School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Parker H. Petit Institute of Bioengineering and Bioscience, Atlanta, GA, USA
Andreas S. Bommarius
School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Parker H. Petit Institute of Bioengineering and Bioscience; School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA, USA
Chen Cao
Department of Bioengineering, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Nagatsuta-cho, Midori-ku, Yokohama, Japan
Pere Clapés
Department Química Biológica y Modelización Molecular, Instituto de Química Avanzada de Cataluña, IQAC-CSIC, Barcelona, Spain
Rodrigo O.M.A. de Souza
Biocatalysts and Organic Synthesis Lab, Organic Chemistry Department, Chemistry Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
Brent D. Feske
Chemistry and Physics Department, Armstrong State University, Savannah, GA, USA
Michael J. Fink
Vienna University of Technology, Institute of Applied Synthetic Chemistry, Vienna, Austria
Animesh Goswami
Chemical Development, Bristol-Myers Squibb, New Brunswick, NJ, USA
Gideon Grogan
Department of Chemistry, University of York, Heslington, York, UK
Harald Gröger
Faculty of Chemistry, Bielefeld University, Universitätsstr, Bielefeld, Germany
Noncovalent complex formation allows chemical reactions between specific amino acids on the enzyme and functional groups on the substrate to occur in an environment that is kinetically equivalent to a unimolecular process. Transforming what would otherwise be bimolecular reactions into effectively intramolecular conversions is a major reason that enzymes can accelerate reactions by up to 23 orders of magnitude over the background (uncatalyzed) reaction [1].
Selectivity is the second benefit from forming a noncovalent enzyme–substrate complex prior to chemical conversion. By focusing the catalytic attention of the enzyme onto a specific area of the substrate, reactions can be restricted to a single portion of the molecule that may or may not be the most reactive portion of the overall substrate structure. This allows enzyme-catalyzed reactions to be selective in many respects: chemoselective (carrying out only one specific transformation while others are possible), regioselective (transforming only one among several possible sites), and stereoselective (producing and/or consuming one stereoisomer in preference to others).
2 DEFINITION OF BIOCATALYSIS
In nature, enzymes catalyze transformations of metabolites that occur within and/or outside of living cells. Although some enzymes accept only a limited variety of substrates, a large fraction is more tolerant and allows conversions of nonnatural starting materials. The field of biocatalysis rests upon this partial promiscuity. If enzymes were truly selective for only a single substrate, it would be impossible to utilize them for synthesizing new molecules from nonnatural substrates. The goal is to identify or engineer enzymes that are sufficiently general to accept a variety of related substrates, but selective enough to yield single products or stereoisomers. We use the term “biocatalysis” to describe the use of enzymes (either native or modified) for synthetic transformations of nonnatural starting materials. Enzymes used for in vitro synthetic transformations are called biocatalysts, and the processes are called biocatalytic transformations.
3 SCOPE OF THIS BOOK
Some enzymes catalyze the reactions that build up large molecules from simple building blocks, for example, complex carbohydrates from carbon dioxide and water or the synthesis of steroids and terpenoids from acetate. Others are involved in the degradation of large assemblies to small molecules, for example, hydrolysis of proteins to amino acids and the oxidative degradation of lignin. Although some of these native conversions are industrially important and practiced on large scales, the use of enzymes to produce their normal primary and secondary products of cells lies outside the scope of this book. Here, our focus is on preparing nonnatural compounds using enzymes since this addresses the need commonly encountered in organic synthesis. However, it should be noted that the native reactions of an enzyme can often be used as starting points for their applications to nonnative reactions.
The field of metabolic engineering also lies outside the scope of this book. These efforts use two or more enzymes to catalyze sequential steps in a pathway that
In nature, most enzyme-catalyzed reactions occur in an aqueous environment and many synthetic applications, therefore, also utilize water as solvent. This is often advantageous with regard to maximizing the sustainability of a chemical process. Moreover, enzymes are usually most stable in water. These benefits, however, must be balanced against two disadvantages of using water as a solvent for biocatalytic processes. Water is a reactant in hydrolytic processes and its concentration must be minimized when such reactions are run in reverse in order to shift the equilibrium, for example, when using enzymes to synthesize esters or amides from carboxylic acids and alcohols or amines, respectively. In such cases, water-organic biphasic systems or completely organic solvents can be used. The second complication associated with aqueous conditions is that many of the starting materials and products of synthetic interest have very limited solubilities in water. This either requires the use of dilute solutions, which lowers space–time yields and also generates large volumes of wastewater that must be treated, or the use of organic solvents as additives or replacements for water.
Biocatalysts are proteins and composed of natural amino acids. In some cases, additional natural ligands (cofactors) are present. This means that biocatalysts are inherently nonhazardous materials, although it should be noted that, because they are proteins, some enzymes may cause allergenic reactions in susceptible individuals. Although such reactions are rare, normal care should be taken when handling solid enzyme powders.
5 MECHANISM AND KINETICS OF ENZYME-CATALYZED REACTIONS
Although it is not necessary to determine kinetic parameters in order to use enzymes for chemical synthesis, this knowledge can often be useful in deciding which avenues offer the best opportunities for process improvements. Likewise, it is not essential – but often highly useful – to understand the mechanism of the enzyme-catalyzed reaction for the same reasons that one often benefits from knowing the mechanisms of any reaction employed in a synthetic route. Although every individual enzymatic reaction has a unique combination of kinetic properties and reaction mechanism, several useful generalizations are summarized in the subsequent section.
5.1 Features of Enzyme Catalyzed Reactions
As noted previously, noncovalent association between the enzyme and its substrate(s) is the essential first step in biocatalytic reactions. Although early theories of enzyme catalysis focused primarily on interactions between the enzyme and substrate, it was later appreciated that maximizing selective, noncovalent interactions between the enzyme and the high-energy transition state(s) that linked enzyme-bound complexes of substrates and products was the key to efficient rate enhancements. A somewhat oversimplified view is that groundstate interactions determine substrate specificity and transition-state interactions yield rate enhancements.
In addition to noncovalent associations (by van der Waals forces, hydrogen bonds, electrostatic and hydrophobic interactions), some enzymes also form covalent bonds between the enzyme and portions of the substrate. Lipases are a well-known example of this phenomenon. These enzymes utilize a specific protein hydroxyl group (most commonly a serine side chain) to form an ester intermediate with the substrate that is subsequently cleaved by an exogenous nucleophile to form the final reaction product and regenerate the free protein hydroxyl group, making the active site suitable for the next catalytic cycle.
5.2 Coenzymes
Although some enzyme-catalyzed reactions utilize only functional groups found in the protein, the limited number and variety of amino-acid side-chain moieties severely limits the range of accessible reactions. For example, there are neither common amino acids with electrophilic side chains nor amino-acid functional groups suitable for redox catalysis.3 Nature has circumvented this problem by evolving a suite of coenzymes (also known as cofactors) that specialize in particular types of chemical conversions. Table 1.1 lists some common cofactors for biocatalytic processes. In some cases, for example, biotin and some flavins, these cofactors are covalently coupled to the enzyme within the active site. Other cofactors such as nicotinamides are bound reversibly by noncovalent forces during the entire catalytic cycle. Finally, a few cofactors such as pyridoxal phosphate form reversible covalent linkages with the resting form of the enzyme that are cleaved during the catalytic cycle, and then re-formed at the end. When needed by specific enzymes, provision for cofactor supply must also be made. Because of their expense, cofactors are normally supplied in substoichiometric quantities (usually ≪ 0.1 mole %). This means that they must be regenerated prior to the start of the next catalytic cycle, and strategies for cofactor regeneration have been developed as an essential adjunct for biocatalytic reactions, particularly for reductions and oxidations.
5.3 Kinetics and Reaction Mechanisms
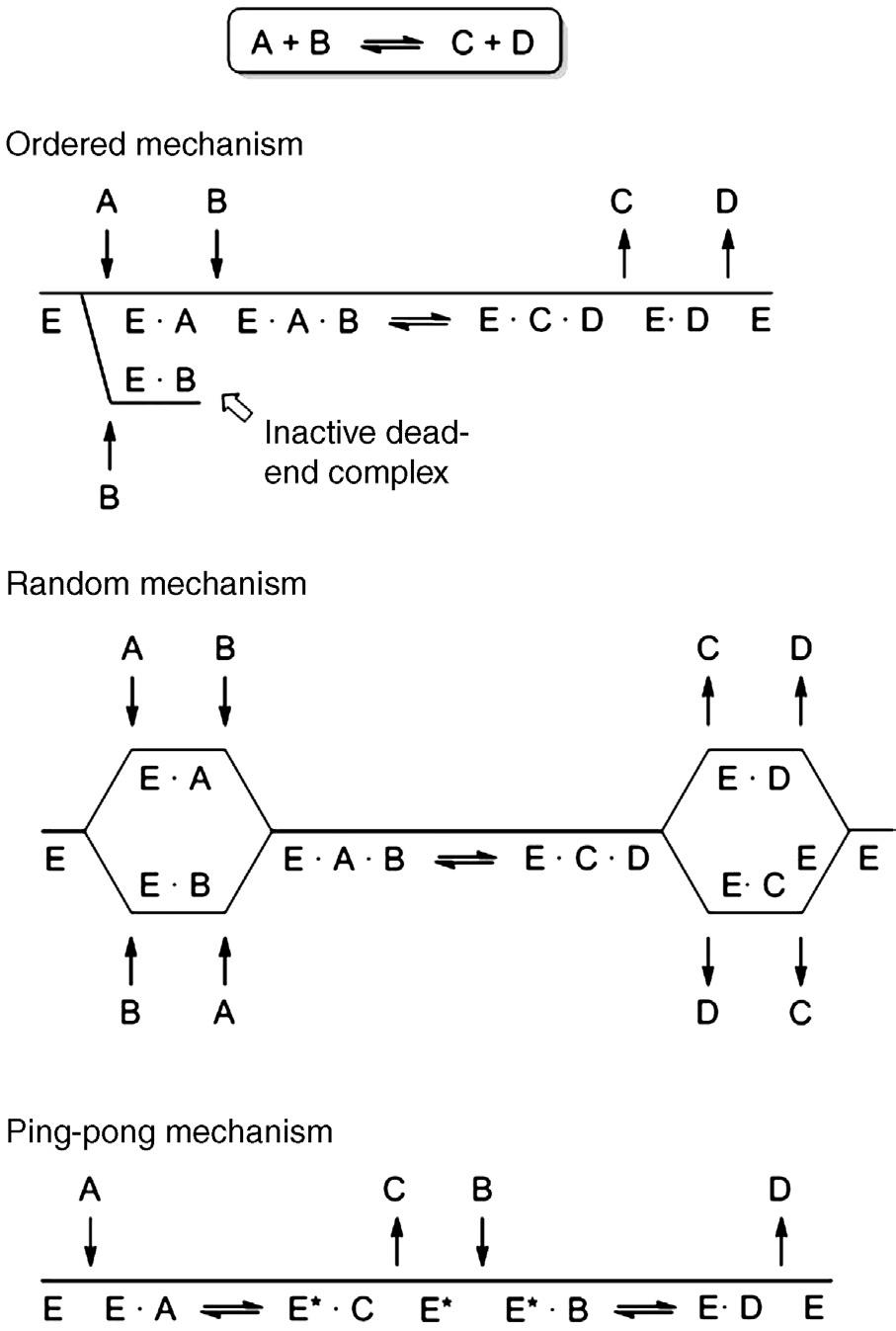
Reaction mechanisms describe the sequence of bond breaking and bond making steps, whereas kinetics are concerned with the nature and timing of the noncovalent complexes that form and break down during the catalytic cycle. Depending on the number of substrates and products, kinetics can be simple or complex. Single-substrate/single-product reactions are the most straightforward schemes, although there are relatively few examples of such reactions in biocatalysis. More commonly, two or more molecules are bound and/or released. This introduces the question of timing with respect to formation and breakdown of enzyme – ligand complexes. Three common reaction mechanisms for a twosubstrate/two-product reaction are illustrated schematically in Figure 1.2. In an ordered mechanism, the enzyme cannot productively bind the second substrate
3 The only exception is disulfide bond formation between a pair of suitably positioned cysteine side-chains.
Introduction, Types of Reactions, and Sources of Biocatalysts
FIGURE 1.2
Three possible kinetic mechanisms for a two-substrate/two-product reaction depicted in Cleland notation. Substrates are designated as “A” and “B”, products as “C” and “D” while “E” represents the enzyme and “E*” is a covalently modified enzyme that forms transiently as part of the normal catalytic pathway in the ping-pong mechanism.
(B) until it previously bound the first (A). Enzymes that follow this scheme often show substrate inhibition when the concentration of B is very high (relative to that of A) since this favors formation of a dead-end E · B complex that must dissociate prior to re-joining the productive pathway. A random mechanism removes this restriction and either substrate can be bound first. Product release steps can be similarly described.4 From a kinetic standpoint, cofactors that do not remain permanently bound to the enzyme are equivalent to substrates and/ or products. Cofactors that remain bound to the enzyme during turnover are kinetically treated as part of the enzyme itself. In either an ordered or random
4 It should be noted that “substrate” and “product” are meaningful only when the direction of the reaction is specified. Like all catalysts, enzymes accelerate both the forward and reverse reactions.
mechanism, the key species is the ternary complex of enzyme with both substrates (E · A · B) since this must be formed in order for the reaction to occur.
Enzymes that form a covalent bond with a part of the substrate often follow a ping-pong mechanism (Figure 1.2). By definition, these must be ordered mechanisms. Ping-pong mechanisms always share two essential features. First, they involve a covalently modified form of the enzyme (or cofactor) as an obligate intermediate.5 In addition, the first product must be released from the active site before the second substrate binds. Together, these two properties of enzymes that follow ping-pong mechanisms open the possibility that the covalent intermediate might be redirected into alternative products by restricting access to the normal second substrate. Employing lipases for synthetic (rather than hydrolytic) acyl transfer reactions is a very common example of this strategy and others can be found in the appropriate sections of later chapters.
Although the methods for determining steady-state kinetic parameters can be complex and the details lie outside the scope of this book, a general understanding of their meaning is useful for applications to biocatalysis. It should be borne in mind that most substrates and products in biocatalytic reactions have limited aqueous solubilities. This complicates kinetic studies since the actual concentration “seen” by the enzyme may not be the same as the concentration added to the reaction mixture unless all of the material is fully dissolved. Therefore, one must be cautious when applying kinetic constants measured under one set of experimental conditions (often dilute aqueous solutions) to reactions run under process conditions that may involve partially dissolved substrates, organic cosolvents, etc.
5.4 Kinetic Constants
Two steady-state kinetic constants are most useful in evaluating biocatalytic reactions. “k cat ” is often known as the turnover number and higher values indicate more catalytically efficient enzymes. This first-order rate constant describes the speed at which an enzyme converts bound substrates to products and re-forms the free enzyme to prepare for the next round of catalysis. It includes both the “chemical” steps (bond making and bond breaking) as well as the product release step(s). Note that it is not uncommon for product release to be the slowest step. As a practical matter, one normally seeks enzymes with k cat ≥ 1 s 1 under the process conditions to ensure reasonable space–time yields along with acceptable catalyst loading levels.
“KM”, also known as the Michaelis constant, is a composite of several microscopic rate constants that primarily describes substrate/product binding and has units of concentration (typically M). Consistent with normal biochemical convention, the binding interaction is considered in the dissociation direction and smaller numerical values of KM therefore signal tighter substrate/product binding. When
5 The covalently modified form of the enzyme has been variously notated as “F,” “E,*” or “ E’ ” in the literature.
Introduction, Types of Reactions, and Sources of Biocatalysts
Table 1.2 Summary of Common Biocatalytic Transformations
Reaction Type
Starting Material(s)
Hydrolysis Esters
Product(s)
Alcohols and carboxylic acids
Amides Amines and carboxylic acids
Common Uses
Synthesis of alcohols and carboxylic acids; resolution of esters, alcohols, acids
Synthesis of amines and carboxylic acids; resolution of amides, acids and amines
Enzyme Types
Lipase, esterase, protease
Lipase, esterase, protease
Nitriles Amides Synthesis of primary amides; resolutions of nitriles Nitrile hydratase
Nitriles Carboxylic acids Synthesis of carboxylic acids; resolutions of acids Nitrilase
Epoxides 1,2-Diols
Esterification Carboxylic acid and alcohol Ester
Amidation Carboxylic acid and amine
Amide
Transesterification Alcohol and ester Alcohol and ester
Transamination Ketone and amine Ketone and amine
Synthesis of diols; resolution of epoxides Epoxide hydrolase
Synthesis of esters; resolutions of carboxylic acids and alcohols
Synthesis of amides; resolutions of carboxylic acids and amines
Synthesis of esters; resolution of esters, alcohols
Lipase, esterase, protease
Lipase, esterase, protease
Lipase, esterase, protease
Dehydrohalogenation
Synthesis of amines Transaminase a-Keto acid and a-amino acid a-Keto acid and amino acid
Synthesis of a-amino acids Amino acid transaminase
Halohydrin Epoxide Synthesis of epoxides; resolution of halohydrins and epoxides
Halohydrin dehalogenase
Carbonyl reduction Ketone Alcohol Synthesis of alcohols Alcohol dehydrogenase/ ketoreductase
Aldehyde Alcohol Synthesis of alcohols Alcohol dehydrogenase/ ketoreductase
(Continued)
Organic Synthesis Using Biocatalysis
Table 1.2 Summary of Common Biocatalytic Transformations (cont.)
Reaction Type
Activated alkene reduction
Reductive amination
Starting Material(s)
a,b-Unsaturated aldehyde, ketone, ester
Ketone, 2-keto acid
Alcohol oxidation Secondary alcohol
Product(s)
Saturated aldehyde, ketone, ester
Amine, 2-amino acid
Ketone
Common Uses
Asymmetric reduction of C ═ C bonds
Synthesis of amines
Synthesis of ketones
Primary alcohol Aldehyde Synthesis of aldehydes
Amine oxidation Amine
Aldehyde, ketone
a-Amino acid a-Keto acid
Hydroxylation
Oxidative dealkylation
Saturated carbon–hydrogen bond
Aromatic carbon-hydrogen bond
Phenyl ethers
Synthesis of aldehydes and ketones; resolutions of amines
Synthesis of a-keto acids; resolutions of amino acids
Alcohols Synthesis of alcohols
Phenols
Phenol, aldehyde
Alkylated anilines Amine, aldehyde
Synthesis of phenols
Synthesis of phenols; dealkylation of methyl or other alkyl ethers
Synthesis of amines; dealkylation of methyl or other secondary amines
Enzyme Types
Alkene reductase (also known as enoate reductase and ene-reductase)
Amino acid dehydrogenase
Alcohol dehydrogenase, alcohol oxidase
Alcohol dehydrogenase, alcohol oxidase
Amine oxidase
Amino acid oxidase, amino acid dehydrogenase
Monooxygenase, peroxidase
Monooxygenase, dioxygenase, peroxidase
Monooxygenase, peroxidase
Monooxygenase, Peroxidase
Baeyer–Villiger oxidation
Cyanohydrin formation
Ketone
Ester
Aldehyde or ketone 2-Hydroxy nitrile
Synthesis of esters and lactones
Synthesis of 2-hydroxy nitriles
Baeyer–Villiger monoxygenase
Oxynitrilase (Hydroxynitrile lyase)
Table 1.2 Summary of Common Biocatalytic Transformations (cont.)
Reaction Type
Aldol condensation
Starting Material(s)
Product(s)
Aldehyde 2-Keto acid
Common Uses
Synthesis of 2-hydroxy ketones
Aldehyde 2-Hydroxy aldehyde
Aldehyde, dihydroxyacetone phosphate
1-Phosphorylated 2-keto-3,4-diol
Aldehyde 2-amino acids
Synthesis of 3,4-dihydroxy aldehydes
Synthesis of polyhydroxylated 2-ketones
Synthesis of 3-hydroxy 2-amino acids
The table is organized by reaction type, rather than by enzyme type to facilitate use in synthetic applications.
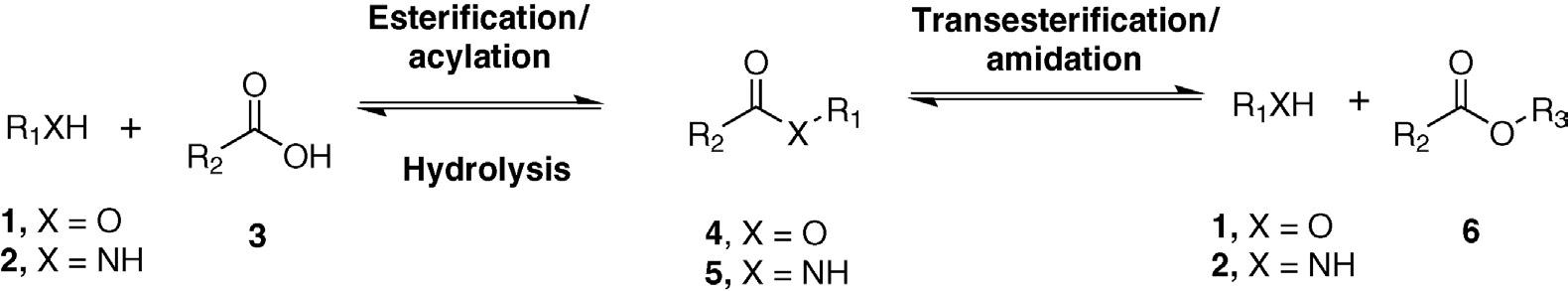
base. The reverse reaction (ester and amide synthesis) cannot be carried out under aqueous conditions due to the large molar excess of water (whose concentration is 55 M in pure water). Enzymes can catalyze the reverse reaction – ester and amide synthesis from acids and alcohols or amines – under nonaqueous conditions, although the yields are limited by thermodynamic constraints unless steps are taken to remove the water formed during the reaction. This limitation can be circumvented by using enzymes to catalyze transesterification and transamidation, using an ester or amide as the starting material and relying on Le Chatêlier’s principle to shift the equilibrium to the desired product (Figure 1.3).
Acyl transferase enzymes have been widely used to synthesize chiral esters, amides, alcohols, and amines. In many cases, these conversions involve kinetic resolutions of alcohols, acids, esters, amines, and amides. Of course, since each enantiomer makes up half of the racemic mixture, kinetic resolutions can provide a maximum 50% yield. This limitation can be overcome by racemizing or inverting the configuration of the unreacted substrate during the enzymatic reaction. Such a scheme is referred to as a dynamic kinetic resolution and theoretically allows complete substrate conversion to product along with 100% chemical yield of a single product enantiomer.
6.2 Carbonyl Reductions to Alcohols
Another common biocatalytic route to alcohols involves enzyme-mediated reductions of the corresponding aldehydes or ketones. Because the starting materials are prochiral, such processes are not kinetic resolutions and are therefore not subject to the 50% yield limitation. Enzymes that catalyze carbonyl
Hydrolysis and synthesis of carboxylic acid derivatives such as esters and amides.
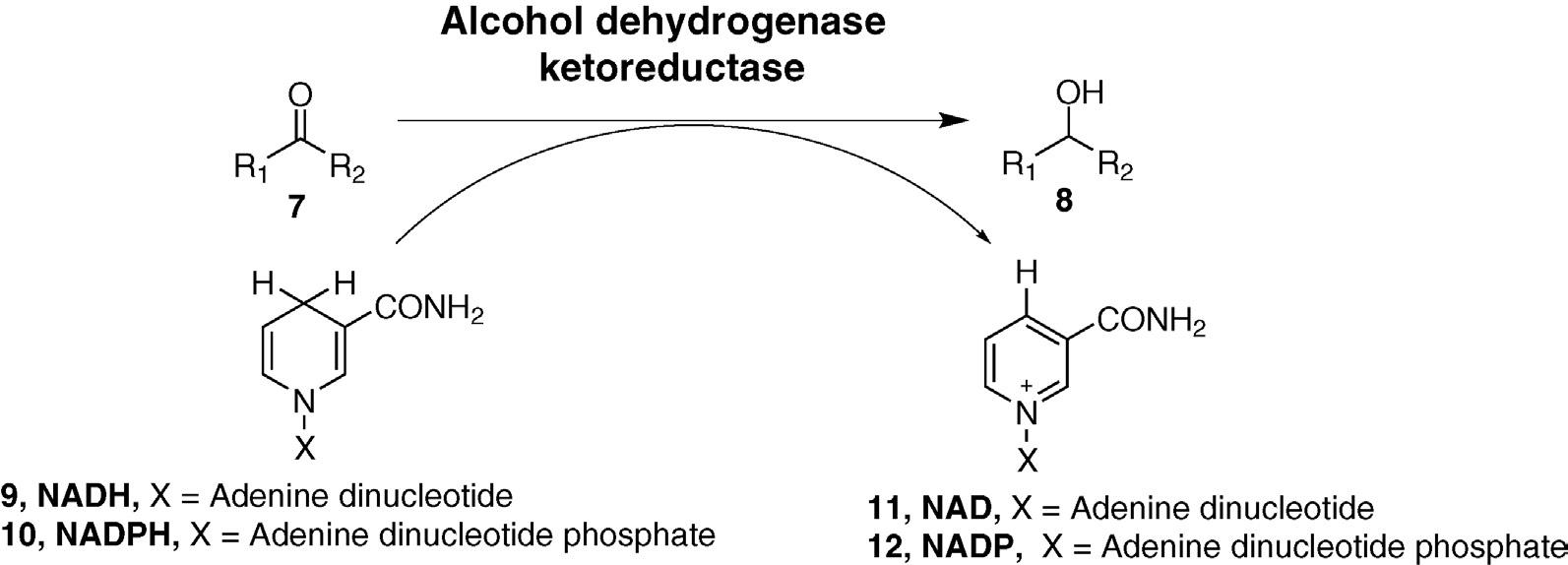
reductions have been variously named as alcohol dehydrogenases or ketoreductases.8 Because carbonyl reductions involve the formal addition of H2 (usually in the form of a hydride ion along with a proton), a second cosubstrate that supplies reducing equivalents must be included along with the aldehyde or ketone of synthetic interest. Nicotinamides are the most common hydride sources for enzymatic reactions and they occur in the form of nicotinamide adenine dinucleotide (NADH) and its phosphorylated analog (NADPH). Some enzymes are highly selective for one cofactor type or the other whereas others accept both NADH and NADPH (a property referred to as dual specificity). Carbonyl reduction converts the nicotinamides into their oxidized forms and these must be reduced back to their original oxidation states prior to reuse in subsequent rounds of catalysis. Several methods for in situ cofactor regeneration have been developed in response to the high cost of nicotinamides and cofactor turnover numbers of more than 1000 are commonly achievable. This usually makes the cofactor contribution to the overall process costs negligible (Figure 1.4).
Alcohol dehydrogenases/ketoreductases are widely distributed in nature where they play many roles in metabolism. Bakers’ yeast is a particularly prolific
FIGURE 1.4
Carbonyl reductions to alcohols.
8 The “alcohol dehydrogenase” nomenclature is more common in the biochemical literature whereas “ketoreductase” is typically used in biocatalysis since this is the synthetically more useful direction for the reaction.
1.6
Enzymatic hydroxylation by C H bond insertion.
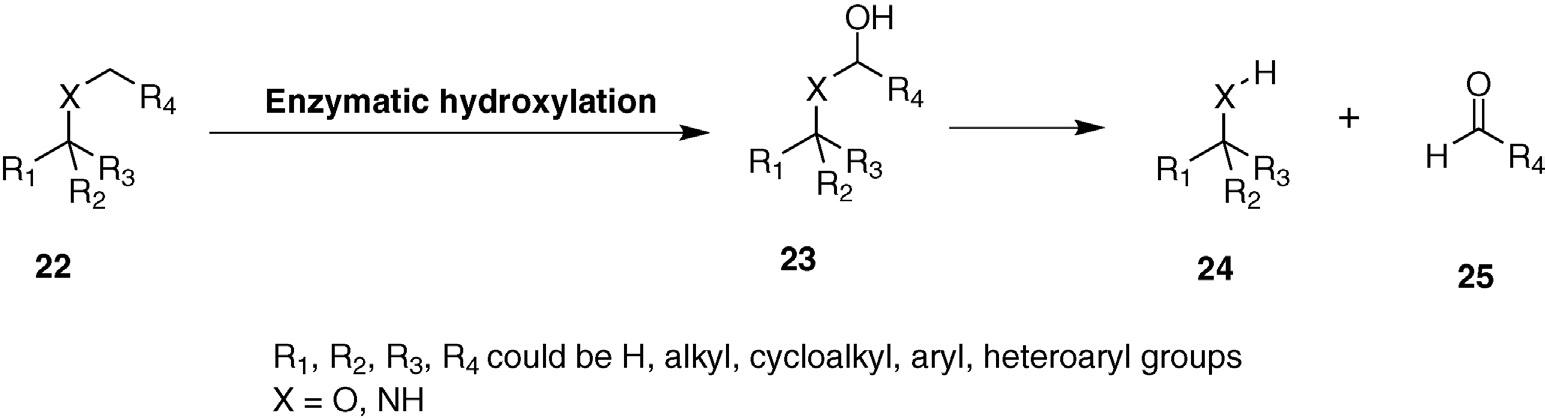
Cytochrome P-450s are the best-known class of hydroxylation enzyme. Their active sites contain a heme iron that forms a highly activated oxygenating species that reacts by a radical mechanism. In higher animals, they function primarily in metabolite degradation as part of pathways that clear unnatural substances such as toxins and drugs. Hydroxylation increases polarity that facilitates further derivatization by other detoxification enzymes or excretion of the hydroxylated products. Other P-450 family members are involved in secondary metabolite biosynthesis, particularly in plants and microbial cells (Figure 1.6).
With few exceptions, most C H hydroxylating enzymes are composed of multiple protein subunits and many are membrane bound and unstable in purified form. All of these properties conspire to make such enzymes difficult to handle as isolated proteins. In addition, oxygen activation requires a pair of electrons that are typically supplied by NADH, which introduces cofactor regeneration as an additional complication. For all of these reasons, most preparative biocatalytic hydroxylations have been carried out by mixing intact microbial cells with the substrate of interest (analogous to the way that Bakers’ yeast cells have been employed in carbonyl reductions). The main difficulty is that many organisms produce more than one P-450 enzyme and this can lead to multiple products from a single reaction. Several groups have therefore created recombinant strains that express single P-450’s in “clean” hosts that minimize side reactions. In addition, by overproducing large quantities of the relevant enzymes, the oxidations are often more efficient with regard to space–time yields.
6.6 O- or N-Dealkylations
Enzymatic hydroxylation of a carbon adjacent to an oxygen or nitrogen usually results in dealkylation by spontaneous hydrolysis of the initial hemiacetal or hemiaminal product. These conversions are commonly employed for two synthetic purposes: cleavage of methyl ethers and oxidative deamination of amines. The latter is particularly useful in amino-acid chemistry. These reactions can be catalyzed by P-450 monooxygenases or by flavin-containing monooxygenases (which are typically metal-free enzymes). As in the previous example, these hydroxylations require two electrons that must be supplied by NADH or NADPH, and most synthetic applications have relied on whole microbial cells rather than the isolated enzymes (Figure 1.7).
FIGURE
FIGURE 1.7
O- or N-dealkylations.
6.7 Oxidative Deamination of Amines to Carbonyl Compounds and the Reverse Reaction
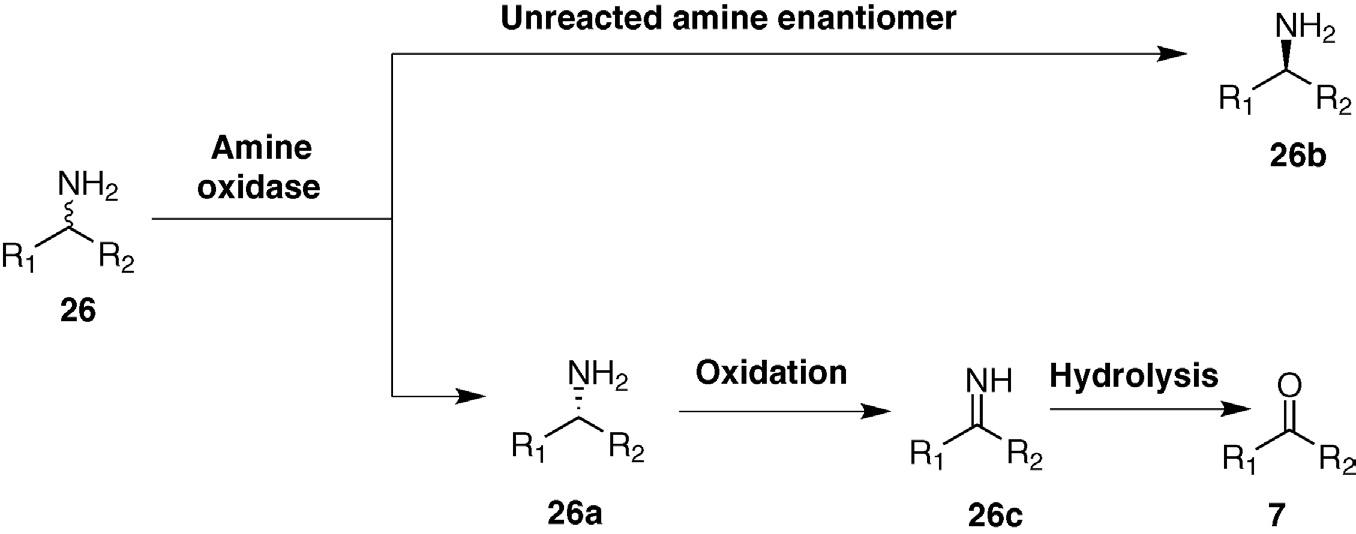
Oxidative deamination by amine oxidases generates ketones or aldehydes. There are two types of amine oxidases. Those of the Type I contain both copper and a covalently attached topaquinone cofactors. In these enzymes, amine oxidation first generates an enzyme bound imine that is subsequently hydrolyzed to a ketone that is finally released from the enzyme. Type II amine oxidases are metal-free enzymes and contain only a flavin cofactor that remains bound to the enzyme throughout the catalytic cycle. Catalysis by these enzymes produces an imine intermediate that is released from the enzyme and subsequently hydrolyzed to the ketone in aqueous medium. To complete the catalytic cycle, the reduced flavin cofactor is oxidized by molecular oxygen generating hydrogen peroxide. In both cases, hydride (or a hydride equivalent) is transferred to the cofactor. Many amine oxidases are highly stereoselective, and this makes them very useful in kinetic resolutions of amines. For example, enantioselective oxidation of one antipode of 26 removes this stereoisomer from the reaction mixture, leaving behind the unreacted enantiomer. As in all kinetic resolutions, however, the maximum yield of the enantiopure product is 50% (Figure 1.8).
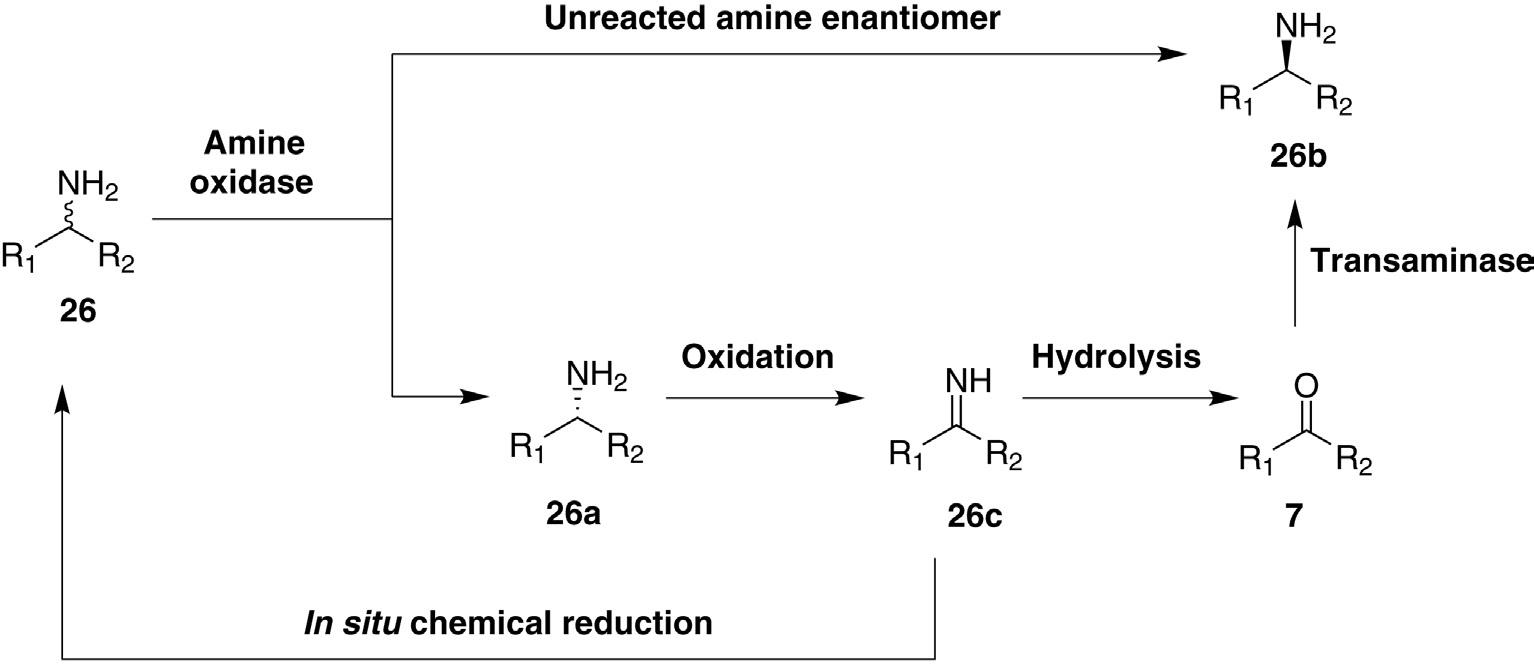
Several ingenious solutions to overcome the 50% yield problem inherent in amine kinetic resolutions have been devised. The most practical pairs are an amine oxidase with another enzyme or in situ chemical reaction that changes the process into a dynamic kinetic resolution with a 100% theoretical yield. The Turner group developed a very successful strategy in which a biocompatible chemical reducing agent such as amino borane was added along with the racemic starting amine and an enantioselective Type II amine oxidase [6]. Enzymatic oxidation depleted the reactive amine enantiomer; the imine product of this step was reduced in a racemic fashion by the amino-borane reagent. The net result was to racemize the starting amine that yielded 50% of the unreactive (desired) amine enantiomer along with another 50% destined for re-oxidation by the enzyme. After several rounds of oxidation/reduction, essentially all of the “wrong” enantiomer had been converted to the desired product. This is a very powerful strategy, particularly when combined with protein engineering to
Introduction, Types of Reactions, and Sources of Biocatalysts
endow amine oxidases with the desired substrate- and stereoselectivities. Since the imine is not released from the Type I amine oxidase enzymes, chemical reduction will reduce the released ketone to the corresponding racemic alcohol. Thus dynamic resolution by combining in situ chemical reduction is possible with Type II amine oxidase only. However, both Type I and II amine oxidases can be combined with transaminase (transferring ketone to the desired amine) to effect dynamic resolution (Figure 1.9).
Amino acid oxidase enzymes are similar to the Type II monoamine oxidase and oxidize amino acids to imino acid and then to keto acid. These enzymes can be used for the kinetic resolution of racemic amino acids.
Rather than using O2 as the ultimate electron acceptor for amine oxidation, amine dehydrogenases transfer hydride from the amine a-carbon to a nicotinamide cofactor. This yields the corresponding imine that undergoes subsequent