EL ESTRES Y LA INTELIGENCIA EMOCIONAL EN MÉDICOS PUERTORRIQUEÑOS



Revista Puertorriqueña de Medicina y Salud Pública 1 www.medicinaysaludpublica.com @RevistaMSP

SPIRIT-P1 ACR20 AT WEEK 2: TALTZ 39% VS PLACEBO=13% SPIRIT-P2 ACR20 AT WEEK 2: TALTZ 38% VS PLACEBO=12%

ACR20 at week 2 was not controlled for type 1 error; therefore, statistical conclusions cannot be made. SPIRIT-P1
NRI of intent-to-treat population through week 24. Inadequate responders (<20% improvement in tender and in swollen joint counts) at week 16 were analyzed as nonresponders after week 16
the primary endpoint1 Primary endpoint=ACR20 response at week 24.
SPIRIT-P1 and -P2 Trial Design3-6
SPIRIT-P1 (N=417) and -P2 (N=363) were phase 3, randomized, doubleblind, placebo-controlled trials to evaluate the efficacy and safety of Taltz compared with placebo in patients with active psoriatic arthritis. Patients in SPIRIT-P1 were biologic-naive. Patients in SPIRIT-P2 were tumor necrosis factor inhibitor (TNFi)- experienced, having had an inadequate response and/or intolerance to 1 or 2 prior TNFis. In both trials, the primary efficacy endpoint was the proportion of patients achieving ACR20 response at week 24. All patients were ≥18 years of age and had ≥3 swollen and ≥3 tender joints. Patients were randomized to placebo or Taltz 80 mg every 2 or 4 weeks following a 160 mg starting dose. In SPIRIT-P1, an active reference arm of adalimumab 40 mg every 2 weeks was included. Patients in all study
arms were allowed to continue taking stable background medications during the trial. Inadequate responders (as defined by blinded criteria of <20% improvement in tender and in swollen joint counts) at week 16 received rescue therapy and were analyzed as nonresponders after week 16 until the primary endpoint. After receiving rescue therapy, inadequate responders in the placebo and adalimumab arms were re-randomized to Taltz 80 mg every 2 or 4 weeks. NRI methods were used for categorical efficacy analyses during the double-blind treatment period.
ACR=American College of Radiology; TNFi=tumor necrosis factor inhibitor; NRI=nonresponder imputation.
VAS Injection Site Pain Score Immediately Following Injection7
25.2
3.5¶
Taltz 80 mg original formulation (n=61) Taltz 80 mg Citrate-Free formulation (n=63)
¶P<.0001; based on VAS 0-100
‡Same active ingredient
§Vs original formulation; immediately after injection; based on VAS 0-100
No new National Drug Codes (NDCs)
No new Rx needed for existing Taltz patients
No new PAs to transition existing Taltz patients
No gaps in therapy
Once inventory of Taltz original formulation is depleted, Only citrate-free formulation will be available
The Citrate-Free Bioequivalence Study (N=245) was a 2-arm, subjectblind, parallel-design study in healthy subjects age 18-75 years to evaluate bioequivalence of Taltz citrate-free (CF) formulation compared to the original formulation of Taltz. Subjects were stratified into 1 of 3 weight categories (low: <70.0 kg; medium: 70.0-80.0 kg; high: >80.0 kg). Participants were then randomized within the 3 weight categories 1:1 to a single subcutaneous dose of either 80 mg Taltz original formulation (n=126) or 80 mg Taltz CF formulation (n=119). Subjects in each group were sub-randomized 1:1:1 to injection site (arm, thigh, or abdomen). Injections were administered by a medical professional using an autoinjector. The primary endpoint was bioequivalence as measured by maximum concentration (Cmax) of serum ixekizumab and area under the concentration versus time curve (AUC) of ixekizumab from time of injection through day 85 and time of injection through infinity.
Citrate-Free Injection Pain Study (N=70) was a subject-blind, randomized, crossover study in healthy subjects age 18-75 years to determine injection site pain differences between Taltz citrate-free formulation compared to the original formulation of Taltz. The primary endpoint was pain intensity on injection, as measured by VAS Pain 0-100. Subjects were randomized 1:1:1 to receive a single 1 mL subcutaneous injection of 80 mg Taltz original formulation, 80 mg Taltz citrate-free formulation 1 (CF1), or 80 mg Taltz citrate-free formulation 2 (CF2) in 1 of 3 possible treatment sequences on Days 1, 8, and 15 in a 3-period cross-over design. Injections were administered in the abdomen by a medical professional using a prefilled syringe. CF2 is not an approved formulation. Only data on the commercially available CF1 will be presented.
Taltz is indicated for adult patients with active ankylosing spondylitis, for adult patients with active psoriatic arthritis (PsA), and for adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation. Taltz is also indicated for adult patients and pediatric patients aged 6 years or older with moderate-to-severe plaque psoriasis (PsO) who are candidates for systemic therapy or phototherapy.
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients.
Taltz may increase the risk of infection. In clinical trials of adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). A similar increase in risk of infection was seen in placebocontrolled trials of adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis. Serious infections have occurred. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a serious infection develops, discontinue Taltz until the infection resolves.
Pre-Treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Closely monitor patients receiving Taltz for signs and symptoms of active TB during and after treatment.
Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz. If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than the placebo group. During Taltz treatment, monitor patients for onset or exacerbations of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations
Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz.
Most common adverse reactions (≥1%) associated with Taltz treatment are injection site reactions, upper respiratory tract infections, nausea, and tinea infections. Overall, the safety profiles observed in adult patients with psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and pediatric patients with plaque psoriasis were consistent with the safety profile in adult patients with plaque psoriasis, with the exception of influenza and conjunctivitis in psoriatic arthritis and conjunctivitis, influenza, and urticaria in pediatric psoriasis.
Please see Brief Summary of Prescribing Information on the following pages. Please see Instructions for Use included with the device.
IX HCP ISI 07MAY2020
References: 1. Data on file. Lilly USA, LLC. DOF-IX-US-0304. 2. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79-87.
3. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327.
4. Taltz. Prescribing information. Lilly, USA. LLC. 5. Mease PJ, van der Heijde D, Ritchlin CT, et al; on behalf of SPIRIT-P1 Study Group. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebocontrolled and active (adalimumab)-controlled period of the phase 3 trial SPIRIT-P1. Ann Rheum Dis. 2017;76(suppl):1-30. 6. Nash P, Kirkham B, Okada M, et al; on behalf of SPIRIT-P2 Study Group. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317-2327. Supplementary appendix. 7. Chabra S, Gill BJ, Gallo G, et al. Ixekizumab citrate-free formulation: results from two clinical trials. Adv Ther. 2022;Epub (Incl Suppl Inf):1-11, 1-4. https://doi.org/10.1007/s12325-022-02126-0.
Taltz® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-IX-US-5471 05/2022 ©Lilly USA, LLC 2022. ALL RIGHTS RESERVED.
Brief Summary: Consult the package insert for complete prescribing information.
Plaque Psoriasis—Taltz is indicated for the treatment of patients aged 6 years and older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Psoriatic Arthritis—Taltz is indicated for the treatment of adult patients with active psoriatic arthritis. Ankylosing Spondylitis—Taltz is indicated for the treatment of adult patients with active ankylosing spondylitis.
Non-radiographic Axial Spondyloarthritis—Taltz is indicated for the treatment of adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients (Warnings and Precautions)
Infections—Taltz may increase the risk of infection. In clinical trials in adult patients with plaque psoriasis, the Taltz group had a higher rate of infections than the placebo group (27% vs 23%). Upper respiratory tract infections, oral candidiasis, conjunctivitis and tinea infections occurred more frequently in the Taltz group than in the placebo group. A similar increase in risk of infection was seen in placebo-controlled trials in patients with pediatric psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis (Adverse Reactions) Instruct patients treated with Taltz to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a serious infection or is not responding to standard therapy, monitor the patient closely and discontinue Taltz until the infection resolves. Pre-treatment Evaluation for Tuberculosis—Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Taltz. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering Taltz. Consider anti-TB therapy prior to initiating Taltz in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Patients receiving Taltz should be monitored closely for signs and symptoms of active TB during and after treatment.
Hypersensitivity—Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the Taltz group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post-marketing use with Taltz (Adverse Reactions). If a serious hypersensitivity reaction occurs, discontinue Taltz immediately and initiate appropriate therapy.
Inflammatory Bowel Disease—Patients treated with Taltz may be at an increased risk of inflammatory bowel disease. In clinical trials, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the Taltz group than in the control group (Adverse Reactions). During Taltz treatment, monitor for onset or exacerbation of inflammatory bowel disease and if IBD occurs, discontinue Taltz and initiate appropriate medical management.
Immunizations—Prior to initiating therapy with Taltz, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with Taltz. No data are available on the response to live vaccines.
The following adverse drug reactions are discussed in greater detail in other sections of the label:
• Infections (Warnings and Precautions)
• Hypersensitivity Reactions (Contraindications and Warnings and Precautions)
• Inflammatory Bowel Disease (Warnings and Precautions)
Clinical Trials Experience—Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Plaque Psoriasis
Weeks 0 to 12: Three placebo-controlled trials in subjects with plaque psoriasis were integrated to evaluate the safety of Taltz compared to placebo for up to 12 weeks. A total of 1167 subjects (mean age 45 years; 66% men; 94% White) with plaque psoriasis received Taltz (160 mg at Week 0, 80 mg every 2 weeks [Q2W] for 12 weeks) subcutaneously. In two of the trials, the safety of Taltz (use up to 12 weeks) was also compared with an active comparator, U.S. approved etanercept. In the 12-week, placebo-controlled period, adverse events occurred in 58% of the Taltz Q2W group (2.5 per subject-year of follow-up) compared with 47% of the placebo group (2.1 per subject-year of follow-up). Serious adverse events occurred in 2% of the Taltz group (0.07 per subject-year of follow-up), and in 2% of the placebo group (0.07 per subject-year of follow-up). Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the Taltz group than the placebo group during the 12-week placebo-controlled period of the pooled clinical trials.
Table 1: Adverse Reactions Occurring in ≥1% of the Taltz Group and More Frequently than in the Placebo Group in the Plaque Psoriasis Clinical Trials through Week 12 Adverse Reactions Taltz 80 mg Q2W (N=1167) (n%) Etanerceptb (N=287) (n%) Placebo (N=791) (n%)
Injection site reactions 196 (17) 32 (11) 26 (3)
Upper respiratory tract infectionsa 163 (14) 23 (8) 101 (13)
Nausea 23 (2) 1 (<1) 5 (1) Tinea infections 17 (2) 0 1 (<1)
a Upper respiratory tract infections cluster includes nasopharyngitis and rhinovirus infection.
b U.S. approved etanercept.
Taltz® (ixekizumab) injection
Adverse reactions that occurred at rates less than 1% in the Taltz group and more frequently than in the placebo group during the 12-week induction period included rhinitis, oral candidiasis, urticaria, influenza, conjunctivitis, inflammatory bowel disease, and angioedema.
Weeks 13 to 60: A total of 332 subjects received the recommended maintenance regimen of Taltz 80 mg dosed every 4 weeks. During the maintenance period (Weeks 13 to 60), adverse events occurred in 80% of subjects treated with Taltz (1.0 per subject-year of follow-up) compared to 58% of subjects treated with placebo (1.1 per subject-year of follow-up). Serious adverse events were reported in 4% of subjects treated with Taltz (0.05 per subject-year of follow-up) and none in the subjects treated with placebo.
Weeks 0 to 60: Over the entire treatment period (Weeks 0 to 60), adverse events were reported in 67% of subjects treated with Taltz (1.4 per subject-year of follow-up) compared to 48% of subjects treated with placebo (2.0 per subject-year of follow-up). Serious adverse events were reported in 3% of subjects treated with Taltz (0.06 per subject-year of follow-up), and in 2% of subjects treated with placebo (0.06 per subject-year of follow-up).
Specific Adverse Drug Reactions: Injection Site Reactions: The most frequent injection site reactions were erythema and pain. Most injection site reactions were mild-to-moderate in severity and did not lead to discontinuation of Taltz.
Infections: In the 12-week, placebo-controlled period of the clinical trials in plaque psoriasis, infections occurred in 27% of subjects treated with Taltz (1.2 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.4% of subjects treated with Taltz (0.02 per subject-year of follow-up) and in 0.4% of subjects treated with placebo (0.02 per subject-year of follow-up) (Warnings and Precautions)
During the maintenance treatment period (Weeks 13 to 60), infections occurred in 57% of subjects treated with Taltz (0.70 per subject-year of follow-up) compared to 32% of subjects treated with placebo (0.61 per subject-year of follow-up). Serious infections occurred in 0.9% of subjects treated with Taltz (0.01 per subject-year of follow-up) and none in the subjects treated with placebo.
Over the entire treatment period (Weeks 0 to 60), infections were reported in 38% of subjects treated with Taltz (0.83 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.7% of subjects treated with Taltz (0.02 per subject-year of follow-up), and in 0.4% of subject treated with placebo (0.02 per subject-year of follow-up).
Inflammatory Bowel Disease: In adult subjects with plaque psoriasis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ 80 mg Q2W group (Crohn’s disease 0.1%, ulcerative colitis 0.2%) than the placebo group (0%) during the 12-week, placebo-controlled period in clinical trials (Warnings and Precautions).
Laboratory Assessment of Cytopenia: Neutropenia—Over the entire treatment period (Weeks 0 to 60), neutropenia occurred in 11% of subjects treated with Taltz (0.24 per subject-year of follow-up) compared to 3% of subjects treated with placebo (0.14 per subject-year of follow-up). In subjects treated with Taltz, the incidence rate of neutropenia during Weeks 13 to 60 was lower than the incidence rate during Weeks 0 to 12.
In the 12-week, placebo-controlled period, neutropenia ≥ Grade 3 (<1,000 cells/mm3) occurred in 0.2% of the Taltz group (0.007 per subject-year of follow-up) compared to 0.1% of the placebo group (0.006 per subject-year of follow-up). The majority of cases of neutropenia were either Grade 2 (2% for Taltz 80 mg Q2W versus 0.3% for placebo; ≥1,000 to <1,500 cells/mm3) or Grade 1 (7% for Taltz 80 mg Q2W versus 3% for placebo; ≥1,500 cells/mm3 to <2,000 cells/mm3). Neutropenia in the Taltz group was not associated with an increased rate of infection compared to the placebo group.
Thrombocytopenia—Ninety eight percent of cases of thrombocytopenia were Grade 1 (3% for Taltz 80 mg Q2W versus 1% for placebo; ≥75,000 cells/mm3 to <150,000 cells/mm3). Thrombocytopenia in subjects treated with Taltz was not associated with an increased rate of bleeding compared to subjects treated with placebo.
Active Comparator Trials: In the two clinical trials that included an active comparator, the rate of serious adverse events during weeks zero to twelve was 0.7% for U.S.-approved etanercept and 2% for Taltz 80 mg Q2W, and the rate of discontinuation from adverse events was 0.7% for U.S. approved etanercept and 2% for Taltz 80 mg Q2W. The incidence of infections was 18% for U.S. approved etanercept and 26% for Taltz 80 mg Q2W. The rate of serious infections was 0.3% for both Taltz 80 mg Q2W and U.S. approved etanercept.
Taltz was evaluated in a placebo-controlled trial in pediatric subjects with moderate-to-severe psoriasis 6 to less than 18 years of age. A total of 171 subjects were studied (115 subjects on Taltz and 56 subjects on placebo). Overall, the safety profile observed in pediatric subjects with plaque psoriasis treated with Taltz every 4 weeks is consistent with the safety profile in adult subjects with plaque psoriasis with the exception of the frequencies of conjunctivitis (2.6%), influenza (1.7%), and urticaria (1.7%).
In this clinical trial, Crohn’s disease occurred at a greater frequency in the Taltz group (0.9%) than the placebo group (0%) during the 12-week, placebo-controlled period. Crohn’s disease occurred in a total of 4 Taltz treated subjects (2.0%) in the clinical trial (Warnings and Precautions).
Taltz was studied in two placebo-controlled trials in patients with psoriatic arthritis. A total of 678 patients were studied (454 patients on Taltz and 224 on placebo). A total of 229 patients in these trials received Taltz 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with psoriatic arthritis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis with the exception of the frequencies of influenza (1.3%) and conjunctivitis (1.3%).
IX HCP BS 06JAN2022 Taltz® (ixekizumab) injection
IX HCP BS 06JAN2022
Taltz was studied in two placebo-controlled trials in patients with ankylosing spondylitis. A total of 566 patients were studied (376 patients on Taltz and 190 on placebo). A total of 195 patients in these trials received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with ankylosing spondylitis treated with Taltz Q4W is consistent with the safety profile in adult patients with plaque psoriasis.
In adult patients with ankylosing spondylitis, Crohn’s disease and ulcerative colitis, including exacerbations, occurred in 2 patients (1.0%) and 1 patient (0.5%), respectively, in the Taltz 80 mg Q4W group and 1 patient (0.5%) and 0%, respectively, in the placebo group during the 16-week, placebo-controlled period in clinical trials. Of these patients, serious events occurred in 1 patient in the Taltz 80 mg Q4W group and 1 patient in the placebo group (Warnings and Precautions).
Non-radiographic Axial Spondyloarthritis
Taltz was studied in a placebo-controlled trial in patients with non-radiographic axial spondyloarthritis. A total of 303 patients were studied (198 patients on Taltz and 105 on placebo). A total of 96 patients in this trial received Taltz 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with non-radiographic axial spondyloarthritis treated with Taltz 80 mg Q4W up to Week 16 is consistent with the previous experience of Taltz in other indications.
Immunogenicity—As with all therapeutic proteins, there is the potential for immunogenicity with Taltz. The assay to test for neutralizing antibodies has limitations detecting neutralizing antibodies in the presence of ixekizumab; therefore, the incidence of neutralizing antibodies development could be underestimated.
By Week 12, approximately 9% of adult subjects treated with Taltz every 2 weeks developed antibodies to ixekizumab. Approximately 22% of subjects treated with Taltz at the recommended dosing regimen developed antibodies to ixekizumab during the 60-week treatment period. The clinical effects of antibodies to ixekizumab are dependent on the antibody titer; higher antibody titers were associated with decreasing drug concentration and clinical response.
Of the adult subjects who developed antibodies to ixekizumab during the 60-week treatment period, approximately 10%, which equates to 2% of subjects treated with Taltz at the recommended dosing regimen, had antibodies that were classified as neutralizing. Neutralizing antibodies were associated with reduced drug concentrations and loss of efficacy.
In pediatric psoriasis subjects treated with ixekizumab at the recommended dosing regimen up to 12 weeks, 21 subjects (18%) developed anti-drug antibodies, 5 subjects (4%) had confirmed neutralizing antibodies associated with low drug concentrations. No conclusive evidence could be obtained on the potential association of neutralizing antibodies and clinical response and/or adverse events due to small number of pediatric subjects in the study.
For subjects treated with Taltz 80 mg every 4 weeks for up to 52 weeks (PsA1), 11% developed anti-drug antibodies, and 8% had confirmed neutralizing antibodies.
For patients treated with Taltz 80 mg every 4 weeks for up to 16 weeks (AS1, AS2), 5.2% developed anti-drug antibodies, and 1.5% had neutralizing antibodies.
Of patients treated with Taltz 80 mg every 4 weeks for up to 52 weeks (nr-axSpA1), 8.9% developed anti-drug antibodies, all of which were low titer. No patient had neutralizing antibodies. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to Taltz across indications or with the incidences of antibodies to other products may be misleading.
Postmarketing Experience—The following adverse reactions have been identified during postapproval use of Taltz. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Taltz exposure.
Immune system disorders: anaphylaxis (Contraindications and Warnings and Precautions)
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Pregnant women should be encouraged to enroll themselves in the registry by calling 1-800-284-1695.
Risk Summary—There are no available data on Taltz use in pregnant women to inform any drug associated risks. Human IgG is known to cross the placental barrier; therefore, Taltz may be transmitted from the mother to the developing fetus. An embryofetal development study conducted in pregnant monkeys at doses up to 19 times the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus. When dosing was continued until parturition, neonatal deaths were observed at 1.9 times the MRHD [see Data]. The clinical significance of these nonclinical findings is unknown.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data Animal Data—An embryofetal development study was conducted in cynomolgus monkeys administered ixekizumab. No malformations or embryofetal toxicity were observed in fetuses
from pregnant monkeys administered ixekizumab weekly by subcutaneous injection during organogenesis to near parturition at doses up to 19 times the MRHD (on a mg/kg basis of 50 mg/ kg/week). Ixekizumab crossed the placenta in monkeys.
In a pre- and post-natal development toxicity study, pregnant cynomolgus monkeys were administered weekly subcutaneous doses of ixekizumab up to 19 times the MRHD from the beginning of organogenesis to parturition. Neonatal deaths occurred in the offspring of two monkeys administered ixekizumab at 1.9 times the MRHD (on a mg/kg basis of 5 mg/kg/week) and two monkeys administered ixekizumab at 19 times the MRHD (on a mg/kg basis of 50 mg/kg/ week). These neonatal deaths were attributed to early delivery, trauma, or congenital defect. The clinical significance of these findings is unknown. No ixekizumab-related effects on functional or immunological development were observed in the infants from birth through 6 months of age.
Risk Summary—There are no data on the presence of ixekizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Ixekizumab was detected in the milk of lactating cynomolgus monkeys. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Taltz and any potential adverse effects on the breastfed infant from Taltz or from the underlying maternal condition.
Pediatric Use—The safety and effectiveness of Taltz have been established in pediatric subjects aged 6 years to less than 18 years with moderate-to-severe plaque psoriasis. The safety and effectiveness of Taltz in other pediatric indications and for pediatric subjects less than 6 years of age have not been established.
Geriatric Use—Of the 4204 psoriasis subjects exposed to Taltz, a total of 301 were 65 years or older, and 36 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects.
PATIENT COUNSELING INFORMATION—Advise the patient and/or caregiver to read the FDAapproved patient labeling (Medication Guide and Instructions for Use) before the patient starts using Taltz and each time the prescription is renewed, as there may be new information they need to know.
Instructions on Self-Administration: Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the autoinjector or prefilled syringe correctly (Instructions for Use)
Infection: Inform patients that Taltz may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider, and contacting their healthcare provider if they develop any symptoms of infection (Warnings and Precautions).
Allergic Reactions: Advise patients to seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions (Warnings and Precautions).
Pregnancy: Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Taltz during pregnancy. Advise patients to contact the registry at 1-800-284-1695 to enroll (Use in Specific Populations)
Additional information can be found at www.Taltz.com
See Instructions for Use accompanying the product device.
Marketed by: Lilly USA, LLC, Indianapolis, IN 46285, USA
Copyright © 2016, 2017, 2019, 2020, 2021, 2022 Eli Lilly and Company. All rights reserved.
PP-IX-US-5359
DISCAPACIDAD ENTRE ADULTOS CON DIAGNÓSTICO DE VIH EN ESTADOS UNIDOS
TRATAMIENTO DE LA PSORIASIS ERITRODÉRMICA CON BIOLÓGICOS: UNA REVISIÓN SISTEMÁTICA
DIAGNOSTICO INESPERADO EN PACIENTE CON SOSPECHA DE GIST: INFILTRACIÓN GÁSTRICA POR MIELOMA MÚLTIPLE
OBESIDAD E HIPERTENSIÓN EN ESCOLARES DE PUERTO RICO
EDITOR FUNDADOR Juan Carlos Orengo Valverde, MD, MPH, PhD EDITOR Alberto Santiago Cornier, MD, PhD CONSEJO ASESOR Oscar Soto Raíces, MD, Ahmed Morales, MD, FACP, FACG, FASGE, AGAF, Lcda. Wanda González PRINCIPAL OFICIAL EJECUTIVO Pedro Carlos Lugo Hernández III, P.A.C. PRESIDENTA Y FUNDADORA Glorybelle Hernández Figueroa, MBA VICEPRESIDENTA Y FUNDADORA Laila Paloma Lugo, MBA CONTABILIDAD Julio Soto ADMINISTRACIÓN Marta Ivelisse Vélez Ramos, MBA, MARKETING Y SERVICIOS 360 Alexelena Cayere, Yasmin Morell, Belinda Burgos PERIODISTAS Grenda Rivera, Mayra Acevedo, Luis Penchi, Limarys Suárez, Yolimarian Torres DIRECCIÓN GRÁFICA Natalia Zoé Rivera Torres ARTISTA GRÁFICO Jhorman González DIRECTORA AUDIOVISUAL Fabiola Plaza REALIZADORA AUDIOVISIAL Salomé Mateus, Duban Valencia FOTOS Revista Medicina y Salud Pública DIRECCIÓN GENERAL / FUNDADOR Carlos Alexis Lugo Marrero DISTRIBUCIÓN OFICINAS Y TORRES MÉDICAS Editorial Mundo ENVÍO DE REVISTAS Y DISTRIBUCIÓN A GRUPOS MÉDICOS Servicio de correo postal/Comunicación Inteligente Para ventas y otros servicios pueden comunicarse al 787.848.3333, msp@editorialmundo.com o www.medicinaysaludpublica.com Revista Puertorriqueña de Medicina y Salud Pública ISSN 1937-8521
COMITÉ EDITORIAL Olga Rodríguez, MD - Decana Escuela de Medicina de Ponce (Puerto Rico), Vivian Green, LND, MS, PhD, Sub editora y fundadora (Puerto Rico), José Cordero, MD, MPH - Exdecano Escuela Graduada Salud Pública Recinto de Ciencias Médicas UPR (Puerto Rico), Ángeles Rodríguez, MD, MPH (Puerto Rico), Simón Carlo, MD (Puerto Rico), Bárbara Rosado, MD (Puerto Rico), Idhaliz Flores PhD (Puerto Rico), Jesús Cruz-Correa, MD, FACOG (Puerto Rico), Rafael Bredy, MD, LicMTo, MBE, MS (Puerto Rico), David Caseida, MD, FACOG, (Puerto Rico), José Capriles, MD, MHSA (Puerto Rico) Joaquín Laboy, MD, FACOG (Puerto Rico), Luis Adrian Rivera Pomales, MD, PEMBA, MPH, CMQ (Puerto Rico), Juan Fernández, MS, PhD (Puerto Rico), Nuria Sebate, MD (Puerto Rico), Pedro Amador, MD, MPH (Puerto Rico), Nydia Cappas, PsyD (Puerto Rico), Luis Franco, MD (Puerto Rico), Federico Montealegre, DVM, PhD, Msc (Puerto Rico), Nydia Ortiz, PsyD (Puerto Rico), José Pons, PhD, FPPR (Puerto Rico), Esdrás Vélez, JD, MPH (Puerto Rico), Diego Zavala, MSc, PhD, (Puerto Rico), Ana Torres-Martín, MD (Puerto Rico), Julio Cádiz, MD, MPH (Puerto Rico), Rafael Gómez-Cuevas (Colombia), José Javier Orengo, PhD(c) (España), Cesar A. Del Rey, MD (Panamá), Pedro Serrano, MD, PhD (España), Luis Serra-Majem, MD, PhD (España), José Ramón Calvo, MD, PhD (España).

Síguenos en www.medicinaysaludpublica.com, www.facebook.com/revistamsp, en Twitter @revistamsp, en LinkedIn como Revista Puertorriqueña de Medicina y Salud Pública. Las normas editoriales de la Revista Puertorriqueña de Medicina y Salud Pública para la publicación de artículos originales y cartas al editor pueden ser accesadas en la página web: www.medicinaysaludpublica.com, y solicitadas a través de msp@editorialmundo.com.
Medicina y Salud Pública es propiedad de publicaciones mundo. Medicina es una publicación de la REVISTA PUERTORRIQUEÑA DE MEDICINA Y SALUD PÚBLICA. Medicina y Salud Pública tiene como política corregir y aclarar cualquier información incorrecta que pueda ser publicada en su revista. Medicina y Salud Pública no asume responsabilidad alguna por los anuncios, artículos y otros servicios anunciados en nuestra publicación.
LA ELASTOGRAFÍA DE ONDAS DE CORTE DIRECTA CAMBIOS ASINTOMÁTICOS DE HÍGADO EN PACIENTES CON DIABETES MELLITUS TIPO 2
La salud pública comprende todas las acciones que se puedan ejecutar para proteger la salud y prevenir la aparición de enfermedades. El objetivo principal es fomentar el bienestar de las personas, aumentar la longevidad y mejorar la calidad de vida.
Sin embargo, en muchos países se acentúan las disparidades cuando se trata de salud pública. Una muestra de ello es que la esperanza de vida, por ejemplo, puede llegar a tener una diferencia de hasta 18 años, dependiendo de si se vive en un país rico o uno pobre.
Si algo dejó claro la pandemia por covid-19 es que el sistema de salud, a nivel mundial, es mucho más frágil de lo que se pensaba y que todavía queda mucho por mejorar.
Son muchos los retos en materia de salud pública a los que se debe hacer frente, y ello incluye derribar los mitos de la vacunación que aún proliferan entre la población, así como informar sobre los productos alimenticios que son nocivos y cobran la vida de millones de personas.
Estos retos no solo atañen al campo médico, sino a la política de las naciones, puesto que se trata de unos desafíos enormes que requieren de grandes inversiones para la elaboración de planes eficientes.
La crisis climática es otro problema al cual se deben destinar suficientes fondos y voluntades, ya que la contaminación del aire mata millones de personas al año, en tanto que el cambio climático genera desastres naturales, aumenta la desnutrición y facilita la propagación de enfermedades infecciosas.
Las emisiones producidas por el calentamiento global ocasionan decesos por ataques cardíacos, cánceres de pulmón, accidentes cerebrovasculares y enfermedades respiratorias crónicas.
Por otro lado, es imperativo que la atención médica sea justa, especialmente la materna y la infantil. En cuanto al acceso a los medicamentos, son muchas las personas en Puerto Rico y el el mundo que no cuentan con ellos y esto pone en peligro sus vidas.
Las dietas poco saludables, la carencia de alimentos, la inseguridad alimentaria, la escasez y el hambre continúan acechando al planeta.
En ciertos lugares hay quienes consumen alimentos e ingieren bebidas con un contenido alto de azúcar, grasas saturadas y sal, lo cual aumenta las enfermedades, el sobrepeso y la obesidad.
Aunque la tecnología y el progreso avanzan a pasos vertiginosos, miles de millones de personas en todo el mundo viven sin agua potable y no cuentan con servicios de saneamiento, los cuales son imprescindibles para detener las enfermedades.
Las causas de estos males son principalmente la falta de financiamiento, la debilidad de los sistemas de salud y el poco compromiso por parte de países ricos. Todo esto forma parte de los grandes retos a los que se enfrenta la salud pública en la actualidad y cuya atención es impostergable.
Desde la Revista de Medicina y Salud Pública todos estos desafíos nos obligan a preguntarnos qué podemos hacer mejor para avanzar en materia de salud y, sin duda, además del esfuerzo genuino de nuestros héroes médicos, necesitamos del empeño de los líderes gubernamentales y de los avances de la ciencia para redoblar fuerzas.

La Revista de Medicina y Salud Pública continuará informando a la población y garantizando el acceso al conocimiento científico al servicio de todos para alcanzar la meta de tener una población saludable. Avanzar juntos es la mejor forma de cambiar el curso de la historia y encaminarnos hacia una mejor humanidad.
Dr. Oscar Soto Raices Presidente Asociación Enfermedades Reumáticas de Puerto Rico (FER).
a SELECT-EARLY (RA-I; MTX-naïve) [primary endpoint at Week 12: ACR50 response vs MTX, select ranked secondary endpoint at Week 24: Δ mTSS vs MTX]; SELECT-MONOTHERAPY (RA-II; MTX-IR) [primary endpoint at Week 14: ACR20 response vs MTX, select ranked secondary endpoints at Week 14: DAS28-CRP<2.6 vs MTX, DAS28-CRP≤3.2 vs MTX]; SELECT-NEXT (RA-III; csDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD]; SELECT-COMPARE (RA-IV; MTX-IR) [RINVOQ + MTX; primary endpoint at Week 12: ACR20 response vs placebo + MTX, select ranked secondary endpoints at Week 26: Δ mTSS vs placebo + MTX]; SELECT-BEYOND (RA-V; bDMARD-IR) [RINVOQ + csDMARD; primary endpoint at Week 12: ACR20 response vs placebo + csDMARD, select ranked secondary endpoints at Week 12: DAS28-CRP≤3.2 vs placebo + csDMARD.]


RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
Limitation of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Serious Infections: Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. These infections include tuberculosis (TB), invasive fungal, bacterial, viral, and other infections due to opportunistic pathogens. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids.
Mortality: A higher rate of all-cause mortality, including sudden cardiovascular (CV) death, was observed with a Janus kinase (JAK) inhibitor in a study comparing another JAK inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years of age with at least one CV risk factor.
Malignancies: Lymphoma and other malignancies have been observed in RINVOQ-treated patients. A higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with another JAK inhibitor when compared with TNF blockers in RA patients. Patients who are current or past smokers are at additional increased risk.
Major Adverse Cardiovascular Events: A higher rate of CV death, myocardial infarction, and stroke was observed with a JAK inhibitor in a study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years of age with at least one CV risk factor. Current or past smokers are at additional increased risk.
Thrombosis: Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. A higher rate of thrombosis was observed with another JAK inhibitor when compared with TNF blockers in RA patients.
Hypersensitivity: RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients.

Other Serious Adverse Reactions: Hypersensitivity Reactions (anaphylaxis and angioedema), Gastrointestinal Perforations, Laboratory Abnormalities (neutropenia, lymphopenia, anemia, lipid elevations, liver enzyme elevations), and Embryo-Fetal Toxicity.


Please see additional Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, on the previous page of this advertisement.
Please see Brief Summary of full Prescribing Information on previous pages of this advertisement.
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis (TB), which may present with pulmonary or extrapulmonary disease. Test patients for latent TB before RINVOQ use and during therapy. Consider treatment for latent TB infection prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
Carefully consider the risks and benefits of treatment with RINVOQ prior to initiating therapy in patients with chronic or recurrent infection. Monitor patients closely for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy.
In a large, randomized, postmarketing safety study comparing another Janus kinase (JAK) inhibitor with tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients ≥50 years old with at least one cardiovascular (CV) risk factor, a higher rate of all-cause mortality, including sudden CV death, was observed with the JAK inhibitor. Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Lymphoma and other malignancies have been observed in patients treated with RINVOQ.
In a large, randomized, postmarketing safety study comparing another JAK inhibitor with TNF blockers in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]), lymphomas, and lung cancer (in current or past smokers) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk.
With RINVOQ, consider the benefits and risks for the individual patient prior to initiating or continuing therapy, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers. NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer. Advise patients to limit sunlight exposure by wearing protective clothing and using sunscreen.
In a large, randomized, postmarketing study comparing another JAK inhibitor with TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke) was observed with the JAK inhibitor. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other CV risk factors. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing study comparing another JAK inhibitor to TNF blockers in RA patients ≥50 years old with at least one CV risk factor, a higher rate of thrombosis was observed with the JAK inhibitor. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated.
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients. Serious hypersensitivity reactions, such as anaphylaxis and angioedema, were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy.
Gastrointestinal (GI) perforations have been reported in clinical trials with RINVOQ. Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Promptly evaluate patients presenting with new onset abdominal pain for early identification of GI perforation.
Treatment with RINVOQ was associated with an increased incidence of neutropenia (absolute neutrophil count [ANC] <1000 cells/mm3). Treatment with RINVOQ is not recommended in patients with an ANC <1000 cells/mm3. Evaluate neutrophil counts at baseline and thereafter according to routine patient management.
Absolute lymphocyte counts (ALC) <500 cells/mm3 were reported in RINVOQtreated patients. Treatment with RINVOQ is not recommended in patients with an ALC <500 cells/mm3. Evaluate at baseline and thereafter according to routine patient management.
Decreases in hemoglobin levels to <8 g/dL were reported in RINVOQ-treated patients. Treatment should not be initiated or should be interrupted in patients with hemoglobin levels <8 g/dL. Evaluate at baseline and thereafter according to routine patient management.
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Manage patients according to clinical guidelines for the management of hyperlipidemia. Evaluate patients 12 weeks after initiation of treatment and thereafter according to the clinical guidelines for hyperlipidemia.
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevation compared to placebo. Evaluate at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. If increases in aspartate aminotransferase (AST) or alanine aminotransferase (ALT) are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose. Verify pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ.
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, patients should be brought up to date on all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
There are no data on the presence of RINVOQ in human milk, the effects on the breastfed infant, or the effects on milk production. Available data in animals have shown the excretion of RINVOQ in milk. Advise patients that breastfeeding is not recommended during treatment with RINVOQ and for 6 days after the last dose.
RINVOQ is not recommended for use in patients with severe hepatic impairment.
The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenzalike illness, elevated liver enzymes, and rash.
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.
Dosage Forms and Strengths: RINVOQ is available in 15 mg, 30 mg, and 45 mg extended-release tablets.
References: 1. RINVOQ [package insert]. North Chicago, IL: AbbVie Inc; 2022. 2. Data on file, AbbVie Inc. ABVRRTI68885. 3. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139): 2513-2524. 4. Smolen JS, Emery P, Rigby W, et al. Upadacitinib as monotherapy in patients with rheumatoid arthritis and prior inadequate response to methotrexate: results at 84 weeks from the SELECTMONOTHERAPY study. Poster presented at: The European Congress of Rheumatology; June 3-6, 2020; E-Congress. 5. Smolen JS, Pangan AL, Emery P, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303-2311. Erratum in: Lancet. 2019;393(10191):2590. 6. Genovese MC, Combe B, Hall S, et al. Upadacitinib in patients with rheumatoid arthritis and inadequate response or intolerance to biological DMARDs: Results at 60 weeks from the SELECT-BEYOND study. Poster presented at: The American College of Rheumatology; November 8-13, 2019. 7. Cohen SB, van Vollenhoven R, Curtis JR, et al. Integrated safety profile of upadacitinib with up to 4.5 years of exposure in patients with rheumatoid arthritis. Poster presented at: The European Congress of Rheumatology; June 2-5, 2021; E-Congress. 8. Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383(16):1511-1521.
Please see Brief Summary of full Prescribing Information on previous pages of this advertisement.
RINVOQ® and its design are registered trademarks of AbbVie Biotechnology Ltd. ©2022 AbbVie Inc. North Chicago, IL 60064 US-RNQR-220149 April 2022 Printed in Puerto Rico
Revista Puertorriqueña de Medicina y Salud Pública
Patients treated with RINVOQ are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions, Adverse Reactions]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt RINVOQ until the infection is controlled.
Reported infections include:
• Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ use.
• Invasive fungal infections, including cryptococcosis and pneumocystosis.
• Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with RINVOQ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RINVOQ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions]
In a large, randomized, postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another Janus kinase (JAK) inhibitor to tumor necrosis factor (TNF) blockers, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed with the JAK inhibitor [see Warnings and Precautions]
Lymphoma and other malignancies have been observed in patients treated with RINVOQ. In RA patients treated with another JAK inhibitor, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk [see Warnings and Precautions].
In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke [see Warnings and Precautions].
Thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis have occurred in patients treated with JAK inhibitors used to treat inflammatory conditions. Many of these adverse events were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of thrombosis was observed when compared with TNF blockers. Avoid RINVOQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ and be promptly evaluated [see Warnings and Precautions]
Rheumatoid
RINVOQ® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs (DMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is indicated for the treatment of adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
RINVOQ is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for ulcerative colitis, or with potent immunosuppressants such as azathioprine and cyclosporine.
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers.
• Limitations of Use: Use of RINVOQ in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine, is not recommended.
RINVOQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients [see Warnings and Precautions].
Serious and sometimes fatal infections have been reported in patients receiving RINVOQ. The most frequent serious infections reported with RINVOQ included pneumonia and cellulitis [see Adverse Reactions]. Among opportunistic infections, tuberculosis, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis, were reported with RINVOQ.
Avoid use of RINVOQ in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating RINVOQ in patients:
• with chronic or recurrent infection
• who have been exposed to tuberculosis
• with a history of a serious or an opportunistic infection
• who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
• with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ. Interrupt RINVOQ if a patient develops a serious or opportunistic infection.
A patient who develops a new infection during treatment with RINVOQ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and RINVOQ should be interrupted if the patient is not responding to antimicrobial therapy. RINVOQ may be resumed once the infection is controlled.
Evaluate and test patients for latent and active tuberculosis (TB) infection prior to administration of RINVOQ. Patients with latent TB should be treated with standard antimycobacterial therapy before initiating RINVOQ. RINVOQ should not be given to patients with active TB. Consider anti-TB therapy prior to initiation of RINVOQ in patients with previously untreated latent TB or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but who have risk factors for TB infection.
Consultation with a physician with expertise in the treatment of TB is recommended to aid in the decision about whether initiating anti-TB therapy is appropriate for an individual patient.
During RINVOQ use, monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster) and hepatitis B virus reactivation, were reported in clinical trials with RINVOQ [see Adverse Reactions]. The risk of herpes zoster appears to be higher in patients treated with RINVOQ in Japan. If a patient develops herpes zoster, consider temporarily interrupting RINVOQ until the episode resolves. Screening for viral hepatitis and monitoring for reactivation should be performed in accordance with clinical guidelines before starting and during therapy with RINVOQ. Patients who were positive for hepatitis C antibody and hepatitis C virus RNA, were excluded from clinical trials. Patients who were positive for hepatitis B surface antigen or hepatitis B virus DNA were excluded from clinical trials. However, cases of hepatitis B reactivation were still reported in patients enrolled in the Phase 3 trials of RINVOQ. If hepatitis B virus DNA is detected while receiving RINVOQ, a liver specialist should be consulted.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed in patients treated with the JAK inhibitor compared with TNF blockers.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ.
Malignancy and Lymphoproliferative Disorders
Malignancies, including lymphomas, were observed in clinical trials of RINVOQ [see Adverse Reactions]
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. In this study, current or past smokers had an additional increased risk of overall malignancies.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Non-Melanoma Skin Cancer NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen.
Major Adverse Cardiovascular Events
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of major adverse cardiovascular events (MACE) defined as cardiovascular death, non-fatal myocardial infarction (MI), and non-fatal stroke was observed with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients
should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur. Discontinue RINVOQ in patients that have experienced a myocardial infarction or stroke.
Thrombosis, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death.
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers.
If symptoms of thrombosis occur, patients should discontinue RINVOQ and be evaluated promptly and treated appropriately. Avoid RINVOQ in patients that may be at increased risk of thrombosis.
Serious hypersensitivity reactions such as anaphylaxis and angioedema were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ and institute appropriate therapy [see Adverse Reactions].
Gastrointestinal perforations have been reported in clinical trials with RINVOQ.
Monitor RINVOQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis or taking NSAIDs). Evaluate promptly patients presenting with new onset abdominal pain for early identification of gastrointestinal perforation.
Treatment with RINVOQ was associated with an increased incidence of neutropenia (ANC less than 1000 cells/mm3).
Evaluate neutrophil counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation and interrupt RINVOQ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3).
Lymphopenia
ALC less than 500 cells/mm3 were reported in RINVOQ-treated patients in clinical trials.
Evaluate lymphocyte counts at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3).
Decreases in hemoglobin levels to less than 8 g/dL were reported in RINVOQ-treated patients in clinical trials.
Evaluate hemoglobin at baseline and thereafter according to routine patient management. Avoid RINVOQ initiation or interrupt RINVOQ treatment in patients with a low hemoglobin level (i.e., less than 8 g/dL).
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol [see Adverse Reactions] Elevations in LDL cholesterol decreased to pre-treatment levels in response to statin therapy. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assess lipid parameters approximately 12 weeks after initiation of treatment, and thereafter according to the clinical guidelines for hyperlipidemia. Manage patients according to clinical guidelines for the management of hyperlipidemia.
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevations compared to treatment with placebo.
Evaluate liver enzymes at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ should be interrupted until this diagnosis is excluded.
Based on findings in animal studies, RINVOQ may cause fetal harm when administered to a pregnant woman. Administration of upadacitinib to rats and rabbits during organogenesis caused increases in fetal malformations. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with RINVOQ and for 4 weeks following completion of therapy [see Use in Specific Populations]
Avoid use of live vaccines during, or immediately prior to, RINVOQ therapy. Prior to initiating RINVOQ, it is recommended that patients be brought up to date with all immunizations, including varicella zoster or prophylactic herpes zoster vaccinations, in agreement with current immunization guidelines.
The following clinically significant adverse reactions are described elsewhere in the labeling:
• Serious Infections [see Warnings and Precautions]
• Mortality [see Warnings and Precautions]
• Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions]
• Major Adverse Cardiovascular Events [see Warnings and Precautions]
• Thrombosis [see Warnings and Precautions]
• Hypersensitivity Reactions [see Warnings and Precautions]
• Gastrointestinal Perforations [see Warnings and Precautions]
• Laboratory Abnormalities [see Warnings and Precautions]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 3833 patients with rheumatoid arthritis were treated with upadacitinib in the Phase 3 clinical trials of whom 2806 were exposed for at least one year.
Patients could advance or switch to RINVOQ 15 mg from placebo, or be rescued to RINVOQ from active comparator or placebo from as early as Week 12 depending on the trial design.
A total of 2630 patients received at least 1 dose of RINVOQ 15 mg, of whom 1860 were exposed for at least one year. In trials RA-I, RA-II, RA-III and RA-V, 1213 patients received at least 1 dose of RINVOQ 15 mg, of which 986 patients were exposed for at least one year, and 1203 patients received at least 1 dose of upadacitinib 30 mg, of which 946 were exposed for at least one year.
Table 1: Adverse Reactions Reported in ≥ 1% of Rheumatoid Arthritis Patients Treated with RINVOQ 15 mg in Placebo-controlled Trials
Adverse Reaction
Placebo RINVOQ 15 mg n=1042 (%) n=1035 (%)
Upper respiratory tract infection (URTI)* 9.5 13.5 Nausea 2.2 3.5
Cough 1.0 2.2 Pyrexia 0 1.2
*URTI includes: acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
Four integrated datasets are presented in the Specific Adverse Reaction section:
Placebo-controlled Trials: Trials RA-III, RA-IV, and RA-V were integrated to represent safety through 12/14 weeks for placebo (n=1042) and RINVOQ 15 mg (n=1035). Trials RA-III and RA-V were integrated to represent safety through 12 weeks for placebo (n=390), RINVOQ 15 mg (n=385), and upadacitinib 30 mg (n=384). Trial RA-IV did not include the 30 mg dose and, therefore, safety data for upadacitinib 30 mg can only be compared with placebo and RINVOQ 15 mg rates from pooling trials RA-III and RA-V.
MTX-controlled Trials: Trials RA-I and RA-II were integrated to represent safety through 12/14 weeks for MTX (n=530), RINVOQ 15 mg (n=534), and upadacitinib 30 mg (n=529).
12-Month Exposure Dataset: Trials RA-I, II, III, and V were integrated to represent the long-term safety of RINVOQ 15 mg (n=1213) and upadacitinib 30 mg (n=1203).
Exposure adjusted incidence rates were adjusted by trial for all the adverse events reported in this section.
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, infections were reported in 218 patients (95.7 per 100 patient-years) treated with placebo and 284 patients (127.8 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, infections were reported in 99 patients (136.5 per 100 patient-years) treated with placebo, 118 patients (164.5 per 100 patient-years) treated with RINVOQ 15 mg, and 126 patients (180.3 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Infections were reported in 127 patients (119.5 per 100 patient-years) treated with MTX monotherapy, 104 patients (91.8 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 128 patients (115.1 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Infections were reported in 615 patients (83.8 per 100 patient-years) treated with RINVOQ 15 mg and 674 patients (99.7 per 100 patient-years) treated with upadacitinib 30 mg.
Serious Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, serious infections were reported in 6 patients (2.3 per 100 patient-years) treated with placebo, and 12 patients (4.6 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, serious infections were reported in 1 patient (1.2 per 100 patientyears) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 7 patients (8.2 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Serious infections were reported in 2 patients (1.6 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 8 patients (6.4 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Serious infections were reported in 38 patients (3.5 per 100 patient-years) treated with RINVOQ 15 mg and 59 patients (5.6 per 100 patient-years) treated with upadacitinib 30 mg.
The most frequently reported serious infections were pneumonia and cellulitis.
Placebo-controlled Trials and MTX-controlled Trials: In the placebo-controlled period, there were no active cases of tuberculosis reported in the placebo, RINVOQ 15 mg, and upadacitinib 30 mg groups. In the MTX-controlled period, there were no active cases of tuberculosis reported in the MTX monotherapy, RINVOQ 15 mg monotherapy, and upadacitinib 30 mg monotherapy groups.
12-Month Exposure Dataset: Active tuberculosis was reported for 2 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg. Cases of extra-pulmonary tuberculosis were reported.
Opportunistic Infections (excluding tuberculosis)
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, opportunistic infections were reported in 3 patients (1.2 per 100 patient-years) treated with placebo, and 5 patients (1.9 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, opportunistic infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (7.1 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Opportunistic infections were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 0 patients
treated with RINVOQ 15 mg monotherapy, and 4 patients (3.2 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Opportunistic infections were reported in 7 patients (0.6 per 100 patient-years) treated with RINVOQ 15 mg and 15 patients (1.4 per 100 patient-years) treated with upadacitinib 30 mg.
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, malignancies excluding NMSC were reported in 1 patient (0.4 per 100 patient-years) treated with placebo, and 1 patient (0.4 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, malignancies excluding NMSC were reported in 0 patients treated with placebo, 1 patient (1.1 per 100 patient-years) treated with RINVOQ 15 mg, and 3 patients (3.5 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Malignancies excluding NMSC were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 0 patients treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Malignancies excluding NMSC were reported in 13 patients (1.2 per 100 patient-years) treated with RINVOQ 15 mg and 14 patients (1.3 per 100 patient-years) treated with upadacitinib 30 mg.
Placebo-controlled Trials: There were no gastrointestinal perforations (based on medical review) reported in patients treated with placebo, RINVOQ 15 mg, and upadacitinib 30 mg.
MTX-controlled Trials: There were no cases of gastrointestinal perforations reported in the MTX and RINVOQ 15 mg group through 12/14 weeks. Two cases of gastrointestinal perforations were observed in the upadacitinib 30 mg group.
12-Month Exposure Dataset: Gastrointestinal perforations were reported in 1 patient treated with RINVOQ 15 mg and 4 patients treated with upadacitinib 30 mg.
Placebo-controlled Trials: In RA-IV, venous thrombosis (pulmonary embolism or deep vein thrombosis) was observed in 1 patient treated with placebo and 1 patient treated with RINVOQ 15 mg. In RA-V, venous thrombosis was observed in 1 patient treated with RINVOQ 15 mg. There were no observed cases of venous thrombosis reported in RA-III. No cases of arterial thrombosis were observed through 12/14 weeks.
MTX-controlled Trials: In RA-II, venous thrombosis was observed in 0 patients treated with MTX monotherapy, 1 patient treated with RINVOQ 15 mg monotherapy and 0 patients treated with upadacitinib 30 mg monotherapy through Week 14. In RA-II, no cases of arterial thrombosis were observed through 12/14 weeks. In RA-I, venous thrombosis was observed in 1 patient treated with MTX, 0 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg through Week 24. In RA-I, arterial thrombosis was observed in 1 patient treated with upadacitinib 30 mg through Week 24.
12-Month Exposure Dataset: Venous thrombosis events were reported in 5 patients (0.5 per 100 patient-years) treated with RINVOQ 15 mg and 4 patients (0.4 per 100 patient-years) treated with upadacitinib 30 mg.
Arterial thrombosis events were reported in 0 patients treated with RINVOQ 15 mg and 2 patients (0.2 per 100 patient-years) treated with upadacitinib 30 mg.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, alanine transaminase (ALT) and aspartate transaminase (AST) elevations ≥ 3 x upper limit of normal (ULN) in at least one measurement were observed in 2.1% and 1.5% of patients treated with RINVOQ 15 mg, and in 1.5% and 0.7% of patients treated with placebo, respectively. In RA-III and RA-V, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 1.0% of patients treated with RINVOQ 15 mg, 1.0% and 0% of patients treated with upadacitinib 30 mg and in 1.3% and 1.0% of patients treated with placebo, respectively.
In MTX-controlled trials, for up to 12/14 weeks, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 0.4% of patients treated with RINVOQ 15 mg, 1.7% and 1.3% of patients treated with upadacitinib 30 mg and in 1.9% and 0.9% of patients treated with MTX, respectively.
Upadacitinib treatment was associated with dose-related increases in total cholesterol, triglycerides and LDL cholesterol. Upadacitinib was also associated with increases in HDL cholesterol. Elevations in LDL and HDL cholesterol peaked by Week 8 and remained stable thereafter. In controlled trials, for up to 12/14 weeks, changes from baseline in lipid parameters in patients treated with RINVOQ 15 mg and upadacitinib 30 mg, respectively, are summarized below:
• Mean LDL cholesterol increased by 14.81 mg/dL and 17.17 mg/dL.
• Mean HDL cholesterol increased by 8.16 mg/dL and 9.01 mg/dL.
• The mean LDL/HDL ratio remained stable.
• Mean triglycerides increased by 13.55 mg/dL and 14.44 mg/dL.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related increases in creatine phosphokinase (CPK) values were observed. CPK elevations > 5 x ULN were reported in 1.0%, and 0.3% of patients over 12/14 weeks in the RINVOQ 15 mg and placebo groups, respectively. Most elevations >5 x ULN were transient and did not require treatment discontinuation. In RA-III and RA-V, CPK elevations > 5 x ULN were observed in 0.3% of patients treated with placebo, 1.6% of patients treated with RINVOQ 15 mg, and none in patients treated with upadacitinib 30 mg.
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in neutrophil counts, below 1000 cells/mm3 in at least one measurement occurred in 1.1% and <0.1% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in neutrophil counts below 1000 cells/mm3 in at least one measurement occurred in 0.3% of patients treated with placebo, 1.3% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg. In clinical trials, treatment was interrupted in response to ANC less than 1000 cells/mm3
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.9%
and 0.7% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.5% of patients treated with placebo, 0.5% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg.
Anemia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, hemoglobin decreases below 8 g/dL in at least one measurement occurred in <0.1% of patients in both the RINVOQ 15 mg and placebo groups. In RA-III and RA-V, hemoglobin decreases below 8 g/dL in at least one measurement were observed in 0.3% of patients treated with placebo, and none in patients treated with RINVOQ 15 mg and upadacitinib 30 mg.
Adverse Reactions in Patients with Psoriatic Arthritis
A total of 1827 patients with psoriatic arthritis were treated with upadacitinib in clinical trials representing 1639.2 patient-years of exposure, of whom 722 were exposed to upadacitinib for at least one year. In the two Phase 3 trials, 907 patients received at least 1 dose of RINVOQ 15 mg, of whom 359 were exposed for at least one year.
Two placebo-controlled trials were integrated (640 patients on RINVOQ 15 mg once daily and 635 patients on placebo) to evaluate the safety of RINVOQ 15 mg in comparison to placebo for up to 24 weeks after treatment initiation.
Overall, the safety profile observed in patients with active psoriatic arthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis. During the 24-week placebo-controlled period, the frequencies of herpes zoster and herpes simplex were ≥1% (1.1% and 1.4%, respectively) with RINVOQ 15 mg and 0.8% and 1.3%, respectively with placebo. A higher incidence of acne and bronchitis was also observed in patients treated with RINVOQ 15 mg (1.3% and 3.9%, respectively) compared to placebo (0.3% and 2.7%, respectively).
Three Phase 3 (AD-1, AD-2, and AD-3) and one Phase 2b (AD-4) randomized, double-blind, placebo-controlled, multicenter trials evaluated the safety of RINVOQ in patients with moderate-to-severe atopic dermatitis. The majority of patients were White (68%) and male (57%). The mean age was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. In these 4 trials, 2612 patients were treated with RINVOQ 15 mg or 30 mg orally once daily, with or without concomitant topical corticosteroids (TCS).
In the Phase 3 clinical trials (AD-1, AD-2, and AD-3), a total of 1239 patients received RINVOQ 15 mg, of whom 791 were exposed for at least one year and 1246 patients received RINVOQ 30 mg, of whom 826 were exposed for at least one year.
Trials AD-1, AD-2, and AD-4 compared the safety of RINVOQ monotherapy to placebo through Week 16. Trial AD-3 compared the safety of RINVOQ + TCS to placebo + TCS through Week 16.
Weeks 0 to 16 (Trials AD-1 to AD-4)
In RINVOQ trials with and without TCS (Trials AD-1, 2, 3 and 4) through Week 16, the proportion of patients who discontinued treatment because of adverse reactions in the RINVOQ 15 mg, 30 mg and placebo groups were 2.3%, 2.9% and 3.8%, respectively. Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% in the RINVOQ 15 mg or 30 mg groups during the first 16 weeks of treatment.
Table 2: Adverse Reactions Reported in ≥ 1% of Patients with Atopic Dermatitis Treated with RINVOQ 15 mg or 30 mg
Adverse Reaction Placebo RINVOQ 15 mg RINVOQ 30 mg n=902 (%) n=899 (%) n=906 (%)
Upper respiratory tract infection (URTI)* 17 23 25
Acne** 2 10 16
Herpes simplex*** 2 4 8 Headache 4 6 6
Increased blood creatine phosphokinase 2 5 6 Cough 1 3 3
Hypersensitivity**** 2 2 3
Folliculitis 1 2 3 Nausea 1 3 3
Abdominal pain***** 1 3 2 Pyrexia 1 2 2
Increased Weight 1 2 2
Herpes zoster****** 1 2 2
Influenza <1 2 2 Fatigue 1 1 2
Neutropenia <1 1 2 Myalgia 1 1 2 Influenza like illness 1 1 2
* Includes: laryngitis, laryngitis viral, nasopharyngitis, oropharyngeal pain, pharyngeal abscess, pharyngitis, pharyngitis streptococcal, pharyngotonsillitis, respiratory tract infection, respiratory tract infection viral, rhinitis, rhinolaryngitis, sinusitis, tonsillitis, tonsillitis bacterial, upper respiratory tract infection, viral pharyngitis, viral upper respiratory tract infection
** Includes: acne and dermatitis acneiform
*** Includes: genital herpes, genital herpes simplex, herpes dermatitis, herpes ophthalmic, herpes simplex, nasal herpes, ophthalmic herpes simplex, herpes virus infection, oral herpes
**** Includes anaphylactic reaction, anaphylactic shock, angioedema, dermatitis exfoliative generalized, drug hypersensitivity, eyelid oedema, face oedema, hypersensitivity, periorbital swelling, pharyngeal swelling, swelling face, toxic skin eruption, type I hypersensitivity, urticaria
***** Includes abdominal pain and abdominal pain upper ****** Includes herpes zoster and varicella
Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg and/or 30 mg group and at a higher rate than in the placebo group through Week 16 included anemia, oral candidiasis, pneumonia, and the adverse event of retinal detachment.
The safety profile of RINVOQ through Week 52 was generally consistent with the safety profile observed at Week 16.
Overall, the safety profile observed in patients with AD treated with RINVOQ was similar to the safety profile in patients with RA. Other specific adverse reactions that were reported in patients with AD included eczema herpeticum/Kaposi’s varicelliform eruption.
Eczema Herpeticum/Kaposi’s Varicelliform Eruption
Placebo-controlled Period (16 weeks): Eczema herpeticum was reported in 4 patients (1.6 per 100 patient-years) treated with placebo, 6 patients (2.2 per 100 patient-years) treated with RINVOQ 15 mg and 7 patients (2.6 per 100 patient-years) treated with RINVOQ 30 mg.
12-Month Exposure (Weeks 0 to 52): Eczema herpeticum was reported in 18 patients (1.6 per 100 patient-years) treated with RINVOQ 15 mg and 17 patients (1.5 per 100 patient-years) treated with RINVOQ 30 mg.
Adverse Reactions in Patients with Ulcerative Colitis
RINVOQ was studied up to 8 weeks in patients with moderately to severely active ulcerative colitis in two randomized, double-blind, placebo-controlled induction studies (UC-1, UC-2) and a randomized, double-blind, placebo controlled, dose-finding study (UC-4; NCT02819635). Long term safety up to 52-weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (UC-3) and a long-term extension study.
In the two induction studies (UC-1, UC-2) and a dose finding study (UC-4), 1097 patients were enrolled of whom 719 patients received RINVOQ 45 mg once daily.
In the maintenance study (UC-3), 746 patients were enrolled of whom 250 patients received RINVOQ 15 mg once daily and 251 patients received RINVOQ 30 mg once daily.
Adverse reactions reported in ≥2% of patients in any treatment arm in the induction and maintenance studies are shown in Tables 3 and 4, respectively.
Table 3. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (UC-1, UC-2 and UC-4)
Adverse Reaction
Placebo RINVOQ 45 mg Once Daily N= 378 (%) N = 719 (%)
Upper respiratory tract infection* 7 9
Acne* 1 6
Increased blood creatine phosphokinase 1 5
Neutropenia* <1 5
Rash* 1 4
Elevated liver enzymes** 2 3
Lymphopenia* 1 3 Folliculitis 1 2
Herpes simplex* <1 2
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury and cholestasis.
Other adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 8 included herpes zoster and pneumonia.
Table 4. Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (UC-3)1
Adverse Reaction Placebo RINVOQ 15 mg Once Daily RINVOQ 30 mg Once Daily n = 245 (%) n = 250 (%) n = 251 (%)
Upper respiratory tract infection* 18 16 20
Increased blood creatine phosphokinase 2 6 8
Neutropenia* 2 3 6
Elevated liver enzymes** 1 6 4 Rash* 4 5 5
Herpes zoster 0 4 4
Folliculitis 2 2 4
Hypercholesterolemia* 1 2 4 Influenza 1 3 3
Herpes simplex* 1 2 3
Lymphopenia* 2 3 2
Hyperlipidemia* 0 2 2
1 Patients who were responders to 8 weeks induction therapy with RINVOQ 45 mg once daily
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury, and cholestasis.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Overall, the safety profile observed in patients with ulcerative colitis treated with RINVOQ was generally similar to the safety profile in patients with RA and AD.
Induction Studies: In UC-1, UC-2, and UC-4, serious infections were reported in 5 patients (8.4 per 100 patient-years) treated with placebo and 9 patients (8.4 per 100 patient-years) treated with RINVOQ 45 mg through 8 weeks.
Placebo-controlled Maintenance Study: In UC-3, serious infections were reported in 8 patients (6.3 per 100 patient-years) treated with placebo, 8 patients (4.5 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (3.1 per 100 patient-years) treated with RINVOQ 30 mg through 52 weeks.
In studies UC-1, UC-2, and UC-4, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 1.5% of patients treated with RINVOQ 45 mg, and 0% of patients treated with placebo for 8 weeks. AST elevations to ≥ 3 x ULN occurred in 1.5% of patients treated with RINVOQ 45 mg, and 0.3% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN occurred in 0.4% of patients treated with RINVOQ 45 mg and 0% of patients treated with placebo.
In UC-3, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 4% of patients treated with RINVOQ 30 mg, 2% of patients treated with RINVOQ 15 mg, and 0.8% of patients treated with placebo for 52 weeks. Elevations of AST to ≥ 3 x ULN in at least one measurement were observed in 2% of patients treated with RINVOQ 30 mg, 1.6% of patients treated with RINVOQ 15 mg and 0.4% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN were observed in 0.8% of patients treated with 30 mg, 0.4% of patients treated with 15 mg, and 0.4% of patients treated with placebo.
Overall, laboratory abnormalities observed in patients with ulcerative colitis treated with RINVOQ were similar to those described in patients with RA.
Adverse Reactions in Patients with Ankylosing Spondylitis
A total of 596 patients with ankylosing spondylitis were treated with RINVOQ 15 mg in the two clinical trials representing 577.3 patient-years of exposure, of whom 228 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active ankylosing spondylitis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis and psoriatic arthritis.
During the 14-week placebo-controlled period in Trial AS-I, the frequency of headache was 5.4% with RINVOQ 15 mg and 2.1% with placebo. During the 14-week placebo-controlled period in Trial AS-II, the frequency of headache was 3.3% with RINVOQ 15 mg and 1.4% with placebo.
Strong CYP3A4 Inhibitors
Upadacitinib exposure is increased when RINVOQ is co-administered with a strong CYP3A4 inhibitor (such as ketoconazole and clarithromycin), which may increase the risk of RINVOQ adverse reactions. Monitor patients closely for adverse reactions when co-administering RINVOQ 15 mg once daily with strong CYP3A4 inhibitors.
For patients with atopic dermatitis, coadministration of RINVOQ 30 mg once daily with strong CYP3A4 inhibitors is not recommended.
For patients with ulcerative colitis taking strong CYP3A4 inhibitors, reduce the RINVOQ induction dosage to 30 mg once daily. The recommended maintenance dosage is 15 mg once daily.
Strong CYP3A4 Inducers
Upadacitinib exposure is decreased when RINVOQ is co-administered with strong CYP3A4 inducers (such as rifampin), which may lead to reduced therapeutic effect of RINVOQ. Coadministration of RINVOQ with strong CYP3A4 inducers is not recommended.
Available data from the pharmacovigilance safety database and postmarketing case reports on use of RINVOQ in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. Based on animal studies, RINVOQ has the potential to adversely affect a developing fetus. Advise patients of reproductive potential and pregnant patients of the potential risk to the fetus.
In animal embryo-fetal development studies, oral upadacitinib administration to pregnant rats and rabbits at exposures equal to or greater than approximately 1.6 and 15 times the 15 mg dose, 0.8 and 7.6 times the 30 mg dose, and 0.6 and 5.6 times the maximum recommended human dose (MRHD) of 45 mg (on an AUC basis) resulted in dose-related increases in skeletal malformations (rats only), an increased incidence of cardiovascular malformations (rabbits only), increased post-implantation loss (rabbits only), and decreased fetal body weights in both rats and rabbits. No developmental toxicity was observed in pregnant rats and rabbits treated with oral upadacitinib during organogenesis at exposures approximately 0.29 and 2.2 times the 15 mg dose, 0.15 times and 1.1 times the 30 mg dose, and at 0.11 and 0.82 times the MHRD (on an AUC basis).
In a pre- and post-natal development study in pregnant female rats, oral upadacitinib administration at exposures approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and the same as the MRHD (on an AUC basis) resulted in no maternal or developmental toxicity (see Data)
The background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages are 2-4% and 15-20%, respectively.
Report pregnancies to the AbbVie Inc.’s Adverse Event reporting line at 1-888-633-9110, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
In an oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 5, 25, and 75 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that consisted of misshapen humerus and bent scapula) at exposures equal to or greater than approximately 1.7 times the
15 mg dose, 0.9 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 5 mg/kg/day and higher). Additional skeletal malformations (bent forelimbs/hindlimbs and rib/vertebral defects) and decreased fetal body weights were observed in the absence of maternal toxicity at an exposure approximately 84 times the 15 mg dose, 43 times the 30 mg dose, and 31 times the MRHD (on an AUC basis at a maternal oral dose of 75 mg/kg/day).
In a second oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 1.5 and 4 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that included bent humerus and scapula) at exposures approximately 1.6 times the 15 mg dose, 0.8 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 4 mg/kg/day). No developmental toxicity was observed in rats at an exposure approximately 0.29 times the 15 mg dose, 0.15 times the 30 mg dose, and 0.11 times the MRHD (on an AUC basis at a maternal oral dose of 1.5 mg/kg/day).
In an oral embryo-fetal developmental study, pregnant rabbits received upadacitinib at doses of 2.5, 10, and 25 mg/kg/day during the period of organogenesis from gestation day 7 to 19. Embryolethality, decreased fetal body weights, and cardiovascular malformations were observed in the presence of maternal toxicity at an exposure approximately 15 times the 15 mg dose, 7.6 times the 30 mg dose, and 5.6 times the MRHD (on an AUC basis at a maternal oral dose of 25 mg/kg/day). Embryolethality consisted of increased post-implantation loss that was due to elevated incidences of both total and early resorptions. No developmental toxicity was observed in rabbits at an exposure approximately 2.2 times the 15 mg dose, 1.1 times the 30 mg dose, and 0.82 times the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
In an oral pre- and post-natal development study, pregnant female rats received upadacitinib at doses of 2.5, 5, and 10 mg/kg/day from gestation day 6 through lactation day 20. No maternal or developmental toxicity was observed in either mothers or offspring, respectively, at an exposure approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and at approximately the same exposure as the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
There are no data on the presence of upadacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Available pharmacodynamic/toxicological data in animals have shown excretion of upadacitinib in milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with RINVOQ, and for 6 days (approximately 10 half-lives) after the last dose.
A single oral dose of 10 mg/kg radiolabeled upadacitinib was administered to lactating female Sprague-Dawley rats on post-partum days 7-8. Drug exposure was approximately 30-fold greater in milk than in maternal plasma based on AUC0-t values. Approximately 97% of drug-related material in milk was parent drug.
Females and Males of Reproductive Potential Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ [see Use in Specific Populations]
Based on animal studies, upadacitinib may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations]. Advise female patients of reproductive potential to use effective contraception during treatment with RINVOQ and for 4 weeks after the final dose.
Juvenile Idiopathic Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The safety and effectiveness of RINVOQ in pediatric patients with juvenile idiopathic arthritis, psoriatic arthritis, and ankylosing spondylitis have not been established.
The safety and effectiveness of RINVOQ in pediatric patients 12 years of age and older weighing at least 40 kg with atopic dermatitis have been established. A total of 344 pediatric patients aged 12 to 17 years with moderate to severe atopic dermatitis were randomized across three trials (AD-1, AD-2 and AD-3) to receive either RINVOQ 15 mg (N=114) or 30 mg (N=114) or matching placebo (N=116) in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the pediatric patients and adults. The adverse reaction profile in the pediatric patients was similar to the adults [see Adverse Reactions].
The safety and effectiveness of RINVOQ in pediatric patients less than 12 years of age with atopic dermatitis have not been established.
The safety and effectiveness of RINVOQ in pediatric patients with ulcerative colitis have not been established.
Rheumatoid Arthritis and Psoriatic Arthritis
Of the 4381 patients treated in the five clinical trials, a total of 906 rheumatoid arthritis patients were 65 years of age or older, including 146 patients 75 years and older. Of the 1827 patients treated in the two psoriatic arthritis Phase 3 clinical trials, a total of 274 patients were 65 years of age or older, including 34 patients 75 years and older. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of overall adverse events, including serious infections, in patients 65 years of age and older.
Of the 2583 patients treated in the three Phase 3 clinical trials, a total of 120 patients with atopic dermatitis were 65 years of age or older, including 6 patients 75 years of age. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of serious infections and malignancies in those patients 65 years of age or older in the 30 mg dosing group in the long-term trials.
Of the 1097 patients treated in the controlled clinical trials, a total of 95 patients with ulcerative colitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ulcerative colitis to determine whether they respond differently from younger adult patients.
For patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), or severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2).
For patients with atopic dermatitis, the maximum recommended dosage is 15 mg once daily for patients with severe renal impairment. No dosage adjustment is needed in patients with mild or moderate renal impairment. For patients with ulcerative colitis, the recommended dosage for severe renal impairment is 30 mg once daily for induction and 15 mg once daily for maintenance. No dosage adjustment is needed in patients with mild or moderate renal impairment.
RINVOQ has not been studied in patients with end stage renal disease (eGFR <15 mL/min/1.73m2). Use in patients with atopic dermatitis or ulcerative colitis with end stage renal disease is not recommended.
The use of RINVOQ has not been studied in patients with severe hepatic impairment (Child Pugh C), and therefore not recommended for use in patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ulcerative colitis, or ankylosing spondylitis.
For patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, and ankylosing spondylitis, no dosage adjustment is needed in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment.
For patients with ulcerative colitis, the recommended dosage for mild to moderate hepatic impairment is 30 mg once daily for induction and 15 mg once daily for maintenance
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients that they may be more likely to develop infections when taking RINVOQ. Instruct patients to contact their healthcare provider immediately during treatment if they develop any signs or symptoms of an infection [see Warnings and Precautions]
Advise patients that the risk of herpes zoster is increased in patients taking RINVOQ and in some cases can be serious [see Warnings and Precautions]
Inform patients that RINVOQ may increase their risk of certain cancers and that periodic skin examinations should be performed while using RINVOQ. Advise patients that exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen [see Warnings and Precautions]
Major Adverse Cardiovascular Events
Inform patients that RINVOQ may increase their risk of major adverse cardiovascular events (MACE) including myocardial infarction, stroke,
and cardiovascular death. Instruct all patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see Warnings and Precautions]
Inform patients that events of deep venous thrombosis and pulmonary embolism have been reported in clinical trials with RINVOQ. Instruct patients to seek immediate medical attention if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions]
Hypersensitivity Reactions
Advise patients to discontinue RINVOQ and seek immediate medical attention if they develop any signs and symptoms of allergic reactions [see Warnings and Precautions].
Inform patients that gastrointestinal perforations have been reported in clinical trials with RINVOQ and that risk factors include the use of NSAIDS or history of diverticulitis. Instruct patients to seek medical care immediately if they experience new onset of abdominal pain, fever, chills, nausea, or vomiting [see Warnings and Precautions].
Retinal Detachment
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ [see Adverse Reactions]
Inform patients that RINVOQ may affect certain lab tests, and that blood tests are required before and during RINVOQ treatment [see Warnings and Precautions]
Vaccinations
Advise patients to avoid use of live vaccines with RINVOQ. Instruct patients to inform their healthcare practitioner that they are taking RINVOQ prior to a potential vaccination [see Warnings and Precautions]
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to RINVOQ during pregnancy may result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions and Use in Specific Populations]
Advise females of reproductive potential that effective contraception should be used during treatment and for 4 weeks following the final dose of upadacitinib [see Use in Specific Populations]
Advise females patients who are exposed to RINVOQ during pregnancy to contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Advise women not to breastfeed during treatment with RINVOQ and for 6 days after the last dose [see Use in Specific Populations]
Administration
Advise patients not to chew, crush, or split RINVOQ tablets.
Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd. ©2019-2022 AbbVie Inc.
Ref: 20071734 Revised: April 2022
LAB-7082 MASTER
US-RNQR-220149

Ivette S. Rivera Martínez Ph.D. Psicología Industrial Organizacional Pontificia Universidad Católica de Puerto Rico, Ponce

PALABRAS
Estrés, Inteligencia Emocional, Médicos

Stress, Emotional Intelligence, Physicians
Esta investigación pretendió analizar la relación entre el estrés y la inteligencia emocional en una muestra de médicos de la región sur de Puerto Rico. La investigación cuantitativa quedó constituida por 115 participantes. Se encontró una relación estadísticamente significativa entre la inteligencia emocional y el estrés. Igualmente se encontró diferencia estadísticamente significativa entre los médicos de ambos géneros y el estrés. Contrario a lo esperado, no se halló diferencia estadísticamente significativa entre los médicos de ambos géneros y la inteligencia emocional. Se estableció un nivel de significancia de .05, encontrándose niveles de correlación rho de Spearman en .001. El valor obtenido producto de la correlación fue de -.335. El estudio sirve como herramienta para poder comprender mejor las variables estudiadas en la población médica puertorriqueña. Se evidencia la importancia de un buen control de las emociones para mantener reducidos los niveles de estrés percibido. Para futuras investigaciones se recomienda entre otras cosas, proveer una mayor representatividad de la muestra.
This research aimed to analyze the relationship between stress and emotional intelligence in a sample of physicians from the southern region of Puerto Rico. The quantitative research was made up of 115 participants. A statistically significant relationship was found between emotional intelligence and stress. Likewise, a statistically significant difference was between the physicians of both genders and stress. Contrary to expectations, no statistically significant difference was found between physicians of both genders and emotional intelligence. A significance level of .05 was established, finding Spearman’s rho correlation levels at .001. The value obtained as a product of the correlation was -.335. The study serves as a tool to better understand the variables studied in the Puerto Rican medical population. The importance of good control of emotions to keep levels of perceived stress low is evidenced. For future research it is recommended among other things, to provide a greater representativeness of the sample.
El propósito de esta investigación fue analizar la relación entre el estrés y la inteligencia emocional en una muestra de médicos. El estudio conllevó la oportunidad de que los individuos pudiesen identificar con qué frecuencia se han sentido estresados, particularmente durante el último mes. Igualmente, permitió conocer la frecuencia con la cual el individuo siente que ha sido capaz de afrontar sus problemas.

La investigación cuantitativa, contó con la participación de 115 médicos afiliados a una organización sin fines de lucro de la región sur de Puerto Rico que brinda educación continua a médicos con licencia vigente. Los criterios de inclusión fueron: ser mayor de 21 años, de ambos géneros y llevar al menos tres meses o más ejerciendo la profesión de la Medicina en Puerto Rico entre los cuales podían encontrarse médicos/médicos residentes. Recibida la aprobación de la Junta de Revisión Institucional (Institutional Review Board, IRB), se inició la investigación. El proceso fue realizado de manera virtual
entre octubre de 2020 a febrero de 2021 utilizando la plataforma PsychData. En primer lugar, se encontraba la Hoja de Consentimiento Informado, documento que explicaba a los posibles participantes el propósito de la investigación, la voluntariedad de su participación y la garantía de su anonimato a tenor con el Código de Ética que rige la profesión de la Psicología en Puerto Rico. Igualmente, se informó a los participantes acerca del respeto a la dignidad y la confidencialidad que se mantendría durante el estudio y posterior al mismo. Luego se encontraba la Hoja de Datos Sociodemográficos, donde se recopiló
información de variables relacionadas con los participantes en el estudio. A este instrumento le seguía la Escala de Estrés Percibido (Versión Adaptada, EEP-14) de Cohen et al. en 1983 y traducida al español por Remor en 2006. Por último, se encontraba el Inventario de Inteligencia Emocional (Versión Corta, IIE-20) de Andújar en 2005.
Para el análisis estadístico de resultados se utilizó la base de datos IBM SPSS Statistics versión 27. Los resultados de esta investigación están disponibles en la Biblioteca Encarnación Valdés de la Pontificia Universidad Católica, Ponce.
Análisis Sociodemográfico. Los hallazgos reflejaron que 61 (53%) de los participantes fueron mujeres, mientras 54 (47%) fueron hombres. Además, 69 (60%) de los participantes tienen más de 51 años. Un total de 83 (72%) de los participantes indicó estar casado (a). De los cuales, 85 (74%) de los participantes opinó tener hijo (a) o dependientes. En cuanto a la especialidad de los participantes, se obtuvo que 38 (33%) de los médicos que participaron de la investigación ejercen la medicina general. De igual manera 58 (50%) de los médicos reportaron poseen otra especialidad. Según los hallazgos 78 (68%) de los participantes tienen más de 18 años de servicio en la Medicina. Además, 67 (58%) de los participantes cuenta con oficina privada para ejercer sus funciones en Medicina. Finalmente, se encuestó si es catedrático en la universidad. El resultado fue que 71 (62%) de los médicos opinó que no.
Escala de Estrés Percibido. Con relación a las preguntas de la 1 a la 7, se observó que el resultado de mayor porcentaje ante muy a menudo fue: en el último mes, ¿con qué frecuencia se ha sentido nervioso o estresado? (12%, ƒ = 13). Sin embargo, hay que resaltar que los participantes presentaron nunca estar estresado en cuanto en el último mes, ¿con qué frecuencia ha estado seguro sobre su capacidad para manejar sus problemas personales? (66%, ƒ = 61). Las preguntas 8 a la 14 completaron con la indagación sobre el estrés percibido que presentaron
los participantes. Entre los resultados de mayor porcentaje se destacan, en el último mes, ¿con que frecuencia ha pensado sobre las cosas que le quedan por hacer? (44%, ƒ = 47). Además, se observó que los médicos nunca perciben estrés en el último mes en la frecuencia para controlar las dificultades de su vida (49%, ƒ = 53).
Inventario de Inteligencia Emocional. Con relación a los resultados relacionados con la escala de Autoconocimiento, la premisa de mayor porcentaje en totalmente de acuerdo fue las personas deben ser capaces de conocer sus emociones (68%, ƒ = 70). Sobre la escala de Automanejo, la premisa de mayor porcentaje en totalmente de acuerdo fue uno debe tratar de dar el máximo en lo que hace (87%, ƒ = 90). En cuanto a la escala de Conciencia Emocional, los resultados reflejaron que la premisa de mayor porcentaje en totalmente de acuerdo fue disfruto ayudar a que otros crezcan (86%, ƒ = 89). Los resultados relacionados a la escala de Manejo de las Relaciones, mostraron que la premisa de mayor porcentaje en totalmente de acuerdo fue un líder respeta a su gente y NO abusa de su poder (91%, ƒ = 94).
Preguntas de Investigación. La primera pregunta tuvo como propósito discutir la relación entre el estrés y la inteligencia emocional. De acuerdo con los resultados de la prueba estadística, Spearman (N = 108) = -.335, p = .001. Este valor de p, si se compara a un nivel de significancia de 0.05 (p < 0.05) establece que se rechaza la hipótesis nula. Por tanto, existe relación estadísticamente significativa entre el estrés y la inteligencia emocional. Con relación a la segunda pregunta de investigación, la misma tuvo como propósito discutir la diferencia entre los médicos de ambos géneros y el estrés que perciben de las situaciones de la vida cotidiana, mediante la prueba no paramétrica Mann-Whitney. Los resultados indicaron un rango promedio en los masculinos de 45.52 y en las féminas un rango promedio de 62.24. El valor de la significancia de la prueba fue de p = .006 < 0.05, por lo tanto, se determinó rechazar la hipótesis nula, y se encontró
que existe diferencia estadísticamente significativa entre los médicos de ambos géneros y el estrés que perciben de las situaciones de la vida cotidiana a un nivel de .05 de significancia. Por último, la tercera pregunta de investigación, tuvo como propósito discutir la diferencia entre los médicos de ambos géneros y las competencias de inteligencia emocional (autoconocimiento, automanejo, conciencia emocional y manejo de las relaciones) que poseen, mediante la prueba no paramétrica de Mann-Whitney. Los resultados indicaron que los rangos promedios en las competencias de inteligencia emocional no difieren significativamente. El valor de la significancia de la prueba en las cuatro competencias fue > 0.05, por lo tanto, se determinó no rechazar la hipótesis nula, y se encontró que no existe diferencia estadísticamente significativa entre los médicos de ambos géneros y las competencias de inteligencia emocional que poseen a un nivel de .05 de significancia.
Se encontró que mientras más fortalecida está la inteligencia emocional existe mayor probabilidad de que los niveles de estrés sean bajos. Esto evidencia la importancia de que las organizaciones puedan establecer programas de control de emociones que vayan dirigidos a fomentar una buena salud mental entre sus empleados, en este caso en particular, los médicos. Para esto, se pueden establecer protocolos que vayan dirigidos a crear alianzas con entidades que trabajen el aspecto de la salud mental y creen una línea de apoyo 24/7 con la intención de que los médicos puedan manifestar incomodidades ante situaciones que le provoquen estrés. Lo anterior pudiese representar una apertura para que desde la disciplina de la Psicología Industrial Organizacional se pueda ayudar a atender la complicada situación del éxodo de médicos fuera del País, buscando alternativas que colaboren a retener el talento médico puertorriqueño en las organizaciones.
Revista Puertorriqueña de Medicina y Salud
by Gastroenterologists, Primary Care Physicians, Nurse Practitioners, and Physician Assistants5
Based on the FDA-approved labels, CREON is the only PERT specifically indicated for patients with EPI due to chronic pancreatitis and pancreatectomy, in addition to cystic fibrosis and other conditions.
Based on the FDA-approved labels, CREON is the only PERT specifically indicated for patients with EPI due to chronic pancreatitis and pancreatectomy, in addition to cystic fibrosis and other conditions.
Nearly 9 out of 10 Gastroenterologists prescribe CREON more than other PERTs (pancreatic enzyme replacement therapies)5*
Nearly 9 out of 10 Gastroenterologists prescribe CREON more than other PERTs (pancreatic enzyme replacement therapies)5*
References: 1. CREON [package insert]. AbbVie Inc. 2. Whitcomb DC, Lehman GA, Vasileva G, et al. Pancrelipase delayed-release capsules (CREON) for exocrine pancreatic insuf
References: 1. CREON [package insert]. AbbVie Inc. 2. Whitcomb DC, Lehman GA, Vasileva G, et al. Pancrelipase delayed-release capsules (CREON) for exocrine pancreatic insuf ciency due to chronic pancreatitis or pancreatic surgery: a double-blind randomized trial. Am J Gastroenterol. 2010;105(10):2276-2286. 3. IMS Health Xponent Prescribing Data. November 2021. 4. Data on le. AbbVie Inc. Managed Markets Insight & Technology LLC. November 2019 5. IMS Health, IMS National Prescription Audit, 10/02/2020
• Porcine-derived pancreatic enzyme products contain purines. Caution should be exercised when prescribing CREON to patients with gout, renal impairment, or hyperuricemia.
• Porcine-derived pancreatic enzyme products contain purines. Caution should be exercised when prescribing CREON to patients with gout, renal impairment, or hyperuricemia.
• There is theoretical risk of viral transmission with all pancreatic enzyme products including CREON.
• There is theoretical risk of viral transmission with all pancreatic enzyme products including CREON.
• Exercise caution when administering pancrelipase to a patient with a known allergy to proteins of porcine origin.
• Exercise caution when administering pancrelipase to a patient with a known allergy to proteins of porcine origin.
• were vomiting, dizziness, and cough.
• were vomiting, dizziness, and cough.
• Adverse reactions that occurred in at least 1 chronic pancreatitis or pancreatectomy patient (greater than or equal bowel movements, and nasopharyngitis.
• Adverse reactions that occurred in at least 1 chronic pancreatitis or pancreatectomy patient (greater than or equal bowel movements, and nasopharyngitis.
• CREON is not interchangeable with any other pancrelipase product.
• CREON is not interchangeable with any other pancrelipase product.
Please see the following pages for Brief Summary of Full Prescribing Information.
Please see the following pages for Brief Summary of Full Prescribing Information.
´
conditions.
DOSAGE AND ADMINISTRATION
Fibrosing Colonopathy
CREON is not interchangeable with other pancrelipase products.
CREON is orally administered. Therapy should be initiated at the lowest recommended dose and gradually increased. The dosage of CREON should be individualized based on clinical symptoms, the degree of steatorrhea present, and the fat content of the diet as described in the Limitations on Dosing below [see Dosage and Administration and Warnings and Precautions]
Administration
Infants (up to 12 months)
CREON should be administered to infants immediately prior to each feeding, using a dosage of 3,000 lipase units per 120 mL of formula or prior to breastfeeding. Contents of the capsule may be administered directly to the mouth or with a small amount of applesauce. Administration should be followed by breast milk or formula. Contents of the capsule should not be should be taken to ensure that CREON is not crushed or chewed or retained in the mouth, to avoid irritation of the oral mucosa.
Children and Adults
CREON
capsules and capsule contents should not be crushed or chewed. Capsules should be swallowed whole.
For patients who are unable to swallow intact capsules, the capsules may be carefully opened and the contents added to a small amount of acidic soft food with a pH of 4.5 or less, such as applesauce, at room temperature. The CREON-soft food mixture should be swallowed immediately without crushing or chewing, and followed with water or juice to ensure complete ingestion. Care should be taken to ensure that no drug is retained in the mouth.
Dosage recommendations for pancreatic enzyme replacement therapy were published following the Cystic Fibrosis Foundation Consensus Conferences. CREON should be administered in a manner consistent with the recommendations of the Cystic Fibrosis Foundation Consensus Conferences (also known as Conferences) provided in the following paragraphs, except for infants. Although the Conferences recommend doses of 2,000 to 4,000 lipase units in infants up to 12 months, CREON is available in a 3,000 lipase unit capsule. Therefore, the recommended dose of CREON in infants up to 12 months is 3,000 lipase units per 120 mL of formula or per breastfeeding. Patients may be dosed on a fat ingestion-based or actual body weight-based dosing scheme.
Additional recommendations for pancreatic enzyme therapy in patients pancreatectomy are based on a clinical trial conducted in these populations.
Infants (up to 12 months)
CREON is available in the strength of 3,000 USP units of lipase thus infants may be given 3,000 lipase units (one capsule) per 120 mL of formula or per breastfeeding. Do not mix CREON capsule contents directly into formula or breast milk prior to administration [see Administration]
Children Older than 12 Months and Younger than 4 Years
Enzyme dosing should begin with 1,000 lipase units/kg of body weight units/kg of body weight per meal (or less than or equal to 10,000 lipase units/kg of body weight per day), or less than 4,000 lipase units/g fat ingested per day.
Children 4 Years and Older and Adults
Enzyme dosing should begin with 500 lipase units/kg of body weight per meal
body weight per meal (or less than or equal to 10,000 lipase units/kg of body weight per day), or less than 4,000 lipase units/g fat ingested per day.
Usually, half of the prescribed CREON dose for an individualized full meal
approximately three meals plus two or three snacks per day.
Enzyme doses expressed as lipase units/kg of body weight per meal should be decreased in older patients because they weigh more but tend to ingest less fat per kilogram of body weight.
The initial starting dose and increases in the dose per meal should be individualized based on clinical symptoms, the degree of steatorrhea present, and the fat content of the diet.
In one clinical trial, patients received CREON at a dose of 72,000 lipase units per meal while consuming at least 100 g of fat per day]. Lower starting doses recommended in the literature are consistent with the 500 lipase units/kg of body weight per meal lowest starting dose recommended for adults in the Cystic Fibrosis Foundation Consensus Conferences Guidelines. Usually, half of the prescribed CREON dose for an individualized full meal should be given with each snack.
Dosing should not exceed the recommended maximum dosage set forth by the Cystic Fibrosis Foundation Consensus Conferences Guidelines. If symptoms and signs of steatorrhea persist, the dosage may be increased by the healthcare professional. Patients should be instructed not to increase the dosage on their own. There is great inter-individual variation in response to enzymes; thus, a range of doses is recommended. Changes in dosage may require an adjustment period of several days. If doses are to exceed 2,500 lipase units/kg of body weight per meal, further investigation is warranted. Doses greater than 2,500 lipase units/kg of body weight per meal (or greater than 10,000 lipase units/kg of body weight per day) should be used with caution and only if they are documented to be effective by of fat absorption. Doses greater than 6,000 lipase units/kg of body weight
colonopathy, in children less than 12 years of age [see Warnings and Precautions]. Patients currently receiving higher doses than 6,000 lipase units/kg of body weight per meal should be examined and the dosage either immediately decreased or titrated downward to a lower range.
CONTRAINDICATIONS
None.
Fibrosing colonopathy has been reported following treatment with different pancreatic enzyme products. Fibrosing colonopathy is a rare, serious adverse reaction initially described in association with high-dose pancreatic enzyme use, usually over a prolonged period of time and most commonly
products exceeding 6,000 lipase units/kg of body weight per meal have been associated with colonic stricture in children less than 12 years of age.
some patients may be at risk of progressing to stricture formation. It is recommended, unless clinically indicated, that enzyme doses should be less than 2,500 lipase units/kg of body weight per meal (or less than 10,000 lipase units/kg of body weight per day) or less than 4,000 lipase units/g fat ingested per day [see Dosage and Administration].
Doses greater than 2,500 lipase units/kg of body weight per meal (or greater than 10,000 lipase units/kg of body weight per day) should be used with caution and only if they are documented to be effective by 3-day fecal fat
Patients receiving higher doses than 6,000 lipase units/kg of body weight per meal should be examined and the dosage either immediately decreased or titrated downward to a lower range.
Potential for Irritation to Oral Mucosa
Care should be taken to ensure that no drug is retained in the mouth. CREON should not be crushed or chewed or mixed in foods having a pH greater than 4.5. These actions can disrupt the protective enteric coating resulting in early release of enzymes, irritation of oral mucosa, and/or loss of enzyme activity [see Dosage and Administration and Patient Counseling Information]
For patients who are unable to swallow intact capsules, the capsules may be carefully opened and the contents added to a small amount of acidic soft food with a pH of 4.5 or less, such as applesauce, at room temperature. The CREON-soft food mixture should be swallowed immediately and followed with water or juice to ensure complete ingestion.
Potential for Risk of Hyperuricemia
Caution should be exercised when prescribing CREON to patients with gout, renal impairment, or hyperuricemia. Porcine-derived pancreatic enzyme products contain purines that may increase blood uric acid levels.
Potential Viral Exposure from the Product Source
CREON is sourced from pancreatic tissue from swine used for food consumption. Although the risk that CREON will transmit an infectious agent to humans has been reduced by testing for certain viruses during manufacturing and by inactivating certain viruses during manufacturing, there is a theoretical risk for transmission of viral disease, including diseases
of transmission of an infectious illness associated with the use of porcine pancreatic extracts have been reported.
Caution should be exercised when administering pancrelipase to a patient with a known allergy to proteins of porcine origin. Rarely, severe allergic reactions including anaphylaxis, asthma, hives, and pruritus, have been reported with other pancreatic enzyme products with different formulations of
CREON treatment in patients with severe allergy should be taken into consideration with the overall clinical needs of the patient.
The most serious adverse reactions reported with different pancreatic enzyme products of the same active ingredient (pancrelipase) that are described
allergic reactions [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly the rates observed in practice.
The short-term safety of CREON was assessed in clinical trials conducted in
or pancreatectomy were treated with CREON.
Cystic Fibrosis
Studies 1 and 2 were randomized, double-blind, placebo-controlled, crossover studies of 49 patients, ages 7 to 43 years, with EPI due to CF. Study 1 included 32 patients ages 12 to 43 years and Study 2 included 17 patients ages 7 to 11 years. In these studies, patients were randomized to receive CREON at a dose of 4,000 lipase units/g fat ingested per day or matching placebo for 5 to 6 days of treatment, followed by crossover to the alternate treatment for an additional 5 to 6 days. The mean exposure to CREON during these studies was 5 days.
In Study 1, one patient experienced duodenitis and gastritis of moderate severity 16 days after completing treatment with CREON. Transient neutropenia without clinical sequelae was observed as an abnormal
In Study 2, adverse reactions that occurred in at least 2 patients (greater than or equal to 12%) treated with CREON were vomiting and headache. Vomiting occurred in 2 patients treated with CREON and did not occur in patients treated with placebo; headache occurred in 2 patients treated with CREON and did not occur in patients treated with placebo.
The most common adverse reactions (greater than or equal to 4%) in Studies 1 and 2 were vomiting, dizziness, and cough. Table 1 enumerates adverse reactions that occurred in at least 2 patients (greater than or equal to 4%) treated with CREON at a higher rate than with placebo in Studies 1 and 2. than or equal to 4%) in Cystic Fibrosis (Studies 1 and 2)
Adverse Reaction CREON Capsules n = 49 (%) Placebo n = 47 (%) 3 (6) 1 (2) 2 (4) 1 (2) 2 (4) 0
An additional open-label, single-arm study assessed the short-term safety and tolerability of CREON in 18 infants and children, ages 4 months to pancreatic enzyme replacement therapy (mean dose of 7,000 lipase units/kg/day for a mean duration of 18.2 days) followed by CREON (mean dose of 7,500 lipase units/kg/day for a mean duration of 12.6 days). There were no serious adverse reactions. Adverse reactions that occurred in patients during treatment with CREON were vomiting, irritability, and decreased appetite, each occurring in 6% of patients.
Chronic Pancreatitis or Pancreatectomy
A randomized, double-blind, placebo-controlled, parallel group study was conducted in 54 adult patients, ages 32 to 75 years, with EPI due to chronic pancreatitis or pancreatectomy. Patients received single-blind placebo treatment during a 5-day run-in period followed by an intervening period of up to 16 days of investigator-directed treatment with no restrictions on pancreatic enzyme replacement therapy. Patients were then randomized to receive CREON or matching placebo for 7 days. The CREON dose was 72,000 lipase units per main meal (3 main meals) and 36,000 lipase units per snack (2 snacks). The mean exposure to CREON during this study was 6.8 days in the 25 patients that received CREON.
The most common adverse reactions reported during the study were related to glycemic control and were reported more commonly during CREON treatment than during placebo treatment.
Table 2 enumerates adverse reactions that occurred in at least 1 patient (greater than or equal to 4%) treated with CREON at a higher rate than with placebo.
Table 2: Adverse Reactions in at Least 1 Patient (greater than or equal to 4%) in the Chronic Pancreatitis or Pancreatectomy Trial
Adverse Reaction CREON Capsules n = 25 (%) Placebo n = 29 (%)
2 (8) 2 (7)
1 (4) 1 (3)
1 (4) 1 (3)
1 (4) 0 1 (4) 0 1 (4) 0 1 (4) 0
Postmarketing data from this formulation of CREON have been available post approval use of this formulation of CREON. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
constipation and nausea), skin disorders (including pruritus, urticaria and rash), blurred vision, myalgia, muscle spasm, and asymptomatic elevations of liver enzymes have been reported with this formulation of CREON.
Delayed- and immediate-release pancreatic enzyme products with different formulations of the same active ingredient (pancrelipase) have been used for
intestinal obstruction syndrome (DIOS), recurrence of pre-existing carcinoma, and severe allergic reactions including anaphylaxis, asthma, hives, and pruritus.
been conducted.
Pregnancy Risk Summary
Published data from case reports with pancrelipase use in pregnant women or other adverse maternal or fetal outcomes. Pancrelipase is minimally absorbed systematically; therefore, maternal use is not expected to result in fetal exposure to the drug. Animal reproduction studies have not been conducted with pancrelipase.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
There are no data on the presence of pancrelipase in either human or animal milk, the effects on the breastfed infant or the effects on milk production. Pancrelipase is minimally absorbed systemically following oral administration; therefore, maternal use is not expected to result in clinically relevant exposure
breastfeeding should be considered along with the mother’s clinical need for CREON and any potential adverse effects on the breastfed infant from CREON or from the underlying maternal condition.
The short-term safety and effectiveness of CREON were assessed in two randomized, double-blind, placebo-controlled, crossover studies of 49 Study 1 included 8 adolescents between 12 and 17 years of age. Study 2
in pediatric patients in these studies were similar to adult patients [see Adverse Reactions and Clinical Studies]
An open-label, single-arm, short-term study of CREON was conducted in 18 infants and children, ages 4 months to six years of age, with EPI due to
therapy (mean dose of 7,000 lipase units/kg/day for a mean duration of 18.2 days) followed by CREON (mean dose of 7,500 lipase units/kg/day for a mean duration of 12.6 days). The mean daily fat intake was 48 grams during treatment with usual pancreatic enzyme replacement therapy and 47 grams
during treatment with CREON. When patients were switched from their usual pancreatic enzyme replacement therapy to CREON, they demonstrated similar spot fecal fat testing results; the clinical relevance of spot fecal fat testing has not been demonstrated. Adverse reactions that occurred in patients during treatment with CREON were vomiting, irritability, and decreased appetite [see Adverse Reactions]
formulations of pancrelipase consisting of the same active ingredient (lipases, proteases, and amylases) for treatment of children with exocrine pancreatic literature and through clinical experience.
Dosing of pediatric patients should be in accordance with recommended guidance from the Cystic Fibrosis Foundation Consensus Conferences [see Dosage and Administration]. Doses of other pancreatic enzyme products exceeding 6,000 lipase units/kg of body weight per meal have been than 12 years of age [see Warnings and Precautions]
Geriatric Use
65 and over to determine whether they respond differently from younger responses between the elderly and younger patients.
There have been no reports of overdose in clinical trials or postmarketing surveillance with this formulation of CREON. Chronic high doses of pancreatic colonic strictures [see Dosage and Administration and Warnings and Precautions]. High doses of pancreatic enzyme products have been associated with hyperuricosuria and hyperuricemia, and should be used with caution in patients with a history of hyperuricemia, gout, or renal impairment [see Warnings and Precautions]
Carcinogenesis, Mutagenesis, Impairment of Fertility Carcinogenicity, genetic toxicology, and animal fertility studies have not been performed with pancrelipase.
Dosing and Administration
• Instruct patients and caregivers that CREON should only be taken as directed by their healthcare professional. Patients should be advised that the total daily dose should not exceed 10,000 lipase units/kg body weight/ day unless clinically indicated. This needs to be especially emphasized for patients eating multiple snacks and meals per day. Patients should be informed that if a dose is missed, the next dose should be taken with the next meal or snack as directed. Doses should not be doubled [see Dosage and Administration (2)]
• Instruct patients and caregivers that CREON should always be taken with food. Patients should be advised that CREON delayed-release capsules and the capsule contents must not be crushed or chewed as doing so could cause early release of enzymes and/or loss of enzymatic activity. Patients should swallow the intact capsules with adequate amounts of liquid at mealtimes. If necessary, the capsule contents can also be sprinkled on soft acidic foods [see Dosage and Administration]
Advise patients and caregivers to follow dosing instructions carefully, as doses of pancreatic enzyme products exceeding 6,000 lipase units/kg of body weight per meal have been associated with colonic strictures in children below the age of 12 years [see Dosage and Administration]
Advise patients and caregivers to contact their healthcare professional immediately if allergic reactions to CREON develop [see Warnings and Precautions]
Manufactured by: Abbott Laboratories GmbH Hannover, Germany Marketed by: AbbVie Inc. North Chicago, IL 60064, U.S.A. © 2009-2020 AbbVie Inc.
Ref: 03-C054-R4 Revised March, 2020
LAB-3604 MASTER US-CREO-220088
En los Estados Unidos, uno de cada cuatro adultos vive con una discapacidad. Los cambios relacionados con la edad, la patología relacionada con la enfermedad y los tratamientos pueden poner a una persona con VIH en riesgo de sufrir una discapacidad. Analizamos datos representativos a nivel nacional para describir el estado de discapacidad entre adultos ≥18 años con diagnóstico de VIH en los Estados Unidos y Puerto Rico por características demográficas, conductas de salud, calidad de la atención, resultados clínicos y estado de salud mental. Informamos porcentajes ponderados y razones de prevalencia con medias marginales predichas para evaluar diferencias significativas entre grupos (P < 0,05). En general, el 44,5% informó alguna discapacidad; las discapacidades reportadas con mayor frecuencia estaban relacionadas con la movilidad (24,8%) y la cognición (23,9%). Las personas que vivían en hogares en o por debajo del nivel de pobreza o que habían estado sin hogar en los últimos 12 meses informaron una mayor prevalencia de cualquier discapacidad que las personas que no eran pobres o no estaban sin hogar (60,2 % frente a 33,4 % y 61,8 % frente a 42,8 % , respectivamente). La prevalencia de depresión y ansiedad fue mayor entre las personas con alguna discapacidad en comparación con aquellas sin discapacidad (32,8 % y 26,6 % frente a 10,1 % y 7,0 %, respectivamente). Mejorar el apoyo de los médicos y proveedores auxiliares puede ayudar a optimizar los resultados de salud a largo plazo entre las personas con discapacidad que viven con el VIH.
In the United States, one in four adults is living with a disability. Age-related changes, disease-related pathology and treatments can place a person with HIV at risk for a disability. We analyzed nationally representative data to describe disability status among adults ≥18 years with diagnosed HIV in the United States and Puerto Rico by demographic characteristics, health behaviors, quality of care, clinical outcomes and mental health status. We reported weighted percentages and prevalence ratios with predicted marginal means to evaluate significant differences between groups (P < .05). Overall, 44.5% reported any disability; the most frequently reported disabilities were related to mobility (24.8%) and cognition (23.9%). Persons who lived in households at or below the poverty level or who experienced homelessness in the last 12 months reported a higher prevalence of any disability than persons who were not poor or not homeless (60.2% vs. 33.4%and 61.8% vs. 42.8%, respectively). Prevalence of depression and anxiety was higher among persons with any disability compared with those with no disability (32.8% and 26.6% versus 10.1% and 7.0%, respectively). Enhancing support from clinicians and ancillary providers may help optimize long-term health outcomes among HIV-positive persons with disabilities.
The Centers for Disease Control and Prevention (CDC) defines a disability as a condition that makes it more difficult for a person to do certain activities and interact with the world around them.
In the United States, 61 million adults are living with some type of disability (Okoro et al., 2018) and the three most frequently reported disabilities were related to mobility (13.7%), cognition (10.8%) and independent living (6.8%). Prevalence of disability increases with age (Courtney-Long et al., 2015). With the advances in antiretroviral therapy (ART) treatments, people with HIV are living longer (Antiretroviral Therapy Cohort Collaboration, 2008). Persons with HIV infection may be more likely to have disabilities because nearly half of the people with HIV are over 50 years (CDC, 2018) and many older adults with HIV experience health-related deterioration from aging (Pathai et al., 2014), HIV-related comorbidities (Leveille & Thapa, 2017), as well as negative long term effects of HIV treatments (Onen & Overton, 2011). It is important to know how disability can affect people with HIV and understanding who is disproportionately affected by disability may be helpful for public health prevention. The purpose of this analysis was to use Medical Monitoring Project (MMP) data to describe the prevalence of disability status overall and by selected characteristics, among adults with diagnosed HIV living in the United States and Puerto Rico.
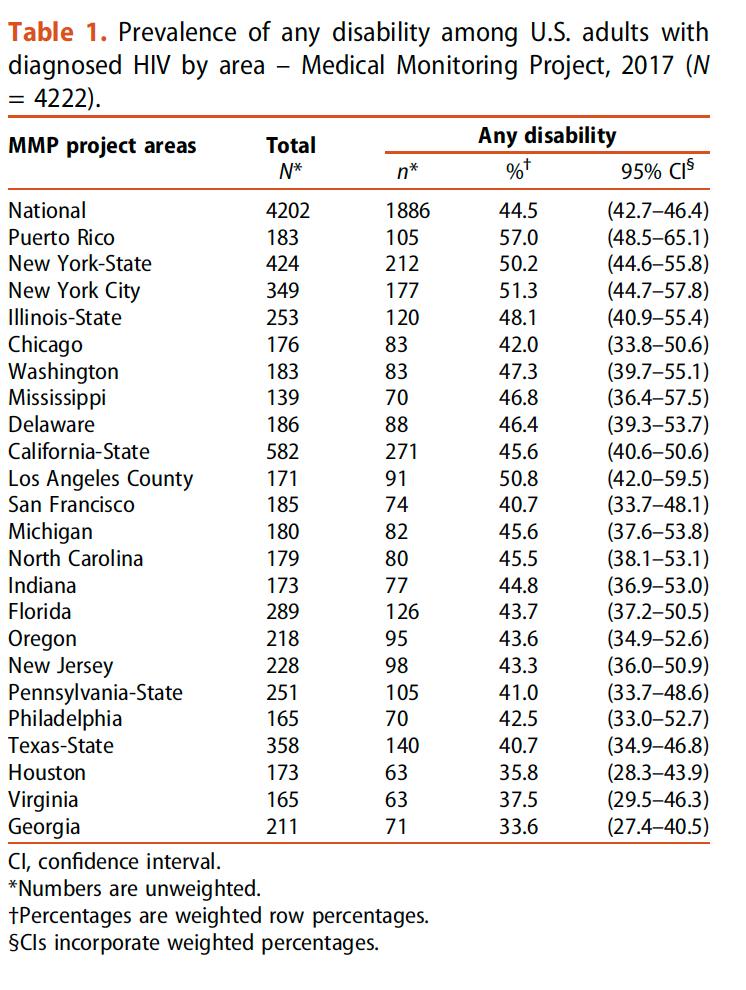
MMP is a surveillance system that produces nationally representative estimates of behavioral and clinical characteristics of adults aged ≥18 years with diagnosed HIV living in the United States and Puerto Rico. We used interview and medical record data collected from 4222 adults living with HIV collected from June 2017 through May 2018. Details about MMP sampling, data collection and weighting processes were described previously (Beer et al., 2019). All analyses were conducted using SAS callable SUDAAN version 11.03 (RTI International, Research Triangle Park, NC) to account for the complex survey design and weights. We estimated the weighted prevalence and 95% confidence interval (CI) of reporting at least one disability overall and by variables capturing socio demographics, any Ryan White HIV/ AIDS Program (RWHAP) assistance, unmet needs, quality of care, clinical outcomes, health behaviors and mental health. To compare groups, unadjusted prevalence ratios (PR) with CIs were calculated using logistic regression with predicted marginal means (Bieler et al., 2010).
MMP included six questions about disabilities related to hearing, vision, cognition, mobility, selfcare and independent living (HHS, 2011). Respondents were asked
“Are you deaf or do you have serious difficulty hearing?” (hearing disability); “Are you blind or do you have serious difficulty seeing, even when wearing glasses?” (vision disability); “Because of a physical, mental, or emotional condition, do you have serious difficulty concentrating, remembering, or making decisions?” (cognition disability); “Do you have serious difficulty walking or climbing stairs?” (mobility disability); “Do you have difficulty dressing or bathing?” (self-care disability) and “Because of a physical, mental or emotional condition, do you have difficulty doing errands alone such as visiting a doctor’s office or shopping?” (indepen-dent living disability). Respondents could report more than one disability. Persons who responded “yes” to at least one of these questions were identified as having any disability and those who responded “no” to all six questions were identified as having no disability. Socio-demographics, RWHAP assistance, unmet needs, mental health, adherence to ART, and health behaviors were selfreported for the past 12 months unless otherwise indicated. Mental health status included symptoms of depression or anxiety in the past two weeks prior to the interview. ART prescription and viral suppression measures were abstracted from medical records.
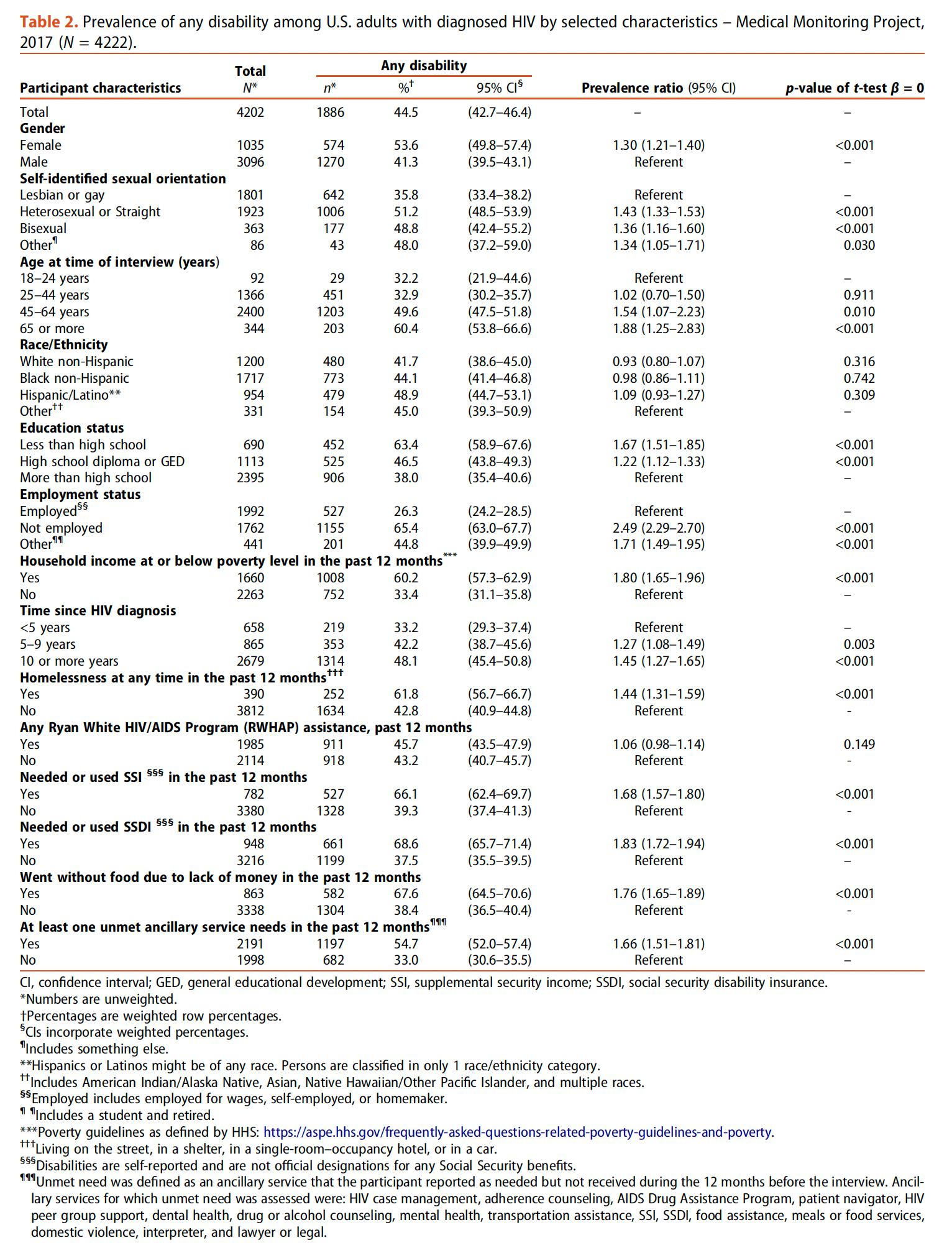
Overall, 44.5% (CI 42.7–46.4) of adults with diagnosed HIV reported any disability; 13% reported three or more disabilities. The most frequently reported disability was difficulty related to mobility (24.8%, CI 23.2–26.4), followed by cognition (23.9%, CI 22.2–25.6), indepen-dent living (12.5%, CI 11.3–13.8), vision (12.5%, CI 11.0–14.1), hearing (9.7%, CI 8.5–11.1) and self-care (7.0%, CI 6.1–8.1) (not in tables). The prevalence of any disability differed across MMP project areas, ranging from 33.6% in Georgia to 57.0% in Puerto Rico (Table 1). Women reported a higher prevalence of any disability than men (53.6% vs. 41.3%) (Table 2). Among age groups, the prevalence of any disability was highest among adults aged 65 years or older (60.4%) and lowest among those aged 18–24 years (32.2%). Among racial/ ethnic groups, Hispanic/Latino adults reported the highest prevalence of any disability (48.9%). Persons who lived in households at or below the poverty level were 80% (PR = 1.80, CI 1.65–1.96) and who were homeless in the last 12 months were 44% (PR = 1.44, CI 1.31–1.59) more likely to report any disability than
persons who were not poor or not homeless. Persons who went without food due to lack of money or who had at least one unmet ancillary service need were 76% (PR = 1.76, CI 1.65–1.89) and 66% (PR = 1.66, CI 1.51–1.81) more likely to report any disability than their counterparts, respectively.
There was no association between disability status and ART prescription, adherence to HIV medications, and viral suppression (Table 3). Persons with any dis-ability were 34% (PR = 1.34, CI 1.24–1.46) as likely to be current smoker than person with no disability. Simi-larly, compared with persons who did not have a disability, person with disability were over three times (PR = 3.27, CI 2.87–3.27) as likely to report depression and nearly four times (PR = 3.81, CI 3.12–4.65) as likely to report anxiety. When we stratified the analysis by num-ber of disabilities (data not presented in table), com-pared with persons who did not have a disability, persons with three or more disabilities were 10% (PR = 0.90, CI 0.83–0.98) less likely to be adherent to HIV medicine, 57% (PR = 1.57, CI 1.40–1.75) more likely to be a current smoker, over four times (PR = 4.36, CI 3.73–5.11)
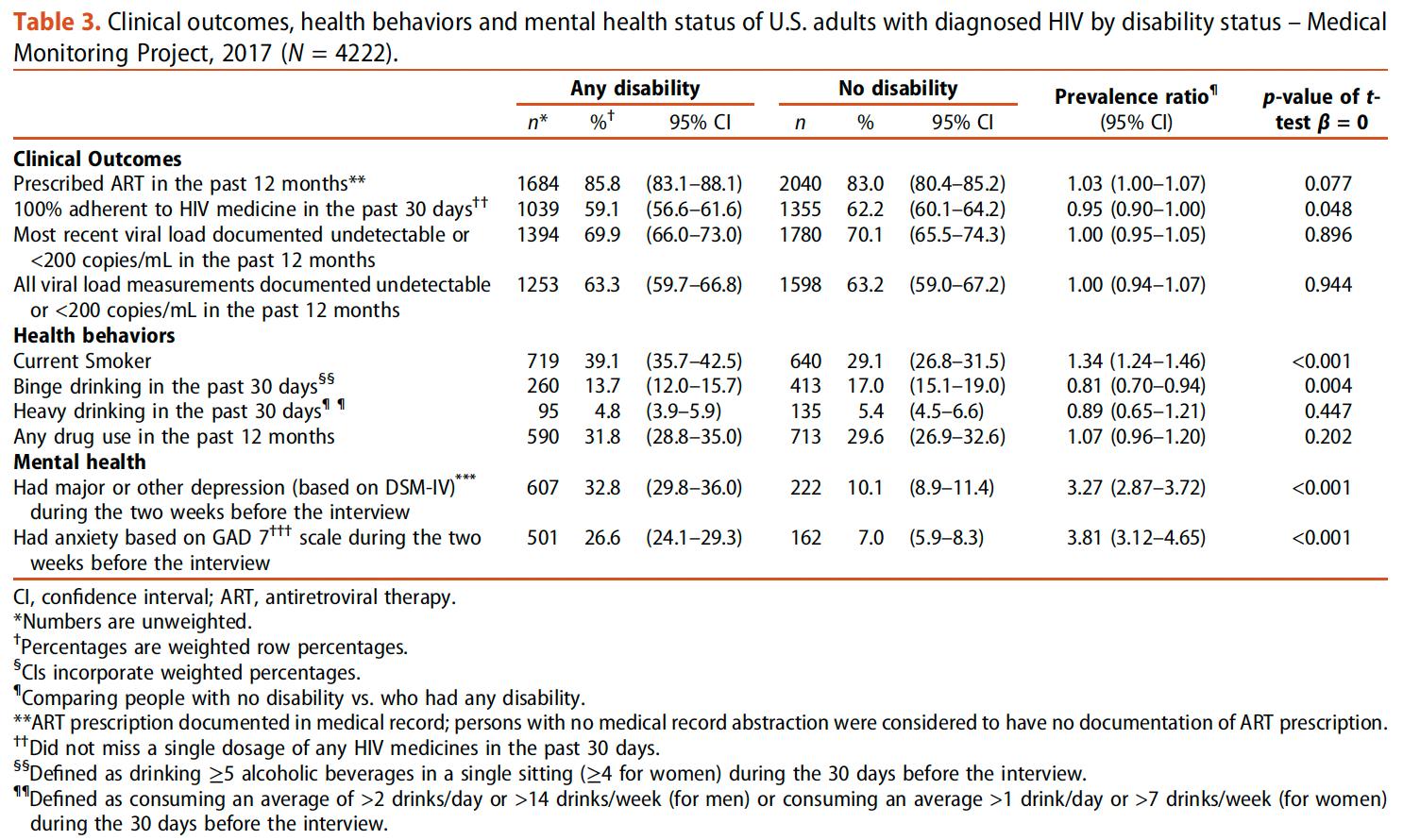
as likely to report depression, and over five times (PR = 5.31, CI 4.08–6.91) as likely to report anxiety. There was no association between number of disabilities and ART prescription or viral suppression.
The prevalence of any disability among adults with diagnosed HIV is higher than in the general population (44.5% vs. 25.7%) and is also higher among those aged 45–64 years (49.6% vs. 28.6%) and 65 or more years (60.4% vs. 41.7%) – indicating the substantial burden of disability among persons with HIV (Okoro et al., 2018). Like previous studies among the general popu-lation (Courtney-Long et al., 2015), the wide variation in the prevalence of disabilities among adults with diagnosed HIV across U.S. jurisdictions may reflect geo-graphic differences in demographic factors, health behaviors, health care access or combinations of these factors, and highlights the importance of monitoring of disability status in this population by area.
With advances in efficacy and tolerability of ART, people diagnosed with HIV are living longer; as the number of older persons living with HIV increases (Pathai
et al., 2014), the prevalence of disabilities may also increase. Similar to what has been found among the general population (Okoro et al., 2018), disabilities related to difficulties with mobility and cognition were the most frequently reported. The CDC-recommended self-management intervention, “Living Well with a Dis-ability” (Ravesloot et al., 2016), may be an effective tool to improve the health and quality of life among adults with HIV who are living with disabilities.
Our findings indicate that adults with HIV who are living with any disability may need enhanced access to ancillary services that can support their needs for hous-ing, food or nutrition, substance abuse and mental health services to improve their health outcomes (Con-viser & Pounds, 2002). We did not find any difference in RWHAP assistance between persons with and without disability, although persons with a disability may have a higher need for services available through Ryan White. The federally funded Ryan White HIV/AIDS Program provides access to medical and support ser-vices for nearly half a million persons living with HIV in the USA (Kaiser, 2019) and expanding its patient-centered medical home model may improve access to care for persons with disabilities (Pappas et al., 2014).
The findings of this analysis are subject to limitations. First, self-reported information may be subject to biases that may lead to measurement error. Second, disability measures were self-reported and are not official designa-tions for any Social Security benefit. However, self-report is the most commonly used method to assess disability for surveillance purposes. A strength of this analysis was the use of the Department of Health and Human Service’s standard self-reported six-question measure of any disability, which facilitates monitoring of disability status among people with HIV and enables comparisons of disability prevalence across different population-based studies. Third, we cannot assess causality due to MMP’s cross-sectional design.
Successful treatment and management of HIV infec-tion have transformed HIV into a chronic disease. However, optimizing health outcomes among persons with HIV with a disability may require enhanced support from clinicians and anci-
llary providers.
Participating Medical Monitoring Project respondents, advi-sory boards, and project areas.
No potential conflict of interest was reported by the authors.
This work was supported by National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention.
The findings and conclusions in this article are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Antiretroviral Therapy Cohort Collaboration. (2008). Life expectancy of individuals on combination antiretroviral therapy in high-income countries: A collaborative analysis of 14 cohort studies. Lancet, 372(9635), 293–299. https://doi.org/10.1016/S01406736(08)61113-7
Beer, L., Johnson, C. H., Fagan, J. L., et al. (2019). A national behavioral and clinical surveillance system of adults with diagnosed HIV (The Medical Monitoring Project): protocol for an annual cross-sectional interview and medical record abstraction survey. JMIR Research Protocols, 8(11), e15453. https://doi. org/10.2196/15453
Bieler, G. S., Brown, G. G., Williams, R. L., & Brogan, D. J.(2010). Estimating model-adjusted risks, risk differences, and risk ratios from complex survey data. American Journal of Epidemiology, 171(5), 618–623. https:// doi.org/ 10.1093/aje/kwp440 CDC, Centers for Disease Control and Prevention (2018). HIV Surveillance Report, 2017, vol. 29. https://www.cdc. gov/hiv/ pdf/library/reports/surveillance/cdc-hiv-surveillance-report-2017-vol-29.pdf
Conviser, R., & Pounds, M. B. (2002). The role of ancillary services in client-centered systems of care. AIDS Care, 14 (sup1), 119–131. https://doi.org/10.1080/ 09540120220150018
Courtney-Long, E. A., Carroll, D. D., Zhang, Q. C., Stevens, A. C., Griffin-Blake, S., Armour, B. S., & Campbell, V. A.(2015). Prevalence of disability and disability type among adults–United States, 2013. MMWR Morbidity and Mortality Weekly Report, 64(29), 777–783. https://doi.org/ 10.15585/mmwr. MM6429a2
HHS, U.S Department of Health and Human Services. (2011). Implementation guidance on data collection standards for race, ethnicity, sex, primary language, and disability status. http://aspe.hhs.gov/datacncl/standards/ ACA/4302
Kaiser Family Foundation. (2019). The Ryan White HIV/ AIDS Program: The basics. https://www. kffi.org/hivaids/fact-sheet/the-ryan-whitehivaids-program-the-basics Leveille, S. G., & Thapa, S. (2017). Disability among persons aging with HIV/AIDS. In M. Brennan-Ing, & R. F. DeMarco (Eds.), HIV and aging. Interdisciplinary topics in gerontology and geriatrics, vol. 42 (pp. 101–118). Karger. doi:10.1159/000448547.
Okoro, C. A., Hollis, N. D., Cyrus, A. C., & Griffin-Blake, S. (2018). Prevalence of disabilities and health care access by disability status and type among adults – United States, 2016. MMWR Morbidity and Mortality Weekly Report, 67 (32), 882–887. https:// doi.org/10.15585/mmwr.mm6732a3
Onen, N. F., & Overton, E. T. (2011). A review of premature frailty in HIV-infected persons; another manifestation of HIV-related accelerated aging. Current Aging Science, 4 (1), 33–41. https://doi. org/10.2174/1874609811104010033
Pappas, G., Yujiang, J., Seiler, N., Malcarney, M. B., Horton, K., Shaikh, I., … Hidalgo, J. (2014). Perspectives on the role of patient-centered medical homes in HIV care. American Journal of Public Health, 104(7), e49–e53. https://doi.org/10.2105/ AJPH.2014.302022
Pathai, S., Bajillan, H., Landay, A. L., & High, K. P. (2014). Is HIV a model of accelerated or accentuated aging? The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 69(7), 833–842. https://doi.org/10.1093/gerona/glt168
Ravesloot, C., Seekins, T., Traci, M., Boehm, T., White, G., Witten, M. H., … Mayer, M. (2016). Living well with a dis-ability, a self-management program. MMWR Morbidity and Mortality Supplements, 65(01), 61–67. https://doi.org/ 10.15585/mmwr.su6501a10




MCS te brinda beneficios EXTRA para que tengas salud completa. Con más de 13,000 proveedores de salud alrededor de toda la isla y varios programas de bienestar, porque tú mereces estar extra querido.

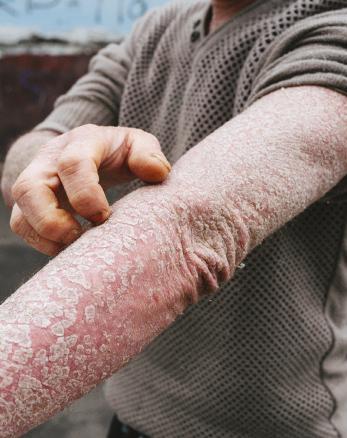 Revista Puertorriqueña de Medicina y Salud Pública
Revista Puertorriqueña de Medicina y Salud Pública
1
3
2
Antecedentes: Los medicamentos biológicos para la psoriasis en placas se han utilizado para tratar la psoriasis eritrodérmica (EP). Desde que se publicaron las guías para el manejo de la EP, se han aprobado nuevos productos biológicos para el tratamiento de la psoriasis en placas.
Objetivo: Analizar la evidencia de medicamentos biológicos en el tratamiento de la PE en base a respuesta y tolerabilidad.
Métodos: se realizó una búsqueda exhaustiva en las bases de datos PubMed, Cochrane Library, EMBASE y Scopus hasta el 31 de diciembre de 2018. Estudios que informaron uno o más casos de EP, definida como >75% de compromiso del área de superficie corporal, en pacientes ≥18 años Se incluyeron los tratados con biológicos. Se documentaron la puntuación PASI inicial, la mejora de la puntuación PASI y los eventos adversos. Se definió respuesta adecuada al tratamiento como PASI≥ 50.
Resultados: Se incluyeron 43 artículos, arrojando un total de 179 pacientes. La mayoría de los pacientes respondieron en algún momento del tratamiento, con un mayor nivel de evidencia para infliximab, ustekinumab, ixekizumab y guselkumab. La infección fue el evento adverso más común (n=35).
Limitaciones: los datos se limitan a informes de casos, series de casos y estudios no controlados. Conclusiones: Los pacientes con EP tratados con biológicos demostraron respuestas positivas y el tratamiento fue bien tolerado con recomendación débil y calidad de evidencia limitada a favor de infliximab, ustekinumab, ixekizumab y guselkumab.
Background: Biologic medications for plaque psoriasis have been used to treat erythrodermic psoriasis (EP). Since the guidelines for management of EP were published, new biologics have been approved for the treatment of plaque psoriasis.
Objective: To analyze the evidence of biologic medications in the treatment of EP based on response and tolerability.
Methods: A comprehensive search was conducted with the PubMed, Cochrane Library, EMBASE, and Scopus databases through December 31, 2018. Studies reporting one or more cases of EP, defined as >75% body surface area involvement, in patients ≥18 years old treated with biologics were included. Baseline PASI score, PASI score improvement, and adverse events were documented. Adequate response to treatment was defined as PASI≥ 50.
Results: Forty-three articles were included, yielding a total of 179 patients. Most patients responded at some point during treatment, with a higher level of evidence for infliximab, ustekinumab, ixekizumab, and guselkumab. Infection was the most common adverse event (n=35).
Limitations: Data is limited to case reports, case series, and uncontrolled studies. Conclusions: Patients with EP treated with biologics demonstrated positive responses and treatment was well-tolerated with a weak recommendation and limited quality of evidence in favor of infliximab, ustekinumab, ixekizumab, and guselkumab.
Psoriasis is a chronic inflammatory condition of the skin affecting approximately 2% of the world’s population.1 Several clinically distinct subtypes of psoriasis include chronic plaque psoriasis, nail psoriasis, pustular psoriasis, erythrodermic psoriasis (EP), and guttate psoriasis.2 EP is a rare and severe variant with a prevalence of less than 3% of all cases.3 This condition generally develops in patients with poorly controlled psoriasis. Other factors that may trigger EP are abrupt withdrawal of systemic medications like corticosteroids, drug reactions to medications such as lithium, and underlying systemic infections.4 EP is characterized by generalized erythema and scaling affecting more than 75% of body surface area.3,5 Affected patients can present numerous systemic symptoms such as fever, tachycardia, lymphadenopathy, arthralgia, and fatigue.5 Without appropriate treatment, high output cardiac failure, malabsorption, anemia, and sepsis could occur resulting in increased morbidity, and in some cases, death.2,3
Treatment can be very challenging. The latest National Psoriasis Foundation Consensus Guidelines recommend cyclosporine or infliximab as first-line therapy due to their rapid onset of action. Other recommended first line agents include acitretin and methotrexate.6 However, these recommendations were made before evidence of efficacy and safety in the treatment of EP was available for most biologic agents recently approved to treat psoriasis. Levin et al. reviewed the safety and efficacy of ustekinumab, adalimumab, etanercept, and infliximab in the treatment of EP.7 Since then, additional biologic medications, like ixekizumab, secukinumab, and guselkumab have been approved for the treatment of moderate to severe plaque psoriasis.
The objective of this study is to perform a systematic review to determine the response and tolerability of biologic medications used in the treatment of EP, including adalimumab, etanercept, infliximab, ixekizumab, golimumab, guselkumab, secukinumab, and ustekinumab. We aim to make treatment recommendations based on level of evidence, onset of action, adverse effects, and sustained response with these medications.
This review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).8 A primary literature search was conducted with the databases PubMed, Scopus, EMBASE, and Cochrane Library. We identified eligible articles from database inception to December 31, 2018. The search terms were “biologics” AND “erythrodermic psoriasis”, “biologic” AND “erythrodermic psoriasis”, “[drug name]” AND “erythrodermic psoriasis”. Drug names included: secukinumab, ixekizumab, golimumab, guselkumab, infliximab, etanercept, ustekinumab, and adalimumab. Articles written in Spanish and English were included. There were no limitations on article type. After the selection process, the references of all included articles were assessed for missing publications. The review protocol is registered with PROSPERO (CRD42019133413) (https://www. crd.york.ac.uk/prospero/).
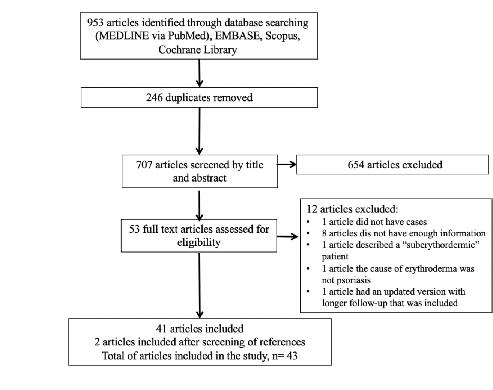
Independent assessment of the titles and abstracts was carried out independently by two of the authors (O.Y.C. and K.J.C.). Disagreements were resolved through discussion with a third author (R.F.M.). All studies reporting one or more cases of EP treated with biologics were included. EP was defined
as >75% of body surface area involvement with inflammatory erythema and scaling at baseline. The following criteria were used to exclude articles: pediatric patients (<18 years); <75% of body surface area involvement; erythroderma caused by a condition other than psoriasis; patient was treated with biologics for a different condition that was not EP; article failed to mention response to treatment. If the full text was not available online, it was ordered at the University of Puerto Rico School of Medicine library.
Data extraction and statistical analysis
The following variables were gathered as available: age, sex, body mass index, duration of disease, comorbidities, prior therapies, adjuvant treatments, Psoriasis Area and Severity Index (PASI) score, total follow-up time, PASI score improvement, and adverse events. Adequate response to treatment was defined as PASI≥ 50. Statistical analysis was done using IBM SPSS version 25.
Risk of bias, level of evidence, and grade of recommendation assessment
Due to the nature of the condition, we did not expect to find comparative studies. To assess the risk of bias of studies, we used a tool developed by Haffar et al. derived from the New-Castle Ottawa scale.9,10 This tool has been used previously.9,11-14 It consists of 5 criteria in the form of questions with a binary response (yes/no) to indicate whether or not the item is suggestive of bias. The tool is composed of the following questions:
1.Did the patient(s) represent the whole case(s) of the medical center?
2.Was the diagnosis correctly made?
3.Were other important diagnoses excluded?
4.Were all important data cited in the report?
5.Was the outcome correctly ascertained?
Quality of the report was considered good (low risk of bias) when all 5 criteria were fulfilled, moderate (moderate risk of bias) when 4 were fulfilled, and poor (high risk of bias) when ≤ 3 were fulfilled. Two of the authors (O.Y.C. and G.P.) assessed the risk of bias of the studies with discussion with a third author (R.F.M.) in case of disagreement. Recommendations for each biologic were given based on criteria by Robinson and colleagues.15
Forty-three articles were included, yielding a total of 179 patients of EP treated with biologic medications (Figure 1). The majority of the articles were case reports (65.1%), followed by case series (14.0%) and open label studies (11.6%). There were 3 retrospective and 1 open prospective study. Sixteen articles reported treatment with infliximab16-31, 10 ustekinumab32-41, 4 etanercept42-45, 5 secukinumab46-50, and 3 adalimumab51-53. Ixekizumab54, golimumab55, and guselkumab56 were each found in one study. One article reported cases treated with either infliximab, adalimumab, etanercept or ustekinumab, and another article reported cases treated with infliximab or etanercept.57,58
The mean age at presentation was 46.4 years with a male-to-female ratio of 3.6:1. Table 1 shows baseline characteristics of patients. After assessing all the selected articles, the most common medications reported were ustekinumab and infliximab with 54 and 41 cases, respectively.16-41,58 They were followed by secukinumab (n=19), etanercept (n=14), guselkumab (n=11), ixekizumab (n= 8), adalimumab (n=3), and golimumab (n=1).42-56,58 One article described 42 flares in 28 patients treated with multiple biologic agents (infliximab, adalimumab, etanercept, and ustekinumab) and was not included in the aforementioned analy-
sis as data for individual patients could not be extracted.57 Supplemental Table I shows a summary of the studies where EP was treated with biologics and risk of bias assessment.
The majority of patients showed adequate response in their condition at some point during treatment intervention. Out of 122 patients that reported PASI scores following treatment with biologics, 9.0% of patients failed to achieve PASI 50.26,34,39,46 Furthermore, in a multicenter retrospective study with multiple biologics, Viguier and colleagues57 reported PASI 75 at 10-14 weeks in 40% of those treated with infliximab, and equal improvement in 67%, 67%, and 0% of patients treated with etanercept, adalimumab, and ustekinumab, respectively. A smaller proportion of patients maintained PASI 75 at weeks 22-24 (infliximab 20%; etanercept 50%; adalimumab 60%). Below there is a summary of the efficacy of each biologic medication in the treatment of EP based on reduction of PASI score. Viguier’s study was excluded from the following analysis as the article reported flares instead of patients and thus, we were unable to extract the individual data from the study. Recommendations for each biologic are depicted on Table 2.
A total of 19 patients were administered secukinumab of whom 16 responded to treatment. Four of 8 cases that reported improvement between weeks 2-6 achieved PASI 75.47-49 One of these patients demonstrated continuous improvement and reached PASI 100 by weeks 8-12. Two additional patients showed PASI 100 within this time frame.47,50 Mateu-Puchades et al., also reported PASI scores during weeks 16-20 with 5/5 patients
reaching PASI 90.48 Weng et al., described that 70% of patients responded to treatment showing evident clearing of psoriasis (PASI> 75) by week 16. One of these patients experienced relapse by week 24. Two patients demonstrated a sustained response after approximately 6 months.46
Forty-one patients were treated with infliximab. Seventeen had no PASI score reported but described marked improvement of erythroderma 2 weeks after infliximab infusion.20,22-25,31 One patient evaluated using a modified PASI score achieved significant response.18 In a study by Torii et al., eight patients were treated with infliximab and the median PASI improvement reported was 67.9% at week 10.27 Thus, we were unable to assess if any patient from the aforementioned study did not respond to therapy. Arroyo-Tridico and colleagues reported a patient who maintained PASI 100 for eleven years.17 Another study reported more than 90% improvement at week 6 in all 7 patients but did not specify which instrument was used to measure this response.19 Six other studies reported significant improvement in 10 patients by weeks 2-12 (PASI≥ 75).16,21,28-30,58 Poulalhon et al. reported PASI score-based outcomes by week 14: three patients achieved PASI≥ 75 and 2 patients did not respond.26
Only three patients received adalimumab for EP. One patient showed remission at week 3 and the other two at week 12. None of them reported PASI scores.51-53
Characteristic
Total no. reported cases
Mean age at presentation, years 46.4 174
Mean psoriasis duration, years 17.2 140
Male cases, n (%) 134 (78.4%) 171
Mean body mass index (BMI), kg/cm2 24.8 68
Mean baseline PASI score 40.5 152
Mean total follow-up, months 12.3 135
Biologic Level of evidence
Grade
Secukinumab 4 2B
Source
Weng et al.46, MateuPuchades et al.48, Mugheddu et al.47, 5 2B Galluzo et al.49, Rongioletti et al.50
Ustekinumab 3 2A Pescitelli et al.34, 4 2B Wang et al.39, Concha5 2B Garzón et al.41 Saraceno et al.32, Stinco et al.33,Kim et al.35, Koutsoukou et al.36, Castiñeiras et al.37, SantosJuanes et al.38, Errichetti et al.40
Infliximab 5 2B Arroyo-Tridico et al.17, Rongioletti et al.18, Takahashi et al.19, Lisby et al.20,O’Quinn et al.22, Fiehn et al.23,
Fourteen patients were treated with etanercept. One patient did not report PASI scores but described significant improvement. Nevertheless, this patient relapsed during therapy.42 Twelve patients’ PASI scores were reported on weeks 8-12: seven achieved PASI≥ 75, three achieved PASI 50, and two did not respond.44,45,58 Romero-Mate et al. reported a patient with PASI 100 over 34 months.43
Fifty-four patients were administered ustekinumab. A total of sixteen cases reported PASI score improvement at weeks 2-6. Eight of these patients were not responsive during that time frame; all except one of the patients eventually responded to therapy.32,33,38-40 There were 44/46 cases with PASI≥ 50 16-24 weeks after starting medication.33-36,39,41 One patient did not respond to treatment; another patient initially responded but relapsed 28 weeks after intervention.39,41 Another case reported PASI≥ 90 at week 114 of treatment.37 Only 7.4% of patients failed to respond to treatment.34,39,41
Eight patients were treated with ixekizumab as part of an open-label study. By
week 12, all patients achieved PASI 75, 5/8 achieved PASI 90, and 2/8 achieved PASI 100. By week 24, 100% of patients reached PASI 75, 7/8 reached PASI 90, and 1/8 reached PASI 100. PASI scores were sustained by week 52.54
One patient was treated with golimumab and responded to treatment by week 4 and continued to improve with PASI≥ 75 by week 12.55
Guselkumab was reported in eleven patients and all responded. By week 8, all patients achieved PASI> 50. Fifty-two weeks after treatment, 10 patients demonstrated sustained responses (mean PASI≥ 75). One patient was lost to follow-up.56
Sixty (37.3%) adverse events (AE) were reported secondary to biologic therapy among studies that recorded their occurrence. Infectious events (35/60) were the most common. Further details can be found on Table III.
We aim to update the recommendations for treating EP with biologics published by Levin and colleagues in 2012.7 Since their
publication, new biologics have been approved for the treatment of plaque psoriasis. There is a lack of randomized, double-blind, controlled trials, and head-to-head comparisons. Therefore, it is challenging to recommend any of the biologics as a first-line treatment for the management of EP. Recommendations for each biologic are depicted on Table II. Overall, ustekinumab was the most frequent biologic reported. It demonstrated a slower onset of action than infliximab. Nevertheless, patients treated with this medication reported longterm clinical improvement (follow-up period range 3-29 months) with few adverse events. These findings support the important role of IL-12 and IL-23 inflammatory pathways in the pathogenesis of EP.33 As reported by Stinco and colleagues, ustekinumab seems to be a promising candidate for the long-term control of EP, especially when taking into consideration the well-known possibility of developing tachyphylaxis while on infliximab.7,33
The second most common biologic used was infliximab, demonstrating excellent efficacy with few serious AE that cannot be fully attributed to the medication. The drug had a rapid onset of action, with responses achieved as early as 48 hours after initiation of treatment.22,24. Most patients demonstrated sustained improvement after 6-10 weeks.17,20,21,26-28 Infliximab was used in combination with methotrexate or acitretin in several cases but these patients failed to respond faster than those on monotherapy. 17,19-21,25,27 For these reasons, we agree with Rosenbach et al. and Levin et al., who suggested that infliximab should be considered a first-line therapeutic agent in the treatment of acute, severe, and/or unstable cases of EP.6,7
Secukinumab, an IL-17A inhibitor, was administered to 19 patients, of which 84.2% showed improvement by ≥8 weeks with no significant AE.46-48,50. However, 31.6% of patients suffered a relapse. Weng et al. attributed these recurrences to history of prior biologic failure in their patients.46 Although this may be a possibility, a proportion of the cases responsive to such agents had also been previously treated with alternate biologic medications. This implies that another mechanism could be responsible for relapses after treatment with secukinumab. Due to its high relapse rate, we recommend using secukinumab as second line therapy for EP based on individual patient characteristics.
Most patients treated with etanercept and adalimumab responded to therapy. Nevertheless, it is important to mention that 21 and 18 cases, respectively, who received other biologics reported prior failure to etanercept or
adalimumab. Etanercept was discontinued in some cases mainly due to its lack of efficacy and AE.44,57 Consequently, we recommend considering etanercept and adalimumab as second-line treatments for EP.
Guselkumab (IL-23 inhibitor) and ixekizumab (IL-17 inhibitor) achieved significant improvement after 52 weeks of follow-up.54 Minor infections were a common AE of both medications. However, it is important to note that these studies had longer follow-up time periods when compared to most case reports and case series that reported on the response of other biologics. Due to their level of evidence both guselkumab and ixekizumab can be considered first line treatments for EP.
Regardless of which medication is eventually selected for the treatment of EP, adjuvant measures including medium-potency topical steroids, moisturizers, wet dressings, oatmeal baths, and continued supportive care are important.6
Unfortunately, data is limited to case reports, case series, and uncontrolled studies. This could contribute to publication bias, as evidenced by the risk of bias assessment in which most studies had a moderate risk of bias. Furthermore, it is difficult to recommend a specific medication, and despite the suggested therapies it is important to treat every case on an individual basis.
In conclusion, biologic therapy in patients with EP seems to be well-tolerated and demonstrated positive response. Recommendations for biologic agent therapy in EP are based on limited evidence. We recommend infliximab or ustekinumab in acute, severe cases of EP as first-line biologic agents. IL-23 and IL-17 inhibitor agents seem to be a promising category of biologic treatment of EP and can also be considered as first-line therapy based on their level of evidence. Etanercept and adalimumab can be used in milder, biologically naïve cases. Therapy of patients with EP should be individualized and based on each patient’s disease characteristics, history of previous therapies, and comorbidities. Increased understanding of pathogenic mechanisms in EP and the continued development of newer biologic agents for the treatment of psoriasis will contribute to improve the quality of life in these seriously affected patients.
1.Christophers E. Psoriasis--epidemiology and clinical spectrum. Clin Exp Dermatol. 2001 Jun;26(4):314-20.
2.Ladizinski B, Lee KC, Wilmer E, Alavi A, Mistry N, Sibbald RG. A review of the clinical variants and the management of psoriasis. Adv Skin Wound Care. 2013 Jun;26(6):271-84.
3.Lebwohl M. Psoriasis. Lancet. 2003 Apr 5; 361: 1197–204.
4.Raychaudhuri SK, Maverakis E, Raychaudhuri SP. Diagnosis and classification of psoriasis. Autoimmun Rev. 2014 AprMay;13(4-5):490-5.
5.Singh RK, Lee KM, Ucmak D et al. Erythrodermic psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:93-104.
6.Rosenbach M, Hsu S, Korman NJ et al. Treatment of erythrodermic psoriasis: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010 Apr;62(4):655-62.
7.Levin EC, Debbaneh M, Koo J, Liao W. Biologic therapy in erythrodermic and pustular psoriasis. J Drugs Dermatol. 2014 Mar;13(3):342-54.
8.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Open Med. 2009;3:e123-e130.
9.Haffar S, Bazerbachi F, Prokop L, Watt KD, Murad MH, Chari ST. Frequency and prognosis of acute pancreatitis associated with fulminant or non-fulminant acute hepatitis A: A systematic review. Pancreatology. 2017 Mar-Apr;17(2):166–175.
10.Wells G, Shea B, O’Connell D et al; The Newcastle-Ottawa scale (NOS) for assessing the quality of nonrandomized studies in meta-analysis. Ottawa, Ontario: The Ottawa Health Research Institute, 2011.
11. Bazerbachi F, Haffar S, Szarka LA et al. Secretory diarrhea and hypokalemia associated with colonic pseudo-obstruction: A case study and systematic analysis of the literature. Neurogastroenterol Motil. 2017 Nov 29(11).
12.Bazerbachi F, Sawas T, Vargas EJ et al. Metal stents versus plastic stents for the management of pancreatic walled-off necrosis: a systematic review and meta-analysis. Gastrointest Endosc. 2018 Jan;87(1):3042.
13.Bazerbachi F, Leise MD, Watt KD, Murad MH, Prokop LJ, Haffar S. Systematic review of mixed cryoglobulinemia associated with hepatitis E virus infection: association or causation?Gastroenterol Rep.
2017;5:178–84.
14.Bazerbachi F, Haffar S, Hussain MT et al. Systematic review of acute pancreatitis associated with interferon-α or pegylated interferon-α: possible or definitive causation? Pancreatology. 2018 Oct;18(7):691699.
15.Robinson JK, Dellavalle RP, Bigby M, Callen JP. Systematic reviews: grading recommendations and evidence quality. Arch Dermatol. 2008 Jan;144(1):97-9.
16.Valdés AM del P, Schroeder HF, Roizen GV, Honeyman MJ, Sánchez ML. [Efficacy of infliximab in patients with moderate and severe psoriasis treated with infliximab (Remicade)]. Rev Med Chil. 2006 Mar;134(3):326-31.
17.Arroyo-Trídico L, Antonio JR, Mathias CE, Pozetti EMO. Effectiveness and safety of infliximab for 11 years in a patient with erythrodermic psoriasis and psoriatic arthritis. An Bras Dermatol. 2017 SepOct;92(5):743-745.
18.Rongioletti F, Borenstein M, Kirsner R, Kerdel F. Erythrodermic, recalcitrant psoriasis: clinical resolution with infliximab. J Dermatolog Treat. 2003 Dec;14(4):222-5.
19. Takahashi MD, Castro LG, Romiti R. Infliximab, as sole or combined therapy, induces rapid clearing of erythrodermic psoriasis. Br J Dermatol. 2007 Oct;157(4):82831.
20.Lisby S, Gniadecki R. Infliximab (Remicade) for acute, severe pustular and erythrodermic psoriasis. Acta Derm Venereol. 2004;85(3):247-8.
21.Yip L, Harrison S, Foley P. From biologic to biologic to biologic: lessons to learn for erythrodermic and recalcitrant chronic plaque psoriasis. Australas J Dermatol. 2008 Aug;49(3):152-5.
22.O’Quinn RP, Miller JL. The effectiveness of tumor necrosis factor alpha antibody (infliximab) in treating recalcitrant psoriasis: a report of 2 cases. Arch Dermatol. 2002 May;138(5):644-8.
23.Fiehn C, Andrassy K. Case number 29: hitting three with one strike: rapid improvement of psoriatic arthritis, psoriatic erythroderma, and secondary renal amyloidosis by treatment with infliximab (Remicade). Ann Rheum Dis. 2004 Mar;63(3):232.
24.Lewis TG, Tuchinda C, Lim HW, Wong HK. Life-threatening pustular and erythrodermic psoriasis responding to infliximab. J Drugs Dermatol. 2006 Jun;5(6):546-8.
25.Heikkilä H, Ranki A, Cajanus S, Karvonen SL. Infliximab combined with methotrexate as long-term treatment for erythro
dermic psoriasis. Arch Dermatol. 2005 Dec;141(12):1607-10.
26.Poulalhon N, Begon E, Lebbé C et al. A follow-up study in 28 patients treated with infliximab for severe recalcitrant psoriasis: evidence for efficacy and high incidence of biological autoimmunity. Br J Dermatol. 2007 Feb;156(2):329-36.
27.Torii H, Nakagawa H. Long-term study of infliximab in Japanese patients with plaque psoriasis, psoriatic arthritis, pustular psoriasis and psoriatic erythroderma. J Dermatol. 2011 Apr;38(4):321-34.
28. Kurokawa R, Hagiwara A, Niijima Y, Kojima K. Computed tomography imaging findings in erythrodermic psoriasis treated with infliximab: A case report. Radiol Case Rep. 2018 Mar 2;13(2):460-463.
29.Belinchon I, Lucas A, Ballester I, Betlloch I, Pérez-Crespo M: Successful treatment of life-threatening erythrodermic psoriasis with infliximab. J Am Acad Dermatol. 2009, 60(3):AB171.
30.Yip L, Harrison S, Foley P: Infliximab rescue of efalizumab withdrawal flare and psoriasis-precipitated depression. Australas J Dermatol. 2008, 49(4):250-251.
31.Suárez Pedreira I, Santos Juanes J, Caminal Montero L, Trapiella L: Infliximab: An alternative in refractory erythrodermic psoriasis. Piel. 2006, 21(6):317-318.
32.Saraceno R, Talamonti M, Galluzzo M, Chiricozzi A, Costanzo A, Chimenti S. Ustekinumab treatment of erythrodermic psoriasis occurring after physical stress: a report of two cases. Case Rep Dermatol. 2013 Sep 26;5(3):254-9.
33.Stinco G, Piccirillo A, Errichetti E, Bergamo S, Patrone P. Treatment of Recalcitrant Erythrodermic Psoriasis With Ustekinumab. Eur J Dermatol. 2014 MayJun;24(3):387-90.
34.Pescitelli L, Dini V, Gisondi P et al. Erythrodermic psoriasis treated with ustekinumab: an Italian multicenter retrospective analysis. J Dermatol Sci. 2015 May;78(2):149-51.
35.Kim YS, Kim HJ, Lee S, Park YL. Erythrodermic Psoriasis Improved by Ustekinumab: A Report of Two Cases. Ann Dermatol. 2016 Feb;28(1):121-2.
36.Koutsoukou XA, Papadavid E, Theodoropoulos K, Rigopoulos D. Ustekinumab in severe complicated erythrodermic psoriasis: rapid clearing, safety, and sustained remission. Dermatol Ther. 2014 SepOct;27(5):257-9.
37. Castiñeiras I, Fernández-Diaz L, Juárez Y, Lueiro M. Sustained efficacy of ustekinu-
mab in refractory erythrodermic psoriasis after failure of antitumor necrosis factor therapies. J Dermatol. 2012 Aug;39(8):730-1.
38.Santos-Juanes J, Coto-Segura P, Mas-Vidal A, Galache Osuna C. Ustekinumab induces rapid clearing of erythrodermic psoriasis after failure of antitumour necrosis factor therapies. Br J Dermatol. 2010 May;162(5):1144-6.
39.Wang TS, Tsai TF. Clinical experience of ustekinumab in the treatment of erythrodermic psoriasis: a case series. J Dermatol. 2011 Nov;38(11):1096-9.
40.Errichetti E, Piccirillo A: Latent tuberculosis reactivation in a patient with erythrodermic psoriasis under treatment with ustekinumab and a low dose steroid, Despite isoniazid chemoprophylaxis. Eur J Dermatol. 2014, 24(4):508-509.
41.Concha-Garzón MJ, Godoy-Trapero A, Daudén E, et al. Short- and long-term treatment of erythrodermic psoriasis with ustekinumab: A national and multicenter case series. J Am Acad Dermatol. 2014, 70(5):AB189.
42.Talat H, Wahid Z, Feroz F, Sajid M. Erythrodermic Psoriasis and Hepatitis C Infection Treated with Pegylated Interferon and Anti-TNFα α(Etanercept) Therapy. J Coll Physicians Surg Pak. 2017 Sep;27(9):S77-S79.
43.Romero-Maté A, García-Donoso C, Martinez-Morán C, Hernández-Núñez A, Borbujo
J.Long-term management of erythrodermic psoriasis with anti-TNF agents. Dermatol Online J. 2010 Jun 15;16(6):15.
44. Esposito M, Mazzotta A, de Felice C, Papoutsaki M, Chimenti S. Treatment of erythrodermic psoriasis with etanercept. Br J Dermatol. 2006 Jul;155(1):156-9.
45. Piqué-Duran E, Pérez-Cejudo JA. [Psoriatic erythroderma treated with etanercept]. Actas Dermosifiliogr. 2007 Sep;98(7):508-10.
46.Weng HJ, Wang TS, Tsai TF. Clinical experience of secukinumab in the treatment of erythrodermic psoriasis: a case series. Br J Dermatol. 2018 Jun;178(6):1439-1440.
47.Mugheddu C, Atzori L, Lappi A, Pau M, Murgia S, Rongioletti F. Successful Secukinumab treatment of generalized pustular psoriasis and erythrodermic psoriasis. J Eur Acad Dermatol Venereol. 2017 Sep;31(9):e420-e421.
48.Mateu-Puchades A, Santos-Alarcón S, Martorell-Calatayud A, Pujol-Marco C, Sánchez-Carazo JL. Erythrodermic psoriasis and secukinumab: Our clinical experience. Dermatol Ther. 2018 Jul;31(4):e12607.
Revista Puertorriqueña de Medicina y Salud Pública
49.Galluzzo M, D’Adamio S, Campione E, Mazzilli S, Bianchi L, Talamonti M. A clinical case of severe disease burden: an erythrodermic psoriatic patient treated with secukinumab. J Dermatolog Treat. 2018 Sep 26:1-11.
50.Rongioletti F, Mugheddu C, Murgia S: Repigmentation and new growth of hairs after anti–interleukin-17 therapy with secukinumab for psoriasis. JAAD Case Rep. 2018 Jun; 4(5): 486–488.
51.Mumoli N, Vitale J, Gambaccini L, Sabatini S, Brondi B, Cei M. Erythrodermic psoriasis. QJM. 2014 Apr;107(4):315.
52.Richetta AG, Maiani E, Carlomagno V et al. Treatment of erythrodermic psoriasis in HCV+ patient with adalimumab. Dermatol Ther. 2009 Nov;22 Suppl 1:S16-8.
53.Vidal D, Peramiquel L, Olivella R, Villar M, Petiti G, González A: Erythrodermic psoriasis successfully treated with adalimumab: A case study. J Eur Acad Dermatol Venereol. 2013, 27(S4):68.
54.Saeki H, Nakagawa H, Nakajo K et al. Efficacy and safety of ixekizumab treatment for Japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (UNCOVER-J). J Dermatol. 2017 Apr;44(4):355-362.
55.Lee WK, Kim GW, Hyun-Ho Cho et al. Erythrodermic Psoriasis Treated with Golimumab: A Case Report. Ann Dermatol 2015;27(4): 446-449.
56.Sano S, Kubo H, Morishima H, Goto R, Zheng R, Nakagawa H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: Efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J Dermatol. 2018 May;45(5):529-539.
57.Viguier M, Pagès C, Aubin F et al. Efficacy and safety of biologics in erythrodermic psoriasis: a multicentre, retrospective study. Br J Dermatol. 2012 Aug;167(2):41723.
58.Sahel H, Otsmane F, Bouadjar B: Treatment of erythrodermic psoriasis with biological therapies in two cases. J Eur Acad Dermatol Venereol. 2016, 30(S6):102.
DUPIXENT es un medic amento de vent a con recet a que se utiliza par a tr at ar a adultos y niños de 6 meses o m á s co n de r m ati ti s ató pi c a (e c ze m a) m o de r a d a a g r a v e , q u e n o pu e d e c o n t r o l a r s e c o r r e c t a me nt e c o n t e r a p i a s r e c e t a d a s q u e s e a p l i c a n s o b r e l a p i e l (tópic as) o en c asos en los que no se pue dan utiliz ar terapias tópicas DUPIXENT puede utilizarse con o sin cor ticos teroides tópicos. Se desconoce si DUPIXENT es seguro y efic az en niños menores de 6 meses que tengan der matitis atópic a
INFORMACIÓN IMPORTANTE DE SEGURIDAD No uses este medicamento si eres alérgico al dupilumab o a alguno de los ingredientes en DUPIXENT®
Antes de usar DUPIXENT, infórmale a tu proveedor de atención médica sobre todas tus afecciones médicas, incluyendo: Si tienes problemas oculares Si tienes una infección parasitaria (helmintos) Si tienes programado recibir alguna vacuna No deb es recibir una “ vacuna viva” antes o dur ante el t r at amiento con DUPIXENT Si estás embarazada o planeas quedar embarazada Se desconoce si DUPIXENT dañará a tu bebé en gestación Un registro de embarazo para mujeres que usan DUPIXENT durante el embarazo recopila información sobre tu salud y la d e t u b e b é. P a r a ins cr ibir te o p a r a o bte n e r más información, llama al 1-877-311-8972 o visit a https:// mother tobaby.org /ongoing-study/dupixent /. Si est ás amamant ando o planeas amamant ar Se desconoce si DUPIX ENT se tr ansmite a tr avés de la leche materna Infór male a t u p rove e d or d e ate n ción m é dic a s ob re t o d o s l o s m e d i c a me n t o s q u e t o m a s , i n c l ui d o s l o s medic amentos de vent a libre y de vent a con recet a , las vit aminas y los suplementos a base de hier bas .
Especialmente, infórmale a tu proveedor de atención médica si estás tomando medicamentos corticosteroides orales, tópicos o inhalados, o si tienes dermatitis atópica y asma y usas un medicamento para el asma. No cambies
ni suspendas tu medic amento cor ticos teroide u otro medic amento par a el asma sin hablar primero con tu proveedor de atención médic a Esto puede hacer que reaparezcan otros síntomas que fueron controlados por el medic amento cor ticos teroide u otro medic amento par a el asma
DUPIXENT puede c ausar efec tos secundarios graves, que incluyen:
R e a c c i o n e s a l ér g i c a s . D U P I X E N T pu e d e c au s a r r e a c c i on e s a l ér g i c a s qu e a lg un a s v e c e s p u e d e n s e r g r ave s . D e j a d e u s a r DU P IX E N T e i n f ó r m a l e a t u p r ove e d o r d e ate n ci ó n m é di c a o b u s c a ate n ci ó n d e em e r ge n cia d e in m e diato si tien e s a lg u n o d e l os siguientes signos o síntomas: Problemas respiratorios o sibilancias, inflamación del rostro, los labios, la boc a, la lengua o la gargant a , desmayos , mareos , sens ación d e ma re o, p uls o a ce le r a d o, fie b re, ur tic a r ia , d olo r e n las ar ticulaciones, malestar general, picazón, erupción c u t á n e a , g a ng l io s l i n f á ti c o s i n fl a m a d o s , n á u s e a s o vó mitos , o c alam b re s e n e l á re a d e l e s tó mago P r o b l e ma s o c u l a r e s . I n f ó r ma l e a t u p r o v ee d o r d e atención médic a si tienes problemas oculares nuevos o si n ot as u n e m pe o r a mie nto d e l os p ro b l e m as q u e te n ías , i n c l ui d o d olo r o cul a r o c am b io s e n l a v isió n , como visión borrosa Tu proveedor de atención médica puede referir te a un oft almólogo par a que te realicen un examen de la vis t a , si es neces ario Dolores en las ar ticulaciones A lgunas per sonas que usan DUPIXENT han tenido dificultades para caminar o moverse debido a los síntomas en sus ar ticulaciones y, en algunos casos, debieron ser hospitalizados Infórmale a t u p rove e d o r d e ate n ció n m é di c a s o b re c u a l q u ie r síntoma nuevo en las ar ticulaciones o si empeoran los síntomas que tenías Tu proveedor de atención médica pu e d e inte r r um pir e l t r at a mie nto co n D U PI XENT si des ar rollas sínto mas en las ar ticulaciones Los efec tos secundarios más comunes en pacientes con dermatitis atópica son reacciones en el lugar de la
inyección, inflamación de los ojos y los párpados, que incluyen enrojecimiento, hinchazón y picazón, a veces con visión borrosa, herpes en la boca o en los labios, y un alto recuento de ciertos glóbulos blancos (eosinofilia)

Infórmale a tu proveedor de atención médic a si sufres algún efecto secundario que te cause malestar o que no desaparezca Estos no son todos los efectos secundarios posibles de DUPIXENT Llama a tu médico para obtener consejos mé dicos sob re los efe c tos se cun dar ios Te recomendamos informar sobre los efectos secundarios negativos de los medic amentos de vent a con recet a a la Adminis t r ación de A limentos y Me dic amentos de los E s t a d os Unid os (Fo o d an d D r ug Adminis t r ation , F D A ) V is i t a ww w f d a g o v/m e d w a t c h o l l a m a a l 1-8 0 0 -FDA-10 8 8
Us a DUPIXENT exac t amente según lo prescrito p or tu proveedor de atención médic a DUPIXENT es una inyección que se administra debajo de la piel (inyección subcutánea) Tu proveedor de atención médica decidirá si t ú o t u cuid a d o r pu e de n inye c t a r DU PIX EN T N o intentes preparar e inyectar DUPIXENT hasta que tú o tu cuidador hayan sido c apacit ados por tu proveedor d e ate n ci ó n m é di c a En ni ñ o s d e 12 a ñ o s o m á s , s e recomienda que DUPIXENT sea adminis tr ado p or un a d ul to o b a j o l a s u p e r v is i ó n d e u n a d ul to En ni ñ o s menores de 12 años , un cuidador deb er á adminis tr ar DUPIXENT
Resumen breve de información importante para pacientes sobre DUPIXENT® (dupilumab) Inyección para uso subcutáneo
¿Qué es DUPIXENT?
• DUPIXENT es un medicamento con receta utilizado para lo siguiente:
–
moderada a grave, que no puede controlarse correctamente con terapias recetadas que se aplican sobre la piel (tópicas) o en casos en los que no se puedan utilizar terapias tópicas DUPIXENT puede utilizarse con o sin corticosteroides tópicos.
• DUPIXENT actúa bloqueando dos proteínas que contribuyen a un tipo de
• que tengan dermatitis atópica ¿Quiénes no deberían usar DUPIXENT? No utilices DUPIXENT si eres alérgico al dupilumab o a cualquiera de los para conocer la lista completa de ingredientes de DUPIXENT
¿Qué debo informarle a mi proveedor de atención médica antes de usar DUPIXENT? Antes de usar DUPIXENT, infórmale a tu proveedor de atención médica sobre todas tus afecciones médicas, lo que incluye:
• Si tienes problemas oculares
• Si tienes una infección parasitaria (helmintos)
• Si tienes programado recibir algún tipo de vacuna No debes recibir ninguna “vacuna viva” justo antes o mientras recibes tratamiento con DUPIXENT
• Si estás embarazada o tienes planes de quedar embarazada Se desconoce si DUPIXENT puede dañar a tu bebé en gestación – Registro de exposición durante el embarazo. Hay un registro de exposición para mujeres que toman DUPIXENT durante el embarazo El objetivo de este registro es reunir información sobre tu salud y la de tu bebé Tu proveedor de atención médica puede inscribirte en este registro También puedes inscribirte por tu cuenta u obtener más información sobre el registro llamando al 1-877-311-8972 o ingresando en https://mothertobaby.org/ongoing-study/dupixent/.
• Si estás amamantando o tienes planes de hacerlo Se desconoce si DUPIXENT se transmite a través de la leche materna
Informa a tu proveedor de atención médica todos los medicamentos que tomas, incluidos los medicamentos de venta libre y de venta con receta, las vitaminas
Informa a tu proveedor de atención médica en especial en los
• Si estás tomando corticosteroides orales, tópicos o inhalados
• Si tienes dermatitis atópica y asma y usas un medicamento para el asma
No cambies ni interrumpas la administración del medicamento corticosteroide
médica Esto puede hacer que reaparezcan otros síntomas que fueron controlados con el medicamento corticosteroide u otro medicamento para el asma.
• Consulta las “Instrucciones de uso” detalladas que se proporcionan con DUPIXENT para obtener información sobre cómo preparar
• Utiliza DUPIXENT exactamente como te lo recetó tu proveedor de atención médica
• Tu proveedor de atención médica te dirá cuánto DUPIXENT debes inyectar
• DUPIXENT viene en una jeringa precargada de dosis única con protector
– La pluma precargada de DUPIXENT es solo para uso en adultos y niños
– La jeringa precargada de DUPIXENT es solo para uso en adultos y niños
•
• Si tu proveedor de atención médica decide que tú o un cuidador pueden administrar las inyecciones DUPIXENT, tú o tu cuidador deben recibir capacitación sobre la manera correcta de preparar e inyectar DUPIXENT No intentes inyectar DUPIXENT hasta que tu proveedor de atención médica te haya mostrado la adulto coloque o super vise la administración de DUPIXENT. A niños menores de
• Si tu cronograma de dosis es semana de por medio y omites una dosis de DUPIXENT: a la dosis omitida y, luego, continúa con el cronograma original Si la dosis omitida para administrar la inyección de DUPIXENT
• Si tu cronograma de dosis es cada cuatro semanas y omites una dosis de DUPIXENT: posteriores a la dosis omitida y, luego, continúa con el cronograma original
administrarte la inyección de DUPIXENT
• Si inyectas más DUPIXENT de lo que se recetó (sobredosis), busca ayuda médica o comunícate inmediatamente con un experto del Centro de Toxicología llamando al 1-800-222-1222
• Es posible que tu proveedor de atención médica te recete otros medicamentos para utilizar con DUPIXENT Utiliza los otros medicamentos recetados exactamente como te lo indique tu proveedor de atención médica ¿Cuáles son los efectos secundarios posibles de DUPIXENT? DUPIXENT puede provocar efectos secundarios graves, incluidos
• Reacciones alérgicas DUPIXENT puede causar reacciones alérgicas que algunas veces pueden ser graves. Deja de usar DUPIXENT emergencia de inmediato si tienes alguno de los siguientes síntomas:
la boca, la lengua o la garganta, urticaria, picazón, náuseas o vómitos, desmayos, mareos, sensación de mareo, dolor en las articulaciones, erupción
• Problemas oculares. Informa a tu proveedor de atención médica si tienes problemas oculares nuevos o si notas un empeoramiento de los problemas que tenías, incluido el dolor ocular o cambios en la visión, como visión borrosa Tu proveedor de atención médica puede referirte a un oftalmólogo para que te realicen un examen de la vista, si es necesario • Dolores en las articulaciones Las personas que utilizan DUPIXENT pueden experimentar dolores en las articulaciones Algunas personas han articulaciones y, en algunos casos, debieron ser hospitalizados Informa a tu proveedor de atención médica sobre cualquier síntoma nuevo en las articulaciones o si empeoran los síntomas que tenías Tu proveedor de atención médica puede interrumpir el tratamiento con DUPIXENT si desarrollas síntomas en las articulaciones Los efectos secundarios más comunes de DUPIXENT en pacientes con dermatitis atópica son: los ojos y los párpados (que incluye enrojecimiento, hinchazón y picazón) a veces con visión borrosa, herpes en la boca o en los labios y un alto recuento de ciertos
Se han informado los siguientes efectos secundarios adicionales con el uso de DUPIXENT: Erupción facial o enrojecimiento Informa a tu proveedor de atención médica si sufres algún efecto secundario que te cause malestar o que no desaparezca Estos no son todos los efectos secundarios posibles de DUPIXENT Llama a tu médico para obtener consejos médicos sobre los efectos secundarios Puedes informar los efectos secundarios a la FDA Visita www.fda.gov/medwatch o llama al 1-800-FDA-1088
en un prospecto de Información para el paciente No uses DUPIXENT para una afección para la que no se recetó No administres DUPIXENT a otras personas, incluso si tienen los mismos síntomas que tú Es posible que les haga daño Este es un resumen breve de la información más importante sobre DUPIXENT para este uso Si deseas recibir más información, habla con tu proveedor de atención médica Puedes pedirle a tu farmacéutico o proveedor de atención médica más información sobre DUPIXENT dirigida a profesionales de atención médica. Para obtener más información sobre DUPIXENT, visita www.DUPIXENT.com o llama al 1-844-DUPIXENT (1-844-387-4936)
¿Cuáles son los ingredientes de DUPIXENT?
Ingrediente activo: dupilumab Ingredientes inactivos: acetato de sodio, sacarosa y agua para inyección
Fecha de publicación: Junio de 2022 DUP.22.06.0030
apendicitis, absceso, intraabdominal, colección, post-apendectomía, niños
appendicitis, abscess, intraabdominal, collection, post-appendectomy, children
La apendicitis es la emergencia quirúrgica mas común en un niño.Hasta un 20% de los niños que se operan con apéndices complicados desarrollan una colección o absceso intraabdominal. El diagnostico de un absceso intraabdominal después de una apendectomía se corrobora con un ultrasonido y/o CT-Scan. El manejo consiste en antibióticos intravenosos y drenar el absceso de forma percutánea utilizando radiología intervencional o cirugía laparoscópica/ abierta. El curso de antibióticos intravenosos luego de haber drenado el absceso se debe ajustar a la clínica del paciente y su recuperación inmediata.

Appendicitis is the most common surgical emergency in the pediatric age. Almost 20% of children that undergo an appendectomy due to complicate appendicitis develop an intraabdominal infected fluid collection. The diagnosis of an intraabdominal abscess after appendectomy is establish using ultrasound and/ or CT-Scan. The management of an intraabdominal abscess in a child consist of intravenous antibiotics and drainage of the abscess using percutaneous interventional radiology or open/laparoscopic surgery. The timing of antibiotic regime after drainage of the abscess is guided by the clinical systemic response and immediate recovery of the patient.
La apendicitis es la emergencia quirúrgica mas común en un niño. Es causada en la mayoría de los casos por obstrucción del lumen del apéndice por material fecal. La oclusión produce oclusión arterial e infarto del apéndice. Comienza con dolor referido en el dermatomo T10 que se manifiesta como un dolor periumbilical no especifico. Puede estar acompañado de anorexia, nausea y vómitos. A medida que la obstrucción del lumen del apéndice progresa, la circulación del área aumenta y llegan las células blancas a soltar su citoquinas y substancias inflamatorias en el área. El dolor migra al cuadrante inferior derecho en 4 a 6 horas y se hace mas especifico y agudo. La migración del dolor es una de las constantes diagnosticas mas certeras de que el paciente podría tener apendicitis. Los blancos suben, específicamente los neutrófilos en la mayoría de los casos. El pulso cardiaco también. Usando un Ultrasonido (US) se puede diagnosticar la inflamación del apéndice si se observa que el órgano esta recrecido, es no compresible, y hay liquido alrededor de su área. El CT-Scan Abdomino-pélvico, que se debería hacer con contraste por boca e intravenoso, hace el diagnostico de apendicitis en mas 98% de las veces al demostrar el órgano hinchado de mas de 6.6 mm de su diámetro normal, grasa inflamada, liquido, o hasta la presencia de un absceso intraabadominal. Se comienza antibióticos, hidrata el paciente por un periodo de 18-24 horas y se le practica una apendectomía laparoscópica o abierta, dependiendo de la inhabilidad de insuflar un paciente que tiene obstrucción abdominal por ileo reflejo. Una vez se remueve el apéndice se decide continuar o nolos antibióticos dependiendo si el apéndice estaba agudamente inflamado, gangrenoso o perforado.
Los apéndices agudos y simples se pueden manejar con una sola dosis de antibióticos preoperatorio. No necesitan de antibióticos posoperatorios. Una vez el paciente este comiendo, deje de tener fiebre y síntomas de dolor abdominal se puede enviar para la casa. Tampoco necesita antibióticos orales.
Los pacientes con apéndices gangrenosos o perforados deben recibir antibióticos intravenosos por al menos tres a cinco días posoperatorios en lo que el crecimiento local de bacterias disminuye. Una vez están sin fiebre, usando el tracto gastrointestinal, sin dolor abdominal, sin taquicardia y el contaje de células blancas (WBC) este menos de 11000, se pueden ir. Es a discreción del cirujano usar una terapia adicional de antibióticos orales por los próximos siete días de forma ambulatoria. Dependiendo me muchos factores como el tiempo de progresión de la enfermedad, la presencia de gangrena o perforación del apéndice, la situación inmunológica del huésped y otras condiciones comorbidas, el paciente puede desarrollar una infección en la herida o colección intraabdominal que se convierta en un absceso y necesite tratamiento adicional. Hasta un 20% de los pacientes que se operan con apéndices gangrenosa o perforada desarrollan una colección o absceso intraabdominal. La técnica que se utiliza para remover el apéndice sea laparoscópica o abierta, no tiene un impacto en el desarrollo de una colección intraabdominal.
El niño que desarrolla una colección intraabdominal posoperatoria desarrolla fiebre sostenida o en picos, dolor abdominal cerca de la región del absceso, dolor al orinar y frecuencia causado por que el absceso esta cerca de la vejiga, diarrea y tenesmos si el absceso este pegado al recto. Sus blancos, CRP y pulsos se elevan. Linfopenia causada por supresión del sistema inmunológico luego de la infección siguiere la presencia de un absceso y se puede utilizar para determinar el largo de la terapia de antibióticos. Todo comienza después de el tercer día posoperatorio cuando las bacterias remanentes en el lugar comienzan a dividirse y causar los síntomas de una colección intraabdominal posoperatoria. Los blancos se elevan, se libera citoquinas, se forma fibrina alrededor de la colección cuyas paredes son el intestino, vejiga, u otro órgano solido, dependiendo de la localización de los abscesos intraabdominales. Se crea neovascularización en esa capa de fibrina o pared del absceso.
La primera imagen que se debe utilizar para determinar, en el paciente que no mejora con antibióticos, la posibilidad de que tenga un absceso intraabadominal es un Ultrasonido abdomino-pél -
vico debido a su ventaja de no causar daño por radiación. Si el absceso mide menos de 3 a 5 centímetros, o tiene un volumen total de menos de 100 centímetros cúbicos se puede manejar prolongando el curso de antibióticos intravenosos. Si después de 48 horas que se toma esta decisión el paciente no mejora y sigue con signos de infección (fiebre, leucocitosis, linfopenia, taquicardia, dolor) se debe hacer un CT-Scan abdomino-pélvico con contraste oral e intravenoso buscando la localización del absceso, tamaño y la posibilidad de una ventana para drenaje percutáneo por radiología intervencional.
El manejo de un absceso intraabdominal posapendectomía puede necesitar antibióticos intravenosos, drenaje percutáneo, drenaje abierto o laparoscópico. Abscesos mayores de 5 cm en diámetro se recomienda que se drenen de forma menos invasiva usando radiología intervencional y dejando un catéter de drenaje por su efectividad y baja morbilidad. Para que eso ocurra el radiólogo determina que existe una ventana de acceso al absceso donde no se le cause trauma a otras estructuras con la aguja, alambre y catéter que se utiliza en el drenaje. Abscesos entre los intestinos y la proximidad del absceso a estructuras vasculares o solidas, podrían ser causa para no hacer el procedi
"EL MANEJO DE UN ABSCESO INTRAABDOMINAL POSAPENDECTOMÍA
PUEDE NECESITAR ANTIBIÓTICOS INTRAVENOSOS, DRENAJE PERCUTÁNEO, DRENAJE ABIERTO O LAPAROSCÓPICO"
miento de forma percutánea. De forma que si no se puede drenar el absceso usando radiología intervencional hay que tomar la decisión de prolongar los antibióticos o drenar el absceso con una cirugía abierta o laparoscópica dependiendo de su deterioro clínico y sepsis. El uso prolongado de antibióticos luego de cirugía para apendicitis perforada o gangrenosa no reduce la incidencia de producirse un absceso intraabdominal postoperatorio.
La decisión de usar un abordaje laparoscópico o abierto para drenar un absceso intraabdominal depende de deterioro clínico del paciente en esta era de antibióticos potentes. Hay que tener en cuenta que cualquier de estos abordajes pueden causar problemas mas serios como dolor en la incisión, infección de herida, hernias incisionales, fistulas enterocutáneas, enterotomías traumáticas necesitando reparo, sangrado profuso, sepsis prolongada y resultados cosméticos pobres. El riesgo debe ser menor que el riesgo asociado a prolongar la terapia con antibióticos esterilizando la colección que esta causando el problema.
El drenaje se debe quedar mientras este teniendo residuos de liquido sanguino-serosos o pus significativos. Si una vez se deja un drenaje percutáneo, el drenaje se tapa, se podría irrigar con terapia fibrinolítica usando el activador de plasma tPA. Pero en términos generales un absceso bien drenado se le remueve el drenaje en tres a cinco días después de puesto.
Una vez el absceso se drena de forma percutánea, laparoscópica o abierta, hay que tomar la decisión de cuanto tiempo adicional se debe dar de antibióticos intravenosos. La decisión de usar antibióticos prolongados no puede estar justificada con la presencia del drenaje, pero en términos generales lo antibióticos se mantienen mientras el drenaje
este puesto. La decisión de continuar antibióticos intravenosa debe estar sostenida por la clínica de recuperación del paciente. Dar de tres a cinco días de antibióticos una vez puesto el drenaje es mas que suficiente siempre y cuando el paciente deje de tener síntomas abdominales, normalice su contaje de células blancas a menos de 11000, tenga un contaje de linfocitos normales, un CRP normal oen vías de ir bajando, este sin taquicardia, sin taquipnea y sin fiebre.
No existe ninguna razón para hacer imágenes de seguimiento si la clínica del paciente demuestra que se ha recuperado completamente. Si tiene duda por que algún signo o síntomas de sepsis persiste, el estudio que se debe llevar a cabo, de nuevo es un ultrasonido del abdomen y pelvis ya que no produce radiación. Los hallazgos de las imágenes, ya sea US y CT, deben correlacionarse con la clínica y laboratorios del paciente, y no deben usarse como razón para prolongar el uso de antibióticos intravenosos.
En términos clínicos la ausencia de fiebre, dolor, leucocitosis, linfopenia, taquicardia, taquipnea, restauración del sistema gastrointestinal son los mejores índices para finalizar la terapia de antibióticos intravenosos. Algunos profesionales usan un régimen corto de 5-7 días adicionales de antibióticos orales ambulatorios una vez dan de alta al paciente. Pero muchos han dejado de llevar esta costumbre que ha demostrado no ser eficaz desde el punto de vista clínico.
Un paciente de apéndice perforado o gangrenoso necesita un seguimiento de 2 a 5 años cada seis meses para otras complicaciones de su recuperación como formación de adherencias u obstrucción intestinal parcial o completa.
1. Santini I, Pacheco R, Lugo-Vicente HL: Perforated Appendicitis in Children: Evaluation of a delayed diagnosis (Spanish). PR Health Science J 14(4):263-267, 1995
2. Dilley A, Wesson D, Munden M, Hicks J, Brandt M, Minifee P, Nuchtern J: The impact of ultrasound examinations on the management of children with suspected appendicitis: a 3-year analysis. J PediatrSurg 36(2):303-8, 2001
3. Gwynn LK: The diagnosis of acute appendicitis: clinical assessment versus computed tomography evaluation. J EmergMed 21(2):119-23, 2001
4. Meguerditchian AN, Prasil P, Cloutier R, Leclerc S, Peloquin J, Roy G: Laparoscopic appendectomy in children: A favorable alternative in simple and complicated appendicitis. J PediatrSurg 37(5):695-8, 2002
5. Chen C, Botelho C, Cooper A, Hibberd P, Parsons SK: Current practice patterns in the treatment of perforated appendicitis in children. J Am Coll Surg 196(2):212-21,
2003
6. Emil S, Laberge JM, Mikhail P, Baican L, Flageole H, Nguyen L, Shaw K: Appendicitis in children: A ten-year update of therapeutic recommendations. J PediatrSurg 38(2):236-42, 2003
7. Newman K, Ponsky T, Kittle K, Dyk L, Throop C, Gieseker K, Sills M, Gilbert J: Appendicitis 2000: Variability in practice, outcomes, and resource utilization at thirty pediatric hospitals. J PediatrSurg 38(3):372-9, 2003
8. Clark JJ, Johnson SM: Laparoscopic drainage of intraabdominal abscess after appendectomy: an alternative to laparotomy in cases not amenable to percutaneous drainage. J Pediatr Surg. 46(7): 1385-1389, 2011
9. Nataraja RM, Teague WJ, Galea J, et al: Comparison of intraabdominal abscess formation after laparoscopic and open appendicectomies in children. J Pediatr Surg. 47: 317-321, 2012
10. -Gorter RR, Meiring S, van der Lee JH, Heij HA: Intervention not always necessary in post-appendectomy abscess in children; clinical experience in a tertiary surgical centre and an overview of the literature. Eur J Pediatr 175: 1185-1191, 2016
11. Lodwick DL, Cooper JN, Kenney B, et al: Lymphocyte depression as a predictor of postoperative intraabdominal abscess after appendectomy in children.JPediatr Surg. 52(1):93-97, 2017
12. Delgdo-Miguel C, Munoz-Serrano AJ, Nunez V, et al: Neutropthil-to-Lymphocyte Ratio as a Predictor of Postsurgical Intraabdominal Abscess in Children Operated for Acute Appendicitis. Front Pediatr. 7:424, 2019
13. van Rossem CC, Schreinemacher MH, van Geloven AA, Bemelman WA; Snapshot Appendicitis Collaborative Study Group: Antibiotic Duration After Laparoscopic Appendectomy for Acute Complicated Appendicitis. JAMA Surg. 151(4):323-9, 2016
14. van Wijck K, de Jong JR, van Heurn LW, van der Zee DC: Prolonged antibiotic treatment does not prevent intra-abdominal abscesses in perforated appendicitis. World J Surg. 34(12):3049-53, 2010
Por: Laura Martí-Gelonch*, José I. Asensio-Gallego, Emma Eizaguirre-Letamendia, Francisco J. Murgoitio-Lazkano, Íñigo Arana-Iñiguez, José M. Enríquez-Navascués
Palabras clave Estómago. GIST. Hemorragia. Mieloma.
Stomach. GIST. Hemorrhage. Mieloma.
,
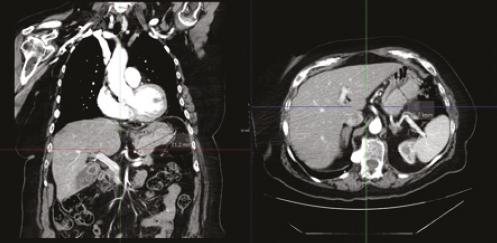
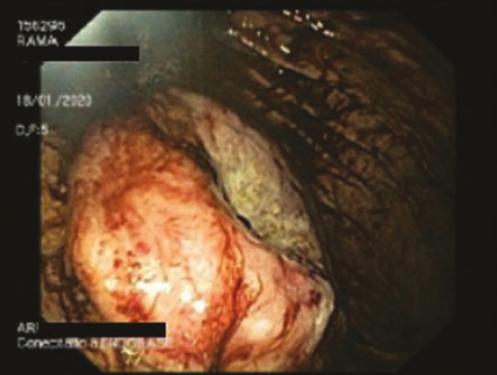
El mieloma múltiple se caracteriza por la proliferación neoplásica medular de células plasmáticas productoras de inmunoglobulina monoclonal. Un porcentaje pequeño de pacientes presenta compromiso extramedular en forma de plasmocitoma, siendo la localización más habitual las vías respiratorias altas. La afectación gastrointestinal es rara y la clínica asociada dependerá de la localización, la extensión y el mecanismo de infiltración. La afectación gástrica en forma de tumoración tiene un aspecto y una sintomatología similares a los de otras lesiones, por lo que es necesario realizar un diagnóstico histológico para un adecuado tratamiento. A continuación se presenta el caso de un mieloma múltiple con afectación gástrica.
Multiple myeloma is characterized by medullary neoplastic proliferation of plasma cells producing monoclonal immunoglobulin. A small percentage of patients have extramedullary involvement in form of plasmacytoma, the most common location being the upper respiratory tract. Gastrointestinal involvement is rare and the associated symptoms will depend on the location, extent and mechanism of infiltration. Gastric involvement presents an appearance and symptoms similar to other lesions, so a histological diagnosis is necessary for proper treatment. This article presents the case of multiple myeloma with gastric involvement.
El mieloma múltiple se caracteriza por la proliferación neoplásica de células plasmáticas productoras de inmunoglobulina (Ig) monoclonal. Aunque la mayoría de los pacientes presentan únicamente compromiso intramedular, hasta en un 20% puede haber compromiso extramedular en forma de plasmocitoma, siendo la localización más habitual las vías respiratorias altas (orofaringe, nasofaringe, senos paranasales y laringe)1. La afectación del tracto digestivo es rara; representa menos del 5% de los plasmocitomas y menos del 1% de los casos de mieloma múltiple2. La localización gastrointestinal más habitual es el intestino delgado, seguido del estómago, el colon y el esófago1. La mayoría de los pacientes con afectación gastrointestinal son diagnosticados durante el seguimiento de la enfermedad o en recidivas, siendo excepcional su diagnóstico como primera manifestación. A continuación se presenta el caso de un plasmocitoma gástrico que debuta con hemorragia digestiva, y se realiza una revisión de la literatura.
Mujer de 76 años con antecedentes de hipertensión y diabetes tipo 2, diagnosticada de mieloma múltiple IgG-kappa en febrero de 2018, en estadio IIIA ISS-3, con afectación únicamente intramedular con múltiples lesiones óseas y tratada con nueve ciclos de bortezomib-ciclofosfamida-dexametasona (VCD) con muy buena respuesta parcial en diciembre de 2018. En seguimiento en consultas de hematología, en el último control, en diciembre de 2019, presenta estabilidad clínica pero con una lenta progresión de la gammapatía, por lo que se plantea reiniciar el tratamiento antimieloma.
Previo al inicio del tratamiento, ingresa en el servicio de digestivo en enero de 2020 por melenas junto con anemización hasta una hemoglobina de 7,9, sin presentar clínica digestiva ni repercusión hemodinámica. Se inicia el estudio realizando una gastroscopia preferente (Fig. 1), con hallazgo de una lesión de aspecto submucoso en la curvatura mayor de unos 50 mm de diámetro, con gran úlcera central que sugiere tumoración del estroma gas -
trointestinal (GIST, gastrointestinal stromal tumor); se realizan biopsias de la lesión. Se completa el estudio con tomografía computarizada toracoabdominopélvica (Fig. 2), en la que se objetiva, además de la masa en el fundus gástrico de 7 × 2,6 cm, un nódulo sólido paravertebral izquierdo de 2,4 cm y múltiples lesiones óseas líticas. Se realiza también una resonancia magnética de cuerpo entero solicitada por el servicio de hematología, y se objetivan los mismos hallazgos que en la tomografía computarizada.
umbilicada y muy friable. Presenta adenopatías aumentadas de tamaño en el tronco celíaco. Dado que se desconoce todavía la naturaleza de la lesión, y ante los hallazgos intraoperatorios sospechosos de malignidad, se decide realizar una gastrectomía total con linfadenectomía D2 y reconstrucción en Y de Roux.
El posoperatorio transcurre sin incidencias y se reinicia la dieta oral al séptimo día, tras realizar un estudio baritado que no objetivaba fugas. Al décimo día de posoperatorio presenta fiebre y dolor en el hipocondiro derecho secundario a un hematoma intraabdominal infectado que requiere drenaje percutáneo, con mejoría posterior. La paciente es dada de alta en el día 21 de posoperatorio.
Figura 1. Imagen endoscópica de la lesión.
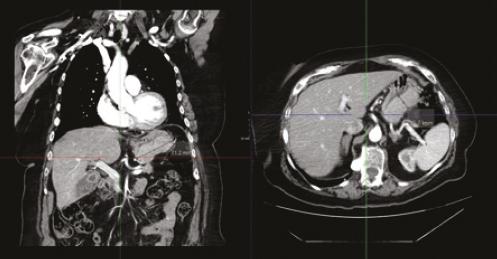
El estudio anatomopatológico de la pieza reveló una tumoración de 12, 5 × 9 cm correspondiente a una proliferación neoplásica de células plasmáticas con restricción de cadenas ligeras Kappa. Cinco ganglios linfáticos del grupo 1 presentaban proliferación de células plasmáticas; el resto eran reactivos. Se hallaron además tres implantes en el tejido adiposo del tronco celíaco. Las biopsias realizadas preoperatoriamente también mostraban infiltración gástrica por mieloma de células plasmáticas kappa.
Actualmente la paciente se encuentra a la espera de iniciar el tratamiento antimieloma.
Figura 2. Imagen de tomografía computarizada de la lesión.
Se presenta el caso en el comité de tumores y se decide realizar cirugía resectiva de la lesión gástrica, sospechosa de GIST, por hemorragia digestiva, a pesar de no tener los resultados de anatomía patológica y previo a reiniciar el tratamiento del mieloma. Antes de la fecha programada para la intervención, la paciente reingresa por melenas y anemización hasta una hemoglobina de 4,8, por lo que se decide realizar una cirugía preferente.
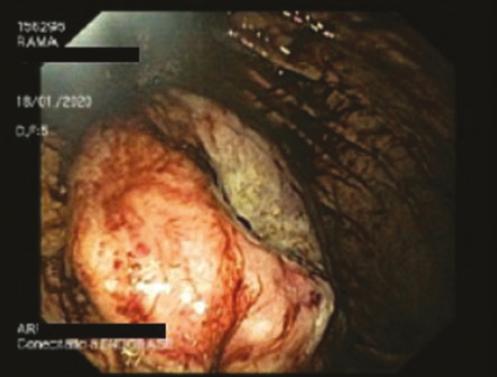
Se interviene a principios de febrero de 2020 mediante abordaje laparoscópico, que muestra una tumoración de gran tamaño en el cuerpo gástrico, cara posterior, de unos 12 cm de diámetro máximo,
El plasmocitoma extramedular es poco frecuente, puede aparecer de forma solitaria o asociado a mieloma múltiple, representando una progresión extramedular de la enfermedad3. La localización más habitual es las vías aéreas altas, y la afectación gastrointestinal rara. En un estudio publicado por Talamo, et al.2 en el que se revisaban 2584 casos de mieloma múltiple, solo 24 (0,9%) presentaban afectación gastrointestinal. Además, se observó que la afectación gástrica al manifestarse la enfermedad es mucho más infrecuente que cuando se presenta en el curso de una enfermedad avanzada o en progresión, como en nuestra paciente, asocián
Endoscópicamente se presentan como múltiples ulceraciones mucosas o bien como una masa única ulcerada, similar a otro tipo de tumores con los cuales debe hacerse un diagnóstico diferencial.
dose en estos casos a mal pronóstico.
La sintomatología asociada a la afectación gastrointestinal depende de la extensión y se produce por tres mecanismos: invasión directa de un órgano, efecto masa o ascitis mielomatosa2. Cuando afecta al estómago, el mecanismo suele ser por invasión directa y los síntomas son anorexia, pérdida de peso, náuseas, vómitos, sangrado digestivo oculto y rara vez sangrado activo1,3-6. En nuestra paciente destacaban la importante anemización y la hemorragia digestiva en forma de melenas, sin presentar inestabilidad hemodinámica que requiriese realizar una intervención de urgencia.
Endoscópicamente se presentan como múltiples ulceraciones mucosas o bien como una masa única ulcerada, similar a otro tipo de tumores con los cuales debe hacerse un diagnóstico diferencial. Dentro de los diagnósticos hay que considerar el adenocarcinoma, los GIST, los tumores neuroendocrinos, los linfomas (en particular el linfoma MALT [mucosa associated lymphoid tissue]) y la amiloidosis gastrointestinal, por lo que la realización de biopsias para el estudio anatomopatológico e inmunohistoquímico es crucial3,4,7 Desde el punto de vista inmunológico, un 27% expresan IgG, un 33% IgA y un 40% IgM1. En cuanto a su estudio histológico, es importante diferenciarlo del linfoma de células B de bajo grado asociado a las mucosas (linfoma MALT), ya que el estómago es su localización más habitual1,4
Cuando se manifiesta como plasmocitoma solitario, tanto la cirugía o exéresis endoscópica como la radioterapia se han descrito como opciones terapéuticas3,6,7 En ocasiones, cuando no es posible conseguir márgenes libres de tumor, se puede asociar radioterapia. La única excepción serían los plasmocitomas solitarios localizados en la cabeza y el cuello. En estos casos, la radioterapia es el tratamiento de elección6. Cuando se manifiesta asociado a mieloma múltiple se han descrito diversas opciones terapéuticas según su forma de presentación. Si se presentan en forma de hemorragia digestiva se puede realizar una endoscopia terapéutica (habitualmente con malos resultados y resangrado por tratarse de un tejido tumoral)
o radioterapia local. Siddique, et al.8 reportan un caso de plasmocitoma duodenal tratado mediante embolización de la arteria gastroduodenal en un paciente con hemorragia digestiva recurrente y alto riesgo quirúrgico. La cirugía se suele indicar en los pacientes que presentan un sangrado incontrolable o recurrente, como en nuestro caso, y en aquellos que presentan clínica obstructiva3. La quimioterapia con esquemas como bortezomib + dexametasona se suele reservar para los casos más avanzados; es bien tolerada, con pocos efectos adversos y consigue remisiones completas, además de evitar los efectos adversos de la radiación abdominal9
En nuestro caso, las biopsias preoperatorias confirmaban el diagnóstico definitivo, pero su resultado no se obtuvo hasta después de la intervención. Nuestra paciente presentaba un sangrado persistente, por lo que la cirugía resectiva estaba indicada. El conocimiento de la naturaleza de la lesión preoperatoriamente podría haber modificado la técnica quirúrgica y haber realizado un procedimiento menos agresivo, por lo que creemos importante realizar un diagnóstico histológico.
Los autores declaran que no existe conflicto de intereses.
Los autores declaran que no existen fuentes de financiación.
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
1. Chim CS, Wong WM, Nicholls J, Chung LP, Liang R. Extramedullary sites of involvement in hematologic malignancies:case 3. Hemorrhagic gastric plasmacytoma as the primary presentation in multiple myeloma. J Clin Oncol. 2002;20:344-7.
2. Talamo G, Cavallo F, Zangari M, Barlogie B, Lee CK, Pineda-Roman M, et al. Clinical and biological features of multiple myeloma involving the gastrointestinal system. Haematologica. 2006;91:964-7.
3. Telakis E, Tsironi E, Tavoularis G, Papatheodorou K, Tzaida O, Nikolaou A. Gastrointestinal involvement in a patient with multiple myeloma:a case report. Ann Gastroenterol. 2009;22:287-90.
4. Vicuña Arregui M, Borobio Aguilar E, Vila Costas JJ, Cruz Viguria Alegría M, Arrechea Irigoyen M, Borda Celaya F, et al. Plasmocitoma gástrico como causa infrecuente de hemorragia digestiva alta. Gastroenterol Hepatol. 2008;31:217-20.
5. Comba IY, Torres Luna NE, Cooper C, Crespo MW, Carilli A. A rare case of extramedullary plasmacytoma presenting as massive upper gastrointestinal bleeding. Cureus. 2019;11:e3993.
6. Honkisz BJ. Multiple myeloma with the primary gastric manifestation. J Clin Case Rep. 2015;05(04). (COnsultado el 24 de marzo de 2020.) Disponible en:http://www.omicsgroup.org/journals/ multiple-myeloma-with-the-primary-gastric-manifestation-2165-7920-1000525. php?aid=55637
7. Park CH, Lee SM, Kim TO, Kim DU, Jung WJ, Kim GH, et al. Treatment of solitary extramedullary plasmacytoma of the stomach with endoscopic submucosal dissection. Gut Liver. 2009;3:334-7.
8. Siddique I, Papadakis KA, Weber DM, Glober G. Recurrent bleeding from a duodenal plasmacytoma treated successfully with embolization of the gastroduodenal artery. Am J Gastroenterol. 1999;94:1691-2.
9. Katodritou E, Kartsios C, Gastari V, Verrou E, Mihou D, Banti A, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone:first reported case. Leuk Res. 2008;32:339-41.
Revista Puertorriqueña de Medicina y Salud Pública
She needs a treatment shown to reduce risk of recurrence in high-risk early breast cancer (EBC) 1

The first FDA-approved addition to adjuvant ET in nearly 2 decades 1-9
ET=endocrine therapy; HER2−=human epidermal growth factor receptor 2–negative; HR+=hormone receptor–positive.
VERZENIO® (abemaciclib) is indicated in combination with endocrine therapy (tamoxifen or an aromatase inhibitor) for the adjuvant treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, node-positive, early breast cancer at high risk of recurrence and a Ki-67 score ≥20% as determined by an FDA-approved test.1


Severe diarrhea associated with dehydration and infection occurred in patients treated with Verzenio. Across four clinical trials in 3691 patients, diarrhea occurred in 81 to 90% of patients who received Verzenio. Grade 3 diarrhea occurred in 8 to 20% of patients receiving Verzenio. Most patients experienced diarrhea during the first month of Verzenio treatment. The median time to onset of the first diarrhea event ranged from 6 to 8 days; and the median duration of Grade 2 and Grade 3 diarrhea ranged from 6 to 11 days and 5 to 8 days, respectively. Across trials, 19 to 26% of patients with diarrhea required a Verzenio dose interruption and 13 to 23% required a dose reduction.
Instruct patients to start antidiarrheal therapy, such as loperamide, at the first sign of loose stools, increase oral fluids, and notify their healthcare provider for further instructions and appropriate follow-up. For Grade 3 or 4 diarrhea, or diarrhea that requires hospitalization, discontinue Verzenio until toxicity resolves to ≤Grade 1, and then resume Verzenio at the next lower dose.
Neutropenia, including febrile neutropenia and fatal neutropenic sepsis, occurred in patients treated with Verzenio. Across four clinical trials in 3691 patients, neutropenia occurred in 37 to 46% of patients receiving Verzenio. A Grade ≥3 decrease in neutrophil count (based on laboratory findings) occurred in 19 to 32% of patients receiving Verzenio. Across trials, the median time to first episode of Grade ≥3 neutropenia ranged from 29 to 33 days, and the median duration of Grade ≥3 neutropenia ranged from 11 to 16 days. Febrile neutropenia has been reported in <1% of patients exposed to Verzenio across trials. Two deaths due to neutropenic sepsis were observed in MONARCH 2. Inform patients to promptly report any episodes of fever to their healthcare provider.
Monitor complete blood counts prior to the start of Verzenio therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, or delay in starting treatment cycles is recommended for patients who develop Grade 3 or 4 neutropenia. Severe, life-threatening, or fatal interstitial lung disease (ILD) or pneumonitis can occur in patients treated with Verzenio and other CDK4/6 inhibitors. In Verzenio-treated patients in EBC (monarchE), 3% of patients experienced ILD or pneumonitis of any grade: 0.4% were Grade 3 or 4 and there was one fatality (0.1%). In Verzenio-treated patients in MBC (MONARCH 1, MONARCH 2, MONARCH 3), 3.3% of Verzenio-treated patients had ILD or pneumonitis of any grade: 0.6% had Grade 3 or 4, and 0.4% had fatal outcomes. Additional cases of ILD or pneumonitis have been observed in the postmarketing setting, with fatalities reported.
Monitor patients for pulmonary symptoms indicative of ILD or pneumonitis. Symptoms may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams. Infectious, neoplastic, and other causes for such symptoms should be excluded by means of appropriate investigations. Dose interruption or dose reduction is recommended in patients who develop persistent or recurrent Grade 2 ILD or pneumonitis. Permanently discontinue Verzenio in all patients with Grade 3 or 4 ILD or pneumonitis.
monarchE was a phase III clinical trial that enrolled 5,637 peri- and postmenopausal adult women and men with HR+, HER2−, node-positive EBC at high risk of recurrence. High risk was defined as 4+ positive nodes, or 1-3 positive nodes with Grade 3 disease or tumor size ≥5 cm (central Ki-67 testing was conducted retrospectively for patients with untreated breast tissue samples), or 1-3 positive nodes with Ki-67 ≥20%. All patients completed primary treatment prior to 1:1 randomization to receive either 150-mg, twice-daily Verzenio plus SoC ET or SoC ET alone for 2 years. ET continued through 5-10 years as clinically indicated. The primary endpoint was IDFS.1,2

IDFS=invasive disease–free survival; SoC=standard of care.


Grade ≥3 increases in alanine aminotransferase (ALT) (2 to 6%) and aspartate aminotransferase (AST) (2 to 3%) were reported in patients receiving Verzenio. Across three clinical trials in 3559 patients (monarchE, MONARCH 2, MONARCH 3), the median time to onset of Grade ≥3 ALT increases ranged from 57 to 87 days and the median time to resolution to Grade <3 was 13 to 14 days. The median time to onset of Grade ≥3 AST increases ranged from 71 to 185 days and the median time to resolution to Grade <3 ranged from 11 to 15 days.
Monitor liver function tests (LFTs) prior to the start of Verzenio therapy, every 2 weeks for the first 2 months, monthly for the next 2 months, and as clinically indicated. Dose interruption, dose reduction, dose discontinuation, or delay in starting treatment cycles is recommended for patients who develop persistent or recurrent Grade 2, or any Grade 3 or 4 hepatic transaminase elevation.
Please see Select Important Safety Information throughout and Brief Summary of full Prescribing Information for Verzenio on the following pages.

At 3 years, Verzenio reduced the risk of recurrence by more than a third1


HR=hazard ratio; OS=overall survival.
HR=hazard ratio; OS=overall survival.
See the breakthrough results at VerzenioData.com/EBC
See the breakthrough results at VerzenioData.com/EBC
At 3 years, Verzenio reduced the risk of recurrence by more than a third1 86.1% of patients remained recurrence-free with Verzenio plus ET vs 79.0% with ET alone.1





86.1% of patients remained recurrence-free with Verzenio plus ET vs 79.0% with ET alone.1
The number of events at the time of analysis was 104 with Verzenio plus ET vs 158 with ET alone.1
The number of events at the time of analysis was 104 with Verzenio plus ET vs 158 with ET alone.1
OS was immature. A total of 95 (4.7%) patients had died. Long-term follow-up is planned.1,2
OS was immature. A total of 95 (4.7%) patients had died. Long-term follow-up is planned.1,2
This post hoc efficacy analysis was performed at a median follow-up of 27.1 months. Additional exploratory analyses were performed at this time; efficacy results for the subpopulation with high-risk clinicopathological features and Ki-67 ≥20% are provided.3*
This post hoc efficacy analysis was performed at a median follow-up of 27.1 months. Additional exploratory analyses were performed at this time; efficacy results for the subpopulation with high-risk clinicopathological features and Ki-67 ≥20% are provided.3*
* Statistical significance was achieved for this subpopulation earlier at the final IDFS analysis. The result in this post hoc analysis cannot be interpreted as statistically significant.1
Statistical significance was achieved for this subpopulation earlier at the final IDFS analysis. The result in this post hoc analysis cannot be interpreted as statistically significant.
Venous thromboembolic events (VTE) were reported in 2 to 5% of patients across three clinical trials in 3559 patients treated with Verzenio (monarchE, MONARCH 2, MONARCH 3). VTE included deep vein thrombosis, pulmonary embolism, pelvic venous thrombosis, cerebral venous sinus thrombosis, subclavian and axillary vein thrombosis, and inferior vena cava thrombosis. In clinical trials, deaths due to VTE have been reported in patients treated with Verzenio.
Venous thromboembolic events (VTE) were reported in 2 to 5% of patients across three clinical trials in 3559 patients treated with Verzenio (monarchE, MONARCH 2, MONARCH 3). VTE included deep vein thrombosis, pulmonary embolism, pelvic venous thrombosis, cerebral venous sinus thrombosis, subclavian and axillary vein thrombosis, and inferior vena cava thrombosis. In clinical trials, deaths due to VTE have been reported in patients treated with Verzenio.
Verzenio has not been studied in patients with early breast cancer who had a history of VTE. Monitor patients for signs and symptoms of venous thrombosis and pulmonary embolism and treat as medically appropriate. Dose interruption is recommended for EBC patients with any grade VTE and for MBC patients with a Grade 3 or 4 VTE.
Verzenio has not been studied in patients with early breast cancer who had a history of VTE. Monitor patients for signs and symptoms of venous thrombosis and pulmonary embolism and treat as medically appropriate. Dose interruption is recommended for EBC patients with any grade VTE and for MBC patients with a Grade 3 or 4 VTE.

Verzenio can cause fetal harm when administered to a pregnant woman, based on findings from animal studies and the mechanism of action. In animal reproduction studies, administration of abemaciclib to pregnant rats during the period of organogenesis caused teratogenicity and decreased fetal weight at maternal exposures that were similar to the human clinical exposure based on area under the curve (AUC) at the maximum recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Verzenio and for 3 weeks after the last dose. Based on findings in animals, Verzenio may impair fertility in males of reproductive potential. There are no data on the presence of Verzenio in human milk or its effects on the breastfed child or on milk production. Advise lactating women not to breastfeed during Verzenio treatment and for at least 3 weeks after the last dose because of the potential for serious adverse reactions in breastfed infants.
Verzenio can cause fetal harm when administered to a pregnant woman, based on findings from animal studies and the mechanism of action. In animal reproduction studies, administration of abemaciclib to pregnant rats during the period of organogenesis caused teratogenicity and decreased fetal weight at maternal exposures that were similar to the human clinical exposure based on area under the curve (AUC) at the maximum recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Verzenio and for 3 weeks after the last dose. Based on findings in animals, Verzenio may impair fertility in males of reproductive potential. There are no data on the presence of Verzenio in human milk or its effects on the breastfed child or on milk production. Advise lactating women not to breastfeed during Verzenio treatment and for at least 3 weeks after the last dose because of the potential for serious adverse reactions in breastfed infants.
The most common adverse reactions (all grades, ≥10%) observed in monarchE for Verzenio plus tamoxifen or an aromatase inhibitor vs tamoxifen or an aromatase inhibitor, with a difference between arms of ≥2%, were diarrhea (84% vs 9%), infections (51% vs 39%), neutropenia (46% vs 6%), fatigue (41% vs 18%), leukopenia (38% vs 7%), nausea (30% vs 9%), anemia (24% vs 4%), headache (20% vs 15%), vomiting (18% vs 4.6%), stomatitis (14% vs 5%), lymphopenia (14% vs 3%), thrombocytopenia (13% vs 2%), decreased appetite (12% vs 2.4%), ALT increased (12% vs 6%), AST increased (12% vs 5%), dizziness (11% vs 7%), rash (11% vs 4.5%), and alopecia (11% vs 2.7 %).
The most frequently reported ≥5% Grade 3 or 4 adverse reaction that occurred in the Verzenio arm vs the tamoxifen or an aromatase inhibitor arm of monarchE were neutropenia (19.6% vs 1%), leukopenia (11% vs <1%), diarrhea (8% vs 0.2%), and lymphopenia (5% vs <1%).
Lab abnormalities (all grades; Grade 3 or 4) for monarchE in ≥10% for Verzenio plus tamoxifen or an aromatase inhibitor with a difference between arms of ≥2% were increased serum creatinine (99% vs 91%; .5% vs <.1%), decreased white blood cells (89% vs 28%; 19.1% vs 1.1%), decreased neutrophil count (84% vs 23%; 18.7% vs 1.9%), anemia (68% vs 17%; 1% vs .1%), decreased lymphocyte count (59% vs 24%; 13.2 % vs 2.5%), decreased platelet count (37% vs 10%; .9% vs .2%), increased ALT (37% vs 24%; 2.6% vs 1.2%), increased AST (31% vs 18%; 1.6% vs .9%), and hypokalemia (11% vs 3.8%; 1.3% vs 0.2%).
Strong and moderate CYP3A inhibitors increased the exposure of abemaciclib plus its active metabolites to a clinically meaningful extent and may lead to increased toxicity. Avoid concomitant use of ketoconazole. Ketoconazole is predicted to increase the AUC of abemaciclib by up to 16-fold. In patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the Verzenio dose to 100 mg twice daily with concomitant use of strong CYP3A inhibitors other than ketoconazole. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the Verzenio dose to 50 mg twice daily with concomitant use of strong CYP3A inhibitors. If a patient taking Verzenio discontinues a strong CYP3A inhibitor, increase the Verzenio dose (after 3 to 5 half-lives of the inhibitor) to the dose that was used before starting the inhibitor. With concomitant use of moderate CYP3A inhibitors, monitor for adverse reactions and consider reducing the Verzenio dose in 50 mg decrements. Patients should avoid grapefruit products.
Avoid concomitant use of strong or moderate CYP3A inducers and consider alternative agents. Coadministration of strong or moderate CYP3A inducers decreased the plasma concentrations of abemaciclib plus its active metabolites and may lead to reduced activity.
With severe hepatic impairment (Child-Pugh C), reduce the Verzenio dosing frequency to once daily. The pharmacokinetics of Verzenio in patients with severe renal impairment (CLcr <30 mL/min), end stage renal disease, or in patients on dialysis is unknown. No dosage adjustments are necessary in patients with mild or moderate hepatic (Child-Pugh A or B) and/or renal impairment (CLcr ≥30-89 mL/min).
Please see Select Important Safety Information throughout and Brief Summary of full Prescribing Information for Verzenio on the following pages.
AL HCP ISI_mE 12OCT2021
REFERENCES: 1. Verzenio [package insert]. Indianapolis, IN: Eli Lilly and Company. 2. Johnston SRD, Harbeck N, Hegg R, et al; monarchE Committee Members and Investigators. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, nodepositive high-risk, early breast cancer (monarchE). J Clin Oncol. 2020;38(34):3987-3998. doi:10.1200/JCO.20.02514. 3. Harbeck N, Rastogi P, Martin M, et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: updated efficacy and Ki-67 analysis from the monarchE study. Ann Oncol. 2021. doi:10.1016/j.annonc.2021.09.015. 4. Breast International Group (BIG) 1-98 Collaborative Group, Thürlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353(26):2747-2757. doi:10.1056/NEJMMoa052258. 5. Baum M, Buzdar A, Cuzick J, et al; ATAC (Arimidex, Tamoxifen Alone or in Combination) Trialists’ Group. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer. 2003;98(9):1802-1810. doi:10.1002/cncr.11745. 6. Howell A, Cuzick J, Baum M, et al; ATAC Trialists’ Group. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365(9453):60-62. doi:10.1016/SO140-6736(04)17666-6. 7. Mayer EL, Dueck AC, Martin M, et al. Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2021;22(2):212222. doi:10.10160/S1470-2045(20)30757-9. 8. A trial to evaluate efficacy and safety of ribociclib with endocrine therapy as adjuvant treatment in patients with HR+/HER2− early breast cancer (NATALEE). https://clinicaltrials. gov/ct2/show/ NCT03701334. Accessed February 12, 2021. 9. Loibl S, Marmé F, Martin M, et al. Palbociclib for residual high-risk invasive HR-positive and HER2-negative early breast cancer—the Penelope-B trial. J Clin Oncol. 2021;39(14):1518-1530. doi:10.1200/JCO.20.03639.
PP-AL-US-3237 03/2022 ©Lilly USA, LLC 2022. All rights reserved. Verzenio® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries or affiliates.

therapy (tamoxifen or an aromatase inhibitor) alone. Patients were randomly assigned to receive 150 mg of VERZENIO orally, twice daily, plus tamoxifen or an aromatase inhibitor, or tamoxifen or an aromatase inhibitor, for two years or until discontinuation criteria were met. The median duration of VERZENIO treatment was 24 months.
The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, leukopenia, diarrhea, and lymphopenia.
Fatal adverse reactions occurred in 0.8% of patients who received VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor), including: cardiac failure (0.1%), cardiac arrest, myocardial infarction, ventricular fibrillation, cerebral hemorrhage, cerebrovascular accident, pneumonitis, hypoxia, diarrhea and mesenteric artery thrombosis (0.03% each).
Permanent VERZENIO treatment discontinuation due to an adverse reaction was reported in 19% of patients receiving VERZENIO, plus tamoxifen or an aromatase inhibitor. Of the patients receiving tamoxifen or an aromatase inhibitor, 1% permanently discontinued due to an adverse reaction. The most common adverse reactions leading to VERZENIO discontinuations were diarrhea (5%), fatigue (2%), and neutropenia (0.9%).
Dose interruption of VERZENIO due to an adverse reaction occurred in 62% of patients receiving VERZENIO plus tamoxifen or aromatase inhibitors. Adverse reactions leading to VERZENIO dose interruptions in ≥5% of patients were diarrhea (20%), neutropenia (16%), leukopenia (7%), and fatigue (5%).
Dose reductions of VERZENIO due to an adverse reaction occurred in 44% of patients receiving VERZENIO plus endocrine therapy (tamoxifen or an aromatase inhibitor). Adverse reactions leading to VERZENIO dose reductions in ≥5% were diarrhea (17%), neutropenia (8%), and fatigue (5%).
The most common adverse reactions reported (≥20%) in the VERZENIO, plus tamoxifen or an aromatase inhibitor, arm and ≥2% higher than the tamoxifen or an aromatase inhibitor arm were: diarrhea, infections, neutropenia, fatigue, leukopenia, nausea, anemia, and headache. Adverse reactions are shown in Table 1 and laboratory abnormalities are shown in Table 2.
Table 1: Adverse Reactions (≥10%) of Patients Receiving VERZENIO Plus Tamoxifen or an Aromatase Inhibitor [with a Difference between Arms of ≥2%] in monarchE VERZENIO Plus Tamoxifen or an Aromatase Inhibitor N=2791
Tamoxifen or an Aromatase Inhibitor N=2800
All Gradesa % Grade 3 % Grade 4 % All Gradesb % Grade 3 % Grade 4 %
Gastrointestinal Disorders
Diarrhea 84 8 0 9 0.2 0 Nausea 30 0.5 0 9 <0.1 0 Vomiting 18 0.5 0 4.6 0.1 0 Stomatitisc 14 0.1 0 5 0 0
Infections and Infestations
Infectionsd 51 4.9 0.6 39 2.7 0.1
General Disorders and Administration Site Conditions
Fatiguee 41 2.9 0 18 0.1 0
Headache 20 0.3 0 15 0.2 0 Dizziness 11 0.1 0 7 <0.1 0
Metabolism and Nutrition Disorders
Decreased appetite 12 0.6 0 2.4 <0.1 0
Skin and Subcutaneous Tissue Disorders
Rashf 11 0.4 0 4.5 0 0 Alopecia 11 0 0 2.7 0 0
a Includes the following fatal adverse reactions: diarrhea (n=1), and infections (n=4)
b Includes the following fatal adverse reactions: infections (n=5)
c Includes mouth ulceration, mucosal inflammation, oropharyngeal pain, stomatitis.
d Includes all reported preferred terms that are part of the Infections and Infestations system organ class. Most common infections (>5%) include upper respiratory tract infection, urinary tract infection, and nasopharyngitis.
e Includes asthenia, fatigue.
f Includes exfoliative rash, mucocutaneous rash, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculo-papular, rash maculovesicular, rash morbilliform, rash papular, rash papulosquamous, rash pruritic, rash vesicular, vulvovaginal rash.
Clinically relevant adverse reactions in <10% of patients who received VERZENIO in combination with tamoxifen or an aromatase inhibitor in monarchE include:
• Pruritus-9%
• Dyspepsia-8%
• Nail disorder-6% (includes nail bed disorder, nail bed inflammation, nail discoloration, nail disorder, nail dystrophy, nail pigmentation, nail ridging, nail toxicity, onychalgia, onychoclasis, onycholysis, onychomadesis)
• Lacrimation increased-6%
• Dysgeusia-5%
• Interstitial lung disease (ILD)/pneumonitis-3% (includes pneumonitis, radiation pneumonitis, interstitial lung disease, pulmonary fibrosis, organizing pneumonia, radiation fibrosis – lung, lung opacity, sarcoidosis)
• Venous thromboembolic events (VTEs)-3% (includes catheter site thrombosis, cerebral venous thrombosis, deep vein thrombosis, device related thrombosis, embolism, hepatic vein thrombosis, jugular vein occlusion, jugular vein thrombosis, ovarian vein thrombosis, portal vein thrombosis, pulmonary embolism, subclavian vein thrombosis, venous thrombosis limb)
Table 2: Laboratory Abnormalities (≥10%) in Patients
Receiving VERZENIO Plus Tamoxifen or an Aromatase Inhibitor [with a Difference between Arms of ≥2%] in monarchE
VERZENIO Plus Tamoxifen or an Aromatase Inhibitor N=2791
Tamoxifen or an Aromatase Inhibitor N=2800
All Grades % Grade 3 % Grade 4 % All Grades % Grade 3 % Grade 4 %
Creatinine increased 99 0.5 0 91 <0.1 0 White blood cell decreased 89 19 <0.1 28 1.1 0 Neutrophil count decreased 84 18 0.7 23 1.6 0.3
Anemia 68 1.0 0 17 0.1 0 Lymphocyte count decreased 59 13 0.2 24 2.4 0.1
Platelet count decreased 37 0.7 0.2 10 0.1 0.1 Alanine aminotransferase increased 37 2.5 <0.1 24 1.2 0 Aspartate aminotransferase increased
31 1.5 <0.1 18 0.9 0 Hypokalemia 11 1.2 0.1 3.8 0.1 0.1
CYP3A Inhibitors
Strong and moderate CYP3A4 inhibitors increased the exposure of abemaciclib plus its active metabolites to a clinically meaningful extent and may lead to increased toxicity.
Avoid concomitant use of ketoconazole. Ketoconazole is predicted to increase the AUC of abemaciclib by up to 16-fold.
In patients with recommended starting doses of 200 mg twice daily or 150 mg twice daily, reduce the VERZENIO dose to 100 mg twice daily with concomitant use of strong CYP3A inhibitors other than ketoconazole. In patients who have had a dose reduction to 100 mg twice daily due to adverse reactions, further reduce the VERZENIO dose to 50 mg twice daily with concomitant use of strong CYP3A inhibitors. If a patient taking VERZENIO
VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021 VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021
discontinues a strong CYP3A inhibitor, increase the VERZENIO dose (after 3-5 half-lives of the inhibitor) to the dose that was used before starting the inhibitor. Patients should avoid grapefruit products.
With concomitant use of moderate CYP3A inhibitors, monitor for adverse reactions and consider reducing the VERZENIO dose in 50 mg decrements, if necessary.
Coadministration of strong or moderate CYP3A inducers decreased the plasma concentrations of abemaciclib plus its active metabolites and may lead to reduced activity. Avoid concomitant use of strong or moderate CYP3A inducers and consider alternative agents.
Based on findings in animals and its mechanism of action, VERZENIO can cause fetal harm when administered to a pregnant woman. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus. In animal reproduction studies, administration of abemaciclib during organogenesis was teratogenic and caused decreased fetal weight at maternal exposures that were similar to human clinical exposure based on AUC at the maximum recommended human dose (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
In an embryo-fetal development study, pregnant rats received oral doses of abemaciclib up to 15 mg/kg/day during the period of organogenesis. Doses ≥4 mg/kg/day caused decreased fetal body weights and increased incidence of cardiovascular and skeletal malformations and variations. These findings included absent innominate artery and aortic arch, malpositioned subclavian artery, unossified sternebra, bipartite ossification of thoracic centrum, and rudimentary or nodulated ribs. At 4 mg/kg/day in rats, the maternal systemic exposures were approximately equal to the human exposure (AUC) at the recommended dose.
There are no data on the presence of abemaciclib in human milk, or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed infants from VERZENIO, advise lactating women not to breastfeed during VERZENIO treatment and for 3 weeks after the last dose.
Based on animal studies, VERZENIO can cause fetal harm when administered to a pregnant woman.
Verify pregnancy status in females of reproductive potential prior to initiating treatment with VERZENIO.
Advise females of reproductive potential to use effective contraception during VERZENIO treatment and for 3 weeks after the last dose.
Males
Based on findings in animals, VERZENIO may impair fertility in males of reproductive potential.
The safety and effectiveness of VERZENIO have not been established in pediatric patients.
Of the 2791 VERZENIO-treated patients in monarchE, 15% were 65 years of age or older and 2.7% were 75 years of age or older.
Of the 900 patients who received VERZENIO in MONARCH 1, MONARCH 2, and MONARCH 3, 38% were 65 years of age or older and 10% were 75 years of age or older. The most common adverse reactions (≥5%) Grade 3 or 4 in patients ≥65 years of age across MONARCH 1, 2, and 3 were: neutropenia, diarrhea, fatigue, nausea, dehydration, leukopenia, anemia, infections, and ALT increased.
No overall differences in safety or effectiveness of VERZENIO were observed between these patients and younger patients.
No dosage adjustment is required for patients with mild or moderate renal impairment (CLcr ≥30-89 mL/min, estimated by Cockcroft-Gault [C-G]). The pharmacokinetics of abemaciclib in patients with severe renal impairment (CLcr <30 mL/min, C-G), end stage renal disease, or in patients on dialysis is unknown.
No dosage adjustments are necessary in patients with mild or moderate hepatic impairment (Child-Pugh A or B).
Reduce the dosing frequency when administering VERZENIO to patients with severe hepatic impairment (Child-Pugh C).
Additional information can be found at www.verzenio.com
VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021 VERZENIO® (abemaciclib) tablets, for oral use AL HCP BS_MonE 12OCT2021
Revista Puertorriqueña de Medicina y Salud Pública
Palabras clave Obesidad, Hipertensión, Niños, Puerto Rico, Prevalencia
Keywords Obesity, Hypertension, Children, Puerto Rico, Prevalence

Objective: To determine the prevalence of obesity and hypertension and the association of hypertension with obesity in an island-wide sample of school children in Puerto Rico.
Methods: The quantitative descriptive study included 3,145 children, 5 to 17 years of age, from Puerto Rico; they were examined once during a 3-year (2014−2017) period for weight (lbs) and height (cm) to calculate their body mass index (BMI) based on their age and sex. Children with BMIs in or above the 95th percentile were considered obese. The systolic and diastolic blood pressures (mm Hg) were measured once to determine the prevalence of hypertension based on age, height, and sex. Children with blood pressures in or above the 95th percentile were considered hypertensive. The blood pressures of obese and non-obese children were compared
using the independent samples t-test. The association between obesity status (obese/non-obese) and hypertensive status (hypertensive/non-hypertensive) was analyzed using the chi-square test. Results: A total of 25.7% of the children were obese. Boys were 1.38 times as likely to be obese as girls were. 9.9% of the children were hypertensive. Obese children were 2.82 times as likely to be hypertensive as non-obese children were. Conclusion: About 1 of every 4 children in the sample was obese; about 1 of 10 was hypertensive. Obese children were at a significantly higher risk for hypertension than non-obese children were. The study indicates the need for public health strategies that promote prevention and parental education to reduce the prevalence of obesity and the sequelae of hypertension. [P R Health Sci J 2021;40:45-49]
Objetivo: Determinar la prevalencia de la obesidad y la hipertensión y la asociación de la hipertensión con la obesidad, en una muestra de escolares de toda la isla, en Puerto Rico. Métodos: El estudio descriptivo cuantitativo incluyó a 3,145 niños, de 5 a 17 años de edad, de Puerto Rico; fueron examinados una vez durante un período de 3 años (2014-2017) para medir el peso (libras) y la altura (cm) para calcular su índice de masa corporal (IMC) en función de su edad y sexo. Los niños con IMC en o por encima del percentil 95 se consideraron obesos. Las presiones arteriales sistólica y diastólica (mm Hg) se midieron una vez para determinar la prevalencia de hipertensión en función de la edad, la estatura y el sexo. Los niños con presiones arteriales en o por encima del percentil 95 se consideraron hipertensos. Las presiones arte

riales de niños obesos y no obesos se compararon mediante la prueba t de muestras independientes. La asociación entre el estado de obesidad (obeso/no obeso) y el estado hipertenso (hipertenso/no hipertenso) se analizó mediante la prueba de chi-cuadrado.
Resultados: Un total de 25.7% de los niños eran obesos. Los niños tenían 1.38 veces más probabilidades de ser obesos que las niñas. El 9.9% de los niños eran hipertensos. Los niños obesos tenían 2.82 veces más probabilidades de ser hipertensos que los niños no obesos.
Conclusión: Aproximadamente 1 de cada 4 niños de la muestra era obeso; aproximadamente 1 de cada 10 era hipertenso. Los niños obesos tenían un riesgo significativamente mayor de hipertensión que los niños no obesos. El estudio indica la necesidad de estrategias de salud pública que promuevan la prevención y la educación de los padres para reducir la prevalencia de la obesidad y las secuelas de la hipertensión. [PR Health Sci J 2021;40:45-49
Realizamos el estudio con la aprobación de la junta de revisión institucional de la Universidad Interamericana de Puerto Rico y la División de Investigación del Departamento de Educación de Puerto Rico. El presente estudio fue parte de un proyecto más amplio que incluyó un examen ocular, que exploró, entre otras cosas, el error de refracción, la desviación ocular, la acomodación y las patologías oculares. Los padres firmaron consentimientos por escrito que permiten la participación de sus hijos y la adquisición de información de salud identificable e individual de sus hijos.
La población de estudio fue de 374,521 estudiantes de 5 a 17 años del sistema escolar público de Puerto Rico (4). La muestra incluyó a 3,145 niños de 5 a 17 años de edad de 30 escuelas en los 7 distritos escolares del Estado Libre Asociado de Puerto Rico. Un total de 1,584 de los niños (50.4%) eran varones y 1,561 (49.6%) eran féminas. Al menos 1 escuela primaria, intermedia y superior fueron escogidas al azar de cada distrito por el Director de Servicios de Enfermería de Salud del Departamento de Educación de Puerto Rico. Cada escuela tenía un máximo de 300 niños. Una vez que se seleccionó una escuela, todos los niños dentro de esa escuela eran elegibles para participar. Los padres de los niños recibieron un formulario de consentimiento informado para decidir si permitían la participación de su hijo en el estudio.
Se visitó cada escuela con anticipación, al menos 1 mes antes de los exámenes programados para esa institución, para discutir los objetivos y requisitos del proyecto con el director de la escuela, el trabajador social, la enfermera y otro personal asignado y para distribuir los formularios de consentimiento.
Se seleccionó un lugar dentro de cada escuela que pudiera acomodar el equipo, las estaciones de examen y el personal de examen.
Cada escuela requería de 2 a 4 visitas para examinar a todos los niños. El personal examinador en cada escuela visitada incluía 2 ó
3 doctores en optometría con licencia y de 6 a 8 estudiantes de optometría avanzada, quienes trabajaban bajo la supervisión de los optómetras de la Escuela de Optometría de la Universidad Interamericana de Puerto Rico. Todos los miembros del equipo de examen fueron entrenados en el protocolo del estudio. Cada niño cuyos padres o tutores dieron su consentimiento para su participación recibió una descripción del procedimiento de prueba y, si estaba dispuesto a participar, se le pidió que diera su consentimiento. Los exámenes de los niños participantes en las 30 escuelas involucradas en el estudio comenzaron en agosto de 2014 y terminaron en diciembre de 2017.
Usamos un estadiómetro portátil para medir la estatura de cada niño, sin zapatos, al milímetro más cercano. El peso (al décimo de libra más cercano) de cada niño se determinó utilizando una báscula electrónica. La báscula se calibró con pesos estándar antes de cada día de prueba. Las presiones arteriales sistólica y diastólica de cada niño se midieron una vez usando el siguiente protocolo: El manguito de un esfigmomanómetro se envolvió alrededor de la parte superior del brazo del niño sentado y el codo del niño se flexionó al nivel del corazón. El tamaño del manguito se eligió para adaptarse al brazo. Se pidió a los niños que se sentaran durante 3 minutos con los pies en el suelo y las piernas sin cruzar antes de tomar las medidas de la presión arterial. Las lecturas de la presión arterial se tomaron desde las 9:00 a.m. hasta el mediodía (5,6).
Las medidas de estatura, peso y edad se utilizaron para calcular el índice de masa corporal (IMC) de cada niño. Los niños con un IMC igual o mayor al percentil 95 fueron considerados obesos (7). Se consideró hipertensos a los niños con presión arterial (sistólica, diastólica o ambas) igual o superior al percentil 95 para su sexo, talla y edad (8). Se utilizó una aplicación de Estadísticas EBMcalc, que aplica las normas vigentes para la identificación de niños obesos e hipertensos (9).
Los resultados en cuanto a las prevalencias de obesidad e hipertensión fueron ponderados para cada grupo de escolares por edad y género. Los pesos se obtuvieron al dividir el número de niños (en el año 2014) en la población, entre el número de niños en la muestra de estudio. Realizamos análisis estadísticos utilizando los datos ponderados (10).
Determinamos para cada grupo de edad (5 a 17 años), el intervalo de confianza del 95% de la proporción de niños obesos y/o hipertensos con respecto al total de niños (11).
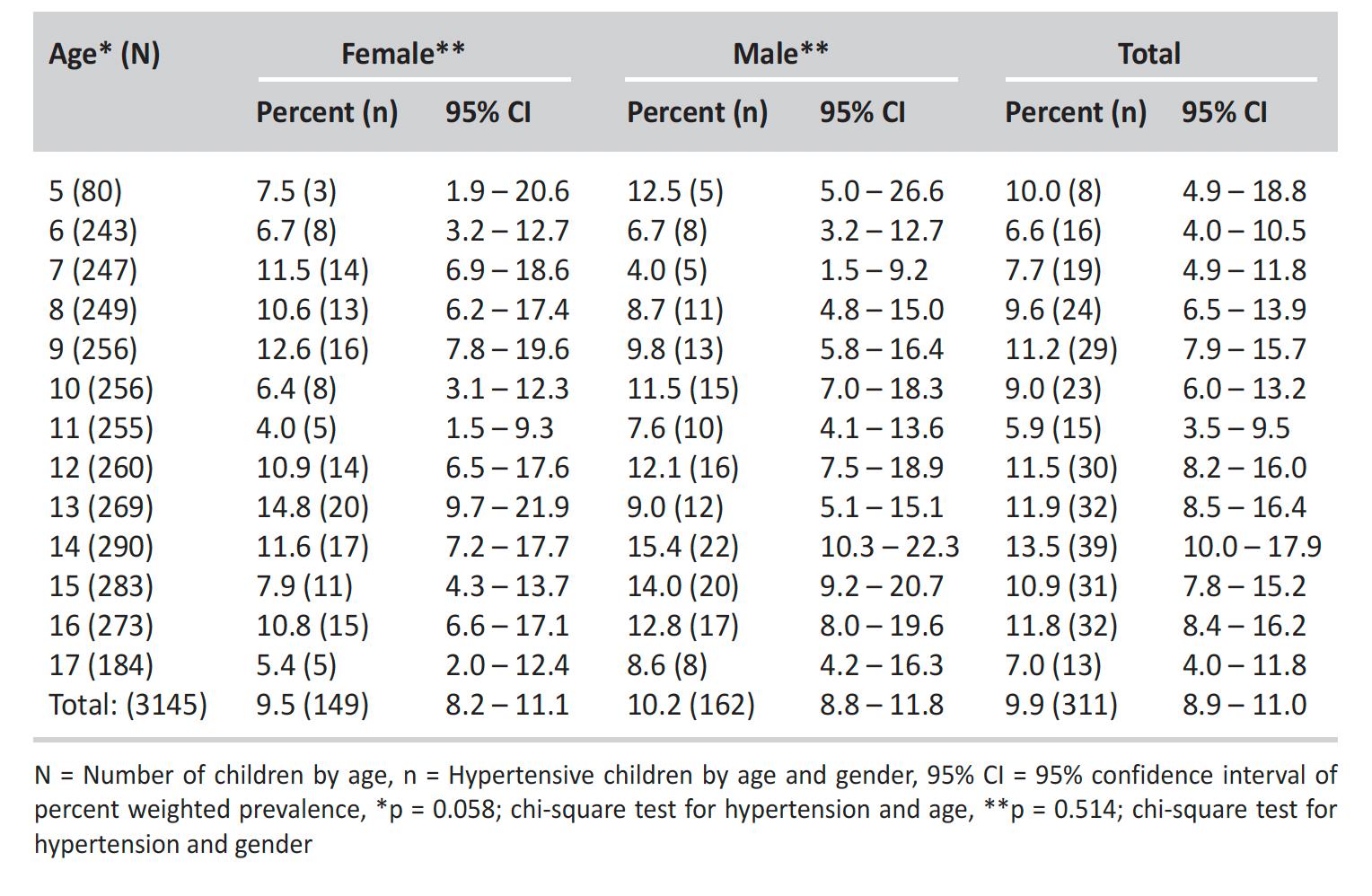
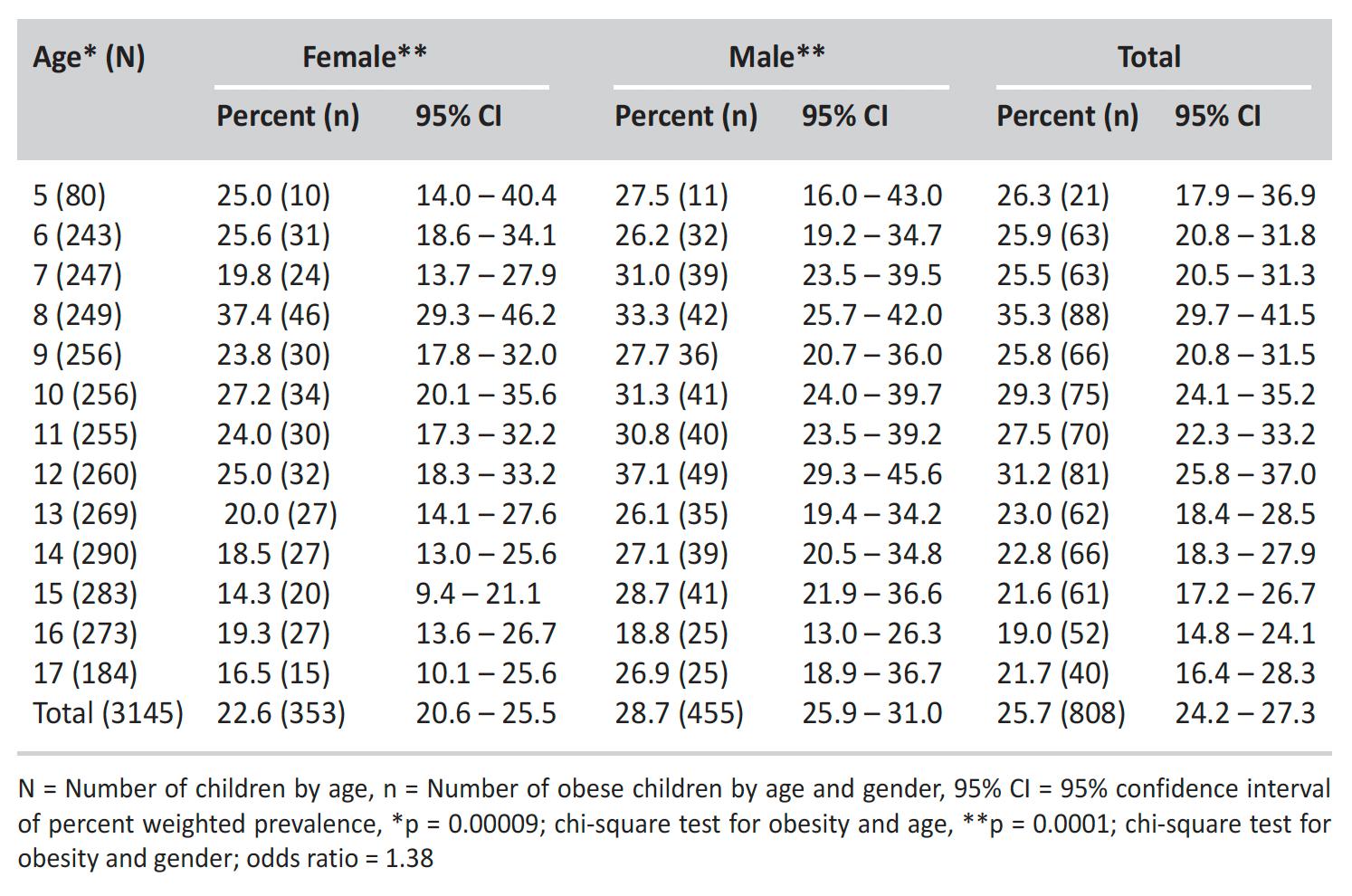
Comparamos las presiones arteriales sistólica y diastólica media de los niños obesos y no obesos utilizando la prueba t de muestras independientes (12).
Utilizamos pruebas de chi-cuadrado para determinar la asociación entre las variables nominales (obesidad e hipertensión) y la variable grupo de edad (5 a 17 años) de los niños. La fuerza de la asociación se determinó usando el Phi (valor x) para las comparaciones de la tabla de contingencia 2 X 2 (estado de obesidad y género o estado de hipertensión y género). El valor de la V de Cramer se utilizó para tablas de contingencia más grandes (estado de obesidad y edad y estado de hipertensión y género) (12). Los residuos ajustados de la prueba de chi-cuadrado se utilizaron para realizar pruebas post-hoc con el fin de determinar las categorías de edad en las que la obesidad y la hipertensión eran significativamente diferentes entre sí (13). Finalmente, realizamos una prueba de chi-cuadrado para determinar la razón de posibilidades y la asociación entre obesidad e hipertensión (14). Todos los análisis estadísticos se realizaron con el software IBM SPSS Statistics para Windows, versión 25 (15).
Tabla 1.
Prevalencia ponderada de obesidad en escolares de Puerto Rico, por edad y sexo
Tabla 2.
Prevalencia ponderada de hipertensión en escolares de Puerto Rico, por edad y sexo
Un total de 25.7% (IC 95%: 24.2 − 27.3%) de los niños eran obesos. De los varones, el 28.7 % (IC 95 %: 25.9 − 31.0 %) eran obesos. Un total de 22.6% (IC 95%: 20.6 − 25.5%) de las féminas eran obesas. Hubo asociación significativa (x2 = 15.24; p = 0.0001) entre el estado de obesidad (obeso, no obeso) y el sexo (masculino, femenino) del niño. Sin embargo, la fuerza de la asociación fue baja (x= 0.07; p = 0.0001). El cociente de probabilidades fue de 1.38 (IC del 95 %: 1.17 - 1.62), lo que indica que los niños tenían aproximadamente 1.4 veces más probabilidades de ser obesos que las niñas. Hubo una asociación significativa entre el estado de obesidad y la edad (x2 = 31.71; p = 0.002). La fuerza de la asociación fue baja (V de Cramer = 0.10). Una prueba post-hoc utilizando los residuos ajustados mostró que la obesidad era significativamente mayor en la categoría de edad de 8 años (35.3%) que en las otras categorías de edad, como se muestra en la Tabla 1.
En total, el 9.9 % (IC 95 % = 8.9 –11.0 %) de los niños eran hipertensos.
La asociación entre el estado hipertenso (hipertensión, no hipertensión) y el sexo (masculino, femenino) no fue significativa (x2 = 0.426; p = 0.514). La asociación del estado hipertenso con la edad del niño (5 a 17 años) tampoco fue significativa (x2 = 20.51; p = 0.058). La Tabla 2 muestra la prevalencia ponderada de hipertensión arterial por edad y sexo de los escolares.
De los niños obesos, el 17.8% (IC 95%: 15.4 − 20.6%) eran hipertensos y el 82.2% (IC 95%: 79.4 − 84.7%) no eran hipertensos. De los niños no obesos, el 7.1% (IC 95%: 6,2-8,2%) eran hipertensos y el 92.9% (IC 95%: 91.7-93,9%) no eran hipertensos. Una prueba de chi-cuadrado para la asociación entre obesidad e hipertensión mostró una asociación estadísticamente significativa entre las 2 variables (x2 = 77.244; p<0.001). La fuerza de la asociación entre hipertensión y obesidad fue baja (x = 0.157; p<0.001).
La razón de posibilidades (OR) indicó que los niños obesos tenían 2.82 veces más probabilidades de ser hipertensos que los niños no obesos (IC del 95% = 2.23 - 3.59).
La presión arterial sistólica media (mm
Hg) de los sujetos fue significativamente mayor en los niños obesos (107.4 ± 16.6) que en los niños no obesos (100.6 ± 14.6). La presión arterial diastólica media fue significativamente mayor en los niños obesos (68.4 ± 10.7) que en los no obesos (63.9 ± 9.9). La diferencia en las presiones arteriales sistólica y diastólica también fue significativamente mayor en las féminas y los varones obesos que en las niñas y los niños no obesos, respectivamente (Tabla 3).
Tabla 3.
Niveles medios (±DE) de presión arterial sistólica y diastólica, por género y nivel de obesidad, en escolares de Puerto Rico
Aproximadamente 1 de cada 4 niños en nuestra muestra de escolares de 5 a 17 años (25.7%) que vivían en Puerto Rico era obeso. Un estudio previo de 352 niños residentes en Puerto Rico de 12 a 16 años de edad de una sola escuela en San Juan (la ciudad capital de Puerto Rico) realizado en 1999 y 2000 encontró una prevalencia de obesidad de 14.2% (95%IC total: 10.9 − 18.3%) (16). La prevalencia de obesidad entre los niños de 12 a 16 años en nuestro estudio fue del 23.4 % (IC del 95 %: 21.3 − 25.8 %). Una diferencia fue que nuestra muestra en este grupo de edad era más grande (1,375 niños) e incluía niños de 7 regiones educativas de Puerto Rico, no solo de la región de San Juan.
El estudio de los niños de San Juan también se completó casi una década antes que el nuestro. La prevalencia de la obesidad ha ido en aumento en los niños de los Estados Unidos (incluidos los 50 estados y el Distrito de Columbia), y podrían estar ocurriendo tendencias similares en los niños de Puerto Rico (17). Nuestra prevalencia de obesidad es similar a la del 25.8% en hispanos de 2 a 19 años en los Estados Unidos (17). En nuestro estudio, la prevalencia en niños (28.7%) fue significativamente mayor que la prevalencia en niñas (22.6%). En hispanos de 2 a 19 años en Estados Unidos (incluidos los 50 estados y el Distrito de Columbia), la prevalencia también fue mayor en niños (28.0%) que en niñas (23.6%),
aunque esta diferencia no fue estadísticamente significativa (17).
El grupo de 8 años de nuestro estudio presentó la mayor prevalencia de obesidad, con un 35.3% (IC 95%: 29.7 − 41.5%). Este hallazgo concuerda con la prevalencia de obesidad en niños hispanos en los Estados Unidos (incluyendo los 50 estados y el Distrito de Columbia), en el grupo de edad media (6 a 11 años), que es superior a la encontrada en los otros grupos de edad (18). Entre 1971 y 2016, inclusive, el grupo de edad media también ha mostrado el mayor incremento de obesidad (4X), en comparación con los demás grupos de edad (19). Futuros estudios deberían abordar los factores que hacen que este grupo de edad sea más vulnerable al aumento de la obesidad.
Como descubrimos con nuestra prueba única, 1 de cada 10 (9.9 %) de los niños de nuestra muestra era hipertenso. Esta prevalencia es comparable a la prevalencia del 9.5 % a la que se llegó después de 2 exámenes de detección en un estudio
de niños multiétnicos en Houston, Texas (3). La prevalencia de hipertensión en los niños obesos de nuestro estudio (17.8%) fue similar a la prevalencia del 18.0% en los niños hispanos con sobrepeso en Texas. Reprodujimos el hallazgo de una asociación significativa entre la hipertensión infantil y la obesidad encontrada en algunos estudios (3,20). Nuestros resultados mostraron que un niño obeso tenía 2.82 veces más probabilidades de ser hipertenso que un niño no obeso; ligeramente superior a las probabilidades de 2.10 para los niños hispanos de Texas con sobrepeso (20).
La American Heart Association tiene recomendaciones para la determinación óptima de la presión arterial en niños (5), incluido el uso de la media de 2 medidas de presión arterial por sesión. Además, el Instituto Nacional del Corazón, los Pulmones y la Sangre recomienda que se tomen medidas en el transcurso de 3 sesiones separadas para el diagnóstico de presión arterial alta en niños (8,21). Aunque
seguimos la mayoría de las recomendaciones, los resultados de nuestro estudio estuvieron limitados por el uso de una sola medida de presión arterial tomada en 1 sesión. Nuestras conclusiones también se vieron limitadas por la caída del 12 % en la población de niños en edad escolar en Puerto Rico que ha ocurrido desde el inicio del estudio en 2014, caída que puede afectar potencialmente los valores de prevalencia actuales (4,22).
Aunque la esperanza de vida ha aumentado constantemente durante muchos años, la epidemia de obesidad puede anular o incluso revertir esta tendencia. Como resultado, algunos investigadores concluyen que la generación actual de niños y adolescentes puede tener una esperanza de vida más corta que la de sus padres (23). Los resultados de nuestro estudio indican que existe una necesidad urgente de establecer políticas en toda la isla para limitar el crecimiento de la prevalencia de la obesidad y la hipertensión e los niños. Dichas políticas deben incluir la
promoción de estilos de vida saludables, la educación de los padres y la identificación temprana de niños obesos e hipertensos.
Objetivo: Determinar la prevalencia de la obesidad y la hipertensión y la asociación de la hipertensión con la obesidad en una muestra de escolares de toda la isla de Puerto Rico.
Métodos: El estudio descriptivo cuantitativo incluyó a 3.145 niños de 5 a 17 años de Puerto Rico. Fueron examinados una vez durante un período de 3 años (2014-2017) para determinar el peso (lb) y la altura (cm) para calcular su índice de masa corporal (IMC) en función de su edad y sexo. Los niños con IMC en o por encima del percentil 95 se consideraron obesos. Las presiones arteriales sistólica y diastólica (mm Hg) se midieron una vez para determinar la prevalencia de hipertensión según la edad, la altura y el sexo. Los niños con presión arterial dentro o por encima del percentil 95 se consideraron hipertensos. La presiones arteriales de niños obesos y no obesos se compararon utilizando la prueba t de muestras independientes. La asociación entre el estado de obesidad (obeso / no obeso) y el estado de hipertensión (hipertenso / no hipertenso) se analizó mediante la prueba de chi-cuadrado.
Revista Puertorriqueña de Medicina y Salud Pública
Resultados: El 25.7% de los niños eran obesos. Los niños tenían 1.38 veces más probabilidades de ser obesos que las niñas. El 9.9% de los niños eran hipertensos. Los niños obesos tenían 2.82 veces más probabilidades de ser hipertensos que los niños no obesos.
Conclusión: Aproximadamente 1 de cada 4 niños de la muestra era obeso; aproximadamente 1 de cada 10 era hipertenso. Los niños obesos tenían un riesgo significativamente mayor de hipertensión que los niños no obesos. El estudio indica la necesidad de estrategias de salud pública que promuevan la prevención y educación de los padres para reducir la prevalencia de la obesidad y las secuelas de la hipertensión.
Reconocemos el apoyo financiero de nuestro proyecto por parte de la Fundación Internacional de Clubes de Leones (LCIF). También reconocemos la colaboración del personal del Departamento de Educación
de Puerto Rico (Secretaría de Educación, directores de escuela y enfermeras).
1. Caballero B. The global epidemic of obesity: an overview. Epidemiol Rev. 2007;29:15. doi:10.1093/epirev/mxm012
2. Obesity and overweight. World Health Organization. February 16, 2018. Accessed October 13, 2019. http://www.who.int/mediacentre/fact-sheets/fs311/en/
3. Sorof JM, Lai D, Turner J, Poffenbarger T, Portman RJ. Overweight, eth-nicity, and the prevalence of hypertension in school-aged children. Pedi-atrics. 2004;113(3 Pt 1):475482. doi:10.1542/peds.113.3.475
4. Disdier-Flores OM, Jara-Castro AG. Anuario estadístico del sistema edu-cativo año escolar 2014-2015. Instituto de Estadísticas de Puerto Rico. 2017. Accessed October 18, 2018 https://estadisticas.pr/files/Publica-ciones/ Anuario_Estadistico_Educativo_2014-2015.pdf
5. Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pres-sure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circula-tion. 2005;111(5):697-716. doi:10.1161/01. CIR.0000154900.76284.F6
6. Corrado C. Blood pressure measurement in children. Ital J Pediatr. 2015;41(Suppl 2):A19. Published 2015 Sep 30. doi:10.1186/18247288-41-S2-A19
7. Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11. 2002;(246):1-190.
8. National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2 Suppl 4th Report):555-576.
9. EBMcalc. Foundation Internet Services; 2019.
10. Bethlehem J. Applied survey methods –A statistical perspective: weight-ing. Wiley; 2009. Accessed October 10, 2019. http:// www.applied-sur-vey-methods.com/weight.html#weg1
11. QuickCalcs: Confidence interval of a proportion or count. GraphPad. 2019. Accessed October 10, 2019. https://www.graphpad. com/quick-calcs/ConfInterval1.cfm
12. Statistical tutorials and software guides. Laerd Statistics. 2019. Accessed October 10, 2019. https://statistics.laerd.com
13. Gignac GE. Non-Parametrics. In: How2statsbook (Online Edition 1). 2019. Accessed October 1, 2019. http://www.how2statsbook.com
14. Lang TA, Secic M. How to Report Statistics in Medicine. American Col-lege of Physicians; 2006.
15. IBM. SPSS. Version 25. International Business Machines. https://www. ibm.com/ support/knowledgecenter/SSLVMB_25.0.0/ statistics_kc_ ddita/spss/product_landing.html
16. Venegas HL, Pérez CM, Suárez EL, Guzmán M. Prevalence of obesity and its association with blood pressure, serum lipids and selected lifestyles in a Puerto Rican population of adolescents 12-16 years of age [published correction appears in P R Health Sci J. 2003 Dec;22(4):417]. P R Health Sci J. 2003;22(2):137-143.
17. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity Among Adults and Youth: United States, 2015-2016. NCHS Data Brief. 2017;(288):1-8. Accessed October 9, 2019. https://www.cdc.gov/nchs/data/databriefs/db288.pdf
18. Ogden CL, Carroll MD, Lawman HG, et al. Trends in Obesity Preva-lence Among Children and Adolescents in the United States, 1988-1994 Through 2013-2014. JAMA. 2016;315(21):2292-2299. doi:10.1001/ jama.2016.6361
19. Overweight children and youth. 2018. November 4, 2018. Child Trends. Accessed October 10, 2019. https://www.childtrends. org/indicators/overweight-children-and-youth.
20. Rosner B, Cook N, Portman R, Daniels S, Falkner B. Blood pressure dif-ferences by ethnic group among United States children and adolescents. Hypertension. 2009;54(3):502508. doi:10.1161/HYPERTENSIO-NAHA.109.134049
21. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents. Summary Report. National Heart, Blood and Lung Institute. October 2012. Accessed October 8, 2019. https://www.nhlbi.nih.gov/files/docs/peds_guidelines_sum.pdf
22. Irizarry-Alvarez F. Educación tendrá 41,000 estudiantes menos para este año escolar. Primera Hora. July 19, 2018. Accessed October 2, 2018. Available at: Url: http://www.primerahora.com/noticias/ puerto-rico/nota/educaciontendra41000estudiantesmenosparaesteanoescolaryc-uidaosimas-1293421/
23. Olshansky SJ, Passaro DJ, Hershow RC, et al. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. 2005;352(11):1138-1145. doi:10.1056/ NEJMsr043743

2021 ABBOTT LABORATORIES ANAPRGLU202115698
© 2021 ABBOTT LABORATORIES ANAPRGLU202115698
Diseñado como un reemplazo de merienda o comida con CARBSTEADY mezcla única de carbohidratos de liberación lenta para ayudarte a manejar el azúcar* en la sangre.
Diseñado como un reemplazo de merienda o comida con CARBSTEADY mezcla única de carbohidratos de liberación lenta para ayudarte a manejar el azúcar* en la sangre.
Diseñado como un reemplazo de merienda o comida con CARBSTEADY mezcla única de carbohidratos de liberación lenta para ayudarte a manejar el azúcar* en la sangre.


PARA PERSONAS CON DIABETES
PARA PERSONAS CON DIABETES
APD afiliado de Abbott. Diseñado para ayudar a minimizar los picos de azúcar en la sangre.
*Diseñado para ayudar a minimizar los picos de azúcar en la sangre.
APD afiliado de Abbott. *Diseñado para ayudar a minimizar los picos de azúcar en la sangre.
APD afiliado de Abbott.
Revista Puertorriqueña de Medicina y Salud Pública 61
*Diseñado para
*Diseñado

Por: Dr. Bárbara L. Riestra-Candelaria α†, Juan Carlos Jorge σ, Miriam Rodríguez ρ, Gory Ballester-OrtizѠ & Loida A. González-Rodríguez¥

Autor α: PhD Departmento de Medicina, Endocrinología, Diabetes, División de Metabolismo, Escuela de Medicina, Universidad de Puerto Rico, San Juan, PR. † Dirección: Departamento de Anatomía y Biología y Biología Celular, Universidad Central del Caribe. Bayamón, PR.
Autor σ: Doctorado Departamento de Anatomía y Neurobiología, Facultad de Medicina, Universidad de Puerto Rico, San Juan, PR.
Autor ρ: Departamento de Imágenes Médicas de BSD, Facultad de Ciencias y Tecnología. Universidad Central del Caribe. Bayamón, PR.
Autor Ѡ: MD Departamento de Ciencias Radiológicas, Escuela de Medicina, Universidad de Puerto Rico. San Juan, PR.
Autor para correspondencia ¥: MD Departamento de Medicina, Endocrinología, División de Diabetes y Metabolismo, Facultad de Medicina, Universidad de Puerto Rico. San Juan, PR. correo electrónico: loida.gonzalez@upr.edu
GJMR-D
Classification: NLMC
Code: WK 810
Estrictamente de acuerdo con el cumplimiento y las regulaciones de:
© 2020. Dra.
Gory
&
A.
Rodríguez. Este es un artículo de investigación / revisión, distribuido bajo los términos de la Licencia de Reconocimiento-No Comercial 3.0 Unported de Creative Commons http://creativecommons.org/ licenses/by-nc/3.0/), que permite todo uso, distribución y distribución no comercial. reproducción en cualquier medio, siempre que la obra original esté debidamente citada.
El daño al hígado es una consecuencia clínica común de la diabetes mellitus crónica tipo 2 (DM2). Este estudio evalúa si la elastografía de ondas de corte por ultrasonido y la hemodinámica de la vena porta y la arteria hepática pueden complementar los datos de estudios clínicos tradicionales para el seguimiento de la salud del hígado en pacientes con DM2.
Se reclutaron sesenta y cuatro (64) participantes (31 controles y 33 pacientes con diabetes mellitus tipo 2 confirmada) entre 21 y 74 años de edad. El tamaño del hígado, la rigidez y la hemodinámica de la vena porta y la arteria hepática fueron evaluado. Se controlaron la hemoglobina glucosilada (HbA1c), el aspartato aminotransferasa (AST), la alanina a minotransferasa (ALT) y la fosfatasa alcalina (ALP). Se empleó la prueba t de Student con
alcanzada ap <0.05.
Se detectaron diferencias significativas asintomáticas entre los pacientes con DMT2: (1) Tamaño del hígado más grande (p = 0,04); (2) Mayor rigidez del hígado (p = 0.04); (3) Niveles más altos de fosfato alcalino (p = 0,03); (4) Niveles más altos de HbA1c (<0,001) y (7) presencia de fibrosis hepática de moderada a grave. El estadio DM2 F1 tiene mayor rigidez hepática (0,006) y niveles de HbA1c (<0,001).
El uso de la elastografía de ondas transversales proporciona una valiosa evaluación clínica de primera línea de la salud del hígado en pacientes con DM2.
Damage to the liver is a common clinical consequence of chronic diabetes mellitus type 2 (DM2). This study evaluates whether ultrasound shear wave elastography and hemodynamics of the portal vein and the hepatic artery can complement traditional clinical work-up data for the monitoring of liver health among DM2 patients.
Sixty-four (64) participants (31 controls and 33 patients with confirmed type 2 diabetes mellitus) between 21 to 74 years of age were recruited. Liver size, stiffness and hemodynamics of the portal vein and the hepatic artery were evaluated. Glycated hemoglobin (HbA1c), aspartate aminotransferase (AST), alanine a minotransferase (ALT), and alkaline phosphatase (ALP) were monitored. Student’s t-test was employed with significance attained at p <0.05.
Asymptomatic significant differences were detected among DMT2 patients: (1) Largest Liver size (p=0.04); (2) Higher liver stiffness (p=0.04); (3) Higher alkaline phosphate levels (p=0.03); (4) Higher HbA1c levels (<0.001) and (7) presence of moderate to severe liver fibrosis. DM2 F1 stage has higher liver stiffness (0.006) and HbA1c levels (<0.001).
TABLA 1
CONTROL DM2 P VALUE
(KPA) 6.6 (1.5) 7.9 (2.1) 0.01
TAMAÑO
14.3 (1.6) 15.5 (2.8) 0.04
HBA1C (%) 5.4(0.3) 7.7 (1.7) < 0.001
ALT (UNIDADES / L) 25.7 (14.4) 34.2 (27.1) 0.12
AST (UNIDADES / L) 21.8 (6.0) 25.5 (15.2) 0.21
ALK PHOSP (UNIDADES / L) 71.8 (15.2) 82.7 (23.9) 0.03
DM2 = DIABETES MELLITUS TIPO 2
Según la Federación Internacional de Diabetes, en 2019, la diabetes afecta a 463 millones de personas en todo el mundo. Es motivo de gran preocupación que se espera que esta prevalencia aumente a 700 millones para 2045.1
Efectos perjudiciales sobre la salud del hígado, como la esteatosis o el hígado graso, son consecuencias habituales de la diabetes mellitus crónica tipo 2 (DM2). Más del 70% de los pacientes adultos con DM2 pueden desarrollar esteatosis o enfermedad del hígado graso no alcohólico (EHGNA), con importantes efectos perjudiciales anatómicos y fisiológicos.2-4 De hecho, la EHGNA es una epidemia mundial de gran impacto financiero, con una prevalencia estimada de hasta el 30% de la población mundial, muy probablemente debido al hecho de que la obesidad y la diabetes son factores de riesgo de esta condición fatal, si no se tratan.5-7
La esteatohepatitis no alcohólica (NASH) suele preceder a la NAFLD, ya que el hígado se inflama y se desarrolla fibrosis8-9. La fibrosis se caracteriza por un exceso de tejido conectivo que produce un aumento de la densidad hepática, que a su vez, eventualmente conduce a una
disfunción orgánica. La fibrosis hepática puede empeorar y convertirse en cirrosis y cáncer. Desafortunadamente, como es el caso de NAFLD, la fibrosis hepática también puede ser asintomática. Por lo tanto, una laguna en los algoritmos clínicos estándar para el manejo a largo plazo de la DM2, es poder monitorear la salud del hígado con un enfoque rentable.
A pesar de que las enzimas hepáticas se utilizan como detección de enfermedades hepáticas, es posible que no se correlacionen con la gravedad de la enfermedad.10 La biopsia hepática se considera el estándar de oro para confirmar la patología hepática, pero es una herramienta de diagnóstico costosa. Además, es un procedimiento invasivo de alto riesgo con posibles efectos secundarios injustificados.11 Por el contrario, la elastografía hepática de ondas de corte es una herramienta de diagnóstico no invasiva y rentable que mide la elasticidad y el endurecimiento del tejido hepático en todo el órgano.
12 -14 Nuestro objetivo es determinar si el uso de la elastografía hepática de ondas de corte, puede complementar los datos de estudios clínicos tradicionales para el seguimiento de la salud del hígado en pacientes con DM2.
Se reclutaron sesenta y cuatro (64) participantes (31 controles y 33 pacientes con diabetes mellitus tipo 2 confirmada) entre 21 y 74 años de edad en dos centros clínicos: una clínica hospitalaria de endocrinología universitaria en Puerto Rico y una clínica de endocrinología, asociada con una escuela de medicina puertorriqueña. El reclutamiento se realizó con los siguientes criterios de exclusión: enfermedad hepática previa, hiperlipidemia, traumatismo en cuadrante superior derecho, enfermedad renal crónica, obesidad mórbida, alcoholismo y enfermedad cardíaca. El estudio adherido al protocolo de investigación aprobado por la Oficina de Protección de Participantes Humanos en Investigación del Recinto de Ciencias Médicas, Universidad de Puerto Rico (número de protocolo A9000113). Todos los participantes firmaron y proporcionaron su consentimiento informado por escrito antes del reclutamiento. Todas las imágenes ecográficas fueron realizadas por uno de los autores (BLRC) que es un ecografista experimentado; y fueron evaluados de forma independiente por el mismo radiólogo de diagnóstico que es el presidente de la Sociedad Radiológica de Puerto Rico (GBO).
b) Resultados de las pruebas de laboratorio
Los resultados de las pruebas de laboratorio se obtuvieron de los registros médicos: niveles en sangre de hemoglobina glucosilada (HbA1c), enzimas hepáticas (aspartato aminotransferasa [AST], alanina aminotransferasa [ALT] y fosfatasa alcalina [ALP]. Los informes de las pruebas de laboratorio se obtuvieron dentro de una ventana de tiempo de 6 meses de la sesión de ecografía. Para el propósito de este estudio, se utilizaron los niveles de HbA1c para confirmar la diabetes, mientras que los niveles de ALT, AST y ALP se utilizaron como indicadores de la función hepática.
Se realizó un estudio de ecografía abdominal en tiempo real para evaluar la anatomía hepática y la hemodinámica de la vena porta y la arteria hepática, con un equipo de ultrasonido Logiq E9 (GE Healthcare, Milwaukee, Wisconsin, EE. UU.) con una sonda convexa C1-6-VN 2D. Se obtuvieron imágenes de ecografía hepática y medidas craneocaudales con el paciente en posición oblicua anterior izquierda (15º - 20º) con el brazo derecho colocado por encima de la cabeza. La exploración se realizó en la región axilar anterior (AAR). La medición craneocaudal
del lóbulo hepático derecho (RLL) se trazó desde el hemidiafragma derecho más alto visualizado en la imagen de ultrasonido hasta el extremo inferior del lóbulo derecho, lo más paralelo posible a la pared anterior del hígado.15 También se obtuvieron Imágenes de ultrasonido de la vena porta principal (VPM) y la arteria hepática (HA) en posición oblicua para evaluar el diámetro de la vena VPM (cm), la velocidad de la VPM (cm / seg), el índice de pulsatilidad de la VPM (PI = V2 / V1), la velocidad de la HA ( cm / seg) e índice resistivo HA (RI = V1 - V2 / V1).
Para la rigidez del hígado, se obtuvieron imágenes de RLL con los participantes del estudio colocados en una posición oblicua anterior izquierda (15 ° -20 °), con el brazo derecho colocado por encima de la cabeza y con la piel expuesta desde la cadera hasta la apófisis xifoides. Se escaneó el cuadrante superior derecho intercostal para obtener una imagen longitudinal de una determinada región de interés (ROI) en el segmento VIII del hígado, a una profundidad de <8 cm debajo de la piel para evitar vasos sanguíneos, áreas de sombra y límites anatómicos entre órganos. Se pidió a los pacientes que contuvieran la respiración y evitaran la inspiración profunda mientras se tomaban las medidas de la elastografía. Se informan los valores
14.2 (1.7) 14.8 (1.8) 0.57 14.2 (1.7) 15.8 (3.1) 0.13 14.6 (0.7 13.7 (1.0) 0.24
HBA1C (%) 5.4 (0.4) 7.7 (2.0) 0.01 5.4 (0.3) 7.6 (1.9) < 0.001 5.3 (0.3) 7.8 (2.4) 0.06
ALT (UNIDADES/L) 22.1 (11.0) 27.3 (12.3) 0.56 25.5 (16.2) 37.6 (31.3) 0.11 37.0 (7.5) 28.0 (12.4) 0.32
AST (UNIDADES/L) 21.3 (6.2) 23.0 (6.8) 0.66 21.3 (6.0) 27.3 (16.6) 0.20 27.7 (3.8) 22.0 (5.3) 0.18
ALK PHOSP (UNIDADES/L) 68.6 (17.3) 92.5 (12.4) 0.04 72.4 (14.0) 83.3 (28.0) 0.14 82.3 (17.1) 67.0 (18.8) 0.32
C= CONTROL, DM2=DIABETES MELLITUS TIPO 2
Revista Puertorriqueña de Medicina y Salud Pública
CONTROL
DIÁMETRO DE LA VENA PORTA (CM)
VELOCIDAD DE LA VENA PORTA (CM / SEG)
1.1 (0.2) 1.0 (0.2) 0.05
26.1 (5.7) 25.7 (7.3) 0.80
VENA PORTA ÍNDICE PULSÁTIL 0.3 (0.13) 0.2 (0.07) 0.002
VELOCIDAD DE LA ARTERIA HEPÁTICA (CM / SEG)
92.6 (22.0) 82.0 (19.9) 0.06
0.7 (0.07) 0.8 (0.09) 0.002 DM2 = DIABETES MELLITUS TIPO 2
medios (en kPa). La presencia y el grado de fibrosis se identificaron siguiendo la clasificación de la escala METAVIR para GE LogiqE9: hígado sano F0 (<5,48 kPa), fibrosis normal a leve F1 (5,48 - 8,29 kPa), fibrosis leve a moderada F2 (8,29 - 9,40 kPa), moderada a fibrosis grave F3 (9,4011,9 kPa) y cirrosis F4 (> 11,9 kPa).
d) Análisis estadístico
Los datos mostrados se expresan como media ± desviación estándar a menos que se especifique lo contrario. Los análisis se realizaron con el software XLSTAT-Biomed (Versión 2018.5, Add in soft, Nueva York, Nueva York, EE. UU.). La normalidad se evaluó mediante la prueba de ShapiroWilk y la homogeneidad de la varianza se evaluó de acuerdo con los resultados de normalidad.16 Los datos distribuidos normalmente se analizaron con una prueba “t de Student”; de lo contrario, se utilizó la prueba de Mann-Whitney. La significación estadística se alcanzó ap <0,05. Cuando la significación alcanzó los cuatro puntos decimales, el valor de p se informa como <0,001; de lo contrario, se informa el valor específico.
Se observaron diferencias significativas en la ecografía del hígado entre los controles y los pacientes con diabetes mellitus tipo 2 (Tabla 1). Los pacientes con DM2 mostraron hígados más grandes (p = 0,04) y más rígidos (p = 0,01) en comparación con los pacientes controles. También se midieron HbA1c, ALT, AST y, como era de esperar, los niveles de HbA1 confirmaron el estado de diabetes
(p <0,001). Con respecto a la función hepática, el fosfato alcalino (p = 0,03) fue significativamente mayor entre los pacientes con DM2 (Tabla 1).
Se detectó una distribución distinta de pacientes cuando se estratificó por categoría de fibrosis. Específicamente, la distribución de los pacientes control fue la siguiente: F0 (n = 6), F1 (n = 19), F2 (n = 3), F3 (n = 1) y F4 (n = 0), mientras que la distribución de los pacientes con DM2 fueron los siguientes: F0 (n = 4), F1 (n = 18), F2 (n = 4), F3 (n = 7), F4 (n = 1). La Tabla 2 muestra los valores de HbA1c, enzimas hepáticas y rigidez cuando se estratifica por categoría de fibrosis. Es de interés que se observó una diferencia significativa entre los grupos F1 para la rigidez del hígado (p = 0,006) y los niveles de HbA1 (p <0,001). En cuanto a la hemodinámica de los vasos sanguíneos, no se encontró diferencia estadística en la velocidad de la vena porta principal (VPM) y la velocidad de la arteria hepática (VHA) entre los controles y los pacientes con DM2 (tabla 3). Por el contrario, se observó una diferencia significativa entre el diámetro de MPV (p = 0,05), el índice de pulsatilidad (PI) de MPV (p = 0,002) y el índice de resistencia de la arteria hepática (HARI; p = 0,002).
La ecografía diagnóstica con elastografía de ondas de corte del hígado, muestra algunas diferencias asintomáticas en pacientes con DM2. Este estudio informó que el tamaño del hígado era mayor y la rigidez del hígado
era mayor en los grupos de DM2 en comparación con los controles. Aunque el mayor número de pacientes de nuestra cohorte mostró estar en una categoría de estadio temprano (F1), el grupo de diabéticos mostró una mayor proporción de pacientes en estadios avanzados (F2 a F4) de fibrosis hepática. De acuerdo con estudios previos, no se detectaron diferencias significativas en los niveles de las enzimas hepáticas AST o ALT, lo que respalda aún más la opinión emergente de que las enzimas hepáticas pueden no siempre correlacionarse con la gravedad de la enfermedad hepática10. Se necesitan herramientas de diagnóstico para el seguimiento a largo plazo de la salud del hígado.
Entre los parámetros hemodinámicos de interés, encontramos un mayor índice de resistencia de la arteria hepática (IRAH) en los pacientes diabéticos, lo que concuerda con los hallazgos de mayor rigidez hepática en este grupo. Este hallazgo es similar a otros estudios que encontraron una correlación positiva entre el IRA y el grado de fibrosis17-18. Nuestro estudio también encontró un índice de pulsatilidad de la vena porta (IPVP) más bajo entre los pacientes con DM2. Existe evidencia de disminución del índice de pulsatilidad venosa en pacientes con NAFLD19-20. Tomados en conjunto; estos hallazgos sugieren un mecanismo compensatorio de la distensibilidad vascular que es secundario a la infiltración grasa del hígado. Esta hipótesis justifica una mayor investigación.
Durante la última década, la EHNA se ha convertido en uno de los principales indicadores para el trasplante de hígado.21-22 Nuestro estudio detectó cambios significativos en la rigidez hepática en pacientes diabéticos en etapas tempranas (F1), donde los cambios pueden ser potencialmente reversibles con un tratamiento temprano para evitar más complicaciones clínicas. Esto es de gran importancia en la atención preventiva, ya que las etapas avanzadas de la fibrosis hepática se han asociado con un mayor riesgo cardiovascular y mortalidad23. Si los cambios observados en los parámetros hemodinámicos se correlacionan con la enfermedad cardiovascular en nuestra cohorte de pacientes amerita una evaluación adicional.
Hay una serie de limitaciones de este estudio. En primer lugar, se trata de un estudio transversal que no controló por
el momento con diagnóstico de DM2. Por lo tanto, es interesante realizar un estudio longitudinal en el que se debe monitorear el momento de la enfermedad. En segundo lugar, este estudio no incluyó muestras de biopsia hepática, aunque sigue siendo la confirmación estándar de oro del daño hepático. En tercer lugar, podría haber sido valioso recopilar datos sobre los niveles de plaquetas y albúmina como parte del panel de sangre para evaluar aún más los cambios bioquímicos a largo plazo entre los pacientes con DM2.
Este estudio apoya la noción de que la ecografía hepática con elastografía de ondas de corte es una herramienta útil para el diagnóstico y clasificación de la fibrosis hepática en pacientes con DM212-14. Una de las principales ventajas de este abordaje clínico es la capacidad de evaluar la elasticidad del tejido mientras se obtiene una imagen visual del área de interés en tiempo real. Además, permite la evaluación de diferentes áreas del órgano dentro de una sola sesión de imágenes. Como procedimiento no invasivo, es clínicamente factible realizar un seguimiento del paciente a lo largo del tiempo para evaluar la salud del hígado e implementar una intervención terapéutica temprana cuando sea necesario. Tomados en conjunto, creemos que el uso de la elastografía de ondas de corte en entornos de escasos recursos y de ritmo rápido proporciona una evaluación clínica de primera línea y perspicaz de la salud del hígado entre los pacientes con DM2.
El Dr. Luis E. Vázquez-Quiñones (Facultad de Ciencias y Tecnología, Universidad Ana G. Méndez, Cupey, Puerto Rico) brindó asesoría en análisis estadísticos. Este estudio fue apoyado por la concesión de subvención Título V del Departamento de Educación de los EE. UU. # P031S160068 y por el Centro Hispano de ExcelenciaFacultad de Medicina de la Universidad de Puerto Rico, Número de subvención: D34HP24463 Departamento de Salud y Servicios Humanos de los EE. UU. Administración de Recursos y Servicios de Salud Oficina de la Fuerza Laboral de la Salud.
©2020. Dra. Bárbara L. RiestraCandelaria, Juan Carlos Jorge, Miriam Rodríguez, Gory Ballester-Ortiz & Loida A. González-Rodríguez. Este es un artículo de investigación / revisión, distribuido
bajo los términos de la licencia Creative Commons Reconocimiento-No comercial 3.0 Unported http://creativecommons. org/licenses/by-nc/3.0/), que permite todo uso, distribución y distribución no comercial. reproducción en cualquier medio, siempre que la obra original esté debidamente citada.
1. International Diabetes Federation. (2019). IDF Diabetes Atlas. 9th edition. https://www. diabete satlas.org/en/ 2. Portillo-Sanchez, P., Bril, F., Maximos, M., Lomonaco, R., Biernacki, D., Orsak, B., Subbarayan, S., Webb, A., Hecht, J., & Cusi, K.(2015). High Prevalence of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. The Journal of clinical endocrinology and metabolism, 100(6): 2231–2238.
3. Hazlehurst Jonathan M, Woods Conor, Marjot Thomas, Cobbold Jeremy F., Tomlinson Jeremy W.(2016). Non-alcoholic fatty liver disease and diabetes. Metabolism, 65(8): 1096-1108.
4.Pappachan JM, Antonio FA, Edavalath M, Mukherjee A. (April 2014). Non-alcoholic fatty liver disease: a diabetologist's perspective. Endocrine. 45(3):344-53.
5.Tanaka, N., Kimura, T., Fu jimori, N., Nag aya, T., Komatsu, M., & Tanaka, E. (2019). Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World journal of gastroenterology, 25(2): 163–177.
6.Shehab M Abd El-Kader and Eman M Salah El-Den Ashmawy. (2015). Non-alcoholic fatty liver disease: The diagnosis and management. World J Hepatol. 7(6): 846–858.
7.Lomonaco R, Bril F, Portillo-Sanch ez P, et al. (201 6). Metabolic Impact of Nonalcoholic Steatohepatitis in Obese Patients with Type 2 diabetes. Diabetes Ca re.39(4):6 32-8.
8.Xu Li , Yuan Jiao, Yunlong Xing, Pujun Gao. (2019). Diabetes Mellitus and Risk of Hepatic Fibrosis/Cirrhosis. BioMed Research International.
9.Mantovani A, Petracca G, Beatrice G, et al. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: an updated meta-analysis of 501 022 adult in dividuals. Gut Published Online First: 16 September 2020.
10.Ahmed, Z., Ahmed, U., Walayat, S., Ren, J., Martin,D.K., Moole, H., Koppe, S., Yong, S., & Dhillon, S.(2018). Liver function tests in identifying patients with liver disease. Clinical and experimental gastroenterology, 11, 301–307. https://doi.org/ 10.2147/CEG. S160537.
11.Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. American Association for the Study of Liver Diseases. Liver biopsy. Hepatology. 2009; 49(3):1017-44.
12.Sande, J. A., Verjee, S., Vinayak, S., Amersi, F., & Ghesani, M. (2017). Ultrasound shear wave elastography and liver fibrosis: A
Prospective Multicenter Study. World journal of hepatology, 9(1), 38–47.
13.Gharibvand, M. M., Asare, M., Motamedfar, A., Alavinejad, P., & Momeni, M. (2020). Ultrasound shear wave elastography and liver biopsy to determine liver fibrosis in adult patients. Journal of family medicine and primary care, 9(2), 943–949.
14.Fu, J., Wu, B., Wu, H. et al. (2020). Accuracy of real-time shear wave elastography in staging hepatic fibrosis: a meta-analysis. BMC Med Imaging 20, 16.
15.Riestra-Candelaria BL, Rodríguez-Mojica W, Jorge JC. (2018). Anatomical criteria to measure the adult right liver lobe by ultrasound. Sonography; 5(4): 181–186.
16. Razali NM, Wah YB. Power Comparisons of Shapiro-Wilk, Kolmogorov-Smirnov, Lilliefors and Anderson-Darling Tests. Journal of Statistical Modeling and Analytics. 2011; 2: 21-33.
17. Tana C, Schiavone C, Ticinesi A, Lauretani F, Nouvenne A, Dietrich CF, Meschi T. (2018). Hepatic artery resistive index as surrogate marker for fibrosis progression in NAFLD patients: A clinical perspective. Int J Immunopathol Pharmacol. Jan-Dec; 32:2058738418781373.
18.Stulic M, Culafic D, Obrenovic R, Jankovic G, Alempijevic T, Lalosevic MS, Dostanic N, Kovacevic SV, Culafic M. (2018 May). The Clinical Importance of Cystatin C and Hepatic Artery Resistive Index in Liver Cirrhosis. Medicina (Kaunas). 28;54(3):37.
19.Solhjoo E, Mansour-Ghanaei F, MoulaeiLangorudi R, Joukar F. Comparison of portal vein doppler indices and hepatic vein doppler waveform in patients with nonalcoholic fatty liver disease with healthy control. Hepat Mon. 2011 Sep;11(9):740-4.
20.Baikpour M, Ozturk A, Dhyani M, Mercaldo ND, Pierce TT, Grajo JR, Samir AE. (2020 Apr). Portal Venous Pulsatility Index: A Novel Biomarker for Diagnosis of High-Risk Nonalcoholic Fatty Liver Disease. AJR Am J Roentgenol. 214(4):786-791.
21.Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, Setiawan VW, Tran T, Ayoub WS, Lu SC, Klein AS, Sundaram V, Nissen NN. (2018 Nov). NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am J Gastroenterol.; 113(11): 1649-1659.
22.Mikolasevic, I., Filipec-Kanizaj, T., Mijic, M., Jakopcic, I., Milic, S., Hrstic, I., Sobocan, N., Stimac, D., & Burra, P. (2018). Nonalcoholic fatty liver disease and liver transplantation - Where do we stand?. World journal of gastroenterology, 24(14), 1491–1506.
23.Mangla N, Ajmera VH, Caussy C, Sirlin C, Brouha S, Bajwa-Dulai S, Madamba E, Bettencourt R, Richards L, Loomba R. (2020 Mar). Liver Stiffness Severity is Associated with Increased Cardiovascular Risk in Patients With Type 2 Diabetes. Clin Gastroenterol Hepatol. 18(3):744-746.e1.
Revista Puertorriqueña de Medicina y Salud Pública

Mounjaro—the first and only approved single molecule that activates GIP and GLP-1 receptors in the body.1
Mounjaro is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use
• Mounjaro has not been studied in patients with a history of pancreatitis.
• Mounjaro is not indicated for use in patients with type 1 diabetes mellitus.
In both male and female rats, tirzepatide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2, and in patients with known serious hypersensitivity to tirzepatide or any of the excipients in Mounjaro.
Please see Important Safety Information here and on the following page, including Boxed Warning about possible thyroid tumors, including thyroid cancer, and Brief Summary on the following pages.
Risk of Thyroid C-cell Tumors: Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. Pancreatitis has been reported in Mounjaro clinical trials. Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. Observe patients for signs and symptoms, including persistent severe abdominal pain sometimes radiating to the back, which may or may not be accompanied by vomiting. If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management.
Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Concomitant use with an insulin secretagogue (e.g., sulfonylurea) or insulin may increase the risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by reducing the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions, sometimes severe, have been reported with Mounjaro in clinical trials. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity to Mounjaro. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown if such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury. In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, sometimes requiring hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of reported events occurred in patients who had experienced nausea, vomiting,
diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: In clinical trials, acute gallbladder disease was reported by 0.6% of Mounjarotreated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated.
The most common adverse reactions reported in ≥5% of Mounjaro-treated patients in placebo-controlled trials were nausea, diarrhea, decreased appetite, vomiting, constipation, dyspepsia, and abdominal pain.
Drug Interactions: When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (such as sulfonylureas) or insulin to reduce the risk of hypoglycemia. Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications, so caution should be exercised.
Pregnancy: Limited data on Mounjaro use in pregnant women are available to inform on drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide. Use only if potential benefit justifies the potential risk to the fetus.
Lactation: There are no data on the presence of tirzepatide in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females of Reproductive Potential: Advise females using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established and use is not recommended in patients less than 18 years of age.
Please see the Brief Summary on the following page, including the Boxed Warning about possible thyroid tumors, including thyroid cancer, and Medication Guide. Please see Instructions for Use included with the pen. TR HCP ISI MAY2022
Reference: 1. Mounjaro. Prescribing Information. Lilly USA, LLC.
Mounjaro™ and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-TR-US-0193 05/2022 ©Lilly USA, LLC 2022. All rights reserved.
Revista Puertorriqueña de Medicina y Salud Pública 73
Mounjaro™ (tirzepatide) Brief Summary: Consult the package insert for complete prescribing information.
INDICATIONS AND USAGE Mounjaro™ is a glucose-dependent insulinotropic polypeptide (GIP) receptor and glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use: Mounjaro has not been studied in patients with a history of pancreatitis. Mounjaro is not indicated for use in patients with type 1 diabetes mellitus.
In both male and female rats, tirzepatide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined.
Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro.
CONTRAINDICATIONS: Mounjaro is contraindicated in patients with: A personal or family history of medullary thyroid carcinoma (MTC), in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) and in patients with a known serious hypersensitivity to tirzepatide or any of the excipients in Mounjaro.
Risk of Thyroid C-Cell Tumors: In both sexes of rats, tirzepatide caused a dose-dependent and treatment-duration-dependent increase in the incidence of thyroid C-cell tumors (adenomas and carcinomas) in a 2-year study at clinically relevant plasma exposures. It is unknown whether Mounjaro causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as human relevance of tirzepatide-induced rodent thyroid C-cell tumors has not been determined. Mounjaro is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of Mounjaro and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with Mounjaro. Such monitoring may increase the risk of unnecessary procedures, due to the low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin values may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Pancreatitis: Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists. In clinical studies, 14 events of acute pancreatitis were confirmed by adjudication in 13 Mounjarotreated patients (0.23 patients per 100 years of exposure) versus 3 events in 3 comparator-treated patients (0.11 patients per 100 years of exposure). Mounjaro has not been studied in patients with a prior history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on Mounjaro. After initiation of Mounjaro, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, discontinue Mounjaro and initiate appropriate management. Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin: Patients receiving Mounjaro in combination with an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia, including severe hypoglycemia. The risk of hypoglycemia may be lowered by a reduction in the dose of sulfonylurea (or other concomitantly administered insulin secretagogue) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in clinical trials (e.g., urticaria and eczema) and were sometimes severe. If hypersensitivity reactions occur, discontinue use of Mounjaro; treat promptly per standard of care, and monitor until signs and symptoms resolve. Do not use in patients with a previous serious hypersensitivity reaction to tirzepatide or any of the excipients in Mounjaro.
Anaphylaxis and angioedema have been reported with GLP-1 receptor agonists. Use caution in patients with a history of angioedema or anaphylaxis with a GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to these reactions with Mounjaro.
Acute Kidney Injury: Mounjaro has been associated with gastrointestinal adverse reactions, which include nausea, vomiting, and diarrhea. These events may lead to dehydration, which if severe could cause acute kidney injury.
In patients treated with GLP-1 receptor agonists, there have been postmarketing reports of acute kidney injury and worsening of chronic renal failure, which may sometimes require hemodialysis. Some of these events have been reported in patients without known underlying renal disease. A majority of the reported events occurred in patients who had experienced nausea, vomiting, diarrhea, or dehydration. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe gastrointestinal adverse reactions.
Severe Gastrointestinal Disease: Use of Mounjaro has been associated with gastrointestinal adverse reactions, sometimes severe. Mounjaro has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Diabetic Retinopathy Complications in Patients with a History of Diabetic Retinopathy: Rapid improvement in glucose control has been associated with a temporary worsening of diabetic retinopathy. Mounjaro has not been studied in patients with non-proliferative diabetic retinopathy requiring acute therapy, proliferative diabetic retinopathy, or diabetic macular edema. Patients with a history of diabetic retinopathy should be monitored for progression of diabetic retinopathy.
Acute Gallbladder Disease: Acute events of gallbladder disease such as cholelithiasis or cholecystitis have been reported in GLP-1 receptor agonist trials and post marketing. In Mounjaro placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic, and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Pool of Two Placebo-Controlled Clinical Trials: The data in Table 1 are derived from 2 placebo-controlled trials (1 monotherapy trial [SURPASS-1] and 1 trial in combination with basal insulin with or without metformin [SURPASS-5]) in adult patients with type 2 diabetes mellitus. These data reflect exposure of 718 patients to Mounjaro and a mean duration of exposure to Mounjaro of 36.6 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 54% were male. The population was 57% White, 27% Asian, 13% American Indian or Alaska Native, and 3% Black or African American; 25% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.1%. As assessed by baseline fundoscopic examination, 13% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 53%, 60 to 90 mL/min/1.73 m2 in 39%, 45 to 60 mL/min/1.73 m2 in 7%, and 30 to 45 mL/min/1.73 m2 in 1% of patients.
MOUNJAROTM (tirzepatide) Injection, for subcutaneous use
TR HCP BS MAY2022
Pool of Seven Controlled Clinical Trials: Adverse reactions were also evaluated in a larger pool of adult patients with type 2 diabetes mellitus participating in seven controlled clinical trials which included two placebo-controlled trials (SURPASS-1 and -5), three trials of Mounjaro in combination with metformin, sulfonylureas, and/or SGLT2 inhibitors (SURPASS-2, -3, -4), and two additional trials conducted in Japan. In this pool, a total of 5119 adult patients with type 2 diabetes mellitus were treated with Mounjaro for a mean duration of 48.1 weeks. The mean age of patients was 58 years, 4% were 75 years or older and 58% were male. The population was 65% White, 24% Asian, 7% American Indian or Alaska Native, and 3% Black or African American; 38% identified as Hispanic or Latino ethnicity. At baseline, patients had type 2 diabetes mellitus for an average of 9.1 years with a mean HbA1c of 8.3%. As assessed by baseline fundoscopic examination, 15% of the population had retinopathy. At baseline, eGFR was ≥90 mL/min/1.73 m2 in 52%, 60 to 90 mL/min/1.73 m2 in 40%, 45 to 60 mL/min/1.73 m2 in 6%, and 30 to 45 mL/min/ 1.73 m2 in 1% of patients.
Common Adverse Reactions: In the pool of placebo-controlled trials, the following adverse reactions were reported in ≥5% of Mounjaro-treated adult patients with type 2 diabetes mellitus (excluding hypoglycemia): (frequencies listed, respectively, as: placebo [N=235]; 5 mg [N=273]; 10 mg [N=240]; 15 mg [N=241]): nausea (4%, 12%, 15%, 18%), diarrhea (9%, 12%, 13%, 17%), decreased appetite (1%, 5%, 10%, 11%), vomiting (2%, 5%, 5%, 9%), constipation (1%, 6%, 6%, 7%), dyspepsia (3%, 8%, 8%, 5%), and abdominal pain (4%, 6%, 5%, 5%). Percentages reflect the number of patients who reported at least 1 occurrence of the adverse reaction. In the pool of seven clinical trials, the types and frequency of common adverse reactions, not including hypoglycemia, were similar.
Gastrointestinal Adverse Reactions: In the pool of placebo-controlled trials, gastrointestinal adverse reactions occurred more frequently among patients receiving Mounjaro than placebo (placebo 20.4%, Mounjaro 5 mg 37.1%, Mounjaro 10 mg 39.6%, Mounjaro 15 mg 43.6%). More patients receiving Mounjaro 5 mg (3.0%), Mounjaro 10 mg (5.4%), and Mounjaro 15 mg (6.6%) discontinued treatment due to gastrointestinal adverse reactions than patients receiving placebo (0.4%). The majority of reports of nausea, vomiting, and/or diarrhea occurred during dose escalation and decreased over time.
The following gastrointestinal adverse reactions were reported more frequently in Mounjaro-treated patients than placebo-treated patients (frequencies listed, respectively, as: placebo; 5 mg; 10 mg; 15 mg): eructation (0.4%, 3.0%, 2.5%, 3.3%), flatulence (0%, 1.3%, 2.5%, 2.9%), gastroesophageal reflux disease (0.4%, 1.7%, 2.5%, 1.7%), abdominal distension (0.4%, 0.4%, 2.9%, 0.8%).
Other Adverse Reactions: Hypoglycemia: Hypoglycemia adverse reactions in placebo-controlled trials in adults with type 2 diabetes mellitus were as follows. For the 40-week monotherapy study (frequencies listed, respectively, as: placebo [N=115]; 5 mg [N=121]; 10 mg [N=119]; 15 mg [N=120]): blood glucose <54 mg/dL (1%, 0%, 0%, 0%), severe hypoglycemia (0%, 0%, 0%, 0%). For the 40week add-on to basal insulin with or without metformin study (placebo [N=120], 5 mg [N=116], 10 mg [N=119], 15 mg [N-120]): blood glucose <54 mg/dL (13%, 16%, 19%, 14%), severe hypoglycemia (0%, 0%, 2%, 1%). Data include events occurring during 4 weeks of treatment-free safety follow up. Events after introduction of a new glucose-lowering treatment are excluded. Severe hypoglycemia was defined as episodes requiring the assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions.
Hypoglycemia was more frequent when Mounjaro was used in combination with a sulfonylurea. In a clinical trial up to 104 weeks of treatment, when administered with a sulfonylurea, hypoglycemia (glucose level <54 mg/dL) occurred in 13.8%, 9.9%, and 12.8%, and severe hypoglycemia occurred in 0.5%, 0%, and 0.6% of patients treated with Mounjaro 5 mg, 10 mg, and 15 mg, respectively.
Heart Rate Increase: In the pool of placebo-controlled trials, treatment with Mounjaro resulted in a mean increase in heart rate of 2 to 4 beats per minute compared to a mean increase of 1 beat per minute in placebo-treated patients. Episodes of sinus tachycardia, associated with a concomitant increase from baseline in heart rate of ≥15 beats per minute, also were reported in 4.3%, 4.6%, 5.9% and 10% of subjects treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. For patients enrolled in Japan, these episodes were reported in 7% (3/43), 7.1% (3/42), 9.3% (4/43), and 23% (10/43) of patients treated with placebo, Mounjaro 5 mg, 10 mg, and 15 mg, respectively. The clinical relevance of heart rate increases is uncertain.
Hypersensitivity Reactions: Hypersensitivity reactions have been reported with Mounjaro in the pool of placebo-controlled trials, sometimes severe (e.g., urticaria and eczema); hypersensitivity reactions were reported in 3.2% of Mounjaro-treated patients compared to 1.7% of placebo-treated patients.
In the pool of seven clinical trials, hypersensitivity reactions occurred in 106/2,570 (4.1%) of Mounjaro-treated patients with antitirzepatide antibodies and in 73/2,455 (3.0%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Injection Site Reaction: In the pool of placebo-controlled trials, injection site reactions were reported in 3.2% of Mounjarotreated patients compared to 0.4% of placebo-treated patients.
In the pool of seven clinical trials, injection site reactions occurred in 119/2,570 (4.6%) of Mounjaro-treated patients with antitirzepatide antibodies and in 18/2,455 (0.7%) of Mounjaro-treated patients who did not develop anti-tirzepatide antibodies.
Acute Gallbladder Disease: In the pool of placebo-controlled clinical trials, acute gallbladder disease (cholelithiasis, biliary colic and cholecystectomy) was reported by 0.6% of Mounjaro-treated patients and 0% of placebo-treated patients.
Laboratory Abnormalities: Amylase and Lipase Increase: In the pool of placebo-controlled clinical trials, treatment with Mounjaro resulted in mean increases from baseline in serum pancreatic amylase concentrations of 33% to 38% and serum lipase concentrations of 31% to 42%. Placebo-treated patients had a mean increase from baseline in pancreatic amylase of 4% and no changes were observed in lipase. The clinical significance of elevations in lipase or amylase with Mounjaro is unknown in the absence of other signs and symptoms of pancreatitis.
Concomitant Use with an Insulin Secretagogue (e.g., Sulfonylurea) or with Insulin
When initiating Mounjaro, consider reducing the dose of concomitantly administered insulin secretagogues (e.g., sulfonylureas) or insulin to reduce the risk of hypoglycemia.
Oral Medications:
Mounjaro delays gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications. Caution should be exercised when oral medications are concomitantly administered with Mounjaro. Monitor patients on oral medications dependent on threshold concentrations for efficacy and those with a narrow therapeutic index (e.g., warfarin) when concomitantly administered with Mounjaro. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro. Hormonal contraceptives that are not administered orally should not be affected.
Pregnancy: Risk Summary: Available data with Mounjaro use in pregnant women are insufficient to evaluate for a drug-related risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy. Based on animal reproduction studies, there may be risks to the fetus from exposure to tirzepatide during pregnancy. Mounjaro should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. In pregnant rats administered tirzepatide during organogenesis, fetal growth reductions and fetal abnormalities occurred at clinical exposure in maternal rats based on AUC. In rabbits administered tirzepatide during organogenesis, fetal growth reductions were observed at clinically relevant exposures based on AUC. These adverse embryo/fetal effects in animals coincided with pharmacological effects on maternal weight and food consumption. The estimated background risk of major birth defects is 6–10% in women with pre-gestational diabetes with an HbA1c >7% and has been reported to be as high as 20–25% in women with an HbA1c >10%. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively. Clinical Considerations: Disease-Associated Maternal and/or
MOUNJAROTM (tirzepatide) Injection, for subcutaneous use
TR HCP BS MAY2022
Embryo/Fetal Risk: Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia-related morbidity. Data: Animal Data: In pregnant rats given twice weekly subcutaneous doses of 0.02, 0.1, and 0.5 mg/kg tirzepatide (0.03-, 0.07-, and 0.5-fold the MRHD of 15 mg once weekly based on AUC) during organogenesis, increased incidences of external, visceral, and skeletal malformations, increased incidences of visceral and skeletal developmental variations, and decreased fetal weights coincided with pharmacologically-mediated reductions in maternal body weights and food consumption at 0.5 mg/kg. In pregnant rabbits given once weekly subcutaneous doses of 0.01, 0.03, or 0.1 mg/kg tirzepatide (0.01-, 0.06, and 0.2-fold the MRHD) during organogenesis, pharmacologically-mediated effects on the gastrointestinal system resulting in maternal mortality or abortion in a few rabbits occurred at all dose levels. Reduced fetal weights associated with decreased maternal food consumption and body weights were observed at 0.1 mg/kg. In a pre- and post-natal study in rats administered subcutaneous doses of 0.02, 0.10, or 0.25 mg/kg tirzepatide twice weekly from implantation through lactation, F1 pups from F0 maternal rats given 0.25 mg/kg tirzepatide had statistically significant lower mean body weight when compared to controls from post-natal day 7 through post-natal day 126 for males and post-natal day 56 for females.
Lactation: Risk Summary: There are no data on the presence of tirzepatide in animal or human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Mounjaro and any potential adverse effects on the breastfed infant from Mounjaro or from the underlying maternal condition.
Females and Males of Reproductive Potential: Contraception: Use of Mounjaro may reduce the efficacy of oral hormonal contraceptives due to delayed gastric emptying. This delay is largest after the first dose and diminishes over time. Advise patients using oral hormonal contraceptives to switch to a non-oral contraceptive method, or add a barrier method of contraception for 4 weeks after initiation and for 4 weeks after each dose escalation with Mounjaro.
Pediatric Use: Safety and effectiveness of Mounjaro have not been established in pediatric patients (younger than 18 years of age).
Geriatric Use: In the pool of seven clinical trials, 1539 (30.1%) Mounjaro-treated patients were 65 years of age or older, and 212 (4.1%) Mounjaro-treated patients were 75 years of age or older at baseline. No overall differences in safety or efficacy were detected between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Renal Impairment: No dosage adjustment of Mounjaro is recommended for patients with renal impairment. In subjects with renal impairment including end-stage renal disease (ESRD), no change in tirzepatide pharmacokinetics (PK) was observed. Monitor renal function when initiating or escalating doses of Mounjaro in patients with renal impairment reporting severe adverse gastrointestinal reactions.
Hepatic Impairment: No dosage adjustment of Mounjaro is recommended for patients with hepatic impairment. In a clinical pharmacology study in subjects with varying degrees of hepatic impairment, no change in tirzepatide PK was observed.
OVERDOSE: In the event of an overdosage, contact Poison Control for latest recommendations. Appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms. A period of observation and treatment for these symptoms may be necessary, taking into account the half-life of tirzepatide of approximately 5 days.
PATIENT COUNSELING INFORMATION: Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use)
Missed Doses: Inform patients if a dose is missed, it should be administered as soon as possible within 4 days after the missed dose. If more than 4 days have passed, the missed dose should be skipped and the next dose should be administered on the regularly scheduled day. In each case, inform patients to resume their regular once weekly dosing schedule.
Additional information can be found <at www.Mounjaro.com/at lillymedical.com/by calling The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979)/ on Twitter @MounjaroHCP/at Click to Chat/etc.>.
See Brief Summary of Prescribing Information on adjacent page. See User Guide accompanying the product device.


Siguiendo los postulados de SPED, sus miembros participan activamente en la investigación clínica que conduce al uso seguro y eficaz de nuevas alternativas terapéuticas. SPED ha fomentado el aumento de endocrinólogos en la isla al subsidiar plazas de adiestramiento en Endocrinología, Diabetes y Metabolismo. Los endocrinólogos han adiestrado y servido de modelo para generaciones de nuevos endocrinólogos. Al cumplirse los 45 años de la fundación de SPED celebramos los logros del crecimiento de su membresía de 27 fundadores a sobre 130 miembros. SPED auspicia y celebra múltiples actividades educativas que permiten a estos miembros mantener sus conocimientos actualizados y vigentes. Su participación en actividades comunitarias ha sido constante para mantener educados a nuestros pacientes en la prevención y manejo de la Diabetes Mellitus, enfermedad cardiovascular y enfermedades del tiroides.
La Sociedad Puertorriqueña de Endocrinología y Diabetología, SPED, se fundó hace 45 años. Un grupo de médicos endocrinólogos, pediatras y una geneticista se reunieron y organizaron una sociedad que trabajara en pro de la excelencia en la práctica clínica, brindando educación a médicos y a la comunidad. Los fundadores querían fomentar la investigación médica y la confraternización entre sus miembros. Tras varias reuniones incorporaron a SPED como entidad sin fines de lucro en 1976, se eligió una directiva y se estableció un reglamento que ha sido enmendado y sigue vigente. A partir del 1976 los trabajos y el crecimiento de SPED ha ido en forma ascendente. Hoy por hoy SPED agrupa casi la totalidad de los endocrinólogos en Puerto Rico y es reconocida entre la comunidad médica como la entidad a emular por la calidad de sus actividades académicas y la implementación de los estándares de calidad en la práctica clínica. SPED ha fomentado la educación sobre las condiciones endocrinas a médicos, público y otros profesionales de la salud.
Apesar de la pandemia de COVID 19, SPED continuó ofreciendo asambleas anuales, el curso postgraduado de Diabetes en honor al Dr. Manuel Paniagua (fundador), el curso en honor al Dr. Agustín Martínez de Andino (fundador), el curso postgraduado de Osteoporosis en honor a la Dra. Lillian Haddock (fundadora); el foro de investigación en honor al Dr. Francisco Aguiló (fundador), el curso de enfermedades del tiroides, la actualización de Endocrinología Clínica, además de muchos otros ofrecimientos de alto nivel académico.
SPED es dirigida por una junta que se elige cada dos años y que ha trabajado manteniendo los valores, misión y visión de la sociedad. Coincidiendo con el aniversario zafiro de SPED se plasma su historia en un capítulo del libro La Endocrinología en Puerto Rico: Anécdotas, Pinceladas y Recuerdos. El libro recoge la trayectoria de la sociedad desde sus comienzos hasta el año 2021.
Estoy segura de que la Sociedad Puertorriqueña de Endocrinología y Diabetología permanecerá fuerte llevando en alto el estandarte de la práctica clínica de excelencia en endocrinología.
Sus miembros han dicho presente en tiempos de normalidad, así como en tiempos de pandemia, desastres naturales, terremotos y huracanes.
“
• NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for NSCLC recommend molecular testing and strongly advise broad molecular profiling for, at minimum, RET rearrangements, EGFR mutations, BRAF mutations, METex14 skipping mutations, ALK fusions, and ROS1 fusions in all appropriate patients with metastatic NSCLC to assess whether patients are eligible for targeted therapies2*†
• Next-generation sequencing (NGS) testing early in metastatic NSCLC diagnosis provides a comprehensive, tissue-efficient way to test for actionable alterations, including RET fusions3,4
An FDA-approved test for the detection of RET gene fusions and RET gene mutations is not currently available.1
NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
*The NCCN Guidelines ® for NSCLC provide recommendations for individual biomarkers that should be tested and recommend testing techniques but do not endorse any specific commercially available biomarker assays.
† It is recommended at this time that when feasible, testing be performed via a broad, panel-based approach, most typically performed by NGS. For patients who, in broad panel testing don’t have identifiable driver oncogenes (especially in never smokers), consider RNA-based NGS if not already performed, to maximize detection of fusion events.
ALK=anaplastic lymphoma kinase; BRAF=v-raf murine sarcoma viral oncogene homolog B; EGFR=epidermal growth factor receptor; METex14=mesenchymalepithelial transition exon 14 skipping; NCCN=National Comprehensive Cancer Network; NSCLC=non-small cell lung cancer; RET=rearranged during transfection; RNA=ribonucleic acid; ROS1=reactive oxygen species 1.
Retevmo is a kinase inhibitor indicated for the treatment of:
• adult patients with metastatic RET fusion-positive non-small cell lung cancer (NSCLC)
• adult and pediatric patients 12 years of age and older with advanced or metastatic RET-mutant medullary thyroid cancer (MTC) who require systemic therapy
• adult and pediatric patients 12 years of age and older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate)
These indications are approved under accelerated approval based on overall response rate (ORR) and duration of response (DoR). Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials.
Hepatotoxicity: Serious hepatic adverse reactions occurred in 2.6% of patients treated with Retevmo. Increased aspartate aminotransferase (AST) occurred in 51% of patients, including Grade 3 or 4 events in 8% and increased alanine aminotransferase (ALT) occurred in 45% of patients, including Grade 3 or 4 events in 9%. The median time to first onset for increased AST was 4.1 weeks (range: 5 days to 2 years) and increased ALT was 4.1 weeks (range: 6 days to 1.5 years). Monitor ALT and AST prior to initiating Retevmo, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose or permanently discontinue Retevmo based on the severity.
Hypertension occurred in 35% of patients, including Grade 3 hypertension in 17% and Grade 4 in one (0.1%) patient. Overall, 4.6% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications. Do not initiate Retevmo in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating Retevmo. Monitor blood pressure after 1 week, at least monthly thereafter, and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue Retevmo based on the severity.
References: 1. Retevmo (selpercatinib) [package insert]. Indianapolis, IN: Eli Lilly and Company; 2021. 2. Referenced with permission from The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.6.2020. © National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed June 15, 2020. To view the most recent and complete version of the guidelines, go online to https://www.nccn.org. 3. Gregg JP, Li T, Yoneda KY. Molecular testing strategies in non-small cell lung cancer: optimizing the diagnostic journey. Transl Lung Cancer Res. 2019;8(3):286-301. 4. Suh JH, Schrock AB, Johnson A, et al. Hybrid capture-based comprehensive genomic profiling identifies lung cancer patients with well-characterized sensitizing epidermal growth factor receptor point mutations that were not detected by standard of care testing. Oncologist. 2018;23(7):776-781.
Please see Important Safety Information and Brief Summary of Prescribing Information for Retevmo on subsequent pages.
Retevmo® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates. PP-SE-US-0591 04/2021 ©Lilly USA, LLC 2021. All rights reserved.
SAFETY INFORMATION FOR RETEVMO™ (selpercatinib 40 mg, 80 mg capsules) (CONT’D)
Retevmo can cause concentration-dependent QT interval prolongation. An increase in QTcF interval to >500 ms was measured in 6% of patients and an increase in the QTcF interval of at least 60 ms over baseline was measured in 15% of patients. Retevmo has not been studied in patients with clinically significant active cardiovascular disease or recent myocardial infarction. Monitor patients who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, and severe or uncontrolled heart failure. Assess QT interval, electrolytes and TSH at baseline and periodically during treatment, adjusting frequency based upon risk factors including diarrhea. Correct hypokalemia, hypomagnesemia and hypocalcemia prior to initiating Retevmo and during treatment. Monitor the QT interval more frequently when Retevmo is concomitantly administered with strong and moderate CYP3A inhibitors or drugs known to prolong QTc interval. Withhold and dose reduce or permanently discontinue Retevmo based on the severity.
Serious, including fatal, hemorrhagic events can occur with Retevmo. Grade ≥3 hemorrhagic events occurred in 2.3% of patients treated with Retevmo including 3 (0.4%) patients with fatal hemorrhagic events, including one case each of cerebral hemorrhage, tracheostomy site hemorrhage, and hemoptysis. Permanently discontinue Retevmo in patients with severe or lifethreatening hemorrhage.
Hypersensitivity occurred in 4.3% of patients receiving Retevmo, including Grade 3 hypersensitivity in 1.6%. The median time to onset was 1.7 weeks (range 6 days to 1.5 years). Signs and symptoms of hypersensitivity included fever, rash and arthralgias or myalgias with concurrent decreased platelets or transaminitis. If hypersensitivity occurs, withhold Retevmo and begin corticosteroids at a dose of 1 mg/kg prednisone (or equivalent). Upon resolution of the event, resume Retevmo at a reduced dose and increase the dose of Retevmo by 1 dose level each week as tolerated until reaching the dose taken prior to onset of hypersensitivity. Continue steroids until patient reaches target dose and then taper. Permanently discontinue Retevmo for recurrent hypersensitivity.
Tumor lysis syndrome (TLS) occurred in 1% of patients with medullary thyroid carcinoma receiving Retevmo. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, Retevmo has the potential to adversely affect wound healing. Withhold Retevmo for at least 7 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of Retevmo after resolution of wound healing complications has not been established.
Based on data from animal reproduction studies and its mechanism of action, Retevmo can cause fetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with Retevmo and for at least 1 week after the final dose. There are no data on the presence of selpercatinib or its metabolites in human milk or on their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with Retevmo and for 1 week after the final dose.
Severe adverse reactions (Grade 3-4) occurring in ≥15% of patients who received Retevmo in LIBRETTO-001, were hypertension (18%), prolonged QT interval (4%), diarrhea (3.4%), dyspnea (2.3%), fatigue (2%), abdominal pain (1.9%), hemorrhage (1.9%), headache (1.4%), rash (0.7%), constipation (0.6%), nausea (0.6%), vomiting (0.3%), and edema (0.3%).
Serious adverse reactions occurred in 33% of patients who received Retevmo. The most frequently reported serious adverse reaction (in ≥ 2% of patients) was pneumonia.
Fatal adverse reactions occurred in 3% of patients; fatal adverse reactions which occurred in >1 patient included sepsis (n=3), cardiac arrest (n=3) and respiratory failure (n=3).
Common adverse reactions (all grades) occurring in ≥15% of patients who received Retevmo in LIBRETTO-001, were dry mouth (39%), diarrhea (37%), hypertension (35%), fatigue (35%), edema (35%), rash (27%), constipation (25%), nausea (23%), abdominal pain (23%), headache (23%), cough (18%), prolonged QT interval (17%), dyspnea (16%), vomiting (15%), and hemorrhage (15%).
Laboratory abnormalities (all grades; Grade 3-4) ≥20% worsening from baseline in patients who received Retevmo in LIBRETTO-001, were AST increased (51%; 8%), ALT increased (45%; 9%), increased glucose (44%; 2.2%), decreased leukocytes (43%; 1.6%), decreased albumin (42%; 0.7%), decreased calcium (41%; 3.8%), increased creatinine (37%; 1.0%), increased alkaline phosphatase (36%; 2.3%), decreased platelets (33%; 2.7%), increased total cholesterol (31%; 0.1%), decreased sodium (27%; 7%), decreased magnesium (24%; 0.6%), increased potassium (24%; 1.2%), increased bilirubin (23%; 2.0%), and decreased glucose (22%; 0.7%).
Concomitant use of acid-reducing agents decreases selpercatinib plasma concentrations which may reduce Retevmo anti-tumor activity. Avoid concomitant use of proton-pump inhibitors (PPIs), histamine-2 (H2) receptor antagonists, and locally-acting antacids with Retevmo. If coadministration cannot be avoided, take Retevmo with food (with a PPI) or modify its administration time (with a H2 receptor antagonist or a locally-acting antacid).
Concomitant use of strong and moderate CYP3A inhibitors increases selpercatinib plasma concentrations which may increase the risk of Retevmo adverse reactions including QTc interval prolongation. Avoid concomitant use of strong and moderate CYP3A inhibitors with Retevmo. If concomitant use of a strong or moderate CYP3A inhibitor cannot be avoided, reduce the Retevmo dosage as recommended and monitor the QT interval with ECGs more frequently.
Concomitant use of strong and moderate CYP3A inducers decreases selpercatinib plasma concentrations which may reduce Retevmo anti-tumor activity. Avoid coadministration of Retevmo with strong and moderate CYP3A inducers.
Concomitant use of Retevmo with CYP2C8 and CYP3A substrates increases their plasma concentrations which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of Retevmo with CYP2C8 and CYP3A substrates where minimal concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for CYP2C8 and CYP3A substrates provided in their approved product labeling.
The safety and effectiveness of Retevmo have not been established in pediatric patients less than 12 years of age The safety and effectiveness of Retevmo have been established in pediatric patients aged 12 years and older for medullary thyroid cancer (MTC) who require systemic therapy and for advanced RET fusion-positive thyroid cancer who require systemic therapy and are radioactive iodine-refractory (if radioactive iodine is appropriate). Use of Retevmo for these indications is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients aged 12 years and older. Monitor open growth plates in adolescent patients. Consider interrupting or discontinuing Retevmo if abnormalities occur.
No dosage modification is recommended for patients with mild to severe renal impairment (estimated Glomerular Filtration Rate [eGFR] ≥15 to 89 mL/min, estimated by Modification of Diet in Renal Disease [MDRD] equation). A recommended dosage has not been established for patients with end-stage renal disease. Reduce the dose when administering Retevmo to patients with severe hepatic impairment (total bilirubin greater than 3 to 10 times upper limit of normal [ULN] and any AST). No dosage modification is recommended for patients with mild or moderate hepatic impairment. Monitor for Retevmo-related adverse reactions in patients with hepatic impairment.
Please see Brief Summary of Prescribing Information for Retevmo on subsequent pages.
SE HCP ISI All_25MAR2021
RETEVMO® (selpercatinib) capsules, for oral use
Initial U.S. Approval: 2020
BRIEF SUMMARY: Consult the package insert for complete prescribing information.
RETEVMO (selpercatinib) is a kinase inhibitor indicated for the treatment of:
• Adult patients with metastatic RET fusion-positive non-small cell lung cancer (NSCLC)
• Adult and pediatric patients 12 years of age and older with advanced or metastatic RET-mutant medullary thyroid cancer (MTC) who require systemic therapy
• Adult and pediatric patients 12 years of age and older with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy are radioactive iodine-refractory (if radioactive iodine is appropriate)
These indications are approved under accelerated approval based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials.
CONTRAINDICATIONS: None
Serious hepatic adverse reactions occurred in 2.6% of patients treated with RETEVMO. Increased AST occurred in 51% of patients, including Grade 3 or 4 events in 8% and increased ALT occurred in 45% of patients, including Grade 3 or 4 events in 9%. The median time to first onset for increased AST was 4.1 weeks (range: 5 days to 2 years) and increased ALT was 4.1 weeks (range: 6 days to 1.5 years).
Monitor ALT and AST prior to initiating RETEVMO, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose or permanently discontinue RETEVMO based on the severity.
Hypertension occurred in 35% of patients, including Grade 3 hypertension in 17% and Grade 4 in one (0.1%) patient Overall, 4.6% had their dose interrupted and 1.3% had their dose reduced for hypertension. Treatment-emergent hypertension was most commonly managed with anti-hypertension medications.
Do not initiate RETEVMO in patients with uncontrolled hypertension. Optimize blood pressure prior to initiating RETEVMO. Monitor blood pressure after 1 week, at least monthly thereafter and as clinically indicated. Initiate or adjust anti-hypertensive therapy as appropriate. Withhold, reduce dose, or permanently discontinue RETEVMO based on the severity.
Hypersensitivity occurred in 4.3% of patients receiving RETEVMO, including Grade 3 hypersensitivity in 1.6%. The median time to onset was 1.7 weeks (range: 6 days to 1.5 years). Signs and symptoms of hypersensitivity included fever, rash and arthralgias or myalgias with concurrent decreased platelets or transaminitis.
If hypersensitivity occurs, withhold RETEVMO and begin corticosteroids at a dose of 1 mg/kg prednisone (or equivalent). Upon resolution of the event, resume RETEVMO at a reduced dose and increase the dose of RETEVMO by 1 dose level each week as tolerated until reaching the dose taken prior to onset of hypersensitivity. Continue steroids until patient reaches target dose and then taper. Permanently discontinue RETEVMO for recurrent hypersensitivity.
Tumor lysis syndrome (TLS) occurred in 1% of patients with medullary thyroid carcinoma receiving RETEVMO. Patients may be at risk of TLS if they have rapidly growing tumors, a high tumor burden, renal dysfunction, or dehydration. Closely monitor patients at risk, consider appropriate prophylaxis including hydration, and treat as clinically indicated.
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, RETEVMO has the potential to adversely affect wound healing.
Withhold RETEVMO for at least 7 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of RETEVMO after resolution of wound healing complications has not been established.
Based on data from animal reproduction studies, and its mechanism of action, RETEVMO can cause fetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for at least 1 week after the final dose.
RETEVMO can cause concentration-dependent QT interval prolongation An increase in QTcF interval to >500 ms was measured in 6% of patients and an increase in the QTcF interval of at least 60 ms over baseline was measured in 15% of patients. RETEVMO has not been studied in patients with clinically significant active cardiovascular disease or recent myocardial infarction.
Monitor patients who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, and severe or uncontrolled heart failure. Assess QT interval, electrolytes and TSH at baseline and periodically during treatment, adjusting frequency based upon risk factors including diarrhea. Correct hypokalemia, hypomagnesemia and hypocalcemia prior to initiating RETEVMO and during treatment.
Monitor the QT interval more frequently when RETEVMO is concomitantly administered with strong and moderate CYP3A inhibitors or drugs known to prolong QTc interval. Withhold and dose reduce or permanently discontinue RETEVMO based on the severity.
Serious including fatal hemorrhagic events can occur with RETEVMO. Grade ≥ 3 hemorrhagic events occurred in 2.3% of patients treated with RETEVMO including 3 (0.4%) patients with fatal hemorrhagic events, including one case each of cerebral hemorrhage, tracheostomy site hemorrhage, and hemoptysis.
Permanently discontinue RETEVMO in patients with severe or life-threatening hemorrhage.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS and PRECAUTIONS and below reflects exposure to RETEVMO as a single agent at 160 mg orally twice daily evaluated in 702 patients in LIBRETTO-001. Among 702 patients who received RETEVMO, 65% were exposed for 6 months or longer and 34% were exposed for greater than one year. Among these patients, 95% received at least one dose of RETEVMO at the recommended dosage of 160 mg orally twice daily.
The median age was 59 years (range: 15 to 92 years); 0.3% were pediatric patients 12 to 16 years of age; 52% were male; and 69% were White, 22% were Asian, 5% were Hispanic/ Latino, and 3% were Black. The most common tumors were NSCLC (47%), MTC (44%), and non-medullary thyroid carcinoma (5%).
Serious adverse reactions occurred in 33% of patients who received RETEVMO. The most frequent serious adverse reaction (in ≥ 2% of patients) was pneumonia. Fatal adverse reactions occurred in 3% of patients; fatal adverse reactions which occurred in > 1 patient included sepsis (n = 3), cardiac arrest (n = 3) and respiratory failure (n = 3).
Permanent discontinuation due to an adverse reaction occurred in 5% of patients who received RETEVMO. Adverse reactions resulting in permanent discontinuation included increased ALT (0.4%), sepsis (0.4%), increased AST (0.3%), drug hypersensitivity (0.3%), fatigue (0.3%), and thrombocytopenia (0.3%).
Dosage interruptions due to an adverse reaction occurred in 42% of patients who received RETEVMO. Adverse reactions requiring dosage interruption in > 2% of patients included ALT increased, AST increased, hypertension, diarrhea, pyrexia, and QT prolongation.
RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021
RETEVMO, SE HCP BS 25MAR2021 - 7.5 x 10
Dose reductions due to an adverse reaction occurred in 31% of patients who received RETEVMO. Adverse reactions requiring dosage reductions in > 2% of patients included ALT increased, AST increased, QT prolongation and fatigue.
The most common adverse reactions, including laboratory abnormalities, (≥ 25%) were increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), increased glucose, decreased leukocytes, decreased albumin, decreased calcium, dry mouth, diarrhea, increased creatinine, increased alkaline phosphatase, hypertension, fatigue, edema, decreased platelets, increased total cholesterol, rash, decreased sodium, and constipation.
Table 1 summarizes the adverse reactions in LIBRETTO-001.
Table 1: Adverse Reactions (>15%) in Patients Who Received RETEVMO in LIBRETTO-001
Adverse Reaction RETEVMO (n=702)
Grades 1-4 (%) Grade 3-4 (%)
Clinically relevant adverse reactions in <15% of patients who received RETEVMO include hypothyroidism and tumor lysis syndrome.
Table 2 summarizes the laboratory abnormalities in LIBRETTO-001.
Table 2: Select Laboratory Abnormalities (≥ 20%) Worsening from Baseline in Patients Who Received RETEVMO in LIBRETTO-001
Laboratory Abnormality
Chemistry
Gastrointestinal
Dry Mouth 39 0 Diarrhea1 37 3.4* Constipation 25 0.6* Nausea 23 0.6* Abdominal pain2 23 1.9* Vomiting 15 0.3* Vascular Hypertension 35 18 General Fatigue3 35 2* Edema4 35 0.3* Skin Rash5 27 0.7*
Nervous System Headache6 23 1.4* Respiratory Cough7 18 0 Dyspnea8 16 2.3
Investigations
Prolonged QT interval 17 4* Blood and Lymphatic System Hemorrhage9 15 1.9
1Diarrhea includes diarrhea, defecation urgency, frequent bowel movements, and anal incontinence
2Abdominal pain includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal discomfort, gastrointestinal pain
3Fatigue includes fatigue, asthenia, malaise.
4Edema includes edema, edema peripheral, face edema, periorbital edema, eye edema, eyelid edema, orbital edema, localized edema, lymphedema, scrotal edema, peripheral swelling, scrotal swelling, swelling, swelling face, eye swelling
5Includes rash, rash erythematous, rash macular, rash maculopapular, rash morbilliform, rash pruritic
6Headache includes headache, sinus headache, tension headache,
7Includes cough, productive cough
8Includes dyspnea, dyspnea exertional, dyspnea at rest
9Hemorrhage includes epistaxis, hematuria, hemoptysis, contusion, rectal hemorrhage, vaginal hemorrhage, ecchymosis, hematochezia, petechiae, traumatic hematoma, anal hemorrhage, blood blister, blood urine present, cerebral hemorrhage, gastric hemorrhage, hemorrhage intracranial, spontaneous hematoma, abdominal wall hematoma, angina bullosa hemorrhagica, diverticulum intestinal hemorrhagic, eye hemorrhage, gastrointestinal hemorrhage, gingival bleeding, hematemesis, hemorrhagic anemia, intraabdominal hemorrhage, lower gastrointestinal hemorrhage, melena, mouth hemorrhage, occult blood positive, pelvic hematoma, periorbital hematoma, pharyngeal hemorrhage, pulmonary contusion, purpura, retroperitoneal hematoma, subarachnoid hemorrhage, subdural hemorrhage, upper gastrointestinal hemorrhage, vessel puncture site hematoma
*Only includes a grade 3 adverse reaction.
RETEVMO1
Grades 1-4 (%)Grades 3-4 (%)
Increased AST 51 8
Increased ALT 45 9
Increased glucose 44 2.2
Decreased albumin 42 0.7
Decreased calcium 41 3.8
Increased creatinine 37 1.0
Increased alkaline phosphatase 36 2.3
Increased total cholesterol 31 0.1
Decreased sodium 27 7
Decreased magnesium 24 0.6
Increased potassium 24 1.2
Increased bilirubin 23 2.0
Decreased glucose 22 0.7
Hematology
Decreased leukocytes 43 1.6
Decreased platelets 33 2.7
1 Denominator for each laboratory parameter is based on the number of patients with a baseline and post-treatment laboratory value available, which ranged from 675 to 692 patients.
In healthy subjects administered RETEVMO 160 mg orally twice daily, serum creatinine increased 18% after 10 days. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
Concomitant use of RETEVMO with acid-reducing agents decreases selpercatinib plasma concentrations, which may reduce RETEVMO anti-tumor activity. Avoid concomitant use of PPIs, H2 receptor antagonists, and locally acting antacids with RETEVMO. If coadministration cannot be avoided, take RETEVMO with food (with a PPI) or modify its administration time (with a H2 receptor antagonist or a locally acting antacid).
Concomitant use of RETEVMO with a strong or moderate CYP3A inhibitor increases selpercatinib plasma concentrations, which may increase the risk of RETEVMO adverse reactions, including QTc interval prolongation. Avoid concomitant use of strong and moderate CYP3A inhibitors with RETEVMO. If concomitant use of strong and moderate CYP3A inhibitors cannot be avoided, reduce the RETEVMO dosage and monitor the QT interval with ECGs more frequently.
Concomitant use of RETEVMO with a strong or moderate CYP3A inducer decreases selpercatinib plasma concentrations, which may reduce RETEVMO anti-tumor activity. Avoid coadministration of strong or moderate CYP3A inducers with RETEVMO.
CYP2C8 and CYP3A Substrates
RETEVMO is a moderate CYP2C8 inhibitor and a weak CYP3A inhibitor. Concomitant use of RETEVMO with CYP2C8 and CYP3A substrates increases their plasma concentrations, which may increase the risk of adverse reactions related to these substrates. Avoid coadministration of RETEVMO with CYP2C8 and CYP3A substrates where minimal
RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021
RETEVMO, SE HCP BS 25MAR2021 - 7.5 x 10
Puertorriqueña de Medicina y Salud Pública
concentration changes may lead to increased adverse reactions. If coadministration cannot be avoided, follow recommendations for CYP2C8 and CYP3A substrates provided in their approved product labeling.
RETEVMO is associated with QTc interval prolongation. Monitor the QT interval with ECGs more frequently in patients who require treatment with concomitant medications known to prolong the QT interval.
Based on findings from animal studies, and its mechanism of action, RETEVMO can cause fetal harm when administered to a pregnant woman. There are no available data on RETEVMO use in pregnant women to inform drug-associated risk. Administration of selpercatinib to pregnant rats during the period of organogenesis resulted in embryolethality and malformations at maternal exposures that were approximately equal to the human exposure at the clinical dose of 160 mg twice daily. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Selpercatinib administration to pregnant rats during the period of organogenesis at oral doses ≥ 100 mg/kg [approximately 3.6 times the human exposure based on the area under the curve (AUC) at the clinical dose of 160 mg twice daily] resulted in 100% postimplantation loss. At the dose of 50 mg/kg [approximately equal to the human exposure (AUC) at the clinical dose of 160 mg twice daily], 6 of 8 females had 100% early resorptions; the remaining 2 females had high levels of early resorptions with only 3 viable fetuses across the 2 litters. All viable fetuses had decreased fetal body weight and malformations (2 with short tail and one with small snout and localized edema of the neck and thorax).
Lactation
There are no data on the presence of selpercatinib or its metabolites in human milk or on their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with RETEVMO and for 1 week after the final dose.
Based on animal data, RETEVMO can cause embryolethality and malformations at doses resulting in exposures less than or equal to the human exposure at the clinical dose of 160 mg twice daily.
Verify pregnancy status in females of reproductive potential prior to initiating RETEVMO
Advise female patients of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the final dose.
Advise males with female partners of reproductive potential to use effective contraception during treatment with RETEVMO and for 1 week after the final dose.
RETEVMO may impair fertility in females and males of reproductive potential.
The safety and effectiveness of RETEVMO have been established in pediatric patients aged 12 years and older for medullary thyroid cancer (MTC) who require systemic therapy and for advanced RET fusion-positive thyroid cancer who require systemic therapy and are radioactive iodine-refractory (if radioactive iodine is appropriate). Use of RETEVMO for these indications is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients aged 12 years and older. The safety and effectiveness of RETEVMO have not been established in these indications in patients less than 12 years of age.
The safety and effectiveness of RETEVMO have not been established in pediatric patients for other indications.
In 4-week general toxicology studies in rats, animals showed signs of physeal hypertrophy and tooth dysplasia at doses resulting in exposures ≥ approximately 3 times the human exposure at the 160 mg twice daily clinical dose. Minipigs also showed signs of minimal to marked increases in physeal thickness at the 15 mg/kg high dose level (approximately 0.3 times the human exposure at the 160 mg twice daily clinical dose). Rats in both the 4- and 13-week toxicology studies had malocclusion and tooth discoloration at the high dose levels (≥1.5 times the human exposure at the 160 mg twice daily clinical dose) that persisted during the recovery period.
Monitor growth plates in adolescent patients with open growth plates. Consider interrupting or discontinuing therapy based on the severity of any growth plate abnormalities and based on an individual risk-benefit assessment.
Of 702 patients who received RETEVMO, 34% (239 patients) were ≥ 65 years of age and 10% (67 patients) were ≥ 75 years of age. No overall differences were observed in the safety or effectiveness of RETEVMO between patients who were ≥65 years of age and younger patients.
No dosage modification is recommended for patients with mild to severe renal impairment [estimated Glomerular Filtration Rate (eGFR) ≥15 to 89 mL/min estimated by Modification of Diet in Renal Disease (MDRD) equation]. The recommended dosage has not been established for patients with end-stage renal disease.
Reduce the dose when administering RETEVMO to patients with severe [total bilirubin greater than 3 to 10 times upper limit of normal (ULN) and any AST] hepatic impairment No dosage modification is recommended for patients with mild (total bilirubin less than or equal to ULN with AST greater than ULN or total bilirubin greater than 1 to 1.5 times ULN with any AST) or moderate (total bilirubin greater than 1.5 to 3 times ULN and any AST) hepatic impairment. Monitor for RETEVMO-related adverse reactions in patients with hepatic impairment.
Rx only.
Additional information can be found at www.retevmo.com.
Eli Lilly and Company, Indianapolis, IN 46285, USA Copyright ©2021, Eli Lilly and Company. All rights reserved.
SE HCP BS 25MAR2021
RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021 RETEVMO® (selpercatinib) capsules, for oral use SE HCP BS 25MAR2021
Revista Puertorriqueña de Medicina y Salud Pública
La época festiva es momento de alegría, compartir y celebración para muchos. Para otros, es una época de recordar pérdidas, enfrentar situaciones familiares o de enfrentar el reto de manejar su condición crónica, entre un banquete y otro. También, un tiempo de grandes retos para los profesionales de la salud.


La evidencia científica demuestra que durante y luego de la época festiva, hay un deterioro en la salud poblacional— desde la salud cardiovascular hasta la mental y emocional. Vemos una interrupción en el manejo adecuado de líquidos en pacientes con fallo congestivo, un alza en presión arterial, colesterol y azúcar en sangre, un aumento en sentimientos de soledad y ansiedad y patrones de alimentación desordenados.
Es por esto que la recomendación del profesional de la salud va en la línea de: ¡goza, pero no te descuides! Para el experto en temas de salud es una recomendación lógica y clara, pero para el paciente, posiblemente una recomendación paradójica y difícil de aplicar.
Entonces, ¿cómo guiamos a los pacientes en el cuidado de su salud y nutrición durante las celebraciones navideñas? Los siguientes hábitos y comportamientos antes, durante y después de la celebración apoyan el mantenimiento de metas de salud y previenen sentimientos de culpa y arrepentimiento al cierre de las festividades.
1No saltar comidas ni dejar los carbohidratos para más tarde. Saltar comidas dificulta el manejo del azúcar en sangre. Mientras que, mantener una ingesta consistente de comida y carbohidratos a través del día ayuda a evitar atracones y picos altos de azúcar en sangre y previene el terminar incómodamente lleno.
2Llevar consigo meriendas ricas en fibra y balanceadas e intentar mantener el horario de las comidas y meriendas. En esta temporada habrá cambios en rutina, viajes largos y listas interminables de compras. Al organizarse, se evita llegar al punto de necesitar comer lo primero que se encuentre y se podrán hacer las selecciones que mejor se ajusten a las necesidades.
3Recordar que, porque algunos estén comiendo o bebiendo, no significa que todos tienen que hacerlo. La clave es prepararse mentalmente para estar rodeado de más comida y personas comiendo más de lo usual y seguir el propio ritmo.
Comenzar a hidratarse desde temprano en el día. No esperar a estar sediento.
Visualizar cómo se manejarán emociones que puedan surgir con familia o amigos. Tener de ante mano un plan para canalizar emociones ayuda a no acudir automáticamente a la comida para sentir alivio.
Revista Puertorriqueña de Medicina y Salud Pública
1No prohibirse alimentos. Preparar una lista mental de las comidas típicas favoritas y permitirse disfrutarlas de forma balanceada cuando estén disponibles. Se puede integrar ese arroz con gandules que tan rico hace la tía utilizando el método del plato. Mantener el plato balanceado ayudará a cuidar el cuerpo y sentirse bien física y mentalmente al finalizar el día. Las comidas que no se logren integrar se pueden guardar para más tarde o para el próximo día.
2Si no le encanta o no tiene hambre, déjelo a un lado. Tenga conciencia de no comer por comer. Se puede socializar en áreas donde pueda evitar picar inconscientemente.
3Comer despacio y escuchar las señales internas de hambre y saciedad. El cerebro tarda al menos 20 minutos en darse cuenta de que está cómodamente lleno.
5Si elige beber alcohol, recuerde hacerlo en moderación. El alcohol puede resultar en episodios no deseados de hipoglicemia, elevar triglicéridos y colesterol y deshidratar. En caso de consumirlo, siempre se debe beber con comida y no con el estómago vacío.
4Respetar el cuerpo y sus necesidades. Si encuentra que un alimento no se alinea con la condición de salud, como quizás uno muy alto en sodio o grasa saturada, se debe intentar no hacerlo parte focal de la comida. Luego de la fiesta, se sentirá orgulloso de las selecciones.
1Si aún es temprano, se puede bailar, caminar alrededor del bloque e integrar movimiento después de la comida.
3Atender con diligencia síntomas de advertencia. Monitorear los síntomas, utilizar el tratamiento recomendado y comunicarse con el médico según necesario.
Prepararse para retomar la rutina. La meta no es ser perfecto. En ocasiones se comerá más de lo que se desea o alimentos que no le harán sentir bien física o mentalmente. Recuerde hablarse con compasión y volver a caer en ritmo sin autocastigarte.
4Disfrutar de los sobrantes una vez regresen las señales internas de hambre. Es decir, saborear el placer de comer con hambre.
LA PREPARACIÓN, EL BALANCE Y LA COMPASIÓN SON PILARES PARA DISFRUTAR LA ÉPOCA FESTIVA Y NAVEGAR LOS BANQUETES QUE LA CARACTERIZAN, SIN CULPAS O ARREPENTIMIENTOS Y, SOBRE TODO, SIN PERDER DE VISTA NUESTRA SALUD COMPLETA.
1. Vedel-Krogh, S., Kobylecki, C. J., Nordestgaard, B. G., & Langsted, A. (2019). The Christmas holidays are immediately followed by a period of hypercholesterolemia. Atherosclerosis, 281, 121–127. https:// doi.org/10.1016/j.atherosclerosis.2018.12.011 Accessed 11/2/22.
2. Navigating the winter holiday blues. National Alliance on Mental Illness website. https://www.nami.org/Blogs/ NAMI-s-Ask-the-Expert/2021/ NAMI-Ask-the-Expert-Navigating-the-Winter-Holiday-Blues Accessed 11/2/22.
3. How the holidays shape Americans’ diets. Ipsos website. https://www.ipsos.com/en-us/ news-polls/how-holidays-shape-americans-diets Accessed 11/2/22.

La Revista Medicina y Salud Pública, en alianza con diferentes organizaciones de salud de Puerto Rico, lideraró un importante encuentro dedicado a los pacientes con psoriasis y otras condiciones en la piel, en el que importantes especialistas en reumatología, dermatología, inmunología y endocrinología, abordaron todo lo relacionado con el manejo de diferentes afecciones dermatológicas que afectan a la población puertorriqueña. Los especialistas que integraron el evento desde las instalaciones de la Universidad Central del Caribe, fueron;

Dr. Samuel Sánchez, presidente de la Sociedad Dermatológica de Puerto Rico: Dra. Marely Santiago, dermatóloga y miembro de la Sociedad Dermatológica de Puerto Rico:Dr. Anardi Agosto, presidente de la Asociación Puertorriqueña de Médicos Alergistas: Dra. Eneida de la Torre, dermatóloga y catedrática auxiliar de la Universidad Central del Caribe: Dr. Luis Ortiz Espinosa, dermatólogo y director médico de Nova Derm Puerto Rico y la Dra. Paloma Alejandro, reumatóloga
Danilo Beauchamp, productor y paciente de Psoriasis junto al Dr. Anardi Agosto, alergista pediátrico.

De izquierda a derecha: Nelson González, paciente de artritis y psoriasis, Dra. Paloma Alejandro, reumatóloga, Dr. Luis Espinosa, dermatólogo, Dra. Marely Santiago, dermatóloga, Danilo Beauchamp, comediante y productor, Dra. Eneida de la Torre, dermatóloga y Dra. Mariely Sierra, endocrinóloga, Dr. Anardi Agosto, alergista pediátrico.


Foto: Revista Medicina y Salud Pública.
En el evento, los especialistas resolvieron dudas de los pacientes sobre los tratamientos para determinadas condiciones como la hidradenitis supurativa y el cáncer de piel.
Danilo Beauchamp, productor y paciente de Psoriasis junto a la Dra. Marely Santiago, dermatóloga.

La trasmisión del evento se realizó en vivo junto a los especialistas, pacientes y profesionales de la salud.
Revista Puertorriqueña de Medicina y Salud Pública




Con el impacto que tiene la Dermatitis Atópica en la población puertorriqueña, la Revista de Medicina y Salud Pública, junto a un grupo de destacados especialistas, presentaron una alianza médica y educativa en Plaza Las Américas, que contó con un panel de destacados médicos en dermatología, entre ellos, el Dr. José González Chávez, dermatólogo y pionero en investigación de las enfermedades de la piel: Dra. Marely Santiago, dermatóloga en representación de la Sociedad Dermatológica de Puerto Rico: Dr. Hiram Ruíz, dermatólogo y cirujano de Mohs: Dr. Wilfredo Cosme, alergista; Dra. Elena Nogales, dermatóloga; Brenda Gerena, Fundación Cross The Goal; Ketsy Román, paciente Dermatitis Atópica y Lee Nieves, paciente de Dermatitis Atópica. En el espacio pacientes y médicos compartieron valiosa información y promovieron estrategias para diagnosticar a tiempo la condición y que obtuvieran un tratamiento oportuno.


La



El
El





En el mes de sensibilización sobre el cáncer de mama, la Revista Medicina y Salud Pública, en alianza con el Hospital Auxilio Mutuo y la organización Susan G. Komen Puerto Rico, realizaron el primer encuentro de pacientes con cáncer de seno, un evento que tuvo como protagonistas a las guerreras sobrevivientes de esta enfermedad y la intervención de un importante panel multidisciplinario de especialistas


en cáncer de mama. En la actividad desarrollada en el salón de conferencias del Hospital Auxilio Mutuo, se abordaron temas relacionados con la detección temprana del cáncer de mama, el manejo adecuado de esta enfermedad, los avances más recientes en el diagnóstico y las novedosas alternativas de tratamientos para las pacientes que padecen esta condición.













La Revista de Medicina y Salud Pública, junto al Centro Comprensivo de Cáncer de la Universidad de Puerto, VOCES y un grupo de destacados especialistas del País, realizaron un conversatorio en alianza donde profundizaron sobre los retos, logros y estrategias para la prevención, diagnóstico, tratamiento y los esfuerzos por erradicar el virus de la isla para el año 2030 y reunió a los siguientes especialistas; Dr. Humberto Guiot, presidente de la Sociedad de Enfermedades Infecciosas de Puerto Rico (SEIPR); Dr. Juan Carlos Lemos, infectólogo especializado en
trasplante adscrito al Centro Comprensivo de Cáncer; Dr. José Rivera, gastroenterólogo y representante de la Asociación Puertorriqueña de Gastroenterología; Dra. Bárbara Rosado, gastroenteróloga y hepatóloga; Marta Torres, especialista en avances clínicos II representando ASES; Kiani Canales, coordinadora del proyecto de prevención de Hepatitis del Departamento de Salud; Senador Rubén Soto, presidente de la Comisión de Salud del Senado; Lilliam Rodríguez, Directora Ejecutiva de la Organización VOCES; Licenciado Jonathan Meléndez, administrador de NeoMed.









Durante el mes de noviembre, Mes de la Concienciación sobre el Cáncer del Pulmón y Gástrico, la biofarmacéutica Bristol Myers Squibb, la Revista de Medicina y Salud Pública y un grupo de profesionales de la salud se unieron en una actividad efectuada en el Senado de Puerto Rico para dialogar y conocer la información más actualizada sobre la importancia del diagnóstico temprano, terapias de tratamiento y apoyo al paciente de estos dos tipos de cánceres. La iniciativa contó con la participación de un panel compuesto por: Dr.Oscar Soto Raíces, Editor de la Revista Medicina y Salud Pública; Dr. Carlos Micames, pasado presidente de la Asociación Puertorriqueña de Gastroenterología; Dra. Daphne Delgado, presidenta de la Sociedad Puertorriqueña de Neumología; el hematólogo oncólogo, Dr. Luis Báez Vallecillo de PR Oncology Inc.; Dra. Marivelisse Soto, investigadora adscrita al Centro Comprensivo de Cáncer y la Sra. María Cristy, vicepresidenta de Servicio a los Pacientes y Control de Cáncer de la Sociedad Americana Contra el Cáncer de Puerto Rico.



El Salón de la Fama de la Medicina Puertorriqueña celebró una conferencia de prensa junto a su junta directiva, compuesta por importantes médicos boricuas, donde exaltaron la loable labor y grandiosa contribución que han realizado nueve especialistas en beneficio de la salud pública del país. Entre los reconocidos se encontraron; Dr. Isaac González Martínez; Dr. Óscar Costa Mandry: Dr. Manuel Pila Iglesias: Dr. Bailey K. Ashford: Dr. Guillermo Picó Santiago: Dra. Ana Judith Román: Dra. Amalia Martínez Picó: Dra. Liliam Haddock y el Dr. Norman Maldonado.


Los galenos recbieron un importante reconocimiento por su incansable labor en el bienestar de los pacientes.
La trayectoria de los médicos ha permitido mejorar la salud de Puerto Rico.
Son el pilar de las nuevas generaciones de médicos.


La Revista Medicina y Salud Pública, en alianza con la Coalición de Inmunización y Promoción de la Salud (VOCES), llevó a cabo el evento XpoKids, en Plaza las Américas, un espacio donde padres y pacientes pudieron conocer de la mano de especialistas las principales condiciones que más afectan a la población infantil puertorriqueña desde un enfoque de salud preventiva en el que abordarán los cuidados y tratamientos necesarios para los más
pequeños. De este evento formaron parte; Dr. Antonio del Valle, gastroenterólogo pediátrico; Dr. David de Ángel Solá, neumólogo pediátrico; Dra. Sheila Pérez Colón, endocrinóloga pediátrica; Dra. Jinette Santos Rodríguez, pediatra adscrita al Hospital Metropolitano; Dra. Wanda Torres, infectóloga; Dr. Gerardo Tosca, presidente de la Sociedad Puertorriqueña de Pediatría; Licenciada Wanda González, nutricionista, dietista y fisióloga del ejercicio.
Revista Puertorriqueña de Medicina y Salud Pública


La Fundación Puertorriqueña de Enfermedades Reumáticas en alianza con la Revista de Medicina y Salud Pública celebró un foro educativo para pacientes con artritis reumatoide en la Universidad Central del Caribe, con el objetivo principal de proveer la información y herramientas más actualizadas que ayuden a sospechar de esta condición a tiempo. El foro, moderado por la Directora Ejecutiva, Griselle Lugo de FER, contó con sesiones de conversatorio, mesa de expertos en diferentes temas de salud y un grupo de médicos especialistas en reumatología tales como: Dr. Oscar Soto Raíces, reumatólogo, Miembro Fundador, Presidente de FER; la Dra. Amarilis Pérez de Jesús, reumatóloga, del Centro de Reumatológico de Caguas y Miembro de la Junta de FER; Dra. Noemí Varela, reumatóloga, Vicepresidente de FER; Lcda. Wanda González, Nutricionista y Delia Román, “coach” de Mindfulness.




ExpoSalud-MSP llegó a Salinas el pasado sábado 8 de octubre, como la feria más grande de salud organizada por la Revista Medicina y Salud Pública, dirigida a la población afectada por el paso del huracán Fiona, que dejó a la población sin energía eléctrica, muchas zonas del país inundadas, ciudadanos sin hogar y afectó la situación la salud pública de toda la Isla. A la iniciativa de la Revista de Medicina y Salud Pública, se unieron entidades como la Alcaldía de Salinas, la Administración de Seguros de Salud (ASES), el Departamento de Salud de Puerto Rico, el programa WIC, la Administración de Servicios de Salud Mental y Contra la Adicción (ASSMCA) y entidades como Abbvie, Voces, la Asociación Puertorriqueña de Diabetes, MMM (caminamos juntos), SurMed Medical Center, Heroes para Campeones, Sistema de Salud Episcopal San Lucas y la Sociedad Puertorriqueña de Endocrinología y Diabetología.

Multitudinaria asistencia en #ExpoSalud Salinas.
Foto: Revista Medicina y Salud Pública.

Parte del grupo de médicos y residentes de la Ponce Health Sciences University y el Recinto de Ciencias Médicas encabezado por el Dr. José García Mateo.
Durante la feria de salud se hicieron cientos de pruebas a pacientes que aprovecharon para poner sus vacunas al día.

Jornadas de acompañamiento a los damnificados del huracán Fiona, por parte del Colegio de Médicos Cirujanos de Puerto Rico y MSP.



La atención a la niñez fue unas de las prioridades en los servicios de salud que se ofrecieron.
Abbvie fue uno de los principales auspiciadores de la Feria de Salud.
La actriz y comediante Suzette Bacó con su personaje de Doña Soto.
La feria ofreció diversas atenciones en el área de salud incluyendo psiquiatras.

Unos 15 profesionales de la salud fueron reconocidos por su labor y contribuciones en la salud femenina en Puerto Rico durante una actividad que se llevó a cabo en el salón de audiencias José Joaquín “Yiye” Ávila del Edificio Baltazar Corrada en el Capitolio de Puerto Rico. La revista Medicina y Salud Pública se encargó de otorgar estas distinciones a las salubristas con el objetivo de visibilizar los diferentes problemas de salud que sufren las mujeres en Puerto Rico. Los reconocidos que representan diversas disciplinas fueron, la Dra. Idhaliz Flores, investigadora en endometriosis, Dra. Ángeles Rodríguez, infectóloga y ex epidemióloga del estado, Dra. Elivette Zambrana, reumatóloga pediátrica y presidenta de la Asociación de Reumatólogos de Puerto Rico, Dra. Esther
Torres, gastroenteróloga y la directora del Centro de Enfermedades Inflamatorias del Intestino de la UPR y presidenta de la Fundación, Esther A Torres. Igualmente, se reconoció la faena de otras profesionales, como la Dra. Damaris Torres Paoli, actualmente pasada presidenta de la Sociedad Dermatológica de Puerto Rico, Dra. María Ramos, cardióloga y presidenta de la Sociedad Puertorriqueña de Cardiología, Dra. Leticia Hernández, presidenta de la Sociedad Puertorriqueña de Endocrinología y Diabetología, Dra. Eva Cruz Jove, MD, radióloga con especialidad en Imágenes de la Mujer, Dra. Franchesca Fiorito, MD, neuróloga especialista en cefalea, Dra. Bárbara Rosado Carrión, gastroenteróloga hepatóloga.