damaris Torres Paoli, presidenta de la sociedad dermatológica de Puerto Rico; Dra. Eva Cruz, radióloga, especialista en imágenes de la mujer; Dr. Bolívar Arboleda, director médico y director del Instituto de Seno, del Hospital HIMA, San Pablo, Caguas; Dra. Franchesca Fiorito, neuróloga especialista en cefaleas; Dra. Ángeles Rodríguez, infectóloga y exepidemióloga del Estado; Dra. Bárbara Rosado, gastroenteróloga hepatóloga; Dra. Elivette Zambrana, reumatóloga pediátrica y presidenta de la Asociación de Reumatólogos de Puerto Rico; Lcda. Linette Sánchez, directora del programa de vacunación del colegio de médicos cirujanos de Puerto Rico; Adria García, enfermera oncóloga.
de la doctora Carmen Zorrilla, ella se mantiene activa en varios proyectos, pues actualmente, está trabajando en respuesta a la pandemia de COVID-19 en PR, con un centro de pruebas diagnósticas moleculares, el estudio de la vacuna Novavax para COVID, un centro de vacunación de COVID en el RCM y futuros estudios de estrategias de vacunación de COVID en embarazadas. Zorrilla, al recibir su premio, señaló que falta que la Organización Mundial de la Salud reconozca que Puerto Rico fue el primer país en eliminar la infección perinatal del VIH desde 1994.
de la doctora Carmen Zorrilla, ella se mantiene activa en varios proyectos, pues actualmente, está trabajando en respuesta a la pandemia de COVID-19 en PR, con un centro de pruebas diagnósticas moleculares, el estudio de la vacuna Novavax para COVID, un centro de vacunación de COVID en el RCM y futuros estudios de estrategias de vacunación de COVID en embarazadas. Zorrilla, al recibir su premio, señaló que falta que la Organización Mundial de la Salud reconozca que Puerto Rico fue el primer país en eliminar la infección perinatal del VIH desde 1994.
En este sentido, se destaca la labor de: Lilliam Rodríguez, principal oficial y fundadora de VOCES; Dra. María Ramos, cardióloga y presidenta de la sociedad de cardiología; Dra. Leticia Hernánez, endocrinóloga y presidenta de la sociedad puertorriqueña de endocrinología y diabetología; Dra. Idhaliz Flores, Investigadora en endometriosis; Dra. Esther Torres, gastroenteróloga, directora del centro de enfermedades inflamatorias del intestino de la UPR, presidenta de la fundación Esther A. Torres; Dra. Carmen Zorrilla, catedrática del recinto de ciencias médicas y miembro de la coalición científica de Puerto Rico; Dr. Nabal Bracero, ginecólogo, presidente de Progyn, especialista en infertilidad y endocrinología reproductiva; Dra. Damaris Torres Paoli, presidenta de la sociedad dermatológica de Puerto Rico; Dra. Eva Cruz, radióloga, especialista en imágenes de la mujer; Dr. Bolívar Arboleda, director médico y director del Instituto de Seno, del Hospital HIMA, San Pablo, Caguas; Dra. Franchesca Fiorito, neuróloga especialista en cefaleas; Dra. Ángeles Rodríguez, infectóloga y exepidemióloga del Estado; Dra. Bárbara Rosado, gastroenteróloga hepatóloga; Dra. Elivette Zambrana, reumatóloga pediátrica y presidenta de la Asociación de Reumatólogos de Puerto Rico; Lcda. Linette Sánchez, directora del programa de vacunación del colegio de médicos cirujanos de Puerto Rico; Adria García, enfermera oncóloga.
La Revista Medicina y Salud Pública agradece y reconoce el valor y contribución para la salud de todas las mujeres puertorriqueñas, por parte de los profesionales mencionados.
La Revista Medicina y Salud Pública agradece y reconoce el valor y contribución para la salud de todas las mujeres puertorriqueñas, por parte de los profesionales mencionados.
Los profesionales homenajeados recibieron un premio en el marco del Día Internacional de Acción por la Salud de las Mujeres, destacando que todos cuentan con una preparación académica y una experiencia laboral formidable. En el caso
Los profesionales homenajeados recibieron un premio en el marco del Día Internacional de Acción por la Salud de las Mujeres, destacando que todos cuentan con una preparación académica y una experiencia laboral formidable. En el caso


La doctora Elivette Zambrana indicó que es necesario trabajar de la mano en políticas públicas que promuevan un estilo de vida saludable para paliar muchas condiciones que están asociadas a la obesidad y que afectan a las mujeres. El Dr. Nabal Bracero también resaltó la preocupación que existe sobre la educación reproductiva, que esta sea de calidad, tangible y que permite defender a la mujer de la violencia sexual o reproductiva.
La doctora Elivette Zambrana indicó que es necesario trabajar de la mano en políticas públicas que promuevan un estilo de vida saludable para paliar muchas condiciones que están asociadas a la obesidad y que afectan a las mujeres. El Dr. Nabal Bracero también resaltó la preocupación que existe sobre la educación reproductiva, que esta sea de calidad, tangible y que permite defender a la mujer de la violencia sexual o reproductiva.
La Revista Medicina y Salud Pública agradece y reconoce el valor y contribución para la salud de todas las mujeres puertorriqueñas, por parte de los profesionales mencionados. Los profesionales homenajeados recibieron un premio en el marco del Día Internacional de Acción por la Salud de las Mujeres, destacando que todos cuentan con una preparación académica y una experiencia laboral formidable. En el caso de la doctora







lo que aboga por el derecho y el acceso a la salud, felicitando a la Revista Medicina y Salud Pública por los años que han dedicado a la educación.
Dra.
Carmen Zorrilla, ella se mantiene activa en varios proyectos, pues actualmente, está trabajando en respuesta a la pandemia de COVID-19 en PR, con un centro de pruebas diagnósticas moleculares, el estudio de la vacuna Novavax para COVID, un centro de vacunación de COVID en el RCM y futuros estudios de estrategias de vacunación de COVID en embarazadas. Zorrilla, al recibir su premio, señaló que falta que la Organización Mundial de la Salud reconozca que Puerto Rico fue el primer país en eliminar la infección perinatal del VIH desde 1994.
La doctora María Ramos es cardióloga y la primera presidenta de la Sociedad Puertorriqueña de Cardiología, en 72 años, lo cual la hace sentir honrada. La gastroenteróloga Bárbara Rosado se pronunció sobre el honor que significa servir a su país.
lo que aboga por el derecho y el acceso a la salud, felicitando a la Revista Medicina y Salud Pública por los años que han dedicado a la educación.
La doctora María Ramos es cardióloga y la primera presidenta de la Sociedad Puertorriqueña de Cardiología, en 72 años, lo cual la hace sentir honrada. La gastroenteróloga Bárbara Rosado se pronunció sobre el honor que significa servir a su país.
Finalmente, se honró la labor que realizan las enfermeras 7 enfermeros, quienes cumplen un rol importante como enlace entre los médicos y pacientes, en especial a la licenciada Adria García.
Finalmente, se honró la labor que realizan las enfermeras 7 enfermeros, quienes cumplen un rol importante como enlace entre los médicos y pacientes, en especial a la licenciada Adria García.
La doctora Elivette Zambrana indicó que es necesario trabajar de la mano en políticas públicas que promuevan un estilo de vida saludable para paliar muchas condiciones que están asociadas a la obesidad y que afectan a las mujeres. El Dr. Nabal Bracero también resaltó la preocupación que existe sobre la educación reproductiva, que esta sea de calidad, tangible y que permite defender a la mujer de la violencia sexual o reproductiva.
El doctor Bolívar Arboleda mencionó lo importante que ha sido la Revista y Salud Pública en la difusión de temas científicos que permiten la actualización de conocimientos, acotando que el cáncer de seno continúa siendo la primera causa de mortalidad por cáncer en la mujer, por lo que queda mucho trabajo para avanzar.
El doctor Bolívar Arboleda mencionó lo importante que ha sido la Revista y Salud Pública en la difusión de temas científicos que permiten la actualización de conocimientos, acotando que el cáncer de seno continúa siendo la primera causa de mortalidad por cáncer en la mujer, por lo que queda mucho trabajo para avanzar.
La doctora Esther Torres reconoce que su inspiración han sido sus pacientes, por lo que aboga por el derecho y el acceso a la salud, felicitando a la Revista Medicina y Salud Pública por los años que han dedicado a la educación.
El doctor Bolívar Arboleda mencionó lo importante que ha sido la Revista y Salud Pública en la difusión de temas científicos que permiten la actualización de conocimientos, acotando que el cáncer de seno continúa siendo la primera causa de mortalidad por cáncer en la mujer, por lo que queda mucho trabajo para avanzar.
La doctora Esther Torres reconoce que su inspiración han sido sus pacientes, por
La doctora Esther Torres reconoce que su inspiración han sido sus pacientes, por
Finalmente, Belinda Z. Burgos González, gerente de proyectos, indicó que “a nivel editorial reconocemos la importancia que ha tenido para nosotros el poder ser testigo del trabajo de mujeres científicas desde los laboratorios del País, precisamente en estudios que pudieran beneficiar a otras mujeres en un futuro, como lo es el caso de enfermedades como el cáncer triple negativo y otros tipos de enfermedades. Igualmente hemos sido testigos de la empatía de distintas profesionales de la salud que dan la milla extra por esas mujeres trabajadoras y jefas de familia, quienes en muchas ocasiones se tienen como última prioridad. Expresamos nuestro agradecimiento a este selecto grupo de profesionales de la salud que se han hecho eco de las atenciones necesarias hacia las mujeres puertorriqueñas”.
La doctora María Ramos es cardióloga y la primera presidenta de la Sociedad Puertorriqueña de Cardiología, en 72 años, lo cual la hace sentir honrada. La gastroenteróloga Bárbara Rosado se pronunció sobre el honor que significa servir a su país.
Finalmente, se honró la labor que realizan las enfermeras 7 enfermeros, quienes cumplen un rol importante como enlace entre los médicos y pacientes, en especial a la licenciada Adria García.
Finalmente, Belinda Z. Burgos González, gerente de proyectos, indicó que “a nivel editorial reconocemos la importancia que ha tenido para nosotros el poder ser testigo del trabajo de mujeres científicas desde los laboratorios del País, precisamente en estudios que pudieran beneficiar a otras mujeres en un futuro, como lo es el caso de enfermedades como el cáncer triple negativo y otros tipos de enfermedades. Igualmente hemos sido testigos de la empatía de distintas profesionales de la salud que dan la milla extra por esas mujeres trabajadoras y jefas de familia, quienes en muchas ocasiones se tienen como última prioridad. Expresamos nuestro agradecimiento a este selecto grupo de profesionales de la salud que se han hecho eco de las atenciones necesarias hacia las mujeres puertorriqueñas”.
Durante la convención anual de la Asociación de Médicos Alergistas de Puerto Rico, destacados especialistas concientizaron sobre diferentes afecciones alérgicas que son prevalentes en la población puertorriqueña, los avances en tratamientos y terapias novedosas enfocadas en el manejo de estas condiciones, como por ejemplo, el asma, las alergias y la dermatitis atópica. Su presidente, Dr. Anardi Agosto, destacó la combinación de esteroides inhalados, anticolinérgicos y el beneficio de medicamentos biológicos en el manejo de estas afecciones y sus complicaciones. Por su parte, el Dr. Wilfredo Cosme, vicepresidente de la Asociación de Médicos Alergistas, enfatizó la importancia de cuidar la piel frente al agua o la exposición a diferentes agentes infecciosos Dr. Anardi Agosto, alergista pediátrico y presidente de la Asociación de Médicos Alergistas de Puerto Rico, Dr. Wilfredo Cosme, vicepresidente de la Asociación de Médicos Alergistas y la Dra. Verónica Díaz, alergista e inmunóloga. Foto: Revista Medicina y Salud Pública
En la quinta edición de los VOCES Awards liderada por la Coalición de Inmunización y Promoción de la Salud de Puerto Rico, se reconocieron y premiaron los esfuerzos de centenares de profesionales de la salud, médicos y especialistas por su compromiso son la atención de la pandemia por COVID-19 en el territorio y la vacunación de la población, incluyendo a la Revista de Medicina y Salud Pública. Lilliam Rodríguez Capó, CEO y fundadora de VOCES, destacó que “ estoy más que orgullosa de todas aquellas personas que, sin precedentes, con valentía y esmero han realizado una labor titánica. Personas extraordinarias que, sin importar las circunstancias ante el impacto de esta pandemia, han trabajado arduamente, arriesgando su vida y sus familias, en solidaridad con su gente, su comunidad y nuestro país”


Reconocimiento a los héroes de la pandemia del COVID-19. Foto: suministrada por la Coalición de Inmunización y Promoción de la Salud de Puerto Rico.
Convención anual de la Asociación de Gastroenterología y Hepatología Pediátrica de Puerto Rico

El Dr. Carlos Díaz, presidente del Colegio de Médicos Cirujanos de Puerto Rico, denunció en rueda de prensa que las aseguradoras están llevando a un colapso al sistema de salud de la Isla y solicitó al Gobierno supervisar la función de estas empresas y los planes médicos. “(...) le estamos exigiendo al Gobierno abrazar la calidad y excelencia para sacar del colapso al sistema de salud. Anticipo una ola de escándalos en torno a la deprimente estructura de aseguramiento de la salud y la peor supervisión de la historia por parte de las agencias de gobierno, que no exigen, no vigilan los incumplimientos, y no fiscalizan a las aseguradoras de salud, las cuales violentan los derechos de los pacientes y de los proveedores y son premiadas con mayores contrataciones”, sostuvo.

Dr. Alexis Cruz Chacón, hematólogo oncólogo y presidente de AHOMPR. Foto: Revista Medicina y Salud Pública.
Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública




El presidente de la Asociación de Gastroenterología y Hepatología Pediátrica de Puerto Rico, Dr Leonardo Hormaza, enfatizó durante su evento anual la necesidad de más especialistas en gastroenterología pediátrica, ya que en total Puerto Rico cuenta con unos 16 profesionales de la salud de este campo, en su mayoría concentrado en prácticas en el área metropolitana. Igualmente, aprovechó para resaltar la importancia de que este grupo continúe de la mano de los pediatras para detectar a tiempo condiciones gastrointestinales en niños y adolescentes, sobre todo aquellas de origen infeccioso.

Héroes de la pandemia del COVID-19 recibieron reconocimiento en la quinta edición de los VOCES Awards
“En Puerto Rico, las aseguradoras nos están llevando a una crisis en salud”, denuncia Colegio de Médicos
Un grupo de científicos en Puerto Rico busca proteger a la población de la variante Ómicron y sus sub-linajes a través de una vacuna intranasal, según reportó en exclusiva el infectólogo Dr. Javier Morales a la Revista de Medicina y Salud Pública. La misma funciona con DNA, y fue conceptualizada para atacar la espiga y otra parte del virus ORF3A, el cual es el responsable del ensamblaje de que la proteína del virus salga a invadir células nuevas. La investigación se encuentra en su Fase 1.

Puerto Rico tendrá por primera vez un fondo dedicado exclusivamente a fomentar la investigación científica sobre las condiciones urológicas más prevalentes en la población, con apoyo de la American Urological Association y destacadas organizaciones de urólogos de los Estados Unidos, según lo confirmó el Dr. Marcos Pérez Brayfield, urólogo pediátrico y pasado presidente de la Puerto Rico Urological Association, durante la convención anual. El especialista también destacó que los fondos estarán disponibles para estudiantes e investigadores, no solo para los urólogos del territorio y que “es un fondo que se va a usar para hacer investigación dedicada a los problemas urológicos que afectan a los pacientes de la Isla”.
La Asociación de Hematología y Oncología Médica de Puerto Rico en su convención anual enfatizaron sus educaciones continuas en los últimos avances terapéuticos para la atención del cáncer en la comunidad boricua, y además, conmemoraron su aniversario número 50. La agenda de la convención incluirá exposiciones abiertas con el Dr. Cristian Rodríguez, presidente del Comité de Educación de AHOMPR; el manejo y evolución del mieloma múltiple y el papel de la enfermedad residual mínima con el Dr. Rachid Baz; las actualizaciones en el manejo del cáncer de mama avanzado y la terapia dirigida con BCMA para el mieloma múltiple.

Galardonan a Dra. Cabanillas con destacado reconocimiento por su contribución científica al cáncer
La Dra. María Cabanillas, quien desde 2014 realiza estudios científicos y ensayos clínicos sobre el cáncer anaplásico de tiroides (ATC en inglés), de crecimiento rápido y agresividad, fue distinguida con el prestigioso galardón Jack and Beverly Randall Prize, premio que otorga $100,000 y honra la excelencia en el tratamiento de cáncer y el cuidado de los pacientes, y consigo las innovaciones en la investigación contra con el cáncer. El premio usualmente se le otorga a un oncólogo, pero como endocrinóloga, Cabanillas ha trascendido con un proyecto que buscaba reducir el tiempo que transcurre desde que se detecta el tumor hasta la primera cita con un especialista.

Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública

Convención de la Puerto Rico Urological Association y la creación fondo para investigación en urologíaDr. Marcos Pérez Brayfield, urólogo pediátrico y pasado presidente de la Puerto Rico Urological Association. Fotomontaje: Revista Medicina y Salud Pública

RECIBE
DE LA FDA COMO LA PRIMERA Y
(IL_23) ESPECÍFICA PARA TRATAR LA ENFERMEDAD DE CROHN ACTIVA DE MODERADA A SEVERA EN ADULTOS Eli Lilly and Company (NYSE: LLY) and Incyte (NASDAQ:INCY) announced today that the U.S. Food and Drug Administration (FDA) has approved OLUMIANT® (baricitinib), a once-daily pill, as a first-in-disease systemic treatment for adults with severe alopecia areata (AA), available as 4-mg, 2-mg and 1-mg tablets. The recommended dose is OLUMIANT 2- mg/day, with an increase to 4-mg/day if treatment response
is inadequate. For patients with nearly complete or complete scalp hair loss, with or without substantial eyelash or eyebrow hair loss, consider treating with 4- mg/day. Once an adequate response is achieved on 4-mg/day, the dosage is to be decreased to 2-mg/day. OLUMIANT is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, cyclosporine or other potent immunosuppressants.
AbbVie (NYSE: ABBV) hoy anunció que la Administración Federal de Drogas y Alimentos (FDA, por sus siglas en inglés) de los Estados Unidos aprobó SKYRIZI® (risankizumab-rzaa) como el primer y único inhibidor de interleucina-23 (IL-23) específico para el tratamiento de adultos con enfermedad de Crohn (EC) activa de moderada a severa. En dos estudios clínicos de inducción y uno de mantenimiento, SKYRIZI demostró mejoras significativas en la respuesta endoscópica
(definida como una disminución de más del 50% del Puntaje Endoscópico Simple en EC [SES-CD, por sus siglas en inglés] inicial o en pacientes con enfermedad ileal aislada y SES-CD de 4, una reducción de al menos 2 puntos desde el inicio) y en la remisión clínica (definida como un Índice de Actividad de la Enfermedad de Crohn [CDAI, por su siglas en inglés] de menos de 150) en comparación con placebo, tanto en terapia de inducción como de mantenimiento.

A través de un comunicado, el Departamento de Salud informó que se prepara para vacunar a los niños de seis meses a cuatro años con las vacunas de Pfizer y Moderna. El proceso de inoculación debe comenzar la próxima semana, una vez, se reciban las recomendaciones puntuales por parte de los científicos asesores de los Centros para el Control y Prevención de Enfermedades (CDC, siglas en inglés).
La Fundación Puertorriqueña de Enfermedades Reumáticas en alianza con la Revista de Medicina y Salud Pública celebró un foro educativo para pacientes con artritis reumatoide en la Universidad Central del Caribe, con el objetivo principal de proveer la información y herramientas más actualizadas que ayuden a sospechar de esta condición a tiempo. El foro, moderado por la Directora Ejecutiva, Griselle Lugo de FER, contó con sesiones de conversatorio, mesa de expertos en diferentes
En ese contexto, la agencia, estableció un plan de trabajo que incluye adiestrar a los más de 200 proveedores pediátricos de la vacuna contra el COVID-19. En Puerto Rico fueron asignadas 22,000 vacunas para atender esa población.
Dr. Carlos Mellado, secretario del Departamento de Salud. Foto: Revista de Medicina y Salud Pública.
temas de salud y un grupo de médicos especialistas en reumatología tales como: Dr. Oscar Soto Raíces, reumatólogo, Miembro Fundador, Presidente de FER; la Dra. Amarilis Pérez de Jesús, reumatóloga, del Centro de Reumatológico de Caguas y Miembro de la Junta de FER; Dra. Noemí Varela, reumatóloga, Vicepresidente de FER; Lcda. Wanda González, Nutricionista y Delia Román, “coach” de Mindfulness.


Diseñado para ayudar a minimizar los picos de azúcar en la sangre.
Diseñado como un reemplazo de merienda o comida con CARBSTEADY®, una mezcla única de carbohidratos de liberación lenta para ayudarte a manejar el azúcar* en la sangre.

El Salón de la Fama de la Medicina Puertorriqueña tiene el noble propósito de reconocer a los médicos y entidades que con su aportación han realizado una magna labor en favor de la medicina. Su desempeño y legado en el desarrollo social de Puerto Rico quedará preservado y servirá de inspiración para las futuras generaciones.

Este momento histórico en que se exaltarán éstas figuras ilustres, quienes son los cimientos de la medicina puertorriqueña, queremos igualmente reconocer en una actividad fraternal a los que han realizado tan ardua y arriesgada gesta por la salud de nuestra isla durante la Pandemia, época que nos ha hecho atravesar un período sumamente difícil y lleno de grandes retos para la medicina, la clase médica y la industria de la salud en general.
Un prestigioso grupo de profesionales de la medicina conformó un Comité de Selección, el cual tuvo como principal objetivo, seleccionar a los “Miembros Exaltados” al Salón de la Fama de la Medicina. Luego de un proceso de investigación, nominaciones y evaluación, se escogió mediante votación, a las figuras y entidades que formarán parte de la Primera Edición.

La idea de fundar una asociación médica respondía a una necesidad. Los doctores Manuel Quevedo Báez, Rafael Vélez López y Mariano Ramírez circularon una carta entre los médicos citándolos a una reunión en julio de 1902, la cual se realizó el 21 de septiembre de 1902, en la Cámara de Delegados de la Antigua Diputación Provincial. Concurrieron 15 médicos: Dres. José M. Amadeo, José N. Carbonell, José María Cueto, Narciso Dobal, Manuel Figueroa, P. Janer, M. Fernández Vizcarrondo, Francisco Seín, Rafael Vélez López, M. Fernández Náter, J. Reguero Feliú, Mariano Ramírez, José Esteban Saldaña, Ramón Ruiz Arnau y Manuel Quevedo Báez. Estos actuaron a la vez como delegados de otros médicos de la Isla que no pudieron asistir a la asamblea. En sus comienzos la Asociación Médica quedó integrada por 76 médicos. Esta asamblea eligió el siguiente cuerpo directivo: Dr. Manuel Quevedo Báez, Presidente; Dr. José N. Carbonell, Secretario-Tesorero; y el Dr. Rafael Vélez López, Vocal.
En 1903 se hizo un registro completo de los médicos asociados. También se edita, bajo la dirección del Dr. Ruíz Arnau, el primer Boletín Médico, órgano muy importante de la AMPR, en el que se han publicado artículos de carácter científico y comunicaciones oficiales de la Asociación respecto a decisiones de carácter directivo y administrativo.

Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública

Internista y endocrinóloga, especialista en Diabetes y Metabolismo. En los años 60, lse une a la facultad de la Escuela de Medicina de la Universidad de Puerto Rico. Asimismo, en los años 70, fue nombrada Jefa de la Sección de Endocrinología, Diabetes y Metabolismo y directora del Comité de Educación que llevó a todo Puerto Rico el programa regional sobre “Tratamiento, Complicaciones y Detección de la Diabetes”. Con este programa se proveyó educación a médicos, enfermeras, técnicos, dietistas y administradores de la salud, obteniendo el reconocimiento de la American Diabetes Association; para luego ser Decana de Asuntos Académicos del Recinto de Ciencias Médicas de la UPR. Para la década de los 80, En 1980 fue Presidente de la Sociedad Puertorriqueña de Endocrinología y Diabetología, y en 1990 fue la Presidente del Capítulo Puerto Rico del American College of Physicians, recibiendo el grado de Master y Laureate Award. Entre múltiples honores fue nombrada ´Mujer Destacada en la Educación Médica´ por la Cámara de Comercio de Puerto Rico y ´Profesor Emeritus´ de la Universidad de Puerto Rico.
Dr. Isaac González: médico puertorriqueño que se especializó en investigación y tratamientos para el cáncer.
En 1900, el Dr. González se inmiscuyó por completo en el mundo de la parasitología. Logrando así, en 1904, descubrir un nuevo parásito en Puerto Rico, del que nunca se había escrito y no existía en la literatura médica, llamado Visalia, que causó estragos en la Isla durante aquella época. Asmimismo, ante los brotes de peste bubónica en la Isla durante los inicios del siglo XX, el Dr. González, en 1912, logró evitar la propagación de la infección recluyendo a las personas en sus hogares. En 1938, fundó la Liga Puertorriqueña contra el Cáncer, y en 1953, se construye el primer hospital subespecializado en cáncer que lleva su nombre, como homenaje a los años de trabajo y labor comunitaria con la población puertorriqueña.
y vida del doctor Bailey K. Ashford, líder de la medicina en Puerto Rico del siglo XIX

Fue gran responsable de la creación de la Escuela de Medicina Tropical en San Juan, graduado como Médico en la Universidad de Georgetown, en Washington, donde siempre mostró interés por la investigación. En el año de 1897 se enlistó en el Cuerpo de Médicos del Ejército de Estados Unidos, al que ingresó ostentando el cargo de teniente. Motivado por las cifras de mortalidad por anemia, y con el respaldo de la Asociación Médica, Ashford logró en 1904 crear la “Comisión de Anemia”, que permitió atender a casi 1,000 pacientes diarios, a quienes además se les orientaba, recomendando el uso de zapatos y la construcción de letrinas para evitar re-infectarse. Hablar de su labor, y de su paso por Puerto Rico, es destacar ese ser investigador y visionario que pudo observar más allá, donde la gente veía menos, y que expandió la medicina de la Isla, de forma que aún se sigue practicando su teoría, y se sigue su labor y su entereza a la hora de defender la profesión.


Hospital Auxilio Mutuo es exaltado por 140 años al servicio de los puertorriqueños
Con el deseo de ayudar a todas las personas que necesitaban atención médica de calidad y para todos, se tuvo la idea de crear el Hospital Auxilio Mutuo, liderado por españoles que se encontraban en Puerto Rico, quienes tenían como objetivo brindar atención médica, no solo a sus compatriotas, sino a todo aquél que la necesitara. Los valores como el servicio, la ayuda mutua, la beneficencia, fueron trascendentales para el cubrimiento de la salud en Puerto Rico, sobre todo para aquellos que no tiene muchos recursos, o son de otra nacionalidad. Por ello, el Plan de Socios, cubre a más de 25 mil personas que pueden acceder a un plan de salud complementaria, donde la prevención y el compromiso de ayuda están por delante en todo momento. Asimismo, la entidad se ha caracterizado por tener líderes con los pies en la tierra, pero con una capacidad de innovación increíble, por eso, desde el primer momento en el que se creó una casa de servicio, fueron aumentando las ideas y las necesidades de crear otras edificaciones para atender las enfermedades de todos los puertorriqueños.
Revista Puertorriqueña de Medicina y Salud Pública 97 Revista Puertorriqueña de Medicina y Salud Pública 3
Laboraporte
Se convirtió en parte del primer grupo de residentes admitidos a un programa de entrenamiento de tres años, cuando el Dr. Luis Sánchez Longo dio inicio al fellowship en Neurología. Una vez terminó ese tiempo de la especialización en Neurología fue becada por la Universidad de Harvard y viajó a la ciudad de Bostón, para trabajar en el prestigioso hospital Massachusetts General Hospital, donde realizó una subespecialidad en Electroencefalografía. Se hizo merecedora de una beca para realizar rotaciones en centros de electroencefalograma del mayor prestigio internacional en las principales ciudades de Europa, como Londres, Bélgica, París, Múnich, Salzburgo y Barcelona y en la universidad de Harvard como consultora. A su regreso a Puerto Rico, fundó la Fundación de Parkinson y comenzó a dirigir el Laboratorio de Encefalografía del Centro Médico, del cual estuvo a cargo hasta 1995, siendo no solo la directora, sino catedrática del centro.
Dr. Norman Maldonado: una vida al servicio de la comunidad de Puerto Rico El 3 de noviembre de 1935, nace en Adjuntas, el doctor Norman Maldonado, de padre puertorriqueño (agricultor de café) y madre de Jumaguiña. Es médico interno con especialización en Hematología-Oncología e Inmunología así como ex-presidente de la Universidad de Puerto Rico y de la Escuela de Medicina. Sus estudios los realizó en la escuela elemental en Adjuntas y Escuela Superior en Ponce, destacando como un estudiante comprometido. Obtuvo con honores el bachillerato en Artes en el Politécnico de San Germán y un doctorado en Medicina en la Escuela de Medicina de la UPR, labrado con el máximo honor de ser el primero de su clase. Se mudó a Washington, donde realizó su internado en el D.C. Washington General Hospital (1959 a 1960), dirigió el Centro de Ayuda a Víctimas de Violación durante 11 años, quien por más de 30 años, también ha sido líder en la Casa Julia de Burgos para víctimas de violencia doméstica. Ha sido un gran investigador, ha publicado más de 70 artículos científicos en las áreas de hematología, oncología y anemia.Como Rector del Recinto de Ciencias Médicas, creó las clínicas ambulatorias, activó el plan de práctica de la Facultad, logró la compensación diferida y que se mantuvieran todas las acreditaciones de los programas educativos. Logró la compra del Hospital de Carolina.



Dr. Manuel de la Pila Iglesias, el médico que logró aprobar el primer plan médico en Puerto Rico
Fue especialista en varias áreas médicas y estudió en diferentes países del mundo, lo que le permitió obtener un amplio conocimiento en el tratamiento de enfermedades y la atención de los enfermos. Es experto en técnica quirúrgica, electrocardiografía, otorrinolaringología, cardiología e incluso alergología. Fue el primer médico que trajo los electrocardiógrafos a la Isla para que las personas pudieran conocer estas novedosas máquinas que examinan el corazón. Fundó varios hospitales en Ponce, entre los cuales se destacan el Hospital San Lucas de Ponce, el Hospital Tricoche o el Albergue Caritativo Tricoche, y fue director médico del Hospital Damas, antiguamente llamado el Santo Asilo de Damas. Logró que se aprobara el primer plan de medicina prepagada en Puerto Rico. También fue presidente de la Asociación Médica de Puerto Rico, delegado representante ante la American Medical Association y junto al Dr. Costa Mandry, y el Dr. Ramón Suárez Calderón, el reconocido especialista integró la comisión designada para la creación de la primera escuela de medicina en la Isla, que fue parte de la formación de los médicos, sentando así todas las bases para la educación de las nuevas generaciones.
El valioso
de la Dra. Ana Judith Román, primera neuróloga de Puerto Rico
Dra. Amalia Martínez Picó, la primera cardióloga pediátrica de Puerto Rico

Cardióloga pediátrica, la primera graduada de esta especialidad médica en Puerto Rico, también es profesora de pediatría y decana de asuntos académicos del Recinto de Ciencias Médicas. La especialista estableció el servicio clínico de Cardiología Pediátrica del Departamento de Pediatría de la Escuela de Medicina de la Universidad de Puerto Rico. Además, creó un programa educativo e investigativo sobre el SIDA en el Recinto de Ciencias Médicas y fundó el Compucentro, un valioso centro educativo dotado de computadoras, disponibles al personal docente y estudiantes de la Universidad. Además, fue directora de la Sección de Cardiología pediátrica del Hospital Pediátrico de la Universidad de Puerto Rico entre los años 1959 y 1985. Allí estableció un cronograma de servicio dental exclusivo para pacientes cardiovasculares pediátricos y lideró un importante un programa preventivo de enfermedades cardiovasculares, siendo el primero que se realizaba en la Isla. Nacido en Yabucoa el 17 de septiembre de 1898, fue el mayor de cuatro hermanos. Sus padres tomaron la decisión de mudarse a Patillas y luego a San Juan, en búsqueda de una mejor calidad para sus hijos y el hogar. Ingresó en la Escuela de Medicina de la Universidad de Maryland, de donde se graduó en 1922, siendo reconocido con los mayores honores y como el primero de su clase. Empezó como interno en el Mercy Hospital, y regresó a Puerto Rico para trabajar en el Departamento de Salud en bacteriología. Estuvo trabajando como residente en el Hospital Presbiteriano, luego fue a la Universidad de Columbia en Nueva York para especializarse en patología y bacteriología. Llegó el año de 1944, el Rector de la Universidad de Puerto Rico, Don Jaime Benítez, lo integró a la Comisión para crear la Escuela de Medicina. Gracias a su recomendación, el “Informe Costa Mandry”, permitió que se creará la Escuela de Medicina de la Universidad de Puerto Rico.
Dr. Óscar Costa Mandry y su gran aporte a la medicina de emergencias en Puerto Rico
Fue Director de Educación Médica del Departamento de Salud y creó la Residencia en Patología Clínica en el Hospital de Distrito de Bayamón. Se destaca porque identificó la relación entre los huracanes y los brotes epidémicos de disentería en los pueblos directamente afectados por estos desastres naturales en la isla.
Dr. Guillermo Picó: El oftalmólogo que puso sus ojos en la educación internacional

Los primeros años de estudio, Guillermo Picó dedicó su vida a aprender sobre medicina, hasta que en 1940 se gradúa como médico de la Universidad de Maryland. Estos años de servicio, lo llevaron hasta la comunidad Fanguito, siendo integrante del Departamento de Salud, para entender y estudiar las causas de las enfermedades como la malaria y la tifoidea, que por aquella época ya estaban causando muchos estragos en la salud de los puertorriqueños. En 1948, se instauró como Jefe de Oftalmología del Departamento de Cirugía de la Escuela de Medicina de la Universidad de Puerto Rico. En 1964, creó el curso básico para médicos Latinoamericanos, que fue impulsado con los recursos donados de los Institutos Nacionales de Salud. Publicó más de 50 artículos especializados en oftalmología. Fue Presidente de la Asociación Médica de Puerto Rico, y Vicepresidente de la Academia Americana de Oftalmología.


Durante el mes de noviembre, Mes de la Concienciación sobre el Cáncer del Pulmón y Gástrico, la biofarmacéutica Bristol Myers Squibb, la Revista de Medicina y Salud Pública y un grupo de profesionales de la salud se unieron en una actividad efectuada en el Senado de Puerto Rico para dialogar y conocer la información más actualizada sobre la importancia del diagnóstico temprano, terapias de tratamiento y apoyo al paciente de estos dos tipos de cánceres. La iniciativa contó con la participación de un panel compuesto por: Dr.Oscar Soto Raíces, Editor de la Revista Medicina y Salud Pública; Dr. Carlos Micames, pasado presidente de la Asociación Puertorriqueña de Gastroenterología; Dra. Daphne Delgado, presidenta de la Sociedad Puertorriqueña de Neumología; el hematólogo oncólogo, Dr. Luis Báez Vallecillo de PR Oncology Inc.; Dra. Marivelisse Soto, investigadora adscrita al Centro Comprensivo de Cáncer y la Sra. María Cristy, vicepresidenta de Servicio a los Pacientes y Control de Cáncer de la Sociedad Americana Contra el Cáncer de Puerto Rico.
Especialistas puertorriqueños y representantes de organizaciones de salud en el Capitolio de Puerto Rico.





Foto: Yamirar Jiménez
Lic. Jesús Manuel-Laboy, Representante del Hon. Rubén Soto Rivera, presidente de la Comisión de Salud y Pedro Lugo, Vicepresidente de Grupo Editorial Mundo. Foto: Yarimar Jiménez
Durante la ceremonia se condecoraron a instituciones, fundaciones y personas, que han contribuido de alguna manera en la educación, divulgación, para la prevención y tratamiento del cáncer de pulmón y gástrico.


Foto: Yarimar Jiménez
Represetantes
Especialistas
• NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC recommend molecular testing and strongly advise broad molecular profiling for, at minimum, RET rearrangements, EGFR mutations, BRAF mutations, METex14 skipping mutations, ALK fusions, and ROS1 fusions in all appropriate patients with metastatic NSCLC to assess whether patients are eligible for targeted therapies2*†
• Next-generation sequencing (NGS) testing early in metastatic NSCLC diagnosis provides a comprehensive, tissue-efficient way to test for actionable alterations, including RET fusions3,4
An FDA-approved test for the detection of RET gene fusions and RET gene mutations is not currently available.1
NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
*The NCCN Guidelines ® for NSCLC provide recommendations for individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays.
† It is recommended at this time that when feasible, testing be performed via a broad, panel-based approach, most typically performed by NGS. For patients who, in broad panel testing don’t have identifiable driver oncogenes (especially in never smokers), consider RNA-based NGS if not already performed, to maximize detection of fusion events.
ALK=anaplastic lymphoma kinase; BRAF=v-raf murine sarcoma viral oncogene homolog B; EGFR=epidermal growth factor receptor; METex14=mesenchymalepithelial transition exon 14 skipping; NCCN=National Comprehensive Cancer Network; NSCLC=non-small cell lung cancer; RET=rearranged during transfection; RNA=ribonucleic acid; ROS1=reactive oxygen species 1.
Retevmo is a kinase inhibitor indicated for the treatment of:
• adult patients with metastatic RET fusion-positive non-small cell lung cancer (NSCLC)
• adult and pediatric patients 12 years of age and older with advanced or metastatic RET-mutant medullary thyroid cancer (MTC) who require systemic therapy
• adult and pediatric patients 12 years of age and older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate)
These indications are approved under accelerated approval based on overall response rate (ORR) and duration of response (DoR). Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials.
IMPORTANT SAFETY INFORMATION
Hepatotoxicity: Serious hepatic adverse reactions occurred in 2.6% of patients treated with Retevmo. Increased aspartate aminotransferase (AST) occurred in 51% of patients, including Grade 3 or 4 events in 8% and increased alanine aminotransferase (ALT) occurred in 45% of patients, including Grade 3 or 4 events in 9%. The median time to first onset for increased AST was 4.1 weeks (range: 5 days to 2 years) and increased ALT was 4.1 weeks (range: 6 days to 1.5 years). Monitor ALT and AST prior to initiating Retevmo, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose or permanently discontinue Retevmo based on the severity.
Hypertension occurred in 35% of patients, including Grade 3 hypertension in 17% and Grade 4 in one (0.1%) patient. Overall, 4.6% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications. Do not initiate Retevmo in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating Retevmo. Monitor blood pressure after 1 week, at least monthly thereafter, and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue Retevmo based on the severity.
References: 1. Retevmo (selpercatinib) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2021. 2. Referenced with permission from The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.6.2020. © National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed June 15, 2020. To view the most recent and complete version of the guidelines, go online to https://www.nccn.org. 3. Gregg JP, Li T, Yoneda KY. Molecular testing strategies in non-small cell lung cancer: optimizing the diagnostic journey. Transl Lung Cancer Res. 2019;8(3):286-301. 4. Suh JH, Schrock AB, Johnson A, et al. Hybrid capture-based comprehensive genomic profiling identifies lung cancer patients with well-characterized sensitizing epidermal growth factor receptor point mutations that were not detected by standard of care testing. Oncologist. 2018;23(7):776-781.
Please see Important Safety Information and Brief Summary of Prescribing Information for Retevmo on subsequent pages.
Retevmo® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-SE-US-0591 04/2021 ©Lilly USA, LLC 2021. All rights reserved.
IMPORTANT SAFETY INFORMATION FOR RETEVMO™ (selpercatinib 40 mg, 80 mg capsules) (CONT’D)
Retevmo can cause concentration-dependent QT interval prolongation. An increase in QTcF interval to >500 ms was measured in 6% of patients and an increase in the QTcF interval of at least 60 ms over baseline was measured in 15% of patients. Retevmo has not been studied in patients with clinically significant active cardiovascular disease or recent myocardial infarction. Monitor patients who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, and severe or uncontrolled heart failure. Assess QT interval, electrolytes and TSH at baseline and periodically during treatment, adjusting frequency based upon risk factors including diarrhea. Correct hypokalemia, hypomagnesemia and hypocalcemia prior to initiating Retevmo and during treatment. Monitor the QT interval more frequently when Retevmo is concomitantly administered with strong and moderate CYP3A inhibitors or drugs known to prolong QTc interval. Withhold and dose reduce or permanently discontinue Retevmo based on the severity.
Serious, including fatal, hemorrhagic events can occur with Retevmo. Grade ≥3 hemorrhagic events occurred in 2.3% of patients treated with Retevmo including 3 (0.4%) patients with fatal hemorrhagic events, including one case each of cerebral hemorrhage, tracheostomy site hemorrhage, and hemoptysis. Permanently discontinue Retevmo in patients with severe or lifethreatening hemorrhage.
Hypersensitivity occurred in 4.3% of patients receiving Retevmo, including Grade 3 hypersensitivity in 1.6%. The median time to onset was 1.7 weeks (range 6 days to 1.5 years). Signs and symptoms of hypersensitivity included fever, rash and arthralgias or myalgias with concurrent decreased platelets or transaminitis. If hypersensitivity occurs, withhold Retevmo and begin corticosteroids at a dose of 1 mg/kg prednisone (or equivalent). Upon resolution of the event, resume Retevmo at a reduced dose and increase the dose of Retevmo by 1 dose level each week as tolerated until reaching the dose taken prior to onset of hypersensitivity. Continue steroids until patient reaches target dose and then taper. Permanently discontinue Retevmo for recurrent hypersensitivity.
Tumor lysis syndrome (TLS) occurred in 1% of patients with medullary thyroid carcinoma receiving Retevmo. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, Retevmo has the potential to adversely affect wound healing. Withhold Retevmo for at least 7 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of Retevmo after resolution of wound healing complications has not been established.
Based on data from animal reproduction studies and its mechanism of action, Retevmo can cause fetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with Retevmo and for at least 1 week after the final dose. There are no data on the presence of selpercatinib or its metabolites in human milk or on their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with Retevmo and for 1 week after the final dose.
Severe adverse reactions (Grade 3-4) occurring in ≥15% of patients who received Retevmo in LIBRETTO-001, were hypertension (18%), prolonged QT interval (4%), diarrhea (3.4%), dyspnea (2.3%), fatigue (2%), abdominal pain (1.9%), hemorrhage (1.9%), headache (1.4%), rash (0.7%), constipation (0.6%), nausea (0.6%), vomiting (0.3%), and edema (0.3%).
Serious adverse reactions occurred in 33% of patients who received Retevmo. The most frequently reported serious adverse reaction (in ≥ 2% of patients) was pneumonia.
Fatal adverse reactions occurred in 3% of patients; fatal adverse reactions which occurred in >1 patient included sepsis (n=3), cardiac arrest (n=3) and respiratory failure (n=3).
Common adverse reactions (all grades) occurring in ≥15% of patients who received Retevmo in LIBRETTO-001, were dry mouth (39%), diarrhea (37%), hypertension (35%), fatigue (35%), edema (35%), rash (27%), constipation (25%), nausea (23%), abdominal pain (23%), headache (23%), cough (18%), prolonged QT interval (17%), dyspnea (16%), vomiting (15%), and hemorrhage (15%).
Laboratory abnormalities (all grades; Grade 3-4) ≥20% worsening from baseline in patients who received Retevmo in LIBRETTO-001, were AST increased (51%; 8%), ALT increased (45%; 9%), increased glucose (44%; 2.2%), decreased leukocytes (43%; 1.6%), decreased albumin (42%; 0.7%), decreased calcium (41%; 3.8%), increased creatinine (37%; 1.0%), increased alkaline phosphatase (36%; 2.3%), decreased platelets (33%; 2.7%), increased total cholesterol (31%; 0.1%), decreased sodium (27%; 7%), decreased magnesium (24%; 0.6%), increased potassium (24%; 1.2%), increased bilirubin (23%; 2.0%), and decreased glucose (22%; 0.7%).
Concomitant use of acid-reducing agents decreases selpercatinib plasma concentrations which may reduce Retevmo anti-tumor activity. Avoid concomitant use of proton-pump inhibitors (PPIs), histamine-2 (H2) receptor antagonists, and locally-acting antacids with Retevmo. If coadministration cannot be avoided, take Retevmo with food (with a PPI) or modify its administration time (with a H2 receptor antagonist or a locally-acting antacid).
Concomitant use of strong and moderate CYP3A inhibitors increases selpercatinib plasma concentrations which may increase the risk of Retevmo adverse reactions including QTc interval prolongation. Avoid concomitant use of strong and moderate CYP3A inhibitors with Retevmo. If concomitant use of a strong or moderate CYP3A inhibitor cannot be avoided, reduce the Retevmo dosage as recommended and monitor the QT interval with ECGs more frequently.
Concomitant use of strong and moderate CYP3A inducers decreases selpercatinib plasma concentrations which may reduce Retevmo anti-tumor activity. Avoid coadministration of Retevmo with strong and moderate CYP3A inducers.
Concomitant use of Retevmo with CYP2C8 and CYP3A substrates increases their plasma concentrations which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of Retevmo with CYP2C8 and CYP3A substrates where minimal concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for CYP2C8 and CYP3A substrates provided in their approved product labeling.
The safety and effectiveness of Retevmo have not been established in pediatric patients less than 12 years of age
The safety and effectiveness of Retevmo have been established in pediatric patients aged 12 years and older for medullary thyroid cancer (MTC) who require systemic therapy and for advanced RET fusion-positive thyroid cancer who require systemic therapy and are radioactive iodine-refractory (if radioactive iodine is appropriate). Use of Retevmo for these indications is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients aged 12 years and older. Monitor open growth plates in adolescent patients. Consider interrupting or discontinuing Retevmo if abnormalities occur.
No dosage modification is recommended for patients with mild to severe renal impairment (estimated Glomerular Filtration Rate [eGFR] ≥15 to 89 mL/min, estimated by Modification of Diet in Renal Disease [MDRD] equation). A recommended dosage has not been established for patients with end-stage renal disease.
Reduce the dose when administering Retevmo to patients with severe hepatic impairment (total bilirubin greater than 3 to 10 times upper limit of normal [ULN] and any AST). No dosage modification is recommended for patients with mild or moderate hepatic impairment. Monitor for Retevmo-related adverse reactions in patients with hepatic impairment.
Please see Brief Summary of Prescribing Information for Retevmo on subsequent pages.
SE HCP ISI All_25MAR2021
RETEVMO® (selpercatinib) capsules, for oral use
Initial U.S. Approval: 2020
BRIEF SUMMARY: Consult the package insert for complete prescribing information.
RETEVMO (selpercatinib) is a kinase inhibitor indicated for the treatment of:
• Adult patients with metastatic RET fusion-positive non-small cell lung cancer (NSCLC)
• Adult and pediatric patients 12 years of age and older with advanced or metastatic RET-mutant medullary thyroid cancer (MTC) who require systemic therapy
• Adult and pediatric patients 12 years of age and older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy are radioactive iodine-refractory (if radioactive iodine is appropriate)
These indications are approved under accelerated approval based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials.
CONTRAINDICATIONS: None
Serious hepatic adverse reactions occurred in 2.6% of patients treated with RETEVMO. Increased AST occurred in 51% of patients, including Grade 3 or 4 events in 8% and increased ALT occurred in 45% of patients, including Grade 3 or 4 events in 9%. The median time to first onset for increased AST was 4.1 weeks (range: 5 days to 2 years) and increased ALT was 4.1 weeks (range: 6 days to 1.5 years).
Monitor ALT and AST prior to initiating RETEVMO, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose or permanently discontinue RETEVMO based on the severity.
Hypertension occurred in 35% of patients, including Grade 3 hypertension in 17% and Grade 4 in one (0.1%) patient Overall, 4.6% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications.
Do not initiate RETEVMO in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating RETEVMO. Monitor blood pressure after 1 week, at least monthly thereafter and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue RETEVMO based on the severity.
RETEVMO can cause concentration-dependent QT interval prolongation An increase in QTcF interval to >500 ms was measured in 6% of patients and an increase in the QTcF interval of at least 60 ms over baseline was measured in 15% of patients. RETEVMO has not been studied in patients with clinically significant active cardiovascular disease or recent myocardial infarction.
Monitor patients who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, and severe or uncontrolled heart failure. Assess QT interval, electrolytes and TSH at baseline and periodically during treatment, adjusting frequency based upon risk factors including diarrhea. Correct hypokalemia, hypomagnesemia and hypocalcemia prior to initiating RETEVMO and during treatment.
Monitor the QT interval more frequently when RETEVMO is concomitantly administered with strong and moderate CYP3A inhibitors or drugs known to prolong QTc interval. Withhold and dose reduce or permanently discontinue RETEVMO based on the severity.
Serious including fatal hemorrhagic events can occur with RETEVMO. Grade ≥ 3 hemorrhagic events occurred in 2.3% of patients treated with RETEVMO including 3 (0.4%) patients with fatal hemorrhagic events, including one case each of cerebral hemorrhage, tracheostomy site hemorrhage, and hemoptysis.
Permanently discontinue RETEVMO in patients with severe or life-threatening hemorrhage.
Hypersensitivity occurred in 4.3% of patients receiving RETEVMO, including Grade 3 hypersensitivity in 1.6%. The median time to onset was 1.7 weeks (range: 6 days to 1.5 years). Signs and symptoms of hypersensitivity included fever, rash and arthralgias or myalgias with concurrent decreased platelets or transaminitis.
If hypersensitivity occurs, withhold RETEVMO and begin corticosteroids at a dose of 1 mg/kg prednisone (or equivalent). Upon resolution of the event, resume RETEVMO at a reduced dose and increase the dose of RETEVMO by 1 dose level each week as tolerated until reaching the dose taken prior to onset of hypersensitivity. Continue steroids until patient reaches target dose and then taper. Permanently discontinue RETEVMO for recurrent hypersensitivity.
Tumor lysis syndrome (TLS) occurred in 1% of patients with medullary thyroid carcinoma receiving RETEVMO. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, RETEVMO has the potential to adversely affect wound healing.
Withhold RETEVMO for at least 7 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of RETEVMO after resolution of wound healing complications has not been established.
Based on data from animal reproduction studies, and its mechanism of action, RETEVMO can cause fetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for at least 1 week after the final dose.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS and PRECAUTIONS and below reflects exposure to RETEVMO as a single agent at 160 mg orally twice daily evaluated in 702 patients in LIBRETTO-001. Among 702 patients who received RETEVMO, 65% were exposed for 6 months or longer and 34% were exposed for greater than one year. Among these patients, 95% received at least one dose of RETEVMO at the recommended dosage of 160 mg orally twice daily.
The median age was 59 years (range: 15 to 92 years); 0.3% were pediatric patients 12 to 16 years of age; 52% were male; and 69% were White, 22% were Asian, 5% were Hispanic/ Latino, and 3% were Black. The most common tumors were NSCLC (47%), MTC (44%), and non-medullary thyroid carcinoma (5%).
Serious adverse reactions occurred in 33% of patients who received RETEVMO. The most frequent serious adverse reaction (in ≥ 2% of patients) was pneumonia. Fatal adverse reactions occurred in 3% of patients; fatal adverse reactions which occurred in > 1 patient included sepsis (n = 3), cardiac arrest (n = 3) and respiratory failure (n = 3).
Permanent discontinuation due to an adverse reaction occurred in 5% of patients who received RETEVMO. Adverse reactions resulting in permanent discontinuation included increased ALT (0.4%), sepsis (0.4%), increased AST (0.3%), drug hypersensitivity (0.3%), fatigue (0.3%), and thrombocytopenia (0.3%).
Dosage interruptions due to an adverse reaction occurred in 42% of patients who received RETEVMO. Adverse reactions requiring dosage interruption in > 2% of patients included ALT increased, AST increased, hypertension, diarrhea, pyrexia, and QT prolongation.
Dose reductions due to an adverse reaction occurred in 31% of patients who received RETEVMO. Adverse reactions requiring dosage reductions in > 2% of patients included ALT increased, AST increased, QT prolongation and fatigue.
The most common adverse reactions, including laboratory abnormalities, (≥ 25%) were increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), increased glucose, decreased leukocytes, decreased albumin, decreased calcium, dry mouth, diarrhea, increased creatinine, increased alkaline phosphatase, hypertension, fatigue, edema, decreased platelets, increased total cholesterol, rash, decreased sodium, and constipation.
Table 1 summarizes the adverse reactions in LIBRETTO-001.
Table 1: Adverse Reactions (>15%) in Patients Who Received RETEVMO in LIBRETTO-001
Adverse Reaction RETEVMO (n=702) Grades 1-4 (%) Grade 3-4 (%)
Gastrointestinal
Dry Mouth 39 0 Diarrhea1 37 3.4* Constipation 25 0.6* Nausea 23 0.6* Abdominal pain2 23 1.9* Vomiting 15 0.3* Vascular Hypertension 35 18 General Fatigue3 35 2* Edema4 35 0.3* Skin Rash5 27 0.7*
Nervous System Headache6 23 1.4* Respiratory Cough7 18 0 Dyspnea8 16 2.3 Investigations Prolonged QT interval 17 4* Blood and Lymphatic System Hemorrhage9 15 1.9
1Diarrhea includes diarrhea, defecation urgency, frequent bowel movements, and anal incontinence
2Abdominal pain includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal discomfort, gastrointestinal pain
3Fatigue includes fatigue, asthenia, malaise.
4Edema includes edema, edema peripheral, face edema, periorbital edema, eye edema, eyelid edema, orbital edema, localized edema, lymphedema, scrotal edema, peripheral swelling, scrotal swelling, swelling, swelling face, eye swelling
5Includes rash, rash erythematous, rash macular, rash maculopapular, rash morbilliform, rash pruritic
6Headache includes headache, sinus headache, tension headache,
7Includes cough, productive cough
8Includes dyspnea, dyspnea exertional, dyspnea at rest
9Hemorrhage includes epistaxis, hematuria, hemoptysis, contusion, rectal hemorrhage, vaginal hemorrhage, ecchymosis, hematochezia, petechiae, traumatic hematoma, anal hemorrhage, blood blister, blood urine present, cerebral hemorrhage, gastric hemorrhage, hemorrhage intracranial, spontaneous hematoma, abdominal wall hematoma, angina bullosa hemorrhagica, diverticulum intestinal hemorrhagic, eye hemorrhage, gastrointestinal hemorrhage, gingival bleeding, hematemesis, hemorrhagic anemia, intraabdominal hemorrhage, lower gastrointestinal hemorrhage, melena, mouth hemorrhage, occult blood positive, pelvic hematoma, periorbital hematoma, pharyngeal hemorrhage, pulmonary contusion, purpura, retroperitoneal hematoma, subarachnoid hemorrhage, subdural hemorrhage, upper gastrointestinal hemorrhage, vessel puncture site hematoma
*Only includes a grade 3 adverse reaction.
Clinically relevant adverse reactions in <15% of patients who received RETEVMO include hypothyroidism and tumor lysis syndrome.
Table 2 summarizes the laboratory abnormalities in LIBRETTO-001.
Table 2: Select Laboratory Abnormalities (≥ 20%) Worsening from Baseline in Patients Who Received RETEVMO in LIBRETTO-001
Laboratory Abnormality
Chemistry
RETEVMO1
Grades 1-4 (%)Grades 3-4 (%)
Increased AST 51 8
Increased ALT 45 9
Increased glucose 44 2.2
Decreased albumin 42 0.7
Decreased calcium 41 3.8
Increased creatinine 37 1.0
Increased alkaline phosphatase 36 2.3
Increased total cholesterol 31 0.1
Decreased sodium 27 7
Decreased magnesium 24 0.6
Increased potassium 24 1.2
Increased bilirubin 23 2.0
Decreased glucose 22 0.7
Hematology
Decreased leukocytes 43 1.6
Decreased platelets 33 2.7
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available, which ranged from 675 to 692 patients.
In healthy subjects administered RETEVMO 160 mg orally twice daily, serum creatinine increased 18% after 10 days. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
Concomitant use of RETEVMO with acid-reducing agents decreases selpercatinib plasma concentrations, which may reduce RETEVMO anti-tumor activity. Avoid concomitant use of PPIs, H2 receptor antagonists, and locally acting antacids with RETEVMO. If coadministration cannot be avoided, take RETEVMO with food (with a PPI) or modify its administration time (with a H2 receptor antagonist or a locally acting antacid).
Concomitant use of RETEVMO with a strong or moderate CYP3A inhibitor increases selpercatinib plasma concentrations, which may increase the risk of RETEVMO adverse reactions, including QTc interval prolongation. Avoid concomitant use of strong and moderate CYP3A inhibitors with RETEVMO. If concomitant use of strong and moderate CYP3A inhibitors cannot be avoided, reduce the RETEVMO dosage and monitor the QT interval with ECGs more frequently.
Concomitant use of RETEVMO with a strong or moderate CYP3A inducer decreases selpercatinib plasma concentrations, which may reduce RETEVMO anti-tumor activity. Avoid coadministration of strong or moderate CYP3A inducers with RETEVMO.
CYP2C8
RETEVMO is a moderate CYP2C8 inhibitor and a weak CYP3A inhibitor. Concomitant use of RETEVMO with CYP2C8 and CYP3A substrates increases their plasma concentrations, which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of RETEVMO with CYP2C8 and CYP3A substrates where minimal
RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021
RETEVMO, SE HCP BS 25MAR2021 - 7.5 x 10
Revista Puertorriqueña de Medicina y Salud Pública
concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for CYP2C8 and CYP3A substrates provided in their approved product labeling.
RETEVMO is associated with QTc interval prolongation. Monitor the QT interval with ECGs more frequently in patients who require treatment with concomitant medications known to prolong the QT interval.
Based on findings from animal studies, and its mechanism of action, RETEVMO can cause fetal harm when administered to a pregnant woman. There are no available data on RETEVMO use in pregnant women to inform drug-associated risk. Administration of selpercatinib to pregnant rats during the period of organogenesis resulted in embryolethality and malformations at maternal exposures that were approximately equal to the human exposure at the clinical dose of 160 mg twice daily. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Selpercatinib administration to pregnant rats during the period of organogenesis at oral doses ≥ 100 mg/kg [approximately 3.6 times the human exposure based on the area under the curve (AUC) at the clinical dose of 160 mg twice daily] resulted in 100% postimplantation loss. At the dose of 50 mg/kg [approximately equal to the human exposure (AUC) at the clinical dose of 160 mg twice daily], 6 of 8 females had 100% early resorptions; the remaining 2 females had high levels of early resorptions with only 3 viable fetuses across the 2 litters. All viable fetuses had decreased fetal body weight and malformations (2 with short tail and one with small snout and localized edema of the neck and thorax).
There are no data on the presence of selpercatinib or its metabolites in human milk or on their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with RETEVMO and for 1 week after the final dose.
Based on animal data, RETEVMO can cause embryolethality and malformations at doses resulting in exposures less than or equal to the human exposure at the clinical dose of 160 mg twice daily.
Verify pregnancy status in females of reproductive potential prior to initiating RETEVMO
Advise female patients of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the final dose.
Advise males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the final dose.
RETEVMO may impair fertility in females and males of reproductive potential.
The safety and effectiveness of RETEVMO have been established in pediatric patients aged 12 years and older for medullary thyroid cancer (MTC) who require systemic therapy and for advanced RET fusion-positive thyroid cancer who require systemic therapy and are radioactive iodine-refractory (if radioactive iodine is appropriate). Use of RETEVMO for these indications is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients aged 12 years and older. The safety and effectiveness of RETEVMO have not been established in these indications in patients less than 12 years of age.
The safety and effectiveness of RETEVMO have not been established in pediatric patients for other indications.
In 4-week general toxicology studies in rats, animals showed signs of physeal hypertrophy and tooth dysplasia at doses resulting in exposures ≥ approximately 3 times the human exposure at the 160 mg twice daily clinical dose. Minipigs also showed signs of minimal to marked increases in physeal thickness at the 15 mg/kg high dose level (approximately 0.3 times the human exposure at the 160 mg twice daily clinical dose). Rats in both the 4- and 13-week toxicology studies had malocclusion and tooth discoloration at the high dose levels (≥1.5 times the human exposure at the 160 mg twice daily clinical dose) that persisted during the recovery period.
Monitor growth plates in adolescent patients with open growth plates. Consider interrupting or discontinuing therapy based on the severity of any growth plate abnormalities and based on an individual risk-benefit assessment.
Of 702 patients who received RETEVMO, 34% (239 patients) were ≥ 65 years of age and 10% (67 patients) were ≥ 75 years of age. No overall differences were observed in the safety or effectiveness of RETEVMO between patients who were ≥65 years of age and younger patients.
No dosage modification is recommended for patients with mild to severe renal impairment [estimated Glomerular Filtration Rate (eGFR) ≥15 to 89 mL/min estimated by Modification of Diet in Renal Disease (MDRD) equation]. The recommended dosage has not been established for patients with end-stage renal disease.
Reduce the dose when administering RETEVMO to patients with severe [total bilirubin greater than 3 to 10 times upper limit of normal (ULN) and any AST] hepatic impairment No dosage modification is recommended for patients with mild (total bilirubin less than or equal to ULN with AST greater than ULN or total bilirubin greater than 1 to 1.5 times ULN with any AST) or moderate (total bilirubin greater than 1.5 to 3 times ULN and any AST) hepatic impairment. Monitor for RETEVMO-related adverse reactions in patients with hepatic impairment.
Rx only.
Additional information can be found at www.retevmo.com.
Eli Lilly and Company, Indianapolis, IN 46285, USA Copyright ©2021, Eli Lilly and Company. All rights reserved. SE HCP BS 25MAR2021
RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO, SE HCP BS 25MAR2021 - 7.5 x 10
Revista Puertorriqueña de Medicina y Salud Pública
En aras de seguir promoviendo la concienciación del cáncer de pulmón y el cáncer gástrico, desde el Capitolio de Puerto Rico se realizó una ceremonia donde representantes de la rama Ejecutiva, la Asamblea Legislativa y del sector Salud, se comprometieron a realizar esfuerzos concertados para prevenir y tratar el cáncer y mejorar la calidad de los pacientes, sobrevivientes y cuidadores. Por tal razón, el Senado condecoró a la Revista de Medicina y Salud Pública por estar siempre pendiente de las necesidades médicas de la población, así como en la contribución de llevar un mensaje y educar tanto a los médicos como a los pacientes y familiares de diferentes enfermedades.