10 Review of the 12ᵗʰ Edition of the European Conference on Rare Diseases and Orphan Products (ECRD) 2024, 15th-16th May 2024
Congress Feature
19 How Can We Bridge the Rare Disease Treatment Gap?
Darcy Richards
Symposium Review
23 Protecting and Preserving Dystrophic Muscle: The Balance Between Exercise and Contraction-Induced Muscle Injury
Interviews
31 Climate Change and Air Pollution: How Healthcare Providers Can Help Mitigate the Risks to Respiratory Health
42 Role of Nebulisers in the Treatment of Patients with Severe and Very Severe Chronic Obstructive Pulmonary Disease
49 Enhancing Treatment Success in Osteoporosis: Optimising the Use of Teriparatide
56 Smeeta Sinha
56 Nina Gold Infographics
64 Muscle Matters: Protein Requirements for Muscle Preservation During Ageing
66 Importance of Timely and Accurate Diagnosis of Myotonic Disorders: Role of Electromyography Feature
68 Lost in the System: The Labyrinth of Rare Disease Diagnosis Fish
Articles
73 Editor's Pick: Genetics and Pathophysiology of Co-occurrence of Congenital Heart Disease and Autism Spectrum Disorder
Ong
84 Zinc for Wilson’s Disease: What We Know and What We Don’t Know
Di Dato and Hedera
96 Fever, Sore Throat, and Abdominal Pain –Connecting the Dots to a ‘Forgotten’ Disease: A Case Report of Atypical Lemierre’s Syndrome
Yasmin et al.
104 Treatment of Dermatitis Artefacta: A Systematic Review
Estill and Jafferany
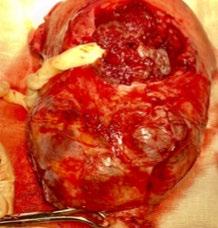
116 Caesarean Hysterectomy for Placenta Accreta Spectrum in a Single Centre: A Series of 19 Cases
Bhanumathy et al.
123 Nipah Virus in Kerala, India – Unravelling the Local Outbreak and Assessing Global Threats: A Narrative Review
Gopika et al.
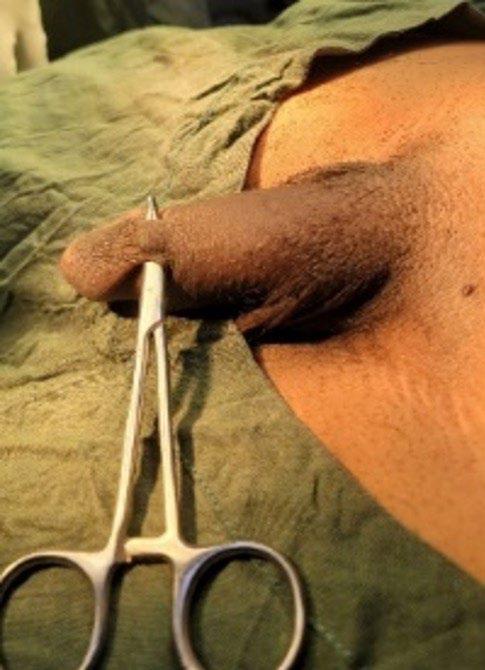
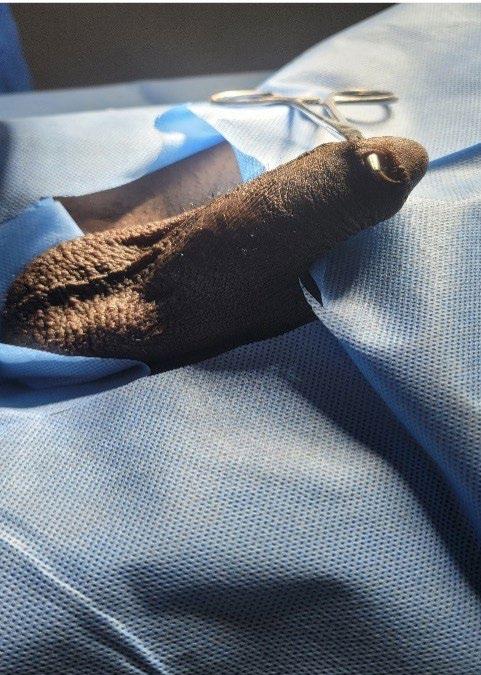
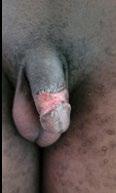
133 Penile Skin Bridge: Uncommon Cause of Painful Spontaneous Erection in Young Males
Takure
138 Frequency Determination of Central Line
Blood Stream Infection at a Renal Care Centre
Moin et al.
145 Caregiver Burden, Resilience, and Wellbeing in Cases of Severe Cutaneous Adverse Drug Reactions
Mukherjee et al.
154 Endovascular Stenting for Superior Vena Cava Syndrome - A Systematic Review
Anyagwa et al.
"Together we can achieve transformative advancements in research, policy, and patient care."
Editorial Board
Editor-in-Chief
Prof Markus Peck-Radosavljevic
Klinikum Klagenfurt am Wörthersee, Austria
Current Chairman and Head of the Department of Gastroenterology and Hepatology, Endocrinology, Rheumatology and Nephrology at Klinikum Klagenfurt am Wörthersee, with expertise in portal hypertension, hepatocellular carcinoma, and HIV-HCV coinfection.
Prof Ahmad Awada
Jules Bordet Institute, Belgium
Prof Sorin T. Barbu
“Iuliu Hațieganu” University of Medicine and Pharmacy, Romania
Dr Abdullah Erdem Canda
Yildirim Beyazit University, Türkiye
Prof Ian Chikanza
Harley Street Clinic, UK
Prof Lászlo Vécsei
University of Szeged, Hungary
Dr Pierfrancesco Agostoni
St. Antonius Hospital, Nieuwegein, the Netherlands
Dr Fernando Alfonso
Hospital Universitario de La Princesa, Madrid, Spain
Dr Emanuele Angelucci
IRCCS Ospedale Policlinico San Martino, Genoa, Italy
Dr George Anifandis University of Thessaly, Greece
Dr Riccardo Autorino
Virginia Commonwealth University, Richmond, USA
Dr Mátyás Benyó University of Debrecen, Hungary
Prof Andrew Bush Imperial College London, UK
Dr Hassan Galadari
United Arab Emirates University, Al Ain, United Arab Emirates
Dr Amir Hamzah Abdul Latiff
Pantai Hospital, Kuala Lumpur, Malaysia
Dr Lorenz Räber
Bern University Hospital, Switzerland
Aims and Scope
EMJ, the flagship journal of the EMJ portfolio, is an openaccess, peer-reviewed eJournal, committed to elevating the quality of healthcare globally by publishing high-quality medical content across the 18 clinical areas covered in our portfolio. The journal is published quarterly and showcases the latest developments across these clinical areas.
EMJ publishes peer-reviewed research papers, review articles, and case reports across all therapy areas of the EMJ portfolio. In addition, the journal publishes features and opinion pieces create a discussion around key topics in the field and broaden readers’ professional interests. The journal also features interviews with leading experts in various clinical disciplines.
The journal covers advances within the pharmaceutical arena by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ endeavours to increase knowledge, stimulate discussion, and contribute to the delivery of world-class updates in the clinical realm. We do not publish veterinary science papers or laboratory studies that are not linked to patient outcomes. Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
• A team of internal and independent medical writers.
Peer Review
Every review article, case report, feature, and research article published in EMJ undergoes peer review by at least two independent experts.
On submission, all manuscripts are assessed and undergo a technical check by the EMJ Editorial staff to determine their suitability for the journal and appropriateness for peer review. Editorial staff identify appropriate reviewers who are selected based on their specialist knowledge in the relevant area. All peer review is double-blind.
Following review, manuscripts are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected. Editorial staff are responsible for ensuring that necessary amendments to the manuscript have been made, with input from our Editorial Board or the original reviewers where necessary. The Editor of EMJ has final discretion over any proposed amendments. Manuscripts authored by members of the Editorial Board are subjected to the same double-blind process. Short opinion pieces are published following internal review and publication is at the discretion of the Editor. Congress-associated content authored by the EMJ Editorial staff undergoes internal quality control checks. Congress-related content sponsored or funded by our industry partners undergoes quality control checks independently. Industry-supported content that falls into any of
the categories that are eligible for peer review, undergoes the same peer review process.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields globally.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®.
EMJ is available through the websites of our leading partners and collaborating societies.
EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. EMJ is completely independent of the review event (ECRD 2024) and the use of the organisations does not constitute endorsement or media partnership in any form whatsoever. The cover photo is of Brussels, Belgium, the location of ECRD 2024.
Victoria Antoniou, Helena Bradbury, Ada Enesco, Laith Gergi, Katrina Thornber, Aleksandra Zurowska
Creative Director
Tim Uden
Design Manager
Stacey Rivers
Senior Designer
Roy Ikoroha, Steven Paul
Designer Owen Silcox
Junior Designers
Dillon Benn Grove, Shanjok Gurung
Head of Sales
Robert Hancox
Marketing Director
Kristina Mestsaninova
Chief Content Officer
Justin Levett
Chief Commercial Officer
Dan Healy
Founder and Chief Executive Officer
Spencer Gore
Welcome
Dear Readers,
I am delighted to welcome you to the summer issue of EMJ, with a refreshed look for our journal and a more reader-friendly layout, helping to bring out the key takeaways from the content. With clarity and transparency as a high priority, we have ensured that the captivating contributions from our pharmaceutical partners are clearly highlighted, and we hope that you enjoy this new look.
One of the highlights of this issue is our comprehensive coverage of the 2024 European Conference on Rare Diseases and Orphan Products (ECRD), which provides a great overview of discussions ranging from the epidemiology of rare diseases to specific conditions such as familial hypercholesterolaemia and Lynch syndrome. This issue also features a thought-provoking article on the diagnostic challenges faced by people living with rare diseases, which captures the essence of discussions in the field.
Be sure not to miss our Editor’s pick, a cross-disciplinary article discussing the genetics of autism spectrum disorder and congenital heart disease, and highlighting the importance of early diagnosis and interventions for autism spectrum disorder in children with congenital heart disease. There is, of course, a plethora of other content to explore, spanning microbiology, radiology, and numerous other disciplines.
I would like to thank everyone who has provided insights of immense value: our authors, interviewees, Editorial Board, and peer reviewers. As we continue on our mission of elevating the quality of healthcare globally, I kindly ask you, our readers, to share your ratings and comments on the quality of our content via our website; to shape the future direction of EMJ, ensure that we continue to deliver valuable content, and help you elevate the standard of care for your patients.
Editorial enquiries: editor@emjreviews.com
Sales opportunities: salesadmin@emjreviews.com
Permissions and copyright: accountsreceivable@emjreviews.com
Editor
Reprints: info@emjreviews.com
Media enquiries: marketing@emjreviews.com
Evgenia Koutsouki
Foreword
Welcome to the latest issue of EMJ, exploring the management of rare diseases across a diverse range of topics. Specifically, our articles consider the management of dermatitis artefacta, Nipah virus, Lemierre's syndrome, and severe cutaneous adverse reactions.
Our coverage of the European Conference on Rare Diseases (ECRD) also provides updates on recent advancements in the field, spotlighting a session discussing strategies for bridging the gaps in access to rare disease treatment. Affordability, regulatory pathways, and research and development in clinical trials are among the challenges discussed.
My Editor’s Pick, ‘Genetics and Pathophysiology of Co-Occurrence of Congenital Heart Disease and Autism Spectrum Disorder’, summarises the genetics and pathophysiology underlying both conditions. Ong concludes by advising that children with congenital heart disease should undergo regular neurodevelopmental assessment for autism spectrum disorder to aid with early diagnosis and intervention.
This issue also includes a feature entitled ‘Lost in the System: The Labyrinth of Rare Disease Diagnosis’. This fascinating article highlights the paradox of rare diseases, affecting one in 10 people despite each disease being rare. Fish outlines the
challenges associated with this group of diseases, before offering solutions such as screening, education, and AI.
This second EMJ issue of 2024 also includes insightful interviews with Nina Gold and Smeeta Sinha. Gold discusses her work focusing on newborn sequencing and its impact on rare disease outcomes, while Sinah outlines her career in rare renal diseases such as calciphylaxis.
Affordability, regulatory pathways, and research and development in clinical trials are among the challenges discussed
I would like to extend thanks to all the authors, reviewers, interviewees, and Editorial Board members for their continued dedication and commitment to EMJ.
I hope this issue proves to be an illuminating and informative read for all healthcare professionals.
Lászlo Vécsei Head of Neuroscience Research Group, Department of Neurology, University of
Szeged, Hungary
Congress Review ECRD 2024
‘ACTION Within Reach: Pioneering Solutions for Rare Diseases’ was the theme of the 12th edition of the European Conference on Rare Diseases and Orphan Products (ECRD) 2024, which took place in Brussels, Belgium, between the 15th–16th May 2024.
ECRD, the leading patient-shaped, patientled, and patient-organised space, is co-organised by Orphanet and partner organisations of the European Organisation for Rare Diseases (EURORDIS-Rare Diseases Europe). The 2024 hybrid event saw approximately 300 in-person delegates and 400 attendees from around the world joining online to address the complex challenges faced by people living with rare diseases (PLWRD).
Considered the heart of the European Union (EU), Brussels provided the ideal backdrop for ECRD 2024, which set to focus on driving transformative change through policy, in light of the impending European election. Spanning the course of 2 days, attendees including patients and caregivers, policy makers, healthcare professionals, researchers, and representatives from both industry and the private sector, had the opportunity to attend a plethora of sessions delivered by 50 expert speakers.
In a pre-conference video, Virginie BrosFacer, newly appointed Chief Executive Officer of EURORDIS, gave a short and impactful speech, highlighting how the conference provides a unique opportunity to influence the rare disease landscape. Whilst the challenges discussed during the event would be numerous and complex, BrosFacer stated: “Together we can achieve transformative advancements in research, policy, and patient care.”
A key focus for ECRD 2024 was priority needs for PLWRD. To help make the event as accessible and inclusive as possible, all speakers introduced themselves by giving a short visual description of their appearance, and multilanguage closed captions were available for the online audience.
To officially open the conference, BrosFacer and Avril Daly, EURORDIS President, joined the stage. Daly welcomed delegates
Together we can achieve transformative advancements in research, policy, and patient care”
to the first in-person event since ECRD 2018, which took place in Vienna, Austria. Daly highlighted the special nature of the event, organised under auspices of the Belgian presidency of the EU council, and taking place in the heart of the EU. Daly commented that rare disease policy is innovative and dynamic, and emphasised the desire to shape goal-driven rare disease policies. She addressed the audience, stating: “It’s critical for your voices to be heard.”
Bros-Facer noted that the conference was taking place at a crucial time of change, with the approaching European election, and stated that the rare disease community looks to new European leaders to bring the Rare 2030 Foresight Study recommendations into fruition. Her hopes for the conference were to continue to build on momentum and comprehensive review of strategy for rare diseases; and shape the thinking of policy makers to encourage a more streamlined and proactive approach to addressing the unmet needs of PLWRD, and persisting inequalities across Europe. She discussed the need to harness this momentum for change, and to take bold decisions. She then drew attention to the Open Letter, informed by sessions at the 12th ECRD, with the aim of calling the next European leaders to develop a European action plan for rare diseases that bridges
diverse policy areas, and streamline existing efforts with clear, measurable objectives. She reminded the audience that policies need to address the real needs of PLWRD, and encouraged attendees to get involved, commenting: “We can only be the change we want to see in the world.”
Video messages from both Frank Vandenbroucke, Minister of Health and Social Affairs for Belgium; and Stella Kyriakides, European Commissioner for Health and Food Safety, echoed the sentiments around the steps that have been taken to improve patient access to diagnosis, treatment, and care. Vandenbroucke stressed that it is crucial that rare diseases are kept on the European health agenda, that collaboration beyond borders will be pivotal, and a European action plan for rare diseases is needed to improve the situation for the approximately 30 million European PLWRD.
Kyriakides highlighted that integration of European Reference Networks (ERN) into national health systems; the new Horizon Europe Programme to advance research and innovation; and closing the distance to bring the European health data space to reality, to ensure PLWRD have access to, and control of, their health data in Europe, are key to improving access and care for patients, and will be crucial for research.
300
in-person delegates
400
Additionally, Kyriakides emphasised that PLWRD must have access to the best and most innovative treatments, irrespective of their disease, or where they live. She discussed the need for inclusion of patient organisations to better shape European policies on rare diseases, commenting: “In the field of rare diseases, collaboration is the key to success, and patient voices are essential.” Kyriakides concluded her
address with an empowering statement: “Together, I truly believe we can deliver a brighter future for patients living with rare disease.” At the closing plenary, individuals were invited to the stage to formally sign the ECRD Open Letter, starting with Daly signing on behalf of EURORDIS.
In the field of rare diseases, collaboration is the key to success, and patient voices are essential
Bros-Facer closed ECRD 2024 with a powerful reminder of the strength that lies within the collective voices of attendees, and how collaboration can drive change for the 30 million PLWRD in Europe.
The 12th edition of the ECRD provided a platform for collaboration amongst multiple stakeholders in the rare disease space to discuss solutions to challenges, advocate for change, and champion the need for access and equity in European policies, with PLWRD at the forefront of the discussions.
attendees from around the world joining online to address the complex challenges faced by PLWRD
Defining Healthcare Experiences for Patients with Rare Diseases
PEOPLE living with rare diseases (PLWRD) face unique healthcare challenges that are often not captured by standard quality measures. With an increasing emphasis on patient-centred care, understanding their experiences is essential for improving service delivery and patient outcomes.
A scoping review, presented at the 12th ECRD, identified key healthcare experience domains for PLWRD, using patient reported experience measures. The review aimed to extract and collect categories and domains of patient healthcare experiences included in empirical studies that explored the views of PLWRD, and to study and assess existing questionnaires to establish whether they capture PLWRD healthcare experiences in a comprehensive manner.
Notably, safety and trust were less frequently addressed at 13% and 23%, respectively
The review utilised comprehensive searches on Medline, Embase, and Cumulative Index to Nursing and Allied Health Literature (CINAHL) with keywords related to rare diseases and patient experiences. Papers from 2005–2022 were retrieved, and a total of 61 studies were analysed. The Organization for Economic Cooperation and Development (OECD) PatientReported Indicator Surveys (PaRIS) domain
was adapted and utilised for data extraction and analysis.
Results showed that quantitative methods dominated the study designs, with a substantial portion also employing qualitative and mixed methods. The review revealed that current instruments variably cover essential domains such as access (69%), co-ordination (72%), and peoplecenteredness (87%). Notably, safety and trust were less frequently addressed at 13% and 23%, respectively. The study also mapped 42 questionnaires against the domains and categories, and identified the Cystic Fibrosis Patient Experience and Satisfaction with Care Services questionnaire as the only one that covers all domains investigated as part of this review (11), with the highest number of categories (31/71).
This scoping review provides crucial insights into the healthcare experiences of PLWRD, utilising patient-reported experience measures to delineate the complexities and nuances of their interactions within secondary and tertiary healthcare settings.
The Evolving Experience of Patients with Rare Conditions
FEEDBACK
from patients is crucial to improve healthcare delivery for rare conditions. A recent study presented at the 12th ECRD analysed changes in patient experience at the Royal Hospital For Children, Glasgow, UK, between the periods of 2018–2020 and 2021–2023.
%
of patients reported being told little or no information at diagnosis between 2021–2023
A total of 130 questionnaire-based surveys were completed by patients and parents of children from 2018–2020, and 62 completed from 2021–2023. The survey explored six key themes: diagnosis, provision of information, availability of support, satisfaction with healthcare team, awareness and support for life-limiting conditions, and participation in research.
Results showed that 68% of patients reported being told little or no information at diagnosis between 2021–2023, compared with 46% in 2018–2020 (P=0.19), and the proportion of current adequate understanding also decreased from 66% to 53% (P=0.09). A total of 72% of respondents were aware of support groups, compared to 59% prior (P=0.09), and membership increased from 55% to 67% (P=0.13). Overall satisfaction with the healthcare team also increased from 60% to 70% (P=0.18).
Support for children with life-limiting conditions increased from 8% to 23% (P=0.01). In the latter survey, 73% of patients had experienced remote consultations, with only 10% and 20% satisfied with video and telephone consultations, respectively. Hospital appointments were cancelled in 13%, therapy appointments in 17%, investigations in 9%, and surgeries in 13% of respondents during the pandemic.
The study provides insights into areas of improvement and concern in the management of patients with rare conditions. Addressing the decrease in information provision observed since the pandemic will enable healthcare providers to deliver patient-centred care, enhancing their quality of life. Further surveys will be required to track changes in these benchmarks and improve patient services.
Lynch Syndrome and Thyroid Nodules: A Single Centre Experience
LYNCH Syndrome (LS) is a genetic condition linked to mutations in MLH1, MSH2, MSH6, PMS2, and EPCAM genes, increasing the risk of colorectal cancer and other malignancies, with few reported cases of thyroid cancer in patients with LS.
Research presented at the 12th ECRD aimed to investigate the presence of thyroid nodules in patients with LS and explore the association with genetic features of the disease.
A retrospective and descriptive analysis was conducted on patients with LS at the Center for the Diagnosis, Treatment and Prevention of Digestive System Diseases (CEMAD), Agostino Gemelli IRCCS University Hospital Foundation, Rome, Italy. The study evaluated LS disease characteristics, gene mutations, and previous thyroid disease history. Most patients underwent thyroid ultrasound, with nodule cytology performed when necessary.
Among 139 patients with LS (52 male; 87 female), 103 (74%) underwent thyroid ultrasound, with seven patients (5%) having a history of thyroid disease. Thyroid nodules were found in 62 patients (60%) who had ultrasound exams, and nine of these (14%) had suspicious features leading to fineneedle aspiration biopsy. Cytologic analysis showed seven cases (78%) of benign nodules (TIR2) and two cases (22%) of lowrisk indeterminate lesions (TIR3a). Among patients with nodular thyroid disease, the majority were MSH6 mutation carriers (36%), followed by MSH2 (32%), MLH1 (24%), and PMS2 (7%).
Overall, there was a high prevalence of thyroid nodules in patients with LS, especially in those with the MSH6 mutation. It is recommended that patients with LS undergo at least one thyroid ultrasound examination to detect nodular thyroid disease. Further systematic investigations are needed to understand the prevalence, features, and malignancy risk of thyroid nodules in this population.
Unveiling the Epidemiological Landscape of Rare Diseases: A Demographic Analysis by Medical Domain
ACCORDING to new findings presented at the 12th ECRD, 4.40–5.28% of the general population is affected by rare diseases.
The scarcity of population-based and epidemiological studies examining rare diseases delays diagnosis and the early implementation of evidence-based care. Therefore, an enhanced understanding of the demographics of rare diseases based on medical specialty is required to improve patient care.
Researchers used Orphanet, an epidemiological database that collects information on rare diseases, including medical specialty, age of onset, and epidemiological data, from publicly available medical literature and collaborations with medical experts. The analysis of rare diseases from the Orphanet database included 6,349 rare diseases until February 2024, of which 3,733 were included in the study. The research group established exclusion criteria, which included if the disease’s medical specialty fell under cancer, infectious disease, or poisoning; assigned “unknown” point prevalence; no assigned epidemiological data available; and epidemiological indicator is not point prevalence. The study analysed the global
point prevalence and age of disease onset for every included medical category in the Orphanet database.
Results from the analysis showed that of the 3,733 rare diseases, rare developmental defects originating during embryogenesis represented the most significant single category, with a prevalence of 37.1%, followed by rare neurological diseases at 20.2%, and rare inborn errors of metabolism at 7.6%. Furthermore, the onset of rare diseases occurred frequently during early development, most commonly in the neonatal period (30.7%), followed by infancy (25.3%), and childhood (14.7%).
The findings from this study revealed a narrower global point prevalence of rare diseases compared to previous studies. However, this study demonstrated more precise demographics of rare diseases. Nevertheless, the insights into the epidemiology of rare diseases remain limited. A more detailed understanding of rare diseases across medical specialties can lead to targeted allocation of resources and enhanced intervention of specific diseases.
4.40–
5.28 of the general population is affected by rare diseases.
Prevalence of Rare Diseases on the Rise in
Geriatric Populations
THE global population of older individuals living with rare diseases (RD) is on the rise, a trend driven by both an ageing population and advancements in medical science. This growing demographic presents unique challenges for patients and healthcare providers alike, necessitating a greater awareness of the complexities involved in caring for this particularly vulnerable group.
A recent study presented at the 12ᵗʰ ECRD utilised data from the Veneto Region Rare Disease Registry (VRRDR) to explore the epidemiology of older individuals living with RD. The study focused on the number of patients diagnosed with RD in old age, and those who transitioned from adulthood into old age, estimating the prevalence of older patients with RD as of December 31ˢᵗ 2022. Additionally, the study examined the composition of therapeutic plans for these patients.
There is a pressing need for geriatricians and general practitioners to be actively involved in RD care
The findings revealed that during the study period, 8,975 patients were diagnosed with an RD after the age of 65 years. Furthermore, 4,214 individuals who were diagnosed with RD in their childhood or adulthood transitioned into old age. As of December 31ˢᵗ 2022, the study area had 9,508 residents with RD aged ≥65 years, accounting for 20.8% of all patients with RD in the Veneto region.
The most prevalent RD groups among elderly patients included systemic or rheumatologic diseases, neurologic disorders, and skin diseases, affecting 27%, 25%, and 9% of the geriatric RD population, respectively. Among the prevalent cases, 1,519 patients aged ≥65 years had therapeutic plans specifically related to their rare conditions. The most commonly prescribed medications were those targeting the nervous system (27.8%), the alimentary tract and metabolism (12.8%), and anti-neoplastic and immunomodulating agents (11.6%).
The study concluded that, as the number of older patients with RD continues to grow, there is a pressing need for geriatricians and general practitioners to be actively involved in RD care, and for specialised training programmes. Further research is essential to identify the unmet care needs of older patients with RD, and to develop health policies that effectively address the unique challenges faced by this emerging patient group within the RD population.
Gaps in Awareness and Adherence in Patients with Homozygous Familial Hypercholesterolaemia
A STUDY presented at the 12th ECRD revealed substantial gaps in awareness among patients with homozygous familial hypercholesterolaemia (FH), a rare genetic disorder that dramatically elevates cardiovascular complications due to a defect in cholesterol metabolism, leading to premature mortality.
Awareness of the disease was low, with only of patients correctly identifying their condition as FH
22.5 %
Effective management can prevent adverse outcomes. However, despite the availability of effective therapies, individuals with homozygous FH can face challenges in managing their condition due to lack of awareness.
The study assessed awareness and knowledge among patients with homozygous FH regarding FH and its treatment options, with a particular focus on the patient’s understanding of the disease, its severity, and their adherence to therapy.
A 48-item survey was conducted face-to-face with 40 patients with homozygous FH at the outpatient clinic of Ege Üniversitesi Department of Cardiology, Izmir, Türkiye. Results showed that awareness of the disease was low, with only 22.5% of patients correctly identifying their condition as FH. The majority of patients referred to their condition under the generalised term of “high cholesterol,” indicating a significant lack of understanding regarding the genetic and clinical specificity of FH. Additionally, while two-thirds of patients acknowledged having a genetic disorder, only 22.5% were aware of the low-density lipoprotein (LDL) cholesterol target of <55 mg/dL recommended to mitigate the heightened cardiovascular risks associated with homozygous FH. Furthermore, only 55% of patients were aware of their latest LDL
levels, indicating a disconnection from ongoing treatment monitoring.
Therapeutic adherence rates were also reportedly low, with 15% of patients indicating poor adherence to pharmacotherapy, 17% to prescribed physical activity, and 69% to dietary modifications. Additionally, 21% acknowledged non-compliance with broader lifestyle modifications, including smoking cessation, which is critical for cardiovascular risk reduction in FH. In terms of patient engagement and advocacy, 83% were not engaged with, or aware of, patient advocacy groups, and nearly half showed substantial reluctance to participate in randomised clinical trials for novel and effective LDL-lowering therapies, despite previous involvement.
These findings highlight significant gaps in patient awareness and adherence to treatment for homozygous FH. This calls for targeted educational interventions to raise awareness, enhance patient engagement in their own care, and promote understanding of the disease severity and its implications. Further research should explore the barriers to effective patient education, promote trial involvement, and encourage participation in patient advocacy groups, as a resource that could significantly enhance patient support and education.
COLLABORATION was the common thread during an illuminating session titled ‘Innovative therapies, unequal access: bridging the gap for rare disease treatments’, which took place during the 12ᵗʰ edition of the European Conference on Rare Diseases and Orphan Products (ECRD) 2024, in Brussels, Belgium, from the 15ᵗʰ–16ᵗʰ May. Stakeholder representatives from multiple domains came together to discuss the challenges and potential solutions to close this gap, with a clear and unified message that joining forces at both the national and European level will be needed moving forward.
SETTING THE SCENE
Jo De Cock, Former CEO, National Institute of Health and Disability Insurance (NIHDI); and World Health Organization (WHO) Consultant, introduced the two key factors implicated in rare disease treatment gaps: access and affordability.
Access, meaning getting the right treatment to the right patient at the right time, and at the right price, presents a multidimensional challenge influenced by numerous factors, including availability and the added value of the therapeutic, e.g., does it provide the right clinical benefit and does it address both patient and societal needs. Affordability on the other hand, encompasses the cost of the therapy and the ability for the cost to be paid.
De Cock noted that, according to the European Medicines Agency (EMA), 240 orphan medicines had market authorisation between 2003–2023, but there is no guarantee of equal access to these medications across Europe. He noted that almost 6,500 rare diseases have been identified, but there is a long road ahead to provide the appropriate treatments for the up to 36 million people living with a rare disease in Europe. He stressed that improved approaches and policies are
needed to overcome this, before inviting Christine Leopold, Utrecht University, the Netherlands, to the stage to present from the drug regulatory science perspective.
ACCESS AND AFFORDABILITY: WHAT ARE THE CHALLENGES?
Leopold explored the factors that impact accessibility and affordability, explaining that research and development, drug authorisation, and the preparedness of health systems affect accessibility; and that health technology assessments (HTA), payment models, and out-of-pocket payments affect affordability.
Delving into these in greater detail, the main challenges associated with research and development in clinical trials that impact accessibility are the small numbers of patients, single-arm designs, and short duration compared with the potential lifetime benefit. This means that the trials performed are comparable to investigational drugs, and as such, regulatory decisions are often based on insufficient safety and efficacy data. Leopold noted that between 2013–2023, according to IQVIA data, the number of clinical trials for rare diseases has not increased in the same way as clinical trials for other diseases.
Furthermore, there is a clear disparity in that most of the rare disease trials, across all phases, occur in the field of oncology. This unmet need will need to be addressed to improve accessibility in the future.
With respect to health system preparedness, Leopold explained that many new drugs for rare diseases require special administration. This generates a need for appropriate training of staff and requires appropriate infrastructure and access to, and availability of, testing, as well as service redesigns. Whilst this does pose a challenge, Leopold explained that since 2017, the first 24 European Reference Networks (ERN) were launched, involving >900 highly specialised healthcare units from >300 hospitals in 26 member states, and that these will be the way forward to increasing access to treatments and ensuring skilful drug administration.
In terms of affordability, most orphan drugs are impacted by budget and potential uncertainty regarding their effectiveness due to mismatches between regulatory and HTA evidentiary requirements. Leopold highlighted that the time to the first HTA decision is longer for orphan medicines
than standard medicines. In addition to the budget impact, innovative payment models can often be too complex to implement. Leopold also drew attention to out-of-pocket payments, which appear to disparately affect Central and Eastern European countries, compared to higherincome countries, which is concerning.
When discussing future hopes, Leopold put the spotlight on the WHO and Europe’s Access to Novel Medicines Platform, a neutral platform unifying all stakeholders to reshape political discourse through the aims of establishing collaboration, improving transparency, strengthening voluntary collaborations that focus on solidarity, developing principles that recognise the need for sustainability, and identifying policy options for sustainable innovation and access to novel antimicrobials. This could help create partnerships to build momentum for change. Leopold commented that she hopes to see concrete outcomes from this by 1 year.
She concluded that whilst regulatory pathways have seen movement in 10 years, resulting in more products on the market, there remains a lot of work to do
around paying for these drugs. There is a need to invest into horizon scanning and early scientific advice; strengthen health care systems through ERNs; and establish multidisciplinary team processes involving patients, clinicians, payer organisations, and HTA organisations to collect the evidence needed for the reimbursement process. Leopold stressed that this needs to be done at the European level, not just at the national level.
Following this, a roundtable discussion with key stakeholders took place to gain perspectives on the challenges across different domains, including industry, patients, and reference networks. This highlighted discrepancies between market access to treatment and effective patient access to treatment. When considering access to treatment and market authorisation, Mariangela Pellegrini, APHP Hospital Saint-Louis, Paris; and ERN-EuroBloodNet Educational & Patient Program Manager, Paris, France, emphasised the need to contemplate the full pathway, including post-production elements, such as the burden on patients and healthcare providers. She also spoke on the need to consider mapping availability and accessibility for rare disease treatments and including both societal and patient burdens in these pathways. Additionally, Pellegrini raised the issue of distributive justice, noting that access to treatment for all patients and all rare diseases should be given, but not all countries are able to afford orphan medicines or have the healthcare infrastructure to provide them. She suggested that cross-border health directives and social security regulations, as well as national plans for rare diseases, are needed.
From the patient perspective, Daniel De Vincente, Asociación de pacientes ASMD España, Madrid, Spain, stressed the importance of involving patients from the very start of treatment development, highlighting that patients can help design trial endpoints as they know the unmet needs they face. Trial endpoints are not always aligned with the impact on quality of life for patients.
PROPOSED SOLUTIONS
All stakeholders discussed the need for greater solidarity and collaboration to provide access to patients across borders.
The utilisation of ERNs was discussed as the common ground for harmonising data that are accessible, interpretable, searchable, and reusable. Pellegrini highlighted that by collecting data in registries such as Patient Reported Outcome Measures and realworld data registries, and epidemiological platforms, valuable insights for research and policy makers could be provided. This could aid the evaluation and estimation of pay-for-performance schemes to review the risks and benefits of developing a treatment, and provide information about the right centre to develop a clinical trial. She further emphasised that platforms collecting Patient Reported Outcome Measures and real-world data would provide a valuable tool for developing repurposed drugs, which is key for treating rare and ultra-rare diseases. ERNs have the structure for providing, collecting, and sharing these data, alongside the appropriate ethical and legal frameworks. Moreover, registries allow for the tracking of data, including clinical, quality of life, and cost of administration at the healthcare provider level, which would enable accurate resource allocation.
Leopold echoed the need to strengthen ERNs and focus on joint negotiations and collaboration. She highlighted that even outside of rare diseases, studies on price negotiations have shown that different hospitals at the local level do not
6,500 rare diseases have been identified disease
36
240 orphan medicines had market authorisation between 2003–2023 million people living with a rare disease in Europe
communicate and negotiate for medicines independently. This challenge also occurs at the national level and indicates that trust is an issue that needs to be addressed. She noted that the awareness that researchers and policy makers need to collaborate is increasing, with some good initiatives already underway.
The stakeholders encouraged crossborder health directives and discussed that national rare disease plans should provide standard-of-care lists. In this way, cohorts of member states could be developed to match needs, economical systems, and infrastructure to create an analysis of the unmet medical needs, what can be offered, and where groups of countries could propose joint procurements when buying a treatment. De Vincente spoke on the fact that patients often bear invisible costs, such as impacts on their ability to work, environment, and education. He stressed that equitable access between different countries is required and that this is a global
responsibility, noting that joint negotiations would be a good idea, particularly for ultrarare disease advanced therapies, where there are very small numbers of patients.
Other potential solutions discussed included joint regulatory and HTA assessments, with mutual recognition and facilitation across borders, streamlining the time from clinical proof of concept to market, sustainable drug development, and pricing transparency.
CONCLUSION
Overall, the unambiguous message delivered was that of a need for unity and collaboration at both the national and European levels, with input from all stakeholders, in order to effect meaningful change in policy, accessibility, and affordability, and work towards the goal of bridging the treatment gap for people living with a rare disease.
Protecting and Preserving Dystrophic Muscle: The Balance Between Exercise and Contraction-Induced Muscle Injury
This symposium took place on 23rd April 2024, as part of the 8th International Myology Congress in Paris, France
Speakers: John Vissing,1 Tanja Taivassalo,2 Joanne Donovan3
1. University of Copenhagen, Denmark
2. University of Florida, Gainesville, USA
3. Edgewise Therapeutics, Boulder, Colorado, USA
Disclosure:
Vissing has been a consultant on advisory boards for Edgewise Therapeutics, Roche, Sanofi Genzyme, Sarepta Therapeutics, Novartis Pharma AG, Fulcrum Therapeutics, Biogen, Lupin, Amicus, Zogenix, Regeneron, Argenx BVBA, UCB Biopharma SPRL, Arvinas, ML Biopharma, Atamyo, Horizon Therapeutics, and Dyne Therapeutics; has received research, travel support, and/or speaker honoraria from Sanofi Genzyme, Alexion Pharmaceuticals, Edgewise Therapeutics, Fulcrum Therapeutics, and UCB Biopharma SPRL; and has been principal investigator in clinical trials for Edgewise Therapeutics, Sanofi Genzyme, Roche, Horizon Therapeutics, Argenx BVBA, Novartis Pharma AG, Alexion Pharmaceuticals, UCB Biopharma SPRL, Genethon, ML Biopharma, Reneo Pharma, Pharnext, Janssen Pharmaceutical, Khondrion, Regeneron, and Dynacure SAS. Taivassalo has been a consultant for CFD Research Corporation; has received research, travel support, and/or speaker honoraria from Edgewise Therapeutics; and been principal investigator in clinical trials sponsored by the Department of Defence MD. Donovan is the chief medical officer at Edgewise Therapeutics.
Acknowledgements: Medical writing assistance was provided by Amanda Barrell, Brighton, UK.
Disclaimer: The opinions expressed in this article belong solely to the named speakers.
Support: The publication of this article was supported by Edgewise Therapeutics.
Meeting Summary
During a symposium at the 8th International Myology Congress in Paris, France, key opinion leaders discussed the need to address the balance between exercise and contraction-induced muscle injury in Duchenne muscular dystrophy (Duchenne) and Becker muscular dystrophy (Becker). They explained the significance of dystrophin in enabling healthy muscle repair, the detrimental impact of contraction-induced muscle
injury in the absence of dystrophin, and the safety and potential benefits of exercise in these patients. Sharing data challenging the notion that individuals with Becker and Duchenne are "untrainable," they emphasised the importance of tailored exercise regimens in these patients. The symposium also provided an overview of the ongoing trials of sevasemten, an investigational drug which is not approved in any territory. Unpublished 24-month data suggest it can preserve function in Becker, giving it potential as a useful adjuvant in disease management.
INTRODUCTION
The rare diseases Duchenne and Becker are X-linked recessive neuromuscular disorders.1 They are characterised by progressive, irreversible weakness and atrophy of the skeletal muscles and of the heart.1,2 With an incidence of between one in every 3,500–5,000 male births, Duchenne is the more common phenotype. It is also the more severe. Diagnosis tends to occur before the age of 5 years, and boys are usually dependent on a wheelchair by age 13.1 Life expectancy is 20–30 years, with typical causes of mortality being respiratory or heart failure.1,2 Becker, which affects between 0.1–1.8 per 10,000 males,3 is also a debilitating and degenerative neuromuscular disorder, usually developing around the age of 12 years.2 Functional decline can begin at any age, while ambulation tends to be preserved until the male’s 30s; once that muscle loss occurs, the decline in function is irreversible and continues throughout the individuals life.
Duchenne and Becker are caused by mutations in the Dystrophin gene.1 Containing 79 exons, this is the largest human gene, and is responsible for producing dystrophin,1 the central protein of the dystrophin-glycoprotein complex in skeletal and heart muscle cells.4 Dystrophin connects the actin cytoskeleton to the extracellular matrix, providing stability to the sarcolemma, as well as facilitating mechanotransduction.4,5
Joanne Donovan, chief medical officer at Edgewise Therapeutics, Boulder, Colorado, USA, explained that dystrophin plays a critical role in enabling the repair of healthy muscle following wear and tear, by helping to stabilise the membrane during
contraction-induced damage. “When you lack the ability to crosslink muscle fibres, contraction-induced injury starts a repetitive cycle that ultimately leads to fat and scar and fibrosis in the muscle, and loss of function,” she said.
PHYSICAL EXERCISE AND MUSCLE DAMAGE IN BECKER MUSCULAR DYSTROPHY
Animal studies have shown the potentially deleterious effect of exercise in X-chromosome-linked muscular dystrophy (mdx) mice. However, John Vissing, Director of the U niversity of Copenhagen’s Neuromuscular Center, Denmark, said it was important to remember the limitations of these investigations, i.e., that they evaluated only eccentric exercise, in which the muscle lengthens, or electrical stimulation of the musculature, and that the effect has not been demonstrated in a clinical setting.6,7 While there are data to suggest that people with dystrophies are more susceptible to muscle damage than the general population,8 that does not mean they cannot derive similar benefits from exercise, if carefully planned.
In 2008, Vissing’s team conducted the first in-human study of endurance exercise in Becker.9 Eleven patients moderately affected by Becker and seven matched healthy controls cycled for 30 minutes, five times a week for 3 months. Six patients continued, training two or three times a week, for 1 year. Over the 12 weeks, fitness, as measured by maximal oxygen uptake and achieved wattage, increased in all patients. This improvement was sustained in those who continued for the 12 months. “Physiologically, this looks very reassuring.
They get the same response as the healthy person,” said Vissing. The study also showed that cycling resulted in a mean 40% increase in knee extension strength among the patient group. “This is something you won’t see in a healthy person, and probably relates to the fact that they are really deconditioned when they start this exercise.” The regime also appeared to be safe, with levels of the muscle damage biomarker creatine kinase (CK) remaining stable across all phases of the study.
Muscle biopsies performed in five patients found no signs of increased degeneration or satellite cell depletion, in line with the CK measurement findings. The paper concluded that it is feasible to train persons with BECKER. “It appears to be safe, the patients develop more endurance, they get stronger, and it seems to be long lasting, at least for a year,” said Vissing.9
Exercise in Weaker Patients with Duchenne Muscular Dystrophy
Vissing next debunked the general thinking that people with muscle strength of <10% of normal are “untrainable.” He pointed to a 2012 study of resistance training in people with limb‐girdle muscular dystrophies (LGMD) and Becker.10 It showed that even those with <20% of normal knee extension strength achieved significant improvements following 6 months of strength training.10 “This is something we have seen in a number of cases,” he added.
There are ways clinicians can help this patient group to take part in exercise training, such as anti-gravity treadmills, which use a lifting effect to support up to 80% of the person’s body weight and alleviate the impact of training on the muscles.11 In one study, eight patients performed 10 weeks of aerobic and strength training on an anti-gravity treadmill in Vissing’s laboratory. Six-minute walking distance, dynamic postural balance, and plasma CK levels were assessed 10 weeks prior to training, immediately before, and 10 weeks after the training. The study recorded an 8% increase in walking distance and a 13% increase in dynamic postural balance, suggesting improvements
in physical function.11 Analysis showed strength training (squats, calf raises, lunges) demonstrated increases in closed-kineticchain leg muscle strength over 10 weeks.11
Assisted cycling may be useful for people who are wheelchair bound, Vissing added. He highlighted as yet unpublished data from a study of 19 people with muscular dystrophies at his centre. It suggests this model can improve cardiovascular fitness, and increase strength, though probably with minor functional importance, while alleviating the back and buttock pain associated with sitting, and the gastrointestinal symptoms related to immobilisation.
Dystrophies, Exercise, and Biomarkers
Vissing also spoke about biomarkers of muscle-injury response, and their potential role in the evaluation of disease progression. In a study by his team and Edgewise, nine people with Becker, eight with LGMD 2I, nine with LGMD2L, and nine healthy controls underwent a high intensity, bimodal aerobic and strength exercise regimen. Blood was taken before, during, and after exercise, and analysed for around 7,000 proteins. In the Becker and LGMD groups, there were 32 common elevated proteins, and one commonly decreased protein, at baseline. Of the 32 that were elevated, which included CK, all were expressed in the muscle tissue, and 25 increased further during exercise. The results, Vissing said, were validated using Becker samples from the Newcastle Tissue Bank Dataset. “We have found a common signature for these muscular dystrophies,” said Vissing.12
Upon further analysis, the researchers found proteins that responded to exercise appeared to decrease with age, while the non-responsive proteins followed the opposite trend.12 Vissing said: “Maybe the non-responsive biomarkers can be used for disease progression long term, whereas the responsive proteins can be used acutely, if you give a treatment here and now.”
PHYSICAL EXERCISE IN DUCHENNE MUSCULAR DYSTROPHY
Focusing on the appropriate type of exercise to prescribe to boys with Duchenne could help to balance the negative impact of muscle damage with the positive impact of physical activity, said Tanja Taivassalo, Associate Professor in the Department of Physiology and Aging at the University of Florida, Gainesville, USA.
Muscle adaption to exercise, she explained, relates to two factors: the intensity and the frequency of muscle contractions. “If a muscle is subjected to higher intensity, lower frequency contractions, it will elicit a signalling cascade that leads to hypertrophy and a stronger muscle. Conversely, if a muscle has more frequent, less intense muscle contractions, it will become much more fatigue resistant,” she explained. Aerobic exercise induces a signalling cascade that leads to a slow oxidative phenotype that results in more mitochondria, better antioxidant capacity, better calcium handling, more capillaries, and an increased utrophin expression along the sarcolemma. Preclinical studies have shown this signalling cascade is intact in the mdx mouse model.13 “Promoting this slow oxidative phenotype through strategies like aerobic training is a promising and physiologically relevant strategy. However, it hasn’t been widely accepted or applied to Duchenne.” This, she believes, is because most preclinical research has focused on downhill treadmill running, which elicits eccentric muscle contractions, or lengthening, known to be damaging to mdx muscle.14 Indeed, a study comparing muscle damage in mdx mice running on a horizontal treadmill to those running on a downhill treadmill found the former did not generate the same increases in MRI T2 as the latter.14 This, Taivassalo said, suggests that contraction-induced injury is dependent on the type of muscle contraction. “A consequence of these preclinical studies is that we understand that boys with Duchenne should not do eccentricallyfocused exercise,” said Taivassalo.
Exercise Type
Isometric exercise, in which force is generated without a change in length of the muscle, does not expose the muscle to damaging eccentric contractions.15 In one study, eight boys with Duchenne completed a 12-week, remotely-supervised, mild-to-moderate intensity strengthening programme. There was no change in serum CK levels 48-hours after one ‘bout’ of the activity, or after multiple sessions. In addition, there was no increase in MRI T2 signal or pixel intensity, either 48 hours after the session, or after 12 weeks. At the end of the programme, there were improvements in peak knee extension and knee flexor strength. This translated to an improvement in function, with a decrease in the time taken to ascend/descend four stairs.15 “These results are really exciting because they show, for the first time, that dystrophic muscle in boys with Duchenne is capable of adapting to an appropriate overload,” said Taivassalo.
The 2013 ‘No Use is Disuse’ study was the first randomised controlled trial of assisted bicycle training in Duchenne.16 Ambulatory and recently wheelchair-dependent boys with Duchenne were allocated to an intervention or control group. Those in the former took part in 15 minutes of upper and lower limb assisted bicycle training five times a week for 6 months, while the control group did no exercise. After 24 weeks, the control group received the same intervention. The researchers recorded a 6% decline in motor function among the control group, which did no exercise, while function measures were stable in the intervention group.16 “They concluded that this type of exercise is safe and has the potential to delay functional deterioration, but they did not include any measures of muscle damage. Importantly, there was no information about how much work the muscles actually did, versus what the motor did in the ergometer,” said Taivassalo.
Meaningful Change
To address these questions, her team collaborated with the University of Florida’s engineering department, developing “an active cycling paradigm.” Feedback sensors
in the peddles of the cycle ergometer allowed them to track the cadence and adjust the level of motor-assistance in realtime, depending on the patient’s abilities. Taivassalo said: “The boys were looking at a computer screen with a target intensity, dictated by their heart rate. If they were not able to reach the target, the motor kicked in to add some assistance, but if they were above the target, it applied some resistance. We were able to then capture how much work was done passively, by the motor, and how much work the boys did themselves.”
Six boys with Duchenne took the device home for a 6-month, remotely-monitored endurance training pilot study. The length of each session increased over time, and the intensity was set at 50–60% of their peak heart rate. Presenting the results ahead of publication, Taivassalo said there was no evidence of muscle damage after 6 months, as measured by CK at rest, or T2 signal. Fitness increased, with the level of assistance provided to each boy by the machine decreasing over the period of the study. Comparing baseline to 6-month data, each participant had an average 15–20 beats per minute lower heart rate at the same submaximal workload. Cardiopulmonary exercise testing showed improvements in time to fatigue, peak work, and peak aerobic capacity. Bone density, as measured by DEXA, was maintained, and the team is now planning further studies to explore impact on the rate of muscle fat accumulation.
Importantly, “pretty much every single parent” said quality of life had “somewhat” or “very much” improved as a result of the training. “The boys were much more independent, they engaged in activities more, more active in gym class, and they were just much more confident in things they were doing,” said Taivassalo, highlighting the story of one boy who “didn’t want to stop exercising.” The team installed the feedback system on his own tricycle, and he has since gone from only being able to cycle for 2 minutes to taking part in a 5 km run, on his tricycle, with his family. “He says this is a whole new world, because this is so meaningful to him.”
TARGETING PROTECTION AGAINST CONTRACTION-INDUCED INJURY IN BECKER: AN OVERVIEW OF THE SEVASEMTEN (EDG-5506) CLINICAL PROGRAMME
Controlled exercise can be beneficial in preserving and improving muscle function in Becker and Duchenne, and could be used as an adjunct to emerging treatments. Pharmacological modulation of fast muscle myosin, for example, “could protect against contractioninduced muscle injury,” said Donovan.
While no such agents are currently available, sevasemten, an investigational drug that is not approved in any territory, is currently in clinical development. Designed to protect the muscle fibres against contraction-induced damage, it is an orally administered, allosteric, selective, fast myofiber (Type II) myosin inhibitor.17 Donovan presented 24-month data of the ARCH study, ahead of publication.
History and Measures
ARCH is an open-label, single-centre study assessing the safety, tolerability, and pharmacokinetics of sevasemten, as well as its impact on muscle biomarkers. It used the North Star Ambulatory Assessment (NSAA), a well-established and validated 17-item rating scale used to measure functional motor abilities,18,19 as an outcome measure (Figure 1). Published data that was augmented by additional unpublished data from the Padova Becker Natural History Study20, presented by Luca Bello at the 2022 Muscular Dystrophy Association (MDA) conference, demonstrated that NSAA decline is consistent in people with BECKER who are already progressing, with individuals with a baseline score of between 10–32 experiencing an estimated decline of -1.22 NSAA points each year (unpublished data, Bello. L). These results were further validated by two natural history studies, which recorded a -2.5 point decline over 18 months and over 2 years.21,22
Putting this into context, Donovan said: “At high NSAA scores, there are some things people are compensating for, like getting
Figure 1: North Star Ambulatory Assessment (NSAA) and its real world implications.18,19
Composite evaluation of motor function across 17 tests with increasing difficulty
Real-world implications for Becker individuals
Each activity is scored on whether it can be completed
off the floor or going on tiptoes, but they can do all the NSAA tasks. In a few years, when they reach a NSAA score in the 20s, they are starting to compensate. The natural history would predict that in another 8 years or so their NSAA would be in the teens. Those with a NSAA in the teens have
Stand on heels Walking on uneven ground, cycling, difficulty getting out of a chair, striding, cycling
Rise from floor Getting up after falling, playing on the floor with children
Climb box steps Independent outdoor mobility particularly easy tasks like stairs and sidewalk curbs
Stand on one leg
Dressing oneself, putting on shoes/socks while standing, reaching high shelves
Gets to sitting Sitting up in bed, adjust to falls
Rise from chair Using a toilet independently, getting out of bed, using public transportation to get around
Walk
Walking to mailbox to pick up mail, hiking, everyday mobility
Stand Grooming, preparing meals, adapting to mobility device, transferring to chair
started to lose meaningful functions such as being able to climb a step or rise from the floor.” In another 8 years, she said, natural history would predict that many NSAA functions will be completely lost. “What we need to do is find something that will stabilise them,” she added.
Figure 2: ARCH study data presented ahead of publication: North Star Ambulatory Assessment (NSAA) stabilised, diverging from natural history at 24 months.
NSAA stabilized, diverging from natural history at 24 months NSAA change
mean change in NSAA with sevasemten at 24 months 2.4 natural history average change
*All data through 24 months, including patients recovering from meniscus surgery.
Natural history based on unpublished data presented by Bello at MDA and van de Velde et al.22
Mean ±95% confidence intervals.
NSAA: North Star Ambulatory Assessment.
24-Month Data
ARCH was an open-label, singlecentre study assessing the safety and pharmacokinetics of sevasemten in adults with BECKER. The study also assessed the agent’s longer-term functional affect. At baseline, the 12 ambulatory males, all aged 18–55 years, had an average NSAA of 15, increased serum CK, and decreased lean muscle mass. They were already functionally impaired, and “you would expect them to decline,” Donovan said.
The 24-month data showed that sevasemten was well tolerated at all doses, with no dose reductions or adjustments, no treatment discontinuations due to adverse events, and no serious adverse events. Over the 2 years, 11 of the 12 patients remained stable with regard to their NSAA score. “Whether they were at the top of the NSAA at 31, or were barely ambulatory, starting out with a score of four, they maintained
function. In some cases, they even gained some function,” said Donovan. This contrasts significantly, she went on, with the approximately 2.4 point decrease that would be expected from the natural history studies (Figure 2). The one patient who experienced a decline in NSAA had suffered a meniscal tear that required surgery, and immobilised them post-operatively. Donovan said that this spoke to the role of continued exercise in Becker management.
Biomarkers of muscle damage that are elevated in people with Becker, including CK, fast skeletal muscle troponin I, and myoglobin, decreased rapidly after treatment initiation, and this decrease was maintained over the 24-month period. Other measures of muscle function, such as the 100 m timed walk test and maximum grip strength, remained stable, suggesting that inhibiting the fast myofiber (Type II) myosin inhibitor does not negatively impact muscle strength.
Maximal biomarker response was recorded at a 10 mg dose, and this finding has now been taken forward into a pivotal clinical trial (NCT05291091/ Grand Canyon).
Edgewise is aiming to recruit 120 adult males diagnosed with Becker across the USA and Europe in their GRAND CANYON study. Key inclusion criteria are: aged between 18–50, a mutation in the dystrophin gene with Becker phenotype, and ambulatory with NSAA of between 5–32. The company is also looking at the agent’s safety and efficacy in Duchenne, Donovan added.
References
1. Mohammed F et al. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: the era of personalized medicine. PloS one. 2018;13(5):e0197205.
2. Wilson K et al. Duchenne and Becker muscular dystrophies: a review of animal models, clinical end points, and biomarker quantification. Toxicol Pathol. 2017;45(7):961-76.
3. Darras BT et al., Dystrophinopathies [Internet] (2022) GeneReviews. Available at: https://www.ncbi.nlm.nih. gov/books/NBK1119/ Last accessed: 23 May 2024.
4. Wilson DG et al. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun Biol. 2022;27;5(1):1022.
5. Kiriaev L et al. Eccentric contractioninduced strength loss in dystrophindeficient muscle: preparations, protocols, and mechanisms. J Gen Physiol. 2023;155(2):e202213208.
6. Sacco P et al. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin Sc. 1992;82(2):227-36.
7. Carter GT et al. Adaptations to exercise training and contractioninduced muscle injury in animal models of muscular dystrophy. Ame J Phys Med Rehabil. 2002;81(11 Suppl):S151-61.
8. Dahlqvist JR et al. A pilot study of muscle plasma protein changes after exercise. Muscle Nerve. 2014;49(2):261-6.
CONCLUSION
While contraction-induced injury underlies disease progression in Becker and Duchenne, this does not mean that exercise is not beneficial, and clear benefits have been observed. However, avoiding the type of exercise that is most associated with damage, i.e., eccentric contraction, is important in guiding exercise in muscular dystrophy. Additionally, approaches to pharmacologically protect against contraction-induced injury hold promise.
9. Sveen ML et al. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain. 2008;131(Pt 11):2824-31.
10. Sveen ML et al. Resistance training in patients with limb‐girdle and Becker muscular dystrophies. Muscle Nerve. 2013;47(2):163-9.
11. Berthelsen MP et al. Anti-gravity training improves walking capacity and postural balance in patients with muscular dystrophy. Neuromuscul Disord. 2014;24(6):492-8.
12. Barthel B et al. Use of an exercise challenge system to define a universal proteomic signature of muscle injury in a diverse set of adult individuals with inherited myopathy. Abstract FP.06. World Muscle Society meeting, 11–15 October, 2022.
13. Baltgalvis KA et al. Exercise training improves plantar flexor muscle function in mdx mice. Med Science Sports Exerc. 2012;44(9):1671-9.
14. Mathur S et al. Changes in muscle T2 and tissue damage after downhill running in mdx mice. Muscle Nerve. 2011;43(6):878-86.
15. Lott DJ et al. Safety, feasibility, and efficacy of strengthening exercise in Duchenne muscular dystrophy. Muscle Nerve. 2021;63(3):320-6.
16. Jansen M et al. Assisted bicycle training delays functional deterioration in boys with Duchenne muscular dystrophy: the randomized controlled trial “no use is disuse”. Neurorehabil Neural Repair. 2013;27(9):816-27.
17. Russell AJ et al. Modulating fast skeletal muscle contraction protects
skeletal muscle in animal models of Duchenne muscular dystrophy. Journal Clin Investig. 2023;133(10):e153837.
18. Mazzone ES et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul Disord. 2009;19(7):458-61.
19. Great Ormond Street Hospital NHS Foundation Trust & The Newcastle upon Tyne Hospitals NHS Foundation Trust. North Star Ambulatory Assessment Manual. 2020. Available at: https://community. musculardystrophyuk.org/wp-content/ uploads/2017/06/North-star-PTManual_MAY_2017.pdf Last accessed: 23 May 2024.
20. Bello L et al. Functional changes in Becker muscular dystrophy: implications for clinical trials in dystrophinopathies. Sci Rep. 2016;6(1):32439.
21. De Wel B et al. Lessons for future clinical trials in adults with Becker muscular dystrophy: Disease progression detected by muscle magnetic resonance imaging, clinical and patient‐reported outcome measures. Eur J Neurol. 2024:e16282. [Epub ahead of print].
22. van de Velde NM et al. Selection approach to identify the optimal biomarker using quantitative muscle MRI and functional assessments in Becker muscular dystrophy. Neurology. 2021;97(5):e513-22
Climate Change and Air Pollution: How Healthcare Providers Can Help Mitigate the Risks to Respiratory Health
Interviewees:
1. Imperial College London, UK
2. Chinese University of Hong Kong, Shatin, Hong Kong
3. Symbiosis International University, Pune, India
4. University of British Columbia, Vancouver, Canada
Disclosure: The authors have declared no conflicts of interest.
Acknowledgements: Medical writing assistance was provided by Amanda Barrell, Brighton, UK.
Support: This article was written by Advisory Board members of The Clean Breathing Institute and funded by Haleon.
Interview Summary
The intricate relationship between climate change and air pollution is having a significant impact on public health. Increases in global temperatures are leading to extreme weather events, changes in plant growth patterns, and higher levels of aeroallergens and air pollution; all of which can exacerbate pre-existing respiratory conditions and increase the risk of developing respiratory and other diseases. In this article, four world leaders in the field of respiratory health outline the evidence linking climate change and air pollution to poor respiratory health outcomes. They highlight that people living with lung conditions, such as asthma and chronic obstructive pulmonary disease (COPD), as well as pregnant people, children, older people, and those living in low- and middle-income countries (LMIC), are the most at risk. They emphasise the need for greater awareness among the public and healthcare professionals alike, talk about the role of healthcare teams in helping people to recognise and mitigate the risks, and share practical ways people can help to minimise the health impacts of climate change and pollution.
PHARMA
Fan Chung,1 Gary Wong,2 Sundeep Salvi,3 Christopher Carlsten4
INTRODUCTION
Climate change is happening, with the evidence clearly demonstrating an increase in global temperatures; extreme weather events; and levels of air pollution, aeroallergens, and airborne pathogens.1,2 The complex interplay between these various factors is changing the quality and composition of the air we breathe, directly aggravating pre-existing respiratory diseases and increasing everyone’s exposure to their risk factors.3 “This is happening and we need to prepare for it,” said Fan Chung, Professor of Respiratory Medicine and Head of Experimental Studies Medicine at the National Heart and Lung Institute, Imperial College London, UK; and Consultant Physician at the Royal Brompton and Harefield NHS Trust, London, UK.
It is now widely accepted the Earth’s climate is changing, due to the burning of fossil fuels resulting in the generation of greenhouse gases (GHG).4 The global atmospheric CO2 concentration, for example, has increased from pre-industrial levels (1850–1900) of approximately 280 parts per million, to 415 parts per million in 2021.2 Such GHGs become trapped in the planet’s atmosphere, and create a warming effect. Chung said: “There is already evidence that this is happening. The World Meteorological Organization (WMO) has said that 2023 was another record year for temperatures.”5 Higher global temperatures mean more extreme weather events, such as flooding, drought, and wildfires, as well as changes to how plants grow.3 Professor of Paediatrics at the Chinese University of Hong Kong, Shatin, Hong Kong, Gary Wong, explained: “When we burn large amounts of fossil fuels, it releases GHGs that change the atmospheric composition and leads to gradual increases in the overall temperature. This has subtle trickle-down effects on plant growth: they have longer growing seasons and they generate more pollen.3 Taken together, it means we have more pollutants and more pollen in the atmosphere.” The changing composition of air also influences the way airborne pathogens, such as viruses and bacteria, behave in the air and access human bodies.
CLIMATE CHANGE AND HEALTH
The complex interplay between increasing global temperatures and air pollution is having an important impact on public health. The Intergovernmental Panel on Climate Change has projected a significant increase in climate-related ill health and premature deaths in the coming years, estimating excess deaths of 250,000 a year by 2050.2 However, there is clear evidence to show that decreasing air quality and climate change are already impacting the world’s respiratory health.1 Christopher Carlsten, Director of the Centre for Lung Health and Legacy for Airway Health (LAH), Vancouver, Canada; and Professor of Respiratory Medicine at the University of British Columbia, Vancouver, Canada, said there was no one way to quantify the impact of climate change on health. Though imperfect, he said that the easiest way is to “break it up into its parts,” by looking at the “effects of particulate matter (PM), toxic gases such as ozone, temperature, and allergens, to name but a few.” Each of these generally increase as the climate warms, and their interaction can have compounding effects.
Pollution and Particulate Matter
Sundeep Salvi, Director of the Pulmocare Research and Education (PURE) Foundation in Pune, India, and winner of the 2024 American Thoracic Society (ATS) World Lung Health Award, explained that climate change and air quality were “very intricately connected.” “A human being breathes 10,000 litres of air a day,”6 he said. “The air we breathe comes into close contact with a very large surface area in the lung, around the size of a tennis court, where the gas exchange takes place,” he said. “The gas exchange portion of the lung surface is very sensitive and very thin, so if polluted air, hot air, or cold air reaches that, it is going to have an adverse effect.”
An estimated 50% of all childhood pneumonia deaths, for example, are connected to air pollution, with the highest concentrations of mortality being found in LMIC.7 In addition, air pollution has been found to be the most important driver of COPD in India, being responsible for more
burden than smoking.8 While asthma is largely recognised to be a genetic disease, the environment has a “very important and perhaps equal contribution,” Salvi said.9 “If you live in a polluted environment, you’re at risk of developing these conditions.9 If you have underlying asthma or COPD and live in a polluted environment, you’re going to get repeated exacerbations,” he added.9
With the burning of fossil fuels producing both GHGs and pollutants, climate change and air pollution are intrinsically linked.1 PM refers to everything in the air that is not gas, and consists of a wide variety of chemical compounds, organic matter, and other materials.10 Scientists tend to classify these particles according to their size.10 PM10, for example, are particles of less than 10 µm in diameter, PM2.5 are particles of less than 2.5 µm, and PM1 are particles of less than 1 µm.10 Chung said: “The size is important because the small ones can travel deep into the lungs.” Particles >5 μm reach and are deposited mainly in the proximal large airways, and then removed via mucociliary clearance; whereas, those <5 μm will reach, and are deposited, in the small airways and alveoli.11 Studies have suggested that 83% of PM2.5 are deposited in the lung, compared to just 31% of PM with a diameter of less than 11.5 μm.11 “PM can contain various carbon-like substances, metals, organic
material, and plastic nanoparticles, and many can have a toxic-type effect,” Chung explained. In 2019, PM2.5 was responsible for an estimated 6.7 million deaths worldwide, including 373,000 in Europe.1
PM has many known effects across the respiratory system. “You can visualise it going from the nose to the bottom of the lungs: from the nasal passages at the beginning of the upper airway, through the pharynx, the larynx, through the bronchial tree, and all the way down to the alveoli,” Carlsten said. “Every known respiratory condition or disorder along that pathway has been demonstrated, in one way or another, to be affected by PM.” 7,8,9,12-14 (see Table 1)
It does not stop, however, with the respiratory system, Salvi explained. “A lot of the pollutants that enter the lungs go on to enter the circulation, and get transported to all the organs of the body.15 They get deposited in the brain, in the kidney, the bowel, and so on,” he said, highlighting his recent study that teenagers living in the polluted city of Delhi, India, had a high prevalence of not only asthma and respiratory symptoms, but also allergic rhinitis, eczema, and high BMI.16 This, he went on, was a new and important finding that warrants further investigation.
Table 1: Potential health impact of particulate matter by respiratory tract feature.7,8,9,12-14
As well as PM, air pollution can also contain gases, including nitrogen oxides (NOx), carbon monoxide, sulphur dioxide, and ground-level ozone, an irritant that is promoted by sunlight exposure and higher temperatures.1,17 “All of these tend to irritate the airways,” Wong said. “Without underlying disease, it can result in respiratory symptoms, such as cough and phlegm.3 But for those living with, for example, asthma or COPD, it can irritate the airway so much that it makes the pre-existing inflammation worse. That’s why, when the level of pollutants rise, we see exacerbations.”18
Where Does Particulate Matter Come From?
Various factors are associated with higher levels of PM. Carlsten explained that diesel exhaust particles (DEP) have typically been the most common contributor to ultrafine particles (<0.05–0.10 μm), at least in urban environments. DEP contains a complex mixture of particles and gases including carbon monoxide, NOx, sulphur dioxide, hydrocarbons, formaldehyde, transition metals, and carbon particles.11 It is the size of PM found in DEP, however, that scientists believe accounts for its systemic effects, which include increased carcinogenicity, the potentiation of autoimmune disorders, alterations in blood coagulability, and increased cardiovascular disorders.11 “In some ways, DEP are the most concerning of the particles because of how deep they get into the lungs,” Carlsten said, but extreme weather events, such as wildfires and sandstorms, also generate high levels of dangerous PM that can travel thousands of kilometres from their source over days or even weeks.1,19,20
Indoor pollution is another important area for consideration, particularly in areas of the world that use coal or biomass fuel. Worldwide, more than three billion people use traditional cookstoves that burn wood, animal dung, or crop residues to cook food or heat water.21 Wong said: “When you are biomass fuel in an enclosed area, all the pollutants are trapped inside the home environment. It is very common in India, Africa, and Latin America.” In addition, many people in tropical parts of the world
routinely use products such as mosquito coils and agarbatti incense, Salvi said. “Our research has shown that burning one mosquito coil for 6 or 7 hours exposes people to the same amount of pollution as smoking 100 cigarettes,” he said.22
In high-income countries, much indoor pollution, Chung said, was “leakage” of outdoor pollution into the home, while the rest was the result of cooking and cleaning products.17 “We might not necessarily think of this as a climaterelated phenomenon, but it’s more related than one might imagine,” Carlsten said. “The higher the outdoor air pollution, the higher the indoor air pollution because homes are far from being a complete barrier. The challenge of indoor air pollution becomes linked to climate change by virtue of the connection between the outdoor and the indoor worlds.”17
Temperatures
Increasing temperatures also have an adverse impact on mortality and morbidity.23 “The impact of temperature on health has been less extensively studied than that of PM,” Carlsten said, “but heat has been associated with increased mortality.” A recent study in England found that the risk of COPD hospitalisation increased by 1.5% per 1 °C increase in temperature, and attributed 1,851 events per year to temperatures above 23 °C.24 According to France’s public health body, Santé Publique France, the country recorded almost 33,000 heat-related deaths between 2014–2022, 28% of which occurred during heatwaves.25
While the mechanisms of this connection are still under investigation, the doctors said it was likely related to the impact of impaired thermoregulation.23 Wong explained: “We are warm-blooded animals and we need to keep our body temperature in a very narrow range. When we get too hot, we reduce our body temperature by breathing faster and drinking more water. If someone already has borderline lung function, this will likely stress the respiratory and cardiovascular systems.”
Aeroallergens and Allergies
Climate change has also been associated with an increase in allergic manifestations, including allergic rhinitis and asthma.2 Higher temperatures mean longer pollen and fungal spore seasons, and, when combined with increasing CO2 concentrations, higher levels of airborne allergens and higher pollen allergenicity.2 The interactions between aeroallergens and air pollutants, including PM, also play a role with some pollutants being able to alter the immunogenicity of allergenic proteins.2 “Climate change means longer summers, so high pollen levels for longer, and there is evidence to suggest the allergenicity of pollen may increase,”2 Chung said. “It means there is a possibility that more people will develop a pollen allergy, because more people will be exposed to it.” Studies have also suggested that diesel exhaust contributes to an increasing prevalence of allergies, with DEPs being shown to act as adjuvants that increase the sensitisation response in some experimental models.11
“Allergens have a huge impact on quality of life, through rhinitis and other allergic conditions,” Carlsten said. “We often think of exacerbations of allergic disease as things that are inconvenient, but not deadly, like a runny nose, but they’re also a risk factor for mortality.” Thunderstorm asthma, or asthma exacerbations linked to high winds drawing higher levels of pollens and pollution particles into the air, is one example of how climate change can have a potentially devastating impact.26 “Pollen particles are usually quite big, so they can’t get into the body, but thunderstorms break them down to the PM2.5 size, enabling them to get deep into the lungs,” Chung said.27 Thunderstorm asthma, Wong went on, is particularly dangerous because it can affect people who do not have asthma, with one study finding 65% of people with seasonal allergic rhinitis experienced it.28 They have never learnt to recognise the symptoms, meaning they do not necessarily understand how to manage the symptoms with an inhaler, or when to seek help,” he explained. In 2016, a thunderstorm asthma event in Victoria, Australia, led to 3,365 people presenting to an emergency department with acute respiratory symptoms.29 From these presentations, 476
asthma-related hospital admissions occurred, equating to a 992% increase in typical asthma-related hospital admissions.29 Thirtyfive people were admitted to an intensive care unit and 10 died.29
Climate Change and Infections
In addition, climate change can enhance the spread of vector-borne diseases. Rising temperatures and higher precipitation can directly affect the life cycles of pathogens, and change their habitats and environments. While the climate change-related increase in infectious diseases has not yet been quantified, studies have shown, for example, that the incidence of water and vectorborne diseases such as malaria and dengue fever are very much influenced by changes in the climate and environment.30
As temperatures and rain levels continue to rise, this could pose a challenge worldwide. Wong said: “Singapore is a tropical country, with the same temperature, humidity, and so on throughout the year. In that atmosphere, there are various bacteria and fungi that we know could cause trouble if inhaled into the lungs. If we are moving towards a climate that is more like Singapore’s in London, for example, we might move towards that same kind of ecology.” The consequences, he went on, could be felt by people with preexisting respiratory disease, as well as the general population.
CLIMATE CHANGE AND SUSCEPTIBLE POPULATIONS
Taken all together, then, climate change is changing the air that we breathe. Chung said that increased humidity, higher temperatures, and increased ozone is affecting the way PM, pollen, and organisms behave in the air, and how they access and act in our bodies. “Anything that travels with the PM2.5, including micro-organisms, can ‘piggyback’ on the particles and get deep into the lungs,” he explained.31
Everyone’s health, at least to some extent, is at risk from the health impact of climate change, but those with pre-existing respiratory issues are in the greatest danger.1
“By definition, those with pre-existing COPD, asthma, and interstitial lung disease, etc., are the ones at greatest risk of exacerbations. That is obvious,” Carlsten said. “But who is at risk for new disease? That is a much more complicated question than most people appreciate.”
Air pollution causes 8.34 million excess deaths every year.32 “What characterises these people who are at risk of dying from pollution, and makes them different to the other 30 or 40 billion people in the world who do not die from pollution?” Chung asked. The answer, he said, is likely to be linked to personal factors. His team has just completed a study in which people wore sensors to measure their own personal levels of exposure to PM10, PM2.5, and PM1. “We found that it really depended on lifestyle, and the level of exposure varied from person to person. Even two people living at the same address can have very different exposure levels because of the different ways they travel, their workplace, the places they go to, etc.,” he said (unpublished data; Chung, F).
In general, however, the current literature shows that, aside from those with preexisting medical conditions including cardiorespiratory disease, it is pregnant people, children, and older people who are most at risk. Socioeconomic factors can also play a role.1
Pregnant People
Prenatal exposure to air pollution has been linked to infant and child lung function impairment, an increase in respiratory symptoms, and the development of childhood asthma.33 It may also be indirectly linked to early birth, low birth weight, and impaired development of the immune system.34 “It is important to appreciate that the health effects of air pollution start even before you are born,” Salvi said. “The quality of air the mother breathes is going to decide the quality of nutrients the baby receives in the womb.”
Children
Children are at both acute and chronic risk of the effects of air pollution, due to their behaviour, their physiology, and their environment. Their lungs and immune systems are still developing, they tend to spend more time being physically active outside, and they breathe two to three times faster than adults; meaning they can inhale more polluted, hot, or aeroallergencontaining air, Salvi explained.1 There are data to suggest, for example, that children exposed to higher levels of PM2.5 are more likely to develop asthma and persistent wheezing than children who are not.34 Such asthma exacerbations can be extremely dangerous. In December 2020, the UK became the first country to list air pollution exposure as a cause of death after a 9-year-old girl who lived on a busy road in London, UK, died following an asthma attack. An inquest heard she had experienced a lifetime of exposure to excessively high levels of a traffic-derived mix of nitrogen dioxide and PM10, including non-exhaust sources of PM such as tyre and brake wear.35
Air pollution can also negatively impact the normal growth of lung function, and this can leave children vulnerable to respiratory disease later in life.36 Wong pointed to a study that showed that children who lived in more polluted areas of California, USA, had more impaired lung function, compared to those who lived in less polluted areas.36 “A small percentage reduction in lung function may not cause a big problem in terms of daily activity, but our lung function improves as we get older, and then starts to decline,” he said. “If you start with a lower peak, you are going to start feeling symptoms easier. Impaired lung growth reduces the maximum lung capacity of a healthy individual,”36 Salvi agreed. “There is a stunting of the lung growth that happens because of air pollution. Not only does that give rise to asthma or upper and lower respiratory tract infections, but it also makes children more vulnerable to developing COPD when they become adults.”
Older People
Older people are at increased risk from climate change in several ways. They are, for example, more likely to be trapped after extreme events such as flooding or drought, due to a lack of mobility, disability, or frailty.37 Their propensity for dehydration puts them at increased risk of heat-related illnesses, such as cardiac events, and they are particularly vulnerable to the pollutants and pathogens in the air.38 Salvi said: “As the immune system ages, the defences wear off, and the air pollutants they inhale will have a bigger impact.” Wong agreed, adding: “Older people with pre-existing lung disease or cardiovascular disease are much more prone to the effects of pollution, global warming, and climate change.”
Female Sex
There is also some evidence to suggest female sex plays a role in risk. A 2017 literature search, for example, suggested that females over the age of 65 years were more likely to suffer heat-related mortality than their male counterparts.39
Carlsten said the relationship between female sex and risk in the context of climate change “has not been adequately studied.” Increased risk among females, he went on, was likely to be related at least in part to their smaller respiratory tree, as compared to males, which could affect the deposition of pollution, he added.
Low- and Middle-Income Countries
People living in LMICs will “bear the brunt” of climate change and its health impact, Chung said. “A lot of people cannot pay for medical care, and they often live in places more affected by climate change; they experience more flooding, more forest fires, and so on,” he said. According the 2023 World Air Quality Report, Bangladesh, Pakistan, and India have the highest concentrations of PM2.5 of all 134 countries that collect data.40 Of his native country, India, Salvi said: “There is rapid urbanisation and industrialisation, and the number of motor vehicles has shot up.” In developed parts of the world, he went on, systems and policies are being put in
place to combat climate change. “But in developing parts of the world, there is no way we can halt it because a country’s economic growth depends on how much they can manufacture.” In addition, females in LMICs are thought to be at high risk from the adverse respiratory effects of indoor pollution, as they tend to be the homemakers and therefore have high exposure to pollution generated by the burning of biomass fuel.41
PROTECTION AND MITIGATION
The evidence base is still evolving, and much more research is needed on the intricate interplay between climate change, pollution, and respiratory health, as well as the possible mitigation and resiliencebuilding strategies. “There is a lack of research in the field of practical solutions: on things like wearing a mask, using nasal filters, saline nasal washes, and the use of antioxidants and dietary supplements,” Salvi said. “There are papers that seem to suggest they are effective, but we need more evidence to support that. The space is not mature.”21
However, all four doctors agreed that we understand enough to know that urgent action is needed. Chung said: “The problem is not in the distant future, the problem is here, now, on our doorstep, and it is just getting worse.” Greater awareness and education among the public, healthcare professionals (HCP), policymakers, and researchers alike is the “first and the most important intervention,” Salvi said. “Once that knowledge base is established, it’s a matter of disseminating and using it to offer advice to patients.” Raising awareness of the issues among hospital and healthcare systems is another important task, Carlsten added. “Broadly, healthcare systems tend to focus on the traditional challenges of capacity. Everything gravitates to the immediate crisis of the day. Yet, over time, these issues will impact those systems on a very obvious level, costing more money, and contributing to the busyness of emergency departments,” he said.
THE ROLE OF HEALTHCARE PROVIDERS
Asked if HCPs had a responsibility to help patients mitigate the risks of climate change, global warming, and pollution, Carlsten said it would be better described as an opportunity. “Healthcare professionals are supposed to do everything they reasonably can to promote health. They are also considered high on the spectrum of trust, so there’s reason to believe that their patients, and the community in general, will respect and listen to what they say,” he went on. Due to the “intuitive relationship between the air we breathe and the lungs,” respiratory specialists have been at the forefront of educating patients so far, but with pollution and climate change affecting every system in the body, there is an opportunity for all HCPs to become involved.
HCPs are already aware of, and have contact with, those people who are at greater risk from climate change and pollution. They are “at the forefront of the health battle,” Chung said. “I think it’s something that needs to be raised at each visit, particularly for those people suffering from chronic conditions. Pregnant people, children, older people, and so on, need to be informed of the risks.” While it is not the job of physicians to tackle climate change, Wong said that they were in the perfect position to identify at-risk individuals, and provide them with the information they need to mitigate the effects of climate change and pollution.
Healthcare providers can:
• identify at-risk populations;
• educate patients on the risks climate change poses to their health;
• educate patients on the mitigation strategies people can follow to protect their health from climate change; and
• support patients to implement the necessary changes.
Offering Solutions
While we wait for the evidence base to mature, there are certain “working solutions” that HCPs can recommend, Salvi said. In 2020, he, along with Carlsten, Wong, and Chung, published ‘Personal strategies to minimise effects of air pollution on respiratory health: advice for providers, patients and the public’.21 The paper reviewed the literature on a number of personal strategies aimed at reducing exposure to outdoor or indoor air pollution, and made recommendations based on the strength of the evidence (Table 2).
It also argued that HCPs need to receive the most up-to-date evidence and information.21 “HCPs need to be aware of the depth of knowledge on air pollution, climate change, and human health. Next, they need to give potential simple, practical, affordable, sustainable, and effective solutions to the patients they see in the clinic,” Salvi said. Wearing a mask on polluted days and using saline nasal washes to clear pollutants that become trapped in the nose, for example, may be of some benefit.21
Preparedness was an important theme, and the doctors recommended HCPs advise patients to learn how to access local air pollution information. They can then use these data to plan where and when they exercise, or plot alternative, less polluted routes when commuting or travelling. Ensuring all patients have an up-to-date action plan that considers the impact of air pollution, and explains what to do when they experience an exacerbation, is important. Likewise, in areas at high risk of extreme weather events, whether that’s wildfires, flooding, or heatwaves, HCPs can work with patients to ensure they are prepared. Carlsten’s team, principally Emily Brigham, University of British Columbia, Vancouver, Canada, has developed an action plan checklist for wildfire smoke and extreme heat event readiness.42 Designed to be completed during clinic, it ensures people know how to check their local air quality, how to recognise potentially dangerous levels of heat and pollutant exposure, when to act, and how to minimise the risks.42,43
Table 2: Personal strategies to mitigate the impact of air pollution on respiratory health.21
Strategy Level of Evidence
Using facemasks when anticipating unavoidable exposure to air pollution exceeding recommended levels
Using active rather than motorised transport where possible
Travelling via routes that minimise near-road air pollution
Optimise driving conditions
Optimise outside exercising conditions
Grade C
Monitor air pollution
Grade C
Grade C
Minimise exposure to indoor pollution
Grade D
Grade C
Treat and manage respiratory conditions
Grade D
Grade C
Modify diet and supplement with antioxidants or antiinflammatory agents
Recommendations
• Consider close-fitting N95 facemasks
• Follow manufacturers’ guidance on correct usage, maintenance, and fit, including a user seal check
• People with chronic healthcare conditions that make breathing difficult should check with their healthcare provider before using an N95 facemask
• Healthcare providers can encourage cycling or walking wherever possible
• Avoid major intersections, roads with heavy traffic, etc.
• Select routes with open spaces
• Use off-road versus on-road bicycle paths where possible
• Utilise up-to-date information on local air quality, including mobile phone apps, newsfeeds, and websites, when planning routes
• Drive with windows closed and keep the air on internal circulation when in areas of high pollution
• Avoid rapid accelerations and decelerations, avoid engine idling, and maintain vehicles correctly
• Avoid exercising in places and at times of extreme air pollution
• Exercise away from traffic wherever possible
• Use local air quality information to plan outside exercise
• Decrease or stop exercising if coughing, chest tightness, or wheezing develops
• Healthcare providers should teach patients how to check the air quality forecast and act to minimise exposure
• Patients with underlying susceptibility should be aware of air quality alerts and learn how to protect themselves on days of high air pollution
• Users of personal pollution monitors should to be aware of their highly variable accuracy
Use clean fuels
Grade C Use portable air cleaners
Grade C
Grade D
Grade D
Grade D
Optimise household ventilation
Adopt efficient cookstoves
• Maximise control of airway diseases: optimise medical care through symptom and airflow monitoring, medication, and vaccination
• Promote primary, secondary, and tertiary interventions, such as reducing obesity, increasing physical activity, smoking cessation, etc., which may attenuate the burden of cardio-pulmonary disease associated with air pollution exposure
• A healthy, balanced diet is associated with reductions in the risk of chronic lung diseases known to be compounded by air pollution
• There is not enough evidence to show any dietary supplement can counteract the detrimental effect of air pollution on respiratory health
Grade A: Rich body of evidence based on randomised controlled trials and/or meta-analyses; Grade B: Limited body of evidence based on randomised controlled trials and meta-analyses; Grade C: Body of evidence based only on non-randomised trials or observational studies; Grade D: Panel consensus judgement only.
At the same time, however, HCPs need to be careful not to add to anxiety, the doctors said. “We don’t want everything to be doom and gloom, and for people to feel like they have to lock themselves up during any kind of pollution event. We have to balance the very real evidence about the serious health effects with the need to exercise, be outside, socialise, and be connected to nature,” Carlsten said. The next phase of research, he went on, will be to understand how to build resilience to the changes that are already happening. “Scientifically, we’ve got the methodologies and resources to do the analyses and publish the papers on all the bad things that are happening, but there’s a lot less work going on looking at how we can manage these, and not be so anxious about it all the time. Part of what I am trying to focus on is what we can do together to build the strength and capacity we will need to withstand this threat.”
References
1. Vicedo-Cabrera AM et al. Climate change and respiratory health: a European Respiratory Society position statement. Eur Respir J. 2023;62(2):2201960.
2. Beggs PJ et al. Climate change, airborne allergens, and three translational mitigation approaches. EBioMedicine. 2023;93:104478.
3. D’Amato G et al. Climate change and respiratory diseases. Eur Resp Rev. 2014;23(132):161-9.
4. Lynas M et al. Greater than 99% consensus on human caused climate change in the peer-reviewed scientific literature. Environ Res Lett. 2021;16(11):114005.
5. World Meteorological Organization (WMO). State of the global climate 2023. WMO-No.1347. 2024. Available at: https://library.wmo.int/records/ item/68835-state-of-the-globalclimate-2023. Last accessed: 11 April 2024.
6. Neupane AS et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell. 2020;183(1):110-25.
7. UNICEF. Pneumonia. 2023. Available at: https://data.unicef.org/topic/childhealth/pneumonia/. Last accessed: 11 April 2024.
8. Salvi S; India State-Level Disease Burden Initiative CRD Collaborators. The burden of chronic respiratory
TIME TO ACT
The doctors concluded that the complex interplay between climate change, air pollution, and respiratory health presents a significant challenge, and that urgent action is needed to address the impact of these factors, particularly on vulnerable populations. While the evidence base is still evolving, there are practical solutions and mitigation strategies that HCPs can recommend to minimise exposure to outdoor and indoor air pollution, as well as extreme-weather-related ill health. Greater awareness, education, and preparedness are essential, they agreed.
“It is absolutely crucial” to take action, Salvi concluded. “I am worried the next generation is going to be more vulnerable, so we need to be very, very serious about this.”
diseases and their heterogeneity across the states of India: the Global Burden of Disease Study 1990–2016. Lancet Glob Health. 2018;6(12):e136374.
9. Bronte-Moreno O et al. Impact of air pollution on asthma: a scoping review. Open Respir Arch. 2023;5(2):100229.
10. Department for Environment, Food, and Rural Affairs (Defra). National statistics: Emissions of air pollutants in the UK – particulate matter (PM10 and PM2.5). 2024. Available at: https://www.gov.uk/government/ statistics/emissions-of-air-pollutants/ emissions-of-air-pollutants-in-the-ukparticulate-matter-pm10-and-pm25. Last accessed: 11 April 2024.
11. Sydbom A at al. Health effects of diesel exhaust emissions. Eur Respir J. 2001;17(4):733-46.
12. Mariani J et al. Particulate matter exposure and allergic rhinitis: the role of plasmatic extracellular vesicles and bacterial nasal microbiome. Int J Environ Res Public Health. 2021;18(20):10689.
13. Ziou M et al. Outdoor particulate matter exposure and upper respiratory tract infections in children and adolescents: a systematic review and meta-analysis. Environ Res. 2022;210:112969.
14. Singh N, Singh S. Interstitial lung diseases and air pollution: narrative review of literature. Pulm Ther. 2021;7(1):89-100.
15. Schraufnagel DE et al. Air pollution and noncommunicable diseases: a review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: damaging effects of air pollution. Chest. 2019;155(2):409-16.
16. Salvi SS et al. Association between air pollution, body mass index, respiratory symptoms, and asthma among adolescent school children living in Delhi, India. Lung India. 2021;38(5):408-15.
17. Leung DYC. Outdoor-indoor air pollution in urban environment: challenges and opportunity. Front Environ Sci. 2015;2:69.
18. Chou CH et al. Environmental pollutants increase the risks of acute exacerbation in patients with chronic airway disease. Front Public Health. 2023;11:1215224.
19. Jaffe DA et al. Wildfire and prescribed burning impacts on air quality in the United States. J Air Waste Manag Assoc. 2020;70(6):583-615.
20. Jones BA. Dust storms and human well-being. Resour Energy Econ. 2023;72:101362.
21. Carlsten C et al. Personal strategies to minimise effects of air pollution on respiratory health: advice for providers, patients and the public. Eur Respir J. 2020;55(6):1902056.
22. Salvi D et al. Indoor particulate matter < 2.5 μm in mean aerodynamic diameter and carbon monoxide levels
during the burning of mosquito coils and their association with respiratory health. Chest. 2016;149(2):459-66.
23. Arsad FS et al. The impact of heatwaves on mortality and morbidity and the associated vulnerability factors: a systematic review. Int J Environ Res Public Health. 2022;19(23):16356.
24. Konstantinoudis G et al. Ambient heat exposure and COPD hospitalisations in England: a nationwide case-crossover study during 2007–2018. Thorax. 2022;77(11):1098-104.
25. Pascal M. Estimation of the mortality attributable to heat in France between 2014 and 2022. Available at: https:// www.researchsquare.com/article/rs3085843/v1. Last accessed: 11 April 2024.
26. Asthma and Lung UK. Thunderstorms and asthma. 2024. Available at: https://www.asthmaandlung.org.uk/ conditions/asthma/asthma-triggers/ thunder. Last accessed: 11 April 2024.
27. Reinmuth-Selzle K et al. Air pollution and climate change effects on allergies in the anthropocene: abundance, interaction, and modification of allergens and adjuvants. Environ Sci Technol. 2017;51(8):4119-41.
28. Douglass JA et al. Thunderstorm asthma in seasonal allergic rhinitis: the
29. Beggs PJ. Thunderstorm asthma and climate change. JAMA. 2024;331(10):878-9.
30. Uwishema O et al. Impacts of environmental and climatic changes on future infectious diseases. Int J Surg. 2023;109(2):167-70.
31. Soumana IH, Carlsten C. Air pollution and the respiratory microbiome. J Allergy Clin Immunol. 2021;148(1):67-9.
32. Lelieveld J et al. Air pollution deaths attributable to fossil fuels: observational and modelling study. BMJ. 2023;383:e077784.
33. Rani P, Dhok A. Effects of pollution on pregnancy and infants. Cureus. 2023;15(1):e33906.
34. Holst GJ et al. Air pollution and family related determinants of asthma onset and persistent wheezing in children: nationwide case-control study. BMJ. 2020;370:m2791.
35. Whitehouse A, Grigg J. Air pollution and children’s health: where next? BMJ Paediatr Open. 2021;5(1):e000706.
36. Gauderman WJ et al. Association between air pollution and lung function growth in southern California children. Am J Respir Crit Care Med. 2000;162(4):1383-90.
37. Harper S. The implications of climate change for the health of older adults. J Popul Ageing. 2023;16(3):565-8.
38. Kriebel-Gasparro A. Climate change: effects on the older adult. J Nurse Pract. 2022;18(4):372-6.
39. van Steen Y et al. Sex differences in mortality after heat waves: are elderly women at higher risk? Int Arch Occup Environ Health. 2019;92(1):37-48.
40. IQAir. 2023 World Air Quality Report. 2024. Available at: https://www.iqair. com/dl/2023_World_Air_Quality_ Report.pdf. Last accessed: 11 April 2024.
41. Wankar RL, Deo DS. Impact of biomass fuels on the respiratory functions of women in Rural India. J Family Med Prim Care. 2022;11(11):7212-6.
42. Center for Lung Health. Wildfire smoke and extreme heat action plan (landing page). Available at: https:// centreforlunghealth.ca/wildfiresmoke-and-extreme-heat-actionplan/. Last accessed: 11 April 2024.
43. Center for Lung Health. Wildfire smoke and extreme heat action plan. 2023. Available at: https:// centreforlunghealth.ca/wp-content/ uploads/2023/04/BC-Wildfire-ActionPlan_2023June08_EB.pdf. Last accessed: 11 April 2024.
Role of Nebulisers in the Treatment of Patients with Severe and Very Severe Chronic
Obstructive Pulmonary Disease
Interviewee: Omar Usmani1
1. National Heart and Lung Institute (NHLI), Imperial College London, UK
Disclosure: Usmani reports receiving grants from AstraZeneca, Boehringer Ingelheim, Chiesi, and GlaxoSmithKline (GSK); consulting fees from AstraZeneca, Cipla, and Mereo BioPharma; and personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Covis Pharma, Deva, GSK, Kamada, Menarini, Mundipharma, Novartis, Orion, Sandoz, Takeda, Trudell Medical, and UCB.
Acknowledgements: Medical writing assistance was provided by Jennifer Taylor, London, UK.
Disclaimer: The views and opinions expressed are those of the interviewee based on their clinical and scientific expertise.
Support: The publication of this article has been supported by PARI GmbH. PHARMA
Interview Summary
Chronic obstructive pulmonary disease (COPD) is a condition for which there is a portfolio of devices that can be used for treatment. These include pressurised metered dose inhalers (pMDI), pMDIs with a spacer, soft mist inhalers (SMI), dry powder inhalers (DPI), and nebulisers. In this interview with EMJ, Omar Usmani, Professor of Respiratory Medicine at the National Heart and Lung Institute (NHLI), Imperial College London, UK, summarised how nebuliser therapy works, the reasons for using these devices, and the characteristics of patients who typically benefit.
The importance of peak inspiratory flow (PIF) was discussed, and how suboptimal PIF (sPIF) should be managed. Usmani explained the concept of nebuliser efficiency and how it can be objectively measured, illustrating that not all nebulisers are the same. A case study was described, showing the need to assess inhaler technique, and the benefit of switching to nebuliser therapy when the patient demonstrates poor engagement with inhaler devices. Usmani concluded by emphasising that nebulisers have a crucial role to play in the everyday management of patients with COPD.
PARTNERSHIP
INTRODUCTION
Nebulisers are devices used to rapidly deliver high doses of medication to the lungs in people with respiratory disease. They convert liquid medication into an aerosol that is then inhaled through a facemask or mouthpiece. Nebulisers provide a rapid onset of action of the administered drug, proven efficacy, a favourable safety profile, and ease of use.1 They are suitable for patients of all ages, with various underlying respiratory diseases, and for those with cognitive disorders.1,2 Physicians can influence efficacy by choosing an efficient nebuliser which maximises the amount of medication reaching the lungs.
A growing body of evidence supports using nebulisers for maintenance therapy of respiratory disease in the outpatient setting.3
The management of patients with COPD can involve a range of inhalation devices, including pMDIs, DPIs, SMIs, and nebulisers.4 While pMDIs, DPIs, and SMIs are appropriate for many patients, they may not be the optimal delivery method in certain patients, such as those with sPIF and impaired physical and/or cognitive capabilities.4 Nebulised treatment may be a beneficial alternative in these populations.4,5 For example, nebuliser therapy may be preferred in patients with COPD, who are unable to effectively use pMDIs or DPIs despite instruction and training (Table 1).3
1 Patients with cognitive impairment, e.g., Alzheimer's dementia, intellectual disability, or altered consciousness, that precludes effective use of handheld inhalers;
2 Patients with impaired manual dexterity due to arthritis, Parkinsonism, or stroke;
3 Patients who have severe pain or muscle weakness due to neuromuscular disease;
4
Patients who are unable to use pMDIs or DPIs in an optimal manner despite adequate instruction and training, such as those patients who are generally debilitated after hospitalisation or by chronic illness and are unable to coordinate their breathing with a pMDl, or cannot generate adequate inspiratory flow for effective aerosol delivery from a DPI;
5 Patients with inadequate symptom relief with appropriate use of pMDIs/DPIs;
6 Patients who do not comply with the use of MDIs and DPIs or who prefer to use nebulisers;
7 Patients who need respiratory medications that are not available in pMDI or DPI formulations, e.g., in the USA, SABAs are not available with DPIs, and LABAs are not available with pMDIs;
8 Patients who need higher bronchodilator or corticosteroid doses for optimal control of their disease; or when multiple agents need to be co-administered;
9 Patients who cannot afford therapy with MDIs or DPIs.†
*Based on the consensus of the authors’ expert opinion and clinical experiences.
Table 1: Clinical scenarios where maintenance nebuliser therapy may be preferred in patients with chronic obstructive pulmonary disease.*3
This can include patients who are unable to coordinate their breathing with a pMDI, or who have sPIF, as discussed above, and cannot produce sufficient inspiratory flow for effective aerosol delivery from a DPI.3 Nebulisers avoid the need for manual dexterity and complex hand–breath coordination.4 Potential disadvantages that have been reported for some jet nebuliser devices include the many parts that need to be cleaned and maintained, as well as a treatment time which is typically relatively long.6 Jet nebulisers are also heavy and bulky (compressor/tubes/nebuliser), and loud when operating.6 Their performance furthermore depends on compressor/ nebuliser pairing, with a large variability in dose delivery depending on their combination, and a residual volume is to be expected.6
SUBOPTIMAL PEAK INSPIRATORY FLOW
Usmani discussed the importance of PIF, its prevalence in patients with COPD, and how patients with sPIF should be managed.
What is Peak Inspiratory Flow and Why is it Important?
PIF refers to the maximal airflow obtained during an inspiratory manoeuvre.5 The delivery of drugs via DPIs depends on PIF, since they require an initial fast inhalation.7,8 The optimal PIF for DPIs is generally ≥60 L/ min or ≥30 L/min, although this depends on the type of device.5 Studies have shown that sPIF is associated with worse COPDrelated symptom burden, and a greater likelihood of COPD-related hospital readmissions.5,7
How Common is Suboptimal
Peak Inspiratory Flow in Patients with Chronic Obstructive Pulmonary Disease?
A study by Mahler et al.9 found that the overall prevalence of sPIF was 44.6% in patients with COPD, who were hospitalised for any cause, and were using at least one DPI at admission.9 In this study, sPIF was defined as <60 and <30 L/min for low-to-
medium high-resistance DPIs and highresistance DPIs, respectively.9
How are Patients with Suboptimal Peak Inspiratory Flow Generally Managed?
In the UK, many patients with severe and very severe COPD and sPIF are still managed with DPIs. However, DPIs require an initial fast inhalation to break down the medicine correctly, so that it reaches far enough down into the lungs.7 Patients with sPIF can therefore struggle with DPIs.
How Should Patients with Suboptimal Peak Inspiratory Flow who Struggle to Inhale Sufficient Medicine with Dry Powder Inhalers Be Managed?
Studies have shown that patients with sPIF do respond to nebulised therapy.5,7 For example, a study by Loh et al.7 demonstrated that in patients with sPIF, all-cause and COPD 30- and 90-day readmission rates were significantly lower for those discharged with nebuliser compared with DPI therapy.7
THE ROLE OF EFFICIENCY IN NEBULISER THERAPY: NOT ALL NEBULISERS ARE THE SAME
Usmani outlined the UK recommendations on nebuliser efficiency, its relevance to patients, and how efficiency can be objectively measured.
What Do UK National Institute for Health and Care Excellence Guidelines Say?
The UK National Institute for Health and Care Excellence (NICE) guidelines recommend using a nebuliser system that is known to be efficient.10
How Important is it That Patients Have Access to Efficient Nebulisers?
Mistakes can occur when using inhaler devices, for example, due to poor hand–breath coordination.11 A meta-analysis of 10
studies including 1,360 patients found that more than three in four adults with COPD used pMDIs incorrectly.11 Patient errors in using pMDIs or DPIs can lead to a poorlycontrolled disease status.12 Nebulisers are an effective alternative to inhalers, and can be prescribed for patients with COPD who are unwilling or unable to use inhaler devices.12 Efficiency is a key factor when selecting a nebuliser system. An efficient nebuliser rapidly delivers a high amount of medication to the lungs, which can result in a positive effect on patient adherence.13
How Can Nebuliser Efficiency be Objectively Measured?
Nebuliser efficiency can be objectively measured using the respirable drug delivery rate (RDDR), which is calculated as the product of respirable fraction (representing the proportion of particles <5 μm) and aerosol output rate, defined by the amount of salbutamol deposited on the inspiratory filter within the first minute of nebulisation. Research by Fischer et al.13 has demonstrated major differences in the efficiency of nebuliser systems.13 The study evaluated 15 types of jet nebuliser systems, which were filled with 2 mL of 0.1% (w/v) salbutamol.13
The nebuliser system with the highest RDDR had a value that was approximately threefold higher than the system with the lowest RDDR.13 PARI devices demonstrated superior performance over other systems (Figure 1).
CASE STUDY
Usmani discussed the case of a patient in the UK who had chronic severe COPD, and was stabilised after using an efficient nebuliser.
While being treated at a previous healthcare centre, the patient had described increasing breathlessness over the last 3–6 months.
The patient had been tried on different inhalers during that period, starting with a pMDI and a spacer, followed by an SMI, and then a DPI with a long-acting β2 agonist, and a long-acting muscarinic antagonist as a combination therapy. The patient had attended due to the lack of
clinical benefit. The healthcare professionals involved in the case had not assessed the patient’s ability to engage with the inhaler device. Instead, they had assumed that the patient was using the device correctly, but the drugs were ineffective. Dosages of the long-acting β2 agonist, or the longacting muscarinic antagonist combination were therefore increased, but with no improvement in efficacy.
At that point, the patient was referred to Usmani’s clinic, where their inhaler technique was assessed. This revealed that the patient had early-onset dementia and poor coordination, which meant they were unable to correctly use a pMDI with a spacer, an SMI, or a DPI. They also had sPIF, which was inhibiting their use of a DPI.
Usmani emphasised that when a patient is unresponsive to treatment, healthcare professionals need to assess their engagement with the inhaler to confirm that they are using the device properly, rather than assuming that the medicine is incorrect, or that the patient is non-adherent or non-compliant.
The patient was switched to an efficient nebuliser with ipratropium and salbutamol, a short-acting muscarinic antagonist, and a short-acting β2 agonist. The prescription was to use the device up to four times a day, an increase from their inhaler prescriptions of once or twice daily. However, it was found that the patient received sufficient drugs to obtain clinical benefit by using the nebuliser just twice a day. In addition to alleviating their breathlessness, nebuliser therapy led to improved abilities to perform activities of daily living. The patient was able to walk around the house longer than previously, and undertake more housework than before, all leading to an overall improvement in quality of life.
Usmani stressed that nebulisers are part of the armamentarium of devices that can be used in the everyday management of patients with COPD, not just in acute situations, with the full portfolio also including pMDIs, pMDIs with a spacer, DPIs, and SMIs.
Figure 1: Mean respirable drug delivery rate across 15 jet nebuliser types.13
140
PARI BOY® Junior (yellow NA)
PARI BOY® Classic/Pro (blue NA)
PARI BOY® Pro/Junior (red NA)
Atomisor® Classic Aerodjinn+ NL9MP
Phillips InnoSpire Elegance
Omron NE C900/COMP AIR Pro
aponorm® Compact Kids
BRM-085II
PARI COMPACT 2
Philips InnoSpire Deluxe Yuwell 403H
Omron C28P/Omron X105 Advanced
Atomisor® Classic Aerodjinn+ 28NLOMU
MPV MicroDrop® Family2
aponorm® Compact PLUS
Error bars indicate 95% confidence interval.
Reproduced with permission from PARI GmbH.
aponorm® Compact Kids: Microlife, Widnau, Switzerland; aponorm® Compact PLUS: Microlife; Atomisor® Classic Aerodjinn+ 28NL0MU: DTF Medical, Saint-Etienne, France; Atomisor® Classic Aerodjinn+ NL9MP: DTF Medical; BRM-085II: Bairui Medicine Co, Guangzhou, China; MVP MicroDrop® Advanced: OMRON Healthcare, Kyoto, Japan; Omron NE C900/COMP AIR Pro: OMRON Healthcare; PARI BOY® Classic/Pro (blue NA): PARI GmbH; PARI BOY® Junior (yellow NA): PARI GmbH; PARI BOY® Pro/Junior (red NA): PARI GmbH; PARI COMPACT2: PARI GmbH; Philips InnoSpire Deluxe: Philips, Amsterdam, the Netherlands; Philips InnoSpire Elegance: Philips; Yuwell 403H: Yuwell, Shanghai, China.
NA: nozzle attachment; RDDR: respirable drug delivery rate.
FUTURE DIRECTIONS AND CONCLUSIONS
Usmani provided his perspectives on the future of nebuliser therapy. and his takehome messages on the use of this therapy in the everyday management of patients with COPD.
What is Your Opinion on the Development and Future Availability of Additional Medications for Nebuliser Treatment of Patients with Chronic Obstructive Pulmonary Disease?
It is an exciting time for nebuliser therapy, with the development of novel drugs and
devices specifically for patients with COPD. Efficient nebulisers have become more portable, with handheld devices that can be battery operated, making them more convenient for the patient to use. Coupled with that, work is underway to deliver inhaled α1 antitrypsin to patients with COPD. α1 deficiency is a genetic disorder that predisposes to COPD, and currently, treatments are administered intravenously. In addition, a phosphodiesterase ¾ combination is currently under the U.S. Food and Drug Administration (FDA) review for approval in the USA, to be given via the nebulised route for patients with COPD. Standalone PDE3 and PDE4 nebulised drugs are also in development.
What is Your Take-Home Message Regarding the Role of Nebuliser Therapy in the Management of Patients with Chronic Obstructive Pulmonary Disease?
As shown in the case, nebulisers are part of the portfolio of devices that can be used in the everyday management of patients with COPD.
The inhalation technique is really important, and nebulisers play a role in helping patients with COPD achieve clinical benefit for the very reason that they may not be able to use other devices.
Biography
References
Another key point is that, often, patients do not realise that they are using a device incorrectly. Instead, they attribute the lack of clinical benefit to ineffective medication. Engaging with patients, and observing how they use a device, will show whether or not there are errors. If the patient is still unable to use a device properly after training, then another device, such as a nebuliser, should be considered.
When choosing a nebuliser, it is important to note that not all devices are the same. An efficient nebuliser will ensure that patients receive a sufficient quantity of medication in the lungs, and make it more likely that they will adhere to the treatment.
Omar S. Usmani
Omar S. Usmani, MBBS, PhD, FHEA, FRCP, FERS, is a Professor of Respiratory Medicine and Consultant Physician at the National Heart and Lung Institute (NHLI), Imperial College London, UK. He serves as the Co-Deputy Director of the Phase 1 Imperial MBBS Programme, Clinical Lead for the CSI MBBS program, Director of the Imperial College Respiratory Research Unit (ICRRU) at St. Mary's Hospital, and Head of Assembly 5 - Asthma, COPD, Chronic Cough at the European Respiratory Society (ERS). His expertise in respiratory diseases has earned him fellowships from the Higher Education Academy (FHEA) and the Royal College of Physicians (FRCP).
1. Chinese College of Emergency Physicians (CCEP), Emergency Committee of PLA, Beijing Society for Emergency Medicine, and Chinese Emergency Medicine. Expert consensus on nebulization therapy in pre-hospital and in-hospital emergency care. Ann Transl Med. 2019;7(18):487.
2. Katiyar SK et al. Indian guidelines on nebulization therapy. Indian J Tuberc. 2022;69(Suppl 1):S1-191.
3. Dhand R et al. The role of nebulized therapy in the management of COPD: evidence and recommendations. COPD. 2012;9(1):58-72.
4. Barjaktarevic IZ, Milstone AP. Nebulized therapies in COPD: past, present, and the future. Int J Chron Obstruct Pulmon Dis. 2020;15:1665-77.
5. Ghosh S et al. Peak inspiratory flow rate in chronic obstructive pulmonary disease: implications for dry powder
inhalers. J Aerosol Med Pulm Drug Deliv. 2017;30(6):381-7.
6. Pritchard JN et al. Mesh nebulizers have become the first choice for new nebulized pharmaceutical drug developments. Ther Deliv. 2018;9(2): 121-36.
7. Loh CH et al. Suboptimal inspiratory flow rates are associated with chronic obstructive pulmonary disease and allcause readmissions. Ann Am Thorac Soc. 2017;14(8):1305-11.
8. Dhand R et al. Considerations for optimal inhaler device selection in chronic obstructive pulmonary disease. Cleve Clin J Med. 2018;85 (2 Suppl 1):S19-27.
9. Mahler DA et al. High prevalence of suboptimal peak inspiratory flow in hospitalized patients with COPD: a real-world study. Chronic Obstr Pulm Dis. 2022;9(3):427-38.
10. National Institute for Health and Care Excellence (NICE). Chronic obstructive pulmonary disease in over 16s: diagnosis and management. 2019. Available at: https://www.nice.org.uk/ guidance/ng115/resources/chronicobstructive-pulmonary-disease-inover-16s-diagnosis-and-managementpdf-66141600098245. Last accessed: 6 February 2024.
11. Cho-Reyes S et al. Inhalation technique errors with metered-dose inhalers among patients with obstructive lung diseases: a systematic review and meta-analysis of U.S. studies. Chronic Obstr Pulm Dis. 2019;6(3):267-80.
12. Rogliani P et al. Optimizing drug delivery in COPD: the role of inhaler devices. Respir Med. 2017;124:6-14.
13. Fischer R et al. Efficiency assessment of 15 nebuliser systems by the respirable drug delivery rate: a comparable quality parameter. EMJ Respir. 2023;11(1):69-73.
Enhancing Treatment Success in Osteoporosis: Optimising the Use of Teriparatide
2. Orthopedic and Traumatology Surgery Department, Julius-Maximilians-Universität, Würzburg, Germany
Disclosure:
Casado has received funding for travel or speaker fees from Amgen, Stada, Gedeon Richter, GP Pharm, Theramex, Rubió, and UCB. Seefried has received honoraria for lectures and advice from AstraZeneca, Alexion, Amgen, BioMarin, Chiesi, Gedeon Richter, GlaxoSmithKline, Inozyme, Ipsen, Kyowa Kirin, medi, STADA, Theramex, and UCB; and funding for scientific projects to the institution from AstraZeneca, Alexion, Chiesi, Kyowa Kirin, and Novartis.
Acknowledgements: Medical writing assistance was provided by Amanda Barrell, Brighton, UK.
Disclaimer: The views and opinions expressed in this article belong solely to the named interviewees, and do not necessarily reflect those of STADA AG.
Keywords: Bone mineral density (BMD), fracture, osteoporosis, sequential therapy, teriparatide.
Support: The publication of this article was funded by STADA AG. STADA AG suggested the topic and authors, and carried out full medical approval on all materials to ensure compliance with regulations. The sponsorship fee included honoraria for the authors.
Interview Summary
Osteoporosis, a chronic metabolic bone disease affecting over 200 million people globally, is characterised by low bone mass and microarchitectural deterioration, leading to an increased risk of fractures. The prevalence and associated healthcare costs of osteoporosis are substantial, with an estimated 27.6 million individuals in the European Union (EU) living with the condition in 2010. Fractures, particularly vertebral and hip fractures, are linked to reduced health-related quality of life and increased mortality risk.
While several efficacious osteoporosis treatments are available, none are suitable for permanent use. Instead, clinicians employ a sequential therapy approach in which patients transition between treatments, based on their individual risk factors. Here, Enrique Casado, rheumatologist at Parc Taulí Hospital Universitari, Sabadell, Spain; and Lothar Seefried, orthopaedic surgeon at Julius-Maximilians-Universität, Würzburg, Germany, explain the importance of early identification of at-risk individuals and personalised management strategies. They look at the evolving understanding of the osteoanabolic teriparatide, and what the last two decades of data tell us about its risks and benefits. The growing evidence base, they say, has
PHARMA
PARTNERSHIP
influenced regulatory and clinical changes regarding the use of teriparatide. They emphasise the agent’s safety profile, and talk about its potential to reduce fracture risk within a short timeframe; as well as its place in sequential therapy.
OSTEOPOROSIS OVERVIEW
Osteoporosis is the most common chronic metabolic bone disease globally. Characterised by low bone mass, deterioration of bone tissue, and disruption of the bone microarchitecture, it affects more than 200 million people.1
The bone remodelling cycle involves the removal of older bone and its replacement with new bone. Via this process, which involves the absorption of obsolete and/ or physiologically useless bone and reconstruction of viable and mechanically competent bone, bone tissue is continuously restructured. If the resorption rate is higher than the formation rate, or the process is not well orchestrated, it leads to bone loss and reduced bone mass or compromised bone quality, predisposing people to an increased risk of fractures.1
Hereditary traits, age, and lifestyle factors all impact on the risk of osteoporosis and fractures. The most frequent risk factors include menopause, and the associated loss of bone-protective sex hormones; ageing and the natural ‘slowing down’ of bone remodelling; and the use of glucocorticoids, which initially increase bone loss as well as compromising bone formation in the long term.1 Other common risk factors for osteoporosis and fracture include excessive alcohol consumption; smoking; and low calcium intake, either through diet or disease-related malabsorption (potentially due to inflammatory diseases such as rheumatoid arthritis).1
In the EU, an estimated 32 million people, or 5.6% of the total population, were living with osteoporosis in 2019. Of these, 25.5 million were females and 6.5 million were males, and together they experienced 4.3 million fragility fractures each year. Such fractures can be painful and cause functional disability, as well as reduced health-related
quality of life and increased risk of mortality. Every year, nearly 250,000 people in Europe die as a direct consequence of hip or spine fractures.2
Casado said: “As a rheumatologist, I mainly see patients with vertebral fractures. They suffer from pain, from deformities of the spine, and some of them even can have pulmonary restrictions. However, hip fracture is the most dangerous fracture. These patients tend to be older and they can suffer severe complications or even can die in the first year following the fracture.3 Those who, fortunately, survive can suffer from health-related quality of life impairment because they can’t work or do the things they used to do, and may need a wheelchair or other assistance.”
In addition, fractures are associated with high healthcare costs. A 2020 systematic review found the average direct annual cost of treating osteoporotic fractures in Canada, Europe, and the USA alone to be between 5,000–6,500 billion USD. This figure does not include indirect costs, such as those related to disability and loss of productivity.4
ACT EARLY. REDUCE FRACTURES.
With several osteoporosis therapies available, clinicians have the opportunity to identify and manage the condition, and help prevent fractures before they occur, noted Seefried. “We now have agents and regimens that significantly reduce fracture risk, and the prognosis is really good, much better than what it was in the past. There is no need for people to be scared or concerned because we can anticipate fracture risk and manage osteoporosis so that patients can live a normal and, at least with regard to osteoporosis, untroubled life.” Identifying patients before they experience fractures “is the most challenging and
intriguing aspect” of osteoporosis care, he went on, adding that there is a “large degree of individuality” around the risk factors.
Casado and Seefried agreed that all people deemed to be at risk of developing or having osteoporosis should undergo bone mineral density (BMD) testing and individualised fracture risk assessment, which may include the use of established tools, such as the Fracture Risk Assessment Tool (FRAX), according to applicable guidelines.5 The next step would be the creation of an individualised management strategy, tailored to the patient’s age and risk of fracture.
The various types of available osteoporosis treatment include anabolic agents and antiresorptive drugs. Anabolic agents, such as teriparatide, abaloparatide, and romosozumab, increase bone formation over time by targeting osteoblasts, while antiresorptive drugs, including bisphosphonates, selective oestrogen receptor modulators, and denosumab, primarily reduce bone resorption.5 Unlike in most chronic diseases, however, the use of approved treatments tends to be limited to a single drug at a fixed dose and frequency.6 As such, international guidelines recommend sequential therapy, or initiating treatment with one class of drug before transitioning to another, to capitalise on their respective benefits.5
As the different agents have different modes of action, and some can impact on the efficacy of those that follow,5 it is important to plan the sequence early, said Casado and Seefried. “The long-term perspective is critical,” observed Seefried. “We have a lot of treatment options for osteoporosis, but none are suitable to continue for 25 years. We have to change the medication and the drug regimen over time, and that means establishing individualised sequences of treatment.” His aim when treating patients with osteoporosis, he explained, was to keep the bone resilient and avoid supressed bone turnover, so that treatment could still be adjusted 10 or 15 years in the future.
RISK-BASED SEQUENTIAL THERAPY PLANNING
Current clinical guidelines recommend using individual fracture risk as the starting point for sequential therapy.5 Following diagnosis of osteoporosis, clinicians can use various algorithms to approximate anticipated future fracture risk as much as possible. It has become common practice to not only define thresholds for treatment initiation, but to further differentiate between patients at high and very high fracture risk, the doctors explained.5 Fracture risk assessment tools, such as FRAX, Qfracture, Garvan, and others, consider a variety of risk factors for fracture, like age and sex, BMD, prior fractures, smoking and alcohol use, as well as various secondary causes of osteoporosis.1 People who have particularly critical risk factors, such as a recent (within 1–2 years) sentinel fracture, or a particularly low BMD and glucocorticoid intake, are considered to be at imminent risk of subsequent fracture.7 Internationally, the majority of current guidelines recommend that patients at very high or imminent risk of fracture be initiated on an osteoanabolic agent, such as the fast-acting teriparatide, followed by an antiresorptive medication.5
“We have to look at the individual risk, which may be high, very high, or at the borderline of high to very high, to guide the individual treatment. Where there are indicators of elevated imminent risk, we have to start a treatment that is not just effective over time, but that also has rapid efficacy so as to preclude and to limit that immediate risk,” said Seefried.
However, while risk is an important factor, it is not the sole consideration. As Casado explained: “We have to know the characteristics of the patients: not just their age but also their comorbidities.” For example, bisphosphonates should be used selectively and with caution in people living with chronic kidney disease,8 and needle phobia often precludes the use of subcutaneous treatments.
TERIPARATIDE
Teriparatide is a parathyroid hormone analogue. The osteoanabolic increases the formation of new bone tissue.9 It initiates bone growth in days or weeks by stimulating osteoblast activity, decreasing osteocyte sclerostin expression, and inducing osteoblastogenesis. It also increases osteoblast receptor activator of nuclear factor κΒ ligand production, activating osteoclasts and resorbing bone.10
Casado said: “Teriparatide is very fast acting. It increases BMD and changes bone microarchitecture, increasing the cortical and trabecular thickness, offering benefit in a very short time.”11 Seefried agreed, calling it a “fast approach” to mitigating increased fracture risk at short notice. “Teriparatide offers the option of an enhanced remodelling process, which includes the reduction of deteriorated old bone, replacing it with newly structured bone, which is more sustainable, and that makes a good starting point for subsequent treatment options.”11
The use of teriparatide has evolved since it was first approved by the U.S. Food and Drug Administration (FDA) in 2002,12 and then the European Medical Agency (EMA) the following year.13 These approvals were based on Phase III clinical trials that were originally designed to monitor 36 months of treatment, but which were halted early after preclinical studies found an increased risk of osteosarcoma in rats treated with high doses for almost their full lifespan. The trial data, which covered an average 19-month treatment course, however, showed efficacy and tolerability. The FDA approved teriparatide for the treatment of osteoporosis in patients at very high risk for fracture, with a black box warning regarding the potential risk of osteosarcoma, and a 2-year lifetime limitation of use. Manufacturers were also required to assess for risk of osteosarcoma in humans.14
Since then, the scientific community has amassed more than two decades of data that provide a much better understanding of the risks and benefits of teriparatide. For example, two large cohort studies
that linked pharmacy claims data with data from cancer registries showed no increase in osteosarcoma among patients using teriparatide when compared with unexposed groups, or against the expected background incidence of the disease.14 As such, in 2020 the FDA removed the black box warning for osteosarcoma.4,15
“After 20 years of teriparatide use, and a very accurate system of pharmacovigilance, there is no evidence of these risks. The risk of tumours, including osteosarcoma, in rats does not appear to be a concern in humans,” said Casado. Seefried added that, while there was “no need to be concerned,” this is an issue that often looms large in the minds of patients. “It’s interesting because when you instruct patients about the treatment and they look it up themselves, osteosarcoma is always the first question they bring up,” he said. It is important, then, for clinicians to mitigate these concerns, and explain that the vast majority of patients are “really doing well on the treatment” and “hardly feel any side effects,” he said. Casado explained that some of his patients on teriparatide did experience minor side effects, such as headache or leg cramps.16 In his experience, these effects tend to occur at the start of the treatment course, and normally decrease over time. “Some patients do decide to change treatment because of this, but not many,” he said. The doctors also said that while some patients do develop hypercalciuria, it was relatively rare, and was not often clinically relevant.16
Turning to the increase in maximum treatment length, both doctors agreed that a full 24-month course of treatment was the most beneficial. Studies have shown ‘highly significant’ increases in BMD between 18–24 months of therapy,17 and a 2015 review of the available evidence found that patient outcomes and skeletal health appeared to be improved by the full 24-month continuous course, as opposed to 18 months of treatment.9 The review authors noted that the biochemical and histological data suggest ongoing bone formation throughout the 24 months, resulting in increases in bone mass and strength, and a decreased fracture risk. While the
review highlighted that no randomised controlled trial comparing the efficacy of 18- and 24-month treatment has yet been performed, “the available information suggests that the full 24-month treatment course is important to achieve the best clinical outcomes.”9 Casado said: “During the first months of treatment, patients can show a decrease in hip BMD, because teriparatide can lead to some degree of cortical porosity. However, after the first 12 months of treatment, this porosity is filled with new bone that will go on to become mineralised, reflected in BMD gains.”9
TERIPARATIDE AND SEQUENCING THERAPIES
The literature has shown the best outcomes are achieved when teriparatide is used before antiresorptives.5,17,18 As such, clinical practice guidelines recommend osteoanabolics as a first-line treatment in very high-risk patients.5
“Teriparatide is a perfect starting point to enhance the remodelling process initially, and rebuild and restructure the bone before continuing with antiresorptives,” said Seefried. Casado added that, based on the science, the best approach would be teriparatide followed by antiresorptives. He said: “To treat with osteoanabolics, particularly teriparatide, only after antiresorptive treatment failure, i.e., when the patient has suffered one or two fractures in high- or very high-risk patients, is simply too late.”
In routine practice, however, antiresorptives are more readily available due to their lower cost, while their oral administration means that some patients find them easier to use. Casado went on: “We cannot stay with the idea we can only use osteoanabolics as a first-line in very high-risk patients, or after antiresorptives when the patient experiences treatment failure or an increase in fracture risk.” It is important to note, though, that starting therapy with antiresorptives does not preclude the use of teriparatide later on. Clinicians should be aware that pre-treatment with antiresorptives will merely ‘blunt’
teriparatide’s effect on BMD, but not reduce its antifracture effect.17,18
“When we have to start a patient on an antiresorptive for whatever reason,” said Seefried, “we should always keep in mind that sooner or later they will require an osteoanabolic, so we should always have an approach that facilitates switching.” Using a short half-life bisphosphonate, for example, may limit the antiresorptive activity that inhibits teriparatide efficacy. This is one way to “pave the way for osteoanabolic treatment later on,” he observed.19
Particular care needs to be taken when switching from denosumab, due to a decrease in BMD, and the risk of rebound vertebral fracture after treatment discontinuation.20 “The longer patients are on denosumab, the more challenging it becomes to transition them to teriparatide,” said Seefried.21 “No one should be so selfconfident as to claim that this was easygoing, and wouldn’t require diligent followup. There will always be a certain risk, which brings us back to the importance of planning the sequence early.”
TERIPARATIDE CONTRAINDICATIONS
Asked about teriparatide contraindications, Seefried said it was important to be mindful of the patient’s individual history and characteristics. Clinicians should, for example, be cautious in recommending the agent to patients who have experienced malignant disease, and previous radiation therapy is a clearly specified contraindication. Despite there being no evidence that the osteosarcoma risk seen in preclinical trials applies to humans, “we should be smart enough not to expose these patients at increased risk to teriparatide,” he said. Any laboratory findings suggesting another metabolic bone condition, such as hyperparathyroidism or Paget’s bone disease, should be evaluated mindfully before considering or initiating teriparatide.16
Casado said that clinicians also need to look at other patient characteristics. Teriparatide
is administered as a subcutaneous daily injection and requires self-administration, so may not be suitable for people with needle phobia, or those with limited dexterity, for example.
Although older age is not a formal contraindication, teriparatide is often avoided for these patients in routine practice. Both doctors, however, said they would not withhold the treatment on the basis of age alone. “I’m very open to prescribing osteoanabolics, specifically teriparatide, to elderly patients, even if they are 85 or older. If they are self-sustained and want to keep their lifestyle, there’s no reason to deny them teriparatide,” said Seefried. These are very high-risk patients, with the highest risk of fracture, said Casado. “It means that the number needed to treat to reduce fragility fracture risk is lower than in any other group,” he went on. “If they have a fracture risk that justifies teriparatide, of course it is smart to give them this 2-year treatment, improve their bone quality, make it sustainable, and then continue with antiresorptives for the rest of their lifetime.”
BIOSIMILARS, HEALTH ECONOMICS, AND FUTURE DIRECTIONS
Casado and Seefried explained the decision to restrict the use of teriparatide as a firstline therapy for very high-risk patients had, at least in part, been influenced by its cost.
“The reasons for it being used so cautiously and conservatively may have to do with costs,” said Seefried. “I consider this highly relevant, because the cost difference between teriparatide and the antiresorptives, as compared with the costs of inappropriate treatment and the fractures that can happen otherwise, is negligible.”
Casado agreed, adding that changing matrix architecture and increasing bone strength can avoid future fractures and the associated comorbidities. “This is good for patients, but it is also good for governments and administrations because, by avoiding fractures, we can also save money.”
Both doctors said they used teriparatide early in the sequence, with Seefried adding that, “from a scientific perspective, everyone would think that this is appropriate.” While guidelines are now moving in this direction, “they are not there yet,” and though the FDA has lifted the once-per-lifetime limitation on teriparatide treatment, the European label remains more restrictive, said Seefried.
The emergence of biosimilars in recent years has widened access, the doctors went on. Biosimilars are biological medicines which are highly similar to their ‘reference medicine’, or a previously approved biological medicine. Due to the ‘living’ nature of such products, there may be minor natural variability between the biosimilar and the reference medicine, but regulators have strict controls in place to ensure there are no clinically meaningful differences.22
Biosimilars are approved according to the same standards of pharmaceutical quality, safety, and efficacy that apply to all biological medicines. Biosimilar manufacturers are obliged to demonstrate that their products have a high similarity, in terms of structure, biological activity and efficacy, safety, and immunogenicity profile, to the reference medicine.22
“Biosimilars have changed our clinical practice, and I think some guidelines have changed thanks to them. They have lowered the price, and expanded the pool of patients who can benefit from teriparatide,” said Casado. Seefried described biosimilar teriparatide as a “tremendous step forward.” “Sick funds, funding agencies, and insurance companies all around the globe have developed an open-minded approach to patients being treated with teriparatide since we have had biosimilars on the market, and that is being reflected in the common practice,” he said.
The updated guidelines, allowing for the use of osteoanabolics in all very high-risk patients from diagnosis, “is a very beneficial development,” he went on, concluding that “everyone should be made aware of this opportunity to improve patient care.”
References
1. Sözen T et al. An overview and management of osteoporosis. Eur J Rheumatol. 2017;4(1):46-56.
2. International Osteoporosis Foundation (IOF). Scorecard for osteoporosis in Europe: Scope 2021 summary report. 2021. Available at: https:// www.osteoporosis.foundation/ sites/iofbonehealth/files/2022-01/ SCOPE%20Summary%20Report.pdf. Last accessed: 9 April 2024.
3. Clynes MA et al. The epidemiology of osteoporosis. Br Med Bull. 2020;133(1):105-17.
4. Kemmak AR et al. Economic burden of osteoporosis in the world: a systematic review. Med J Islam Repub Iran. 2020;34:154.
5. Mondo I et al. Using sequential pharmacotherapy for the treatment of osteoporosis: an update of the literature. Expert Opin Pharmacother. 2023;24(18): 2175-86.
6. Leder BZ et al. Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA-Switch study): extension of a randomised controlled trial. Lancet. 2015;386(9999):1147-55.
7. Javaid MK et al. Assessment and management of imminent fracture risk in the setting of the fracture liaison service. Osteoporos Int. 2022;33(6):1185-9.
8. Toussaint ND et al. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc
Nephrol. 2009;4(1):221-33.
9. Lindsay R et al. Teriparatide for osteoporosis: importance of the full course. Osteoporos Int. 2016;27(8):2395-410.
10. Sauhta R et al. The sequential therapy in osteoporosis. Indian J Orthop. 2023;57(Suppl 1):150-62.
11. Guelman R et al. Effect of teriparatide on bone mineral density and bone markers in real-life: Argentine experience. Int J Endocrinol. 2023;DOI:10.1155/2023/9355672.
12. U.S. Food and Drug Administration (FDA). Drug approval package: forteo [teriparatide (rDNA origin)] injection. 2002. Available at: https://www. accessdata.fda.gov/drugsatfda_ docs/nda/2002/21-318_Forteo. cfm#:~:text=Approval. Last accessed: 25 March 2024.
13. European Medicines Agency (EMA). Forsteo. 2022. Available at: https:// www.ema.europa.eu/en/medicines/ human/EPAR/forsteo. Last accessed: 25 March 2024.
14. Krege JH et al. Teriparatide and osteosarcoma risk: history, science, elimination of boxed warning, and other label updates. JBMR Plus. 2022;6(9):e10665.
15. European Medicines Agency (EMA). Forsteo: procedural steps taken and scientific information after the authorisation. 2022. Available at: https://www.ema.europa.eu/en/ documents/procedural-steps-after/ forsteo-epar-procedural-steps-takenand-scientific-information-afterauthorisation_en.pdf. Last accessed:
25 March 2024.
16. European Medicines Agency (EMA). Movymia, INN-teriparatide. Annex I summary of product characteristics. 2021. Available at: https://www.ema. europa.eu/en/documents/productinformation/movymia-epar-productinformation_en.pdf.
Last accessed: 25 March 2024.
17. Obermayer‐Pietsch BM et al. Effects of two years of daily teriparatide treatment on BMD in postmenopausal women with severe osteoporosis with and without prior antiresorptive treatment. J Bone Miner Res. 2008;23(10):1591-600.
18. Miller PD et al. Teriparatide: label changes and identifying patients for long-term use. Cleve Clin J Med. 2021;88(9):489-93.
19. Leder BZ. Optimizing sequential and combined anabolic and antiresorptive osteoporosis therapy. JBMR Plus. 2018;2(2):62-8.
20. Anastasilakis AD et al. Denosumab discontinuation and the rebound phenomenon: a narrative review. J Clin Med. 2021;10(1):152.
21. Tay WL, Tay D. Discontinuing denosumab: Can it be done safely? A review of the literature. Endocrinol Metab (Seoul). 2022;37(2):183-94.
22. European Medicines Agency (EMA). Biosimilars in the EU: information guide for healthcare professionals. 2023. Available at: https://www.ema.europa. eu/en/documents/leaflet/biosimilarseu-information-guide-healthcareprofessionals_en.pdf.
Last accessed: 3 April 2024.
Interviews
EMJ spoke with Smeeta Sinha and Nina Gold, who shared their experiences and insights as experts in rare diseases. The topics explored in these interviews include calciphylaxis and the challenges associated with performing rare disease research, and genomic newborn screening and its potential impact on rare genetic disease outcomes.
Featuring Smeeta Sinha and Nina Gold
Smeeta Sinha
National Clinical Director for Renal Medicine, NHS England, UK; Consultant Nephrologist, Northern Care Alliance NHS Foundation Trust, Salford, UK; Honorary Professor, University of Manchester, UK; Visiting Professor, Manchester Metropolitan University, UK
Citation:
Q1
Over 25% of adults, and over half of all children with kidney failure have a rare disease
What initially attracted you to a career in nephrology, and more specifically in rare renal diseases?
I qualified as a doctor in 2000, and in my first year I worked on a renal transplant unit for 6 months, which sparked my interest in nephrology. Obviously, the surgical side was interesting, but what really intrigued me was the fact that the function of these organs can be replaced. There was also so much variety in the specialty, including immunology, and managing people with long term-conditions. It was the wide scope of practice, and the fact that I didn't need to be shoehorned into one particular thing. It was also the enthusiasm of individuals, as everyone I met who did renal seemed to love it. And I can’t say I regret it; I love my job, so it was definitely a wise choice.
Q2 You are recognised as an international leader in the field of calciphylaxis. What sparked your interest in this area of nephrology?
I think it came from two avenues. When I was going through my
training to be a consultant nephrologist, I wanted to take time to do something different to complement my practice. So, I went back to university to do a laboratory-based PhD in basic science, looking at smooth muscle cells and how they calcified.
While this research had clinical relevance, as individuals with kidney disease often suffer from heart and vascular issues due to vessel calcification, applying my PhD findings in practice wasn't quite as easy as I thought. Then, sadly, I came across a patient who had calciphylaxis, a rare condition that didn’t have any treatments or cures. In the organisation I was working at, they had established a registry to try and collect data on people with this rare disease. Since so few people are affected by it, you can’t find out what's happening very quickly, which makes it hard to do research, and understand the disease. It was an opportunity to combine something that I had seen somebody suffer with, and try to do something better for them with research. Calciphylaxis is a disorder of vascular calcification, so it allowed me to bring together my PhD and
experience with patients, and gave me the opportunity to do translational research.
I do think we need to be looking at rare diseases as a whole, not just calciphylaxis. Although rare diseases are, by definition, rare, if you add them all up, then over 25% of adults, and over half of all children with kidney failure have a rare disease.1 To complicate matters, patients don't always get access to the treatments or experts that may help.
Q3
You are currently actively involved in calciphylaxis research. Could you tell us if there are any exciting developments in the field?
Calciphylaxis research has changed a lot over the course of the last 15 years. Back then, it was very much about trying to collect data, and encouraging patients who had calciphylaxis to share their experiences, allow us to collect samples, and follow their progress. That was done via the UK Calciphylaxis Registry, or the UK Calciphylaxis Study that has
been running for over a decade; there are similar studies running in Australia and the USA. That allowed us to collect samples, and to do observational research. Simultaneously, there were basic scientists in laboratories who were trying to understand the processes that cause calciphylaxis at a cellular level, and then starting to test potential treatments for it.
In the last 5 years, those therapies have emerged from the lab. They have gone through basic laboratory testing, and are now being evaluated in people. So, we've moved from observational research to doing interventional studies with new therapies. The global, multicentred, randomised controlled trial, the CALCIPHYX study,2 reported its headline data last November, and I'm sure there will be other studies now that one has been completed.
It's hard to do studies in rare diseases, because there are not enough patients, but I think what the CALCIPHYX study2 showed was that no disease is
too rare; trials can be done, and we shouldn't give up on doing them. My take-home message would be that we've moved from observational studies to interventional studies with new therapies, and the trials are feasible, so we should continue to do them.
It is also good to be able to offer patients something. In the early part of my career, it was very much about pain management, and trying to look after their wounds and dialysis. Patients would ask me, “What's going to happen to me?” I'd have to be honest, and try and explain that, sadly, many people don’t get better, and will die from the condition, but we would try to manage their pain and wounds. At least now, whilst I still need to tell them that, I can say, “We have a clinical trial; I don’t know what it will show, but there’s something I can offer you.” Hopefully, in 10 years’ time, I'll be having a different conversation, where I could offer them treatments that could cure, or at least significantly improve, their condition.
Q4
In July 2023, you received funding from Kidney Research UK and Kidney Wales to gain a deeper understanding of the lived experiences of patients with calciphylaxis, their family members, and caregivers. Is this research now underway, and have any key themes emerged from the data collected so far?
This is a really important study. I've talked about smooth muscle cells, epidemiology, observational studies, and investigational trials of new therapies, but there are patients who are experiencing the disease throughout this. The research community is rightly placing a much greater emphasis on the importance of patients’ experiences and outcomes, as reported by them. We shouldn’t be congratulating ourselves if the numbers on a lab report get better, but the patient doesn’t feel any better. Patient groups and voices are now actively involved in designing research.
The research grant was funded by Kidney Research UK, to whom we are very grateful, but the idea was very much driven by our patients, their caregivers, and a fantastic researcher called Sharon Hewish in Exeter, UK, who is a postdoctoral dietitian. Again, this shows that it's not just medical doctors and university-based scientists who can do research. All healthcare professionals can shape research, and deliver it.
Hewish is now interviewing patients and their carers. Hopefully we'll have some data by 2025, which will then shape the information we give to patients, and this can inform future trials so that they're patient-focused. The other thing about rare disease patient advocates is that they are often the experts in their
disease, so they have a huge amount of information to impart to us as researchers, as well as clinicians. It would be a real missed opportunity not to listen to our experts.
Q5As a rare disease expert, what have been the main challenges associated with conducting calciphylaxis research, and do you see any additional barriers impairing future research?
In addition to the small numbers of patients with calciphylaxis, people make assumptions about which treatments do and don’t work, and some of those treatments don’t have an evidence base to support them. However, I do understand why people use them, they want to do something good, and want to try anything based on biological plausibility. However, we now live in a scenario where we can do clinical trials, so I think one of the barriers is clinicians, or researchers not wanting to put their patient forward for a trial. We need to have a research-positive approach, which a lot of my colleagues and I have, but I don't think that's necessarily universal. We can learn from oncology, where clinical trials are embedded into routine practice. Patients will go through their standard treatment, and if they're not responding, or if they're slightly different, they will go into a trial as part of their treatment. I would like to see this in rare diseases, and in kidney research in general.
In the calciphylaxis field, I would like to see clinicians saying, “This is your disease, this is what we're going to do, and a clinical trial is one of the things we can offer you.” If we embed that, it gives patients more options and more choice, and hopefully leads to more treatments for other patients in the future.
Q6 What advances have been made in the understanding and treatment of calciphylaxis over the course of your career thus far?
In addition to the evolution from observational to lab-based data, scientists around the world have been busy trying to understand the pathophysiology of vascular calcification as a whole. Vascular calcification can present with coronary artery calcification and cardiovascular disease, or peripheral artery disease calcification, but in skin, it's calciphylaxis. Our understanding of what makes vessels calcify has increased significantly, and there are a whole range of pathways that are involved. We now know it's not just calcium and phosphate getting stuck in the blood vessels, it's actually a process whereby the cells are changing and becoming more bone-like, and depositing calcium and phosphate. We now understand the pathophysiology of vascular calcification far better than we did, which means we have potential targets for therapeutic intervention.
Q7 What further research do you feel is necessary to improve outcomes for patients with calciphylaxis?
Sadly, despite the fact that kidney disease affects so many people, it doesn't have the same degree of research investment as you’d expect, not just in the UK, but globally. Investment into kidney research as a whole is disproportionately low, so we're starting off at a low baseline.
When we get into rare renal diseases, it’s even lower. We have looked to cardiovascular research groups to try and help us, but if you think about what happens in cancer and
cardiovascular disease, you have academic centres, and the pharmaceutical industry investing and trying to improve things. I would really like to see industry, as well as policymakers and governments, really prioritising kidney disease research as a whole. We know there's a World Health Organization (WHO) report that has indicated that kidney disease will be the fifth biggest cause of lives lost. We need more investment in kidney research, and then hopefully that will come to rare renal diseases, and looking at pathways and therapeutics that we need to move into trials.
Q8
You are the Rare Disease Group (RDG) Lead for the UK Calciphylaxis Rare Diseases Group. What are the aims of this group, and what does your role as the RDG Lead entail?
The UK has the National Registry of Rare Kidney Diseases (RaDaR). This is an initiative of the UK Kidney Association, which is our professional society, and the UK Renal Registry. The UK Renal Registry collects data from UK renal units every year. This enables us to audit how we're doing, compare against each other, and look for opportunities to improve. The establishment of RaDaR was an opportunity to look at rare diseases across the UK systematically. On the 1st of September 2022, there were over 29,500 UK patients recruited into RaDaR, from 107 sites.
It's huge, and there's nothing like it in the world. RaDaR has various rare disease groups, and calciphylaxis was accepted by the RaDaR group as a rare disease. This allowed us to establish a rare disease group, which I lead. It's made up of doctors, dieticians such as Hewish, and also patients
and laboratory scientists. It's an open group for people who have an interest in rare diseases, but specifically for calciphylaxis. Currently, RaDaR for calciphylaxis is being used to collect data and understand the disease burden in the UK, but in the future, it could allow us to find patients and give them an opportunity to participate in trials, and hopefully receive treatments.
Q9
What other rare renal diseases do you think require a greater research focus or increased awareness amongst the wider spectrum of healthcare professionals?
Not just rare diseases, but kidney disease as a whole needs more awareness. Historically, it is poorly taught in medical school, and is sometimes seen as being complicated. It affects a lot of people, so we need to raise awareness, right from medical school, and all the way through training, so that people detect the kidney disease in the first place. That has been our priority nationally, to raise awareness of kidney disease as a whole, whether that be chronic kidney disease that develops slowly, or the acute type (acute kidney injury).
I believe that once we establish a strong foundation in our understanding of common diseases, it's crucial to shine a spotlight on rare diseases as well. These conditions often fall within the realm of nephrology healthcare professionals, given their rarity. However, raising awareness of rare diseases within the nephrology community is a significant step forward. From what I've observed, the renal community in the UK is proactive in considering and addressing rare renal diseases.
The other thing that is helping us identify rare disease is our increasing awareness of the importance of our genes. The UK is a leader in genomic testing, and there is a specific renal section in our UK National genomic test directory.3 This means that frontline nephrologists can order genetic tests to identify rare diseases, and this is all available within the National Health Service (NHS). I think having early access to genomic testing means we should be able to diagnose patients with certain rare diseases a lot earlier.
Q10Do you think there is a role for artificial intelligence (AI) in improving earlier diagnosis and awareness of rare renal diseases?
We can't ignore AI, can we? I think the short answer is, there is probably going to be a role for some sort of AI and machine learning in renal medicine.
Research groups are looking into it, but this is harder in rare disease, because AI needs a large amount of data to learn effectively, and rare diseases often lack sufficient data. With this in mind, I suspect AI will largely focus on diseases which have clear characteristics in imaging or pathology. It is still a very early stage for AI in the renal world.
In the future, it could allow us to find patients and give them an opportunity to participate in trials
Q11Toconclude, are there any innovations or developments on the horizon, in either the calciphylaxis or rare renal disease space, that you are excited about?
The way we are delivering trials is exciting. For many years, the standard trial design involved half of the patients receiving a drug, and the other half receiving a placebo, or dummy drug, in placebo-controlled, randomised trials. COVID-19 did teach us the value of doing trials in a slightly different and more pragmatic way, which is really relevant for rare diseases. A good example of a pragmatic trial is a ‘platform trial’, which enables you to test multiple interventions in a given patient, over a period of time. As something is proven
References
1. Wühl E et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA–EDTA Registry. Nephrol Dial Transplant. 2014;29(Suppl 4):iv1-8.
2. Sanfit Therapeutics S. A. Phase 3
to be effective, it becomes standard care, and is offered to everyone. The platform trial also enables new interventions to be introduced as they become ready for testing. The BEAT-Calci trial4 is a platform trial that is running in Australia and New Zealand for calciphylaxis, and we hope to be able to bring that to the UK in the future. Sticking with trials, the way we evaluate trials and the endpoints are changing a lot. Again, we're moving towards pragmatic trials that are easier to deliver, but still give us an answer, and I think we'll see more pragmatic calciphylaxis trials in the future. The big development has been the CALCIPHYX trial,2 which showed us that randomised controlled trials in people with calciphylaxis are possible in the first place.
Transitioning from clinical trials into actual treatments or therapies, we are getting better at broadening our approach from solely relying on medications. In the case of calciphylaxis, recent efforts have explored alternative avenues, such as wound treatments, including topical therapies, and enhancing the efficiency of dialysis itself. It is important to widen our scope beyond medications, and to consider factors like wound care, dialysis optimisation, and even patient-centred aspects, such as pain management, as these directly impact the wellbeing and concerns of patients.
study of SNF472 for calciphylaxis (Calciphyx). NCT04195906. https:// clinicaltrials.gov/study/NCT04195906.
3. National Health Service (NHS) England. National genomic test directory. 2024. Available at: https:// www.england.nhs.uk/publication/ national-genomic-test-directories/. Last accessed: 3 February 2024.
4. University of Sydney. Better Evidence and Translation for Calciphylaxis (BEAT-Calci). NCT05018221. https:// clinicaltrials.gov/study/NCT05018221.
Nina B. Gold
Division of Medical Genetics and Metabolism, Department of Pediatrics, Massachusetts General Hospital for Children, Boston; Director of Prenatal Medical Genetics and Associate Director of Research, Massachusetts General Brigham Personalized Medicine, Boston; Harvard Medical School, Boston, Massachusetts, USA
After completing your medical degree at Harvard Medical School, Boston, Massachusetts, USA, you undertook a combined residency in paediatrics and genetics. Is this a field you had always wanted to specialise in, or was there a pivotal moment in your education and training that inspired you?
I always knew that I wanted to work with children. My mother is a paediatric nurse practitioner, and my father is a child psychiatrist. Their careers brought them joy, and allowed them to make tremendous contributions to our community.
and has guided me toward new and fascinating projects.
Q2
Your research has recently focused on genomic newborn sequencing (NBSeq). What are the expert opinions on the feasibility of performing this for all newborn babies, and what infrastructure would be needed to achieve this?
I am collecting data to inform the sensitivity and specificity of genome-first approaches
My interest in genetics began after college. Before beginning medical school, I worked as a clinical research assistant to Lewis Holmes, Chief Emeritus of Medical Genetics at Massachusetts General Hospital in Boston. My job involved interviewing new mothers of infants born with congenital anomalies, and creating a new classification system for limb deficiencies. I loved how medical genetics combined family-centred care with cutting-edge science.
During medical school, I was mentored by Vamsi Mootha, an international expert in mitochondrial disorders, and Robert Green, who is renowned for his work in the implementation of genomic sequencing. These experiences led me to develop a research niche at the intersection of inherited metabolic disorders and genomic testing implementation. Over 10 years later, Green continues to be my primary research and career mentor. He is extraordinarily supportive of my academic goals,
To borrow an analogy from retail, I think NBSeq is currently experiencing a ‘last mile delivery’ problem. When you order a package online, the final phase of shipping from the delivery truck to your doorstep is the slowest, and most expensive. We have travelled nearly all the way toward NBSeq; we have the infrastructure in place to collect blood samples from newborns at birth; DNA can be sequenced and interpreted at increasingly reasonable costs; and genomics is being more widely integrated into clinical care.
However, here we are, stuck at the last mile before implementation. Which genes should we sequence and report in children? Can newborn screening laboratories perform and possibly interpret sequencing results? Are genetics and paediatrics workforces prepared to care for the children who receive positive screening results? Finally, although both parents of children with rare diseases and parents of apparently healthy children have demonstrated enthusiasm for genomic newborn screening, some critics remain concerned about how such testing might affect familial stress and parent–child relationships.
I hope my work will provide a little bit of fuel to help us reach the goal of equitable, scalable genomic newborn screening. I am collecting data to inform the sensitivity and specificity of genome-first approaches. These findings might eventually be used to counsel families about the meaning of positive genomic findings in their infants.
Q3 What potential clinical impact would routine newborn genomic screening have on rare genetic disease outcomes?
exams that lead to an early cancer diagnosis, and spare the child’s sight.
Several genetic disorders, such as lysosomal storage disorders, can be treated before birth
Genomic newborn screening has the potential to save lives. You can imagine a young boy born with haemophilia A. Instead of experiencing a mysterious and life-threatening bleeding episode as a child, he might be identified as an infant through genomic screening, monitored for bleeding symptoms, and promptly receive a factor VIII infusion when needed. A child at risk for retinoblastoma, a hereditary eye tumour, might receive early and routine eye
Genetic information is nuanced, however, and many genetic variants do not directly lead to disease symptoms. For every success, there will be a challenge, such as the variable symptoms of many genetic disorders, or the lack of curative treatments for some disorders. The longer I am in this field, the more research questions and quandaries I see ahead of us. These challenges should not stop us, however, from implementing genomic sequencing as a tool to screen for several highly treatable disorders now.
Q4 In 2021 you received an Eleanor and Miles Shore Faculty Development Award. What research was this award based on, and have you been able to build on this since 2021?
I was very fortunate to receive an Eleanor and Miles Shore Faculty Development Award, which provides research funding for
early career physician-scientists at Harvard Medical School. With my frequent collaborator, Jessica Gold, I collected data on adults in two hospital-based biobanks who had genomic variants associated with two undiagnosed metabolic disorders. Our work was published, and I used these pilot data to apply for a K08 mentored clinical scientist award from the National Human Genome Research Institute (NHGRI).
Q5 You are currently the Director of Prenatal Medical Genetics at the Massachusetts General Brigham hospital-based biobank. How did you come to take on this position, and what does the role entail?
I took on this role in 2020, shortly after I became a mother. Although I am primarily trained in paediatrics, I see prenatal genetic care on a continuum with newborn genetic care. I also feel privileged to be a part of patients’ care during pregnancy. I regularly care for pregnant people who have genetic conditions, and counsel those
whose fetuses are at risk for genetic conditions.
Q6 Are there any innovations in either genomic screening or gene therapy that you are excited to see develop further?
I am fascinated by prenatal therapies for genomic conditions. Several genetic disorders, such as lysosomal storage disorders, cobalaminopathies, and even cystic fibrosis, can be treated before birth. The next step will be to determine how best to identify the fetuses who may benefit from these therapies.
Q7
Alongside your clinical work, research, and involvement in teaching, you are now undertaking a Master of Medical Sciences in Biomedical Informatics. What does this course entail, and how does it apply to your day-to-day practice?
I am a first-year student in the part-time accelerated Master of Medical Sciences in Biomedical
Informatics Program at Harvard Medical School, directed by Nils Gehlenborg. It's not easy keeping up with weekly homework, but I’ve learned so much from my courses! I’m working hard to improve my skills in coding, biostatistics, and data visualisation. I have expanded my own research skillset, and look forward to sharing what I’ve learned with my mentees.
Q8 To conclude, what advice would you give to medical students or early-career physicians aspiring to a career in medical genetics?
Above all, caring for patients continues to be the most rewarding part of my job. I think that clinical care reminds us why our research matters. Find an area that fascinates you, and make time for your life outside of work.
Muscle Matters: Protein Requirements for Muscle Preservation During Ageing
The publication of this infographic was supported by Nestlé Health Science. EMJ. 2024;9[2]:64-65. https://doi.org/10.33590/emj/CIBI3508.
High Prevalence of Suboptimal Protein Intake Among Older Adults
Protein intake recommendations minimum intake for healthy older people
Current RDA for all adults2
0.8 g/kg BW/d for all adults
1.0–1.2 g/kg BW/d 1.2–1.5 g/kg BW/d Up to 2.0 g/kg BW/d
Expert recommendation for older adults (>65 years)3 in acute or chronic disease in severe illness* or injury or marked malnutrition
*Patients with severe kidney disease not on dialysis may need to limit protein intake3
Consumption reality of older adults† do not meet the basic recommendation of 0.8 g/kg BW/d1 of older adults† do not meet the higher recommendation of 1.2 g/kg BW/d1
Higher prevalence of suboptimal
Female sex
†Data derived from surveys in community-dwelling (94% of participants aged ≥65 years)
Take Action Now to Preserve Mobility and Quality of Life Later: Seven Steps to Support
Look and listen for red flags suggesting malnutrition or risk 1 2 3 4
Perform a nutritional assessment to capture potential low protein intake
Educate about importance of muscle health
Recommend adjustments to optimise protein
Visual Indicators
• Unintentional weight loss
• Visible fat or muscle loss
• Other visual signs of poor nutrition
Clinical indicators
• Loss of appetite
• Swallowing difficulty
• Poor dentition
• GI or bowel issues
• Medication side effects
• Polypharmacy
• Low mood
• Chronic disease
Abbreviations
BMI: body mass index; GI: gastrointestinal; g/kg BW/d: grams per kilogram of body weight per day; RDA: recommended dietary allowance.
Social indicators
• Poor food access
• Food insecurity
• Career stress
• Social isolation
• Bereavement
• Limited nutrition or cooking skills
• Fixated eating
• Unnecessary food restrictions
Spread protein intake across the day, aiming for 25–30 g per meal5,6
Consume high-quality protein (e.g., supplement) immediately after to maximise muscle protein synthesis
Introduction Aim
Muscle health plays a vital role in maintaining overall well-being and quality of life as individuals age.1
suboptimal protein intake was associated with:1
Higher BMI Poor appetite
community-dwelling adults aged ≥55 years years)1
This infographic aims to raise awareness among healthcare providers about the importance of protein intake for muscle preservation in older adults, highlighting that ‘muscle matters’.
Healthcare
Professionals Can Help Fill the Gaps in Patient Knowledge
In a European survey of 1,825 adults aged ≥65 years:4
Over 35%
did not know what dietary protein is
Support Patients in Achieving Adequate Protein Intake
5 6
Among those who did indicate awareness of dietary protein (n=1,180):
were aware that having just one meal per day with a good protein source is insufficient Only ~25%
7 adjustments protein intake
Provide tangible examples of nutrition with higher protein content (including suggested quantities)7
(e.g., 20 g protein exercise sessions synthesis2
10 g of protein are in:
Supplement, e.g., recommend high-protein drinks7
Vegetable-based products
• 2 handfuls of nuts
• 16 tablespoons of oatmeal
• 400 g of cooked rice
• 250 g of cooked pasta
• 125 g of cooked pulses
• 3 slices of bread
• 1.5 slices of cooked tofu
Meat
• 33 g cooked beef
• 33 g cooked liver
• 33 g cooked chicken breast
• 3 slices of ham
• 2 slices of roast beef
• 4 slices of chicken breast
Dinner Fish
Cheese
• 0.5 bowl of cottage cheese
• 2 slices mozzarella
• 1.5 slices Gouda cheese
meta-analysis across cohorts from the PROMISS consortium. J Cachexia dietary reference values for protein. EFSA Journal. 2012;10(2):2557. from the PROT-AGE Study Group. J Am Med Dir Assoc. 2013;14(8):542-59. 2021;13(3):1006. Clin Nutr Metab Care. 2009;12(1):86-90. adults of the NuAge study. Am J Clin Nutr. 2016;104(3):694-703. https://www.promiss-vu.eu/community/health-professionals/. Last protein distribution
• 50 g smoked salmon
• 4 canned sardines
• 45 g baked trout
Other
• 2 eggs
• 1.5 glasses of milk
• 1.5 bowls of yoghurt
Call to Action
Over 75% of all participants indicated that they would increase protein intake if recommended by a healthcare professional (i.e., physician or dietician)
Demonstrate easy ways to be active and reduce sedentary time
Balance exercises/ gait training Power exercises
Aerobic exercises/ walking Resistance training
Together, let’s recognise that ‘muscle matters’, and take action to ensure that our ageing patients receive optimal care for maintaining muscle health.
By implementing evidence-based recommendations, enhancing patient knowledge, and employing practical tips, we can make a significant impact on the well-being and quality of life of our ageing population.
Importance of Timely and Accurate Diagnosis
Myotonic Disorders: Role of Electromyography
Myotonic Dystrophy and Non-dystrophic Myotonias
•Myotonic disorders are a heterogeneous group of interited neuromuscular disorders1
•Myotonia is a symptom that is a common feature of several types and subtypes of myotonic disorders, including myotonic dystrophy (DM) and non-dystrophic (NDM) myotonias.
•Myotonia presents clinically as delayed muscle relaxation after voluntary contraction, leading to muscle stiffness or cramping, and/or electrophysiologically as spontaneous discharge of muscle fibres1
DM subtypes: DM1; DM2
Prevalence: 1 in 8,000 for DM1; DM2 rarer 2,3
Diagnostic delay: typically >5 years (DM1) and >14 years (DM2)2
2.Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myologica. 2013;32(3):154-65.
3.Lehmann-Horn F et al. Diagnostics and therapy of muscle channelopathies – guidelines of the Ulm Muscle Centre. Acta Myol. 2008;27(3):98-113.
4.Díaz-Manera J et al. Understanding the impact of non-dystrophic myotonia on patients and caregivers: results from a burden of disease healthcare survey. EMJ. 2021;6.2:37-46.
5.Stunnenberg BC et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. 2020;62(4):430-44.
EU-NDM-2403-00010
6.Matthews E et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. 2010;133:9-22.
7.Hilbert JE et al. Diagnostic odyssey of patients with myotonic dystrophy. J Neurol. 2013;260(10):2497-504.
8. Trip J. Redefining the non-dystrophic myotonic syndromes: phenotypic characterisation based on genetic testing. Available at: http://www.equipware.nl/mcbecker/ myotonie%20van%20jeroen%20trip.pdf: Last accessed: March 2024
9.Vereb N et al. Non-dystrophic myotonias: clinical and mutation spectrum of 70 German patients. J Neurol. 2021;268(5):1708-20.
Myotonia
Citation: EMJ. 2024;9[2]:66-67. https://doi.org/10.33590/emj/XZMG9439. This content was funded by Lupin Neurosciences. The content of this infographic is based on satellite symposia hosted by Lupin Neurosciences at the 13th International Congress of Paediatric EMG (La Baule, France, 13th–15th November, 2023), and the Myology 2024 International Congress (Paris, France, 22nd–25th April, 2024), recordings of which can be accessed here. Symposium content was developed by Yann Péréon, Nantes, France; Emma Matthews, London, UK; and Valeria Sansone, Milan, Italy.
Diagnostic delays
Variable, non-specific symptoms 4-7
• Muscles affected
• Disease severity
•Age of onset
• Warm-up phenomenon
• Cold phenomenon
•Overlap with other diseases
Delays seeking medical help9
• Patients don’t ask
•Non-specialists don’t refer on
Coping 8
•A “family problem” (inherited disease) people manage by themselves
There are numerous reasons for delays in diagnosing DM and NDM
Under-recognised disease burden 4
•Underestimation of impact on patients and carers
Lack of disease recognition 4
•Rare diseases, not frequently encountered by non-specialist HCPs
•Diagnostic delays have a negative impact on patients’ wellbeing,2 as they have to learn to cope with their condition, often by limiting what they do, instead of being offered treatment to ameliorate symptoms
•Timely and accurate diagnosis is important for genetic counselling and screening of systemic features in DM, as well as determining appropriate management1
• Using EMG can help provide timely confirmation of a diagnosis
Role of EMG in DM
Low need for EMG when there is a clear DM phenotype and clear signs of clinical myotonia
Greater need for EMG when:
Genetic screening is required
Myotonia fluctuates or is difficult to find
Role of EMG in NDM
?
Useful when clinical myotonia is uncertain
Helpful when interpreting relevance of a gene variant, and ensuring correct diagnosis
Can prevent delay to appropriate medical management
Can prevent erroneously offering inappropriate medical management
Key Learnings
•EMG, while not necessary in every case, plays an important role in the timely diagnosis of both DM and NDM
•EMG, alongside genetic testing, can facilitate accurate differential diagnosis of disease subtypes
Lost in the System: The Labyrinth of Rare Disease Diagnosis
Authors: Peter Fish1
1. Mendelian, London, UK *Correspondence to peter@mendelian.co
Disclosure: Fish is the CEO and a shareholder of Mendelian, London, UK, a company that generates revenue from life science companies for projects aimed at shortening the diagnostic odyssey of rare diseases.
One in 10 people may be affected by a rare disease (RD) in their lifetime.1 This is the surprising paradox of RDs. While each disease is individually rare, when considered collectively, they affect a staggering portion of the population. This prevalence is expected to be even higher in those who frequently seek care. The full burden is often hidden and poorly understood. As a result, their impact on patients and healthcare systems is profoundly difficult to quantify, and expected to be substantially under-estimated.
In Europe, a disease is classified as rare if it affects less than one in 2,000 people. RDs that affect less than one in 50,000 people are termed ultra-rare. Most sources suggest there are 6,000–7,000 known RDs.1 Recently, RARE-X estimated the number of distinct RDs at almost 11,000, noting that 2,000 of these are vaguely defined.2 Around five new RDs are discovered and added to these lists every week.1 This will accelerate as knowledge expands, and as we move to stratified and personalised care.
Most RDs lack an approved drug treatment, although the number of RD treatments is expected to increase rapidly.3 Orphan drug (drugs developed to treat RDs) trials represent half of the active trials. Approved treatment or not, an accurate diagnosis is
essential for optimal disease management, empowering patients to understand their condition and facilitating access to supportive care, advocacy groups, and clinical trials.
Unfortunately, the diagnostic process remains an odyssey for most: long, challenging, and rife with misdiagnoses and inefficiencies, allowing diseases to progress and irreversible complications to accumulate. Unnecessary tests and procedures are carried out, and inappropriate medication is prescribed.
Early and accurate diagnosis is noted as a priority by almost all relevant organisations, including the European Organisation for Rare Disorders (EURORDIS-Rare Diseases Europe),4 and in various RD-focused government frameworks. Diagnostic odysseys appear to have been amplified by the strain that the COVID-19 pandemic placed on our healthcare systems.5
Our exposure to RDs has increased in recent years, driven by greater patient advocacy, novel diagnostic technologies, our expanded understanding, and the rise of orphan drugs. This aside, routine medical practice, in Europe and the rest of the world, still fails patients with RDs. We need to change this.
WHY THE LABYRINTH?
RDs are a vast and heterogeneous group of diseases. This leads to information complexity beyond simply the sheer number of diseases, which, in turn, delays recognition and diagnosis.
We Only Recognise What We Know
Unsurprisingly, most clinicians have limited experience with RDs, having rarely encountered patients with these conditions, particularly the ultra-rare RDs. Just 150 RDs account for 80% of all patients diagnosed with a RD.3 Some, like cystic fibrosis, occur near the one in 2,000 threshold, while 85% of these diseases affect less than one in 1,000,000 people.3 Some diseases have only ever been recorded in one or two patients worldwide. We generally recall some of the important or intriguing RDs that are more common, linked to famous people, or that present with unique or pathognomonic signs. Cystic fibrosis, sickle cell disease, haemophilia, Duchenne muscular dystrophy, achondroplasia, amyotrophic lateral sclerosis (Lou Gehrig’s and Stephen Hawking’s disease), fibrodysplasia ossificans progressiva (‘stone man syndrome’), and progeria might all ring a bell.
Diverse Aetiologies
Over 70% of all RDs are genetic.6 Others are infectious, autoimmune-driven, or rare cancers. Some RD databases even include diseases arising from toxin exposure. Wide diversity exists within each group. Genetic RDs can be the result of single nucleotide variants, insertions/deletions, structural variants, repeat expansions, and more.7 These may occur in one of 20,000 genes, or numerous, now known to be important, noncoding sites.
This variation opens a huge search space for diagnosis, which requires diverse clinical expertise and testing methods. Frequently, a specific suspected RD, perhaps the most common, or one that was top of mind, turns out not to be the culprit, while another related RD is. This shifts the way we need to diagnose; instead of requesting a highly
targeted test, it often pays to explore more broadly from the start.
New diagnostic technologies generally allow a greater ‘catch’ with a single test. As the costs decrease and availability improves, guidelines are swiftly updated to recommend tests covering a greater range of conditions. With genetic RDs, shortread whole genome sequencing should be considered in most circumstances,7 while promising advances in long-read sequencing means it may supersede this in the years to come. Sadly, the diagnostic yield, even from the most advanced tests, may still leave most complex patients undiagnosed, or with a variant of unknown significance, which may or may not be pathogenic.7 These modern tests generate huge amounts of data, and interpreting the results is often a substantial feat.
Population Background
Different populations are affected disproportionately. The discrepancies in disease prevalence may make a disease rare in certain populations, while fairly common in others. Thalassaemia is rare in Northern European patients, yet common in Mediterranean patients. Clinicians may miss diseases in populations they are only occasionally exposed to.
Temporal and System Fragmentation
RDs can present at any life stage, from birth to far later in life. Some present acutely, while many have insidious, progressive presentations. Almost 70% are known to start in childhood.6 Not all these patients present clinically at this young age, yet on deeper evaluation or enquiry, early signs may have been there. As RDs progress, patients present with additional pathologies over time. Clinicians often only have the capacity to focus on the current presenting complaint in isolation. The temporal fragmentation of patient data obscures clinical patterns, highlighting the importance of comprehensive longitudinal reviews.
Some RDs affect isolated organ systems (retinal diseases), while others are multisystemic (metabolic diseases). Specialist
care can be notoriously siloed, and as a result, clinicians may miss an underlying multi-systemic disease. A dermatologist may manage a skin rash; a month or two later, a hepatologist reviews abnormal liver function: both pathologies driven by a single, undiagnosed RD. In some settings, primary care electronic health records may house more complete data on the patient’s journey.
The Wide Range of Severity
Some RDs have mild presentations, but many present acutely and severely. Various RDs are incompatible with life, leading to miscarriages and stillbirths during the antenatal and early neonatal periods. These may be diagnosed retrospectively after death, which may bring closure to some parents, useful knowledge for future pregnancies, and academic value. Sadly, many children with an RD will not survive to their fifth birthday.
RDs often exhibit significant inter-patient variability. Patients with the same genotype manifest with varied phenotypes, ranging from no signs at all to markedly severe. Diseases may have early- or late-onset subtypes. Less severely affected patients, and those with later-onset disease, often go undiagnosed until later in life.
HOW DO WE SOLVE IT?
Undiagnosed patients can be split into three stages based on symptom presence, patient engagement with the healthcare system, and whether an RD is suspected or not. There are mechanisms and levers that can be lent on to expedite diagnosis at each stage. Not every patient will pass through every stage.
Stage 1: Pre-symptomatic (or Subclinical Symptoms), No Healthcare System Engagement, Rare
Disease Is Not Suspected
These patients may be diagnosed via cascade testing (testing of relatives when a family member tests positive with a relevant RD), or newborn blood spot (NBS) screening programmes.
NBS varies notably across the world. In Europe, the UK and Ireland screen for nine diseases, while Italy screens for over 40. Advocacy groups are calling for a more harmonious approach. Advances in mass spectrometry and molecular methods now allow far more diseases to be included, yet the downsides, mostly associated with false positives, need to be carefully weighed. The International Society for Neonatal Screening (ISNS) collaborates closely with various RD organisations, including the European Reference Network for Hereditary Metabolic Diseases and Rare Endocrine disorders (metabERN/EndoERN) and EURORDIS, to drive NBS forward via focused programmes like the first pillar of the Screen4Rare project.8
Genomic tests are being evaluated as screening tools. A research project being conducted in England, UK, called The Generation Study, looks to sequence the genomes of 100,000 newborns by March 2025. Pathogenic mutations will be reported on for around 200 carefully selected diseases, all of which present, and can be treated, in the early years of life.9 Genomic screening programmes, if proven clinically useful, cost-effective, and ethically sound, may be routine in the future.
Additionally, early phenotypic detection, through routine health checks, and automated, somewhat objective, biomarker detection via patient wearables and other related technologies, may hold future promise in surfacing these hidden patients, and moving them into Stage 2 or 3.
Stage 2: Symptomatic, Healthcare System Engagement, Rare Disease Is Not Suspected Patients may seek care for seemingly unrelated conditions over the course of years. Too often, clinicians focus on managing only the acute issue, rather than searching for a possible unifying root cause, a natural outcome of time pressure and limited knowledge of RDs.
Education programmes may yield some effect; yet due to the expansive (and expanding) list of RDs,
educating most patients and clinicians about individual RDs may be futile. Two approaches can be considered:
1. General ‘Think Rare’ education challenges patients and clinicians to always explore the potential that a patient’s clinical pattern may be the result of an undiagnosed RD (hoof beats do not always mean a horse: it may be a zebra). Hopefully this article raises awareness in this way.
2. Phenotype-specific education integrates an RD consideration into day-to-day care, by including a ‘Consider an RD’ branch into diagnostic or management guidelines and algorithms (for instance, patients with early-onset, treatmentresistant epilepsy may warrant further investigation for an underlying, undiagnosed RD).
Health informatic and artificial intelligence approaches, including electronic health record case-finding technologies, and automated differential diagnosis generators, can identify patients who warrant a review, or suggest alternative diagnoses. Screen4Care’s second pillar focuses on this aspect.
Additionally, we can ensure we break down silo walls, enhance health data standardisation and sharing, and ensure clinicians have a comprehensive view of every patient’s health. The European Health Data Space (EHDS) initiative strives to do just this.
Stage 3: Symptomatic, Healthcare System Engagement, Rare Disease Is Suspected (Not Yet Diagnosed)
As previously noted, even when a RD is suspected, a successful and accurate
References
1. Haendel M et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19(2):77-8.
diagnosis is far from guaranteed. Between 50–95% patients with suspected RDs who are put forward for whole genome sequencing remain undiagnosed.7 Multimodal diagnostic pathway enhancements, diagnostic hardware and software improvements, and data-driven phenotype to disease and genotype search engines are levers in this phase.
Specialist units and organisations focused on these undiagnosed patients are purposefully being set up, such as Syndrome Without a Name (SWAN) clinics and Undiagnosed Disease Networks, with European organisations like Solve-RD driving this agenda forward.
MOVING FORWARD
Much of the diagnostic odyssey is the result of a massive information challenge. Clinicians (and humans in general) lack the tools to effectively integrate and interpret the vast amounts of disease and patient information required for earlier RD diagnoses. To address this, it would be beneficial to continue to invest and support coordinated European initiatives, harmonise and expand genomic and health data sharing, ramp-up cross-border research, and develop robust tech-enabled diagnostic technologies and decision support tools. Adopting new technologies is challenging, and these bring notable changes, so political support is an imperative. Continually evaluating diagnostic pathways, identifying and widening bottlenecks, and optimising resoource allocation, will maximise the impact of new technologies and specialised care.
I urge clinicians to ‘Think Rare’, and utilise the valuable RD resources at your disposal, to change patients’ lives for the better.
2. RARE-X. The power of being counted. 2022. Available at: https://rare-x.org/ case-studies/the-power-of-beingcounted/. Last accessed: 15 March 2024.
3. Tambuyzer E et al. Therapies for rare diseases: therapeutic modalities, progress and challenges ahead. Nat Rev Drug Discov. 2022;19(2):93-111.
4. EURORDIS Rare Diseases Europe. Earlier, faster and more accurate diagnosis. Available at: https://www. eurordis.org/our-priorities/diagnosis/. Last accessed: 15 March 2024.
5. Hampson C et al. Measuring the impact of the COVID-19 pandemic on
diagnostic delay in rare disease. EMJ. 2022; DOI/10.33590/emj/21-00181.
6. Nguengang Wakap S et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2022;28(5):165-73.
7. Souche E et al. Recommendations for whole genome sequencing in diagnostics for rare diseases. Eur J Hum Genet. 2022;30(9):1017-21.
8. Loeber JG et al. Neonatal screening in Europe revisited: an ISNS perspective on the current state and developments since 2010. Int J Neonatal Screen.
2021;7(1):15.
9. Genomics England. Newborn Genomes Programme. 2024. Available at: https://www.genomicsengland.co.uk/ initiatives/newborns. Last accessed: 15 March 2024.
Genetics and Pathophysiology of Co-occurrence of Congenital Heart Disease and Autism Spectrum Disorder
Editor's Pick
My Editor’s pick for this issue is a comprehensive review article discussing the genetics and pathophysiology underlying autism spectrum disorder and congenital heart disease. Ong concludes that children with congenital heart disease are at an increased risk of neurodevelopmental disabilities due to genetic mutations and haemodynamic changes. Early diagnosis and interventions for autism spectrum disorder in children with congenital heart disease are crucial, with genetic testing the key to this.
Lászlo Vécsei
Head of Neuroscience Research Group, Department of Neurology, University of Szeged, Hungary
Author: *Leong Tung Ong1
Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia *Correspondence to leotungong@gmail.com
Disclosure: The author declares no conflict of interest. No funding was received for the publication of this article.
There is increasing evidence demonstrating that children with congenital heart disease (CHD) have a greater risk of developing autism spectrum disorder (ASD) in later life. This review aims to summarise the genetics and pathophysiology underlying both conditions. A PubMed search was performed to identify relevant studies exploring the comorbidities of ASD and CHD. The comorbidities of ASD and CHD can be explained by the influence of common and rare variants that contribute to genetic risks. De novo mutations in chromatin remodelling genes, and common genetic loci in the development of brain and heart in utero, can lead to the co-occurrence of ASD and CHD. Furthermore, there are several cases of syndromic ASD with concurrent CHD presentation. Foetuses with CHD may have abnormal haemodynamic changes and alteration of brain circulation in utero, resulting in impaired development of the brain, and increased risk of ASD. Abnormal brain development or brain injury as observed in MRI studies of infants with CHD may also contribute to the risk of ASD. Children with CHD should have regular neurodevelopmental assessment to screen for ASD symptoms for early diagnosis and intervention.
Key Points
1. De novo mutations in chromatin remodelling genes and common genetic loci in the development of brain and heart in utero can lead to co-occurrence of autism spectrum disorders (ASD) and congenital heart disease (CHD). The presence of CHD has also been reported in patients with syndromic ASD such as Fragile X syndrome, Coffin–Siris syndrome, DiGeorge or velocardiofacial syndrome, Phelan–McDermid syndrome, 16p11.2 deletion syndrome, and CHARGE syndrome.
2. Foetuses with CHD may have abnormal haemodynamic changes and alteration of brain circulation in utero, which result in impaired brain development, leading to an increased risk of ASD.
3. Children with CHD should receive regular follow-up to assess neurodevelopment and screen for symptoms of ASD.
INTRODUCTION
Children diagnosed with congenital heart disease (CHD) have an increased risk of developing neurodevelopmental disabilities encompassing language, motor, cognitive and social communications in the longterm impairments.1 There is increasing evidence demonstrating that the deficits in social communication among children with CHD may be potentially linked to the diagnosis of autism spectrum disorders (ASD).2 ASD is a heterogenous neurodevelopmental condition often cooccurring with other congenital disorders such as CHD, significantly impacting social communication and behavior.3 A recent meta-analysis has demonstrated a rapid increasing trend in ASD prevalence over recent decades with an average prevalence of 0.6% worldwide.4 The core features of ASD in children include a deficit in social communication and restrictive and repetitive behaviors.5 The severity of CHD has a positive correlation with the risk of neurodevelopmental disorders, including ASD, attributed to presence of de novo mutations in chromatin remodelling genes, Notch signalling, and cilia function.3
Comorbidities of ASD and CHD may arise due to the underlying genetic or haemodynamic changes during in utero or post-natal cardiac surgery.6,7 Additionally, brain development in infants with CHD may be impaired due to intrinsic epigenetic factors.8 The development of both heart and brain occurs simultaneously, sharing common genetic pathways during foetal
development.9 Therefore, abnormal heart development may precipitate neurodevelopmental disorders due to these shared pathways.9 Several studies have established that common and rare variants in ASD and CHD may contribute to the concurrent existence of these conditions.10 Mutations in specific genetic loci associated with both ASD and CHD may explain the associations between the two conditions.11 Notably, previous studies have identified a mere five genes that are overlapped between the 65 ASD risk genes and 66 CHD risk genes.12,13
Potential insult to the development of the brain in infants with CHD may occur during intrauterine life.14 Impairment in cerebral blood flow during in utero, post-natal, or post-operative repair also plays a significant role in the comorbidity of ASD and CHD.15 Anomalous development of cardiac structures with intracardiac or extracardiac mixing can lead to a decrease in cerebral oxygen levels or substrate changes, due to alterations in blood oxygen saturation, thus increasing the risk of impaired brain development in foetuses.15 Neuroimaging studies of infants born with CHD often showed abnormalities of the brain structures which may lead to poor neurodevelopmental outcomes.11 Moreover, infants with complex CHD also exhibit neurobehavioral abnormalities, even prior to undergoing cardiac surgery.16 The variability in brain development alterations among infants with CHD can be attributed to haemodynamic instability during cardiac procedures, potentially
leading to intraoperative brain injuries arising from ischaemia or hypoxia.7
As the medical management of CHD and cardiothoracic surgical techniques progress significantly, survival of CHD patients has increased, and many patients are now able to survive to adulthood.17 Therefore, the research direction has shifted towards the long-term neurodevelopmental outcomes to improve patient quality of life.18 Understanding the association between ASD and CHD may aid the clinician in early diagnosis and intervention to tackle neurodevelopmental difficulties. Therefore, the aim of this review is to summarise the genetics and pathophysiology underlying both ASD and CHD.
METHODS
A PubMed search was conducted on 31st November 2023 using the search terms “autism spectrum disorder” OR “Asperger’s Syndrome” AND “congenital heart disease” OR “congenital heart malformation”. Additional studies were identified from the bibliographies of relevant articles.
The abstracts and titles of the articles were screened, and the full paper was examined if the abstracts did not provide sufficient information.
In total, nine articles were included in this review. Table 1 shows the epidemiology of the co-occurrence of ASD and CHD.19–27
GENETICS OF AUTISM SPECTRUM DISORDER AND CONGENITAL HEART DISEASE
Multiple studies have shown that the comorbidity of ASD and CHD may be attributed to common and rare variants, contributing to the genetic risks.10-12,28,29 Genome-wide association studies (GWAS) of FinnGen identified a complex risk locus for developmental left-sided CHD on chromosome 17 near BAHCC1, and associated with LRRC37A2,WNT9B, WNT3, and MYL4 genes.30 Another GWAS study conducted
on patients at the German Heart Center, Munich, Germany found a single nucleotide polymorphism located on chromosome 5q22.2 that is shared among all CHD phenotypes.31 Additionally, genes such as MACROD2, GOSR2, WNT3, and MSX1, which are known to play essential roles in embryonic heart development, were implicated in the study.31 On the other hand, GWAS of ASD found that de novo and rare variants of genes such as NEGR1, PTBP2, CADPS, KCNN2, KMT2E, and MACROD2, which are located in the identified loci, were linked to an increased risk of ASD.32
In a study by Rosenthal et al.,10 101 genes with shared genetic risk for both ASD and CHD were identified within a convergent molecular network through network genetics analysis.10 Seven of these identified genes contribute to the genetic pathways of both conditions, including the sodium voltage-gated channel α subunit 2 (SCN2A) which is an ion transport gene.10 These convergent pathways encompass chromatin modifications, mitogen-activated protein kinase/Notch signalling, and ion transport, all integral to the development of brain and heart.10 The rare and common variants genes in these convergent pathways are interconnected within molecular networks.10 In contrast, the study by Satterstorm et al.33 suggests that ASD risk genes lack enrichment in GWAS, implying that common and variant alleles do not significantly influence individual genes.
The association between ion channels and the development of ASD and CHD becomes apparent as different ion-channel genes exhibit genetic signalling via de novo variants within a molecular network.10 In a Xenopus model, a genetic disruption in SCN2A, an ion transport gene, results in major structural defects in both heart and brain development.10 Ion-channel genes may be involved in the regulation of leftright patterning, impacting the left-right patterning in the heart development.34 Other than regulating embryonic development of the heart, SCN2A is also involved in the regulation of cardiac rhythm and post-natal neuron action potential.35 Furthermore, loss-of-function mutations of SCN2A are associated with an increased rate of ASD
Table 1: Studies exploring the association between autism spectrum disorder and congenital heart disease.
Study Design
Wier et al., 200619
Neufeld et al., 200820
Dawson et al., 200921
Davidson et al., 201522
Razzaghi et al., 201523
Tsao et al., 201724
Bean Jaworski et al., 201725
Sigmon et al., 201926
Werninger et al., 202027
Population
Case-control study 417 children with ASD and 2,067 controls
Prospective cohort study 69 neonates who underwent ASO for repair of TGA
Case–control study 465 children diagnosed with ASD and 1,313 controls
Retrospective cohort study 58 patients with HLHS and 44 patients without HLHS
Retrospective cohort study 374 children with CHD and 158,243 children without reports of CHD
Retrospective case–control study 3,552 children (<18 years of age) diagnosed with CHD and 14,208 controls
Prospective cohort study 381 children with a history of CHD
Case-control study 8,760 cases of ASD and 26,280 controls
Prospective cohort study 125 children with CHD at 10 years of age
Prevalence Data
3.1% (n=13) of the ASD cohort had CHD, and 1.9% (n=39) of controls had CHD.
5.8% (n=4) of children were diagnosed with ASD.
1.3% (n=6) of children with ASD had CHD, and 0.8% (n=11) of controls had CHD.
7% (n=4) of patients with HLHS were diagnosed with ASD, and no patients without HLHS were diagnosed with ASD.
2.6% (n=6) of children with CHD were diagnosed with ASD, and 0.6% (n=883) of children without CHD were diagnosed with ASD.
0.84% (n=30) of the CHD cohort were diagnosed with ASD, and 0.17% (n=24) of the non-CHD cohort were diagnosed with ASD.
56.69% (n=216) of children with CHD met the criteria for ASD.
4.6% (n=401) of children with ASD had CHD, and 2.5% (662) of controls had ASD.
0.8% (n=1) of children with CHD had a diagnosis of ASD.
ASD: autism spectrum disorder; ASO: arterial switch operation; CHD: congenital heart disease; HLHS: hypoplastic left heart syndrome; TGA: transposition of great arteries.
by 50%.36 SCN2A encodes the voltagegated sodium channel NaV1.2, expressed in cortical pyramidal neurons and cerebellar granule neurons, which are essential for supporting action potential firing and brain development.37,38 SCN2A mutations result in NaV1.2 haploinsufficiency, due to the production of premature stop codons, contributing to the development of ASD.30 Besides that, de novo mutations of SCN2A variants are also associated with epilepsy and intellectual disability, other than ASD.36
Chromatin remodelling represents a main pathogenic mechanism of ASD.40 ATP-dependent chromatin remodelling involves epigenetic regulation, impacting gene expression and histone-modifying pathways that are essential for embryonic
neurogenesis and cardiogenesis.41 Loss-offunction mutations affecting the regulatory functions of histone have implications for both ASD and CHD.42 Mutations in the BAF complex, which alters histone-DNA interactions, have been identified in both ASD and CHD.43,44 The neuronal version of BAF plays a role in the expression of synaptic genes during development of the brain, and its mutation is implicated in ASD.43 Additionally, Smarcd3, which encodes for Baf60c, a subunit of the BAF complexes, is expressed in the heart and is essential for heart morphogenesis.45 Mutation of this gene in a mouse model is associated with impaired development of anterior and secondary heart field and abnormal cardiac muscle development.45 Furthermore, methylation of the H3K4
gene, involved in transcriptional activation, may result in a monogenic form of both ASD and CHD.11,46 Remodelling of H3K4 in the cortex is most active during late gestation to early adolescence, potentially contributing to the development of ASD in CHD patients.47 Another histone-modifying pathway, involving methylation of H3K7 in transcriptional deactivation, is also noteworthy.11 The regulatory protein SMAD2 controls the transcriptional activation process through the removal of a methyl group during embryonic development.39 Furthermore, mutations in CHD8, which are frequently associated with ASD, have also been shown to be mutated in CHD.49,50 Moreover, mutation of CHD7 is also commonly associated with CHD.51 De novo mutations in the H3K4me pathway are involved in the pathogenesis of both ASD and CHD, which lead to the downregulation of transcription of developmental genes in the heart and brain.49,52 A Xenopus model further demonstrated that chromatin remodelling genes such as KANSL1, KAT6A, KMT2A, and KMT2D are involved in the development of both heart and brain.12,33
The study conducted by Assimopoulos et al.53 demonstrated that deletion of 16p11.2 results in more pronounced differences in the structures and functions of the heart and brains when compared to the wild type. This heightened impact is attributed to the presence of multiple genes within 16p11.2 that play significant roles in the development of both heart and brain, such as BCKDK, MAPK3, and SRCAP.53 Additionally, the study also demonstrated the involvement of the Wnt/β-catenin canonical pathway in a convergent signalling pathway crucial for both cardiac and brain development through transcriptional activation.53 Loss of FMR1 protein contributes to increased bone morphogenetic protein signalling, primarily through elevated levels of bone morphogenetic protein type II receptor, affecting Wnt signaling.54 Furthermore, loss-of-function mutations in ARID1B, a target of FMR1 in neural progenitor cells and neurons, result in the upregulation of Wnt/β-catenin target genes.55 β-catenin, in turn, regulates the fibroblast growth factor
signalling which is a key element in the regulation of second heart field progenitors during the development of atrial, ventricular, and outflow tract structures.56 In addition, Wnt can also activate a non-canonical pathway involving signalling proteins such as Jun N-terminal kinase, responsible for the regulation the cytoskeleton and cell polarity.57 Both canonical and non-canonical pathways are integral to embryonic cardiac and brain development.11 Furthermore, Assimopoulos et al.53 demonstrated that mutations in the SHANK3 gene cause thickening of the left ventricle anterior wall, which can be explained as they can be effects of CHD, such as tetralogy of Fallot (TOF), pulmonary atresia, or aortic valve abnormalities.
Recurrent microduplications and microdeletions in the distal region of chromosome 1q21.1 are susceptible to variable phenotypes of ASD and CHD.58 Deletion of 1q21.1 is more commonly observed in cases of non-TOF CHD, microcephaly, and schizophrenia, while duplication of 1q21.1 is frequently associated with TOF, macrocephaly, and ASD.59 The duplication of GJA5 in this region results from non-allelic homologous recombination of 1q21.1, leading to an increased risk TOF.59 GJA5 is regulated by Hand2, a cardiac transcription factor, and is expressed in cells of secondary heart field during the development of outflow tract.60 Furthermore, GJA5 also encodes for protein connexin-40, a cardiac gap junction protein responsible for cell adhesion and cell-cell communication.59 Experimental studies in mice with knockout connexin protein showed increased rates of CHD and brain malformations.61 Furthermore, several genes in the 1q21 critical region are expressed in the brain, including HYDIN2 and three NOTCH2NL genes.62 NOTCH2NL genes are essential for cortical development, where overexpression of the genes delays neuronal differentiation, while deletion of the genes accelerates it.63 Loss of genomic stability of these genes may lead to neurodevelopmental disorders.63 Copy number variants of other genes located in the 1q21.1 region, including CHDIL, PRKAG2, and GJA8, also contribute to the risk of both ASD and CHD.59,64
SYNDROMIC AUTISM SPECTRUM DISORDER WITH CONGENITAL HEART DISEASE
The presence of CHD has also been reported in patients with syndromic ASD.11 Various syndromic ASD conditions have shown concurrent CHD, including Fragile X syndrome, Coffin-Siris syndrome, DiGeorge or velocardiofacial syndrome, Phelan–McDermid syndrome, 16p11.2 deletion syndrome, and CHARGE syndrome.53 Fragile X syndrome, primarily caused by FMR1 mutation, exhibits a CHD prevalence of less than 10%.65 This can encompass aortic root dilation, mitral valve prolapses, and arrhythmias.65,66 Coffin–Siris syndrome, a result of loss-offunction mutations in ARID1B, leads to the upregulation of the Wnt/β-catenin signalling pathway.67 This syndrome is associated with a variable degree of neurodevelopmental disability, including ASD.66 Approximately 20% of patients with Coffin–Siris syndrome also present with CHD, which can include mitral valve insufficiency, septal defects, and atrioventricular block.66 DiGeorge, or velocardiofacial syndrome, stemming from the deletion of 22q11.2, has a high CHD prevalence of 75%.68 This may involve conditions such as TOF, pulmonary atresia, truncus arteriosus, and ventricular septal defect.68 Phelan–McDermid Syndrome is associated with terminal deletions of the chromosomal region 22q13.3, which contains the SHANK3 gene.69 CHD is prevalent in this syndrome, with a prevalence of more than 25%.69,70 CHD manifestations can include atrial septal defect, aortic arch anomalies, tricuspid valve regurgitation, patent ductus arteriosus, and total anomalous pulmonary return.69,70 16p11.2 deletion syndrome has a CHD prevalence of 33%, including conditions such as TOF and associated anomalies, and pulmonary atresia.66,71 CHARGE syndrome, attributed to genetic mutations in the CHD7 gene, has ASD prevalence of 15–50%, and a high CHD prevalence of 75–80%.72,73 CHD presentations can include ventricular septal defect, atrioventricular canal defect, TOF, and aortic arch anomalies.72
PATHOPHYSIOLOGY OF CONGENITAL HEART DISEASE LEADING TO AUTISM SPECTRUM DISORDER
The risk factors contributing to the development of ASD in patients with CHD encompass various elements. These risk factors include patient-specific factors, prenatal and perinatal conditions, brain abnormalities, the timing of surgical intervention, and cognitive impairments.2
De novo mutations in shared genetic loci contribute to the overlap between CHD and ASD.40 However, additional patientspecific risk factors can further increase the likelihood of developing ASD in individuals with CHD.2 Foetal and perinatal conditions play a significant role, with levels and duration of hypoxaemia being a crucial stressor in infants with CHD.74 Studies have shown that foetuses with CHD experience abnormal haemodynamic changes that result in alterations in brain circulation.14 This is evidenced by reduced middle cerebral artery pulsatility index and cerebroplacental ratio, leading to reduced brain volume and delay in brain maturation.14 Shunting of blood in foetuses with CHD occurs at atrial, ductal, or isthmic levels to maintain adequate cerebral blood flow.11 The shunting of blood in CHD leads to significant reductions of blood-flow in the major cerebral vessels.75 Studies also showed an increase in cerebral resistance indices using Doppler ultrasound, due to the alterations of blood flow caused by the cardiac defect.76 Furthermore, disruption in pathways that deliver oxygen and substraterich blood to the brain, due to intrinsic autoregulatory mechanisms, may lead to an increase in oxygen delivery to the brain to meet the cerebral energy requirements.15 Therefore, the development of ASD in CHD may be due to the burden of pulmonary and circulatory compromise, resulting from brain-sparing circulatory shift changes.77 Additionally, the time to corrective surgery is also an important determining factor, as more injury to the brain occurs when there is a longer wait to the corrective cardiac surgery in CHD infants.78 Longer wait to surgery results in higher extent and duration of cerebral desaturation, which may lead to poor neurodevelopmental outcomes.11
Infants with CHD have a high incidence of brain abnormalities, even with an absence of genetic syndrome.79,80 These abnormalities may arise from abnormal brain development in utero, or from brain injury after delivery.79,80 MRI studies of newborns with CHD showed reduced brain volume for gestational age, decreased metabolism, and delayed cortical folding and development compared to healthy newborns.81-83 Besides that, MRI brain of full term infants also revealed similar findings to preterm infants, including diffuse white-matter injury and focal injury of periventricular leukomalacia, suggestive of hypoxic-ischaemic encephalopathy.8,84
Furthermore, term infants with cyanotic CHD demonstrated similar white matter anomalies and delays in myelination, which are observed in preterm infants.82 Diffusion tensor imaging MRI of infants with CHD has shown a decrease in the white-matter fractional anisotropy and ratio of N-acetylaspartate to choline, which is indicative of disorganisation of white matter structure.79 Damage to the oligodendrocyte progenitor cells and subplate neurons in CHD infants results in impairment of the myelination process and development of white matter track, potentially affecting neural connectivity.80 These structural anomalies can result in emotional processing and social impairments in ASD, associated with atypical cortical and sub-cortical structures, such as the medial prefrontal cortex, orbitofrontal cortex, the right temporo-parietal junction, and amygdala.85 Furthermore, children with ASD have an estimated prevalence of 12–26% of epilepsy.86,87 This increased risk of epilepsy in CHD may be attributed to cerebral hypoperfusion and brain injury during in utero development, and perioperative corrective cardiac surgery.88 Early onsets of seizures after corrective cardiac surgery are associated with poor neurodevelopmental outcomes.89 Therefore, development of epilepsy syndrome in children with CHD may increase the risk of development of ASD.79
Moreover, infants with CHD have a higher rates of adverse perinatal outcomes, including preterm delivery, low birth weight, and perinatal or neonatal infection,
all of which can contribute to the risk of developing ASD.90,91 There are two distinct chronological risk factors for the increased risk of ASD in children with CHD, which are during birth and early childhood.92 Furthermore, children and adolescents with CHD often display some extent of social cognition impairments, even if they do not fulfil the diagnostic criteria for ASD.76 For instance, a study by Calderon et al.76 reported that CHD patients who underwent open-heart corrective surgery displayed significant impairments in complex affective mental state understanding, processing social cues, and theory of mind. Notably, the risk of ASD appears to be higher in children with milder forms of CHD, such as atrial septal defects and ventricular septal defects, possibly due to the limited number of cases among children with severe forms of CHD.88
CLINICAL APPLICATIONS, LIMITATIONS AND FUTURE DIRECTIONS
Understanding the genetic links between CHD and ASD may facilitate the comprehensive care and management strategies for affected children. Around 30% of children diagnosed with CHD exhibit syndromic phenotypes, wich include extracardiac manifestations.93 Shikany et al.94 found that 26% of the infants with critical CHD had abnormal genetics test. More widespread genetic testing of patients with CHD, especially in the presence of syndromic phenotypes, should be advocated to identify presence of neurodevelopmental disabilities such as ASD, or extracardiac malformations.95 However, genetic testing in children with isolated CHD may have limited clinical utility due to the high heterogeneity of genetic panels associated with CHD.95 On the other hand, the neurodevelopmental specialist who diagnoses the children with ASD should refer them to a genetic service to identify the presence of underlying medical conditions, single gene defects, or cytogenetic abnormalities.96 However, research shows that only 3% of patients diagnosed with ASD undergo suggested clinical genetic testing, underscoring the discrepancy between expert recommendations and clinical
practice.97 Therefore, cardiologists and neurodevelopmental paediatricians should refer the patients with co-occurrence of ASD and CHD for genetic testing and counselling to identify potential underlying genetic mutations.
The existing studies confront several limitations. CHD encompasses a spectrum of severity, ranging from mild left-toright shunts, such as atrial septal defects, to severe right-to-left shunts such as TOF. Consequently, the risk of ASD may vary depending on the severity of the underlying CHD. Current studies include patients with diverse types of CHD, which might not accurately reflect the true risk of ASD. Moreover, both CHD and ASD result from multifactorial causes, complicating efforts to identify specific genetic mutations implicated in both conditions. Furthermore, the predominant reliance on animal models in genetic studies investigating the co-occurrence of CHD and ASD poses challenges, as the findings may not necessarily translate to human populations. Given the current limitations in our understanding of the neurobiology of ASD, and its obscure pathophysiological mechanisms, elucidating causal relationships between these two conditions presents a formidable challenge. While genetic syndromes provide valuable insights into specific genetic pathways implicated in both ASD and CHD, it may not generalise to non-syndrome cases, due to the phenotypic variability and variable penetrance of associated genetic mutations of the syndromes.
Future research into the genetic associations between CHD and ASD may further improve our understanding of the pathophysiology underpinning both conditions, and inform clinical practice. Firstly, investigating the functional consequences of genetic variants via multi-omics approaches, such as transcriptomics and proteomics, may improve the genetic models of CHD
and ASD comorbidity by molecular phenotype profiling.98 Advanced technologies, such as single-cell genomics and spatial transcriptomics, enable high-resolution profiling of cellular heterogeneity and spatial organisation of tissues in children with CHD and ASD comorbidity, thereby improving our understanding of pathogenesis, and allowing the identification of novel therapeutic strategies.99,100 Studies focusing on the gene-environment interactions of CHD and ASD comorbidity will provide insights into the influence of phenotype expression on disease susceptibility.101,102 Furthermore, studies exploring polygenic risk scores and genetic modifiers will be essential to elucidate the complex genetic architecture underlying disease heterogeneity and susceptibility.103
CONCLUSION
There is an increased risk of development of ASD in children with CHD. Therefore, children with CHD should undergo regular follow-up to assess neurodevelopmental outcomes, and screen for ASD symptoms for early diagnosis and intervention.1 Furthermore, family members of children with CHD should be counselled regarding the increased risk of ASD and other developmental disabilities. Further research on the genetics and pathophysiology underlying the cooccurrence of ASD and CHD may aid in understanding the aetiology of the disease, and contribute to the development of novel treatments. Future research should also focus on different prognostic factors, such as genetics, brain abnormalities, haemodynamic changes, and time to surgery, to better elucidate the risk of ASD in children with CHD. This will contribute to the development of more precise risk assessment, and personalised intervention strategies.
References
1. Marino BS et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126(9):1143-72.
2. Calderon J, Bellinger DC, Newburger JW. Autism and congenital heart disease: evidence and unresolved questions. Pediatrics. 2019;144(5):e20192752.
3. Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. 2017;120(6):923-40.
4. Salari N et al. The global prevalence of autism spectrum disorder: a comprehensive systematic review and meta-analysis. Italian Journal of Pediatrics. 2022;48(1):112.
5. American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders (2013) 5th edition, American Psychiatric Association.
6. Homsy J et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies Science.2015;350(6265):1262-6.
7. Gaynor JW et al. Neurodevelopmental outcomes after cardiac surgery in infancy. Pediatrics. 2015;135(5):816-25.
8. Martinez-Biarge M et al. Neurodevelopmental outcome in children with congenital heart disease. Semin Fetal Neonatal Med. 2013;18(5):279-85.
9. McQuillen PS, Goff DA, Licht DJ. Effects of congenital heart disease on brain development. Prog Pediatr Cardiol. 2010;29(2):79-85.
10. Rosenthal SB et al. A convergent molecular network underlying autism and congenital heart disease. Cell Syst. 2021;12(11):1094-107.e6.
11. Nattel SN et al. Congenital heart disease and neurodevelopment: clinical manifestations, genetics, mechanisms, and implications. Can J Cardiol. 2017;33(12):1543-55.
12. Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49(11):1593-1601.
13. Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237-41.
14. Khalil A et al. Prevalence of prenatal brain abnormalities in fetuses with congenital heart disease: a systematic review. Ultrasound Obstet Gynecol. 2016;48(3):296-307.
15. Donofrio MT et al. Impact of congenital heart disease on fetal brain development and injury. Curr Opin
Pediatr. 2011;23(5):502-11.
16. Limperopoulos C et al. Neurodevelopmental status of newborns and infants with congenital heart defects before and after open heart surgery. J Pediatr. 2000;137(5):638-45.
17. Jortveit J et al. Trends in mortality of congenital heart defects. Congenit Heart Dis. 2016;11(2):160-8.
18. Andonian C et al. Current research status on the psychological situation of adults with congenital heart disease. Cardiovasc Diagn Ther. 2018;8(6):799804.
19. Wier ML et al. Congenital anomalies associated with autism spectrum disorders. Dev Med Child Neurol. 2006;48(6):500-7.
20. Neufeld RE et al. Five-year neurocognitive and health outcomes after the neonatal arterial switch operation. J Thorac Cardiovasc Surg. 2008;136(6):1413-21, 1421.e1-1421.e2.
21. Dawson S et al. Birth defects in children with autism spectrum disorders: a population-based, nested case-control study. Am J Epidemiol. 2009;169(11):1296-303.
22. Davidson J et al. Physical and neurodevelopmental outcomes in children with single-ventricle circulation. Arch Dis Child. 2015;100(5):449-53.
23. Razzaghi H et al. Long-term outcomes in children with congenital heart disease: National Health Interview Survey. J Pediatr. 2015;166(1):119-24.
24. Tsao PC et al. Additive effect of congenital heart disease and early developmental disorders on attentiondeficit/hyperactivity disorder and autism spectrum disorder: a nationwide population-based longitudinal study. Eur Child Adolesc Psychiatry. 2017;26(11):1351-1359.
25. Bean Jaworski JL et al. Rates of autism and potential risk factors in children with congenital heart defects. Congenit Heart Dis. Jul 2017;12(4):421-429.
26. Sigmon ER et al. Congenital Heart Disease and Autism: A Case-Control Study. Pediatrics. 2019;144(5).
27. Werninger I et al. Social and Behavioral Difficulties in 10-Year-Old Children With Congenital Heart Disease: Prevalence and Risk Factors. Front Pediatr. 2020;8:604918.
28. Fakhro KA et al. Rare copy number variations in congenital heart disease patients identify unique genes in leftright patterning. Proc Natl Acad Sci U S A. 2011;108(7):2915-20.
29. Glessner JT et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and
30. Broberg M et al. Genome-wide association studies highlight novel risk loci for septal defects and leftsided congenital heart defects. BMC Genomics. 2024;25(1):256.
31. Lahm H et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J Clin Invest. 2021;131(2):e141837.
32. Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51(3):431-44.
33. Satterstrom FK et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568-84.e23.
34. Jain S et al. Community-acquired pneumonia requiring hospitalization among U.S. adults. The New England journal of medicine. 2015;373(5):415-27.
35. Spratt PWE et al. The Autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron. Aug 21 2019;103(4):673-85.e5.
36. Kruth KA et al. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: a role for pluripotent stem cells? Mol Autism.2020;11(1):23.
37. Yamagata T et al. Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: mutually exclusive distributions of Nav1.1 and Nav1.2. Biochem Biophys Res Commun. 2017;491(4):1070-76.
38. Martínez-Hernández J et al. Polarised localisation of the voltage-gated sodium channel Na(v)1.2 in cerebellar granule cells. Cerebellum.2013;12(1):16-26.
39. Planells-Cases R et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J. 2000;78(6):2878-91.
40. Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry. 2013;18(10):1054-6.
41. Morrison AJ. Chromatin-remodeling links metabolic signaling to gene expression. Mol Metab. 2020;38:100973.
42. Shen E et al. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369(1652): :20130514.
43. Rowland ME et al. Insights into the emerging role of Baf53b in autism spectrum disorder. Front Mol Neurosci. 2022;15:805158.
44. Han P et al. Chromatin remodeling
in cardiovascular development and physiology. Circ Res. 2011;108(3): 378-96.
45. Lickert H et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432(7013):107-12.
46. Neale BM et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242-5.
47. Shulha HP et al. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013;9(4):e1003433.
48. Dahle Ø et al. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci Signal. 2010;3(127):ra48.
49. Zaidi S et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220-3.
50. O’Roak BJ et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619-22.
51. Corsten-Janssen N et al. The cardiac phenotype in patients with a CHD7 mutation. Circ Cardiovasc Genet. 2013;6(3):248-54.
52. Wang G et al. Epigenetics in congenital heart disease. J Am Heart Assoc. 2022;11(7):e025163.
53. Assimopoulos S et al. Genetic mouse models of autism spectrum disorder present subtle heterogenous cardiac abnormalities. Autism Res. 2022;15(7):1189-208.
54. Kumar S et al. Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J Neurodev Disord. 2019;11(1):10.
55. Li M et al. Identification of FMR1regulated molecular networks in human neurodevelopment. Genome Res. 2020;30(3):361-74.
56. Hubert F et al. FGF10 signaling in heart development, homeostasis, disease and repair. Front Genet. 2018;9:599.
57. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136(19):3205-14.
58. Mefford HC et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359(16):1685-99.
59. Soemedi R et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease
patients and 6760 controls. Hum Mol Genet. 2012;21(7):1513-20.
60. Holler KL et al. Targeted deletion of Hand2 in cardiac neural crest-derived cells influences cardiac gene expression and outflow tract development. Dev Biol. 2010;341(1):291-304.
61. Simon AM et al. Heart and head defects in mice lacking pairs of connexins. Dev Biol. 2004;265(2):369-83. Erratum in: Dev Biol. 2004;271(2):517-8.
62. Linden SC. The psychiatric phenotypes of 1q21 distal deletion and duplication. Translational Psychiatry. 2021;11(1):105. Erratum in: Transl Psychiatry. 2021;11(1):372.
63. Fiddes IT et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell. May 31 2018;173(6):1356-1369.e22.
64. Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Human Molecular Genetics. 2010;19(R2):R176-87.
65. Ciaccio C et al. Fragile X syndrome: a review of clinical and molecular diagnoses. Ital J Pediatr. 2017;43(1):39.
66. Pierpont ME et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the american heart association. Circulation. 2018;138(21):e653-e711. Erratum in: Circulation. 2018;138(21):e713.
67. Vasileiou G, Ekici AB, Uebe S, et al. Chromatin-remodeling-factor ARID1B represses Wnt/β-catenin signaling. Am J Hum Genet. 2015;97(3):445-56.
68. Digilio M et al. Clinical manifestations of deletion 22q11.2 syndrome (DiGeorge/ Velo-Cardio-Facial syndrome). Images Paediatr Cardiol. 2005;7(2):23-34.
70. Cusmano-Ozog K et al. 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. Am J Med Genet C Semin Med Genet. 2007;145c(4):393-8.
71. Ghebranious N et al. A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. Am J Med Genet A. 2007;143a(13):1462-71.
73. Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53(10):852-73.
74. Jenabi E et al. The association between
congenital heart disease and the risk of autism spectrum disorders or attentiondeficit/hyperactivity disorder among children: a meta-analysis. The European Journal of Psychiatry. 2021;36(3):71-6.
75. Ruiz A et al. Longitudinal changes in fetal biometry and cerebroplacental hemodynamics in fetuses with congenital heart disease. Ultrasound Obstet Gynecol. 2017;49(3):379-86.
76. Donofrio MT et al. Autoregulation of cerebral blood flow in fetuses with congenital heart disease: the brain sparing effect. Pediatr Cardiol. 2003;24(5):436-43.
77. Chu PY et al. Congenital heart disease in premature infants 25-32 weeks’ gestational age. J Pediatr. 2017;1 81:37-41.e1.
78. Lynch JM et al. Time to surgery and preoperative cerebral hemodynamics predict postoperative white matter injury in neonates with hypoplastic left heart syndrome. J Thorac Cardiovasc Surg. 2014;148(5):2181-8.
79. Miller SP et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357(19):1928-38.
80. McQuillen PS, Miller SP. Congenital heart disease and brain development. Ann N Y Acad Sci. 2010;1184:68-86.
81. Ortinau CM et al. Prenatal to postnatal trajectory of brain growth in complex congenital heart disease. Neuroimage Clin. 2018;20:913-22.
82. Licht DJ et al. Brain maturation is delayed in infants with complex congenital heart defects. J Thorac Cardiovasc Surg. 2009;137(3):529-36.
83. Clouchoux C et al. Quantitative in vivo MRI measurement of cortical development in the fetus. Brain Struct Funct. 2012;217(1):127-39.
84. Miller SP et al. Early brain injury in premature newborns detected with magnetic resonance imaging is associated with adverse early neurodevelopmental outcome. J Pediatr. 2005;147(5):609-16.
85. Calderon J et al. Facial expression recognition and emotion understanding in children after neonatal open-heart surgery for transposition of the great arteries. Dev Med Child Neurol. 2014;56(6):564-71.
86. Viscidi EW et al. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS One. 2013;8(7):e67797.
87. Lai MC et al. Autism. Lancet. 2014;383(9920):896-910.
88. Sigmon ER et al. Congenital heart disease and autism: a case-control study. Pediatrics. 2019;144(5):e20184114.
89. Wernovsky G, Licht DJ. Neurodevelopmental outcomes in children with congenital heart diseasewhat can we impact? Pediatr Crit Care Med. 2016;17(8 Suppl 1):S232-42.
90. Almeida LF et al. Epidemiological risk factors and perinatal outcomes of congenital anomalies. Rev Bras Ginecol Obstet. 2016;38(7):348-55.
91. Story L et al. Influence of birthweight on perinatal outcome in fetuses with antenatal diagnosis of congenital heart disease. J Obstet Gynaecol Res. 2015;41(6):896-903.
92. Tsao PC et al. Additive effect of congenital heart disease and early developmental disorders on attentiondeficit/hyperactivity disorder and autism spectrum disorder: a nationwide population-based longitudinal study. Eur Child Adolesc Psychiatry. 2017;26(11):1351-9.
93. Chaix MA et al. Genetic testing in congenital heart disease: a clinical approach. World J Cardiol.
2016;8(2):180-91.
94. Shikany AR et al. A comprehensive clinical genetics approach to critical congenital heart disease in infancy. J Pediatr. 2020;227:231-38.e14.
95. De Backer J, Muiño Mosquera L. Genetic testing in patients with congenital heart disease: you do no harm when using the right tools! circulation: Genomic and Precision Medicine. 2023;16(2):e004104.
96. Çöp E et al. Genetic testing in children with autism spectrum disorders. Anadolu Psikiyatri Derg. 2015;16(6): 426-32.
97. Moreno-De-Luca D et al. Clinical genetic testing in autism spectrum disorder in a large community-based population sample. JAMA Psychiatry. 2020;77(9):979-81.
98. Ristori MV et al. proteomics and metabolomics approaches towards a functional insight onto AUTISM spectrum disorders: phenotype stratification and biomarker discovery.
Int J Mol Sci. 2020;21(17):6274.
99. Bearden CE, Glahn DC. Cognitive genomics: searching for the genetic roots of neuropsychological functioning. Neuropsychology. 2017;31(8):1003-19.
100. Rodriques SG et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363(6434):1463-67.
101. Lipkin WI et al. Cohort-guided insights into gene–environment interactions in autism spectrum disorders. Nat Rev Neurol. 2023;19(2):118-25.
102. Chapman G et al. Functional genomics and gene-environment interaction highlight the complexity of congenital heart disease caused by Notch pathway variants. Hum Mol Genet. 2020;29(4):566-79.
103. Cirnigliaro M et al. The contributions of rare inherited and polygenic risk to ASD in multiplex families. Proc Natl Acad Sci USA. 2023;120(31):e2215632120.
Zinc for Wilson’s Disease: What We Know and What We Don’t Know
Authors: *Fabiola Di Dato,1 Peter Hedera2
1. Department of Translational Medical Science, University of Naples Federico II, Italy
2. Department of Neurology, University of Louisville, Kentucky, USA *Correspondence to fabiola.didato@unina.it
Disclosure: The authors have declared no conflicts of interest.
Acknowledgements: Medical writing was provided by Eleanor Roberts, Beeline Science Communications, UK.
Disclaimer: The article content and views expressed herein are those of the authors.
Support: The publication of this independently commissioned article was funded through a grant by Orphalan.
Summary
Wilson’s disease (WD) occurs due to excess copper leading to hepatic or neurologic symptoms, or a combination of both. Treatment for symptomatic WD typically includes initial therapy with a chelator, such as D-penicillamine or trientine, with zinc salts recommended in some guidance documents for neurologic WD, although this is not universally accepted. For presymptomatic patients, guidelines suggest either chelators or zinc as initial therapy. Zinc therapy has been used for over 60 years, with effectiveness demonstrated in a number of studies or retrospective case reviews. However, there are few head-to-head controlled trials of zinc compared to chelators. Accordingly, more such trials are needed to understand the use of zinc for WD. The main adverse event associated with zinc therapy is gastrointestinal issues, which can be a limiting factor in the usefulness of zinc. Copper deficiency may occur in a minority of patients taking zinc, with early warning signs including leukopenia and pancytopenia. Further studies are needed to understand the mechanism of action of zinc for WD, and long-term studies are also needed to understand the adverse event profile.
Zinc can be an effective and safe choice for WD treatment; however, further studies are needed to fully understand the mechanism of action and use.
INTRODUCTION
Zinc salts are one of three approved treatments for the autosomal recessive condition Wilson’s disease (WD). Their use was first described in 1961, and further developed from the 1970s.1,2
However, while in some countries zinc is increasingly used to treat WD, in others, use is less evident. Along with the discussion about zinc’s current use for WD, the authors explore whether the reluctance to recommend or use this therapy may in part be due to a paucity of scientific
PHARMA
PARTNERSHIP
knowledge, as well as inexperience in using zinc compared to chelators. Knowledge gaps in the understanding of the use of zinc for patients with WD, and where there may be study opportunities, will be assessed.
WD onset is typically before age 40 years,3 with prevalence varying, depending on ethnicity, from 1:17,000−50,000 people.4,5 In paediatric patients, WD more typically presents as liver disease (hepatic Wilson’s disease [hWD]), which can range from asymptomatic cytolysis to acute liver failure, and includes elevated liver enzymes, splenomegaly, fatty liver, acute hepatitis, and cirrhosis.6,7 In adolescents and adults, initial presentation is commonly neurologic/ psychiatric (neurologic Wilson’s disease [nWD]), with symptoms including dysarthria, seizures, sleep disorders, dystonia, ataxia, hypokinesia, and tremor; and in some, depression, anxiety, personality changes, and psychosis.7-9 Around 95% with nWD and 50% with hWD show corneal copper Kayser–Fleischer rings.10
In WD,11 copper accumulation in the liver, brain, and other organs7,12,13 occurs due to functional deficits of copper ATPase, attributable to variants in ATP7B.14,15 This enzyme is involved in the binding of excess copper in hepatocytes by intracellular metallothioneins. Copper is then excreted in bile or incorporated into plasma ceruloplasmin. Metallothioneins can also bind dietary copper, which is retained within intestinal cells until they are sloughed off and excreted.7
WD can be fatal, so early diagnosis and treatment are vital.16 Symptomatic WD is typically first treated with chelators to promote copper excretion, then in-range copper levels are maintained with a chelator or zinc.7,12,17-21 The chelator D-penicillamine (DPA) mobilises and binds copper from tissues; trientine promotes copper excretion and decreases intestinal copper absorption. Ammonium tetrathiomolybdate may also be used, but clinical experience and drug availability is limited.7,9,12,17-21 Adverse events (AE) associated with DPA include neurological worsening, cutaneous eruptions, lymphadenopathy, neutropenia, thrombocytopenia, and proteinuria; as well
as later, rarer, nephrotoxicity, lupus-like syndrome, Goodpasture syndrome, and elastosis perforans serpiginosa9,17-19,21,22
AEs with trientine include early- or lateronset hypersensitivity reactions as well as rare lupus-like reactions, pancolitis, and sideroblastic anaemia.9,17-19,21
ZINC MECHANISM OF ACTION
The mechanism of action of zinc in WD is not fully elucidated. It is known that on administration, intestinal zinc can greatly increase intracellular metallothionein expression. As metallothioneins preferentially bind copper over zinc, this action can prevent intestinal and hepatic copper absorption/reabsorption.2,7,23-25 Additionally, zinc may directly reduce oxidant liver injury, which can occur due to excess copper.26 However, while the time lag from first zinc administration, typically around 3 weeks, is hypothesised to be due to the time needed to increase metallothionein levels and restore zinc balance,1 it is not clear why this lag varies between patients,1 and why in some patients zinc has no measurable effect.27
Animal models show that high copper levels may directly disrupt zinc levels and associated mechanisms, such as zinc’s role in lipid metabolism.28 Zinc itself may also affect other metals, such as iron, with which it shares a metal transporter into duodenal enterocytes.29 As such, zinc supplementation may both decrease copper levels, and restore zinc balance and zinc-dependent functions.28 Further work is needed to fully understand these mechanisms.
USE OF ZINC THERAPY
Zinc salts include acetate (ZA), sulphate (ZS), gluconate (ZG), or picolinate.17 The recommended dose is 50 mg 3x /day in adults, or 25 mg 2−3x /day in children, ≥1 hour before and ≥2 hours after eating for best absorption. This should maintain inrange copper levels while limiting effects if a dose is missed.30-32 While there is little perceived difference in copper control between zinc salts, ZS may be associated
with more gastrointestinal (GI) AEs, although there is wide inter-individual variability that merits investigation.19,27,33,34
Only ZA is approved by the U.S. Food and Drug Administration (FDA) for WD maintenance therapy,32,35 and by the European Medicines Agency (EMA) for all patients.36,37 Studies are needed to directly compare the efficacy and safety of all zinc salts, for them to be regulated similarly to ZA, and thus available for prescribing. These are important, as in a retrospective review of 31 patients who were recommended zinc, only 45% were taking ZA, with 42% taking non-prescription ZG, and the remainder taking ZS or picolinate. Additionally, it was found that 51% purchased their zinc salt without a prescription.38 These findings are of concern as problems may occur with regard to content and quality when buying unregulated zinc salts intended as dietary supplements, and not otherwise tested.38 Of concern too, non-prescription medication use is typically not formally recorded by a healthcare professional, leaving ‘ghost patients’ with WD who are taking zinc without any record of the type, dose, effects, or AEs. Studies are needed to understand how patients access zinc, what salts they use, and why they access it without a prescription, which may be for financial, availability, or AE issues.
Studies of Zinc for Wilson’s Disease
EMA/FDA approvals for ZA31,32,35 were based on uncontrolled clinical trials and case studies (Table 1). These, along with post-authorisation reviews, show that zinc therapy leads to adequate copper control and WD symptom stabilisation or improvement in both pre-symptomatic and symptomatic patients, and those initiated on chelation therapy and then switched later. Disease progression, in a minority of patients, was typically due to non-adherence.31,35,38-42
Zinc as Initial Therapy and Monotherapy
The use of zinc for initial therapy has gained attraction, with one study showing it is used in around 50% of Polish National Reference
Centre patients.52 While the EMA states that, “in principle, ZA is not recommended for initial therapy of symptomatic patients because of its slow onset of action,”31 it is noted that full symptom control with chelators may also be delayed due to the recommendation for titrated initiation to avoid neurological worsening.20,21 As such, investigation is needed into the time of onset of action comparing zinc to titrated chelators, to ascertain if there are differences that may affect the patient.
Although studies have been carried out with at least some patients initiated on zinc (Table 1),31,35,38-40,43-46 few provide direct comparison to chelators. In one that did, no significant differences were found between ZS or DPA in percentages achieving ‘therapeutic success’, although a significantly higher percentage of patients with hWD receiving DPA achieved ‘complete enzymatic success’. The authors concluded that “ZS may be considered a reasonable alternative to DPA as first-line therapy in all patients with WD.”44 A 2019 review by National Health Service (NHS) England concluded that “the best available evidence suggests zinc salt monotherapy or penicillamine is effective for treating most people with symptoms of WD.”53 However, in another, the conclusion regarding zinc monotherapy was that, “results for symptomatic neurologic disease are highly satisfactory,” but that “clinical outcome was less satisfactory for hepatic disease.”45
To aid the understanding of zinc as initial therapy, more prospective, randomised studies that investigate zinc versus chelators in newly diagnosed patients, as well as in patients with a wide range of presentations, are needed. Previously published retrospective studies evaluated zinc primarily in stable patients. Further prospective studies could help elucidate if there are differences in WD symptom control between patients initiated on zinc or chelator therapy, or those switching from a chelator to zinc. Longitudinal studies are also needed to understand how well WD symptom control is maintained with zinc.
Table 1: Selected studies on the use of zinc therapy for Wilson’s disease.
Initial treatment choice: presentation, potential AEs, costs; initial ZS: 59% hWD; 38% nWD. Median treatment: 4 yrs. nWD: 20.0% DPA to ZS switch, 23.8% ZS to DPA switch; hWD: 30.6% DPA to Z switch, 11.8% Z to DPA switch
ZS monotherapy after diagnosis (5 switched to ZA, 1 to ZG due to GI AEs). Follow-up: 1 yr and final (median 14 [range 2−30] yrs)
36% initiated on Zn, 86% continued; 64% initiated on DPA, and 70% switched to Zn monotherapy; 2% on combination therapy. Treatment duration: median 12 yrs, range 1.6−25.2 yrs
Symptomatic: 85% initially received chelation therapy. Presymptomatic: 63% ZA monotherapy, 37% DPA then ZA. Mean treatment: symptomatic: 3.2 yrs; presymptomatic: 3.1 yrs
150−1200 mg/day ZS; 68% previously on DPA. Z mean/ median treatment duration: 9.1/6.5 yrs
65% had prior chelation therapy. Zn treatment duration: 8 m−52 yrs; ZA (53%), ZG (37%), alternative Zn (ZS or ZP; 15%) (some switched). 46% symptomatic, 54% asymptomatic completed online survey
Outcomes
Symptom improvement/no change/deterioration: ZS: 42%/10%/0%; DPA: 32%/5%/10%; 44% on DPA switched to ZS (10: AEs; 5: inadequate symptom improvement); 12% on ZS switched to DPA (2: AEs; 2: inadequate symptom improvement)
hWD: significantly more on DPA (87.5%) achieved ‘complete enzymatic success’ compared to ZS (68.1%), mostly due to treatment compliance (80.4% with ZS, 97.1% with DPA; p=0.024). nWD: 20.6% on DPA, 14.3% on ZS gained independence, 2.9%/4.8%, respectively, lost independence (NSD between treatments)
Significant serum NCBC decrease; 80% with nWD improved, typically over median 12 m, some complete resolution. 80% of 10 with baseline brain abnormalities improved/no change, 20% worsened. 90% with hWD stabilised or improved; 2 required liver transplant
Treatment response: 74% on DPA, 87% on Zn. More treatment switches for AEs with DPA (p=0.03) or combination therapy (p=0.04) than Zn. Treatment failure in 12%, mainly due to noncompliance; switch higher with combination therapy compared to Zn (p=0.03) only
11% discontinued maintenance therapy, mostly non-compliant. Copper control: symptomatic adults (n=100), Yr 1: 91%; Yr 4: 97%; presymptomatic (n=23), Yr 2: 100%. In nWD patients, speech scores stable in most
Of 28 patients on a dose of up to 150 mg/day zinc, 82% ‘improved’, ‘stabilised’, or ‘asymptomatic’
In-range urinary copper levels: ZA 81%, ZG 73%, alternative Zn 57%. 24-hour urinary copper levels <25 µg/>100 µg: ZA 8%/12%, ZG 11%/10%, alternative Zn 29%/14%. 77% survey respondents: no ongoing WD symptoms, 23% symptomatic, no symptom worsening
Gupta, India, 2018,47 N=31 (68% M) with hWD (32% also with nWD)
Sinha, India, 2008,48 N=45 (62% M; 88.8% with nWD)
Stabilised with DPA, switched to ZS. Treatment duration: median 506 w (range 112−1030). Baseline WD severity scores: mild−severe. Switching mostly due to cost
DPA and ZS for 107.4±67.3 m, then ZS monotherapy for 27.2±8.5 m
Overall mean severity/WD scores decreased; complications in minority (DPA or ZS). Neurological worsening: 16% on ZS, mostly non-compliant, improved with further therapy. Decompensated liver disease improvements with DPA (n=17) maintained with ZS
97.7% remained stable or improved
Table 1 continued.
Author, country, year, patients
Paediatric studies
Brewer, USA, 2001,49
N=34 (2.3−17.6 yrs at diagnosis; 56% M; 15% <10 yrs of age; 30% nWD, 20% hWD, 50% presymptomatic)
Abbassi, Morocco, 2022,50 N=46 (48% F; mean age at diagnosis: 10.9±2.8 yrs; 8.7% asymptomatic; 47.8% nWD, 43.5% hWD)
Marcellini, Italy, 2005,51 N=22 (55% F; mean age 6.2 [range 3−12] yrs); presymptomatic
Treatment
Dose: ZA 50−150 mg/day
Treatment duration: 1.3−7.6 years
43 initially treated with DPA, 30 on maintenance treatment: 70% DPA; 26.7% ZA; 3.3% ZS
ZS monotherapy+low copper diet
Outcomes
Significant decreases from baseline copper levels, majority obtained target by Yr 1. Neurological symptoms, liver function: significant improvement from baseline, no deterioration
DPA switched to Zn (n=9) due to AEs; ZS discontinued (n=1) due to GI AEs. Symptoms improved in majority, marked clinical improvement in fewer with nWD than hWD. 38% symptomatic patients died: fulminant hepatitis, advanced WD, or stopping treatment
Hepatic histologic scores, enzyme levels, copper content significantly decreased; latter remained above normal. Normal psychomotor development and growth. One AE: temporary epigastric pain
Although many patients initiated on zinc or DPA continue the treatment, in others, the treatment is switched.31,43,44,46 In one study, while for nWD switching was at a similar rate between treatments (around 22%), for hWD, more patients initially treated with DPA (30.6%) were switched to zinc than vice versa (11.8%), especially in the first 6 months. Therapy switch because of AEs occurred in 15.3% on DPA, mostly due to albuminuria, and 2.6% on ZS (p=0.008). Switch due to lack of improvement was 7.7% for ZS, and 2.8% for DPA (p=0.279).44
DPA may also be switched to zinc due to cost.48 However, two Indian studies concluded that “in resource-constrained settings, sequential DPA followed by zinc therapy may be safe and effective across all degrees of baseline disease severity,”47 and that “withdrawal of DPA from DPA/ZS maintenance therapy was effective, safe, and economic for almost all patients.”48 There are problems though, that when, or how, to switch therapies is not well defined in most guidelines.
Combination zinc/chelator therapy is off-label and rarely studied.18 A pooled analysis54 showed that effectiveness was lower, and AEs higher than with DPA,55 trientine,56 or zinc57 alone, especially for hWD. The mortality rate was highest with a DPA/ZS combination.54 While the Indian Society, American Association for the Study of Liver Diseases (AASLD), and European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) guidelines do suggest combination therapy use (Table 2), especially for severe disease, drugs should be given at widely spaced time intervals to prevent zinc chelation.19,20,33 However, evidence levels for these recommendations are low, and the efficacy and safety of zinc/chelator combinations need further investigation.
Paediatric Cases
In children, starting doses of all treatments are typically lower, and age- and weightdependent. In symptomatic patients, zinc can significantly decrease copper levels, and for most, significantly improve neurological symptoms and liver
Table 2: Pharmacological treatment guidance for the use of zinc for Wilson’s disease (by date of issue). Initial Maintenance
Notes
DPA (1st line), TETA (2nd line except where AEs with DPA), Zn (3rd line)
BASL: A practical guide21 (2022)
DPA, TETA, Zn N/A
Chelation therapy can be continued throughout pregnancy; no advise against breastfeeding should be given to females on chelation therapy Zn (adults) only initiated in specialist centres, not as monotherapy in cirrhosis unless other treatments unavailable/ contraindicated. No strong recommendation for Zn in children due to inadequate data
AASLD practice guidance20 (2022)
DPA, TETA DPA, TETA, Zn DPA, TETA, Zn
Dose decrease of DPA and TETA in pregnancy. Pros and cons should be weighed in each case of breastfeeding
Indian society (INASL, ISPGHN, MDSI) clinical practice guidelines19 (2019)
Dose decrease of DPA and TETA in pregnancy. Breastfeeding under chelation therapy not recommended
TETA may be better tolerated; presymptomatic and maintenance treatments may be at lower doses than initial therapy. Combined therapy can be used for decompensated cirrhosis and acute liver injury
Suggested that zinc can be started from age 2 years in presymptomatic patients. DPA or TETA±Zn can be used for acute hWD
Starting dose dependent on age, and for Zn, also weight. Occurrence of DPA-related AEs should prompt discontinuation, and switching to TETA or Zn according to liver disease severity
Advantages of combination therapy are not known
AASLD: American Association for the Study of Liver Diseases; AE: adverse event; BASL: British Association for the Study of the Liver; DPA: D-penicillamine; EASL: European Association for the Study of the Liver; ESPGHAN: European Society for Paediatric Gastroenterology, Hepatology and Nutrition; hWD: hepatic Wilson’s disease; INSAL: Indian National Association for Study of the Liver; ISPGHN: Indian Society of Pediatric Gastroenterology, Hepatology, and Nutrition; MDSI: Movement Disorders Society of India; nWD: neurologic Wilson’s disease; TETA: trientine; Zn: zinc salts.
function,49-51 although it was noted in one study that marked improvement was found in fewer with nWD compared to hWD50 (Table 1). Further studies are needed to understand if there are differences in symptom control with zinc therapy between pre-symptomatic and symptomatic children with WD. Longitudinal comparison studies are needed to evaluate drug-related longterm AEs and efficacy, and to understand which therapy is most suitable for lifelong treatment.
Guidance Documents for the Use of Zinc Therapy
The positioning of zinc therapy varies between guidance documents (Table 2). For example, for initial therapy in symptomatic patients, the AASLD does not recommend zinc,20 the British Association for the Study of the Liver (BASL) places zinc third-line,21 and the European Association for the Study of the Liver (EASL)18 and relevant Indian societies19 guidelines recommend zinc as initial therapy in patients with nWD only. Guidelines also vary with regard to recommendations for pre-symptomatic patients, and for maintenance therapy.18-21,33
As the guidelines are drawn up by experts in the field, the reasons for the differences may be due to more experience, and thus confidence in chelators and low level of available evidence. Indeed, the Indian joint societies guidelines state that “each centre uses a protocol based on their experience and patient compliance.”19 Robust, comparative trials of zinc and chelators that include a homogenous group of patients on which to base recommendations are needed.
SAFETY OF ZINC
Few studies directly compare treatments with regard to safety, but there are
indications that while zinc is more likely to be discontinued than DPA due to treatment failure,57 it may have a better safety profile, and be more tolerated than DPA.41-43
Gastrointestinal Symptoms
Overall, the EMA concludes that “gastric irritation is the only AE for which there is good evidence of a causal link to zinc.”31 Across studies, gastric irritation with zinc occurred in 9−65% of patients, with symptoms including a burning sensation in the throat, cough, abdominal pain, nausea, and vomiting.31,38,49,58,59 Where endoscopic evaluation has been warranted, gastric mucosal alterations, including redness, erosions, and for some, ulcers, have been noted with ZA. These were resolved following therapy cessation.59 In one study, ZS administration significantly increased the odds of gastropathy compared to DPA (p=0.01) or no treatment (p=0.02).58 Even though GI AEs are those most associated with zinc,31 there is a lack of distinct recommendations for how to best track all drug-related AEs if these occur, or if mild AEs become serious. Studies are needed to find if AEs are tracked; for example, by using a patient diary or phone application, this could lead to earlier intervention, and better-informed treatment decisions.
As GI symptoms are primarily shown with the first dose of zinc prior to breakfast, patients who experience such may be advised to take zinc between breakfast and lunch (unless this impairs their ability to take zinc 3x /day), or with a small amount of a high protein food.60 Of note, as zinc absorption can be decreased by certain foods,61 co-administration with food is not advised, and further studies are needed to fully ascertain the relationship between zinc absorption and food types in WD. In some studies, switching the zinc salt led to symptom resolution.38 However, this is not recommended in guidelines, and
Figure 1: Knowledge gaps and study opportunities with regard to zinc use in Wilson's disease.
Knowledge gaps
Mechanism of action/efficacy
Zinc mechanism of action, absorption, time lag to action
Mechanism of copper deficiency and neurological worsening
Patient genetic/ metabolic differences that can affect zinc processing
Mechanism of hepatic/pancreatic enzyme increase and cholesterol changes
Efficacy of over-thecounter zinc salts manufactured to food standards only
Monitoring/ adherence
Best ways to monitor zinc and copper levels
Monitoring compliance and reasons for nonadherence
Accessing/ dosing zinc
Why patients switch zinc types and how issues are communicated
How often is zinc taken <3x /day by patients?
comparison studies of salts are needed to ascertain if GI symptoms, or any AEs occur to a greater extent according to salt, and if an AE does occur, does switching to a different salt lessen such effect.
Copper Deficiency
Development of copper deficiency (CD), typically over months or years, is rare in patients receiving WD therapy (0.9% in one study),62 and predominantly only occurs when zinc is administered alone or with a chelator.63 It occurs potentially due to metallothionein overproduction, and can occur in both symptomatic and presymptomatic patients.62
Study opportunities
Efficacy and safety of zinc in combination with chelators
Efficacy and safety comparing different zinc salts
Head-to-head comparison studies between treatments
CD may result in anaemia, neutropenia, or leuconeutropenia, or in more severe cases, lower limb paraesthesia, progressive sensory ataxic gait disorder, and irreversible neurological symptoms.62,64-66 Zinc-induced CD is best predicted by plasma copper levels ≤6 µmol/L combined with plasma zinc levels ≥18 µmol/L (indicating potential overtreatment),67 with warning signs including severely reduced urinary copper excretion, combined with low levels of total serum copper and ceruloplasmin.63 CD usually resolves completely following dose decrease or treatment switching.62 The role of individual metabolism and/or genotype merits investigation.
Further studies regarding zinc’s mechanism of action are needed to understand the relationship with CD, why CD only occurs in some patients, or if occurrence differs between zinc salts. Subclinical CD may not be recognised if proper attention is not paid to the aforementioned warning signs capable of guiding early recognition and prompt reduction or discontinuation of zinc.
Neurological Worsening
Guidance documents point to early neurological deterioration in 8.8−35.3% of patients following WD therapy initiation.18-21,44,57,68 Neurological deterioration is more common in nWD and less common in pre-symptomatic WD.70 While in some studies neurological worsening more often occurred with DPA,44,68,69 in others, occurrence was similar between DPA, zinc, or trientine.57 As treatment switching can lead to gradual neurological improvement,69 it is presumed that this AE is directly associated with therapy; however, there are unknowns that need investigating, including why neurological deterioration occurs only in some patients, the relationship between copper control and neurological worsening, and whether it is greater according to zinc salt.
Liver Enzymes, Pancreatic Enzymes, and Cholesterol
Zinc administration can lead to alanine aminotransferase levels >1−2x the upper level of normal;2,38,70 however, this may not be accompanied by cholestasis31,70 or disease progression.71 While elevated liver enzymes may be due to treatment non-adherence,31 a study showed this even when urinary copper was within target.38 Further studies are needed to investigate the relationship between zinc and hepatic enzymes.
Zinc dose may also influence pancreatic amylase and lipase levels, although this may not lead to pancreatitis symptoms.31,70 One study suggested that dosing normalised for weight or body surface area may limit potentially damaging pancreatic enzyme increases; however, this may not be possible with set-dose tablets.72
Zinc therapy has also been associated with significant reductions in high density lipoprotein (HDL) cholesterol (by around 20%) and total cholesterol (by around 10%) in males only. These reductions have not been found to influence cardiovascular risk,73 and no such risk was found in the overall studies presented to the EMA.31 In a paediatric cohort, the mean HDL/total cholesterol ratio significantly decreased by around 10%.49 This was hypothesised to indicate potential CD due to over-treatment.31 Lower serum cholesterol levels in hWD have been reported regardless of treatment.74 Again, further studies are needed to ascertain the relationship between zinc therapy and HDL, and cholesterol changes.
THE IMPORTANCE OF MONITORING AND TREATMENT ADHERENCE
Following treatment initiation, lifetime monitoring includes WD symptoms, serum and urinary copper, ceruloplasmin, liver enzymes, and complete blood count.18-21 With zinc therapy, 24-hour urinary copper monitoring is used to ascertain if levels are within limits (typically 30−100 µg).7,18-21,33,37,60 However, as copper is also excreted in faeces and sweat,75 urinary copper may not be an entirely accurate measure of copper levels. It remains to be ascertained if this is the best way to monitor WD treatment, why some patients develop neurological worsening or CD, and why some do not respond to treatment.
Hepatic copper levels can be obtained via liver biopsy, which is impractical beyond clinical studies,2 or via less invasive tomography/CT scans following ingestion of copper-64.27 Copper metabolism control can also be evaluated via assay of nonceruloplasmin-bound plasma copper (typically 5−25 µg/dL); however, it is of debate as to which is the best method for determining this.20 More formal evaluation of the relationship between urinary zinc and urinary copper levels is needed, including between symptomatic and presymptomatic populations, and those with nWD or hWD.
Treatment adherence is of paramount importance in WD, with overall survival in treatment-compliant patients comparable to the general population.76 Non-adherence in pre-symptomatic patients, with either zinc or a chelator, is associated with the development of WD symptoms, including hepatic failure, and even death.76 It is thus troubling that non-adherence to zinc may be high.31,35,38-40,57
According to AASLD guidelines, nonadherence may occur in previously symptomatic patients due to ‘a false sense of security after prolonged treatment’, and in pre-symptomatic patients due to ‘incomplete buy-in for the diagnosis’.20 As such, monitoring should include 24-hour zinc excretion (typically >2,000 µg/day), along with serum levels (typically >125 µg/dL), to assess compliance.7,18-21,33,37,60 Of note, while zinc levels can help detect compliance, they may not provide a true picture, as a patient who has lapsed with taking their required zinc dose may revert to correct dosing if they know a monitoring visit is imminent, then lapse post-visit. Further investigation is needed to ascertain better ways to monitor and aid dose compliance, especially as 24-hour urine collection may be difficult for a patient.
Adherence to zinc may be hampered by gastritis,31,38,49,58-60 especially with the morning dose, treatment costs, supply issues, and treatment schedule as not all patients can take a tablet at work or school at a time, remote from their lunch break. While 50 mg 3x /day is recommended, this is partially to negate missed doses. However, in a study of healthy participants, reductions in hepatic copper content were achieved with either 150 mg 1x /day or 50 mg 3x /day ZA. Although the latter was superior to the former overall, high inter-individual differences were shown.27 Studies are needed to investigate whether such dosing is adequate to maintain copper balance and symptom control in patients with WD without increasing AEs. It may also be that fewer doses are missed with once- or twice-daily dosing, leading to better adherence and subsequent outcomes. Better guidance is needed as to how to individually ascertain what zinc dosing schedule best fits with an individual’s needs regarding symptom control, AEs, and their lifestyle.
REPRODUCTION IN PEOPLE WITH WILSON’S DISEASE
WD is associated with ovulatory and menstrual dysfunction,77,78 but for patients taking zinc, while there may be alterations in some fertility-related hormones, fertility is not necessarily affected.79 AASLD guidelines advise advanced pregnancy planning, with treatment stabilised before conceiving.20 Guidance documents point to lowering of DPA and trientine doses during pregnancy, but not of zinc.18-20 Miscarriage rates for patients with WD may be higher than the general population.80,81 However, while one study found this occurred despite zinc or DPA therapy,80 another found the relative risk to be significantly lower with these drugs.81 Offspring may be more likely to have a low birth weight, with no difference according to therapy type.80
The EMA, and some guidance documents, advise against breastfeeding while receiving zinc therapy due to potential zinc-induced CD in the infant.17,20,31 However, while zinc levels may increase in breastmilk when taking this therapy, this was apparently without consequences in one study, leading the authors to conclude that breastfeeding while taking zinc was safe.82 Despite all these findings, there are differences in guidance documents regarding zinc use during pregnancy and breastfeeding, (Table 2) and there is a need for updates, to help guide prescribers.18-21
CONCLUSION
Zinc salts have an established role in WD treatment;18-21,33 however, the lack of head-to-head studies and randomised clinical trials limits recommendations as to where zinc should be positioned compared to chelators. With the above considerations in mind, Figure 1 highlights where there are knowledge gaps in the understanding of the use of zinc for patients with WD, and where there may be study opportunities to enhance this understanding and inform guidance documents.
References
1. Hoogenraad TU et al. Oral zinc sulphate as long-term treatment in Wilson's disease (hepatolenticular degeneration). Eur Neurol. 1979;18(3):205-11.
2. Brewer GJ et al. Oral zinc therapy for Wilson's disease. Ann Intern Med. 1983;99(3):314-9.
3. Poujois A, Woimant F. Challenges in the diagnosis of Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S67.
4. Wallace DF, Dooley JS. ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum Genet. 2020;139(8):1065-75.
5. Sandahl TD et al. The prevalence of Wilson's disease: an update. Hepatology. 2020;71(2):722-32.
6. Trocello J-M et al. [Wilson disease]. La Presse Médicale. 2009;38(7):1089-98. (In French).
7. Brewer GJ, Yuzbasiyan-Gurkan V. Wilson disease. Medicine (Baltimore). 1992;71(3):139-64.
8. Starosta-Rubinstein S et al. Clinical assessment of 31 patients with Wilson's disease. Correlations with structural changes on magnetic resonance imaging. Arch Neurol. 1987;44(4):365-70.
9. Kerkar N, Rana A. Wilson disease in children. Clin Liver Dis. 2022;26(3):473-88.
10. Kasztelan-Szczerbinska B, CichozLach H. Wilson's disease: an update on the diagnostic workup and management. J Clin Med. 2021;10(21):5097.
11. Frydman M et al. Assignment of the gene for Wilson disease to chromosome 13: linkage to the esterase D locus. Proc Natl Acad Sci U S A. 1985;82(6):1819-21.
12. Trocello J-M et al. Wilson's disease, 100 years later… Rev Neurol (Paris). 2013;169(12):936-43.
13. Lutsenko S et al. Function and regulation of human coppertransporting ATPases. Physiol Rev. 2007;87(3):1011-46.
14. Terada K et al. Restoration of holoceruloplasmin synthesis in LEC rat after infusion of recombinant adenovirus bearing WND cDNA. J Biol Chem. 1998;273(3):1815-20.
15. Bull PC et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327-37.
16. Daniel-Robin T et al. Epidemiology, treatment and burden of Wilson
disease in France: a 10-year analysis of the national health insurance database. Clin Res Hepatol Gastroenterol. 2022;46(10):101992.
17. Aggarwal A, Bhatt M. Advances in treatment of Wilson disease. Tremor Other Hyperkinet Mov (N Y). 2018;8:525.
18. European Association for the Study of the Liver (EASL). EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56(3):671-85.
19. Nagral A et al. Wilson's Disease: clinical practice guidelines of the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India. J Clin Exp Hepatol. 2019;9(1):74-98.
20. Schilsky ML et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2022;DOI:10.1002/hep.32801.
21. Shribman S et al. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022;7(6):560-75.
22. Atzori L et al. D-penicillamine elastosis perforans serpiginosa: description of two cases and review of the literature. Dermatol Online J. 2011;17(4):3.
23. Schilsky ML et al. Hepatocellular copper toxicity and its attenuation by zinc. J Clin Invest. 1989;84(5):1562-8.
24. Yuzbasiyan-Gurkan V et al. Treatment of Wilson's disease with zinc: X. Intestinal metallothionein induction. J Lab Clin Med. 1992;120(3):380-6.
25. Sturniolo GC et al. Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson's disease patients. Am J Gastroenterol. 1999;94(2):334-8.
26. Farinati F et al. Zinc treatment prevents lipid peroxidation and increases glutathione availability in Wilson's disease. J Lab Clin Med. 2003;141(6):372-7.
27. Munk DE et al. Effect of oral zinc regimens on human hepatic copper content: a randomized intervention study. Sci Rep. 2022;12(1):14714.
28. Meacham KA et al. Altered zinc balance in the Atp7b(-/-) mouse reveals a mechanism of copper toxicity in Wilson disease. Metallomics. 2018;10(11):1595-606.
29. Szabo R et al. Iron, copper, and zinc homeostasis: physiology,
physiopathology, and nanomediated applications. Nanomaterials (Basel). 2021;11(11):2958.
30. Hill GM et al. Treatment of Wilson's disease with zinc. I. Oral zinc therapy regimens. Hepatology. 1987;7(3):5228.
31. European Medicines Agency (EMA). Wilzin: EPAR - scientific discussion. 2005. Available at: https://www.ema. europa.eu/en/documents/scientificdiscussion/wilzin-epar-scientificdiscussion_en.pdf. Last accessed: 21 May 2024.
32. Teva Pharmaceuticals USA. GALZIN (zinc acetate) capsules. 2020. Available at: https://www.galzin.com/ galzin.pdf. Last accessed: 21 May 2024.
33. Socha P et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018;66(2):334-44.
34. Wiernicka A et al. Gastrointestinal side effects in children with Wilson's disease treated with zinc sulphate. World J Gastroenterol. 2013;19(27):4356-62.
35. Center for Drug Evaluation and Research (CDER). Approval package for GALZIN capsules. 1997. Available at: https://www.accessdata.fda.gov/ drugsatfda_docs/nda/97/020458ap. pdf. Last accessed: 21 May 2024.
36. European Medicines Agency (EMA). Wilzin. 2019. Available at: https://www. ema.europa.eu/en/medicines/human/ EPAR/wilzin. Last accessed: 21 May 2024.
37. European Medicines Agency EMA). Wilizin: Summary of product characteristics. 2019. Available at: https://www.ema.europa.eu/en/ documents/product-information/ wilzin-epar-product-information_ en.pdf. Last accessed: 21 May 2024.
38. Camarata MA et al. Zinc maintenance therapy for Wilson disease: a comparison between zinc acetate and alternative zinc preparations. Hepatol Commun. 2019;3(8):1151-8.
39. Hoogenraad TU et al. Management of Wilson's disease with zinc sulphate. Experience in a series of 27 patients. J Neurol Sci. 1987;77(2-3):137-46.
40. Hoogenraad TU, Overeynder E. ‘Treatment’, Wilson's disease (2001) 2nd edition, Amsterdam: Intermed Medical Publishers, pp.138-178.
41. Tang S et al. Comparison of the effectiveness and safety of d-Penicillamine and zinc salt treatment for symptomatic Wilson disease: a
systematic review and meta-analysis. Front Pharmacol. 2022;13:847436.
42. Appenzeller-Herzog C et al. Comparative effectiveness of common therapies for Wilson disease: a systematic review and meta-analysis of controlled studies. Liver Int. 2019;39(11):2136-52.
43. Członkowska A et al. Effects of longterm treatment in Wilson's disease with D-penicillamine and zinc sulphate. J Neurol. 1996;243(3):269-73.
44. Członkowska A et al. D-penicillamine versus zinc sulfate as first-line therapy for Wilson's disease. Eur J Neurol. 2014;21(4):599-606.
45. Linn FHH et al. Long-term exclusive zinc monotherapy in symptomatic Wilson disease: experience in 17 patients. Hepatology. 2009;50(5):1442-52.
46. Ranucci G et al. Zinc monotherapy is effective in Wilson's disease patients with mild liver disease diagnosed in childhood: a retrospective study. Orphanet J Rare Dis. 2014;9:41.
47. Gupta P et al. Maintenance zinc therapy after initial penicillamine chelation to treat symptomatic hepatic Wilson's disease in resource constrained setting. Indian J Gastroenterol. 2018;37(1):31-8.
48. Sinha S, Taly AB. Withdrawal of penicillamine from zinc sulphatepenicillamine maintenance therapy in Wilson's disease: promising, safe and cheap. J Neurol Sci. 2008;264(12):129-32.
49. Brewer GJ et al. Treatment of Wilson's disease with zinc XVI: treatment during the pediatric years. J Lab Clin Med. 2001;137(3):191-8.
50. Abbassi N et al. Epidemiology, clinical features, and mortality rate of Wilson disease in Moroccan children: a pediatric case series. Arch Pediatr. 2022;29(6):453-8.
51. Marcellini M et al. Treatment of Wilson's disease with zinc from the time of diagnosis in pediatric patients: a single-hospital, 10-year follow-up study. J Lab Clin Med. 2005;145(3):139-43.
52. Członkowska A et al. Seven decades of clinical experience with Wilson's disease: report from the national reference centre in Poland. Eur J Neurol. 2022;DOI:10.1111/ene.15646.
53. National Health Service (NHS) England. Evidence review: zinc salts for Wilson disease. 2018. Available at: https://www.england.nhs.uk/wpcontent/uploads/2018/12/Evidencereview-Zinc-salts-for-Wilson-disease. pdf. Last accessed: 21 May 2024.
54. Chen J-C et al. Combination therapy using chelating agent and zinc for Wilson's disease. J Med Biol Eng. 2015;35(6):697-708.
55. Bruha R et al. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int. 2011;31(1):83-91.
56. Weiss KH et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-35.e1-2.
57. Weiss KH et al. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140(4):118998.e1.
58. Antczak-Kowalska M et al. Gastropathy in patients with Wilson disease. Scand J Gastroenterol. 2020;55(1):14-7.
59. Iwamuro M et al. Zinc acetate dihydrate tablet-associated gastric lesions. Intern Med. 2022;61(13):19318.
60. Brewer GJ. Zinc and tetrathiomolybdate for the treatment of Wilson's disease and the potential efficacy of anticopper therapy in a wide variety of diseases. Metallomics. 2009;1(3):199-206.
61. Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12(3):762.
62. Chevalier K et al. Can patients with Wilson's disease develop copper deficiency? Mov Disord Clin Pract. 2023;10(9):1306-16.
63. Litwin T et al. Copper deficiency as Wilson's disease overtreatment: a systematic review. Diagnostics (Basel). 2023;13(14):2424.
64. Halfdanarson TR et al. Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol. 2008;80(6):523-31.
65. Gabreyes AA et al. Hypocupremia associated cytopenia and myelopathy: a national retrospective review. Eur J Haematol. 2013;90(1):1-9.
66. Litwin T et al. Brain metal accumulation in Wilson's disease. J Neurol Sci. 2013;329(1-2):55-8.
67. Duncan A et al. The predictive value of low plasma copper and high plasma zinc in detecting zinc-induced copper deficiency. Ann Clin Biochem. 2016;53(Pt 5):575-9.
68. Antos A et al. Early neurological deterioration in Wilson's disease: a systematic literature review and meta-analysis. Neurol Sci. 2023;44(10):3443-55.
69. Kumar M et al. Management of children and adolescents with Wilson disease and neurological worsening following D-Penicillamine therapy: a single centre experience. Ann Indian Acad Neurol. 2022;25(4):698-702.
70. Yuzbasiyan-Gurkan V et al. Treatment of Wilson's disease with zinc. V. Changes in serum levels of lipase, amylase, and alkaline phosphatase in patients with Wilson's disease. J Lab Clin Med. 1989;114(5):520-6.
71. Iorio R et al. Serum transaminases in children with Wilson's disease. J Pediatr Gastroenterol Nutr. 2004;39(4):331-6.
72. Di Dato F et al. Towards tailored zinc therapy in Wilson disease patients. European Society for Paediatric Gastroenterology, Hepatology and Nutrition; 17-20 May, 2022.
73. Brewer GJ et al. Treatment of Wilson's disease with zinc. IX: response of serum lipids. J Lab Clin Med. 1991;118(5):466-70.
74. Seessle J et al. Alterations of lipid metabolism in Wilson disease. Lipids Health Dis. 2011;10:83.
75. Kuan W-H et al. Excretion of Ni, Pb, Cu, As, and Hg in sweat under two sweating conditions. Int J Environ Res Public Health. 2022;19(7):4323.
76. Dzieżyc K et al. Compliant treatment with anti-copper agents prevents clinically overt Wilson's disease in presymptomatic patients. Eur J Neurol. 2014;21(2):332-7.
77. Kaushansky A et al. Endocrine studies of the ovulatory disturbances in Wilson's disease (hepatolenticular degeneration). Fertil Steril. 1987;47(2):270-3.
78. Tarnacka B et al. Procreation ability in Wilson's disease. Acta Neurol Scand. 2000;101(6):395-8.
79. Iorio GG et al. Reproductive function of long-term treated patients with hepatic onset of Wilson's disease: a prospective study. Reprod Biomed Online. 2021;42(4):835-41.
80. Roseira J et al. Gynecological history to diagnosis and pregnancy outcomes in diagnosed Wilson's disease patients under therapy - a bicentric matchedcontrol cohort study. Rev Esp Enferm Dig. 2022;114(4):198-203.
81. Pfeiffenberger J et al. Pregnancy in Wilson's disease: management and outcome. Hepatology. 2018;67(4):1261-9.
82. Kodama H et al. Copper and zinc concentrations in the breast milk of mothers undergoing treatment for Wilson's disease: a prospective study. BMJ Paediatr Open. 2021;5(1):e000948.
Fever, Sore Throat, and Abdominal Pain – Connecting the Dots to a ‘Forgotten’ Disease: A Case Report of Atypical Lemierre’s Syndrome
Authors: Taniya Yasmin,1 Sara Glazer,2 Breanna A. Chen,3 *Farah Abdulsatar3
1. Department of Epidemiology and Biostatistics, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Canada
2. Department of Paediatrics, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Canada
3. Children's Health Research Institute, Lawson Health Research Institute, University of Western Ontario, London, Canada
*Correspondence to fabduls@uwo.ca.
Disclosure: The authors have declared no conflicts of interest. Written patient consent was obtained. There were no funders to report for this submission. There are no financial relationships relevant to this article to disclose from all the identified authors.
Acknowledgements: The authors would like to express their gratitude to the patient and his family, for recognising the importance of sharing their clinical case, thereby contributing to the expansion of knowledge among healthcare providers. Yasmin has contributed towards conceptualisation (equal), writing the original draft (lead), writing-review, and editing (equal). Abdulsatar has contributed towards conceptualisation (equal), writing the original draft (supporting), writing-review, and editing (equal). Chen has contributed towards conceptualisation (equal), writing the original draft (supporting), writing-review, and editing (equal). Glazer has contributed towards conceptualisation (equal), writing the original draft (supporting), writing-review, and editing (equal).
Received: 16.11.23
Accepted: 08.05.24
Keywords: Atypical, case report, Fusobacterium, Lemierre’s syndrome.
Lemierre's syndrome (LS) is a rare but potentially life-threatening condition resulting from oropharyngeal infections. It is characterised by septic thrombophlebitis of the internal jugular vein and disseminated metastatic abscesses. However, atypical presentations with unusual sites of thrombosis and spread have been reported. The authors present a case of LS with an atypical presentation in a previously healthy 17-year-old male. This case highlights the need for a high level of suspicion, and comprehensive investigation in cases of unexplained sepsis following oropharyngeal infections, as LS can have atypical presentations and potentially lifethreatening complications. The traditional definition of LS may need to be re-evaluated in light of such atypical manifestations.
Key Points
1. This case report highlights Lemierre's syndrome (LS), a rare but serious condition arising from oropharyngeal infections, highlighting the necessity of increased awareness among clinicians to prevent severe outcomes.
2. The article details a case of atypical LS, characterised by unusual thrombosis sites and septic complications, underscoring the importance of comprehensive investigations and an extensive differential in cases of unexplained sepsis.
3. Clinicians should consider LS in differential diagnoses of sepsis following pharyngitis, particularly with atypical presentations. Early recognition and intensive treatment are crucial to improving patient outcomes in such potentially fatal conditions.
CASE HISTORY
A previously healthy 17-year-old male was transferred from a community hospital to the authors’ tertiary paediatric emergency department due to concern for sepsis. On presentation to the community hospital, the patient was febrile, tachycardic, and hypotensive. Before transfer, he was resuscitated with intravenous fluid boluses and was given a dose of ceftriaxone. Further history revealed that in the preceding 5 days, the patient had fevers with chills, sore throat, odynophagia with associated trismus, emesis, and rightsided groin pain. His past medical history was unremarkable, with no recent travel or sick contacts. He disclosed recent marijuana and tobacco use, and ongoing contact with four Rottweilers. His immunisations were complete up until 12 years of age; however, he was unimmunised for COVID-19. Upon admission to the authors’ hospital, the patient remained mildly tachycardic but was haemodynamically stable. General examination revealed a few small and tender lymph nodes in the anterior cervical chain (<1.5 cm), along with an area of tenderness at the left angle of the mandible. Oropharyngeal examination showed Grade 1 tonsils (tonsils just outside of the tonsillar fossa and occupying ≤25% of the oropharyngeal width) bilaterally.1 The patient had a normal cervical range of motion, but limited jaw opening of two to three finger breadths. Abdominal examination revealed mild tenderness in the epigastric and right inguinal region with no associated masses. The neurological examination was unremarkable.
DIFFERENTIAL DIAGNOSIS AND INVESTIGATIONS
Laboratory examinations showed a normal leukocyte count (9.5x109 cells/L), elevated c-reactive protein (270 mg/L), thrombocytopenia (36x109 /L), elevated D-dimer assay (3,226 µg/L), hyponatraemia (126 mEq/L), and high creatinine (147 µmol/L). Broad-spectrum intravenous antibiotics were initiated empirically including ceftriaxone, clindamycin, and vancomycin due to the concern for sepsis and potential toxic shock syndrome. Methicillin-resistant Staphylococcus aureus screen; sexually transmitted infection screen including HIV, syphilis, gonorrhoea, chlamydia, and hepatitis B; and serology assays for COVID-19, hepatitis B virus, hepatitis C, cytomegalovirus, Epstein–Barr virus, toxoplasmosis, Lyme, and parvovirus were all negative. Initial neck ultrasound and chest radiograph on presentation were unremarkable. Blood, throat, and urine cultures (collected after starting antibiotics) were negative.
Considering the patient’s initial presentation with neck stiffness and trismus, there was a strong suspicion of a potential deep neck pathology. On admission, a head and neck CT scan revealed left retromandibular vein thrombosis and infiltrative densities, suggestive of thrombophlebitis. A repeat ultrasound and Doppler study of the neck also revealed similar findings (Figure 1 and Figure 2). Based on these findings, a presumptive diagnosis of Lemierre’s syndrome (LS) was made, and further imaging was initiated. MRI of the head
1: Occlusive retromandibular vein on ultrasonography.
Figure
Figure 2: Doppler image shows lack of flow through the retromandibular vein.
showed punctate diffusion restriction of the right frontal–parietal subcortical white matter, suggestive of cerebral infarction. Persistent right groin pain prompted further radiological investigations. CT abdomen/ pelvis showed a moderate amount of non-specific ascites. Subsequent MRI of the pelvis identified a right iliacus abscess measuring 2.7 cm x 3.8 cm, confined to the iliacus musculature without extension to adjacent muscles or bones (Figure 3 and Figure 4). Culture of the fluid drained from the abscess via ultrasound-guided aspiration, while on antibiotics, was sterile. 16S ribosomal RNA (16S rRNA) PCR was also performed and confirmed the presence of Fusobacterium necrophorum. A cardiac bubble study revealed normal cardiac anatomy with no shunting. This constellation of clinical sepsis, a history of pharyngotonsillitis, thrombus of left retromandibular vein, acute cerebral infarct, and F. necrophorum pelvic abscess confirmed the authors’ working clinical diagnosis of LS.
TREATMENT, OUTCOME, AND FOLLOW-UP
Following the clinical diagnosis of LS, antibiotic therapy was adjusted to ceftriaxone and metronidazole. Anticoagulation therapy with enoxaparin was initiated and later transitioned to rivaroxaban. The patient experienced a favourable recovery throughout his hospital stay, with significant clinical and biochemical improvement during his admission. Despite the radiologically significant cerebral infarction on MRI, the patient did not present with any neurological deficits clinically. Paediatric neurology assessed the patient during admission, reviewed the MRI findings, and confirmed that imaging findings were clinically nonsignificant. He was discharged on the 12th day of admission with a prescription for intravenous ceftriaxone, administered via a peripherally inserted central catheter line, to complete a total course of 6 weeks. Additionally, the patient received anticoagulation therapy for a duration of 3 months. At the 2-month follow-up, a neck ultrasound revealed a stable appearance of the thrombus, and the patient exhibited
satisfactory clinical progress. Other followup laboratory investigations, including complete blood count and c-reactive protein, were normal.
Three months post-discharge, the patient re-presented to the emergency department with fever, neck pain, sore throat, and emesis. A repeat neck ultrasound revealed a new left internal jugular vein (IJV) thrombus. He underwent a septic workup, including blood cultures and a CT neck, which were negative for an infectious cause of his symptoms. It was determined that he likely had a viral illness in addition to the new left IJV thrombus. Additionally, it was discovered that the patient had not been adherent to his medication prior to this presentation. Two months after the second hospitalisation, and with consistent adherence to his anticoagulation therapy, a repeat ultrasound showed complete resolution of the left IJV thrombus. Consequently, the patient was discharged from the paediatric haematology clinic.
DISCUSSION
LS, often regarded as the ‘forgotten’ disease, is a rare but potentially life-threatening illness that primarily affects healthy young adults and adolescents.2 It typically arises from oropharyngeal infections, leading to thrombophlebitis of the IJV and distant metastatic abscesses. LS impacts the pulmonary system with pulmonary abscesses in 95% of the patients, which is often considered a key diagnostic criterion.3,4 The most common causative organism in LS is F. necrophorum, although other pathogens such as β-haemolytic streptococci have also been rarely reported.2,5 Atypical sites of infection associated with LS have been reported in the abdomen, often in association with urogenital infections,6 or with pelvic infections.7 While rare, CNS complications in LS such as meningitis and cerebral abscess have been reported.8,9 In the authors’ case, the progression of the disease had atypical features. The site of thrombosis was the left retromandibular vein, and the patient developed a right iliacus abscess and cerebral infarction as metastatic complications.
Figure 3: Axial view on MRI pelvis showing right iliacus abscess.
Figure 4: Coronal view on MRI pelvis showing right iliacus abscess.
In the case the authors presented, the occurrence of an iliacus abscess away from the primary cervical infection site is hypothesised to result from hematogenous spread rather than local extension. This hypothesis is supported by the absence of pelvic or abdominal infections that have been found to precede such abscesses,10 coupled with the identification of septic emboli in cervical veins, suggesting a possible route for pathogens to reach distant sites such as the iliacus muscle. This scenario is consistent with other rare reports in the literature where distant muscular abscesses developed in the absence of adjacent primary infections, highlighting the potential for hematogenous spread in cases of LS. Such cases highlight the importance of considering atypical infectious pathways in systemic infections, especially when clinical presentations seem to deviate from expected norms.11 Also, in the authors’ patient, the occurrence of cerebral infarction raises the question of its aetiology in the context of LS. While typically associated with septic emboli,9 there was no definitive evidence suggesting bacterial seeding, or abscess formation in the brain. Instead, the authors hypothesise that the hyper-coagulable state induced by systemic
inflammation and infection could have contributed to ischaemic stroke.12 This view is supported by the elevated inflammatory markers during the course of the patient’s illness and absence of venous-arterial shunts on imaging.
A search on PubMed for the past 20 years using the following terms: “cerebral infarction+Lemierre’s syndrome” and “iliacus muscle abscess+Lemierre’s syndrome,” yielded only four case reports of cerebral infarction, with only one case reported in the paediatric population. Additionally, only one case report discussed an iliacus muscle abscess in relation to LS. Table 1 reports the findings. The literature review was deliberately focused on using specific search terms to correlate directly with the atypical manifestations observed in the authors’ case. This targeted approach was chosen to ensure a thorough exploration of the most relevant studies, enabling a detailed and appropriate discussion on the rare occurrences of cerebral infarction and iliacus muscle abscess in LS.
Bird et al.13 conducted a systemic review on 78 cases of LS, challenging the traditional
Author Age in years, sex Infection sites
Laurence et al.19 58, male
Halawa et al.20 61, female
Yoko et al.9 59, male
Ratnasingham et al.11 14, female
Thrombosis of the two iliac veins, abscesses in muscles, pleural effusion complicating a pneumonia, and lumbar spine (L4-L5) spondylodiscitis
Thrombosis of the right internal jugular vein and sigmoid sinus, both cavernous sinuses, and superior ophthalmic veins
Thrombosis of the left external jugular vein and peritonsillar vein
Internal jugular vein thrombophlebitis
Organism Manifestations
Fusobacterium nucleatum
Site of abscesses: iliac and psoas muscle
Streptococcus anginosus
Fusobacterium necrophorum
Fusobacterium necrophorum
Congestive orbitopathy, and bilateral retinal, optic nerve, and cerebral infarctions
Table 1: Case reports of atypical Lemierre’s syndrome with cerebral infarction and/or iliacus muscle abscess.
understanding of the condition. Their findings revealed that approximately 21% of cases had primary infections in non-oropharyngeal sites, 31% of cases did not exhibit IJV thrombosis, and 30% lacked any pulmonary involvement. Based on these observations, the authors proposed redefining LS as F. necrophorum sepsis, incorporating thrombosis as an additional diagnostic criterion. While septic metastases are commonly associated with LS, and display significant anatomical variation, they are not essential for diagnosing the condition. The mainstay of LS treatment is prolonged broad-spectrum antibiotic therapy, typically administered for around 6 weeks, and guided by clinical response.14 Early diagnosis and antimicrobial therapy have significantly improved the prognosis of LS compared to the pre-antibiotic era, when fatality rates were as high as 5%.1517 The use of anticoagulant therapy in LS remains controversial.18 Due to the rarity of the condition, and the lack of randomised controlled studies, there are currently no definitive recommendations for the use of anticoagulation in LS. However, in highrisk patients with severe IJV thrombosis or septic spread despite antibiotic therapy, anticoagulation therapy can be considered.18 Given that the patient developed complications that were likely due to septic emboli, the authors opted to initiate anticoagulation alongside antimicrobial therapy.
Diagnosing LS can be challenging as it often occurs when F. necrophorum is incidentally isolated in patients, leading to significant delays in appropriate care.21 As witnessed in the authors’ patient, a high index of clinical suspicion is crucial for the prompt diagnosis and treatment of LS. Early signs of acute pharyngitis may not be sufficient to identify septicaemia and other potentially life-threatening sequelae.2 While documenting a positive culture with a classical pathogen is an important diagnostic feature of LS, F. necrophorum cultures can take 6–8 days to grow.21 Blood cultures in LS have limited sensitivity, particularly when abscesses are present.22 In such cases, molecular diagnostics, such as 16S rRNA testing, can aid in identifying bacterial pathogens. This technique is increasingly used as a promising supplemental diagnostic test.23 This molecular diagnostic tool is not
specifically targeted to F. necrophorum, and has broad applications in detecting a variety of bacterial pathogens from different sample types, including serum and directly from abscesses. Results from this method are generally available within a few days.23 Although not routinely used, 16S rRNA might be particularly helpful in identifying the causative organism where cultures remain negative, as was observed in the authors’ patient, confirming the diagnosis and guiding antimicrobial therapy.24
CONCLUSION
The case discussed here emphasises the importance of considering rare complications of common oropharyngeal infections. Unexplained sepsis in a previously healthy young adult, with a recent history of pharyngitis or tonsillitis, should raise suspicion of LS, despite the nonspecific clinical presentation and rarity. Therefore, maintaining a high index of suspicion is crucial. Although uncommon, the identification of septic cervical venous thrombosis in locations other than IJV should prompt a comprehensive investigation for additional metastatic infections. LS carries the potential to cause fatality, and because of this, healthcare providers should be aware of it, and be prompted to recognise it. Furthermore, the presence of atypical metastatic disease, and the frequently reported absence of IJV thrombosis underscores the need for a re-evaluation of the traditional definition of LS, considering these variations in presentation.
KEY CLINICAL MESSAGE
This article aims to educate readers about Lemierre’s Syndrome (LS), emphasising its clinical features, including oropharyngeal infections and thrombophlebitis, as well as challenges in diagnosis. It also highlights uncommon LS presentations like cerebral infarctions and non-typical abscess locations, underlining their significance. Additionally, it discusses the limitations of traditional culture methods, and the potential value of 16S rRNA testing for identifying causative pathogens when cultures yield no results.
References
1. Ng SK et al. Reproducibility of clinical grading of tonsillar size. Arch Otolaryngol Head Neck Surg. 2010;136(2):159-62.
2. Lee WS et al. Lemierre's syndrome: a forgotten and re-emerging infection. J Microbiol Immunol Infect. 2020;53(4):513-7.
3. Sacco C et al. Lemierre syndrome: clinical update and protocol for a systematic review and individual patient data meta-analysis. Hamostaseologie. 2019;39(1):76-86.
4. Moreno S et al. Lemierre's disease: postanginal bacteremia and pulmonary involvement caused by Fusobacterium necrophorum. Rev Infect Dis. 1989;11(2):319-24.
5. Soumya A, Aditya R. Lemierre's syndrome: a lethal complication of acute tonsillitis. Cureus. 2022;14(10):e30072.
6. Bonny V et al. A prostatic Lemierre syndrome. Int J Infect Dis. 2019;84:734.
7. Yamamoto S et al. Fusobacterium necrophorum septic pelvic thrombophlebitis after intrauterine device insertion. Int J Gynecol Obstet. 2019;145(1):122-3.
8. Dasari SP et al. A challenging case of Lemierre's syndrome with central nervous system involvement and a comprehensive review. Cureus. 2020;12(8):e10131.
9. Warabi YS et al. Cerebral infarctions and brain abscess due to Lemierre syndrome. Intern Med. 2005;44(6):653-6.
10. Hsieh MS et al. Features and treatment modality of iliopsoas abscess and its outcome: a 6-year hospital-based study. BMC Infect Dis. 2013;13:578.
11. Ratnasingham Y et al. Arterial ischemic stroke as a complication to disseminated infection with Fusobacterium necrophorum. Neuropediatrics. 2014;45(2):120-2.
12. Elkind MSV et al. Infection as a stroke risk factor and determinant of outcome after stroke. Stroke. 2020;51(10):315668.
13. Bird NT et al. Lemierre’s disease: a case with bilateral iliopsoas abscesses and a literature review. World J Emerg Surg. 2014;9:38.
14. Patel PN et al. Lemierre's syndrome in the pediatric population: trends in disease presentation and management in literature. Int J Pediatr Otorhinolaryngol. 2020;136:110213.
15. Valerio L et al. Management of Lemierre syndrome. Minerva Med. 2021;112(6):726-39.
16. Johannesen KM, Bodtger U. Lemierre’s syndrome: current perspectives on diagnosis and management. Infect Drug Resist. 2016;9:221-7.
17. Gore MR. Lemierre syndrome: a metaanalysis. Int Arch Otorhinolaryngol.
2020;24(3):e379-85.
18. Schulman S. Lemierre syndrome–treat with antibiotics, anticoagulants or both? J Intern Med. 2021;289(3):4378.
19. Laurencet ME et al. Atypical presentation of Lemierre’s syndrome: case report and literature review. BMC Infect Dis. 2019;19(1):868.
20. Halawa A et al. Retinal, optic nerve, and cerebral infarction in odontogenic Lemierre syndrome. J Neuroophthalmol. 2022;42(1):e443-5.
21. Valerio L et al. Lemierre syndrome: current evidence and rationale of the Bacteria-Associated Thrombosis, Thrombophlebitis and LEmierre syndrome (BATTLE) registry. Thromb Res. 2020;196:494-9.
22. Denes E, Barraud O. Fusobacterium nucleatum infections: clinical spectrum and bacteriological features of 78 cases. Infection. 2016;44(4):475-81.
23. Fida M et al. Diagnostic value of 16S ribosomal RNA gene polymerase chain reaction/Sanger sequencing in clinical practice. Clin Infect Dis. 2021;73(6):961-8.
24. Heney C et al. Use of 16S rRNA gene sequencing on direct culture-negative clinical specimens for the diagnosis of bacterial infections, including 2 case reports. Int J Infect Dis. 2014;21(S1):367.
Treatment of Dermatitis Artefacta: A Systematic Review
Authors: *Mariah C. Estill,1 Mohammad Jafferany2
1. Georgetown University School of Medicine, Washington, D.C., USA
2. Department of Psychiatry and Behavioral Sciences, College of Medicine, Central Michigan University, Saginaw, USA *Correspondence to mce66@georgetown.edu
Disclosure: The authors have declared no conflicts of interest.
Dermatitis artefacta (DA) is a rare psychocutaneous disorder characterised by self-inflicted skin lesions that the patient denies producing. DA poses a complex clinical challenge to clinicians as patients often are resistant to the diagnosis, and can be hesitant to follow up with psychiatric or psychological services. There is a need to understand the optimal approach for management of patients with DA. This systematic review was undertaken to address this gap in knowledge.
A search was conducted on PubMed and Embase using the following search strategy: Dermatitis artefacta OR factitious dermatitis OR factitial dermatitis OR artefactual skin AND treatment OR management OR therapy OR psychotherapy OR pharmacotherapy. Included studies were published from inception to 5ᵗʰ April 2023 in peer-reviewed journals, and discussed the treatment and management of DA. Studies were excluded if they were published in a language other than English.
A total of 11 retrospective or prospective studies were included in this systematic review. They all found DA to be challenging to treat, with every study reporting patients experiencing a comorbid psychiatric condition or associated psychosocial stressor. Overall, there were better reported outcomes among follow-up patients treated in psychodermatology clinics with multidisciplinary teams of dermatologists, psychiatrists, and psychologists.
Medical treatments can help with symptom control and promote wound healing, while psychological and psychiatric treatments can help address underlying psychosocial stressors for the condition. Further research is needed to evaluate optimal management and long-term treatment outcomes in patients with DA.
Key Points
1. Dermatitis artefacta is a complex psychocutaneous condition that poses a diagnostic and therapeutic challenge for clinicians. The lesions are bizarre, angular, or geometrical, not conforming to any recognised skin disease.
2. In this systematic review, 11 studies were reviewed with the aim of evaluating the treatment of dermatitis artefacta.
3. Clinicians should consider a multidisciplinary approach with a team of dermatologists and mental health professionals in the treatment of dermatitis artefacta.
INTRODUCTION
Dermatitis artefacta (DA), also known as artefactual skin disease, is a rare psychocutaneous disorder characterised by self-inflicted skin lesions that the patient denies producing. DA is considered a primary psychiatric disorder with a dermatological presentation, as patients create lesions on the skin to satisfy an underlying psychological need.1,2 DA occurs more commonly in females, and is mainly seen in older children and adolescents, although all age groups may be affected.2,3
The lesions of DA can mimic recognised skin conditions; however, in most cases, the morphology and distribution of these lesions do not align with conventional dermatological patterns. DA is typically identified by its irregularly shaped lesions with linear or geometric outlines, predominantly found in areas of the body that are easily reachable by the patient.4 Patients with DA typically report the appearance of lesions, but are vague about their condition, and cannot clearly explain how the lesions developed. This is termed a ‘hollow history’, which is characteristic of patients with DA.5,6
In many cases, the onset of DA is associated with the presence of an identifiable psychosocial stressor or a coexisting psychiatric condition. Depression, anxiety disorders, obsessivecompulsive disorder, and borderline personality disorder are common psychiatric diagnoses in patients with DA. Skin lesions in DA can arise either consciously or during a dissociated state.4,7,8 When patients are unable to recognise their involvement in the
development of the skin lesions, this leads to a classic denial of any responsibility for the pathogenesis of DA, and inability or unwillingness to disclose the origin of the lesions. Patients and their families are often resistant to the diagnosis initially, and can be hesitant to follow up with psychiatric or psychological services.9–11 As a result, dermatologists often find it difficult to approach treating patients with this condition.
DA poses a complex clinical challenge to clinicians, and there is a need to determine the optimal approach for management and treatment. Although there are many case reports published on DA, the review of published literature has revealed a limited number of case series, retrospective, or prospective studies on the topic. The aim of this systematic review is to evaluate the treatment and management of this complex condition.
METHODOLOGY
To analyse literature published up until the 5th of April 2023, the following search strategy was used on PubMed and Embase: Dermatitis artefacta OR factitious dermatitis OR factitial dermatitis OR artefactual skin AND treatment OR management OR therapy OR psychotherapy OR pharmacotherapy. Article reference lists were also examined. Studies were included if they met the following inclusion criteria: discussed the treatment and management of dermatitis artefacta; case series, systematic reviews, cohort studies, randomised controlled trials, cross-sectional studies, or case-controlled studies; and publication in peer-reviewed
journals. Studies were excluded if published in a non-English language, or the wrong publication type (for example, case reports or letter to the editor). Included studies discuss the management of dermatitis artefacta, which differs from other types of self-inflicted cutaneous diseases such as excoriation disorder, trichotillomania, delusional parasitosis, and Munchausen syndrome. The initial review of studies was conducted in chronological order by one reviewer. Figure 1 provides a flow diagram illustrating the search process. Table 1 summarises the findings of the included studies.
RESULTS
The authors' search found 11 retrospective or prospective studies that discussed the treatment of patients with DA. Larger studies analysing the characteristics and management of DA date back to the 1980s. One retrospective case series published in 1986 reviewed the records of patients with psychogenic skin disease, seen in a clinical practice, over a 3-year period. Eight patients with DA were identified. All eight patients were females, with a mean age of 29.5 years (range: 10–67 years). Three of the eight patients were treated with occlusion. One patient was seen by psychiatry, two were referred to psychologists, three refused psychiatric help, and one self-discharged before psychiatric evaluation. All patients were under significant psychological stress at the time their symptoms began. Four patients had a formal comorbid psychiatric diagnosis, two patients had depression, one patient had trichotillomania, and one patient had ‘hysterical conversion’. All lesions healed after occlusion, close observation of the patient, or confrontation.12
A retrospective chart review from a 19-year period between 1980–1999 was conducted at the National Institute of Paediatrics in Mexico City, Mexico. There were 29 patients (25 females and four males) with a mean age of 11.17 years (range: 1.8–17.9). Out of these patients, 21 were recommended for psychiatric evaluation, but seven of them refused. Fourteen patients received a psychiatric diagnosis (seven had anxiety;
four had depression; and three had personality disorders, aggressive behaviour, and irritability). Twelve of these patients were treated with psychotherapy, and two received pharmacologic treatment for their comorbid depression and anxiety disorders. Of the 14 patients who underwent psychiatric evaluation, only 10 patients were followed up for a period of 6 months. Of these 10 patients at follow-up, four (40%) were considered cured, three (30%) showed improvement, and three (30%) had worsening of their condition. The most commonly prescribed dermatologic treatment was topical antibiotics. Additionally, two patients treated with occlusion showed improvement in their lesions.9
There have been two retrospective studies published looking at subsets of 201 patients with DA, treated over a 30-year period, from 1976–2006, at the Department of Dermatology of the Virgen Macarena Hospital in Seville, Spain. The first study by Rodríguez-Pichardo et al.11 reviewed patients diagnosed with DA of the breast. Out of 201 patients diagnosed with DA, 27 had lesions affecting the breast. These patients were all female, with a mean age of 34.33 years. The skin lesions were treated with occlusive bandages, at times utilising zinc paste or plaster splints. Every patient was offered psychiatric evaluation, but only 13 patients (48.1%) agreed to be seen by a psychiatrist. Out of these patients, five received a diagnosis of depression treated with antidepressants, and three received a diagnosis of a personality disorder treated with psychotherapy. The other 14 patients (51.9%) declined psychiatric evaluation and were subsequently lost to follow-up.11
In the second study looking at a subset of the 201 patients with DA, Alcántara Luna et al.13 identified paediatric patients diagnosed with DA over the same 30-year time period. There were 44 children and adolescents, with a mean age of 12.9 years. On average, patients were evaluated four times before receiving a diagnosis of DA, and were also seen at numerous nursing visits for bandage treatments. All patients were given the opportunity for psychiatric referral; however, 41 patients (93.2%), the vast majority,
Identification of studies via databases
Records (n=527) identified from: Pubmed (n=192) Embase (n=335)
Duplicate records removed before screening (n=125)
Records screened (n=402) Reports sought for retrieval (n=246)
Records excluded by title/abstract (n=156) Reports not retrieved (n=14)
Reports assessed for eligibility (n=232)
Reports excluded: No discussion of treatment (n=4) Wrong publication type (n=173) Non-English language (n=44)
Studies included in review (n=11)
declined to undergo psychiatric evaluation. Only three patients (6.8%) agreed to undergo evaluation. Out of these patients, two had psychiatric diagnoses, including a personality disorder and history of suicide attempt.13
A prospective study was carried out to investigate DA among members of the Israel Defence Force (IDF). Over a 3-year period, from 1997–2000, 14 soldiers were
treated. There were 13 males and one female between the ages of 19–26 included in this study. For all patients, DA was characterised by an acute rash that was distributed in a linear pattern on the limbs and abdomen, and was associated with fever and systemic symptoms. The patients all denied deliberately causing the lesions, although eight patients acknowledged exposure to possible inflicting factors. All
Figure 1: Flow diagram of search process.
patients received topically administered corticosteroids, and orally administered antihistamines. Antibiotics were administered to four patients (two topically and two systemically). Three patients were given a short course of corticosteroids, administered systemically. In all cases, the rash resolved completely within a span of 1–3 weeks.14
In a study, Ugurlu et al.15 identified patients with factitious dermatitis at Mayo Clinic, Rochester, Minnesota, USA, treated between 1985–1997. Eighteen patients were seen for factitious dermatitis involving the periocular skin and face. Nine of these patients had received the specific diagnosis of DA. The majority of patients were provided reassurance, and received advice to avoid manipulating the lesions. Seven of the 18 patients were prescribed topical antibiotics, corticosteroids, or warm compressions. Eight of the 18 patients were seen by psychiatry at Mayo Clinic, but the rest turned down a referral for psychiatric evaluation, or expressed a preference to seek psychiatric care outside of Mayo Clinic.15
A retrospective study reviewed the records of 57 patients, over 20 years, from 1982–2002, in Denmark. DA was found to be 2.8 times more common in females compared to males, with 42 (74%) patients being female. The median age was 39 years (range: 12–86 years). A total of 61% of patients were treated with either anxiolytic or anti-depressive drugs. Ten patients (18%) had a psychiatric diagnosis. For patients who received occlusive dressings, lesions demonstrated improvement in 30 out of 32 patients. Among 30 patients where ‘selfinfliction’ of the lesions was addressed with the patient, one-third acknowledged involvement in the development of the lesions. The rest of the patients either denied it, or discontinued the follow-up. Only a single patient agreed to be evaluated by psychiatry. The researchers concluded that psychological intervention appeared unhelpful because of the patient denying their involvement in creating the lesions.16
Recently, a retrospective study in Türkiye was performed to review the records of patients diagnosed with DA during a 10-
year time period, from 2002–2012. Out of the 25 patients, there were 23 females and two males, with a mean age of 25.64 years (range: 11–54 years). Psychiatric evaluation was performed for all patients, and seven patients were found to have additional psychiatric conditions (four with anxiety disorders and three with depression). Nine of the 25 patients were able to be contacted by phone for follow-up. Five patients had resolution of lesions, while four continued to have lesions. Despite referral for treatment and further psychiatric evaluation, all of the patients avoided follow-up. The authors concluded that their patients perceived engaging in psychiatric treatment as ‘shameful’, and thus avoided follow-up and treatment.3
There were three studies included in this systematic review where patients were treated at a formal psychodermatology clinic or conference. Chung et al.10 performed a retrospective case series to review the records of patients seen in a bi-monthly psychodermatology conference jointly conducted by the Departments of Dermatology and Psychological Medicine at Changi General Hospital, Singapore, from November 2009–July 2011. The majority of patients were diagnosed with either a psychophysiologic disorder or a primary psychiatric disorder. One patient diagnosed with DA had comorbid major depression. The patient was started on fluvoxamine to treat the associated depression, but was lost to follow-up.10
A retrospective study by Rogers et al.17 reviewed patients referred to paediatric dermatologists at the Royal Alexandra Hospital for Children in Sydney, Australia, over a 15-year period. Thirty-two paediatric patients diagnosed with DA were identified, including 24 females and eight males, ranging from 8–16 years in age. Most patients were treated at the hospital’s formal psychodermatology clinic that had opened 3 years prior. Patient data were incomplete or unavailable for many patients prior to this time. For the 12 cases of patients treated at the psychodermatology clinic, eight cases showed symptom resolution or improved functioning. Four patients declined to be evaluated by psychiatry or to follow up
with dermatology, after being diagnosed with DA. The researchers suspected that DA in these patients were secondary to life difficulties such as medical illness, or stress at home or school. Most parents were hesitant to pursue additional psychological care once there was an improvement in the symptoms, suggesting that patients can have notable improvement due to changes in life situations, or the development of maturation, instead of directly from treatment directly.17
Similarly, Mohandas et al.2 performed a retrospective study to review charts of 28 patients diagnosed with DA, who had received care at a formal psychodermatology clinic in England, from 2003–2011. Most patients seen were female (24 female and four male), and ages ranged from 13–70 years. The vast majority of patients (n=26) endorsed psychosocial stressors. Thirteen patients (46%) presented with comorbid psychiatric conditions, which included diagnoses of anxiety, depression, and personality disorders. Patients were seen in the clinic by a multidisciplinary team with a dermatologist and psychiatrist, both evaluating the patient at the same visit. This collaborative approach also involved psychologists, nurses, and social workers. Twenty-six patients (93%) were considered successfully managed (defined as DA resolved or in remission), with only two patients lost to follow-up.2
DISCUSSION
The main strategies for DA management include building strong patient relationships, addressing skin symptoms, and providing extended psychiatric care with consideration of any relevant underlying psychosocial factors. The preferred initial treatment for patients is cognitive behavioural therapy or psychotherapy.6 Medical management of DA focuses on wound healing, preventing infection, and symptom control. The use of emollients, antibiotics, antifungals, and analgesia paired with suitable dressings or bandaging can promote the healing of lesions.
Psychopharmacological treatment options include selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, tricyclic antidepressants, and antipsychotics.6 Most patients do not require surgery, although debridement may be beneficial in some cases of wound colonisation or abscess formation.18
Management should be personalised to each patient, with an emphasis on exploring any psychosocial stressors that may be contributing to the development of DA. Direct confrontation of the patient behaviours that led to the formation of the lesions is counterproductive, often leading to patients disengaging with medical care, and becoming lost to follow-up.
One approach to addressing the patient’s problems could be avoiding discussion of the exact mechanisms behind the development of the lesions, and instead emphasising the impact of stress on the development of the condition, as this may be more easily accepted by the patient. It also may be beneficial to acknowledge that the condition can create stress for the patient, and this can be used as a justification for introducing psychological therapy as a treatment option. Regardless of the approach, it remains crucial to address both treatment of the skin and the psychological aspects of DA.
The use of a multidisciplinary psychodermatology team for DA is important because many patients require psychological intervention to address the underlying cause of their behaviour, and psychiatric evaluation to manage any comorbid psychiatric conditions. The opening of formal psychodermatology clinics has allowed dermatologists to play a crucial role in diagnosing DA, excluding other diseases, and establishing effective communication with mental health providers. This approach is essential for building and maintaining patient trust because it minimises the likelihood of losing patients to follow-up, and allows for continuity of building and maintaining patient trust.
The prognosis of DA is influenced by various factors, such as the age of presentation, identification of any triggering stressors, pre-existing state of health, and any underlying psychological disorders. Positive long-term outcomes are seen in paediatric patients, who were diagnosed and treated at an early stage.9 In contrast, older patients, or those with significant comorbid psychological or medical conditions, may experience a more relapsing clinical course. To achieve sustained remission, ongoing follow-up is important for patients with DA, particularly among those who experience a pattern of relapse. The establishment of psychodermatology multidisciplinary teams can play a vital role in streamlining care for patients with DA.
LIMITATIONS
This review is subject to some important limitations. Despite some of the included retrospective studies having larger sample sizes, examining a rare psychiatric disorder with a low prevalence can be challenging. DA is diagnosed clinically without welldefined diagnostic criteria. In some cases, clinicians who diagnosed DA may not have excluded similar conditions, such as skinpicking disorder, malingering, or intentional self-harm. All included studies relied on either clinician interviews or medical chart reviews, without standardisation in patient evaluations. It is possible that certain comorbid psychiatric diagnoses may have been missed. Some studies included in this review have limited detail on treatments provided or patient outcomes. In particular, limited retrospective studies focused on psychopharmacology. In some studies, a significant number of patients were lost to
follow-up, or had short follow-up intervals, giving limited data on long-term treatment outcomes for patients with DA. Finally, each study included in this review differs in design and outcome measures, which makes it challenging to establish definitive statistical conclusions.
CONCLUSION
DA is a complex psychocutaneous condition that can be challenging for clinicians to diagnose and manage. This is the first systematic review published on DA that highlights the treatment and management of this condition. The cornerstone of DA management is creating a safe and trusting atmosphere during the patient visit, where clinicians can focus on understanding why a patient has the condition, rather than delving into the mechanisms of how the lesions were created by the patient. A nonjudgmental and non-confrontational attitude on the part of the physician is warranted in seeing these patients.
Medical treatments can promote the healing of skin lesions, and facilitate the management of symptoms. Psychological and psychiatric treatments can help address underlying psychosocial stressors for the condition. This systematic review found better reported outcomes among followup patients treated in psychodermatology clinics. This suggests that multidisciplinary teams comprised of dermatologists, psychiatrists, and psychologists working together can offer optimal care for patients. Further research is needed to better evaluate the management options and long-term treatment outcomes for patients with DA.
Table 1: Included studies in chronological order.
Source, Time Frame, Location
Sheppard et al.,12 3-year period, Ireland
Study Design
Participants Adults, Children, or Both
Retrospective n=8
Both n=8
females mean: 29.5 years; range: 10–67 years
Comorbid Psychiatric Conditions Life Event or Psychosocial Precipitant Treatment Outcome
All patients were under significant psychological stress at the time of production of their symptoms n=3 occlusion, n=1 seen by psychiatry, n=2 referred to psychologist or for psychotherapy, n=3 refused psychiatric help, and n=1 self-discharged before psychiatric evaluation.
All lesions healed after occlusion, close observation of the patient, or confrontation
Ugurlu et al.,15
1985–1997, USA
Retrospective n=9 with facial lesions; n=18 factious or self-inflicted dermatitis with facial lesions
Both n=8 females; n=1 males mean: 37.89 years; range: 13–66 years
Saez-deOcariz et al.,9 1980–1999, Mexico
Retrospective n=29 Children n=25 females; n=4 males mean: 11.17 years; range: 1.8–17.9 years
n=5; n=1 delusion of parasitosis, n=1 folie à deux, n=2 la belle indifference, n=1 conversion reaction, n=1 depression, n=1 somatoform disorder, n=1 diuretic and laxative abuse
Most patients were offered reassurance and advised to avoid manipulating the lesions. Topical antibiotics, corticosteroid preparations, or warm compresses were prescribed in seven patients. Eight of the 18 patients were evaluated by a psychiatrist. Remaining patients either declined psychiatric referrals, or preferred to undergo a psychiatric evaluation elsewhere.
Dermatologic treatment was given to all but one patient. Topical antibiotics were the most frequently administered medication; 21 patients were recommended for psychiatric evaluation, and seven refused. Twelve of the 14 patients (86%) who underwent psychiatric evaluation received both family and individual psychotherapy. Two of them (14%) also required pharmacologic treatment for anxiety or depression.
N/A
Out of the 14 seen by psychiatry, 10 attended follow-up for 6 months with four cured (40%), three improved (30%), three worsened (30%); 12 (41%) attended for their second follow-up appointments.
Table 1 continued.
Source, Time Frame, Location
Cohen and Vardy,14 1997–2000, Israel
Study Design Participants Adults, Children, or Both Sex
Comorbid Psychiatric Conditions Life Event or Psychosocial Precipitant
N/A
Children n=24 females; n=8 males mean: 11.59 years; range: 8–16 years
N/A
Treatment
Military service All patients were treated with topically administered corticosteroids and orally administered antihistamines. Four patients were treated with antibiotics (topically administered antibiotics for two patients, and systemically administered antibiotics for two patients). Three patients were treated with a short course of systemically administered corticosteroids.
School stress, discord at home, and recent physical illness
Patients diagnosed and referred by dermatologists are assessed and managed by a psychiatrist and a psychologist.
All patients’ symptoms resolved completely within 1–3 weeks.
Eight out of 12 patients treated in the psycho-dermatology clinic improved (66%), four (33%) refused follow-up after given a diagnosis of DA. There is no recorded outcome for the remaining 20 cases due to incomplete or unavailable data.
Table 1 continued.
Source, Time Frame, Location
Nielsen et al.,16 1982–2002, Denmark
Study Design Participants Adults, Children, or Both Sex Age
Comorbid Psychiatric Conditions Life Event or Psychosocial Precipitant
n=10; n=8 neurosis hysteriformis, anxiosa, or characterogenes; n=2 depression and schizophrenia Majority of the patients (75%) were unemployed or on sick leave
RodríguezPichardo et al.,11 1976–2006, Spain
Retrospective n=27 with breast lesions Both n=27 females mean: 34.33 years; range: 12–77 years
n=5 depression, n=3 personality disorder
N/A
A total of 61% were treated with anxiolytic or antidepressive drugs. Only one female patient agreed to meet a psychiatrist. 32 patients were treated with occlusive dressings.
Among 32 patients occlusive dressing could be administered, and the lesions all showed improvement except in two patients. Among 30 patients who were confronted with ‘self-infliction’ as the cause for their symptoms, onethird admitted that self-infliction was part of the cause, but two-thirds either denied this or stopped coming for follow-up.
The cutaneous lesions were treated with occlusive bandages using zinc paste or plaster splint when necessary. All patients were offered to be seen by a psychiatrist, but only 13 patients (48.1%) accepted to be evaluated. Out of these, five patients were diagnosed with depression, and received treatment in the form of antidepressants. Three patients were diagnosed with a personality disorder, and were treated with psychotherapy.
The 14 patients who declined to be evaluated by psychiatry were lost to medical follow-up.
1 continued.
Source, Time Frame, Location
Alcántara Luna et al.,13 1976–2006, Spain
Study Design Participants Adults, Children, or Both
Retrospective
n=44
Children n=32 females; n=12 males mean: 12.9 years; range: 4–17 years
n=2 (4.5%); n=1 personality disorder, n=1 history of suicide attempt
Event or Psychosocial Precipitant
N/A
Chung et al.,10 2009–2011, Singapore
Mohandas et al.,2 2003–2011, UK
Retrospective n=1 Both n=1 female 54 years Major depression N/A
Retrospective n=28 Both n=24 females; n=4 males mean: 36.96 years; range: 13 –70 years
n=13 (46%); n=1 schizophrenia, n=1 ADHD, n=11 anxiety, depression, or bereavement, n=1 alcohol dependency, n=1 intravenous drug use
Majority of patients (n=26) had psychological stressors present in their lives. Most adult patients were either single or divorced, or were in unhappy relationships
All patients were observed on multiple follow-up visits until other diagnoses were excluded. Patients were seen an average of four times in consultation, as well as additional uncountable visits to the Dermatology Nursing Unit for treatment with occlusive bandages. The possibility of psychiatric referral was offered to all patients, but only three (6.8%) accepted the evaluation. The remaining 41 patients (93.2%) refused to be evaluated by a psychiatrist.
Fluvoxamine (to treat the associated depression).
All patients were treated by the psychodermatology multidisciplinary team. This involved an initial assessment with a dermatologist and psychiatrist seeing the patient concurrently. A referral was made if necessary to other psychiatric or psychological services. n=5 counselling, n=4 CBT, n=9 SSRIs, n=1 olanzapine, n=2 antibiotics, n=1 OCP, n=1 isotretinoin, n=3 psychiatric care, n=1 CPS, n=2 phototherapy, n=1 patch testing, n=1 lidocaine, n=4 emollients, occlusions or dressings, n=2 steroids, n=5 biopsy.
N/A
Defaulted on follow-up.
Of the 28 case notes reviewed, 20 (72%) patients had resolution or great improvement of their condition, six (21%) remained under follow-up with stable or intermittent lesions, and two patients (7%) failed to attend follow-up appointments.
Table 1 continued.
Source, Time Frame, Location
Uçmak et al.,3 2002–2012, Türkiye
Retrospective
n=25 Both n=23 females; n=2 males mean: 25.64 years, range: 1–54 years
n=7; n=4 anxiety, n=3 depression
N/A
Psychiatric evaluation was performed for all patients but many refused treatment or did not continue psychiatric follow-up.
None of the patients were coming regularly for control visits. Out of nine patients who researchers were able to contact, none continued psychiatric treatment. Lesions had healed in five patients, but continued to appear in the remaining four. Researchers concluded that, because most patients considered the undergoing psychiatric therapy ‘shameful’, they avoid psychiatric evaluation or treatment, although they have been referred.
1. Koblenzer CS. Dermatitis artefacta. Clinical features and approaches to treatment. Am J Clin Dermatol. 2000;1(1):47-55.
2. Mohandas P et al. Dermatitis artefacta and artefactual skin disease: the need for a psychodermatology multidisciplinary team to treat a difficult condition. Br J Dermatol. 2013;169(3):600-6.
3. Uçmak D et al. Dermatitis artefacta: a retrospective analysis. Cutan Ocul Toxicol. 2014;33(1):22-7.
5. Lyell A. Cutaneous artifactual disease. A review, amplified by personal experience. J Am Acad Dermatol. 1979;1(5):391-407.
6. Nagesh NM et al. Dermatitis artefacta. Clin Dermatol. 2023;41(1):10-5.
7. Gupta MA, Gupta AK. Dermatitis artefacta
and sexual abuse. Int J Dermatol. 1993;32(11):825-6.
8. Gupta MA et al. Posttraumatic stress disorder (PTSD) and the dermatology patient. Clin Dermatol. 2017;35(3):260-6.
9. Saez-de-Ocariz M et al. Dermatitis artefacta in pediatric patients: experience at the national institute of pediatrics. Pediatr Dermatol. 2004;21(3):205-11.
10. Chung WL et al. A review of patients managed at a combined psychodermatology clinic: a Singapore experience. Singapore Med J. 2012;53(12):789-93.
11. Rodríguez-Pichardo A et al. Dermatitis artefacta of the breast: a retrospective analysis of 27 patients (1976–2006). J Eur Acad Dermatol Venereol. 2010;24(3):2704.
12. Sheppard NP et al. Psychogenic skin disease: a review of 35 cases. Br J Psychiatry. 1986;149:636-43.
13. Luna SAet al. Dermatitis Artefacta in childhood: A retrospective analysis of 44 patients, 1976-2006. Pediatr Dermatol. 2015;32(5):604-8.
14. Cohen AD, Vardy DA. Dermatitis artefacta in soldiers. Mil Med. 2006;171(6):497-9.
15. Ugurlu S et al. Factitious disease of periocular and facial skin. Am J Ophthalmol. 1999;127(2):196-201.
16. Nielsen K et al. Self-inflicted skin diseases. a retrospective analysis of 57 patients with dermatitis artefacta seen in a dermatology department. Acta Derm Venereol. 2005;85(6):512-5.
17. Rogers M et al. Artefactual skin disease in children and adolescents. Australas J Dermatol. 2001;42(4):264-70.
18. Woolf RT et al. A difficult case of dermatitis artefacta requiring surgical intervention. Br J Dermatol. 2013;168(4):889-91.
Caesarean Hysterectomy for Placenta Accreta Spectrum in a Single Centre: A Series of 19 Cases
1. Department of Obstetrics and Gynaecology, Lifeline Multispeciality Hospital, Adoor, Pathanamthitta, India
2. Lifeline Multispeciality Hospital, Adoor, Pathanamthitta, India
3. Government Medical College, Thiruvanathapuram, India
*Correspondence to divyamb90@gmail.com
Disclosure: The authors declare no conflicts of interest. The authors received no financial support for the research, authorship, or publication of this article. Bhanumathy conceived the study. The institution gives approval for case reports and does not mandate a separate ethical approval for reporting cases. All authors contributed to the study design. Bhanumathy and Balachandran wrote the first draft. Cleetus, Hassan, Pappachan, and G.K. revised the manuscript. Bhanumathy conducted the final approval of the manuscript. Balachandran accepts responsibility for the work.
The objective of this study is to assess obstetric complications, blood loss, blood transfusion requirement, and maternal and fetal outcomes associated with the authors’ approach of Caesarean hysterectomy for placenta accreta spectrum disorders. Data were collected from case records of all females who underwent Caesarean hysterectomy between August 2013–August 2023 at Lifeline Multispeciality Hospital, Adoor, Kerala, India. There were 19 cases of Caesarean hysterectomy. The mean age was 33.63±2.90 years. Mean blood loss during surgery was 1.11±0.16 L, and the mean packed red blood cells transfused was 2.00±0.38 units. The mean gestational age of termination was 33 weeks and 5 days, and mean birth weight was 2.28±0.21 kg. There were no maternal or neonatal deaths. Placenta accreta spectrum should be managed in a multidisciplinary setup with the involvement of a senior experienced obstetrician. Early, careful bladder dissection before proceeding with hysterectomy will help in reducing haemorrhage, and in accelerating hysterectomy.
Key Points
1. Whilst rare, the incidence of placenta accreta spectrum disorders (PAS) is rising. Life-threatening haemorrhage can occur as a result of PAS; therefore, optimised understanding and management is crucial for patient outcomes.
2. This case series reviews records from patients with PAS who were managed with Caesarean hysterectomy, to review complications and maternal and fetal outcomes. The study highlights that correct pre-operative diagnosis by ultrasound or, if needed, MRI, is important for informed surgical planning, which should be performed in a multidisciplinary setting by an experienced team to reduce morbidity and mortality.
3. Intra-operative blood loss is a complication that can be greatly reduced by initial meticulous dissection of the bladder prior to hysterectomy. This also reduces the duration of procedure, especially in the region of the lower segment.
INTRODUCTION
Placenta accreta spectrum (PAS) is rare, but its incidence is increasing due to a rise in Caesarean section rates.1,2 PAS is an abnormal trophoblastic invasion into the myometrium, up to the serosa, or more.2 This condition can lead to lifethreatening haemorrhage, and usually requires hysterectomy. The severity of invasion can be graded according to the International Federation of Gynaecology and Obstetrics (FIGO) classification.3 Primary risk factors include prior uterine surgery, prior surgical termination, and in vitro fertilisation.2 Caesarean hysterectomy (CH) is the definitive treatment, and should be performed in a multidisciplinary care centre by a senior experienced obstetrician. However, there are various methods to reduce blood loss, such as aortic balloon occlusion, aortic clamp, and pre-operative insertion of embolisation catheter.2,4 Even then, the blood loss and need for blood transfusions are found to be high. The objective of this study is to assess the demographic parameters, obstetric complications, blood loss, blood transfusion requirement, and maternal and fetal outcomes associated with the authors’ approach of CH for PAS disorders, in a single centre.
MATERIALS AND METHODS
This is a case series of all females who underwent CH between August 2013–August 2023 at Lifeline Multispeciality
Hospital, Adoor, Kerala, India. The authors collected data from all case records of patients who had CH during this period. Nineteen cases of CH were retrieved, informed patient consent was taken from these females, and their data were analysed. Maternal demographic variables, comorbidities, gestational age, obstetric score, risk factors, pre-operative and post-operative haemoglobin levels, blood loss, blood transfusion details, surgery details, maternal outcome, fetal outcome, and maternal and fetal complications were collected from case records. A data abstraction sheet was used to extract details, and data was entered in an Excel sheet (Microsoft, Redmond, Washington, USA). Data were summarised as numbers and mean with standard deviation and percentages.
CH was performed by vertical midline skin incision (Figure 1A), followed by a vertical incision in the upper segment of the uterus, above the placental edge. After the fetus was delivered (Figure 1B), they were handed over to the paediatrician. The uterus was exteriorised, and uterotonics, including oxytocin and methergine, were used. Tranexamic acid was used to reduce bleeding.5 Methergine was not given to four patients who had hypertension. Since the edges did not bleed significantly after delivery of the fetus, they were not sutured. The maximum blood loss occurred when the uterus was severed in the region of the lower segment and cervix, where the placenta is attached. Hence, it was important to accomplish this suturing step
Figure 1: Caesarean hysterectomy steps.
A) Midline vertical incision. B) Delivery of fetus. C) Uterus after delivering the fetus, showing the lower segment bulged with placenta. D) Bladder dissection. E) Lower segment with bulging placenta after dissecting and pushing down the bladder. F) Exposed internal iliac artery. G) Hysterectomy. H) Hysterectomy specimen with placenta in situ
as fast as possible. With this in mind, the authors routinely dissected the bladder in small steps, and brought the bladder below the lower edge of the placenta. In cases where bleeding was higher during bladder dissection, bilateral internal iliac artery ligation was performed (Figure 1F), followed by total hysterectomy (Figure 1A–H). Blood loss was assessed by weighing the mops used and measuring the amount of blood in the suction apparatus. All procedures were performed by the same senior obstetrician, in a multidisciplinary setup.
RESULTS
Over the 10-year study period, there were 19 cases of CH performed for PAS disorders. There were four referred cases from other hospitals. Seventeen cases were diagnosed antenatally by ultrasound, and two cases were diagnosed intraoperatively, which were referred in labour without diagnosing PAS. These patients underwent CH in the authors’ centre, where PAS was diagnosed. The mean age of patients was 33.63±2.90 years. Other demographic variables are given in Table 1. Previous Caesarean sections, in vitro fertilisation, previous myomectomy, and curettage were the common risk factors. The
Demographic and Clinical Variables
mean gestational age of termination was 33 weeks and 5 days, and a total hysterectomy was performed for all cases. Internal iliac artery ligation was performed for eight cases (Table 2). One case was FIGO 3C, for which intraoperative stenting was performed. Two cases were FIGO 3A, and bladder injury occurred in these cases. Hysterotomy followed by hysterectomy was performed in one case at 23 weeks and 2 days, which was terminated for multiple congenital anomalies, and the fetus passed away soon after birth. Mean blood loss during surgery was 1.11±0.16 L, and mean packed red blood cells transfused was 2.00±0.38 units (Table 2). The mean pre-operative haemoglobin was 11.36±0.42 g/dL, and mean postoperative haemoglobin was 11.18±0.69 g/dL. There were no neonatal deaths, and mean birth weight was 2.28±0.21 kg. In total, 57.89% of neonates required neonatal intensive care unit admission for low birth weight and preterm care. There were no maternal deaths, and none of these cases required intensive care management or massive blood transfusion. Only 21% of cases required more than two blood transfusions. The mean duration of postoperative stay was 11.94±2.79 days, as patients stayed until sutures were removed. All specimens were sent for histopathological examination, and
Mean Values and Percentages
1. Mean age (±SD) 33.63±2.90 years
2. Mean gestational age at diagnosis 30 weeks and 6 days
3. Referred 4 patients (21.05%)
4. Antenatally diagnosed with ultrasound 17 patients (89.47%)
5. Presented with antepartum haemorrhage 5 patients (26.31%)
6. Twin pregnancy 1 patient (5.26%)
7. Comorbidities
i) Anaemia
ii) GD
iii) Hypertensive disorders
iv) Hypothyroid
v) APLA
7 patients (36.84%)
6 patients (31.58%)
4 patients (21.05%)
3 patients (15.79%)
2 patients (10.52%)
2 patients (10.52%)
APLA: anti-phospholipid antibody; GD: gestational diabetes; SD: standard deviation.
Table 1: Patient demographic and clinical variables.
Table 2: Patient obstetric and anaesthetic management.
Obstetric and Anaesthetic management
Mean gestational age of termination
Mean pre-operative Hb (±SD)
Internal iliac artery ligation
Mean blood loss (±SD)
Mean PRBC transfused (±SD)
Mean postoperative Hb (±SD)
Mean duration of surgery (±SD)
Anaesthesia
i) Spinal with epidural anaesthesia
ii) Spinal anaesthesia
iii) General anaesthesia
Mean duration of postoperative stay (±SD)
Complications
Bladder injury
Intraoperative ureteric stenting
Mean Values/Percentages
33 weeks and 5 days
11.36±0.42 g/dL
8 patients (42.1%)
1.115±0.166 L
2.00±0.38 units
11.189±0.692 g/dL
2.815±0.294 hours
8 patients (42.1%)
8 patients (42.1%)
3 patients (15.8%)
11.94±2.79 days
2 patients (10.52%)
1 patient (5.26%)
Maternal deaths 0
Hb: haemoglobin; PRBC: packed red blood cells; SD: standard deviation.
increta was confirmed. A summary of cases is included in Table 3
DISCUSSION
Over the 10-year study period, there were 19 cases of PAS disorders, for which CH was performed. The mean age group in this study was 33.63 years, which was comparable to the mean age of 32.70 years reported by Dandolu et al.6 There is an increase in the risk of PAS with advanced maternal age.2 The mean gestational age in the study by Ansar et al.7 was 33.84 weeks, comparable to the mean gestational age of 33 weeks and 6 days in this study.
The most common risk factor in the study by Dandolu et al.6 was previous Caesarean (47.37%), followed by prior history of curettage (15.80%). The authors’ study identified multiple risk factors, and the most common was previous Caesarean section (68.42%). The risk factors for each case are mentioned in Table 3. In this study, one female had no risk factors.
She was a primigravida, and hysterectomy specimen histopathology found placenta increta. In this study, 13 cases were FIGO Grade 2 (84.2%), followed by FIGO Grade 3 (15.8%). Blood loss and requirement for blood transfusion were lower in this study compared to the study by Ansar et al.7 Mean blood loss and mean packed red blood cells transfused in this study were 1.11±0.16 L and 2.00±0.38 units, respectively, compared to 2.60 L and 7.76 L in the study by Ansar et al.,7 which are much higher. In the study by Paily et al.,8 aortic clamp was used, and the mean blood loss was 1,000±1,500 mL. The correct surgical technique involves a right plane of bladder separation, without touching the placenta; this is a core step to reduce haemorrhage and associated complications. Bladder dissection should be initially performed, so that hysterectomy at the lower portion, where the placenta is situated, can be accomplished more quickly to reduce haemorrhage.
Methods to control haemorrhage include internal iliac artery ligation, aortic balloon, embolisation, and aortic clamp.2,4 In the
Table 3: Summary of patient cases.
CS: caesarean section; D&C: dilation and curettage; FIGO: International Federation of Gynaecology and Obstetrics; G,P,L,A,VM: gravida, parity, live, abortion, vesicular mole; IVF: in vitro fertilisation; N/A: not applicable; PRBC: packed red blood cells; PRIMI: primigravida.
authors’ study, internal iliac artery ligation was performed in eight cases (42.11%).
The mean birth weight in this study was 2.289±0.217 kg, which was comparable to the mean birth weight of 2.40±0.95 kg in the study by Ansar et al.7 In this study, 57.89% of newborns required neonatal intensive care unit admission, which was for pre-term and low birth weight care. None required ventilator support.
Panaitescu et al.9 stated that, in the case of conservative management of PAS, one in three patients will still require hysterectomy after expectant management. Expectant management requires long term followup, carries risk of complications, and at
any time can become an emergency, with patients requiring lengthy hospital stays.
There were no maternal or neonatal deaths in the authors’ study, none required intensive care monitoring, and there were no postoperative complications. Early diagnosis and management of PAS disorders in a planned multidisciplinary setup, and hysterectomy by an experienced obstetrician, will reduce maternal morbidity and mortality.
CONCLUSION
CH is the standard approach for PAS disorders. The most common risk factor for
PAS is previous Caesarean section. Proper pre-operative diagnosis and management in a multidisciplinary setup by a senior skilled obstetrician reduces morbidity and mortality. Early bladder dissection before proceeding to hysterectomy will reduce blood loss, and hence allow faster completion of hysterectomy.
References
1. Higgins MF et al. Real increasing incidence of hysterectomy for placenta accreta following previous caesarean section. Eur J Obstet Gynecol Reprod Biol. 2013;171(1):54-6.
2. Cahill AG et al.; Society of Gynecologic Oncology; American College of Obstetricians and Gynecologists and the Society for MaternalFetal Medicine. Placenta accreta spectrum. Am J Obstret Gynecol. 2018;219(6):B2-16.
3. Jauniaux E et al.; FIGO Placenta Accreta Diagnosis and Management Expert Consensus Panel. FIGO classification for the clinical diagnosis
of placenta accreta spectrum disorders. Int J Gynaecol Obstet. 2019;146(1):20-4.
4. Kingdom JC et al. Minimizing surgical blood loss at Cesarean hysterectomy for placenta previa with evidence of placenta increta or placenta percreta: the state of play in 2020. Am J Obstet Gynecol. 2020;223(3):322-9.
5. Huls CK. Cesarean hysterectomy and uterine-preserving alternatives. Obstet Gynecol Clin North Am. 2016;43(3):517-38.
6. Dandolu V et al. Obstetrical hysterectomy, Cesarean delivery and abnormal placentation. J Matern Fetal
Neonatal Med. 2012;25(1):74-7.
7. Ansar A et al. Hysterectomy as a management option for morbidly adherent placenta. J Coll Physicians Surg Pak. 2014;24(5):318-22.
8. Paily VP et al. Managing placenta accreta spectrum in low‐resource settings using a novel dissection‐free aorta clamp: operative technique. Int J Gynaecol Obstet. 2022;158(2):469-75.
9. Panaitescu AM et al. Risk of subsequent hysterectomy after expectant management in the treatment of placenta accreta spectrum disorders. Medicina (Kaunas). 2022;58(5):678.
Nipah Virus in Kerala, India – Unravelling the Local Outbreak and Assessing Global Threats: A Narrative Review
Authors: M.G. Gopika,1 *Raj Mohan,2 Sayan Roy3
1. School of Public Health, SRM Institute of Science and Technology, Chennai, Tamil Nadu, India
2. Department of Respiratory Medicine, Gloucestershire Royal Hospital, Gloucester, UK
3. Jan Chetna Manch Bokaro, Jharkhand, India *Correspondence to raj2007.mohan@gmail.com
Disclosure: The authors declare no conflicts of interest. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgements: The authors extend their gratitude to Dr Jissa Vinoda Thulaseedharan from the Sree Chitra Tirunal Institute for Medical Sciences and Technology for her valuable suggestions that significantly improved the paper.
Received: 24.01.24
Accepted: 23.04.24
Keywords: Henipavirus, India, Kerala, Nipah virus (NiV), prevention.
The recent emergence of the Nipah virus in the same district of Kozhikode, India, following its outbreak in 2018 and 2021, has elicited heightened apprehension among the public amidst the era of COVID-19. The potential fatality associated with this virus has been effectively mitigated through mass awareness, community and healthcare involvement, and stringent implementation of preventive measures. Nevertheless, the rate of transmission has consistently escalated over a span of several years, suggesting an emerging potential for global public health implications. Although the transmission rate remains low, the elevated mortality linked to the Nipah virus constitutes a potential threat, accentuated by the absence of vaccines and dependable treatments, thereby underscoring the risk to public health and emphasising the necessity for proactive measures to protect the wellbeing of the community. This narrative review provides an overview of the Nipah outbreaks in Kerala, India, and its global impact by conducting a thorough search of databases such as PubMed, Google Scholar, and ResearchGate using the following keywords: “Nipah virus,” “Henipavirus,” and “Kerala.”
Key Points
1. Recurrent Nipah outbreaks in Kerala, India, emphasise the critical need for preparedness due to its high fatality rate and potential for human transmission, underscoring the urgency of proactive measures to prevent it from emerging as a pandemic.
2. This article comprehensively analyses Nipah virus outbreaks, focusing on epidemiology, clinical features, preventive measures, treatment, public health responses, and the ‘One Health' approach, from Kerala-specific data.
3. Healthcare professionals must prioritise early recognition of Nipah virus symptoms, enhance surveillance efforts, engage in interdisciplinary collaboration, and advocate for public awareness to mitigate future outbreaks and safeguard public health.
BACKGROUND
Nipah virus disease is a zoonotic infection caused by the Nipah virus (NiV) of the Genus Henipavirus of the Paramyxoviridae family, an enveloped ribonucleic acid virus. The natural host of the virus is supposed to be the Pteropus bats (fruit-eating species, popularly known as flying foxes).1
According to the World Health Organization (WHO), the fatality rate of NiV is estimated to be high, ranging from 40–75%. This indicates that NiV is significantly more lethal compared to COVID-19, which exhibits a mortality rate ranging from 0.1–19.0%, depending on the local capacity for epidemiological monitoring and healthcare management.2,3 Unlike the previous viral outbreaks, the incidence of NiV infection has been comparatively limited, with a reduced occurrence of human-to-human transmission. Yet, it is intriguing that NiV infection exhibits a significantly higher fatality rate, suggesting the potential for future outbreaks to occur.4
METHODS
The authors conducted an extensive search across PubMed, Google Scholar, and ResearchGate, focusing on articles written in English by using keywords such as “Nipah virus,” “Henipavirus,” and “Kerala,” primarily targeting studies published from 2018 onwards, coinciding with the first reported case in Kerala, India. The authors gathered information on various aspects of NiV, including its epidemiology; outbreaks in countries such as Malaysia, Singapore, Bangladesh, India, and other countries; as well as covering clinical manifestations, diagnostic methods, preventive measures, treatment options, vaccine development, public health interventions, surveillance
strategies, and the economic impact associated with NiV infection. The findings regarding NiV were compiled from reputable scholarly journals and authoritative public health sources.
EPIDEMIOLOGY
NiV was initially reported in Malaysia and Singapore between 1998–1999 and caused severe febrile encephalitis among pig farmers. The transmission of the virus to humans occurs through direct contact, inhalation, or ingestion of contaminated food with NiV, which was facilitated by infected bats and infected pigs as intermediate animal hosts. The outbreaks resulted in the deaths of 105 individuals, leading to mass panic and notable socioeconomic upheaval.5 After the initial occurrences, subsequent outbreaks were documented in Meherpur, Bangladesh, followed by Siliguri city, West Bengal, India, in 2001.3 The mode of transmission, clinical manifestations, and mortality rates were different in Indo-Bangladesh when compared to the outbreak in Malaysia. The significant aspects of this outbreak were the transmission of the virus between individuals, and nosocomial infections. Furthermore, the Indo-Bangladesh outbreak exhibited rapid progression of the disease with high secondary attack rates and severity of the disease. The probable reason for the high mortality could be acute respiratory distress syndrome and respiratory failure accompanied by multiorgan dysfunction syndrome, along with neurological manifestations.3,6 Multiple small outbreaks have been reported in Cambodia, Indonesia, Ghana, Madagascar, the Philippines, and Thailand. However, outbreaks in Malaysia (43%), Bangladesh (42%), and India (15%) contributed to
the overall global incident cases.7 The outbreaks reported in Cambodia provide limited insight into the circulation of batborne diseases, including NiV. Although the virus was isolated from a roost of Pteropus lylei bats in Western Cambodia in 2000, this discovery remains unverified, and there have been no documented cases of NiV infection in humans within the country to date.8 Similarly, in Thailand, where NiV circulates in P. lylei populations, no human or domestic animal cases have been reported.9 Epidemiological evidence from the Philippines in 2014 indicates that the primary mode of NiV transmission to humans involved direct contact with infected horses, exposure to their contaminated bodily fluids during the slaughtering process, and/or ingestion of inadequately cooked meat from
Thomas et al.12
Sudeep et al.,13 Warrier14
Kozhikode, May 2018 Fruit bats (reservoir), unknown primary case transmission. Human-to-human and nosocomial transmission
Kochi, June 2019 Unknown transmission (probable source: fruit bats)
Pragya et al.15
Srivastava et al.,16 WHO11
diseased horses. However, clinical and epidemiological data suggest instances of direct human-to-human transmission in at least five cases.10
Nipah Outbreak in Kerala
Over the last 5 years, Kerala has grappled with four Nipah outbreaks, the most recent occurring in 2023. These incidents, spanning 2018, 2021, and 2023, have primarily affected the Kozhikode district and its surrounding areas, with an additional occurrence in 2019 in the Ernakulam district.11 Table 1 shows the major findings during the Nipah outbreaks in Kerala, and Figure 1A and 1B illustrate the geographical distribution of Nipah outbreaks, detailing cases and deaths in Kerala. The strain of NiV found in Kerala exhibited a genomic
Table 1: Major findings during the Nipah outbreaks in Kerala.
sequence sharing a resemblance ranging from 85.14–96.15% with the M and B genotypes of NiV found in Malaysia and Bangladesh.17 In 2018, all cases in Kerala, excluding the index case, experienced human-to-human transmission, with exposure potentially occurring during activities such as cleaning a bat-infested unused well or visiting the nearby forest. The mapping of fruit bats in the area and specimen studies confirmed the presence of the virus in the bats captured by animal husbandry and national agencies.12 The National Institute of Virology, Pune, India, confirmed in 2021 that antibodies detected in bat samples belonged to the Rousettus genus, in addition to the previously identified Nipah strains in the Pteropus genus.18 The State Government of Kerala documented six laboratory-confirmed cases of NiV infection, in two fatalities, among males aged 40 and 49 years, from 12th–15th
September 2023. The present cases are epidemiologically connected to the initial case with an unknown source of infection, focusing on the Kozhikode district.11 The expeditious urbanisation and environmental alterations in Kerala may contribute to Nipah outbreaks by disrupting natural habitats, which prompts bats to migrate closer to human populations.11,16 The region’s climate change, evident through rising temperatures and altered rainfall patterns, is thought to amplify viral epidemics by fostering conditions conducive to increased virus transmission. Other contributing factors to the recurrent spillover of NiV include extensive forest fragmentation, elevated livestock density, industrialisation, and concentrated human settlement in Kerala.7,19
Figure 1: A) Geographical distribution of the Nipah outbreaks in Kerala, India; and B) year-wise Nipah virus cases and deaths in Kerala.
Kozhikode, 2018
Kozhikode, 2023
Kozhikode, 2021 Kochi, 2019
CLINICAL FEATURES OF NIPAH VIRUS AND ITS DIAGNOSIS
Variations in the incubation period of NiV have been observed across multiple geographical regions. Generally, the incubation period of NiV in Kerala ranges between 4–21 days.20 However, prolonged incubation periods have also been noticed, particularly the outbreak in Malaysia, where the incubation period was reported to be up to 2 months.21 Individuals affected with the viral infection show a spectrum of conditions, ranging from asymptomatic subclinical infection to the development of acute respiratory disease, and potentially deadly encephalitis. Contrary to Malaysia, the outbreaks in Bangladesh and India do not include any information about asymptomatic NiV infection.6 The major clinical manifestations commonly observed in individuals infected with NiV include respiratory distress, nausea, vomiting, headache, fever, and severe encephalitis. Other symptoms reported were behavioural distortion, confusion, pneumonia, reduced consciousness, muscle pain, diarrhoea, cough, and disruptions in the nervous system.3,20 Detecting NiV infection in its initial phases can pose challenges due to the existence of non-specific early symptoms linked to the illness. Nevertheless, the timely identification and assessment of cases assume paramount importance in enhancing the likelihood of survival among affected individuals, curbing the spread of infection to others, and effectively coordinating response measures during an outbreak. Various diagnostic tests are available for the identification of NiV infection. Diagnostic procedures like real-time PCR (RT-PCR) on samples obtained from throat and nasal swabs, urine, cerebrospinal fluid, and blood can be employed during the initial stage of the disease. Adequate and proper infection control measures should be adopted during sample collection, transportation, storage, and processing of the samples of the suspected patients to avoid further risks. The enzyme-linked immunosorbent assay (ELISA) is used to test for antibodies later in the course of the illness and following recovery.3,20,22
PREVENTIVE MEASURES
Although past NiV outbreaks were confined to localised areas, their inherent lethality elicits apprehension among researchers expressing concerns about the prospect of increased human-to-human transmission augmenting the virus’ contagiousness.16 Hence, it is imperative to implement suitable preventative and control strategies to prevent any further transmission of NiV, which includes practising regular handwashing, employing 70% ethanol sanitisation, and refraining from sharing things with infected individuals.16,23 As bats are the natural reservoir hosts of NiV, vigilant inspection of fruit from bat-inhabited trees, and surveillance in affected areas are critical for early outbreak detection. Strict adherence to handwashing, usage of personal protective equipment like gloves and face masks, and the isolation of the affected patients can mitigate health worker transmission.11,16,20,23 Prompt isolation of symptomatic individuals and dedicated NiV wards with discharge contingent on negative RT-PCR results are essential. Discharged patients should be isolated for 21 days from infection detection due to the unclear incubation period.11,20 Community awareness campaigns and utilising various media platforms can help educate the public about the risk factors and stress the importance of strict preventive measures to contain virus spread, particularly within socioeconomically disadvantaged populations.11,20,24
TREATMENT
The Nipah viral infection exhibits distinct characteristics compared to other viral diseases, due to its ability for humanto-human transmission, with no specific treatment or vaccines available to date. Therefore, effective management primarily encompasses the implementation of infection control measures, the process of triaging, the practice of isolation, and the comprehensive management of patients, which includes providing intense supportive care.11,20 During previous outbreaks, the administration of ribavirin and acyclovir
has been employed as a therapeutic intervention for the treatment of NiV infection. The mortality rate was reduced to 36% when ribavirin was administered orally or intravenously during the Malaysian outbreak, whereas acyclovir was utilised during the outbreak in Singapore. However, the effectiveness of these treatments remains uncertain.25,26 Evidence-based studies on the efficacy of the antiviral drug favipiravir (T-705) in hamsters infected with NiV have exhibited positive outcomes, along with investigations into vaccines using the hamster models.27 It is essential to develop targeted antiviral therapeutics to tackle the deleterious impact of NiV on the public as early as possible.
PUBLIC HEALTH RESPONSE IN KERALA
In response to the outbreak, national and state authorities, including the Department of Health and Family Welfare (Government of Kerala), the National Centre for Disease Control (NCDC), the Indian Council of Medical Research (ICMR), the National Institute of Epidemiology (NIE), the National Institute of Virology (NIV), the National Institute of High Security Animal Disease (NIHSAD), and the State Animal Husbandry, promptly implemented a comprehensive multisectoral coordination and response strategy with support from the Ministry of Health and Family Welfare (MoHFW).11,28,29 This involved intensified surveillance, contact tracing, and laboratory testing for suspected cases and high-risk contacts, ensuring hospital readiness for case management, and robust risk communication and community engagement. Containment measures in the affected area included the enforcement of restricted zones, movement restrictions, social distancing, and mandatory maskwearing. The state government meticulously followed a standardised protocol for managing and transferring deceased individuals, utilising dedicated ambulances with skilled medical personnel. Additionally, the implementation of point-of-care assays, monoclonal antibody administration, bio-risk management, and hospital infection control training for healthcare workers significantly
contributed to the effective control and containment of the NiV outbreak.2,11,28,29
NIPAH AS A POTENTIAL THREAT
The global impact of COVID-19 serves as a poignant reminder of the necessity for proactive and collaborative efforts in addressing global health crises. The longterm consequences of the pandemic have been felt across various sectors, including health, education, and the economy. This highlights the critical lesson that a coordinated and pragmatic approach is essential to manage and mitigate the repercussions of future outbreaks effectively.30
Drawing parallels to historical outbreaks, the 1998 NiV outbreak in Malaysia serves as a pertinent example. Beyond the immediate costs associated with culling livestock, this event triggered extensive economic repercussions. The long-term aftermath encompassed disruptions in international trade, tourism, agriculture, and travel. The collapse of the pig farming industry led to widespread unemployment and underemployment. The overall economic impact of the 1998 Nipah outbreak in Malaysia was estimated at a staggering 582 million USD.31,32 While the transmission modes of NiV in India may differ from those in Malaysia, the lessons learned from historical outbreaks emphasise the need to prevent and manage such events effectively. The mortality, morbidity, and economic losses associated with outbreaks make it crucial to establish global collaborative mechanisms to respond swiftly and comprehensively.
Moreover, Kerala has been susceptible to a spectrum of health threats, including mpox and COVID-19, stemming from its global connectivity, international trade, and a significant diaspora. The state’s extensive international links and a substantial overseas healthcare workforce contribute to its vulnerability.33 Additionally, zoonotic diseases such as rabies, leptospirosis, Kyasanur forest disease, and bird flu (avian influenza) continue to pose significant risks in Kerala.34 Unlike COVID-19, which
easily spreads among humans, zoonotic diseases often have a specific animal host before infecting humans. This limited host range reduces their pandemic potential if adequate precautious measures are taken. Transmission modes, such as direct contact with infected animals or their fluids, can hinder widespread human-to-human spread, unlike respiratory viruses like COVID-19. Environmental factors like climate and habitat also influence the spread of zoonotic diseases. Some pathogens are confined to specific ecological niches or regions, limiting their global impact.35
The recent Nipah outbreak in Kerala exemplifies the risks, with unknown exposure in the initial case, higher incidents of human-to-human transmission, limited understanding of virology, and a lack of specific treatment and vaccination. NiV demonstrates a notable propensity for mutation, enabling it to adeptly acclimate to varied environments, infiltrate new hosts, develop novel virulence mechanisms, evade human immune responses, and potentially amplify its transmissibility. If the virus were to mutate in a way that enhances its ability to transmit between humans or evade the immune system, it could increase the likelihood of a pandemic.36 Therefore, extreme precautions should be taken to safeguard against future outbreaks in Kerala, and mitigate risks beyond its borders by enhancing surveillance systems, early detection, developing vaccines and specific antiviral therapies, fostering enhanced public awareness, and leveraging advanced technologies like digital health.
ONE HEALTH CONCEPT
The cross-species transmission of the disease, extending to humans, emphasises the critical need for a ‘One Health’ approach, demanding inter-sectoral collaboration at the global, national, state, and district levels. This strategy, especially in the context of zoonotic diseases like that of NiV, seeks to institute an integrated coordination mechanism with consideration of the human-animal-environment interface, while leveraging the extant surveillance systems of multiple diverse stakeholders for
the early detection of potential outbreaks (Figure 2). This collaborative framework enables a prompt and efficient public health response, fostering the exchange of pertinent information among stakeholders to facilitate necessary actions.29 Kerala was able to confine the further spread of NiV by adopting a comprehensive ‘One Health’ approach, which facilitated seamless coordination among health, veterinary, environmental, and public health sectors.
CLINICAL RELEVANCE AND RECOMMENDATIONS
The emergence of NiV outbreaks in Kerala and its recurring nature underscores the urgent need for heightened clinical awareness, and preparedness among healthcare professionals. Given the high fatality rate associated with NiV infection, early recognition of the symptoms, and prompt diagnostic testing are imperative for timely intervention and containment of the disease spread. Clinicians must remain vigilant, particularly in regions with a history of NiV outbreaks, to promptly identify suspected cases and initiate appropriate infection control measures.
Furthermore, the diverse clinical manifestations of NiV infection, ranging from respiratory distress to neurological complications, necessitate a comprehensive approach to patient management. Healthcare providers should be equipped with the necessary resources and expertise to provide intensive supportive care tailored to the individual’s needs. Additionally, the absence of specific treatments or vaccines for NiV highlights the importance of symptom management and supportive therapy in improving patient outcomes.
RECOMMENDATIONS
Enhanced surveillance: implementing robust surveillance systems for early detection and monitoring of NiV outbreaks is paramount. This includes regular monitoring of bat populations, conducting serological surveys in high-risk areas, and maintaining vigilance
Public awareness
Personal hygiene
Health promotion and prevention
Access to healthcare
Early diagnosis and treatment
Disease surveillance
Development of vaccines & medicines
Environmental health
Ecosystem conservation
Pollution control
Climate change mitigation
Water and food safety
Human health
Communication Collaboration Coordination
Animal health
Veterinary care and surveillance
Wildlife conservation
Livestock health management
Vector control
for unusual clusters of febrile illness or encephalitis cases.
Capacity building: healthcare facilities should be adequately equipped and staffed to manage NiV cases effectively. This includes training healthcare workers in infection control protocols, case management, and the safe handling of laboratory specimens.
Public awareness: engaging in community outreach and education initiatives is essential for raising awareness about NiV
transmission, symptoms, and preventive measures. Empowering communities with accurate information can facilitate early recognition of symptoms, and encourage adherence to preventive practices.
Research and development: continued investment in research, aimed at developing vaccines and specific antiviral therapies for NiV, is crucial. Collaborative efforts between government agencies, research institutions, and pharmaceutical companies can accelerate progress towards effective prevention and treatment strategies.
Figure 2: One Health approach against Nipah virus.
One Health approach: adopting a ‘One Health’ approach that integrates human, animal, and environmental health considerations is essential for mitigating the risk of future NiV outbreaks. This interdisciplinary approach facilitates early detection, rapid response, and coordinated interventions across sectors.
CONCLUSION
The resurgence of the Nipah outbreak, in the same location, in Kerala, should be monitored to avoid potential causes of future occurrences, by implementing preventive measures, enhancing medical preparedness, and protecting public health against the deadly virus. The knowledge acquired from past outbreaks provides insights into formulating an effective approach. This not only protects the affected area but also serves as a paradigm for global health security in response to emerging infectious threats.
References
1. Kulkarni DD et al. Nipah virus infection: current scenario. Indian J Virol 2013;24(3):398.
2. Uwishema O et al. A short communication of Nipah virus outbreak in India: an urgent rising concern. Ann med surg. 2022;82:104599.
3. World Health Organization (WHO). Nipah virus. 2023. Available at: https:// www.who.int/news-room/fact-sheets/ detail/nipah-virus. Last accessed: 29 November 2023.
4. Devnath P, Masud HMAA. Nipah virus: a potential pandemic agent in the context of the current severe acute respiratory syndrome coronavirus 2 pandemic. New Microbes New Infect. 2021;41:100873.
5. Sai KL, Kaw BC. Nipah virus encephalitis outbreak in Malaysia. Clin Infect Dis 2002;34(Supplement 2):S48-51.
6. Banerjee S et al. Nipah virus disease: a rare and intractable disease. Intractable Rare Dis Res. 2019;8(1):1.
7. Pillai VS et al. Nipah virus: past outbreaks and future containment. Viruses. 2020;12(4).
8. Reynes JM et al. Nipah virus in Lyle’s flying foxes, Cambodia. Emerg Infect Dis. 2005;11(7):1042-7.
9. Wacharapluesadee S et al. Bat Nipah virus, Thailand. Emerg Infect Dis. 2005;11(12):1949-51.
10. Ching PKG et al. Outbreak of henipavirus infection, Philippines, 2014. Emerg Infect Dis. 2015;21(2):328.
11. World Health Organization (WHO). Nipah virus infection – India. 2023. Available at: https://www.who.int/ emergencies/disease-outbreak-news/
item/2023-DON490. Last accessed: 29 November 2023.
12. Thomas B et al. Nipah virus infection in Kozhikode, Kerala, South India, in 2018: epidemiology of an outbreak of an emerging disease. Indian J Community Med. 2019;44(4):383.
13. Sudeep AB et al. Detection tablof Nipah virus in Pteropus medius in 2019 outbreak from Ernakulam district, Kerala, India. BMC Infect Dis. 2021;21(1):1-7.
14. Warrier A. A single case outbreak of Nipah Encephalitis from India in May-June 2019. Int J Infect Dis. 2020;101(S1):247.
15. Yadav PD et al. Nipah virus outbreak in Kerala State, India amidst of COVID-19 pandemic. Front Public Health. 2022;10:818545.
16. Srivastava S et al. Recent Nipah virus outbreak in India: lessons and imperatives. Ther Adv Infect Dis. 2023;10:20499361231208535.
17. Yadav PD et al. Nipah virus sequences from humans and bats during Nipah outbreak, Kerala, India, 2018. Emerg Infect Dis. 2019;25(5):1003.
18. Kaliappan A et al. Nipah amidst Covid-19 pandemic, another reemerging infectious disease of pandemic potential - a narrative review. Maedica (Bucur). 2022;17(2):464.
19. Rulli MC et al. Land-use change and the livestock revolution increase the risk of zoonotic coronavirus transmission from rhinolophid bats. Nat Food. 2021;2(6):409-16.
20. Government of Kerala Department of Health and Family Welfare. Annexure Nipah virus infection-treatment guidelines-revised September 2023. 2023. Available at: https://dhs.kerala. gov.in/wp-content/uploads/2023/09/
GO-Rt-No-2363-2023-HFWDdated-16-09-2023-with-AnnexureNipah-Tratment-Guidelines-2023September-.pdf. Last accessed: 13 November 2023.
21. Chua KB. Nipah virus outbreak in Malaysia. J Clin Virol. 2003;26(3):26575.
22. Centers for Disease Control and Prevention (CDC). Diagnosis, Nipah virus (NiV). 2022. Available at: https:// www.cdc.gov/vhf/nipah/diagnosis/ index.html. Last accessed: 25 September 2023.
23. Sazzad HMS et al. Nipah virus infection outbreak with nosocomial and corpse-to-human transmission, Bangladesh. Emerg Infect Dis. 2013;19(2):210-7.
24. Dhillon J, Banerjee A. Controlling Nipah virus encephalitis in Bangladesh: policy options. J Public Health Policy. 2015;36(3):270-82.
25. Broder CC. Henipavirus outbreaks to antivirals: the current status of potential therapeutics. Curr Opin Virol. 2012;2(2):176-87.
26. Chong HT et al. Treatment of acute Nipah encephalitis with ribavirin. Ann Neurol. 2001;49(6):810-3.
27. Dawes BE et al. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Scientific Reports. 2018;8(1):1-11.
28. Sahay RR et al. Experiential learnings from the Nipah virus outbreaks in Kerala towards containment of infectious public health emergencies in India. Epidemiol Infect. 2020;148:e90.
29. Singhai M et al. Nipah virus disease: recent perspective and One Health approach. Ann Glob Health. 2021;87(1):102.
30. Stenseth NC et al. Lessons learnt from
the COVID-19 pandemic. Front Public Health. 2021;9:694705.
31. Smith KM et al. Infectious disease and economics: the case for considering multi-sectoral impacts. One Health. 2019;7:100080.
32. Food and Agriculture Organization of the United Nations Regional Office for Asia and the Pacific Animal Production and Health Commission for Asia and the Pacific (APHCA). Manual on the diagnosis of Nipah Virus infection in animals. 2002. Available at: https://
coin.fao.org/coin-static/ cms/edia/1/13170263993060 /2002_01_high.pdf. Last accessed: 25 September 2023.
33. Firstpost. From monkeypox to COVID-19, why Kerala is a hotbed of diseases. 2022. https://www.firstpost. com/health/from-monkeypox-tocovid-19-why-kerala-is-a-hotbedof-diseases-10915761.html. Last accessed: 17 December 2023.
34. Sukumaran A, Pradeepkumar AS. One Health approach: a platform for
intervention in emerging public health challenges of Kerala state. Int J One Health. 2015;1:14-25.
35. Filho WL et al. Climate change and zoonoses: a review of concepts, definitions, and bibliometrics. Int J Environ Res Public Health. 2022;19(2):893.
36. Kulkarni S et al. Nipah virus edits its P gene at high frequency to express the V and W proteins. J Virol. 2009;83(8):3982-7.
Penile Skin Bridge: Uncommon Cause of Painful Spontaneous Erection in Young Males
Authors: *A.O. Takure1,2
1. Department of Surgery, College of Medicine, University of Ibadan, Nigeria
2. Division of Urological Surgery, Department of Surgery, University College Hospital (UCH), Ibadan, Nigeria
*Correspondence to augusturoendo@gmail.com
Disclosure: The author has declared no conflicts of interest. The patients consented to their pictures being published without revealing their identities. In line with the World Medical Association (WMA) Declaration of Helsinki 2013 on ethical principles for medical research involving human subjects, the author ensured that there was no patient-identifying information. The institutional review board (IRB) number is UI/EC/24/0307.
Received: 23.03.24
Accepted: 03.05.24
Keywords: Painful erection, skin bridge, young male.
Erection is a neurovascular response to visual, emotional, and tactile stimuli. It is classified as psychogenic, reflexogenic, and nocturnal. The causes of painful erections are Peyronie’s disease, penile fracture, penile tumours, penile lichen sclerosis, and a rare disorder called sleep-related painful erection syndrome. Here, the authors report six cases of painful spontaneous erection due to penile skin bridges in young males, seen between 2021–2023. They analysed the age, clinical features, treatment offered, and outcome at 3 months postsurgery. Patients’ mean age was 20.5 years (range: 13–35 years), and the haemoglobin genotype was AA. Two males had associated yellowish discharges under the skin bridges. They all had excision of the skin bridges with satisfactory post-operative appearance, and no pain with erection. Penile skin bridge is an uncommon cause of painful penile erection, and the authors recommend that all general practitioners be aware of this entity, and keep it in mind during a thorough physical examination.
Key Points
1. Penile skin bridge is a complication of neonatal circumcision. Therefore, meticulous circumcision must be practised.
2. Penile skin bridges should be considered an uncommon cause of painful spontaneous penile erection in young males.
3. Surgical excision of skin bridges can provide a satisfactory cosmetic and functional outcome.
INTRODUCTION
Erection is a neurovascular response accompanied by the release of neurotransmitters. The penis is innervated by autonomic (sympathetic and parasympathetic) and somatic (sensory and motor) nerves. The neurons in the spinal cord and peripheral ganglia give rise to the sympathetic and parasympathetic nerves, which form the cavernous nerves that innervate the corpora cavernosa and corpus spongiosum, to stimulate the neurovascular activity during erection and detumescence.1,2
Stimulation of the pelvic parasympathetic nerves and cavernous nerves causes the release of nitric oxide (NO) and acetylcholine. NO mediates the activation of cyclic guanosine monophosphate that results in the downstream activation of protein kinases leading to sinusoidal relaxation, arterial dilatation, and venous compression. The stimulation of the sympathetic nerves inhibits norepinephrine release. Furthermore, stimulation of the somatic nerves releases acetylcholine, which reenforces the penile contraction. The somatic nerve is responsible for sensation, and the contraction of the bulbocavernous (needed for ejaculation) and ischiocavernous muscles (produces rigid erection). Both the autonomic and somatic nerves are activated at the spinal cord level (T11–L2, and S2–S4), and at tactile stimulation of the external genitalia. Nocturnal erection takes place during rapideye-movement (REM) sleep. Therefore, erection requires an intact brain with three components: visual stimuli (inferior temporal cortex), emotional/motivational component (paralimbic areas), and physiological component (controls endocrine and autonomic function in the left anterior cingulate cortex). There are three types of erection: psychogenic, reflexogenic, and nocturnal.1
Anatomically, the penis comprises three erectile tissues: right and left corpora cavernosa, and corpus spongiosum. The corpora cavernosa are surrounded by a rigid, tough bilaminar tunica albuginea. The three erectile tissues are covered by a
thin fibrous tissue called the Bucks fascia, and all these structures are surrounded by the dartos fascia (subcutaneous areolar tissue) and penile skin. The corpus spongiosum expands distally to form the glans penis. The vascular supply to the erectile tissues is the cavernous artery and veins, which are branches of the internal pudendal vessels. The penile skin is supplied by the dorsal artery of the penis that is derived from the external pudendal artery. The cavernous vein communicates with the emissary veins through the subtunica venous plexus, and these veins are compressed during erection.3
Abnormality with adequate arterial and venous return during erection may result in persistent painful erection, especially in ischaemic priapism;2,4 as well as penile fracture that occurs during sexual intercourse, when the penis slips outside the vagina, and hits the inferior pubic rami bone.5,6 There is an association between lichen sclerosus et atrophicus and penile cancer. Lichen sclerosus is a benign, chronic, sclerosing atrophy of the glans and foreskin. It was reported in a 36-year-old male with whiteish atrophic patch on the glans penis, telangiectasia, and superficial ulcer associated with painful erection, which turned out to be an aggressive squamous cell tumour of the penis.7 Painful nocturnal erections, or sleep-related painful erections that occur during REM sleep, have been reported. This is found in young males, and has a common mean age of onset of 40 years.8
Skin bridges, a complication of circumcision, which commonly accompanies childhood circumcision, have been reported to cause tethering of the penis and occasional pain.9 The literature search in Nigeria, North Africa, East Africa, and South Africa did not reveal any previous publication on the relationship between penile skin bridge and erection in young males.
The authors present cases of postcircumcision penile skin bridge as an uncommon cause of painful erection in young males.
METHOD
Following the first case of a 17-yearold male who presented with painful spontaneous erection and yellowish discharge around the junction between distal penis and glans penis in 2021, the authors subsequently collected data from patients who presented with painful spontaneous erection, in the absence of sexual stimulation, and were managed over a 3-year period (2021–2023). The data included the age, clinical features, treatment offered, and outcome of treatment.
RESULTS
Six young males were managed for painful spontaneous penile erection. They were ages 13, 16, 17, 19, 23, and 35 years. The mean age was 20.5 years, with a range of 13–35 years. They all presented with erection, without sexual arousal, that was associated with pain, and two had yellowish discharge around the corona of
the penis. There was no significant medical or surgical history apart from the neonatal circumcision that they all had; neither was there any genital trauma. They were not sexually active, except the 35-year-old who was afraid of engaging in sexual intercourse because of the painful erection.
The examination confirmed a circumcised male penis with multiple skin bridges around the dorsal, lateral, and ventral aspect of the penile corona (Figure 1A and 1B). Discharges (smegma) around the penis were seen in two patients (Figure 2A). Their haemoglobin genotype was AA. Preoperative full blood count and urinalysis were essentially within the normal range. All patients had excision of the skin bridges under general intravenous anaesthesia, spinal anaesthesia, and local anaesthesia with sedation (Figure 2B). At 3 months postsurgery, the cosmetic appearance of the penis was satisfactory (Figure 3), and there was no pain with subsequent spontaneous penile erection.
Figure 1: Pre-operative A) dorsal and B) ventral penile skin bridges.
Figure 2: Intra-operative A) smegma and B) excised penile skin bridges.
A B
3: Post-operative cosmetic functional penis.
DISCUSSION
Painful penile erection in adults implies persistence of venous obstruction and disruption of tunica albuginea, manifesting as acute priapism and penile fracture. Priapism is the sudden, persistent, often painful,
purposeless penile erection that is not related to sexual arousal or ejaculation. The commonest cause of ischaemic priapism in 50% of cases is sickle cell disease. Other causes are the use of aphrodisiacs (19.2%), leukaemia (7.7%), and idiopathic (23.1%).4 Recurrent ischaemic priapism is another terminology
Figure
for stuttering priapism, which is defined as repeated ischaemic episodes over time, and is self-limiting.2
Penile fracture is a sudden disruption of the tunica albuginea of the penis that often follows sexual intercourse in 48% of cases, where the penis slips out of the vagina and hits the pubic bone, or during masturbation and forced flexion of an erect penis in 39% of cases.5,6
Burnett8 described two rare causes of painful penile erection: painful nocturnal erection and sleep-related painful erection (SRPE), and idiopathic stuttering priapism. SRPE occurs during REM sleep, and is classified as parasomnia, which is a group of undesirable physical phenomena, such as movements/ behaviours, or experiences of emotions, perceptions, and dreams that are seen while falling asleep, being asleep, or while waking up from sleep. SRPE affects males of all ages, with an average age of onset of 40 years.8
A separate subcategory of ischaemic priapism that is recurrent and idiopathic is termed idiopathic stuttering priapism. It is of short duration, recurrent, and believed to be stimulated by psychic centres, and manipulation of external genitalia.8
In this study, the authors report spontaneous penile erection in young males, due to skin bridges on the corona of the penis. These act as band-like vascularised tissue, and are a complication of neonatal circumcision, seen in all patients who were circumcised.
References
1. Dean RC, Lue TF. Physiology of penile erection and pathophysiology of erectile dysfunction. Urol Clin North Am. 2005;32(4):379.
2. Joice GA et al. Medical treatment of recurrent ischaemic priapism: a review of current molecular therapeutics and a new clinical management paradigm. BJU Int. 2021;127:498-506.
3. Hsu GL et al. Anatomy of the human penis: relationship of the architecture between skeletal and smooth muscles. J Androl. 2013;25(3):426-31.
These areas of adhesion between the glans and skin of the penile shaft prevent adequate elongation of the penis, and ultimately result in an abnormal curvature of the penis that is painful in these patients, as also previously reported by Kampouroglou et al.9 Kamal et al.10 documented the management of 57 adult males in Saudi Arabia who presented with penile skin bridges following childhood circumcision.
Two patients in this series presented with a yellowish discharge under the penile skin bridge. This was smegma, which is commonly found in uncircumcised males, but Almutawa et al.11 reported this in a 36-yearold, circumcised male. Smegma is an opaque white or yellowish substance composed of dead cells and skin oils that is malodorous.11
The penile skin bridge, when thick, is often vascularised. The treatment in this series was surgical excision.8,10,11 All patients were satisfied with the cosmetic appearance and function of their penis, with disappearance of pain with subsequent erection. Meticulous suturing and dressing at the time of circumcision can reduce the occurrence of penile skin bridge.
CONCLUSION
Penile skin bridges are an uncommon cause of painful erection, among others, and the authors recommend that all general practitioners be aware of this entity, and to keep it in mind during a thorough physical examination.
4. Mohammad AS et al. Pattern and management of priapism in a tertiary hospital of North-Western Nigeria. ECAJS. 2017;22:66-71.
5. Pandler et al. Presentation, management, and outcome of penile fractures. JOMH. 2022;18(11):215.
6. Ouattara A et al. Penile fracture in Burkina Faso: our experience on the management of 21 cases. Afr J Urol. 2023;29:38.
7. Lei MA et al. Rapid onset of squamous cell carcinoma in a short-standing lichen sclerosus et atrophicus penis.
Background: Central line-associated bloodstream infection (CLABSI) is a serious infection typically increasing morbidity and mortality in patients with chronic kidney disease (CKD). It can be prevented through proper insertion techniques and management of the central line (CL). However, the first step in reducing the CLABSI rate is to define the extent of the problem through proper surveillance. This study aimed to determine the frequency of CLABSI in patients with CKD at a specialised renal care centre.
Methods: The authors conducted a retrospective observational study to determine the frequency of CLABSI in patients with CKD between November 2021–September 2022 at their institute. They included all patients with CLs registered at their institute. Primary CLABSI was defined as CLABSI attributable to their hospital, while secondary CLABSI was defined as those not attributed to their hospital.
Results: Fifty-nine incidences of CLABSI were identified in a total of 310 patients with CL and 1,413 CL days, giving a total of 42 CLABSI incidences per 1,000 CL days. Primary CLABSI was more common (n=36 [61%]) than secondary CLABSI (n=23 [39%]). Most of the patients recovered (53 [89.8%]); however, four (6.9%) patients expired. Most of the patients who recovered had permanent vascular access (n=32 [60.4%]), internal jugular placement (n=44 [83%]), and primary CLABSI (n=33 [62.3%]), although the p-values were non-significant.
Conclusion: Strict implementation of CLABSI prevention bundles for line insertion and its maintenance and regular surveillance using laboratory confirmed cases is needed to reduce the rates of CLABSI.
Key Points
1. Central line bloodstream infection (CLABSI) is a leading cause of high morbidity and mortality in patients with chronic kidney disease (CKD) who are dialysis dependent.
2. This was a retrospective observational study to determine the frequency of CLABSI in patients with CKD at a renal care centre.
3. Appropriate surveillance of the disease, followed by strict aseptic measures during central line insertion and maintenance, are the key steps to reduce CLABSI in patients with CKD.
BACKGROUND
Infection leads to an increased rate and duration of hospitalisation, increased treatment costs, and high morbidity and mortality in patients with chronic kidney disease (CKD) with end-stage renal disease. Its major risk factor in patients on chronic haemodialysis (HD) is vascular access, especially central line (CL),1-7 which leads to central line-associated bloodstream infection (CLABSI), resulting in lifethreatening complications.8-10
Appropriate infection prevention and control (IPC) measures taken at the time of insertion and maintenance can prevent CLABSI.11 A focus on education and strict adherence to IPC measures is associated with reducing its rate.12,13 However, the first step in reducing the CLABSI rate is to define the extent of the problem through proper surveillance.14,15 There is a high burden of CLABSI in patients with CKD, as evident by the past data.1,7,16,17 This study aimed to determine the frequency of CLABSI in patients with CKD at a specialised renal care centre, to add to the previous existing literature from the authors’ country.
METHODS
The authors conducted a retrospective observational study to determine the frequency of CLABSI in patients with CKD between November 2021–September 2022 at their institute, which is a not-for-profit specialised hospital providing care to patients suffering from kidney disease.
An exemption was obtained from the institute’s ethical review committee (ref. no. 152-IDC-102022(EXEMPTION).
Using the Centers for Disease Control and Prevention (CDC) surveillance definition,14,18 CLABSI was defined as a laboratoryconfirmed bloodstream infection (LCBI) that was not secondary to an infection at another body site, where an eligible bloodstream infection organism was identified and a CL had been in place for more than two consecutive calendar days on the LCBI date of an event or the day before.
CL was defined as an intravascular catheter that terminated at or close to the heart, or in one of the great vessels, used for dialysis, infusion, withdrawal of blood, or haemodynamic monitoring.
The authors included all patients with CLs registered at their institute; these included patients admitted with them, those routinely receiving dialysis at the institute, and those who presented to the emergency room. They excluded nonregistered patients and registered patients without any CL. CL days were assessed to determine if an LCBI was a CLABSI. The count of CLs was recorded in the monthly IPC hospital acquired infections data.
The authors modified the CDC criteria and also identified CLABSI not attributed to their institute. Primary CLABSI was defined as CLABSI attributable to them, while secondary CLABSI was defined as those not attributed to their hospital.
STATISTICAL ANALYSIS
The data were analysed on SPSS version 26 (IBM, Armonk, New York, USA). Continuous variables were evaluated using mean with standard deviation, while categorical data were evaluated using frequency with percentage. The ꭓ2 test observed association among variables. A p-value of ≤0.05 was considered statistically significant.
RESULTS
Fifty-nine incidences of CLABSI were identified in a total of 310 patients with CL and 1,413 CL days, giving a total of 42 CLABSI incidences per 1,000 CL days. Primary CLABSI was more common (n=36 [61%]) than secondary CLABSI (n=23 [39%]), and there were more male cases (36 [61%]) than female ones (n=23 [39%]).
The mean age was 53±17.8 years, and the median age was 57, with an interquartile range of 27 years. Most patients had permanent vascular access (n=36 [61%]). The internal jugular was the major site of choice (n=49 [83.1%]; Table 1). Most CLs (n=37 [63%]) were inserted using ultrasound guidance. Most of the patients recovered (53 [89.8%]); however, four (6.9%) patients expired (Figure 1). Most of the patients who recovered had permanent vascular access (n=32 [60.4%]), internal jugular placement (n=44 [83%]), and primary CLABSI (n=33 [62.3%]), although the p-values were non-significant (Table 2).
Staphylococcus aureus; Enterobacterales, including Enterobacter spp. (such as Enterobacter cloacae and other species), Klebsiella spp., Escherichia Coli, and Citrobacter freundii; Candida spp., including Candida tropicalis, Candida parapsilosis,
CLABSI: central line-associated bloodstream infection.
Table 1: Baseline characteristics of the patients with central line-associated bloodstream infection (n [%] /mean±standard deviation).
Figure 1: Outcome of the patients.
Table 2: Association of clinical characteristics of the patients with the outcome.
and other Candida spp.; Enterococcus spp.; Acinetobacter spp.; Pseudomonas aeruginosa; Stenotrophomonas maltophilia; and other Pseudomonas spp., including Ralstonia pickettii, Shewanella putrefaciens, and Chryseobacterium spp., were identified in decreasing order of their occurrence (Figure 2).
DISCUSSION
The authors’ institute is a specialised hospital providing care to patients with CKD. The majority of these patients receive HD, for which temporary or permanent vascular access is required.16 The authors aim for an arteriovenous fistula (AVF) or arteriovenous graft for better clinical outcomes and lower infection rate;19,20 however, temporary access is needed in cases where permanent access is either not available or not ready to be used. It is achieved with a CL, which can be permanent (tunnelled/implanted), or temporary (non-tunnelled), but chances of infection are generally higher in these.8 In a previous study from the authors’ institute, Qureshi et al.16 concluded that out of a total of 429 non-tunnelled CLs, 50 (17%) were removed due to CLABSI. Similar to the authors’ data, S. aureus was the most
common organism responsible for CLABSI.16 Another previous study from the authors’ institute also shows a high burden of S. aureus CLABSI in patients with CKD.7 This is consistent with the previous data from the authors’ country, which shows a high burden of S. aureus CLABSI in patients with CKD.21 However, this differs from the National Healthcare Safety Network (NHSN) data from January 2006–October 2007, where an estimated 250,000 bloodstream infections occur annually, and most are related to the presence of intravascular devices. The order of selected pathogens associated with CLABSI was as follows: 34.1% coagulase-negative Staphylococcus spp.; 16.0% Enterococcus spp.; and 9.9% S. aureus; followed by Gram-negatives with 5.8% Klebsiella spp., 3.9% Enterobacter spp., 3.1% Pseudomonas spp., 2.7% E. coli, and 2.2% Acinetobacter spp.; Candida species (11.8%); and others (10.5%). 22
In the authors’ study, out of a total of 59 incidences of CLABSI, 36 (61%) were tunnelled (permanent) catheters, and 23 (39%) were non-tunnelled ones. The internal jugular vein was used in 50 (85%) cases, while the femoral vein was used in nine (15%) cases.
Staphylococcus aureus
Enterococcus spp.
Enterobacterales
Pseudomonas aeruginosa
Stenotrophomonas maltophilia
Pseudomonas spp.
Acinetobacter spp.
Candida spp.
Figure 2: Frequency of organisms isolated.
These findings contrast with Zanoni et al.,8 who concluded that as compared to tunnelled jugular CL, a higher CLABSI rate was associated with non-tunnelled jugular and non-tunnelled femoral CL use. They also concluded that non-tunnelled and femoral CL were associated with higher CLABSI rates.8 The possible reason for more internal jugular lines can be the fact that in the authors’ hospital, most lines are inserted into these, as also supported by the findings of Qureshi et al.16 Likewise, the practice of promoting tunnelled catheters instead of non-tunnelled ones at the authors’ institute could be a reason for more CLABSI cases in tunnelled ones.
In an Irish cohort, while determining the outcomes of access-related bloodstream infections in patients on HD, Mohamed et al.3 concluded that access-related bloodstream infection was more associated with CLs than AVF in HD, and AVF preference over CL should be emphasised.
There was a high rate of CLABSI. Proper surveillance of CLABSI is essential for determining the frequency of its incidence, and thus for preventive measures. The majority of CLABSI incidences were attributable to the authors’ centre. However, the authors also identified cases where the line was inserted/maintained at other institutes, and these were considered as secondary CLABSI.
In addition to adding to the existing local data of CLABSI, the authors’ data emphasise that IPC practices, including preventive bundles during line insertion and maintenance, need to be strengthened, together with proper surveillance of rates of infection. Major areas of emphasis include strict adherence to aseptic measures, i.e., CLABSI prevention
References:
1. Anser F et al. Comparison between the yield of different number of blood cultures in chronic kidney disease patients with suspected septicemia. Cureus. 2021;13(12):e20381.
2. Kumbar L, Yee J. Current concepts in hemodialysis vascular access infections. Adv Chronic Kidney Dis. 2019;26(1):16-22.
insertion and maintenance bundles, especially focusing on proper sterile barrier precautions, together with skin antisepsis with >0.5% chlorhexidine preparations. Focus should also lie on pre-procedure patient shower or bed sponge with chlorhexidine. Previous studies support that adequate surveillance followed by strict adherence to CLABSI prevention insertion and maintenance bundles, which include, but are not limited to, hand hygiene, continuous training of healthcare providers, chlorhexidine bath and skin antisepsis, proper sterile barrier precautions, use of ultrasound guidance to prevent multiple pricks, chlorhexidine dressings, minocycline/rifampin catheters, and removal of the line as soon as possible, lead to a reduction in the rate of CLABSIs.13,23
The authors are emphasising on these prevention bundles, with proper hand hygiene as the key to all IPC measures. Regular teaching, on the spot reminders, and frequent and surprise audits are being practised to reduce the CLABSI rates.
A small sample size is a major limitation of the authors’ study. Secondly, many samples yielded multiple organisms, though they fulfilled the CDC criteria for CLABSI, and thus were included in the study. Still, these data help in monitoring the frequency of CLABSI at the institute through surveillance and focusing on the education of healthcare providers in aseptic line insertion and maintenance bundles.
CONCLUSION
Strict implementation of CLABSI prevention bundles for line insertion and its maintenance and regular surveillance using laboratory confirmed cases is needed to reduce the rates of CLABSI.
3. Mohamed H et al. Determinants and outcomes of access-related blood-stream infections among Irish haemodialysis patients; a cohort study. BMC Nephrol. 2019;20(1):68.
4. Allon M et al; HEMO Study Group. The spectrum of infection-related morbidity in hospitalized haemodialysis patients. Nephrol Dial Transplant. 2005;20(6):1180-6.
5. James MT et al; Alberta Kidney Disease Network. Risk of bloodstream infection in patients with chronic kidney disease not treated with dialysis. Arch Intern Med. 2008;168(21):2333-9.
6. Sarnak MJ, Jaber BL. Mortality caused by sepsis in patients with endstage renal disease compared with the general population. Kidney Int. 2000;58(4):1758-64.
7. Moin S et al. Staphylococcus aureus bacteraemia in patients with chronic kidney disease: single-centre data from Pakistan. EMJ. 2024;9(1):68-76.
8. Zanoni F et al. Catheter-related bloodstream infections in a nephrology unit: analysis of patient- and catheterassociated risk factors. J Vasc Access. 2021;22(3):337-43.
9. Rojas-Moreno CA et al. Catheterrelated bloodstream infections in patients on emergent hemodialysis. Infect Control Hosp Epidemiol. 2016;37(3):301-5.
10. Gupta V, Yassin MH. Infection and hemodialysis access: an updated review. Infect Disord Drug Targets. 2013;13(3):196-205.
11. O’Grady NP et al; Healthcare Infection Control Practices Advisory Committee (HICPAC) (Appendix 1). Summary of recommendations: guidelines for the prevention of intravascular catheterrelated infections. Clin Infect Dis. 2011;52(9):1087-99.
12. Mermel LA et al. Clinical practice guidelines for the diagnosis and management of intravascular catheterrelated infection: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49(1):1-45.
13. Marschall J et al.; Society for Healthcare Epidemiology of America. Strategies to prevent central line–associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol. 2014;35(7):753-71.
14. Centers for Disease Control National Healthcare Safety Network. Bloodstream infection event (central line-associated bloodstream infection and non-central line associated bloodstream infection). 2024. Available at: https://www.cdc.gov/nhsn/pdfs/ pscmanual/4psc_clabscurrent.pdf. Last accessed: 17 February 2024.
15. Sulong A et al. Surveillance of centralline associated bloodstream infections in ICU at a Malaysian medical centre. BMC Proceedings. 2011;5(Suppl 6):P212.
16. Qureshi R et al. Reasons for removal of non-tunneled double lumen catheters in incident dialysis patients. J Coll Physicians Surg Pak. 2018;28(4):284-7.
17. Jamil B et al. Bacteremia: prevalence and antimicrobial resistance profiling in chronic kidney diseases and renal transplant patients. J Pak Med Assoc. 2016;66(6):705-9.
18. National Healthcare Safety Network (NHSN). Bloodstream infection event
(central line-associated bloodstream infection and non-central line associated bloodstream infection). 2024. Available at: https://www.cdc. gov/nhsn/pdfs/pscmanual/4psc_ clabscurrent.pdf. Last accessed: 1 February 2024.
19. Vascular Access Work Group. Clinical practice guidelines for vascular access. Am J Kidney Dis. 2006;48(Suppl 1):S248-73.
20. Drew DA, Lok CE. Strategies for planning the optimal dialysis access for an individual patient. Curr Opin Nephrol Hypertens. 2014;23(3):314-20.
21. Javaid S et al. Clinical features and outcome of Staphylococcus aureus bacteremia from a tertiary care hospital in Pakistan. Infect Dis J Pak. 2022;31(3):89-95.
22. Haddadin Y et al., Central Line–Associated Blood Stream Infections [Internet] (2017) Treasure Island: StatPearls. Available at: https:// pubmed.ncbi.nlm.nih.gov/28613641/. Last accessed 13 May 2024.
23. Walz JM et al.; CCOC Research Group. The bundle “plus”: the effect of a multidisciplinary team approach to eradicate central line-associated bloodstream infections. Anesth Analg. 2015;120(4):868-76.
Caregiver Burden, Resilience, and Wellbeing in Cases of Severe Cutaneous Adverse Drug Reactions
Background: Severe cutaneous adverse reactions (SCAR) can be traumatic and emotionally distressing for both the patients and their families. However, caregivers must also take care of themselves to prevent burnout. They should seek respite when needed, and prioritise self-care activities that maintain their own wellbeing.
Aim: This study aimed to explore the caregiver’s burden and resilience in patients experiencing SCARs.
Methods: A cross-sectional observational study included patients experiencing SCARs who presented with their caregivers. Patients and their caregivers were enquired about their sociodemographic variables, and were administered the Brief Resilience Scale (BRS). Caregivers were further given the Burden Scale for Family Caregivers (BSFC) and the World Health Organization Quality of Life Brief (WHOQOL-BREF).
Results: Quality of life assessment suggested diminished physical and psychological health among the caregivers. Burden grade conferred 27.1% of caregivers experiencing severe burden, while 56.5% and 16.5% of caregivers experienced mild and moderate burden, respectively. Furthermore, 85.9% of caregivers showed low resilience. Increase in reaction severity was associated with greater caregiver burden and low resilience (p=0.001). Higher age and lower socioeconomic strata were also associated with increased burden and lesser caregiver resilience (p<0.001).
Conclusion: Providing care for individuals with SCARs can be physically and emotionally demanding, requiring assistance with daily activities, wound care, and medication management. Caregivers may face challenges as they navigate the complexities of the condition; hence, understanding and addressing the challenges faced by caregivers is of utmost importance.
Key Points
1. Severe cutaneous adverse reactions (SCAR) can be traumatic and emotionally distressing for both patients and their families. Alongside the delivery of support and care, it is important that caregivers also look after themselves to prevent burnout.
2. This study explored the caregiver burden and resilience in 85 patients experiencing SCARs using relevant questionnaires. The findings showed that 27.1% of caregivers experience severe burden.
3. Besides prioritising patient outcomes, disease management, and treatment efficacy, a compassionate understanding of caregiver burnout and informed communication can help in the holistic management of patients with SCAR.
INTRODUCTION
Severe cutaneous adverse drug reactions (SCAR) are a group of rare but potentially life-threatening conditions that can occur in response to certain medications. These reactions include conditions such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reactions with eosinophilia and systemic symptoms (DRESS). Resilience, in the context of SCARs, refers to a patient’s ability to cope with and recover from these severe reactions, both physically and emotionally. Knowledge about SCARs, their causes, treatment options, and potential outcomes can empower patients and their families to actively participate in their recovery process.1 Understanding the condition can alleviate fear and anxiety. Adhering to medical treatments, such as wound care, medications, and follow-up appointments, is crucial for recovery. It is important to note that every individual’s experience with SCARs, and their recovery journey, will be unique. Healthcare providers, mental health professionals, and support groups can all play crucial roles in helping patients build resilience, and navigate the challenges posed by SCARs.2 Caregivers play a pivotal role in supporting these patients. These reactions can be physically and emotionally demanding for patients, often requiring significant medical attention and support. Caregivers provide essential assistance, comfort, and advocacy throughout the patient’s recovery process.3
SCARs can be traumatic and emotionally distressing for both patients and their families.4 Caregivers offer emotional support
by providing a listening ear, offering words of encouragement, and being empathetic to the patient’s feelings and concerns. They ensure that the patient’s medical needs are met. SCARs can significantly limit a patient’s ability to perform basic activities like bathing, dressing, eating, and mobility.4 Caregivers may provide physical assistance with these tasks, helping the patient maintain their dignity and comfort. They help manage the patient’s medication regimen, ensuring that medications are taken as prescribed, tracking dosages and schedules, and noting any adverse effects or improvements. In severe cases, these reactions can cause extensive skin damage and open wounds. Caregivers may be responsible for assisting with wound care, including cleaning, dressing changes, and following medical instructions for proper care.5 They further ensure that the patient maintains proper nutrition and hydration, as these factors play a crucial role in the healing process. They might prepare meals, provide water, and monitor the patient’s intake; and may be responsible for transporting the patient to medical appointments, follow-up visits, and therapy sessions as needed, helping ensure that the patient receives timely medical care and support.
Caregivers often serve as a bridge of communication between the patient and healthcare providers. They help relay important information, questions, and concerns to the medical team and vice versa. Caregivers also help create a positive and supportive environment that contributes to the patient’s emotional wellbeing. They engage in activities that bring comfort and
joy to the patient, and they help manage any anxiety or emotional distress. Caregivers learn about the patient’s condition, treatment plan, and potential complications in order to provide informed care. This education helps them make informed decisions, and offer explanations to the patient and family. However, caregivers must also take care of themselves to prevent burnout.6 They should seek respite when needed, and prioritise self-care activities that maintain their own wellbeing.
The prevalence of SCARs varies widely depending on the specific type of reaction and the population studied. The annual incidence of SJS and TEN7 combined is estimated to be between one and six cases per million people worldwide, and DRESS syndrome is estimated to occur in one in 1,000 to one in 10,000 drug exposures.8 Acute generalised exanthematous pustulosis, considered a rare occurrence, has an estimated incidence of one to five cases per million people per year.9 However, these incidences can be higher in certain populations with specific drug exposures. Healthcare research often prioritises patient outcomes, disease management, and treatment efficacy. While these are crucial, there is a growing recognition of the importance of understanding and addressing the challenges faced by caregivers. Despite having a significant prevalence with a known impact of SCARs on the physical and emotional wellbeing of the patient, research on the aspects of caregiver burden has not been explored. The present study thus tried to address this dearth, and explore the caregiver burden and resilience in patients experiencing SCARs.
METHODS
Study Design and Setting
The study had a cross-sectional observational design. It was carried out for a period of 2 years, in a tertiary care teaching hospital in Eastern India. The study was approved by the Institutional Ethics Committee of the School of Tropical Medicine, Kolkata, India (Approval No: CREC-STM/385).
All the patients provided written informed consent after a full explanation of the protocol design, and the study was conducted according to the Declaration of Helsinki.
Participants
The study included patients experiencing SCARs who presented with their caregivers on the first follow-up post-discharge, and who consented to be a part of the study. Consenting caregivers, as well as the patient, were required to provide written informed consent before participation. Patients with documented psychiatric comorbidities, or any other psychological conditions that could potentially impede their understanding of the questionnaire and accurate responses, were excluded from the study.
Variables and Data Measurement
Patients and their caregivers were enquired about their sociodemographic variables, and were given brief resilience scale (BRS) questionnaires.10 Caregivers were given assessment scales, namely the Burden Scale for Family Caregivers (BSFC),11 and the World Health Organization Quality of Life Brief (WHOQOL-BREF).12
The BRS10 is a self-report assessment tool used to measure an individual’s ability to bounce back from stress and adversity. It is designed to assess resilience, which is the capacity to adapt and recover from challenging situations. The BRS is a short and simple questionnaire, often used in research and clinical settings to quickly evaluate an individual’s resilience level. It typically consists of six items, and respondents rate their agreement with each item on a scale, such as a 5-point Likert scale (ranging from strongly disagree to strongly agree). The questions on the BRS are meant to capture various aspects of resilience, including the ability to cope with stress, adapt to change, and maintain a positive outlook in the face of adversity. The scoring is obtained by adding the responses, and then dividing the total sum by the total number of questions answered. A score of 1.00–2.99 suggests low
resilience, 3.00–4.30 suggests normal, and 4.30–5.00 suggests high resilience.
BSFC11 is a 28-item scale measuring subjective burden, with 4 points on scale, from strongly agree to strongly disagree. The BSFC helps assess the emotional, physical, and social burden experienced by family caregivers, providing insights into the challenges they face. This information can be valuable in healthcare settings for care planning, and in research studies to understand the impact of caregiving on family caregivers’ wellbeing. A cumulative score of 0–41 suggests no to mild burden, 42–55 suggests moderate burden, and 56–84 suggests severe-to-very-severe burden.11 Quality of life was adjudged using WHOQOLBREF,12 a self-administered questionnaire comprising 26 questions on the individual’s perceptions of their health and wellbeing. The WHOQOL-BREF covers four domains, each with specific facets: physical health, psychological health, social relationships, and environment. There are also two separate questions which ask specifically about the individual’s overall perception of their health and quality of life.
Each of these generic instruments were pilot-tested in a representative population that was not included in the final study. Each instrument was used in the study population only if it yielded acceptable reliability measures. Caregivers were interviewed during their facility visit along with the patient for the scheduled (first) follow-up post-discharge.
Study Size
Considering the prevalence of cutaneous adverse drug reactions among hospitalised patients was 5% as reported,13 with a 5% allowable margin of error and a 95% confidence interval (CI), the estimated sample size of the study was 73. However, the study included a total of 85 patients, and analysed the data collected statistically.
Statistical Methods
Data collected were statistically analysed. Descriptive statistics were used to analyse the data, and the results were represented
as mean, standard deviation, frequency, and percentages, as applicable. p<0.05 was considered significant. The normality test using Shapiro–Wilk test showed that the measures were normally distributed; hence, a parametric approach was undertaken. Multiple linear logistic regression analyses were performed to find predictors for burden and resilience. The variables with variance inflation factor greater than 10 were omitted from the analysis to avoid multi-collinearity. All statistical tests were conducted on standard statistical software like SPSS Windows version 23.0 (IBM, Armonk, New York, USA) and Microsoft Excel (Microsoft, Redmond, Washington, USA).
RESULTS
Patient Demographics and Characteristics
The study included a total of 85 patients with SCARs, whose caregivers were interviewed. Of the 85 patients, the majority were males (61.2%). The most represented age group was that of 31–50 years, followed by 51–70 years, and 18–30 years. The mean age of the patients was recorded as 41.85±8.90 (95% CI: 39.93–43.77). A total of 42.4% of patients belonged to the lower-middle class of socioeconomic strata, followed by 32.9% belonging to the upperlower strata, and 24.7% belonging to the upper-middle socioeconomic class. Only 23 patients (27.05%) had comorbidities.
Among the 85 caregivers interviewed for this study, the majority belonged to the age group of 31–50 years, followed by 51–70 years. The mean caregiver age was 45.64±11.96 (95% CI: 43.06–48.22).
A total of 52.9% of caregivers were females. Caregivers were mostly spouses (54.1%), followed by parents (27.1%) and children (18.8%; Table 1)
Main Results
A total of 35.3% of patients experienced SJS, followed by 21.2% of patients experiencing DRESS and TEN. Erythema multiforme was experienced by 18 (21.25%)
Table 1: Patient and caregiver characteristics.
patients, and 8.20% of patients experienced acute generalised exanthematous pustulosis. No cases of SJS-TEN overlap were encountered. As per the Hartwig Siegel scale,14 68.2% of cases were assessed to be ‘severe’, followed by 29.4% moderate cases, and 2.4% mild cases. A total of 92.9% of SCARs were nonpreventable as per Schumock Thornton Preventability Scale (Table 2).15 Post-SCAR sequelae were noted in two patients with TEN, and one patient with SJS. The noted sequelae in all three cases were ocular complaints, including eye pain and photophobia. Conjunctival hyperaemia was noted in one case of TEN. Quality of life was assessed using WHOQOLBREF and EuroQol Visual Analogue Scale (EQVAS). Mean domain scores of WHOQOLBREF revealed diminished physical and
psychological health among caregivers of patients with SCAR. Burden grade using the BSFC scale estimated that 27.1% (n=23) of the caregivers experienced severe burden, and 56.5% (n=48) and 16.5% (n=14) experienced mild and moderate burden, respectively. Using the BRS measure, 85.9% (n=73) showed low resilience, with the rest exhibiting normal resilience (Table 3).
Furthermore, the severity of the SCARs was correlated with the burden and resilience score. A significant association was observed with both measures, suggesting that the increase in reaction severity was associated with greater caregiver burden and low resilience among patients (p=0.001). Other factors, like age and socioeconomic class, were also probed for association.
AGEP: acute generalised exanthematous pustulosis; DRESS: drug reactions with eosinophilia and systemic symptoms; SCAR: severe cutaneous adverse reaction.
Table 2: Characteristics of severe cutaneous adverse reactions.
Table 3: Quality of life, burden, and resilience in severe cutaneous adverse reaction cases.
BRS: Brief Resilience Scale; BSFC: Burden Scale for Family Caregivers; EQVAS: EuroQol Visual Analogue Scale; N/A: not applicable; SCAR: severe cutaneous adverse reaction; WHOQOL-BREF: World Health Organization Quality of Life Brief.
Higher age and lower socioeconomic strata were associated with increased caregiver burden and lesser resilience among patients (p<0.001). However, no significant association of caregiver burden was noted with the patient’s age (p=0.326) and comorbidities (p=0.214).
Multivariable linear regression was conducted to examine the best combination of factors for predicting caregiver burden and resilience. Multivariate linear regression analysis showed that BRS score in SCAR cases was negatively affected by severity of adverse drug reaction (β: -5.588; standard error [SE]: 0.527; p=0.000; 95% CI: -6.623–4.553) and patient’s age (β: -1.246; SE: 0.225; p=0.000; 95% CI: -1.688–-0.805).
BSFC score was positively affected by the adverse drug reaction severity (β: 0.468; SE: 0.579; p=0.000; 95% CI: -0.669–1.605), and caregiver’s age (β: 0.178; SE: 0.207; p=0.000; 95% CI: -0.061–0.132). Age and reaction severity were the only strong predictors for both higher caregiver burden and lower resilience.
DISCUSSION
The impact of SCARs extends beyond the immediate medical consequences. Besides affecting mental health, quality of life, and economic wellbeing of the individual, it can also affect interpersonal relationships. SCARs can strain personal relationships as individuals cope with the physical and emotional aftermath of the condition. Family members and caregivers may also experience stress and emotional burden while providing support; therefore, a comprehensive and holistic approach to care, including medical, psychological, and social support, is essential for individuals affected by SCARs.16
The present study tried to assess the quality of life, burden, and resilience among caregivers of patients affected by SCARs. Quality of life assessment suggested diminished physical and psychological health among the caregivers. Burden grade conferred 27.1% of caregivers experiencing severe burden, while 56.5% and 16.5% of caregivers experienced mild and moderate burden, respectively, and 85.9% of patients showed low resilience. An increase in reaction severity was
associated with greater caregiver burden and low resilience. Also, higher age and lower socioeconomic strata were associated with increased burden and lesser caregiver resilience.
Providing care for individuals with SCARs can be physically demanding, requiring assistance with daily activities, wound care, and medication management. The need for specialised care, including hospitalisation and frequent medical appointments, can disrupt caregivers’ daily routines and responsibilities. The economic burden of SCARs may also affect caregivers, especially if they need to take time off work, or incur additional expenses related to medical care and support.17 Balancing caregiving responsibilities with other family or work commitments can create tension and stress. Caregivers often take on the responsibility of learning about the condition, treatment options, and potential complications. They may need to advocate for the patient’s needs within the healthcare system, ensuring they receive appropriate and timely care.18
Supporting a patient through SCARs can be emotionally challenging, and caregivers may face challenges as they navigate the complexities of the condition. Caregivers need to have access to resources, support groups, and healthcare professionals who can offer guidance and assistance. Open communication, collaboration with the medical team, and a strong support network are all key components of a successful caregiving role in patients with SCARs.19 Caregiver burden can vary significantly depending on the specific disease or condition being cared for. A study by Cheng et al.20 examined caregiver burden and depression in dementia caregivers. Neuropsychiatric symptoms were found to be most predictive of caregiver burden and depression, regardless of the specific dementia diagnosis. Disruptive behaviours (such as agitation, aggression, and disinhibition) had a significant impact on caregivers’ emotional wellbeing. These symptoms strain the emotional connection between caregivers and care recipients. A study by Mwinbam et al.21 highlighted family caregivers’ experience and barriers in caregiving children with cerebral palsy,
in a resource-limited context, in Northern Ghana. Caregiving in the context of chronic diseases has also been researched. Luttik et al.22 explored the determinants of caregivers’ burden in cases of patients with heart failure. They commented that the burden assessment should focus on the mental strength of the partners, and highlighted the challenges caregivers face in managing complex medication regimens, symptom monitoring, and lifestyle modifications. According to a study by Onyeneho et al.23 on family carers for patients with cancer, providing care has an impact on the patients’ social, physical, psychological, and financial spheres of life. The majority of carers, the study found, were only somewhat burdened. This is easily explained by the fact that, despite the burden being there, most people are reluctant to voice their opinions due to their relationship with the care recipient. The authors’ results support this opinion.
The concept of a caregiver support system is evolving, with a growing recognition of the importance of supporting caregivers, to ensure the wellbeing of both the care recipients and the caregivers themselves.24, 25 Primarily, family plays a significant role in caregiving in culture. In many cases, family members, especially spouses, children, or extended family, take on the responsibility of caregiving. This support can include emotional, physical, and financial assistance. Apart from this, community support networks, such as religious or community groups, can provide valuable assistance to caregivers, and may offer respite care, support groups, educational resources, and counselling services.26 Several non-governmental organisations in India focus on providing support and services to caregivers. These organisations often offer training programmes, counselling, support groups, and advocacy efforts to raise awareness about caregiver needs and challenges. The Indian government has various schemes and programmes aimed at supporting caregivers, especially for elderly care and persons with disabilities.
These initiatives may include financial assistance, caregiver training, and access to healthcare services. Hospitals, clinics, and
healthcare providers also offer caregivers’ education, counselling services, and respite care options to alleviate caregiver burden. With the advancement of technology, digital platforms, mobile applications, and telehealth services are increasingly being used to provide support and resources to caregivers. These platforms offer information, communication tools, and virtual support groups.
To the best of the authors’ knowledge, this is the first study highlighting the caregiver’s health when handling patients with SCARs. This study definitely has its own limitation of being a single-centre, small sample study, but it undoubtedly paves the path for caregiver-related research works in this domain. Due to the cross-sectional design
References
1. Tempark T et al. Drug-induced severe cutaneous adverse reactions: insights into clinical presentation, immunopathogenesis, diagnostic methods, treatment, and pharmacogenomics. Front Pharmacol. 2022;13:832048.
2. Committee on Family Caregiving for Older Adults, Family Caregiving Roles and Impacts [Internet] (2016) Treasure Island: StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/ books/NBK396398/. Last accessed: 2 December 2023.
3. Peacock SC et al. The journey with dementia from the perspective of bereaved family caregivers: a qualitative descriptive study. BMC Nursing. 2014;13(1):42-52.
4. Verma R et al. Severe cutaneous adverse drug reactions. Med J Armed Forces India. 2013;69(4):375-83.
5. Reinhard SC et al., Patient safety and Quality: An Evidence-Based Handbook for Nurses [Internet] (2008) Treasure Island: StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/ books/NBK2665/. Last accessed: 2 December 2023.
6. Demirbağ BC et al. Caregiver burden and responsibilities for nurses to reduce burnout. Caregiving and Home Care. InTech. 2018;DOI:10.5772/ intechopen.68761.
7. Yang MS et al. Incidence of StevensJohnson syndrome and toxic epidermal necrolysis: a nationwide populationbased study using national health insurance database in Korea. PLoS One. 2016;11(11):e0165933.
8. Castellazzi ML et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome in two young children: the importance of an early diagnosis. Ital
of this study, the authors were unable to evaluate the longitudinal effects on caregiver burden and resilience. Therefore, a longitudinal impact assessment is necessary to address this gap in understanding.
CONCLUSION
SCARs have a physical, mental, and social toil on the patient as well their caregivers. Besides prioritising patient outcomes, disease management, and treatment efficacy, a compassionate understanding of caregiver burnout, and informed communications can help in the holistic management of the patient with better patient outcomes.
J Pediatr. 2018;44(1):93.
9. Creadore A et al. Clinical characteristics, disease course, and outcomes of patients with acute generalized exanthematous pustulosis in the US. JAMA Dermatol. 2022;158(2):176-83.
10. Smith BW et al. The brief resilience scale: assessing the ability to bounce back. Int J Behav Med. 2008;15(3):194-200.
11. Jamali AR et al. Translation, face and content validity of burden scale for family caregivers. Middle East J Rehabil Health Stud. 2018;5(1):e62424.
12. World Health Organization (WHO). WHO-BREF: Introduction, administration, scoring and generic version of the assessment. 1996. Available at: http:// www.who.int/mental_health/media/en/76. pdf. Last accessed: 10 December 2023.
13. Guzman AI, Paliza AC. Epidemiology of severe cutaneous adverse drug reactions in a University Hospital: a five-year review. Journal of Medicine, University of Santo Tomas. 2018;2(1):171-84.
14. Hartwig SC et al. Preventability and severity assessment in reporting adverse drug reactions. Am J Hosp Pharm. 1992;49(9):2229-32.
15. Schumock GT, Thornton JP. Focusing on the preventability of adverse drug reactions. Hosp Pharm. 1992;27(6):538.
16. Sharif L et al. An Exploration of family caregiver experiences of burden and coping while caring for people with mental disorders in Saudi Arabia—a qualitative study. Int J Environ Res Public Health. 2020;17(17):6405.
17. Gibbons SW et al. Liminality as a conceptual frame for understanding the family caregiving rite of passage: an integrative review. Res Nurs Health. 2014;37(5):423-36.
18. McLennon SM et al. Task difficulty
and life changes among stroke family caregivers relationship to depressive symptoms. Arch Phys Med. 2014;95(12):2484-90.
19. Buljac-Samardzic M et al. Interventions to improve team effectiveness within health care: a systematic review of the past decade. Hum Resour Health. 2020;18(1):2.
20. Cheng ST. Dementia caregiver burden: a research update and critical analysis. Curr Psychiatry Rep. 2017;19(9):64.
21. Mwinbam MM et al. Family caregivers' experience of care with a child with cerebral palsy: the lived experiences and challenges of caregivers in a resourcelimited setting in northern Ghana. BMJ Paediatr Open. 2023;7(1):e001807.
22. Luttik ML et al. Caregiver burden in partners of heart failure patients; limited influence of disease severity. Eur J Heart Fail. 2007;9(6-7):695-701.
23. Onyeneho CA, Ilesanmi RE. Burden of care and perceived psycho-social outcomes among family caregivers of patients living with cancer. Asia Pac J Oncol Nurs. 2021;8(3):330-6.
24. Beach B et al. Caring for the caregiver: why policy must shift from addressing needs to enabling caregivers to flourish. Front Public Health. 2022;10:997981.
25. Midgette AJ, Ferreira JM. Conceptualizing care: US and Finnish caregivers’ reflections on caregiving within the family. J Child Fam Stud. 2024;33:253-70.
26. Agarwal D et al. Scaling a group intervention to promote caregiver mental health in Uttarakhand, India: a mixedmethods implementation study. Glob Ment Health (Camb). 2023;10:e85.
Endovascular Stenting for Superior Vena Cava Syndrome – A Systematic Review
Superior vena cava syndrome (SVCS) results from the obstruction or narrowing of the superior vena cava, causing venous congestion and various symptoms such as facial and upper limb swelling, shortness of breath, chest pain, coughing, and, in severe cases, dizziness and headache. The primary treatment for SVCS is balloon angioplasty with endovascular stenting. Post-procedural complications are influenced by factors such as SVCS aetiology, comorbidities, and the presence of arteriovenous fistulas. This review examined eight clinical studies to assess the effectiveness of percutaneous endovascular stenting and associated complications, focusing on improving patient prognosis. The research, conducted through internet search engines and reputable databases, revealed that percutaneous endovascular stenting demonstrated efficacy ranging from 95–100% in addressing SVCS. Common complications post-procedure included SVC narrowing recurrence, airway constriction, and mortality, often linked to malignancy. The findings emphasise the need to refine therapeutic approaches, especially in addressing the root cause of SVCS, which is frequently malignancy. Consequently, implementing additional protocols to reduce the risk of SVCS development is crucial. This comprehensive review provides insights into the effectiveness of endovascular stenting in treating SVCS, highlighting the importance of tailored approaches and ongoing efforts to enhance patient outcomes.
Key Points
1. Superior vena cava syndrome (SVCS) presents as clinical manifestations of obstructed venous blood flow to the heart and is often caused by malignancy. Endovascular stenting effectively treats this, yet post-surgery cardiac complications are documented. The treatment's success remains debatable despite its clinical significance.
2. An analysis of eight research papers evaluated the efficacy and complications of endovascular stenting for SVCS. Most studies reported 95–100% efficacy, yet participants often experienced post-operative complications such as airway stenosis, stent dysfunction, and, in some cases, mortality.
3. Whilst endovascular stenting effectively treats SVCS, current literature highlights the necessity for further research into tailored interventions to minimise associated complications. Furthermore, additional investigation to address the primary cause, often malignancy, is needed.
INTRODUCTION
Superior vena cava syndrome (SVCS) is a group of clinical manifestations, caused by an obstruction of venous blood flow to the heart. Normally, the superior vena cava (SVC) drains deoxygenated blood from the upper part of the body into the right atrium of the heart. This disease has been observed and diagnosed amongst many people presenting with characteristic symptoms, with an estimated incidence globally ranging from 1/650–1/3,100 patients.1 The symptoms of SVCS vary, depending on the degree of vasculature compromise by a given cause of obstruction, the rate at which venous blood flow through the SVC is impaired, and the specific underlying cause. Common symptoms include facial swelling, visible enlargement of neck and chest veins, shortness of breath, cough and hoarseness, swelling of the arms, and rarely headache and dizziness indicating central nervous system involvement due to cerebral oedema.
The underlying pathology for SVCS is venous flow obstruction and venous congestion. This can be caused by external compression, and internal occlusion or stenosis of the SVC (most commonly by a malignancy). While malignancy is the predominant factor responsible for approximately 80% of cases of SVC obstruction, there has been a recent rise in the occurrence of cardiac devicerelated SVCS caused by central venous catheters, as well as pacemaker or
defibrillator leads.2,3 Interventions that can lead to SVCS include procedures related to the chest or mediastinum, such as lung resection; mediastinal tumour removal; catheter placement for large or bulky cardiac devices, pacemakers, and implantable cardioverter-defibrillators; or lead placement, which may compress the SVC and contribute to SVCS symptoms. The increasing use of cardiac devices accentuates the need for careful patient selection, monitoring, and prompt evaluation of symptoms suggestive of SVCS.4
The SVC is a crucial part of our lowpressure venous system, and its walls are relatively thin, making them vulnerable to damage by various pathological processes. These processes can be categorised into three main types: compromised vessel anatomy, impaired venous flow, and reduced vessel wall integrity. It is common for patients with SVCS to experience a combination of these three mechanisms at one given moment in time.1 Recognising the significance of SVCS, one can observe and categorise the extent of damage that is placed upon the SVC. Thus, the Stanford and Doty classification for SVC obstruction, a widely recognised system used to categorise different types of SVC obstructions based on anatomical localisation and predominant aetiology, is an important tool.4
A comprehensive clinical assessment is often conducted to diagnose SVCS, beginning with a physical examination to evaluate the degree of venous distention
and swelling. Furthermore, diagnostic imaging techniques including chest X-rays, CT scans, or MRI are often utilised to identify the underlying cause of SVCS and assess the extent of the obstruction.1 Neglecting SVCS can lead to other complications that may further hinder a patient’s condition and life expectancy. These complications include, but are not limited to, cerebral oedema, which may often lead to various neurologically associated symptoms such as headaches, confusion, seizures, or even coma. Additionally, oedema of the larynx and upper respiratory tract may concomitantly occur, leading to difficulty in swallowing, voice hoarseness, and, potentially, a compromised airway. Furthermore, reported complications often arising in patients with SVCS include altered respiration, which leads to breathing difficulties and pleural effusion.5
Persistent SVCS may also impede the efficiency of central venous access for post-transplant care and monitoring, potentially leading to further complications or delays in management.6 These complications highlight the importance of a thorough assessment and a multidisciplinary approach involving cardiologists, radiologists, and surgeons to ensure timely diagnosis and appropriate management of this rare but potentially severe complication. Despite the clinical significance of persistent SVCS in patients receiving cardiac transplant, the literature on this specific topic remains limited and fragmented.6
Treatment of SVCS involves both supportive and definitive therapy. Supportive measures include elevating the patient’s head to reduce hydrostatic pressure and oedema, although the effectiveness of this manoeuvre is not well-documented. Glucocorticoid therapy, such as dexamethasone, is commonly prescribed, but its effects have not been extensively studied. Loop diuretics may also be used, but their impact on venous pressure is unclear.6 This systematic review seeks to address the dearth of knowledge regarding cardiac transplant complications in patients with persistent SVCS. By consolidating the
available evidence, this review hopes to contribute to the understanding of this complex condition and inform clinical decision-making for improved patient outcomes.
METHODOLOGY
In April 2023, the objectives in this document were provided utilising the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 (Figure 1) and Preferred Reporting Items for Overviews of Reviews (PRIOR) guidelines, which were followed rigorously to ensure the transparency and reproducibility of this investigation. This systematic review aims to provide valuable insights into this complex, understudied area, and analyse the effectiveness of endovascular stenting in patients with SVCS, as well as postprocedural complications.
Information Sources and Search Strategies
The studies in this review were selected to provide comprehensive information regarding the results of treating patients with SVCS using endovascular stenting. Specific keywords were interpolated, such as “Superior Vena Cava, SVC, Superior Vena Cava Syndrome, Angioplasty, and endovascular Stenting,” into designated search engines, including Cochrane Library, Google Scholar, and Medline/PubMed. The target population for each study comprised adults aged 18–75 years, with all participants being either male or female.
Several different study designs were considered, including randomised clinical trials (RCT), cohort studies, and retrospective studies. The reviewers of this paper specifically selected papers pertaining to research conducted between 2016–2023, providing a comprehensive and up-to-date pool of relevant studies for the review process. This careful and thorough methodology allowed for a robust evaluation of long-term complications in patients with SVCS treated by endovascular stenting.
1: Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flowchart for data analysis.
Identification of studies by databases and registers
Records identified from:
Databases (n=117)
Cochrane Library (n=25)
Medline/PubMed (n=20)
Google Scholar (n= 72)
Records screened (n=114)
Records removed before screening: Duplicate records removed (n=3)
Records excluded (n=53)
Reports sought for retrieval (n=61)
Reports assessed for eligibility (n=59)
Reports not retrieved due to data silos (n=2)
Reports excluded: 50
Patient age below 18 (n=2)
Patient age above 75 (n=14)
Pre-existing cardiac disease (n=8) etc.
Studies included in review (n=8)
Reports of included studies (n=8)
Limitations
To evaluate the long-term outcomes of stenting and problems linked to SVCS, adequate follow-up data should have been additionally provided, with there being either a minimum follow-up duration or a predetermined number of follow-up appointments.
Inclusion Criteria
Studies included in this review had patients with a confirmed clinical diagnosis of SVCS, validated by suitable imaging methods. Medical records for patients had to be thorough and well-documented, including pertinent clinical data, imaging results, lab results, and cardiac studies.
The authors decided to include studies discussing SVCS without discriminating based on the underlying aetiology, including malignancy and non-malignant reasons, such as thrombosis, aortic aneurysm, vasculitis, arteriovenous fistulas, and infections (histoplasmosis, tuberculosis, syphilis, and actinomycosis).
Exclusion Criteria
Patients with pre-existing cardiac diseases or conditions unrelated to SVCS were not included in this systematic review to ensure that the research specifically addressed the authors’ research question. To ensure that complete information was available, studies with insufficient follow-up data were disregarded. Additionally, to avoid
Figure
the introduction of additional confounding variables, patients with comorbidities that significantly influence cardiac function, such as severe congestive heart failure or valvular disease, were also excluded.
Studies published in languages other than the primary language of the review team (English), or missing translation resources, were omitted in order to reduce linguistic bias and maintain the feasibility and integrity of the review process. Furthermore, to avoid the inclusion of redundant data, duplicate studies or multiple publications of the same study were excluded.
Selection and Data Collection Process
Review selection
The selection process for this systematic review on the effectiveness of endovascular stenting in patients with SVCS followed a rigorous approach. A systematic approach was adopted to ensure dependability and accuracy whilst obtaining data. The reviewers independently screened all the titles and abstracts. Initially, a total of 117 studies were collected: 72 from Google Scholar, 25 from Cochrane, and 20 from Medline/Pubmed. The disagreements were resolved by discussion. Three duplicate studies were found and omitted. The process was repeated for full text article screening to determine the relevant studies, and as a result, 53 studies were excluded for lack of relevance. Two studies were not retrieved due to data silos. The remaining 59 studies were assessed for eligibility, and 50 of them were excluded because they did not adhere to the inclusion and exclusion criteria.
Data extraction
Three independent reviewers used a standardised protocol for data extraction from each study. The data included:
• Study design, sample size, patient demographic data (sex and age of patients)
• Procedural techniques used to treat patients with SVCS
• Reasonable data collection methods and medical reports of patient outcomes post-operatively
• Long-term follow-up outcomes
Any discrepancies among reviewers were resolved through discussion and consultation.
Outcomes
Primary outcome
The primary outcome of this systematic review was to comprehensively assess the efficacy of endovascular revascularisation of SVCS (technical and clinical success rate). Technical success of revascularisation was defined as suggested by the Society of Interventional Radiology (SIR): complete coverage of the lesion (stent overlapping the margins of the stenosis by 1 cm on either side) and residual stenosis of <30% as assessed by visual estimate. Clinical success was defined as relief of symptoms 48 hours after the procedure.7
Secondary outcome
The secondary outcomes included possible complications of endovascular stenting for SVCS. These complications include recurrence, headaches, confusion, seizures, or coma. Additionally, oedema of the larynx and upper respiratory tract may concomitantly occur, leading to difficulty in swallowing, voice hoarseness, and, potentially, a compromised airway. Further reported complications often arising in patients with SVCS include altered respiration, which leads to breathing difficulties, pleural effusion,8 and mortality.
Preparation for synthesis
In synthesising the data, a narrative summary of the included studies was created and presented in a table, emphasising the success rate after 48 hours, and common clinical complications in patients with SVCS.
Tabulation and Graphical Methods
Clinical trials found were grouped and tabulated according to year and category.
For the production of efficient data analysis in a concise manner in this systematic review, clinical trials were presented in tabular format.
The data collected from all studies were collated and structured within a summary table. This table played a crucial role in categorising the data, forming the foundation for the results of the systematic review of all eight studies included in the review. To ensure transparency and adherence to inclusion and exclusion criteria, a PRISMA flow chart was created.
Methods to Explore Heterogeneity
The presence of heterogeneity in the systematic review was examined by analysing data collected through tabulation. The findings identified three to four distinct types of clinical trials. Despite this variation, the results of endovascular revascularisation in patients with SVCS were consistent across all trial types.
Assessment of Bias Risk
To reduce bias and enhance the reliability of the systematic review, stringent inclusion and exclusion criteria were used, with a particular emphasis on clinical trials. Furthermore, specific tools for assessing the likelihood of bias in systematic reviews, such as the one given by the Agency for Healthcare Research and Quality (AHRQ), were utilised.9 This technique allowed the reviewers of this systematic review to rigorously examine and address potential sources of bias in the included papers, improving the overall quality and validity of this review.
Reporting bias
To prevent and minimise the potential for bias, predefined criteria outlined in the methodology section were adhered to. Furthermore, findings were supported with robust statistical evaluation to provide objective evidence. Any observed discrepancies were explicitly acknowledged and addressed in both the results and discussion sections of this systematic review.
RESULTS
In conducting this systematic review, the authors focused on the investigative methods used to treat those with SVCS and the complications that follow. The utilisation of internet-based search tools proved instrumental in identifying a total of eight studies that shared a common focal point, presented in Table 1. This comprehensive analysis aimed to explore and evaluate the various approaches employed in managing this specific cohort of patients. By harnessing the power of online resources, this review gathered a diverse range of studies, allowing for a comprehensive synthesis of findings and evidence-based insights into the treatment strategies for SVCS.
Number of Patients Assessed
Across the eight studies listed within this systematic review, a diverse number of subjects volunteered to participate in each. In the study by Takeuchi et al.,13 32 patients were incorporated into the Phase III clinical trial, of whom 16 were categorised into a ‘control’ group and the remaining 16 into a ‘test’ group. The RCT conducted by Ma et al.16 consisted of 158 patients, similarly to the study by Takeuchi et al.,13 and the subjects were divided into two subgroups: 79 were placed into an ‘observation’ group and the remainder into a ‘control’ group. The treatment of malignant SVC blockage, which was the main priority of the retrospective study conducted by Wang et al.,14 showed that the patients within this study were divided accordingly; however, they were additionally categorised by the type of cardiac treatment being utilised within the study. For instance, 30 patients received a covered stent and 34 patients received an uncovered stent.15 Furthermore, initial screening of patients for potential access to stent treatment in the study by Haddad et al.12 resulted in 59 patients being selected overall. With more vigorous screening, 49 of the patients were selected following a secondary clinical diagnosis and a followup consultation.16
Technical Methods Utilised
Table 1: Variables identified across the eight studies collated for the analysis for this systematic review on the success rate of endovascular revascularisation and complications arising in patients with superior vena cava syndrome.
Treatment Protocol for Patients Experiencing Complications
stent insertion Catheterdirected thrombolysis
Takeuchi et al.13 Phase III randomised controlled trial
Grade 4 aspartate aminotransferase elevation and serum bilirubin elevation
None reported
(Covered group)
(Uncovered group)
stent insertion Wei et al.15
Ma et al.16
group=79; control group=79)
internal jugular vein; N/A: not applicable; pt: patient.
Catheter exit site
(P=0.477)
Catheter-related thrombosis (P=0.129)
Catheter-related obstruction (P=0.199)
Catheter-related infection
Catheter displacement Catheters un-obstruction
for Revascularisation
Optimal outcomes were achieved in the study by Wang et al.,14 in which airway insertion was achieved using either a covered stent (e.g., Fluency™, Becton, Dickinson and Company [BD], Franklin Lakes, New Jersey, USA) or an uncovered stent (e.g., E-Luminexx™, BD), both of which are highly efficient tools exceeding the overall diameter of the SVC, and which are essential for the treatment of SVCS. Similar cardiac instruments were chosen within the retrospective study conducted by Haddad et al.,12 in which the covered (e.g., GORE® VYBAHN® , Gore Medical, Newark, Delaware, USA; iCAST™, Getinge, Gothenburg, Sweden) and uncovered (e.g., WALLSTENT™, Boston Scientific, Marlborough, Massachusetts, USA; Protégé™, Medtronic, Watford, UK; S.M.A.R.T. ®, Cordis, Miami Lakes, Florida, USA) stents were used to treat the complications of SVCS. Next, according to the Haddad et al.12 clinical study, the balloon angioplasty procedure was effective in revamping specific types of stents (WALLSTENT, Protégé, and S.M.A.R.T.), which were used to correct any obstruction leading to SVCS. The majority of studies in this paper adopted the use of endovascular stenting as the procedure of choice to treat SVCS. Although, clinical femoral catheterisation was proven to be a more cost-effective and efficient, as well as accurate, technique to reach the targeted anatomical location (SVC), with reports of a low complication rate, specifically in the study by Ma et al.16
Revascularisation Success Rate
Among the various studies reviewed, the outcomes achieved through the utilisation of balloon angioplasty and endovascular stenting for the management of SVCS were favourable. Notably, in four of the eight studies, including those by Anton et al.10 and Niu et al.,11 a technical success rate of 100% was observed, signifying that all participants in these studies underwent successful initial treatment for SVCS. While the study conducted by Ma et al.16 reported a slightly lower success rate of 97.5%, in comparison with the perfect score of 100%, this figure still demonstrated a high degree of efficacy,
indicating that a significant proportion of participants in that study were effectively treated using the endovascular stenting revascularisation technique. Unfortunately, data regarding the success rates of treatment in the Phase III trial by Takeuchi et al.13 and the retrospective study by Wei et al.15 were not obtainable.
Cardiac Complications Observed After Revascularisation
The reported complications observed in patients with SVCS after revascularisation varied across all studies. Since various techniques were implemented to treat patients with SVCS, complications cannot be attributed to endovascular revascularisation in general. Instead, the different treatment modalities are responsible for the diverse complications. In a study by Anton et al.,10 restenosis of the newly operated SVC was reported leading to secondary facial and neck swelling. Restenosis correspondingly was reported in the study conducted by Haddad et al.,12 where additional reported symptoms of headaches and upper extremity swelling were observed amongst patients. In many of the clinical studies selected for analysis, complications pertaining specifically to endovascular stent placement were reported. As exhibited in the two retrospective studies conducted by Niu et al.11 and Wang et al.,14 both provided reports of patients experiencing airway stenosis, secondary to endovascular stenting.
Furthermore, the study conducted by Ma et al.16 utilised a different surgical procedure than those reported in other literature; although complications pertaining to femoral catheterisation, such as catheter-related thrombosis, infection, and displacement were noted. Within three of the reported studies, death was a common complication arising amongst the reported population of patients. For instance, the Phase II trial and Phase III RCT conducted by Takeuchi et al.13 reported the deaths of five patients and 17 patients, respectively. Death was further reported in the study by Niu et al.;11 however, this was solely due to malignancy and not due to surgical treatment. Only one study out of the eight, that being by
Wei et al.,15 failed to report any significant complications among patients after the procedure had begun to be used to treat the SVCS in its population of participants. All exhibited complications can be viewed below in Table 2.
Post-operative Treatment Protocol for Patients with Complications
Various treatment protocols were utilised by different studies to treat the cardiacrelated complications that arose postoperatively in patients possessing SVCS. For instance, studies by Niu et al.11 and Wang et al.14 both similarly selected airway stent insertion to treat airway stenosis, secondary to endovascular stenting. Additionally, catheter-directed thrombolysis was proposed for patients partaking in the study by Niu et al.,11 a treatment regime replicated in a study of Haddad et al.12 Though the two studies shared similar post-operative treatment protocols, the study by Haddad et al.12 concomitantly offered angioplasty as a source of treatment for patients with SVCS experiencing complications. Treatment regimens specific for restenosis of the SVC were provided for patients in the study by Anton et al.10 through stent recanalisation and balloon angioplasty.12 Moreover, two of the studies conducted by Takeuchi et al.13 and the retrospective study carried out by Wei et al.15 did not report any significant treatment protocols for patients experiencing adverse complications from operative treatment administered for the correction of SVCS.
Follow-Up Period
The systematic review data reveal diverse patterns in the choice of post-procedural follow-up duration for patients. The Anton et al.10 retrospective study involved followups for up to 184 days, whereas Niu et al.11 implemented a 6-month follow-up, leading to all patients’ unfortunate demise due to malignancy progression. In their retrospective study, Haddad et al.12 adopted a 3-month, 6-month, and yearly followup protocol, using the Kishi score >4 as a guide, along with digital subtraction venogram and contrast-enhanced CT venogram.17 Takeuchi et al.,13 in their
prospective, multi-institutional Phase II trial, employed a 14-day follow-up by CTCAE score, continuing evaluation for 4 weeks. In their Phase III RCT, they utilised a 28-day follow-up after treatment, also using CTCAE score, and conducted weekly follow-ups with individual patients.13 Lastly, Wang et al.,14 in their retrospective study, employed a 14-month follow-up protocol, during which all patients succumbed to tumour progression or respiratory failure.
Prognosis for Patients With Superior Vena Cava Syndrome After the Procedure (Mortality, Survival Rate, Recurrence)
After undergoing repair of the impacted SVC, patients exhibited a diverse range of prognostic outcomes, as shown in Table 1. In both studies conducted by Takeuchi et al.,13 patients undergoing treatment had a calculated mortality rate of 17.9% and 25.0%. Regarding the median survival rate of all patients within the study by Anton et al.,10 patients treated by endovascular stenting had a median survival rate (MSR) of 180±248 days, while patients in the study by Niu et al.11 reported a similar MSR of 167 days.
Upon the comparison of both studies utilising the 2x2 factorial design method of grouping subjects for analysis, the study by Wang et al.14 reported an MSR of 175 days in the group of subjects receiving an uncovered stent, while those receiving a covered stent had a reported MSR of 159 days. Although a similar study design was utilised in the Phase III RCT by Takeuchi et al.,13 MSR of 67 days was reported in the test group, while the included control group reported an MSR of 93 days.13 Among all the studies, the retrospective study by Wei et al.15 had the largest documented number for MSR amongst its patients, with a total varying between 1–18 months. Only one out of the eight studies reported a proportion of patients experiencing recurrent episodes of SVCS, with 12.8% of all patients within the study by Niu et al.11 having recurrent episodes.
Table 2: Types of complications observed in patients with superior vena cava syndrome by data collation.
DISCUSSION
Age of Patients
Endovascular stent-based revascularisation was performed in patients with a mean age of 67±8 years in the study by Anton et al.,10 and patients with a mean age of 60 were utilised for a similar retrospective study conducted by Wei et al.15 Patients with a mean age of 58 years were in the observation group, and patients with a mean age of 57 years were in the control group in the Ma et al.16 randomised controlled clinical trial; whereas, in the Takeuchi et al.13 RCT, patients in the control group were of an average age of 57 years, and the patients in the observation group were of an average age of 63. Overall analyses of information acquired through web search engines suggest that the mean age of patients possessing SVCS, and who experience cardiac complications, lies around 59 years old. As malignancy tends to increase exponentially with age, the prevalence of SVCS among those over the age of 50 can be linked to malignancy, the main cause of SVCS. Thus, an increase in one’s age enhances one’s chance of developing SVCS.17
Sex of the Patients with Superior Vena Cava Syndrome
Despite the category of the study undertaken, there is a clear difference in the presented number of males and females studied within each clinical trial, with the population of males being significantly higher than the females. For example, the retrospective study by Anton et al.10 utilised 21 males and 10 females, while 27 males and 20 females were selected to partake in the retrospective study by Niu et al.11 Other studies exhibited a similar pattern of difference between the number of male and female participants included for review. For example, in the studies by Wang et al.14 and Wei et al.,15 more male participants diagnosed with SVCS were included in these studies, compared to female participants.11,15 Based on the available literature, SVCS is diagnosed when there is a disruption or obstruction in the transportation of blood through the SVC. This condition
is considered a medical emergency and typically occurs in patients with a malignant disease process, primarily affecting the thorax.18 According to a study by Stabellini et al.,18 lung cancer is the leading cause of cancer-related deaths and the second most often diagnosed malignancy worldwide, with males having a higher incidence of lung cancer and a higher rate of mortality.19 Thus, there is a high probability that the elevated incidence rate of SVCS amongst male patients could be linked to the higher incidence of lung cancer seen in the male population. Generally, it was evident that more males were utilised in the selected studies used for this analysis of the use of endovascular stenting as a method of treating SVCS.
Methods Utilised for Revascularisation
Through analysis of all nine clinical studies conducted on the populous treatment type for patients diagnosed with SVCS, it was clear that endovascular stenting was the primary surgical technique employed as a form of first-line therapy. For example, in the studies by Anton et al.10 and Niu et al.,11 endovascular stenting was the primary form of treatment option utilised amongst the participants. This was due to supportive factors such as optimal procedural safety, lower cost, lower rate of complications, and better quality of life outcome that the surgical technique delivered.20 In the studies conducted by Haddad et al.12 and Wang et al.,14 covered stent use normalised all patients’ conditions, unlike uncovered stents. This was owed to the fact that covered stents had higher gross stent patency rates and lower stent occlusion rates than uncovered stents, thereby delivering a 100% technical success rate and 92% clinical success rate. However, the diameter of the covered stents used was kept 10–15% greater than the SVC diameter in order to prevent stent migration.21
Nonetheless, endovascular stent implantation was often required in patients who underwent balloon angioplasty, due to below par results and persistent stenosis outcomes. Moreover, SVC stent implantation provides a substantial 6-month remission from relapse of stenosis.22
Clinical femoral catheterisation was yet another surgical method that proved to be effective in revitalising patients with SVCS, according to the study by Ma et al.16 When conducted with the help of 2D-ultrasound guidance, puncture of the femoral vein, pseudoaneurysm formation, arteriovenous fistula formation, and further procedural complications were decreased by 49%, and first attempt success was increased by 42%.23
Types of Complications After Revascularisation
The adopted endovascular techniques outlined within this systematic review all had various levels of effectiveness, although complications arose with use. In the longterm follow-up trial conducted by Anton et al.,10 both endovascular stenting and balloon angioplasty were the preferred methods of treatment used on patients. However, associated complications, such as intrastent thrombosis and death, were observed. Intra-stent thrombosis has been commonly recognised as a relative complication in approximately 16% of all patients poststent deployment. The aetiology of intrastent thrombosis can often be attributed to haemodynamic factors resulting from stent expansion within a vessel, as well as genetically classified or acquired hypercoagulability disorders in patients. These include antithrombin deficiency, protein S/C deficiency, or aplastic anaemia.24 Similarly, the use of endovascular stenting in the study conducted by Haddad et al.12 led to restenosis of the impacted SVC following surgery.10 Commonly, restenosis arises due to the pathophysiological process of hypertrophic wound healing, occurring as a regulatory mechanism to override vascular inflammation, which arises due to stent implantation and vascular injury.25
Studies by Niu et al.11 and Wang et al.14 both presented reports of airway stenosis as a secondary complication arising from the utilisation of endovascular stenting to treat persistent SVCS.14,15 With the use of tracheal intubation during surgical procedures, patients often have a high probability of developing airway stenosis as the overriding cuff pressure rises above that of the normal
capillary pressure of the trachea, resulting in the ischaemia and long-term ulceration of the tracheal cartilages. This, in turn, induces fibrotic healing mechanisms that progressively damage the tracheal tube.26 Lasty, amongst several studies, including both clinical trials conducted by Takeuchi et al.,13 a small proportion of patients died as a result of cardiac transplantation treatment methods. Death can arise as a late complication following the treatment of SVCS, mainly as a consequence of anticoagulation. Thus, anticoagulant therapy by the use of warfarin, clopidogrel, or aspirin, is given as a source of prophylactic treatment to patients prior to surgery and following a given risk assessment.27
Prognosis for Patients With Superior Vena Cava Syndrome After Revascularisation
As observed acoss several studies highlighted in this systematic review, many of the patients treated with endovascular revascularisation either experienced adverse side effects, acute recurrences of SVCS , or, consequently, did not survive. For example, in the studies by Anton et al.10 and Niu et al.,11 a large proportion of patients died following treatment.10,14 All of the patients presented in these two studies were initially diagnosed with various forms of malignancy or mass effect, both of which cause shear stress on the venous intimal layer and damage to the SVC.28 Malignant causes of SVCS include oesophageal cancer, mediastinal tumours, and small-cell lung carcinoma, with the latter accounting for 50% of all diagnosed patient cases.13 Recurrent episodes of SVCS were observed in both studies by Niu et al.11 and Haddad et al.12 Episodes of SVCS arising amongst medically treated populations can be accounted for by several factors; for instance, the increased incidence of pacemakers, and their associated leads, used to treat alternate cardiovascular diseases such as those listed in the paper by Wei et al.15,27
Treatment Protocol for Patients with Superior Vena Cava Recurrence
From the studies sourced for this systematic
review, Takeuchi et al.13 utilised endovascular techniques to treat patients with SVCS experiencing complications post-surgery; however, such were not specified within the paper.13 According to a paper by Ponti et al.,29 successful endovascular therapy ensues in more than 95% of patients, and over 90% of them report symptom relief.29 Further analyses state that within the study by Anton et al.,10 stent recanalisation (by balloon angioplasty) technique was chosen to fix specific complications experienced, including in-stent stenosis or restenosis of the SVC.10 In this regard, a report by Volpi et al.20 indicates that this technique does not interfere with subsequent antitumour treatments and provides urgent relief of symptoms, as well as additionally improving the quality of life in patients with benign causes of SVCS.20 In the studies by Niu et al.11 and Wang et al.,14 airway stent insertion was used to rectify all complications associated with airway stenosis following surgical treatment, though all patients died due to tumour progression or respiratory failure.10,15 Consequently, no definitive conclusion can safely be made on the authority of which treatment protocol is suitable for patients being treated for
References
1. Seligson MT, Surowiec SM, Superior Vena Cava Syndrome [Internet] (2022) Treasure Island: StatPearls. Available at: https://www.ncbi.nlm.nih.gov/ books/NBK441981/. Last accessed: 19 August 2023.
2. Gabriels J et al. Percutaneous management of superior vena cava syndrome in patients with cardiovascular implantable electronic devices. Heart Rhythm. 2021;18(3):392-8.
3. Locke AH et al. Lead-associated superior vena cava syndrome. J Innov Card Rhythm Manag. 2021;12(4):445965.
4. Mumoli N et al. Superior vena cava syndrome after pacemaker implantation treated with direct oral anticoagulation. Thromb J. 2021;19(1):84.
5. Lepper PM et al. Superior vena cava syndrome in thoracic malignancies. Respir Care. 2011;56(5):653-66.
6. Seo M et al. Central venous catheterrelated superior vena cava syndrome
complications, secondary to the treatment of persistent SVCS.
CONCLUSION
Significant advancements have been observed in the developed therapeutic approaches for managing patients diagnosed with SVCS. However, further comprehensive research is imperative to enhance patient survival rates and mitigate morbidity arising from common complications subsequent to therapeutic intervention for this condition. Malignancy continues to emerge as the primary causative factor in the development of this medical disorder. Hence, the implementation of additional protocols, including screening measures and the utilisation of prevailing diagnostic modalities, is warranted to ensure timely identification and treatment of cancer, thereby reducing the predisposition to SVCS. Additionally, alternative endovascular approaches and anticoagulation therapy show promise in mitigating primary causes of SVCS, warranting further investigation to enhance outcomes for the general population.
following renal transplantation -A case report-. Korean J Anesthesiol. 2012;63(6):550-4.
7. De Raet JM et al. Surgical management of superior vena cava syndrome after failed endovascular stenting. Interact Cardiovasc Thorac Surg. 2012;15(5):915-7.
8. Kvale PA et al. Palliative care in lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition). Chest. 2007;132(3 Suppl):368S-403S.
9. Viswanathan M et al., Assessing the risk of bias of individual studies in systematic reviews of health care interventions. In Methods Guide for Effectiveness Reviews [Internet] (2012). Rockville (MD): Agency for Healthcare Research and Quality (US);2008. Available at: https:// pubmed.ncbi.nlm.nih.gov/22479713/. Last accessed: 4 September 2023.
10. Anton S et al. Endovascular stentbased revascularization of malignant superior vena cava syndrome with concomitant implantation of a port device using a dual venous approach. Support Care Cancer.
2018;26(6):1881-8.
11. Niu S et al. Stent insertion for malignant superior vena cava syndrome: effectiveness and long-term outcome. Radiol Med. 2017;122(8):633-8.
12. Haddad MM et al. Comparison of covered versus uncovered stents for benign superior Vena Cava (SVC) obstruction. Cardiovasc Intervent Radiol. 2018;41(5):712-7.
13. Takeuchi Y et al. Evaluation of stent placement for vena cava syndrome: phase II trial and phase III randomized controlled trial. Support Care Cancer. 2019;27(3):1081-8.
14. Wang ZS et al. Covered versus uncovered stent insertion for malignant superior vena cava obstruction. Minim Invasive Ther Allied Technol. 2020;29(6):353-8.
15. Wei S et al. A retrospective stenting study on superior vena cava syndrome caused by lung cancer. Thorac Cancer. 2020;11(7):1835-9.
16. Ma M et al. The application of intracavitary electrocardiogram for tip
location of femoral vein catheters in chemotherapy patients with superior vena cava obstruction. J Vasc Access. 2021;22(4):613-22.
17. Estapé T. Cancer in the elderly: challenges and barriers. Asia Pac J Oncol Nurs. 2018;5(1):40-2.
18. Stabellini N et al. Sex differences in lung cancer treatment and outcomes at a large hybrid academiccommunity practice. JTO Clin Res Rep. 2022;3(4):100307.
19. Nickloes TA et al. Superior Vena Cava Syndrome (SVCS). 2024. Available at: https://emedicine.medscape. com/article/460865-overview. Last accessed: 23 September 2023.
20. Volpi S et al. Superior Vena Cava (SVC) endovascular reconstruction with implanted central venous
catheter repositioning for treatment of malignant SVC obstruction. Front Surg. 2018;5:4.
21. Cho Y et al. Covered stent placement for the treatment of malignant superior vena cava syndrome: is unilateral covered stenting safe and effective? Korean J Radiol. 2014;15(1):87-94.
22. Aldoss O et al. Endovascular stent provides more effective early relief of SVC obstruction compared to balloon angioplasty. Catheter Cardiovasc Interv. 2014;83(7):E272-6.
23. Sobolev M et al. Ultrasound-guided catheterization of the femoral artery: a systematic review and meta-analysis of randomized controlled trials. J Invasive Cardiol. 2015;27(7):318-23.
24. Modi K et al., Stent Thrombosis [Internet] (2023) Treasure Island:
StatPearls. Available at: https://www. ncbi.nlm.nih.gov/books/NBK441908/. Last accessed: 24 September 2023.
25. Schillinger M, Minar E. Restenosis after percutaneous angioplasty: the role of vascular inflammation. Vasc Health Risk Manag. 2005;1(1):73-8.
26. De S, De S. Post intubation tracheal stenosis. Indian J Crit Care Med. 2008;12(4):194-7.
27. Sourgounis A et al. Coronary stents and chronic anticoagulation. Circulation. 2009;119(12):1682-8.
28. Cohen R et al. Superior vena cava syndrome: a medical emergency? Int J Angiol. 2008;17(1):43-6.
29. Ponti A et al. Insights into endovascular management of superior vena cava obstructions. Front Cardiovasc Med. 2021;8:765798.