08 Review of the 18th Annual Congress of the European Association for Haemophilia and Allied Disorders
Symposium Review
25 Entering a New Era in Protection in Haemophilia A
Congress Interview
33 Flora Peyvandi
Infographic
36 Gene Therapy: The New Frontier in Haemophilia Treatment
"Each day was packed with an array of insightful sessions, spotlighting the latest
advancements in the diagnosis, treatment, and prevention of haematological conditions"
MOVING TOWARDS NORMALISED HAEMOSTASIS IN HAEMOPHILIA A WITH ALTUVOCT®
FEATURING
Florian Langer & Martin Olivieri
3rd April 2025
12:30 - 13:30 CET
*ALTUVOCT® is indicated for use in patients of all ages for the treatment and prophylaxis of bleeding in patients with haemophilia A (congenital FVIII deficiency)1 .
For full details of ALTUVOCT® , please see the Summary of Product Characteristics by scanning this QR Code.
This is a promotional webinar intended for healthcare professionals only.
For Medical Information
Please contact your local Sobi office or medical.info@sobi.com.
This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 of SmPC for how to report adverse reactions.
1. ALTUVOCT® EU SmPC. Available at: https://www.ema.europa.eu/ en/documents/product-information/altuvoct-epar-product-information_ en.pdf (accessed March 2025).
Editor
Evgenia Koutsouki
Managing Editor
Darcy Richards
Copy Editor
Noémie Fouarge
Editorial Leads
Helena Bradbury, Ada Enesco
Editorial Co-ordinators
Victoria Antoniou, Jenna Lorge, Bertie Pearcey
Editorial Assistants
Katie Wright, Katrina Thornber, Aleksandra Zurowska
Creative Director
Tim Uden
Design Manager
Stacey White
Creative Artworker
Dillon Benn Grove
Designers
Owen Silcox, Fabio van Paris, Shanjok Gurung
Senior Performance & Insight Lead
Darren Brace
Commercial Strategy
Manager
Nabihah Durrani
Marketing Director
Kristina Mestsaninova
Chief Content Officer
Justin Levett
Chief Commercial Officer
Dan Healy
Founder and Chief
Executive Officer
Spencer Gore
Welcome
Dear Readers,
I am pleased to introduce our comprehensive coverage of the European Association for Haemophilia and Allied Disorders (EAHAD) Annual Congress 2025, a landmark event covering pertinent topics in haemophilia and bleeding disorders. From next-generation sequencing to big data, and the latest clinical trials, this year’s event showcased cutting edge research in the field. With the treatment of bleeding disorders in an ageing population being among key topics, experts addressed the unique challenges this presents.
Contact us
From next-generation sequencing to big data, and the latest clinical trials, this year’s event showcased cutting edge research in the field
In the presidential interview, Flora Peyvandi discussed with us the most pressing challenges in the field, as well as the potential of gene therapy and AI in haemophilia treatment. As we look ahead to the publication of EMJ Hematology in July, I hope you enjoy this timely insight into the latest advancements in haemophilia care that sets the stage for future innovations in the field.
Editorial enquiries: editor@emjreviews.com
Sales opportunities: salesadmin@emjreviews.com
Evgenia Koutsouki Editor
Permissions and copyright: accountsreceivable@emjreviews.com Reprints: info@emjreviews.com
Mission & Aims
The European Association for Haemophilia and Allied Disorders (EAHAD) is a multidisciplinary association of healthcare professionals who provide care for individuals with haemophilia and other bleeding disorders. Its members include haematologists, internists, paediatricians, nurses, physiotherapists, laboratory scientists and researchers from across Europe. Since its establishment in 2007, EAHAD has worked to improve the situation of people living with haemophilia and other bleeding disorders.
Goals
2.
1. Educate healthcare professionals and the public on haemophilia and its treatment.
Ensure the provision of the highest quality of clinical care for people with haemophilia and allied disorders in Europe.
3.
Support and share research on causes, prevention, and management of haemophilia.
Aims and Scope
EMJ Hematology is an open access, peer-reviewed eJournal committed to publishing the highest quality medical research concerning all aspects of diseases of the blood and bone marrow to help advance the development of this field.
The journal is published annually, approximately six weeks after the European Hematology Association (EHA) Congress, and features highlights from this congress, alongside interviews with experts in the field, reviews of abstracts presented at the congress, as well as in-depth features on congress sessions. The journal also covers advances within the clinical and pharmaceutical arenas by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ Hematology also publishes peer-reviewed research papers, review articles, and case reports in the field. In addition, the journal welcomes the submission of features and opinion pieces intended to create a discussion around key topics in the field and broaden readers’ professional interests. The journal is managed by a dedicated editorial team that adheres to a rigorous double-blind peer-review process, maintains high standards of copy editing, and ensures timely publication.
EMJ Hematology endeavours to increase knowledge, stimulate discussion, and contribute to a better understanding of blood disorders. Our focus is on research that is relevant to healthcare professionals in this field. We do not publish veterinary science papers or laboratory studies not linked to patient outcomes. We have a particular interest in topical studies that advance research and inform of coming trends affecting clinical practice in haematology.
Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
Peer Review
On submission, all articles are assessed by the editorial team to determine their suitability for the journal and appropriateness for peer review.
Editorial staff, following consultation with a member of the Editorial Board if necessary, identify three appropriate reviewers, who are selected based on their specialist knowledge in the relevant area.
All peer review is double blind. Following review, papers are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected.
Editorial staff have final discretion over any proposed amendments.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields across Europe.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®
EMJ is available through the websites of our leading partners and collaborating societies. EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
This Publication
Publication Date: April 2025 Online ISSN: 2053-6631
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. Although EMJ is independent of the EAHAD 2025 review event, EMJ and EAHAD have signed a media partnership The cover photo is of Milan, Italy the location of EAHA D2025.
The 18th Annual Congress of the European Association for Haemophilia and Allied Disorders (EAHAD) was held this year in Milan, Italy, from 4th–7th February. Each day was packed with an array of insightful sessions, spotlighting the latest advancements in the diagnosis, treatment, and prevention of haematological conditions.
A valuable opportunity for professionals from diverse fields to come together and exchange knowledge on this rapidly evolving technology
DAY 1
AHP
Professionals Day
THE ALLIED Healthcare Professionals (AHP) Day, which took place on 4th February 2025, was a multi-disciplinary event designed for nurses, physiotherapists, and psychosocial professionals in the greater field of bleeding disorders. It was an opportunity to celebrate and raise awareness of the pivotal role AHPs play in the management of bleeding disorders for patients.
Nurse-led education for female haemophilia carriers throughout their lives’ was commended as the best
The theme for this year’s AHP Day was ‘bleeding disorders through life’, and began with a collaborative, joint session between the Nurses’, Physiotherapists and Psychosocial Professionals Committees. Fitting in with the theme, three case studies were presented that followed bleeding disorders through different stages of one’s life: adolescence, menopause, and mature age. This was then followed by 'SLAM sessions', in which submitted research abstracts were awarded. The abstract ‘Nurse-led education for female hemophilia carriers throughout their lives’, presented by Nuria Caballero, Hospital Sant Joan de Déu (SJD), Barcelona, Spain, was commended as the best. This abstract presented a programme, developed by a multidisciplinary team, that created five leaflets designed to provide tailor education to female haemophilia carriers at distinct stages of their lives.
Other noteworthy abstracts presented at these SLAM sessions, include ‘Psychometric Evaluation of the Timed Up and Go (TUG) Test in Adults with Haemophilia’ by Fabian Tomschi, University of Wuppertal, Germany, and 'Thermographic change and muscle strength examination in children with haemophilia who had at least one time history of bleeding at lower extremity' by Hande Güney Deniz, Hacettepe University Faculty of Physical Therapy and Rehabilitation, Ankara, Türkiye, from the physiotherapists SLAM.
It was an opportunity to celebrate and raise awareness of the pivotal role AHPs play in the management of bleeding disorders for patients
DAY 2
The second day of the EAHAD 2025 Annual Congress offered an insightful multidisciplinary educational session, led by members of the executive committee, followed by a series of engaging satellite symposia.
Multidisciplinary Session
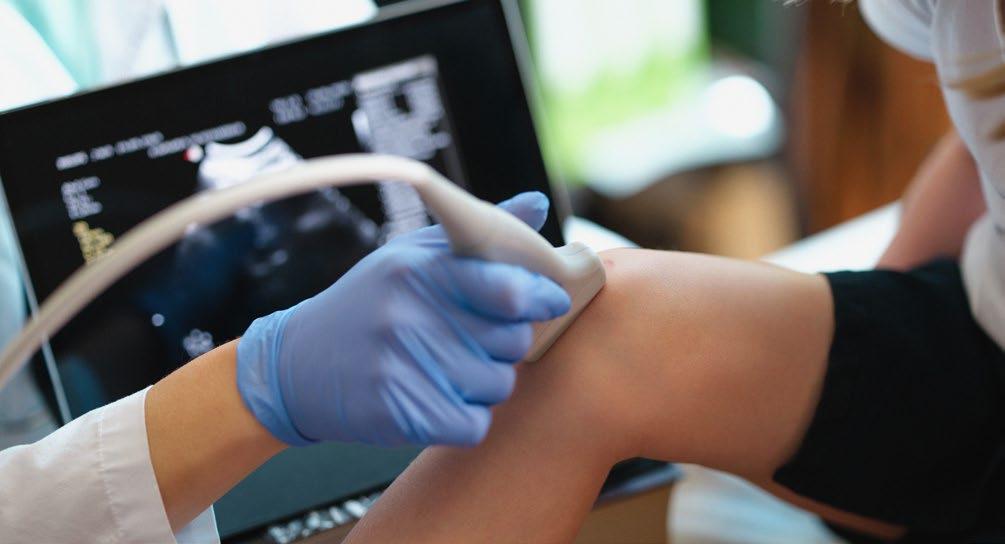
CHAIRED by Jan Blatný, EAHAD Executive Committee President, and Roberta Gualtierotti, University of Milan, Italy, this year’s multidisciplinary session focused on the topic of ultrasound.
The session featured an engaging panel discussion, where the audience had the opportunity to pose questions, vote on them, and hear the panellists’ insights and responses. A variety of thought-provoking questions were raised, covering key issues such as the importance of regular ultrasound screening for early detection, the growing role of AI in the analysis of ultrasound data, and the numerous benefits ultrasound examinations offer in clinical practice. Between each voting session, presentations were given by the
following experts: Carlo Martinoli, Cattedra di Radiologia - DISC, Università di Genova, Italy; Roberta Gualtierotti, Università degli Studi di Milano, Italy; Maj Friberg Birkedal, Chair of the EAHAD Nurses Committee; Roberto Cairoli, ASST Grande Ospedale Metropolitano Niguarda, Milano, Italy; and finally Merel Timmer, University Medical Center Utrecht, the Netherlands. The session was a valuable opportunity for professionals from diverse fields to come together and exchange knowledge on this rapidly evolving technology.
Symposia Summary
THE SATELLITE symposia covered key aspects of bleeding disorder treatments.
> Sanofi focused on thrombin generation in haemostasis, exploring its role in haemophilia treatments and the clinical use of thrombin generation assays.
> F. Hoffman-La Roche explored the evidence on physical activity in haemophilia A and key considerations for shared decision-making and management strategies.
> CSL Behring presented real-world gene therapy experiences in haemophilia B, providing insights into clinic preparations for gene therapy.
> LFB Biomedicaments looked at redefining treatment options for haemophilia patients with inhibitors.
> Takeda focused on advancements in von Willebrand disease care, emphasising how genetic insights enhance early diagnosis.
> Novo Nordisk reviewed innovations in non-factor therapies for haemophilia and the role that these treatments play in modern haemophilia care.
> Pfizer explored practical considerations in haemophilia care, especially in the new era of marstacimab therapy.
> BioMarin discussed emerging treatment strategies in haemophilia care, namely valoctocogene roxaparvovec.
> Sobi explored the impact of normalised haemostasis in people with haemophilia A across their lives, using clinical data and real-world experiences.
> Octapharma highlighted the need for equity in treating severe von Willebrand disease and severe haemophilia A.
> Kedrion focused on hereditary factor X deficiency, addressing disease management and clinical challenges.
DAY 3
The third day of EAHAD 2025 was filled with a diverse range of sessions, highlighting milestones such as the 100th anniversary of the discovery of von Willebrand disease and the presentation of the prestigious EAHAD Lifetime Achievement Awards.
Modern Era of Diagnosis and Treatment
THIS WAS an insightful session featuring three talks; firstly, Wolfram Ruf, Johannes Gutenberg University Mainz, Germany, addressed novel concepts to know in haemostasis; followed by Rosanna Asselta, Humanitas University, Milan, Italy, who looked at the innovative use of next generation sequencing in bleeding disorders; and finally, Sergio Mascetti, Department of Computer Science, Università degli Studi di Milano, Italy, highlighted the potential of AI in improving early diagnosis in bleeding disorders.
Coagulation revisited
Within his talk, Wolfram Ruf, drew on several insightful papers, including a 2024 paper by Johannes Taler and colleagues.1 Bodily fluids, such as saliva, contain extracellular vesicles that can stimulate blood coagulation by exposing extrinsic tenase complexes of tissue factor and activated factor VII (FVIIa). In this study, researchers demonstrated that the saliva of patients with severe haemophilia A, that is people with FVIII deficiency, still contain these extracellular vesicles. Therefore, extracting the vesicles from the blood and adding them to the FVIII-deficient blood of haemophilia A patients, resulted in salivaryinduced blood coagulation. In people with haemophilia A, the deficiency in FVIII leads to a deficiency of intrinsic tenase complexes (FVIIIa/FIXa) and thus reduction in FXa activation and clot formation. This study was significant as it demonstrated a way to bypass the limiting FVIII factor and still generate FX. The authors were then able to
reproduce similar results in patients with FVII deficiency; where introduction of saliva EVs from the patients induced FXa generation.1
This study was significant as it demonstrated a way to bypass the limiting FVIII factor and still generate FX
Citing a 2013 study by Schuijt TJ et al.,2 in which a recombinant tick salivary protein (TIX-5) was added to inhibit FXa-mediated activation of FV, it was concluded that FX plays a pivotal role in the generation of the first amount of activated FVa, which then binds with FXa to produce thrombin. In a follow-up study from 2017, Ruf and colleagues found that the extrinsic complex, TF-FVIIa, not only generates FIXa but also the first amounts of FXa.3 Additionally, they concluded that by inhibiting the naturally occurring TPFI inhibitory pathway you are able to generate more FXa, which in combination with FVa in turn generates more thrombin. Overall, this research deepens our understanding of the coagulation cascade pathway, and provides possible therapeutic uses in haemophilia, where patients are deficient in essential clotting factors such as FVIII. Finally, he explained that glycoprotein V (GPV) cleavage is required for limiting fibrin formation, and that GPV supresses GPIbα -thrombin dependent platelet activation, as demonstrated in a 2023 publication.4 GPV is a surface membrane protein found on the surface of platelets, that helps adhere to exposed collagen in the initial stages of haemostasis.
Next generation sequencing in bleeding disorders
Asselta took the stage to discuss the role of next-generation sequencing in bleeding disorders. Bleeding disorders are a group of conditions resulting from an impairment of the clotting process, associated either with defects in coagulation factors or platelets, both of which can be inherited or acquired. She highlighted the incidence versatility of bleeding disorders. The most
common bleeding disorders include Von Willebrand disease, haemophilia A (which is a deficiency of FVIII), and haemophilia B (a deficiency of FIX). Less common bleeding disorders encompass fibrinogen deficiency, prothrombin deficiency, and deficiencies of various clotting factors, including FV, FVII, FX, FXI, and FXIII. Platelet disorders also fall under this category. Extremely rare conditions include α2-antiplasmin deficiency, α1-antitrypsin deficiency, and combined factor deficiencies.
As explained by Asselta, genetic diagnosis is traditionally achieved through PCR amplification of the exons and splice sites followed by Sanger sequencing. Additionally, multiplex ligation-dependent probe amplification (MLPA) analysis is a genetic technique that can be used to identify mutations or structural variations in the genes associated with bleeding disorders. Asselta highlighted the advantages and disadvantages of traditional diagnostic approaches. While these methods are easy to use and standardised, they are also time-consuming, costly, and, by design, fail to provide insights into intronic or regulatory regions.
Next-generation sequencing (NGS) is a cutting-edge technology that rapidly and accurately determines the sequence of DNA or RNA, providing valuable insights into genetic variation and its role in diseases and other biological processes. This technique has revolutionised genomics by enabling the sequencing of hundreds of human genomes in a single run. In stark contrast to the original Human Genome Project, which took from 1990–2003 and cost approximately 2.7 billion USD, NGS today can sequence genomes in just a few days at a fraction of the cost, typically between $500–700 USD.
Asselta summarised the evolution of genetic techniques used to study disorders, specifically bleeding disorders. She began by discussing the linkage analysis approach, popular in the late 20th century, which involved genotyping large families and calculating LOD scores to identify genomic regions linked to diseases. She highlighted the success of this method in identifying genes responsible for haemophilia. Moving
The most common bleeding disorders include Von Willebrand disease, haemophilia A (which is a deficiency of FVIII), and haemophilia B (a deficiency of FIX)
to more modern techniques, she described genome-wide association studies (GWAS), which use SNPs to identify genetic markers associated with bleeding phenotypes. Asselta also introduced polygenic risk scores (PRS), which estimate an individual’s likelihood of developing a disorder based on numerous genetic variants.
The presentation then focused on the use of whole-exome sequencing, which allows for the sequencing of all protein-coding genes. Asselta also discussed unexplained bleeding disorders (UBD), suggesting that they may have a polygenic or non-coding genetic basis. Finally, she shared her lab’s research on UBDs, using multi-omics approaches, like whole genome sequencing and methylation analysis, to investigate potential genetic causes. She concluded by emphasising the importance of integrating data and AI in advancing genetic research.
How can AI improve early diagnosis in bleeding disorders?
Mascetti introduced a groundbreaking telemedicine solution aimed at enhancing the self-management of patients with haemophilia. The solution, called GAJA, is a tablet application paired with a portable ultrasound probe that allows patients to self-acquire ultrasound images for remote evaluation. It provides step-by-step guidance throughout the imaging process, helping patients properly position the probe and apply ultrasound gel. The system
incorporates AI to ensure that images are taken correctly by comparing the real-time positioning of key anatomical landmarks to a set of reference images acquired by the medical practitioner during a brief in-person training session. This approach is designed to address the challenges of patient training, which is often long and prone to patient forgetfulness.
Mascetti also discussed the CADET system; a web application designed to assist medical practitioners in evaluating these remotely acquired images. The CADET system leverages AI-based tools to automatically order images by suitability, suggest diagnoses, and generate semiautomatic medical reports to streamline the evaluation process. The system’s deep learning algorithms offer suggestions based on image analysis, including detecting joint effusion levels, which aids in quicker and more accurate diagnoses.
The GAJA and CADET systems have already demonstrated their effectiveness for knee joint assessments, with plans to expand to other joints, such as the elbow and ankle. Future work will focus on further refining the AI models, improving image acquisition and annotation standards, and evaluating the impact of these systems in longitudinal studies.
The GAJA and CADET systems have already demonstrated their effectiveness for knee joint assessments, with plans to expand to other joints, such as the elbow and ankle
A Century of Von Willebrand Disease
VON WILLEBRAND disease (VWD) is a type of clotting disorder characterised by the deficiency or defect in von Willebrand factor (VWF), a protein involved in the aggregation of platelets and thus blood clot formation.
First discovered by Erik von Willebrand in 1924, the disease has now reached its centenary anniversary and was spotlighted at the EAHAD 2025 meeting.
Diagnosis and optimal treatment
Giancarlo Castaman, Careggi University Hospital, Florence, Italy, opened this session providing an informative summary of the disease diagnosis and optimal treatment. VWF plays a significant role in haemostasis; it mediates platelet adhesion to the exposed sub-endothelium via collagen and GLP1 interactions, and as a carrier protein of FVIII, VWF also protects FVIII from rapid proteolysis. There are three main types of the disease: Type 1, the most common and mildest form, in which the patient produces little VWF; Type 2, which is divided in different subtypes (2A,2B,2M,2N) and is characterised by a defect in the protein; and Type 3, which the rarest and most severe. People with Type 3 produce no VWF.
Looking to the diagnosis, Castaman highlighted the ASH ISTH NFH WFH 2021 guidelines, created to provide a standardised approach in the diagnosis of the disease. In summary, the guidelines recommend for an initial screening test using the Bleeding Assessment tool (BAT), followed by laboratory tests with distinct cut-offs for Type 1 and 2 VD, as well as the use of genetic testing versus phenotypic assays for types for distinguishing between the subtypes of Type 2. He highlighted the diverse array of phenotypic tests available for VD diagnosis, such as VWF:Ag, (antigen test), VWF:Act (testing functional activity of the factor), and VWF:CB (collagen binding assay) to assess the adhesion capabilities of VWF, or PFA-100 to assess platelet function. Moving from diagnosis to treatment, the key determinants of
treatment efficacy for VWD is FVIII levels, which are in turn effected by the deficiency of VWF, and the function of VWF itself. The current main treatment strategies include desmopressin, VMF concentrates or tranexamic acid.
The current main treatment strategies include desmopressin, VMF concentrates or tranexamic acid
Demographics and epidemiology of VWD using big data
Omid Seidizadeh, University of Milan, Italy, subsequently took the stage to provide a comprehensive overview of the epidemiology of VWD. Understanding the prevalence of a disease is incredibly important, as it can impact research focus, resource allocation, healthcare planning, and public health initiatives. There are several traditional methods of determining disease prevalence, such as national/international registries, population surveys or screening programs. However, as highlighted by Seidizadeh, VWD is a complex disease, and these traditional methods carry risks of underreporting, sampling bias and data inaccuracy. Alternatively, genetic prevalence, defined as ‘the estimated proportion of a population that has a causal genotype for a genetic disorder’, allows for a larger population scale analysed across diverse ethnicities and more precise estimation.
It was estimated that the final global prevalence of VWD in 1,000 individuals was 74 for Type 1, three for Type 2A, three for Type 2B, six for Type 2M, 0.31 for Type 2N, and 0.7 for Type 3.
So how is VMD inherited? For Types 1, 2A, 2B, and 2M, it follows autosomal dominant inheritance, meaning a person needs only one copy of the mutated gene to express the disease phenotype, whilst Type 2N and Type 3 follow autosomal recessive inheritance, meaning both copies of the mutated gene must be inherited for expression. In 2023, Seidizadeh co-authored a paper where exome and genome data, gathered by the genome Aggregation Database (gnomAD) were analysed and global prevalence of both dominant and recessive forms of VMW were estimated.5 Based on Seidizadeh’s research, it was estimated that the final global prevalence of VWD in 1,000 individuals was 74 for Type 1, three for Type 2A, three for Type 2B, six for Type 2M, 0.31 for Type 2N, and 0.7 for Type 3.5
Novel therapies for VWD
To conclude the session, Jeroen Eikenboom, Leiden University, the Netherlands, discussed future therapies for VWD, emphasising the limitations of current treatments. Desmopressin, commonly used to treat bleeding, is an analogue of vasopressin that stimulates the release of endogenous FVIII and VWF. In people with Type 2 VWD, where VWF is abnormal, desmopressin only increases the production of faulty protein, limiting its effectiveness. Alternatively, exogenous VWF concentrate can be used for all VWD patients, but it requires frequent injections, has a short half-life, and carries the risk of alloantibody formation.
So, what new treatment approaches are on the horizon for VWD? Firstly, new treatments are looking to tackle the root cause of the disease by increasing the VWF levels directly. BT200, also known as rondoraptivon pegol, for example, is a pegylated aptamer that binds to the A1 domain of VWF. Eikenboom also drew on the nanobody KB-V13A12, a bi-functional molecule that binds both VWF and human albumin, effectively prolonging the halflife of endogenous VWF in circulation. An alternative to targeting VWF directly, is to target FVIII levels, as these are affected in the disease. For instance, emicizumab is a bispecific antibody that binds both activated FIX and FX, helping to facilitate coagulation. Elfanescotocog alfa is also currently under investigation in a Phase I open label study to assess the pharmacokinetics and safety and tolerability in adults with Type 2N and Type 3 VWD.6
RNA interference and gene editing is also an incredibly exciting area of research for treating VWD, by targeting the mutant gene itself and silencing the mutant VMF alleles. One promising method involved silencing the mutant allele responsible for producing faulty VWF. The research explored the use of small interfering RNAs (siRNA) to degrade mutant VWF messenger RNA and reduce abnormal protein production. This targeted approach improved VWF’s multimer formation, particularly in Type 2A and 2B patients, where mutations caused the protein to be retained in the endoplasmic reticulum.
Current and Rapid Developments Around Novel Therapies
IN A collaboration between the EAHAD and the International Society on Thrombosis and Haemostasis (ISTH), this session featured two renowned experts: Peter Lenting, Director of Research Inserm, Le Kremlin-Bicentre, France, who discussed the opportunities and challenges of novel therapies; and Armando Tripodi, Hemophilia and Thrombosis Center, IRCCS Ca’ Granda Maggiore Hospital Foundation in Milan, Italy, who explored advancements in monitoring haemophilia treatments.
Expanding haemophilia therapies to other bleeding disorders
Lenting started by introducing the different therapeutic options for patients with haemophilia A and B, like the traditional replacement therapies, bypassing agents, bispecific antibodies, monoclonal antibodies, siRNA, and gene therapy.
Lenting described the need to shift the focus in haemophilia treatment to consider the different patient groups affected by other bleeding disorders, such as VWD, women with haemophilia, and deficiencies in FV, FII, FVII, and FX. He explained how treatments for these conditions could be applied beyond haemophilia. Providing two examples, Lenting first discussed VWD Type 3, where it remains unclear to what extent VWF and FVIII each contribute to the bleeding tendency in patients with a combined deficiency of both proteins. This raised the question of whether improving FVIII-like activity could increase the haemostatic potential of a patient with VWD. Lenting referenced the current literature, noting a few studies on patients with VWD Type 3 who also had anti-VWF antibodies. In these cases, VWF concentrate, and surgical interventions were ineffective, but continuous FVIII infusions showed good
haemostatic efficiency. Lenting concluded that correcting FVIII levels in VWD Type 3 improved the haemostatic potential, though the short half-life of rFVIII made it unsuitable for prophylactic treatment.
This raised the question of whether improving FVIII-like activity could increase the haemostatic potential of a patient with VWD
Lenting then highlighted the potential of non-factor therapies in addressing these challenges. However, he stressed that while these therapies are effective for FVIII deficiency, they are not suitable for VWF deficiency. Nonetheless, they could be used regardless of whether the patient has VWF and/or FVIII deficiencies. Two examples of such treatments were emicizumab and ETX148. Lenting explained that emicizumab could significantly reduce bleeding episodes by correcting FVIII deficiencies, even without addressing the underlying VWF deficiency. Meanwhile, ETX-148, an siRNA targeting protein Z-dependent protease inhibitor (ZPI), demonstrated enhanced
Tripodi advocated for using chromogenic assays with bovine reagents, which are insensitive to emicizumab’s effects, allowing accurate measurement of endogenous or infused FVIII levels
thrombin generation and reduced blood loss in mouse models, making it a promising adjunct therapy for VWD.
Despite the promising data, Lenting acknowledged several challenges. One major concern was therapeutic balance, as there was a risk that repurposed therapies could excessively increase thrombin generation, leading to thrombosis. Additionally, he pointed out that most supporting data for these novel therapies came from animal studies, which may not fully replicate human coagulation processes. Lastly, Lenting highlighted the significant financial and regulatory hurdles in bringing new treatments for rare disorders to market.
Innovations in monitoring haemophilia treatments
In the following session, Tripodi explored advancements in monitoring haemophilia treatments. Tripodi began by highlighting the disparities between different assays used to measure FVIII and FIX activities, particularly for long-acting concentrates. He explained that these inconsistencies arise from differences in how modified and native coagulation factors are recognised by these assays. Long-acting factors being modified to extend half-life do not always behave identically to native factors, leading to variance in assay results. Tripodi also discussed the challenges in monitoring non-replacement therapies like emicizumab, a bispecific antibody
that mimics FVIII, and fitusiran, an siRNA targeting antithrombin. Traditional APTT and one-stage clotting assays proved ineffective, often producing misleading results. Even low concentrations of emicizumab can normalise APTT, masking underlying deficiencies. Similarly, one-stage assays produced paradoxically highFVIII activity results, and Tripodi advised against using these for emicizumab monitoring.
To address these issues, Tripodi advocated for using chromogenic assays with bovine reagents, which are insensitive to emicizumab’s effects, allowing accurate measurement of endogenous or infused FVIII levels. Additionally, he emphasised the importance of measuring emicizumab concentration directly using certified standards for reliable monitoring.
For fitusiran, which enhances thrombin generation by reducing antithrombin, the primary challenge lies in the lack of sensitive assays to measure low levels of antithrombin effectively. Current commercial kits, designed for congenital antithrombin deficiencies, are inadequate for monitoring fitusiran-treated patients. Tripodi highlighted the need for the development of new, more sensitive assays to accurately assess residual antithrombin levels and ensure safe and effective dosing.
Hematology Department, Hospital Sant Joan de Déu, Barcelona, Spain
Poster title: ‘Nurse-led education for female hemophilia carriers throughout their lives’
3rd Poster Prize
Alessandra Loureiro Prezotti
HEMOES, Vitoria, Brazil
Poster title: ‘Reclassification of hemophilia carriers and analysis of their hemorrhagic phenotype: experience from a center in Brazil’
2nd Poster Prize
Roberta Gualtierotti
Angelo Bianchi Bonomi Haemophilia and Thrombosis Center, Milan, Italy
Poster title: ‘Circulating miRNA Landscape in Hemophilic Arthropathy: Distinguishing Disease Conditions and Identifying Potential Biomarkers’
1st Poster Prize
Claire Kelly
National Coagulation Center, St. James Hospital, Dublin, Ireland
Poster title: ‘Spinal Stenosis: An Emerging Complication of Aging in People with Haemophilia’
DAY 4
The final day of the EAHAD 2025 Annual Congress was truly exceptional, featuring a dynamic SLAM session showcasing cutting-edge research in the field, followed by a discussion on EAHAD’s ongoing activities. The day also included an insightful session on the ageing population with rare bleeding disorders, as well as a presentation of the latest clinical trial results.
Ageing Population with Rare Bleeding Disorders
AGAINST a backdrop of an ageing population, EAHAD 2025 put the spotlight on managing rare bleeding disorders in older persons in a session chaired by EAHAD Honorary Member, Pier Mannucio Mannucci, and EAHAD Committee Member, Fariba Baghaei Sahlgrenska University Hospital, Gothenburg, Sweden. In the era of modern medicine, where people are living longer with disease, these insights are crucial for empowering healthcare professionals to optimise care for this patient cohort.
Defining frailty
The session started with a presentation by William McKeown, Antrim Area Hospital, Northern Ireland, who discussed frailty in people with haemophilia. After explaining that frailty is an age-associated condition, he emphasised that secondary to effective therapeutic interventions for haemophilia, physicians will see an increasing number of older patients with this condition.
Frailty is a state of increased vulnerability due to accumulation of deficits which lead to a poor resolution of homeostasis after a stress event. The impact of a stressor is much more significant in those with frailty and recovery may take longer and/or the
individual may not recover back to their original baseline. Spotlighting research by Sangha et al.7 that looked at frailty in the haemophilia population, McKeown noted the study found that frailty levels were higher, and also more likely to be of greater severity in these individuals, with a frailty incidence of 28.6% in the study population compared to just 10% in the general population.7
Frailty syndromes
Frailty syndromes, defined as the presence of ≥1 of falls, immobility, delirium, incontinence, or susceptibility to medication side effects, should raise suspicion for frailty, according to McKeown. He noted
The prevalence of falls, cognitive impairment, and polypharmacy in older people with haemophilia are approximately
32.4%, 73.0%, and 40.8%, respectively.
that whilst data on these syndromes are limited, best estimates show that the prevalence of falls, cognitive impairment, and polypharmacy in older people with haemophilia are approximately 32.4%, 73.0%, and 40.8%, respectively. All of these have an impact on loss of dependence and mental health.
In terms of a solution, the gold standard intervention for frailty is the comprehensive geriatric assessment (CGA), which includes a multidisciplinary team (MDT) assessment to identify problems and propose solutions.
World Federation of Hemophilia (WFH) guidelines recommend coordinated delivery of comprehensive care by an MDT with expertise and experience in haemophilia. McKeown made recommendations that pharmacists, dieticians, speech and language therapists, occupational therapists, audiologists, and opticians should also be included in this MDT. He stated that the haemophilia MDT for older people should be expanded urgently to help meet their needs.
Venous thrombosis in haemophilia
Whilst the concept may seem paradoxical, Cihan Ay, Medical University of Vienna, Austria, spoke on management of venous thrombosis in older people with haemophilia.
Similarly to those without haemophilia, major orthopaedic surgery is a risk factor for venous thromboembolism in those with haemophilia. Ay also noted that venous thromboembolism can potentially occur during factor replacement therapy, and with non-factor therapies and after gene therapy. Furthermore, thrombosis risk increases significantly with age, and Ay drew attention to evidence from a small study which showed those with haemophilia exhibit an accelerated biological ageing.
Whilst there is limited evidence, Ay spotlighted a recent joint EHA-ISTHEAHAD-ESO initiative, which suggests treating venous thromboembolism in haemophilia with a limited duration of anticoagulation, maintaining trough factor
levels >20 IU/dL, and eliminating the potential trigger.
He stressed that approaches should be individualised, and clinicians should look to balance the risk of VTE progression/ recurrence without anticoagulation against the risk of bleeding whilst receiving anticoagulation and the severity of haemophilia. He noted that for those with haemophilia and venous thromboembolism, direct oral anticoagulants are recommended over vitamin K antagonists. However, Ay recommended considering an alternative approach of full dose heparin whilst maintaining a FVIII/FIX trough level >20 IU/ dL for 5–7 days, and if no bleeding, then switching to a direct oral anticoagulant at a standard dose.
The important role haemophilia centres and national member organisations have in supporting the ageing community to improve their quality of life
Psychosocial aspects of ageing with haemophilia
Christina Burgess, Haemophilia and Bleeding Disorders Counselling Association, UK, closed the session by discussing the psychosocial aspects ageing with haemophilia. Emphasising how haemophilia impacts quality of life as we age, Burgess highlighted severe joint damage, longterm chronic pain, HIV and/Hepatitis C infection, unresolved psychological trauma, missed educational or employment opportunities due to long hospital stays, and comorbidities as key factors influencing psychosocial wellbeing. She concluded the important role haemophilia centres and national member organisations have in supporting the ageing community to improve their quality of life.
The Latest Clinical Trial Updates
A HIGHLYANTICIPATED session covering recent data from six clinical trials took place at EAHAD 2025, and was chaired by the Congress President, Flora Peyvandi, and Executive Committee President, Jan Blatny. These presentations provided experts with key updates into long-term safety and efficacy data for therapeutic agents used in bleeding disorders, as well as insights into new interventions currently in early-phase trials.
Trial updates for haemophilia
Mike Makris, University of Sheffield, UK presented the 15-year European Haemophilia Surveillance System (EUHASS) data on thrombotic rates in individuals with bleeding disorders treated with bispecific antibody or concentrate.
Diving into the data, Makris compared the thrombosis rates per 1,000 treatment years for individual product classes and different bleeding disorders. The thrombosis rate per 1,000 treatment years for those with haemophilia A treated with plasma FVIIII/VWF was 1.19 (95% CI: 0.90–1.54), compared with a rate of 0.70 (95% CI: 0.54–0.90) for those who received standard half-life recombinant FVIII, and a rate of 0.50 (95% CI: 0.20–1.02) for those who received extended half-life recombinant FVIII.
For those with haemophilia B, the thrombosis rates per 1,000 treatment years were 0.98 (95% CI: 0.39–2.01), 0.73 (95% CI: 0.35–1.35), and 0.52 (95% CI: 0.11–1.52) for those treated with plasma derived
FIX, standard half-life recombinant FIX, and extended half-life recombinant FIX, respectively. Makris also drew attention to the fact that risk of thrombosis may be higher when treating patients with FXIII deficiency, FXI deficiency, or afibrogenemia.
Following this, Lynn Malec, Versiti Blood Research Institute, Wisconsin; and Medical College of Wisconsin, USA, presented 2-year data on the outcomes of onceweekly efanesoctocog alfa prophylaxis in children with severe haemophilia A, as part of a second interim analysis of the XTENDed Phase III study. The primary endpoint of this study is the occurrence of inhibitor development.
After covering the trial design and safety data, Malec concluded that FVIII inhibitors did not develop over the course of the extension study, mean annualised bleed rates remained low at <1 with median annualised bleed rates of 0, the percentage of patients with zero bleeding episodes remained high, and that the
2-year results have shown that onceweekly efanesoctocog alfa in previously treated children with severe haemophilia A continues to be well tolerated and effective at bleed protection.
Davide Matino, McMaster University, Ontario, Canada, presented the long-term efficacy data for marstacimab in adults and adolescents with severe haemophilia A or B without inhibitors who completed the BASIS trial. He summarised the study design, marstacimab exposure in both BASIS and the open-label extension, and safety data.
HMB-001 showed doseproportionate pharmacodynamics and pharmacokinetics with peak FVIIa accumulation 4–8 days postdose in part A and B of the study
From the trial findings, Matino reported that once-weekly, subcutaneous marstacimab demonstrated sustained/improved efficacy for treated and total annualised bleeding rates in the patient cohort, without inhibitors. This was found to be consistent in those who received on-demand or routine prophylaxis factor replacement therapy at baseline. Matino concluded that overall, marstacimab was safe and well tolerated.
Steven Pipe, University of Michigan, Ann Arbor, USA, discussed long-term safety and efficacy data of etranacogene dezaparvovec from the Phase III HOPE-B trial in adult males with severe/moderately severe haemophilia B. The trial looked at durability of FIX expression, bleed data, exogenous FIX consumption, and safety over 4 years after treatment.
After summarising key components of the trial design, participant demographics, annualised bleeding rates, endogenous FIX levels over time, exogenous FIX use during the study period, and safety data, Pipe concluded that treatment-related adverse events were almost absent after the first 6 months following gene therapy, FIX replacement use decreased from baseline by 95%, that mean FIX activity levels were stable and in the near-normal range over the duration of follow-up, and that etranacogene dezaparvovec resulted in significant annualised bleeding rate reduction across the 4-year follow-up compared with the lead-in FIX prophylaxis period.
Trial updates for glanzmann thromboasthenia
Suthesh Sivapalaratnam, Queen Mary University of London; and Barts Health NHS Trust, UK, presented the interim analysis of
a Phase I/II study on safety and efficacy of HMB-001 as a prophylactic treatment for Glanzmann Thromboasthenia. He explained that there are currently no approved prophylactic therapies for this rare genetic bleeding disorder.
HMB-001 showed doseproportionate pharmacodynamics and pharmacokinetics with peak FVIIa accumulation 4–8 days postdose in part A and B of the study
Sivapalaratnam explored the mechanism of HMB-001 bispecific antibody and explained the three study parts. He went on to discuss patient demographics and safety data, before concluding that HMB-001 showed dose-proportionate pharmacodynamics and pharmacokinetics with peak FVIIa accumulation 4–8 days post-dose in part A and B of the study, and that at lower doses there were no thromboses or serious adverse events. However, he did highlight
References
1. Thaler J et al. Saliva of persons with hemophilia A triggers coagulation via extrinsic tenase complexes. Blood. 2024;144(25):2666-77.
2. Schuijt TJ et al. Factor Xa activation of factor V is of paramount importance in initiating the coagulation system: lessons from a tick salivary protein. Circulation. 2013;128(3):254-66.
3. Kamikubo Y et al. Selective factor
one serious adverse event and D-dimer increase at a higher dose.
To conclude his presentation, Sivapalaratnam explained that there were clinically meaningful reductions in treated bleeds across all dose levels and that the phase II study is ongoing to further investigate the lower doses to confirm their safety and efficacy as a prophylactic for individuals with this condition.
Trial updates for von willebrand disease
Carolyn M. Millar from Imperial College London and Imperial College Healthcare NHS Trust, UK, presented the findings from a Phase I study of VGA039 in individuals with von Willebrand disease (VWD). VWD is a genetic bleeding disorder caused by a deficiency or dysfunction of VWF, essential for blood clotting. The Phase I study aimed to evaluate the safety, tolerability, and pharmacokinetics of VGA039, a novel therapeutic candidate designed to address this condition.
VIII activation by the tissue factor–factor VIIa–factor Xa complex. Blood. 2017;130(14):1661-70.
4. Beck S et al. Platelet glycoprotein V spatio-temporally controls fibrin formation. Nature Cardiovascular Research. 2023;2(4):368-82.
5. Seidizadeh O et al. Population-based prevalence and mutational landscape of von Willebrand disease using largescale genetic databases. NPJ Genom Med. 2023;8(1):31.
6. Bioverativ. To Assess the Pharmacokinetics and Safety and Tolerability of Efanesoctocog Alfa (BIVV001)in Adults With Type 2N and 3 Von Willebrand Disease (VWD). NCT04770935. https://clinicaltrials. gov/study/NCT04770935.
7. Sangha G et al. Frailty and haemophilia; speaking the language of geriatricians. Haemophilia. 2023;29(5):1371-5.
Entering a New Era in Protection in Haemophilia A
This symposium took place on 5th February as part of the Annual Congress of the European Association for Haemophilia and Allied Disorders (EAHAD) 2025, held in Milan, Italy, between 4th–7th February 2025.
Chairperson: Jan Astermark1,2
Speakers: Maria Elisa Mancuso,3,4 Christoph Königs,5 Robert Klamroth6,7
1. Department of Translational Medicine, Lund University, Sweden
2. Department of Haematology, Oncology and Radiation Physics, Skåne University Hospital, Malmö, Sweden
3. Centre for Thrombosis and Haemorrhagic Diseases, IRCCS Humanitas Research Hospital, Milan, Italy
4. Humanitas University, Milan, Italy
5. Department of Paediatrics and Adolescent Medicine, Clinical and Molecular Haemostasis, Goethe University, University Hospital, Frankfurt, Germany
6. Department of Internal Medicine, Vascular Medicine and Haematology, Vivantes Klinikum Friedrichshain, Berlin, Germany
7. Haemophilia Treatment Centre, Vivantes Klinikum Friedrichshain, Berlin, Germany
Disclosure: Astermark was principal investigator (PI) or has received research support or grant funding from CSL Behring, SobiTM, and Takeda; been a consultant for Bayer, BioMarin, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Sanofi, SobiTM, and Takeda; received speaker’s fees from Bayer, BioMarin, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, and SobiTM; and has participated on scientific Advisory Boards for Bayer, BioMarin, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, SobiTM, and Takeda. Mancuso was PI or has received research support or grant funding from Bayer, CSL Behring, Novo Nordisk, and Takeda; been a consultant for Bayer, BioMarin, CSL Behring, Kedrion, Novo Nordisk, Octapharma, Pfizer, Regeneron, Roche, Sanofi, and SobiTM; received speaker’s fees from Bayer, CSL Behring, Kedrion, Novo Nordisk, Octapharma, Pfizer, Regeneron, Roche, Sanofi, and SobiTM; and has participated on scientific Advisory Boards for Bayer, BioMarin, CSL Behring, Kedrion, Novo Nordisk, Octapharma, Pfizer, Regeneron, Roche, Sanofi, SobiTM, and Takeda. Königs was PI or has received research support or grant funding from Bayer, Biotest, CSL Behring, Intersero, Novo Nordisk, Pfizer, Roche/Chugai, Sanofi, SobiTM, and Takeda; received speaker’s fees from BFSH, Bayer, CSL Behring, Novo Nordisk, Roche/Chugai, SobiTM, and Takeda; and has participated on scientific Advisory Boards for CSL Behring, Novo Nordisk, Pfizer, Roche/Chugai, and SobiTM. Klamroth was PI or has received research support or grant funding from Novo Nordisk, Octapharma, and SobiTM; been a consultant for Bayer, Biotest, BioMarin, CSL Behring, Grifols, Kedrion, LFB, Novo Nordisk, Octapharma, Pfizer, Roche/Chugai, Sanofi, SobiTM, and Takeda; received speaker’s fees from Bayer, Biotest, BioMarin, CSL Behring, Grifols, Kedrion, Leo, LFB, Novo Nordisk, Octapharma, Pfizer, Roche/Chugai, Sanofi, SobiTM, Takeda, and Viatris; and has participated on scientific Advisory Boards for Bayer, Biotest, BioMarin, CSL Behring, Grifols, Kedrion, Leo, LFB, Novo Nordisk, Octapharma, Pfizer, Roche/ Chugai, Sanofi, SobiTM, and Takeda.
Acknowledgements:
Medical writing assistance was provided by Leonie Glasson, AMICULUM, Bollington, UK.
Disclaimer The views and opinions expressed are those of the authors and not necessarily of SobiTM
Keywords: Factor VIII (FVIII), haemophilia A (HA), haemostasis, health equity, normalisation.
Support: The symposium and the publication of this article were funded by SobiTM. Approval number: NP-39929 - March 2025.
Meeting Summary
Haemophilia A (HA), defined by factor VIII (FVIII) levels ≤40 IU/dL, is a chronic condition with consequences beyond bleeding complications. Jan Astermark (Professor of Clinical Coagulation Medicine, Senior Consultant, and Head of Department of Translational Medicine, Lund University; and Department of Haematology, Oncology and Radiation Physics, Skåne University Hospital, Malmö, Sweden) outlined the burden of HA on the quality of life (QoL) of patients, including bleeding, joint damage, pain, psychosocial wellbeing, and physical activity. He shared real-world evidence showing that current prophylactic regimens with FVIII or non-factor therapy (NFT) are not sufficient to eliminate all types of bleeds and that many challenges remain. Astermark presented several analyses highlighting that FVIII levels in the non-haemophilia range may be necessary to prevent residual bleeding. These analyses have informed recent treatment goals that transcend historical targets of converting severe HA (SHA) into moderate or mild forms and aim towards normalised haemostasis to eliminate bleeds.
Maria Elisa Mancuso (Senior Consultant in Haematology, Centre for Thrombosis and Haemorrhagic Diseases, IRCCS Humanitas Research Hospital and Humanitas University, Milan, Italy) presented a patient case to illustrate the challenges that people with HA (PwHA) face over their lives and the evolution of treatment strategies to address unmet needs. Christoph Königs (Head of Clinical and Molecular Haemostasis at the Department of Paediatrics and Adolescent Medicine, Clinical and Molecular Haemostasis, Goethe University, University Hospital, Frankfurt, Germany) emphasised the unique challenges faced by children with HA and their caregivers, including restrictions in daily activities, regular evaluations for subclinical and evident bleeds, long-term joint protection, delayed inhibitor development, self-injection skills, and suboptimal adherence. He discussed how standard and extended half-life (SHL, EHL) therapies have improved care in children with HA but highlighted how prophylaxis with existing therapies is not sufficient to eliminate evident and subclinical bleeds. He concluded by sharing data on novel therapies that offer the potential to maintain FVIII levels in the non-haemophilia range (≥40 IU/dL) to help address these unmet needs. Robert Klamroth (Head of the Department of Internal Medicine, Vascular Medicine and Haematology, and Director of Haemophilia Treatment Centre, at Vivantes Klinikum Friedrichshain, Berlin, Germany) focused on the evolving challenges of HA in adulthood, including surgery and the need for anticoagulant or antiplatelet therapy for the management of comorbidities. Recent clinical data were shown to demonstrate how high sustained FVIII levels could minimise bleeding risk and improve joint health, surgical management, and overall QoL in adults with HA. In the panel discussion, two patient cases were reviewed to consider unmet needs in people with mild HA and in elderly people with HA, and the panel summarised how sustaining FVIII levels in the non-haemophilia range could help address these needs. The panel concluded by reviewing the evolution of treatment strategies and the importance of targeting normalised haemostasis in a new era of protection in HA.
PHARMA PARTNERSHIP
Welcome and Introduction
Jan Astermark
Despite regular and early prophylactic treatment, PwHA at all ages still experience bleeds that impact their daily lives. In a realworld cross-sectional survey of 244 people with severe or moderate HA treated with prophylaxis in the USA, 25% of patients had joint problems and 43% experienced at least one bleed in the year prior to the survey.1 Bleeding and joint problems, in turn, have a significant burden on patients’ QoL, with 39% of patients experiencing disruptions to daily living, 28% being unable to work full time, and 84% sometimes or often avoiding physical activity because of HA.1
Recent analyses indicate that sustained time in the non-haemophilia range (FVIII levels ≥40 IU/dL) may be a necessary first step to elevating the standard of care in PwHA (Figure 1).2-6 Targeting FVIII levels ≥40 IU/dL could improve clinical outcomes and the QoL of PwHA by reducing the risk of clinical and subclinical bleeds, improving long-term joint health, and reducing synovitis, joint damage, and pain.2-5 This symposium discussed this new era in HA protection and explored how novel therapies may help achieve normalised haemostasis to improve the physical and mental well-being of PwHA.
Evolving Goals in Haemophilia A: From Concept to Reality
Maria Elisa Mancuso
Mancuso illustrated the challenges associated with HA and the evolution of treatment strategies via the example case of a male patient who was diagnosed with SHA at 14 months old following a gluteus haematoma.
Childhood
To manage the gluteus haematoma, the patient was treated with SHL-FVIII and started on regular prophylaxis to minimise the risk of future bleeds. In children, venous access can be a challenge; for this child, it led to skipped infusions and suboptimal protection. He experienced recurrent joint bleeds and so began prolonged treatment with a central venous line to help optimise prophylaxis. At 8 years old, he experienced a central venous line infection that necessitated a temporary stop in SHL-FVIII, but his regular treatment was resumed once the infection subsided.
Adolescence
When he was 11 years old, the first EHLFVIII became available, but his parents were reluctant to switch his treatment in fear of inhibitor development. He continued on
1: Normalisation of haemostasis as the beginning of the journey to elevate standards of care2–6
1: Normalisation of haemostasis as the beginning of the journey to elevate standards of care.2-6
FVIII: factor VIII; PwHA: people with haemophilia A; QoL: quality of life.
Figure
Figure
FVIII, factor VIII; PwHA, people with haemophilia A; QoL, quality of life
PwHA can
SHL-FVIII for a further 7 years until, after recurrent target joint bleeds, he decided to switch to EHL recombinant FVIII (rFVIII) at 15 years old.
When playing basketball at school, he noticed mild ankle discomfort that he managed with injections prior to training. However, an ultrasound evaluation identified synovial hypertrophy at his right ankle and left elbow as well as initial chondral changes at his left knee. His trough level at 72 hours was 3–6%. To his disappointment, he was advised to change hobby.
Adulthood
The patient continued his treatment of EHL-rFVIII but variable venous access and a reluctance to inject at inconvenient times resulted in suboptimal adherence. To minimise treatment burden, he switched to a once-weekly NFT, which lessened his joint pain to a degree that he felt able to return to the gym. However, over the next few years, he experienced several spontaneous bleeds in his left knee, and subsequent ultrasound evaluations showed synovial hypertrophy at his right ankle and evidence of progressive osteochondral changes at his left knee. He decided to switch back to EHL-rFVIII to intensify his prophylaxis and started selective Cox-2 inhibitors and regular physiotherapy to help manage his pain.
Ageing
Despite his treatment programme, his knee pain continued, and the patient was indicated for knee replacement surgery when he was 42 years old. The surgery was successfully managed with perioperative FVIII management, and he subsequently entered a trial with a rebalancing agent administered once every 2 months. This regimen minimised his treatment burden, but ongoing pain limited his activity leading to gradual weight gain and the development of hypertension and mild dyslipidaemia. In his sixties, he underwent surgery for prostate cancer that necessitated antagonism of the NFT effect. EHL-rFVIII was used and FVIII activity levels were closely monitored to ensure adequate haemostasis during and after his prostatectomy.
Mancuso concluded her presentation by summarising the evolution of treatment strategies. Treatments for HA historically focused on converting severe disease (FVIII <1 IU/dL) into moderate forms (FVIII ~1–5 IU/dL) to minimise life-threatening bleeds. Improvements in care were observed with EHL therapies and NFTs; however, these are not sufficient to fully protect patients from bleeds, including subclinical and joint bleeds.7 With the development of novel treatment options, including ultra-long FVIII replacement therapy, it is feasible to now target more ambitious goals of normalised haemostasis.2
Realising New Possibilities for Children with Haemophilia A
Christoph Königs
Königs emphasised the unique challenges faced by children with HA and their caregivers, including the risk of inhibitor development, lack of self-injection skills, suboptimal adherence, the need for regular evaluations of subclinical and joint bleeds, and limitations in physical activities (Figure 2).
Children with HA can be affected by significant joint and muscle bleeding. In a UK cohort of 237 PwHA (including 80 children <18 years) selected for high compliance with EHL or SHL prophylaxis, 32.5% were affected by joint bleeds.8 Similarly, in a real-world, retrospective, multi-institutional cohort of 314 children and young adults with HA receiving NFT, 15 patients (including four with inhibitors) experienced 19 muscle bleeds that required intensive and prolonged factor treatment.9 Impaired joint health in the absence of reported bleeding was also documented in a cross-sectional study of 106 children (636 joints) with SHA on prophylaxis, where 50% scored >0 in Hemophilia Early Arthopathy Detection with Ultrasound (HEAD-US) and 13% of joints with no bleeding history were scored ≥1 in HEAD-US.10
2: Challenges for children with SHA and their caregivers 10,20–22
Among the current treatment approaches for children, emicizumab is an NFT indicated in the European Union as routine prophylaxis in PwHA of all ages with FVIII inhibitors and in those without FVIII inhibitors if they have SHA or moderate HA with a severe bleeding phenotype.11 In the Phase IIIb HAVEN 7 trial, emicizumab prophylaxis demonstrated efficacy and a favourable safety profile in 55 infants aged ≤1 year with SHA without inhibitors.12 After a median efficacy period of 101.9 weeks, the model-based mean (95% CI) annualised bleeding rate (ABR) was 0.40 (0.30–0.63) for treated bleeds, and 54.5% of patients experienced no treated bleeds.12 Emicizumab had an acceptable safety profile with no intracranial haemorrhages or new safety signals identified during the study.12 Prior studies have reported that emicizumab maintains FVIII-equivalent levels in animal models of ~9–20 IU/dL and FVIII-like activity in clinical trials of ~15–20 IU/dL.1316 However, FVIII-like activity reported for PwHA treated with emicizumab cannot be compared with, or interpreted as equivalent to, FVIII activity reported in participants treated with FVIII.12,14
Advances to sustain FVIII levels within the non-haemophilia range could help to prevent evident and subclinical bleeding in PwHA. Efanesoctocog alfa is an FVIII replacement therapy indicated in the European Union for the treatment and prophylaxis of bleeding in PwHA of all ages.17 In the Phase III XTEND-Kids trial in people with SHA (PwSHA) <12 years, efanesoctocog alfa provided high sustained FVIII activity in the non-haemophilia range (>40 IU/dL) for 3 days and >10 IU/dL for almost 7 days after administration, leading to effective bleeding treatment and prevention.18 The mean (95% CI) ABR for treated bleeds was 0.61 (0.42–0.90) for overall bleeding and 0.16 (0.0–0.30) for spontaneous bleeding (sensitivity population; n=73), and 88% of participants had zero treated spontaneous bleeds.18 Efanesoctocog alfa was well tolerated, with no adverse events leading to discontinuation. No inhibitors or antidrug antibodies were reported.18 Low bleed rates were maintained for children (n=71) throughout the XTEND-ed extension study, where over a median (range) cumulative treatment duration of 119.1 (88.1–152.6) weeks with efanesoctocog alfa, a mean (95% CI) ABR of 0.67 (0.48–0.93), and 83% (44/53) of patients with zero bleeds at Months 12–18 were reported.19
Figure
FVIII: factor VIII; ICH: intracranial haemorrhage; IV: intravenous; NFT: non-factor therapy; SHA: severe haemophilia A.
Figure 2: Challenges for children with SHA and their caregivers.10,20-26
Achieving sustained FVIII levels in the nonhaemophilia range makes the ambition of a haemophilia-free mind for children with HA and their caregivers more feasible.2,5
Transforming Clinical Perspectives in Adults with Haemophilia A
Robert Klamroth
Adults living with SHA face multiple challenges, including healthcare dependency, evident and subclinical bleeds, physical activity limitations, surgeries, and comorbidities requiring concomitant medications, including anticoagulants and antiplatelets.9,20,26-36
Pain, particularly joint pain, is a significant burden to PwHA and is often associated with a higher use of pain, depression, and anxiety medications compared with population controls.37,38 In a German survey including adults with HA (N=513), pain prevalence increased with age, and over two-thirds of patients aged >40 years reported frequent joint pain.38 In a Phase III, open-label, multicentre study of PwSHA, patients reporting no pain were significantly more likely to have zero bleeds (57%) than patients reporting pain (43%; p<0.05).39
Despite prophylaxis, adults with HA can still experience bleeding, including spontaneous joint bleeds. In a 12-month observational study in the UK including 237 PwSHA (157 adults), 60% of adults were affected by joint bleeds despite FVIII prophylaxis.8 Similarly, in an observational cohort of 40 PwSHA in Italy, 25% developed spontaneous joint bleeding despite receiving NFT. In the latter study, thrombin generation assays failed to differentiate patients by risk of spontaneous joint bleeding, but synovitis and higher HEAD-US score were strong predictive factors for spontaneous joint bleeds.28
Targeting FVIII levels in the non-haemophilia range is supported by data from a Phase III gene therapy trial, which found that 100% of patients (n=11) with FVIII levels ≥40 IU/ dL had zero treated bleeds after Year 2 (Figure 3).40 Once-weekly efanesoctocog
alfa provides high sustained FVIII activity, with a mean steady-state half-life (CI) of 47.0 (42.3–52.2) hours, thus enabling FVIII levels ≥40% for 4 days and >15% for 7 days in adults.41 In the first 2 years of the XTENDed extension trial, efanesoctocog alfa demonstrated sustained bleed protection in adults and adolescents (N=146), with a mean (standard deviation [SD]) overall ABR of 0.64 (1.0) and mean (SD) spontaneous ABR of 0.24 (0.5), after a median (range) cumulative treatment duration of 170.5 (46.3–192.6) weeks.41,42 The proportion of patients with zero bleed rates remained high and stable over the full efficacy period, from 74.3–80.4% for overall bleeds and from 87.5–93.2% for spontaneous bleeds.41,42 Improvements were also observed in haemophilia joint health scores (particularly in patients >50 years) and Haem-A-QoL (mean [SD] change in physical health: -7.5 [20.05]; total score: -5.8 [13.35]).41-43 Efanesoctocog was overall well tolerated. No FVIII inhibitors were detected. Two patients experienced thromboembolic events: one was a deep vein thrombosis following corrective surgery for a femur fracture (in the setting of treatment with another FVIII product), and the other was a cerebral infarction in a patient with pre-existing atrial fibrillation and other risk factors; neither event was related to efanesoctocog alfa treatment. Two related treatment-emergent adverse events were reported (facial paralysis and locally measured coagulation FVIII level decrease), and both events resolved.42,44
In PwHA, surgery requires careful management of bleeding risk by maintaining high perioperative FVIII levels. Over 4 years in the XTEND clinical programme, 45 patients underwent 62 major surgeries (31 orthopaedic). A good or excellent haemostasis response was reported in 95.2% of patients, with low mean (SD) perioperative efanesoctocog alfa consumption (Days -1 to 14) of 176.2 (54.6) IU/kg across an average (SD) of 4.1 (1.7) injections.45 Few injections and low perioperative consumption was also observed for minor surgeries, of which 47 patients (56 surgeries) had a mean (SD) perioperative consumption (Days -1 to 7) of 94.3 (31.8) IU/kg, across an average (SD) of 1.9 (0.8) injections.45
FVIII: >5 to <15 IU/dL
74% patients with zero bleeds
FVIII: >15 to <40 IU/dL
92% patients with zero bleeds
Transitioning From Clinical Trials to Real-World Practice
Speaker presentations were followed by a panel discussion of two patient cases to demonstrate the challenges of PwHA.
The first patient was a 15-year-old boy living with mild HA. He was receiving ondemand EHL-rFVIII that fit well around his active lifestyle and had no recent history of joint bleeds or obvious symptoms of his disease. In a routine ultrasound, however, he was found to have synovitis in both ankle joints. In an audience poll, long-term joint health and the ability to be physically active/play with friends were voted to be the most important outcomes to consider for a child/adolescent like this patient. The panel agreed that regular prophylaxis was important to maintain high FVIII levels, even in patients living with mild HA. Furthermore, despite low evident bleed rates, it is important to maintain optimal adherence and regular ultrasound monitoring to ensure adolescents do not experience clinical or subclinical bleeds that could contribute to long-term joint damage.2,4,46
References
1. Malec L et al. Unmet needs among people with hemophilia A in the United States: a real-world analysis of the Adelphi hemophilia disease specific programme. Oral 322. ASH Annual Meeting, 7-10 December, 2024.
FVIII: ≥40 IU/dL Non-haemophilia range
100% patients with zero bleeds
The second case introduced a 77-yearold man living with SHA and concomitant cardiovascular disease. In an audience poll, approximately half of the audience thought the optimal required FVIII level to permit dual antiplatelet therapy without bleeding risk in PwHA was 20%, with approximately one-third choosing FVIII levels of 30%. Based on the current available recommendation, the panel agreed on a baseline FVIII coagulation >20% for dual antiplatelet therapy or oral anticoagulation, and recommendations suggest the duration of such therapies should be as short as possible.35,47
The panel concluded that treatment goals for PwHA are evolving to optimise health and well-being.3,7 Haemophilia therapies that can sustain FVIII levels in the non-haemophilia range could help overcome the challenges that children and adults with HA face and improve QoL by reducing the emotional burden caused by bleeds, pain, and limitations in everyday life.3,4,6,48
2. Skinner MW et al. Achieving the unimaginable: health equity in haemophilia. Haemophilia. 2020;26(1):1724.
3. Srivastava A et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
4. Malec L, Matino D. Targeting higher factor VIII levels for prophylaxis in haemophilia A: a narrative review. Haemophilia. 2023;29(6):1419-29.
5. Hermans C, Pierce GF. Towards achieving a haemophilia-free mind. Haemophilia. 2023;29(4):951-3.
Figure 3: Maintaining FVIII levels in the non-haemophilia range could help achieve the goal of zero bleeds.40
FVIII: factor VIII.
FVIII,
6. Holme PA et al. Moving towards normalization of haemostasis and health equity: evolving treatment goals for haemophilia A. Haemophilia. 2024;30(5):1109-14.
7. den Uijl IE et al. Turning severe into moderate haemophilia by prophylaxis: are we reaching our goal? Blood Transfus. 2013;11:364-9.
8. Wilkins RA et al. Twelve-month prevalence of haemarthrosis and joint disease using the Haemophilia Joint Health score: evaluation of the UK National Haemophilia Database and Haemtrack patient reported data: an observational study. BMJ Open. 2022;12(1):e052358.
9. Batsuli G et al. Severe muscle bleeds in children and young adults with hemophilia A on emicizumab prophylaxis: real-world retrospective multi-institutional cohort. Am J Hematol. 2023;98(10):E285-7.
10. Daffunchio C et al. The hidden joint in children with haemophilia on prophylaxis. Thromb Res. 2023;226:86-92.
11. European Medicines Agency. Hemlibra. Available at : https://www.ema.europa. eu/en/medicines/human/EPAR/hemlibra. Last accessed: 3 March 2025.
12. Pipe SW et al. Emicizumab prophylaxis in infants with hemophilia A (HAVEN 7): primary analysis of a phase 3b openlabel trial. Blood. 2024;143(14):1355-64.
13. Lenting PJ. Laboratory monitoring of hemophilia A treatments: new challenges. Blood Adv. 2020;4(9):2111-8.
14. Schmitt C et al. Pharmacokinetics and pharmacodynamics of emicizumab in persons with hemophilia A with factor VIII inhibitors: HAVEN 1 study. Thromb Haemost. 2021;121(3):351-60.
15. Pipe SW et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6(6):e295-305.
16. Ferriere S et al. A hemophilia A mouse model for the in vivo assessment of emicizumab function. Blood. 2020;136(6):740-8.
17. European Medicines Agency. Altuvoct. Available at: https://www.ema.europa.eu/ en/medicines/human/EPAR/altuvoct. Last accessed: 3 March 2025.
18. Malec L et al. Efanesoctocog alfa prophylaxis for children with severe hemophilia A. N Engl J Med. 2024;391:235-46.
19. Malec L et al. Clinical outcomes over 2 years of once-weekly efanesoctocog alfa treatment in children with severe hemophilia A: second interim analysis from the Phase 3 XTEND-ed long-term extension study. Blood. 2024;144(Suppl 1):5495.
20. Mahajerin A et al. Channeling effects in the prescription of new therapies: the case of emicizumab for hemophilia A. J Comp Eff Res. 2021;11:717-28.
21. Schrijvers L et al. Achieving self-
management of prophylactic treatment in adolescents: the case of haemophilia. Patient Educ Couns. 2016;99(7):1179-83.
22. Hoefnagels JW et al. The perspectives of adolescents and young adults on adherence to prophylaxis in hemophilia: a qualitative study. Patient Prefer Adherence. 2020;14:163-71.
23. Andrade PEA et al. Emicizumab : the hemophilia A game-changer. Haematologica. 2024;109:1334-47
24. Hermans C et al. Disruptive technology and hemophilia care: the multiple impacts of emicizumab. Res Pract Thromb Haemost. 2021;5:e12508
25. Andersson N et al. Intracrnial hemorrhage before start of prophylaxis in children with hemophilia: incidence, timing, and potential for prevention. Haematologica. 2024;109:doi: 10.3324/ haematol.2024.285874
26. Malec L et al. Unmet needs in people with hemophilia receiving prophylaxis treatment: a real-world survey. Blood. 2024;144:2314-5.
27. van Bergen EDP et al. Subclinical synovial proliferation in patients with severe haemophilia A: the value of ultrasound screening and biochemical markers. Haemophilia. 2023;29(6):1580-8.
28. Arcudi S et al. Predictive parameters for spontaneous joint bleeding during emicizumab prophylaxis. Blood Adv. 2024;8(11):2901-7.
29. Castaman G et al. Safe and successful surgical outcome in persons with hemophilia A with and without inhibitors treated with emicizumab: a large, single center, real-world experience. J Clin Med. 2023;12(6):2317.
30. Hassan E, Motwani J. Management and outcomes of paediatric patients on emicizumab prophylaxis undergoing surgical procedures: experience from a large haemophilia centre in the UK. Haemophilia. 2021;27(5):e620-3.
31. McCary I et al. Real-world use of emicizumab in patients with haemophilia A: bleeding outcomes and surgical procedures. Haemophilia. 2020;26(4):631-6.
32. Cohen O et al. Management of surgery in persons with hemophilia A receiving emicizumab prophylaxis: data from a national hemophilia treatment center. Res Pract Thromb Haemost. 2023;7(6):102178.
33. Swan D et al. Non-factor therapies for bleeding disorders: a primer for the general haematologist. EJHaem. 2022;3(3):584-95.
34. Guillet B et al. Long-term antithrombotic treatments prescribed for cardiovascular diseases in patients with hemophilia: results from the French registry. Thromb Haemost. 2021;121(3):287-96.
35. Escobar M et al. Use of antithrombotic therapy in patients with hemophilia: a selected synopsis of the European Hematology Association - International Society on Thrombosis and Haemostasis - European Association for Hemophilia and Allied Disorders - European Stroke Organization Clinical Practice Guidance document. J Thromb Haemost.
2025;23:745-9.
36. Geller D et al. The impact of emicizumab prophylaxis on hospitalizations and emergency department visits among hemophilia A patients is age related. Pediatr Blood Cancer. 2025;72(2):e31456.
37. Steen CK et al. High use of pain, depression, and anxiety drugs in hemophilia: more than 3000 people with hemophilia in an 11-year Nordic registry study. Res Pract Thromb Haemost. 2023;7(2):100061.
38. Kalnins W et al. Pain therapy in haemophilia in Germany. Patient survey (BESTH study). Hamostaseologie. 2015;35(2):167-73.
39. Pasi J et al. Improvement in pain-related quality of life in patients with hemophilia A treated with rFVIIIFc individualized prophylaxis: post hoc analysis from the A-LONG study. Ther Adv Hematol. 2022;13:20406207221079482.
40. Madan B et al. Three-year outcomes of valoctocogene roxaparvovec gene therapy for hemophilia A. J Thromb Haemost. 2024;22:1880-93.
41. von Drygalski A et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A. N Engl J Med. 2023;388(4):310-8.
42. Klamroth R et al. Clinical outcomes over 3 years of once-weekly efanesoctocog alfa treatment in adults and adolescents with severe hemophilia A: second interim analysis from the Phase 3 XTENDed long-term extension study. Oral presentation. ASH Annual meeting, 7-10 December, 2024.
43. von Drygalski A et al. Interim analysis of joint outcomes in adult and adolescent patients with severe hemophilia A receiving efanesoctocog alfa during the Phase 3 XTEND-ed long-term extension study. Oral presentation OC 01.4. ISTH, 22-26 June, 2024.
44. Klamroth R et al. Clinical outcomes over 3 Years of once-weekly efanesoctocog alfa treatment in adults and adolescents with severe hemophilia a: second interim analysis from the Phase 3 XTEND-ed long-term extension study. Blood. 2024;144:717-8.
45. Chan A KC et al. Efanesoctocog alfa for the perioperative management of patients with severe haemophilia A: 4 years of experience in the XTEND Clinical program. Abstract 1243. EAHAD, 4-7 February, 2025.
46. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125:2038-44.
47. Schutgens REG et al. Antithrombotic treatment in patients with hemophilia: an EHA-ISTH-EAHAD-ESO Clinical Practice Guidance. Hemasphere. 2023;7(6):e900.
48. Hermans C, Pierce GF. Ultra-long factor VIII: a major step forward toward a hemophilia-free mind. J Thromb Haemost. 2024;22(7):1844-6.
NP-39929
March 2025
Congress Interview
Flora Peyvandi discusses her journey into haematology, advancements in haemophilia treatment, and the role of gene therapy. She also shares insights on the upcoming European Association for Haemophilia and Allied Disorders (EAHAD) Annual Congress, covering topics like AI in healthcare, gene therapy, and the care of elderly patients, while stressing the importance of engaging young haematologists in the field's future.
Flora Peyvandi
Professor of Internal Medicine, University of Milan, Italy; European Association for Haemophilia and Allied Disorders (EAHAD) Congress President
Can you share what initially drew you to the field of haematology, and what continues to drive you today?
I love to do medicine, and I always wanted to do internal medicinehaematology, because I thought haematology was just facing up to blood, and blood is everywhere in every organ. That was one of the main reasons. I was very interested in the process of clotting and the complexity of clotting, and then I ended up being interested in the bleeding area, which is part of haematology.
Q2
It’s amazing to see how much progress has been made and how much it has improved patients’ lives, allowing them to live differently and enjoy a better quality of life
You’ve earned degrees and conducted research at prestigious institutions across Italy, the Netherlands, the UK, and the USA. How have these diverse academic experiences shaped your approach to haematology research and patient care?
I had the opportunity to be in different institutions and different haematological research areas, some of which are very much focused on patients’ clinical evaluation. In Milan, I started doing a lot of activity in internal medicine and haemostasis out-
patient clinic, and then I moved to the Royal Free Hospital at the University College London (UCL), UK, as part of my PhD. There, I was focused on the characterisation of genes that are involved in rare bleeding disorders. At that time, I was working on factor seven (FVII) deficiency, so was predominantly doing basic research in molecular characterisation.
Then I moved to Harvard, Boston, USA, and worked on the recombinant wild-type and mutant proteins by in vitro production of FVII cDNA to mimic the disorder, and that was also very much basic research. I returned to Milan and took on the responsibility of managing the bleeding clinic. This role involved a busy schedule with clinical activities in the mornings and research in the afternoons. At that time, I was also responsible for overseeing the entire haemostasis and thrombosis research team, which allowed me to combine both clinical and research work.
I have been the head of the Internal Medicine unit and the Angelo Bianchi Bonomi Hemophilia and Thrombosis Centre at the Fondazione IRCCS
Ca’ Granda Ospedale Maggiore Policlinico for more than 15 years now, with several clinicians, biologists, biotechnologists, and geneticists in the team.
Q3 What do you believe are the most promising advancements in haemophilia treatment today, and how do you see these evolving in the coming years?
I think haemophilia treatment has seen a significant evolution in the last 10 years. We’ve moved from substitutive therapy to nonreplacement therapies, such as mimic therapies, and now to gene therapy, with promising results. However, this is not a cure; it’s a long-term response to a single infusion. Soon, gene therapies will likely be able to cure patients. It’s amazing to see how much progress has been made and how much it has improved patients’ lives, allowing them to live differently and enjoy a better quality of life.
Q4
The European Association for Haemophilia and Allied Disorders (EAHAD) Congress has become a landmark event for professionals in haemophilia and allied disorders. In your opinion, what sets this congress apart from other medical conferences in the field of haematology?
The size of EAHAD and the multidisciplinary participation make it special. While the meeting is large, with nearly 2,000 attendees, it’s still smaller compared to events like the American Society of Hematology (ASH) Congress, which hosts 30,000 people. The size is perfect, and it allows for a focused approach to various aspects of bleeding disorders and patient needs. Being together in the same room and listening to everything happening creates a unique experience. It feels like a family.
Being together in the same room and listening to everything happening creates a unique experience
Q5 This year’s EAHAD Congress took place in the vibrant city of Milan, Italy. Can you share why this location was chosen, and how it enhances the congress experience for attendees?
It was the right time to come to Milan, as the meeting had been held in Italy before but not in Milan. Many Italian haematologists and experts in the bleeding area regularly participate in EAHAD, and it is a great honour and pleasure for all of us to invite physicians, physiotherapists, and other multidisciplinary
professionals working in the field of bleeding disorders, as well as all pharmaceutical companies who bring novel data on novel therapeutics in bleeding disorders, to come to Milan. We hope they can enjoy both the scientific aspects of the event and the cultural experiences, food, and everything else the city has to offer.
Q6
The upcoming EAHAD 2025 Congress will explore cutting-edge areas such as AI in healthcare, gene therapy, and the care of elderly patients with haemophilia. Gene therapy has been hailed as a potential game-changer for haemophilia care. What challenges remain in making gene therapy widely accessible, and how can we ensure its long-term safety?
Gene therapy is the future; there is no doubt about that. However, several points need to be clarified, and during the EAHAD meeting we will have the chance to address them. First, the shortterm safety aspects need to be better understood and managed. From an educational standpoint, people need to better understand the pros and cons of this long-term treatment, as well as the next steps and the future of gene therapy.
Additionally, the accessibility of gene therapy is an important point, and I hope experts will contribute by submitting abstracts to bring this issue into the discussion during the meeting.
Q7 The use of AI in healthcare is expanding rapidly. How do you see AI being implemented specifically in the diagnosis and management of bleeding disorders?
As you mentioned, AI in healthcare is rapidly expanding, including in the field of haemophilia. This applies to both diagnostics using ultrasounds and very accessible sound technology, which we use in our centre. This will be discussed in detail during the Pre-Congress Day. Additionally, AI plays a role in early treatment and in understanding the efficacy of treatments. As this area is improving so quickly, we will have the opportunity to explore these developments in more detail during the meeting, and explain how much needs to be done for its use during our daily activity on management of people with haemophilia.
Q8 As the population of elderly patients with haemophilia grows, what unique challenges are being faced in their care, and how is the healthcare system adapting to meet the needs of this ageing demographic?
Yes, that’s another important point. Fortunately, as we mentioned, the treatment of patients is improving, and they are living longer. As a result, we are seeing more elderly patients who are beginning to live a lifestyle closer to the general population. This means they may develop other comorbidities, such as cardiovascular disorders or conditions that require
treatment with anticoagulants or antiaggregant drugs. Fortunately, with the availability of newer drugs that have a longer half-life and higher protection level, we can better manage these patients with a lower risk of bleeding. This needs to be explained and discussed, and we must focus on educating healthcare professionals and collecting data on this type of patient management.
Q9
In your view, what are the most pressing and persistent challenges currently facing the field of haematology, and what strategies do you believe will be crucial in addressing them?
I think the most important challenge is involving young haematologists in the field of haemostasis and encouraging their interest in bleeding disorders. It’s essential to engage them in shaping the future of patient management and treatment. I hope that during the EAHAD Congress, we will see more and more young people attending and joining our meetings. It would also be great for them to have the opportunity to connect with one another, enhance their knowledge, and take advantage of the science and culture that this city has to offer.
Gene therapy is the future; there is no doubt about that
1. The Haemophilia Society. Available at: https://haemophilia.org.uk/bleedingdisorders/haemophilia-a-and-b/what-causes-haemophilia/. Last accessed: 18 February 2025.
2. Cleveland Clinic. Available at: https://my.clevelandclinic.org/health/
Carrier daughter Affected son Unaffected daughter Unaffected son
Carrier daughter Unaffected son = Unaffected = Affected
First trial reporting successful results of AAV-based gene therapy for haemophilia B. The six patients treated showed elevated FIX levels after intravenous administration of the therapy.7
Valoctocogene roxaparvovec approved in the EU and UK in August 20229 and in the USA in June 2023.10
diseases/14083-hemophilia. Last accessed: 18 February 2025.
3. NHS. Available at: https://www.nhs.uk/conditions/haemophilia/. Last accessed: 18 February 2025.
4. Mayo Clinic. Available at: https://www.mayoclinic.org/diseases-conditions/ hemophilia/diagnosis-treatment/drc-20373333. Last accessed: 18 February 2025.
5. International Society on Thrombosis and Haemostasis (ISTH). 2019. Available at: https://genetherapy.isth.org/content/isth-module02/ story_html5.html?endpoint=https://saas.learninglocker.net/data/ xAPI/&auth=Basic%20ZWJkNmEzYjVkMjllYjljZjRhNmM2ZDVhODgzMW
Etranacogene deszaparvovec, based gene therapy was approved in the 2022,11 and in the UK
American Society of Hematology (ASH). hematology.org/about/ history/50-years/milestones-hemophilia. 18 February 2025. 7. Nathwani AC et al. Adenovirus-associated hemophilia B. N Engl J Med 2011;365:2357-65. 1982
Haemophilia Treatment
General population
Affected father
AAV gene therapy
1. Receptor-mediated uptake
Carrier daughter Unaffected son
deszaparvovec, an AAVfor haemophilia B, USA in November UK and EU in 2023.12, 13
DQ2ZGY3Yzg2MGI5ZTcwMzRiZDU2 _id=https://genetherapy.isth.org/99150002&actor={%22name%22:%20[%22anon%20user%22], %20%22mbox%22:% 20%22mailto:5261926@genetherapy.com%22}. Last accessed: 18 February 2025. (ASH). 2008. Available at: https://www. history/50-years/milestones-hemophilia. Last accessed: Adenovirus-associated virus vector–mediated gene transfer in 2011;365:2357-65.
F8 gene (~180kb)
F9 gene (~34kb) Functional FVIII protein Functional FIX protein
Patients with haemophilia
F8 gene (~180kb)
F9 gene (~34kb) Absent or deficient FVIII protein Absent or deficient FIX protein
Advantages14
Reduction of bleeding events.
Reduced burden of prophylactic infusion therapy.
Improved quality of life by reducing joint bleeding, pain and arthopathy is a major impact quality of life.
8. Rangarajan S et al. AAV5–Factor VIII gene transfer in severe hemophilia A. N Engl J Med 2017;377:2519-30. 9. EMA. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/ roctavian#authorisation-details. Last accessed: 18 February 2025.
10. FDA. 2023. Available at: https://www.fda.gov/news-events/press-announcements/ fda-approves-first-gene-therapy-adults-severe-hemophilia. Last accessed: 18 February 2025. 11. FDA. Available at: https://www.fda.gov/media/163466/download?attachment. Last accessed: 18 February 2025.
2. Internalisation
3. Nuclear import
4. Uncoating
DNA
Therapeutic protein
5. Protein synthesis and secretion Receptor
Disadvantages14
Little is known on the suitability of AAV-based gene therapy for children and infants with haemophilia, or its use in symptomatic female carriers.
Liver toxicity has been reported as a side effect from gene therapy. Additionally, side effects from corticosteroids often given to treat transaminitis have a negative impact.
Pre-existing anti-AAV antibodies limit eligibility, while treatment effects vary and long-term durability remains uncertain.
12. EME. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/ hemgenix. Last accessed: 18 February 2025.
13. MHRA. Available at: https://mhraproducts4853.blob.core.windows.net/ docs/8dcbd83799317a10cfe5a2551123b649e6541609. Last accessed: 18 February 2025.
14. Ay C et al. Haemophilia. 2024;30:5-15.
15. Gualtierotti R et al. Hemophilic arthropathy: Current knowledge and future perspectives. J Thromb Haemost. 2021;19(9):2112-21.
Haemophilia Articles
Interview:
Niamh O’Connell
• Breakthroughs in rare bleeding disorders: Discover the power of global networks like EAHAD.
• Discover the future of treatment: Learn about exciting advancements in gene therapy and beyond.
• Advice for aspiring haematologists: Get invaluable guidance on mentorship, research, and building a global network.
Read the full interview here
Article:
Acute
Flank Pain from May-Thurner Syndrome: A Case Report
Discover valuable insights on maintaining a high index of suspicion for May Thurner syndrome and implementing personalised management strategies.
Read the full case report here
Article: Immunoglobin D Multiple Myeloma: A Single Centre Experience
Gain insights into IgD myeloma, an uncommon subtype of multiple myeloma, from a recent examination of 12 cases, treated at a single centre.
Read the full article here
EMJ - Elevating the Quality of Healthcare Globally