1 Introduction
The ideal chemical process is that which a one-armed operator can perform by pouring the reactants into a bath tub and collecting pure product from the drain hole. Sir John Cornfortha
1.1 Introduction
1.1.1 Multicomponent reactions (MCRs)
Chemistry as a fundamental and essential science in our daily life is facing a progressively accumulative demand in different disciplines, covering a broad range, as vast as, material sciences to life sciences. Organic chemistry is one of the subdisciplines of chemistry that studies the structure, properties, and reactions of organic compounds. They contain carbon in covalent bonding leading to synthesis of organic complex molecules, using commercially available or easily accessible starting materials. Organic chemistry is frequently known as a central science which is fronting an increasingly demand for the synthesis of new chemical entities (NCE). Nevertheless, NCE not only demand an accurate structural space but also their practical and economic feasibility must be contemplated. Most importantly, for development of NCE, organic synthetic chemists are obliged to comply with the principles of green chemistry in order to preserve environment as healthy and safe as possible. Nowadays, interest in carrying out sustainable chemistry has significantly overgrown [1], thus, design and selecting the best synthetic pathway is an important and great challenge to synthetic organic chemists. In another word a main and vital question and consideration, is how to design and conduct organic transformations. The answer is, designing and selecting a superior reaction pathway, before performing it, making sure that the selected pathway will proceed smoothly to completion under an ideal reaction condition, especially being in agreement with principles of green chemistry. The next question which arises is, what parameters should be taken to consideration for approaching to an ideal organic synthesis [2,3]? Rationally, an ideal organic synthesis should be straightaway, safe, facile, concise, selective, high yielding, being completed in short reaction time, environmentally benign, being commenced from commercially available or easily accessible starting materials and being performed with high diversity [4]. In addition, the principle of selectivity has to be harmonized with increasing significance from economic and ecological points of view [5,6]
Nowadays, among synthetic organic chemists community “MULTICOMPONERT REACTIONS” (MCRs) are considered as masterpieces of synthetic efficiency and state of art for designing an ideal synthetic pathway. MCRs have experienced an extraordinary progression in status and practice, particularly in recent decades, becoming an unprecedented and powerful synthetic tool, standing in a very projecting position.
It is worthwhile to mention that historically, MCRs have actually gone along with the field of organic chemistry, since the early days of its development. Indeed, it goes back to 1838 when Lauren reacted bitter almond oil and ammonia. It used to be recognized as the first MCR [7,8]. This mixture was expected being involved of hydrogen cyanide, and benzaldehyde present in almond oil which upon condensation with ammonia, generates an α-aminonitrile as intermediate that once created, reacts with another molecule of benzaldehyde to afford the corresponding Schiff base. Nevertheless, in the compositions reported, none of the scrutinized products aligned with the MCR’s expected products. Neither the α-aminonitrile nor its consequent Schiff base was detected. Thus, historically, the Strecker reaction, involving amino acids, aldehydes, potassium cyanide, and ammonium chloride leading to the formation of α-amino acids, reported in 1850, is recognized as the first MCR, in contemporary organic chemistry. Remarkably,
aSir John Warcup “Kappa” Cornforth, was an Australian-British and a Nobel Prize laureate in Chemistry in 1975 for his brilliant research on the stereochemistry of enzyme-catalyzed reactions, J. Hanson, J. Cornforth, Nature, 506 (2014) 35
Recent Advances in Applications of Name Reactions in Multicomponent Reactions. http://dx.doi.org/10.1016/B978-0-12-818584-1.00001-X
Introduction
Strecker reaction is not only important for being recognized as the first classical MCR, but it is extremely renowned for the synthesis of biologically important α-amino acids (so-called, stuff of life).
At the beginning of their growth, the common MCRs were based on typical condensations between carbonyl compounds and different nucleophiles. The descriptive example is the first known MCR, the Strecker reaction. Since the discovery of the Strecker reaction [9], a plethora of interesting MCR such as the Mannich [10], Ugi [11], Passerini [12], Kabachnik-Fields [13], Bucherer-Bergs [14–17], van Leusen [18], Pauson-Khand [19–21], Gewald [22,23], Staudinger [24–26] were discovered and reported.
A host of transformation was also planned for the synthesis of nitrogen-containing heterocycles based on MCR. Examples are the Biginelli [27], Hantzsch [28], and Asinger [29] MCRs. In spite of popularity of MCR, it had not been acknowledged as a fundamental principle in synthetic organic chemistry, till Ugi’s revolutionary extension lead of the Passerini reaction and his brilliant conclusions, he described from his research group achievement. The introduction of the isocyanide-based Ugi MCR [11], allowed the extension of this approach to the synthesis of a broad range of important accessible biologically active molecules, bearing both reactive amide and ester functionalities. Applications of Ugi-type MCRs dominated the field for an extended period of time, as shown by the massive numbers of published reviews [30], and monographs [31]
Ugi reaction is actually a four-component reaction achieved and reported by I. K. Ugi and coworkers in 1962 for the first time. It involves the reaction of aldehydes, amines, isonitriles, and carboxylic acids in one pot fashion, resulting in the formation of corresponding α-acylamino carboxamide adduct. This MCR particularly attracted much attention, when it was shown that the post-Ugi products can be converted into a wide range of heterocycles [32].
The ideal MCRs are commonly defined as reactions in which three or more components are added to a one pot fashion at the same time, resulting in a desired target which contains most of the atoms from the starting reagents [33]. Consequently, these reactions incorporate a sequence of more than one chemical transformation without the necessity of changing the reaction media and reaction vessel after each conversion. Moreover, in comparison with step-by-step procedures, MCRs are generally necessitating less time and effort [34,35]
Thus, it is not surprising that MCRs results in high molecular diversity and permit for the design of libraries of small organic molecules. Design of resourceful MCRs, by combining molecular diversity [36] under eco-friendly conditions [37] is currently the major focus of many research groups for an ideal synthesis of complex backbones and poly- substituted molecules. Indeed, the rational design of such reactions which convert commercially available or easily accessible substrates into more complex structures in a one pot fashion is one of the contemporary main challenges in the art of organic synthesis. In this perspective, MCRs have become one of the best recognized strategies for attaining this goal, since they imply atom economy [38] and bond-forming efficiency [39].
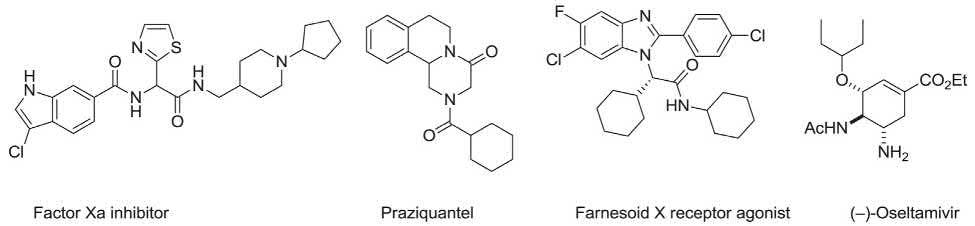
This is particularly striking for the pharmaceutical research centers and industries, in which the facile formation of large libraries of compounds with possible biological activity is a priority and importance. The implication of these processes can be vividly witnessed in a plethora of publications being found when “MCRs” as key word is searched in the chemical literature, especially over the last decade. The screened biological efficacy of compounds synthesized via MCRs has also been established by discovery of several molecules with significant biological potency (Fig. 1.1) [40–44].
Presently, MCRs, manipulating almost all the mechanistic pathways recognized in contemporary organic synthesis being reported in chemical literature, comprising organocatalyzed-transformations, cycloadditions, and transition metal-catalyzed or radical-mediated strategies [45]. The important merits of MCRs trail from the fast increase of molecular complexity in the products which remarkably improves the efficiency and economy of the planned syntheses. The modularity of MCRs permits one to diverse any one of the multiple starting materials, independently
FIGURE 1.1 Typical molecules with significant biological potency synthesized via MCRs.
to generate the effective assemblages of compounds having a common structural core decorated with miscellaneous substituents.
Particularly, MCRs were cordially welcomed by medicinal chemists in quest of general large combinatorial libraries of compounds for biological screening, leading to drug discovery [46]. Very recently, the introduction of diversityoriented synthesis (DOS) [47] commanded further advances in the design of novel MCRs. Currently, the development and application of MCRs get hold of a copiously mature state, as designated by a plethora of recent hot review focusing on the key roles played by MCRs in specific zones, involving the synthesis of bioactive molecules [44], via DOS [48], in the synthesis of heterocycles [49], natural product-like polycyclic structures [50], and etc.
In 2011, Ruijter and co-workers [35] underlined the necessity to maximize scaffold complexity and diversity in libraries of small organic products of MCRs, as well as the discovery of asymmetric MCRs as two major encounters for design of novel MCRs, in future [35]
1.1.2 General conception
In general, organic reactions can be categorized according to the number of partaking of startingmaterials. In this regard, they can be classified as one-component reactions, two-component reactions, and multicomponent reactions (MCRs) as well as polymerizatons. For example, the classical Claisen rearrangement [51] is classified as a one-component reaction. Thus, the one-component reactions comprise one starting material and a catalyst, if required, leading to the desired products. In a two-component reaction two starting materials are reacted to give the anticipated single product [52,53]
On the other hand, those reactions including three and more starting materials are classified as MCRs. As mentioned above, multicomponent reactions (MCRs) are nowadays considered as tools of paramount importance and remarkable approach to reach ecofriendly and sustainable transformations in the art of organic synthesis.
Some of the basic concepts associated to MCRs are one-pot, domino/cascade, tandem sequential reaction which concisely are described in the following section in order to make the readers acquaint with this ground and its characteristics. Conceptually, the terms, MCRs, are more clarified as well as being justified.
The familiar terminologies, “one-pot, domino/cascade, tandem, and MCRs” are probably looks like the same for most synthetic organic chemists from the practical point of view, but they have critical differences which are important to know in order to differentiate each term from the others. The terminology ”one-pot synthesis“ involves reactions that comprise multiple chemical conversions between, substrates, reagents and catalysts (if required) which are performed in a single vessel. Therefore, MCRs (involving, multiple chemical transformations) state into the type of one-pot reactions or being performed in the sole reactor. In addition, various types of catalyzed-MCR in one-pot fashion was classified in 2004 [54] while afterward, Tietze defined domino reactions, clearly [39].
In this classification, one pot catalyzed-MCRs were differentiated from one pot catalyzed-domino/cascade and catalyzed-domino tandem reactions depending on few parameters, such as the instant, when the (pre)catalysts are added to the reaction mixture, different numbers of mechanistic pathways which may be comprised. In general, catalyzed-domino/cascade and dimino/ tandem reactions are also one-pot reactions where all the components are bring together at the same time at the beginning of the reaction, whereas in catalyzed-MCR, all of the components are not added at the same time. One more requisite for catalyzed- domino/cascade and tandem reaction is that all sequential transformations must occur as a result of the generation of intermediate in the prior reaction step. Thus, a classification can be established, based on that catalyzed domino/cascade and tandem reactions upon differentiation made by the number of mechanistic pathways present in the reaction. Considering all the above-mentioned perceptions demarcated, it is obvious that MCRs are also one-pot reactions that might well fall under the type of domino/ cascade or domino/tandem reactions. A reaction is a domino/cascade or domonio/tandem MCR when it has the physical appearance of one of these classes of reactions in addition to comprising three or more starting materials that react to form a desirable target.
As mentioned before, MCRs are identified when more than two starting materials react in one pot fashion. Since the impact of three or more self-determining molecules is highly not prospective. MCRs, characteristically include a number of basic reactions with discrete mechanisms, each one typically necessitating altered reaction conditions.
In 1997, Karl Ugi suggested a classification system for MCRs relied on the reversibility of their specific reactions [30,55]. These reversibility factors play a key role in the results, attained from MCRs, and usually are changed depending on the reaction conditions.
After, design and successful performance of catalyzed-MCRs, their optimization conditions is indeed puzzling and challenging issues since the best condition for a particular single reaction are not usually the ideal condition for the development of other MCRs. Then, reasonably and professionally, a conciliation should be found via the
1. Introduction
investigation and optimization of different reaction factors, such as kind and even concentration of solvent, temperature, source of energy (conventional heating, ultrasonic irradiation or MWI) and sort of catalyst and its optimized quantity. Therefore, optimization of MCR conditions denote one of the most problematic and challenging jobs for emerging novel MCRs. Worthy to mention that recent advances in MCRs along with other fields of chemistry, such as instrumental chemistry and/or computational chemistry have delivered respected tools for saving time and assets for optimization of MCRs. Nowadays, well-examined computational programs, is employed along with the analytical and reaction preparation systems as already stated, which can speed up the optimization process of a MCR and increase the yields of the obtained products, significantly [56]
This combination of computational chemistry with mechanized systems has also established to be very beneficial in drug discovery, since they can be employed to optimize the principles of a specific biological potency of products, achieved via MCRs [57]
The increase in the utilization of these techniques and use of green solvents, in particular water as the most abundant and safest solvent in MCRs show that these reactions will advance along with green chemistry.
1.1.3 Green chemistry
Anastas and Warner in 1998 set several basic perceptions for MCRs which were found being in agreement with the basic principles of green chemistry [58].
Synthesis of complex final products via MCRs in one vessel fashion is convenient as well as environmentally benign. Some attractive merits of designing MCR as a method of choice are the formation of a slighter quantity of discarded and waste, the maintenance of assets and resources and the decrease of the essential energy for proceeding reaction to completion. The use of water as a solvent in MCRs offers several “green chemistry” benefits. In addition, conducting MCRs in water increases the rate of reaction significantly, compared with organic solvents. This rate enhancement can be attributed to several factors, involving the hydrophobic effect [59,60], generation of hydrogen bonding in the transition state [61], and the high cohesive energy density of water (550.2 cal‚mL 1 at 25 °C) [62].
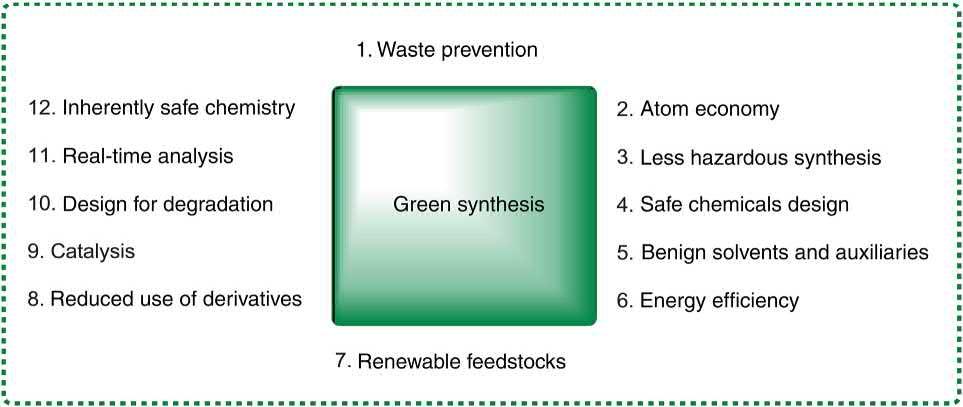
All these benefits and merits have conveyed MCRs into the attention of synthetic organic chemists whose aim is evolving innovative green chemistry procedures. Conventionally, efficacy is prearranged in the synthetic organic chemist’s attention, typically in terms of number of steps, selectivity and yields of the reaction. However, the complying with the green chemistry principles is also in their mind. Nowadays, they are more concerned about measures of discarded and waste generation, utilization of safe substrates, reagents and solvents, avoiding the use of hazardous or poisonous chemicals, sort of energy and even its strength and broad-spectrum of safety and general security. All these conscience and ethics are in agreement with 12 principles, conveyed by Anastas and Warner in 1998 (Fig. 1.2) [58].
Different MCRs have previously being employed for the synthesis of several biologically active products with fruitful outcomes. However, the increasing awareness in performing chemical reactions under green and mild reaction conditions, cleaner MCRs also must be designed and conducted under mild, energy-efficient, and atom-economical
FIGURE 1.2 Principles of Green chemistry.
processes. Other methods, being conducted in the pharmaceutical sector, for the syntheses of medicines, if applicable, should be improved by designing new MCRs under green conditions. Although, several progresses have been made, but further efforts and attempts are prerequisite to adapt these processes into competitive procedures which can be executed in the synthesis of a broad collection of biologically potent compounds and prescribed drugs.
Atom economy is a perception that was presented by Trost in 1991. As a matter of fact, it is a measure of the efficacy and productivity of a reaction by comparing the quantity of the desired final product to the amount of other possible formed products [38].
This notion discloses the necessity of designing reactions where the mainstream of the reactants are merged into the targeted desired product. This strategy shows several merits by comparison with others, such as breaking a complex reactant down to attain a product. Owing to this point, in the latter strategy, although the reaction yields are 100%, the rest of the substrates and reagents are typically wasted. By this definition, MCRs can be recognized as perfect cases of magnificent practical atom economy. In MCRs, diverse molecules are transformed into a complex product in perfectly efficient manner. Thus, the expansion of MCRs also infers the advance of atom-economical reactions.
Although, MCRs exemplify a fundamental stage in the progress of modern organic chemistry, regrettably, most of the above-mentioned reactions, when practically performed, results in the formation of the desired products but in their racemic forms. In organic chemistry, a racemic mixture, or racemate is one that has equal amounts of left- and right-handed enantiomers of a chiral molecule. A sample with only a sole enantiomer is an enantiomerically pure or enantiopure compound. In contrast to the two pure enantiomers, which have identical physical properties except for the direction of rotation of plane-polarized light, either of the pure enantiomers frequently shows different biological activities. Thus, pharmaceuticals may be available as a racemate or as the pure enantiomer, which might have different biological activities; one of them exhibits biological potency, while the other one shows no potency. However, in some racemates, one enantiomer being useful medicines while the other may be very dangerous (for example, thalidomide, a sedative drug discovered at the end of the 50s, which triggered a global catastrophe. Thalidomide was first explored and marketed in West Germany in 1957 where it was available over-the-counter. The drug in its racemic form has been prescribed to thousands of pregnant women, worldwide in order to relieve pregnancy nausea. However, it was resulted in thousands of babies being born with malformed limbs. The damage was revealed in 1962 when a research group isolated the drug’s metabolites and identified a compound, CPS49 that causes severe limb defects and blocks growth of new blood vessels. Researchers finally figured out the mechanism of the tragic birth defects caused by thalidomide. It was prescribed to pregnant women in the late 1950s as a remedy for nausea, actually was manufactured and sold as a racemic mixture. It was found out that only one of the isomer was useful in treatment of nausea in pregnant women while the other inhibited development of new blood vessels at a crucial time in the pregnancy [63].
A part from thalidomide, several other prescribed drugs, manufactured and sold in racemate form with no problems, since the other enantiomer is biologically inactive or has synergic effect, for example, the antidepressant prescribed drug, fluoxetine [64].
In nature, asymmetric synthesis is a definite, thus, natural products provided via biosynthesis are optically pure. Even, those natural products bear several stereogenic centers, can be obtained as a single steroisomer from a natural source thus, frequently exhibit high biological potency. Examples are notorious morphine, and antitussive, codeine.
The high interest in MCRs lies not only in their promising and green features but also in their products biological potencies, frequently perceived for the products obtained, straightaway. Many MCR adducts exhibit prominent biological potencies even in their racemic forms, thus the attention to such compounds and their production in easier and less costly, approach have been increased, in last decades.
Although, some biologically active compounds obtained via MCRs are also potent in their racemic form but pure enantiomers frequently show much more activity. In several cases, the other enantiomer present in racemic mixture may have opposite biological effects, thus should be used as single enantiomer form when used as a drug.
For this imperative and proven issue by screening, development of dependable asymmetric multicomponent reactions (AMCRs) is in much demand. To have reliable protocols for AMCRs in hands, straightforward and sophisticated access to libraries of biologically active compounds is possible. In the last decade, development of AMCRs has attracted much attention of synthetic organic chemists since the necessity for continuous discovery and developments in the synthesis of biologically active compounds, especially for their stereoselective synthesis for libraries of novel optically active organic compounds were well recognized. Biological screening of enantiomerically pure compounds is of enormous reputation [46], since each optically active isomer may show discrete bioactivity. Having, a series of optically active organic compounds obtained via AMCRs permits the use of high-quantity of biological measurements (genetics, transcripts, and chemogenetic interactions) and the computational methods assist to further combinatorial drug design (CDD) [65]
1. Introduction
Delightfully, over the years, during the progress of MCRs, new approaches have come to the line by development of asymmetric variant of catalyzed-MCRs, enabling, synthetic organic chemists to conduct these hitherto racemic MCRs with high stereocontrol. The importance of optically pure compounds, especially in pharmaceutical and agrochemical industries has stimulated extensive research toward more stereoselective synthetic approaches toward these treasured molecules. In this regard, advances in asymmetric variant of known MCRs or totally novel asymmetric MCRs for the construction of optically pure compounds are highly and still in demands.
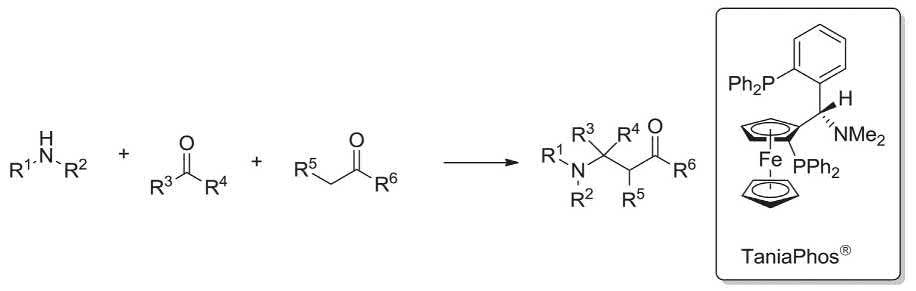
Asymmetric MCRs (AMCRs) can be distributed into two classes, that is, the catalyzed enantioselective and catalyzed-diastereoselective MCRs. A catalyzed-enantioselective MCRs can be demarcated as a reaction between three or more achiral substrates and reagents in the presence of a catalytic quantity of a chiral catalyst via a one fashion. While, the reaction proceeds, the two or more bonds are formed as well as at least one newly stereogenic center is generated which should be well-stereocontrolled. In diastereoselective MCRs, the new stereogenic center is distereoselectively controlled by one or more optically pure starting materials or a chiral auxiliary. Interestingly, some chiral catalysts such as TaniaPhos® are nowadays commercially available, notably, it has successfully been applied in the asymmetric Mannich-MCR [66]
Another chiral ligand for preparation of a chiral catalyst was synthesized from the (R)-Ugi amine which can be effectively utilized as effective chiral organo-catalyst in asymmetric hydrogenation [67–71].
Several reviews and monographs, have been published, focused on different reaction kinds and features of AMCRs. They mainly concentrated on the applications of the chiral organo-catalysts [72], 1,3-carbonyl-based [73], and isocyanide-based [42], AMCRs. In addition, a comprehensive review was published by Ramón and Yus in 2005. It deals with the complete scope and features of AMCRs, particularly, focused on both catalyzed- enantioselective and catalyzed-diastereoselective AMCRs [74]
Although, AMCR variants and the role of chiral inductors in asymmetric Hantzsch and Biginelli reactions have been more studied and found being readily applicable (Chapter 4). Several developments in asymmetric cycloaddition-based multicomponent reactions, for example, AMCRs based on Diels–Alder, Tietze and 1,3-dipolar cycloaddition reactions, have been introduced and found applications in the art of organic synthesis [75,76]. This progress was broken up with recent advances in Michael addition-based AMCRs, including the asymmetric Michael/Michael/ aldol domino sequential reaction [77]
To consider the specific step of chiral induction and the effectiveness of stereo-controlling of chiral element, in fact are the most important issue in AMCRs [78]. In this regard, in spite of several insights understanding of AMCRs, worthy to mention that catalyzed asymmetric versions of MCRs is highly debatable, particularly because actual stereocontrol falls short. In general, chiral induction steps in AMCRs are actually uncertain [77].
Nowadays, MCRs are undoubtedly considered as a treasured approach in the “Green Chemistry and Sustainability Toolbox.” When synthetic organic chemists intend to design a synthetic pathway, choosing a strategy to program combinatorial chemistry, in drug discovery and in the total synthesis of natural products as well as synthesis of polymers [80]. AMCRs are also ideally suited for diversity oriented synthesis and library generation [81].
MCRs, enjoy different merits. Water is the most favorite solvent in MCRs It is initially being examined and used as solvent of choice. Water is preferred solvent in MCRs due to its advantages from different points of view. (1) It is the most abundant and greenest solvent, (2) it is highly polar, (3) it accelerates most common MCRs. The rate of Passerini [82] and Ugi reactions [83] are increased under high pressure which supports and justify the acceleration of MCRs in water. Another important profit of MCRs is that they conceptually bring at least three reactants together in one-pot vessel, providing an effective and inherently atom economical reaction (forming a product that includes basically all the atoms of the substrates), chiefly, under mild reaction conditions and as mentioned earlier, commonly utilizing water as the greenest solvent [80].
The other advantage of MCRs is waste lessening. Noticeably, due to their great convergent nature, MCRs decrease waste formation by using resources which every so often shortening the overall number of steps in a multistep synthesis. Due to their exceptional chemo-and regio-selectivity, the formation of unwanted side-products and byproducts are minimized in MCRs. The by-products in most MCRs are usually simple small molecules such as water, alcohols, amines, or common salts, leading to not only a compact quantity of waste, but reduced waste itself, since they commonly are ecofriendly. That evades complications related with the regaining and discarding of dangerous and unsafe waste. The work-up procedures for MCRs are habitually straightforward. Their products are often precipitated in the reaction mixture, circumventing to consume longer time and resource for exhaustive recovery and purification of the products. Solvent used in MCRs are often environmentally benign and also generally used, in remarkable minimized volumes.
According to literature survey for multiple-bond-forming efficacy, MCRs in fact can be branded as the most promising approach, to touch an exceptional arrangement of efficacy, atom economy and sustainability. The future of MCR
most apparently relies on the advance of techniques that save time, energy, effort, and resources, as well as diminishing the quantity of waste and unwanted by-products, generated during the course of reactions.
In spite of all merits and gifted features as mentioned-earlier for MCRs, as important synthetic tools in organic chemistry, some of them suffer from some disadvantages such as requiring harsh reaction conditions, needing reagent excesses, demanding high temperatures, sometimes should be performed in toxic solvents, catalyzed by expensive and hazardous metal catalysts, purification of their products by column chromatography, giving low yields, experiencing low selectivity, and requiring long-reaction times. After all, the earlier-mentioned drawbacks for some MCRs, are compensated by their merits and useful features, observed, and experienced for MCRs. Appropriate and order addition of stating materials, more sensible designs, inventive applications, and profound understanding and knowledge of the mechanisms (catalyzed or non-catalyzed), can consequently overcome to the earlier mentioned drawbacks, leading to improvement of MCRs. Generated intermediates in MCR are not normally isolated. Although some intermediates of MCRs are isolated and their structures are accurately elucidated, which allows to a better and more accurate mechanistic pathway for the organic transformations occur during MCRs. What is actually surprising about MCR mechanistic pathway is that, regardless of the favored and more rational mechanisms, it resulted in the same final desired product.
Alternatively, it can be proposed that MCRs proceed in an “all roads lead to Rome” manner. For example the chief product may be constructed through various but convergent reaction pathways. Among all MCRs previously introduced, the Biginelli, Hantzsch, Mannich, Passerini, and Ugi reactions are the kind of special. These MCRs in fact are the most prevalent, investigated and extensively employed in the synthesis of biologically active compounds, thus dictating their prominence, expediency and promising styles. All these earlier-mentioned MCR kinds have been extensively investigated by different analytical methods as well as spectroscopic/spectrometric techniques which sometimes propose dissimilar mechanism schemes for some MCR, based on the data obtained [84–86].
As anticipated, all these MCRs have at least two potential reaction routes. The knowledge and understanding the exact mechanistic pathway is therefore crucial for sensible design and selection of catalysts, ligands, reagents, stereoand electronic controls. They also give the researchers an idea about the nature of by-products and most prominently an expectation of new products which after elucidation of their structures can be screened for their possible biological activities or being used as intermediates in total syntheses of natural products. MCRs mechanism, commonly induce a cascade of sequential bimolecular reactions, though sometimes termolecular stages, needing the involvement of at least three chemical entities in the transition state. Thus, for designing MCRs, it is advised to collect precise information about generation of various possible intermediate, leading to the formation of final desired products.
1.1.4 Name reactions
The name reaction is a type of shorthand indication that evades the necessity to give a longer description of the features of a specific transformation of concern. Writing or saying the name reaction permits to bring to the mind of a well-informed reader or listener a conceivable idea about substrates, reagents, catalysts and reaction conditions, or possible events which may occur during the MCRs, thus gaining their detailed mechanistic pathways [87,88].
Name reactions in organic chemistry are those that named after its discoverers or developers. Among the tens of thousands of organic reactions that are well- known and acknowledged, only about hundreds of them are wellestablished enough and reached to such status to be named after people who discovered and devloped them [89].
Well-known examples in organic chemistry involve the Grignard reaction, the Sabatier reaction, the Wittig reaction, the Claisen condensation, the Friedel-Crafts acylation, and the Diels-Alder reaction, etc. There significant name reactions in organic chemistry, explored and well-established during years or even centuries. Notably, among tens of thousands of already known organic reactions, only about hundreds of them have reached to such position and status were deserved, being named after their discoverers or developers. Accordingly among about thousands of reported MCRs only few of them reached to such eminence and grade being merited to be named after their explorer. Expectedly, several of these discoverers are “Noble Prize Laureates in Chemistry.” They are, Victor Grignard and Paul Sabatier (Joint winners in 1912), Sir Robert Robinson (1947), Diels and Kurt Alder (Joint winners in 1950), Hermann Staudinger (1953), Karl Ziegler and Giulio Natta (Joint winners in 1963), Robert Burns Woodward (1965), Ryōji Noyori and Barry K Sharpless (Joint winners in 2001), Richard F. Heck, Ei-ichi Negishi and Akira Suzuki, (Joint winners in 2010).
Several typical MCRs which are name reactions have a broad range of applicability in organic synthesis. Although systematic approaches for naming reactions based on the reaction mechanism or the overall transformation exist (such as the IUPAC Nomenclature for Transformations), the more descriptive names are often unwieldy or not specific enough, so people names are often more practical for efficient communication [90], the Diels-Alder reaction,
1. Introduction
Grignard reaction [91]. Other well-known examples are, the Strecker reaction [9], the Passerini reaction [12], the Claisen condensation [92], the Friedel-Crafts acylation [93], the Sabatier reaction [94], the Biginelli reaction [27], the Hantzsch reaction [28], the Reimer-Timen reaction [95], the Pummerer rearrangement [96,97], the Pinnick oxidation [98], the Birch reduction [99,100], and the Huisgen 1,3-dipolar cycloaddtion reaction [101], etc.
In some cases MCRs were not actually named after namesakes. Examples involve Aldol condensation [102], Click reaction [103,104], A3 Coupling reactions [105], etc. As organic chemistry advanced during the 20th century, chemists began relating synthetically useful and common reactions with the names of the discoverers or developers; in many cases, the name is merely a memory aid.
Several books and monographs devoted exclusively to name reactions have been published, [105–108], the Merck Index, a chemical encyclopedia, also includes an appendix on name reactions.
Synthesis of several biologically important compounds for screening and even production of few prescribed drugs has been successfully accomplished via different MCRs. Some important examples are underlined as follow.
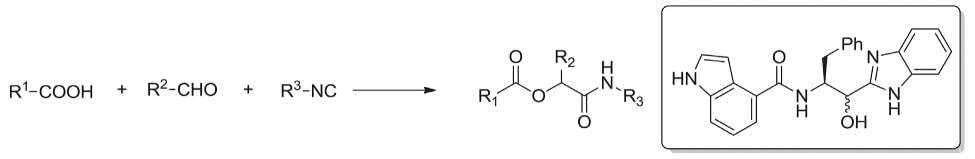
An Italian chemist, M. Passerini in 1921 successfully achieved and reported a three-component reaction involving, carboxylic acids, aldehydes, and isonitriles, to obtain the corresponding α-acyloxy amides [109]. The Passerini reaction was employed in the synthesis of several pharmaceuticals. As an example, Hulme and co-workers successfully achieved library synthesis of novel norstatine derivatives bearing benzimidazole moieties via Passerini MCR (Scheme 1.1) [110]
The Mannich MCR is one of the most disciplined MCRs with well-established mechanism. It is an organic reaction which involves of an amino alkylation of an acidic proton located next to a carbonyl group of say formaldehyde and a primary or secondary amine or ammonia. The final product of this three-component reaction is a β-amino-carbonyl compound so-called a Mannich base. Reactions between aldimines and α-methylene carbonyls are also deliberated as Mannich type reactions since the imines are generated from reaction of amines and aldehydes.
Commercially available TaniaPhos® is a chiral ligand which is used as chiral catalyst in the asymmetric hydrogenation in the Mannich three-component reaction. TaniaPhos® Ligand in turn has been asymmetrically synthesized from the (R)-Ugi amine in two steps. Interestingly, the (R)-Ugi amine itself can be synthesized via a Mannich MCR, involving ferrocene, dimethyl amine and acetaldehyde (Scheme 1.2) [10,67–71]
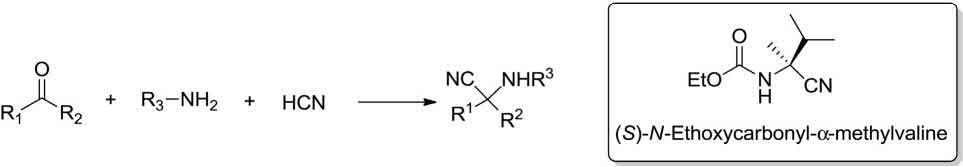
Strecker in 1850 accomplished and reported the synthesis of α-amino acids via MCR. It involves, aldehydes, hydrogen cyanide, and ammonia as substrates, and is renowned as the world‘s first MCR. α, α-Disubstituted amino acids
1.2 Mannich three-component reaction leading to formation of the (R)-Ugi amine.
SCHEME 1.1 Passerini reaction.
SCHEME
as unnatural amino acid analogues are biologically important compounds that have stirred up much attention of synthetic organic chemists, due to their important uses in peptide-mimics and in the de novo design of proteins. The Strecker MCR was employed in the synthesis of ((S)-N-ethoxycarbonyl-α-methylvaline) when 3-methyl-2-butanone and NaCN were treated with NH4Cl in the presence of MgSO4 in NH3/MeOH at 30 °C. Further steps includes the generation of the tartrate salt and the fruitful synthesis of (S)-2-ethoxycarbonylamino-2,3-dimethylbutyric acid dicyclohexylamine salt (Scheme 1.3) [9].
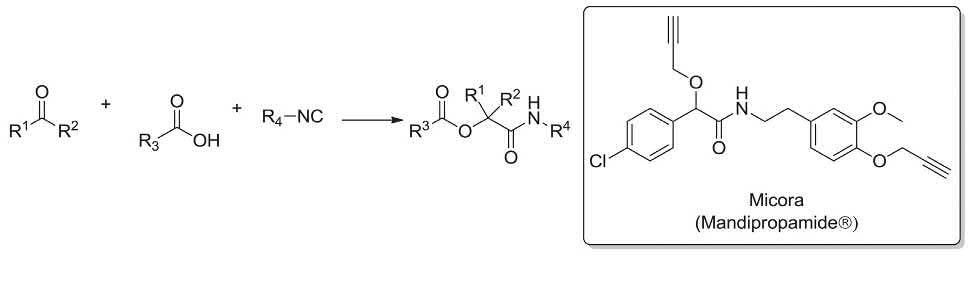
The Passerini reaction affords the fungicidal compound Mandipropamid in just two steps. Initially, the Passerini reaction of an in situ provided isocyanide, an aldehyde and a carboxylic acid gave the corresponding α-acyloxycarboxamide. Next, the latter was alkylated by propargyl bromide to afford Micora (Mandipropamid®) (Scheme 1.4) [111–114]
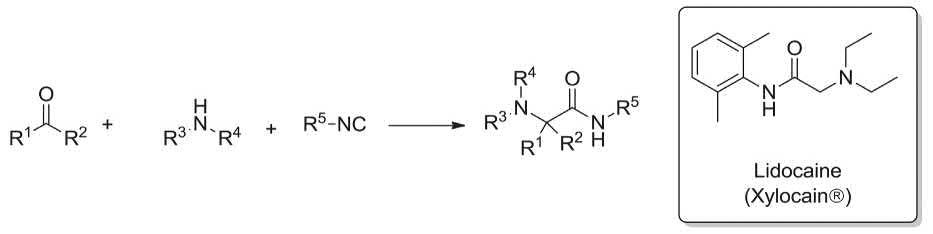
Lidocaine (Xylocain®) is a very common strong local anesthetic. It was synthesized via the Ugi three-component reaction involving, formaldehyde, diethyl amine and 2,6-dimethyl-phenylisocyanide. As a matter of fact this strategy for the synthesis of Lidocaine (Xylocain®) as a prescribed drug, isonitrile-based multicomponent reaction (IMCR) (Scheme 1.5) [115]
SCHEME 1.5 Ugi MCR for the synthesis of Lidocaine (Xylocain®).
SCHEME 1.3 Synthesis of α-amino acids by Strecker MCR.
SCHEME 1.4 Passerini 3CR.
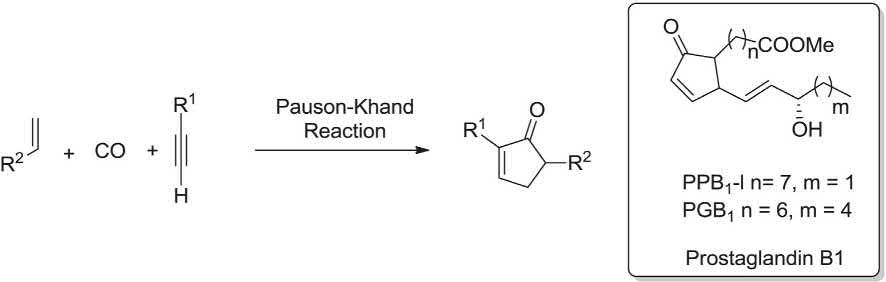
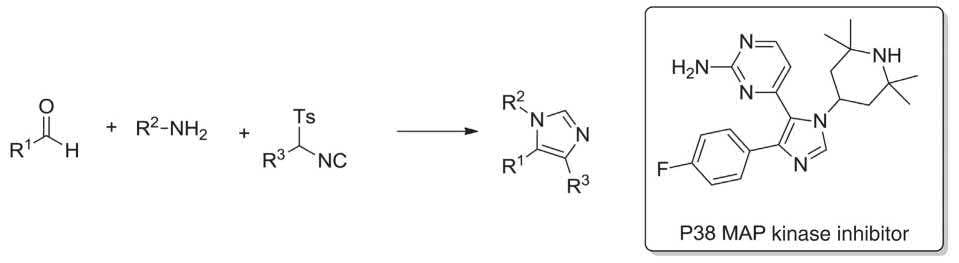
Prostaglandins exhibited antioxidant and ionophoric potencies. The Pauson-Khand MCR was employed as the vital step for the regio- and stereoselective synthesis of prostaglandin B1. This MCR comprised a silyl- protected propargyl acetylene, ethylene and octacarbonyl dicobalt (carbon monoxide source) to give the 3-tert-butyldimethylsilyloxymethyl-2-substtuted-cyclopent-2-en-1-one at ambient temperature in satisfactory yield (Scheme 1.6) [19–21,116] p38 MAP kinase exhibited the inflammatory route, acting as inhibitors of the p38 MAP kinase were extensively screened and found to be biologically active. 1,4,5-Trisubsttuted imidazoles were prepared as p38 MAP kinase inhibitors via the van Leusen MCR involving an α-substituted tosylmethyl isocyanide, a primary amine and aldehdye in a basic medium. Worthy to mention that p38 MAP kinase was prepared on a 500 kg batch scale to afford adequate amount of drug for phase III clinical trials (Scheme 1.7) [117,118].
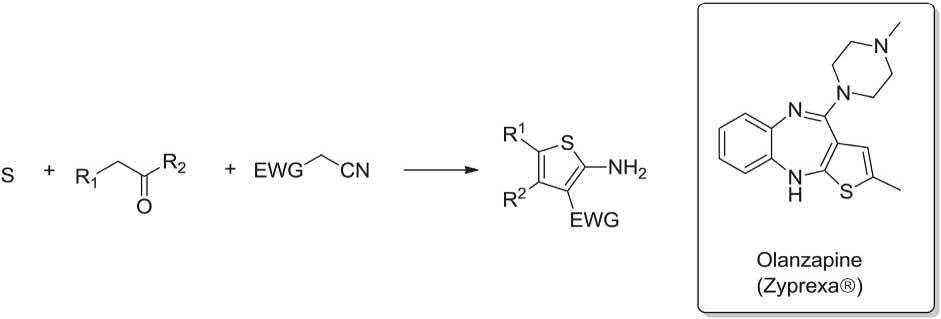
In General, the Gewald MCR provides bioisosteres of anthranilic acids. Noticeably, 2-amino-3-carbonyl thiophene is used as starting material for the synthesis of several drugs for example, Olanzapine (Zyprexa®), an atypical antipsychotc drug. This thiophene-phenol bioisostere can be easily prepared by the Gewald-3CR using cyanoacetamides, α-methylene active aldehydes or ketones, and sulfur (Scheme 1.8) [22,23,119,120]
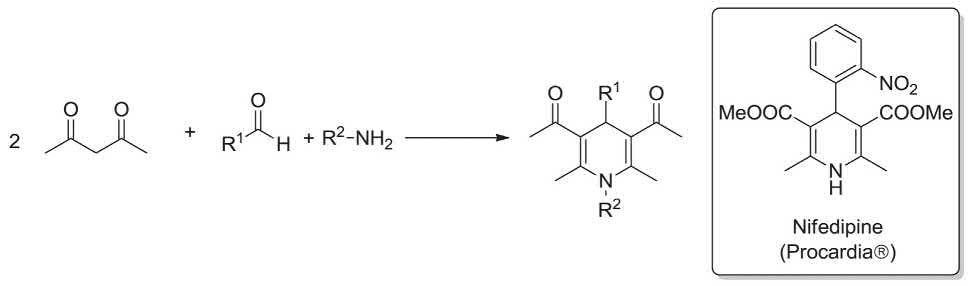
The Hantzsch-MCR was reported by A. R. Hantzsch in 1881. It is the well-known three-component reaction, which affords 1,4-dihydropyridine derivatives using β-ketoesters, aldehydes, and ammonia. Remarkably, it is employed for
SCHEME 1.6 Pauson-khand MCR.
SCHEME 1.7 Van Leusen MCR for the synthesis of p38 MAP kinase.
SCHEME 1.8 Gewald MCR.
SCHEME 1.10 Biginelli MCR leading to anti-tubercular agents.
the synthesis of the calcium channel blocker Nifedipine (Procardia®) (Adalat, Hoffmann La Roasch). Synthesis of this unique prescribed dihydropyridine derivative, involves condensation of 2-nitro benzaldehyde with 2 equivalents of methyl acetoacetate and ammonia as a source of nitrogen (Scheme 1 9) [28,121,122]
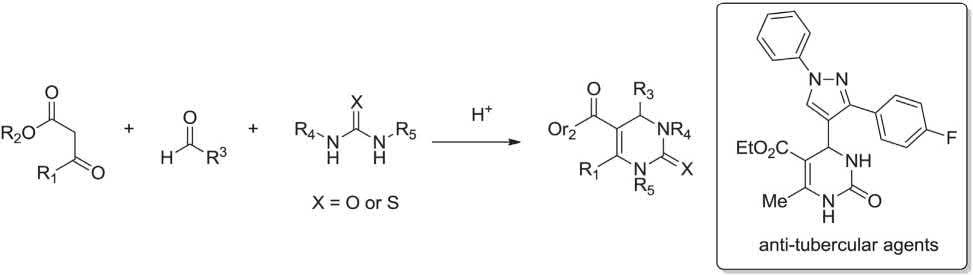
An Italian chemist, P. Biginelli in 1891 achieved and reported a three-component reaction, using β-ketoesters such as ethyl acetoacetate, aromatic aldehydes such as benzaldehyde, and ureas (or thioureas) prompted by acid catalyst (Brönsted or Lewis acids), which gave dihydropyrimidinone derivatives. Dihydropyrimidinones are important compounds because of their various biological activities such as anti-inflammatory or anti-bacterial potencies. Several anti-tubercular agents have been synthesized via Biginelli MCR as below (Scheme 1.10) [27].
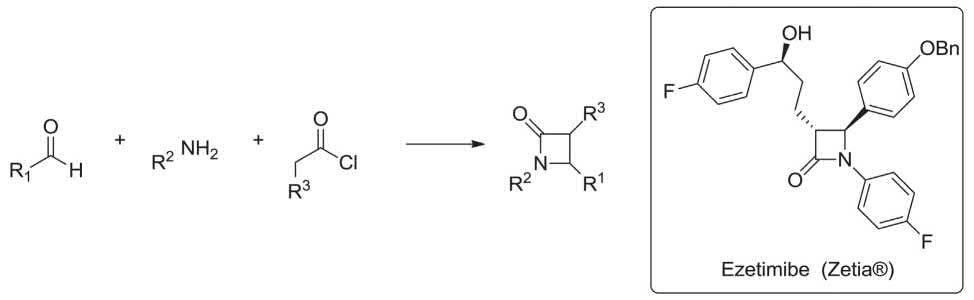
Ezetmibe (Zeta®) is a lipid-lowering compound which selectively inhibits the intestinal absorption of cholesterol. It is prepared via multi-step synthesis by employing the Staudinger-MCR as a vital step. The imine generated from p-fluoroaniline and benzyloxybenzaldehyde was reacted with methyl 5-chloro-5-oxopentanoate mediated by tributylamine in toluene to construct the β-lactam ring. This reaction includes the generation of an intermediate ketene which is subjected to a [2 + 2] cycloaddition reaction with the imine to construct the β-lactam ring, regioselectively, affording the trans isomer as the chief product (Scheme 1.11) [123–125].
SCHEME 1.11 Staudinger-MCR for construction of β-lactam ring.
SCHEME 1.9 Hantzsch MCR leading to synthesis of nifedipine (Procardia®).
1.2 Summary and outlook
Undoubtedly, MCRs as, supreme and beneficial tool combined with the other accessible arsenal in the modern organic synthetic toolbox are employed by synthetic organic chemists, worldwide. Modern diversity-oriented synthesis is somehow interiorly related with MCRs. Considering green chemistry necessities, MCRs verified to have, at least in concept, all the structures required for sustainable synthesis. Regrettably, the widely held of the existing reports just practice these values as mottos. Utilization of ‘catchwords’ such as environmentally benign, eco-friendly, sustainable and green chemistry should be strongly circumvented, except for those reports, providing actual developments for MCRs as green tools. It is vital to begin an era of more difficult and worthwhile investigations on MCRs and not being forced to more apparently accidental catalyst selections. In this case, MCRs actually occupy the projecting position, predicted for this sort of reaction. Indeed MCRs do not have much more to deal on this subject.
Actual developments definitely need profound information on the mechanism of the MCR transformation and generally, fine details of MCR mechanisms are only nowadays beginning to develop. Hypothetical methods have, thus, much to deal, and several actual developments are due to some extraordinary theoretical assistances. Just a few kinetic data are accessible in the MCR can be found in chemical literature; and this is frequently due to the high level of problem related with real-time observing (and quantification) of all potentials of intermediates and reaction trails comprised in multicomponent transformations. NMR, IR, and MS data have verified to be the most efficient techniques smeared so far in explaining MCR mechanisms, with distinctive importance of ESI-MS(/MS).
The Biginelli MCR is feasibly the most prevalent MCR. This imperative conversion, show real, deficiencies to improve its experimental condition. Requirement for reagent excesses, needing high temperatures and large amounts of catalysts to proceed are serious limitations for this MCR. In addition just a few efficient asymmetric variant with serious limitations for the enantioselective syntheses DHPMs is reported. It is quiet not to think how to select a reaction route for the Biginelli reaction and therefore much effort is necessary to attain a profound understanding of the vital required factors for an efficient conversion. However, the iminium-based mechanism is preferred over the other prospects, so far is reported.
The Hantzsch MCR is suffering from similar disadvantages already mentioned for the Biginelli reaction. The option for dissimilar reaction route is adding more complication. Much is still needed to understand how to select a mechanistic pathway for the synthesis of DHPs using the Hantzsch MCR, especially for asymmetric versions applied in the synthesis of bioactive DHPs. Although many developments in diastereoselective MCRs have been reported, the field of catalytic enantioselective MCRs has just started to flourish. Expressively, extended scopes, novel techniques, more eco-friendly strategies and totally new MCRs imitate the progressively creative pathways that synthetic organic chemists trail in this ground. Until recently, enantioselective transition metal-catalyzed MCRs exemplified the main stream of enantioselective MCRs. Nevertheless, metal contamination is highly objectionable for drug synthesis. The development of organocatalysis highly affects the expedition for novel AMCRs. Indeed, possibility for advance and creative applications of AMCR is almost unlimited. A wide-ranging avenue for new discoveries regarding mechanisms and applications of AMCRs is just waiting to be journeyed.
References
[1] P. Anastas, N. Eghbali, Chem. Soc. Rev. 39 (2010) 301
[2] P.A. Wender, S.T. Handy, D.L. Wright, Chem. Ind. 765 (1997) 767
[3] T. Gaich, P.S. Baran, J. Org. Chem. 75 (2014) 657.
[4] I.T. Horvath, P.T. Anastas, Innovations and green chemistry. (2007) 2169
[5] M.O. Simon, C.J. Li, Chem. Soc. Rev. 41 (2012) 1415
[6] T. Kitanosono, K. Masuda, P. Xu, S. Kobayashi, Chem. Rev. 118 (2017) 679
[7] A. Laurent, Ann. Chim. Phys. 66 (1837) 181.
[8] A. Laurent, C.F. Gerhard, Ueber einige Stickstoffverbindungen des Benzoyls, Ann. Pharm. 28 (1838) 265
[9] A. Strecker, Justus Liebigs Ann, Chem. 75 (1850) 27
[10] C. Mannich, W. Krösche, Archiv der Pharmazie. 250 (1912) 647
[11] I. Ugi, Angew Chem. 71 (1959) 386
[12] L. Banafi, R. Riva, Org. React. 65 (2005) 1
[13] G. Keglevich, E. Bálint, Molecules. 17 (2012) 12821
[14] H.T. Bücherer, H.T.J. Fischbeck, Adv. Synth. Catal. 140 (1934) 69
[15] H.T. Bucherer, W. Steiner, J. Prakt, Chem. 140 (1934) 291
[16] H. Bergs, Ger. pat. 566 (1929) 094.
[17] E. Ware, Chem. Rev. 46 (1950) 403
[18] O.H. Oldenziel, D. Van Leusen, A.M. Van Leusen, J. Org. Chem. 42 (1977) 3114
[19] P.L. Pauson, I.U. Khand, N. Y. Ann. Acad. Sci. 295 (1977) 2
[20] J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez, J. Pérez-Castells, Chem. Soc. Rev. 33 (2004) 32 [21] H. Werner, Angew. Chem. Int. Ed. 53 (2014) 3309. [22] K. Gewald, E. Schinke, H. Böttcher, Ber. 99 (1966) 94 [23] R.W. Sabnis, Sulfur Rep. 16 (1994) 1 [24] Y.G. Gololobov, Tetrahedron 37 (1981) 437
[25] Y.G. Gololobov, L.F. Kasukhin, Tetrahedron 48 (1992) 1353
[26] H. Staudinger, J. Meyer, Helv. Chim. Acta 2 (1919) 635. [27] P. Biginelli, Ber. Dtsch. Chem. Ges. 24 (1891) 2962
[28] A. Hantzsch, Chem Ber. 14 (1881) 1637
[29] F. Asinger, Angew. Chem. 68 (1956) 413 [30] A. Dömling, I. Ugi, Angew. Chem. Int. Ed. Engl. 39 (2000) 3168.
[31] V. Nenajdenko (Ed.), Isocyanide Chemistry: Applications in Synthesis and Material Science, Wiley-VCH, Weinheim, Germany, 2012 [32] M. M. Heravi, L. Mohammadkhani. Synthesis of various N-heterocycles using the four-component Ugi reaction, Advances in Heterocyclic Chemistry, 2019, doi.org/10.1016/bs.aihch.2019.04.001.
[33] I. Ugi, A. Dömling, W. Hörl, Endeavour. 18 (1994) 115
[34] H. Bienaymé, C. Hulme, G. Oddon, P. Schmitt, Chem. Eur. J. 6 (2000) 3321
[35] E. Ruijter, R. Scheffelaar, R.V.A. Orru, Angew. Chem. Int. Ed. 50 (2011) 6234
[36] T.E. Nielsen, S.L. Schreiber, Angew. Chem. Int. Ed. 47 (2008) 48–56
[37] Y. Coquerel, T. Boddaert, M. Presset, D. Mailhol, J. Rodriguez in Ideas in chemistry and molecular sciences: advances in synthetic chemistry. B. Pignataro (Ed.) Wiley-VCH Verlag GmbH, Weinheim, Germany, 2010, pp. 187-202.
[38] B.M. Trost, Science 254 (1991) 1471
[39] L.F. Tietze, Chem. Rev. 96 (1996) 115
[40] L. Weber, Curr. Med. Chem. 9 (2002) 2085
[41] C. Hulme, V. Gore, Curr. Med. Chem. 10 (2003) 51. [42] A. Dömling, Chem. Rev. 106 (2006) 17 [43] I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol. 12 (2008) 324 [44] A. Dömling, W. Wang, K. Wang, Chem. Rev. 112 (2012) 3083 [45] J. Zhu, H. Bienaymé (Eds.), Multicomponent Reactions, Wiley-VCH, Weinheim, Germany, 2005 [46] P. Slobbe, E. Ruijter, R.V.A. Orru, Med. Chem. Comm. 3 (2013) 1189 [47] J.M. Knapp, M.J. Kurth, J.T. Shaw, A. Younai, Strategic applications of multicomponent reactions in diversity-oriented synthesis, in: A. Trabocchi (Ed.), Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology, John Wiley & Sons, Inc, Hoboken, N. J, 2013, pp. 29–57
[48] J.E. Biggs-Houck, A. Younai, J.T. Shaw, Curr Opin Chem Biol. 14 (2010) 371.
[49] J.D. Sunderhaus, S.F. Martin, Chemistry. 15 (2009) 1300
[50] A. Ulaczyk-Lesanko, D.G. Hall, Curr. Opin. Chem. Biol. 9 (2005) 266 [51] L. Claisen, Ber. Dtsch. Chem. Ges. 45 (1912) 3157
[52] V.R. Annamalai, E.C. Linton, M.C. Kozlowski, Org. Lett. 11 (2009) 621.
[53] X. Li, S.J. Danishefsky, J. Am. Chem. Soc. 130 (2008) 5446
[54] D.E. Fogg, E.N. dos Santos, Coord. Chem. Rev. 248 (2004) 2365
[55] I. Ugi, J. Prakt, Chem. 339 (1997) 499 [56] L. Weber, K. Illgen, M. Almstetter, Synlett (1999) 366 [57] L. Weber, S. Wallbaum, C. Broger, K. Gubernator, Angew. Chem. Int. Ed. Engl. 34 (1995) 2280. [58] P.T. Anastas, J.C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, (1998) [59] R. Breslow, Acc. Chem. Res. 24 (1991) 159 [60] S. Otto, J.B.F.N. Engberts, Org. Biomol. Chem. 1 (2003) 28090 [61] J. Chandrasekhar, S. Shariffskul, W.L. Jorgensen, J. Phys. Chem. B 106 (2002) 8078. [62] J. Lubineau, Y. Augé, Queneau, Synthesis (1994) 742 [63] T. Eriksson, S. Björkman, B. Roth, A. Fyge, P. Höglund, Chirality. 7 (1995) 44 [64] A.C. Pinto, J.B.D. Andrade, J. Braz. Chem. Soc. 23 (2012) 2131 [65] X. Sun, S. Vilar, N.P. Tatonetti, Sci. Transl. Med. 5 (2013) 205rv1 [66] C. de Graaff, E. Ruijter, R.V. Orru, Chem. Soc. Revi. 41 (2012) 3969 [67] W. Chen, W. Mbafor, S.M. Roberts, J. Whitall, J. Am. Chem. Soc. 128 (2006) 3922 [68] W. Chen, S.M. Roberts, J. Whitall, A. Steiner, Chem. Commun. 27 (2006) 2916 [69] D. Marquarding, H. Klusacek, G. Gokel, P. Hofmann, I. Ugi, J. Am. Chem. Soc. 18 (1970) 5389 [70] L.F. Batelle, R. Bau, G.W. Gokel, R.T. Oyakawa, I. Ugi, Angew. Chem. Int. Ed. 11 (1972) 138. [71] L.F. Batelle, R. Bau, G.W. Gokel, R.T. Oyakawa, I.K. Ugi, J. Am. Chem. Soc. 95 (1973) 482
[72] G. Guillena, D.J. Ramon, M. Yus, Tetrahedron: Asymmetry 18 (2007) 693
[73] D. Bonne, Y. Coquerel, T. Constantieux, J. Rodriquez, Tetrahedron: Asymmetry 21 (2010) 1085
[74] D.J. Ramón, M. Yus, Angew. Chem. Int. Ed. 44 (2005) 1602.
[75] H.B. Kagan, O. Riant, Chem. Rev. 92 (1992) 1007
[76] T. Oh, M. Reilly, Org. Prep. Proc. Int. 26 (1994) 129
[77] M. Kumar, P. Chauhan, S.J. Bailey, E. Jafari, C. von Essen, K. Rissanen, D. Enders, Org. Lett. 20 (2018) 1232
[78] D.J. Cram, F.A.A. Elhafez, J. Am. Chem. Soc. 74 (1952) 5828
[79] T.P. Yoon, Beilstein J Org Chem. 13 (2017) 63
[80] H. Cao, H. Liu, A. Dömling, Chem. Eur. J. 16 (2010) 12296
[81] R.C. Cioc, E. Ruijter, R.V. Orru, Green Chem. 16 (2014) 2958
Introduction
[82] G. Jenner, Tetrahedron Lett. 43 (2002) 1235
[83] T. Yamada, T. Yanagi, Y. Omote, T. Miyazawa, S. Kuwata, M. Sugiura, K.J. Matsumoto, Chem. Soc. Chem. Commun. (1990) 1640.
[84] H.G. Alvim, E.N. da Silva Junior, B. A. Neto, Rsc Adv. 4 (2014) 54282
[85] G.J. Cheng, X.M. Zhong, Y.D. Wu, X. Zhang, Chem. Commun. 55 (2019) (2019) 12749
[86] C. Iacobucci, S. Reale, J.F. Gal, F. De Angelis, Eur. J. Org. Chem. 2014 (2014) 7087
[87] T. Lazar, Synthesis 2006 (2006) 1390
[88] A. Hassner, C. Stumer, Organic Syntheses based on Name Reactions and Unnamed Reactions, vol. 11, Elsevier, Pergamon, New York, (2013)
ISBN: 9781483287348
[89] J.W. Suggs. Organic Chemistry. Barron’s, 2002, p. 109. ISBN 0-7641-1925-7.
[90] R. Arman, Organic Name Reactions. A contribution to the terminology of organic chemistry, biochemistry, and theoretical organic chemistry. Helmut Krauch and Werner Kunz. Translated from the second revised German edition by John M. Harkin. Wiley, New York, 1964.
[91] M.B. Smith, J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 6th ed., Wiley-Interscience, New York, (2007)
ISBN 978-0-471-72091-1
[92] F.A. Carey, Organic Chemistry, 6th ed., McGraw-Hill, New York, NY, (2006) ISBN 0-07-111562-5
[93] C. Friedel, J.M. Crafts, Compt. Rend. 84 (1877) 1392
[94] S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran, S. Bajohr, Fuel. 166 (2016) 276
[95] K. Reimer, F. Tiemann, Ber. Dtsch. Chem. Ges. 9 (1876) 1268
[96] O. De Lucchi, U. Miotti, G. Modena, (1991). The Pummerer Reaction of Sulfinyl Compounds. Organic Reactions. Vol. 40. pp. 157-184. ISBN 978-0471264187.
[97] A. Padwa Jr., D.E. Gunn, M.H. Osterhout, Synthesis. 1997 (1997) 1353.
[98] B.O. Lindgren, T. Nilsson, S. Husebye, Ø. Mikalsen, K. Leander, C.-G. Swahn, Acta Chem. Scand. 27 (1973) 888
[99] P.W. Rabideau, Z. Marcinow, Org. React. 42 (1992) 1
[100] L.N. Mander, Compr. Org. Synth. 8 (1991) 489
[101] R. Huisgen, Angew. Chem. 75 (1963) 604.
[102] F. A. Carey, R. J. Sundberg, (1993). Advanced Organic Chemistry Part B Reactions and Synthesis (3rd ed.). 233 Spring Street, NY: Plenum. p. 55. ISBN 0-306-43440-7.
[103] H. C. Kolb, M. G. Finn, K. B. Angew. Chem. Int. Ed. 40 (2001) 2004.
[104] R.A. Evans, Aust. J. Chem. 60 (2007) 384–395
[105] W.-J. Yoo, L. Zhao, C.-J. Li, Aldrichimica Acta 44 (2011) 43
[106] A. Hassner, C. Stumer, Organic Syntheses based on Name Reactions, vol. 22, Elsevier, Pergamon, New York, (2002) ISBN 0-08r-r043260-3
[107] J. Jack, A. Li, Name Reactions: A Collection of Detailed Reaction Mechanisms, Springer-Verlag, Berlin Heidelberg, (2003) ISBN 3-540-40203-9
[108] B.P. Mundy, M.G. Ellerd, F.G. Favaloro, Name reactions and reagents in organic synthesis, Wiley, Hoboken, NJ, (2005) ISBN 0-471-22854-0
[109] M. Passerini, Gazz. Chim. Ital. 51 (1921) 181.
[110] A.Y. Shaw, F. Medda, C. Hulme, Tetrahedron Lett. 53 (2012) 1313 [111] M. Passerini, L. Simone, Gazz. Chim. Ital. 51 (1921) 126 [112] M. Passerini, G. Ragni, Gazz. Chim. Ital. 61 (1931) 964
[113] L. Banf, R. Riva, Org. React. 65 (2005) 1. [114] C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H.-J. Kempf, R. Zeun, Bioorg. Med. Chem. 16 (2008) 1531 [115] I. Ugi, C. Steinbrückner. ”DE-B 1 1959, 103, 337.(b) (c) I. Ugi, R. Meyr, U. Fetzer, Angew. Chem., Int. Ed 71 (1959) 386. [116] A. Vazquez-Romero, L. Cardenas, E. Blasi, X. Verdaguer, A. Riera, Org. Lett. 11 (2009) 3104 [117] A.M. Van Leusen, J. Wildeman, O.H. Oldenziel, J. Org. Chem. 42 (1977) 1153 [118] J. Sisko, J. Org. Chem. 63 (1998) 4529. [119] J. K. Chakrabart, T. M. Hoten, D. E. Tupper, 1991, EP 454436. [120] K. Wang, D. Kim, A. Dömling, J. Comb. Chem. 12 (2010) 111 [121] F. Bossert, W. Vater, Naturwissenschafen 58 (1971) 578 [122] F. Bossert, H. Meyer, E. Wehinger, Angew. Chem. Int. Ed. 20 (1981) 762. [123] H. Staudinger, Justus Liebigs Ann, Chem. 356 (1907) 51 [124] S.B. Rosenblum, T. Huynh, A. Afonso, H.R. Davis, N. Yumibe, J.W. Clader, D.A. Burnet, J. Med. Chem. 41 (1998) 973 [125] C. Palomo, J.M. Aizpurua, I. Ganboa, M. Oiarbide, Eur. J. Org. Chem. 12 (1999) 3223
2 Direct synthesis of heterocycles via MCRs, using a name reaction
2.1 Biginelli reaction
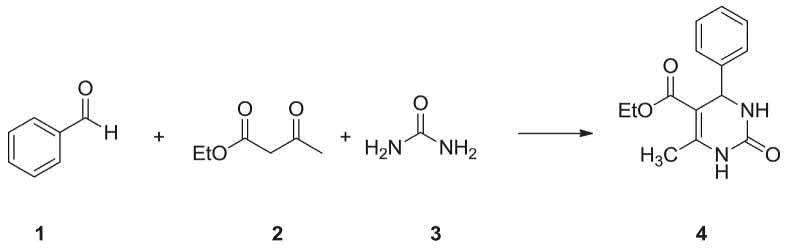
The Biginelli reaction is an MCR involving, ethyl acetoacetate 2, an aryl aldehyde (i.e., benzaldehyde 1, and urea 3 giving the corresponding 3,4-dihydropyrimidin-2(1H)-ones 4) (Scheme 2.1) [1–4]. This reaction is named after its explorer, the Italian chemist Pietro Biginelli [5,6], who introduced it 1891. The Biginelli reaction can be catalyzed by Brønsted acids and/or by Lewis acids such as copper(II) trifluoroacetate hydrate [7], boron trifluoride [8], and several solid-phase catalysts using different linker combinations [9,10]. The products of the Biginelli reaction, such as dihydropyrimidinones, are extensively utilized as prescribed drug as calcium channel blockers antihypertensive, and alpha-1-a-antagonists [11]
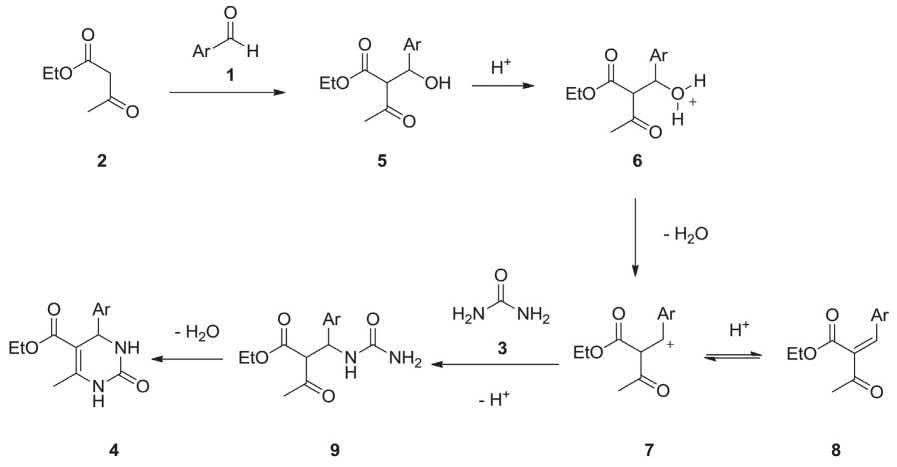
The reaction mechanism of the Biginelli reaction is a series of bimolecular reactions leading to the desired dihydropyrimidinone [12]. A plausible mechanism for this MCR was suggested by Sweet in 1973 as illustrated in Scheme 2.2. Initially, aldol condensation of ethylacetoacetate 2 and the aryl aldehyde takes place resulting in the generation of the carbenium ion 7 that is the rate-determining step. Then, the nucleophilic addition of urea to the carbenium ion 7 generates the intermediate 9, which rapidly subjected to dehydration to afford the desired product 4 [13].
The MCR Biginelli reaction was achieved using different catalysts such as glutamic acid [14], [Hmim][Tfa] [15], in aqueous NaOH using Aliquat-336 as a phase transfer agent [16], citric acid [17], β-cyclodextrin [18], acidic ionic liquid [19], H5PW10V2O40/Pip-SBA-15 [20], cellulose sulfuric acid [21], sulfonated-phenylacetic acid treated with Fe3O4 nanoparticles [22], sulfonated-mercaptopropanoic acid embedded Fe3O4 nanoparticles as a magnetic acid [23], acidic choline-based ionic liquids [24], PEG1000-DAIL/toluene [25], vanadatesulfuric acid [26], bis[(L)prolinato-N,O]Znwater [27], Brønsted acidic ionic liquids [28], Fe-Al/clay under solvent-free conditions [29], sulfonic acid-functionalized polypropylene fiber as Brønsted acid [30], pyridine dicarboxylic acid guanidine–cobalt complex (PDAG-Co) [31], iron(III)tosylate [32], silica gel-immobilized onto l-pyrrolidine-2-carboxylic acid-4-hydrogen sulfate [33], iodine in solvent-less system under MWI [34], imidazolium-tagged recyclable iron [35], bovine serum albumin (BSA) [36], nanosilica-supported tin(II) chloride [37], solvent-free and catalyst-free [38], poly(4-vinylpyridine)-supported CuI NPs [39], Zn (1 x) NixAl2O4 spinels [40], nano-ZnO and ZnO [41], Brønsted and Lewis acid catalysts with ionic tags under homogeneous and heterogeneous conditions [42], Psychotria douarrei and Geissois pruinosa known as a hypernickelophore plants [43], Fe3O4-MWCNT nanocomposite under ultrasound irradiation [44], poly(ethylene)glycol/ AlCl3 [45], silica-supported ionic liquid Si-[SbSipim][PF6] [46], NaHSO4·H2O under ultrasound system under reflux [47], Two eco-friendly organosulfonic acid-functionalized silica-coated magnetic nanoparticle (Fe3O4@SiO2@Et-PhSO3H) and (Fe3O4@SiO2@Me&Et-PhSO3H) [48], cupric acetate under solvent-less system [49], carboxylic acid functionalized mesoporous organic polymer [50], graphite under solvent free conditions [51], carbon nanotubes immobilized onto titanium dioxide nanoparticles [52], nanomagnetic-immobilized onto sulfonic acid [53], p-dodecylbenzene sulfonic acid [54], lanthanum oxide under solvent-less system [55], silica-bonded N-propyl sulfamic acid [56], cerium(III) trislaurylsulfonate (Ce(LS)3), as a Lewis acid combined with surfactant [57], alumina supported Mo catalysts (Mo/γ-Al2O3) [58], N-sulfonic acid poly(4-vinylpyridinium) chloride [59], magnetic nanoparticles immobilized imidazolium-based ionic liquids [60], CuS quantum dots as heterogeneous catalyst [61], boehmite nanoparticle catalyst [62], Fe3O4 nanoparticle immobilized onto Ni(II) complexes [63], phytic acid as a biogenic organocatalyst [64], Brønsted acidic ionic liquids of aza-crown ether complex cations [65], ionic liquid and sulfonic acid based bifunctional periodic mesoporous organosilica (BPMO-IL-SO3H) [66], titanium dioxide immobilized onto MWCNTs under
Recent Advances in Applications of Name Reactions in Multicomponent Reactions. http://dx.doi.org/10.1016/B978-0-12-818584-1.00002-1
SCHEME 2.1 The Biginelli reaction.
SCHEME 2.2 Suggested probable mechanism for Biginelli reaction.
MWI [67], nafion-Ga, as green Lewis acid [68], polyacrylonitrile fiber-supported Brønsted acid [69], ethyl lactate (EL) with trimethylsilyl chloride (TMSCl) [70], ZnO nanoparticles as a solid acid catalyst [71], solid silica-based sulfonic acid under MWI [72], magnetically reusable niobium nanocatalyst [73], erbium trichloride hexahydrate under solvent-free conditions[74], SiO2-CuCl2 [75], Mg-containing periodic mesoporous organosilica with ionic-liquid framework (Mn@PMO-IL) [76], molybdenum oxide NPs under MWI [77], ytterbium-(III) triflate hydrate in acetic acid and water as solvent [78], indium(III) trifluoromethane sulfonate under solvent-less system [79], Li(glycine)(CF3SO3) in solvent-less system [80], l-tyrosine [81], phosphonic acid functionalized well-ordered mesoporous material [82], tungstate sulfuric acid under solvent free conditions [83], Brønsted acidic ionic liquid-based magnetic nanoparticles [84], nanosilica chloride [85], a perchloric acid-modified PEG-6000 (PEG-HClO4) under solvent-free protocol [86], solid acids involving 12-tungstophosphoric acid and 12-tungstosilicic acid attached on to metal oxide (ZrO2), zeolites (Hβ and HZSM-5), and mesoporous substance (MCM-41) [87], ionic liquid-based well-ordered mesoporous organosilica-supported copper [88], preyssler heteropolyacid immobilized onto silica coated NiFe2O4 noparticles [89], nanocobalt manganese oxide [90], heteropolyanion-based ionic liquids in solvent-less system under MWI [91], ZnO nanoparticles [92], di-DACH-pyridylamide ligands, symmetrical bridged bis-Schiff base, and spiro pyrrolizines [93], B(C6F5)3 [94], three Brønsted acid-based ionic liquids, so-called, 1-ethyl-1,2,4-triazolium triflate, 1-propyl-1,2,4-triazolium triflate and 1-butyl-1,2,4-triazolium triflate [95], phthalimide-N-sulfonic acid [96], MCM-41 supported perchloric acid [97], supported phosphomolybdic acid nanoparticles on imidazole functionalized Fe3O4@SiO2:(Fe3O4@SiO2imid-H3PMo12O40) [98], Brønsted acidic ionic liquid [Btto][p-TSA] in solvent-less system [99], Ti-grafted polyamidoamine dendritic silica hybrid [100], Zn(ClO4)2 [101], bismuth vanadate nanocatalyst [102], copper complex (PhNH3)2CuCl4 [103], Two coordination polymers (CPs) (both CPs and the Zn-based material) [104], three metal coordination polymers, so-called, [Co(DPP)2(H2O)2]·(BS)2·2H2O, [Co(DPP)2(H2O)2]·(ABS)2·2H2O and [Co(DPP)2(MBS)2],
2.1 Biginelli reaction 17
[DPP = 1,3-di (pyridin-4-yl) propane, BS = phenyl sulfonic acid, ABS = p-aminobenzene sulfonic acid, MBS = pmethylbenzene sulfonic acid] in solvent-less system [105], perlite as natural support for immobilization of sulfonic acid as a heterogeneous solid acid [106], natural organic acids under solvent-free conditions [107], phenylboronic acid in acetonitrile [108], zeolite ZSM-5 [109], β-cyclodextrin-propyl sulfonic acid [110], free or MCM-41 immobilized onto ZnNO3 [111], ZnO nanoparticles [112], nickel oxide immobilized onto multi-walled carbon nanotubes [113], heteropoly anion-based acidic ionic liquids, [TMAPS]H2PMo12O40, [TEA PS]H2PMo12O40, and [TBAPS] H2PMo12O40 [114], nano-Fe3O4@silica sulfuric acid [115], N-propylcarbamothioyl benzamide complex of Bi(III)immobilized onto superparamagnetic Fe3O4/SiO2 nanoparticles [116], aurivillius nanostructures of Bi2ZnAl2O9 [117], halogenated macroporous sulfonic resins [118], lanthanum oxide under MWI [119], bismuth nitrate in CH3CN or PPh3 under solventfree conditions [120], calix[8]arene sulfonic acid under ultrasonic irradiation [121], graphene-supported NiBr2 [122], zirconia sulfuric acid in solvent-less system [123], PEG-SANM nanocomposite (as the solid acid nanocatalyst in solvent-less system) [124], Cu(NO3)23H2O [125], polymer-supported benzimidazolium-based ionic liquid [126], nanoZrO2 sulfuric acid: a heterogeneous solid acid [127], benzotriazolium-based ionic liquids in solvent-less system [128], nanocrystalline mullite synthesized from monophasic precursor gel [129], n-glass-waste-supported sulfonic acid (n-GW-SA) [glass waste materials] [130], magnetically BiFeO3 nanowire-reduced graphene oxide [131], Cu–EDTAmodified APTMS-Fe3O4@SiO2 core–shell nanocatalyst [132], molybdenum(VI) dichloride dioxide (MoO2Cl2) [133], utilizing ball milling technique under solvent-free and catalyst-free [134], partially fluorinated, angular tetracarboxylic acid linker (H4L) incorporating a pendant amine moiety in form of a three-dimensional Zn(II) framework [135], 1,4-diazabicyclo[2.2.2]octaniumdiacetate [136], quaternary ammonium ionic liquid as a dual solvent-catalyst [137], triphenylphosphine as Lewis base under solvent-free conditions [138], sulfated polyborate as a Brønsted as well as Lewis acid [139], l-proline N-sulfonic acid-functionalized magnetic nanoparticles [140], Keggin heteropoly acid (H4SiMo12O40) [141], 1-methyl-3-nitro-1H-imidazol-3-ium trinitromethanide {[MIM-NO2]C(NO2)3} as a green nano structure ionic liquid (NIL) [142], un-catalyzed and under solvent-free conditions [143], nano-isopolyoxomolybdate: [in the presence of a keplerate type giant nanoporous isopolyoxomolybdate, (NH4)42[MoV7I2MoV60O372(CH3COO)30(H2O)72] [144], a heterogenized hybrid substance of NaY zeolite immobilized onto 1-sulfonic acid-3-methyl imidazolium ferric chloride [Msim][FeCl] [145], Keggin and Dawson-type heteropoly acids [146], [bmim(SO3H)][OTf]/[bmim][X] and Zn(NTf2)2/[bmim][X] (X = PF6 and BF4) [147], amine-functionalized titania as an inorganic–organic hybrid heterogeneous basic nanocatalyst [148], aliovalent ion substituted fluorapatite with formula Bi0.5Na0.5Ca4(PO4)3F [149], sulfamic acid supported magnetic Fe3O4 nanoparticles [150], Co@imine-Na+-MMT (a heterogeneous catalyst based on Cofunctionalized Na+-montmorillonite) [151], polyaniline supported FeCl3 [152], polyethylene immobilized onto Fe/ ionic liquid complex (PEt@Fe/IL) [153], 3-[(3-(trimethoxysilyl)propyl)thio]propane-1-oxy-sulfonic acid [154], triethylammonium acetate ionic liquid ([Et3-NH] [CH3COO ] [155], nickel chloride hexahydrate [156], 3-sulfonic acid-1-imidazolopyridinium hydrogen sulfate in solvent-less system [157], apatite like oxyphosphate {an oxyphosphate with formula of BiCa4 (PO4)3O}, [158], Punica granatum peel as an organocatalyst in solvent-less system [159], catalyst-free and solvent-free conditions [160], metallophthalocyanines (MPcs) [161], Au nanoparticle [162], 1,3,5-tris(2-hydroxyethyl)isocyanurate functionalized graphene oxide [163], Hierarchical zeolite using a ball mill technique [164], Fe+3montmorillonite K10 under grinding condition [165], Fe3O4@SiO2-APTMS-Fe(OH)2 [166], calcium, barium, and strontium apatites [167], nanoparticles of organosilane-based NaHSO4 ionic liquid supported on silica [168], an amine functionalized metal-organic framework (anionic Zn(II)-framework, {[(CH3)2NH2+]2[Zn3((µ3-O))(L)2(H2O)]·4DMF·2H2 O}n, having exposed metal sites and pendant amine groups) [169], ferric citrate [170], in acetic acid under reflux condition [171], propylene carbonate as solvent and molecular iodine as the catalyst [172], graphene oxide [173], cobalt immobilized on alumina [174], 3D printed α-Al2O3 woodpile [175], magnetic nanoparticles immobilized onto Schiffbase/copper complex [176], acid activated mesoporous montmorillonite clay (AT-Mont.) [177], l-proline nitrate [178], salt (NMSMSA) as two nanostructures [179], (C5H6N4O)(C5H5N4O)3(C5H4N4O)[Bi2Cl11]Cl2 [180], 1-hexadecyl-1H-imidazol-3-iumoxalte as highly effective Brønsted acidic type ([C16Im][Oxa] as an elastic organocatalyst) [181], copper oxide NPs [182], copper-doped mesoporous silica immobilized onto dual acidic ionic liquid [183], nano Fe3O4@meglumine sulfonic acid [184], dendrimer-PWAn as a nanocatalyst under ultrasonic irradiation (Ultrasonic [185], ZnO nanoparticles surrounded in SBA-15 [186], Cu(II) supported onto mesoporous organosilica [187], bis(p-sulfoanilino) triazine-functionalized silica-supported onto magnetite nanoparticles [188], Fe3O4-halloysite-SO3H [189], [P4-VP]Fe3O4MNP [cross-linked poly (4-vinyl-pyridine) immobilized onto Fe3O4 nanoparticle] immobilized onto Brønsted acid ionic liquid [190], white marble [191], NHC copper(I) complexes [192], mesoporous graphitic carbon nitride (mpg-C3N4) [193], heteropoly acid supported on activated natural clay in solventless system [194], N-alkylated sulfamic acids [195], Ti(IV) species [(MeO)2Ti(NHCONH2)]+ [196], transition metal-doped heteropoly acid (PMoV, FePMoV, and CuPMoV) [197], sulfonated highly ordered mesoporous graphitic carbon nitride as solid acid [198], and urease immobilization on magnetic micro/nano-cellulose dialdehydes [199]
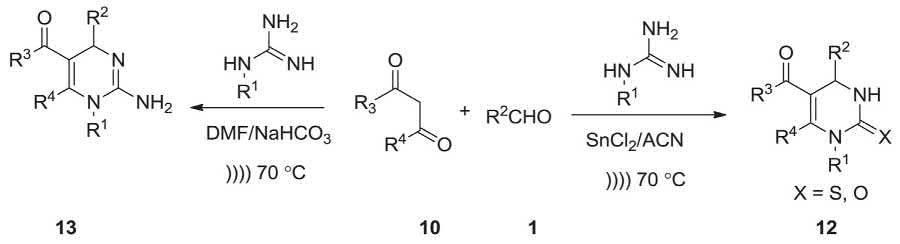

Monastrol has been recognized as the initial hit in SBVS experiment, and thus a series of the corresponding DHPM derivatives were prepared via Biginelli MCR [200] for examination of SAR around all the six diverse points of DHPM backbone. Nowadays, these nonplanar heterocycles have attracted much attention of synthetic organic chemists as well as the pharmacist and pharmaceutical industry due to their interesting multifaceted pharmacological profiles. Biginelli MCR involving, various aldehydes, dicarbonyle compound and guanidine in the presence of SnCl2 in acetonitrile or in the presence of NaHCO3 in DMF under ulterasonic irradiation afforded different elements of diversity at C-2, C-4, C-5, and C-6 as illustrated in Scheme 2.3 [201]
Chlorotrimethylsilane-catalyzed Biginelli MCR, involving benzaldehyde, acetoacetic acid derivatives, and different carboxyl-containing ureas were studied. It was concluded that the steric hindrance of the urea substituents highly affected the reaction consequence. Notably, this methodology was particularly effective only in the case of unbranched mono-substituted ureas bearing either aliphatic or aromatic groups. This strategy permits conducting a one-pot, protecting group free synthesis of dihydropyrimidines possessing carboxylic moiety. Practically, the reactions of benzaldehyde with ureas 14 and different acetoacetic acids 10 were conducted by maintaining the reagents in the presence of Me3SiCl-DMF system at room temperature for 3–4 days (Scheme 2.4). Steric loading in the urea 14 seemed being a principal factor that define the reaction outcome. Particularly, reactions of benzaldehyde, acetoacetic acid derivatives 10, and ureas 14 led to construction of the desired target, dihydropyrimidines 15 in satisfactory yields [202]
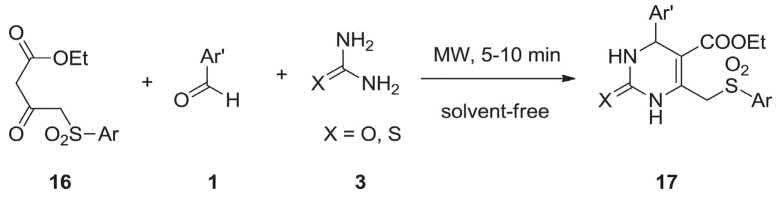
A collection of ethyl 2-oxo/thio-4-aryl-6-(arylsulfonylmethyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylates 17 were synthesized via the Biginelli MCR including ethyl 3-oxo-4-(arylsulfonyl)butanoate 16, differently substituted benzaldehydes 1 and diamide (urea/thiourea) 3 under catalyst- and solvent-free conditions and using MWI as a source of energy (Scheme 2.5) [203].
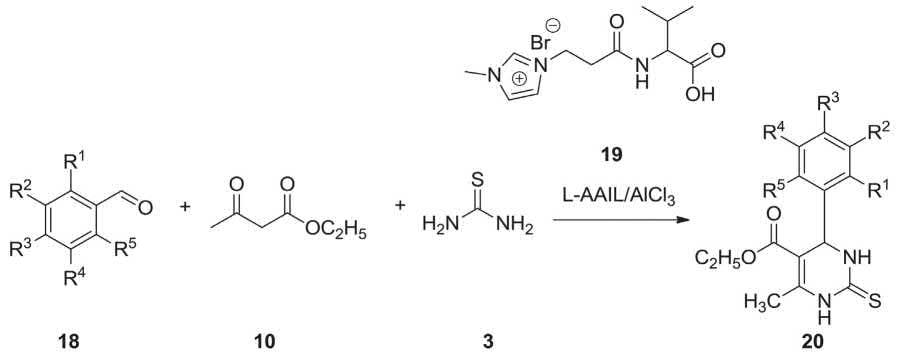
An eco-friendly, inexpensive commercially available and recyclable l-amino acid functionalized ionic liquid [LAAIL]/AlCl3 was found being an efficient catalyst for the high yielding synthesis of 3,4-dihydropyrimidine-2-(1H)thiones via Biginelli MCR in solvent-less system. Comparing with the classical Biginelli reactions, this strategy reliably enjoys the merits of mild reaction conditions, easy work-up procedure, and being completed in relatively shorter reaction time. In addition, due to heterogeneous nature of the catalyst it could be separated easily from the reaction mixture, and being reused several times without appreciable loss in its catalytic activity. Under already secured optimal reaction conditions, a collection of 3,4-dihydropyrimidine-2(1H)-thiones were synthesized in the presence of L-AAIL/AlCl3. Differently substituted benzaldehydes, ethyl acetoacetate and thiourea were reacted in the presence of [L-AAIL]/AlCl3 under solvent-free conditions to give the corresponding 3,4-dihydropyrimidine-2(1H)-thiones in excellent yields (Scheme 2.6) [204].
SCHEME 2.3 Synthesis of products 12.
SCHEME 2.4 Synthesis of dihydropyrimidines 15 via Biginelli MCR.
SCHEME 2.5 Preparation of ethyl 2-oxo/thio-4-aryl-6-(arylsulfonylmethyl)-1,2,3,4-tert-rahydropyrimidine-5-carboxylates 17 Biginelli MCR.
SCHEME 2.6 Synthesis of 3,4-dihydropyrimidin-2(1H)-thiones 20 using L-AAIL/ALCl3 via Biginelli MCR.
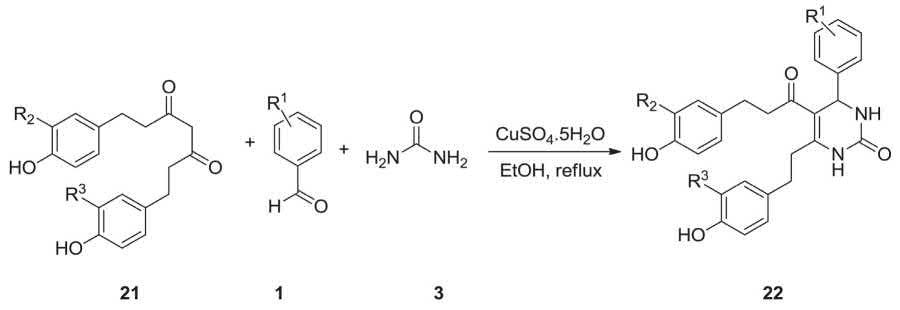
In 2013, Ajabakom and coworkers accomplished and reported the successful synthesis of racemic tetrahydrocurcumi n-(THC-), tetrahydro demethoxycurcumin-(THDC-) and tetrahydrobis demethoxycurcumin-(THBDC-) dihydropyrimidinone (DHPM) analogues via Biginelli MCR. The multicomponent Biginelli reaction was achieved in the presence of copper sulphate as a catalyst. The assessment of acetylcholinesterase inhibitors for Alzheimer’s disease of the above-mentioned compounds exhibited their higher inhibitory activity relative to their parent analogues [205] In this work, after screening several Lewis acids as catalysts for the Biginelli reaction of THC, CuSO4·5H2O gave acceptably promising results (Scheme 2.7). The divalent copper metal may have the coordinating property with the 1,3-dicarbonyl group and stabilizes the enol form generated in situ which prompts to react with other two components to provide the DHPM product.
Biginelli MCR involving, the THC readily accessible by the catalytic hydrogenation reaction curcumin urea and differently substituted benzaldehydes, in the presence of CuSO4·5H2O in EtOH under reflux afforded the respective THC–DHPM derivatives 22 in satisfactory yields. It is worthwhile to mention that all cyclized products were isolated as racemic mixtures [205]
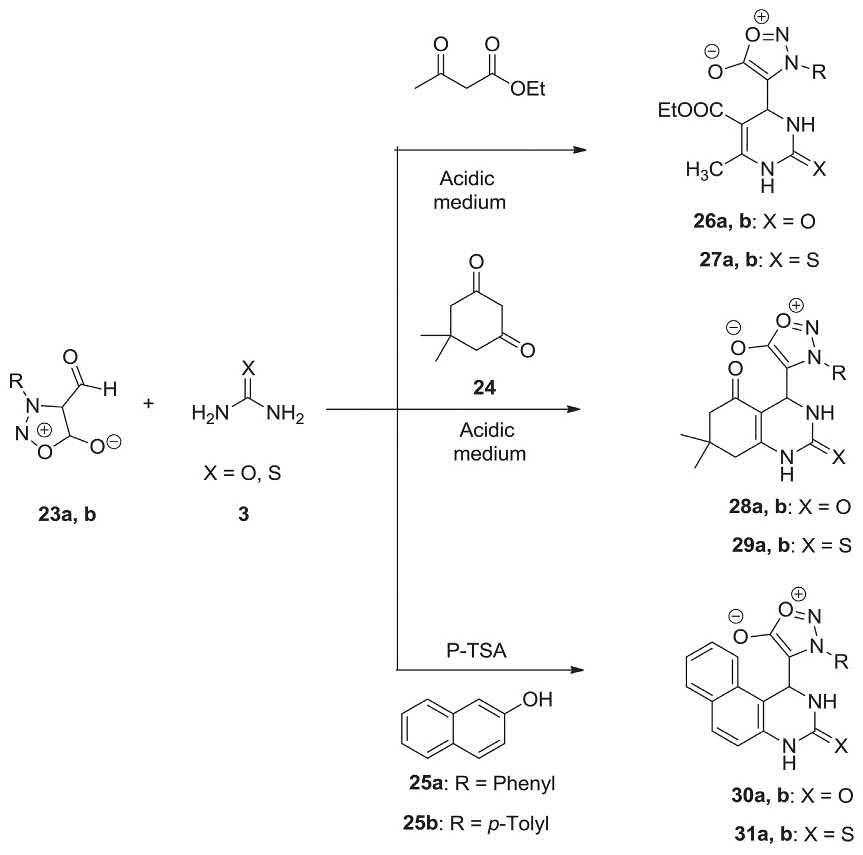
Several aminazolene-2,5-diones 28a, b, 29a, and b were prepared in high yields via Biginelli MCR, involving condensation of compound 23a and 23b, dimedone and urea/thiourea in an acidic medium. The synthesis of naphthalene derivatives 30a, b, 31a, and 23b in satisfactory yields was also achieved by Biginelli MCR of compound 23a and 23b with β-naphthol and urea/thiourea catalyzed by PTSA. Biginelli compounds 26-27a and b and Biginelli-like compounds 28-31a and b were also prepared similarly by using 3-aryl-4-formylsydnone 23a and 23b as a component in Biginelli MCR [206]. 3-Aryl-4-formylsydnone 23a and 23b was provided by formylation of 3-arylsydnone utilizing N-methylformanilide in POCl3 [207]. This reported work involves three sets of reactions for efficient synthesis of dihydropyrimidines 26a, b, 27a, and b via Biginelli MCR (Scheme 2.8).
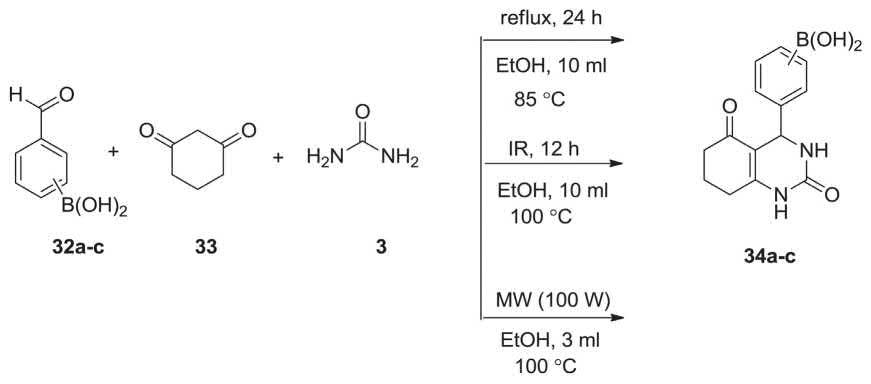
A unique hybrid-boron-containing molecules, were used in Biginelli MCR involving formylphenylboronic acids (ortho, meta and para), 32a-c, dimedone 33 and urea 3 under three different conditions (a. refluxing in EtOH, b. under IR irradiation, c. under MWI) were reacted via MCR to afford the corresponding 3,4-dihydropyrimidinones, in poor yields (Scheme 2.9) [208].
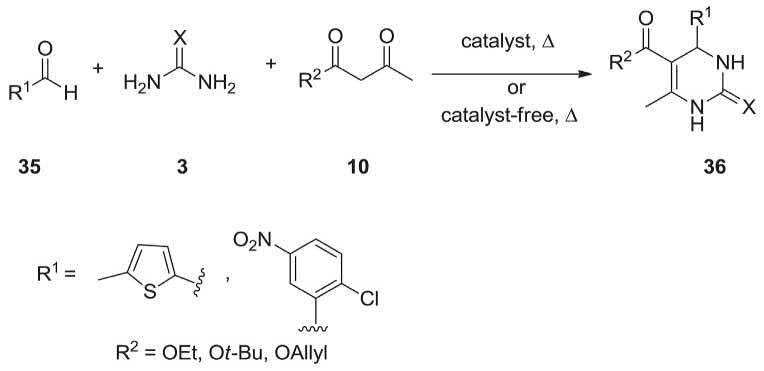
A collection of biologically important 3,4-dihydropyrimidin-2-(1H)-ones (-thiones) 36 were successfully prepared via Biginelli MCR reaction from differently substituted benzaldehydes (5-methyl-2-thiophenecarboxaldehyde and
2.8 Synthesis of compounds 26-31a and b via Biginelli MCR.
SCHEME 2.7 Synthesis of racemic THC–DHPM analogues 22 via MCR Biginelli reaction.
SCHEME
2-chloro-5-nitrobenzaldehyde), β-keto esters (ethylacetoacetate, allylacetoacetate, and t-butylacetoacetate), and urea/thiourea were reacted in one pot fashion in the presence of catalytic quantity of MgBr2 and MgCl2 hexahydrate as safe, inexpensive commercially available at 80 °C and 100 °C. In comparison with the catalyst-free Biginelli MCR conditions, this strategy steadily has the merit of short reaction time period (45–100 min) and giving satisfactory yields (75%–91%) (Scheme 2.10) [209]
A reasonable mechanism for the above-mentioned reaction is depicted in Scheme 2.11. In this rationally proposed mechanism, initially the reaction between aldehyde 35 and urea/thiourea 3 occurs leading to the generation of the acylimine intermediate similar to the Schiff’s base (a conjugated imino-ketone as a Michael acceptor), which is stabilized by Mg ion with subsequent β-carbonyl carbon of the β-carbonyl enolate attack on the imine carbon to give an open-chain ureide followed by cyclization to six-membered heterocyclic system, which upon dehydration results in the formation of DHPMs 36 [209]
A series of biologically active compounds bearing dihydropyrimidones were synthesized via Biginelli MCR [210] The mechanism for the construction of the target compounds comprise the condensation of various aldehydes and urea, which generates an iminium intermediate, acting as electrophile ready for being attacked for the nucleophilic addition of the ketoester in enol form, and the ketone carbonyl of the resulting adduct which undergoes condensation with the amino group of urea to afford the cyclized product. Other heterocycles such as indole, flavone, and benzofuran moieties were successfully employed for the synthesis of dihydropyrimidones. This strategy was particularly efficient with aromatic amines having electron releasing groups (Scheme 2.12) [211]
SCHEME 2.9 Synthesis of 3,4-dihydropyrimidinones 34 via Biginelli MCR.
SCHEME 2.10 Synthesis of DHPMs 36 via Biginelli MCR.