Neuromuscular Disorders-A Symptoms and Signs Approach to Differential Diagnosis and Treatment (April 22, 2016)_(0826171982)_(McGraw-Hill) Nicholas J. Silvestri
For technical assistance: email expertconsult.help@elsevier.com call 1-800-401-9962 (inside the US) call +1-314-447-8300 (outside the US)
NEUROMUSCULAR DISORDERS
TREATMENT AND MANAGEMENT
NEUROMUSCULAR DISORDERS TREATMENT
AND MANAGEMENT
Tulio E. Bertorini, MD
Professor of Neurology and Pathology, and Director of the Clinical Neurophysiology Fellowship University of Tennessee Center for the Health Sciences Director of EMG Laboratory Methodist University Hospital Director of Wesley Neurology Clinic and The Muscular Dystrophy and ALS Clinics, Memphis, Tennessee
Elsevier
3251 Riverport Lane
St. Louis, Missouri 63043
NEUROMUSCULAR DISORDERS, TREATMENT AND MANAGEMENT, SECOND EDITION
No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or any information storage and retrieval system, without permission in writing from the publisher. Details on how to seek permission, further information about the Publisher’s permissions policies and our arrangements with organizations such as the Copyright Clearance Center and the Copyright Licensing Agency, can be found at our website: www.elsevier.com/permissions.
This book and the individual contributions contained in it are protected under copyright by the Publisher (other than as may be noted herein)
Cover image
Top: IBM - Rimmed vacuoles (left) and a ragged red fiber (right) seen in Inclusion Body Myositis stained with Gomri’s Modified Trichrome (200x)
Bottom: Inflammatory - Inflammatory cells stained with hematoxylin and eosin (100x)
Notices
Practitioners and researchers must always rely on their own experience and knowledge in evaluating and using any information, methods, compounds or experiments described herein. Because of rapid advances in the medical sciences, in particular, independent verification of diagnoses and drug dosages should be made. To the fullest extent of the law, no responsibility is assumed by Elsevier, authors, editors or contributors for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any methods, products, instructions, or ideas contained in the material herein.
ISBN: 9780323713177
Content Strategist: Melanie Tucker
Director, Content Development: Ellen Wurm-Cutter
Senior Content Development Specialist: Kathleen Nahm
Publishing Services Manager: Shereen Jameel
Project Manager: Nadhiya Sekar
Design Direction: Renee Duenow
Printed in India
Last digit is the print number: 9 8 7 6 5 4 3 2 1
To my wife, Emma, for her patience, support, and encouragement; my children, Tulio J. Paola and Francisco; my grandchildren, Nicolas, Rafael, and Gabriela; and the memory of my parents, Nico and Queta.
To the Peruvian American Medical Society (PAMS) for their dedication to the care of the indigent and improving medical education in Peru.
To the Ernesto Guevara class of the San Fernando Medical School in Lima on the golden anniversary of their graduation.
To my patients.
To all that died in the front lines fighting the COVID-19 pandemic.
Preface
There have been great advances in the diagnosis and evaluation of patients with segmental and generalized neuromuscular disorders since the first edition of this book. Tests applied to different types of the disorders are discussed in the introduction as well as specific chapters.
The management of these diseases have also improved significantly, particularly in cardiac and pulmonary care, and in the management of autonomic and peripheral neuropathies, and for this reason these chapters have been expanded. We have also expanded chapters in orthopedic surgery and rehabilitation with the use of robotics to manage patients with chronic weakness.
Immunotherapy has improved greatly with the use of new monoclonal antibodies, which increases the armamentarium of clinicians that take care of autoimmune disorders. The basic aspects of immunotherapy are covered in Chapter 7, but also are detailed chapters dedicated to autoimmune neuropathies and myopathies, and disorders of neuromuscular disorders of neuromuscular junction.
The most important development in neuromuscular disorders has been the discovery of pathogenic genes and improved testing in hereditary conditions, which has allowed
us to diagnose patients even without muscle biopsies, and this has led to genetic therapies. We have included a new chapter that introduces the readers to these treatments, which are also discussed in detail in those chapters dedicated to hereditary myopathies and neuropathies.
Some topics are discussed in various chapters, causing some repetition, but the intent is that each reader could consult individual chapters independently.
These have been very difficult times, because of the COVID-19 pandemic, which affects more severely patients with weakening neuromuscular disorders. Medical care has also been complicated by this, and COVID-19 infections are associated with neuromuscular conditions that are discussed in different chapters.
The pandemic has also created difficulties in the completion of this book, and I am most grateful to the Elsevier personnel and the other collaborators that have helped overcome these difficulties. I am also very thankful that they dedicated their time and effort to the completion of this volume, which I believe will be very useful to those taking care of patients with neuromuscular disorders.
We wish to acknowledge the great secretarial help of Cindy Burchfield and Ginger Lindsey, and the excellent artwork of Jason Peck. We also acknowledge the contribution of Joseph Null and Mariallen Shadle for excellent histology work and photography.
We extend our sincere appreciation to Dr. Michael Cartwright and Dr. Francis Walker for providing great ultrasound figures.
Acknowledgments
Finally, we want to acknowledge the excellent and dedicated collaboration from the authors of the different chapters, and express gratitude to Wesley Neurology and UTHSC for their support, and to Kathleen Nahm and Nadhiya Sekar at Elsevier for editorial help.
Contributors
Annas Aljassem, MD, MHSA Assistant Professor
Physical Medicine & Rehab
Oakland University William Beaumont, Rochester Hills
Michigan
United States
Chapter 6
Bassam A. Bassam, MD, FAAN Professor Neurology
University of South Alabama, Mobile Alabama
United States Chapter 10, 21
Tulio E. Bertorini, MD Professor Department of Neurology
University of Tennessee Health Sciences, Memphis Tennessee
United States Chapter 1, 7, 10, 21
Aimee K. Boegle, MD, PhD Instructor Neurology
BIDMC/Harvard Medical School, Boston Massachusetts
United States
Chapter 18
William W. Campbell Jr., MD, MSHA COL, MC, USA (Ret)
Louisiana State University Health Science Center, New Orleans
Louisiana
United States
Chapter 15
Thomas E. Lloyd, MD, PhD
Associate Professor Neurology and Neuroscience
Johns Hopkins University School of Medicine, Baltimore
Maryland
United States
Chapter 14
Catherine Lomen-Hoerth, MD, PhD Professor Neurology
UCSF, San Francisco California
United States
Chapter 12
Carlos A. Luciano, MD Professor Neurology
University of Puerto Rico School of Medicine, San Juan Puerto Rico
Puerto Rico
Chapter 16
Daniel L. Menkes, MD Professor and Chair Neurology
Oakland University William Beaumony, Royal Oak Michigan
United States
Chapter 6
Christopher W. Mitchell, MD Neurologist
Department of Neurology West Tennessee Neuroscience and Spine, Jackson Tennessee
United States
Chapter 7
William Motley, MD, DPhil Director of Translational Medicine
Flare Therapeutics, Cambridge Massachusetts
United States
Chapter 14
Pushpa Narayanaswami, MD
Associate Professor Neurology
Beth Israel Deaconess Medical Center/Harvard Medical School, Boston
Massachusetts
United States
Chapter 11, 18
Shin J. Oh, MD
Distinguished Professor Emeritus Neurology
University of Alabama at Birmingham, Birmingham
Alabama
United States
Chapter 19
Laura Rosow, MD
Assistant Professor Neurology
UCSF, San Francisco
California
United States
Associate Director of ALS Center
Department of Neurology
University of California, San Francisco, California
United States
Chapter 12
Jennifer E. Schramm, MD
Pediatric Cardiology Fellow
Pediatric Cardiology
Children’s National Hospital, Washington District of Columbia
United States
Chapter 3
Shreesh Shrestha, MD
Chief Resident in Patient Safety and Quality Improvement Internal Medicine
UTHSC, Memphis
Tennessee
United States
Chapter 4
Nicholas J. Silvestri, MD
Associate Professor of Neurology Neurology
University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo
New York
United States
Chapter 5
Michael Soliman, MD
Fellow of Neuromuscular Diseases
Louisiana State University, New Orleans Louisiana
United States
Chapter 15
Michael Spickler, MD
Resident
Physical Medicine and Rehabilitation
Beaumont Royal Oak, Royal Oak
Michigan
United States
Chapter 6
Christopher F. Spurney, MD
Associate Professor Cardiology
Children’s National Health System, Washington District of Columbia
United States
Chapter 3
Carolina Tesi Rocha, MD
Associate Professor of Neurology Neurology
Stanford University, Palo Alto
California
United States
Chapter 20
Dorothy Weiss Tolchin, MD, EdM
Instructor, part-time
Physical Medicine and Rehabilitation
Harvard Medical School, Boston
Massachusetts
United States
Director of Medical Student Education
Physical Medicine and Rehabilitation
Spaulding Rehabilitation Hospital/Harvard Medical School, Charlestown
Massachusetts
United States
Chapter 8
Matthias Vorgerd, MD
Adjunct Professor
Ruhr University, Bochum Germany
William C. Warner Jr., MD Professor Orthopedics
University of Tennessee, Memphis
Tennessee
United States
Chapter 9
Saša Živković, MD, PhD Professor
Department of Neurology
University of Pittsburgh School of Medicine, Pittsburgh
Pennsylvania
United States
Chapter 11
PART 1 GENERAL PRINCIPLES IN THE TREATMENT AND MANAGEMENT OF NEUROMUSCULAR DISORDERS
1 Introduction: Evaluation of Patients With Neuromuscular Disorders, 2
Tulio E. Bertorini, MD
2 Respiratory Complications in Neuromuscular Disorders, 40
Jonathan Daniel Finder, MD
3 Cardiac Complications of Neuromuscular Disorders, 52
Christopher F. Spurney, MD | Jennifer E. Schramm, MD
4 Gastrointestinal Complications of Neuromuscular Disorders, 79
Mohammed K. Ismail, MD | Shreesh Shrestha, MD
5 Autonomic Dysfunction in Neuromuscular Disorders, 97
Christopher H. Gibbons, MD, MMSc | Nicholas J. Silvestri, MD
6 A Practical Approach to the Treatment of Painful Polyneuropathies, 118
Annas Aljassem, MD, MHSA | Levi M. Hall, PharmD | Michael Spickler, MD | Daniel L. Menkes, MD
7 Principles and Guidelines of Immunotherapy in Neuromuscular Disorders, 143
Christopher W. Mitchell, MD | Tulio E. Bertorini, MD
8 Rehabilitation in Neuromuscular Disorders, 160
Dorothy Weiss Tolchin, MD, EdM
9 Orthopedic Surgery in Neuromuscular Disorders, 186
William C. Warner Jr., MD
10 Perioperative Management of Patients With Neuromuscular Disorders, 206
Tulio E. Bertorini, MD | Jonathan Daniel Finder, MD | Bassam A. Bassam, MD, FAAN
11 Molecular and Genetic Therapies, 225
Pushpa Narayanaswami, MD | Saša Živković, MD, PhD
PART 2 TREATMENT AND MANAGEMENT OF SPECIFIC NEUROMUSCULAR DISORDERS
12 Treatment and Management of Adult Motor Neuron Diseases, 248
Laura Rosow, MD | Catherine Lomen-Hoerth, MD, PhD
13 Treatment and Management of Spinal Muscular Atrophy and Congenital Myopathies, 261
Diana Castro, MD | Alicia Henriquez, MD
14 Treatment and Management of Hereditary Neuropathies, 278
William Motley, MD, DPhil | Vinay Chaudry, MD, MBA, FRCP | Thomas E. Lloyd, MD, PhD
15 Treatment and Management of Autoimmune Neuropathies, 312
Rima N. El-Abassi, MD, ABPN, ABCN | Michael Soliman, MD | Maxwell Harris Levy, MD | John D. England, MD
16 Treatment and Management of Infectious, Granulomatous, and Toxic Neuromuscular Disorders, 345
Carlos A. Luciano, MD | Sonia Caraballo-Cartagena, MD
17 Treatment and Management of Segmental Neuromuscular Disorders, 380
William W. Campbell Jr., MD, MSHA | Mark Landau, MD, FAAN
18 Treatment and Management of Disorders of Neuromuscular Hyperexcitability and Periodic Paralysis, 414
Aimee K. Boegle, MD, PhD | Pushpa Narayanaswami, MD
19 Treatment and Management of Disorders of the Neuromuscular Junction, 446
Shin J. Oh, MD
20 Treatment and Management of Muscular Dystrophies, 492
Carolina Tesi Rocha, MD | Diana M. Escolar, MD, FAAN
21 Neuromuscular Manifestations of Acquired Metabolic, Endocrine, and Nutritional Disorders, 528
Bassam A. Bassam, MD, FAAN | Tulio E. Bertorini, MD
22 Treatment and Management of Autoimmune Myopathies, 554
Marinos C. Dalakas, MD, FAAN
23 Treatment and Management of Hereditary Metabolic Myopathies, 572
Matthias Vorgerd, MD | Marcus Deschauer, MD Index, 595
PART 1
GENERAL PRINCIPLES IN THE TREATMENT AND MANAGEMENT OF NEUROMUSCULAR DISORDERS
Introduction: Evaluation of Patients with Neuromuscular Disorders
Tulio E. Bertorini, MD
This book is dedicated to the treatment of neuromuscular disorders (NMDs), which include those that affect the anterior horn cells, nerve roots, plexi, peripheral nerves, neuromuscular junction, and muscles (Fig. 1.1) (Dubowitz, 1996); some also affect other areas of the nervous system, such as amyotrophic lateral sclerosis (ALS). These disorders may be caused by genetic defects or may be acquired, as the autoimmune diseases, may be secondary to general medical conditions or may arise as complications of surgery. To make therapeutic decisions about these disorders, clinicians should be able to recognize their clinical presentation and characteristics. This chapter provides a brief introduction to the evaluation of patients with NMDs.
MEDICAL HISTORY AND SYMPTOMS
The evaluation should include obtaining detailed medical and family histories as well as identifying possible complicating factors. In children, information should be obtained on the prenatal period and delivery, especially if the patient was a “floppy baby,” and details of the patient’s developmental milestones should be recorded (Brooke, 1999; Dubowitz, 1996).
Identifying general medical problems is important because some NMDs are associated with other conditions, such as endocrine and connective tissue diseases that might affect other organs. Medications also should be considered, because many are known to produce neurologic complications.
Muscle weakness is a common symptom, except in patients with sensory or autonomic neuropathy or in some radiculopathies and entrapment syndromes. The rate of progression varies, and in some conditions, such as Guillain-Barré syndrome (GBS), electrolyte imbalance, toxic neuropathy, and myopathy associated with rhabdomyolysis, it is rapid (Box 1.1). In disorders of neuromuscular transmission, such as myasthenia gravis (MG), weakness fluctuates during the day. In periodic paralysis, weakness is recurrent (Brooke, 1986), whereas in other disorders, such as muscular dystrophies, or in hereditary and some autoimmune neuropathies, it is subacute or chronic (Box 1.2) (Bertorini, 2002; Brooke, 1986).
The distribution of weakness also is important in diagnosis; for example, it is proximal in spinal muscular atrophies and most myopathies, except for some disorders in which it is more distal, for example, inclusion body myositis (IBM) and Miyoshi myopathy. In myopathies, weakness usually is symmetric, although asymmetry can be seen in some
diseases like fascioscapulohumeral dystrophy and IBM. In polyneuropathies, this characteristically begins in the legs but may initially manifest more prominently in the upper extremities, as in multifocal neuropathy, and also in brachial plexopathies, and cervical spinal canal disorders as well as in ALS. This follows the territory of roots or nerves in radiculopathies, focal neuropathies, (Bertorini, 2002), mononeuritis multiplex, and entrapment neuropathies.
Dysphagia, diplopia, and ptosis also help to identify NMDs because they occur in some myopathies and also in disorders of neuromuscular transmission, such as MG. Symptoms of respiratory difficulty should be recognized and treated promptly because this can be the first manifestation of some disorders such as MG, GBS, ALS, and some myopathies, such as acid maltase deficiency, whereas in other disorders, it appears at later stages (Bertorini, 2002, 2008). During the evaluation, one should always inquire about sleep difficulties, as sleep apnea can be seen in some of these diseases. Difficulty combing the hair and placing objects in high cabinets commonly occurs in patients with shoulder-girdle weakness, whereas difficulty writing and grasping objects indicates involvement of the forearm and hand muscles, as in ALS and IBM. Weakness of the hip extensors usually causes inability to rise from a low chair or a toilet seat, whereas difficulty ascending stairs indicates dysfunction of the hip flexors and quadriceps muscles. More severe weakness of the quadriceps muscles occurs in IBM, causing difficulty descending stairs (Brooke, 1986; Griggs, Mendell, & Miller, 1995). When the distal muscles are affected, foot drop may cause a steppage gait and difficulty negotiating curves or changing courses, as seen in polyneuropathies, distal dystrophies, and ALS.
Muscle stiffness, tightness, and spasms occur as a result of spasticity in disorders affecting the upper motor neuron, but these also occur in patients with motor unit hyperactivity, such as “stiff-person” and Isaac syndromes and the myotonias. Those with inflammatory myopathies, polymyalgia rheumatica, fasciitis, and hypothyroidism also complain of stiff limbs. Cramping at rest or during exercise is a prominent symptom of the cramp-fasciculation syndrome (Masland, 1992) and also some neuropathies. In metabolic myopathies, this usually occurs during or after exercise, or after fasting in some cases. Fatigue is common in disorders of neuromuscular transmission, such as Eaton-Lambert syndrome (ELS) and MG, but also in myopathies, even though weakness is the major symptom. In ELS, there may be temporary improvement after a brief exercise.
Anterior horn cell SMA ALS
Autonomic nerve
Root Radiculopathy
Dorsal root ganglion
Sensory ganglioneuropathy
Root Radiculopathy
Plexus Radiationplexitis
Peripheral nerve
Demyelinatingneuropathies
Axonalneuropathies
Unmyelinated fiber
Myelinated fibers
Demyelinatingneuropathies
CIDP
Neuromuscular junction
Lambert-Eatonsyndrome Myastheniagravis
Muscle Myopathies
Decreased sensation as well as paresthesias and neuropathic pain are symptoms of peripheral neuropathies (Ochoa, 1995). These symptoms are localized in the affected areas in those with radiculopathies, plexopathies, and entrapment neuropathies. Autonomic dysfunction can occur in some neuropathies and also in ELS, and the clinician should ask the patient for dysautonomic symptoms such as orthostatic hypotension and urinary and sexual dysfunction.
PHYSICAL EXAMINATION
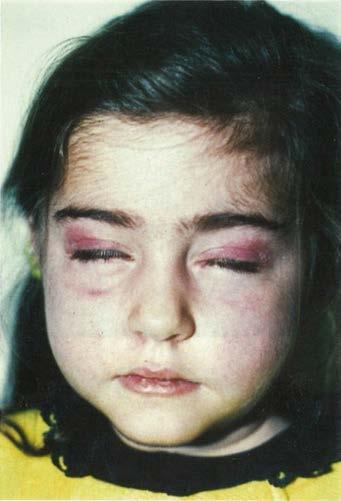
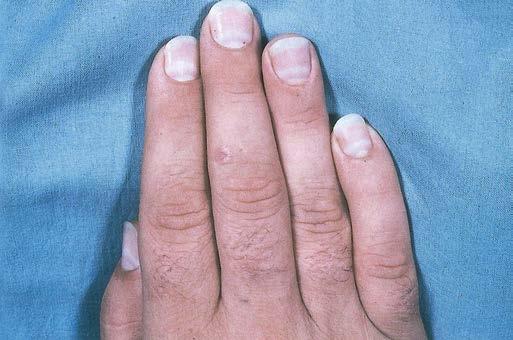
A careful general physical examination is essential to arrive at a diagnosis, and the clinician should assess cardiac and lung function, examine the eyes for cataracts and retinal disease, and check for hearing loss and lipoma, which are often seen in mitochondrial disorders. Visceromegaly and skin changes are present in some patients with neuropathies, for example, those with POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes) syndrome. Skin abnormalities can also be seen in connective tissue disorders, whereas patients with dermatomyositis have a characteristic rash, including the Gottron sign, carpenter
Fig. 1.1 Anatomic elements of the peripheral nervous system and related neurologic disorders. ALS, Amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; SMA, spinal muscular atrophy. (Adapted from Bertorini, T. E. (2002). Overview and classification of neuromuscular disorders. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (pp. 1–13). Woburn, MA: Butterworth-Heinemann.)
fingers, and dilatation of the periungual capillaries (Fig 1.2) (also see figures of patients in Chapter 22) (Bertorini, 2002). High arches of the feet are seen in hereditary motor sensory neuropathy.
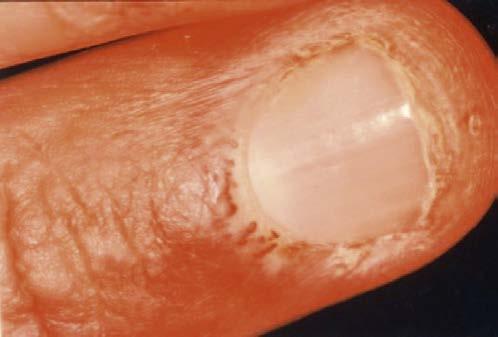
Clubbing of the fingers is seen in some chronic lung disorders, whereas Mees lines are seen in patients with arsenic and other poisoning (Fig 1.3).
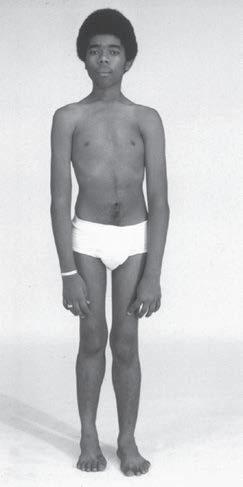
Intellectual function should be assessed because it could be impaired, such as in some cases of ALS and in myotonic dystrophy. Examination of posture and gait is useful to determine if there is hyperextension of the knees, if there is evidence of a waddling gait in myopathies, and if there is a spastic or ataxic gait, or the steppage seen in peripheral neuropathy and some distal dystrophies should test tandem gait which is abnormal in cerebellar disease. Difficulty walking on tiptoes is seen in people with gastrocnemius and soleus weakness, whereas walking on heels cannot be done in persons with foot dorsiflexor weakness. Patients should be asked to get up and down from a stool to determine if there is thigh muscle weakness. The examiner should also notice difficulty arising from the chair or going upstairs, and if the patient has the characteristic Gower maneuver, when arising from the floor, due to proximal muscle weakness (Fig. 1.4), and
Box 1.1 Neuromuscular Disorders That May Present with Acute Generalized Weakness
Motor Neuron Diseases
Poliomyelitis, West Nile virus infection
Amyotrophic lateral sclerosis (rarely)
Neuropathies
Guillain-Barré syndrome and variants
Porphyria, particularly acute intermittent
Dinoflagellate toxins
Diphtheria
Arsenic poisoning and other acute toxic neuropathies
West Nile virus infection
Disorders of Neuromuscular Transmission
Botulism and other biologic toxins (black widow spider bites, snake bites)
Organophosphate poisoning
Eaton-Lambert myasthenic syndrome (rarely)
Hypermagnesemia
Myasthenia gravis
Myopathies
Rhabdomyolysis (from various causes, including metabolic, toxic, and infectious)
Juvenile and adult forms of acid maltase deficiency
Carnitine deficiency
Other metabolic myopathies
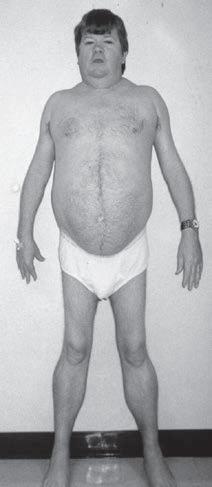
should observe if there is hyperlordosis with proximal atrophy in myopathies and distal atrophy in neuropathies and whether it is symmetric or focal (Figs. 1.5 and 1.6) or whether it affects the upper or lower extremities more prominently. The clinician should examine the patient for muscle hypertrophy, which is seen in some dystrophies and disorders of neuromuscular hyperactivity such as myotonia congenita (Fig. 1.7). Examination of muscle tone also is important to determine whether there is hypotonia, particularly in infants (Fig. 1.8 and Box 1.3), or spasticity and also to determine if there is limitation of passive joint movement like from muscle fibrosis that occurs in various myopathies (Fig. 1.9).
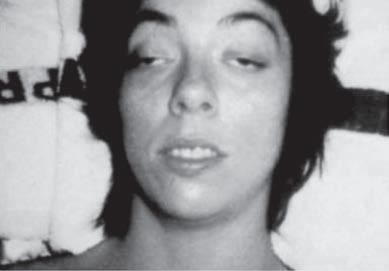
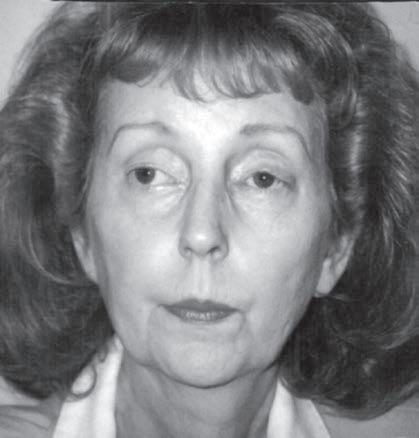
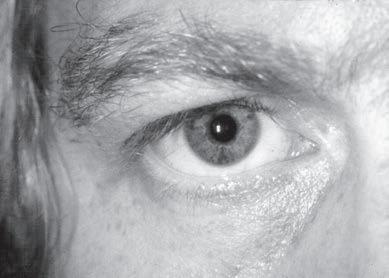
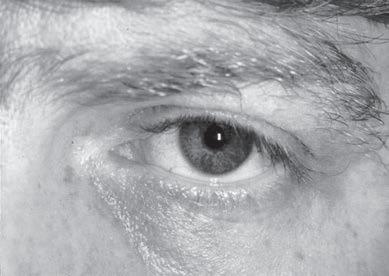
Examination of the eyelids and eye movements is helpful to diagnose acute paralysis in diabetic ophthalmoplegia and Miller-Fisher syndrome or chronic paralysis in mitochondrial myopathy and oculopharyngeal dystrophy (Fig. 1.10). Fluctuating ophthalmoplegia and ptosis are seen in MG (Fig. 1.11) (Barton & Fouladvand, 2000). Assessment of the pupils determines the presence of Horner syndrome (Fig. 1.12), whereas poorly reactive pupils may be seen in some neuropathies (Bertorini, 2002, 2008). The examiner should also assess the presence of eyelid myotonia in the myotonic disorders.
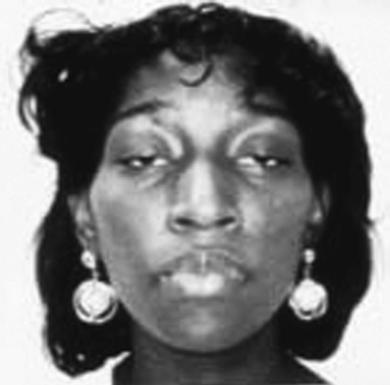
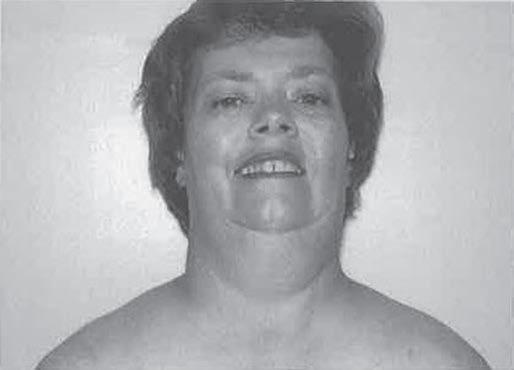
Prominent facial weakness occurs in GBS, but also in MG and some dystrophies (Fig. 1.13). A decreased or hyperactive gag reflex, as in ALS, not only might provide help in the diagnosis but also might determine the risk of aspiration. Tongue atrophy and fasciculations are characteristically seen in motor neuron diseases, whereas a typical forked tongue occurs in MG (Fig. 1.14). Examination of the neck muscles helps to identify neck extensor muscle weakness causing head drop (Fig. 1.15 and Box 1.4) (Narayanaswami & Bertorini, 2000). Rarely, patients with myasthenia can also have a “drop” jaw syndrome.
Manual muscle testing with proper grading helps to determine the distribution and degree of involvement and assess the progression of the disease, as well as diagnose and localize segmental neurologic disorders. For example, focal atrophy in ulnar innervated muscles is seen in ulnar neuropathy, and there is thenar atrophy in median neuropathy or both in C8 radiculopathy and lower plexus lesions. Moreover, dynamometry using different methods, particularly with handheld dynamometers, is used to monitor the progression of muscle weakness.
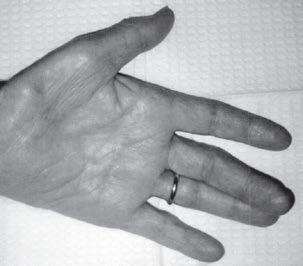
The examination should also include observation for fasciculations, which are more common in motor neuron disorders but also are seen in some neuropathies, such as multifocal motor neuropathy. Increased reflexes with the presence of the Babinski sign indicate involvement of the corticospinal tracts, as in ALS, whereas generalized hypo- or areflexia is seen in peripheral neuropathies, spinal muscular atrophy, and some neuromuscular transmission disorders, such as ELS and botulism. Distal reflexes are lost early in neuropathies and are preserved until the later stages in myopathies (Table 1.1). The examiner also should observe the patient for grip myotonia (Fig. 1.16) and percussion myotonia, myoedema, and slow relaxation of the ankle reflexes, as seen in hypothyroidism.
The sensory examination helps to assess the type and distribution of deficits to determine whether they are distal or symmetric or follow the dermatomes of nerve roots or individual nerves and whether they affect more severely the large myelinated axons (proprioceptive deficits), the unmyelinated axons (dysautonomia, pain, and temperature deficits),
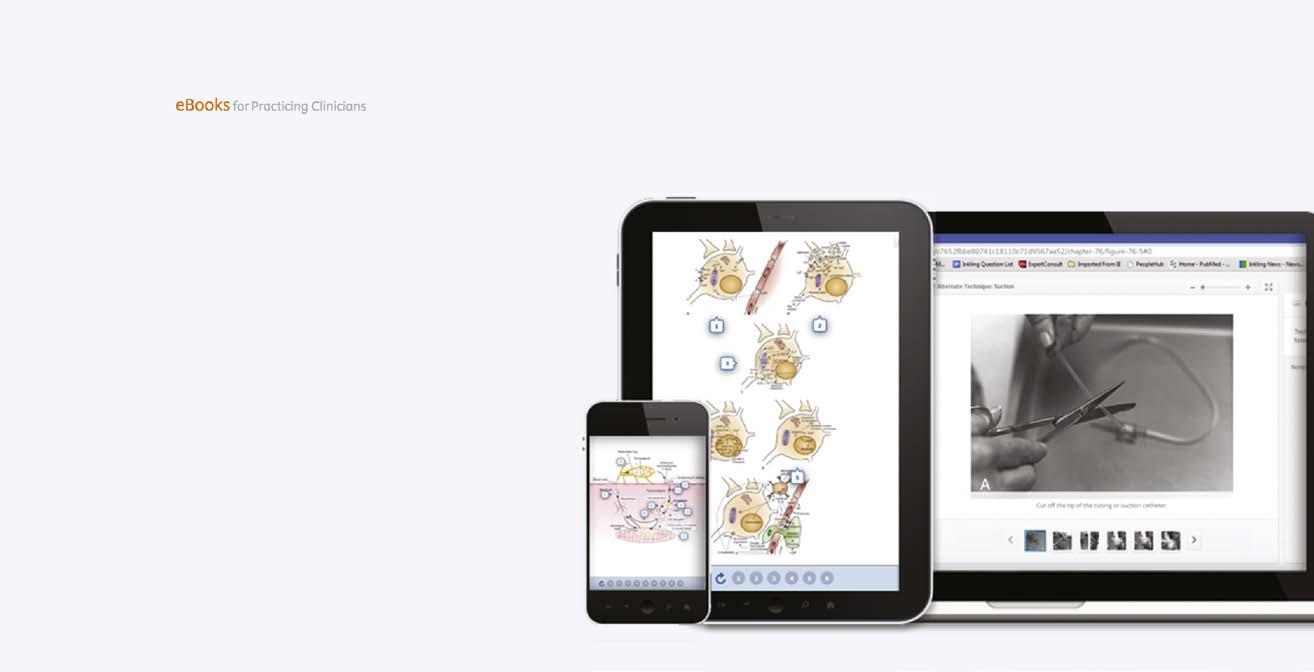
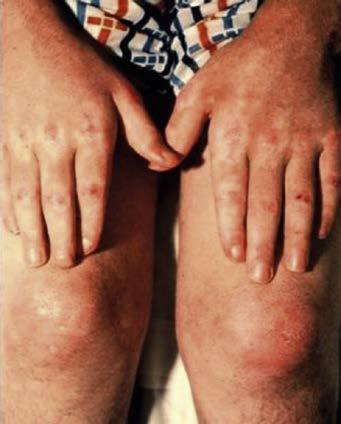
Fig. 1.2 (A) Heliotrope rash in a child with dermatomyositis. (B) Diffuse erythematous facial rash in an adult with dermatomyositis. (Adapted from Bertorini, T. E. (2002). Overview and classification of neuromuscular disorders. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (pp. 1–13). Woburn, MA: Butterworth-Heinemann.) (C) Also see Gottron sign, erythema of the knuckles on dermatomyositis. (2D) Periungual capillary dilatation and carpenter finger in dermatomyositis.)
1.3 White lines (Mees lines) noticed in the fingernails of a patient with arsenic poisoning. (Adapted from Bertorini, T. E. (2002). Overview and classification of neuromuscular disorders. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (pp. 1–13). Woburn, MA: Butterworth-Heinemann.)
and
Fig.
Fig. 1.4 Patient with juvenile acid maltase deficiency showing the Gower sign. Also notice the hyperextension of the elbow while sitting. (From Bertorini, T. E. (2002). Clinical evaluation and clinical diagnostic tests. In T. E. Bertorini (Ed.). Clinical evaluation
diagnostic tests for neuromuscular disorders (p. 31). Woburn, MA: Butterworth-Heinemann.)
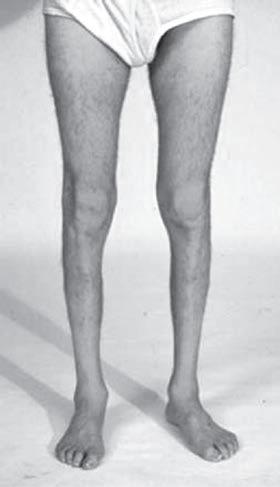
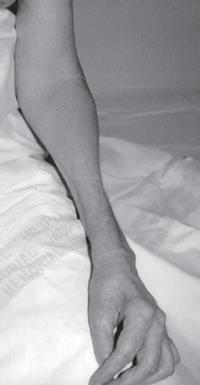
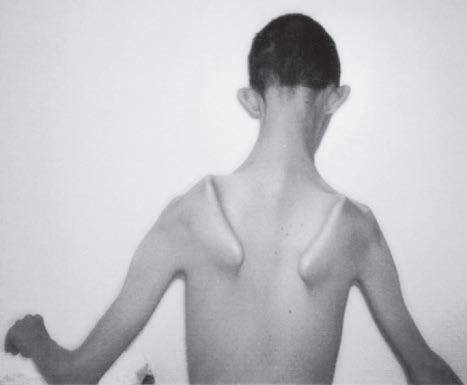
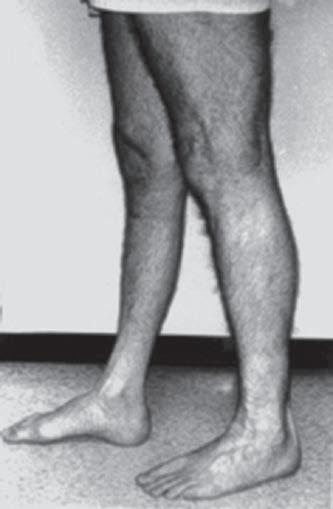
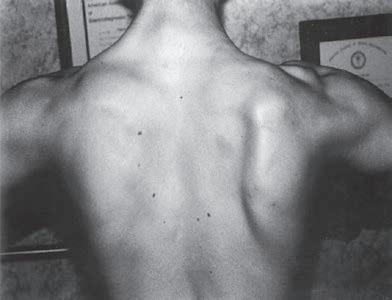
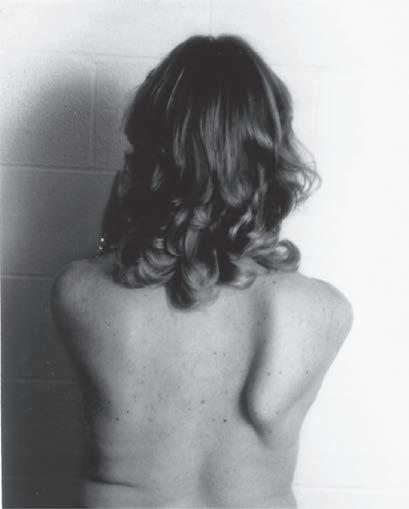
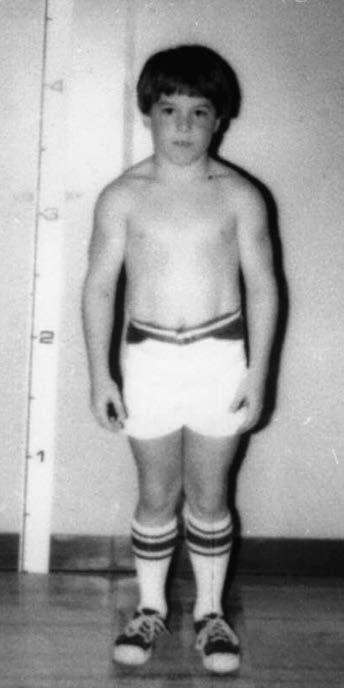
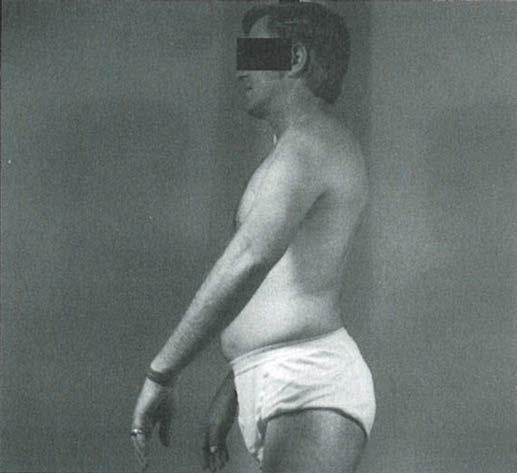
Fig. 1.5 (A) Patient with juvenile spinal muscular atrophy showing pronation of the arms with atrophy of the pectoralis and quadriceps muscles and mild calf hypertrophy. (B) Lordosis, calf hypertrophy, and atrophy of the thigh muscles in a patient with Becker muscular dystrophy. (C) Patient with peripheral neuropathy showing distal leg wasting. (D) Forearm and hand atrophy in a patient with inclusion body myositis. (E) Prominent forearm wasting and wrist extensor weakness in a patient with Welander muscular dystrophy. (F) Patient with congenital myotonic dystrophy with prominent winging and inward rotation of both scapulae.
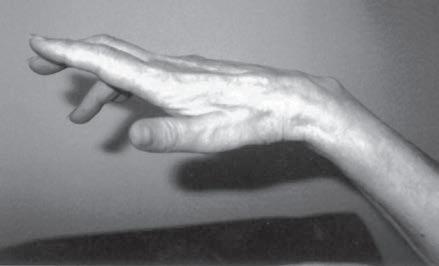
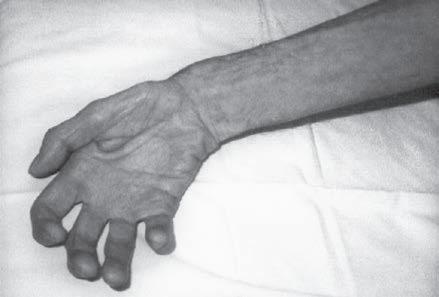
Fig. 1.6 (A) Wasting of the left calf in a patient with postmyelopathy amyotrophy from a conus medullaris lesion involving the anterior horn cells of L5 to S1 segments with chronic denervation on electromyography. (B) Patient with brachial plexopathy and serratus anterior weakness causing winging of the scapula and medial deviation of the bone. (A and B, From Bertorini, T. E. (2008). Neuromuscular case studies. Philadelphia: Butterworth-Heinemann, pp. 247, fig. 11.2.) (C) Patient with scapular winging secondary to spinal accessory neuropathy, showing lateral deviation of the right scapula when raising the arm from upper trapezius weakness; also notice atrophy of the upper trapezius. (From Bertorini, T. E. (2008). Neuromuscular case studies. Philadelphia: Butterworth-Heinemann, pp. 145.) (D) Patient with ulnar neuropathy attempting hand and finger extension, showing partial flexion of the last two digits and atrophy of the first dorsal interosseous muscle. (E) Median neuropathy causing thenar atrophy. (F) Claw hand and atrophy of the median and ulnar innervated muscles. (G) Froment sign. Patient has to flex the distal phalanx to hold the paper from adductor weakness. Note also muscle atrophy in the first dorsal interosseous muscle, left hand. (From Bertorini, T. E. (2008). Neuromuscular case studies. Philadelphia: Butterworth-Heinemann, p. 125.)
Fig. 1.7 A child with autosomal-recessive myotonia congenita. (From Bertorini, T. E. (2008). Neuromuscular case studies. Philadelphia: Butterworth-Heinemann, p. 537.)
Box 1.3 Causes of Floppy Infants
Central Nervous System Disorders
Cerebral palsy
Developmental delay from primary metabolic disorders
Mixed (Central and Peripheral)
Metachromatic leukodystrophy and other lipidosis
Neuroaxonal atrophy
Giant axonal neuropathy
Merosin-deficient muscular dystrophy, other congenital muscular dystrophies (e.g., Fukuyama type)
Anterior Horn Cell Diseases
Infantile spinal muscular atrophy
Neuropathies
Charcot-Marie-Tooth disease, particularly types 3 and 4
Fig. 1.8 Floppy infant with infantile acid maltase deficiency. Note how the limbs hang loosely and the chest is arched when the examiner holds the patient by the thorax. (From Bertorini, T. E. (2002). Clinical evaluation and clinical diagnostic tests. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (p. 68). Woburn, MA: Butterworth-Heinemann.)
Other congenital myopathies (e.g., central core disease, myotubular myopathy, nemaline myopathy)
Congenital myotonic dystrophy
Myopathy from electrolyte and endocrine abnormalities
or both (Bertorini, 2008; Haerer & DeJong, 1992; Ochoa, 1995; Reese, Lovelace-Chandler, & Soderberg, 1999).
Quantitative sensory testing is a subjective test that evaluates perception of temperature, pain, and vibratory sense. This can be used in the assessment of patients with sensory deficits and in clinical trials (Fig 1.17).
DIAGNOSTIC TESTS
Laboratory studies should include a complete chemistry profile, which can help in the diagnosis of several disorders; for example, low or high potassium is seen in the periodic paralyses, whereas hypocalcemia and hypomagnesia are associated with tetany. Hypercalcemia could lead to the diagnosis of hyperparathyroidism. Elevated blood sugar levels could indicate diabetes as a cause of peripheral neuropathy, and if blood sugar levels are normal and the diagnosis is suspected, this testing should be followed by measurement of glucose tolerance test or 2-hour postprandial blood sugar and glycosylated hemoglobin levels. A complete blood count also is helpful to assess for anemia, as seen in connective tissue diseases; B12 deficiency; and leukocytosis, indicating infection or leukopenia from medication effects. An elevated erythrocyte sedimentation rate implies an inflammatory process, although it has low specificity. An increased mean corpuscular volume could suggest pernicious anemia or folate deficiency, and the results of treatment such as with azathioprine (Pruthi & Tefferi, 1994). Testing for serum muscle enzymes is important, particularly serum creatine kinase (CK), aspartate aminotransferase, alanine aminotransferase, and aldolase, which are elevated in myopathies, hypothyroidism, and sometimes to a lesser degree in motor neuron disorders (Welch & Goldberg, 1972). Elevated levels of alanine aminotransferase and aspartate aminotransferase alone suggest liver disease, and when this is considered,
gamma glutamyl transpeptidase should be measured because it is affected only in liver disease. A very high CK level with myoglobulin in plasma and urine is characteristic of rhabdomyolysis (Bertorini, 1997, 2002).
Measurement of a number of antibodies is helpful in the diagnosis. These include, for example, the association of GM1 antibodies in multifocal motor neuropathy or to myelin associated glycoprotein (MAG) in older patients with distal demyelinating neuropathy. In acute autoimmune neuropathy, those with the Miller Fisher variant of Guillain-Barre syndrome usually have antibodies against GQ1b, whereas the pharyngeal brachial variant could be against GT1a and GQ1b antibodies. The acute sensory ataxic neuropathy can be associated GQ1b and GD1b antibodies (see Chapter 15 for details).
Recent data indicate that antibodies against trisulfated heparan disaccharide and fibroblast growth factor receptor 3 can be associated with chronic small fiber sensory neuropathy (Levine et al., 2020).
Assessment of autoimmune myopathies and neuropathies should also include measurement of complement, lupus and rheumatoid arthritis serology, and cryoglobulins (Agnelio, 1995). SSA and SSB antibodies should be tested if Sjögren syndrome is suspected, particularly as the cause of ganglioneuritis and myositis. An acute dysautonomia can be associated with ganglionic acetylcholine receptor antibodies. In chronic inflammatory demyelinating polyneuropathy (CIDP), there are cases with antibodies against adhesion antigens. Others include the Hu antibodies in autoimmune paraneoplastic neuropathies, and those against glutamic acid decarboxylase and antiphysin are seen in stiff person syndrome. Assessment of acetylcholine receptor and MuSK, as well as LRP4 and Agrin antibodies, if necessary, helps in those suspected of having MG, whereas elevation of voltage-gated calcium channel antibodies is seen in patients with ELS, and those against voltage-gated potassium channels (Archelos & Hartung, 2000; Narayanaswami, 2002; Sherer,
Fig. 1.9 (A) Muscle contractures from fibrosis in a patient with Emery-Dreifuss muscular dystrophy. (B) Pentazocine myopathy causing fibrosis resulting in the levitation sign with the arm. (From Bertorini, T. E. (2002). Clinical evaluation and clinical diagnostic tests. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (p. 69). Woburn, MA: Butterworth-Heinemann.)
Fig. 1.10 (A) Patient with diabetic third-nerve palsy with ptosis of the left eye (left). Limitation of adduction of the same eye (right). (B) Ophthalmoplegia and ptosis in a patient with Miller-Fisher syndrome. (C) Ptosis and symmetric limitation of gaze in a patient with mitochondrial myopathy. (D) Astronomer’s posture in a patient with oculopharyngeal dystrophy showing ptosis and contraction of the frontalis muscle to compensate for the ptosis. (A, C, and D, From Bertorini, T. E. (2002). Clinical evaluation and clinical diagnostic tests. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (pp. 15–97). Woburn, MA: Butterworth-Heinemann; B, From Bertorini, T. E. (2008). Neuromuscular case studies. Philadelphia: Butterworth-Heinemann, p. 288.)
Fig. 1.11 (A) Patient with myasthenia gravis. (B) Development of ptosis on sustained upward gaze.
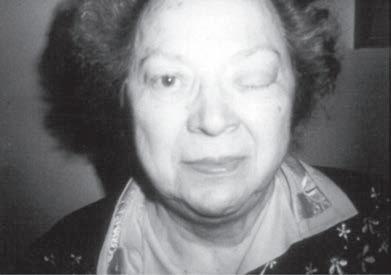
Fig. 1.12 Horner syndrome of the left eye (B) in a patient with lymphoma of the lower brachial plexus showing ptosis and a smaller pupil in the affected eye compared with the normal eye (A).
Fig. 1.13 (A) Horizontal smile in a patient with fascioscapulohumeral dystrophy. (B) Patient with fascioscapulohumeral dystrophy with a weak orbicularis oculi and positive Bell’s palsy (both eyeballs rolling upward). (C) Patient with fascioscapulohumeral muscular dystrophy showing dimples in the upper lip from weakness of the orbicular oris. (From Bertorini, T. E. (2002). Clinical evaluation and clinical diagnostic tests. In T. E. Bertorini (Ed.). Clinical evaluation and diagnostic tests for neuromuscular disorders (p. 37). Woburn, MA: Butterworth-Heinemann.)