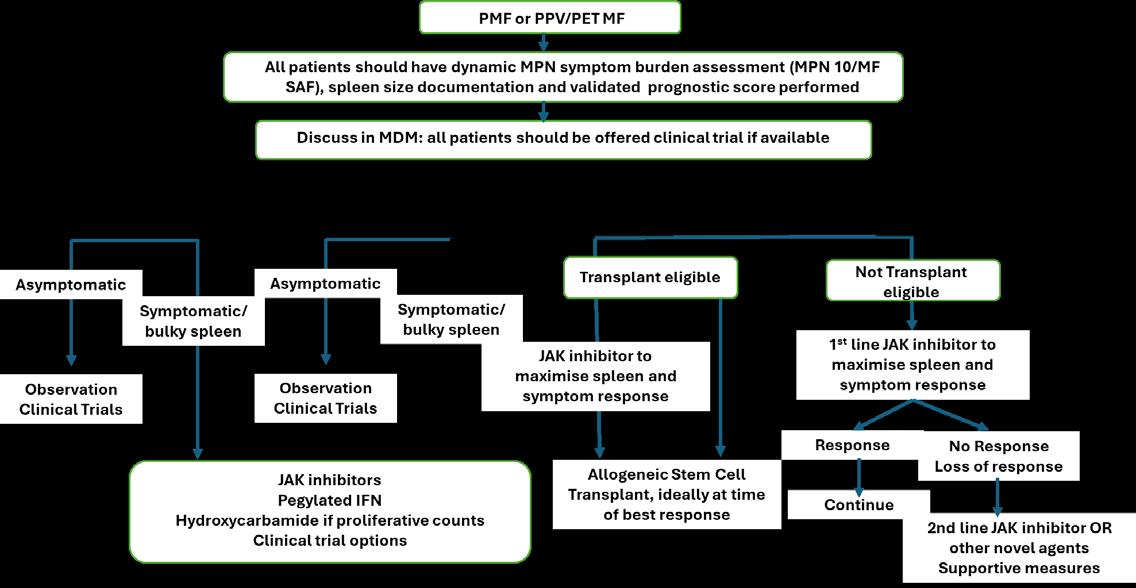
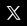Omjjara is indicated for the treatment of disease-related splenomegaly or symptoms in adult patients with moderate to severe anaemia who have primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis and who are Janus Kinase (JAK) inhibitor naïve or have been treated with ruxolitinib. 1
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
ACVR1 = activin A receptor, type 1; JAKi = Janus Kinase inhibitor.
Abbreviated Prescribing Information
148 x 210 mm (W x H)
Learn more about Omjjara:
Omjjara 100, 150 and 200 mg film-coated tablets Abbreviated Prescribing Information (Refer to Summary of Product Characteristics (SmPC) before prescribing). PRESENTATONS: Each 100 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 100 mg of momelotinib and 50.8 mg of lactose monohydrate. Each 150 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 150 mg of momelotinib and 76.1 mg of lactose monohydrate. Each 200 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 200 mg of momelotinib and 101.5 mg of lactose monohydrate. INDICATION: The treatment of disease-related splenomegaly or symptoms in adult patients with moderate to severe anaemia who have primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis and who are Janus Kinase (JAK) inhibitor naïve or have been treated with ruxolitinib. POSOLOGY AND ADMINISTRATION: Treatment should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. Omjjara should not be used in combination with other JAK inhibitors. The recommended dose is 200 mg once daily. Complete blood cell count and liver function tests must be performed before initiating treatment, periodically during treatment, and as clinically indicated. For dose modifications due to adverse reactions see SmPC. Treatment with Omjjara should be discontinued in patients unable to tolerate 100 mg once daily. Duration of use: Treatment may be continued for as long as the benefit-risk remains positive for patients, as assessed by the treating physician. Missed dose: If a dose of Omjjara is missed, the next scheduled dose should be taken the following day. Two doses should not be taken at the same time to make up for the missed dose. Elderly (≥ 65 years): No dose adjustment necessary.
Renal impairment (>15 mL/min): No dose adjustment necessary. Omjjara has not been studied in patients with end-stage renal disease. Hepatic impairment: No dose adjustment is recommended for patients with mild or moderate hepatic impairment. The recommended starting dose is 150 mg once daily in patients with severe hepatic impairment (Child-Pugh Class C). Paediatric population: No data available. Omjjara is for oral use only and can be taken with or without meals. CONTRAINDICATIONS: Hypersensitivity to momelotinib or to any of the excipients. Pregnancy and breast-feeding. WARNINGS/PRECAUTIONS: Omjjara should not be initiated in patients with active infections. Physicians should carefully observe patients for signs and symptoms of infection and initiate appropriate treatment promptly. Patients with chronic HBV infection who receive Omjjara should have their chronic HBV infection treated and monitored according to clinical HBV guidelines. A complete blood count including platelet count should be obtained before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated. Dose interruption or reduction may be required. Liver function tests should be obtained before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated. If increases in ALT, AST or bilirubin related to treatment are suspected, dose interruption or reduction may be required. Prior to initiating or continuing therapy with Omjjara, the benefits and risks for the individual patient should be considered particularly in patients 65 years of age and older, patients who are current or past long-time smokers, and patients with history of atherosclerotic cardiovascular disease or other cardiovascular risk factors. Patients with symptoms of thrombosis should be promptly evaluated and treated appropriately. Lymphoma and other malignancies have been reported in patients receiving JAK inhibitors, including Omjjara. However, a causal association has not been established Women using systemically acting hormonal contraceptives should add a barrier method during treatment and for at least 1 week after the last dose of Omjjara. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take Omjjara. Patients who experience dizziness or blurred vision after taking Omjjara should observe caution when driving or using machines
INTERACTIONS: Co administration of strong CYP3A4 inducers may lead to decreased momelotinib exposure and consequently a risk for reduced efficacy. Therefore, additional monitoring of the clinical signs and symptoms of myelofibrosis is recommended with concomitant use of momelotinib and strong CYP3A4 inducers (including but not limited to carbamazepine, phenobarbital, phenytoin, and St John’s wort [Hypericum perforatum]). Caution and monitoring for adverse reactions are advised with concomitant use of OATP1B1/1B3 inhibitors, including ciclosporin. Momelotinib may increase exposure to other sensitive BCRP substrates, including sulfasalazine; monitor for adverse reactions. Caution is advised when administering momelotinib with P-gp substrates with a narrow therapeutic index. Caution is advised when administering momelotinib with sensitive substrates of OCT1, MATE1 and MATE2-K (e.g., metformin). Narrow therapeutic index or sensitive substrate medicinal products of CYP1A2 (e.g., theophylline, tizanidine) or CYP2B6 (e.g., cyclophosphamide) should be co-administered with momelotinib with caution. Fertility, pregnancy and lactation: Fertility: No clinical data. Omjjara is contraindicated during pregnancy. If Omjjara is used during pregnancy, or if the patient becomes pregnant while taking this medicinal product, the patient should discontinue treatment and be advised of the potential hazard to the foetus. Omjjara is contraindicated during breast-feeding. UNDESIRABLE EFFECTS: Very common (≥ 1/10): Thrombocytopenia, dizziness, headache, cough, diarrhoea, abdominal pain, asthenia, fatigue. Common (≥ 1/100, < 1/10) Urinary tract infection, upper respiratory tract infection, pneumonia, nasopharyngitis, COVID19, cystitis, bronchitis, oral herpes, sinusitis, herpes zoster, cellulitis, respiratory tract infection, sepsis, lower respiratory tract infection, oral candidiasis, rash, skin infection, gastroenteritis, neutropenia, Vit B1 deficiency, syncope, peripheral neuropathy, paraesthesia, blurred vision, vertigo, hypotension, haematoma, flushing, vomiting, constipation, arthralgia, pain in extremity, pyrexia, ALT increased, AST increased, contusion. For more details on undesirable effects, see SmPC. Marketing Authorisation (MA) Holder: GlaxoSmithKline Trading Services Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland. MA Nrs: EU/1/23/1782/001- 003. Legal category: POM A. Date of preparation of API: April 2025. Code: PI-14515. Further information available on request from GlaxoSmithKline, 12 Riverwalk, Citywest Business Campus, Dublin Tel: 01-4955000.
Adverse events should be reported directly to the Health Products Regulatory Authority (HPRA) on their website: www.hpra.ie Adverse events should also be reported to GlaxoSmithKline on 1800 244 255. Start with
Call for papers: make your contribution to Hospital Professional News
Articles
Research Papers
Reviews
Programme Descriptions
Reports
Case Reports
Letters to Editor
Support fellow hospital professionals as well as aspiring junior professionals and early-year hospital pharmacists
Practice reports share innovations on any area of practice, including delivering clinical services, pharmacy administration, or new approaches to inform and engage with patients
Perspective articles focus on a specific field or discipline and discuss current advances or future directions, and may include original data as well as expert insight and opinions
Contents Foreword
New Critical Care Facility at St Vincent’s University Hospital P4
New Framework Agreement positive step for Medicines Access P6
Irish Pharmacy Researchers
Spotlight Global Gaps in Clozapine Prescribing P10
Ireland’s chance to better connect healthcare data P13
Cancer Patients to Receive new Treatments following ¤10m Investment P16
Persistent Gaps in Cardiovascular Care for Women P17
New Frontier in Early Lung Cancer Detection P34 REGULARS
Editor
February’s issue comes at an important time in the healthcare calendar as we mark World Cancer Day, a global moment to reflect on progress in cancer care while recognising the work that still lies ahead. Hospital pharmacists remain central to that effort, supporting complex treatment regimens, ensuring safe and timely access to medicines, and contributing to multidisciplinary care across the cancer pathway. In this issue, we feature a series of expert contributions covering breast cancer, lung cancer, and the evolving science behind mature B-cell neoplasia, offering both clinical insight and a look at the rapidly advancing therapeutic landscape.
Feature: Diabetes Mellitus P19
CPD: Lithium Therapy P29
Oncology Focus: Lung Cancer
Oncology Focus:
P55
Hospital Professional News is a publication for Hospital Professionals and Professional educational bodies only.
All rights reserved by Hospital Professional News. All material published in Hospital Professional News is copyright and no part of this magazine may be reproduced, stored in a retrieval system or transmitted in any form without written permission.
IPN Communications Ltd have taken every care in compiling the magazine to ensure that it is correct at the time of going to press, however the publishers assume no responsibility for any effects from omissions or errors.
We are also pleased to include a contribution from Professor Naidoo, an Irish consultant and internationally recognised leader in thoracic oncology, who writes with colleagues about The Lung Health Check and its role in shifting the paradigm in lung cancer care. Early detection remains one of the most powerful tools in improving outcomes, and initiatives such as this highlight how screening, prevention and targeted interventions can reshape survival rates in the years ahead.
Alongside clinical advances, policy developments continue to influence how medicines reach patients. This month, we report on the new four-year Framework Agreement welcomed by the Irish Pharmaceutical Healthcare Association. The agreement is expected to support faster access to innovative therapies while sustaining Ireland’s life sciences sector. For hospital pharmacists, timely reimbursement and access to new treatments remain key issues, and this development signals a continued commitment to improving patient outcomes through collaborative policy.
Professional development also remains a major focus. With the 2025/26 ePortfolio Review underway, approximately 1,800 pharmacists will submit their CPD records to the Irish Institute of Pharmacy. In this issue, Áine Barrett outlines the process, key deadlines and available supports, reinforcing the importance of ongoing education in maintaining high standards of practice.
We also feature important research led by Irish pharmacy professionals, including a major international study examining global clozapine use in treatment-resistant schizophrenia. The findings highlight persistent under-prescribing worldwide and underscore the role of pharmacy-led research in identifying gaps between evidence and practice.
Together, the stories in this issue reflect a profession at the centre of clinical innovation, policy change, research and education— continuing to shape safer, more effective care for patients across the hospital setting.
Expanding Critical Care Capacity
St. Vincent’s University Hospital (SVUH) has officially opened ICUSouth, a new critical care facility adding six beds to the hospital’s intensive care capacity. The new ward was opened by Minister for Health, Jennifer Carroll MacNeill last month.
ICU-South represents a significant expansion in SVUH’s ability to care for critically ill patients requiring complex specialist care and life support. The new unit forms part of Phase 1 of the national strategic plan to increase critical care capacity across Ireland.
The development has been delivered through close collaboration between HSE Estates and the St. Vincent’s Healthcare Group Estates team, led by Peter Mortell and Nick Andrews, with support from the Perioperative Directorate and ICU clinical and support teams.
Cancer Care Hub for Pharmacists
Minister for Health Jennifer Carroll MacNeill pictured with members of the ICU clinical Peri-Operative Directorate and support teams during the opening of ICU-South at St. Vincent’s University Hospital
The original 20-bed ICU at SVUH cares for approximately 154 patients per month. With the addition of six beds, the hospital expects to create capacity for an estimated 46 additional patients monthly, based on current throughput. This expansion will enable more complex surgical cases to proceed, support national specialty services such as transplant and trauma and strengthen the resilience of the hospital system.
To mark World Cancer Day, the Irish Institute of Pharmacy is highlighting their Cancer Care Hub which was created for pharmacists in collaboration with the National Cancer Control Programme (NCCP).
This resource signposts pharmacists to the information that you need to be confident in supporting your patients in the prevention, early detection, treatment and navigation of life after cancer.
You can access the hub by visiting www.iiop.ie/content/cancer-care-hub.
Since the series launched, the IIOP have delivered a range of webinars on the topic of cancer - from prevention and early diagnosis, genomics in cancer care, cervical cancer and cancer in men.
The Hub is divided into six colourcoded categories or “pillars” below. Within each of the pillars you will find sections for the following types of information:
• Pharmacist information (immediate) – for information that you require quickly, to support your practice in real time.
• Pharmacist further education opportunities – for courses, webinars and supplemental information to increase your knowledge over time.
• Information for patients –supports to provide to patients at the relevant stage of their cancer journey.
Doctors Prepare to Vote
Nominations have closed for doctors seeking election to the Medical Council for the next term, which will commence in June 2026.
The Medical Council is the regulatory body for doctors and has a statutory role in protecting the public by promoting the highest professional standards amongst doctors practising in Ireland. The Council, which consists of 25 members, has a majority of non-medical members, with 13 non-medical members and 12 medical members.
Of these, six members are elected by their fellow registered medical practitioners, five are appointed by the Minister for Health, one being nominated by the Minister for Further and Higher Education, Research, Innovation and Science and the remaining 13 are appointed by nominating bodies.
The current term of office of one of the directly elected members of the Medical Council is due to expire on 31st May 2026. This position, along with two existing
vacancies on the Medical Council, are subject to an election process in accordance with the Medical Council’s election regulations.
The three members to be appointed following this election process must fall under the following categories:
• One medical practitioner registered or able to be registered in the Specialist Division in relation to anaesthesia
• One medical practitioner registered or able to be registered in the Specialist Division in relation to public health medicine
• One medical practitioner registered or able to be registered in the Specialist Division in relation to pathology or radiology
To be eligible for election a medical practitioner must be registered on the Specialist Division and practising medicine in the State (but excluding any visiting EEA practitioner) on the day preceding
the last day for receiving nominations (13 January 2026) and must be in one in of three categories above.
Additionally, a registered medical practitioner must be nominated by 10 registered medical practitioners practising medicine in the State and meet the eligibility requirements set out in the election regulations
Following the close of the nominations period, the independent returning officer, Mr Fergus Gallagher, Dublin County Sheriff, has announced the candidates for election to the Medical Council.
“As the Returning Officer, I have reviewed the nominations received and have deemed the following candidates eligible for election in the following categories, in accordance with the Medical Practitioners Act 2007:
Specialist Division - Anaesthesia
Dr. Brian Patrick Joseph O’Brien –Registration Number: 018422
Dr. David Michael Honan –Registration Number: 013213
Dr. Ehtesham Izhar Khan –Registration Number: 049848
Dr Georgina Flood - Registration Number: 321315
Dr. Khalid Rasheed – Registration Number: 021642
Specialist Division – Public Health Medicine
Dr Mai Mannix, Registration Number: 011309
Dr Ina Mary Kelly, Registration Number: 011181
All doctors who were on the Medical Council Register as at 12 noon on 23rd January 2026 are eligible to vote. The voting opens today, 6th February, and closes at noon on 26th February 2026. All doctors are encouraged to make sure they have their say in the formation of the next Medical Council by voting.
Faculty of Occupational Medicine Admission Ceremony
50 doctors were awarded at a special ceremony held in No. 6 Kildare Street during RCPI Faculty of Occupational Medicine Smiley Symposium.
At the faculty’s annual Admission Ceremony, doctors are awarded Honorary Fellowship of RCPI, Fellowship of RCPI (Occupational Medicine), Membership of RCPI Faculty of Occupational Medicine, Certificate of Satisfactory Completion of Specialist Training, and Licentiateship of RCPI Faculty of Occupational Medicine.
The college welcomed two esteemed Honorary Fellows – the highest award conferred by RCPI, and reserved for world leaders in medical science and those who have made an exceptional contribution to medicine or healthcare. Each college faculty and institute may admit two Honorary Fellows per year.
Accelerating Access to Medicines
The Irish Pharmaceutical Healthcare Association (IPHA) has welcomed the new four-year Framework Agreement which we believe will allow more innovative medicines to reach patients faster; and is a positive step in supporting the life sciences industry in Ireland. This Agreement on the pricing and supply of medicines reflects a continued desire on all sides to invest in treatments that improve outcomes for patients in Ireland and speed up the process of reimbursing new medicines in compliance with the law.
The negotiations and outcome were critically enabled by assurances from the Minister for Health that the Agreement should address “the explicit aim of achieving the timelines set out in legislation. This marked the
first time a Minister for Health confirmed a policy commitment to achieving the timelines in the 2013 legislation and is now the basis for this new Agreement. We pay tribute to her for that decision.
In a paper published last February, Faster and Fairer Access to Medicines, IPHA called on the State to reform the reimbursement system so that it is resourced, governed, and designed to operate within the legal 180-day timeline for HSE decisions set by the Oireachtas in the Health Act 2013.
This Agreement establishes a practical framework of process reforms to drive measurable improvement over its lifetime. Looking ahead, consistent delivery of timely decisions will strengthen patient access to innovative
Shane Ryan, IPHA President
treatments, improve predictability for clinicians and the health service, and reinforce Ireland’s position as an attractive environment for life sciences investment and research while supporting better outcomes for patients.
Throughout these negotiations, our aim was to support the conditions for sustained investment in innovative medicines in Ireland, ensuring that we continue to be considered a pro-innovation economy and society. The Agreement will allow for efficiencies to be achieved in medicines expenditure by the State that will support investment in innovation. On-going review mechanisms allow for continued dialogue between industry and State on all matters.
Shane Ryan, IPHA President, said, “Today’s Agreement is a critical step forward in supporting patients in Ireland gain faster access to innovative and life-changing medicines, whilst empowering clinicians to provide the best care available. Through this Agreement, we have the opportunity to significantly enhance patient care and drive benefits across the healthcare system, which is vital if we are to improve patient
Cervical Cancer Awareness Week
This Cervical Cancer Prevention Week (19 - 25 January 2026), the HSE reminded everyone that cervical cancer is one of the most preventable cancers, and that the actions which can be taken to stop it developing in the future. By preventing HPV, finding it, and treating abnormal cells, we can prevent cervical cancer before it develops. The HPV vaccine is the first line of protection against cervical cancer. It protects against the main types of HPV that cause most cervical cancers.
Getting vaccinated now means lower risk of cervical cancer in the future. It also protects against genital warts and other cancers caused by HPV.
Irish research shows that girls who were vaccinated in school have a 60% lower rate of serious pre-cancer changes at their first screening test at age 25clear evidence that the vaccine prevents disease.
The free HPV vaccine is offered to girls and boys in their first year
of secondary school through the HSE National Immunisation Programme. It’s a once-off vaccine. Research shows that it is safe, highly effective, and longlasting. It protects you for life from the virus that causes most cases of cervical cancer.
From January 2026, the Laura Brennan HPV vaccine catch-up programme in secondary schools is giving students in fifth and sixth year another chance to get the vaccine if they didn’t get it in first year. Students aged from 16 to 19
outcomes and advance our ambitions as a leader for the life sciences sector.
Today’s announcement is the result of significant collaboration and engagement across Government and the sector, and we look forward to continuing to work together to ensure the Agreement delivers for patients, the health system, and industry.”
Oliver O’Connor, IPHA Chief Executive, said:
“This Agreement is a turning point for patients and also an important outcome for Ireland and the pharmaceutical industry. It underscores a sustained commitment to investing in medicines that deliver better patient outcomes, while signalling that increased investment in innovation will not only strengthen Ireland’s leadership in healthcare and life sciences but also reinforce our economic growth and global competitiveness. Faster access means better outcomes for patients and a stronger healthcare system overall. IPHA and our members are proud to play our part in making this happen with our stakeholders representing the State.”
• New four-year deal designed to deliver faster patient access to innovative medicines, within 180 days to HSE decisions on reimbursement and a stable policy framework.
• Agreement supports growing investment in innovative medicines, reflecting the vital economic contribution of the pharmaceutical industry in Ireland.
can make their own choice to have the vaccine.
Everyone should still be aware of possible symptoms of cervical cancer, because not all cases of cervical cancer are caused by HPV, and screening won’t find every abnormality.
The symptoms can include abnormal vaginal bleeding (between periods, after sex or after menopause), unusual vaginal discharge and pelvic or lower back pain.
KISQALI® is the only CDK4/6i approved for the broadest range of HR+/ HER2- patients including those with high-risk N0 or N+ disease 2,15,16
KISQALI® demonstrated a predictable, manageable and reversible safety profile. Most AEs are asymptomatic and QoL was maintained compared to baseline. The most common grade 3/4 AEs were neutropenia, abnormal liver function tests and leukopenia2,8-11 NATALEE1 iDFS ACHIEVED
KISQALI® reduced the relative risk of invasive disease recurrence by 28.4% vs NSAI alone1
Fictional healthcare professional and patient.
AEs, adverse events; aBC, advanced breast cancer; CDK4/6i, cyclin-dependent kinase 4 and 6 inhibitor.
ABBREVIATED PRESCRIBING INFORMATION
Please refer to Summary of Product Characteristics (SmPC) before prescribing. Kisqali (ribociclib) 200 mg film-coated tablets
Presentation: Film coated tablets (FCT) containing 200 mg of ribociclib and 0.344 mg soya lecithin. Indications: Early breast cancer - Kisqali in combination with an aromatase inhibitor is indicated for the adjuvant treatment of patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative early breast cancer at high risk of recurrence (see section 5.1 for selection criteria). In pre- or perimenopausal women, or in men, the aromatase inhibitor should be combined with a luteinising hormone-releasing hormone (LHRH) agonist. Advanced or metastatic breast cancer - Kisqali is indicated for the treatment of women with HR-positive, HER2-negative locally advanced or metastatic breast cancer in combination with an aromatase inhibitor or fulvestrant as initial endocrine-based therapy, or in women who have received prior endocrine therapy. In pre- or perimenopausal women, the endocrine therapy should be combined with a LHRH agonist. Dosage and administration: Patient selection for treatment with Kisqali based on the tumour expression of HR and HER2 should be assessed by a CE-marked in vitro diagnostic (IVD) medical device with the corresponding intended purpose. If the CE-marked IVD is not available, an alternative validated test should be used. Adults: Early breast cancer - The recommended dose is 400 mg (two 200 mg FCT) taken orally, once daily for 21 consecutive days followed by 7 days off treatment, resulting in a complete cycle of 28 days. In patients with early breast cancer, Kisqali should be taken until completion of 3 years of treatment or until disease recurrence or unacceptable toxicity occur. When Kisqali is used in combination with an aromatase inhibitor (AI), the AI should be taken orally once daily continuously throughout the 28-day cycle. Please refer to the Summary of Product Characteristics (SmPC) of the AI for additional details. In pre- or perimenopausal women, or in men, the aromatase inhibitor should be combined with a LHRH agonist. Advanced or metastatic breast cancer - The recommended dose is 600 mg (3 x 200 mg FCT) taken orally, once daily for 21 consecutive days followed by 7 days off treatment, resulting in a complete cycle of 28 days. When Kisqali is used in combination with an AI, the AI should be taken orally once daily continuously throughout the 28 day cycle. Please refer to the Summary of Product Characteristics (SmPC) of the AI for additional details. When Kisqali is used in combination with fulvestrant, fulvestrant is administered intramuscularly on days 1, 15 and 29, and once monthly thereafter. Please refer to the SmPC of fulvestrant for additional details. Treatment of pre and perimenopausal women with the approved Kisqali combinations should also include an LHRH agonist in accordance with local clinical practice. Management of severe or intolerable adverse reactions (ARs) may require temporary dose interruption, reduction or discontinuation of Kisqali. Please see section 4.2 of SmPC for recommended dose modification guidelines. Kisqali can be taken with or without food (see section 4.5 of SmPC). The tablets should be swallowed whole and should not be chewed, crushed or split prior to swallowing. Special populations: ♦Renal impairment: Mild or moderate: No dose adjustment is necessary. Severe: A starting dose of 200 mg is recommended in patients with severe renal impairment. Kisqali has not been studied in breast cancer patients with severe renal impairment. Caution should be used in patients with severe renal impairment with close monitoring for signs of toxicity. ♦Hepatic impairment: No dose adjustment is necessary in patients with early breast cancer with hepatic impairment (see section SmPC 5.2). In patients with advanced or metastatic breast cancer, no dose adjustment is necessary in patients with mild hepatic impairmentModerate or severe: Dose adjustment is required, and the starting dose of 400 mg once daily is recommended. ♦Elderly (>65 years): No dose adjustment is required. ♦Pediatrics(<18 years): Safety and efficacy have not been established. Contraindications: Hypersensitivity to the active substance or to peanut, soya or any of the excipients. Warnings/Precautions: ♦Neutropenia was most frequently reported AR. A complete blood count (CBC) should be performed before initiating treatment. CBC should be monitored every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, then as clinically indicated. Febrile neutropenia was reported in 1.7% of patients exposed to Kisqali in the phase III clinical studies. Patients should be instructed to report any fever promptly. Based on the severity of the neutropenia, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 2 (see section 4.2 of SmPC). ♦Hepatobiliary toxicity increases in transaminases have been reported. Liver function tests (LFTs) should be performed before initiating treatment. LFTs should be monitored every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, then as clinically indicated. If grade ≥2 abnormalities are noted, more frequent monitoring is recommended. Recommendations for patients who have elevated AST/ALT grade ≥ 3 at baseline have not been established. Based on the severity of transaminase elevations, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 3 (see section 4.2). ♦QT interval prolongation has been reported with Kisqali. The use of Kisqali should be avoided in patients who have already or who are at significant risk of developing QTc prolongation. This includes patients with long QT syndrome, with uncontrolled or significant cardiac disease, including recent myocardial infarction, congestive heart failure, unstable angina and bradyarrhythmias and patients with electrolyte abnormalities. The use of Kisqali with medicinal products known to prolong QTc interval and/or strong CYP3A4 inhibitors should be avoided as this may lead to clinically meaningful prolongation of the QTcF interval (see SmPC sections 4.2, 4.5 and 5.1). If co-administration of Kisqali with a strong CYP3A4 inhibitor cannot be avoided, the Kisqali dose should be changed as described in SmPC section 4.2. QT interval prolongation in early breast cancer –study O12301C (NATALEE), a QTcF interval increase >60 msec from baseline was observed in 19 (0.8%) patients receiving Kisqali plus AI. The ECG should be assessed prior to initiation of treatment. Treatment with Kisqali should be initiated only in patients with QTcF values <450 msec. The ECG should be repeated at approximately Day 14 of the first cycle then as clinically indicated. In case of QTcF prolongation during treatment, more frequent ECG monitoring is recommended. Appropriate monitoring of serum electrolytes (including potassium, calcium, phosphorous, and magnesium) should be performed prior to initiation of treatment, at the beginning of the first 6 cycles, and then as clinically
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the
indicated. Any abnormality should be corrected before the start of Kisqali treatment. Based on the observed QT prolongation during treatment, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 4 (see section 4.2 of SmPC). Based on the E2301 study QTcF interval data, Kisqali is not recommended for use in combination with tamoxifen. ♦Critical visceral disease. The efficacy and safety of ribociclib have not been studied in patients with critical visceral disease. ♦Severe cutaneous reactions Toxic epidermal necrolysis (TEN) has been reported with Kisqali treatment. If signs and symptoms suggestive of severe cutaneous reactions (e.g. progressive widespread skin rash often with blisters or mucosal lesions) appear, Kisqali should be discontinued immediately. ♦Interstitial lung disease/pneumonitis ILD/pneumonitis has been reported with CDK4/6 inhibitors including Kisqali. Patients should be monitored for pulmonary symptoms indicative of ILD/pneumonitis which may include hypoxia, cough and dyspnoea and dose modifications should be managed in accordance with Table 5 (see section 4.2 of SmPC) Based on the severity of the ILD/pneumonitis, which may be fatal, Kisqali may require dose interruption, reduction or discontinuation as described in Table 5 (see section 4.2 of SmPC). ♦Blood creatinine increase ribociclib may cause blood creatinine increase – if this occurs it is recommended that further assessment of the renal function be performed to exclude renal impairment. ♦CYP3A4 substrates ribociclib may interact with medicinal products which are metabolised via CYP3A4, which may lead to increased serum concentrations of CYP3A4 substrates (see section 4.5 of SmPC). Caution is recommended in case of concomitant use with sensitive CYP3A4 substrates with a narrow therapeutic index and the SmPC of the other product should be consulted for the recommendations regarding co administration with CYP3A4 inhibitors. Pregnancy, Fertility and Lacation ♦Pregnancy: Pregnancy status should be verified prior to starting treatment as Kisqali can cause foetal harm when administered to a pregnant woman. Kisqali is not recommended during pregnancy and in women of childbearing potential not using contraception. ♦Women of childbearing potential who are receiving Kisqali should use effective contraception (e.g. double-barrier contraception) during therapy and for at least 21 days after stopping treatment with Kisqali. ♦Breast‑feeding: Patients receiving Kisqali should not breast feed for at least 21 days after the last dose. ♦Fertility: There are no clinical data available regarding effects of ribociclib on fertility. Based on animal studies, ribociclib may impair fertility in males of reproductive potential. ♦Effects on ability to drive and use machines Patients should be advised to be cautious when driving or using machines in case they experience fatigue, dizziness or vertigo during treatment with Kisqali. Interactions: ♦Concomitant use of strong CYP3A4 inhibitors should be avoided, including, but not limited to, clarithromycin, indinavir, itraconazole, ketoconazole, lopinavir, ritonavir, nefazodone, nelfinavir, posaconazole, saquinavir, telaprevir, telithromycin, verapamil, and voriconazole. Alternative concomitant medicinal products with less potential to inhibit CYP3A4 should be considered. Patients should be monitored for ARs. If concomitant use of a strong CYP3A4 inhibitor cannot be avoided, the dose of Kisqali should be reduced (see section 4.2 of SmPC). ♦Grapefruit or grapefruit juice should be avoided. ♦Concomitant use of strong CYP3A4 inducers should be avoided, including, but not limited to, phenytoin, rifampicin, carbamazepine and St John’s Wort (Hypericum perforatum). An alternative medicinal product with no or minimal potential to induce CYP3A4 should be considered. ♦Caution is recommended when Kisqali is administered with sensitive CYP3A4 substrates with narrow therapeutic index (including, but not limited to, alfentanil, ciclosporin, everolimus, fentanyl, sirolimus, and tacrolimus), and their dose may need to be reduced. ♦Concomitant administration of Kisqali with the following CYP3A4 substrates should be avoided: alfuzosin, amiodarone, cisapride, pimozide, quinidine, ergotamine, dihydroergotamine, quetiapine, lovastatin, simvastatin, sildenafil, midazolam, triazolam. ♦Caution and monitoring for toxicity are advised during concomitant treatment with sensitive substrates of drug transporters P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 and BSEP which exhibit a narrow therapeutic index, including but not limited to digoxin, pitavastatin, pravastatin, rosuvastatin and metformin. ♦Co-administration of Kisqali with medicinal products with known potential to prolong the QT interval should be avoided such as anti-arrhythmic medicinal products (including, but not limited to, amiodarone, disopyramide, procainamide, quinidine and sotalol) and other medicinal products known to prolong the QT interval including, but not limited to, chloroquine, halofantrine, clarithromycin, ciprofloxacin, levofloxacin, azithromycin, haloperidol, methadone, moxifloxacin, bepridil, pimozide and intravenous ondansetron. Kisqali is not recommended for use in combination with tamoxifen. Adverse reactions – advanced or metastatic breast cancer: ♦Very common: Infections, neutropenia, leukopenia, anaemia, lymphopenia, decreased appetite, headache, dizziness, dyspnoea, cough, nausea, diarrhoea, vomiting, constipation, stomatitis, abdominal pain, dyspepsia, alopecia, rash, pruritus, back pain, fatigue, peripheral oedema, asthenia, pyrexia, abnormal liver function tests. ♦Common: thrombocytopenia, febrile neutropenia, hypocalcaemia, hypokalaemia, hypophosphataemia, vertigo, lacrimation increased, dry eye, syncope, dysgeusia, hepatotoxicity, erythema, dry skin, vitiligo, dry mouth, oropharyngeal pain, blood creatinine increased, electrocardiogram QT prolonged, interstitial lung disease (ILD)/pneumonitis. ♦Rare: Erythema multiforme ♦Not known: Toxic epidermal necrolysis (TEN) ♦ Please refer to SmPC for a full list of adverse reactions. Adverse reactions - early breast cancer: ♦Very common: Infections, neutropenia, leukopenia, headache, Cough, Nausea, diarrhoea, constipation, abdominal pain, alopecia, fatigue, asthenia, pyrexia, abnormal liver function tests ♦Common: Anaemia, thrombocytopenia, lymphopenia, hypocalcaemia, hypokalaemia, appetite decreased, dizziness, dyspnoea, interstitial lung disease (ILD) / pneumonitis, vomiting, stomatitis, hepatotoxicity, rash, pruritus, peripheral oedema, oropharyngeal pain, blood creatinine increased, electrocardiogram QT prolonged ♦Uncommon: Febrile neutropenia ♦ Please refer to SmPC for a full list of adverse reactions. Legal Category: POM. Pack sizes: Unit packs containing 21, 42 or 63 FCTs. Not all pack sizes may be marketed. Marketing Authorisation Holder: Novartis Europharm Limited Vista Building
Adverse events can also be reported to Novartis preferably at www.novartis.com/report, by emailing
References 1. Crown JP, et al. Presented at the European Society For Medical Oncology Congress 2025, 17–21 October, Berlin, Germany. 2. KISQALI (ribociclib). Summary of Product Characteristics. 3. Hortobagyi GN, et al. Ann Oncol. 2024:S09237534(24)04064-X. 4. Hortobagyi GN, et al. N Engl J Med. 2022;386(10):942–950. 5. Neven P, et al. Breast Cancer Res. 2023;25:103. 6. Im S-A, et al. N Eng J Med. 2019;381:307–316. 7. Yardley DA, et al. Ann Oncol. 2022; 33(S7):S629. 8. Verma S, et al. Br Cancer Res Treat. 2018;170:535–545. 9. Beck JT, et al. Cancer Res. 2019;79 (4_Supplement):P6-18-14. 10. Fasching PA, et al. Breast. 2020;54:148–154. 11. Harbeck N, et al. Ther Adv Med Oncol. 2020;12:1–8. 12. Fasching PA et al., Annals of Oncology, Volume 34, Issue 10, 2023, Pages 951-953. 13. Lu et al., JCO 42, 2812-2821(2024). 14. Burris et al.Br J Cancer. 2021 Aug;125(5):679-686. 15. Slamon DJ, et al. Ther Adv Med Oncol. 2023;15:1–16. 16. Slamon D, et al. N Engl J Med. 2024; 390:1080-1091. 17. Fasching PA, et al. Oral LBA13. Presented at the European Society for Medical Oncology Congress 2024, 13–17 September, Barcelona, Spain. October 2025 | IE11536045 Novartis Ireland Ltd, Vista Building, Elm Park Green, Merrion Road, Ballsbridge, Dublin 4, D04 A9N6
ePortfolio
2025/26 ePortfolio Review
In October 2025, the Irish Institute of Pharmacy (IIOP) sent an email to approximately 1800 pharmacists who have been selected for the 2025/26 ePortfolio Review. The IIOP also sent a reminder email to pharmacists in early December. In January 2026, these selected pharmacists will be required to submit evidence of their Continuing Professional Development (CPD) to the IIOP via their ePortfolio.
Here, Áine Barrett, ePortfolio Review Project Lead, gives an overview of the process. We talk about the key dates, the process and where you can get more support.
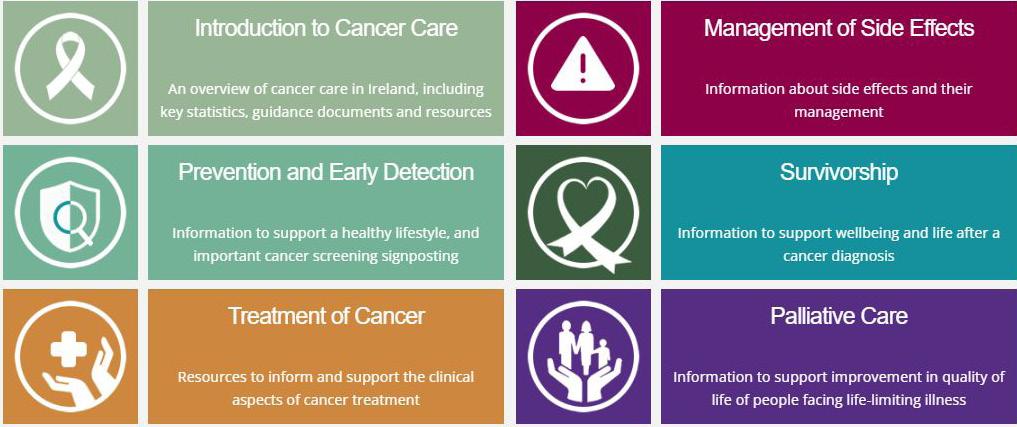
So Áine, how will pharmacists know whether they are selected for the 2025/26 ePortfolio Review?
In addition to the email communications that selected pharmacists have received from the IIOP, at this point selected pharmacists will also notice a Pending sign on their ePortfolio. This sign will remain in place until the submission period is closed (25 January) and will not change when they have submitted cycles. When the first submission period closes this will change from Pending to Active. This means that the review process is happening and is not specific to a particular pharmacist. The Active sign will remain in place until the ePortfolio Review process is complete in early May.
If I’ve been selected, when will I have to submit my ePortfolio extract?
On Monday 5 January 2026 an email will be sent from the ePortfolio System (info@iiop.ie) to your IIOP registered email address inviting you to submit CPD cycles from your ePortfolio for review.
You will have until Sunday 25 January to submit your extract. You may submit cycles at any stage during the three-week submission window, and you may make more than one submission.
Pharmacists must have their current, active email address registered on their IIOP profile in order to ensure they receive communications from the IIOP about the 2025/26 ePortfolio Review. Should you need help in accessing the website, retrieving your password or editing your details contact the IIOP at info@iiop.ie.
How does the ePortfolio Review process work?
The ePortfolio Review process incorporates two elements:
• A System Based ReviewEach extract (i.e. the cycles you choose to submit) will be
automatically reviewed against the System Based Standards which are pre-set within the IIOP ePortfolio system.
• A proportion of the ePortfolio extracts submitted will be reviewed against the Review Standards. This will include:
o All ePortfolio extracts submitted in the first submission period that do not meet the System Based Standards
o A random sample of ePortfolio extracts submitted in the first submission period that do meet the System Based Standards
o All ePortfolio extracts submitted in the second submission period
How will I know which cycles I should submit as part of my ePortfolio Review?
To help selected pharmacists ensure the cycles they submit as part of their ePortfolio extract meet the System Based Standards,
the IIOP developed the Ready Reckoner tool. The Ready Reckoner will be available to pharmacists selected for ePortfolio Review when the submission period opens in January.
Pharmacists can use the Ready Reckoner in two ways;
• Before submissionPharmacists can use the ‘Check readiness’ function before submission to identify cycles to submit which collectively meet all of the System Based Standards.
• After Submission - The Ready Reckoner automatically appears when one or more cycles are submitted for review and uses green and red lights to show progress against the System Based Standards.
In the event that one of the standards has not been met, a red light and a warning icon are shown, and pharmacists can click on the warning icon for an overview of the action they need to take to meet this standard.
I can’t find the “Ready Reckoner” on the IIOP website, where is it?
The Ready Reckoner will only become available to pharmacists included in the 2025/26 ePortfolio Review when the submission period opens on Monday 5 January. It can be found within the Completed Cycles section of the ePortfolio.
I have undertaken CPD but not yet had the opportunity to record it in my IIOP ePortfolio. Can I backdate these records to reflect when the work was completed?
Yes, you can backdate cycles to reflect the date the CPD was undertaken. Simply choose the relevant date when entering the ‘Cycle Start Date’. You can find further information on recording your CPD retrospectively in the How to meet the cycle from previous four years standard support resource which can be found on the 2025/26 ePortfolio Review Support page on the IIOP website.
Since I began using the IIOP ePortfolio, I have been recording entries in my ePortfolio but I haven’t completed all of my CPD cycles. I have been selected for the 2025/26 ePortfolio Review, if I completed a cycle now will it count as CPD from the year I created it?
Yes, it will count from the date it was created e.g. if a cycle is created in 2023 and completed in 2024, the system will count it as a cycle for 2023. We understand that people work in different ways, and the IIOP ePortfolio has been built to reflect this. While some people may like to complete a cycle and finish it right away other people may like to work on them over time.
I understand that the Core Competency Self-Assessment Tool (CCSAT) has been retired. How do I meet the standard for self-assessment against the PSI Core Competency Framework (CCF) in the current year?
Yes, the CCSAT was retired in 2023. The 2025/26 ePortfolio Review standards require that at least one cycle is created and submitted following selfassessment against the Core Competency Framework for Pharmacists in the current year. For the purposes of the 2025/26 ePortfolio Review, the current year is considered to be 2025 and up to 25 January 2026 i.e. the end of the submission period. Having self-assessed against the CCF, you can demonstrate evidence of this by selecting the tick box under the Self-Appraisal stage of the relevant cycle 'Completing self-assessment against the Core Competency Framework'.
I completed the CCSAT in January 2023 and completed a CPD cycle based on the outcome in July 2023. Can I submit this as a cycle which originated from my selfassessment against the PSI Core Competency Framework for assessment in the 2025/26 ePortfolio Review?
The 2025/26 ePortfolio Review Standards require that you submit one cycle created following selfassessment against the PSI’s Core Competency Framework in the current year, therefore the cycle submitted to meet this standard must have a creation date in 2025 to the end of the submission period in January 2026. This is in line with the legislative requirement to regularly self-assess against the Core Competency Framework. A cycle created in 2023 will not meet this standard, however, you may wish to submit this cycle as evidence of one of the cycles that you created in the previous four years.
I have completed some of the CPD training programmes available through Irish Pharmacy News (IPN) and I have also completed IIOP online training programmes, are there other activities that I could record in my ePortfolio?
Many pharmacists are surprised to realise the breadth of what counts as CPD. Traditionally, many pharmacists focus their CPD on their clinical expertise. The PSI Core Competency Framework indicates that pharmacists must be competent across a range of domains. It is important to aim to keep upskilled across all domains – not just those domains relating to medicines. This requires engaging in a breadth of learning. Consequently, there are many different types of activities that can be recorded in your ePortfolio as CPD. Completing the IPN CPD modules, for example, is a very valuable means of undertaking CPD. Other examples of activities that can be recorded in your ePortfolio as CPD include:
• Attending a live learning course
• Reading an article
• Having a discussion with a colleague
• Attending a conference
• Researching a new drug that has been prescribed for a patient It is important to remember that recording any learning activity may be appropriate as long as you can demonstrate how it has contributed to your professional development. It may be helpful to refer to the 2025/26 ePortfolio Review Standards (available on the ePortfolio Review Support Resources page on the IIOP website) to see types of criteria within the scope of the Review.
How much information should I record in my CPD cycles?
There is no right or wrong answer to this. Each pharmacist will have their own style; some choose to use bullet point information, while others prefer to be more detailed. What is important, however, is that you sufficiently outline what you have learned and how that learning has contributed to your understanding, benefited your practice, improved patient outcomes or advanced your continuing professional development. You will find some sample CPD cycles on the IIOP website, in IIOP newsletters or by attending ePortfolio Review information events. You should write in a way that works for you and allows you to reflect on what you have done.
What happens if my ePortfolio extract does not meet the 2025/26 ePortfolio Review Standards?
The ePortfolio Review enables pharmacists to demonstrate evidence of appropriate and ongoing engagement with CPD, in the interest of outcomes-focused professional development. All pharmacists’ ePortfolio extracts are reviewed against the specific standards, mapped to the legislative requirements, which have been set for that year’s ePortfolio Review.
All pharmacists who submit their cycles within the submission period in January 2026 will be provided with feedback on their ePortfolio extract, should one or more of the standards not be met in the first instance. They will then have an opportunity to resubmit cycles based on this feedback to meet the standards.
In the event that a pharmacist does not meet the standards at the end of their first ePortfolio Review process, they are automatically entered into the following year’s ePortfolio Review. The PSI is not informed at this stage. However, if at the end of the second ePortfolio Review the pharmacist does not meet the standard or does not reengage with this process in year 2, the IIOP is obliged under the terms of the ePortfolio Review Policy to refer the pharmacist to the PSI. Will the PSI be aware of how I perform in the ePortfolio Review process?
The IIOP undertakes the ePortfolio Review process to enable pharmacists to demonstrate evidence of CPD, in line with the legislation. Whilst the PSI is responsible for selecting pharmacists for ePortfolio Review, the IIOP undertakes the review process itself, at arm’s length from the PSI. The IIOP will not share any aspect of a pharmacist’s ePortfolio with the PSI.
In the event that the IIOP cannot ascertain that a pharmacist is meeting his or her CPD obligations (i.e. if the standard is not met at the end of two years ePortfolio Review, or if the pharmacist fails to submit an ePortfolio extract for review within the timeframe provided) then the IIOP has a statutory obligation to refer the pharmacist to the PSI. No information from your cycles is communicated to the PSI.
When I was selected for ePortfolio Review, I didn’t apply for an exemption due to extenuating circumstances, but my circumstances have now
changed and I will be unable to submit my ePortfolio. Is it too late to apply for an exemption?
The PSI manages the selection process for ePortfolio Review including all applications for exemptions from ePortfolio Review under its Extenuating Circumstances process. If circumstances apply to you which would have an impact on your ability to submit an extract from your ePortfolio, you should contact the PSI. The PSI's Extenuating Circumstances Policy and the relevant application form are available on the PSI website. All applications submitted will be managed by the PSI on a confidential case by case basis.
Where can I get more information?
The ePortfolio Review Support Page can be accessed via the IIOP homepage and is the main source of information relating to the 2025/26 ePortfolio Review including information on communication from the IIOP, timelines and ePortfolio Review Information Events. The ePortfolio Review support resources are also hosted on this page.
You may also wish to attend an Information Event. The IIOP hosted a series of Information Events from September 2025 until January 2026 to support pharmacists selected for the 2025/26 ePortfolio Review. All events are facilitated by a Peer Support Pharmacist and specifically focus on the key requirements of the 2025/26 ePortfolio Review so that pharmacists will know exactly what to expect at each point in the process. Pharmacists who took part in previous ePortfolio Review processes consistently highlight the IIOP Information Events as a key source of support in their preparation. They felt more confident in participating in ePortfolio Review and had a clear understanding of what the standards were and how to access support if needed, after attending an event. There are two information events (webinars) scheduled on 7 and 20 January 2026. You can book via the IIOP website, under the ‘Courses & Events’ tab. There is also a recorded version of the webinar available to view in your own time on the ePortfolio Review support resources page on the IIOP website.
The IIOP team are always happy to hear from you with any queries you may have, and you can contact the team on info@iiop.ie
Irish Pharmacy Researchers Spotlight Global Gaps in Clozapine Prescribing
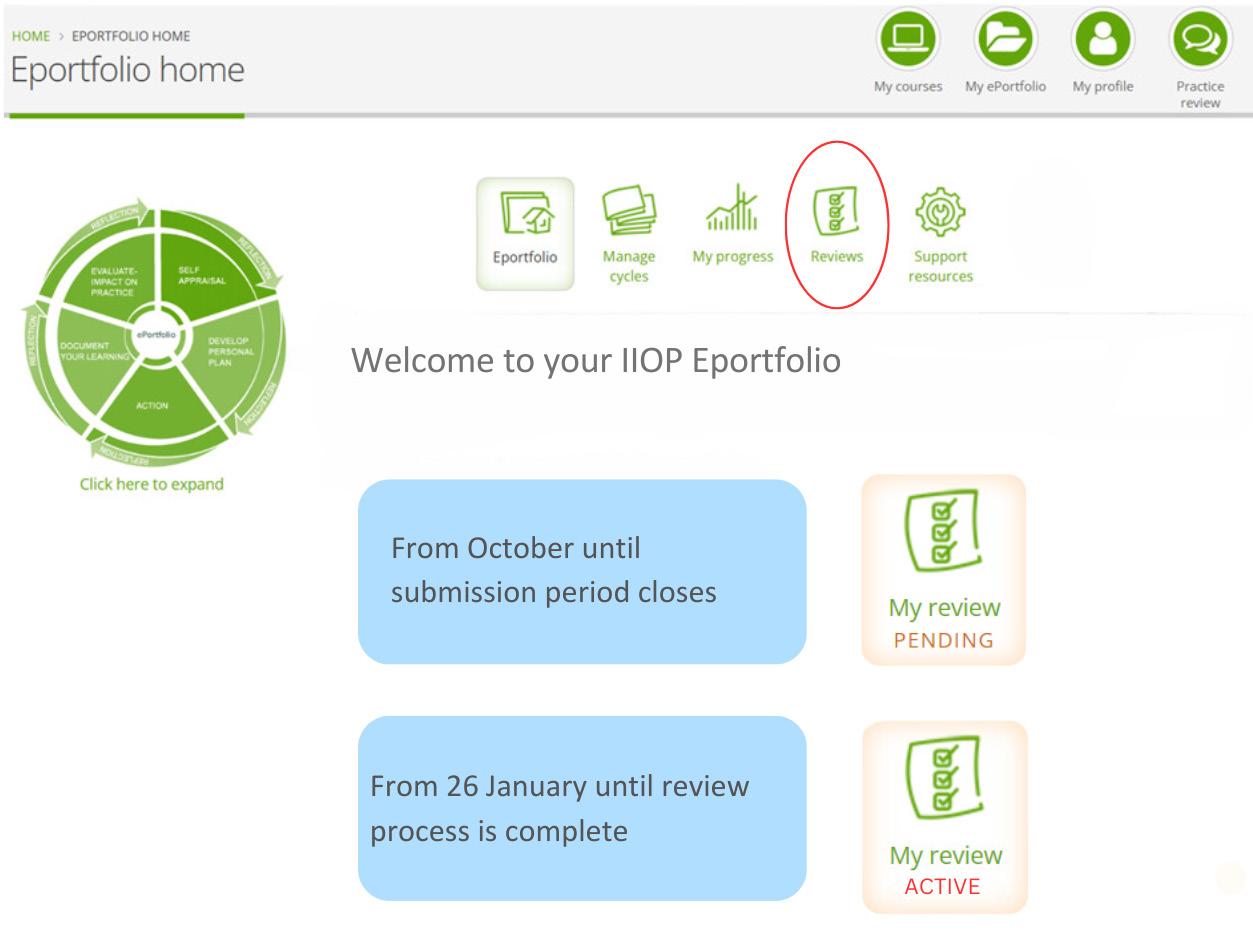
Irish researchers from St Patrick’s Mental Health Services and University College Cork are among those leading in a major international study on clozapine usage for treatment-resistant schizophrenia. The global study is the largest ever assessment of clozapine usage, and included data from 75 countries gathered over a period of 10 years from 2014 to 2024.
As a result of this research, estimates of national clozapine usage are now available in many countries for the first time, including Ireland, where it was estimated that less than 50% of those with treatmentresistant schizophrenia are being prescribed clozapine.
Schizophrenia resistant to typical antipsychotics (treatment-resistant schizophrenia) impacts 30-40% of people diagnosed. International guidelines in psychiatry suggest that the antipsychotic medication clozapine is the most effective treatment in managing schizophrenia unresponsive to other antipsychotics. However, prior research from 2014 has shown that clozapine is chronically under-prescribed globally and this new study aimed to explore if international trends of clozapine prescribing has improved since this time.
Researchers from St Patrick’s Mental Health Services and University College Cork, as well as researchers from the UK, New Zealand, Australia, the US and across Europe, obtained national estimates of clozapine usage from 75 countries through an analysis of national prescribing databases and global pharmaceutical sales data. The study found that in 2024, substantial variation in international use of clozapine remains. While global clozapine usage has increased, this is not uniformly. Although 60% of countries demonstrated an increase in clozapine use, increases were modest. New Zealand was the country identified as having the highest sustained
rates of clozapine use over the study period, followed by Finland. Out of the 75 countries assessed, only New Zealand and Finland achieved rates of clozapine prescribing similar to the population proportion with treatment-resistant schizophrenia.
Speaking about the study, Dr Ita Fitzgerald, lead researcher on the study and Advanced Specialist Pharmacist with St Patrick’s Mental Health Services said, “Pharmacy research plays a critical role in advancing mental healthcare by identifying gaps between available treatments and the real-world needs of service users. It is hoped that this research is the first step in helping to shape future prescribing practice and improving much needed, timely service user access to clozapine.”
Dr Mikkel Højlund, associate professor of psychiatry and psychopharmacology from the University of Southern Denmark added, “Comparing clozapine use across the world is very important because, despite decades of evidence that clozapine can transform outcomes for people with treatmentresistant schizophrenia, access to this treatment still appears to be underutilised. By mapping clozapine use globally and relating it to health-system features such as access to psychiatrists and monitoring policies, we sought to identify structural barriers
which might need more attention to support better access to an essential treatment.”
The research team involved in this study have identified the next steps for clozapine research as identifying how to effectively increase rates of clozapine prescribing. This will include studying systems of clozapine management in countries that have sustained high rates of clozapine prescribing.
The findings of this study have been published in The Lancet Regional Health – Europe.
Other recent research from the Pharmacy Department at St Patrick’s Mental Health Services include:
• From Idealist to RealistDesigning and Implementing Shared Decision-Making Interventions in the Choice of Antipsychotic Prescription in People Living with Psychosis (SHAPE): Part one and part two
• Informing the development of antipsychotic-induced weight gain management guidance: patient experiences and preferences - qualitative descriptive study.
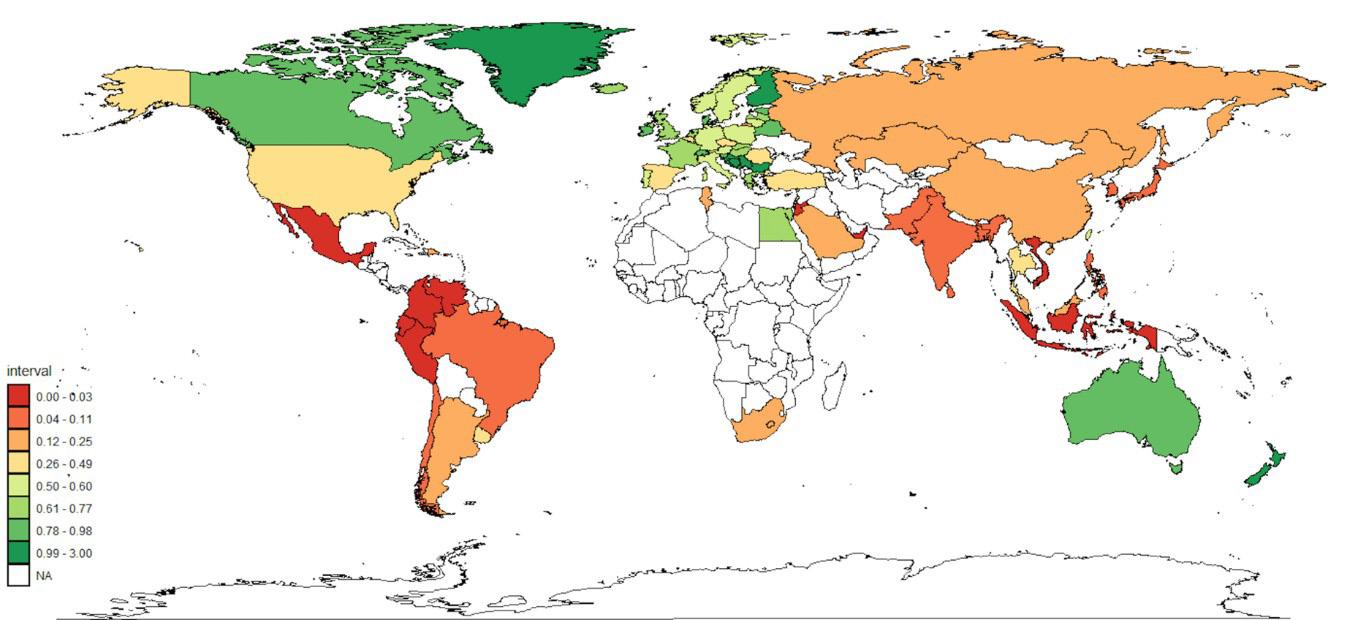
Global utilisation of clozapine in 2024. Utilisation rates are expressed in defined daily doses (DDD)/1,000 inhabitants/day. NA = No data.
Dr Ita Fitzgerald
For healthcare professionals in Ireland only. Abbreviated Prescribing Information can be found below.
Generic Product Launch
Eltrombopag Teva
Film-coated tablets eltrombopag
High Tech Prescription Medicine
Indications
The treatment of adult patients with primary immune thrombocytopenia (ITP) who are refractory to other treatments (e.g., corticosteroids, immunoglobulins).
The treatment of paediatric patients aged 1 year and above with primary immune thrombocytopenia (ITP) lasting 6 months or longer from diagnosis and who are refractory to other treatments (e.g., corticosteroids, immunoglobulins).
In adult patients with chronic hepatitis C virus (HCV) infection for the treatment of thrombocytopenia, where the degree of thrombocytopenia is the main factor preventing the initiation or limiting the ability to maintain optimal interferon-based therapy.
Eltrombopag Teva Film-coated Tablets Abbreviated Prescribing Information. Presentation: Each film-coated tablet contains eltrombopag olamine equivalent to 25mg and 50mg eltrombopag respectively. Indications: Indicated for the treatment of adult patients with primary immune thrombocytopenia (ITP) who are refractory to other treatments (e.g. corticosteroids, immunoglobulins). Indicated for the treatment of paediatric patients aged 1 year and above with primary immune thrombocytopenia (ITP) lasting 6 months or longer from diagnosis and who are refractory to other treatments (e.g. corticosteroids, immunoglobulins). Indicated in adult patients with chronic hepatitis C virus (HCV) infection for the treatment of thrombocytopenia, where the degree of thrombocytopenia is the main factor preventing the initiation or limiting the ability to maintain optimal interferon-based therapy. Dosage and administration: Oral use. Should be initiated by and remain under the supervision of a physician who is experienced in the treatment of haematological diseases or the management of chronic hepatitis C and its complications. Immune (primary) thrombocytopenia: The lowest dose of eltrombopag to achieve and maintain a platelet count ≥50 000/μl should be used. Dose adjustments are based upon the platelet count response. Eltrombopag must not be used to normalise platelet counts. Adults and Children (aged 6 years and above): 50mg once daily. For patients of East-/Southeast-Asian ancestry, eltrombopag should be initiated at a reduced dose of 25mg once daily. Children (aged 1 to 5 years): 25mg once daily. After initiating eltrombopag, the dose must be adjusted to achieve and maintain a platelet count ≥50 000/ μl as necessary to reduce the risk for bleeding. A daily dose of 75mg must not be exceeded. Discontinuation: Treatment with eltrombopag should be discontinued if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4 weeks of eltrombopag therapy at 75mg once daily. Chronic hepatitis C (HCV) associated thrombocytopenia: Eltrombopag should be initiated at a dose of 25mg once daily. No dosage adjustment is necessary for HCV patients of East-/Southeast-Asian ancestry or patients with mild hepatic impairment. If after 2 weeks of eltrombopag therapy at 100mg the required platelet level to initiate antiviral therapy is not achieved, eltrombopag should be discontinued. Children and Adolescents: Eltrombopag is not recommended for use in children under the age of one year with ITP due to insufficient data on safety and efficacy. The safety and efficacy of eltrombopag has not been established in children and adolescents (<18 years) with chronic HCV related thrombocytopenia. Elderly: There are limited data on the use of eltrombopag in ITP patients aged 65 years and older and no clinical experience in ITP patients aged over 85 years. There are limited data on the use of eltrombopag in HCV patients aged over 75 years. Caution should be exercised in these patients. Renal impairment: No dose adjustment is necessary in patients with renal impairment. Patients with impaired renal function should use eltrombopag with caution and close monitoring. Hepatic impairment: Eltrombopag should not be used in ITP patients with hepatic impairment (Child-Pugh score ≥5) unless the expected benefit outweighs the identified risk of portal venous thrombosis. If the use of eltrombopag is deemed necessary for ITP patients with hepatic impairment the starting dose must be 25mg once daily. After initiating the dose of eltrombopag in patients with hepatic impairment an interval of 3 weeks should be observed before increasing the dose. East-/Southeast-Asian patients: For adult and paediatric patients of East-/Southeast-Asian ancestry, including those with hepatic impairment, eltrombopag should be initiated at a dose of 25mg once daily. Contraindications: Hypersensitivity to the active substance or to any of the excipients. Precautions and warnings: Safety and efficacy have not been established in combination with direct-acting antiviral agents approved for treatment of chronic hepatitis C infection. Eltrombopag administration can cause abnormal liver function and severe hepatotoxicity, which might be life-threatening. Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin should be measured prior to initiation of eltrombopag, every 2 weeks during the dose adjustment phase and monthly following establishment of a stable dose. Caution is required when administering eltrombopag to patients with hepatic disease. In ITP and SAA patients a lower starting dose of eltrombopag should be used. Close monitoring is required when administering to patients with hepatic impairment. Chronic HCV patients with liver cirrhosis may be at risk of hepatic decompensation when receiving alfa interferon therapy. In patients with low albumin levels (≤ 35g/l) or with a MELD score ≥10 at baseline, there was a 3-fold greater risk of hepatic decompensation and an increase in the risk of a fatal adverse event compared to those with less advanced liver disease. In addition, the benefits of treatment in terms of the proportion achieving SVR compared with placebo were modest in these patients (especially for those with baseline albumin ≤35g/l) compared with the group overall. Eltrombopag should only be administered to such patients after careful consideration of the expected benefits in comparison with the risks. Patients with these characteristics should be closely monitored for signs and symptoms of hepatic decompensation. In controlled studies in thrombocytopenic patients with HCV receiving interferon-based therapy (n=1 439), 38 out of 955 patients (4%) treated with eltrombopag and 6 out of 484 patients (1%) in the placebo group experienced thrombotic/thromboembolic events (TEEs). Reported thrombotic/thromboembolic complications included both venous and arterial events. The majority of TEEs were non-serious and resolved by the end of the study. The risk of TEEs has been found to be increased in patients with chronic liver disease (CLD) treated with 75mg eltrombopag once daily for 2 weeks in preparation for invasive procedures. Eltrombopag should not be used in ITP patients with hepatic impairment (Child-Pugh score ≥5) unless the expected benefit outweighs the identified risk of portal venous thrombosis. When treatment is considered appropriate, caution is required when administering eltrombopag to patients with hepatic impairment. Thrombocytopenia is likely to reoccur in ITP patients upon discontinuation of treatment with eltrombopag. Following discontinuation of eltrombopag, platelet counts return to baseline levels within 2 weeks in the majority of patients, which increases the bleeding risk and, in some cases, may lead to bleeding. This risk is increased if eltrombopag treatment is discontinued in the presence of anticoagulants or anti-platelet agents. Platelet counts must be monitored weekly for 4 weeks following discontinuation of eltrombopag. Eltrombopag may increase the risk for development or progression of reticulin fibres within the bone marrow. Prior to initiation of eltrombopag, the peripheral blood smear should be examined closely to establish a baseline level of cellular morphologic abnormalities. If the patient develops new or worsening morphological abnormalities or cytopenia(s), treatment with eltrombopag should be discontinued and a bone marrow biopsy considered, including staining for fibrosis. The effectiveness and safety of eltrombopag have not been established for the treatment of
Teva Pharmaceuticals Ireland, Digital Office Centre Swords, Suite 101 - 103, Balheary Demesne, Balheary Road, Swords, Co Dublin, K67E5AO, Ireland.
Freephone: 1800 - 201 700 | Email: info@teva.ie
Product subject to prescription which may not be renewed (A)
thrombocytopenia due to myelodysplastic syndrome (MDS). Cytogenetic abnormalities are known to occur in SAA patients. It is not known whether eltrombopag increases the risk of cytogenetic abnormalities in patients with SAA. Bone marrow examination with aspirations for cytogenetics is recommended prior to initiation of eltrombopag, at 3 months of treatment and 6 months thereafter. If new cytogenetic abnormalities are detected, it must be evaluated whether continuation of eltrombopag is appropriate. Routine ophthalmologic monitoring of patients is recommended. In controlled studies in thrombocytopenic patients with HCV receiving interferon therapy (n=1 439), progression of pre-existing baseline cataract(s) or incident cataracts was reported in 8% of the eltrombopag group and 5% of the placebo group. Retinal haemorrhages, mostly Grade 1 or 2, have been reported in HCV patients receiving interferon, ribavirin and eltrombopag (2% of the eltrombopag group and 2% of the placebo group. Haemorrhages occurred on the surface of the retina (preretinal), under the retina (subretinal), or within the retinal tissue. QTc interval prolongation has been reported in clinical studies of patients with ITP and thrombocytopenic patients with HCV. A loss of response or failure to maintain a platelet response with eltrombopag treatment within the recommended dosing range should prompt a search for causative factors, including an increased bone marrow reticulin. Eltrombopag is highly coloured and so has the potential to interfere with some laboratory tests. Serum discolouration and interference with total bilirubin and creatinine testing have been reported in patients taking eltrombopag. Interactions: Administration of eltrombopag 75mg once daily for 5 days with a single 10mg dose of the OATP1B1 and BCRP substrate rosuvastatin to 39 healthy adult subjects increased plasma rosuvastatin Cmax 103%. Interactions are also expected with other HMG-CoA reductase inhibitors, including atorvastatin, fluvastatin, lovastatin, pravastatin and simvastatin. When co-administered with eltrombopag, a reduced dose of statins should be considered and careful monitoring for statin adverse reactions should be undertaken. Concomitant administration of eltrombopag and OATP1B1 (e.g. methotrexate) and BCRP (e.g. topotecan and methotrexate) substrates should be undertaken with caution. eltrombopag (up to 100mM) showed no in vitro inhibition of the CYP450 enzymes 1A2, 2A6, 2C19, 2D6, 2E1, 3A4/5, and 4A9/11 and was an inhibitor of CYP2C8 and CYP2C9. No clinically significant interactions are expected when eltrombopag and CYP450 substrates are co-administered. Dose adjustment is not required when eltrombopag is co-administered with either telaprevir or boceprevir. A decrease in eltrombopag exposure was observed with co-administration of 200mg and 600mg ciclosporin (a BCRP inhibitor). Eltrombopag chelates with polyvalent cations such as iron, calcium, magnesium, aluminium, selenium and zinc. Eltrombopag should be taken at least two hours before or four hours after any products such as antacids, dairy products or mineral supplements containing polyvalent cations to avoid significant reduction in eltrombopag absorption. Co-administration of eltrombopag with lopinavir/ritonavir may cause a decrease in the concentration of eltrombopag. Platelet count should be closely monitored to ensure appropriate medical management of the dose of eltrombopag when lopinavir/ritonavir therapy is initiated or discontinued. Eltrombopag is metabolised through multiple pathways including CYP1A2, CYP2C8, UGT1A1, and UGT1A3. Medicinal products that inhibit or induce a single enzyme are unlikely to significantly affect plasma eltrombopag concentrations, whereas medicinal products that inhibit or induce multiple enzymes have the potential to increase (e.g. fluvoxamine) or decrease (e.g. rifampicin) eltrombopag concentrations. Co-administration of repeat doses of boceprevir 800mg every 8 hours or telaprevir 750mg every 8 hours with a single dose of eltrombopag 200mg did not alter plasma eltrombopag exposure to a clinically significant extent. Platelet counts should be monitored when combining eltrombopag with other medicinal products for the treatment of ITP in order to avoid platelet counts outside of the recommended range. Pregnancy and lactation: Eltrombopag Teva is not recommended during pregnancy and is not recommended in women of childbearing potential not using contraception. A decision must be made whether to discontinue breastfeeding or to continue/abstain from eltrombopag therapy, considering the benefit of breast-feeding for the child and the benefit of therapy for the woman. Effects on ability to drive and use machines: negligible influence on the ability to drive and use machines. The clinical status of the patient and the adverse reaction profile of eltrombopag, including dizziness and lack of alertness, should be borne in mind when considering the patient’s ability to perform tasks that require judgement, motor and cognitive skills. Adverse reactions: Pneumonia, rectosigmoid cancer, leukocytosis, thrombocytopenia, haemolytic anaemia, hypersensitivity, toxic neuropathy, retinal haemorrhage, acute myocardial infarction, electrocardiogram QT prolonged, deep vein thrombosis, haematoma, embolism, pulmonary embolism, pulmonary infarction, hyperbilirubinaemia, hepatic function abnormal, cholestasis, hepatic lesion, hepatitis, drug-induced liver injury, thrombotic microangiopathy with renal failure, renal failure, leukocyturia, lupus nephritis, hepatic neoplasm (malignant), hepatic encephalopathy, neutropenia, splenic infarction, syncope. Very Common: Nasopharyngitis, upper respiratory tract infection, cough, nausea, diarrhoea, back pain, headache, dizziness, anaemia, decreased appetite, pruritus, myalgia, pyrexia, fatigue, influenzalike illness, asthenia, chills, oropharyngeal pain, rhinorrhoea. Common: Pharyngitis, influenza, oral herpes, sinusitis, tonsillitis, respiratory tract infection, gingivitis, Anaemia, eosinophilia, haemoglobin decreased, white blood cell count decreased, hypokalaemia, decreased appetite, blood uric acid increased, sleep disorder, depression, paraesthesia, hypoaesthesia, somnolence, migraine, dry eye, vision blurred, eye pain, visual acuity reduced, ear pain, vertigo, hot flush, oropharyngeal pain, rhinorrhoea, mouth ulceration, toothache, vomiting, abdominal pain, mouth haemorrhage, flatulence, rash, alopecia, hyperhidrosis, pruritus generalised, petechiae, myalgia, muscle spasm, musculoskeletal pain, bone pain, menorrhagia, pyrexia, chest pain, asthenia, hypoglycaemia, anxiety, epistaxis. Consult the Summary of Product Characteristics in relation to other side effects. Overdose: In the event of overdose, platelet counts may increase excessively and result in thrombotic/thromboembolic complications. In case of an overdose, consideration should be given to oral administration of a metal cation-containing preparation, such as calcium, aluminium, or magnesium preparations to chelate eltrombopag and thus limit absorption. Platelet counts should be closely monitored. Because eltrombopag is not significantly renally excreted and is highly bound to plasma proteins, haemodialysis would not be expected to be an effective method to enhance the elimination of eltrombopag. Legal category: POM. Marketing Authorisation Number: PA22579/003/001-002. Marketing Authorisation Holder: TEVA GmbH, Graf-Arco-Str. 3, 89079 Ulm, Germany. Job Code: MED-IE-00103. Date of Preparation: November 2025
Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie.
Adverse events should also be reported to Teva UK Limited on +44 (0) 207 540 7117 or medinfo@tevauk.com
Date of Preparation: December 2025 | Job Code: GEN-IE-00167
Further information is available on request or in the SmPC. Product Information also available on the HPRA website.
Artificial Intelligence in Health and Social Care Services
The Health Information and Quality Authority (HIQA) has launched a six-week public consultation, seeking feedback from the public on its Draft National Guidance for the Responsible and Safe use of Artificial Intelligence in Health and Social Care Services. This guidance was commissioned by the Department of Health and is informed by an evidence review conducted by HIQA which was also published today.
The purpose of this draft guidance is to build awareness and good practice among services and staff around the responsible and safe use of artificial intelligence (AI) to ensure safer, better care for people using health and social care services. It is underpinned by four principles: accountability, a human rights-based approach, safety and wellbeing, and responsiveness,
and aims to educate and empower people using services on what to expect when AI tools are used in their care.
The development of this draft guidance comes at a time when the health and social care system in Ireland is facing rising costs, increased demand due to an ageing population, increased prevalence of chronic and complex conditions and a shortage of healthcare staff to meet demand.
AI has already begun to be used across health and social care in Ireland, with its role expected to grow significantly. Uses which can positively impact on the delivery of care include streamlining administrative tasks, supporting diagnostics and predicting medical outcomes to enable preventative measures. These might include
helping clinicians to detect illness earlier, flagging patterns in scans and lab results more accurately, or supporting with note taking to enable clinicians to focus directly on the patient during consultations. Although AI has the potential to enhance health and social care, there are potential risks, therefore, it is important that human oversight is maintained.
Commenting on the draft guidance, HIQA’s Director of Health Information and Standards, Rachel Flynn, said, “The draft national guidance for the responsible and safe use of AI in health and social care services has been developed against the backdrop of a dynamic and evolving AI landscape. Innovative solutions are needed to meet the challenges in our health
and social care system, and AI is a promising tool that can be integrated to help address some of these challenges. As the role of AI in health and social care service delivery continues to grow, the development of national guidance for the responsible and safe use of AI in a health and social care context is timely, and will also support the implementation of national standards in this area.”
HIQA’s public consultation on the draft national guidance for the responsible and safe use of AI in health and social care is now open until 5 March 2026. All feedback will be carefully considered and used to inform the National Guidance for the Responsible and Safe Use of AI in Health and Social Care which will be published later this year.
¤1m Investment for Pancreatic Cancer Research
University College Dublin is to lead a ¤1 million research programme aimed at improving early detection of pancreatic cancer, one of Ireland’s most difficult to treat cancers.
From accelerating research into biomarkers for earlier detection, to better treatment options, the Breakthrough Cancer Research AllCaN Pancreatic Cancer grant was awarded to a new all-island network led by Professor Gráinne O’Kane, Consultant Medical Oncologist and Pat Smullen Chair in Pancreatic Cancer at the UCD School of Medicine.
The ¤1m investment will fund a multi-year programme that will
bring together senior investigators, early career researchers, clinicians and PPIE representatives from across the island of Ireland to build a genuinely integrated national research effort.
“This All-Ireland network represents a major step forward for pancreatic cancer research,” said Professor O’Kane.
“By combining clinical insight with scientific expertise and embedding public and patient perspectives from the outset, we have an opportunity to address longstanding gaps in early detection, treatment decision-making and care pathways."
Pictured at UCD, Dr Naomi Walsh, Dublin City University; UCD Professor Gráinne O’Kane, Consultant Medical Oncologist at St Vincent’s University Hospital; Professor Richard Turkington, Queen’s University Belfast
Credit: Leon Farrell / Photocall Ireland
Adding, “Pancreatic cancer has not benefited from the same research advances seen in other cancers. This programme allows us to work together at scale, across institutions and disciplines, to generate knowledge that can be translated into real improvements in how this disease is detected and treated.”
The funding announcements comes on World Cancer Day, marked on February 4 each year. Over 800 people die from pancreatic cancer annually across the island of Ireland.
AllCaN is a flagship initiative under Breakthrough Cancer Research’s five-year research strategy Making More Survivors, designed to accelerate progress in cancers with the poorest outcomes through deep, structured collaboration across institutions and jurisdictions.
This is the second All-Ireland Cancer Network the charity has launched. Its first on oesophageal cancer has been in place since 2023.
“Pancreatic cancer is one of the greatest challenges we face in cancer research today,” said Orla Dolan, CEO of Breakthrough Cancer Research. “Outcomes remain devastatingly poor, largely because the disease is so often diagnosed late and treatment options are limited.”
"Through AllCaN, we are creating the conditions needed for real progress by bringing together the very best expertise across the island of Ireland around a shared ambition.”
Professor Richard Turkington, Queen’s University Belfast, CoLead of the AllCaN Pancreatic Cancer added, “Pancreatic cancer outcomes in Northern Ireland are among the poorest, with survival rates even lower than in the Republic of Ireland. That makes the need for coordinated ambitious research even more urgent.
“By working together across jurisdictions, we can accelerate progress in early detection, understand the disease better and develop more effective approaches to treatment that will directly benefit people with pancreatic cancer right across the island.”
European Health Data Space: Ireland’s chance to better connect healthcare data
A long-awaited transformation in European healthcare is now underway. Over the coming years, healthcare professionals across Ireland will benefit from faster, more reliable on-demand access to patient medical data
Now in force, the European Health Data Space (EHDS), will transform how health data is accessed, shared and used across the EU. For Irish clinicians, this is not an abstract EU bureaucratic policy. It’s a practical realistic response to a problem they face every day when having to deal with incomplete, hard-tofind patient information.
Until now, healthcare in Ireland, similar to much of the EU, has been held back by the existence of data silos. Paper records and disconnected IT systems have become the norm rather than the exception. Too often, clinicians are forced to make decisions without a complete view of the patient’s medical history or having to waste valuable time searching for and collecting the critical data they need.
The EHDS is the cornerstone for improving healthcare delivery. Irish doctors, nurses and pharmacists will be able to securely access, update and share essential health information within Ireland and across Europe. Whether a patient is treated in Cork, travels to Donegal, or needs urgent care while abroad, their medical history, prescriptions, allergies, test results and imaging can be available on-demand and accessible at the point of care. The EHDS will streamline the flow of data among IT systems, healthcare facilities, and health systems within the EU, thus enhancing decision-making and patient safety. Healthcare professionals know it best: Faster access to critical health information gives confidence, supports accurate diagnoses and treatment plans, reduces risk and ensures continuity of care.
Less paperwork, better outcomes.
For frontline staff, the EHDS means less time chasing files and more time available to focus on patients. Interoperable systems, connected through the MyHealth@EU infrastructure, will reduce duplicated paperwork
Written by Fulvia Raffaelli, Head of Unit, European Union
and unnecessary repeat tests. Electronic Health Record (EHR) systems will be required to follow EU-wide standards, ensuring they can “talk to each other” to securely and reliably share the required information.
In practice, this means an emergency doctor in Dublin will be able to view a patient’s recent scans from Galway within seconds. A GP in rural Mayo will be able to see hospital discharge notes without waiting days for letters. In emergencies, speed of access to information can save lives. Ultimately, the EHDS will be a support for doctors, rather than an administrative burden.
A strategic opportunity for Irish healthcare
The EHDS is not just about daily clinical care. It is also about strengthening Ireland’s resilience against future health crises. The COVID-19 pandemic exposed how difficult it was to access real-time, reliable health data across Europe. EHDS creates a secure framework for anonymised health data to be used for research, public health planning and innovation.
For Ireland, this opens major opportunities. Irish universities, clinical researchers and medtech innovators will be able to
accelerate clinical trials, advance personalised medicine and bring cutting-edge treatments to patients faster.
Ireland has the talent, the research base and the ambition, what has been missing is the supporting data infrastructure to match.”=
Key
milestones for Ireland
The EHDS will be rolled out in stages:
By 2029: Key parts of the EHDS Regulation will enter into application, including, the exchange of the first group of priority categories of health data (Patient Summaries, ePrescriptions/eDispensations) in all EU Member States.
By 2031: the exchange of the second group of priority categories of health data including medical imaging studies & related imaging reports; medical test results, (including laboratory and other
diagnostic results and related reports); and discharge reports.), should be operational in all EU Member States.
These are practical, timetabled reforms that will directly affect how care is delivered in Irish clinics, GP practices and hospitals.
A turning point for Irish healthcare
For Irish clinicians, the EHDS represents a shift towards more efficient, informed and patientcentred care, with less frustration caused by missing or inaccessible information. For patients, it means greater control over their data and safer treatment wherever they receive care.
The message is clear: the future of healthcare in Ireland and across Europe will be digital, connected, and patient-centred, while keeping patients’ data safe and private. The EHDS is how we get there.
Procurement for a National Electronic Health Record
The Minister for Health, Jennifer Carroll MacNeill TD received Government approval recently for the Health Service Executive (HSE) to begin the procurement phase (shortlisting) for the National Electronic Health Record (EHR), marking a significant milestone in the modernisation of Ireland’s health service.
This investment in digital capability represents a once-in-a-generation opportunity to modernise Ireland’s health infrastructure, delivering safer, smarter and more connected care for decades to come.
The Government’s approval follows completion of the Preliminary
Business Case and independent external assurance processes, confirming compliance with the State’s Infrastructure Guidelines.
The National EHR will be the largest digital transformation project in the history of the health service delivering one secure, integrated digital health record for every patient in Ireland. This will ensure that clinicians have timely access to accurate information, improving safety, reducing duplication, and enabling care that is better coordinated across hospitals, GPs, and community services. Patients will also benefit from greater access to their own health information,
HSE Quit Smoking Campaign
The HSE Tobacco Free Ireland Programme have launched the Financial Incentives to Stop Smoking (FISS) Programme pilot in Kilrush & targeted areas of Limerick city.
This initiative is designed to offer enhanced support and motivation to residents who want to quit smoking, reinforcing the HSE’s commitment to improving health in communities where support is needed most.
The scheme, which began in March 2025, offers individuals who smoke, gift vouchers (financial
incentives) of up to ¤400 over 12 months when they engage with a comprehensive stop smoking support programme and successfully quit smoking.
International evidence clearly demonstrates the power of combining expert support with financial incentives. When paired with intensive support from services like HSE QUIT, financial incentives have been shown to double a person’s chances of successfully quitting smoking long-term.
To be eligible to participate in this pilot, participants need to be
Milestone Event for Hospital Pharmacy
empowering them to manage their care more effectively.
Minister Carroll MacNeill said, “This is a landmark step in delivering a modern, connected health service that puts patients first. The National Electronic Health Record programme will be central to patients receiving safer, faster, and more integrated care, supporting clinicians and improving outcomes for everyone.
“Electronic health records for patients was identified by Sláintecare as a key enabler for the reform and modernisation of the Irish health service and will ensure Ireland meets the highest
international standards for patient safety and data security.”
Minister for Public Expenditure, Infrastructure, Public Service Reform and Digitalisation, Jack Chambers TD said, “Health digitalisation is a key priority. This programme has the potential to positively transform delivery of health care services for patients and healthcare workers. This is another example of the Government’s commitment to digitalisation and to delivering key public infrastructure faster and more cost effectively in the interest of people and communities.”
living in the key target community areas and hold a medical card. "We know most people who smoke want to quit," said Niamh Keating, Health Promotion and Improvement Officer, Health and Wellbeing, HSE Mid West.
“Financial incentives, alongside intensive professional support, are a proven way to make that ambition a reality, especially in communities with higher smoking rates. This programme is about empowering residents in Kilrush and targeted areas of Limerick city with the practical assistance and
FutureRx: Leveraging Advanced Specialist Pharmacists for Progress and Transformation will bring together Ireland’s first cohort of Advanced Specialist Pharmacists (ASPs) for a national, one-day networking and learning event.
The event will be held on Thursday, 5th of March 2026 from 10:00–16:00 (Registration, coffee & pastries from 09:30) at the Radisson Blu Royal Hotel, Golden Lane, Dublin 8.
Bernard Gloster, CEO of the HSE, will deliver the opening address.
This landmark event is designed to strengthen national connections, reduce siloing, and support the continued development of the ASP role as it becomes established across the health system.
The day will feature:
• Keynotes, case studies, and ASP-led presentations
motivation to make this positive, lasting change.”
Tobacco use remains one of the leading causes of preventable illness in Ireland. Smoking rates are higher in disadvantaged areas, resulting in poorer health outcomes for many people. The Tobacco Free Ireland Government Strategy set a target of reducing smoking prevalence to less than 5% nationally. Despite some progress, smoking rates have remained largely unchanged since 2019, with rates up to 25% for some groups and communities.
• Panel discussions exploring opportunities and challenges in developing the ASP role
• Networking with peers and system leaders
• Focus on innovation, digital tools, service redesign, and designing for impact
• Lunch and refreshments provided
FutureRx is about ensuring the ASP role delivers on its full promise, improved patient outcomes, enhanced medication safety, better use of resources, and greater system efficiency.
While this networking day is primarily aimed at ASPs, attendance from other pharmacists within your department is also welcome, based on capacity levels in your hospital as it is recognised that innovation is driven across the profession.
For healthcare professionals in Ireland only. Abbreviated Prescribing Information can be found below.
Generic Product Launch
Pazopanib Teva
Film-coated Tablets
pazopanib
30 film-coated tablets
High Tech Prescription Medicine
Indications
Renal cell carcinoma (RCC)
Pazopanib Teva is indicated in adults for the first-line treatment of advanced renal cell carcinoma (RCC) and for patients who have received prior cytokine therapy for advanced disease.
Soft-tissue sarcoma (STS)
Pazopanib Teva is indicated for the treatment of adult patients with selective subtypes of advanced soft-tissue sarcoma (STS) who have received prior chemotherapy for metastatic disease or who have progressed within 12 months after (neo) adjuvant therapy.
Efficacy and safety has only been established in certain STS histological tumour subtypes.
Presentation: Each film-coated tablet contains pazopanib hydrochloride equivalent to 200mg and 400mg pazopanib respectively. Indications: Indicated in adults for the first-line treatment of advanced renal cell carcinoma (RCC) and for patients who have received prior cytokine therapy for advanced disease. Also, indicated for the treatment of adult patients with selective subtypes of advanced soft-tissue sarcoma (STS) who have received prior chemotherapy for metastatic disease or who have progressed within 12 months after (neo) adjuvant therapy. Dosage and administration: Oral use. Adults: The recommended dose of pazopanib for the treatment of RCC or STS is 800mg once daily. Children and Adolescents (Aged <18 years of age): Not suitable for use. Elderly: There are limited data on the use of pazopanib in patients aged 65 years and older. Overall, no clinically significant differences in safety of pazopanib were observed between subjects aged at least 65 years and younger subjects, but greater sensitivity of some elderly patients cannot be ruled out. Renal impairment: No dose adjustment is required in patients with creatinine clearance above 30ml/min. Caution is advised in patients with creatinine clearance below 30ml/min as there is no experience of pazopanib in this patient population. Hepatic impairment: Administration of pazopanib to patients with mild or moderate hepatic impairment should be undertaken with caution and close monitoring of tolerability. 800mg pazopanib once daily is the recommended dose in patients with mild abnormalities in serum liver tests. A reduced pazopanib dose of 200mg once daily is recommended in patients with moderate hepatic impairment. Pazopanib is not recommended in patients with severe hepatic impairment. Contraindications: Hypersensitivity to the active substance or to any of the excipients. Precautions and warnings: Cases of hepatic failure (including fatalities) have been reported during use of pazopanib and is not recommended in patients with severe hepatic impairment. Serum liver tests should be performed before initiation of treatment with pazopanib, at weeks 3, 5, 7 and 9, then at months 3 and 4, with additional tests as clinically indicated. Periodic testing should then continue after month 4. In clinical studies with pazopanib, events of hypertension including newly diagnosed symptomatic episodes of elevated blood pressure (hypertensive crisis) have occurred, therefore, blood pressure should be well controlled prior to initiating pazopanib. Patients should be monitored for hypertension early after starting treatment (no longer than one week after starting pazopanib) and frequently thereafter to ensure blood pressure control. Pazopanib should be discontinued if there is evidence of hypertensive crisis or if hypertension is severe and persists despite anti-hypertensive therapy and pazopanib dose reduction. Posterior reversible encephalopathy syndrome (PRES)/Reversible posterior leukoencephalopathy syndrome (RPLS) and Interstitial lung disease (ILD)/Pneumonitis have been reported in association with pazopanib and can be fatal. Patients developing PRES/RPLS/ILD/pneumonitis should discontinue treatment with pazopanib. The risks and benefits of pazopanib should be considered before beginning therapy in patients who have pre-existing cardiac dysfunction. The safety and pharmacokinetics of pazopanib in patients with moderate to severe heart failure or those with a below normal left ventricular ejection fraction (LVEF) have not been studied. Interruption of pazopanib and/or dose reduction should be combined with treatment of hypertension in patients with significant reductions in LVEF, as clinically indicated. Patients should be carefully monitored for clinical signs or symptoms of congestive heart failure. Baseline and periodic evaluation of LVEF is recommended in patients at risk of cardiac dysfunction. Pazopanib should be used with caution in patients with a history of QT interval prolongation, in patients taking antiarrhythmics or other medicinal products that may prolong QT interval and in patients with relevant pre-existing cardiac disease. In clinical studies with pazopanib, myocardial infarction, myocardial ischaemia, ischaemic stroke and transient ischaemic attack were observed, with some events being fatal. Pazopanib should be used with caution in patients who are at increased risk of thrombotic events or who have had a history of thrombotic events. In clinical studies with pazopanib, venous thromboembolic events including venous thrombosis and fatal pulmonary embolus have occurred. Thrombotic microangiopathy (TMA) has been reported in clinical studies of pazopanib as monotherapy, in combination with bevacizumab, and in combination with topotecan. Patients developing TMA should permanently discontinue treatment with pazopanib. In clinical studies with pazopanib haemorrhagic events have been reported, including fatal haemorrhagic events. Pazopanib should be used with caution in patients with significant risk of haemorrhage. Before initiating pazopanib, this risk of aneurysm and/or artery dissection formations should be carefully considered in patients with risk factors such as hypertension or history of aneurysms. Pazopanib should be used with caution in patients at risk for gastrointestinal perforation or fistula, as fatal perforation events have occurred in clinical studies. Pazopanib should be discontinued in patients with wound dehiscence. In clinical studies with pazopanib, events of hypothyroidism have occurred. All patients should be observed closely for signs and symptoms of thyroid dysfunction on pazopanib treatment. In clinical studies with pazopanib, proteinuria has been reported. Baseline and periodic urinalysis during treatment is recommended and patients should be monitored for worsening proteinuria. Pazopanib should be discontinued if the patient develops nephrotic syndrome. The occurrence of Tumour lysis syndrome (TLS), including fatal TLS, has been associated with the use of pazopanib. Patients at risk should be closely monitored and treated as clinically indicated. Patients on pazopanib treatment should
Teva Pharmaceuticals Ireland, Digital Office Centre Swords, Suite 101 - 103, Balheary Demesne, Balheary Road, Swords, Co Dublin, K67E5AO, Ireland.
Freephone: 1800 - 201 700 | Email: info@teva.ie
Product subject to prescription which may be renewed (B)
be observed closely for signs and symptoms of pneumothorax. Cases of serious infections (with or without neutropenia), in some cases with fatal outcome, have been reported. Interactions: Pazopanib metabolism is mediated primarily by CYP3A4, with minor contributions from CYP1A2 and CYP2C8, therefore, inhibitors and inducers of CYP3A4 may alter the metabolism of pazopanib. Coadministration of pazopanib with strong inhibitors of the CYP3A4 family (e.g. ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) may increase pazopanib concentrations. Grapefruit juice contains an inhibitor of CYP3A4 and may also increase plasma concentrations of pazopanib. Concomitant use of pazopanib with a strong CYP3A4 inhibitor should be avoided. If no medically acceptable alternative to a strong CYP34A inhibitor is available, the dose of pazopanib should be reduced during concomitant administration. In such cases there should be close attention to adverse drug reaction, and further dose reduction may be considered if possible drug-related adverse events are observed. Combination with strong P-gp or BCRP inhibitors should be avoided, or selection of an alternate concomitant medicinal product with no or minimal potential to inhibit Pgp or BCRP is recommended. CYP3A4 inducers such as rifampin may decrease plasma pazopanib concentrations. Co-administration of pazopanib with potent P-gp or BCRP inducers may alter the exposure and distribution of pazopanib. In vitro studies with human liver microsomes showed that pazopanib inhibited CYP enzymes 1A2, 3A4, 2B6, 2C8, 2C9, 2C19, and 2E1. Concomitant use of pazopanib and simvastatin increases the incidence of ALT elevations. If a patient receiving concomitant simvastatin develops ALT elevations, follow guidelines for pazopanib posology and discontinue simvastatin. Concomitant use of pazopanib and other statins should be undertaken with caution. Administration of pazopanib with a high-fat or low-fat meal results in an approximately 2-fold increase in AUC and Cmax, therefore, pazopanib should be administered at least 1 hour before or 2 hours after a meal. Concomitant administration of pazopanib with esomeprazole decreases the bioavailability of pazopanib by approximately 40%, and co-administration of pazopanib with medicines that increase gastric pH should be avoided. Pregnancy and lactation: Pazopanib should not be used during pregnancy unless the clinical condition of the patient requires treatment with pazopanib. If pazopanib is used during pregnancy, or if the patient becomes pregnant while receiving pazopanib, the potential hazard to the foetus should be explained to the patient. Patients of childbearing potential should be advised to use adequate contraception during treatment and for at least 2 weeks after the last dose of pazopanib and to avoid becoming pregnant while receiving treatment with pazopanib. Male patients (including those who have had vasectomies) should use condoms during sexual intercourse while taking pazopanib and for at least 2 weeks after the last dose of pazopanib to avoid potential exposure to the medicinal product for pregnant partners and female partners of reproductive potential. Breast-feeding should be discontinued during treatment with pazopanib, as a risk to the breastfed child cannot be excluded. Effects on ability to drive and use machines: No or negligible influence on the ability to drive and use machines. A detrimental effect on such activities cannot be predicted from the pharmacology of pazopanib. The clinical status of the patient and the adverse event profile of pazopanib should be borne in mind when considering the patient’s ability to perform tasks that require judgement, motor or cognitive skills. Patients should avoid driving or using machines if they feel dizzy, tired or weak. Adverse reactions: Thrombocytopenia, neutropenia, leukopenia, thrombotic microangiopathy, hypothyroidism, tumour lysis syndrome, peripheral sensory neuropathy, transient ischaemic attack, cerebrovascular accident, ischaemic stroke, posterior reversible encephalopathy/reversible posterior leukoencephalopathy syndrome, bradycardia, cardiac dysfunction, myocardial ischaemia, venous thromboembolic event, hypertensive crisis, haemorrhage, aneurysms and artery dissections, interstitial lung disease/pneumonitis, pancreatitis, rectal haemorrhage, gastrointestinal haemorrhage, melaena, anal haemorrhage, large intestine perforation, mouth haemorrhage, upper gastrointestinal haemorrhage, enterocutaneous fistula, haematemesis, haemorrhoidal haemorrhage, ileal perforation, oesophageal haemorrhage, retroperitoneal haemorrhage, hepatotoxicity, jaundice, hepatic failure, palmar-plantar erythrodysaesthesia syndrome, haemorrhage urinary tract, vaginal haemorrhage, metrorrhagia. Very Common: Decreased appetite, dysgeusia, headache, hypertension, diarrhoea, nausea, vomiting, abdominal pain, hair colour change, alopecia, rash, proteinuria, fatigue. Common: Infections, hypophosphataemia, dehydration, insomnia, dizziness, lethargy, paraesthesia, vision blurred, flushing, epistaxis, dysphonia, dyspnoea, haemoptysis, stomatitis, dyspepsia, flatulence, abdominal distension, mouth ulceration, dry mouth, hyperbilirubinaemia, hepatic function abnormal, skin hypopigmentation, dry skin, pruritus, erythema, skin depigmentation, hyperhidrosis, arthralgia, myalgia, muscle spasms, musculoskeletal pain, mucosal inflammation, asthenia, oedema, chest pain. Consult the Summary of Product Characteristics in relation to other side effects. Overdose: Pazopanib doses up to 2000 mg have been evaluated in clinical studies. Grade 3 fatigue (dose-limiting toxicity) and Grade 3 hypertension were each observed in 1 of 3 patients dosed at 2000 mg and 1000 mg daily, respectively. There is no specific antidote for overdose with pazopanib and treatment of overdose should consist of general supportive measures. Legal category: POM. Marketing Authorisation Number: PA22579/006/001- 002. Marketing Authorisation Holder: Teva GmbH, Graf- Arco-Str. 3, 89079 Ulm, Germany. Job Code: MED-IE- 00101. Date of Preparation: October 2025
Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie.
Adverse events should also be reported to Teva UK Limited on +44 (0) 207 540 7117 or medinfo@tevauk.com
Date of Preparation: October 2025 | Job Code: GEN-IE-00153
Further information is available on request or in the SmPC. Product Information also available on the HPRA website.
Cancer Patients to Receive new Treatments following ¤10m Investment
A ¤10 million investment will be made in a cancer care project that is aiming to provide cancer patients in the south of Ireland with greater access to the newest cutting-edge healthcare.
The significant investment in cancer research & care was outlined today in the Aula Maxima during a special event, The Future of Cancer Research. Held to mark World Cancer Day, the event was opened by Minister Jerry Buttimer T.D., Minister of State at the Department of Rural and Community Development and the Gaeltacht and at the Department of Transport.
With one in two people in Ireland facing a cancer diagnosis in their lifetime, the disease remains one of the most significant health challenges facing the community.
Over five years, the ¤10 million investment, composed of clinical trial revenue and philanthropy, will see a project spearheaded by University College Cork (UCC) which will provide a streamlined connection between the latest cancer research and regional cancer centres; providing state of the art clinical trials to patients in the wider Cork region.
Working with the HSE South West and other partners, Cancer Research @UCC will develop a research ecosystem that will bring the latest international cancer research direct to those at risk of or living with a cancer diagnosis.
Decisive step in advancing Ireland’s long-term national ambition
Initiatives will target areas such as cancer therapeutics, the cancer microbiome, and supports for patients to live well with and beyond a cancer diagnosis. Fostering collaboration between scientists, clinicians, industry, charity partners, and patient advocacy organisations, the initiative will improve patient outcomes, reinforcing the region’s position as a leader in healthcare innovation.
It also represents a decisive step in advancing Ireland’s long-term national ambition to transform cancer research, prevention, diagnosis, and treatment at local, national, and international levels.
Welcoming the announcement, Professor John O’Halloran, President of University College Cork, said: “This investment represents a transformative moment for cancer research in Ireland. By backing world-class science, fostering collaboration and having the patient voice central to these plans, we are accelerating progress towards better outcomes for patients and their families.”
A spokesperson for the HSE South West, CEO of Cork University Hospital Ms Jennifer Kearney said, Cancer affects thousands of families across Ireland every year. This commitment will enable our researchers and clinicians
Professor John O’Halloran, President of University College Cork, CEO of Cork University Hospital Ms Jennifer Kearney, Professor Roisin Connolly, Director of Cancer Research @ UCC and Academic Director of the CUH/ UCC Cancer Centre, Minister Jerry Buttimer T.D., Minister of State at the Department of Rural and Community Development and the Gaeltacht and at the Department of Transport, and Professor Helen Whelton, Head of the UCC College of Medicine and Health. (Picture by Daragh McSweeney Provision)
This investment by our region is a step change that demonstrates a long-term commitment by UCC and healthcare partners to innovation, health and people focused outcomes.
to push boundaries, translate discoveries into real-world treatments, and deliver renewed hope to patients nationwide.”
Professor Roisin Connolly, Director of Cancer Research @ UCC and Academic Director of the CUH/ UCC Cancer Centre, added, “Cork and Ireland have the talent, infrastructure, and ambition to lead internationally in cancer research that leads to improved survival and better experience after cancer. This investment by our region is a step change that demonstrates a long-term commitment by UCC and healthcare partners to innovation, health and peoplefocused outcomes. This builds on our regions recent success in developing a Health Research Board funded cancer trials network and attaining European OECI accreditation for the CUH/UCC Cancer Centre; a mark of quality and excellence associated with better cancer outcomes.”
Professor John F. Cryan, UCC Vice President for Research and Innovation, said: “This significant investment will play a major role in transforming cancer care through the delivery of a world-leading cancer research ecosystem at UCC. Cancer Research @UCC is a key UCC Futures Strategic Research Centre, and this investment will enable Professor Roisin Connolly and colleagues to advance new potential cancer treatments and deliver next generation cancer care to patients.”
Professor Helen Whelton, Head of the College of Medicine and Health said: “This is not just an investment in research it is an investment in people, families, and communities affected by cancer across Ireland. It gives real hope for better outcomes, improved care pathways, and ultimately, better lives.”
Inaugural Women’s Heart Summit Examines Persistent Gaps in Cardiovascular Care for Women
Ireland’s first dedicated Women’s Heart Summit took place on Saturday, 24 January 2026, bringing together national and international experts to examine persistent gaps in the diagnosis, treatment and management of cardiovascular disease in women.
Hosted by the Mater Private Network in association with the Cardiovascular Research Institute Dublin, the one-day summit was held at the Westbury Hotel, Dublin, and attracted cardiologists, GPs and healthcare professionals from across Ireland. The programme focused on sex-specific cardiovascular risk, under-recognised clinical presentations and emerging evidence aimed at improving outcomes for women.
Opening the meeting, course director Dr Róisín Colleran, Consultant Cardiologist at the Mater Private Network, emphasised the importance of recognising cardiovascular
disease in women as a distinct clinical challenge rather than a variation of male-pattern disease.
The keynote address was delivered by Prof Roxana Mehran, Professor of Medicine and Director of Interventional Cardiovascular Research and Clinical Trials at Mount Sinai School of Medicine, New York. Her presentation examined heart disease in women as an area that remains under-recognised, underdiagnosed and under-treated worldwide, despite cardiovascular disease being the leading cause of mortality in women globally.
Sessions throughout the day highlighted conditions that disproportionately affect women or present differently compared with men. Spontaneous coronary artery dissection and myocardial infarction with nonobstructive coronary arteries were explored as important but frequently missed causes of myocardial infarction in women.
Dr Fernando Alfonso, Head of the Department of Cardiology at Hospital Universitario de la Princesa in Madrid, addressed the complexities of diagnosing and managing spontaneous coronary artery dissection, while Dr Louise Fitzgerald discussed recognition and follow-up in general practice.
Sex-specific considerations in angina with non-obstructive coronary arteries and ischaemia with non-obstructive coronary arteries were also addressed, with Prof Colin Berry outlining diagnostic and imaging challenges and the need for improved clinical pathways.
Later sessions focused on differences in heart failure outcomes between men and women, with Prof Mark Dayer examining why women experience distinct clinical trajectories and how this should influence treatment strategies. Cardiooncology was another key theme, with Dr Magid Awadalla
discussing breast cancer therapyrelated cardiotoxicity and its growing relevance as cancer survival rates improve.
The programme also included a dedicated focus on cardiovascular disease and pregnancy. Prof Julie De Backer reviewed European guideline-based approaches to managing cardiovascular disease during pregnancy and highlighted the importance of multidisciplinary care for women with complex cardiac conditions.
Across the summit, speakers consistently highlighted the need for greater awareness, improved education and earlier recognition of atypical cardiovascular presentations in women. By bringing together international expertise and Irish clinical experience, the Women’s Heart Summit marked a significant step in advancing discussion and practice around women’s cardiovascular health in Ireland.
Seeking New Treatments for Childhood Dementia
European Rare Diseases Research Alliance funding of ¤440,000 will allow the team to seek new treatments for Multiple Sulfatase Deficiency, an ultra-rare, deadly, inherited disorder where the body can't properly break down fats and sugars.
MSD develops within the first two years of birth and ultimately leads to a devastating and toxic buildup in cells, especially the brain, causing progressive neurological decline. While severity of the condition can vary, the average life expectancy is just 13 years of age, with many children dying much younger.
Here, working in collaboration with an EU consortium led by paediatrician Prof. Lars Schlotawa
at the University Medical Centre Gottingen (UMG), Germany, scientists from the Smurfit Institute of Genetics in Trinity will work to better understand how the disease works and assess potential new treatments.
Trinity’s Dr Gergo Porkolab, postdoctoral research fellow and co-PI on the grant, said: “I’m delighted we have secured this funding to further our understanding of what is a truly devastating disease. We are now well positioned to explore potential therapeutic strategies for patients in the future.”
Professor Matthew Campbell, Head of the Genetics department at Trinity and Chair of Neurovascular Genetics, added: “We are incredibly
Alan Finglas and Professor Matthew Campbell in Trinity's Smurfit Institute of Genetics.
Image: Professor Matthew Campbell
excited to host this project in our lab in the Genetics department at Trinity. Within the next three years, we are going to see some major developments in this field and Trinity scientists will be at the forefront.”
MSD research has been spearheaded in the past by Alan Finglas, a co-founder and research manager of MSD Action Foundation/SavingDylan.com.
Alan’s son Dylan was born with the condition in 2012 and sadly passed away in March 2025.
Speaking today about the new funding Alan Finglas commented: “I’m delighted to be involved in this consortium that will lead one of the largest ever research projects focused on MSD. The coming together of the research teams was years of work, networking and unwavering dedication by Prof. Schlotawa.”
“It gives me great confidence that we may have effective treatments
Under-diagnosis of High Blood Pressure
TILDA’s most recent data shows that 62% of people aged 50 and over have high blood pressure that is not being managed leading to serious potential health risks.
High blood pressure becomes more common after age 40years yet new research from The Irish Longitudinal Study on Ageing (TILDA) at Trinity College shows that many people in Ireland with hypertension are still not optimally diagnosed or treated based on European guidelines. The new study is published in the international journal Open Heart.
High blood pressure (hypertension) is a significant risk factor for cardiovascular disease, stroke, dementia, and chronic kidney disease, yet it often causes no symptoms and can go undetected for years. This 12-year longitudinal study, involving over 8,000 participants, provides a detailed national picture of how high blood pressure is being managed in Ireland and examines adherence to the latest
European Society of Cardiology (ESC) guidelines, including the 2024 recommendations.
Drawing on more than a decade of TILDA data, the researchers show that unmet need in hypertension care is not a new problem, but one that has persisted over time. At the most recent data collection, 62% (approximately 445,000 people aged 50 and over) with high blood pressure were not appropriately managed: they had undiagnosed hypertension, were diagnosed but not receiving treatment, or were on treatment but had blood pressure above recommended targets.
When the lower blood pressure target of <130/80 mmHg, as advised by the 2024 ESC guidelines, is applied, this figure rises to 77% — meaning more than three out of every four older adults with hypertension in Ireland are not optimally managed.
Key findings from the study
• Hypertension prevalence remained consistently high,
increasing from 63% to 71% over 12 years.
• Only 56% of those with hypertension are aware they have it, highlighting substantial ongoing under-diagnosis.
• 71% of those with hypertension were taking a medication, treatment intensity was often suboptimal: only 14% were prescribed a guidelinerecommended dual therapy and only 57% guidelinerecommended monotherapy.
• Among those receiving treatment, just 33% achieved the guideline-recommended blood pressure target of <130/80 mmHg, while 54% were controlled to <140/90 mmHg.
• Overall, 62% (445,000 people) with hypertension in Ireland were not appropriately managed according to the 2018 ESC guidelines, consistent with previous TILDA evidence showing poor long-term control
for patients in the years to come and I am delighted that Trinity are playing a major role. This research represents a tremendous legacy for Dylan and all other patients and families that have been battling against MSD.”
As part of the project in Trinity, a postdoctoral researcher will screen the efficacy of novel drugs to treat the neurological effects of MSD. There was much preliminary research on MSD in recent years that enabled this project. MSD Action Foundation assisted with funding and collaborated to characterise new pre-clinical models that are essential for this study.
Also, in other critical preliminary research, The Health Research Board and MSD Action Foundation co-funded the screening of a drug repurposing library of 5,600 drugs on MSD cells at Fraunhofer and UMG.
Prof. Campbell added: “There are over 50 drug hits from this that must be narrowed down robustly, developed into drug formulations and tested further on MSD models in the lab. We expect that some drugs will translate to real and meaningful therapies for patients in the future, and there is hope that some of the drugs could also help related conditions.”
of modifiable cardiovascular risk factors.
• Forty per cent of the population had elevated blood pressure (SBP value of 120–139mmHg or a DBP value of 70-89mmHg) and 71% of this group had evidence of high cardiovascular risk.
• People aged 85 years and older and those with moderate to severe frailty were less likely to have undiagnosed hypertension and were more likely to be taking guidelinerecommended medications similar rates of blood pressure control when compared to the wider population.
• People with chronic kidney disease (CKD) were more likely to receive guidelinerecommended treatment and achieve blood pressure control to a target of <140/90mmHg, suggesting targeted care can be effective in high-risk groups.
Mitochondrial quality control in diabetes mellitus and complications: molecular mechanisms and therapeutic strategies
Written by Yanling Chen1,2, Xun Liu3, Yixuan Liu1,2, Yujia Li4, Dingxiang Li4, Zhigang Mei1,2 and Yihui Deng4
1School of Integrated Chinese and Western Medicine, Hunan University of Chinese Medicine, Changsha, 410208, China
2Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, 410208, China
3Loudi Central Hospital, Loudi, 417000, China
4School of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha, 410208, China
Diabetes mellitus (DM), a systemic metabolic disease characterised by chronic hyperglycemia and insulin resistance, poses a mounting global health burden, with a prevalence estimated to reach 783.2 million adults by 2045.1 Mitochondria are dynamic organelles essential for cellular energy metabolism.2, 3 Metabolic stress including persistent hyperglycemia and dysregulated glucolipid metabolism could induce pancreatic mitochondrial dysfunction, especially in β cells, which is key to the onset and progression of diabetes, aggravating oxidative stress, inflammation, and glucolipotoxicity, driving diabetic multi-organ damage.4,5,6,7,8
Mitochondrial quality control (MQC), a network encompassing biogenesis, dynamics (fission and fusion), and mitophagy, ensures mitochondrial integrity and metabolic flexibility.9 Emerging evidence positions MQC as a linchpin in DM pathophysiology. MQC imbalance disrupts cellular ATP synthesis, amplifies oxidative damage, and perpetuates metabolic stress across insulinsensitive tissues.10,11,12,13 While the role of MQC in diabetes is welldocumented previously, its tissuespecific regulation in diabetic complications, ranging from cardiac lipotoxicity to renal tubular injury, remains underexplored.14, 15
This review synthesises emerging insights into MQC mechanisms, elucidating their physiological and pathological interplay with DM. We posit that MQC imbalance serves as a co-pathogenic aspect of diabetic complications, and summarise emerging evidence of MQC molecular targets on diabetic complications. What is more, we discuss the therapeutic strategies targeting MQC to break the cycle of mitochondrial
collapse and systemic metabolic decompensation. We searched PubMed for relevant Englishlanguage articles published up to April 2025 (final search date). The query terms related to MQC and diabetic complications: (“mitochondrial quality control” OR “mitochondrial biogenesis” OR “mitochondrial dynamics” OR “mitochondrial fission” OR “mitochondrial fusion” OR “mitophagy”) AND (“diabetes mellitus” OR “insulin resistance”) AND (“pancreas” OR “liver” OR “muscle” OR “adipose” OR “diabetic complications” OR “diabetic heart” OR “diabetic cardiomyopathy” OR “diabetic nephropathy” OR “diabetic kidney” OR “diabetic retinopathy” OR “diabetic retina” OR “diabetic neuropathy” OR “diabetic peripheral neuropathy” OR “diabetic brain”). Studies were included if they investigated mechanistic links between MQC and diabetes or its complications, provided molecular or therapeutic insights, and were peer-reviewed original research. Excluded articles included reviews, editorials, nonEnglish publications, and studies lacking direct relevance to DM and MQC. Two independent reviewers screened titles and abstracts, followed by full-text assessment of eligible articles.
Mitochondrial quality control
To ensure optimal function, mitochondria undergo a finely tuned quality control mechanism involving mitochondrial motility, mitochondrial-derived vesicles (MDVs), the ubiquitinproteasome system, and cellular environmental factors.16
Mitogenesis
Mitogenesis replenishes mitochondrial mass via coordinated nuclear DNA (nDNA)- and mitochondrial DNA
(mtDNA)-encoded programs. The peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) family, including PGC-1α, PGC1β, and PRC, orchestrate this process by activating transcription factors NRF1, NRF2, and ERRα, as well as respiratory complex assembly.17,18,19,20 While PGC-1α drives biogenesis in energydemanding tissues including heart and muscle, PGC-1β compensates during brown adipogenesis.21 PRC regulates NRF1/CREB-dependent transcription but is dispensable for basal biogenesis.22, 23 Upstream regulators and post-translational modifications like PGC-1α monomethylation24 fine-tune biogenesis, while mitochondrial preprotein translocases TOM and TIM complexes ensure nuclearencoded protein import.25, 26 Under stress, mitochondrial also transfer from neighboring cells and can rescue bioenergetic deficits.27, 28
Mitochondrial dynamics
Mitochondrial morphology adapts to metabolic demands via fission and fusion, governed by post-translational modifications (PTMs) of dynamin-related GTPases.29, 30 Fusion merges outer (MFN1/MFN2) and inner (OPA1) membranes, enabling content exchange and cristae stabilisation.31, 32 Transient “kissand-run” fusion events allow selective matrix communication.33 Fission, mediated by Drp1 recruitment to mitochondrial receptors (Mff, Mid49/51, Fis1), partitions damaged regions for degradation.34,35,36,37,38 Imbalanced dynamics including excessive fission and defective fusion disrupt metabolic flexibility and amplify oxidative stress.
Mitophagy
Mitophagy, a specialised form of cellular autophagy, selectively degrades damaged mitochondria
via the Ser/Thr kinase PINK1/ the E3 ubiquitin ligase Parkindependent and -independent pathways. Depolarised mitochondria stabilise PINK1, which phosphorylates ubiquitin to recruit Parkin for OMM protein ubiquitination.39,40,41 Parkinindependent pathways utilise OMM receptors including BCL2 and adenovirus E1B 19-kDainteracting protein3 (BNIP3), BNIP3-like (BNIP3L/NIX), Bcl-2like protein 13 (BCL2L13), FUN14 domain containing 1 (FUNDC1) and tacrolimus (FK506)-binding protein8 (FKBP8), as well as IMM phospholipids cardiolipin to recruit LC3-labeled autophagosomes.42, 43 Regulatory crosstalk involves prohibitin 2 (PHB2), autophagy-beclin1regulator1(AMBRA1), and Myosin VI (MYO6), which promote autophagosomelysosome fusion.44,45,46,47,48
Novel horizons for MQC
Beyond canonical mitophagy, MQC encompasses adaptive mechanisms to preserve homeostasis under stress. MDVs act as a compensatory pathway when LC3-dependent autophagosome formation is impaired, selectively packaging misfolded proteins, oxidised lipids or damaged mtDNA into mitochondrial-derived compartments (MDCs) for lysosomal or peroxisomal degradation.49,50,51,52,53 MDV biogenesis is regulated by microtubule-associated motor proteins MIRO1/2 and fission machinery components (Drp1, Mff, Mid49, Mid51), linking vesicle formation to mitochondrial dynamics.53 Cells unable to recycle dysfunctional mitochondria intracellularly may transfer damaged components via extracellular vesicles
Diabetes
for phagocytic clearance by neighboring cells (e.g., macrophages), a process termed “licensed mitophagy”.27, 28 Recent work also identifies mitocytosis, a migrasome-mediated expulsion of mitochondria during cell migration. Under mild stress, motor proteins direct damaged mitochondria to migrasomes at the cell periphery, enabling extracellular disposal.54
Furthermore, mitochondrialysosome crosstalk enables localised repair through inner mitochondrial membrane-derived vesicles (VDIMs). Cristae damage triggers ROS-dependent activation of lysosomal TRPML1 channels, promoting VDAC1 oligomerisation and herniation pore formation on the OMM. This facilitates extrusion of damaged IMM components, which are engulfed by lysosomes as VDIMs.55
These emerging pathways underscore MQC’s plasticity, offering novel therapeutic entry points to disrupt the cycle of mitochondrial dysfunction and metabolic disease (Fig. 1).
The role of mitochondria in pancreatic beta cells
Mitochondria regulate pancreatic β-cell insulin secretion and peripheral insulin sensitivity through redox balance, metabolic flux, and interorganellar communication.56, 57 Maintaining mitochondria turnover ensures establishment of sufficient mitochondria OXPHOS for islet β cells differentiation and maturation.58 The mitochondrial tRNA-derived fragment interacts with electron transfer system complexes and paly pivotal role in mitochondrial oxidative phosphorylation and its
coupling to insulin secretion of pancreatic β-cells.59
In pancreatic β cells from type 2 diabetic subjects, the impaired secretory response to glucose is associated with a marked alteration of mitochondrial function and morphology. In particular, uncoupling protein-2 expression is increased, which leads to lower ATP, decreased ATP/ADP ratio, with consequent reduction of insulin release.60 Hyperglycemic environment induces metabolic changes in β cells that significantly reduce mitochondrial metabolism and ATP synthesis, which contribute to the progressive failure of β-cells to respond to glucose in diabetes.61
Inactivation of transcriptional factor YY1 impairs mitochondrial oxidative phosphorylation and mitochondrial function, resulting in the reduction of insulin
secretion, β-cell mass, and glucose tolerance.62 Lipotoxicity including saturated free fatty acids (FFAs) accumulation alters β-cell signal transduction,63 induce mitochondrial fragmentation and production of ROS in β cells, thereby promoting β-cell dysfunction and pancreatic insulin sensitivity changes.64,65,66 Effective mitochondrial adaptive responses including mitochondrial redox signaling and mild uncoupling reduce β-cell oxidative stress and dysfunction under metabolic stresses like hypertriglyceridemia.67
MQC in the pathophysiology of insulin resistance and diabetes
Insulin resistance (IR), a hallmark of type 2 diabetes mellitus (T2DM), arises from impaired cellular responsiveness to insulin, disrupting glucose homeostasis and driving hyperglycemia.
Fig 1 caption: Mitogenesis: Nuclear DNA and mitochondrial DNA are responsible for the synthesis of new mitochondria, a process regulated by coactivators and transcription factors through membrane protein transport mechanisms. Mitochondrial dynamics: The fusion is mediated by OMM proteins (MFN1 and MFN2), and the fission is mediated by the interaction between DRP1 and fission factors (Fis1. Mff, Mid49, Mid51). Mitophagy: The process is mainly mediated by the PINK1/Parkin pathway and mitochondrial membrane protein receptor-dependent signaling pathway. MDVs biogenesis: Mitochondria selectively release components and even damaged mtDNA through MDVs, which are degraded through specific transport pathways to lysosomes. Mitocytosis: Mitochondria are translocated to migrasomes, and migrating cells selectively bind damaged mitochondria to motor proteins, localize them to the cell’s periphery, and ultimately discharge them outside the cell to be removed. Licensed mitophagy: A cell transfers its own damaged mitochondrial components to extracellular vesicles to be released and eliminated with the help of mitophagy of another cell. Mitochondria transfer: When endogenous mitogenesis is deficient, the cell replenishes exogenous healthy mitochondria from outside other cells via mitochondrial transfer.
Both insulin insufficiency and IR converge on mitochondrial dysfunction, where defective MQC exacerbates metabolic dysregulation. Targeting MQC pathways offers a strategic avenue to uncouple mitochondrial failure from disease progression.
MQC-insulin signaling crosstalk Enhanced mitochondrial biogenesis improves ATP synthesis, facilitating insulinstimulated glucose uptake via GLUT4 translocation.68
Mitochondrial dynamics proteins directly modulate insulin signaling: Mfn2 stabilises insulin receptor substrate 1 (IRS1) and coordinates mitochondrial-ER crosstalk,69 while phosphorylation of mitochondrial fission factor (MFFS131) promotes hepatic insulin sensitivity by tuning mitochondrial fragmentation.30 Conversely, insulin regulates MQC via Akt/mTOR/FOXO1 signaling, suppressing Drp1/Fis1mediated fission and enhancing PGC-1α-driven biogenesis.70,71,72
Disruption of this bidirectional crosstalk which is evidenced by mitophagy-deficient β-cells with impaired insulin secretion73, 74 fuels a self-perpetuating cycle of IR and mitochondrial collapse.75
MQC dysregulation in metabolic stress
Chronic hyperglycemia and lipid overload impair mitochondrial oxidative phosphorylation, elevating ROS production and depleting ATP synthesis.76 ROS inactivate IRS1/2 via serine phosphorylation, exacerbating β-cell apoptosis and peripheral IR.77 Downregulation of PGC1α and NRF1 in T2DM reduces mitochondrial biogenesis, fatty acid oxidation, and respiratory capacity,78,79,80 while hyperglycemia-driven the upregulation of fission proteins fragments mitochondria, further amplifying oxidative stress.81,82,83 Restoring deficient fusion or inhibiting excessive fission rescues IRS1/Akt signaling and glucose uptake, underscoring MQC’s role in metabolic flexibility.84, 85
Defective mitophagy exacerbates IR by accumulating damaged mitochondria, triggering NLRP3 inflammasome activation and adipose inflammation.13, 86 FUNDC1 knockout mice exhibit worsened obesity and IR under high-fat diets, linking impaired mitophagy to adipose metabolic dysfunction.87 These findings position MQC as a nodal regulator of inflammationmetabolism crosstalk in diabetes.
Impaired MQC in the pathophysiology of metabolic tissues associated with IR
MQC dysregulation induces a retrograde signaling program
that impairs identity and maturity in insulin-sensitive tissues including pancreas, liver and adipose, yielding metabolic disorders and driving systemic IR and diabetes progression.88
Pancreas
The pancreatic endocrine β-cells, central to systemic glucose homeostasis,89 are critically vulnerable to mitochondrial dysfunction in diabetes. The disruption of mitochondrial turnover and function in islets contribute to chronic exposure of pancreatic β-cell to high concentrations of FFAs, aggravating lipotoxicity-mediated suppression of glucosestimulated insulin secretion.90,91,92 Emerging evidence highlights that MQC failure in islets drives β-cell dysfunction through impaired glucose tolerance and exacerbated insulin resistance, accelerating diabetes progression.93, 94 Notably, NRF2 deficiency under metabolic stress disrupts mitogenesis, triggering a marked decline in functional β-cell mass and systemic glucose dysregulation.95 Concurrently, O-GlcNAc transferase (OGT) sustains β-cell physiology by enhancing pancreatic and duodenal homeobox 1 (Pdx1)dependent mitogenesis, thereby preserving insulin secretion.96 Dysregulated mitochondrial dynamics directly underpins β-cell death. For instance, RNA binding protein HuD overexpression rescues diabetic β-cell dysfunction by restoring MFN2-mediated mitochondrial fusion, whereas palmitate (a C16-FFA)- induced activation of TRPM2 elevates mitochondrial Zn2+, potentiating DRP1-driven mitochondrial fission, β-cell apoptosis, and insulin secretory failure.97, 98 The Activation of LRH-1/NR5A2 targeting mitochondrial dynamics attenuates the autoimmune attack coupled to beta cell regeneration and islet survival in type 1 diabetes mellitus (T1DM).99 Furthermore, human islet amyloid polypeptide (hIAPP) aggregation in diabetes exacerbates mitochondrial fragmentation and suppresses mitophagy, leading to ROS overproduction and irreversible β-cell damage.100 Disruption of the PPP3/calcineurin/TFEB axis impairs mitophagy, culminating in β-cell dysfunction and glucose intolerance.101 Under glucotoxic conditions, GRP78 upregulation cause depletion of the antioxidant pool and promote mitophagy in pancreatic cells in T1DM.102 Collectively, MQC impairment contributes to β-cell demise in diabetes, offering potential targets for interventions to preserve pancreatic β-cell mass and function.
Liver
Hepatic MQC directly modulates glucose and lipid homeostasis. Activation of the AMPK/ SIRT1/PGC-1α axis enhances mitogenesis, suppresses gluconeogenesis, and promotes fatty acid oxidation, reversing lipid accumulation and IR in T2DM.80, 103 CerS6-derived sphingolipids, as a kind of hepatic lipid deposition, promote hepatic mitochondrial fragmentation and glucose disorders in a Mff-dependent manner, exacerbating obesity and IR.34 Nutrient-sensing pathways further fine-tune mitochondrial dynamics. AKT-dependent phosphorylation of mitochondrial fission factor MFFS131 induces transient mitochondrial fragmentation, priming the liver for metabolic adaptation to feeding cycles.30 Conversely, Drp1 and Fis1 upregulation disrupts oxidative phosphorylation, exacerbating hepatic steatosis and hyperglycemia.104 As a catabolic process, mitophagy promotes mitochondrial fatty acid oxidation to inhibit hepatic fatty acid accumulation, thereby improving hepatic IR and metabolic syndrome.105
Skeletal muscle
MQC governs insulin-stimulated glucose uptake in skeletal muscle. Upregulation of miR30d-5p undermines glycolipid metabolism in skeletal muscle by targeting SIRT1/PGC-1αdependent mitogenesis.106 Lipid infusion increases the expression of DRP1 and PINK1, regulating MQC networks to suppress insulin sensitivity in human skeletal muscle.75 Nutrient storage stress triggers a BNIP3L-mediated signaling cascade that promotes mitochondrial fission and mitophagy, impairing myocyte glucose uptake with the activation of MTOR-RPS6KB and IRS1 phosphorylation.107 In diabetic sarcopenia, MFG-E8 (lactadherin) is upregulated and induces mitophagy deficiency via the HSPA1L-Parkin pathway, thereby aggravating diabetic muscle atrophy.108 Deubiquitinating enzymes including USP15 and USP30 activates DRP1dependent mitochondrial fission, thereby regulating skeletal muscle MQC and insulin sensitivity in T2DM patients.109
Adipose tissue
Adipocyte MQC regulates systemic energy balance. JMJD1A, a histone demethylase, activates PGC-1-mediated mitogenesis and the formation of beige adipocytes in white adipose tissues (WAT), alleviating obesity and metabolic dysfunction.110 In WAT, activation of small GTPase RalA promotes excessive mitochondrial fission,
suppresses thermogenesis, and drives obesity-associated metabolic dysfunction.111 TMEM135 overexpression promotes brown fat mitochondrial fission, counteracts obesity and insulin resistance, and rescues thermogenesis in adiposespecific peroxisome-deficiency.112 FUNDC1-dependent mitophagy maintains adipose homeostasis, whereas FUNDC1 ablation in WAT disrupts mitochondrial integrity, exacerbates diet-induced obesity and IR.87 Brown adipose tissue (BAT) relies on mitochondrial protein succinylation and malonylation to sustain thermogenic capacity, with defects in these posttranslational modifications impairing mitophagy and glucose homeostasis in T2DM.86, 113
Impaired MQC in diabetic complications
Individuals with diabetes face a significantly elevated risk of macrovascular events, such as stroke and myocardial infarction, as well as microvascular complications, including retinopathy and nephropathy, compared to non-diabetic populations. Emerging evidence underscores MQC dysfunction as a pivotal contributor to diabetic vascular endothelial dysfunction. Hyperglycemia induces mitochondrial fragmentation, impaired fusion, and excessive fission and autophagy across various tissues, including the cardiovascular system, liver, and pancreas.114, 115 FOXO1 inhibition mitigates high glucose (HG)induced DRP1 overexpression and mitochondrial fission in endothelial cells, reducing reactive oxygen species (mtROS) overproduction and ameliorating vascular dysfunction.116 Given that mtROSdriven endothelial injury underlies both macro- and microvascular diabetic complications,117, 118 targeting MQC represents a promising therapeutic avenue to attenuate disease progression.119
Diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM), characterised by myocardial fibrosis and impaired cardiac function independent of coronary artery disease, is linked to MQC defects involving calcium dysregulation, oxidative stress, and lipid accumulation.120 Cardiac mitochondrial damage and biogenesis are induced by oxidative stress in a chronic model of type 1 diabetes.121 While compensatory mitophagy may protect against early lipid overload in high fat diet-induced DCM,122 persistent imbalances in mitochondrial dynamics which is marked by excessive fission and deficient fusion and mitophagy that drive DCM pathogenesis.
Diabetes
Recent research has identified novel targets for DCM that alleviate mitochondrial dysfunction and cellular apoptosis by modulating posttranslational modifications (PTMs) of MQC proteins, including phosphorylation, acetylation, and methylation.
PGAM5 dephosphorylates PHB2 at Ser91, disrupting MQC and exacerbating myocardial injury.123, 124 Downregulated miR144 elevates RAC1, impairing AMPK/ PGC-1α-mediated mitogenesis and promoting apoptosis.125 CaSR enhances ubiquitination of MFN1, MFN2, and CX43 via GP78, disrupting mitochondrial fusion and energy metabolism.126 PFKFB3 deficiency in diabetic hearts destabilises OPA1, whereas NEDD4L-mediated OPA1 ubiquitination preserves mitochondrial integrity.127
MAP4K4 disrupts GPX4dependent redox balance, inducing Drp1 S-nitrosylation (SNO-Drp1), excessive fission, and ferroptosis.128 MST1 inhibits SIRT3/PARKIN mitophagy and promotes DRP1 phosphorylation, accelerating mitochondrial fragmentation.129, 130 By performing targeted metabolomics to characterise the metabolic phenotype of human myocardium of DCM, it is founded that RCAN1 upregulation activates DRP1 Ser616, driving lipid accumulation and metabolic remodeling in cardiomyocytes.131
Epigenetic and epitranscriptomic mechanisms further modulate MQC in DCM. The NOTCH1/ ALKBH5/YTHDF axis, regulated by m6A-mediated phase separation, suppresses NOTCH1 expression, promoting fission and fibrosis.132 Similarly, BRD4 binds H3K27ac at the PINK1 promoter, repressing PINK1/Parkin mitophagy; BRD4 inhibition restores mitophagy and alleviates DCM.133
MQC dysregulation intersects with lipid metabolism, calcium signaling, and ferroptosis in DCM. SFRP2, an adipokine, modulates mitochondrial dynamics via AMPK/PGC-1α and activates calcineurin/TFEB-dependent mitophagy.134, 135 ORAI1mediated Ca2+ influx activates DRP1, exacerbating fission and hypertrophy. Targeting ORAI1/pERK1/2/CnA normalises calcium homeostasis.136 DUSP26 rescues mitochondrial dynamics and lipid metabolism via FAK/ERK signaling.137 LGR6 deficiency impairs STAT3/PGC-1α, aggravating ferroptosis and biogenesis defects.138
Key transcription factors integrate MQC with metabolic stress. STAT3 activation by BNP sustains OPA1-mediated fusion, while FOXO1 competes with STAT3
for 14-3-3 binding, impairing mitochondrial homeostasis under hyperglycemia.139, 140 PPARα downregulation reduces MFN2 expression, contributing to dynamics imbalance.141
ALDH2 preserves mitochondrial function via Akt/GSK3β/Foxo3a signaling and PARKIN-dependent mitophagy.142
DCM pathogenesis involves a complex interplay of MQC defects, PTMs, and transcriptional and epigenetic dysregulation. Targeting nodes such as DRP1 phosphorylation, mitophagy pathways, or redox balance offers therapeutic potential.
Diabetic nephropathy
Diabetic kidney disease (DKD), a chronic complication driven by persistent hyperglycemia and glomerular hyperfiltration, remains a leading cause of endstage renal disease globally.143 Pathologically, DKD is marked by altered glomerular filtration rates, proteinuria, and dysregulation of metabolites and ions. Podocytes, essential components of the glomerular filtration barrier, are particularly vulnerable to hyperglycemia-induced damage, contributing to mitochondrial dysfunction and disease progression. Emerging evidence underscores the centrality of mitochondrial homeostasis in maintaining renal health under diabetic conditions.144
Mitogenesis is critically regulated in DKD through metabolic and signaling pathways. Hyperglycemia suppresses pyruvate kinase M2 (PKM2) tetramerisation and activity via sulfenylation in podocytes, impairing glycolytic flux and amplifying toxic glucose metabolite accumulation. Restoring PKM2 activity enhances glucose metabolism, upregulates PGC-1α expression, and promotes mitochondrial biogenesis, thereby mitigating renal injury.145 Conversely, overexpression of p66SHC, a redox-sensitive adaptor protein, suppresses mitogenesis by downregulating SIRT1, PGC1α, NRF1, and TFAM, exacerbating mitochondrial ROS production and podocyte damage.146 Similarly, mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH) deficiency in diabetic models disrupts PGC-1αdependent mitogenesis, activating the RAGE-S100A10 axis to drive oxidative stress and renal injury.147 Additional regulators include AKAP1, which phosphorylates Larp1 via PKC signaling to inhibit TFAM-mediated mtDNA replication, and adiponectin receptor 1 (AdipoR1), whose deficiency impairs the CREB/ PGC-1α/TFAM axis. Exogenous adiponectin supplementation
rescues mitogenesis and alleviates tubular dysfunction, highlighting therapeutic potential.148, 149 Progranulin (PGRN) depletion further disrupts mitochondrial homeostasis via Sirt1/PGC-1α/ FOXO1 signaling, while the lncRNA PVT1 exacerbates fission and bioenergetic deficits by destabilising AMPKα through TRIM56 interaction.150, 151 Telomerase reverse transcriptase (TERT) also safeguards renal function by sustaining AMPK/ PGC-1α-mediated mitochondrial energy balance.152
Dysregulated mitochondrial dynamics, intertwined with hypoxia, lipid abnormalities, and calcium dyshomeostasis, further propagate DKD. Hypoxia-inducible factor-1α (HIF-1α) upregulation in tubular cells induces heme oxygenase-1 (HO-1) to stabilise mitochondrial dynamics and attenuate hypoxia-driven damage.153 ALCAT1 expression is increased in the glomeruli of DKD patients, accompanying with significant mitochondrial damage.154 Lipidomic perturbations, such as ALCAT1mediated cardiolipin oxidation, impair mitophagy via AMPK signaling, while the ChREBP/ GNPAT/plasmalogen axis links lipid metabolism to mitochondrial fission.154, 155 RIPK3 overexpression exacerbates podocyte injury by activating PGAM5/Drp1-mediated fission through necroptosis, illustrating crosstalk between cell death pathways and organelle dynamics.156
Electron transport chain (ETC) dysfunction exacerbates bioenergetic failure in DKD. NDUFS4 deficiency disrupts complex I integrity, impairing cristae morphology via STOML2 interaction, whereas IGF2BP3 stabilises CAMK1 to inhibit fission and apoptosis through m6A modification.157, 158 Calcium signaling further modulates mitochondrial fitness. CaMKKβ sustains AMPK phosphorylation to support function, but NEDD4L ubiquitination under hyperglycemia destabilises CaMKKβ, promoting fission.159 TRIM22 and WTAP form an m6A-dependent regulatory axis targeting OPA1 ubiquitination, thereby disrupting fusion and amplifying injury.160 HGF maintaining mitochondrial homeostasis in type 1 diabetic podocytes via decreasing ARF6dependent DRP1 translocation.161
Impaired mitophagy, coupled with MAM disruption, ferroptosis, and oxidative stress, is a hallmark of DKD. KCa3.1 channel deficiency impedes TGF-β1/BNIP3-dependent mitophagy, while DsbA-L depletion activates JNK/MFF/ DRP1 signaling to drive fission.
Conversely, DsbA-L preserves MAM integrity via HELZ2/MFN2 to promote mitophagy, illustrating context-dependent roles.162,163,164 TNFAIP8L1/TIPE1 exacerbates tubular injury by degrading the mitophagy receptor PHB2, whereas UHRF1 enhances PINK1-dependent mitophagy and suppresses ferroptosis via TXNIP inhibition.165, 166 MFN2-SERCA2 interactions and VDR activation restore MAMs and mitophagy through the FUNDC1 pathway, while PACS-2 coordinates fission inhibition and BECN1-dependent mitophagosome formation.167, 168 YAP1 inactivation disrupts both mitogenesis and PINK1/Parkinmediated mitophagy, skewing macrophage polarisation via CXCL1, whereas TRPC6/calpain-1 signaling impairs mitophagy to fuel inflammation.169, 170 DLAT, downregulated in diabetes, enhances AMPK-driven mitophagy, offering protection against renal dysfunction.171 N-Acetyl-Cysteine combined with insulin reverses glomerular wrinkling and fibrosis by regulating the mitochondrial dynamics and FUNDC1-mediated mitophagy in type 1 diabetic nephropathy canine.172
Diabetic retinopathy
Diabetic retinopathy (DR), a neurovascular complication of chronic hyperglycemia, is characterised by retinal glial network dysfunction, microvascular damage, and pathological neovascularisation, culminating in vision loss and blindness.173 The retina, composed of neurovascular units integrating endothelial cells, Müller glia, pericytes, ganglion cells, and pigment epithelial cells, undergoes mitochondrial dysregulation under hyperglycemic stress, marked by impaired mitophagy and aberrant mitochondrial dynamics. Emerging evidence highlights MQC as a pivotal therapeutic target to counteract metabolic memory and mitigate DR progression.174, 175
Retinal vascular endothelial cells (RECs), critical for nutrient exchange and barrier integrity, exhibit disrupted mitochondrial homeostasis in diabetes. Hypermethylation of the MFN2 promoter, driven by Dnmt1 binding and diminished SP1 recruitment, suppresses MFN2 expression, while SIRT1 overexpression reduces MFN2 acetylation and GTPase activity, impairing mitochondrial fusion.176, 177 Hyperglycemia further upregulates SENP3, which deSUMOylates Drp1 to promote fission, exacerbating blood-retinal barrier dysfunction.178 Loss of PON2, glycated by N-carboxymethyl lysine (CML), amplifies ER stress, inflammation, and mitochondrial fragmentation, whereas TGR5
activation inhibits Drp1 via Ca2+PKCδ signaling and enhances PINK1/Parkin-mediated mitophagy by displacing mitochondrial hexokinase 2 (HK2).179, 180
Methylglyoxal (MGO)-mediated glycation suppresses glyoxalase 1 (GLO1) and downregulates OPA1/ MFN1, aggravating oxidative injury, while VDAC1 sustains mitophagy and restrains NLRP3 inflammasome activation in RECs.181, 182
Pericytes, essential for vascular stability, succumb to hyperglycemia-induced mitochondrial fission via EPAC1-DRP1 signaling, driving ROS overproduction and microvascular damage.183 In retinal pigment epithelial cells (RPECs), copper transporter 1 (CTR1) overexpression disrupts copper homeostasis, inducing oxidative stress and mitochondrial dysfunction, while TIN2
accumulation impairs PINK1-dependent mitophagy via mTOR, exacerbating cellular senescence and tight junction disruption.184, 185 SIRT3, however, counteracts hyperglycemia by activating AMPK/mTOR/ULK1 and FOXO3a/PINK1/Parkin pathways, restoring mitophagy and attenuating RPEC apoptosis.186, 187
Retinal ganglion cells (RGCs), vulnerable to oxidative stress, are protected by DJ-1, which scavenges ROS and restores mitochondrial homeostasis under hyperglycemic conditions.188 Müller glia, pivotal for retinal repair, exhibit TXNIP-driven mitochondrial fission and Parkin-dependent mitophagy in response to hyperglycemia, alongside reactive gliosis marked by GFAP upregulation.189
Diabetic neuropathy
Diabetic neuropathy,
encompassing central neurocognitive decline and diabetic peripheral neuropathy (DPN), represents a debilitating complication of chronic hyperglycemia, driven by glucotoxicity-induced neuronal and cerebrovascular dysfunction.190,191,192 As mitochondria serve as the primary hubs of energy synthesis, MQC imbalances emerge as critical contributors to diabetic encephalopathy (DE) and peripheral nerve damage, disrupting cellular resilience and metabolic homeostasis.193
Impaired mitogenesis underpins cognitive deficits in diabetes. Phosphorylation of WW domaincontaining oxidoreductase 1 (WWOX) at tyrosine 33 under hyperglycemia exacerbates mitogenesis defects and ROS overproduction, triggering neuronal
apoptosis in neuroblastoma models.194 Conversely, activation of dopamine D1 receptors (DRD1) rescues AMPK/PGC-1α signaling, restoring mitochondrial biogenesis and alleviating cognitive dysfunction in diabetic mice.195 Similarly, sodium butyrate (NaB), a gut-derived metabolite, enhances hippocampal synaptic plasticity by reviving AMPK/PGC1α-driven mitogenesis, highlighting metabolic-epigenetic crosstalk in T2DM neuroprotection.196
Dysregulated mitochondrial dynamics further propagate neuronal injury. Diabetic models reveal diminished OPA1 and MFN1/2 alongside elevated Drp1 and Fis1, reflecting fission-fusion imbalance in hippocampal and peripheral neurons.197 Lipin1 deficiency, by altering mitochondrial membrane phospholipids, disrupts synaptic
Fig 2 caption: Pancreas: MQC failure in islets impairs β-cell mass, insulin secretion and glucose tolerance by harming insulin signaling, which excerbates insulin resistance and elevates blood glucose. Liver: Hepatic MQC imbalance induces hepatic steatosis and fatty acid oxidation. Adipose: Adipocyte MQC imbalance decreases adipocytes thermogenic capacity and influences systemic energy balance. Muscle: MQC failure impairs insulin-stimulated myocyte glucose uptake and promotes muscle atrophy. Retina: MQC damage induces retinal glial network dysfunction and microvascular damage. Heart: The myocardial fibrosis and cardiac dysfunction in DCM are aggravated by an interplay of MQC defects, ferrotopsis and oxidation stress. Kidney: MQC dysfunction is contributed to glomerular injury in diabetic nephropathy through promoting mitochondrial ROS, MAM dysruption and ferrotopsis. Nerve: MQC imbalance induce both central neurocognitive decline and peripheral nerve damage by promoting apoptosis, ROS and calcium metabolism disorder.
Diabetes
mitochondrial dynamics, exacerbating cognitive decline.197 Caveolin-1 (Cav-1) counters these effects by suppressing GSK3β/Drp1-mediated fission and enhancing AMPK/PINK1/ Parkin- and ULK1-dependent mitophagy, though excessive Drp1 inhibition may paradoxically worsen neurodegeneration.198, 199 In peripheral nerves, mitochondrial calcium uniporter (MCU) deletion mitigates neuropathic pain by curbing calcium-dependent fission, preserving small-fiber integrity in diabetic mice.200
Mitophagy dysfunction amplifies diabetic neuropathology. Hyperglycemia and advanced glycation end-products (AGEs) impair hippocampal mitophagy via Keap1/Nrf2/PHB2 suppression, accelerating cognitive decline.201
Brain-derived neurotrophic factor (BDNF) counteracts oxidative stress by activating TrkB/HIF-1α/BNIP3 signaling, restoring mitophagy in cerebral microvasculature.202 NaB further rescues neuronal mitophagy by blocking RELA-HDAC8 repression of Parkin, while the SIRT1 activator piceatannol (PCN) enhances both mitogenesis and mitophagy via SIRT1/PGC1α and SIRT1/ PINK1/Parkin axes, alleviating peripheral neuropathy.203, 204 SIRT3 overexpression attenuates painful DPN through FoxO3a/PINK1/ Parkin-mediated mitophagy, whereas PARP1 hyperactivity drives peripheral nerve injury via mitophagy suppression.205, 206 These mechanisms underscore mitochondrial dysregulation as a nexus of diabetic neuropathies, with therapeutic strategies targeting MQC offering promise to preserve neuronal integrity and function.
Overall, MQC imbalance plays an important role in the pathophysiology of diabetes and diabetic complications (Fig. 2).
Therapeutic potentials for diabetic complications by targeting MQC
Current therapeutic paradigms for diabetic vascular complications, often siloed by organ-specific pathology, inadequately address the systemic MQC dysfunction underpinning these conditions. MQC has emerged as a unifying therapeutic axis across diabetic angiopathies. Aligning with clinical guidelines, repurposing existing therapies and exploring natural compounds to modulate MQC pathways offers transformative potential for multisystem vascular protection.
Clinically approved hypoglycemic agents
Several glucose-lowering agents
exert their pleiotropic effect on diabetic complications by targeting MQC. Metformin, an AMPK activator, efficiently prevents high glucose induced mitochondrial ultrastructural and functional abnormalities in human islet cells.207 It has beneficial clinical implications on promoting mitochondrial function and mitophagy in type 2 diabetic patients, which alleviates retinopathy and nephropathy by restoring AMPK-dependent MQC balance and inhibiting Drp1mediated fission in atherosclerosis models. It further rescues cognitive deficits via PGC-1α-driven mitogenesis and enhances poststroke recovery through mitophagy activation.208,209,210,211,212,213 DPP-4 inhibitors demonstrate classwide vascular benefits. Alogliptin and evogliptin enhance cardiac mitogenesis via PGC-1α/ NRF1/TFAM signaling in DCM, while sitagliptin restores renal mitochondrial dynamics through SDF-1α/CXCR4/STAT3. Vildagliptin attenuates endothelial fission via AMPK/Drp1 inhibition and reduces arrhythmia risk by promoting mitogenesis.214,215,216,217,218,219
SGLT2 inhibitors, including empagliflozin and canagliflozin, mitigate renal and cardiac injury by suppressing fission and enhancing mitophagy. Empagliflozin alleviates tubulopathy and myocardial microvascular damage, whereas canagliflozin activates PINK1/ Parkin-mediated mitophagy in DCM.220,221,222,223,224 GLP-1 receptor agonists exhibit tissuespecific modulation. Exendin-4 reverses cardiac mitochondrial abnormalities and curbs vascular calcification via AMPK-dependent mitophagy, while liraglutide paradoxically suppresses PINK1/ Parkin signaling to protect endothelial function.225,226,227
Pioglitazone and Exendin-4 exert neuroprotective effects by targeting altered brain mitogenesis in T1DM.228
Natural products and herbal compounds
Natural products, rich in bioactive phytochemicals, offer diverse mechanisms to restore MQC. Rosmarinic acid and resveratrol attenuate cardiac dysfunction through SIRT1-mediated PGC-1α deacetylation, while Astragalus-derived astragaloside IV enhances mitogenesis via SIRT1/PGC1α/NRF1 and stabilises mitochondrial dynamics in nephropathy.229,230,231,232,233
Salidroside and astragalin activate AMPK/SIRT3-PGC1α axes, mitigating renal and myocardial fibrosis.234,235,236 Rhein and Gynostemma pentaphyllum combat retinal and cardiac injury
via AMPK/NRF2 signaling, whereas berberine restores podocyte homeostasis by balancing PGC1α-mediated biogenesis and Drp1 inhibition.237,238,239,240,241,242,243 Traditional formulas like Jinlida granules and Si-Miao-Yong-An decoction improve renal and cardiac outcomes through AMPK/PGC-1α activation and PPARα modulation.244, 245
Mitochondrial dynamics are further regulated by compounds such as tanshinone IIa, which activates GLO1 to counteract retinal fission, and paeonol, which enhances OPA1 via CK2α/ JAK2-STAT3 signaling in DCM.181, 246,247,248 Modified Hu-lu-ba-wan and notoginsenoside Fc restore glomerular integrity by modulating PKM2/PGC-1α/OPA1 and HMGCS2-mediated dynamics.249, 250
Natural products also alleviate diabetic complications by targeting mitophagy.251,252,253,254 Notoginsenoside R1 and germacrone enhance PINK1dependent pathways in retinopathy and nephropathy, while Huangqi-Danshen decoction and hyperforin activate STING1/PINK1 or DLAT-AMPK axes to bolster autophagic flux.255,256,257,258,259,260,261 Ginsenoside Ro, a Panax ginsengderived compound, preserves retinal endothelial integrity by restoring EPAC1/AMPK-mediated mitophagy, countering diabetic retinopathy.262 Poricoic acid A, sourced from Poria cocos, mitigates podocyte injury via FUNDC1 activation, a receptordependent mitophagy pathway critical for renal protection.263 Heyingwuzi formulation, a traditional Chinese medicine, alleviates retinal damage by suppressing ROS and apoptosis through HIF-1α/BNIP3/NIX-driven mitophagy, highlighting alternative pathways for vascular rescue.264
Emerging therapeutic strategies
Beyond conventional and natural therapies, exogenous agents like melatonin restore cardiac MQC and myocardial ischemia/ reperfusion injury in T1DM via AMPK/PGC-1αsignaling.265, 266 Photobiomodulation modulates mitochondrial dynamics in the sciatic nerve and in the dorsal root ganglia neurons, alleviating peripheral nervous system in T1DM.267 Immunotherapeutic approach anti-CD3 monoclonal antibodies targets exhausted CD8 + T cells’ energy metabolism and suppresses T cell signaling through regulation of Drp1mediated mitochondrial dynamics in T1DM.268 Finerenone rescues renal mitophagy through PI3K/ Akt/eNOS pathways.269 Placental mesenchymal stem cells rejuvenate
podocyte mitophagy via SIRT1/ PGC-1α/TFAM, highlighting regenerative medicine’s potential.270 In a word, these findings underscore MQC as a nexus for therapeutic innovation in diabetic angiopathy. While repurposed drugs and natural compounds show promise, challenges remain in optimising tissue-specific targeting, resolving mechanistic contradictions, and advancing combinatorial approaches. Translational validation and clinical trials are imperative to harness MQC modulation for multisystem vascular protection.
Conclusions
This review synthesises emerging insights into MQC as a central axis in the pathophysiology of DM and its vascular complications, emphasising the interplay between mitochondrial dysfunction and metabolic dysregulation. Mitochondrial anomalies drive oxidative stress, inflammation, and cellular demise, positioning MQC as a pivotal therapeutic frontier. Despite advances in understanding MQC mechanisms, translational gaps persist, particularly in targeting diabetic complications, which remain understudied compared to primary metabolic defects. Current therapies, including clinical hypoglycemic agents and natural compounds, demonstrate pleiotropic benefits by restoring mitochondrial homeostasis, yet lack specificity for vascular pathologies. Natural products, with their multi-target capacity to modulate MQC pathways, offer promising scaffolds for drug development but require rigorous clinical validation to address challenges such as bioavailability and tissue selectivity. Furthermore, although majority of the therapeutic drugs have proved to be clinical effective, the reported data related to MQC mostly come from experiments with rodent and cell models, while studies conducted in humans (ex vivo and in vivo) are still relatively few and lack higher quality evidence, which provides further direction for our future research. Future research must prioritise combinatorial strategies, precision targeting of MQC nodes, and mechanistic elucidation of organspecific mitochondrial crosstalk. By bridging molecular insights with therapeutic innovation, this review underscores the potential of MQC-centric approaches to redefine the management of diabetic angiopathies, urging interdisciplinary efforts to transform preclinical promise into clinical reality.
References available on request
Prostate Cancer
Recent Patterns and Trends in Global Prostate Cancer Incidence and Mortality: An Update
Written by J. Schafera, Mathieu Laversanneb, Hyuna Sunga, Isabelle Soerjomataramb, Alberto Brigantic,d, William Dahute, Freddie Brayb and Ahmedin Jemala
aSurveillance and Health Equity Science, American Cancer Society, Atlanta, GA, USA
bCancer Surveillance Branch, International Agency for Research on Cancer, Lyon, France
cDivision of Experimental Oncology/Unit of Urology, Urological Research Institute, IRCCS Ospedale San Raffaele, Milan, Italy
dVita-Salute San Raffaele University, Milan, Italy
eOffice of the Chief Scientific Officer, American Cancer Society, Atlanta, GA, USA
Prostate cancer (PC) is the second most commonly diagnosed cancer and the fifth leading cause of cancer death among men worldwide, with more than 1 460 000 estimated cases and 396 000 deaths in 2022.1 It is predicted that by 2040, the PC burden will have increased to approximately 2.4 million cases and 712 000 deaths solely because of the aging and growing global population.2 This increase in mortality translates to approximately 7.5 million life-years lost, with Africa, Asia, and Latin America/Caribbean experiencing the steepest increase in predicted life-years lost.3
Previous studies of trends in international PC incidence and mortality rates4, 5 are based on data up to 2016. There have been recent changes in early detection recommendations in many countries6, 7, as well as improvements in treatment8 and an increase in the number of countries covered by highquality population-based cancer registries.9 Here we examine global patterns and time trends for PC incidence and mortality using incidence data from population-based cancer registries worldwide up to 2021, and national mortality data from vital registration systems up to 2022.
Results
Estimated PC incidence and mortality rates for 2022 across 21 world regions and 185 countries. In 2022, PC incidence and mortality rates varied approximately 13-fold and
Fig. 1. Estimated age-standardised (A) prostate cancer incidence and (B) prostate cancer mortality rates in 2022. ASR = age-standardised rate; IARC = International Agency for Research on Cancer.
Source: GLOBOCAN 2022.
Prostate Cancer
9.5-fold, respectively, across world areas. Northern Europe had the highest estimated incidence rate (82.8 per 100 000 men) and Southern Africa had the highest mortality rate (29.7 per 100 000 men). South-Central Asia had the lowest rates for both incidence (6.4 per 100 000 men) and mortality (3.1 per 100 000 men). At the country level, the highest incidence rates were found in Australia, New Zealand, the USA, Brazil, and several countries in the Caribbean and Northern Europe (Fig. 1). By contrast, mortality rates were highest in many subSaharan African and Caribbean countries, such as Barbados, Jamaica, and Chad, and were lowest across Asia (Fig. 1).
Patterns in recorded incidence and mortality rates for the last 5 yr of available data
Fig. 2, Fig. 3 illustrate PC incidence and mortality rates recorded during the most recent 5-yr period with data available from 52 and 61 populations, respectively. Incidence rates per 100 000 men varied approximately 22-fold across countries, from 7.6 in Thailand to 168.0 in Martinique. Mortality rates per 100 000 men varied approximately ninefold, ranging from 2.9 in Malaysia to more than 24.0 in Cuba and Venezuela. In general, countries in Asia had the lowest incidence and mortality rates, while countries with a higher proportion of men of African ancestry (sub-Saharan Africa and the Caribb ean) and some Eastern European countries (eg, Lithuania and Estonia) had the highest incidence and mortality rates.
Trends in incidence and mortality rates
Figure 4 shows long-term trends for PC incidence and mortality rates, with the observed rates overlaid by linear segments from Joinpoint models for selected countries. By contrast, five countries in Europe and North America (Austria, France, Iceland, Canada, and the USA) experienced increasing or stable incidence rates from the mid-2010s following declines. Trends in 5-yr AAPC significantly increased in 11 of
Fig. 2. Prostate cancer incidence rates in selected countries. a Rates are from 2011–2015. b Rates are from 2012–2016. c Rates are from 2013–2017. d Rates are from 2015–2019. e Rates are from 2017–2021.
Source: Cancer Incidence in Five Continents; Surveillance, Epidemiology and End Results (SEER) program.
the 50 countries examined, with increases ranging from 0.7% per year in Scotland (2013–2017) to 10.0% per year in Poland (2013–2017). By contrast, incidence rates decreased in 15 countries, with decreases ranging from 0.7% per year in New Zealand (2013–2017) to 6.2% per year in Israel (2013–2017). Incidence rates were stable in the remaining 24 countries.
Long-term trends in mortality rates generally decreased or stabilised during the 1990s in many countries following increasing trends
(Fig. 4). However, the pace of the decline slowed or the trend stabilised in 12 of these countries (Australia, Austria, Canada, Colombia, France, Greece, Italy, Israel, Mexico, the Netherlands, Poland, and the USA) since the early 2010s, except in Mexico and the Netherlands, where rates stabilised in 2019. During the most recent 5-yr period, mortality rates decreased in 38 of the 59 countries, with the decrease ranging from 0.6% per year in Hungary (2018–2022) to 7.8%
per year in Estonia (2018–2022). Mortality rates increased in nine countries, with the increase ranging from 0.5% per year in the Philippines (2015–2019) to 7.9% per year in Guatemala (2017–2021), and were stable in the remaining 12 countries.
Rates decreased for both incidence and mortality in 12 countries. By contrast, incidence rates increased while mortality rates remained stable (Austria, Latvia, Poland, and the USA) or decreased (Croatia and Scotland)
4. Trends in prostate cancer incidence and mortality rates for selected countries. Blue represents incidence and red represents mortality. Dots represent observed rates and lines represent fitted values based on joinpoint analyses.
Source: Incidence: Cancer Incidence in Five Continents; Surveillance, Epidemiology, and End Results (SEER) Program. Mortality: World Health Organisation mortality database.
in six countries. Below we describe the trends in incidence and mortality rates by region and country.
Africa
The incidence rate in Uganda was 53.0 per 100 000 men, and rates have been increasing by 2.3% per year since 1993. South Africa had the fourth highest mortality rate among all countries included in the study at 20.3 per 100 000 men, and rates have been increasing by 1.6% per year since 1996.
Asia
Countries throughout Asia had some of the lowest incidence (7.6 to 41.7 per 100 000) and mortality (2.9 to 11.9 per 100 000)
rates during the most recent 5-yr period (Fig. 2, Fig. 3). Incidence rates during the latest 5-yr period continued to increase by approximately 4.0–5.0% per year in China, India, and Thailand, decreased by 2.2–6.2% per year in Turkey and Israel, and remained stable in Bahrain, Philippines, Republic of Korea, and Qatar. However, incidence rates recently stabilised in Japan and Kuwait after increasing up to the early 2010s. By contrast, mortality rates during the most recent 5-yr period increased by 0.5% per year in the Philippines, decreased by 1.5–5.2% per year in Japan, the Republic of Korea, and Armenia, and were stable in the remaining countries.
Central and Eastern Europe
Incidence rates in Central and Eastern Europe ranged from 50.4 to 71.8 per 100 000 men (Fig. 2), while mortality rates ranged from 10.3 to 14.2 per 100 000 (Fig. 3). During the most recent 5-yr period, incidence rates increased in Belarus (4.4% per year) and Poland (10.0% per year) and remained stable in Czechia (Fig. 4.) By contrast, mortality rates decreased by approximately 1.0–3.0% per year in Hungary, Czechia, and Slovakia, increased by approximately 1.0–5.0% per year in Romania, Bulgaria, and Moldova, and were stable in Poland.
Northern Europe
In Northern Europe, incidence rates varied twofold across the 13 countries included in the study, from 63.4 per 100 000 men in Scotland to 128.6 in Lithuania, the third highest rate among all countries (Fig. 2). Similarly, mortality rates varied approximately twofold across the 11 countries, with the highest rate in Latvia at 20.8 per 100 000 men, the third highest rate
among all populations (Fig. 3). Incidence rates during the most recent 5-yr period continued to increase by 4.0% annually in Latvia and by 0.7% per year in Scotland, decreased by approximately 1.0–2.0% per year in five countries (Denmark, Finland, Ireland, Norway, and Sweden), and were stable in the remaining countries. By contrast, mortality rates during the most recent 5-yr period decreased by approximately 1.0–8.0% per year for all 11 Northern European countries except for Latvia, where the rate remained stable.
Southern Europe
Across Southern Europe, incidence rates per 100 000 men ranged from 47.3 in Malta to 79.5 in Slovenia, while mortality rates per 100 000 men ranged from 6.9 in Italy to 15.5 in Slovenia (Fig. 2, Fig. 3). During the most recent 5-yr period, incidence rates increased by 9.2% per year in Croatia, decreased by 3.1% per year in Spain, and remained stable in Cyprus, Italy, Malta, and Slovenia. By contrast, mortality
Fig.
Prostate Cancer
rates decreased by approximately 1.0–2.0% per year in all countries except North Macedonia, where the rate remained stable.
3.3.6. Western Europe
Incidence rates per 100 000 men ranged from 61.4 in Austria to 84.5 in France, while mortality rates per 100 000 men ranged from 8.7 in France to 11.2 in the Netherlands (Fig. 2, Fig. 3). During the most recent 5-yr period, incidence rates decreased by approximately 2.0% per year in Germany and the Netherlands and remained stable in Switzerland. However, incidence rates increased by 3.6% per year in Austria and stabilised in France, a change from the previously declining trends. Mortality rates continued to decrease by approximately 1.0–3.0% per year in Belgium, Germany, Switzerland, and France, and stabilised in Austria and the Netherlands.
Latin America and the Caribbean
Incidence rates per 100 000 men varied more than fourfold across the seven countries located in Latin America and the Caribbean, ranging from 38.2 in Argentina to 168.0 in Martinique (Fig. 2). Mortality rates per 100 000 men ranged from 9.3 in Puerto Rico to 25.8 in Cuba (Fig. 3). The region encompasses four of the top ten populations with the highest mortality rates in the study (Cuba, Venezuela, Uruguay, and Paraguay). During the most recent 5-yr period, incidence rates continued to decrease by approximately 1.0% per year in Argentina, Colombia, and Puerto Rico, and were stable in the four remaining countries. Mortality rates during the most recent 5-yr period decreased by approximately 1.0–4.0% per year in eight of the 14 countries examined, increased by approximately 1.0–8.0% per year in Venezuela, Paraguay, Cuba, and Guatemala, and were stable in Mexico and Nicaragua.
North
America
Incidence rates per 100 000 men were 79.3 in the USA and 64.9 in Canada. Mortality rates were relatively similar between Canada and the USA, at 8.7 and 8.3 per 100 000 men, respectively. Black men in the USA had the second highest incidence rate among all men included in the study at 136.9 per 100 000 men and had a 77% higher incidence rate and a 120% higher mortality rate in comparison to White men (Fig. 2, Fig. 3). During the most recent 5-yr period, the incidence rate increased by 4.2% per year in the USA and stabilised in Canada after rapidly declining in the previous decade. By contrast, the mortality rate during the most recent 5-yr period continued to decline in
Canada, albeit at a slower pace, and stabilised in the USA following two decades of steady decline.
Oceania
Australia and New Zealand had the sixth (97.1 per 100 000 men) and tenth (88.1 per 100 000 men) highest incidence rates, respectively, among all men included in the study. The incidence rate decreased by 3.8% per year in Australia since 2008 and by 0.7% per year in New Zealand since 1995. Similarly, the mortality rate has decreased by 1.0% per year in Australia since 2014 and by 2.7% per year in New Zealand since 1997.
Discussion
The estimated PC incidence rates in 2022 were highest in North America, Australia, New Zealand, Brazil, and in many Northern European countries, probably reflecting higher uptake of prostate-specific antigen (PSA)-based testing in these countries.8, 16 By contrast, mortality rates were highest in many parts of sub-Saharan Africa and Latin America/Caribbean, largely because of the higher proportion of individuals of African ancestry, who are genetically more susceptible to developing PC, and the limited availability of early detection and treatment services.17 During the most recent 5-yr period, incidence rates increased in 11 countries and mortality rates increased in nine, largely confined to countries within Africa, parts of Asia, Latin America/Caribbean, and Europe. In most countries, however, incidence and mortality rates decreased or stabilised.
In general, trends in PC incidence in many high-income countries follow trends in PSA-based screening, with the incidence rate rising when PSA-based screening uptake increases and falling when it decreases.18, 19 For example, the decline in the PC incidence rate since 2008 in Australia coincided with the fall in PSA-based screening prevalence beginning in the same year among men aged 45–84 yr.18 In addition to declines in PSA-based screening uptake, increased use of multiparametric magnetic resonance imaging (mpMRI) to reduce overdiagnosis20 may have contributed to the recent decrease in incidence rate in some countries. For example, in Sweden, where incidence rates are decreasing, the STHLM3-MRI trial found that incorporation of mpMRI in the diagnostic pathway reduced diagnosis of clinically insignificant tumors by 64% and benign findings on biopsy by 74% among men biopsied after a positive MRI test in comparison
to men who underwent transrectal ultrasonography-guided prostate biopsy for elevated PSA.21 Over the past few years, many national or regional organisations, including the European Association of Urology22 and the American Urological Association23, 24, updated their guidelines on early PC diagnosis to recommend mpMRI before prostate biopsy to decrease the number of clinically insignificant PC diagnoses at biopsy.
Incidence rates increased in 11 of the 50 countries examined, including Uganda, the USA, and several countries in Asia and Europe. Such trends may in part reflect increases in incidental detection of tumors via transurethral resection of the prostate (TURP) for lower urinary tract syndrome and benign prostatic hyperplasia25, 26, 27, greater awareness of the disease8, 28, and opportunistic PSA testing.29, 30 A 2022 meta-analysis revealed that the prevalence of incidental PC following TURP was higher in Africa (22.0%) and Asia (9.0%) than in North America (1.0%).31 The rising prevalence of potential risk factors such as obesity, which is associated with advanced-stage disease32, and a Western diet32,33 may have also contributed to the rising incidence of PC. The increase in PC incidence rate since the mid-2010s in the USA, largely driven by advanced-stage disease, was not preceded by a marked increase in PSA-based screening practices.34 Further studies are needed to elucidate reasons underlying this rising trend.
The declining trend for mortality rates in many countries throughout Europe, North America, Oceania, and Latin America/Caribbean are probably the result of improvements in early detection of advanced-stage cancer8, 35 and in treatments.8, 36 Previous modelbased studies estimated that PSA-based screening accounted for 45–70% of the decline in the PC mortality rate in the USA37, and improvements in treatments for 22–33% of the decline by 2005.36 A 2021 study in Lithuania, where a nationwide PSA-based PC screening program in 2006, found that among individuals diagnosed with PC, the PCspecific standardised mortality risk ratio was higher for who did not participate in the national screening program (20.95, 95% CI 20.00–21.94) than for those who did (8.99, 95% CI 8.63–9.37).38
In contrast to the declining or stabilising mortality trends in the majority of the 59 countries included in the study, rates increased in nine countries, including South Africa, the Philippines, Cuba, Moldova,
Bulgaria, and Guatemala. These increasing mortality trends may reflect increasing incidence as well as limited availability of high-quality early detection and treatment services.39 For example, according to WHO, there are fewer than three radiotherapy machines per million population in South Africa and Cuba, compared to more than 10 machines per million population in the USA, Germany, and Sweden.40, 41, 42 Only 28% of PC patients survive for 5 yr in South Africa, compared to more than 90% in high-income countries such as the USA (97.0%)41, France (93.1%)43, and Australia (94.5%)44, where early detection and treatment services are more accessible.
The strengths of this study include the use of high-quality, up-to-date, population-based data to comprehensively examine geographic patterns in PC incidence and mortality rates across five continents.9 The study limitations include subnational coverage in 23 of 50 countries examined for incidence and three of the 59 countries examined for mortality, limiting the degree of national representativeness of the trends, with under-representation of countries in both incidence and mortality data, particularly in Africa and Asia. Furthermore, we could not examine the impact of the COVID-19 pandemic on PC rates and trends owing to the lack of post-COVID-19 data. Future studies should examine the impact of delays in cancer diagnosis and treatment during the COIVD-19 pandemic on PC incidence, survival, and mortality across countries.
Conclusions
Our analysis of high-quality population-based data across the five continents revealed that PC incidence and mortality rates decreased or were stable in the majority of countries examined. However, increasing trends were found in a few countries located in Africa, Asia, Latin America/ Caribbean, and Central and Eastern Europe, possibly because of an increase in detection (incidence) and limited access to and the availability of treatments (mortality only). These findings reinforce the need for improvements in access to early detection and treatment services to mitigate the undue high burden of the disease in these countries. Furthermore, improvements in data availability and quality, particularly in lowand middle- income countries in Asia and Africa, will enhance our understanding of current disparities and prioritisation of early detection and treatment approaches.
References available on request
CPD
CPD
60 Second Summary
This CPD document outlines a national patient-safety initiative to support safer lithium therapy, led by Audrey Purcell in collaboration with multiple healthcare organisations. Lithium remains a key treatment for bipolar disorder, recurrent depressive disorder, and for reducing suicidality. Because it has a narrow therapeutic index and significant toxicity risk, robust baseline assessment and ongoing monitoring are essential. Before initiating therapy, clinicians must complete a full work-up including renal function, thyroid function, calcium level, weight, ECG (when indicated), blood count and pregnancy assessment. Starting doses typically range from 400–800mg nightly in adults, with lower doses advised for older adults, low-weight patients, or those with renal impairment. The document provides detailed guidance on lithium formulations— Priadel and Camcolit tablets, and lithium citrate liquids— highlighting the importance of consistent brands and correct dose conversion when switching between products. Plasma lithium levels should be checked 5–7 days after initiation, dose changes, or interacting medicine changes, with a target range of 0.6–0.8 mmol/L for most patients.
Ongoing safety monitoring includes renal function, thyroid tests, calcium and weight every six months, and lithium levels every three to six months depending on risk factors. The document outlines “alarm bell” interactions—ACE inhibitors, ARBs, NSAIDs, thiazides and sodium fluctuations—which can cause dangerous increases in lithium levels.
Common adverse effects include tremor, gastrointestinal upset, hypothyroidism, hypercalcaemia, polyuria, renal impairment and weight gain. Healthcare professionals should recognise symptoms of toxicity and act urgently.
An updated national lithium patient booklet (“Blue Book”) has been launched, reinforcing the importance of education, consistent monitoring, and patient engagement in therapy.
1. REFLECT - Before reading this module, consider the following: Will this clinical area be relevant to my practice?
3. PLAN - If I have identified a knowledge gap - will this article satisfy those needs - or will more reading be required?
Chief 2 Pharmacist, Saint John of God University Hospital, Stillorgan, Co. Dublin.
2. IDENTIFY - If the answer is no, I may still be interested in the area but the article may not contribute towards my continuing professional development (CPD). If the answer is yes, I should identify any knowledge gaps in the clinical area.
Honorary Senior Clinical Lecturer, Royal College of in Ireland.
4. EVALUATE - Did this article meet my learning needs - and how has my practise changed as a result? Have I identified further learning needs?
5. WHAT NEXT - At this time you may
1. Background and Clinical Information:
Lithium Indications:
Lithium Therapy: a national patient safety and quality improvement initiative
• Bipolar Disorder: mania, hypomania, depression, and prophylaxis of Bipolar Disorder
1. Background and Clinical Information:
• Recurrent Depressive Disorder: used to augment antidepressants
• ECG: if cardiac history, risk factors for QTc prolongation, concomitant medicines that prolong QTc
• Pregnancy test and review of contraception (in women of childbearing age).
Prescribing and dose:
• Thyroid Function Tests (TFTs): include Free T4 and Thyroid Stimulating Hormone (TSH). Patient should euthyroid before initiation
• Full Blood Count
• Weight and height
• Pregnancy test and review of contraception (in women of childbearing age)
• Urea and Electrolytes
Starting dose may usually range from 400mg-800mg OD (nocte) in adults, depending on indication. Elderly patients, those with renal impairment or those below 50kg in weight, often require lower
Switching lithium products: Priadel tablets are recommended for routine use. The 200mg tablets have score- lines therefore they can be divided accurately to provide 100mg dosage requirements. If a patient is unable to swallow tablets a liquid may be prescribed, if essential. Particular care is required with lithium liquid as this can be associated with calculation and administration errors. It is essential that a switch from tablets to liquid is prescribed by the doctor, calculation confirmed by pharmacist, and clear dosage instructions provided to patient by the pharmacist.
Prescribing and dose:
Starting dose may usually range from 400mg-800mg OD (nocte) in adults, depending on indication. Elderly patient those with renal impairment or those below 50kg in weight, often require lower starting dose (eg 200mg), and maintenance doses
30 CPD 122: LITHIUM THERAPY
LITHIUM PRODUCTS
Lithium products:
Product: Formulation: Recommended dosing:
Priadel Tablets:
200mg+400mg
(200mg tablet may be halved to facilitate 100mg dose)
Example: Switching patient from Priadel tablet (Carbonate) 800mg nocte to Priadel liquid nocte.
Equivalent to Lithium Carbonate 200mg/5ml
(Use Carbonate 200mg=5ml for calculations)
Lithium level sampling time:
Release)
Release) 12 hours post dose (acceptable 10-14 hours)
Once daily: at night
Twice daily: morning and night
(Immediate Release)
Once daily: at night
Twice daily: morning and night
(Immediate Release)
Once daily: 12 hours post dose
Twice daily: 12 hours post dose and sample to be taken pre-morning dose (acceptable 10-14 hours)
Once daily: 12 hours post dose
Twice daily: 12 hours post dose and sample to be taken pre-morning dose (acceptable 10-14 hours)
• Last serum lithium level >0.8mmol/L
• Interacting medicines. Key interacting medicines include ACEI, ARBs, NSAIDs and Thiazide diuretics. (See summary below and BNF/ Stockley’s for exhaustive list).
Discontinuation:
If a decision is made to discontinue lithium, the risk of relapse may be reduced by reducing the dose gradually. It is recommended to reduce the dose slowly over at least 4 weeks or longer, and preferably up to 3 months in Bipolar Disorder; except in medical emergency or overdose.
2. ALARM bell medication interactions: Think “ANTS” (TABLE OPPOSITE)
3. Adverse effects: (not an exhaustive list)
Cardiac: Lithium may cause cardiac arrhythmia, including bradycardia, sinoatrial dysfunction (SA block), abnormal T waves on ECG (T-wave inversion), and STsegment depression.
Dermatological: Lithium may cause acne vulgaris and/or psoriasis (including exacerbation of both) in patients with and without either condition at baseline.
Target levels:
Priadel tablets are recommended for routine use. The 200mg tablets have score- lines therefore they can be divided accurately to provide 100mg dosage requirements. If a patient is unable to swallow tablets a liquid may be prescribed, if essential. Particular care is required with lithium liquid as this can be associated with calculation and administration errors. It is essential that a switch from tablets to liquid is prescribed by the doctor, calculation confirmed by pharmacist, and clear dosage instructions provided to patient by the pharmacist.
Calculate Priadel liquid dose in Carbonate: 200mg=5ml; 800mg=20ml.
or every 3 months in at-risk patients.
At risk patients include:
• Elderly (> 65 years)
Example: Switching patient from Priadel tablet (Carbonate) 800mg nocte to Priadel liquid nocte.
Equivalent dose in Priadel liquid is 800mg Carbonate=20ml nocte.
Calculate Priadel liquid dose in Carbonate: 200mg=5ml; 800mg=20ml.
Equivalent dose in Priadel liquid is 800mg Carbonate=20ml nocte.
Lithium level to be checked 5-7 days after switch to liquid formulation.
The minimum effective plasma level for prophylaxis in adults is 0.4mmol/L; optimal range is 0.60.8 mmol/L. A level of 0.4mmol/L may be effective in unipolar depression; 0.6 -1 mmol/L in Bipolar Disorder, and levels at the higher end of the range in mania (0.8-1mmol/L).
Lithium level to be checked 5-7 days after switch to liquid formulation.
Monitoring frequency:
Plasma levels:
Lithium plasma level should be checked 5-7 days after starting, after every dose change, and after addition/discontinuation of medication that can affect level.
• Have received less than 12 months treatment
• Renal impairment (eGFR<60ml/ min)
• Impaired Thyroid function at last test
TFTs, renal function, Calcium level, and weight check, recommended every 6 months; or every 3 months in at-risk patients.
Once stable, serum lithium levels recommended every 3 months for the first year, then every 6 months;
• Raised Calcium level (adjusted) at last test
• Poor symptom control or suspected poor adherence
• Significant change in patient’s sodium or fluid intake
GI: Lithium may cause dyspepsia, diarrhoea, nausea, vomiting, dysgeusia (metallic or salty taste), gastritis and abdominal pain. Some effects (e.g. nausea) may occur early in treatment. Other effects may take longer to develop. Supratherapeutic lithium levels should be suspected with severe nausea, vomiting and diarrhoea. Hypothyroidism: Lithium has varied effects on Thyroid Hormone production and regulation, including inhibition of Iodine uptake in the Thyroid, inhibition of Thyroid Hormone synthesis and release, and hepatic conversion of free Thyroxine. Patient may present with typical hypothyroidism symptoms including lethargy, impaired cognition, weight gain, dry skin, and cold intolerance. Risk factors include females, older adults, family history of hypothyroidism, and presence of anti-thyroid antibodies.
Hyperparathyroidism and hypercalcaemia: Hypercalcaemia has been reported with Lithium
ALARM bell medication interactions: Think “ANTS”
2. ALARM bell medication interactions: Think “ANTS”
From 10% to more than fourfold increase in lithium level
• Develops over several weeks.
• Most likely to cause lithium toxicity within a month of starting
• Variable: a few days to several months
• Sevenfold increased risk of hospitalisation for lithium toxicity in the elderly
• Consider alternative anti-hypertensive.
• If combination necessary, increased monitoring of lithium level and renal function required.
• Examples of increased lithium levels with oral NSAIDs include (but are not limited to): up to 23% increase in lithium level with Diclofenac; up to 25% increase in lithium level with Ibuprofen.
• Patients should be advised to avoid oral OTC NSAIDs (examples include Ibuprofen) and use Paracetamol if OTC analgesic required
Pharmacist should contact doctor if oral NSAID prescribed with lithium.
T: Thiazide Diuretics
Unpredictable
Up to four-fold increase in lithium level
• Thiazides reduce renal clearance of lithium and levels can rise within a few days.
• Usually apparent in the first 10 days
• Any effect will be apparent in the first month.
S: Sodium Excess Sodium can reduce lithium levels
Sodium restriction can lead to lithium toxicity.
therapy, which may or may not be related to drug-induced hyperparathyroidism. While lithium has been observed to affect Parathyroid Hormone levels after a single dose, long -term exposure is required to observe clinically relevant alterations in Calcium homeostasis.
Polydipsia and polyuria: Common adverse effects associated with lithium. Patients may notice
• Thiazide diuretics should only be used where unavoidable and with strict monitoring.
• Loop diuretics may be safer Furosemide is the safest diuretic to use with lithium, but frequent monitoring required.
• Variable • Consider high Sodium content OTC preparations and recommend suitable alternatives.
• Care with Sodium content in effervescent formulations.
• Refer to SPS Pharmacy NHS “Considering Sodium content of medicines;” particular care with products > 17mmol Sodium in maximum daily dose.
High-alert examples: (not an exhaustive list)
1) Sodium Bicarbonate in antacids (eg Gaviscon): recommend Maalox instead.
2) High Sodium content in urinary alkalinising agents (eg Cymalon): avoid
3) High Sodium content in soluble/effervescent products: recommend film-coated tablets.
increased urinary frequency (> 3 L in 24 hours) due to poor urine concentration; and increased thirst, which is independent of dry mouth effects of lithium.
Renal effects: Up to one-third of patients may develop some degree of decreased kidney function during the course of lithium therapy, with approximately 5% developing significant kidney impairment/failure.
Sexual dysfunction: Studies report rates of the various effects of 5-40 %. Effects can include decreased libido, impaired sexual arousal, and erectile dysfunction. Sexual dysfunction can negatively impact a patient’s quality of life.
Tremor: Lithium can cause tremor in up to 25% of patients, making it one of the most common adverse effects. This is commonly a bilateral, symmetrical
hand tremor, which may spontaneously decrease over time as compensatory mechanisms develop within the patient. Course tremor and muscle twitching may be observed in lithium toxicity.
Tremor commonly begins early in treatment, but can develop later in treatment, with or without a dose increase. Risk factors include higher doses/serum levels, medicines that can increase lithium level, medicines known to induce tremor (e.g. antipsychotic, antidepressants), caffeine and older adults.
Weight gain: Increases of 4 to 7 Kg within the first year have been reported in the literature. Effects on central mechanisms related to weight gain, satiety and metabolism are possible. Increased consumption of high-calorie, sugary beverages from increased thirst with lithium could contribute.
Symptoms of toxicity:
Healthcare professionals should be aware of signs and symptoms of lithium toxicity.
For patients with symptoms of toxicity (eg diarrhoea, vomiting, coarse tremor, mental state changes or falls):
Withhold lithium, refer to GP/ Clinic/Hospital for urgent lithium level and U+Es, and seek specialist advice. Referral to secondary care may be required depending on the severity of symptoms.
4. Lithium Therapy: a blue book for safety
• The national patient information booklet has been produced and updated by Audrey Purcell, Chief 2 Pharmacist and the Saint John of God University Hospital Drug and Therapeutics Committee.
• This initiative is intended to provide and promote safer lithium therapy and empower patients to engage with their healthcare professional to discuss all aspects of lithium therapy, monitoring, and side-effects.
• Appropriate information and monitoring are imperative to ensure best outcomes for patients on lithium therapy and reduce likelihood of harm.
• Appropriate information and monitoring are imperative to ensure best therapy and reduce likelihood of harm.
• The HSE National Medication Safety Programme, the Irish Pharmacy Union Network and the College of Psychiatrists of Ireland have collaborated updated booklet in October 2025.
• The HSE National Medication Safety Programme, the Irish Pharmacy Union, the Irish Medication Safety Network and the College of Psychiatrists of Ireland have collaborated to implement a national launch of the updated booklet in October 2025.
If you have too much lithium in your blood, this is called lithium toxicity (or lithium poisoning). This can make you very ill.
Read the following list very carefully. If you get one or more of these problems at any time, talk to your doctor straight away.
• Updates include changes to recommendations for Lithium liquid which daily.
• Updates include changes to recommendations for Lithium liquid which can be prescribed once daily or twice daily.
• Severe hand shake (tremor).
• Vomiting or severe nausea and persistent diarrhoea.
• Muscle weakness.
• An electronic version of the booklet is available online using the hyperlink the QR code below: https://assets.hse.ie/media/documents/Lithium_Therapy_Patient_Information_Booklet.pdf
• An electronic version of the booklet is available online using the hyperlink below or can be accessed using the QR code below: https://assets.hse.ie/ media/documents/Lithium_ Therapy_Patient_Information_ Booklet.pdf.
• Being unsteady on your feet.
• Muscle twitches.
• Slurring of words so that it is difficult for others to understand what you are saying.
• Blurred vision.
• Confusion.
• Feeling unusually sleepy
A small number of people may not have any immediate symptoms of toxicity when the lithium in their blood is too high. This is why it is important to have regular checks. Regular checks can prevent long-term problems.
• This launch aligns with the updated Lithium Guideline produced by the “Best Practice Guideline for Prescribing and Monitoring Lithium Therapy” content/uploads/2025/07/Lithium-v3.pdf
• This launch aligns with the updated Lithium Guideline produced by the Irish Medication Safety Network “Best Practice Guideline for Prescribing and Monitoring Lithium Therapy” available at https://imsn.ie/wp-content/ uploads/2025/07/Lithium-v3.pdf
• Booklets can be ordered from https://surveys.hse.ie/s/EAFX45/ or hospital.pharmacy@sjog.ie
• Booklets can be ordered from https://surveys.hse.ie/s/EAFX45/ or hospital.pharmacy@sjog.ie
Health care professionals provide essential support and are recommended to:
Health care professionals provide essential support and are recommended
• Ensure patients have a lithium booklet
• Reinforce essential information verbally
• Ensure patients have a lithium boo klet
• Refer patient to the booklet to be aware of potential sideeffects and signs of toxicity
• Ensure the patient understands their own programme of monitoring
• Support patients to engage in appropriate blood test monitoring: keep their record book up to date, and have available at consultations with GP, Consultant, Pharmacist, Nurse.
• Reinforce essential information verbally
• Refer patient to the booklet to be aware of potential side-effects
Acknowledgements:
• Ensure the patient understands their own programme of monitoring
Many thanks to the following for article peer-review:
Gilsenan, Senior Pharmacist, St
Margaret Brookes, Senior Pharmacist, St Patrick’s University Hospital
• Support patients to engage in appropriate blood test monitoring: have available at consultations with GP, Consultant, Pharmacist, Nurse.
Call for papers: make your contribution to Hospital Professional News
Articles
Research Papers
Reviews
Programme Descriptions
Reports
Case Reports
Letters to Editor
Support fellow hospital professionals as well as aspiring junior professionals and early-year hospital pharmacists
Practice reports share innovations on any area of practice, including delivering clinical services, pharmacy administration, or new approaches to inform and engage with patients
Perspective articles focus on a specific field or discipline and discuss current advances or future directions, and may include original data as well as expert insight and opinions
Oncology Frontiers
Exhaled Breath Condensate: A New Frontier in Early Lung Cancer Detection and Molecular Profiling
Written by Dr Síle Toland, Specialist Registrar in Respiratory Medicine and an MD candidate at the Royal College of Surgeons in Ireland and Professor Bryan Hennessy, Consultant Medical Oncologist, Beaumont Hospital
Lung cancer remains the leading cause of cancer-related mortality worldwide, accounting for approximately 1.8 million deaths each year. Overall, prognosis remains poor because most patients are still diagnosed at a late stage. Non–small cell lung cancer (NSCLC) represents about 85% of all cases, yet only 20–30% of lung cancers are detected at an early, potentially curable stage. Encouragingly, within screening programmes this early-stage detection rate can rise to nearly 80%, underscoring the transformative potential of timely diagnosis.
Low-dose CT screening is shifting stage at presentation, but it also introduces new challenges for clinical decision-making. In this setting, liquid biopsy is emerging as a valuable adjunct to imaging by detecting early molecular changes that may signal malignancy. While plasma-based analysis of circulating tumour DNA has revolutionised the management of advanced NSCLC, its performance in early-stage disease is limited by low tumour DNA shedding.
Exhaled breath condensate (EBC) has emerged as a promising, noninvasive source of airway-derived nucleic acids, offering the potential to complement existing diagnostic pathways and enhance early detection strategies.
The Clinical Limits of Plasma cfDNA
Plasma cfDNA analysis is now well established in advanced NSCLC. It can be used to identify actionable driver mutations such as EGFR, ALK, ROS1, KRAS and BRAF, to characterise resistance mutations, and to track molecular response during systemic therapy.
However, plasma testing is inherently limited in early-stage disease. When a tumour is small, the fraction of tumour-derived DNA circulating in the bloodstream is often low. Even with highly sensitive sequencing, the signal may be obscured by background cfDNA originating from blood cells and endothelial turnover. Additional interference can arise from clonal haematopoiesis, where age-related mutations in circulating blood cells appear in cfDNA but do not represent true tumour alterations.
As a result, plasma liquid biopsy, though transformative, cannot yet provide consistently reliable molecular detection in patients with low tumour burden.
EBC: A Lung-Specific Liquid Biopsy
Unlike plasma, EBC is organspecific, directly sampling the compartment where lung cancers arise. It is also acellular and entirely non-invasive, requiring only relaxed breathing over 5 to 10 minutes, making it ideal for
repeated monitoring and patientfriendly clinical workflows.
Early studies demonstrated that key mutations associated with NSCLC, such as EGFR and KRAS, can be detected in EBC samples. In early studies, mutations detected in EBC prior to surgery were no longer detectable in some patients following tumour resection, suggesting potential utility for diagnosis and postoperative monitoring. More sensitive molecular techniques now allow detection of actionable mutations, including resistance alterations such as EGFR T790M, directly from EBC. Some findings suggest that EBC may even outperform plasma in certain scenarios, particularly where the tumour is small, localised, or heterogeneous.
Although still investigational, the consistency of these findings highlights EBC as a promising lung-specific form of liquid biopsy. Its organ specificity and lack of background interference from blood-derived DNA make it a particularly attractive complement to plasma testing, especially in early-stage disease or when tissue sampling is limited
How EBC Could Transform Early Detection and Monitoring
The clinical potential of EBC lies not in replacing plasma testing, but in complementing it. Because it samples tumour-proximal airways, EBC could:
1. Enhance molecular detection in early-stage disease
Small tumours may shed detectable DNA into the airway long before it reaches measurable levels in circulation. EBC could therefore improve upon plasma’s sensitivity gap in stage I or screendetected cancers.
2. Assist in evaluating indeterminate pulmonary nodules
Clinicians frequently encounter nodules of uncertain significance on CT. An EBC-based biomarker panel could help determine which patients require urgent evaluation versus interval imaging, potentially reducing unnecessary procedures.
3. Provide a safe option when tissue biopsy is contraindicated
Many patients, including those with frailty, other respiratory conditions such as pulmonary fibrosis or advanced COPD cannot undergo bronchoscopy or CT-guided biopsy. EBC offers a non-invasive alternative that may still yield meaningful molecular insights.
4. Enable real-time, longitudinal surveillance
Because EBC sampling is simple and repeatable, it lends itself to serial monitoring during systemic therapy, after curative surgery or radiotherapy, and throughout survivorship care. Such an approach could ultimately facilitate earlier detection of recurrence or emergence of resistance.
5. Support multi-omic assessment
EBC also contains proteomic, metabolomic, epigenetic and RNA markers. Combined analyses could generate richer, more nuanced information than single-modality plasma testing.
Technological Advances Expanding the Possibilities
Liquid biopsy advances are reshaping minimally invasive diagnostics.
Fragmentomics
Fragmentomic profiling, which is the analysis of cfDNA fragment lengths, cleavage patterns, and nucleosomal footprints can distinguish tumour-derived DNA from normal background with remarkable sensitivity. Because EBC contains airway-origin cfDNA, fragmentomics may provide a lung-specific signal, supporting early detection.
Ultra-sensitive sequencing
Advances in ultra-deep sequencing now allow detection of variant alleles at extremely low frequencies. This is important for EBC, where nucleic acid concentrations can be low. These platforms reduce false positives and expand the range of detectable alterations, including sub-clonal or heterogeneous mutations that may be missed in plasma.
Dr Sile Toland
Professor Bryan Hennessy
Start with OMJJARA
momelotinib
See patients with myelofibrosis and anaemia in a
Omjjara is indicated for the treatment of disease-related splenomegaly or symptoms in adult patients with moderate to severe anaemia who have primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis and who are Janus Kinase (JAK) inhibitor naïve or have been treated with ruxolitinib. 1
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
ACVR1 = activin A receptor, type 1; JAKi = Janus Kinase inhibitor.
Learn more about Omjjara:
Prescribing Information and Important Safety Information Omjjara Summary of product characteristics, available at www.medicines.ie. Accessed June 2025
Abbreviated Prescribing Information
Omjjara 100, 150 and 200 mg film-coated tablets Abbreviated Prescribing Information (Refer to Summary of Product Characteristics (SmPC) before prescribing). PRESENTATONS: Each 100 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 100 mg of momelotinib and 50.8 mg of lactose monohydrate. Each 150 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 150 mg of momelotinib and 76.1 mg of lactose monohydrate. Each 200 mg tablet contains momelotinib dihydrochloride monohydrate equivalent to 200 mg of momelotinib and 101.5 mg of lactose monohydrate. INDICATION: The treatment of disease-related splenomegaly or symptoms in adult patients with moderate to severe anaemia who have primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis and who are Janus Kinase (JAK) inhibitor naïve or have been treated with ruxolitinib. POSOLOGY AND ADMINISTRATION: Treatment should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. Omjjara should not be used in combination with other JAK inhibitors. The recommended dose is 200 mg once daily. Complete blood cell count and liver function tests must be performed before initiating treatment, periodically during treatment, and as clinically indicated. For dose modifications due to adverse reactions see SmPC. Treatment with Omjjara should be discontinued in patients unable to tolerate 100 mg once daily. Duration of use: Treatment may be continued for as long as the benefit-risk remains positive for patients, as assessed by the treating physician. Missed dose: If a dose of Omjjara is missed, the next scheduled dose should be taken the following day. Two doses should not be taken at the same time to make up for the missed dose. Elderly (≥ 65 years): No dose adjustment necessary. Renal impairment (>15 mL/min): No dose adjustment necessary. Omjjara has not been studied in patients with end-stage renal disease. Hepatic impairment: No dose adjustment is recommended for patients with mild or moderate hepatic impairment. The recommended starting dose is 150 mg once daily in patients with severe hepatic impairment (Child-Pugh Class C). Paediatric population: No data available. Omjjara is for oral use only and can be taken with or without meals. CONTRAINDICATIONS: Hypersensitivity to momelotinib or to any of the excipients. Pregnancy and breast-feeding. WARNINGS/PRECAUTIONS: Omjjara should not be initiated in patients with active infections. Physicians should carefully observe patients for signs and symptoms of infection and initiate appropriate treatment promptly. Patients with chronic HBV infection who receive Omjjara should have their chronic HBV infection treated and monitored according to clinical HBV guidelines. A complete blood count including platelet count should be obtained before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated. Dose interruption or reduction may be required. Liver function tests should be obtained before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated. If increases in ALT, AST or bilirubin related to treatment are suspected, dose interruption or reduction may be required. Prior to initiating or continuing therapy with Omjjara, the benefits and risks for the individual patient should be considered particularly in patients 65 years of age and older, patients who are current or past long-time smokers, and patients with history of atherosclerotic cardiovascular disease or other cardiovascular risk factors. Patients with symptoms of thrombosis should be promptly evaluated and treated appropriately. Lymphoma and other malignancies have been reported in patients receiving JAK inhibitors, including Omjjara. However, a causal association has not been established Women using systemically acting hormonal contraceptives should add a barrier method during treatment and for at least 1 week after the last dose of Omjjara. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take Omjjara. Patients who experience dizziness or blurred vision after taking Omjjara should observe caution when driving or using machines INTERACTIONS: Co administration of strong CYP3A4 inducers may lead to decreased momelotinib exposure and consequently a risk for reduced efficacy. Therefore, additional monitoring of the clinical signs and symptoms of myelofibrosis is recommended with concomitant use of momelotinib and strong CYP3A4 inducers (including but not limited to carbamazepine, phenobarbital, phenytoin, and St John’s wort [Hypericum perforatum]). Caution and monitoring for adverse reactions are advised with concomitant use of OATP1B1/1B3 inhibitors, including ciclosporin. Momelotinib may increase exposure to other sensitive BCRP substrates, including sulfasalazine; monitor for adverse reactions. Caution is advised when administering momelotinib with P-gp substrates with a narrow therapeutic index. Caution is advised when administering momelotinib with sensitive substrates of OCT1, MATE1 and MATE2-K (e.g., metformin). Narrow therapeutic index or sensitive substrate medicinal products of CYP1A2 (e.g., theophylline, tizanidine) or CYP2B6 (e.g., cyclophosphamide) should be co-administered with momelotinib with caution. Fertility, pregnancy and lactation: Fertility: No clinical data. Omjjara is contraindicated during pregnancy. If Omjjara is used during pregnancy, or if the patient becomes pregnant while taking this medicinal product, the patient should discontinue treatment and be advised of the potential hazard to the foetus. Omjjara is contraindicated during breast-feeding. UNDESIRABLE EFFECTS: Very common (≥ 1/10): Thrombocytopenia, dizziness, headache, cough, diarrhoea, abdominal pain, asthenia, fatigue. Common (≥ 1/100, < 1/10): Urinary tract infection, upper respiratory tract infection, pneumonia, nasopharyngitis, COVID19, cystitis, bronchitis, oral herpes, sinusitis, herpes zoster, cellulitis, respiratory tract infection, sepsis, lower respiratory tract infection, oral candidiasis, rash, skin infection, gastroenteritis, neutropenia, Vit B1 deficiency, syncope, peripheral neuropathy, paraesthesia, blurred vision, vertigo, hypotension, haematoma, flushing, vomiting, constipation, arthralgia, pain in extremity, pyrexia, ALT increased, AST increased, contusion. For more details on undesirable effects, see SmPC. Marketing Authorisation (MA) Holder: GlaxoSmithKline Trading Services Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland. MA Nrs: EU/1/23/1782/001- 003. Legal category: POM A. Date of preparation of API: April 2025. Code: PI-14515. Further information available on request from GlaxoSmithKline, 12 Riverwalk, Citywest Business Campus, Dublin Tel: 01-4955000.
Adverse events should be reported directly to the Health Products Regulatory Authority (HPRA) on their website: www.hpra.ie Adverse events should also be reported to GlaxoSmithKline on 1800 244 255.
Trademarks are owned by or licensed to the GSK group of companies. @2025 GSK or licensor. PM-IE-MML-JRNA-250001 | July 2025
References: 1. Omjjara Summary of product characteristics, available at www.medicines.ie. Accessed August 2025
Oncology Frontiers
cfRNA detection and fusion analysis
Emerging methodologies have demonstrated that EBC contains detectable RNA species. This opens new possibilities for molecular subtyping and therapy selection without relying on tissue biopsies, a potential advantage for patients who are high-risk, medically frail, or have inaccessible tumours.
Metabolomics and volatile organic compounds
Metabolomic and volatile organic compound (VOC) signatures in breath are recognised as markers of altered tumour biology. These molecular signatures could help differentiate benign from malignant lesions, complementing DNAbased approaches and enriching multi-analyte diagnostic models.
Machine-learning integration
Machine-learning models capable of combining EBC molecular data with imaging and clinical variables may significantly enhance diagnostic accuracy. These models may enhance refine screening and guide surveillance.
Why these developments matter
As assays become more sensitive and computational models more sophisticated, EBC could evolve into a powerful tool for early detection, real-time monitoring, and precision-guided decisionmaking across the lung cancer care continuum.
Challenges to Overcome Before Clinical Implementation
Despite its promise, EBC is not yet ready for routine clinical use. Several challenges
must be addressed before clinical integration.
1. Standardisation of collection and processing
Variability in collection devices, condensation temperatures, tubing materials, and storage conditions can significantly affect sample yield and biomarker stability. Without harmonised protocols, results will remain difficult to compare across studies or institutions. Standardisation is therefore a critical first step toward clinical reliability.
2. Improving nucleic-acid yield
EBC is an inherently dilute biofluid, and cfDNA yield can vary widely. Continued optimisation of extraction protocols, concentration methods, and ultra-sensitive downstream assays is essential to ensure consistent and clinically meaningful detection.
3. Minimising contamination
Oral, salivary, or environmental contamination can compromise the molecular signal. While modern EBC devices have improved physical separation of breath and saliva, meticulous collection technique remain crucial to safeguarding sample integrity.
4. Clinical validation in realworld populations
EBC must be evaluated in large, prospective studies comparing it with plasma, tissue, imaging, and clinical outcomes. These trials are needed to definie sensitivity, specificity, reproducibility, and the added value of EBC to existing pathways such as low-dose CT screening or post-treatment surveillance.
4. Tool for treatment monitoring
Serial EBC sampling may allow early identification of molecular changes in response to therapy, including emerging resistance, without the need for invasive procedures.
5. Surveillance following curative-intent treatment
Because EBC collection is simple and well tolerated, it is well suited to long-term surveillance after surgery or radiotherapy, enabling timely detection of recurrence while minimising patient burden.
Conclusion
5. Economic and regulatory considerations
For EBC-based tests to be adopted, they must be affordable, scalable, and compatible with routine clinical workflows. Device-assay co-development, regulatory approval pathways, and cost-effectiveness analyses will all play central roles in determining feasibility and healthsystem readiness.
The Future – Integrating EBC Into Lung Cancer Pathways
EBC could integrate at several points across the lung cancer care pathway.
1. Adjunct to low-dose CT screening
By adding molecular context to imaging, EBC may improve screening specificity, refine risk stratification, and reduce unnecessary follow-up scans or procedures for indeterminate nodules.
2. Diagnostic support when tissue or plasma is inadequate
In patients for whom biopsy is unsafe or unfeasible or when plasma ctDNA levels are too low, EBC may offer a non-invasive alternative that still captures clinically relevant tumour-derived signals.
3. Complement to plasma-based liquid biopsy
Combining lung-proximal DNA from EBC with systemic DNA from plasma could provide a more comprehensive molecular overview, particularly valuable in early-stage disease where plasma shedding is minimal.
As precision oncology continues to evolve, the concept of a lung-derived, non-invasive liquid biopsy is becoming increasingly compelling. Exhaled breath condensate offers a direct window into the airway microenvironment, with the potential to detect tumour-related signals earlier and more sensitively than plasma alone. While significant work remains particularly in standardisation and large-scale validation, the progress to date suggests that EBC could one day complement existing diagnostic pathways and expand the reach of personalised cancer care. If successfully translated into clinical practice, EBC may bring us closer to a future where early detection is more achievable, where monitoring is more precise, and where lung cancer care becomes increasingly guided by simple, patient-friendly molecular tools.
References available on request
The Authors
Dr Toland’s research focuses on liquid biopsy and the development of non-invasive molecular diagnostics for early-stage lung cancer.
Professor Hennessy is a prominent oncologist and clinical researcher, with a joint academic appointment as a Professor at the Royal College of Surgeons in Ireland (RCSI). His research focuses on oncology translational research, especially kinase signalling, PI3K/AKT pathway biology, and clinical studies in breast, gynaecologic, colorectal and lung cancers, using technologies such as genomics and reverse phase protein arrays to identify predictive and prognostic markers. He has an extensive publication record with hundreds of peer-reviewed articles and an h-index reflecting wide citation impact, and has been coinvestigator on major grants including US NIH SPORE awards. In Ireland he also served as Clinical Lead of Cancer Trials Ireland, contributing significantly to the national clinical trials infrastructure and cancer research enterprise.
Mature B Cell Neoplasia
Unraveling the Epigenetic Landscape of Mature B Cell Neoplasia: Mechanisms, Biomarkers, and Therapeutic Opportunities
Written by Nawar Maher1,2,Francesca Maiellaro2,Joseph Ghanej2,Silvia Rasi2,Riccardo Moia2 and Gianluca Gaidano2,*
1Division of Hematology, Department of Translational Medicine, Università del Piemonte Orientale and Azienda Ospedaliero-Universitaria di Alessandria, 15121 Alessandria, Italy
2Division of Hematology, Department of Translational Medicine, Università del Piemonte Orientale and Azienda Ospedaliero-Universitaria Maggiore della Carità, 28100 Novara, Italy
Mature B-cell neoplasms are a diverse group of lymphoid cancers arising from antigen-experienced B cells, typically at or beyond the germinal center stage.1,2 They include malignancies derived from germinal center B cells (e.g., follicular lymphoma, FL and a fraction of diffuse large B cell lymphomas, DLBCLs), and postGC B cells (e.g., certain DLBCLs), and memory B cells (e.g., chronic lymphocytic leukemia, CLL).1,2 Although these tumors often retain key immunophenotypic traits of their normal counterparts, they exhibit profound dysregulation of gene expression due to disruptions in the regulatory networks that control cellular identity and function.3 This dysregulation is driven by both genetic and epigenetic alterations and leads to aberrant expression patterns, loss of normal differentiation, and malignant features such as uncontrolled growth and immune evasion.4 For instance, the activated B cell-like (ABC) subtype of DLBCL is marked by constitutive activation of the NF-κB pathway, upon which these tumors depend for survival.5 Similarly, certain DLBCL subsets rely on sustained BCL6 expression to repress its target genes, highlighting BCL6 as a central transcriptional driver. However, beyond transcription factors (TFs) like NF-κB and BCL6, gene expression is governed by the epigenetic landscape, that includes chromatin architecture shaped by DNA methylation, histone modifications, nucleosome remodeling and related mechanisms.6
Epigenetics refers to heritable changes in gene function that occur without alterations to the DNA sequence.7 These chromatinbased regulatory layers encode DNA sequence independent information that is essential
for lineage fidelity, cell fate transitions, and the dynamic gene expression programs required for normal B cell development.4 From naïve B cells to germinal center (GC) cells, and ultimately memory or plasma cells, each stage of B cell maturation demands precise coordination of transcriptional networks, signaling pathways and epigenetic machinery. The inherent plasticity of these processes contributes to the molecular heterogeneity seen in B cell malignancies. Notably, large-scale genomic analyses of follicular lymphoma (FL), DLBCL, and chronic lymphocytic leukemia (CLL), among others, consistently reveal a high mutational burden affecting chromatin modifiers, DNA methylation regulators, and TFs that disrupt normal epigenetic control.8
Importantly, the recognition of epigenetic dysregulation as a key oncogenic driver has translated into therapeutic advances. Agents that target the epigenome, such as the DNA methylation inhibitors azacitidine and decitabine, have demonstrated significant clinical efficacy in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).9 These successes provide a proof-of-principle that pharmacologic modulation of epigenetic alterations can produce meaningful therapeutic benefits and offer promising avenues for the treatment of mature B cell neoplasms. As lymphomas often evolve from or hijack normal epigenetic programs, understanding how these regulatory mechanisms are subverted in a subtypespecific manner is essential. While several reviews have examined epigenetic mechanisms in B-cell malignancies, many are either disease-specific or focus on isolated pathways. Our
review presents an integrated analysis across the three most prevalent mature B-cell lymphoid neoplasms, namely DLBCL, FL, and CLL selected for their shared lymphoid origin, overlapping epigenetic features, and the availability of comparable epigenomic data. We explore how aberrant DNA methylation, mutations in chromatin modifiers, and altered histone landscapes contribute to disease pathogenesis, and discuss how these alterations represent potential prognostic biomarkers that are guiding the development of novel epigenetic therapies. Furthermore, by integrating all epigenetic mechanisms with the most recent therapeutic strategies, our review may provide a framework for both research and clinical applications.
2. Epigenetic Mechanisms in B Cell Maturation
B cell maturation is a complex multi-step process strictly regulated by the interplay between different stage-specific TFs and epigenetic complexes, which define specific gene expression programs.10,11 Epigenetic modifications play a crucial role in B cell maturation by using chemical modifications translated into instructions which can regulate gene expression and influence cell growth, apoptosis, development, differentiation, and immune response. The four main epigenetic modifications include DNA methylation, histone modifications, chromatin remodeling, and noncoding RNAs (Figure 1 on page 38).12,13
Epigenetic Landscape in B Cell Neoplasia
3.1. Diffuse Large B Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in the adult
population, arising from mature B cells, and it is characterized by a heterogeneous genetic and epigenetic landscape which influences morphology, immunophenotype, and therapy response. Two principal subtypes can be distinguished based on the cell of origin: the GC B cell-like (GCB) subtype, which is generally associated with a more favorable prognosis, and the activated B cell-like (ABC) subtype, which is typically linked to inferior clinical outcomes.1,2,44
The pathogenesis of DLBCL is characterised by the accumulation of genetic aberrations causing an altered structure and expression of important proto-oncogenes and tumor suppressor genes. Genetic alterations include somatically acquired point mutations, gene copy number variation and translocations. Translocations lead to the placement of a regulatory element (promoter or enhancer), often involving IG loci, in proximity to the coding sequence of an oncogene, resulting in its constitutive expression.45 Among the main aberrations, MYC, BCL2, and BCL6 translocations are assessed by FISH analysis to define the double- or triple- hit status in co-presence of MYC and BCL2 or/and BCL6 alterations.46
3.1.1. DNA Methylation in DLBCL
Aberrant methylation disrupts the normal regulation of critical genes involved in cell cycle control, apoptosis, and differentiation. Among the key genes affected in DLBCL are MYC, SLIT2, KLF4, and CDKN2A, whose inactivation facilitates uncontrolled proliferation and tumor progression. DLBCL shows methylation heterogenicity which defines different molecular subgroups and clinical outcomes. In particular, six molecular subtypes have been identified based on
Mature B Cell Neoplasia
methylation variability, with high grade methylation variability being associated with shorter survival compared to lower grade variability.47,48,49,50 Additionally, higher methylation heterogeneity increases the probability of the appearance of DLBCL clones that are epigenetically programmed to better tolerate chemoimmunotherapy. In fact, patients with higher heterogeneity displayed a poorer outcome with a shorter survival after R-CHOP therapy.51 Consistently, in vitro studies have demonstrated that the hypermethylation of SMAD1, a key transducer of TGF-β signaling, contributes to the development of chemoresistance in DLBCL cells exposed to R-CHOP treatment.52
3.1.2. Histone Modifications and Chromain Remodeling in DLBCL Mutations in genes responsible for histone and chromatin modifications, such as CREBBP, KMT2D, EZH2, and TET2, drive aberrant epigenetic B cell programming and influence DLBCL clinical outcome.53 Loss-offunction mutations in CREBBP are common in GCB-DLBCL, occur in
approximately 30% of cases and lead to reduced acetyltransferase activity. Under normal conditions, CREBBP regulates enhancers that are repressed by BCL6 through cooperation with the co-repressors SMRT and NCOR in the GC.54 This repressor complex also includes HDAC3, which mediates deacetylation of H3K27. Loss of CREBBP function results in unchecked enhancer repression, altering the expression of key genes, including those encoding MHC class II molecules.54 Furthermore, CREBBP deficiency contributes to dysregulated B cell signaling and excessive repression of gene transcription. The CREBBP-mediated deacetylation of H3K27 downregulates the expression of FBXW7, a key negative regulator of NOTCH, and thus activates the NOTCH signaling pathway, which plays a critical role in B cell malignancies.55 CREBBP mutations further disrupt the downstream components of the NOTCH pathway, leading to expression of CCL2 and CSF1, which in turn promote the polarization of tumorassociated macrophages toward the immunosuppressive M2
phenotype.56 These alterations impair normal immune surveillance, facilitating tumor immune evasion and driving disease progression (Figure 2 on page 40).55,56
EZH2 mutations are found in 6–14% of DLBCL overall and in about 20% of GCB-DLBCL, where they are frequently associated with BCL2 translocations.57 Under normal conditions, EZH2 cooperates with BCL6 to mediate gene silencing at the promoters of genes involved in plasma cell differentiation and cell cycle regulation.28 In mouse models, EZH2 knockout leads to accelerated lymphomagenesis.58 EZH2 gain-of-function mutations, most commonly affecting the Tyr641 residue within the SET domain, drive germinal center hyperplasia and contribute to remodeling of the immune microenvironmen.57 Unlike normal GC B cells, where cytosine methylation and H3K27me3 marks are usually mutually exclusive, in DLBCL these modifications often overlap, with hypermethylation partly affecting EZH2 target genes.59
1
KMT2D, also known as MLL2, encodes a methyltransferase essential for the methylation of lysine 4 on histone H3 (H3K4), a key modification that regulates gene enhancer activity.60 Lossof-function mutations in KMT2D are found in approximately 30% of DLBCL.61 These mutations impair H3K4 methylation, leading to the downregulation of FBXW7, a negative regulator of the NOTCH signaling pathway. The resulting activation of NOTCH signaling drives several oncogenic processes, including enhanced cell survival, proliferation, and inhibition of terminal B cell differentiation.62 This aberrant signaling further promotes activation of downstream pathways such as RAS, ERK, and MYC/TGF-β1, contributing to tumor growth and immune evasion.62
The TET family of enzymes plays a key role in active DNA demethylation, particularly at gene enhancers, where this activity is linked to transcriptional activation.63 TET2 mutations occur in approximately 10% of DLBCL patients and result in abnormal DNA hypermethylation at regulatory elements within the GC.64 This hypermethylation leads to the silencing of genes involved in GC exit and B cell receptor signaling, contributing to lymphomagenesis. In TET2 knockout mice, loss of TET2 disrupts GC B cell homeostasis and promotes transformation into aggressive lymphomas. These models also show increased formation of G-quadruplexes and R-loops, that represent structures associated with double-strand DNA breaks at immunoglobulin switch regions.65
Therapeutic Strategies Targeting Epigenetic
Alterations
Over the past decade, our understanding of the epigenetic landscape in B cell malignancies has expanded significantly, revealing how aberrant DNA methylation, histone modifications, and dysregulated chromatin architecture contribute to malignant transformation, disease progression, and therapy resistance.
Figure
IN UNRESECTABLE OR METASTATIC BREAST CANCER1-3
ENHERTU therapeutic indications in mBC include3:
ENHERTU as monotherapy is indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received one or more prior anti-HER2-based regimens.
ENHERTU as monotherapy is indicated for the treatment of adult patients with unresectable or metastatic HER2-low breast cancer who have received prior chemotherapy in the metastatic setting or developed disease recurrence during or within 6 months of completing adjuvant chemotherapy.
ABBREVIATED PRESCRIBING INFORMATION
▼ENHERTU® (trastuzumab deruxtecan) 100 mg powder for concentrate for solution for infusion Consult Summary of Product Characteristics (SmPC) before prescribing.
Indication: • HER2-positive breast cancer: Monotherapy for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received one or more prior antiHER2-based regimens. • HER2-low and HER2-ultralow breast cancer: Monotherapy for the treatment of adult patients with unresectable or metastatic hormone receptor (HR)-positive, HER2low or HER2-ultralow breast cancer who have received at least one endocrine therapy in the metastatic setting and who are not considered suitable for endocrine therapy as next line of treatment. Monotherapy for the treatment of adult patients with unresectable or metastatic HER2low breast cancer who have received prior chemotherapy in the metastatic setting or developed disease recurrence during or within 6 months of completing adjuvant chemotherapy. Presentation: One vial of powder for concentrate for solution for infusion contains 100 mg of trastuzumab deruxtecan. After reconstitution, one vial of 5 mL solution contains 20 mg/mL of trastuzumab deruxtecan. Dosage and Administration: Enhertu should be prescribed by a physician and administered under the supervision of a healthcare professional experienced in the use of anticancer medicinal products. In order to prevent medicinal product errors, it is important to check the vial labels to ensure that the medicinal product being prepared and administered is Enhertu (trastuzumab deruxtecan) and not trastuzumab or trastuzumab emtansine. Enhertu should not be substituted with trastuzumab or trastuzumab emtansine. HER2-positive breast cancer: Patients must have documented HER2-positive tumour status, defined as a score of 3 + by immunohistochemistry (IHC) or a ratio of ≥ 2.0 by in situ hybridization (ISH) or by fluorescence in situ hybridization (FISH) assessed by a CE-marked in vitro diagnostic (IVD) medical device. If a CE-marked IVD is not available, the HER2 status should be assessed by an alternate validated test. HER2-low or HER2-ultralow breast cancer: Patients should have documented HER2-low tumour status defined as a score of IHC 1+ or IHC 2+/ISH-, or HER2-ultralow tumour status, described as IHC 0 with membrane staining (IHC>0<1+), as assessed by a CE-marked IVD medical device. If a CE-marked IVD is not available, the HER2 status should be assessed by an alternate validated test. Recommended dose: Breast cancer: 5.4 mg/kg given as an intravenous (IV) infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. Method of administration: Initial dose should be administered as a 90-minute IV infusion. If well tolerated, subsequent doses may be administered as 30-minute infusions. Infusion rate of Enhertu should be slowed or interrupted if the patient develops infusion-related symptoms. Permanently discontinue in case of severe infusion reactions. Do not administer as an IV push or bolus. Please see SmPC for complete information. Premedication: Enhertu is emetogenic, which includes delayed nausea and/or vomiting. Prior to each dose of Enhertu, patients should be premedicated with a combination regimen of two or three medicinal products (e.g., dexamethasone with either a 5-HT3 receptor antagonist and/or an NK1 receptor antagonist, as well as other medicinal products as indicated) for prevention of chemotherapy-induced nausea and vomiting. Dose modifications: Management of adverse reactions may require temporary interruption, dose reduction, or treatment discontinuation of Enhertu per SmPC guidelines. Dose modifications are required for interstitial lung disease (ILD)/ pneumonitis, neutropenia, febrile neutropenia and decreased left ventricular ejection fraction. Dose should not be re-escalated after a dose reduction is made. Permanently discontinue for symptomatic ILD/pneumonitis (Grade 2 or greater), if LVEF <40% or absolute or absolute decrease from baseline is >20%, or in symptomatic congestive heart failure. Delayed or missed dose: If a dose is delayed or missed, it should be administered as soon as possible. The schedule should be adjusted to maintain a 3-week interval between doses. The infusion should be administered at the dose and rate the patient tolerated in the most recent infusion or at reduced dose if indicated. Elderly: No dose adjustment is required in patients aged 65 years or older. Limited data are available in patients ≥ 75 years of age. Renal impairment: No dose adjustment is required in patients with mild or moderate renal impairment. The potential need for dose adjustment in patients with severe renal impairment or end-stage renal disease cannot be determined due to insufficient data. A higher incidence of Grade 1 and 2 ILD/pneumonitis leading to an increase in discontinuation of therapy has been observed in patients with moderate renal impairment. Patients with moderate or severe renal impairment should be monitored carefully for adverse reactions including ILD/pneumonitis. Hepatic impairment: No dose adjustment is required in patients with total bilirubin ≤ 1.5 times upper limit of normal (ULN), irrespective of aspartate transaminase (AST) value. The potential need for dose adjustment in patients with total bilirubin > 1.5 times ULN, irrespective of AST value, cannot be determined; therefore, these patients should be monitored carefully. Use with caution in patients with moderate and severe hepatic impairment. Contraindications: Hypersensitivity to the active substance or to any of the excipients. Each 100 mg vial contains 1.5 mg of polysorbate 90 (E433). Warnings and Precautions: Traceability: The name and the batch number of the administered product should be clearly recorded. Interstitial lung disease (ILD)/pneumonitis: Cases of ILD, and/or pneumonitis, have been reported with Enhertu. Fatal outcomes have been observed. Patients should be advised to immediately report cough, dyspnoea, fever, and/or any new or worsening respiratory symptoms. Patients should be monitored for signs and symptoms of ILD/
pneumonitis. Evidence of ILD/pneumonitis should be promptly investigated. Patients with suspected ILD/pneumonitis should be evaluated by radiographic imaging, preferably a computed tomography (CT) scan. Consultation with a pulmonologist should be considered. Patients with a history of ILD/ pneumonitis or moderate or severe renal impairment may be at increased risk of developing ILD/ pneumonitis and should be monitored carefully. Please see SmPC for dose modifications in the event of ILD/pneumonitis developing. Neutropenia: Complete blood counts should be monitored prior to initiation of Enhertu and prior to each dose, and as clinically indicated. Based on the severity of neutropenia, dose interruption or reduction may be required. Left ventricular dysfunction: Left ventricular ejection fraction (LVEF) decrease has been observed with anti-HER2 therapies. Standard cardiac function testing (echocardiogram or MUGA scanning) should be performed to assess LVEF prior to initiation of Enhertu and at regular intervals during treatment as clinically indicated. LVEF decrease should be managed through treatment interruption. Enhertu should be permanently discontinued if LVEF of less than 40% or absolute decrease from baseline of greater than 20% is confirmed and in patients with symptomatic congestive heart failure (CHF). Embryo-foetal toxicity: Enhertu can cause foetal harm in pregnant women. In post-marketing reports, use of trastuzumab in pregnancy resulted in cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities and neonatal death. The topoisomerase I inhibitor component of Enhertu, DXd, can also cause embryo-foetal harm if administered to a pregnant woman. Patients with moderate or severe hepatic impairment: There are limited data in patients with moderate hepatic impairment and no data in patients with severe hepatic impairment. As metabolism and biliary excretion are the primary routes of elimination of DXd, Enhertu should be administered with caution in patients with moderate and severe hepatic impairment. Interaction with other medicinal products and other forms of interaction: No dose adjustment is required during coadministration of trastuzumab deruxtecan with medicinal products that are inhibitors of CYP3A or OATP1B or P-gp transporters. Pregnancy and Lactation: Enhertu can cause foetal harm when administered to a pregnant woman. Pregnancy status of women of childbearing potential should be verified prior to initiation. Administration to pregnant women is not recommended and the patient should be informed of the potential risks to the foetus. Women of childbearing potential should use effective contraception during treatment and for at least 7 months following the last dose. Male patients with female partners of reproductive potential should use effective contraception during treatment and for at least 4 months after the last dose. Women who become pregnant must immediately contact their doctor. If a woman becomes pregnant during treatment with Enhertu or within 7 months following the last dose, close monitoring is recommended. It is not known if trastuzumab deruxtecan is excreted in human milk, therefore, women should not breast-feed during treatment with Enhertu or for 7 months after the last dose. No dedicated fertility studies have been conducted with trastuzumab deruxtecan. Before starting treatment, male patients should be advised to seek counselling on sperm storage. Please see SmPC for further details. Ability to Drive and Use Machines: May have a minor influence on the ability to drive and use machines. Patients should be advised to use caution when driving or operating machinery in case they experience fatigue, headache or dizziness during treatment. Undesirable Events: Consult SmPC for further information on side effects. Enhertu 5.4 mg/kg: The most common National Cancer Institute – Common Terminology Criteria for Adverse Events (NCI-CTCAE v.5.0) Grade 3 or 4 adverse reactions were neutropenia, anaemia, fatigue, leukopenia, thrombocytopenia, nausea, lymphopenia, hypokalaemia, transaminases increased, diarrhoea, vomiting, decreased appetite, pneumonia, and ejection fraction decreased. Grade 5 adverse reactions occurred in 1.4% of patients, including ILD/ pneumonitis (1.1%). Very common: Upper respiratory tract infection, anaemia, neutropenia, thrombocytopenia, leukopenia, hypokalaemia, decreased appetite, headache, interstitial lung disease, cough, nausea, vomiting, constipation, diarrhoea, abdominal pain, stomatitis, dyspepsia, transaminases increased, alopecia, musculoskeletal pain, fatigue, pyrexia, ejection fraction decreased, weight decreased. Common: Pneumonia, lymphopenia, febrile neutropenia, pancytopenia, dehydration, dizziness, dysgeusia, dry eye, vision blurred, dyspnoea, epistaxis, abdominal distension, gastritis, flatulence, rash, pruritus, skin hyperpigmentation, oedema peripheral, blood alkaline phosphatase increased, blood bilirubin increased, blood creatinine increased, infusion-related reactions. Legal Category: Product subject to prescription which may not be renewed (A). Marketing Authorisation Number: EU/1/20/1508/001. Marketing Authorisation Holder: Daiichi Sankyo Europe GmbH, Zielstattstrasse 48, 81379 Munich, Germany. Further product information available on request from: Daiichi Sankyo Ireland Ltd, Unit 29, Block 3, Northwood, Dublin 9, Ireland. Date of API preparation: 12/2025. Veeva ID: IE/ADC/12/25/0003.
▼This medicinal product is subject to additional monitoring. Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie. Adverse events should also be reported to Daiichi Sankyo Ireland Ltd Pharmacovigilance by email at pharmacovigilance_ie@daiichisankyo.com or please call +353 1 489 3000. As Enhertu® is a biological medicine, healthcare professionals should report adverse reactions by brand name and batch number.
Reference: 1. Siddiqui T, et al. Ann Med Surg (Lond). 2022;82:104665. 2. Escrivá-de-Romaní S, et al. Cancer Drug Resist. 2023;6(1):45-58. 3. ENHERTU (trastuzumab deruxtecan). Summary of Product Characteristics. Available at www.medicines.ie VEEVA ID: IE/ADC/11/25/0015 | Date of Preparation: December 2025
Mature B Cell Neoplasia
These reversible changes offer promising therapeutic targets, enabling modulation of gene expression without modifying the underlying DNA sequence.
Demethylating Agents
DNMT inhibitors such as 5-azacytidine and decitabine, initially developed for AML, have shown limited efficacy as monotherapy in lymphoid malignancies like DLBCL, FL, and CLL. Clinical trials using these agents alone in B cell malignancies have been largely disappointing, prompting a shift toward combination strategies.130,131,132 While combinations with agents like HDAC inhibitors (e.g., romidepsin) have demonstrated some success in peripheral T-cell lymphoma (PTCL), other regimens, such as azacytidine with immune checkpoint inhibitors, have failed to show sufficient clinical activity.133,134 Efforts to enhance efficacy through co-administration with enzyme cytidine deaminase (CDA) inhibitors or novel DNMTis are ongoing, but robust clinical benefits in B cell malignancies remain unproven (Figure 3 on page 42).
HMT Inhibitors
The development of HMT inhibitors in B cell malignancies
has concentrated on targeting EZH2. Among EZH2 inhibitors, tazemetostat has emerged as the most clinically advanced. Preclinical studies showed that tazemetostat was effective in vitro and in vivo (including in xenograft models) against EZH2mutant lymphoma. In a phase I trial, tazemetostat demonstrated a favorable safety profile and achieved a 38% overall response rate (ORR) in refractory B-NHL.135 A subsequent phase II trial in relapsed/refractory (R/R) FL confirmed higher activity in EZH2-mutant patients (ORR 69%) compared to EZH2-wildtype patients (ORR 35%).136 These data led the FDA to grant tazemetostat accelerated approval for the treatment of R/R FL with EZH2 mutations. In addition, tazemetostat was evaluated in a small DLBCL cohort in combination with R-CHOP, achieving an ORR of 86%.137
However, when combined with the PD-L1 inhibitor atezolizumab in R/R DLBCL, no added benefit was observed (Figure 3).138 This highlights the ongoing challenge of improving outcomes through combination therapies. Further trials are exploring new combinations and strategies to enhance the efficacy of
tazemetostat and other HMT inhibitors, aiming to improve overall survival in patients with these lymphomas.
Other agents have shown more limited efficacy. For instance, GSK126, despite its strong preclinical activity against mutant EZH2, failed to show meaningful clinical responses in a phase I trial in R/R DLBCL, transformed FL, and other NHL, leading to early trial termination.139,140 Valemetostat, a dual EZH1/2 inhibitor, has shown promise in early-phase studies, achieving an ORR of 53% in B- and T-NHL, with particularly high activity (80% ORR) in T-cell lymphoma.141
HDACs Inhibitors
HDACs play a key role in regulating cell proliferation, growth, and angiogenesis, which are often dysregulated in cancer. Overexpression of HDACs promotes tumor development, making HDAC inhibitors (HDACis) a promising anticancer strategy (Figure 3).142 HDACis induce apoptosis, cause cell cycle arrest, and can modulate immune responses, leading to therapeutic effects in various lymphomas.143 Vorinostat, the first FDA-approved HDACi (2006), targets class I and II HDACs and
2
is approved for relapsed/refractory cutaneous T-cell lymphoma (CTCL).144 It has demonstrated benefits as monotherapy in FL and in combination regimens with rituximab, fludarabine, or etoposide for relapsed/refractory B-NHL, CLL, and T-NHL. Belinostat, a pan-HDACi, is approved for relapsed/refractory PTCL and has shown activity in CTCL, although with limited efficacy in B-cell lymphomas. Romidepsin, a class I HDACi, is also approved for relapsed/ refractory CTCL and PTCL and has demonstrated additive or synergistic effects when combined with gemcitabine, 5-azacitidine, or alisertib in T-cell lymphomas and FL.145 Mocetinostat showed limited efficacy as monotherapy in relapsed CLL, with most patients tolerating only two treatment cycles, and no responses observed when combined with rituximab.146 Ricolinostat, a selective HDAC6 inhibitor, exhibited disease stabilization in ~50% of patients with relapsed/refractory B- and T-cell lymphomas in early clinical studies, though no objective responses were recorded; it also showed synergistic effects in preclinical DLBCL models when combined with agents like ibrutinib or carfilzomib.147,148,149 Fimepinostat, a dual HDAC/PI3K inhibitor, achieved response rates of 64% in MYC-altered DLBCL versus 37% in non-MYC-altered cases when combined with rituximab. Panobinostat showed modest activity in relapsed/ refractory DLBCL and limited benefits in Hodgkin lymphoma trials; combination strategies yielded mixed efficacy and notable toxicities.148,150 Abexinostat, another pan-HDACi, demonstrated promising results in phase II trials, with overall response rates ranging from 31% to 56% in FL, PTCL, and DLBCL. These findings underscore the potential of HDACis in lymphoma therapy, particularly in biomarker-selected or combination treatment contexts.151
BET Inhibitor
Early BET inhibitors (BETi) showed promise preclinically, but clinical outcomes have been mixed.
Figure
For healthcare professionals in Ireland only. Abbreviated Prescribing Information can be found below.
Generic Product Launch
Rivaroxaban Teva
film-coated tablets rivaroxaban
Indications
Rivaroxaban Teva 10 mg film-coated tablets
Prevention of venous thromboembolism (VTE) in adult patients undergoing elective hip or knee replacement surgery.
Treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and prevention of recurrent DVT and PE in adults.
Prevention of stroke and systemic embolism in adult patients with non-valvular atrial fibrillation with one or more risk factors, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischaemic attack.
Treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and prevention of recurrent DVT and PE in adults.
Rivaroxaban Teva Film-Coated Tablets Abbreviated Prescribing Information
Presentation: Rivaroxaban Film-coated Tablets contain 10 mg, 15 mg and 20 mg rivaroxaban. Indications: For 10 mg, prevention of venous thromboembolism (VTE) in adult patients undergoing elective hip or knee replacement surgery. For 15 mg and 20 mg, prevention of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation with one or more risk factors, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischaemic attack. For all strengths, treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and prevention of recurrent DVT and PE in adults. Dosage and administration: For oral use. Adults: Prevention of VTE in adult patients undergoing elective hip or knee replacement surgery: The recommended dose is 10 mg rivaroxaban taken orally once daily. The duration of treatment depends on the individual risk of the patient for venous thromboembolism which is determined by the type of orthopaedic surgery. Treatment of DVT, treatment of PE and prevention of recurrent DVT and PE: The recommended dose for the initial treatment of acute DVT or PE is 15 mg twice daily for the first three weeks followed by 20 mg once daily for the continued treatment and prevention of recurrent DVT and PE. When extended prevention of recurrent DVT and PE is indicated (following completion of at least 6 months therapy for DVT or PE), the recommended dose is 10 mg once daily. In patients in whom the risk of recurrent DVT or PE is considered high, such as those with complicated comorbidities, or who have developed recurrent DVT or PE on extended prevention with Rivaroxaban Teva 10 mg once daily, a dose of Rivaroxaban Teva 20 mg once daily should be considered. Children: Rivaroxaban Teva is not recommended for use in children below 18 years of age. Renal impairment: Limited clinical data for patients with severe renal impairment (creatinine clearance 15 - 29 ml/min) indicate that rivaroxaban plasma concentrations are significantly increased. Therefore, Rivaroxaban Teva is to be used with caution in these patients. Use is not recommended in patients with creatinine clearance < 15 ml/min. When the recommended dose is 10 mg once daily, no dose adjustment from the recommended dose is necessary. Hepatic impairment: Rivaroxaban Teva is contraindicated in patients with hepatic disease associated with coagulopathy and clinically relevant bleeding risk including cirrhotic patients with Child Pugh B and C. Contraindications: Hypersensitivity to the active substance or to any of the excipients. Active clinically significant bleeding. Lesion or condition, if considered to be a significant risk for major bleeding. This may include current or recent gastrointestinal ulceration, presence of malignant neoplasms at high risk of bleeding, recent brain or spinal injury, recent brain, spinal or ophthalmic surgery, recent intracranial haemorrhage, known or suspected oesophageal varices, arteriovenous malformations, vascular aneurysms or major intraspinal or intracerebral vascular abnormalities. Concomitant treatment with any other anticoagulants, e.g. unfractionated heparin (UFH), low molecular weight heparins (enoxaparin, dalteparin, etc.), heparin derivatives (fondaparinux, etc.), oral anticoagulants (warfarin, dabigatran etexilate, apixaban, etc.) except under specific circumstances of switching anticoagulant therapy or when UFH is given at doses necessary to maintain an open central venous or arterial catheter. Hepatic disease associated with coagulopathy and clinically relevant
Teva Pharmaceuticals Ireland, Digital Office Centre Swords, Suite 101 - 103, Balheary Demesne, Balheary Road, Swords, Co Dublin, K67E5AO, Ireland.
Freephone: 1800 - 201 700 | Email: info@teva.ie
bleeding risk including cirrhotic patients with Child Pugh B and C. Pregnancy and breast-feeding. Precautions and warnings: Clinical surveillance in line with anticoagulation practice is recommended throughout the treatment period. As with other anticoagulants, patients taking Rivaroxaban Teva are to be carefully observed for signs of bleeding. It is recommended to be used with caution in conditions with increased risk of haemorrhage. Rivaroxaban Teva administration should be discontinued if severe haemorrhage occurs. In patients with severe renal impairment (creatinine clearance < 30 ml/min) rivaroxaban plasma levels may be significantly increased (1.6-fold on average) which may lead to an increased bleeding risk. Rivaroxaban Teva is to be used with caution in patients with creatinine clearance 15 - 29 ml/min. Use is not recommended in patients with creatinine clearance < 15 ml/ min. In patients with moderate renal impairment (creatinine clearance 30 - 49 ml/ min) concomitantly receiving other medicinal products which increase rivaroxaban plasma concentrations Rivaroxaban Teva is to be used with caution. Rivaroxaban is not recommended in patients with an increased bleeding risk. Patients with prosthetic valves: Treatment with Rivaroxaban Teva is not recommended for these patients. Direct acting Oral Anticoagulants (DOACs) including rivaroxaban are not recommended for patients with a history of thrombosis who are diagnosed with antiphospholipid syndrome. Rivaroxaban has not been studied in interventional clinical studies in patients undergoing hip fracture surgery to evaluate efficacy and safety. Rivaroxaban Teva is not recommended as an alternative to unfractionated heparin in patients with pulmonary embolism who are haemodynamically unstable or may receive thrombolysis or pulmonary embolectomy since the safety and efficacy of rivaroxaban have not been established in these clinical situations. To reduce the potential risk of bleeding associated with the concurrent use of rivaroxaban and neuraxial (epidural/spinal) anaesthesia or spinal puncture, consider the pharmacokinetic profile of rivaroxaban. Placement or removal of an epidural catheter or lumbar puncture is best performed when the anticoagulant effect of rivaroxaban is estimated to be low. At least 18 hours should elapse after the last administration of rivaroxaban before removal of an epidural catheter. Following removal of the catheter, at least 6 hours should elapse before the next rivaroxaban dose is administered. If traumatic puncture occurs the administration of rivaroxaban is to be delayed for 24 hours. If an invasive procedure or surgical intervention is required, Rivaroxaban Teva 10 mg should be stopped at least 24 hours before the intervention, if possible and based on the clinical judgement of the physician. If the procedure cannot be delayed the increased risk of bleeding should be assessed against the urgency of the intervention. Rivaroxaban Teva should be restarted as soon as possible after the invasive procedure or surgical intervention provided the clinical situation allows and adequate haemostasis has been established as determined by the treating physician. For the elderly population, increasing age may increase haemorrhagic risk. Serious skin reactions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and DRESS syndrome, have been reported during post-marketing surveillance in association with the use of rivaroxaban. Rivaroxaban should be discontinued at the first appearance of a severe skin rash (e.g. spreading, intense and/or blistering), or any other sign of hypersensitivity in conjunction with mucosal lesions. Interactions: CYP3A4 and P-gp inhibitors: The use
Product subject to prescription which may be renewed (B)
of Rivaroxaban Teva is not recommended in patients receiving concomitant systemic treatment with azole-antimycotics such as ketoconazole, itraconazole, voriconazole and posaconazole or HIV protease inhibitors. The interaction with clarithromycin, erythromycin and Fluconazole is likely not clinically relevant in most patients but can be potentially significant in high-risk patients. Given the limited clinical data available with dronedarone, co-administration with rivaroxaban should be avoided. Anticoagulants: Enoxaparin did not affect the pharmacokinetics of rivaroxaban. Due to the increased bleeding risk care is to be taken if patients are treated concomitantly with any other anticoagulants. NSAIDs/platelet aggregation inhibitors: Care is to be taken if patients are treated concomitantly with NSAIDs (including acetylsalicylic acid) and platelet aggregation inhibitors because these medicinal products typically increase the bleeding risk. SSRIs/SNRIs: As with other anticoagulants the possibility may exist that patients are at increased risk of bleeding in case of concomitant use ith SSRIs or SNRIs due to their reported effect on platelets. Warfarin: No pharmacokinetic interaction was observed between warfarin and rivaroxaban. CYP3A4 inducers: Cncomitant administration of strong CYP3A4 inducers should be avoided unless the patient is closely observed for signs and symptoms of thrombosis. Other concomitant therapies: Rivaroxaban neither inhibits nor induces any major CYP isoforms like CYP3A4. No clinically relevant interaction with food was observed. Laboratory parameters: Clotting parameters (e.g. PT, aPTT, HepTest) are affected as expected by the mode of action of rivaroxaban. Pregnancy and lactation: Rivaroxaban Teva is contraindicated during pregnancy and breastfeeding. Effects on ability to drive and use machines: Minor influence on the ability to drive and use machines. Adverse reactions like syncope and dizziness have been reported. Patients experiencing these adverse reactions should not drive or use machines. Adverse reactions: Thrombocytopenia, angioedema, anaphylactic reactions including anaphylactic shock, cerebral and intracranial haemorrhage, syncope, eye haemorrhage, hypotension, haematoma, cholestasis, hepatitis, Stevens-Johnson syndrome/ Toxic Epidermal Necrolysis, haemarthrosis, muscle haemorrhage, compartment syndrome secondary to a bleeding, urogenital tract haemorrhage, renal impairment, renal failure (including acute renal failure), postprocedural haemorrhage, contusion, wound secretion, vascular pseudoaneurysm. Common: Anaemia, dizziness, headache, epistaxis, haemoptysis, gingival bleeding, gastrointestinal and abdominal pains, dyspepsia, nausea, constipation, diarrhoea, vomiting, increase in transaminases, pruritus (incl. uncommon cases of generalised pruritus), rash, ecchymosis, cutaneous and subcutaneous haemorrhage, pain in extremity, fever, peripheral oedema, decreased general strength and energy (incl. fatigue and asthenia). Consult the Summary of Product Characteristics in relation to other side effects. Overdose: In case of overdose, the patient should be observed carefully for bleeding complications or other adverse reactions. A specific reversal agent (andexanet alfa) antagonising the pharmacodynamic effect of rivaroxaban is available. The use of activated charcoal to reduce absorption in case of rivaroxaban overdose may be considered. Legal category: POM. Marketing Authorisation Number: PA22579/002/001-03. Marketing
Authorisation Holder: TEVA GmbH, Graf-Arco-Str. 3, 89079 Ulm, Germany. Job Code: MED-IE-00077. Date of Preparation: February 2024
Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie.
Adverse events should also be reported to Teva UK Limited on +44 (0) 207 540 7117 or medinfo@tevauk.com
Date of Preparation: October 2025 | Job Code: GEN-IE-00160
Further information is available on request or in the SmPC. Product Information also available on the HPRA website.
Mature B Cell Neoplasia
Birabresib, the first BETi tested in R/R DLBCL, achieved a CR rate of 47% in 17 patients, though only 18% had durable OR (Figure 3).152 Other BETis, including INCB054329 and INCB057643, demonstrated minimal clinical activity with no meaningful responses and significant toxicity, leading to early trial termination. CPI-0610 showed a modest 7% overall response rate (ORR) in B-NHL.153 A combination of the BETi RO6870810 with venetoclax (± rituximab) in R/R DLBCL achieved an ORR of 38.5%, including 20.5% CR and 17.9% partial response (PR), with stable disease in 15.4% of patients, but without clear evidence of synergistic benefits.154 These data underscore the challenges in translating BET inhibition into durable clinical responses in B-cell malignancies.
Discussion and Conclusions
The last decade has witnessed remarkable progress in our understanding of the epigenetic underpinnings of mature B cell neoplasms. Epigenetic dysregulation including aberrant DNA methylation, histone modifications, chromatin remodeling, and noncoding RNA
activity emerges not only as a hallmark of tumor biology but also as a key driver of disease heterogeneity, immune evasion, and therapeutic resistance across DLBCL, FL, and CLL. Mutations in chromatin-modifying enzymes such as EZH2, KMT2D, CREBBP, and TET2 among others, disrupt normal gene expression programs essential for B cell maturation and facilitate malignant transformation. Beyond genetic lesions, many of these epigenetic alterations converge on common oncogenic pathways, leading to the upregulation or downregulation of BCR signaling, NF-κB activity, and apoptosis regulators. The activity of these pathways, whether altered through epigenetic mechanisms or co-existing mutations, can define aggressive disease subsets and influence therapeutic response, underscoring the need to account for their status when designing epigenetic-targeted strategies. Furthermore, the interplay between epigenetic modifiers and transcription factors, along with the contribution of noncoding RNAs, defines disease-specific regulatory networks with clinical relevance. Although current therapeutic strategies targeting the epigenome have shown promise in selected patient subsets, their
clinical impact remains variable and often limited as monotherapy. Emerging evidence underscores the crucial role of epigenetic mechanisms in shaping the tumor microenvironment by modulating immune-related processes and fostering immune evasion. Targeting these epigenetic alterations may offer a promising strategy to reprogram the TME and enhance the efficacy of novel immunotherapies (i.e., chimeric antigen receptor (CAR) T-cells and bispecific antibodies) through synergistic combination approaches. These challenges underscore the necessity of developing rational combination regimens, better biomarkers of response, and subtype-specific approaches guided by the molecular and epigenetic context of each neoplasm. The advent of integrative multi-omics and single-cell epigenetic profiling is poised to unravel intra-tumoral heterogeneity and inform precision medicine strategies. While this review focuses on DLBCL, FL, and CLL as representative mature B-cell neoplasms, epigenomic alterations are also critical in other subtypes. In mantle cell lymphoma, mutations in key regulators such as KMT2D, NSD2, SMARCA4, SP140, and KMT2C
3
contribute to disease biology, with epigenomic profiling distinguishing SOX11-positive and SOX11negative variants of differing aggressiveness.155 In Hodgkin lymphoma, alterations in BCL6 and CREBBP, along with EBV-driven epigenomic modulation, play major pathogenic roles.156,157 Ultimately, a deeper mechanistic understanding of the epigenetic architecture governing B cell malignancies holds promising potential, not only for refining prognostic models and improving risk stratification but also for unlocking new therapeutic avenues that can overcome resistance and induce durable remissions. The convergence of epigenetic insight and clinical innovation stands to redefine the therapeutic landscape for patients with mature B cell neoplasms. Refences available on request
Figure
For healthcare professionals in Ireland only. Abbreviated Prescribing Information can be found below.
Smoking Cessation Medicine
Varenicline Teva
Film-coated Tablets
varenicline
Now reimbursed by the HSE
Indications
Varenicline Teva 0.5 mg and Varenicline Teva 1 mg Film-coated Tablets (initiation pack) and Varenicline Teva 1 mg
Film-coated Tablets
Varenicline Teva is indicated for smoking cessation in adults.
Varenilcine 0.5mg and 1mg Film-Coated Tablets Abbreviated Prescribing Information Presentation: Each film-coated tablet contains varenicline citrate equivalent to 0.5mg and 1mg varenicline. Indications: Varenicline is indicated for smoking cessation in adults. Dosage and administration: Oral use. Adults: The recommended dose is 1mg Varenicline twice daily following a 1-week titration (see SmPC for details). Children: Not recommended for use. Elderly: No dosage adjustment is necessary. Elderly patients are more likely to have decreased renal function, prescribers should consider the renal status of an elderly patient. Renal impairment: No dosage adjustment is necessary for patients with mild (estimated creatinine clearance >50ml/min and ≤80ml/min) to moderate (estimated creatinine clearance ≥30ml/min and ≤50ml/min) renal impairment. For patients with severe renal impairment (estimated creatinine clearance <30ml/min), the recommended dose of Varenicline is 1mg once daily. Hepatic impairment: No dosage adjustment is necessary. Contraindications: Hypersensitivity to the active substance or to any of the excipients. Precautions and warnings: Physiological changes resulting from smoking cessation, with or without treatment with Varenicline, may alter the pharmacokinetics or pharmacodynamics of some medicinal products, for which dosage adjustment may be necessary (examples include theophylline, warfarin and insulin). As smoking induces CYP1A2, smoking cessation may result in an increase of plasma levels of CYP1A2 substrates. Changes in behaviour or thinking, anxiety, psychosis, mood swings, aggressive behaviour, depression, suicidal ideation and behaviour and suicide attempts have been reported in patients attempting to quit smoking with Varenicline. Depressed mood, rarely including suicidal ideation and suicide attempt, may be a symptom of nicotine withdrawal. Clinicians should be aware of the possible emergence of serious neuropsychiatric symptoms in patients attempting to quit smoking with or without treatment. If serious neuropsychiatric symptoms occur whilst on Varenicline treatment, patients should discontinue Varenicline immediately and contact a healthcare professional for re-evaluation of treatment. Smoking cessation, with or without pharmacotherapy, has been associated with exacerbation of underlying psychiatric illness (e.g. depression). In clinical trials and post-marketing experience there have been reports of seizures in patients with or without a history of seizures, treated with Varenicline. Varenicline should be used cautiously in patients with a history of seizures or other conditions that potentially lower the seizure threshold. At the end of treatment, discontinuation of Varenicline was associated with an increase in irritability, urge to smoke, depression, and/or insomnia in up to 3% of patients. In such instances, tapering should be considered. Patients taking Varenicline should seek immediate medical attention if they experience signs and symptoms of myocardial infarction or stroke. Hypersensitivity reactions including angioedema (swelling of the face, mouth neck and extremities) have been reported in patients treated with varenicline. Some rare life-threatening reports required urgent medical attention due to respiratory compromise. Rare and severe cutaneous reactions (Stevens-Johnson-Syndrome and
Teva Pharmaceuticals Ireland, Digital Office Centre Swords, Suite 101 - 103, Balheary Demesne, Balheary Road, Swords, Co Dublin, K67E5AO, Ireland.
Freephone: 1800 - 201 700 | Email: info@teva.ie
Prescription Only Medicine.
Erythema Multiforme) have also been reported in post-marketing reports. Due to the life-threatening nature of these conditions, varenicline should be discontinued and a healthcare provider should be contacted immediately. Interactions: Varenicline has no clinically meaningful drug interactions (see SmPC for further details). No dosage adjustment of Varenicline or co-administered medicinal products listed below is recommended. In vitro studies indicate that Varenicline is unlikely to alter the pharmacokinetics of compounds that are primarily metabolised by cytochrome P450 enzymes. Furthermore, since metabolism of Varenicline represents less than 10% of its clearance, active substances known to affect the cytochrome P450 system are unlikely to alter the pharmacokinetics of Varenicline, therefore a dose adjustment of Varenicline would not be required. Varenilcine is not known to affect the pharmacokinetics of metformin, digoxin, bupropion and warfarin. Co-administration of cimetidine, with Varenicline increased the systemic exposure of varenicline by due to a reduction in varenicline renal clearance. In patients with severe renal impairment, the concomitant use of cimetidine and Varenicline should be avoided. Pregnancy and lactation: As a precautionary measure, it is preferable to avoid the use of varenicline during pregnancy. A decision on whether to continue/discontinue breast-feeding or to continue/ discontinue therapy with varenicline should be made taking into account the benefit of breast-feeding to the child and the benefit of varenicline therapy to the woman. Effects on ability to drive and use machines: Varenicline may have minor or moderate influence on the ability to drive and use machines. Varenicline may cause dizziness, somnolence and transient loss of consciousness, and therefore may influence the ability to drive and use machines. Adverse reactions: Diabetes mellitus, suicidal ideation, depression, hallucinations, psychosis, seizure, cerebrovascular accident, transient loss of consciousness, myocardial infarction, angina pectoris, tachycardia, atrial fibrillation, electrocardiogram ST segment depression, gastritis, haematemesis, severe cutaneous reactions including Stevens Johnson Syndrome and Erythema Multiforme, angioedema. Very Common: Nasopharyngitis, abnormal dreams, insomnia, headache, nausea. Common: Bronchitis, sinusitis, weight increased, decreased appetite, increased appetite, somnolence, dizziness, dysgeusia, dyspnoea, cough, gastrooesophageal reflux disease, vomiting, constipation, diarrhoea, abdominal distension, abdominal pain, toothache, dyspepsia, flatulence, dry mouth, rash, pruritus, arthralgia, myalgia, back pain, chest pain, fatigue, liver function test abnormal. Consult the Summary of Product Characteristics in relation to other side effects. Overdose: In case of overdose, standard supportive measures should be instituted as required. Legal category: POM. Marketing Authorisation Number: 0.5mg PA1986/129/001, 1mg PA1986/129/002, 0.5mg & 1mg Initiation Pack PA1986/129/003. Marketing Authorisation Holder: Teva B.V., Swensweg 5, 2031GA Haarlem, Netherlands. Job Code: MED-IE-00093. Date of Preparation: May 2025.
Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie.
Adverse events should also be reported to Teva UK Limited on +44 (0) 207 540 7117 or medinfo@tevauk.com
Date of Preparation: November 2025 | Job Code: GEN-IE-00165
Further information is available on request or in the SmPC. Product Information also available on the HPRA website.
Evolving practice in Myeloproliferative Neoplasms: Pathways, precision and progress in 2026
This article provides an evidencebased view of MPN practice in 2026, including diagnostics, risk stratification and therapeutic decision-making and looks to idea pathways and the direction of future innovations. The intended audience is all those with an interest in the management of haematology patients.
The myeloproliferative neoplasms (MPNs) are acquired clonal disorders where there is increased cell production associated with thromboembolic events and long-term progression to acute leukaemia and myelofibrosis. The current complete World Health Organisation (WHO) classification of MPNs are listed in Table 1but polycythaemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) are the predominant clinical entities which will be considered in this article. The incidence rates of PV, ET, and PMF are reported as 0.84, 1.03, and 0.47 per 100,000.1 As these are all diseases with long life expectancy which is increasing with
Written
by Mary Frances McMullin, Professor Emerita, Queen’s University, Belfast.
Mary Frances McMullin is now professor emerita of haematology in Queen’s University, Belfast. For the last 35 years she has had a clinical practice in myeloproliferative neoplasms and acute leukaemia. Research and clinical trials have been the focus of her career and she has published extensively on the myeloproliferative neoplasms and congenital erythrocytosis. She was the chair of the British Society for haematology guidelines committee and has been instrumental in the development of myeloproliferative neoplasm management guidelines.
Diagnosis
The diagnostic criteria for MPNs have evolved since the diseases were originally described in the mid twentieth century. They consist of numerical limits for abnormal blood counts, the detection of an acquired clone, and bone marrow abnormalities. The current WHO criteria for PV, ET, and IMF are listed in tables 2 and 3 (on page 46).2 Currently the International Consensus Classification of myeloid neoplasms (ICC)3 has suggested minor variations but the required diagnostic criteria are essentially the same.
to presentation, perhaps most obviously splenomegaly in IMF, may be the clinical feature. Symptoms of MPN which lead to diagnosis are variable. Some patients have non-specific features such as chronic fatigue which initiate further investigation. There are some specific symptoms such as aquagenic itch, night sweats and early satiety which should trigger investigation for an MPN.
Patients with a suspected MPN will be investigated initially with a repeat blood counts screening for JAK2/CALR/MPL clones as indicated and a bone marrow aspirate and trephine (in most cases) in order to confirm the diagnosis.
Risk scoring
treatment advances the prevalence is many times the incidence and often poorly enumerated.
Presentation
Patients with MPNs present by various routes. Many are entirely asymptomatic and an abnormal blood count at routine review suggests an MPN. Abnormal blood counts may also be detected in someone presenting with incidents such as thromboembolic events (TEV). Signs of MPN may also lead
Table 1
The pathway to the diagnosis depends somewhat on the presenting features. It may be from the community for investigation of an abnormal blood count or suggestive symptoms or signs. However, MPNs may be suspected in other areas for instance if an abnormal blood count and evidence of an acquired clone is detected in patients with TEV. Wherever the initial suspicion of MPN arises, the pathway is then referral to haematology for confirmation of diagnosis and ongoing management.
Patients with MPN have reduced life expectancy because of the development of events and progression. All are at risk of development of TEV. PV and ET may progress to myelofibrosis (MF) and in a small minority of cases acute myeloid leukaemia (AML). IMF has a shorter life expectancy as the disease may have be more advanced. Complications even from presentation including cytopenia and transfusion dependency and certainly progression to myelodysplastic syndrome and AML occur.
In order to guide best management MPN patients risk scoring is carried out. These risk scores are aimed at assessing the risk of complications specifically TEV and with some systems the risk of progression and survival.
Table 1: WHO classification of Haematolymphoid tumours: Myeloproliferative neoplasms, 5th edition
Chronic myeloid leukaemia
Polycythaemia vera
Essential thrombocythaemia
Primary myelofibrosis
Chronic neutrophilic leukaemia
Chronic eosinophilia
Juvenile myelomonocytic leukaemia
Myeloproliferative neoplasm, not otherwise specified Table 2
PV has conventionally been divided into low risk and high risk groups based on the risk of TEV. The division is made on age, 60/65 years and prior thrombosis.4 Other similar systems consider other factors such as leucocyte count and stratify survival into low, intermediate and high groups.5 More recently high risk mutations have been considered in the thrombosis risk, and MFPS-PV (Multi-factor prognostic score) score integrates clinical and molecular data for thrombosis prediction.6
WHO classification of Haematolymphoid tumours: Myeloproliferative neoplasms, 5th edition
We are Ireland’s largest producer of cancer information.
• 100 publications
• 770 webpages and 6 patient education video programmes on www.cancer.ie
More to come in 2026
Our work is:
• evidence based
• peer reviewed
• updated regularly
Order our publications straight to your workplace for free. Go to www.cancer.ie
Your patients can speak to our cancer nurses by calling our Support Line on Freephone 1800 200 700, emailing supportline@irishcancer.ie or visiting a Daffodil Centre. Ask us about our Telephone Interpreting Service.
Chronic eosinophilia
Juvenile myelomonocytic leukaemia
Myeloproliferative neoplasm,
Myeloproliferative Neoplasms
Table 2
Diagnostic criteria for PV and ET
1. Hct > 0.49 (men), 0.48 (women) or other evidence of increased red cell mass
2. Presence of a JAK2 mutation
Subnormal EPO level
1.Platelet count ≥ 450 × 109/ L
Presence of a clonal marker or exclusion of reactive thrombocytosis
ET has conventionally been divided into a number of risk groups but currently the most appropriate in the IPSET (International prognostic score for thrombosis) thrombosis score which separates into very low, low intermediate and high risk of thrombosis groups (Figure 1).7
3. Bone marrow showing panmyelosis
Diagnosis all major criteria or 2 major and 1 minor
Table 3
Table 3: Diagnostic criteria for myelofibrosis
Diagnostic criteria for myelofibrosis
PMF, prefibrotic Stage (pre-PMF)
Major criteria
1. Bone marrow biopsy showing megakaryocytic proliferation and atypia, fibrosis grade<2, increased age-adjusted cellularity
2. JAK2, CALR, or MPL mutation or other clonal marker
3. Diagnostic criteria for other myeloid neoplasms not met
Minor criteria
1. Anaemia not attributed to a comorbid condition
2. Leucocytosis > 11 × 109/L
3. Palpable splenomegaly
4. Lactate dehydrogenase level above the normal range
2.Bone marrow histology showing megakaryocyte hyperplasia, hyperlobulated with loose clusters
3.WHO criteria for BCR::ABL1–positive CML, PV, PMF or other myeloid neoplasms not met
JAK2, CALR, or MPL mutation
Diagnosis all major criteria or the first 3 major criteria plus the minor criterion.
IMF progression and survival prediction is more complicated and prognostic scoring systems are outlined in table 4 (on page 48). They have evolved from simple schemes for prediction at diagnosis and dynamically during the course of the disease (IPSS/ DIPSS/DIPSSplus) (International Prognostic Score System /Dynamic IPSS)8, 9 and a scheme specifically for post PV and ET MF (MYSecPM)(Myelofibrosis secondary to PV and ET prognostic model). Molecular studies including NGS (Next Generation Sequencing) have allowed consideration of molecular lesions and lead to scoring systems such as GIPSS (Genetically Inspired Prognostic Scoring System), in the over 70s(MIPSS70) (Mutation Enhanced IPSS) and (MIPSS70+) and for assessing transplant candidates (MTSS)(Myelofibrosis Transplant Scoring System). The response to ruxolitinib in the first months of treatment is also a useful measure for predicting future outcome (RR6).10
There are also more general scoring systems for MPN, in particular, PredictBlood which is based on 63 variables and molecular data.11 All these systems continue to be updated and are widely available.
PMF, overt fibrotic stage
Major criteria
1. Bone marrow biopsy showing megakaryocytic proliferation and atypia, accompanied by reticulin and/or collagen fibrosis grades 2 or 3
2. JAK2, CALR, or MPL mutation or other clonal marker or absence of reactive myelofibrosis
3. Diagnostic criteria for other myeloid neoplasms not met
Minor criteria
1. Anaemia not attributed to a comorbid condition
2. Leucocytosis > 11 × 109/L
3. Palpable splenomegaly
4. Lactate dehydrogenase level above the normal range
5. Leukoerythroblastosis
Diagnosis: The diagnosis of pre-PMF or overt PMF requires all 3 major criteria and at least 1 minor criterion confirmed in 2 consecutive determinations.
Diagnosis: The diagnosis of pre-PMF or overt PMF requires all 3 major criteria and at least 1 minor criterion confirmed in 2 consecutive determinations
All MPN patients should be risk assessed at diagnosis and in some cases, during the course of their treatment, using a system appropriate for their disease and situation.
Molecular testing
The presence of driver mutations is integral to the diagnosis of MPN. Testing for these usually is undertaken in molecular laboratories associated with haematology. The sequence of testing for JAK2, CALR and MPL may be determined by an algorithm determined by blood counts but as new molecular methods come into practice it may be more efficient to test for all driver mutations together on a single panel. NGS allows testing for a range of molecular mutations including JAK-STAT signalling, epigenetic regulation and in the genes associated with leukaemia regulation. The presence of such high risk mutations is now part of some scoring systems. Such extended molecular analysis should be carried out in most MPN patients at diagnosis and in selected patients at follow up.
Table 2: Diagnostic criteria for PV and ET
IPSET thrombosis risk stratification for ET
Current management
A patient with a putative MPN diagnosis should be discussed at an MDM (Multi-disciplinary meeting) which incorporates MPNs. The diagnosis should then be verified and a management plan for the individual patient agreed. There are agreed pathways for patients depending on their individual clinical features and the risk assessment. In PV the use of venesection, low dose aspirin and cytoreductive therapy are the required decisions.4 With ET, again low dose aspirin and cytoreduction are the main considerations. IMF can be more complex as there may be consideration as to whether and when a patient should be considered for potentially curative allogeneic transplant. There are also an increasing number of therapies available. The current BSH (British Society for Haematology) has recently been updated and gives clear guidance and the treatment algorithm in Figure 2 adapted from the guideline gives a pathway for IMF patients.12
All patients should have a symptom assessment using one of the validated tools such as MPN10 carried out at diagnosis and regular intervals thereafter. This is particularly important before starting any new treatment to assess response to treatment. Current pathway in practice
IPSET thrombosis risk stratification for ET
IPSET thrombosis risk stratification for ET
IPSET thrombosis risk stratification for ET
Current practice is that a patient with a potential diagnosis of MPN is referred to haematology for confirmation of diagnosis and long-term management.
In many centres MPN patients are collated in a specific MPN clinic supported by MPN specialist nurses and other support staff. This allows for increased attention and development of expertise in MPNs. In practice MPN patients may be managed in general haematology where they may not get sufficient attention and time. There are also many other pressures in clinic practice such as waiting times, capacity and workload. A dedicated MPN clinic does improve the service for
MPN but there remain resource and capacity issues as with improvements in awareness of the possibility of an MPN diagnosis and the long term nature of these patients, demand for clinic capacity can only expand
Advances Therapy
As treatment for MPNs is not curative there remains a need for new therapies and there are currently many therapies in trial and approaching licensing.
Management pathway for myelofibrosis
Figure1
Figure 2
Figure 2
Figure 1:
Figure 2: Management pathway for myelofibrosis
Figure1
Figure 2
Table 4
Myeloproliferative Neoplasms
Myelofibrosis prognostic scoring systems
Table 4: Myelofibrosis prognostic scoring systems
Scoring system Factors considered Prognostic scoring
IPSS/DIPSS
DIPSS+
MYSEC-PM
GIPSS
MIPSS70MIPSS70+
MTSS
RR6
Ropeginterferon Alfa-2b (ropeg), a long acting pegylated IFN, has been shown to induce significant responses in PV including decreases in the size of the JAK2 clone and is now licenced for the treatment of high risk PV.13 It has also been used in the low PV trial in low risk PV where it was superior to venesection only with less events and deceases in clone size14. This raises questions as to how all PV should be treated in the future. Will the aim of treatment be to eliminate the acquired clone (not just control the counts and reduce the risk of complications)? There are also a number of agents in development, such as the hepcidin
Age >65, symptoms, Hb<100g/L, WCC>25 × 109/L, Blasts>1%
Age >65, symptoms, Hb<100g/L, Blasts>1%, karyotype and pts< 100× 109/L and transfusion dependency
Hb<110g/L, pts< 150× 109/L, Blasts>3%, absence of CALR mutation, symptoms and age
Karyotype, absence of CALR, presence of high risk mutations (ASXL1, SRSF2, U2AF1 Q157)
Karyotype, high risk mutations (ASXL1, SFSF2, EZH2, IDH1/2, U2AF1)
WCC>25 × 109/L, pts< 100× 109/L, BM fibrosis >2, presence on non-CALR type 1 mutation
Age, WCC>25 × 109/L, non MPL/CALR mutation, ASXL1, Karnofsky score, degree of HLA matching
Baseline and response at 3 and 6 months, Spleen length reduction from baseline, ruxolitinib dose, transfusion requirements
For score at diagnosis and during follow-up
Dynamic score which includes more relevant parameters
Specific to post PV and ET myelofibrosis
Complete genetic information may be difficult to obtain
Includes clinical, pathological, mutational and cytogenetic information.
and reimbursed. Other methods of treatment, immunotherapies such as CAR T cell therapy and the afore mentioned CALR antibodies are being trialled, but it remains to be seen which will succeed and provide advances in management.
Molecular testing
Testing for driver mutations is the initial step in diagnosis. These tests are usually under the auspices of haematology. Access to these tests outside haematology would aid the diagnostic process e,g. if any patient presenting to a GP with a raised haemoglobin had access to a JAK2 test then a diagnosis of PV could be confirmed and the patient rapidly referred. Negative tests also help with avoiding unnecessary referral. Other specialities such as hepatology could benefit from direct molecular screening in target patients. However, if assess to driver mutation testing is widened there must be robust processes for follow up and management of positive results. There may be a case in the future to develop specific screening programmes in populations at risk of an underlying MPN for instance all hepatology patients with abdominal thrombosis or in neurological younger patients presenting with cerebrovascular accidents.
Prediction of survival post allograft.
Response to ruxolitinib after 6 months
Molecular investigation is advancing, Currently NGS is widely used and new methodologies such as WGS (whole genome sequencing) are in development. New methodology needs to be developed and utilised in the molecular service.
Currently NGS is frequently carried out at diagnosis as part of the risk assessment. This service needs to be managed judiciously. It is needed in most patients, but does it add anything in the pathway for example in an 80 year old just diagnosed with PV.
Molecular screening may also need repeated at intervals depending on the patient situation.
memetic rusfertide, which control the haematocrit by manipulating iron metabolism and may be useful adjuncts to the therapeutic armoury in PV.15
ET new drugs are also being developed in the ET arena. Bomedemstat, a LSD1 (lysinespecific demethylase) inhibitor, is in phase 3 trials and shows promise in controlling counts and symptoms in ET.16 There are other therapies under investigation but perhaps the most exciting is the use of CALR antibodies in ET and IMF. Early results show rapid control of counts and major reductions in the CALR clone
size.17 These agents may result in a paradigm shift in the treatment of CALR positive disease.
There are at least 4 JAK/STAT pathway inhibitors (ruxolitinib, fedratinib, momelotinib pacritinib) used in the treatment of MF but responses do not last and many issues such as cytopenia limit treatment. Many agents are in development either as single agent or in combination which are considered at various stages of the disease pathway (e.g. Pelabresib a BET (bromodomain and extra terminal) inhibitor.18 It remains to be seen which agents will succeed in trials and then become licensed
As more agents are used which decrease the size of the clone, repeat measurement of the allele burden of the acquired clone over time will advance into routine practice to monitor disease response.
Ideal service
An ideal MPN service would have clear and rapid referral pathways without waiting lists. This pathway would include access to some molecular testing for rapid diagnosis and screening for driver mutations from beyond haematology to allow appropriate referral.
A dedicated MPN clinic in the haematology service is likely to provide the best option for patients. This clinic should be staffed by doctors and nurses with expertise in MPNs. Pharmacists may also be part of the clinic if available as they can also prescribe. New referrals are seen by medical staff, but all clinical staff can see patients for follow up. This provides more time and capacity. Thus, each patient should have sufficient time to discuss their situation and for regular monitoring of symptoms and quality of life.
Telephone follow up clinics is also a useful adjunct. This is particularly beneficial for the elderly and the incapacitated. For these patients we arrange for a district nurse to go to the patient and take blood tests and when the results are available the patient or carer is phoned and medication sent to patient if required.
Other patients may wish to be followed by telephone, but they must be seen face to face at regular intervals (annual as a minimum).
The MPN clinic needs easy access to other specialities such as
hepatology. A dermatology service or rapid access to dermatology is required. Other specialists such as psychology are of great benefit.
Access to a comprehensive clinical trial programme is essential component.
An MPN service should be supported by an MDM with expertise and interest in MPNs (Nice Guidance NG47). All patients should be discussed at diagnosis to confirm diagnosis and agree management plan. They can be discussed further if there are questions about change in treatment or further management.
Conclusion
In conclusion MPN patients should be managed in a specialist MPN service which is resourced to allow access and time for assessment for all patients. New drugs and therapies need to be available and reimbursed as soon as approved and access to clinical trials will also allow rapid access to the newest therapies.
Widening assess to tests including molecular tests will expediate diagnosis and help with management.
References available on request
Oncology News
Joly Cancer Leadership Programme Award for TSJCI surgeon
Mr Conall Fitzgerald, head and neck surgeon at Trinity St James’s Cancer Institute (TSJCI), has become the third awardee to benefit from the philanthropically supported Joly Cancer Leadership Programme.
The competitive Joly programme is designed to enable exceptional academic cancer clinicians to dedicate protected time to focus on advanced research and clinical trials. Its first two awardees were Dr Nina Orfali, Consultant Haematologist and Mr Michael Kelly, Consultant Colorectal Surgeon.
Mr Conall Fitzgerald, MB BCh BAO MCh MSc FRCS, is a Consultant Otolaryngologist (ENT) whose primary clinical interests are oral cavity, oropharyngeal, laryngeal, thyroid, skin, and salivary gland cancers.
Head and neck cancer incidence in Ireland continues to rise and represents 750–800 new cases annually. St James's Hospital is the largest centre in Ireland for these cancers and treats over 400 new patients each year. Growth in incidence reflects both demographic ageing and the increasing prevalence of HPV-associated oropharyngeal carcinoma.
Mr Fitzgerald said: “Through the award, I hope to investigate the immune and metabolic behaviours of head and neck cancers. It is hoped that discoveries from these studies will translate to meaningful clinical insights to guide future clinical decision
making and care for head and neck cancer patients."
Mr Fitzgerald graduated from Trinity College Dublin and completed higher specialist surgical training in ENT through the Royal College of Surgeons in Ireland. During this time, he completed a Masters in Surgery and separate Masters in Medical Education.
Mr Fitzgerald undertook a twoyear fellowship at Memorial Sloan Kettering Cancer Center (MSKCC), New York, USA. He completed a one-year clinical fellowship in Advanced Head & Neck Surgical Oncology and a separate one-year research fellowship in cancer genomics via the Morris Lab of the MSKCC Immunogenomics and Precision Oncology Platform. Mr Fitzgerald was awarded the MSKCC Chairman’s Award for Excellence in Clinical Research.
Professor John Kennedy, co-director of TSJCI, said: "As Ireland's first accredited Comprehensive Cancer Centre, and the country’s largest centre of excellence for managing head and neck cancers, the Trinity St James’s Cancer Institute is ideally placed to nurture and develop the best young talent to serve this important patient population. The Joly Cancer Leadership Programme affords recently appointed cancer specialists at TSJCI the time they need to develop critical research
programmes. We very much appreciate the wonderful support that enables us to do this."
Professor Maeve Lowery, academic director of TSJCI, said: "This award will support the development of a comprehensive translational research programme focused on head and neck cancer at the Trinity St James’s Cancer Institute. With almost 800 people diagnosed with head and neck cancer each year in Ireland, this research can accelerate the discovery of new strategies for cancer prevention, screening, and treatment for the benefit of our patients."
The Joly Cancer Leadership Programme is named in honour of John Joly, a Trinity graduate who pioneered the earliest effective radiation treatment for cancer. He earned international acclaim for developing a method
for extracting radium and, in collaboration with another Trinity graduate, Walter Clegg Stevenson, pioneered its use in cancer treatment. This became known as the ‘Dublin Method’ and has formed the basis for modern radium needle treatment.
TSJCI is an initiative to transform cancer care in Ireland by combining excellent research, patient care and education to pioneer new ways to prevent, detect and treat cancer. It is led by Ireland’s leading university, Trinity College Dublin, and Ireland’s largest public teaching hospital, St James’s Hospital.
In 2025, TSJCI was the first cancer centre in Ireland to be accredited and designated as a Comprehensive Cancer Centre by the Organisation of European Cancer Institutes.
Breast Cancer Brain Metastases: How RNA Splicing and R-Loops Shape DNA Repair and Treatment Response
Written by Dr. Jason McGrath (Postdoc Fellow), Mr. Hamed Alwahaibi (Medical student) and Prof. Leonie Young (Scientific Director) - Department
of Surgery, Royal College of Surgeons in Ireland (RCSI)
Breast cancer remains the most common malignancy affecting women worldwide and the second leading cause of cancer-related death. More than two million new cases and approximately 670,000 deaths were reported globally in 2022.1 In Ireland, one in seven women will develop breast cancer, with roughly 3,600 new diagnoses each year.2 Breast cancers are traditionally classified by the receptors they express, including oestrogen receptorpositive, progesterone receptorpositive, human epidermal growth factor receptor-positive, and triple-negative disease.
While these classifications guide treatment in early disease, they do not fully explain why certain tumours progress aggressively or metastasise to the brain.
Brain metastases occur in approximately 10–30 per cent of patients with metastatic breast cancer and are associated with poor survival and significant neurological morbidity.3 Symptoms such as headaches, seizures, and cognitive impairment are common, and treatment options remain limited. Surgery and radiotherapy are frequently used, but systemic therapies are less effective due to restricted drug
penetration across the blood–brain barrier (Summarized in Figure 1). Crucially, breast cancer brain metastases are biologically distinct from primary tumours, with emerging evidence showing unique molecular alterations that may be exploited therapeutically.4
Brain metastases arise most frequently from HER2-positive and triple-negative breast cancers. HER2-targeted therapies have improved outcomes for patients with HER2-positive brain disease, but triple-negative brain metastases lack established molecular targets and are associated with particularly poor
prognosis. This has driven intense interest in identifying alternative vulnerabilities that can be therapeutically exploited.
One of the most important vulnerabilities identified in recent years involves defects in DNA repair. Cancer cells experience high levels of DNA damage and rely heavily on repair pathways to survive. Double-strand breaks are the most lethal form of DNA damage and are normally repaired through homologous recombination, a high-fidelity process requiring the coordinated action of multiple proteins.5 In many breast cancer brain metastases,
Dr. Jason McGrath
Mr. Hamed Alwahaibi
Figure 1
this pathway is impaired due to alterations in key homologous recombination genes, resulting in a state known as homologous recombination deficiency.
This deficiency has clear clinical relevance. Tumours with homologous recombination deficiency are sensitive to PARP inhibitors, a class of drugs that block the repair of single-strand DNA breaks. When these breaks persist, they are converted into double-strand breaks during DNA replication. In cells that cannot perform homologous recombination, this damage is catastrophic, leading to tumour cell death.6 PARP inhibitors such as rucaparib and niraparib have therefore emerged as promising treatments for selected patients with triple-negative breast cancer brain metastases. However, resistance frequently develops, often through restoration of homologous recombination activity, highlighting the need to understand what drives and sustains this repair defect.7
Increasing attention has turned to RNA splicing as a key contributor to homologous recombination deficiency. RNA splicing is the process by which non-coding sequences are removed from newly transcribed RNA to produce mature messenger RNA. Accurate splicing is essential for generating functional DNA repair proteins. When splicing is disrupted, specific exons may be skipped, leading to truncated or dysfunctional proteins, or to reduced expression of critical repair factors altogether. Importantly, exon skipping does not require permanent genetic mutations; it can arise from altered regulation of the splicing machinery and may therefore be reversible or dynamic.8
In breast cancer brain metastases, aberrant splicing patterns have been shown to affect genes involved in DNA repair, functionally mimicking classical homologous recombination deficiency. From a clinical perspective, this is significant because it suggests that tumours without obvious DNA repair gene mutations may still behave as homologous recombination–deficient and respond to PARP inhibitors.
The PMI Pharma Summit 2026
Disrupted splicing is also closely linked to the formation of RNA:DNA hybrids, known as R-loops. R-loops form when newly synthesised RNA hybridises back to its DNA template, leaving a stretch of single-stranded DNA exposed. Under normal conditions, R-loops are tightly regulated and play physiological roles in gene expression. However, when splicing is inefficient or exons are skipped, RNA can remain associated with DNA for longer periods, promoting abnormal R-loop accumulation.9
Persistent R-loops are a potent source of genomic instability. They interfere with DNA replication and transcription and expose singlestranded DNA to damage. Cells normally rely on homologous recombination proteins to resolve these structures and repair the resulting damage. When homologous recombination is compromised, R-loops accumulate further, creating a vicious cycle of DNA damage and repair failure.
This has direct therapeutic implications. PARP inhibition has been shown to exacerbate R-loop accumulation, increasing DNA damage beyond a tolerable
The Pharmaceutical Managers’ Institute (PMI) Annual Pharma Summit takes place this year at Croke Park on Thursday, March 2026 – an unmissable event that promises to be a cornerstone of industry innovation and collaboration.
The theme for the day is: “Transform & Thrive,” and the team behind the Pharma Summit will be exploring this topic across the backdrop of continuous change, disruption and evolution in across the industry, with stakeholders and . This flagship event will bring together a wide range of attendees, including industry leaders, subject matter experts, and thought leaders.
In February, the PMI will also be hosting a Stakeholder Briefing – CPU update. They will host Linda Fitzharris, Head of the CPU for a breakfast briefing on 5th February. Linda will share some insights into the role of the CPU in managing the complex processes of medicine pricing and reimbursement under the various national health schemes.
threshold in tumour cells.10 In breast cancer brain metastases with splicing defects, exon skipping in repair genes, and elevated R-loops, PARP inhibitors may therefore be particularly effective barrier (Summarized in Figure 2). Clinically, R-loop burden and splicing alterations could serve as functional biomarkers of treatment response, helping to identify patients most likely to benefit from PARP inhibition and to monitor emerging resistance. Taken together, these findings reshape how homologous recombination deficiency is defined in breast cancer brain metastases. Rather than being driven solely by fixed genetic mutations, DNA repair failure can arise from altered RNA processing, exon skipping, and dysregulated transcription. For patients with triple-negative brain metastases, this biology offers a compelling opportunity to refine patient selection, improve therapeutic precision, and extend the benefits of PARP inhibitors to a broader group of patients with urgent unmet clinical need.
References available on request
This will be an essential stakeholder briefing for all PMI members, particularly those involved in market access, commercial and external stakeholder management roles. It takes place from 7.30-9.30am at The Address, Citywest, Dublin 22. Visit www.thepmi.com for more information.
Figure 2
Lung Cancer
The Lung Health Check: Shifting the Paradigm in Lung Cancer Care
As of early 2026, lung cancer remains the leading cause of cancer-related mortality in Ireland. Although 5-year survival rates have improved substantiallydoubling from 12% in 2004–2008 to approximately 24% in recent reports - lung cancer still accounts for 20% of all cancer deaths nationwide, equating to more than 2,000 annual deaths. Data from the National Cancer Registry (NCRI) and the National Cancer Control Programme (NCCP) indicate that approximately 2,600 new cases are diagnosed annually. Alarmingly, incidence is projected to rise sharply, with estimates suggesting an increase of over 100% by 2045. A central message in lung cancer is the profound survival difference by stage at diagnosis. Five-year survival for early-stage disease now routinely exceeds 90%, whereas survival for stage IV disease remains below 10%. Despite advances in diagnostics and treatment, outcomes remain heavily determined by how early the disease is detected.
To streamline the diagnosis, Rapid Access Lung Cancer Clinics (RALCCs) were established in 2009 across eight designated cancer centres. These clinics currently account for approximately 50% of all lung cancer diagnoses in Ireland, with over 95% of referred patients seen within 10 to 15 working days. However, despite access to this streamlined service, the majority of RALCC-diagnosed lung cancers are stage III or IV. A substantial proportion of patients with lung cancer - approximately 25% - still first present through emergency departments with symptomatic disease, the overwhelming majority of whom have advanced-stage disease.
The fundamental challenge is that early-stage lung cancer is typically asymptomatic. Most early cancers are detected incidentally on imaging performed for unrelated reasons. As a result, at a national level, the detection of early lung cancer - when outcomes are much more favourable - is largely left to chance. Consequently, approximately 67% of lung cancer patients in Ireland are diagnosed at stage III or IV, when treatment options, although improving, are significantly less likely to be curative.
The Rationale for Screening: Evidence from Global Trials
Lung cancer screening hinges on its ability to shift the stage at diagnosis from advanced to early. The data supporting the shift to earlier diagnosis comes primarily from two major international randomised controlled trials and recent real-world data from the UK. The NELSON trial (DutchBelgian) compared Low-Dose Computed Tomography (LDCT) screening against a control group with no screening (standard pathways). In the screening arm, 69% of cancers were detected at stage I. In the control arm nearly 70% of cancers were stage III or IV. This “stage shift” translated into a 24% reduction in lung cancer deaths in men and a 33% reduction in women.
The NLST trial (USA) compared LDCT against chest x-ray (rather than no screening). LDCT detected significantly more stage IA lung cancers, compared with chest x-ray. This early detection demonstrated a 20% overall reduction in lung cancer mortality, proving that identifying these cancers early directly saves lives, rather than just finding
Written by Malcolm Herron, Sandra Roche, Ciara Fay, David O’Reilly, Daniel John Ryan and Jarushka Naidoo
Lung Cancer in Ireland: The Current Landscape (2025/2026)
them earlier with the same outcome. Data from the NHS pilot programmes showed that 76% of screening detected cancers were stage I or II. Nationally, less than 30% of cancers are caught at these early stages via standard diagnostic pathways.
Finally, a number of countries in Europe (UK, Croatia) and internationally (Australia) have fully funded public programmes which we unfortunately do not have.
These trials established that screening high-risk populations, typically those aged 55–74 with a significant smoking history, can pivot the diagnostic paradigm from late-stage disease to early-stage surgical resection.
Who Should We Screen? RiskBased Precision
Around 90% of lung cancers occur in current or former smokers, yet most people who smoke will never develop lung cancer. Screening all ever-smokers, while theoretically highly sensitive, is inefficient and economically unsustainable. Such an approach captures a large number of individuals with low cumulative tobacco exposure and low absolute risk, diluting the benefit of screening and increasing relative cost.
To enrich the screened population, eligibility criteria have traditionally relied on age and smoking intensity. The United States Preventive Services Task Force currently recommends annual low-dose CT screening for adults aged 50-80 years with a ≥ 20 pack-year smoking history who either currently smoke or quit within the past 15 years. This was supported by the NLST which originally adopted a ≥ 30 pack-year cut-off, demonstrating a number needed to screen of approximately 320 individuals to prevent one lung cancer death. However, reliance on pack-year history alone may preferentially select older individuals.
In response, multivariable risk prediction models have been developed to estimate an individual’s lung cancer risk rather than relying on pack-year history alone. The Prostate Lung Colorectal Ovarian (PLCO2012) is a validated multivariate regression tool that estimates 6-year lung cancer risk using variables including age, ethnicity, education, BMI, COPD status, personal history of cancer, family history of lung cancer, and detailed smoking history. When applied retrospectively to NLST data, PLCO2012 identified 12.4% more lung cancers while screening 8.8% fewer individuals than the standard pack-year criteria.
Similarly, the Liverpool Lung Project (LLP) risk model was developed using a case-control design comparing 579 lung cancer cases with 1,157 matched controls, identifying additional risk factors beyond smoking alone. The updated LLPV2 model estimates 5-year lung cancer risk and uniquely incorporates asbestos exposure, prior pneumonia or bronchitis, and early-onset family history of lung cancer (diagnosis before 60). The NHS lung cancer screening guidelines currently recommend screening those aged 55-74 and meet the risk threshold on either, LLPV2 ≥ 2.5% or PLCO2012 ≥ 1.51%.
In addition to reducing mortality, targeted lung cancer screening with LDCT has been shown to be cost-effective. Economic evaluations from UK pilot programmes have demonstrated cost-effectiveness comparable or favourable to established national screening programmes such as breast and colorectal cancer screening. The use of risk prediction models to enrich the screened population through community-based models maximises uptake and minimises downstream emergency presentations.
Sustained
*Updated PFS at 6.2-year median follow-up is an investigator-assessed exploratory descriptive ad hoc endpoint. The 5-year PFS rates are based on estimates derived from the 6.2-year median follow-up Kaplan-Meier curves: overall population HR 0.66 (95% CI, 0.55-0.78); HRd population HR 0.51 (95% CI, 0.40-0.66); HRp population HR 0.67 (95% CI, 0.50-0.89). PFS results from the long-term follow-up of the PRIMA study were concordant with the primary analysis. In the PRIMA primary analysis, the primary endpoint was PFS in the HRd population followed by PFS in the overall population, as determined on hierarchical testing. PFS was evaluated by BICR. HRd population HR 0.43 (95% CI, 0.31-0.59, P<0.001); overall population HR 0.62 (95% CI, 0.50-0.76, P<0.001).1,2 †ZEJULA capsules and tablets can be taken at any time of day, at approximately the same time each day.3
1. Monk BJ, Barretina-Ginesta B, Pothuri I, et al. Niraparib first-line maintenance therapy in patients with newly diagnosed advanced ovarian cancer: final overall survival results from PRIMA/ENGOT-OV26/GOG-3012 trial. Ann Oncol. 2024 Sep 14:S0923-7534(24)03762-1. doi:10.1016/j.annonc.2024.08.2241. Online ahead of print.
2. González-Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391-2402.
3. ZEJULA (niraparib). Summary of Product Characteristics. GlaxoSmithKline; 2024. Available at: https://www.medicines.ie/medicines/zejula-100-mg-film-coatedtablets-35495/spc [Accessed: October 2025].
4. González-Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25)(suppl):2391-2402.
Zejula 100 mg film-coated tablets Abbreviated Prescribing Information (Refer to Summary of Product Characteristics (SmPC) before prescribing).
PRESENTATION: Each tablet contains niraparib tosylate monohydrate equivalent to 100 mg of niraparib. Each tablet contains 34.7 mg lactose. INDICATIONS: 1. Monotherapy for the maintenance treatment of adult patients with advanced epithelial (FIGO Stages III and IV) high-grade ovarian, fallopian tube or primary peritoneal cancer who are in response (complete or partial) following completion of first-line platinum-based chemotherapy. 2. Monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed high grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete or partial) to platinum-based chemotherapy. POSOLOGY AND ADMINISTRATION: Treatment should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. Zejula tablets should be taken without food (at least 1 hour before or 2 hours after a meal) or with a light meal. Patients should be encouraged to take their dose at approximately the same time each day. Bedtime administration may be a potential method for managing nausea. First-line ovarian cancer maintenance treatment: 200 mg taken once daily. However, for those patients who weigh ≥ 77 kg and have baseline platelet count ≥ 150,000/μL, the recommended starting dose is 300 mg, taken once daily. Recurrent ovarian cancer maintenance treatment: 300 mg once daily. If patient weighs less than 58 kg a starting dose of 200 mg may be considered. Treatment should be continued until disease progression or toxicity. Missing dose: If patients miss a dose, they should take their next dose at its regularly scheduled time. For dose modifications due to adverse reactions see SmPC. Elderly (≥ 65 years): No dose adjustment necessary; limited clinical data in patients aged >75 years. Renal impairment: No dose adjustment with mild to moderate renal impairment. Use with caution in severe renal impairment or end stage renal disease undergoing haemodialysis. Hepatic impairment: No dose adjustment with mild hepatic impairment (either aspartate aminotransferase (AST) > upper limit of normal (ULN) and total bilirubin (TB) ≤ ULN or any AST and TB > 1.0 x – 1,5 x ULN). For patients with moderate hepatic impairment (any AST and TB > 1.5 x - 3 x ULN) the recommended starting dose of Zejula is 200 mg once daily. Use with caution in patients with severe hepatic impairment (any AST and TB > 3 x ULN) patients. Patients with ECOG performance status 2 to 4: No clinical data. Paediatric population: No data available. CONTRAINDICATIONS: Hypersensitivity to niraparib or to any of the excipients. Breast-feeding. WARNINGS/PRE-
Hypothetical patient representations. HRQoL was preserved in patients receiving ZEJULA throughout treatment (exploratory analysis; data cut-off: primary analysis), while delivering sustained PFS rate benefit vs placebo at 6.2-year median follow-up (per investigator-assessed exploratory descriptive ad hoc analysis).1,2,4*
CAUTIONS: Haematologic adverse reactions: Complete blood counts should be done weekly for the first month, followed by monthly monitoring for the next 10 months and periodically after this time is recommended to monitor for clinically significant changes in any haematologic parameter. If a severe persistent haematologic toxicity develops that does not resolve within 28 days following interruption, Zejula should be discontinued. Myelodysplastic syndrome/acute myeloid leukaemia (MDS/AML): For suspected MDS/ AML or prolonged haematological toxicities the patient should be referred to a haema- tologist. If MDS/AML is confirmed, Zejula should be discontinued, and the patient treated appropriately. Hypertension, including hypertensive crisis: Pre-existing hypertension should be adequately controlled before starting Zejula treatment. Blood pressure should be monitored at least weekly for two months and monitored monthly afterwards for the first year and periodically thereafter during treatment with Zejula. Hypertension should be medically managed as well as adjustment of the Zejula dose if necessary. Zejula should be discontinued in case of hypertensive crisis or if medically significant hypertension cannot be adequately controlled. Posterior Reversible Encephalopathy Syndrome (PRES): PRES is a rare, reversible, neurological disorder which can present with rapidly evolving symptoms including seizures, headache, altered mental status, visual disturbance, or cortical blindness, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). In case of PRES, it is recommended to discontinue Zejula and to treat specific symptoms including hypertension. The safety of reinitiating Zejula therapy in patients previously experiencing PRES is not known. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take Zejula. Patients who take Zejula may experience asthenia, fatigue, dizziness or difficulties concentrating. Patients who experience these symptoms should observe caution when driving or using machines. INTERACTIONS: Caution in combination with vaccines, immunosuppressant agents or with other cytotoxic medicinal products. Caution when combined with active substances the metabolism of which is CYP3A4-dependent and, notably, those having a narrow therapeutic range (e.g. ciclosporin, tacrolimus, alfentanil, ergotamine, pimozide, quetiapine, and halofantrine). Caution when combined with active substances the metabolism of which is CYP1A2-dependent and, notably, those having a narrow therapeutic range (e.g. clozapine, theophylline, and ropinirole). Caution when combined with sub- strates of BCRP (irinotecan, rosuvastatin, simvastatin, atorvastatin, and methotrexate). Increased plasma concentrations of co-administered medicinal products that are substrates of MATE1 and -2 (e.g. metformin) cannot be excluded. Caution when niraparib is combined with active substances that undergo an uptake transport by OCT1 such as metformin. Fertility, pregnancy and lactation: Fertility: No clinical data. Zejula should not be used during pregnancy or in women of childbearing potential not willing to use highly effective contraception during therapy and for 6 month after receiving the last dose. A pregnancy test should be performed on all women of childbearing potential prior to treatment. Pregnancy: Zejula should not be used during pregnancy. Breast-feeding: Contraindicated during administration and for one month after last dose. UNDESIRABLE EFFECTS: Very common (≥ 1/10): UTI, thrombocytopenia, anaemia, neutropenia, leukopenia, decreased appetite, insomnia, headache, dizziness, palpitations, hypertension, dyspnoea, cough, nasopharyngitis, nausea, constipation, vomiting, abdominal pain, diarrhoea, dyspepsia, back pain, arthralgia, fatigue, asthenia. Common (≥ 1/100, < 1/10): Bronchitis, conjunctivitis, MDS/AML, hypersensitivity, hypokalemia, anxiety, depression, cognitive impairment, dysgeusia, tachycardia, epistaxis, dry mouth, abdominal distension, mucosal inflammation, stomatitis, myalgia, photosensitivity, rash, peripheral oedema, increased GGT, AST, ALT, AKK Phos, creatinine, decreased weight. For more details on undesirable effects, see SmPC. Marketing Authorisation (MA) Holder: GlaxoSmithKline (Ireland) Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland. MA Nr: EU/1/17/1235/004. Legal category: POM A. Date of preparation of API: July 2025. Code: PI-12578. Further information available on request from GlaxoSmithKline, 12 Riverwalk, Citywest Business Campus, Dublin Tel: 01-4955000.
Adverse events should be reported directly to the Health Products Regulatory Authority (HPRA) on their website: www.hpra.ie. Adverse events should also be reported to GlaxoSmithKline on 1800 244 255 or via online form https://gsk.public.reportum.com.
Lung Cancer
These data provide reassurance that lung cancer screening is not only clinically effective but a viable and sustainable public health intervention.
The Beaumont RCSI Irish Cancer Society Lung Health Check Pilot
In May 2025, the first mobile Lung Health Check pilot was launched in Ireland, a collaboration between Beaumont Hospital, the Royal College of Surgeons in Ireland (RCSI), with funding from the Irish Cancer Society, and the EU4Health SOLACE consortium.
The aim of this pilot is to assess the feasibility of community-based lung cancer screening incorporating a Lung Health Check.
How it Works:
• Target Population: Individuals aged 55–74 who are current or former smokers.
• Recruitment: Participants are identified through GP records in North Dublin and the North-East region and invited via letter.
• Pre-screening: Eligible individuals undergo a telephone risk assessment (using the PLCO2012 and LLPV2 risk models). High-risk candidates are invited to a mobile scanning unit.
• Community Access: To reduce barriers, mobile units are stationed in familiar locations, such as Croke Park and local GAA clubs.
• Clinical Components: The visit includes a respiratory health assessment, baseline spirometry, smoking cessation support, and an LDCT scan.
The Beaumont RCSI Irish Cancer Society Lung Health Check (LHC) is Ireland’s first lung cancer screening pilot, developed in response to European Union and Irish Government recommendations that all EU member states initiate screening pilots to inform future national policy. The LHC represents the early-detection pillar of a wider lung cancer outreach programme within the Beaumont RCSI Cancer Centre (PI/Chair: Naidoo, Pillar Leads: Glavey, Dowling, Ryan, Redmond), spanning screening, diagnosis, treatment, and survivorship, and is the largest single research grant for lung cancer in Ireland’s history. The goal of the LHC pilot was to determine whether community-based, mobile LDCT screening is feasible within the Irish healthcare system. The pilot operates in North Dublin and the North-East using mobile units equipped with LDCT scanners and clinical assessment facilities, operated
by Alliance Medical Diagnostic Imaging. The units are staffed by nurses, radiographers, and administrative personnel and rotate through four community locations hosted by local GAA clubs, including Fingallians GAA (Swords), An Cumman Uí Thuathail (Ayrfield, D13), O’Raghallaighs CLG (Drogheda), and Croke Park. This enables screening in familiar, accessible settings rather than hospitals. Recruitment is GP-led in collaboration with the Centric Health Network, with patients aged 55-74 years in participating practices invited by letter. Following an initial telephone assessment, participants undergo risk assessment using PLCO2012 and LLPV2 models, ensuring targeted screening of those with the highest risk.
Individuals who meet the screening criteria are invited to attend a LHC appointment, during which they undergo respiratory assessment, spirometry to identify undiagnosed airflow obstruction suggestive of COPD, targeted smoking cessation intervention with referral to HSE services, and a LDCT scan. The programme integrates cancer screening with broader lung health benefits. CT scans are read by trained thoracic radiologists at Beaumont Hospital, supported by artificial intelligence, with followup intervals determined according to established guidelines. Participants attend for follow-up LDCT scan at 1 year if there are no significant findings at baseline LDCT. Suspected lung cancer cases are referred to RALCC in Our Lady of Lourdes Hospital, Drogheda or Beaumont Hospital. Incidental findings and newly diagnosed airflow obstruction are managed according to protocol and referred to a hospital-based specialist or the participant’s GP. The pilot concludes after the second screening round, at which point ongoing clinical care transitions fully into routine hospital services.
Future Directions
The LHC represents a pivotal transition point for lung cancer care in Ireland, moving from a predominantly reactive, incidental or symptom-led model toward proactive, population-based early detection. As Ireland’s first lung cancer screening pilot, LHC is designed not only to detect cancers, but also to evaluate whether a targeted, communitybased screening model can function effectively within the Irish healthcare system.
The immediate future of the trial will focus on demonstrating that high-risk individuals, recruited via GP-led invitation and identified through validated multivariate
risk models, can be successfully engaged outside traditional hospital settings. By embedding mobile CT units within community hubs, integrating spirometry, smoking cessation, and structured pathways for incidental findings, the LHC mirrors best-performing UK programmes while testing their transferability to Ireland. In doing so, the trial will provide real-world evidence on participation rates, diagnostic yield, stage shift, and referral capacity.
Beyond the pilot, the implications for lung cancer care in Ireland are significant. Lung cancer remains the leading cause of cancer death nationally, largely because 70% of patients present with advanced disease and a quarter first present as emergencies. The LHC directly addresses this, detecting early-stage curable disease. If feasibility, acceptability, and costeffectiveness are demonstrated, as observed in the UK, the pilot will provide the evidence for a national lung cancer screening programme aligned with EU policy direction and NHS England’s planned rollout by 2029.
In the longer term, lung cancer care in Ireland should integrate early detection, chronic lung disease identification, smoking cessation, and equal access to a preventative pathway. A transition from pilot to national programme would not only reduce lung cancer mortality and emergency hospital presentations, but also address health inequalities affecting socioeconomically disadvantaged and smoking-exposed populations. The objective is to transition lung cancer from a symptomatic emergency presentation to a screen-detected, manageable condition.
Trial
Progress and Conclusions
To date, we have recruited more than1800 of our intended 2183 participants. This is on account of a remarkably high community engagement of more than 70% which is far in excess of the participation experienced in most lung cancer screening programmes. We hope to complete accrual in February 2026 - ahead of schedule. Once recruitment has been completed, we aim to publish the studyrelated data in late 2026.
Acknowledgements: We would like to acknowledge the pivotal contribution of Centric Health, Alliance Medical and the GAA Clubs generously supporting this pilot - Fingallians GAA (Swords), An Cumman Uí Thuathail (Ayrfield, D13), O’Raghallaighs CLG (Drogheda), and Croke Park. We would also like to acknowledge our primary funders, the Irish Cancer Society.
University Hospital Galway Showcase Global Leadership News
University Hospital Galway (UHG), a recognised leader in interventional radiology, recently showcased the expertise of its Interventional Radiology Department by hosting live endovascularcases for the prestigious LINC (Leipzig Interventional Congress) international conference, which took place in Germany on 27 January.
UHG is the only hospital in Ireland or the UK broadcasting live procedures to LINC, the longestrunning, largest, and most highly respected live endovascular meeting in the world. The hospital has been performing live case broadcasts for LINC since 2013, demonstrating international excellence in deep venous intervention.
During the event, complex endovascular procedures were performed live on camera in Galway and transmitted to an audience of over 2,000 delegates in Leipzig, with thousands more viewing online worldwide. Attendees were able to interact directly with the clinical teams, asking questions as procedures were carried out in real time.
University Hospital Galway is recognised as one of the leading European centres for the treatment of acute deep venous thrombosis, and the broadcast provided an opportunity to share this expertise with an international audience of peers.
Commenting on the event, Professor Gerry O’Sullivan, Consultant Interventional Radiologist at University Hospital Galway, said: “It is an honour and a privilege to showcase the skill, expertise and teamwork demonstrated daily by our Interventional Radiology unit to a worldwide audience.
“This year, our team showcased the Recana Device, the first fully integrated mechanical thrombectomy catheter specifically designed to treat in-stent restenosis, a condition in which veins narrow again after stenting. Last July, UHG became the first hospital in the world to successfully conduct the first patient trial of the Recana Device, which marked a significant advancement in the treatment of chronic venous disease.”
Breast Cancer
Endocrine Therapy for High-Risk
Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor-Negative Breast Cancer: A Case Report
Written by Aftab A. Khan,
Dr Kate Coakley and Professor Catherine M. Kelly - Department of Medical Oncology, Mater Private Hospital, Dublin
Introduction
Estrogen receptor-positive (ER+), human epidermal growth factor receptor-2 negative (HER2-) breast cancer represents 70% of all breast cancers diagnosed. Five years of endocrine therapy (ET) with an aromatase inhibitor or the selective estrogen receptor modulator, tamoxifen substantially reduces the risk of recurrence. However up to 30% of patients with high-risk clinical and/or pathologic features may experience an incurable distant recurrence, many in the first few years. Small molecule inhibitors of the cyclin dependent kinases 4 and 6 (CDK4/6 inhibitors) in combination with ET have consistently improved outcomes in women with metastatic (stage IV) ER+ HER2- breast cancer. Subsequently these agents in combination with ET have been found to be effective in reducing the risk of recurrence in women with high-risk early-stage breast cancer and are now approved by the European Medicines Agency (EMA) in this setting also.
Despite intensive combination therapy with aromatase inhibitors and CDK4/6 inhibitors in high-risk early-stage disease a proportion of patients will experience early relapse due to primary endocrine resistance. Activating mutations in the phosphatidylinositol 3-kinase (PI3K) signaling pathways are central in driving tumour progression in this setting. The PIK3CA inhibitor, inavolisib improved overall survival in women who experience relapse during or within one year of completing adjuvant ET. This agent also has EMA approval however funding is awaited before Irish patients can access this drug.
We present a case report detailing the clinical course of a premenopausal woman referred with symptomatic multifocal, multicentric, node-positive, high-grade, ER+, HER2- breast cancer. She underwent intensive neoadjuvant therapy, followed by a mastectomy and axillary clearance, radiation and intensive endocrine therapy with an aromatase
inhibitor, a CDK4/6 inhibitor, ovarian function suppression and zoledronic acid. Despite this, she developed biopsy-proven locoregional recurrence and lung metastases within two years from diagnosis. Molecular profiling of the primary tumour revealed PIK3CA and AKT mutations, and informed subsequent treatment options. Our case highlights a move away from single agent endocrine therapy for women with disease at high risk of relapse (i.e. larger tumours, lymph node positivity, high tumour grade) and the gradual shift towards earlier molecular profiling of breast cancers to inform treatment decisions at first relapse.
Case Presentation
A 49-year-old woman was referred to breast health with distortion of her right breast. Core biopsies identified invasive ductal carcinoma, grade III that was strongly ER and PR positive and HER2 negative in the breast and axillary lymph nodes. Baseline imaging with mammography and ultrasound demonstrated disease in all quadrants of the breast and at least five abnormal axillary nodes. Staging with a CT chest, abdomen and pelvis and nuclear bone scan showed no distant metastases. She proceeded to neoadjuvant weekly paclitaxel for 12 weeks followed by 4 cycles of doxorubicin and
cyclophosphamide given every two weeks for 4 cycles. On completion of neoadjuvant chemotherapy in December 2023 she had a right mastectomy and axillary lymph node dissection. There was extensive residual disease within the breast and lymph nodes. In February 2024 she began adjuvant endocrine therapy with ovarian function suppression, and letrozole. Abemaciclib was started in July 2024 after it was funded by the HSE. Adjuvant zoledronic acid was given every 6 months to reduce risk of recurrence and to maintain her bone density. She received radiation to the chest wall and regional nodes from April 2024 until May 2024.
Imaging in May and November 2024 demonstrated no metastatic disease; pelvic MRI and breast ultrasound were unremarkable. Clinical examination through early 2025 remained normal. In September 2025, an ultrasound to investigate a different issue identified a new subpectoral lymph node. Biopsy confirmed a local recurrence and she was subsequently restaged with a PET/CT which found multiple sub-centimetre FDG-avid pulmonary nodules and an FDG avid subpectoral node. As the recurrence occurred less than two years after completing neoadjuvant chemotherapy and during active endocrine therapy with letrozole, ovarian function
suppression and abemaciclib, the disease met criteria for primary endocrine resistance.
Molecular Profiling
Oncomine profiling of the primary tumour revealed PIK3CA and AKT mutations. The patient was eligible for a clinical trial in the START phase I unit at the Mater Misericordiae University Hospital where she has been commenced on a PIK3CA inhibitor, fulvestrant and palbociclib. The AKT inhibitor capivasertib is also licensed in this setting and would be an important treatment option for her however it is not funded by the HSE at this time.
Discussion
Endocrine therapy is the cornerstone of care for ER+, HER2- breast cancer. The aromatase inhibitors e.g. letrozole, anastrozole and exemestane and the selective estrogen receptor modulator tamoxifen have been used as single agents. Recently drugs that inhibit the cyclin dependent kinases, CDK4 and CDK6 inducing cell cycle arrest have been approved for use in the early breast cancer setting to reduce the risk of recurrence. Our patient was treated with abemacicib based on the MonarchE phase III trial results. This phase III trial enrolled women with high-risk disease i.e. at least 4 positive lymph nodes, or 1-3
Aftab A. Khan
Dr Kate Coakley
Professor Catherine M. Kelly
Breast Cancer
nodes but high-grade disease or a tumour ≥5cm. In this trial patients received the CDK4/6 inhibitor abemaciclib for 2 years with standard ET versus ET alone. At 5 years follow up almost 1 quarter of women receiving ET alone had experienced a recurrence compared to 16% receiving abemaciclib with ET. The NATALEE phase III trial randomised women to the CDK4/6 inhibitor ribociclib with ET for 3 years versus ET alone. Women with intermediate to high-risk disease were enrolled and at 5 years the absolute reduction in risk of recurrence for the addition of ribociclib was 4.5%.
The CDK4/6 inhibitors in combination with ET are also the standard of care in the first line treatment of relapsed or de novo metastatic ER+, HER2breast cancer. Determining the mechanisms of endocrine resistance and overcoming it in patients who relapse during adjuvant treatment or in those who have a suboptimal response to first line treatment for metastatic disease is being actively researched. Activating mutations in the phosphatidylinositol 3-kinase (PI3K) signaling pathways have been identified in 30-40% of all ER-, HER2- breast cancers and have been shown to play a central role in tumour progression. The INAVO120 phase III, double-
blind placebo-controlled trial compared palbociclib and fulvestrant in combination with either inavolisib or placebo as first-line therapy for patients with PIK3CA-mutated, ER+, HER2metastatic breast cancer. Eligible patients had no prior therapy for advanced disease, had disease recurrence during or within 12 months after completing adjuvant ET. Progression free survival was significantly improved in the inavolisib-containing arm, (17.2 months versus 7.3 months). The objective response rate was 62.7% in the inavolisib arm and 28.0% in the placebo arm, and the median response duration was 19.2 months versus 11.1 months. Overall survival was significantly improved in the inavolisib arm, with a median OS of 34.0 months versus 27.0 months. This drug although approved by the EMA is not currently funded by the HSE so was not available to our patient. Fortunately, she was able to gain access to a clinical trial testing a similar agent.
In conclusion, for patients with high-risk ER+, HER2- breast cancer ET with an aromatase inhibitor plus/minus ovarian function suppression depending on menopausal status and a CDK4/6 inhibitor is now a standard of care. A subgroup of patients like our patient will develop early recurrences despite intensive
Oncology News
adjuvant therapy. Sequencing of the tumour at relapse can guide the first line treatment for metastatic disease. Common driver mutations in PIK3CA or AKT as found in our patient represent important drug targets. These drugs for this indication are approved by the FDA and EMA however they are currently unavailable to Irish patients due to lack of funding. Clinical trials although not a substitute for drug funding can provide a means to access similar agents until funding is available for some patients.
References available on request
The Authors
Aftab A. Khan
Dr Aftab Khan is a medical oncology registrar managing patients with solid tumours and systemic anticancer therapies. His interests include MDT-led care, toxicity management, and evidence-based oncology practice.
Kate Coakley
Medical Oncology Registrar in the Mater Misericordiae University Hospital and Mater Private Hospital. A member of the Royal College of Physicians of Ireland.
Catherine M. Kelly
Consultant Medical Oncologist, UCD Clinical Professor and Head
Transforming Breast Cancer Reduction
Artificial intelligence is set to transform breast cancer detection and treatment in Ireland, with major international trials showing AIsupported screening can detect up to 30% more breast cancers while dramatically reducing pressure on overstretched radiology services. Breast Cancer Ireland, Ireland’s leading breast cancer charity, says the findings mark a turning point for precision oncology, as AI, advanced imaging and targeted therapies reshape cancer care in Ireland in 2026 and beyond.
A recent survey of more than 1500 women attending the symptomatic clinic at the Beaumont Breast Centre, Dublin examined Irish patient’s views on the role of AI in healthcare, and in detection of breast cancer. Almost half of women (46%) agree that the use of AI in healthcare is a good idea, and 61% were comfortable with their mammogram being read by both a radiologist and an AI tool. However, patients remain
of Research Mater Private Hospital. She served as the Head of Breast Medical Oncology at the Mater University Hospital from 20102021. She was elected Chair of the Breast Cancer Group at Cancer Trials Ireland between 2014-2020. Professor Kelly is a member of multiple international steering committees for breast cancer trials. She is a faculty member and serves on the ST Gallen Breast Cancer Consensus Guideline Panel. She is the Irish PI for the pivotal clinical trials. Prof Kelly was the Medical Director of the Mater Clinical Trials Research Unit from 2017 until 2021. Under her leadership the unit was the highest accruing cancer trials centre in Ireland. She has served as international editor for the breast cancer section of the Journal of Clinical Oncology and has written editorials for multiple high impact journals including the JCO, Lancet Oncology and the Journal of the National Cancer Institute. Prof Kelly undertook her specialist medical oncology training in Ireland and the UK. She completed a Fellowship in Breast Medical Oncology in Toronto where she received the Thomas & Edna Naylor Memorial Award for her research examining tamoxifen drug interactions. Prior to returning to Ireland in 2010 she was the Susan G. Komen Interdisciplinary Breast Cancer Fellow, at MD Anderson Cancer Center, Houston, Texas, USA.
somewhat cautious of this new technology with two thirds (66%) claiming that they would still prefer a radiologist to review their mammogram – even if AI was shown to be more accurate.
Two landmark studies – the MASAI trial in Sweden and the AI-STREAM trial in South Korea – show that AI used alongside specialist breast radiologists significantly improves cancer detection without increasing false alarms. Crucially, AI also identifies smaller, earlier-stage cancers, when treatment is most effective.
Professor Arnold Hill, Chairman of Breast Cancer Ireland, and Consultant Breast & General Surgeon at the Beaumont RCSI Cancer Centre, said the implications of this for patients and the health system are profound. “This is one of the most important developments we’ve seen in breast cancer screening in decades,” he said.
“For decades, mammography has been the cornerstone of early breast cancer detection. It’s a powerful tool, but it’s not perfect. Some cancers can be challenging to spot on a mammogram, especially in patients with dense breast tissue. With increasing demand for screening, many countries, including Ireland, have a shortage of breast radiologists – the highly-trained doctors who read mammograms. This puts pressure on the system and can lead to delays in diagnosis.”
According to Dr Prof Nuala Healy, Consultant Radiologist at the Breast Cancer Ireland–funded Beaumont Breast Centre, AI is already reshaping how specialists work. “AI gives breast radiologists an additional pair of expert eyes – prioritising the most challenging mammograms, helping detect subtle early cancers and easing routine workload so that radiologists can focus on complex patient imaging and procedures”
Prof Nuala Healy
Impact of Testicular Cancer on the Socio-Economic Health, Sexual Health, and Fertility of Survivors—A Questionnaire Based Survey
Written by M. Raheel Khan1,2,*, Patrice Kearney Sheehan2, Ashley Bazin2, Christine Leonard2, Lynda Corrigan2 and Ray McDermott1,2,3
1School of Medicine, University College Dublin, D04 V1W8 Dublin, Ireland
2Tallaght University Hospital, D24 NR0A Dublin, Ireland
3St Vincent’s University Hospital, D04 T6F4 Dublin, Ireland
Testicular cancer (TC) is a rare tumour (1%) that is mostly diagnosed at a younger age (15 to 35 years).1 Since the advent of the cisplatin-based chemotherapy regimens, the cure rates are reported to be above 95%.2,3 Cisplatin-based multi-drug regimens, first used in 1974, hugely improved the prognosis compared to the precisplatin era.4,5 Moreover, recent decades have seen an increase in the incidence along with an improvement in the survival rate.1 However, these high cure rates come at a cost of late sideeffects, particularly in the patients who receive chemotherapy and radiotherapy.6,7 These side-effects encompass a list of malignant and non-malignant conditions including late recurrences, second malignant neoplasms, cardiovascular disease, renal insufficiency, pulmonary disease, hypogonadism, infertility, and metabolic syndrome.8,9 On top of these complications, a higher incidence of psychological conditions including anxiety and fear of cancer recurrence has also been reported in this cohort.10,11 As most of the survivors are young at the time of diagnosis, they have to brave these complications for decades to come. These physical and psychological complications have been reported to cause a negative impact on the social, economic, and sexual well-being of TC survivors.12,13 Higher levels of disability, unemployment, and sick leave have been reported in recent studies.12
An estimated half of patients have abnormal semen analysis at the time of diagnosis before any treatments.14 Chemotherapy, radiotherapy, and surgeries can further deteriorate sperm production, motility, and erectile function.15,16 As a result, a reduced
rate of paternity is found in TC survivors compared to the normal population, even with pretreatment semen cryopreservation techniques.15 These consequences of treatment also cause the reentry and readjustment problems for the survivors back into society, as they are nervous and anxious about their fertility.17 Many researchers have reported longterm deterioration in sexual health, including erectile dysfunction, sexual dissatisfaction, low sex drive, and orgasmic dysfunction.18 This long-term deterioration in sexual health results from multiple factors, including hypogonadism, anxiety, systemic diseases, impaired body image perception, and a sense of loss of masculinity.18 These complications, in turn, have a significant effect on the social lives of TC survivors, particularly relationships with their partners. Spouses and partners, especially those who began their
relationship after TC diagnosis, are poorly affected in terms of their quality of life and stress response symptoms.19 In addition, long-term side-effects on career and financial well-being are also reported in TC survivors. Jobs and careers can be affected by late comorbidities like peripheral neuropathy and chronic cancerrelated fatigue.20 A systematic review by Schepisi et al. conveyed a concerning outlook of survivors, revealing a higher incidence of joblessness, absenteeism, and neuroticism, especially in those involved in manual labour.17 TC diagnosis and treatments, mainly chemotherapy, have been found to cause financial distress and financial burden on survivors.13
In the last few years, we have seen growing interest in cancer survivorship, with numerous studies performed to look at different aspects of survivorship care. Previously, we investigated
the prevalence of mortality and morbidity in TC survivors attending Tallaght University Hospital in Dublin, Ireland.17,18 Now, this study was conducted to provide an estimation of the socio-economic, sexual, and fertility-related difficulties faced by TC survivors in Ireland. As we believe a multiteam, collaborative model of care will be key to improving the quality of life in survivors, our study aims to identify areas in need of further research and quality improvement initiatives. Our study may also be helpful for other urban populations in the UK and Western Europe.
This article is a revised and expanded version of a paper entitled “Impact of testicular cancer on socio-economic and sexual health of survivors: A questionnaire-based survey”, which was presented at ESMO Sarcoma and Rare Cancer Congress, held in Lugano, Switzerland, on 14 March 2024.21
Testicular Cancer
Figure 1. Socio-economic impact on testicular cancer survivors. Out of the 83 participants who responded on a 5-point Likert Scale, the chart displays the number of responses in each category
Results
This study was conducted from January 2023 to November 2023 in the testicular cancer survivorship clinic of Tallaght University Hospital, Dublin, Ireland. A total of 83 patients completed our survey questionnaire. All patients agreed to participate in this study. A total of 66 data points were left blank on the questionnaires out of 1262 total, with a response rate of 5%.
Socio-Economic Impact
To investigate the socioeconomic impact on the testicular cancer survivors, we asked the participants about changes in five aspects of their livelihood since the diagnosis of cancer. These five parameters included performance/ productivity at work, job security/ stability, career choices, personal financial goals, and relationship with their partners (Figure 1). When asked about performance or productivity at work, 58% (47) reported no effect, 18% (15) reported a minor effect, 6% (5) reported a moderate effect, and 8% (7) reported a significant and very significant effect. Regarding their job security or stability, the responses showed 72% (59) reporting no effect, 6% reporting a minor effect, 7% reporting a moderate effect, 2% reporting a significant effect, and 10% reporting a very significant effect.
Moreover, we gauged the impact of cancer on their career choices as the majority of patients are diagnosed in the early years of their career. On one hand, 13% (11) reported the effect as moderate and 10% reported as very significant; on the other hand, 71% (58) of patients reported no effect on their careers. With respect to the financial implications of cancer diagnosis and treatment, 57% of responders noted no effect on their personal financial goals. On the contrary, 11% reported very significant, 4%
significant, 14% moderate, and 13% minor effects.
Participants were asked if cancer had any effect on their relationship with their partners. Almost twothirds (64%) reported no effect, while the rest reported some effect. Of those who reported some effect, 27% reported a minor effect, 33% reported a moderate effect, 19% reported a significant effect, and 16% reported a very significant effect. Nearly all (96%) of the participants responded to the question asking about any difficulty in finding a new partner since the diagnosis of cancer. The responses revealed that 80% (66) of respondents did not notice any issues in this regard. Among those who experienced any difference, 43% had a minor effect, 31% had a moderate effect, 31% had a significant effect, while only 12% had a very significant effect.
Sexual Health
In this survey, the impact on sexual health was assessed in five different categories, including libido, erectile function, ejaculation, satisfaction with sexual activity, and body image perception (Figure 2). Almost half of our patients (54%) reported no effect on their libido. Although the rest of the participants stated otherwise, with 6% stating a very significant effect, 14% a significant effect, 12% a moderate effect, and 13% a minor effect. Erectile function reportedly remained intact in more than two-thirds of the respondents (70%), while it was affected very significantly in 6% and significantly in 10%. Moreover, 6% reported a minor and 8% a moderate
effect. Similar to erectile function, ejaculation remained intact in 71% of patients. Conversely, 11% described the impact as very significant, 4% described the impact as significant, 8% described the impact as moderate, and 5% described the impact as minor. Over one-third (39%) of our testicular cancer survivors revealed an adverse impact on satisfaction with sexual activity. Of these, the majority stated only a minor-to-moderate effect (54%), and the rest (46%) experienced a significant or very significant effect. Almost half of patients (53%) reported that cancer no effect on their body image perception. At the same time, it was affected to a minor extent in 18%, a moderate extent in 17%, a significant extent in 10%, and a very significant extent in 2% of participants.
Fertility
One third of the participants experienced biological parenthood after the diagnosis of cancer, whereas two thirds of them reported no children since the testicular cancer diagnosis and treatment. It is important to note that nearly half of them (41%) had children before the diagnosis as per survey responses. Only a minority (17%) of the participants reported having trouble with conception; however, 63% reported no issues and 20% chose not to respond. When asked if the fertility issues stayed for more than a year, 54% of them responded in the negative, but 14% were affirmative. Approximately onethird of the respondents (31%) refrained from providing an answer to the question. We asked our
participants if they sought any medical assistance for fertility issues; only 17% of them stated yes, while 67% stated no and 16% chose not to answer.
Discussion
The majority of our patients reported no major effect on their sexual and socio-economic health. Even the fertility rates after TC diagnosis and treatment were similar to the general population, as discussed below.
In this study, 6% of patients reported a very significant effect on their relationship with their partner. These findings are comparable to 5–10% divorce and break-up rates reported in a literature review by Schepisi et al.17 Another similar study at Yale University, USA, reported on the subjective impact of cancer on personal finances in survivors.22
A total of 8.6% of participants reported impact as “a lot,” compared to the 11% in our study who reported the effect as “very significant”. However, the study at Yale was not confined to TC and included all cancer survivors.
Regarding the effect on career and job, 8–10% of responders revealed a very significant effect. Again, these findings are comparable to similar studies performed previously.12,22 For instance, a major survey-based study conducted in Texas, USA, encompassing 4363 cancer survivors, revealed that 8.5% of survivors considered themselves unable to work since diagnosis.23 This study included all cancer survivors, yet the results are
Figure 2. Impact on sexual health of testicular cancer survivors. Out of the 83 participants who responded on a 5-point Likert Scale, the chart displays the number of responses in each category
similar to those in this study. Another study reported 67% of cancer survivors working fulltime jobs after 5–7 years postdiagnosis, although some of them reported difficulties in carrying out their responsibilities.24 Kerns et al. also reported similar outcomes after studying 1815 TC survivors from the US, the UK, and Canada in 2020.12 As many as 10% of the TC survivors were reported to be out of work, with a higher risk of unemployment compared to the age-adjusted general population (odds ratio: 2.67).12 They also reported a relatively higher impact on survivors who received chemotherapy.12
As most TC survivors are young at the age of diagnosis,1 body image perception has a major influence on their emotional, sexual, and social well-being.25 Rossen et al. reported that 17% of TC survivors in Denmark experienced altered body image, which was associated with all parameters of sexual dysfunction as well.25 However, among our cohort of survivors, only 53% reported no effect, while the rest reported some degree of effect. In the same study, 24.4% of survivors reported reduced sexual interest, which is comparable to our finding of 20% reporting a significant and very significant effect on libido.25 A similar trend was seen with respect to erectile function and satisfaction with sexual activity. Our results show 18% reporting a significant or very significant effect on erectile function compared to the 17% reported by Rossen et. al. With regards to satisfaction with sexual activity, 18% of our participants disclosed a major effect, compared to 14% in their study. Our participants’ responses on the Likert scale regarding ejaculation shows a worse impact than reported in other studies. Among the respondents, 15% described the effect as significant or very significant, whereas only 7% reported
ejaculatory problems in the survey discussed above [25]. However, a systemic review by Schesipi et. al. estimates the proportion as between 29 and 44%.17 In TC survivors, ejaculatory problems are mostly related to retroperitoneal lymph node dissection, and outcomes can be improved with nerve sparing techniques.26 This variation could possibly be due to different trends in the surgical treatments in different countries and cancer centres.
The majority (80%) of patients reported no difficulties in finding a new partner, but unfortunately, our survey failed to identify participants already in stable relationships.
Spermon MD reported paternity after cancer in Norwegian TC survivors as 43% in 2003,27 which is higher than the findings of this study (34%). This difference could be due to the general decline in fertility rates witnessed in Europe in the last couple of decades.28 This study is not able to identify participants who were not planning to have any children. Also, a better overview can be obtained through more studies comparing paternity rates within different treatment modalities. The percentage of patients seeking medical assistance for fertility (17%) in this study is the same as reported by Spermon. The infertility rate (failure to conceive for more than 1 year) reported in our cohort of TC survivors (17%) is closer to the general population (16%).28
Participation was notably comprehensive, as every survivor contacted agreed to be involved. We believe one of the main
reasons for the high recruitment was the complete anonymisation of questionnaires. However, this anonymisation also proved to be a major limitation of our study. Due to anonymisation, we were not able to stratify our data based on age at diagnosis, treatment types, stage of cancer at diagnosis, comorbidities, and social parameters, e.g., employment status, relationship status, and education level. This stratification could have been very useful in identifying the risk factors for each of the major implications in the survivorship phase. The size of the cohort in this study was small; a multicentre study with a larger cohort of patients could give us more significant and impactful data. With the small sample size and single-institute design, the results of this study have limited generalisability.
The participants were asked to reflect on their experiences over a period of several years, which makes them vulnerable to recall bias. This bias could have led to a significant over-estimation of the impact. Also, only the survivors attending the survivorship clinic were included in this study, which may lead to a selection bias.
Another considerable limitation comes from the absence of any comparative cohort as a control arm. A comparable control arm of the general population could help us to better define the effect of testicular cancer on these parameters.
The questionnaire we used in this study was developed on an ad hoc basis without previous
validation. Our questionnaire lacks the authenticity and dependability of a validated questionnaire. We believe a comprehensive validated questionnaire to assess these aspects of survivorship care should be developed and used for future studies. Currently available questionnaires (e.g., EORTC QLQ-TC26) lack the focus on late survivorship care.29,30
Given the small scale of this study and the limitations described above, we strongly believe that more research in this area is needed. We recommend multicentre, pseudonymised, and case–control studies to acquire further insight into the subject of TC survivorship.
Conclusions
In this study, we have highlighted the issues being faced by testicular cancer survivors as a result of the late side-effects of cancer and its treatments. Although most of the TC survivors reported no significant effect on their socio-economic and sexual health, some of them struggled with the negative impacts. Regarding sexual health, most of the repercussions were felt in body image perception and libido, while social sequelae mainly included deterioration in one’s relationship with one’s partner. Economic health was affected in some TC survivors due to job instability and reduced performance at work. Survivorship care should attend to these aspects of well-being and should be designed to provide support where needed.
References available on request
Clinical R&D
DANONE APTAMIL 1 FROM BIRTH FIRST INFANT MILK RECALL
The Food Safety Authority of Ireland (FSAI) today advises that individual packs of Aptamil 1 From Birth First infant milk from a specific batch which was subject to a recall by Danone last week due to the potential presence of cereulide, were sold via the Boots.ie website for online sales. The FSAI is advising parents, guardians and caregivers who may have the recalled product at home not to feed them to their infant or young child. The implicated products were manufactured in Ireland by Danone and exported to a number of EU countries, the UK and third countries. However, due to indirect distribution to Ireland from the UK, the implicated packs were sold via the Boots.ie website for online sales.
The product and batch being recalled is as follows:
• Aptamil 1 From Birth First infant milk; pack size:800g
Expiry date: 31-10-2026
The FSAI is advising parents, guardians and caregivers who may have the recalled product at home not to feed it to their infant or young child. If no symptoms are displayed, nothing further needs to be done. If a parent, guardian or caregiver is concerned about the health of their infant or young child, they should contact a healthcare professional.
The FSAI advises that cereulide toxin may be pre-formed in a food and is extremely heat resistant. Consumption of foods containing cereulide toxin can lead to nausea and severe vomiting. Symptoms can appear within five hours. The duration of illness is usually 6 to 24 hours.
This recall is associated with a contaminated raw ingredient which was also implicated in the recent recalls of some batches of infant formula and follow-on formula. An ingredient, ARA oil, which was manufactured in China, was contaminated with cereulide and added as an ingredient in base powder used to make infant formula and follow-on formula. Cereulide is a toxin produced by some strains of the bacterium Bacillus cereus, which can cause food poisoning.
The FSAI and the Department of Agriculture, Food and the Marine who regulate the Danone manufacturing facilities in Ireland,
continue to engage with Danone to ensure that all food safety measures are being taken to protect consumers.
Danone is advising customers to contact its Aptamil careline team on 1800 22 1234 if they have any queries regarding this recall.
IRISH HEART FOUNDATION CALLS ON GOVERNMENT TO MATCH UK BAN ON JUNK FOOD ADS
The Irish Heart Foundation has called on the Government to match landmark restrictions introduced in the UK this week to tackle saturation levels of junk food marketing that are fuelling crisis rates of childhood obesity.
A blanket ban on all paidfor promotion of unhealthy food online, along with a 9pm watershed for TV advertising of such products, came into effect in the UK as part of measures to improve children’s diets and protect their long-term health.
“This is a crucial measure for children’s future health that stands in stark contrast to lax rules which offer little protection to children in Ireland,” said Irish Heart Foundation director of advocacy, Chris Macey.
“Children in the North will now have greater protection than their counterparts here from unscrupulous online targeting tactics by junk brands that we know are rampant. They result in overconsumption, which in turn causes high rates of overweight and obesity that are damaging children’s long-term health.”
The State’s own research estimates that over 85,000 of today’s children on the island –around one in every 20 – will die prematurely due to overweight and obesity; that children as young as eight are presenting with high blood pressure; and teenagers with a cardiovascular age as high as 60.
Mr Macey said despite this, rules in Ireland restricting TV advertising to children have been so thoroughly undermined that even four and five-year olds see over 1,000 junk food ads per year on average.
And there is no meaningful protection at all for children from online junk food marketing that is more personalised, pervasive and therefore potentially even more damaging.
“Research indicates that children are seeing an average of three junk food ads every 10 minutes they’re online – the equivalent of
over 13,000 ads a year based on a conservative estimate of two hours a day spent on digital platforms.
“To put that into perspective, exposure to just over four minutes of unhealthy food advertising increases consumption by an average of 60.0 kcal. But consumption of an extra 48-71 calories a day depending on age is enough to generate weight gain in children over time.”
Mr Macey said many strong policy recommendations, most notably from the Oireachtas Committee on Children and Youth Affairs here had the potential to curb the impact of marketing on children’s food choices, but were not implemented. A recommendation in the 2020 Programme for Government to introduce a Public Health Obesity Act, including examining restrictions on promotion and advertising aimed at children, was also never acted on.
And just last month (December), the need for action was underlined again in policy options set out as part of the Online Health Taskforce submission to Government which included a ban on paid for and brand advertising online.
“The implementation paralysis of successive Governments, which have been well aware of the need for tough restrictions on junk food marketing has to end. The futures of tens of thousands of today’s children depend on it,” said Mr Macey.
“The UK regulations still allow brand ads and we know that infants as young as 18 months can recognise brands. They also don’t address influencer marketing which is playing a rapidly increasing role in junk food promotion. However, the new law is very much a step in the right direction and an essential part of action urgently needed here to protect the long-term health of our children.”
IRISH CANCER SOCIETY WELCOMES NEXT PHASE OF THE LAURA BRENNAN CATCH UP PROGRAMME
The Irish Cancer Society has today welcomed the next phase of the Laura Brennan Catch Up Programme.
The Government today announced that fifth and sixth year pupils who missed out on the HPV vaccine in first year of secondary school will now have a chance to be vaccinated before the end of the 2025/2026 academic year. The Government also announced that
second to fifth year students will be able to access the vaccination through a second phase of the catch-up programme in the 2026/27 academic year.
Since the catch up programme expired in December 2023, the Irish Cancer Society has been calling for a permanent programme to provide unvaccinated young people aged 24 and younger with additional opportunities to protect themselves against HPV.
Steve Dempsey, Director of Advocacy & Communications at the Irish Cancer Society said:
“This is extremely good news for the pupils in fifth and sixth year who missed out on the HPV vaccination in their first year, and for the younger pupils who will benefit from the programme in 2026/2027. It is a safe and effective vaccine that everyone should avail of.”
The Laura Brennan Catch-Up Programme needs to be made permanent – particularly given the Government’s own stated ambition to eliminate cervical cancer by 2040. Young people post-secondary school up to the age of 24 also need a second chance too. For those who have left the school system, it can cost up to ¤600 for a private HPV vaccination. That’s a high price for a potentially life-saving vaccination, which is effective up to the age of 24 according to the National Immunisation Advisory Committee.
“We want to sincerely thank Tánaiste Simon Harris for his leadership on this matter, and Ministers McNeill and Naughton for delivering this schools-based catch up programme. We also want to acknowledge the work of Deputy Roderic O’Gorman, the Chair of the Oireachtas CrossParty Group on Cancer, in keeping this issue on the political agenda and highlighting the experiences of those who were priced out of vaccination by the lack of a catch-up programme.”
PADCEVTM (ENFORTUMAB VEDOTIN) PLUS KEYTRUDA® SIGNIFICANTLY IMPROVES SURVIVAL FOR PATIENTS WITH MUSCLE- INVASIVE BLADDER CANCER REGARDLESS OF CISPLATIN ELIGIBILITY
Astellas Pharma Co. Ltd. recently announced positive topline results from an interim analysis of the Phase 3 EV-304 clinical trial (also known as KEYNOTE-B15) for enfortumab vedotin, a Nectin-4 directed antibody-drug conjugate, in combination with pembrolizumab, a PD-1 inhibitor.
This pivotal study is evaluating the combination as neoadjuvant and adjuvant treatment (before and after surgery) versus standard of care neoadjuvant chemotherapy (gemcitabine and cisplatin) in patients with muscle-invasive bladder cancer (MIBC) who are eligible for cisplatin-based chemotherapy. The trial met its primary endpoint, demonstrating clinically meaningful and statistically significant improvements in event-free survival (EFS), and overall survival (OS), a key secondary endpoint. An additional secondary endpoint of pathologic complete response (pCR) rate for neoadjuvant enfortumab vedotin plus pembrolizumab versus neoadjuvant chemotherapy was also met, and a clinically meaningful and statistically significant improvement was observed. The safety profile for enfortumab vedotin plus pembrolizumab was consistent with the known profile of the treatment regimen.
RESEARCHERS RESHAPING SEX AND GENDER INCLUSION IN MEDICAL RESEARCH
An international research team has created a roadmap for the integration of sex and gender in medical research.
The PAINDIFF network, led by University of Galway Centre for Pain researchers, brings together 32 international experts from 22 institutions across eight countries to address one of the most persistent gaps in biomedical science with barriers and inconsistencies in how
sex and gender are accounted for in study design, data analysis and reporting.
The results of the project have been published in Nature Neuroscience https://www.nature. com/articles/s41593-025-02164-1
Senior author and consortium coordinator Dr Michelle Roche said: “For too long, medical research often assumed that biological mechanisms and treatment responses are the same for males and females. Historically, males were more commonly used in preclinical research and while clinical research included more balanced participation, data was not routinely analysed or separately by sex.”
The research team noted that increasing evidence now shows meaningful differences between males and females in disease prevalence, biological pathways and responses to treatment.
Dr Roche added: “As medical research moves toward personalised medicine, it is increasingly clear that understanding sex and gender differences and similarities is essential for improving health outcomes. The PAINDIFF network has developed guidelines and recommendations for studies in this field. Widespread adoption and implementation of these recommendations will reduce variability, improve reproducibility, and enhance the translatability of research findings, within and beyond the field of pain.”
Professor David Finn, joint first author on the paper, said: “Chronic
pain is a clear example of a condition where there are important sex and gender differences. It affects one in five people worldwide, with women accounting for 70% of those affected. Our new paper aims to reset the basic requirements for medical research, offering 13 actionable recommendations to guide researchers, reviewers, funders and policymakers, creating a clear and comprehensive roadmap for integrating sex and gender.”
The recommendations include five universal principles applicable across all types of research — such as including both males and females as standard practice, and analysing and reporting data by sex. They also address how gender, distinct from biological sex, should be meaningfully incorporated into research frameworks.
Professor Brian McGuire, joint first author, said: “Historically, there have been deficits, barriers and inconsistencies surrounding the inclusion and study of sex and gender in research. Our paper provides a framework and roadmap for researchers and other stakeholders on how best to include and study sex and gender in research on pain and other biopsychosocial fields going forward.”
New research caption: Professor Seamus Looby, Honorary Associate Professor, RCSI and Consultant Neuroradiologist at Beaumont Hospital, Dr Sophie Sabherwal, NCHD, Beaumont Hospital and Dr Elizabeth Costelloe, Consultant Neurologist, Beaumont Hospital. Pic: Ray Lohan/RCSI
The research was carried out under the ERA-NET NEURON initiative, funded by the European Union and the Health Research Board, and led by Dr Michelle Roche, Professor David Finn and Professor Brian McGuire at the University of Galway’s Centre for Pain Research.
The 13 PAINDIFF recommendations published under three themes are:
• Universal Recommendations
• Include males and females as standard practice unless there is a valid reason not to do so
• Account for sex in randomisation/counterbalancing/ testing order
• Use adequately powered study design to detect sex differences when it is the primary experimental variable or when data suggest sex-specific effects
• Include detailed reporting of experimental design including sex of the experimenter when possible
• Conduct sex-disaggregated analysis and reporting
• Preclinical
• Researchers should be aware of, and report on, the sex of the established cell lines, primary cells and tissues used in their research
• It is not always necessary to test for oestrous cycle stage
• Researchers should include detailed reporting on housing, environmental conditions and experimental design
• Clinical
• Ask for participants’ sex assigned at birth and selfidentified gender
• Include a “prefer/choose not to say” response option when asking about sex and gender
• Include an open textbox response option to capture gender identity followed by a series of tick boxes to aid categorisation
• Report the number of people who hold diverse gender identities and, where possible and permitted, make the raw data accessible for further study (while ensuring anonymity).
• When possible, collect and report on sex-specific variables to allow disaggregated analysis by sex or gender to be better informed by hormonal status, rather than solely by age.
Clinical R&D
PFIZER SELLS VIIV STAKE TO SHIONOGI
GSK plc (LSE/NYSE: GSK) (“GSK”), and Shionogi & Co., Ltd (Head Office: Osaka, Japan; Chief Executive Officer: Isao Teshirogi, Ph.D) (“Shionogi”) today announced that they have reached agreement together with Pfizer Inc. (NYSE: PFE) (“Pfizer”) for the 11.7% economic interest in ViiV Healthcare Limited (“ViiV Healthcare”) currently held by Pfizer to be replaced with an investment by Shionogi. As a result of this transaction, Shionogi will increase its economic interest in ViiV Healthcare to 21.7%. GSK will maintain its 78.3% majority owned economic interest. Shionogi will continue to have one Director position on the ViiV Healthcare Board, and will be represented by Dr John Keller who has been a Director of ViiV Healthcare since 2012.
Under the terms of the agreement, ViiV Healthcare will issue new shares to Shionogi for consideration of $2.125 bn and cancel Pfizer’s holding in ViiV Healthcare. Pfizer will receive $1.875 bn and GSK will receive a special dividend of $0.250 bn (payable in GBP).
ViiV Healthcare, the global specialist HIV company, is dedicated to delivering advances in treatment and care for people living with HIV and for people who could benefit from HIV prevention.
David Redfern, Chair of ViiV Healthcare said: “This agreement simplifies ViiV’s shareholder structure and we look forward to continuing our highly successful collaboration with Shionogi to advance ViiV’s pipeline and portfolio of long-acting injectable HIV treatment and prevention medicines. GSK would also like to thank Pfizer for its longstanding partnership in the development of ViiV since its establishment in 2009.”
GSK ENTERS AGREEMENT TO ACQUIRE RAPT THERAPEUTICS
GSK plc (LSE/NYSE: GSK) has announced that it has entered a definitive agreement to acquire RAPT Therapeutics (“RAPT”) (NASDAQ: RAPT), a California-based, clinical-stage biopharmaceutical company dedicated to developing novel therapies for patients living with inflammatory and immunologic diseases. The acquisition includes ozureprubart, a long-acting anti-immunoglobulin E (IgE) monoclonal antibody, currently in phase IIb clinical development for prophylactic protection against food allergens.
IgE is a clinically validated target and is the only approved systemic therapy shown to protect patients from a harmful allergic and inflammatory immune response. Around 94% of severe food allergies are caused by IgEmediated reactions.1
Current anti-IgE treatment for food allergy involves injections every 2 to 4 weeks, which can be a significant burden, particularly since most patients are children. Ozureprubart’s clinical profile offers the potential for less frequent dosing of every 12 weeks, supporting improved compliance and patient outcomes; as well as providing a new option to approximately 25% of patients currently ineligible for existing therapy. Ozureprubart complements GSK’s extensive commercial footprint and prescriber base in allergy.
Data from the phase IIb trial (prestIgE) assessing use of ozureprubart as monotherapy is expected in 2027, with phase III trials to be focused on both at-risk adult and paediatric populations. In the US, over 17 million people are diagnosed with food allergies, with more than 1.3 million people suffering severe reactions.2,3,4 This results in more than 3 million patient visits each year to hospital and emergency care.5
ASPECT BIOSYSTEMS AND NOVO NORDISK UPDATE ON DIABETES PARTNERSHIP
Novo Nordisk and Aspect Biosystems have announced they are entering a new phase of their partnership to develop advanced cellular medicines for diabetes.
Since 2023, Aspect and Novo Nordisk have collaborated to develop cellular medicines designed to replace, repair or supplement biological functions to deliver truly disease-modifying therapies. This new phase of the partnership builds on the momentum achieved in the existing collaboration. It reflects a shared mission to accelerate the development of potentially curative medicines for serious diseases.
Aspect has acquired rights to stem cell-derived islet cell and hypoimmune cell engineering technologies from Novo Nordisk and will lead development, manufacturing and commercialisation. Novo Nordisk will have defined rights to expand its role in later-stage development and commercialisation.
Novo Nordisk will make an additional equity investment in Aspect and provide research funding to advance these potentially curative therapies. Novo Nordisk will be eligible to
receive royalties and milestone payments on future product sales from Aspect.
“Novo Nordisk was founded on a commitment to improve the lives of people living with diabetes. That commitment is as strong now as it was 100 years ago, and we have a continued focus on bringing innovation to people living with type 1 diabetes through internal and external innovation efforts,” said Jacob Sten Petersen, senior vice president, Global Research, Novo Nordisk.
“Aspect brings tremendous expertise and capabilities in cellular medicines, and we are proud of our partnership with them to progress transformative cell therapies toward clinical development and potentially generating a functional cure for people living with diabetes.”
The agreement between the two companies involves integrating select Novo Nordisk cell therapy research, development, and manufacturing capabilities and expertise from the United States and Denmark into Aspect’s Canada-anchored platform. This integration will strengthen Aspect’s end-to-end capabilities and expand access to highly skilled talent.
“Our partnership with Novo Nordisk combines more than a century of their global leadership in the fight against diabetes with Aspect’s leadership in cell therapy and our biotech speed and agility, creating a powerful force multiplier to deliver clinical impact,” said Tamer Mohamed, founder and CEO, Aspect Biosystems. "By integrating key technologies and capabilities, we are strengthening our full-stack platform,
accelerating our path to curative cell therapies, and advancing our vision to build a generational biotechnology company that delivers global impact for people living with serious diseases.”
Aspect’s platform is being applied to develop a new class of cellular medicines and functional cures for serious metabolic and endocrine diseases, including an islet replacement therapy for type 1 diabetes designed to restore blood glucose control without the need for chronic immune suppression. These cellular medicines are designed to be allogeneic, enabling scalable manufacturing using off-the-shelf cells.
PARALIEF 500 MG FILMCOATED TABLETS
Clonmel Healthcare would like to announce that the name of their 100 pack of Paracetamol filmcoated tablets is changing from Paracetamol Clonmel to Paralief. The pack design has also been updated in line with the consumer 24 pack.
Paralief is indicated for the shortterm symptomatic treatment of moderate pain and/or fever.
Full prescribing information is available on request or alternatively please go to www.clonmel-health. ie. Medicinal product subject to medical prescription.
Please contact Clonmel Healthcare on 01-6204000 if you require any additional information.
PA 126/020/008. PA Holder: Clonmel Healthcare Ltd., Clonmel, Co. Tipperary. Date prepared: January 2026. 0126/NPM/ PAR/002H
WORLD CANCER DAY: INFLUENCER AND BREAST CANCER PATIENT JENNIFER
WRYNNE URGES PUBLIC TO GO ALL IN THIS DAFFODIL DAY
Leitrim-based milliner and influencer Jennifer Wrynne was diagnosed with breast cancer in June 2024 at the age of 36, after discovering a lump in her breast just two months after the birth of her daughter, Amelia Rose.
On World Cancer Day, Jennifer is urging communities in every county in Ireland to turn daffodil yellow on Daffodil Day, March 20th, and to go all in to raise vital funds and give hope to cancer patients.
Speaking ahead of the Irish Cancer Society’s Daffodil Day 2026 launch, Ambassador Jennifer Wrynne said: “Being diagnosed with cancer at 36-years-old is something you never expect, especially when you’re in the middle of raising a young family. Sadly, cancer doesn’t discriminate by age. I had recently welcomed my fourth baby, and cancer was the last thing on my mind. Life was busy and full and then, suddenly, everything stopped. My world was turned upside down overnight.
“A cancer diagnosis brings fear, shock, and huge uncertainty, not just for you, but for the people who depend on you. I quickly learned how much strength you can find when you have the right support around you. Throughout my treatment and recovery, the Irish Cancer Society provided trusted information, reassurance, and a sense of hope at times when I needed it most.
“My diagnosis has highlighted how important awareness, early detection, and support truly are. By sharing my story and supporting Daffodil Day, I want to not only raise awareness and vital funds, but also to remind others that they are not alone, and
that there is strength, hope, and a life beyond a diagnosis. This Spring, every daffodil you see is a reminder that no one has to face cancer alone.”
Today, on World Cancer Day, the Irish Cancer Society is making a further commitment of over ¤1 million for the Young Onset Cancer Pilot Programme for 2026 and 2027, delivered in partnership with the Trinity St James’s Cancer Institute.
This builds on the Irish Cancer Society’s investment of almost ¤2 million in the Research Programme over the past three years, with additional funding in, 2026 and 2027 bringing the figure to ¤3m. The programme is aimed at improving outcomes and providing tailored support for people diagnosed with cancer at a younger age.
Amy Nolan, Director of Clinical Affairs at the Irish Cancer Society said: “Every donation on Daffodil Day helps us be there for cancer patients and their families — and allows us to fund research and initiatives such as the Young Onset Cancer Programme, delivered in partnership with Trinity St James’s Cancer Institute. We are thrilled to continue supporting this critically important programme in the coming years, building on the impact of the work already underway,”
Over the last couple of decades, there has been a concerning rise in cancer cases among patients under 50, most notably cancers of the gastrointestinal tract — including colon, stomach, pancreas, and liver — alongside increasing numbers of breast, lung, and other solid organ tumours.
“None of our free, life-changing services or research would be possible without the incredible generosity of the public, especially on days like Daffodil Day," adds Ms Nolan. "With just 5% of our funding coming from the government, we rely on fundraising to fuel our work. We’re encouraging everyone to get involved and go all in this Daffodil Day — the more we raise, the more care, support, and hope we can provide to people affected by cancer in every corner of Ireland.”
The Young Onset Cancer Pilot Programme addresses the specific unmet needs for people from 25 to 50 affected by cancer (young onset cancer patients). Specific focus areas include the provision of social and psychological support, development of primary care pathways, sexual health, fertility, survivorship, and wellbeing. Other research focus areas include examining the underlying biology and cause of young onset cancer, screening, and early detection.
Daffodil Day takes place on Friday, March 20th. Whether you want to get involved with your school, company, or in your community, there are lots of ways to go all in and support cancer patients across Ireland. Visit cancer.ie to get involved or learn more.
NORDIC PHARMA ANNOUNCES EUROPEAN LAUNCH OF LACRIFILL® CANALICULAR GEL
Nordic Pharma Announces European launch of Lacrifill® Canalicular GelHoofddorp, Netherlands – 6th February 2026 –Nordic Group B.V., an international pharmaceutical company operating as Nordic Pharma, announced today the introduction of Lacrifill®, a novel therapy for dry eye, to the European market. Following CE mark approval in 2025, Lacrifill® is now available to healthcare professionals and patients across a range of European markets,marking a major step in the treatment of dry eye disease.
Lacrifill® is a cross-linked hyaluronic acid gel designed to temporarily block tear drainage by physiologically occluding the canalicular system. By enhancing tear filmpreservation, Lacrifill® helpsmaintain natural lubricating tears on the patients’ ocular surface,offering an individualised approach to dry eye management. The in-office procedure provides a full fill of the canalicular system and delivers long-lasting effects for up to six months.
Dry eye disease is a highly prevalent condition, affecting up to 30% of the European population and represents a particular challenge in patients undergoing ocular surgical procedures. Pre-existing ocular surface disease in candidates for cataract or LASIK surgery can compromise surgical outcomes and impact patient satisfaction.
“Achieving a stable ocular surface is a vital prerequisite for successful surgical outcomes. The introduction of Lacrifill® to the European market is a welcome development, providing us with a new tool for pre-and postoperative ocular surface health optimisation and ensuring our patients have access to the latest advancements in ocular care” said Professor Jose Benitez delCastillo (Complutense University of Madrid, Spain).
“The European launch of Lacrifill® represents a significant milestone for Nordic Pharma” said
Charlotte Phelps, CEO of Nordic Group B.V. “We are proud to bring this innovative dry eye therapy to patients and healthcare professionals across Europe, and we believe it will help many of
those currently living with dry eye disease and become an important addition to dry eyemanagement, particularly around ocular surgery.” By working closely with the ophthalmology community across Europe, Nordic Pharma will continue to support the launch of Lacrifill® through medical education to improve the standard of ocular surface optimisation.
About Nordic Group B.V.Nordic Group B.V. is a privately owned, medium-sized international pharmaceutical company focused on the development and commercialisation of specialty products. Its portfolio includes innovative therapies in Ophthalmology, Rheumatology, and Women’s Health. With strong roots across Europe, NordicGroup B.V. now has affiliates in Canada, the US,and Japan and continues to expand globallythrough strategic acquisitions and partnerships. For more information about Nordic Pharma, visit www.nordicpharma.com.
NOVO NORDISK ANNOUNCES WEGOVY® (SEMAGLUTIDE 1.7MG AND 2.4MG) PRICE REDUCTIONS OF 20% IN
IRELAND
Novo Nordisk Ireland Ltd has announced reductions of the list price of two of its highest doses of Wegovy®, semaglutide 1.7mg and semaglutide 2.4mg, in Ireland. The price reductions are part of a broader European initiative to enhance affordability and access for people living with overweight and obesity.
From February 1st, Novo Nordisk Ireland will implement list price reductions of -20% for the semaglutide 1.7mg dose and -20% for the semaglutide 2.4mg maintenance dose.
General Manager of Novo Nordisk Ireland Dilek Dogan Gurluk, said, “We are very excited to announce that semaglutide 1.7mg and 2.4mg will now be more affordable for the 60% of Irish adults living with overweight and obesity. Novo Nordisk is committed to supporting people to reach and sustain their weight management goals. This is why we are pleased to ensure improved access to our GLP-1 weight management medication.”
Today, one billion people worldwide are living with obesity, which is associated with numerous health complications, making it a ‘gateway’ disease to others, including type 2 diabetes, cancer and cardiovascular disease. For Novo Nordisk, treating obesity is central to our enduring purpose of driving change to defeat serious chronic diseases.
Jennifer Wrynne launches the Irish Cancer Society’s Daffodil Day 2026. Photography by Andres Poveda
KISQALI® is the only CDK4/6i approved for the broadest range of HR+/ HER2- patients including those with high-risk N0 or N+ disease 2,15,16
KISQALI® demonstrated a predictable, manageable and reversible safety profile. Most AEs are asymptomatic and QoL was maintained compared to baseline. The most common grade 3/4 AEs were neutropenia, abnormal liver function tests and leukopenia2,8-11 NATALEE1 iDFS ACHIEVED
KISQALI® reduced the relative risk of invasive disease recurrence by 28.4% vs NSAI alone1
Fictional healthcare professional and patient.
AEs, adverse events; aBC, advanced breast cancer; CDK4/6i, cyclin-dependent kinase 4 and 6 inhibitor.
ABBREVIATED PRESCRIBING INFORMATION
Please refer to Summary of Product Characteristics (SmPC) before prescribing. Kisqali (ribociclib) 200 mg film-coated tablets
Presentation: Film coated tablets (FCT) containing 200 mg of ribociclib and 0.344 mg soya lecithin. Indications: Early breast cancer - Kisqali in combination with an aromatase inhibitor is indicated for the adjuvant treatment of patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative early breast cancer at high risk of recurrence (see section 5.1 for selection criteria). In pre- or perimenopausal women, or in men, the aromatase inhibitor should be combined with a luteinising hormone-releasing hormone (LHRH) agonist. Advanced or metastatic breast cancer - Kisqali is indicated for the treatment of women with HR-positive, HER2-negative locally advanced or metastatic breast cancer in combination with an aromatase inhibitor or fulvestrant as initial endocrine-based therapy, or in women who have received prior endocrine therapy. In pre- or perimenopausal women, the endocrine therapy should be combined with a LHRH agonist. Dosage and administration: Patient selection for treatment with Kisqali based on the tumour expression of HR and HER2 should be assessed by a CE-marked in vitro diagnostic (IVD) medical device with the corresponding intended purpose. If the CE-marked IVD is not available, an alternative validated test should be used. Adults: Early breast cancer - The recommended dose is 400 mg (two 200 mg FCT) taken orally, once daily for 21 consecutive days followed by 7 days off treatment, resulting in a complete cycle of 28 days. In patients with early breast cancer, Kisqali should be taken until completion of 3 years of treatment or until disease recurrence or unacceptable toxicity occur. When Kisqali is used in combination with an aromatase inhibitor (AI), the AI should be taken orally once daily continuously throughout the 28-day cycle. Please refer to the Summary of Product Characteristics (SmPC) of the AI for additional details. In pre- or perimenopausal women, or in men, the aromatase inhibitor should be combined with a LHRH agonist. Advanced or metastatic breast cancer - The recommended dose is 600 mg (3 x 200 mg FCT) taken orally, once daily for 21 consecutive days followed by 7 days off treatment, resulting in a complete cycle of 28 days. When Kisqali is used in combination with an AI, the AI should be taken orally once daily continuously throughout the 28 day cycle. Please refer to the Summary of Product Characteristics (SmPC) of the AI for additional details. When Kisqali is used in combination with fulvestrant, fulvestrant is administered intramuscularly on days 1, 15 and 29, and once monthly thereafter. Please refer to the SmPC of fulvestrant for additional details. Treatment of pre and perimenopausal women with the approved Kisqali combinations should also include an LHRH agonist in accordance with local clinical practice. Management of severe or intolerable adverse reactions (ARs) may require temporary dose interruption, reduction or discontinuation of Kisqali. Please see section 4.2 of SmPC for recommended dose modification guidelines. Kisqali can be taken with or without food (see section 4.5 of SmPC). The tablets should be swallowed whole and should not be chewed, crushed or split prior to swallowing. Special populations: ♦Renal impairment: Mild or moderate: No dose adjustment is necessary. Severe: A starting dose of 200 mg is recommended in patients with severe renal impairment. Kisqali has not been studied in breast cancer patients with severe renal impairment. Caution should be used in patients with severe renal impairment with close monitoring for signs of toxicity. ♦Hepatic impairment: No dose adjustment is necessary in patients with early breast cancer with hepatic impairment (see section SmPC 5.2). In patients with advanced or metastatic breast cancer, no dose adjustment is necessary in patients with mild hepatic impairmentModerate or severe: Dose adjustment is required, and the starting dose of 400 mg once daily is recommended. ♦Elderly (>65 years): No dose adjustment is required. ♦Pediatrics(<18 years): Safety and efficacy have not been established. Contraindications: Hypersensitivity to the active substance or to peanut, soya or any of the excipients. Warnings/Precautions: ♦Neutropenia was most frequently reported AR. A complete blood count (CBC) should be performed before initiating treatment. CBC should be monitored every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, then as clinically indicated. Febrile neutropenia was reported in 1.7% of patients exposed to Kisqali in the phase III clinical studies. Patients should be instructed to report any fever promptly. Based on the severity of the neutropenia, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 2 (see section 4.2 of SmPC). ♦Hepatobiliary toxicity increases in transaminases have been reported. Liver function tests (LFTs) should be performed before initiating treatment. LFTs should be monitored every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, then as clinically indicated. If grade ≥2 abnormalities are noted, more frequent monitoring is recommended. Recommendations for patients who have elevated AST/ALT grade ≥ 3 at baseline have not been established. Based on the severity of transaminase elevations, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 3 (see section 4.2). ♦QT interval prolongation has been reported with Kisqali. The use of Kisqali should be avoided in patients who have already or who are at significant risk of developing QTc prolongation. This includes patients with long QT syndrome, with uncontrolled or significant cardiac disease, including recent myocardial infarction, congestive heart failure, unstable angina and bradyarrhythmias and patients with electrolyte abnormalities. The use of Kisqali with medicinal products known to prolong QTc interval and/or strong CYP3A4 inhibitors should be avoided as this may lead to clinically meaningful prolongation of the QTcF interval (see SmPC sections 4.2, 4.5 and 5.1). If co-administration of Kisqali with a strong CYP3A4 inhibitor cannot be avoided, the Kisqali dose should be changed as described in SmPC section 4.2. QT interval prolongation in early breast cancer –study O12301C (NATALEE), a QTcF interval increase >60 msec from baseline was observed in 19 (0.8%) patients receiving Kisqali plus AI. The ECG should be assessed prior to initiation of treatment. Treatment with Kisqali should be initiated only in patients with QTcF values <450 msec. The ECG should be repeated at approximately Day 14 of the first cycle then as clinically indicated. In case of QTcF prolongation during treatment, more frequent ECG monitoring is recommended. Appropriate monitoring of serum electrolytes (including potassium, calcium, phosphorous, and magnesium) should be performed prior to initiation of treatment, at the beginning of the first 6 cycles, and then as clinically
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the
indicated. Any abnormality should be corrected before the start of Kisqali treatment. Based on the observed QT prolongation during treatment, Kisqali may require dose interruption, reduction, or discontinuation as described in Table 4 (see section 4.2 of SmPC). Based on the E2301 study QTcF interval data, Kisqali is not recommended for use in combination with tamoxifen. ♦Critical visceral disease. The efficacy and safety of ribociclib have not been studied in patients with critical visceral disease. ♦Severe cutaneous reactions Toxic epidermal necrolysis (TEN) has been reported with Kisqali treatment. If signs and symptoms suggestive of severe cutaneous reactions (e.g. progressive widespread skin rash often with blisters or mucosal lesions) appear, Kisqali should be discontinued immediately. ♦Interstitial lung disease/pneumonitis ILD/pneumonitis has been reported with CDK4/6 inhibitors including Kisqali. Patients should be monitored for pulmonary symptoms indicative of ILD/pneumonitis which may include hypoxia, cough and dyspnoea and dose modifications should be managed in accordance with Table 5 (see section 4.2 of SmPC) Based on the severity of the ILD/pneumonitis, which may be fatal, Kisqali may require dose interruption, reduction or discontinuation as described in Table 5 (see section 4.2 of SmPC). ♦Blood creatinine increase ribociclib may cause blood creatinine increase – if this occurs it is recommended that further assessment of the renal function be performed to exclude renal impairment. ♦CYP3A4 substrates ribociclib may interact with medicinal products which are metabolised via CYP3A4, which may lead to increased serum concentrations of CYP3A4 substrates (see section 4.5 of SmPC). Caution is recommended in case of concomitant use with sensitive CYP3A4 substrates with a narrow therapeutic index and the SmPC of the other product should be consulted for the recommendations regarding co administration with CYP3A4 inhibitors. Pregnancy, Fertility and Lacation ♦Pregnancy: Pregnancy status should be verified prior to starting treatment as Kisqali can cause foetal harm when administered to a pregnant woman. Kisqali is not recommended during pregnancy and in women of childbearing potential not using contraception. ♦Women of childbearing potential who are receiving Kisqali should use effective contraception (e.g. double-barrier contraception) during therapy and for at least 21 days after stopping treatment with Kisqali. ♦Breast‑feeding: Patients receiving Kisqali should not breast feed for at least 21 days after the last dose. ♦Fertility: There are no clinical data available regarding effects of ribociclib on fertility. Based on animal studies, ribociclib may impair fertility in males of reproductive potential. ♦Effects on ability to drive and use machines Patients should be advised to be cautious when driving or using machines in case they experience fatigue, dizziness or vertigo during treatment with Kisqali. Interactions: ♦Concomitant use of strong CYP3A4 inhibitors should be avoided, including, but not limited to, clarithromycin, indinavir, itraconazole, ketoconazole, lopinavir, ritonavir, nefazodone, nelfinavir, posaconazole, saquinavir, telaprevir, telithromycin, verapamil, and voriconazole. Alternative concomitant medicinal products with less potential to inhibit CYP3A4 should be considered. Patients should be monitored for ARs. If concomitant use of a strong CYP3A4 inhibitor cannot be avoided, the dose of Kisqali should be reduced (see section 4.2 of SmPC). ♦Grapefruit or grapefruit juice should be avoided. ♦Concomitant use of strong CYP3A4 inducers should be avoided, including, but not limited to, phenytoin, rifampicin, carbamazepine and St John’s Wort (Hypericum perforatum). An alternative medicinal product with no or minimal potential to induce CYP3A4 should be considered. ♦Caution is recommended when Kisqali is administered with sensitive CYP3A4 substrates with narrow therapeutic index (including, but not limited to, alfentanil, ciclosporin, everolimus, fentanyl, sirolimus, and tacrolimus), and their dose may need to be reduced. ♦Concomitant administration of Kisqali with the following CYP3A4 substrates should be avoided: alfuzosin, amiodarone, cisapride, pimozide, quinidine, ergotamine, dihydroergotamine, quetiapine, lovastatin, simvastatin, sildenafil, midazolam, triazolam. ♦Caution and monitoring for toxicity are advised during concomitant treatment with sensitive substrates of drug transporters P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 and BSEP which exhibit a narrow therapeutic index, including but not limited to digoxin, pitavastatin, pravastatin, rosuvastatin and metformin. ♦Co-administration of Kisqali with medicinal products with known potential to prolong the QT interval should be avoided such as anti-arrhythmic medicinal products (including, but not limited to, amiodarone, disopyramide, procainamide, quinidine and sotalol) and other medicinal products known to prolong the QT interval including, but not limited to, chloroquine, halofantrine, clarithromycin, ciprofloxacin, levofloxacin, azithromycin, haloperidol, methadone, moxifloxacin, bepridil, pimozide and intravenous ondansetron. Kisqali is not recommended for use in combination with tamoxifen. Adverse reactions – advanced or metastatic breast cancer: ♦Very common: Infections, neutropenia, leukopenia, anaemia, lymphopenia, decreased appetite, headache, dizziness, dyspnoea, cough, nausea, diarrhoea, vomiting, constipation, stomatitis, abdominal pain, dyspepsia, alopecia, rash, pruritus, back pain, fatigue, peripheral oedema, asthenia, pyrexia, abnormal liver function tests. ♦Common: thrombocytopenia, febrile neutropenia, hypocalcaemia, hypokalaemia, hypophosphataemia, vertigo, lacrimation increased, dry eye, syncope, dysgeusia, hepatotoxicity, erythema, dry skin, vitiligo, dry mouth, oropharyngeal pain, blood creatinine increased, electrocardiogram QT prolonged, interstitial lung disease (ILD)/pneumonitis. ♦Rare: Erythema multiforme ♦Not known: Toxic epidermal necrolysis (TEN) ♦ Please refer to SmPC for a full list of adverse reactions. Adverse reactions - early breast cancer: ♦Very common: Infections, neutropenia, leukopenia, headache, Cough, Nausea, diarrhoea, constipation, abdominal pain, alopecia, fatigue, asthenia, pyrexia, abnormal liver function tests ♦Common: Anaemia, thrombocytopenia, lymphopenia, hypocalcaemia, hypokalaemia, appetite decreased, dizziness, dyspnoea, interstitial lung disease (ILD) / pneumonitis, vomiting, stomatitis, hepatotoxicity, rash, pruritus, peripheral oedema, oropharyngeal pain, blood creatinine increased, electrocardiogram QT prolonged ♦Uncommon: Febrile neutropenia ♦ Please refer to SmPC for a full list of adverse reactions. Legal Category: POM. Pack sizes: Unit packs containing 21, 42 or 63 FCTs. Not all pack sizes may be marketed. Marketing Authorisation Holder: Novartis Europharm Limited Vista Building
Adverse events can also be reported to Novartis preferably at www.novartis.com/report, by emailing
References 1. Crown JP, et al. Presented at the European Society For Medical Oncology Congress 2025, 17–21 October, Berlin, Germany. 2. KISQALI (ribociclib). Summary of Product Characteristics. 3. Hortobagyi GN, et al. Ann Oncol. 2024:S09237534(24)04064-X. 4. Hortobagyi GN, et al. N Engl J Med. 2022;386(10):942–950. 5. Neven P, et al. Breast Cancer Res. 2023;25:103. 6. Im S-A, et al. N Eng J Med. 2019;381:307–316. 7. Yardley DA, et al. Ann Oncol. 2022; 33(S7):S629. 8. Verma S, et al. Br Cancer Res Treat. 2018;170:535–545. 9. Beck JT, et al. Cancer Res. 2019;79 (4_Supplement):P6-18-14. 10. Fasching PA, et al. Breast. 2020;54:148–154. 11. Harbeck N, et al. Ther Adv Med Oncol. 2020;12:1–8. 12. Fasching PA et al., Annals of Oncology, Volume 34, Issue 10, 2023, Pages 951-953. 13. Lu et al., JCO 42, 2812-2821(2024). 14. Burris et al.Br J Cancer. 2021 Aug;125(5):679-686. 15. Slamon DJ, et al. Ther Adv Med Oncol. 2023;15:1–16. 16. Slamon D, et al. N Engl J Med. 2024; 390:1080-1091. 17. Fasching PA, et al. Oral LBA13. Presented at the European Society for Medical Oncology Congress 2024, 13–17 September, Barcelona, Spain. October 2025 | IE11536045 Novartis Ireland Ltd, Vista Building, Elm Park Green, Merrion Road, Ballsbridge, Dublin 4, D04 A9N6