MathematicalPhysics inTheoretical Chemistry
Editedby
S.M.Blinder
UniversityofMichigan, AnnArbor,MIandWolframResearch, Champaign, IL,USA
J.E.House
IllinoisWesleyanUniversity, Bloomington,IL;andIllinoisState University,Normal, IL,USA
Contributors
S.M.Blinder
UniversityofMichigan,AnnArbor,MI,UnitedStates
CailaBruzzese
DepartmentofChemistry,BrockUniversity,St.Catharines,Ontario,Canada
KimberlyJordanBurch
DepartmentofMathematics,IndianaUniversityofPennsylvania,Indiana, PA,UnitedStates
AndrewL.Cooksy
DepartmentofChemistryandBiochemistry,SanDiegoStateUniversity, SanDiego,CA,UnitedStates
GuidoFano
UniversityofBologna,Bologna,Italy
JamesW.Furness
DepartmentofPhysicsandEngineeringPhysics,TulaneUniversity,NewOrleans, LA,UnitedStates
DavidZ.Goodson
DepartmentofChemistryandBiochemistry,UniversityofMassachusetts Dartmouth,NorthDartmouth,MA,UnitedStates
JustinK.Kirkland
DepartmentofChemistry,UniversityofTennessee,Knoxville,TN,UnitedStates
ErrolLewars
DepartmentofChemistry,TrentUniversity,Peterborough,ON,Canada
DevinA.Matthews
InstituteforComputationalEngineeringandSciences,TheUniversityofTexasat Austin,Austin,TX,UnitedStates
EgorOspadov
DepartmentofPhysics,BrockUniversity,St.Catharines;DepartmentofChemistry,TheUniversityofWesternOntario,London,Ontario,Canada
StuartM.Rothstein
DepartmentofPhysics;DepartmentofChemistry,BrockUniversity,St.Catharines, Ontario,Canada
JohnF.Stanton
DepartmentofChemistry,UniversityofFlorida,Gainesville,FL,UnitedStates
JianweiSun
DepartmentofPhysicsandEngineeringPhysics,TulaneUniversity,NewOrleans, LA,UnitedStates
JacobTownsend
DepartmentofChemistry,UniversityofTennessee,Knoxville,TN,UnitedStates
IngaS.Ulusoy
DepartmentofChemistry,MichiganStateUniversity,EastLansing,MI, UnitedStates
KonstantinosD.Vogiatzis
DepartmentofChemistry,UniversityofTennessee,Knoxville,TN,UnitedStates
AngelaK.Wilson
DepartmentofChemistry,MichiganStateUniversity,EastLansing,MI, UnitedStates
YuboZhang
DepartmentofPhysicsandEngineeringPhysics,TulaneUniversity,NewOrleans, LA,UnitedStates
Thedevelopmentsofphysicsinthe20thcenturymadeallofchemistryexplicable, inprinciple,byquantummechanics.AssummarizedbyDirac:“Theunderlying physicallawsnecessaryforthemathematicaltheoryofalargepartofphysicsand thewholeofchemistryarethuscompletelyknown,andthedifficultyisonlythat theexactapplicationoftheselawsleadstoequationsmuchtoocomplicatedtobe soluble”[2].Byitsverynature,quantummechanics is mathematicalphysicsand therebyweestablishtheconnectionwhichisthethemeofthisvolume.However,the loopholenotedbyDirac,theexistenceofchemicalproblemstoomathematically complextobesolvedexactly,justifiesthesurvivalofpartsofchemistryasan empiricalscience.Inthiscategoryaresemiempiricalconceptsofchemicalbonding andreactivity.Thishasalsoledtocomputationalmodelspromotingrationaldrug design.Thesehavealsostimulatedapplicationsofotherbranchesofmathematics, forexample,informationtheoryandgraphtheoryappliedtothedefinitionofvarious chemicalindices.
Theprimaryobjectiveoftheoreticalchemistryistoprovideacoherentaccount forthestructureandpropertiesofatomicandmolecularsystems.Techniques adaptedfrommathematicsandtheoreticalphysicsareappliedinattemptstoexplain andcorrelatethestructuresanddynamicsofchemicalsystems.Inviewofthe immensecomplexityofchemicalsystems,theoreticalchemistry,incontrastto theoreticalphysics,generallyusesmoreapproximatemathematicaltechniques,often supplementedbyempiricalorsemiempiricalmethods.
Thisvolumebeginswithanintroductiontothequantumtheoryforatomsand smallmolecules,expandingupontheoriginalapplicationsofmathematicalphysics inchemistry.Thisfieldisnowlargelysubsumedwithinasubdisciplineknownas computationalchemistry. Chapter1 beginswithanintroductiontotheHartree-Fock method,whichistheconceptualfoundationforcomputationalchemistry. Chapter2 discussesthebasisfunctionsemployedinthesecomputations,nowlargelydominated byGaussianfunctions. Chapter3 describessomepost-Hartree-Fockmethods,which seektoattain“chemicalaccuracy”inatomicandmolecularcomputations,inparticular,configurationinteraction,many-bodyperturbationtheory,andcoupled-cluster theory. Chapter10 discussesdiagrammatictechniquesborrowedfromtheoretical physics,whichcanenhancetheefficiencyofcomputations. Chapter7 isanaccount ofthedevelopmentofpersonalcomputersandtheirapplicationstocomputational chemistry.
Forlargermoleculesandcondensedmatter,alternativeapproaches,including densityfunctionaltheory(Chapter4)andquantumMonte-Carlo(Chapter6),are becomingpopularcomputationalmethods.Someadditionaltopicscoveredinthis volumearevibrationalpartitionfunctions(Chapter5),singularityanalysisofperturbationtheories(Chapter9),andchemicalapplicationsofgraphtheory(Chapter8).
Finally, Chapter11 introducestheprinciplesofthequantumcomputer,whichhas thespeculativepossibilityofexponentialenhancementofcomputationalpowerfor theoreticalchemistry,aswellasmanyotherapplications.
1
S.M.Blinder
UniversityofMichigan,AnnArbor,MI,UnitedStates
Afundamentalbottleneckinbothclassicalandquantummechanicsisthe three-body problem.Thatis,themotionofsystemsinwhichthreeormoremassesinteractcannot besolvedanalytically,sothatapproximationmethodsmustbeutilized.Thischapter introducesthebasicideasoftheself-consistentfield(SCF)andHartree-Fock(HF) methods,whichprovidethefoundationforthevastmajorityofcomputationalwork ontheelectronicstructureofatomsandmolecules.Moreadvancedgeneralizations ofHFarediscussedin Chapter3.ConceptualdevelopmentsbeyondHF,including density-functionalandMonte-Carlomethods,areintroducedinsubsequentchapters.
1 HARTREESELF-CONSISTENTFIELDTHEORY
AprecursorofSCFmethodsmighthavebeentheattemptstostudythemotionsof electronsinmanyelectronatomsinthe1920s,onthebasisoftheOldQuantum Theory.Theenergylevelsofavalenceelectron,suchasthe3s-electroninsodium, couldbereproducedquitecloselyiftheBohrorbitsoftheinnerelectronswere smearedoutintoacontinuoussphericallysymmetricchargedistribution[1–3].After thedevelopmentofwavemechanicsin1926,itwasrecognizedbyHartree[4]that Bohrorbitsmustbereplacedbycontinuouschargecloudsofelectrons,suchthatthe chargedensityofasingleelectronisgivenby ρ(r) =−e|ψ(r)|2 .Here, e isthe magnitudeoftheelectroncharge(1.602 × 10 19 coulomb)andthechargedensity ρ(r) followstheBorninterpretationoftheatomicorbital ψ(r)
Theapproachestoatomicandmolecularstructurethataretobedescribedin thischapterareclassifiedasabinitio(“fromthebeginning”)methods,sinceno experimentalorsemiempiricalparametersareused(otherthanthefundamental physicalconstants).
ThesimplestapplicationofHartree’sSCFmethodistheheliumatom,withtwo electrons.Electron1,whichoccupiestheatomicorbital ψ1 (r1 ),movesinthefield ofthenucleusandelectron2.Thepotentialenergyofanelectronwithcharge e a distance r fromanucleusofcharge +Ze followsdirectlyfromCoulomb’slaw,with MathematicalPhysicsinTheoreticalChemistry.https://doi.org/10.1016/B978-0-12-813651-5.00001-2 Copyright©2019ElsevierInc.Allrightsreserved.
V (r ) =− Ze2 r .(1)
(WeuseGaussianunitstoavoidtheunnecessaryfactors4π 0 ,and,inanyevent,we willsoonbeswitchingtoatomicunits.)Toreview,theSchrödingerequationfora hydrogen-likeatomcanbewritten
),(2)
wheretheenergyforprincipalquantumnumber n isgivenby n =−Z 2 e2 /2a0 n2 , with a0 equaltotheBohrradius h2 /me2 .Theone-electronfunctions ψ(r),whenused inthecontextofamultielectronsystem,arecalled orbitals [5],anadjective,usedas anoun,todenotethequantum-mechanicalanalogofclassical orbits.Foranelectron atpoint r interactingwiththechargedistributionofasecondelectroninanatomic orbital ψ(r ),thepotentialenergyisgivenby
Thus,thetotalpotentialenergyforelectron1isgivenby
wherethenotation V1 [ψ2 ] indicatesthat V1 isa functional of ψ2 ,emphasizingthe dependanceonthechargedistributionofelectron2.InHartree’smethod,electron1 obeystheeffectiveone-particleSchrödingerequation
where 1 istheorbitalenergyofelectron1,negativeforboundstates.Analogously, interchangingthelabels1and2,theorbitalfunctionforelectron2isthesolutionof
Thecoupledintegro-differentialequations(5),(6),knownasthe Hartreeequations, canberepresentedinsymbolicformby
Thesearecoupledinthesensethatthesolutiontothefirstequationentersthesecond equation(viatheeffectiveHamiltonianoperator H eff 2 containing V2 [ψ1 ]),andvice versa.Asolutiontotheseequationscanbefound,inprinciple,byasuccessive approximationprocedure.Aninitial“guess”ofthefunctions ψ1 and ψ2 isusedto
computethepotentialenergies V1 [ψ2 ] and V2 [ψ1 ].EachHartreeequationscanthen besolvedtogive“first-improved”orbitalfunctions ψ (1) 1 and ψ (1) 2 .These,inturn, areusedtorecompute V (1) 1 and V (1) 2 ,andthenewHartreeequationsaresolvedto givesecond-improvedorbitalfunctions.Theiterativeprocedureiscontinueduntil theinputandoutputfunctionsagreetowithinsomedesiredaccuracy.Theorbital functionsandpotentialfieldsarethensaidtobe self-consistent.Theusualquantummechanicalrestrictionsonaboundstatewavefunction—thatitbeeverywheresinglevalued,finite,andcontinuous—applyateachstageofthecomputation.EachHartree equationisthusaneigenvalueproblem,solubleonlyforcertaindiscretevaluesof i (ingeneral,differentineachstage).Fortheheliumatomtheorbitalfunctions ψ1 and ψ2 turnouttobeidentical.ThisdoesnotviolatethePauliprinciplesincethetwo orbitalscanhaveoppositespins.NotethattheHartreemethoddoesnotitselftake spinintoaccount.
ExtensionoftheHartreemethodtoan N -electronatomisstraightforward.Each electronnowmovesinthepotentialfieldofthenucleusplustheoverlappingcharge cloudsof N 1otherelectrons.Now N coupledintegro-differentialequationsareto besolved:
Eachsetoforbitalfunctions ψ1 ...ψN canbeidentifiedwithanelectronicconfiguration,forexample,1s2 2s2 2p6 3s fortheNaatom.Itislefttothegoodsenseoftheuser nottoallowmorethantwooftheorbitals ψ1 ...ψN tobethesame.1 Thedifferent orbitalpairsshouldalsobeconstructedtobemutuallyorthogonal.Theeigenvalues i shouldbenegativeforboundorbitals.Theirmagnitudesareapproximationstothe ionizationenergiesofthecorrespondingelectrons.
Atthispoint,itisconvenienttointroduceatomicunits,whichsimplifiesallofthe previousformulasbyremovingtherepetitivephysicalconstants.Weset
1 Ignoringthisrestrictionhasbeendubbed“inconsistentfieldtheory.”
Theunitoflengthisthe Bohr equaltotheBohrradius a0 = h2 /me2 = 0.529177 × 10 10 m.Theunitofenergyisthe Hartree,equalto e2 /a0 ,correspondingto 27.2114eV.Expressedinatomicunits,theSchrödingerequationforahydrogen-like atom(2)simplifiesto
with n =−Z 2 /2n2 . Hartree’sSCFmethod,asdescribedsofar,followedentirelyfromintuitive considerationsofatomicstructure.Weturnnexttoamorerigorousquantumtheoreticalderivationofthemethod[6,7].Thefirststepistowritedownthe Hamiltonianoperatorforan N -electronatom.Nowusingatomicunits,neglecting magneticinteractionsandotherhigher-ordereffects:
Theone-electronpartsoftheHamiltonian—thekineticenergyandnuclearattraction operators—arecontainedinthefirstsummation.Thesecondsummation,over N (N 1)/2distinctpairs i, j,representstheinterelectronicrepulsiveinteractions.The interelectronicdistancesaredenoted rij =|ri rj |.The N -electronwavefunctionis approximatedbya Hartreeproduct :
where ψ(ri ) aretheone-electronorbitals.Theseshouldconsistofmutuallyorthonormalfunctions
withnonerepeatedmorethantwice(maximumoftwoelectronsperatomicorbital). NotethatwehavenowintroducedDiracnotation,forcompactness.Afullyseparable wavefunctionsuchasEq.(14)wouldbeexactonlyiftheHamiltonianwereasumof one-electronparts.Thisisnotthecasesincetheelectroncoordinatesareinextricably mixedbythe r 1 ij terms,representingmutualelectronrepulsion.Wethereforemust considerapproximatesolutionsofthe N -particleSchrödingerequation,optimized inaccordancewiththevariationalprinciple.Thismeansminimizingtheratioof integrals
Thisgivesanupperlimittotheexactgroundstateenergy E0 : E ≥ E0 WenextgiveaderivationoftheHartreeequations.Usingtheorthonormalized orbitals ψi (r),satisfyingEq.(15),thetotalwavefunctionisfoundtobenormalized aswell:
Thevariationof L [ψ , ψ ∗ ] intermsofvariationsinallthe ψi and ψ ∗ i isgivenby
Sincetheminimumin L isunconditional,thisresultmustholdforarbitrary variationsofallthe δψi and δψ ∗ i .Thisispossibleonlyifeachofthecoefficients ofthesevariationsvanish,thatis,
Letusfocusononeparticularterminthevariation δ L ,namelythetermlinearin δψ ∗ k forsome i = k .Fromthecondition ∂ L ∂ψ ∗ k = 0appliedtoEq.(23),weareledto theHartreeequations2
inagreementwithEqs.(8)–(10).Wehaveusedthefactsthatthefirstsummation i reducestoasingletermwith i = k andthevanishingoftheintegral d 3 r for arbitraryvaluesof δψ ∗ k impliesthattheremainingintegrandisidenticallyequalto0.
2 DETERMINANTALWAVEFUNCTIONS
Theelectronineachorbital ψi (r) isaspin 1 2 particleandthushastwopossiblespin orientationsw.r.t.anarbitraryspatialdirection, ms =+ 1 2 or ms =− 1 2 .Thespin functionisdesignated σ ,whichcancorrespondtooneofthetwopossiblespinstates σ = α or σ = β .Wedefineacompositefunction,knownasa spin-orbital
(27)
denotingby x thefour-dimensionalmanifoldofspaceandspincoordinates.For example,ahydrogen-likespin-orbitalislabeledbyfourquantumnumbers,so a = {n, l, m, ms }.Wewillabbreviatecombinedintegrationoverspacecoordinatesand summationoverspincoordinatesby spin
d 3 r = dx (28)
2 TheHartreeequationsmightappeartodaytohaveonlyhistoricalsignificance,buttheirgeneralization leadstotheKohn-Shamequationsofmoderndensity-functionaltheory.
AHartreeproductofspin-orbitalsnowtakestheform
Ψ(1 ... N ) = φa (1)φb (2)...φn (N ). (29)
Forfurtherbrevity,wehavereplacedthevariables xi simplybytheirlabels i Tobephysicallyvalid,asimpleHartreeproductmustbegeneralizedtoconform totwoquantum-mechanicalrequirements.FirstisthePauliexclusionprinciple, whichstatesthatnotwospin-orbitalsinanatomcanbethesame.Thisallows anorbitaltooccurtwice,butonlywithoppositespins.Second,themetaphysical perspectiveofthequantumtheoryimpliesthatindividualinteractingelectronsmust beregardedasindistinguishableparticles.Onecannotuniquelylabelaspecific particlewithanordinalnumber;theindicesgivenmustbeinterchangeable.Thus eachofthe N electronsmustbeequallyassociatedwitheachofthe N spin-orbitals. Sincewehavenowundonetheuniqueconnectionbetweenelectronnumberand spin-orbitallabel,wewillhenceforthdesignatethespin-orbitallabelsaslowercaseletters a, b, ... , n whileretainingthelabels1,2, ... , N forelectronnumbers. Thesimplestexampleisagainthe1s2 groundstateofheliumatom.Letthetwo occupiedspin-orbitalsbe φa (1) =
2).Tofulfillthe necessaryquantumrequirements,wecanconstructthe(approximate)groundstate wavefunctionintheform
Inclusionofthetermwithinterchangedparticlelabels, φa (2)φb (1),fulfillstheindistinguishabilityrequirement.Thefactor 1 √2 preservesnormalizationforthelinear combination(assumingthat φa and φb areindividuallyorthonormalized).The exclusionprincipleisalsosatisfied,sincethefunctionwouldvanishidenticallyif spin-orbitals a and b werethesame.AgeneralconsequenceofthePauliprincipleis the antisymmetryprinciple foridenticalfermions,whereby Ψ(2,1)
Thefunction(30)hastheformofa2 × 2determinant
Thegeneralizationforafunctionof N spin-orbitals,whichisconsistentwiththe Pauliandindistinguishabilityprinciples,isan N × N Slaterdeterminant3
3 ThedeterminantalformwasfirstproposedbyHeisenberg[8,9]andDirac[10].Slaterfirstuseditin theapplicationtoamany-electronsystem[11].
Thereare N ! possiblepermutationsof N electronamong N spin-orbitals,which accountsforthenormalizationconstant1/√N !.Ageneralpropertyofdeterminantsis thattheyidenticallyequalto0ifanytwocolumns(orrows)areequal;thisconforms tothePauliexclusionprinciple.Asecondpropertyisthat,ifanytwocolumns areinterchanged,thedeterminantchangessign.Thisexpressestheantisymmetry principleforan N -electronwavefunction:
Aclosed-shellconfigurationofanatomormoleculecontains N /2pairsof orbitals,doublyoccupiedwith α and β spins;thiscanberepresentedbyasingle Slaterdeterminant.However,anopenshellconfigurationmust,ingeneral,berepresentedbyasumofSlaterdeterminants,sothat Ψ(1 N ) willbeaneigenfunction oftotalspinandorbitalangularmomenta.Asasimpleillustration,considerthe1s2 and1s2s configurationsofheliumatom.The1s2 groundstatecanberepresentedby asingledeterminant
whichisaneigenfunctionofthespinwitheigenvalues S = 0, MS = 0.The1s2s stateswith S = 1, MS =±1canlikewiseberepresentedbysingledeterminants:
for S = 1, MS =+1and
for S = 1, MS =−1.Thestateswiththesameconfigurationfor MS = 0must, however,bewrittenasasumoftwodeterminants:
The (+) signcorrespondstothe S = 1, MS = 0state,andisthethirdcomponent ofthe1s2s 3 S term,whilethe ( ) signcorrespondsto S = 0, MS = 0andrepresents the1s2s 1 S state.
3 HARTREE-FOCKEQUATIONS
TheHFmethodismostusefullyappliedtomolecules.Wemust,therefore,generalize theHamiltoniantoincludetheinteractionoftheelectronswithmultiplenuclei, locatedatthepoints R1 , R2 , ,withnuclearcharges Z1 , Z2 , :
Ψ(... j i ...) =−Ψ(... i j ...) (34)
Weusetheabbreviation riA =|ri RA |.InaccordancewiththeBorn-Oppenheimer approximation,weassumethatthepositionsofthenuclei R1 , R2 , arefixed. Thustherearenonuclearkineticenergytermssuchas 1 2MA ∇ 2 A .Theinternuclear potentialenergy Vnucl (R) = A,B ZA ZB RAB isconstantforagivennuclearconformation, whichisaddedtotheresultaftertheelectronicenergyiscomputed.Notethatthe totalenergy E (R) aswellastheone-electronenergies i (R) aredependentonthe nuclearconformation,abbreviatedsimplyas R.Itisofmajorcurrenttheoretical interesttoplot energysurfaces,whicharethemolecularenergiesasfunctionsof theconformationparameters R
Wearenowreadytocalculatetheapproximatevariationalenergycorresponding toHFwavefunctions[12,13]
Wewillnowrefertotheone-electronfunctionsmakingupaSlaterdeterminant as molecularorbitals.Toderivetheenergyformulas,itisusefultoreexpressthe determinantalfunctionsinamoredirectlyapplicableform.Recallthatan N × N determinantisalinearcombinationof N ! terms,obtainedbypermutationofthe N electronlabels1,2, ... , N amongthe N molecularorbitals.Whenevernecessary,we willlabelthespin-orbitalsby r , s ... n todistinguishthemfromtheparticlelabels i, j ... N .Wecanthenwrite
where Pp isoneof N ! permutationslabeledby p = 1 N !.Permutationsare classifiedaseither even or odd,accordingtowhethertheycanbecomposedof anevenoranoddnumberofbinaryexchanges.Theproductsresultingfrom anevenpermutationare added,inthelinearcombination,whilethosefroman oddpermutationare subtracted.Evenpermutationsarelabeledbyeven p,odd permutationsbyodd p.Thuseachproductinthesumismultipliedby ( 1)p .Let usfirstconsiderthenormalizationbra-ketof ΨHF
Becauseoftheorthonormalityofthemolecularorbitals φr , φs , ,theonlynonzero termsofthisdoublesummationwillbethosewith x
Therewillbe N ! suchterms,thusthebra-ketreducesto
Thecorecontributionstotheenergyinvolvestermsintheone-electronsumin Eq.(39).Definingthecoreoperator
theexpressionforthecoreintegral Hr reducesto
InanalogywithEq.(43)forcaseofthenormalizationbra-ket,alltheotherfactors φb |φs , s = r areequalto1.ThisisanalogoustoEq.(19),thedefinitionofthe coreintegralintheHartreemethod,exceptthatnowspin-orbitals,ratherthansimple orbitalsarenowused.Actually,thescalarproductsofthespinfunctions σr give factorsof1,sothatonlythespace-dependentorbitalfunctionsareinvolvedinthe computation,justasintheHartreecase.
Weconsidernexttheinterelectronicrepulsions r 1 ij .Followingananalogous calculation,allcontributionsexceptthosecontainingparticlenumbers i or j give factorsof1.Whatremainsis
Theminussignreflectsthefactthatinterchangingtwoparticlelabels i, j multiplies thewavefunctionby 1.ThefirsttermearliercorrespondstoaCoulombintegral (20);againthesearelabeledbyspin-orbitals,butthecomputationinvolvesonly space-dependentorbitalfunctions:
ThesecondterminEq.(46)givesrisetoan exchangeintegral:
Thisrepresentsapurelyquantum-mechanicaleffect,havingnoclassicalanalog,and arisingfromtheantisymmetryprinciple.Intermsoftheorbitals ψ(r),aftercarrying outtheformalintegrationsoverthespin,wecanwrite
Unlike Jij , Kij involvestheelectronspin.Becauseofthescalarproductofthespins associatedwith φi and φj ,theexchangeintegralvanishesif σi = σj ,inotherwords, ifspin-orbitals i and j haveoppositespins, α , β or β , α Theexpressionfortheapproximatetotalenergycannowbegivenbythe summation
Notethat Kii = Jii ,whichwouldcancelanypresumedelectrostaticself-energyof aspin-orbital.Theeffectiveone-electronequationsfortheHFspin-orbitalscanbe derivedbyaprocedureanalogoustothatofEqs.(22)–(26).Anewfeatureisthatthe Lagrangemultipliersmustnowtakeaccountof N 2 orthonormalizationconditions
φi |φj = δij ,leadingto N 2 multipliers λij .Accordingly
TheLagrangemultipliers λij canberepresentedbyaHermitianmatrix.Itshould thereforebepossibletoperformaunitarytransformationtodiagonalizethe λ-matrix. Fortunately,wedonothavetodothistransformationexplicitly;wecanjustassume thatthesetofspin-orbitals φi aretheresultsafterthisunitarytransformationhasbeen carriedout.Thenewdiagonalmatrixelementscanbedesignated i = λii .Again,we seethatthe i willcorrespondtotheone-electronenergiesinthesolutionsoftheHF equations.AsageneralizationofEq.(26),thecontributiontothevariation δ L linear in δφ ∗ i isgivenby
TheeffectiveHFHamiltonian HHF isknownasthe Fockoperator,designated F . Finally,theHFequationscanbewritten
IncontrasttotheHartreeequations(26), F φi (x) alsoproducestermslinearinthe otherspin-orbitals φj , j = i.JustasintheHartreecase,thecoupledsetofHFintegrodifferentialequationscan,inprinciple,besolvednumerically,usingtheanalogous self-consistencyapproach,withiterativelyimprovedsetsofspin-orbitals.
Thesignificanceoftheone-electroneigenvalues i canbefoundbypremultiplyingtheHFequation(53)by φ ∗ i (x) andintegratingover x.Usingthedefinitionsof Hi , Jij ,and Kij ,wefind
E = i Hi +
Kij . (51)
Considernowthedifferenceinenergiesofthe N -electronsystemandthe (N 1)electronsystemwiththespin-orbital φk removed
Therefore,themagnitudesoftheeigenvalues k areapproximationsfortheionization energiesofthecorrespondingspin-orbitals φk .Sincethe k arenegative,IPk =| k |. Thisresultisknownas Koopmans’theorem.Itisnotexactsinceitassumes“frozen” spin-orbitals,whenthe N -electronsystembecomesan (N 1)-electronpositiveion. Inactualfact,theseparatelyoptimizedorbitalsforanatomormoleculeandits positiveionwillbedifferent.
ItcanbeshownthatthemagnitudesoftheCoulombandexchangeintegrals satisfytheinequalities
Ingeneral, Kij isanorderofmagnitudesmallerthanthecorrespondingCoulomb integral Jij .HFexpressionsforthetotalenergycanreadilyexplainwhythetriplet stateof,forexample,the1s2s 3 S configurationofheliumatomislowerinenergy thanthesingletofthesameconfiguration1s2s 1 S.Denotingthetwo-determinant functionsinEq.(38)as Ψ(1,3 S) forthe(+)and( )signs,respectively,wecompute theexpectationvalueofthetwo-electronHamiltonianforhelium(with Z = 2).After somealgebra,thefollowingresultisfound:
Therefore,since K > 0,thetriplet,withthe( )sign,hasthelowerenergy.One caution,however,isagainthefactthatthesingletandtripletstateshavedifferent optimizedorbitals,sothatthevaluesof K1s,2s (aswellas J1s,2s , H1s ,and H2s )arenot equal.Butevenwithseparatelyoptimizedorbitals,theconclusionremainsvalid.
OnecanalsogiveasimpleexplanationofHund’sfirstrulebasedonexchange integrals.Foragivenelectronconfiguration,thetermwithmaximummultiplicity hasthelowestenergy.Themultiplicity2S + 1ismaximizedwhenthenumberof parallelspinsisaslargeaspossible,whileconformingtothePauliprinciple.But moreparallelspinsgivemorecontributionsoftheform Kij ,thuslowerenergy.
4 HARTREE-FOCKEQUATIONSUSINGSECOND QUANTIZATION
InmuchoftherecentliteratureontheoreticaldevelopmentsbeyondtheHFmethod (“post-HF”),ithasbecomecommontoexpressoperatorsandstatevectorsusing secondquantization,whichisbasedon creation and annihilationoperators.This formalismwasoriginallyintroducedtorepresentphysicalprocessesthatinvolved actualcreationordestructionofelementaryparticles,photons,orexcitations(suchas phonons).Inamajorityofapplicationsofsecondquantizationtoquantumchemistry, noelectronsareactuallycreatedorannihilated.Theoperatorsjustserveasa convenientandoperationallyusefuldeviceintermsofwhichquantum-mechanical states,operators,commutators,andexpectationvaluescanberepresented.Tomake thenotationmorefamiliartothereader,wewill,inthissection,reexpresstheHF equationsinthelanguageofsecondquantization.
Acommonwaytointroducecreationandannihilationoperatorsisviaan alternativealgebraicapproachtotheone-dimensionalharmonicoscillator.The Schrödingerequation,inatomicunits,canbewritten
Nowdefinetheoperators
where p =−id /dq isthedimensionlessmomentumoperator.Thecanonical commutationrelation q, p = i implies
andtheHamiltonianoperatorthensimplifiesto
Withthewavefunction ψn (x) writteninDiracnotationas |n ,theSchrödinger equationinEq.(59)becomes
Thisimpliestherelation
Theharmonicoscillatorequationscanbereinterpretedasrepresentinganassembly ofphotons,orotherBose-Einsteinparticles,inwhich |n isthestatewith n particles
Thisiscalledthe occupation-numberrepresentation,the n-representation,or Fock space.Inthelanguageofsecondquantization,onedoesnotask“whichparticleis inwhichstate,”butrather,“howmanyparticlesarethereineachstate.”The vacuum state,inwhichall ni = 0,willbeabbreviatedby
(Anothercommonnotationis |vac .)
Therewillexistcreationandannihilationoperators a† 1 , a† 2 ... , a1 , a2 ....Assumingthatthebosonsdonotinteract, a and a† operatorsfordifferentvarietieswill commute.Thefollowinggeneralizedcommutationrelationsaresatisfied:
Forbosons,theoccupationnumbers ni arenotrestrictedandthewavefunctionofa compositestateissymmetricw.r.t.anypermutationofindices.Thingsare,ofcourse, quitedifferentforthecaseofelectrons,orotherfermions.Theexclusionprinciple limitstheoccupationnumbersforfermions, ni toeither0or1.Also,aswehaveseen, thewavefunctionofthesystemisantisymmetricforanyoddpermutationofparticle indices.
Thebehavioroffermionscanbeelegantlyaccountedforbyreplacingthe bosoncommutationrelations(75)bycorresponding anticommutationrelations.The anticommutatoroftwooperatorsisdefinedby
{A, B}≡ AB + BA,(76)
andthebasicanticommutationrelationsforfermioncreationandannihilation operatorsaregivenby
Theserelationsareintuitivelyreasonable,sincetherelation
isan alternativeexpressionoftheantisymmetryprinciple(34),while
0accords withtheexclusionprinciple.
Thestate(73)canbeconstructedbysuccessiveoperationsofcreationoperators onthe N -particlevacuumstate
Letusnextconsidertherepresentationofmatrixelementsinsecond-quantized notation.Wewishtoreplacetheexpectationvalueofanoperator A forthestate |Ψ withoneevaluatedforthevacuumstate |O :
Weintroducetheconventionthatanannihilationoperator,say ak ,actingonthe N -electronvacuumstate,whichwetemporarilydesignate |ON ,producesthe (N 1)electronvacuumstate |ON 1 ,inwhich0k isdeleted.Foraone-electronoperator,say H (x),suchasthecoreterm(45)intheHFequations,wecanwrite
notingthatthe k summationisoverparticles,whilethe r summationisoverspinorbitallabels.Wecannowshowthattherulefortranscribingasingle-particle operatorintosecond-quantizedformisgivenby
notingthat
sincethe |ON 1 arenormalized.ThisagreeswithEq.(80)andverifiestherule(81). Foratwo-particleoperator,suchastheelectronrepulsion r 1 ij ,wehave
where ar as = ar as or as ar .Withuseofthetruncatedvacuumstate |O
2 ,in analogywiththeabove,wefind
wherewehaveintroducedthenotationforCoulombandexchangeintegrals (47),(48).
5 ROOTHAANEQUATIONS
AsignificantimprovementinthepracticalsolutionoftheHFequationswasintroducedbyRoothaan[14].Almostallcurrentworkonatomicormolecularelectronic structureisbasedonthisandrelatedprocedures.Essentially,theintegro-differential equationsforthe φi (x) aretransformedintolinearalgebraicequationsforasetof
18CHAPTER1 IntroductiontotheHartree-Fockmethod
havingdefinedthe one-electronintegrals overthebasisfunctions
The coreoperator isdefinedby
TheCoulombandexchangeintegralsaregivenby
intermsofthe two-electronintegrals
Inanalternativenotation,moreconsistentwithDiracnotationandfavoredby physicists,
Wecandefinethe Coulomboperator,atermintheFockoperator,by
andthe exchangeoperator K ,suchthat
Formally, K actingon χα (x) givesatermlinearin χβ (x). TheFockoperator,actingonafunction χα (x),cannowbewrittenexplicitlyas
andthematrixelementsas
Wealsoneedanexpressionfortheorthonormalizationbra-kets:
knownas overlapintegrals.Thebasisfunctionsneednotbelongtoanorthonormal set.Oncealltheneededone-andtwo-electronandoverlapintegralsforagivenbasis setarecalculated,nofurtherintegrationsarenecessary.Thustheproblemreducesto linearalgebrainvolvingthecoefficients ciα .
Writingthematrixequation(91)inexplicitform,wehave
Thissystemof n simultaneouslinearequations,usuallycalledthe Roothaanequations,has n nontrivialsolutionsfor i , ci1 , ci2 cin (i = 1 n).Theconditionfora nontrivialsolutionisthevanishingofthedeterminantofthecoefficients,thatis,
Thisisan nthdegreepolynomialequationin .The n rootscorrespondtothe eigenvalues i oftheHFequations.Thecorrespondingeigenvectors ci ,whose elementsarethecoefficients ci1 cin ,arethenfoundbysolutionofthesetof homogeneouslinearequations:
(107)
Thisistypeofgeneralizedmatrixeigenvalueproblem,whichisreadilysolvedby efficientcomputerprogramsparticularlysuitedforvonNeumann-typecomputer architecture.
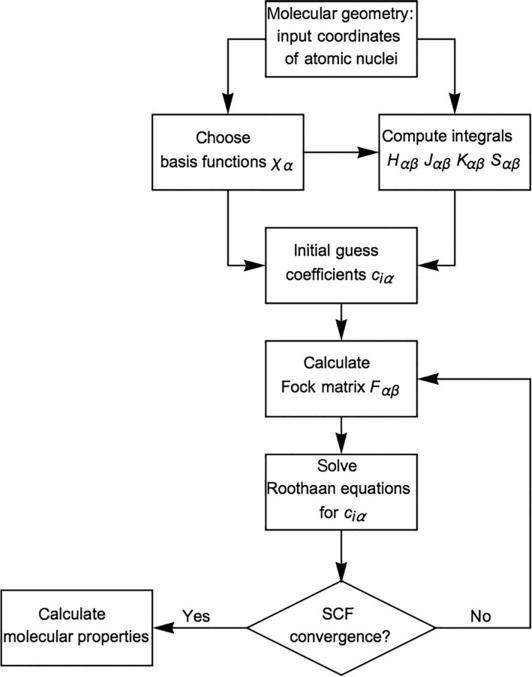
NotethattheelementsoftheFockmatrix Fαβ themselvesdependonthe coefficients ciα ,intheCoulombandexchangeterms.Butthesecoefficientsarenot obtaineduntiltheRoothaanequationsaresolved.So,paradoxically,itseemslikewe needtosolvetheequationsbeforewecanevenwritethemdown!Clearly,however, arecursiveapproachcanbeapplied.Afirst“guess”ofthecoefficients,say c(0) iα ,
FIG.1
FlowchartformolecularSCFcomputation.
isusedtoconstructaFockmatrix.Thesolutionthengivesan“improved”setof coefficients,say c(1) iα .Thesearethenused,inturn,toconstructanimprovedFock matrix,whichcanbeappliedtoobtainasecondimprovedsetofcoefficients c(2) iα .And thisprocedureisrepeateduntil self-consistency isattained,tosomedesiredlevelof accuracy.AalgorithmicflowchartforaHartree-Fock-RoothaanSCFcomputationis shownin Fig.1
Theintegralsinvolvingthebasisfunctions χα ,namely [α |β ], [αβ |γδ ] and Sαβ remainconstantduringthecomputationsinvolvingtheRoothaanequations. Butmoreaccuratecomputationsrequireever-largerbasissetsandincreasingly difficultcomputationsoftheCoulombandexchangeintegrals.Incomputationson molecules,theintegrals [αβ |γδ ] caninvolvebasisfunctionscenteredonasmany asfourdifferentatoms.Formanyyears,thecomputationofthree-andfour-center
integralswasthemostsignificantimpedimenttoprogressinmolecular-structure computations.Thisbarrierhassincebeenlargelysurmountedbytheintroduction ofGaussianbasisfunctions(tobediscussedin Chapter2),aswellasthecontinuing improvementincomputationalspeedandcapacity.
TheHFmethod,asdescribedinthischapter,hasbeenappliedmainlyusingsingle Slater-determinantwavefunctions.Thisisaninstanceofa mean-fieldapproximation, inwhicheachelectronisdescribedasinteractingwiththeaveragednonlocalpotential producedbytheotherelectrons.Theneglectedeffectsofinstantaneousparticlelikeinteractionsareknownas electroncorrelation.Forexample,Londondispersion forcesareduemainlytoelectroncorrelationandarethusnotcorrectlyaccounted forbyHFmethods.The totalenergy computedbytheHFmethodgenerallygives over99%oftheexperimentalvalue.Thismightappearimpressive,butlotsof importantchemistryhappensintheremaining1%.Theerrorduetocorrelation isgenerallyoftheorderof0.04Hartreeperelectronpair.Thisisequivalentto about1eVor100kJ/mol,thesameorderofmagnitudeaselectronexcitationsand molecularbindingenergies.Theserepresentthe differences betweenenergieswith comparableerrors,thusHFcomputationsareofonlylimitedvalueinaccounting forspectroscopicorchemicalparameters.5 Forexample,intheN2 molecule,the correlationenergyrepresents0.5%ofthetotalenergybutabout50%ofthebinding energy.Tobeusefulforchemistry,molecularenergiesmustbecomputedtoa precisionofatleastsixdecimalplaces,equivalenttoapproximately0.1kJ/mol.
Inaddition,HFcomputationsgenerallyneglectmagneticinteractions(e.g.,spinorbitcoupling)andrelativisticeffects.Thesebecomeincreasinglyimportantfor systemscontainingheavyatoms,beginningaround Z > 20.Also,usuallyneglected aretheeffectsoffinitenuclearmasses,leadingtowhatisknownas masspolarization. HFcomputationsare,ontheotherhand,quitegoodforrepresentingtheshapesof atomicandmolecularorbitalsandfordescribingelectronicchargedistributions,as, forexample,radialdistributionfunctionsforatoms.Also,inbiomolecules,reaction sitesanddockinggeometriescanoftenbesuccessfullypredicted.
6 ATOMICHFRESULTS
ThemostfrequentlyusedbasisfunctionsinatomicHFcomputationsare Slatertypeorbitals (STOs),firstintroducedin1930[15].Theseareexponentialfunctions suggestedbyhydrogenicsolutionsoftheSchrödingerequation,butlackingradial nodes,andthusarenotmutuallyorthogonal.STOshavethegeneralform(not normalized):
5 AnanalogyduetoC.A.Coulson:First,weweightheshipwiththecaptainaboardandthenweweigh theshipwiththecaptainashore;thedifferencegivesthecaptain’sweight.
Asaconsequence,four-centerintegralscanbereducedtofinitesumsoftwo-center integrals,withaspeedupofseveralordersofmagnitudecomparedwithSTOs.
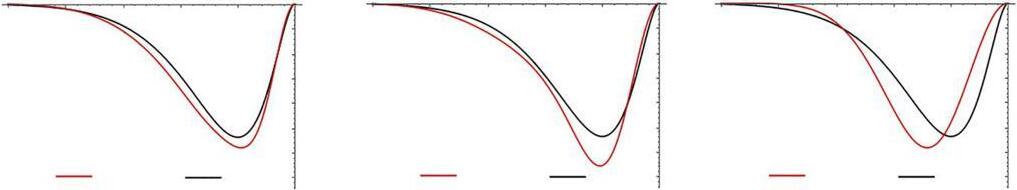
Gaussianfunctions,withtheir e α r 2 exponentialdependence,comparedtothe morerealistic e ζ r dependence,areobviouslynotverygoodphysicalrepresentations ofatomicorbitals.Theyareinaccurateindescribingthecuspbehavior,as r → 0, andtheexponentialdecay,as r →∞.Inordertominimizethesedeficienciesbut stilltakeadvantageofthecomputationalpropertiesofGaussianfunctions,Pople [18]suggestedtheuseof contractedGaussiantypeorbitals (CGTOs).Theseare linearcombinationofGaussian“primitives,”withcoefficientsandexponentsfixed, tosimulatethebehaviorofSTOs. Fig.2 showstheoptimalapproximationsofa1s STObyuptothreeGaussianprimitives.TheapproximationsofSTOsbyasumof threeGaussiansarecalledtheSTO-3Gbasisset,andarethesimplestCGTOset, whichcanproduceHFresultsofusefulaccuracy.Basissetswillbediscussedin muchgreaterdetailinthefollowingchapters.
HFcomputationshavebynowbeencarriedoutonalltheatomsoftheperiodic tableandmostatomicions. Table1 showstheresultsforatomicnumbers Z =2–20, includingtheexperimentalvaluesobtainedfromasumofthe Z ionizationenergies ofeachatom[19].TheenergyvaluesareexpressedinHartrees.Asdiscussedearlier, eventhebestHFcomputationscannotreproducetheexperimentalenergies,largely duetotheexclusionofcorrelationenergy.
TheeigenvaluesoftheRoothaanmatrixrepresenttheenergiesoftheindividual spin-orbitals,which,byKoopmans’theorem,areapproximationstoionization energiesoftheatom.Thefirstionizationenergy,whichisthedifferenceingroundstateenergiesoftheatomanditspositiveion,isofparticularsignificance.The periodicpropertiesoftheseionizationenergiesarewellknownandthesetrendsare verywellaccountedforbyhigh-qualityHFcomputations. Fig.3 showsarecent compendium[20]oftheexperimentalandcalculatedionizationenergiesforelements HthroughXe(Z = 1 54).
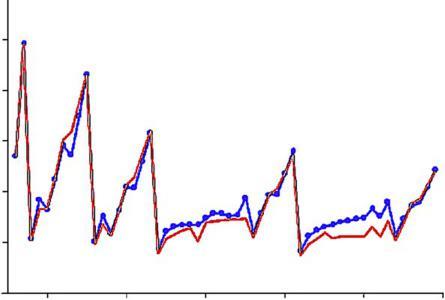
Asmentionedearlier,HFcomputationsarefairlyreliableindeterminingthe shapesofelectronicdistributionsinatomsandmolecules.Inparticular,theshell structureofmany-electronatomscanbeexhibitedinHFresults.Anearlysuccesswas thedetectionoftheradialdistributionfunctionfortheargonatom,asshownin Fig.4. TheSCFcomputationofHartree[21]iscomparedwiththeelectron-diffraction experimentalresultofBartellandBrockway[22].Thisisaveryimpressiveresult, bothfortheoryandforexperiment.
7 POST-HFMETHODS
SeveralextensionsoftheHFmethodhavebeendevelopedtoenablemoreaccurate computationsonatomsandmolecules,inparticular,toaccountfortheeffectsof
RadialdistributionfunctionsforcontractedGaussiansrepresentingthe1s STO.Theoverlapintegral S isshownforeachoftheSTO-nG functions.
FIG.2
Table1 HFComputationsforAtoms
Z
3
5
7
9
12 Mg [Ne]3s 21 S
14 Si [Ne]3s 2 3p 23 P 289.868255
15 P [Ne]3s 2 3p 34 S 341.946219 340.718780
16 S [Ne]3s 2 3p 43 P 399.034923 397.504895
17 Cl [Ne]3s 2 3p 52 P 461.381223 459.482072
18 Ar [Ne]3s 2 3p 61 S 529.112009 526.817512
19 K [Ar]4s 2 S 601.967492 599.164786
20 Ca [Ar]4s 21 S 680.101971 676.758185
FIG.3
ExperimentalandcalculatedIEvalueofatomsH-Xe. (KrishnamohanGP,MathewT,SajuS,JosephJT.WorldJChemEduc2017;5:112–9;licensedundera CreativeCommonsAttribution4.0InternationalLicense.)
Hartree theory
Electron diffraction
Radialdistributionfunctionforargonatom[22].
electroncorrelation.Suchimprovedaccuracyisnecessarytotreatchemicallysignificantpropertiesofmolecules,includingbondenergiesandgeometricparameters— bondlengthsandangles.Inaddition,therearespectroscopicfrequencies,dipole moments,magneticproperties,andNMRcouplingconstants.Post-HFmethodswill bediscussedindetailinthefollowingchapters,butwewillgiveashortintroduction heretothemainideasofconfigurationinteraction(CI),Møller-Plessetperturbation theoryandthecoupled-clustermethod.
AsingleSlater-determinant |Φ0 ,withoccupiedspin-orbitals φ1 , φ2 ...φN can serveasa referenceconfiguration foran N -electronsystem.Basissetsofdimension n > N willalsoproducehigher-energyspin-orbitals φr , φs ...,with r , s > N , knownas virtualspin-orbitals,whichareunoccupiedinthegroundstate.These mighthaveenergies r , s > 0.Nowanexcitedconfigurationcanbeconstructed bypromotingoneoftheelectronsfromanoccupiedspin-orbital,say φi ,tooneofthe virtualorbitals,say φr .WewritethecorrespondingSlaterdeterminantas |Φ r i .We canalsoconstruct doublyexcited configurationsbypromoting two electronsfrom occupiedspin-orbitals φi , φj tovirtualspin-orbitals φr , φs .Suchdeterminantsare denoted |Φ rs ij .Inthejargonofquantumchemistry,thesinglyanddoublyexcited configurationsarecalled singles and doubles,respectively.Thiscanbeextendedto give triples, quadruples,etc.Theimprovedgroundstate,enhancedbyCI,cannow berepresentedbyalinearcombinationsofSlaterdeterminants:
Thecoefficients c0 , cr i areoptimizedusingalinearvariationalmethod.ThisCI functioncan inprinciple approachtheexactsolutionofthe N -electronSchrödinger equation,ascompletenessoftheone-electronbasisset {φi (x)} isenhancedandthe
FIG.4
numberofCIcontributions |Φ isincreased.ButpracticalconsiderationandcomputationallimitationsrequiresignificanttruncationoftheCIspace.Awidelyused approximation,called“CISD,”truncatestheCIexpansiontojustsingleanddouble excitationsrelativetothereferenceconfiguration.SincetheHamiltoniancontains onlyone-andtwo-electronterms,onlysinglyanddoublyexcitedconfigurationscan interactdirectlywith |Φ0 . 7 CISDcomputationscantypicallyaccountforover90% ofthecorrelationenergyinsmallmolecules.Appropriatestrategiesforavarietyof CIcomputationswillbediscussedindetailinsucceedingchapters.
Asecondpost-HFmethodwewilldescribeis Møller-Plessetperturbationtheory [23].RecallingthemolecularHamiltonian(Eq.39),wetreatthecoretermsasan unperturbedHamiltonian
andtheelectronrepulsiontermsasaperturbation
ThesolutionoftheHFequationsfortheground-stateconfigurationwavefunction |Φ0 impliestherelation
ThustheHFenergyformallyrepresentsthesumoftheunperturbedandfirst-order perturbationenergies
InRayleigh-Schrödingerperturbationtheory,thesecond-orderenergyisgivenby
wherethe |Φn areexcitedconfigurations,suchasthoseencounteredinCI.Welimit the |Φn todoubleexcitations,notingthat,byBrillouin’stheorem,singleexcitations donotcontribute.
Toevaluatethematrixelementsof V ,werecalltheresultforasingleSlater determinant
7 Ifthesamebasisfunctions(notindividuallyoptimized)areusedinbuildingallconfigurations,then Brillouin’stheoremstatesthatall Φ0 |Φ r i = 0,sothatsingleexcitationsdonotcontributetothe groundstate.