s6-7


s6-7
SPRAWDŹ
Pierwotna hiperoksaluria może stać się chorobą całego organizmu s4-5
INSPIRACJE
Pacjenci z hipofosfatemią XLH mają szansę na lepsze życie s9
WYZWANIA
SMA: jak terapia genowa zmienia życie pacjentów? s11
EKSPERT
Fenyloketonuria, czyli specjalna dieta przez całe życie s18

Prof. dr hab. n. med. Jerzy Windyga Opieka nad pacjentem z porfirią

Prof.
dr
Zbigniew Żuber
z chorobą Stilla

Prof. dr hab. n. med. Ewa Lech-Marańda
Leczenie chłoniaka DLBCL w Polsce – nowoczesne, ale ciągle są nowe wyzwania

Dr n. med. Tomasz Matuszewski Pacjencie! Sprawdź, czym jest wrodzony obrzęk naczynioruchowy

Zintegrowana opieka dla pacjentów z chorobami rzadkimi
Celem Planu dla Chorób Rzadkich jest poprawa sytuacji polskich pacjentów przez stworzenie kompleksowej i skoordynowanej opieki.

Urszula Demkow Podsekretarz Stanu w Ministerstwie Zdrowia; profesor nauk medycznych, posiada specjalizacje w dziedzinie chorób wewnętrznych (drugiego stopnia), immunologii, alergologii i diagnostyki laboratoryjnej
Chociaż każda z chorób rzadkich występuje naprawdę rzadko, to ich ogromna liczba sprawia, że dotyczą one aż 6-8 proc. populacji (znanych jest już około 8 tysięcy chorób rzadkich). W Polsce żyje 2-3 mln osób z chorobą rzadką. Rozpoznanie choroby rzadkiej jest niezwykle trudne. Diagnoza potrafi trwać latami, zanim dojdzie do ustalenia właściwego rozpoznania. Jest to problem chorych na choroby rzadkie na całym świecie. Mają zazwyczaj przewlekły lub ciężki przebieg i stanowią ogromne wyzwanie zdrowotne i społeczne.
Dlatego Plan dla Chorób Rzadkich zakłada zmiany organizacyjne w sześciu obszarach:
1. Ośrodki Eksperckie Chorób Rzadkich;
2. Diagnostyka chorób rzadkich, w tym dostęp do nowoczesnych metod diagnostycznych z wykorzystaniem wielkoskalowych badań genomowych;
3. Dostęp do leków i środków specjalnego przeznaczenia żywieniowego w chorobach rzadkich;
4. Polski Rejestr Chorób Rzadkich;
5. Karta Pacjenta z Chorobą Rzadką;
6. Platforma Informacyjna „Choroby Rzadkie”. Poprawiamy sytuację pacjentów z chorobami rzadkimi
Kluczową rolę w tym modelu opieki nad pacjentami z chorobami rzadkimi odgrywają krajowe ośrodki eksperckie (OECR), które należą do Europejskich Sieci Referencyjnych Chorób Rzadkich. Wprowadziliśmy dla tych poradni trzy nowe produkty rozliczeniowe w katalogu świadczeń: kompleksowa ocena genetyczna, kompleksowa porada specjalistyczna i kontrolna ocena stanu zdrowia pacjenta.
Ponadto w ramach leczenia szpitalnego pracujemy nad wyodrębnianiem produktu „Kompleksowa diagnostyka i leczenie choroby rzadkiej w Ośrodkach Eksperckich Chorób Rzadkich”.
z obszaru genetyki klinicznej. Planujemy również zmiany w zakresie niegenetycznych badań laboratoryjnych, które mogą być wykorzystywane w diagnostyce chorób rzadkich.
Pracujemy także nad rozszerzeniem koszyka świadczeń gwarantowanych o procedury wykonywane w ośrodkach nerwowo-mięśniowych oraz o badania laboratoryjne wykorzystywane w innych niż metaboliczne i genetyczne chorobach rzadkich.
Podmioty lecznicze uczestniczące w diagnostyce i leczeniu chorób rzadkich zostaną zmodernizowane dzięki środkom pozostającym w dyspozycji ministra zdrowia.
Wprowadzamy nowe narzędzia
W pierwszej kolejności powstała Platforma Informacyjna „Choroby Rzadkie”, która jest kompleksowym źródłem wiedzy klinicznej, naukowej i organizacyjnej dotyczącym chorób rzadkich w Polsce. Celem platformy jest m.in. zwiększenie wiedzy dotyczącej chorób rzadkich i wsparcie opieki zdrowotnej nad chorymi na choroby rzadkie.
Obecnie nie widać chorób rzadkich w medycznych systemach elektronicznych w Polsce. Zamierzamy utworzyć w IV kwartale 2024 r. Polski Rejestr Chorób Rzadkich – ważny instrument poprawy opieki medycznej osób cierpiących na choroby rzadkie.
Zostanie opracowana również w formie elektronicznej tzw. Karta Pacjenta z Chorobą Rzadką. Ma ona umożliwić pacjentowi bezpieczne funkcjonowanie w systemie ochrony zdrowia, w tym udzielenie pomocy w stanach nagłych. Będzie to regularnie aktualizowany zasób informacji na temat pacjenta oraz jego choroby, który będzie mógł zostać udostępniony placówkom medycznym. Udostępnienie do użytku Karty Pacjenta z Chorobą Rzadką jest zaplanowane na IV kwartał 2024 r.
Więcej informacji na stronie:
chorobyrzadkie.com
PATRONAT HONOROWY





W Ministerstwie Zdrowia trwają również prace nad zmianą rozporządzenia w zakresie ambulatoryjnej opieki specjalistycznej, które ma wprowadzić do wykazu nowe metody diagnostyczne






Początkowo działania nad Planem dla Chorób Rzadkich zaplanowano na lata 2021-2023, jednak z uwagi na początkowe opóźnienie wydłużono jego realizację na lata 2024-2025. Wszystkie prace są na zaawansowanym etapie realizacji.



Stowarzyszenie
Nowa Jakość







Dorota Korycińska
Prezeska zarządu Stowarzyszenia Neurofibromatozy Polska, prezeska zarządu Ogólnopolskiej
Federacji Onkologicznej
stałej opieki szytej na miarę
W Centrach Koordynowanej Opieki Medycznej pacjenci z neurofibromatozami i pokrewnymi im rasopatiami otrzymują wsparcie dopasowane do indywidualnych potrzeb, niczym garnitur szyty na miarę. Daje to im poczucie bezpieczeństwa w życiu z nieprzewidywalną chorobą. Owo bezpieczeństwo wzmacniane jest udostępnieniem przez MZ pierwszej celowanej terapii dla małych pacjentów z neurofibromatozą typu 1. Możliwość jej zastosowania działa niczym polisa ubezpieczeniowa – mówi Dorota Korycińska, prezeska zarządu Stowarzyszenia Neurofibromatozy Polska.
Z jakimi trudnościami mierzą się pacjenci z neurofibromatozą (NF)? Wciąż problemem jest uzyskanie prawidłowej diagnozy. Na szczęście, dzięki rozpoczętemu kilka lat temu pilotażowi opieki koordynowanej, pacjenci z NF nie doświadczają już tak długich odysei diagnostycznych jak kiedyś. Niemniej jednak, jak przystało na chorobę rzadką, tak i w tym przypadku pierwsze objawy (np. zmiany na skórze w kolorze kawy z mlekiem) mogą być tak nieoczywiste, że bywają pomijane przez wielu lekarzy. Inne trudności dotyczą kwestii emocjonalnych. Każda osoba, która dowiaduje się, że ona lub jej bliski jest obciążony przewlekłą, nieuleczalną chorobą, zmaga się z długim i trudnym procesem akceptacji.
Dlatego też ten stan niepewności, który jest wyczerpujący emocjonalnie, powoduje ciągły lęk.
Guzki NF, małe i duże, wewnętrzne i zewnętrzne, w różnych częściach organizmu i na skórze – oprócz fizycznych dolegliwości – powodują również zmianę wyglądu, czasami wręcz zniekształcenia skóry. Pacjenci z NF są więc narażeni na ogromny ostracyzm społeczny, którego doświadczają już małe, chore dzieci w przedszkolach i szkołach. To bardzo przykre.
Od stycznia 2024 r. dzieci w Polsce z nieoperacyjnymi nerwiakowłókniakami splotowatymi (najgorszymi z przejawów neurofibromatozy typu 1) mają dostęp do pierwszego leczenia celowanego. Co ta zmiana oznacza dla pacjentów i ich rodzin?
Pacjenci z NF mają wyższe ryzyko zachorowania, szczególnie na raka piersi. Uważam, że powinni być traktowani jak grupa wysokiego ryzyka nowotworowego.
Neurofibromatozy to schorzenia nieprzewidywalne. Medycyna nie potrafi jeszcze jasno wskazać, jakie problemy będą dotyczyć konkretnego przypadku, a wachlarz objawów jest dosyć szeroki (od wspomnianych zmian, przez guzy, po ryzyko zachorowania na nowotwór). Nie wiadomo, kiedy, czy i jakie objawy schorzenia się pojawią, w jakiej części organizmu, jaka będzie ich siła rażenia.
Terapia jest dla chorych jak polisa ubezpieczeniowa – w razie pojawienia się u dziecka nerwiakowłókniaka splotowatego będzie można zastosować leczenie farmakologiczne. Na szczęście tak poważne zmiany dotykają stosunkowo niewielu pacjentów, ale trudno przewidzieć, u kogo się zdarzą i czy będą nieoperacyjne. Te guzy są wyjątkowo ciężkim przypadkiem – nie tylko mogą oszpecać, lecz także powodują silne bóle, niedowłady, świąd, trudności z dopasowaniem ubioru, zniekształcenia. Powodują uciski na narządy wewnętrzne, unieruchamiają kończyny, utrudniają oddychanie czy spożywanie posiłków. Na dodatek niszczą nerwy, z których wyrastają, co wywołuje dodatkowe, negatywne konsekwencje zdrowotne. Dobrze więc, że zyskaliśmy dodatkowe i skutecznie narzędzie do walki z nimi.
Jeszcze dłużej stosowanym narzędziem jest opieka koordynowana, której pilotaż udało się w ubiegłym roku przedłużyć dzięki staraniom Stowarzyszenia. Jakie efekty ten program przyniósł w ciągu 3 lat funkcjonowania?
O korzyściach z pilotażu należy mówić na tle czasów sprzed pilotażu. Brak
systemowej opieki zdrowotnej był przyczyną niezwykle długiej odysei diagnostycznej. Od początku pilotażu pacjenci mają swoje poradnie, które zdjęły z nich ciężar poczucia bycia poza systemem. Trafiają do świetnych specjalistów, którzy mają i wiedzę, i doświadczenie w opiece nad pacjentami z NF. Śmiem twierdzić, że pilotaż to również korzystne rozwiązanie dla płatnika publicznego. Wszak dzięki temu projektowi pacjenci mają postawione prawidłowe diagnozy, otrzymują odpowiednie badania oraz odbywają wizyty kontrolne i diagnostyczne zgodne z dynamiką schorzenia. Poprzednio zdarzały się badania na chybił trafił: czasami zbyt wiele, czasami zbyt mało, czasami nieodpowiednie. W naszych centrach koordynowanej opieki medycznej nad pacjentami z NF opieka jest dopasowana do pacjenta niczym garnitur szyty na miarę. Korzyści z pilotażu, których już teraz doświadczamy, to przesłanki dla ustanowienia opieki koordynowanej jako stałego rozwiązania po jego zakończeniu.
Jakie są dalsze potrzeby pacjentów z neurofibromatozami w Polsce?
O ile potrzeby dzieci z NF zostały zabezpieczone, o tyle niestety brakuje opieki dla dorosłych z NF (6 funkcjonujących centrów opieki koordynowanej jest przeznaczonych wyłącznie dla dzieci), więc szukamy podmiotu medycznego, który się tego zadania podejmie. Dorośli pacjenci często krążą po systemie, odwiedzając wielu specjalistów: neurologa, chirurga, dermatologa, okulistę aż po onkologa. Ci ostatni, jeśli u pacjenta wystąpi nerwiakowłókniak splotowaty, który jest nowotworem niezłośliwym, nie chcą podjąć się leczenia. To powinno się zmienić. Kolejną kwestią jest potrzeba wprowadzenia regularnych badań przesiewowych w kierunku nowotworów złośliwych. Pacjenci z NF mają bowiem wyższe ryzyko zachorowania, szczególnie na raka piersi. Uważam, że powinni być traktowani jak grupa wysokiego ryzyka nowotworowego.
Więcej informacji na stronie: chorobyrzadkie.com


Rozpoznanie pierwotnej hiperoksalurii utrudniają jej niecharakterystyczne objawy, które są tożsame dla chociażby powszechnie występującej kamicy nerkowej. Tymczasem skuteczna diagnostyka znacząco poprawiłaby szansę na walkę z tą ultrarzadką chorobą.
Jakie wyróżniamy postacie pierwotnej hiperoksalurii i jak wygląda profil epidemiologiczny choroby?
Wyróżnia się jej trzy typy, ale zdecydowanie najczęściej, bo w ok. 80 proc. przypadków, występuje typ I. Takich pacjentów w ostatnich dziesięcioleciach rozpoznano w Polsce osiemnastu, z czego trzy to nowe przypadki chorych z Ukrainy. Ponadto znamy w Polsce pięć przypadków hiperoksalurii typu III oraz dwa typu II. Warto zauważyć, że liczby te mogą być niedoszacowane ze względu na możliwość nierozpoznanych przypadków. Sądzę, że może to dotyczyć kilkunastu-, kilkudziesięciu przypadków, w tym, jak się wydaje, głównie typu III, którego przebieg jest łagodniejszy. Jak więc widać po przytoczonej statystyce, pierwotna hiperoksaluria to choroba ultrarzadka. W krajach Europy Zachodniej i USA typ I występuje z częstotliwością jednego przypadku na 1-3 miliony mieszkańców. Jak się wydaje, w Polsce występuje jeszcze rzadziej.
Z czego wynikają trudności w rozpoznaniu tej choroby – ignorowanie objawów przez chorego czy błędna diagnoza lekarza?
Wynika to przede wszystkim z niecharakterystycznego obrazu klinicznego choroby. Objawy początkowe są tożsame z kamicą układu moczowego, która
jest dość powszechną chorobą człowieka uwarunkowaną wieloczynnikowo – począwszy od przyczyn dietetycznych, a na różnorodnych zaburzeniach metabolicznych skończywszy. Biorąc to pod uwagę zrozumiałe jest, że nie wszyscy pacjenci, zwłaszcza dorośli, poddawani są specjalistycznej diagnostyce metabolicznej, w tym w kierunku hiperoksalurii, czyli ocenie wydalania szczawianów z moczem.
Niewątpliwie jednak takie badanie należy wykonać u wszystkich dzieci z rozpoznaną kamicą moczową bądź nefrokalcynozą, natomiast u dorosłych – w wybranych przypadkach, obejmujących nawracającą kamicę moczową bądź nefrokalcynozę. U pacjentów z zaawansowaną niewydolnością nerek pomocne jest oznaczenie poziomu szczawianów w osoczu krwi. Ostatecznym rozpoznaniem pierwotnej hiperoksalurii jest badanie genetyczne i określenie typu mutacji określonego genu.
Jakie właściwie są objawy i przyczyny hiperoksalurii?
Jak już wspomniałem, najczęściej pierwszymi objawami są te związane z kamicą moczową. Są one niecharakterystyczne, a czasami skąpowobjawowe, zwłaszcza u dzieci. Najczęściej jednak
chory cierpi na bóle brzucha, nawracające kolki nerkowe, krwiomocz, a niekiedy wydala z moczem drobiny kamicze. Objawy te mogą wystąpić u pacjentów w różnym wieku, zarówno u niemowląt, dzieci starszych, jak i dopiero u dorosłych. Najczęściej jednak pojawiają się już w wieku dziecięcym, do 10. roku życia. Choroba występuje z podobną częstością u kobiet i mężczyzn, aczkolwiek ze względu na bardzo małą liczbę chorych w populacji relacja ta może być zaburzona. Warto pamiętać, że pierwotna hiperoksaluria jest chorobą metaboliczną, a jej przyczyną jest nadprodukcja szczawianu w wątrobie, co skutkuje zwiększonym wydalaniem szczawianów z moczem (hiperoksalurią), a w konsekwencji uszkodzeniem nerek i utratą ich funkcji. Determinuje to sposób leczenia.
Czy hiperoksaluria może zwiększać ryzyko występowania innych schorzeń?
Pierwotna hiperoksaluria w początkowej fazie jest po prostu chorobą układu moczowego, począwszy od rozwoju kamicy moczowej czy nefrokalcynozy, a na niewydolności nerek skończywszy.
Dlatego pacjenci znajdują się w spektrum działań urologów oraz nefrologów. Później, kiedy niewydolność nerek jest zaawansowana, gromadzący się

Prof. dr hab. n. med.
Przemysław Sikora
Klinika Nefrologii
Dziecięcej, Uniwersytet Medyczny w Lublinie
w organizmie szczawian pod postacią kryształów szczawianowo-wapniowych upośledza większość innych układów i tkanek. Na tym etapie pierwotna hiperoksaluria staje się chorobą wie-
Najczęściej chory cierpi na bóle brzucha, nawracające kolki nerkowe, krwiomocz, a niekiedy wydala z moczem drobiny kamicze.
loukładową. Problem dotyczy głównie kości, co prowadzi między innymi do dolegliwości bólowych i złamań. Pojawiają się też zmiany skórne, choroba atakuje również naczynia krwionośne, serce, oczy, układ nerwowy…
Ostatecznie powikłania z tym związane mogą prowadzić do przedwczesnego zgonu pacjenta.
Jakie terapie dostępne są dla pacjentów z hiperoksalurią?
Zależą one w dużej mierze od typu czy stopnia zaawansowania choroby i dzieli się je na leczenie objawowe i przyczynowe. To pierwsze obejmuje przede wszystkim płynoterapię, czyli przyjmowanie bardzo dużej ilości płynów. Do tego dochodzi podawanie inhibitorów krystalizacji sczczawianów wapnia, czyli alkalicznych cytrynianów, oraz stosowanie odpowiedniej diety.
Natomiast u pacjentów, u których już występuje zaawansowana niewydolność nerek, należy zastosować leczenie nerkozastępcze. Tacy chorzy muszą być dializowani lub poddać się przeszczepowi nerki zazwyczaj z towarzyszącym przeszczepem wątroby. Należy podkreślić, że konieczność przeszczepu wątroby wynika z etiopatogenezy choroby, gdyż przeszczep samej nerki kończy się szybkim zniszczeniem graftu w mechanizmie podobnym do uszkodzenia nerek własnych.
Na szczęście w przypadku hiperoksalurii typu 1 możliwe jest również leczenie przyczynowe. Część takich pacjentów reaguje na leczenie pirydoksyną (witaminą B6), co zmniejsza ilość produkowanego i wydalanego szczawianu. Znakomitą wiadomością dla pozostałych jest pojawienie się w ostatnim czasie nowych leków opartych na technologii interferencji RNA. Mówiąc ogólnie, metoda ta hamuje szlak metabolizmu szczawianów, co doprowadza do zmniejszenia, a nawet normalizacji ich wewnątrzwątrobowej produkcji, a w konsekwencji wydalania z moczem. Obecnie w Polsce leczymy w ten sposób ośmiu pacjentów, a wyniki są bardzo obiecujące. Mamy więc nadzieję, że dzięki temu spowolnimy postęp choroby bądź też w przypadku wczesnego rozpoczęcia leczenia nie dopuścimy do rozwoju niewydolności nerek. Ponadto, co może najważniejsze, u tak leczonych pacjentów nie będzie konieczności wykonania przeszczepu wątroby. Na koniec chciałbym podkreślić podstawową rolę wczesnego rozpoznania choroby dla jej skutecznego leczenia i poprawy rokowania u pacjentów.
Więcej informacji na stronie: chorobyrzadkie.com
PROBLEM
się m.in. nawracającą
Jeśli ataki kolki nerkowej są częste, warto wdrożyć diagnostykę w kierunku hiperoksalurii. Chociaż objawy wskazują na chore nerki, to jej przyczyną jest defekt enzymu w wątrobie.

Prof. dr hab. n. med. Mieczysław Litwin Kierownik Kliniki Nefrologii, Transplantacji Nerek i Nadciśnienia Tętniczego, Instytut „Pomnik – Centrum Zdrowia Dziecka”, Warszawa
Jakie są objawy hiperoksalurii?
U pacjentów występuje nawracająca kamica moczowa, gdyż szczawiany odkładają się w układzie moczowym w postaci tzw. kamieni nerkowych. Istotne jest to, że objawy kamicy rozwijają się bardzo wcześnie, nawet w wieku niemowlęcym. Ponadto hiperoksaluria objawia się też nefrokalcynozą, czyli wapnicą nerek. Schorzenie wynika z odkładania się w nerkach wydalanych w nadmiarze szczawianów, które w tym przypadku łączą się z jonami wapnia, tworząc szczawiany wapnia. Co ważne, hiperoksaluria upośledza funkcjonowanie nerek i im większe upośledzenie ich czynności, tym większe odkładanie szczawianów w układzie krążenia, tj. w sercu i tętnicach. Ten stan nazywa się oksalozą.
Jakie badania należy wykonać w celu postawienia diagnozy?
molekularnych genów odpowiedzialnych za hiperoksalurię. Znane są mutacje trzech genów, które mogą prowadzić do hiperoksalurii. Najczęściej jest to mutacja genu AGXT (powoduje on niedobory aktywności lub funkcji enzymu wątrobowego AGT) odpowiedzialna za najcięższą postać choroby. Na podstawie badania molekularnego można nie tylko potwierdzić chorobę, ale także określić jej typ – hiperoksaluria może być typu I, II lub III.
Czy we wszystkich przypadkach wskazane jest wykonanie łączonego przeszczepu nerki i wątroby?
produkujące szczawiany w nadmiarze, co zwiększa możliwości terapii dla tej grupy chorych i być może w przyszłości przeszczepy nie będą konieczne.
Więcej informacji na stronie: chorobyrzadkie.com
Tak jak w innych przypadkach kamicy moczowej pierwszym badaniem pozwalającym uwidocznić kamienie i rozpoznać wapnice jest badanie ultrasonograficzne układu moczowego. Badanie wydalonego lub usuniętego złogu może wskazać, że składa się on głównie ze szczawianów. Potwierdzeniem nadmiernego wydalania szczawianów jest ocena szczawianów wydalanych w dobowej zbiórce moczu lub w porcji moczu. Obecnie potwierdzeniem rozpoznania jest badanie molekularne, które pozwala na wykazanie wariantów
Zwykle wykonuje się go u osób chorujących na hiperoksalurię typu I, gdyż to właśnie u nich rozwój choroby jest najszybszy. Warto jednak pamiętać, że w każdej z postaci tego schorzenia prędzej czy później dochodzi do nieodwracalnego uszkodzenia nerek. Leczenie polega na wykonaniu przeszczepu łączonego nerki i wątroby, gdyż przyczyną hiperoksalurii jest mutacja genetyczna odpowiadająca za defekt enzymu wątrobowego, w konsekwencji wtórnie uszkadzająca nerki. Tym samym wykonanie przeszczepu samej nerki jest niewystarczające, gdyż defekt enzymatyczny obecny jest w wątrobie. Obecnie widzimy jednak niesamowity postęp medycyny – dostępne są już preparaty, które umożliwiają wyciszenie genów odpowiedzialnych za enzymy
Pierwszym badaniem pozwalającym uwidocznić kamienie i rozpoznać wapnice jest badanie ultrasonograficzne układu moczowego.
Panie profesorze, czy transplantacja narządów u pacjentów z pierwotną hiperoksalurią jest wystarczająca?
U pacjentów stosuje się leczenie typowe dla wszystkich procedur transplantologicznych, które zabezpiecza przed odrzuceniem przeszczepu przez organizm. I warto tu podkreślić, że po przeszczepie oraz otrzymaniu przez pacjenta nowej wątroby i nerki dochodzi do jego wyzdrowienia, chociaż nadal będzie narażony na konsekwencje oksalozy, czyli już wcześniej odłożonych w układzie krążenia złogów szczawianów.

Pacjentki chore na porfirię dzielą się swoimi doświadczeniami związanymi z walką z rzadką chorobą metaboliczną. Opowiedziały o wyzwaniach, jakie napotkały na swojej drodze, podkreślając potrzebę lepszej świadomości i opieki medycznej. Wspólnie ukazują nie tylko trudności, ale także nadzieję i możliwość poprawy.
Pani Kingo, jakie objawy skłoniły panią do zwrócenia się po pomoc medyczną?
KINGA NOWAK: Zaczęło się od zwykłych bóli brzucha. One stopniowo się nasilały, zaczęłam przyjmować leki przeciwbólowe, które jednak w pewnym momencie przestały przynosić ulgę. To dość specyficzny ból, którego centralny punkt trudno dokładnie zlokalizować. Jest „rozlany” po brzuchu. Towarzyszy mu ciemny mocz, mdłości i osłabienie mięśniowe. Z takimi objawami zgłosiłam się do lekarza rodzinnego, który skierował mnie do szpitala na oddział ginekologiczny. Tam przeszłam bardzo dużo badań, ale niczego nieprawidłowego nie stwierdzono. Jednocześnie moje samopoczucie było coraz gorsze, aż wreszcie zapadłam w śpiączkę. Wtedy przewieziono mnie na OIOM i tam po raz pierwszy padło podejrzenie, że cierpię na porfirię wątrobową. Trafiłam do specjalistycznego ośrodka w Warszawie, gdzie otrzymałam właściwe leczenie.
Jak wyglądała ścieżka diagnostyczna ostrej porfirii wątrobowej w pani przypadku, pani Paulino?
PAULINA ROWICKA: Na porfirię cierpiała moja mama, więc jako dziecko zostałam przebadana w tym kierunku i już w wieku 8 lat wiedziałam, że mam tę chorobę w genach. Nie było jednak wiadomo, czy i kiedy ona się ujawni. Nastąpiło to, gdy miałam 19 lat. Początkowo bolał mnie brzuch, który dusiłam paracetamolem i ketonalem. Ból jednak się nasilał, zgłosiłam się więc do szpitala rejonowego, gdzie błędnie wskazano na problemy miesiączkowe. Ja podejrzewałam jednak, że to atak porfirii, co niestety się potwierdziło, gdy trafiłam do warszawskiego Instytutu Hematologii i Transfuzjologii.
Jak porfiria wpływa na pani życie, pani Kingo, i jaką profilaktykę należy stosować, by zminimalizować jej nawroty?


KINGA NOWAK: Obecnie jestem leczona w programie lekowym i moja sytuacja uległa poprawie. Co prawda ataki nie są całkowicie wyeliminowane, ale jeśli się pojawiają, to są rzadsze i mniej dotkliwe. Mogę je przeczekać w domu, bez konieczności pilnej jazdy do szpitala. To znacząca różnica względem ataków wcześniejszych, bo wtedy średnio co dwa tygodnie trafiałam do placówki medycznej. Jeśli chodzi o profilaktykę, to przyjmuję dużo cukrów – glukozy oraz słodyczy. To pomaga w walce z porfirią.
PAULINA ROWICKA: Mój przypadek jest specyficzny, gdyż na skutek porfirii doznałam porażenia czterokończynowego. Bardzo długo leżałam w szpitalu, byłam w bezruchu, potrzebowałam pomocy przy najprostszych codziennych czynnościach. Ból był tak okropny, że ciało piekło mnie przy byle dotyku. Ponadto co chwilę pojawiały się kolejne ataki. Ta dramatyczna sytuacja ciągnęła się przez pół roku, potem nastąpiła pewna poprawa. Rozpoczęłam rehabilitację, powoli wracałam do w miarę normalnego funkcjonowania – mogłam ruszać kończynami, palcami, wstawać z łóżka. Nadal jednak mam problemy z chodzeniem, nie mam czucia w prawej nodze, poruszam się o kulach.
poprawy. Dopiero po zmianie metody leczenia zauważyłam, że glukoza zaczyna działać. Teraz ataki są rzadsze, mniej intensywne. Radzę sobie z nimi znacznie lepiej niż wcześniej.
Pani Katarzyno, jakie są największe potrzeby chorych?
Więcej informacji na stronie: chorobyrzadkie.com
A z jakimi trudnościami w związku z porfirią mierzy się pani, pani Paulino? Jaką profilaktykę pani stosuje?
Borykamy się z brakiem holistycznej opieki ambulatoryjnej dla pacjentów cierpiących na porfirię.
Zanim nadarzyła się możliwość leczenia w programie lekowym, ja również miewałam ataki mniej więcej co dwa tygodnie i wtedy od razu musiałam udać się do szpitala, co było dla mnie ogromnym wyzwaniem ruchowym. Jeśli chodzi o profilaktykę, to ta standardowa, czyli przyjmowanie glukozy, w moim przypadku nie dawała żadnej
KATARZYNA ROMANIUK: Borykamy się przede wszystkim z brakiem holistycznej opieki ambulatoryjnej dla pacjentów cierpiących na porfirię – dotyczy to chorych zarówno na ostrą porfirię przerywaną, ostrą porfirię wątrobową, jak i porfirę skórną. W Polsce nie mamy jednego ośrodka, w którym można uzyskać porady specjalistów z różnych dziedzin. A to choroba tak złożona, że konieczne jest współdziałanie neurologa, gastrologa, ginekologa czy endokrynologa. Ponadto powszechna wiedza wśród lekarzy dotycząca tej jednostki chorobowej jest niewielka. Na podstawie objawów wskazuje się na ogół na przyczyny ginekologiczne, co niestety jest błędnym tropem i znacząco wydłuża rozpoczęcie właściwej terapii. A to naraża chorych na przedłużony ból: zamiast kilku dób – bo po takim czasie następuje znacząca poprawa przy kierunkowym leczeniu – chory cierpi nawet kilka miesięcy.
Gdzie pacjenci mogą szukać wsparcia i informacji o chorobie?
KATARZYNA ROMANIUK: Jeśli tylko ma się podejrzenia występowania tej choroby, to należy bezzwłocznie udać się do specjalistycznego ośrodka, jakim jest Instytut Hematologii i Transfuzjologii w Warszawie, bądź przesłać do niego próbki moczu. To placówka, która udziela profesjonalnej pomocy w trakcie zarówno diagnostyki, jak i terapii. W Instytucie pacjent znajdzie specjalistyczną pomoc, może też uzyskać wszelkie informacje o chorobie oraz wsparcie psychologiczne.
Lepsza znajomość choroby może znacznie poprawić opiekę nad pacjentami z ostrą porfirią wątrobową.
Dr n. med. Robert Wasilewski Hematolog z Kliniki Zaburzeń Hemostazy i Chorób Wewnętrznych, Instytut Hematologii i Transfuzjologii
Czym charakteryzuje się ostra porfiria wątrobowa i jak wygląda jej epidemiologia?
Wyróżniamy trzy rodzaje ostrej porfirii wątrobowej – ostrą porfirię przerywaną (AIP), porfirię mieszaną (VP) i dziedziczną koproporfirię (HC). Są to choroby wrodzone i dziedziczne, wynikają z mutacji w genach kodujących enzymy szlaku syntezy hemu, co prowadzi do zmniejszenia ich aktywności. Efektem tego jest nadmierne gromadzenie się porfiryn lub ich prekursorów w organizmie. U ok. 80 proc. nosicieli mutacji przebieg choroby jest bezobjawowy (tj. postać utajona), a u ok. 70 proc. z nich wydalanie prekursorów porfiryn jest prawidłowe. Napady choroby zazwyczaj pojawiają się między 15. a 45. rokiem życia, a postać jawna częściej występuje u kobiet ze względu na wpływ czynników hormonalnych. W dzieciństwie napady AIP są rzadkie. Najczęściej diagnozowaną formą ostrej porfirii wątrobowej jest ostra porfiria przerywana. W Europie częstość występowania klinicznie jawnych przypadków wynosi 1-2 na 100 000, natomiast w Polsce jest to około 1,5 na 100 000.
Jak objawia się ostra porfiria wątrobowa?
Najczęstsze symptomy ostrej porfirii wątrobowej obejmują silny, narastający ból brzucha, któremu może towarzyszyć ból w obrębie klatki piersiowej, pleców
lub kończyn. Dodatkowo mogą pojawić się objawy neurologiczne, takie jak stopniowe osłabienie siły mięśniowej, zaburzenia czucia, a rzadziej – objawy psychiatryczne, jak pobudzenie, splątanie czy zaburzenia świadomości. Bardzo rzadko napad porfirii przebiega bez bólu, występując wyłącznie z objawami psychicznymi (zaburzenia zachowania, nastroju, pamięci) i neurologicznymi (osłabienie mięśni, zaburzenia czucia). W miarę trwania napadu, bez odpowiedniego leczenia, objawy mogą się nasilać.
Jakie badania mogą zdiagnozować chorobę i dlaczego tak ważne jest wczesne rozpoznanie?
Do rozpoznania zaostrzenia ostrej porfirii wątrobowej konieczne są badania wydalania z moczem prekursorów porfiryn (porfobilinogen i kwas delta-aminolewulinowy) i porfiryn, a także oznaczenie widma fluorescencji porfiryn w osoczu. Badanie wykonuje się w zależności od sytuacji klinicznej z dobowej zbiórki moczu lub z pojedynczej porannej próbki moczu. Oznaczanie wydalania „porfiryn całkowitych w moczu” bez oznaczenia wydalania prekursorów porfiryn jest bezwartościowe i może opóźnić postawienie diagnozy, a bywa oferowane przez niektóre laboratoria. U członków rodzin pacjentów z rozpoznaną ostrą porfirią
wątrobową należy wykonać badania genetyczne wykrywające mutację porfirynogenną w odpowiednich genach. Wczesna diagnoza pozwala na szybkie włączenie leczenia i uniknięcie powikłań neurologicznych. Sposobem na poprawę wykrywalności porfirii jest zwiększenie świadomości choroby wśród lekarzy i łatwiejszy dostęp do badań laboratoryjnych.
Czy istnieją konkretne czynniki wyzwalające, które mogą zwiększać ryzyko wystąpienia napadu porfirii i ich profilaktyka?
Do czynników wywołujących zaostrzenie (porfirynogennych) należą: stosowanie leków przeciwwskazanych w porfirii, kontakt z chemikaliami (farby, lakiery, rozpuszczalniki), zmiany hormonalne występujące w cyklu miesiączkowym, w ciąży lub wywołane leczeniem hormonalnym, spożycie alkoholu, palenie tytoniu, używanie narkotyków, ostre lub przewlekłe zakażenie (szczególnie wirusowe), głodzenie lub przewlekłe niedobory kaloryczne, zaburzenie rytmu dobowego, intensywny wysiłek fizyczny, stres. Aby choroba się nie zaostrzyła, należy unikać powyższych czynników. Bazę leków przebadanych pod kątem bezpieczeństwa w porfirii można znaleźć pod adresami: drugs-porphyria.org i porphyriafoundation.org/drugdatabase
WYZWANIA
System ochrony zdrowia zmierza do poprawy dostępu do specjalistycznej diagnostyki i leczenia porfirii, jednakże nadal istnieją wyzwania związane z brakiem opieki ambulatoryjnej i trudnościami w dostępie do badań laboratoryjnych.

Prof. dr hab. n. med.
Jerzy Windyga Kierownik Kliniki Zaburzeń Hemostazy i Chorób Wewnętrznych, Instytut Hematologii i Transfuzjologii
Do jakich ośrodków mogą się zgłosić pacjenci i lekarze podejrzewający u siebie lub swoich pacjentów objawy porfirii?
Porfiria jest chorobą metaboliczną pozostającą w kręgu zainteresowań lekarzy różnych specjalności, w tym gastroenterologów, hepatologów i hematologów. Klinika Zaburzeń Hemostazy i Chorób Wewnętrznych wraz z Pracownią Genetyki Hemostazy i Porfirii Zakładu Hemostazy i Chorób Metabolicznych Instytutu Hematologii i Transfuzjologii w Warszawie jest jedynym ośrodkiem kompleksowo diagnozującym i leczącym wszystkie typy porfirii w Polsce. Należy zaznaczyć, że w przypadku podejrzenia porfirii nie jest konieczna hospitalizacja pacjenta w naszym ośrodku – wystarczy przesłanie do Pracowni Porfirii próbki moczu. Można też skorzystać z oferty niektórych laboratoriów komercyjnych oferujących diagnostykę w kierunku porfirii pod warunkiem zlecenia odpowiedniego zestawu badań i uwzględniając zwykle długi czas oczekiwania na wyniki badań.
Jakie wyzwania stawia obecnie system ochrony zdrowia w kontekście opieki nad pacjentem z porfirią oraz jak można go poprawić?
Najważniejsze wyzwania to brak opieki ambulatoryjnej dla chorych na porfirię i ich rodzin, co utrudnia sprawną diagnostykę, oraz utrudniony od strony formalno-logistycznej dostęp do badań laboratoryjnych, wymagający rozliczeń między ośrodkami oraz organizowania transportu materiału do badań nierzadko na duże odległości. Rozwiązaniem tych problemów byłoby stworzenie sieci ośrodków zapewniających diagnostykę i leczenie w warunkach ambulatoryjnych i szpitalnych z odpowiednim finansowaniem przez NFZ, np. na wzór ośrodków leczenia chorych na hemofilię.
Jak zmieniła się opieka nad pacjentem z porfirią w ostatnich latach?
W ostatnich latach znacznie wzrosła świadomość choroby zarówno wśród lekarzy, jak i pacjentów. Powstało stowarzyszenie pacjentów, które również pomaga szerzyć wiedzę o chorobie.
Jednak najważniejszą zmianą było dopuszczenie przez EMA do stosowania w Europie terapii zapobiegającej zaostrzeniom choroby u chorych powyżej 12. roku życia z nawracającymi
Więcej informacji na stronie: chorobyrzadkie.com
Najważniejsze wyzwania to brak opieki ambulatoryjnej dla chorych na porfirię i ich rodzin.
zaostrzeniami. Od kwietnia 2022 r. leczenie jest dostępne w Polsce w ramach programu lekowego. Obecnie pacjenci leczeni są w ośrodkach w Warszawie, Bydgoszczy, Krakowie i Słupsku, nic nie stoi jednak na przeszkodzie, aby po spełnieniu warunków i podpisaniu umowy z NFZ leczyć pacjentów również w innych ośrodkach.
Więcej informacji na stronie: chorobyrzadkie.com
Codzienność z hipofosfatemią XLH jest niezwykle uciążliwa – choroba może prowadzić m.in. do niepełnosprawności ruchowej, oddziałującej na całe życie pacjenta. Na szczęście istnieje lek, który może zatrzymać postęp choroby.

Dr hab. n. med. Izabela Michałus Klinika Endokrynologii i Chorób Metabolicznych, Regionalne Centrum Chorób Rzadkich, Instytut Centrum Zdrowia Matki Polki w Łodzi
Czym charakteryzuje się hipofosfatemia XLH i w jaki sposób wpływa na życie pacjentów? Czy wiemy, jak przedstawia się jej epidemiologia?
To rzadka choroba genetyczna, jednak nie na tyle, abyśmy nie mieli pod swoją opieką dużej grupy pacjentów na nią cierpiących. Częstość występowania wynosi 1 na 20 tys. Szacuje się, że w Polsce jest to ponad 100 osób – zarówno dzieci, jak i dorosłych. Przypuszczamy, że wiele przypadków jest jeszcze nierozpoznanych.
Hipofosfatemia polega na utracie fosforanów przez nerki – ich deficyt w surowicy powoduje poważne konsekwencje zdrowotne dla wielu narządów oraz układów, w tym przede wszystkim dla kości. Choroba wpływa negatywnie na ich mineralizację, powoduje deformacje, wywołuje przewlekłe bóle kostne, zaburzenia wzrastania oraz ogólnego rozwoju.
Kiedy najczęściej objawia się choroba i jakie są jej pierwsze symptomy? Jakie badania należy wykonać, aby zdiagnozować chorobę?
Hipofosfatemia ujawnia się, gdy mały pacjent zaczyna chodzić – jej pierwszym symptomem jest szpotawe wygięcie kończyn dolnych, co doprowadza do nieprawidłowego chodu, potykania się i upośledzonej aktywności fizycznej. Wśród innych objawów wymienia się pojawienie się ropni okołozębowych, przedwczesnego zarastania szwów czaszkowych i przewlekłą niewydolność nerek. U dorosłych występują dolegliwości ze strony stawów oraz patologiczne złamania przeciążeniowe, niewynikające z urazu.
Podstawowym badaniem diagnostycznym jest oznaczenie stężenia fosforanów w surowicy krwi, które jest obniżone. Może je zlecić lekarz pierwszego kontaktu. Następnie pacjent powinien być skierowany do poradni specjalistycznej.
Czym charakteryzuje się innowacyjne leczenie? Kto ma do niego dostęp w Polsce? Przez lata terapia oparta była wyłącznie na leczeniu objawowym. Na szczęście od 1 listopada 2023 w Polsce w programie lekowym dostępny jest nowoczesny lek, który trafia w sedno choroby – jest to przeciwciało nerkowej utraty fosforanów. Obecnie trwa kwalifikacja pacjentów do tego leczenia, do którego włączani są pacjenci do 18. roku życia, co oznacza, że jak na razie program obejmuje tylko dzieci.
Jak rozszerzenie dostępu do terapii zmieniło losy małych pacjentów? Jakie konkretne korzyści przynosi im leczenie? Nowoczesne leczenie poprawia komfort życia, dzieci otrzymują iniekcje raz na dwa tygodnie zamiast trudno dostępnej mieszanki fosforanowej kilka razy dziennie. Leczenie celowane zatrzymuje utratę fosforanów, przez co hamuje mechanizm choroby – wycofuje deformacje kostne, zmniejsza ryzyko złamań, przez co przeciwdziała niepełnosprawności i powikłaniom, np. niewydolności nerek. Pośrednio wpływa pozytywnie także na aspekty psychospołeczne dziecka – poprawia jego kontakty z rówieśnikami i zapobiega wykluczeniu spowodowanemu innym wyglądem, ograniczoną wydolnością i sprawnością fizyczną.
Czy jest grupa pacjentów, którzy nie mają dostępu do leczenia?
Choroba trwa całe życie i nie mija wraz z osiągnięciem pełnoletności. Dlatego liczymy na to, że – tak jak w innych krajach – dorośli pacjenci również będą mogli korzystać z tego leczenia. Jak już podkreślałam, hipofosfatemia jest chorobą uwarunkowaną genetycznie i w około 80 proc. przypadków choruje jedno z rodziców naszych pacjentów.
Więcej informacji na stronie: chorobyrzadkie.com
Dla dorosłych dostęp do refundacji jest tak samo ważny, ponieważ ich choroba często prowadzi do nieobecności w pracy, braku możliwości uczestniczenia w aktywności fizycznej.
Jakie znaczenie ma dostęp do nowoczesnej terapii dla tej grupy pacjentów? Jak to leczenie może poprawić komfort ich życia?
Dla dorosłych dostęp do refundacji jest tak samo ważny, ponieważ ich choroba często prowadzi do nieobecności w pracy, braku możliwości uczestniczenia w aktywności fizycznej. Powoduje również obciążenie systemu ochrony zdrowia częstymi wizytami u różnych specjalistów. Dlatego – w dużym skrócie – refundacja nowoczesnego leku stanowiłaby mniejszy koszt dla systemu niż ich leczenie objawowe.

Prof. dr hab. n. med. Maria Mazurkiewicz-Bełdzińska Kierownik Kliniki Neurologii Rozwojowej, Gdański Uniwersytet Medyczny, przewodnicząca Zarządu Głównego Polskiego Towarzystwa Neurologów Dziecięcych
Rdzeniowy zanik mięśni (SMA) powstaje na skutek mutacji w genie SMN1. Jeśli terapia genowa zostanie zastosowana przed wystąpieniem pierwszych objawów, jest w stanie zapobiec pojawieniu się symptomów choroby.
Czym charakteryzuje się rdzeniowy zanik mięśni i jaki jest profil chorego?
30. roku życia). Na SMA w Polsce choruje około 1000 osób. Ponadto rocznie rodzi się tu około 50 dzieci, u których rozpoznawane jest SMA, a u około 35 z nich choroba ma ostry przebieg, czyli rozpoczyna się w okresie niemowlęcym.
Jak wygląda proces diagnostyczny SMA?
Obecnie dzieci rodzące się w Polsce mają wykonywane badanie przesiewowe m.in. w kierunku SMA. Dzięki tak wcześnie wdrożonej diagnostyce możliwe jest zastosowanie leczenia jeszcze przed wystąpieniem pierwszych objawów, co daje chorym szanse na normalne życie.
Czym jest program leczenia SMA? Na czym polega leczenie przyczynowe terapią genową?
Co ubiegły rok przyniósł polskim pacjentom z SMA w obszarze terapii genowej?
Przede wszystkim możemy pochwalić się tym, że program leczenia SMA w Polsce obejmuje wszystkie zarejestrowane na świecie nowoczesne terapie, które w połączeniu z wdrożonym w marcu 2022 r. programem badań przesiewowych noworodków zmienia-
Więcej informacji na stronie: chorobyrzadkie.com
W wyniku wady genetycznej obumierają neurony w rdzeniu kręgowym, co w konsekwencji prowadzi do osłabienia i zanikania mięśni szkieletowych oraz ostatecznie – osłabienia siły mięśniowej do całkowitego porażenia. U większości przypadków pierwsze objawy pojawiają się w okresie niemowlęctwa lub wczesnego dzieciństwa, jednak mogą wystąpić też później – w zależności od momentu pojawienia określany jest typ SMA (wyróżniamy typy opisane liczbami od 1 do 4, gdzie 1 wskazuje na wystąpienie pierwszych objawów przed 6. miesiącem życia, a 4 – powyżej
Od 2019 r. chorzy mogą skorzystać z nowoczesnych terapii w ramach refundowanego programu lekowego. Obecnie leczenie obejmuje stosowanie trzech substancji, w tym także od 2022 r. terapii genowej, która jest najnowocześniejszym osiągnięciem medycyny – działa na przyczynę choroby, poprawiając działanie nieprawidłowych genów. Co ważne, wdrożenie leczenia przed wystąpieniem pierwszych objawów jest w stanie zapobiec pojawieniu się symptomów choroby. Terapie te są bezpłatne, a skorzystanie z nich możliwe po zakwalifikowaniu do programu lekowego.
Dzięki wcześnie wdrożonej diagnostyce możliwe jest zastosowanie leczenia jeszcze przed wystąpieniem pierwszych objawów, co daje chorym szanse na normalne życie.
ją życie chorych i ich rodzin na lepsze. Trzeba podkreślić, że terapia genowa wymaga jedynie jednorazowego podania substancji, a dzięki niej dziecko może być w pełni sprawne i rozwijać się jak jego zdrowi rówieśnicy.
Fundacja SMA: wskazujemy chorym drogę do leczenia, które zmienia życie na lepsze

Dorota Raczek
Prezes Fundacji SMA od 2020 roku, żona i matka syna i dwóch córek – jedna z nich choruje na SMA; PR-owiec z wykształcenia, dyrektor zarządzający szczególnie domowym ogniskiem, kobieta ze Śląska Więcej informacji na stronie: chorobyrzadkie.com
Obecny program lekowy obejmuje trzy terapie, m.in. terapię genową. Leczenie i badania przesiewowe sprawiły, że dzieci rozwijają się jak ich zdrowi rówieśnicy.
Jakie wyzwania na co dzień stawia przed pacjentami i ich opiekunami diagnoza rdzeniowego zaniku mięśni (SMA)?
Do tej pory SMA było chorobą postępującą, dlatego regularną fizjoterapią można było jedynie opóźnić jej skutki, a to sprawiało, że codzienność pacjentów była wymagająca. Jednak po wdrożeniu programu lekowego i zastosowaniu nowoczesnych terapii u dzieci przed wystąpieniem pierwszych objawów leczenie jest w stanie zapobiec pojawieniu się choroby, dzięki czemu chorzy po terapiach programu lekowego mogą rozwijać się jak ich zdrowi rówieśnicy. Czekamy jeszcze na terapie, które mogą
wspomóc mięśnie. Trzeba pamiętać, że nadal jest grupa chorych, których codzienność wymaga m.in. rehabilitacji zwiększającej sprawność mięśni, stosowania wentylacji po utracie umiejętności oddychania, karmienia dojelitowego w przypadku utraty zdolności przełykania czy stosowania sprzętów ułatwiających poruszanie. Musimy pamiętać, że są to osoby potrzebujące specjalistycznej, skoordynowanej opieki.
Gdzie pacjenci i ich opiekunowie mogą szukać wsparcia i informacji o chorobie? Zachęcam do skontaktowania się z naszą fundacją, która działa już od ponad 10 lat, dlatego jesteśmy w stanie odpowiadać na różne potrzeby chorych, a także służyć jako rzetelne źródło wiedzy. Wiemy, że otrzymanie diagnozy SMA jest szokujące, dlatego wspieramy na każdym etapie choroby – zarówno rodziców najmłodszych dzieci, które zdiagnozowano dzięki programowi badań przesiewowych, jak również chorych, u których wykryto chorobę po pojawieniu się objawów.
Jakie są główne trudności i ograniczenia związane z chorobą? Co w tym kontekście
zmienił przełom w dostępie do leczenia SMA w Polsce?
Od końca marca 2022 r. funkcjonuje program badań przesiewowych m.in. w kierunku wykrywania SMA, dzięki czemu diagnoza stawiana jest szybko i tym samym noworodek może być kwalifikowany do nowoczesnego leczenia, w tym np. terapii genowej, które pozwala zatrzymać chorobę. To niewątpliwie odpowiedź na potrzeby tej grupy chorych, bo do 2019 r. trudności i ograniczenia wynikały z braku odpowiedniej diagnostyki i dostępu do refundowanego, innowacyjnego leczenia.
Jak terapia genowa zmieniła życie pacjentów z SMA i ich rodzin?
Zmieniła je zdecydowanie na lepsze, bo po wdrożeniu leczenia widzimy, że te rodziny funkcjonują prawie normalnie. U małych pacjentów rozwój fizyczny jest prawidłowy, czyli potrafią m.in. siadać, raczkować i chodzić, zupełnie tak jak ich zdrowi rówieśnicy, z czego niewątpliwie wszyscy się cieszymy. Poza tym jednorazowe podanie leku w terapii genowej wydaje się najlepszym wyjściem, jeżeli chodzi o noworodki z SMA – pośrednim efektem leczenia jest też to, że zyskuje na nim cała rodzina, której życie toczy się po prostu normalnie.

Od 1 września 2022 r. w ramach programu lekowego B.102 stosowana jest w Polsce terapia genowa, którą otrzymało do tej pory 35 pacjentów.
Kto ma dostęp do terapii genowej SMA?
U jakiej grupy pacjentów można zastosować terapię w Polsce?
W Europie terapia genowa w rdzeniowym zaniku mięśni (SMA) zgodnie z kryteriami rejestracyjnymi jest stosowana u pacjentów z rozpoznaniem genetycznym choroby, w okresie przedobjawowym lub po wystąpieniu objawów SMA typu I i II, którzy mają nie więcej niż 3 kopie genu SMN2 i którzy ważą nie więcej niż 21 kg. Jednak ze względu na to, że w badaniach klinicznych brali udział pacjenci, których masa ciała nie przekraczała 13,5 kg, zaleca się ją jedynie dla dzieci, które tej masy ciała nie przekraczają. W Polsce możliwości zastosowania terapii genowej w ramach programu lekowego B.102 wyznaczają dodatkowe kryteria kwalifikacji. I tak chorzy, u których może być ona zastosowana w momencie podania leku, powinni m.in. być w maksymalnie 6. miesiącu życia (<180 dni), mieć zachowaną zdolność połykania w ocenie lekarza kwalifikującego oraz osiągnąć więcej niż 12 punktów w badaniu funkcjonalnym (ocena w skali CHOP-INTEND). Kryterium koniecznym jest także fakt badania pacjenta w ramach programu przesiewowego noworodków w Polsce.
W jaki sposób kwalifikuje się chorych do terapii genowej w ramach programu lekowego B.102?
Więcej informacji na stronie: chorobyrzadkie.com
W 2022 roku sytuacja w Polsce znacząco się zmieniła, gdyż wszystkie rodzące się w kraju noworodki poddawane
są obecnie badaniom przesiewowym w kierunku SMA, dlatego też w większości przypadków można rozpoznać chorobę właśnie w badaniu przesiewowym. Wynik tego badania dostępny jest około 11.-13. doby życia dziecka. Według ustalonej i dobrze funkcjonującej ścieżki postepowania w przypadku SMA w Polsce w ciągu kilku dni rodzina chorego dziecka pojawia się w ośrodku, który prowadzi terapię SMA. Tam wykonywane jest badanie potwierdzające diagnozę i ocena, czy u dziecka występują objawy choroby. U pacjentów, u których stwierdza się objawy, leczenie można rozpocząć natychmiast, a jeśli objawów nie ma, wówczas czekamy na wynik badania potwierdzającego chorobę. Co istotne u pacjenta z SMA, u którego może zostać zastosowana terapia genowa w ramach programu lekowego, można równocześnie wykonać badanie sprawdzające, czy nie ma on przeciwciał przeciwko wirusowi AAV9. Wirus ten jest nośnikiem terapii genowej i obecność przeciwciał uniemożliwia zastosowanie tego leku. Przeciwciała mogą pochodzić od matki; w takim wypadku najczęściej po czasie znikają. W takiej sytuacji można zastosować od razu inny lek dostępny w programie lekowym albo czekać, aż przeciwciała znikną. Do dyspozycji mamy dla najmłodszych pacjentów w programie lekowym nusinersen. Lek ten może być podany właściwie każdemu dziecku z SMA, także temu, które nie kwalifikuje się do terapii genowej. Co ważne, nie oznacza to mniejszej skuteczności leczenia.
U ilu chorych w Polsce zastosowano terapię genową?
Terapia genowa w ramach programu lekowego B.102 stosowana jest w Polsce od 1 września 2022 r. i do tej pory otrzymało ją 35 pacjentów. Należy jednak pamiętać, że jest też spora grupa pacjentów, którzy otrzymali to leczenie poza programem lekowym, czyli m.in. ze zbiórek społecznych umożliwiających finansowanie terapii genowej. Ponadto mamy też pojedyncze przypadki chorych, którzy otrzymali ją w ramach badań klinicznych lub w ramach darowizn.
W 2022 roku sytuacja w Polsce znacząco się zmieniła, gdyż wszystkie rodzące się w kraju noworodki poddawane są obecnie badaniom przesiewowym w kierunku SMA.

Prof. dr hab. n. med.
Katarzyna Kotulska-Jóźwiak Przewodnicząca Zespołu Koordynacyjnego ds.
Leczenia Chorych na Rdzeniowy Zanik Mięśni, kierownik
Kliniki Neurologii i Epileptologii Instytutu „Pomnik – Centrum Zdrowia Dziecka”
Co dla pacjentów i ich rodzin/opiekunów oznacza możliwość zastosowania terapii genowej? Jakie korzyści odnoszą mali pacjenci leczeni terapią genową? Podanie terapii ma na celu zahamowanie procesu chorobowego, czyli obumierania komórek ruchowych rdzenia kręgowego (motoneuronów). Oznacza to, że terapia daje możliwość uratowania tych komórek, które jeszcze nie zostały nieodwracalnie uszkodzone wskutek choroby. Działanie leku, zarówno terapii genowej, jak i nusinersenu i trzeciej zarejestrowanej w SMA cząsteczki – rysdyplamu, polega na zwiększaniu ilości brakującego białka SMN. W przypadku terapii genowej odbywa się to dzięki dostarczeniu prawidłowej kopii genu produkującego to białko. Gen ten nie zastępuje zmutowanego genu, tylko funkcjonuje obok. Kluczową kwestią dla powodzenia terapii jest czas, rozumiany zarówno jako wiek pacjenta, jak i etap choroby. Dotyczy to wszystkich trzech leków stosowanych w SMA. Terapia jest najbardziej skuteczna u pacjentów, którzy otrzymają ją w okresie przedobjawowym, czyli zanim choroba spowoduje spustoszenie. Wówczas istnieje szansa na to, że objawy SMA w ogóle się nie rozwiną albo jeśli się rozwiną, to będą stosunkowo niewielkie i nie będą wpływały istotnie na jakość życia pacjenta. Wiele tak wcześnie leczonych dzieci rozwija się prawidłowo, tj. zgodnie z kalendarzem rozwojowym dziecka. Ponadto z wyników badań klinicznych już wiemy na pewno, że dzięki wczesnemu leczeniu nie dochodzi do najcięższych powikłań choroby, jakimi są niewydolność oddechowa, głęboka niepełnosprawność czy nawet śmierć z powodu rdzeniowego zaniku mięśni.

Więcej informacji na stronie:
chorobyrzadkie.com
To rzadka choroba zapalna, która dotyczy zarówno dzieci, jak i dorosłych. Wymaga szybkiej interwencji medycznej i leczenia.

Prof. dr hab. n. med.
Brygida Kwiatkowska
Klinika Wczesnego
Zapalenia Stawów, Narodowy Instytut Geriatrii, Reumatologii i Rehabilitacji w Warszawie
Czym charakteryzuje się choroba Stilla?
W trakcie choroby pojawiają się objawy, takie jak: wysoka gorączka utrzymująca się co najmniej tydzień, łososiowa wysypka, bóle stawów trwające co najmniej dwa tygodnie oraz zapalenie stawów z obrzękiem, bólem i zaczerwienieniem. Ponadto mogą pojawić się też symptomy ogólne, np. bóle mięśni, ból gardła, powiększenie węzłów chłonnych czy ogólnie złe samopoczucie.
Kiedy pojawiają się pierwsze objawy?
Czy różnią się one w zależności od wieku pacjenta?
Choroba zwykle zaczyna się między 16. a 25. rokiem życia lub między 36. a 46. rokiem życia. Jej przebieg jest bardziej dramatyczny u dzieci, gdyż u dzieci częściej niż u dorosłych rozwija się zagrażający życiu zespół aktywacji makrofagów. Co ważne, schorzenie może mieć dwa typy przebiegu – pierwszy, w którym dominują objawy stawowe, czyli m.in. obrzęki i bóle stawów, oraz drugi, w którym dominuje postać uogólniona, czyli pojawia się gorączka, zajęcie wielonarządowe i ciężki stan chorego wymagający agresywnego leczenia.
Jak wygląda proces diagnostyczny?
Bardzo istotne jest, by wdrożyć właściwą diagnostykę różnicową, gdyż objawy obserwowane w chorobie Stilla występują również w wielu innych schorzeniach, w tym m.in. w zakażeniach, różnego typu nowotworach czy reumatoidalnym zapaleniu stawów.
Jednak postawienie diagnozy nie jest łatwe – rozpoznanie stawiane jest na postawie obrazu klinicznego chorego, którego uzyskanie umożliwia m.in. przeprowadzenie wywiadu przez specjalistę. Pomocne są także badania laboratoryjne, takie jak: oznaczenie we krwi stężenia ferrytyny, CRP, OB oraz enzymów wątrobowych (AIAT, AspAT, LDH). Ze względu na niespecyficzne, pojawiające się nagle objawy czas w postępowaniu diagnostyczno-terapeutycznym odgrywa istotną rolę. Należy pamiętać, że dynamiczny rozwój choroby i brak zastosowania leczenia lub nieodpowiednia terapia mogą prowadzić do uszkodzenia narządów, a w efekcie nawet do bezpośredniego zagrożenia życia i zgonu pacjenta.
Jak przedstawia się ścieżka terapeutyczna? Z jakimi problemami na jej drodze borykają się polscy pacjenci? Rekomendowana terapia polega na stosowaniu połączenia glikokortykosteroidów (GKS) z nowoczesnymi lekami biologicznymi. I tak chorzy w przypadku postaci stawowej stosują w pierwszym rzucie inhibitor interleukiny IL-6, a w przypadku postaci ogólnonarządowej – inhibitor interleukiny IL-1. Przy czym w Polsce w ramach programu lekowego dostępny dla pacjentów z chorobą Stilla jest inhibitor IL-6 i krótkodziałający inhibitor IL-1. Nie mamy refundacji długodziałającego inhibitora IL-1, który zwiększyłby nasze możliwości terapeutyczne. Leki długodziałające to bardziej komfortowe
rozwiązanie i nie jest to bez znaczenia, jeśli mówimy o terapii przez lata. To równie ważne w przypadku dzieci, dla których psychicznym i fizycznym obciążeniem są codzienne wkłucia. Problematyczna jest kwestia dostępności pacjentów do dobrze ukierunkowanej diagnostyki i terapii, która jest zróżnicowana na terenie całej Polski, gdyż nadal w niektórych
Objawy obserwowane w chorobie Stilla występują również w wielu innych schorzeniach, w tym m.in. w zakażeniach, różnego typu nowotworach czy reumatoidalnym zapaleniu stawów.
województwach brakuje specjalistycznych ośrodków. Istotne jest także to, by każdy chory trafiał do placówki z pełnym zapleczem diagnostyczno-terapeutycznym, tak by jak najszybciej można mu było pomóc. Z tym wiąże się też kolejny problem, który napotykamy, czyli niedostateczna świadomość choroby Stilla wśród specjalistów, przez co nierzadko chorzy wędrują od gabinetu do gabinetu, wykonując kolejne badania diagnostyczne bez właściwej diagnozy.

Dzięki nowatorskiej metodzie leczenia pacjenci z chorobą Stilla mogą powrócić do normalnego trybu życia. Niestety, wciąż pozostaje problem z właściwą diagnozą tego ciężkiego schorzenia i pełną dostępnością do leczenia.
Na czym polega terapia choroby Stilla?
Aby przejść do samej terapii, najpierw trzeba poprawnie zdiagnozować tę chorobę, co niestety jest bardzo trudne. Po pierwsze, jej objawy nie są charakterystyczne – zaczyna się od zwykłej gorączki, bóli stawów czy wysypki. Następnie pojawiają się liczne zmiany narządowe: zaburzenia pracy wątroby i jej powiększenie, wysięki do osierdzia, powiększenie węzłów chłonnych. Obja-
Choroba Stilla ma podłoże genetyczne, nie jesteśmy w stanie całkowicie jej zlikwidować, ale możemy wyeliminować jej objawy.
wy mogą wskazywać na zakażenie uogólnione (sepsę) czy ostrą białaczkę, co utrudnia i opóźnia właściwą terapię w kierunku choroby Stilla. Wielu lekarzy nie spotyka się z chorobami układowymi, jedynie specjaliści, jak reumatolodzy i hematolodzy, mają większą świadomość występowania tej choroby. Jest to schorzenie bardzo poważne, które zagraża życiu. Jeśli chodzi o terapię, to podstawowym zadaniem lekarzy jest
wyeliminowanie objawów, czyli zablokowanie tzw. burzy cytokinowej, która występuje w chorobie Stilla oraz z jeszcze większą gwałtownością w przebiegu zespołu aktywacji makrofagów, będącego powikłaniem choroby Stilla.
Jakie są najlepsze metody leczenia i czy wszystkie możliwe terapie są dostępne dla polskich pacjentów?
Mamy już w Polsce w różnym zakresie dostępne leczenie, które od wielu lat jest praktykowane w innych krajach. Chodzi o blokery interleukiny pierwszej i blokery interleukiny szóstej (IL-1 i IL-6). Od dwóch lat w Polsce możemy leczyć blokerami interleukiny pierwszej o krótkim działaniu i o długim działaniu, z czego się bardzo cieszymy, bo mówimy tu o pewnym przełomie w leczeniu choroby Stilla, choć zmniejszenie ograniczeń do inhibitora IL-1 o długim działaniu byłoby pomocne. Te substancje znakomicie poprawiają sytuację polskich pacjentów. Obecnie jest to podstawowa linia leczenia, od której zaczynamy terapię.
Chorzy wracają do pełni zdrowia i normalnego funkcjonowania?
Choroba Stilla ma podłoże genetyczne, nie jesteśmy w stanie całkowicie jej zlikwidować, ale możemy wyeliminować jej objawy. Trzeba mieć świadomość, że może ona w każdej chwili powrócić. Natomiast dzięki stosowaniu blokerów interleukiny pierwszej i szóstej pacjenci faktycznie mogą normalnie funkcjonować, nie mają żadnych ograniczeń, jeśli

Prof. KAAFM dr hab. n. med. Zbigniew Żuber Katedra Pediatrii KA AFM w Krakowie, Klinika Pediatrii i Reumatologii, Szpital św. Ludwika w Krakowie
chodzi o wysiłek fizyczny czy dietę. Chory wraca do normalnego trybu życia, do pracy czy szkoły. Te leki przywracają całkowicie jego sprawność. Pacjent musi jedynie stawiać się na kontrole, natomiast samo przyjmowanie leków – czy to w formie zastrzyków podskórnych, czy doustnie – może odbywać się w warunkach domowych. To jest właśnie ta główna korzyść z zastosowania innowacyjnej metody leczenia.
Czy wszyscy pacjenci w Polsce kwalifikują się do tego innowacyjnego leczenia?
Choroba Stilla to choroba heterogenna i może mieć różny przebieg, a od niego uzależnia się decyzje, jakiego leczenia wymagają konkretne przypadki. Choroba Stilla może przyjąć formę jednorazowego rzutu, wielu rzutów z regularnymi nawrotami lub utrzymywać się stale. Jeśli przebieg choroby jest łagodny, to w ogóle nie musimy korzystać z tych substancji. Natomiast jeśli objawy są nasilone, to jest to wskazana forma leczenia i wtedy każdy potrzebujący pacjent może być poddany terapii przy zastosowaniu blokerów interleukiny pierwszej i szóstej.
Jakie zmiany powinny nastąpić, aby polskim pacjentom z tym schorzeniem żyło się lepiej?
Więcej informacji na stronie: chorobyrzadkie.com
Problemem w Polsce jest fakt, że pełna oferta terapeutyczna nie jest dostępna dla wszystkich potrzebujących pacjentów. Część leczenia (blokery interleukiny pierwszej o długim działaniu) jest dostępna tylko w ramach programu RDTL, czyli Ratunkowym Dostępie do Technologii Lekowych, który finansuje stosowanie tej terapii, a nie wszystkie ośrodki lecznicze mogą z niej korzystać. To spore utrudnienie dla pacjenta, który na etapie diagnozy, leczenia i późniejszej kontroli może korzystać tylko z wybranych kilku ośrodków w Polsce. Dostęp w ramach programu lekowego znacząco zmieniłby sytuację pacjentów i lekarzy reumatologów. Druga kwestia dotyczy samej diagnostyki – uważam, że potrzebna jest kampania edukacyjna wśród lekarzy. Przebieg choroby Stilla jest nietypowy, niecharakterystyczny, co utrudnia prawidłową diagnozę i szybkie rozpoczęcie terapii.


Choroba Stilla, rzadka choroba autoimmunologiczna, stawia pacjentów przed wieloma wyzwaniami, zarówno w diagnozie, jak i codziennym zarządzaniu zdrowiem. Chorzy potrzebują wsparcia medycznego, psychologicznego oraz społecznego, aby poprawić jakość życia i codzienne funkcjonowanie.

Violetta Zajk Prezes Ogólnopolskiego Stowarzyszenia Młodych z Zapalnymi Chorobami Tkanki Łącznej „3majmy się razem”
Proszę opowiedzieć nam, z czym dokładnie wiąże się choroba Stilla. Jakie są jej główne objawy oraz trudności związane z diagnozą?
Choroba Stilla to rzadka choroba autoimmunologiczna, która charakteryzuje się przewlekłym stanem zapalnym, który może obejmować narządy, takie jak: stawy, skóra, narządy wewnętrzne i układ krwiotwórczy.
Typowe objawy obejmują gorączkę, zmęczenie, utratę wagi oraz bóle stawów i mięśni, u niektórych pacjentów może wystąpić wysypka, często zlokalizowana na tułowiu i kończynach. Choroba może powodować bolesne zapalenie stawów, co prowadzi do sztywności i ograniczenia ruchomości.
Jest to choroba rzadka, co oznacza, że lekarze mogą mieć ograniczone doświadczenie w diagnozowaniu i leczeniu tej choroby. Objawy, takie jak gorączka i bóle stawów, mogą być podobne do objawów innych chorób, co może utrudniać postawienie właściwej diagnozy. Nie ma jednego testu diagnostycznego, który jednoznacznie potwierdzałby obecność choroby Stilla. Diagnoza opiera się na obserwacji objawów oraz wykluczeniu innych możliwych przyczyn.
W związku z tym diagnoza wymaga starannego wywiadu medycznego, obserwacji objawów oraz przeprowadzenia wielu testów w celu wykluczenia innych możliwych przyczyn.
Jakie są główne potrzeby i wyzwania pacjentów dotkniętych chorobą Stilla
w codziennym życiu i zarządzaniu swoim stanem zdrowia?
Pacjenci dotknięci chorobą Stilla mogą napotykać różnorodne potrzeby i wyzwania w codziennym życiu oraz zarządzaniu swoim stanem zdrowia. Często potrzebują skutecznego leczenia objawów, takich jak bóle stawów, gorączka i zmęczenie, aby poprawić komfort życia i funkcjonowanie codzienne.
Choroba może mieć też znaczący wpływ na stan psychiczny pacjentów ze względu na jej przewlekły charakter, objawy fizyczne oraz ograniczenia w codziennym życiu. Wsparcie psychologiczne i psychoterapia mogą być istotne dla radzenia sobie ze stresem, depresją czy lękiem.
Wzrost świadomości na temat choroby może być kluczowy dla pacjentów, aby zrozumieć swoje schorzenie, objawy, leczenie oraz strategie samoopieki.
Objawy w tej chorobie, w tym bóle stawów i ograniczenia ruchomości, mogą utrudniać wykonywanie codziennych czynności, pracy i uczestnictwa w aktywnościach społecznych.
Pacjenci często muszą dokonać zmian w swoim stylu życia, takich jak dostosowanie aktywności fizycznej do swoich możliwości, zmiana diety czy radzenie sobie ze stresem, aby zarządzać swoim stanem zdrowia.
Żeby skutecznie zarządzać chorobą Stilla, pacjenci potrzebują wsparcia medycznego, psychologicznego oraz społecznego, aby poprawić jakość życia i funkcjonowanie codzienne. Regularna opieka lekarska, edukacja pacjentów oraz grupy wsparcia mogą pomóc w radzeniu sobie z wyzwaniami związanymi z tą chorobą.
Gdzie pacjenci mogą szukać wsparcia oraz pomocy?
Oprócz wsparcia od swojego lekarza w tej chwili nie ma wielu miejsc, gdzie pacjenci mogą się dowiedzieć więcej o swojej jednostce chorobowej. Nasze stowarzyszenie rozpoczyna działania edukacyjne na temat choroby Stilla, wkrótce weźmiemy udział w kampanii edukacyjnej dla pacjentów.
W jaki sposób społeczeństwo może lepiej zrozumieć i wesprzeć osoby dotknięte tą rzadką chorobą oraz ich rodziny?
Więcej informacji na stronie:
chorobyrzadkie.com
Organizowanie kampanii edukacyjnych może zwiększyć świadomość na temat chorób rzadkich, w tym choroby Stilla, jej objawów, diagnozy i leczenia. Włączenie informacji na temat chorób rzadkich do programów nauczania w szkołach może pomóc w budowaniu empatii i zrozumienia wśród młodzieży.
Żeby skutecznie zarządzać chorobą Stilla, pacjenci potrzebują wsparcia medycznego, psychologicznego oraz społecznego, aby poprawić jakość życia i funkcjonowanie codzienne.
Tworzenie grup wsparcia dla pacjentów dotkniętych chorobą Stilla i ich rodzin może zapewnić przestrzeń do dzielenia się doświadczeniami, emocjonalnym wsparciem oraz praktycznymi poradami.
Inwestowanie w badania naukowe nad chorobami rzadkimi może prowadzić do lepszego zrozumienia przyczyn, lepszych metod diagnozowania i skuteczniejszych terapii.
Społeczeństwo może aktywnie pracować nad obalaniem stereotypów i uprzedzeń wobec osób z chorobami rzadkimi, aby stworzyć bardziej wspierające i przyjazne dla wszystkich środowisko.
Poprawa zrozumienia i wsparcia społecznego dla osób dotkniętych chorobami rzadkimi wymaga zaangażowania społeczności, instytucji oraz decydentów politycznych na różnych poziomach. Dążenie do większej empatii, lepszej edukacji i większego dostępu do wsparcia może przyczynić się do poprawy życia osób z chorobą Stilla i innych rzadkich schorzeń oraz ich rodzin.
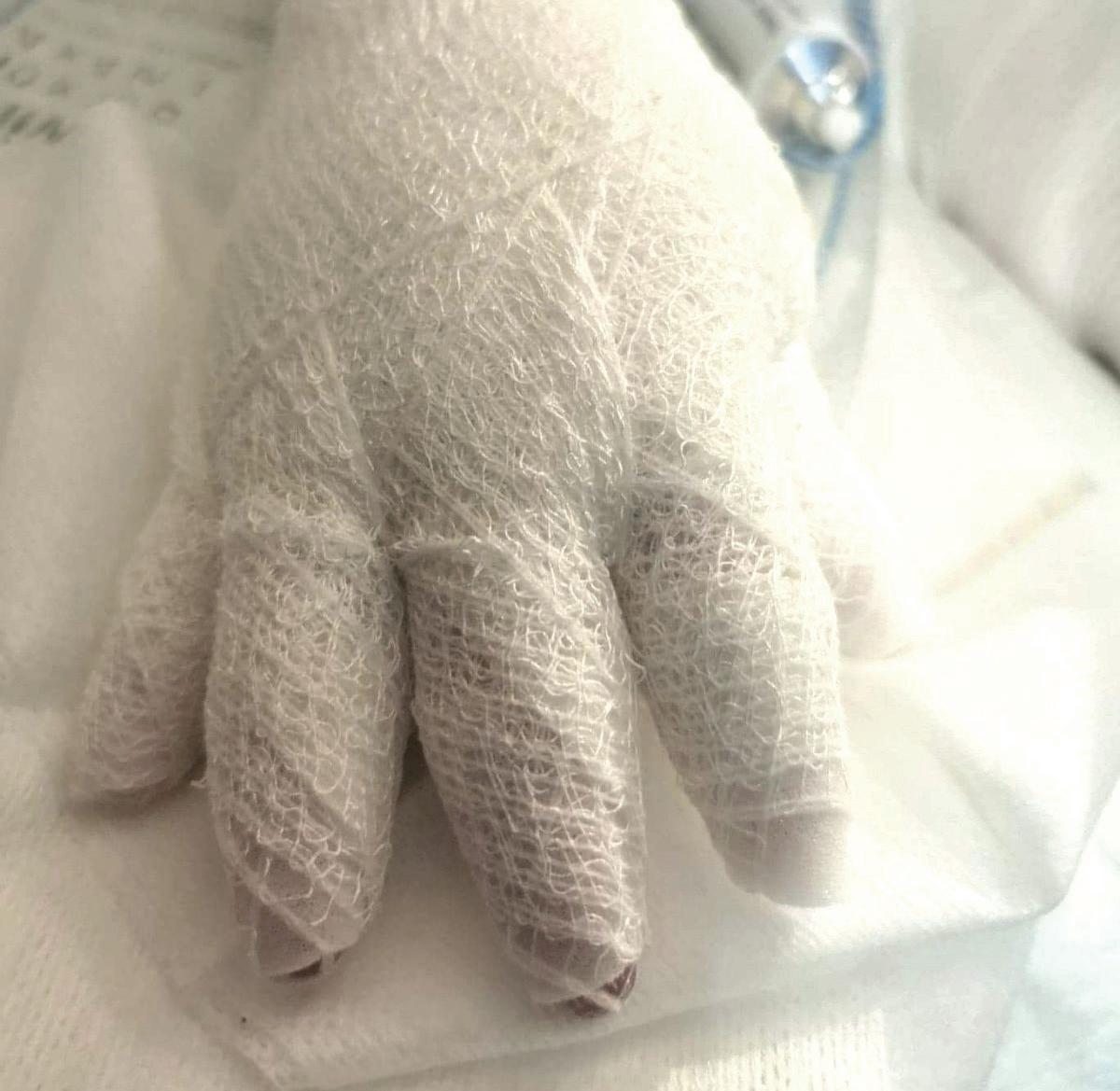
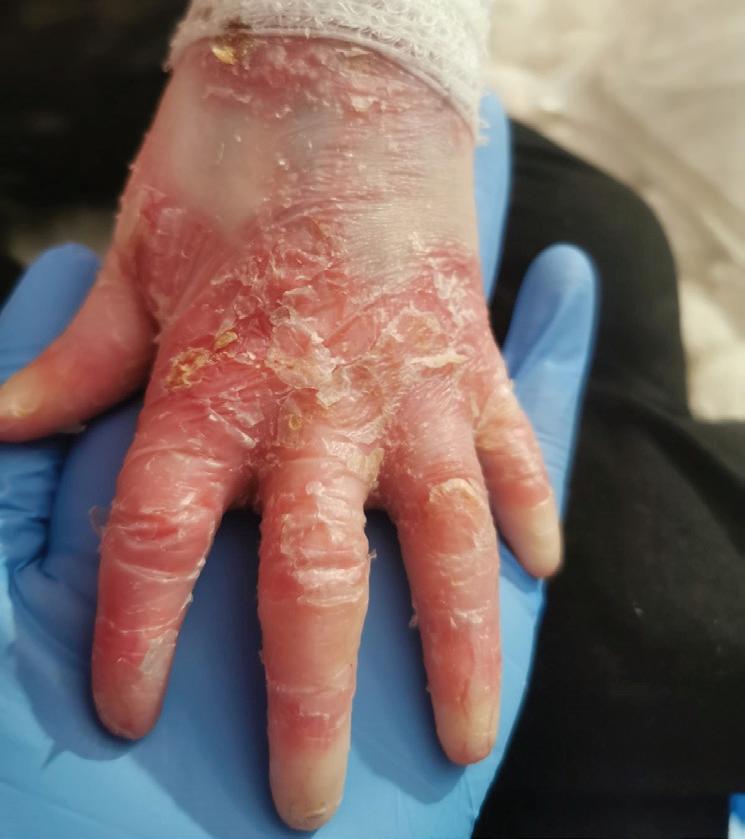
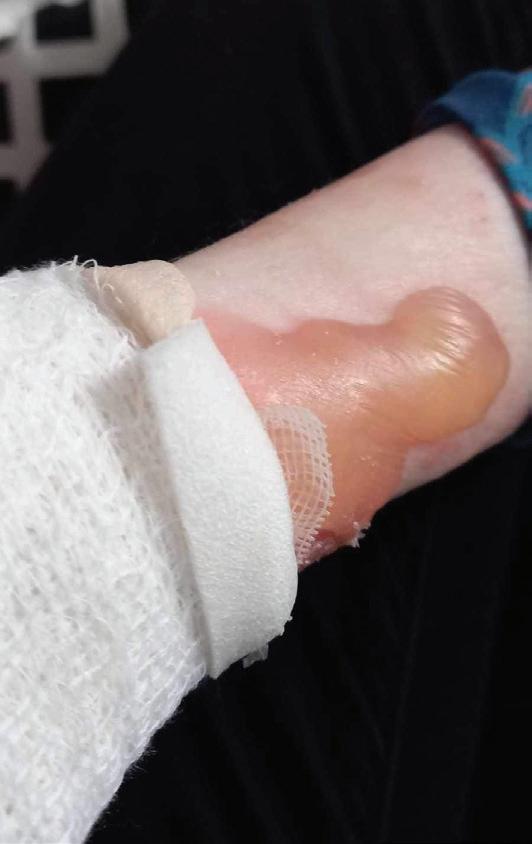
Pęcherzowe oddzielanie się naskórka – EB (epidermolysis bullosa) – to rzadka, dziedziczna choroba skóry charakteryzująca się nadmierną wrażliwością na urazy mechaniczne. Nawet najmniejsze otarcie czy ucisk powodują tworzenie pęcherzy i ran, a w konsekwencji owrzodzeń niegojących się tygodniami, a czasem miesiącami czy latami.

Więcej informacji na stronie: chorobyrzadkie.com
Zespół Fundacji EB Polska
EBokreślane jest jako choroba dzieci motyli, z uwagi na ogromną delikatność i kruchość skóry, ale też jako najboleśniejsza choroba świata. Ból towarzyszący ranom chorzy odczuwają bez przerwy. Każdy z nas zna z życia codziennego ból czy dyskomfort związany z małą raną – a teraz wyobraźcie sobie, że rany zajmują większość waszego ciała… Pacjenci z EB i ich opiekunowie stają przed wieloma wyzwaniami w zmaganiu się z chorobą. Jednym z głównych problemów jest codzienna, kilkugodzinna pielęgnacja ran. Wymaga dużej ostrożności i delikatności, aby uniknąć uszkodzeń skóry, których gojenie jest trudne.
EB powoduje spustoszenie całego organizmu. U wszystkich pacjentów z postacią dystroficzną o ciężkim przebiegu pojawia się niedożywienie, osłabienie odporności, przewlekły stan zapalny, anemia, zrosty przełyku. Pozostałe komplikacje mogą dotyczyć m.in. serca, układu oddechowego, oczu, nerek, uszu. Ryzyko rozwoju raka skóry wzrasta z każdym rokiem – w wieku 35 lat wynosi ponad 65 proc. EB wymaga więc wielospecjalistycznej opieki od najmłodszych lat. Niestety, brakuje ośrodków medycznych, w których
możliwa byłaby opieka kompleksowa. W przypadku EB dodatkowy problem stanowi fakt, iż nawet najprostsze badanie rutynowe może spowodować uraz skóry i przysporzyć dodatkowego cierpienia pacjentowi. Jest to problem w postępowaniu zarówno z dziećmi, jak też z dorosłymi i dotyczy obu stron – tak pacjenta, jak i lekarza przeprowadzającego badanie – i jak wynika z obserwacji Fundacji EB Polska, stanowi istotny problem w dostępie do opieki medycznej.
W wielu krajach funkcjonują specjalne ośrodki medyczne zajmujące się kompleksowo EB. W Polsce nie udało się stworzyć takiego ośrodka. Powołanie go w perspektywie czasowej jest niezbędne. Jednak w chwili obecnej funkcję koordynatora opieki medycznej mogliby pełnić lekarze genetycy kliniczni, którzy planowaliby proces diagnostyczno-kliniczny pacjenta, w tym częstotliwość badań laboratoryjnych i wielospecjalistycznych wizyt kontrolnych. Ważne jest również stworzenie sieci współpracy między lekarzami różnych specjalności. W obecnej chwili nawet osoby działające w Fundacji EB Polska natrafiają często na problem ze znalezieniem ośrodka, który zająłby się monitorowaniem określonego problemu klinicznego u pacjenta (np. zwężenia przełyku, kardiomiopatii, anemii hemolitycznej). Często też wizyta u specjalisty wiąże się z koniecznością wielogodzinnej podróży, co – pomijając

aspekt ogromnego dyskomfortu dla pacjenta – skutkuje powstaniem kolejnych ran wskutek podróży. Pacjenci zmęczeni codziennymi problemami wynikającymi z choroby rzadkiej potrzebują pomocy w środowisku medycznym oraz poczucia, że lekarz specjalista nadzoruje opiekę nad pacjentem, a pacjent i jego rodzina nie pozostają sami z problemem.
W tym miejscu należy nadmienić, iż chorzy na EB, nawet o ciężkim przebiegu, przy prawidłowym prowadzeniu są w stanie zdobywać edukację, prowadzić życie zawodowe, zakładać rodziny. Tym bardziej więc brak należytej opieki medycznej jest odbieraniem szansy na dobrostan nie tyle materialny, ile przede wszystkim psychiczny.
W Polsce pacjenci z EB i ich opiekunowie mogą szukać wsparcia i pomocy w Fundacji EB Polska. Fundacja pomaga m.in. w wyszukiwaniu specjalistów, organizowaniu wizyt lekarskich czy szkoleniu z zakresu leczenia ran (ebpolska.pl).

Wskazania
ostre i przewlekłe: tj. otarcia, owrzodzenia podudzi, zespołu stopy cukrzycowej, odleżyny, owrzodzenia nowotworowe
• Oparzenia termiczne, elektryczne
• Pielęgnacja miejsc wprowadzania cewników urologicznych, gastrostomii (PEG/PEJ), drenów oraz stomii


Chłoniak z dużej komórki B (LBCL) to najczęstszy z chłoniaków tzw. agresywnych, stanowiący 80 proc. rozpoznań w naszym kraju. Dynamiczny rozwój choroby wymaga szybkiej interwencji medycznej.
Czym charakteryzują się chłoniaki z dużych komórek B (LBCL) i jak wygląda przebieg choroby?
Charakteryzują się powiększeniem różnych grup węzłów chłonnych, które często są powiększone powyżej dwóch centymetrów i więcej. Chłoniak LBCL może też zajmować m.in. ośrodkowy układ nerwowy, naciekać płuca, wątrobę, śledzionę i inne narządy, a także szpik kostny. Warto wiedzieć, że agresywny przebieg kliniczny chłoniaka LBCL wiąże się z dynamicznym powiększaniem się węzłów chłonnych, czasami z tygodnia na tydzień, co wymaga szybkiej interwencji medycznej. Należy jednak pamiętać, że chociaż dynamika choroby jest intensywna, to możliwe jest wyleczenie znacznej części chorych.
Więcej informacji na stronie: chorobyrzadkie.com
U kogo najczęściej diagnozowany jest chłoniak LBCL? U ilu chorych w Polsce występuje postać nawrotowa lub oporna na leczenie?
Chorują na niego głównie osoby starsze, zwykle po 60. roku życia. Uważa się, że wyleczyć można około 60 proc. pacjentów, natomiast wydaje się, że chorobę oporną ma 10-20 proc., a chorobę nawrotową 20-30 proc.
Jakie są objawy i jak przebiega diagnostyka chłoniaka LBCL?
Na początku stwierdza się złe samopoczucie, może występować gorączka, nocne poty, utrata masy ciała i uczucie zmęczenia. Ponadto chorzy trafiają do gabinetu lekarza pierwszego kontaktu z powodu powiększonych węzłów chłonnych np. w okolicy szyi lub pod pachami. Ze względu na różnorodną lokalizację chłoniaka objawy też są zróżnicowane – i tak np. jeśli jest zlokalizowany w żołądku, może wywoływać ból brzucha, w klatce piersiowej – duszność, a w migdałku – trudności w przełykaniu. Jeśli chodzi o diagnostykę, to podstawę stanowi biopsja histologiczna. Standardem jest pobranie w całości
lub wycinka powiększonego węzła chłonnego. Następnie określany jest stopień zaawansowania choroby – podstawę stanowi badanie PET/CT. Ponadto w zależności od sytuacji chorego wykonywane są też badania, takie jak: badania genetyczne, rezonans magnetyczny, punkcja płynu mózgowo-rdzeniowego i trepanobiopsja.
Trudnością jest z pewnością szybki dostęp do wyspecjalizowanych ośrodków, w których wykonywana jest diagnostyka i leczenie.

Prof. dr hab. n. med.
Grzegorz W. Basak
Kierownik Kliniki, Katedra i Klinika
Hematologii, Transplantologii i Chorób Wewnętrznych, Warszawski
Uniwersytet Medyczny
Z jakimi problemami borykają się chorzy? Trudnością jest z pewnością szybki dostęp do wyspecjalizowanych ośrodków, w których wykonywana jest diagnostyka i leczenie. Kolejną problemową kwestią jest czas oczekiwania z jednej strony na przeprowadzenie diagnostyki i uzyskanie wyników badań, a z drugiej strony – na leczenie. Należy podkreślić, że wraz ze zwiększeniem dostępności do innowacyjnych, różnorodnych terapii proces leczenia stał się złożony, dlatego konieczna jest właściwa koordynacja, która wpływa na ostateczny efekt terapii. Warto podkreślić, że chorzy uzyskali ostatnio dostęp do nowoczesnych terapii zarówno w pierwszej linii leczenia, jak i w kolejnych. Nadal czekamy na zmiany w kwestii leczenia części chorych, u których występuje postać nawrotowa lub oporna na leczenie. Nie do końca zaopiekowaną grupą są m.in. chorzy, którzy okazali się oporni na terapię limfocytami CAR-T, chorzy, którzy nie odpowiedzieli na wcześniejsze, innowacyjne linie leczenia, jak chociażby schematem Pola-BR. I tutaj pojawia się przestrzeń dla nowoczesnych terapii – są to m.in. bispecyficzne przeciwciała, czyli przeciwciała monoklonalne epkorytamab i glofitamab, które potrafią niszczyć komórki nowotworowe, dając remisję nawet u 40 proc. chorych – żadne z nich nie jest jednak obecnie refundowane w Polsce.

Wdrożenie m.in. przeciwciał bispecyficznych do refundowanego leczenia chorych na chłoniaka DLBCL w Polsce mogłoby istotnie wydłużyć przeżycie wolne od progresji choroby.
Do jakich metod leczenia mają obecnie dostęp polscy pacjenci, u których rozpoznano chłoniaka DLBCL?
Rodzaj wybranej dla konkretnego pacjenta metody leczenia zależy od wielu czynników, w tym m.in. zaawansowania klinicznego choroby, wieku chorego i chorób współistniejących. Jeszcze do niedawna standardem leczenia była immunochemioterapia, którą u chorych z mniej zaawansowaną chorobą (stopień I i II według klasyfikacji z Ann Arbor/Lugano) można było podawać w dwóch-czterech cyklach
Ostatnie lata przyniosły wiele przełomów w leczeniu pacjentów z opornym lub nawrotowym chłoniakiem DLBCL.
w połączeniu z radioterapią, a u chorych w zaawansowanym stadium choroby (stopień III-V) w sześciu cyklach. I tak przez ponad 20 lat stosowano immunochemioterapię według schematu R-CHOP, czyli skojarzenie rytuksymabu z cyklofosfamidem, doksorubicyną, winkrystyną i prednizonem. Od stycznia 2024 r. polscy chorzy na DLBCL z 3-5 czynnikami ryzyka według IPI (International Prognostic Index) w pierwszej linii leczenia uzyskali dostęp do terapii
Pola-R-CHP, czyli skojarzenie polatuzumabu wedotyny z rytuksymabem, cyklofosfamidem i prednizonem, co zgodnie z wynikami randomizowanego badania POLARIX wydłuża przeżycie wolne od progresji choroby w porównaniu ze schematem R-CHOP.
Jakie możliwości terapii mają pacjenci, u których nie powiodło się leczenie I linii? Ostatnie lata przyniosły wiele przełomów w leczeniu pacjentów z opornym lub nawrotowym chłoniakiem DLBCL. U młodszych chorych, do 60.-65. roku życia, standardem jest stosowanie intensywnej immunochemioterapii skonsolidowanej z przeszczepieniem autologicznych, krwiotwórczych komórek macierzystych (auto-HSCT). Z kolei chorzy, którzy nie kwalifikują się do auto-HSCT, mogą obecnie skorzystać z nowoczesnych terapii, takich jak schemat Pola-BR, tj. skojarzenia polatuzumabu wedotyny z bendamustyną i rytuksymabem, lub z terapii tafasytamabem w połączeniu z lenalidomidem. Od 2022 roku dla polskich pacjentów dostępna jest również terapia CAR-T, którą można zastosować w przypadku choroby opornej lub nawrotowej od trzeciej linii leczenia, a więc po niepowodzeniu wcześniejszych dwóch linii leczenia. Ponadto od wielu lat w kolejnych liniach leczenia u chorych na chłoniaka DLBCL mamy do dyspozycji również piksantron, jednak ze względu na niższą w porównaniu z nowoczesnymi terapiami skuteczność obecnie stosowany jest coraz rzadziej. Te wszystkie wymienione przeze mnie terapie są refundowane i mogą być stosowane w ramach programu lekowego B.12FM.

Prof. dr hab. n. med. Ewa Lech-Marańda Konsultant krajowy w dziedzinie hematologii, dyrektor Instytutu Hematologii i Transfuzjologii
Czy istnieją kolejne innowacyjne terapie, które mogą pozytywnie wpłynąć na losy pacjentów?
Wśród najnowszych terapii zarejestrowanych do leczenia chorych na chłoniaka DLBCL należy wymienić przeciwciała bispecyficzne, które angażują własny układ odpornościowy chorego, a konkretnie limfocyty T, do niszczenia komórek chłoniakowych. Taki unikalny mechanizm działania jest możliwy dzięki konstrukcji przeciwciał bispecyficznych, które jednym swoim fragmentem łączą się z antygenem CD20 występującym na komórkach chłoniaka, a drugim fragmentem łączą się z antygenem CD3 obecnym na prawidłowych limfocytach T, umożliwiając tym samym niszczenie komórek nowotworowych. Ten unikalny mechanizm działania przeciwciał bispecyficznych jest zbliżony do terapii CAR-T, która również angażuje własne limfocyty T pacjenta do niszczenia komórek chłoniakowych, choć w nieco odmienny sposób.
Jakie zmiany w terapii DLBCL może przynieść wprowadzenie do terapii przeciwciał bispecyficznych?
Przeciwciała bispecyficzne to nowa grupa leków, wśród których dwie substancje, tj. glofitamab i epkorytamab, zostały zarejestrowane przez EMA (Europejska Agencja Leków) dla pacjentów z opornym lub nawrotowym chłoniakiem DLBCL od III linii leczenia. Leki te nie są jeszcze refundowane w Polsce, ale ich procesy refundacyjne są w toku. Warto również podkreślić, że chociaż działanie przeciwciał bispecyficznych jest podobne do bardzo skutecznej w leczeniu terapii CAR-T, to proces ich uzyskania pod względem technologicznym nie jest tak skomplikowany – przeciwciała bispecyficzne są gotowe do podania od razu.
Jakie zmiany powinny nastąpić, aby polski pacjent mógł być leczony optymalnie i zgodnie z najbardziej aktualnymi światowymi rekomendacjami?
Więcej informacji na stronie: chorobyrzadkie.com
Należy podkreślić, że obecnie dostęp do nowoczesnych terapii dla chorych na chłoniaka DLBCL jest dobry, a rejestracja przeciwciał bispecyficznych przez EMA to jest kwestia ostatniego roku. Obecnie mamy dwie potrzeby refundacyjne dla tej populacji chorych – przeciwciała bispecyficzne oraz rozszerzenie dostępu do terapii CAR-T już od drugiej linii leczenia w przypadku oporności na pierwszą linię leczenia lub szybkiego, to jest do 12 miesięcy, nawrotu po pierwszej linii leczenia.

Fenyloketonuria jest chorobą metaboliczną, której podstawą jest restrykcyjna dieta ograniczająca podaż jednego z aminokwasów – fenyloalaniny. Jak zatem możemy pomóc pacjentom z tą chorobą?

Lek. Dorota Korycińska-Chaaban Pediatra, specjalista pediatrii II stopnia oraz pediatrii metabolicznej, leczy chorych z fenyloketonurią, Poradnia Chorób Metabolicznych w Instytucie „Pomnik – Centrum Zdrowia Dziecka” w Warszawie, sekretarz Polskiego Towarzystwa Fenyloketonurii
Czym charakteryzuje się fenyloketonuria i jak się ją diagnozuje? Jaki jest profil epidemiologiczny choroby?
Fenyloketonuria (PKU) to wrodzona choroba metaboliczna, która nieleczona powoduje niepełnosprawność intelektualną. Rozpoznaje się ją w okresie noworodkowym dzięki badaniom przesiewowym. Polegają one na pobraniu na specjalną bibułę kilku kropel krwi z piętki dziecka w 3. dobie życia w celu oznaczenia m.in. stężenia fenyloalaniny. Jeśli wynik przekracza 132 μmol/l, pacjent wzywany jest do dalszej diagnostyki. Na fenyloketonurię chorują pacjenci na całym świecie. W 2020 roku (dane opublikowane z 64 krajów) było ich ponad 360 tysięcy. W Polsce każdego roku przybywa 30-50 małych pacjentów, choć zdarzają się pojedyncze przypadki chorych rozpoznawanych w późniejszym wieku.
Jakie są metody leczenia fenyloketonurii dostępne w Polsce?
Przyczyną PKU jest deficyt enzymu hydroksylazy fenyloalaniny przekształcającego jeden z aminokwasów – fenyloalaninę – do innego aminokwasu – tyrozyny. Objawy kliniczne (m.in. zaburzenia neurologiczne, w tym padaczka, małogłowie,
Podstawą jest edukacja i dobre nawyki – jeśli dziecko od początku jest właściwie leczone, poznaje zasady diety ubogofenyloalaninowej, to i później jest w stanie zaakceptować ograniczenia.
charakterystyczny „mysi” zapach) stanowią konsekwencję toksycznego działania, m.in. na ośrodkowy układ nerwowy, nagromadzonej w nadmiarze w tkankach chorego nieprzetworzonej fenyloalaniny.
Ponieważ fenyloalanina jest aminokwasem egzogennym, stężenie jej we krwi zależy od ilości przyjętej wraz z pożywieniem. Dlatego też podstawą terapii fenyloketonurii jest zastosowanie diety ubogofenyloalaninowej, z wykluczeniem produktów wysokobiałkowych (wyrobów mlecznych, mięsa, ryb, wędlin, roślin strączkowych, jaj, ziaren zbóż, orzechów) oraz aspartamu. Źródłem białka w diecie pacjentów są specjalne preparaty aminokwasowe, które nie zawierają fenyloalaniny. W Polsce na rynku jest dostępnych ponad 30 takich preparatów, a producenci wciąż wzbogacają swoją ofertę o nowe formy i warianty smakowe. Leczenie dietetyczne pozwala utrzymać stężenia fenyloalaniny we krwi chorego w bezpiecznych granicach, co chroni mózg przed uszkodzeniem i zapewnia chorym prawidłowy rozwój intelektualny. Restrykcyjne ograniczenia w diecie stanowią wyzwanie dla pacjentów, którzy nie zawsze ich przestrzegają, a zdarza się, że całkowicie rezygnują z leczenia.
Dostępne na świecie inne metody leczenia PKU, m.in. farmakoterapia (lekiem zawierającym sapropterynę) czy leczenie enzymatyczne (liaza amonowa fenyloalaniny), w Polsce nie są refundowane, a więc dla polskich pacjentów są niedostępne.
Jak można pomóc pacjentom, którzy mają problem z akceptacją choroby?
Fenyloketonuria jest chorobą na całe życie i wymaga leczenia przez całe życie. Stosowanie diety nie jest proste i wymaga wielu wyrzeczeń – dobrej organizacji i planowania posiłków. Podstawą jest edukacja i dobre nawyki – jeśli dziecko od początku jest właściwie leczone, poznaje zasady diety ubogofenyloalaninowej, to i później jest w stanie zaakceptować ograniczenia. Problem pojawia się, gdy mały pacjent chce próbować nowych produktów od swoich rówieśników, a młody człowiek wchodzi w okres buntu. Konsekwencja, ale i wsparcie ze strony rodziców, stały kontakt i zaufanie do lekarza i dietetyka z poradni chorób metabolicznych ułatwiają pokonanie trudności.
Do czego może doprowadzić niestosowanie specjalistycznej diety? Jak ważna jest ciągłość terapii?
Jeśli stężenie fenyloalaniny we krwi chorego przekracza wartości uznawane za bezpieczne (360 μmol/l do 12. r.ż. i u kobiet w ciąży oraz 600 μmol/l u młodzieży i dorosłych), mogą pojawić się problemy z koncentracją, lęki, agresja, autoagresja, zaburzenia neurologiczne, zachowania autystycze oraz postępująca niepełnosprawność intelektualna, małogłowie. Nasilenie objawów jest tym większe, im wcześniej dojdzie do rezygnacji z diety ubogofenyloalaninowej. Stosowanie właściwego leczenia przez całe życie zapewnia chorym rozwój podobny do rówieśników.
Szczególną grupę chorych stanowią kobiety ciężarne lub planujące ciążę. Kobiety, we krwi których w czasie ciąży stężenia fenyloalaniny przekraczały 360 μmol/l, mogą urodzić dzieci z licznymi wadami wrodzonymi (m.in. małogłowiem, wadami serca i niepełnosprawnością intelektualną), tzw. zespołem fenyloketonurii matczynej (MPKU).
W jaki sposób przeciwdziałać powstaniu zespołu fenyloketonurii matczynej u dziecka? Urodzenie zdrowego dziecka przez kobietę chorą na fenyloketonurię jest możliwe, jeżeli stężenia fenyloalaniny we krwi pacjentki w okresie prekoncepcyjnym i w trakcie trwania całej ciąży będą się utrzymywać między 120 a 360 μmol/l. Każdego roku kilkanaście chorych z PKU rodzi zdrowe dzieci. Planujące ciążę pacjentki powinny zgłosić się do poradni chorób metabolicznych w celu oceny wyrównania metabolicznego, doboru odpowiedniego preparatu bezfenyloalaninowego oraz normalizacji stężeń fenyloalaniny. Należy pozostać w stałym kontakcie z poradnią do czasu zajścia w ciążę, przez cały okres jej trwania i po porodzie. Ciąża u kobiety z fenyloketonurią jest przypadkiem najbardziej priorytetowym. Każda kobieta z fenyloketonurią, która jest lub podejrzewa, że jest w ciąży, powinna pilnie, tj. w najbliższym dniu roboczym, nawiązać kontakt z poradnią chorób metabolicznych i umówić się na wizytę w celu zapewnienia swojemu poczętemu dziecku optymalnych warunków do rozwoju. O swojej chorobie pacjentka powinna poinformować wszystkich specjalistów, pod opieką których się znajduje.

Jesteśmy liderem* w obszarze żywności specjalnego przeznaczenia medycznego stosowanej w postępowaniu dietetycznym, w rzadkich wrodzonych wadach metabolizmu
W naszej ofercie znajdują się produkty do postępowania dietetycznego m.in. w fenyloketonurii (PKU).
Chcąc otoczyć pacjentów kompleksową opieką, już od 8 lat prowadzimy największy** w Polsce serwis edukacyjny i społecznościowy dla chorych na fenyloketonurię oraz osób bliskich ich sercu – nutriciametabolics.pl
Każdego dnia inspirujemy społeczność PKU do podejmowania wyzwań, realizacji marzeń i czerpania z życia pełnymi garściami.


W ramach procedury importu docelowego oferujemy także żywność medyczną stosowaną w chorobie syropu klonowego, tyrozynemii, homocystynurii, kwasicach metabolicznych, zaburzeniach cyklu mocznikowego, zaburzeniach metabolizmu tłuszczów. Oferujemy również serwisy dla specjalistów, aby wesprzeć ich w codziennej pracy m.in. z pacjentami z rzadkimi chorobami metabolicznymi, m.in. akademianutricia.pl.





Nawet pojedynczy napad we wrodzonym obrzęku naczynioruchowym może zagrażać życiu pacjenta. Warto zatem wiedzieć, czym jest ta choroba oraz w jaki sposób można ją skutecznie leczyć.
Czym jest wrodzony obrzęk naczynioruchowy (HAE)? Jak często występuje?
Wrodzony obrzęk naczynioruchowy jest chorobą genetycznie uwarunkowaną, charakteryzuje się niedoborem stężenia inhibitora C1-esterazy bądź jego zaburzoną aktywnością. Objawia się poprzez obrzęk tkanki podskórnej i podśluzówkowej powstającej w wyniku nieprawidłowej, nadmiernej przepuszczalności naczyń krwionośnych pod wpływem działania bradykininy – nazywamy je często obrzękami białymi. Obrzęki w HAE zlokalizowane w obrębie gardła, krtani czy brzucha mogą stanowić bezpośrednie zagrożenie życia.
Pierwsze objawy mogą pojawić się już w wieku od 5 do 10 lat. Wczesne symptomy zazwyczaj zwiastują cięższy przebieg choroby. W Polsce obecnie mamy zarejestrowanych ok. 450 osób chorych na HAE. Są to najczęściej całe rodziny, ponieważ schorzenie jest dziedziczone w sposób autosomalny, dominujący.
Jakie są charakterystyczne objawy tej choroby?
Czy inne schorzenia mogą dawać podobne symptomy? Jak je odróżnić?
Pacjenci często zgłaszają się do przychodni czy do nocnej pomocy lekarskiej z bólem brzucha lub obrzękami różnych części ciała jako jednym z objawów różnych schorzeń. Chorzy z nawracającymi objawami, niepoddającymi się leczeniu typowymi lekami na obrzęki, jak leki przeciwhistaminowe czy sterydy, powinni zwrócić naszą uwagę, czy właśnie u nich nie mamy do czynienia z HAE. Charakterystyczne symptomy to obrzęki, którym nie towarzyszy świąd ani pokrzywka. Są niesymetryczne, utrzymują się od 2 do 5 dni. W przypadku obrzęków brzucha pacjent odczuwa silne dolegliwości bólowe oraz nudności, a przy obrzęku układu oddechowego – duszności i znaczny
niepokój. Przed samym napadem chorzy czują rozpieranie tkanek oraz ogólne złe samopoczucie. Obrzęk może pojawić się samoistnie bądź być indukowany poprzez drobne urazy, wysiłek fizyczny, stres, infekcje czy zabiegi medyczne. Napady występują częściej u kobiet w ciąży i w trakcie miesiączki z powodu zwiększonego stężenia estrogenów.

Nawet pojedynczy epizod takich obrzęków może zagrażać życiu pacjenta.
Czy rozpoznanie HAE jest trudne? Rozpoznanie tej choroby jest trudne, ponieważ objawy mogą pokrywać się z symptomami innych chorób. Dlatego też ważna jest świadomość lekarzy, że takie schorzenie istnieje i gdy tylko mają podejrzenie, że ich pacjent może cierpieć z jego powodu, powinni skierować go do poradni alergologicznej, jak również zlecić badanie stężenia inhibitora C1-esterazy czy C4 komplementu.
Czy nierozpoznany oraz nieleczony wrodzony obrzęk naczynioruchowy może zagrażać życiu pacjenta?
Oczywiście, że tak – obrzęk okolic gardła i układu oddechowego prowadzi do duszności i problemów z oddychaniem, obrzęk brzucha
Dr n. med. Tomasz Matuszewski Wojskowy Instytut Medyczny, Warszawska Poradnia Alergologii i Immunologii Klinicznej, przewodniczący sekcji „obrzęku wrodzonego” przy Polskim Towarzystwie Alergologicznym, konsultant w dziedzinie alergologii dla województwa mazowieckiego
może doprowadzić do perforacji jelita. Wtedy nawet pojedynczy epizod takich obrzęków może zagrażać życiu pacjenta. Od ponad 10 lat zwiększamy świadomość całego personelu medycznego na temat tej rzadkiej choroby, stworzyliśmy sieć ośrodków specjalizujących się w leczeniu HAE (obecnie jest ich 13), by jak najskuteczniej leczyć pacjentów na terenie całego kraju. Prowadzimy dla poszczególnych grup lekarskich liczne wykłady i szkolenia o tej chorobie.
Jakie są możliwości leczenia pacjentów z HAE w Polsce?
Leczenie przy stanach zagrażających życiu obejmuje dożylne podanie osoczopochodnego inhibitora C1-esterazy u wszystkich pacjentów, niezależnie od wieku, jak również u kobiet w ciąży. Rekombinowany inhibitor C1-esterazy również jest podawany dożylnie u osób dorosłych, młodzieży i dzieci powyżej 2. roku życia. Podskórnie podawany ikatybant jest antagonistą receptora bradykininy, służy do podawania osobom dorosłym, młodzieży i dzieciom od 2. roku życia. Pacjenci w większości przypadków są specjalnie przeszkoleni w ośrodkach prowadzących leczenie w celu nabycia umiejętności samodzielnego podania sobie tych preparatów. Każdy z leków działa bardzo szybko – w ciągu od 30 do 60 minut, dzięki czemu pacjent w krótkim czasie wykazuje znaczną poprawę. Dlatego też ważne jest, by chory miał lek zawsze przy sobie. Osoczopochodny inhibitor C1-esterazy można zastosować również w ramach profilaktyki krótkoterminowej, np. przed zabiegami chirurgicznymi, stomatologicznymi czy badaniami z użyciem np. endoskopów. Obecnie dla chorych z najcięższym przebiegiem HAE możemy zastosować profilaktykę długoterminową w ramach leczenia biologicznego.
Więcej informacji na stronie: chorobyrzadkie.com
Do kogo powinien się zgłosić pacjent, u którego wystąpiły objawy podobne do HAE? W pierwszej kolejności pacjent może zgłosić się do lekarza rodzinnego, który zarówno po zebraniu dokładnego wywiadu rodzinnego, jak i braku efektywności ogólnie dostępnych leków powinien skierować pacjenta na badanie w kierunku oznaczenia poziomu stężenia inhibitora C1-esterazy oraz C4 komplementu, może również skierować chorego do specjalisty alergologa. Pacjent może także sam wykonać w laboratorium takie badanie. Kolejnym krokiem przy stwierdzeniu nieprawidłowości powinno być skierowanie takiego chorego do jednego z ośrodków specjalizujących się w leczeniu HAE.
Dziedziczny obrzęk naczynioruchowy (hereditary angioedema, HAE) to rzadka i śmiertelnie niebezpieczna choroba. Objawia się nawracającymi obrzękami, które mogą zagrażać życiu. Jeśli obrzęk dotyczy np. gardła lub krtani, chory może się udusić.
Cierpiący na tę chorobę mają ograniczony dostęp do leczenia – leki do stosowania w ostrych atakach są refundowane w przypadku napadów zagrażająch życiu w obrębie gardła, krtani i brzucha. Istnieje możliwość leczenia profilaktycznego w ramach programu lekowego, jednak ma on swoje ograniczenia. Dostęp i różnorodność terapii powinny zostać rozszerzone, aby umożliwić skuteczną terapię większej grupie pacjentów.
Z jakimi wyzwaniami mierzą się obecnie pacjenci z dziedzicznym obrzękiem naczynioruchowym w Polsce?
Wyzwania te dotyczą ograniczonej wskazaniami refundacyjnymi dostępności zarówno do leczenia doraźnego napadów HAE, jak i do leczenia zapobiegawczego. Zmiana lub rozszerzenie wskazań refundacyjnych umożliwiłyby skuteczne leczenie większej grupy pacjentów oraz dostosowanie leczenia do
praktyki klinicznej zgodnej z międzynarodowymi standardami. W dalszym ciągu problemem pozostaje zapewnienie odpowiedniej jakości życia pacjentkom w okresie ciąży i laktacji, u których częstość i ciężkość napadów znacząco wzrasta. I wreszcie kwestia diagnostyki genetycznej dla pacjentów z HAE typu I, typu II oraz HAE z normalnym poziomem inhibitora C1-esterazy, która w dalszym ciągu nie jest refundowana przez płatnika.
Jakie są aktualne potrzeby polskich chorych z HAE?
Najpilniejsze potrzeby chorych z HAE dotyczą dwóch zagadnień: liberalizacji kryteriów włączenia do programu lekowego B.122 „Leczenie zapobiegawcze chorych z nawracającymi napadami dziedzicznego obrzęku naczynioruchowego o ciężkim przebiegu (ICD-10: D84.1)” poprzez rozszerzenie

Michał Rutkowski jest współzałożycielem i prezesem Zarządu Polskiego Stowarzyszenia Pomocy Chorym z Obrzękiem Naczynioruchowym Pięknie Puchnę. Jest również wiceprezydentem międzynarodowej organizacji parasolowej działającej na rzecz chorych z dziedzicznym obrzękiem naczynioruchowym – HAE International.
wskazań refundacyjnych o napady w obrębie twarzy i genitaliów, co umożliwiłoby większej liczbie pacjentów włączenie do programu lekowego, oraz refundacji kolejnego po lanadelumabie produktu leczniczego stosowanego w leczeniu zapobiegawczym – ludzkiego inhibitora C1-esterazy i zapewnienie jego dostępności w ramach programu lekowego B.122. Umożliwiłoby to włączenie do programu lekowego m.in. pacjentek ciężarnych i karmiących.
CZY WIESZ DLACZEGO PUCHNIESZ pomimo przyjmowania leków przeciwalergicznych?

Więcej informacji na stronie:
chorobyrzadkie.com
6-8% populacji cierpi na choroby rzadkie. Większość nie ma rozpoznania – chorzy nie są widoczni w systemie ochrony zdrowia. Wielu jest skazanych na odyseję diagnostyczną, trwającą nawet wiele lat.
Czasem do rozpoznania w ogóle nie dochodzi. Obecnie przełom w tych chorobach otwiera diagnostyka genetyczna.
W większości są to choroby uwarunkowane genetycznie (80%) , co łączy się ze zwiększonym ryzykiem choroby u członków rodziny.
Dostęp do terapii dla chorób rzadkich wynosi obecnie jedynie 14% (średnia dla krajów Europy to 37%).
W pełni dostępnych – czyli refundowanych w pełni i zgodnie z wytycznymi – jest tylko 2% leków sierocych
Co roku ponad 200 tys. osób otrzymuje diagnozę choroby rzadkiej.
Czas od rejestracji do refundacji – czyli czas dotarcia do pacjenta – dla leków na choroby rzadkie wynosił w latach 2017-2020 średnio 993 dni i był trzecim najdłuższym w Europie.
Choroby ultrarzadkie są przewlekłe, w większości uwarunkowane genetycznie, niejednokrotnie o ciężkim przebiegu. Programy lekowe umożliwiają wdrożenie terapii, która w niektórych przypadkach pozwala chorym normalnie żyć.
Jakie programy lekowe w chorobach ultrarzadkich są dostępne w Polsce? Co umożliwiają pacjentom?
Warto wiedzieć, że obecnie znanych jest około 8 tysięcy chorób określanych jako choroby rzadkie i ultrarzadkie. I chociaż z każdym dniem ta lista się rozszerza, to trzeba pamiętać, że tylko niewielka część tych schorzeń ma jakiekolwiek leczenie. W Polsce w 2009 roku został powołany Zespół Koordynacyjny ds. Chorób Ultrarzadkich, którego członkowie kwalifikują i weryfikują leczenie w ramach 13 programów lekowych. I tak np. dostępne są terapie dla schorzeń takich jak: choroba Fabry’ego, zespół Hurler, choroba Pompego, ciężkie, wrodzone hiperhomocysteinemie, wczesnodziecięca postać cystenozy nefropatycznej, wrodzone zespoły autozapalne i jeszcze kilka innych. Programy są zróżnicowane i dopasowane do potrzeb pacjentów z danymi jednostkami chorobowymi, jednak najważniejsze jest to, że dzięki nim chorzy mają dostęp do bezpłatnego leczenia. Dla
części pacjentów będzie to oznaczało prowadzenie normalnego życia (pomijając konieczność częstych kontaktów z systemem opieki zdrowotnej), a dla części spowolnienie rozwoju choroby. Z reguły całkowite wyleczenie nie jest możliwe z powodu genetycznych przyczyn choroby.
Jakie kroki musi podjąć pacjent i lekarz prowadzący po rozpoznaniu choroby ultrarzadkiej, by chory został zakwalifikowany do leczenia w ramach programu lekowego?
Po zdiagnozowaniu choroby ultrarzadkiej chory lub jego opiekunowie mogą zdecydować się na leczenie w ramach Programu – aby do niego przystąpić, na początku potrzebna jest formalna zgoda na to leczenie. Następnie lekarz prowadzący wypełnia wniosek o zakwalifikowanie chorego do terapii wskazanej w Programie, co po otrzymaniu pozytywnej decyzji będzie wiązało się np. z koniecznością regularnego monitorowania przebiegu leczenia.

Co jaki czas formalności związane z tym procesem muszą być odnawiane? Dlaczego zachowanie terminowości jest tak ważne w procesie leczenia?
W trakcie leczenia w ramach Programu konieczne jest spełnianie wymogów formalnych, czyli wysyłanie do zespołu koordynacyjnego regularnie co 6 miesięcy kart monitorowania terapii. Bez dopełnienia tego obowiązku leczenie nie powinno być kontynuowane. Należy jednak podkreślić, że to obowiązek, który spoczywa na lekarzu prowadzącym, a nie na pacjencie.
Jak ważna jest ciągłość terapii w leczeniu chorób ultrarzadkich? Do czego może doprowadzić przerwa w leczeniu?
W części przypadków przerwa w leczeniu wiążę się z groźnymi konsekwencjami, które skutkują pogorszeniem stanu zdrowia chorego. Dlatego też tak ważne jest realizowanie Programu zgodnie z wymogami formalnymi, co pozwala zachować ciągłość leczenia i tym samym czerpać z niego korzyści.
Więcej informacji na stronie: chorobyrzadkie.com

29 lutego obchodzimy Światowy Dzień Chorób Rzadkich. Czy w tym roku polscy pacjenci będą mogli w końcu świętować wielkie zmiany?
Więcej informacji na stronie: chorobyrzadkie.com
Panie prezesie, dokładnie rok temu rozmawialiśmy o sytuacji polskich pacjentów z chorobami rzadkimi. Co zmieniło się od tego czasu?
Nadal jest wiele do zrobienia. Niepokojące jest to, że Plan dla Chorób Rzadkich, czyli dokument przyjęty uchwałą Rady Ministrów w 2021 roku, zakończył swoją ważność z końcem 2023 roku. Oznacza to, że obecnie jesteśmy bez planu, choć po rozmowie z resortem zauważam sporą determinację, by po aktualizacji przyjąć go do realizacji. Do tego czasu zostało zawarte w nim wiele kluczowych dla pacjentów kwestii, opracowanych przez grono wybitnych specjalistów. Jednak samo ich przygotowanie nie jest tak istotne jak ich wdrożenie, by doprowadzić do konkretnych zmian.
No właśnie, jakie zmiany są zatem potrzebne pacjentom cierpiącym na choroby rzadkie w Polsce?
Najważniejsze jest to, byśmy mieli możliwość uzyskania kompleksowych i koordynowanych świadczeń w potrzebnym czasie. Chodzi przede wszystkim o utworzenie polskiej sieci ośrodków eksperckich należącej do sieci europejskiej. Polskie placówki już spełniają ku temu wszelkie warunki, jednak aby można było stwierdzić, że tworzą one polską sieć, muszą mieć zapewnione stosowne finansowanie. Pacjenci cierpiący na choroby rzadkie to nie są standardowe przypadki medyczne – wymagają oni szczególnego nakładu pracy oraz procedur do ich diagnozowania i leczenia. W związku z tym środki, które są konieczne do ich finansowania, są zdecydowanie większe, niż ma to miejsce w chorobach populacyjnych. Chorzy ci często są niechciani w systemie i odsyłani do placówek referencyjnych – tam ich leczenie jest deficytowe i przynosi zadłużenie. A przecież pieniądze na ten
cel są dostępne – zgodnie z deklaracją premiera m.in. na leczenie chorób rzadkich przeznaczono 3 miliardy.
Pacjenci cierpiący na choroby rzadkie to nie są standardowe przypadki medyczne – wymagają oni szczególnego nakładu pracy oraz procedur do ich diagnozowania i leczenia.
Z jakimi jeszcze wyzwaniami mierzą się pacjenci z chorobami rzadkimi? Jakie są ich potrzeby?
Należy zapewnić chorym i ich opiekunom wsparcie socjalne, dlatego też konieczne jest zaangażowanie do prac przy Planie Ministerstwa Rodziny i Polityki Społecznej. Warto również włączyć do dyskusji Ministerstwo Nauki i Edukacji, aby wdrożyć naukę o chorobach rzadkich na wielu płaszczyznach. Należy postawić na edukację lekarzy, by stworzyć odpowiednią kadrę medyczną oraz stosowne warunki do nauki dla chorych na choroby rzadkie. Bardzo ważny w tej kwestii jest także dostęp do informacji – przy okazji tworzenia Narodowego Planu Chorób Rzadkich zbudowano specjalną platformę internetową Choroby Rzadkie przy stronie Ministerstwa Zdrowia, jednak oczekuje ona na aktualizacje o nowe informacje.

Stanisław Maćkowiak
Prezes Krajowego Forum ORPHAN i Federacji Pacjentów Polskich
29 lutego obchodzimy Światowy Dzień Chorób Rzadkich. Jak będzie wyglądać tegoroczna uroczystość? Jak co roku spotykamy się 29 lutego w Łazienkach Królewskich w Warszawie na konferencji z okazji Światowego Dnia Chorób Rzadkich. 1 marca odbędzie się konferencja merytoryczna „Choroby Rzadkie – diagnostyka i leki dedykowane ich terapii – Wyzwania dla Polski 2024”. Będzie to miejsce, aby omówić aktualną sytuację w tej dziedzinie oraz podkreślić konieczność pilnego przyjęcia i efektywnego wdrożenia Narodowego Planu Dla Chorób Rzadkich. Mam nadzieję, że będą nam towarzyszyć liczni przedstawiciele parlamentu i administracji, w tym Ministerstwa Zdrowia. Nasze wydarzenia zostały objęte patronatem honorowym Ministra Zdrowia, Rzecznika Praw Pacjenta oraz Parlamentarnego Zespołu ds. Chorób Rzadkich.
Swixx BioPharma jest firmą farmaceutyczną, reprezentującą międzynarodowe koncerny z obszaru biotechnologii, preparatów dostępnych bez recepty (OTC) oraz wyrobów medycznych, działając z ich pełnomocnictwa lub w zastępstwie na rynkach, na których nie zdecydowały się one zaangażować lub zdecydowały się z nich wyjść.
Jesteśmy zaangażowani w poprawę zdrowia pacjentów, nasz Zespół niestrudzenie dokłada wszelkich starań, aby zapewnić pacjentom dostęp do nowoczesnych leków naszych Partnerów.
Swixx BioPharma koncentruje się na czterch strategicznych obszarach terapeutycznych, wprowadzając na rynek wysoce innowacyjne, nowe terapie w chorobach rzadkich, onkologii i hematologii, opiece specjalistycznej i szczepionkach.

SWIXX BIOPHARMA SP. Z O.O.
UL. PROSTA 51, 00-838 WARSZAWA
TELEFON: +48 22 460 07 20 E-MAIL: poland.info@swixxbiopharma.com
PM-PL-2023-12-6931
data zatw. materiału: 20.12.2023 r.