10 minute read
VARIAMOLS


Models to find the key to molecular behaviour
Computer-aided methods of investigation play a role of steadily increasing importance in biological research, with sophisticated simulations enabling scientists to understand molecular systems in great detail. Researchers in the VARIAMOLS project are developing in silico approaches to model and study large molecular assemblies, which could open up new avenues in medical research, as Professor Raffaello Potestio explains.
The investigation of biological systems increasingly relies on the use of sophisticated technologies, with researchers applying computer-aided techniques to build a detailed picture of the structure, dynamics, and function of macromolecules and molecular assemblies. As the Principal Investigator of the VARIAMOLS project, Professor Raffaello Potestio is developing new methods to model and study biological molecules, in particular proteins, which could play an important role in medical research. “We mainly focus on single molecules, however this definition encompasses singlechain proteins as well as assemblies of several proteins, as molecular machines are often composed of several distinct units, which are more or less strongly bound together and work cooperatively,” he explains. For example, the capsid of a virus – which is the shell that contains the viral genome – is not a single protein, but rather an assembly of different proteins, like a LEGO toy. “Our aim is to understand how these systems work, that is, what relations exist between their structure, their movements, and their biological function,” says Professor Potestio.
VARIAMOLS project
A class of methods, called coarse-graining, is being used within the project to represent these molecular structures, a way to describe complex systems in a simplified manner that allows researchers to look at large biomolecules. In the most detailed computer
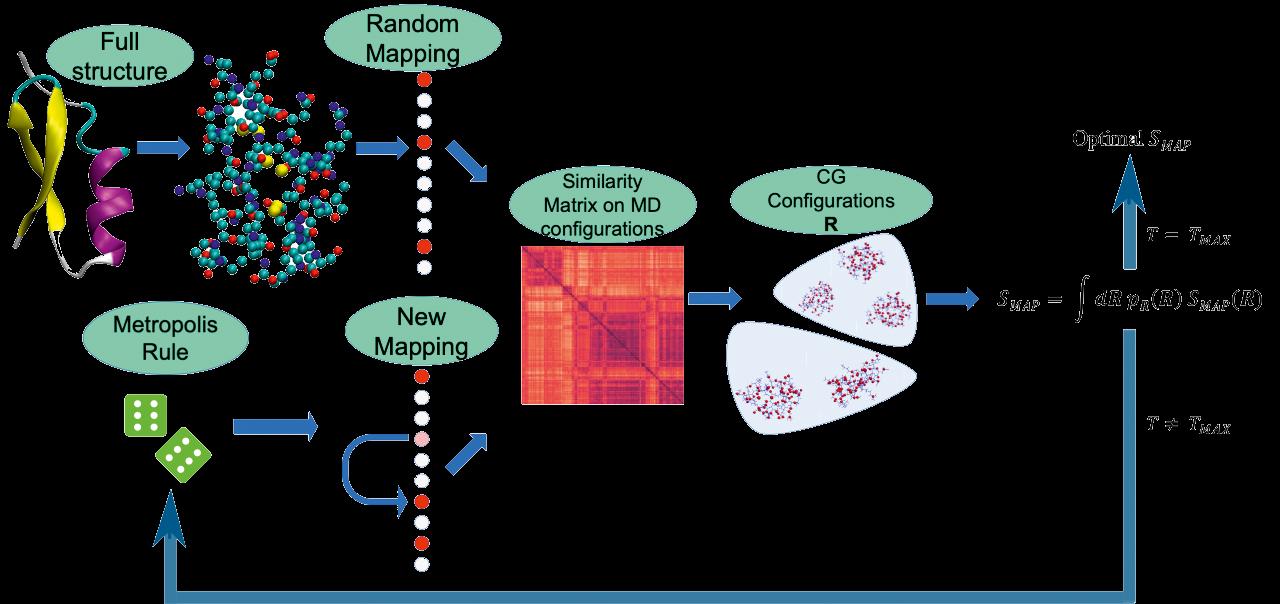
Workflow of one of the methods to identify optimal coarse-grained representations developed in the VARIAMOLS group. From “An information theory-based approach for optimal model reduction of biomolecules”, https://arxiv.org/abs/2004.03988
models of molecules, that is, all-atom ones, an atom is treated as a single, point-like particle; usually, these representations employ classical physics to describe the relationship between them. “Each atom has certain attributes, such as mass, electric charge and atomic radius, which allow the definition of interactions with other atoms,” Professor Potestio outlines. This approach is numerically expensive, as the interaction of each individual atom with the others needs to be accounted for. “At each step of the simulation, forces have to be computed among almost all atoms, and this requires substantial computational effort,” says Professor Potestio. “Nowadays, it is possible to simulate systems up to the tens of millions of atoms, but large computational resources are required, and not everybody has access to these kinds of facilities.”

This is an issue that researchers are addressing by using these relatively complex simulations as a starting point to devise methods through which simpler representations of the system can be constructed. Professor Potestio and his colleagues in the project develop and employ coarse-grained models in which a single particle does not represent just an atom, but rather several atoms together. “There are fewer interactions, so they are easier to compute. This improves the efficiency of the simulation,” he explains. This means efficiency in terms of time scale attained as well as number of calculations that need to be performed. “It could be that I would need a week rather than two months in order to observe the same process, if the coarsegrained model is sufficiently accurate,”
A view from the Physics department of the University of Trento. The city hosting the VARIAMOLS group offers the perfect environment for both thrilling academic work and relaxing walks in alpine landscapes.
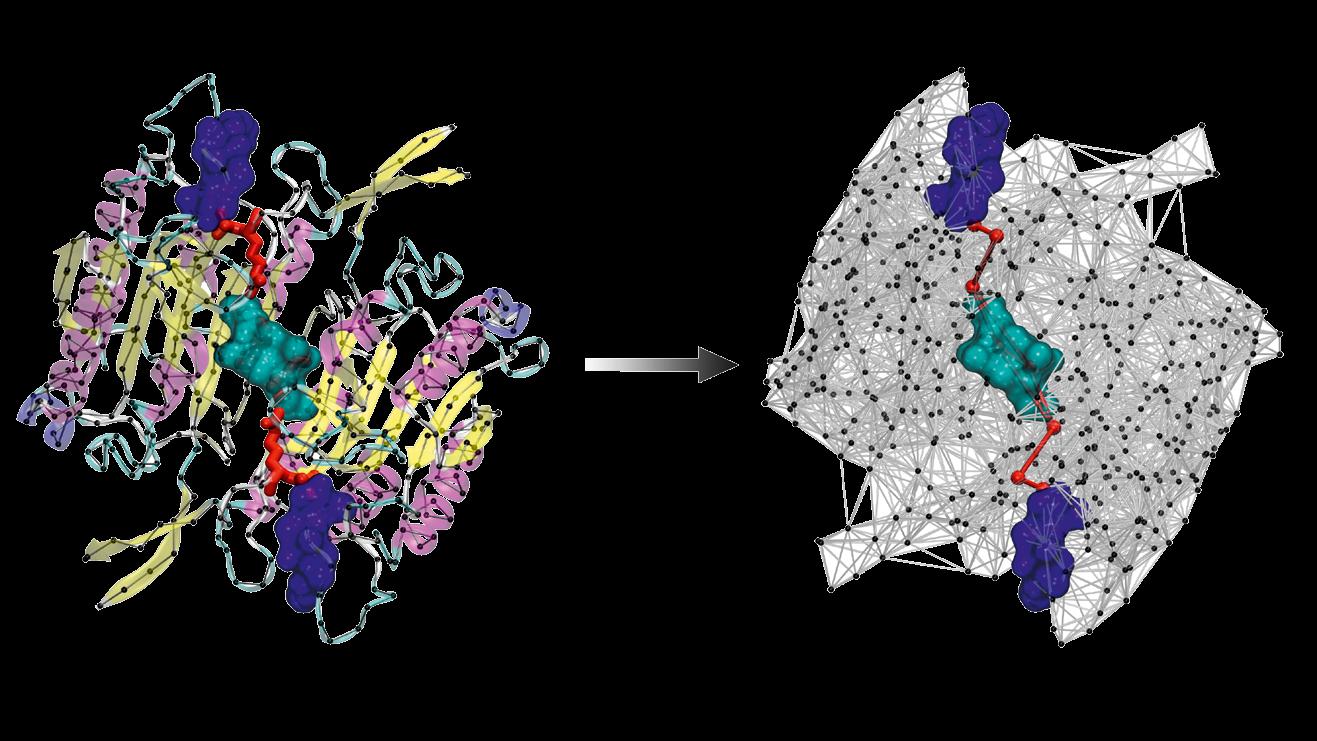
Distal parts of the protein communicate via allosteric pathways. Researchers in the VARIAMOLS group employ coarse-grained models to identify them.
continues Professor Potestio. “However, our main aim is to understand the system better in the process of constructing the coarsegrained model.”
By definition, a coarse-grained model is a simplified representation of a molecular system, so a lot of detail is not included in the description. Yet, important insights can still be gained: if a lower-resolution model reproduces a particular property of the molecular system under examination, e.g. a specific pattern of fluctuations or a global change of its structural arrangement, then this itself is highly informative. “This means that the particular features that the model entails are those that determine the process of interest,” explains Professor Potestio. Coarse-graining can be thought of as a process of selection, of identifying those pieces of information that need to be retained in a model and those that can be treated differently – incorporated in the effective interactions or just discarded. “If this process is performed correctly, then the expected system behaviour emerges from the model as it does from the more accurate description. This means that you have retained the appropriate detail and made the right choices in assuming certain features to be important,” says Professor Potestio. “We try to identify what is determinant in a system with respect to a certain interaction, property, or behaviour – more importantly, we try to perform this identification in an automated manner.”
However, the model still has to retain sufficient detail for it to be a realistic representation of the reference system. By providing different levels of resolution within the same system, Professor Potestio aims to strike the right balance between accuracy and efficiency. “Our strategy is to employ different degrees of detail, or resolution, in different regions of the system. Where it is possible, we simplify the structure, but where it is necessary, we retain the high-level detail,” he outlines. Researchers work from the bottom-up, in the sense that the starting point is a more detailed, all-atom simulation, from which the important regions of a protein can then be identified. “We try to infer from a high-resolution model which regions can be coarse-grained and which cannot,” explains Professor Potestio.
It is also important to consider the local properties of the molecule when developing a coarse-grained model. In conventional coarse-graining approaches, the same groups of atoms are typically associated with the same ‘flavour’ of effective sites. “For example, in many models proteins are described as chains of beads; each amino acid is represented as one of these beads, and a given amino acid type is associated with a specific type of bead and effective interactions,” outlines Professor Potestio. Two chemically identical amino acids,
If we can efficiently figure out how these molecular systems behave, then we can also potentially uncover the flanks that can be attacked from the pharmaceutical point of view. We can then develop new strategies to deal with various diseases at the molecular level.
however, may experience very different forces and fluctuations depending on their location and environment, and hence describing them as the same effective interaction unit might not be the right choice, an issue Professor Potestio is addressing. “We aim to provide different representations, even for the same group of atoms, according to the particular environment that these find themselves in,” he says.
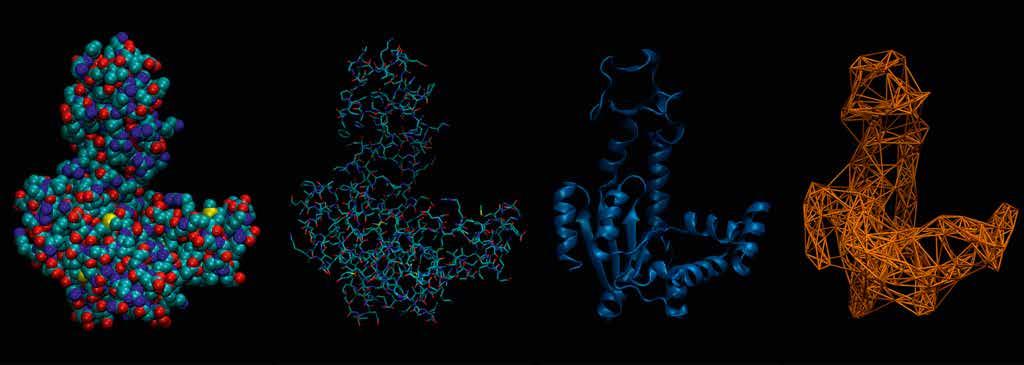
As in a tailor shop for proteins. Different representations of the same system provide different levels of insight.
VARIAMOLS VAriable ResolutIon Algorithms for macroMOLecular Simulations Project Objectives
The main goal of the VARIAMOLS project is to develop and apply novel computer-aided methods for the study of large molecular assemblies and their dynamics. The research will unfold along two intertwined lines: 1.) The development of non-uniform resolution models of the system, which optimize the balance between detail and efficiency 2.) The study of dynamics-mediated properties of protein assemblies.
Project Funding
ERC Starting Grant project € 1 339 351
Project Collaborators
• M. Scott Shell, Chemical Engineering,
University of California Santa Barbara (USA) • Robinson Cortes Huerto, Max Planck
Institute for Polymer Research (Germany) • Flavio Vella, Lab for Advanced Computing and Systems, Free University of Bozen (Italy) • Markus Deserno, Department of Physics,
Carnegie Mellon University (USA)
Contact Details
Project Coordinator, Raffaello Potestio Assistant professor Physics Department University of Trento via Sommarive, 14 - 38123 Trento (Italy) T: +39 0461 282912 E: variamols@unitn.it W: http://variamols.physics.unitn.eu/ W: sites.google.com/g.unitn.it/sbp W: https://eutopia.unitn.eu
Raffaello Potestio

Raffaello Potestio is tenure track Assistant Professor in the physics department at the University of Trento. His main research interests are the development and application of coarse-grained models and coarse-graining strategies for soft matter, in particular biologically relevant systems.

The VARIAMOLS group in January 2020. From left to right: Roberto Menichetti, Raffaello Potestio, Marta Rigoli, Thomas Tarenzi, Marco Giulini.

Photo credit: Lorenzo Petrolli.
Machine learning
The methods that Professor Potestio and his colleagues are developing to identify the appropriate level of resolution for the different parts of a system and to parameterise the interactions are algorithmic procedures. Overall, the process of constructing a model can be time-consuming, so researchers are keen to harness the power of artificial intelligence to try and reduce the amount of time required. “Our aim is to exploit the advantages that machine learning offers as a method in order to speed up the process of constructing these models,” outlines Professor Potestio. Researchers in the VARIAMOLS group are developing these algorithms and producing training sets for a deep neural network-based approach, so that it is capable of then performing the same task without having to run a reference, all-atom molecular dynamics simulation beforehand. “We aim to use deep learning approaches as a kind of trick to accelerate the process, but only at the end of a pipeline of which we understand each step. We tend to avoid black boxes,” explains Professor Potestio.
These tools are designed primarily for simulating macromolecules, and Professor Potestio is keen to make them available to the wider biocomputing research community, where they could prove to be a valuable instrument. The methods have been developed in such a way that they can be distributed as free software, and Professor Potestio is looking towards their practical application. “As soon as we have tested and aptly documented our codes, we will provide them for other researchers to use,” he says. The programs developed in the project will be of interest to a broad audience, believes Professor Potestio. “On the one hand we have the physics-oriented community, which might be interested in the computational
statistical mechanical aspects of our work. On the other hand, researchers from different fields may be interested in employing our tools for biochemical and pharmaceutical applications,” he outlines.
A researcher in biochemistry may not necessarily have deep knowledge of statistical mechanics or how these codes work, so accessibility is an important issue in terms of the wider application of these tools. “Our ideal is to provide programs that are accessible also to people who are not in the modelling field, or the statistical mechanics field, but would like to make use of our research in order to better understand their proteins of interest for example, and possibly devise better ways to interface with them,” says Professor Potestio. An improved understanding of the physics of biological macromolecules could open up exciting new possibilities in research. “If we can efficiently figure out how these systems behave, then we can also potentially uncover the flanks that can be attacked from the pharmaceutical point of view,” explains Professor Potestio. “We can then develop new strategies to deal with various diseases at the molecular level, for example testing several possible drugs at a higher speed and lower computational cost.”
The main focus for Professor Potestio and his group is on fundamental research at this stage, and on developing a deeper understanding of the physics of biomolecules. However, once researchers have a clearer picture of a molecular system, e.g. the relationship between its structure, dynamics and thermodynamics, that then holds wider relevance. “We can go to a biochemist and say: ‘this is what we see. Do you think you can make use of that in order to prevent the functioning of this protein? Might this be beneficial in terms of inhibiting a pathogenic process?’ This is the ultimate objective,” says Professor Potestio.








