Director of the Headache Center Professor of Neurology University of California, San Francisco
San Francisco, California
OTHER VOLUMES IN THE SERIES
Headache and Facial Pain
Epilepsy
Neuro-Ophthalmology
Pain
Emergency Neurology
Neuroinfections
Neurogenetics
Neurotology
Pediatric Neurology, Second Edition
Neurocritical Care, Second Edition
Stroke, Second Edition
Peripheral Nerve and Muscle Disease, Second Edition
Cerebrovascular Disease, Second Edition
Movement Disorders, Second Edition
Women’s Neurology
Neuroimmunology
SECOND EDITION
Edited by Aaron E. Miller, MD
Medical Director
Corinne Goldsmith Dickinson Center for Multiple Sclerosis
Professor and Vice-Chair for Education
Department of Neurology
Icahn School of Medicine at Mount Sinai New York, NY
Tracy M. DeAngelis, MD
Adjunct Assistant Professor of Neurology
The Mount Sinai Hospital New York, NY
Michelle Fabian, MD
Assistant Professor of Neurology
Corinne Goldsmith Dickinson Center for Multiple Sclerosis
The Mount Sinai Hospital New York, NY
Ilana Katz Sand, MD
Assistant Professor of Neurology
Icahn School of Medicine at Mount Sinai
Associate Medical Director
Corinne Goldsmith Dickinson Center for Multiple Sclerosis
The Mount Sinai Hospital
New York, NY
1
Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and certain other countries.
Published in the United States of America by Oxford University Press 198 Madison Avenue, New York, NY 10016, United States of America.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by license, or under terms agreed with the appropriate reproduction rights organization. Inquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above.
You must not circulate this work in any other form and you must impose this same condition on any acquirer.
Library of Congress Cataloging-in-Publication Data
Names: Miller, Aaron E., author. | DeAngelis, Tracy M., author. | Fabian, Michelle, author. | Sand, Ilana Katz, author.
Title: Neuroimmunology / by Aaron E. Miller, Tracy M. DeAngelis, Michelle Fabian, Ilana Katz Sand. Other titles: What do I do now?
Description: Second edition. | Oxford ; New York : Oxford University Press, [2018] | Series: What do I do now? | Includes bibliographical references and index. Identifiers: LCCN 2018009729 | ISBN 9780190693190 (paperback : alk. paper)
Subjects: | MESH: Demyelinating Diseases—diagnosis | Demyelinating Diseases—therapy | Neurologic Manifestations | Vasculitis, Central Nervous System | Nervous System—immunology | Case Reports
Classification: LCC QP356.47 | NLM WL 141 | DDC 616.97/8—dc23 LC record available at https://lccn.loc.gov/2018009729
This material is not intended to be, and should not be considered, a substitute for medical or other professional advice. Treatment for the conditions described in this material is highly dependent on the individual circumstances. And, while this material is designed to offer accurate information with respect to the subject matter covered and to be current as of the time it was written, research and knowledge about medical and health issues is constantly evolving and dose schedules for medications are being revised continually, with new side effects recognized and accounted for regularly. Readers must therefore always check the product information and clinical procedures with the most up-to-date published product information and data sheets provided by the manufacturers and the most recent codes of conduct and safety regulation. The publisher and the authors make no representations or warranties to readers, express or implied, as to the accuracy or completeness of this material. Without limiting the foregoing, the publisher and the authors make no representations or warranties as to the accuracy or efficacy of the drug dosages mentioned in the material. The authors and the publisher do not accept, and expressly disclaim, any responsibility for any liability, loss or risk that may be claimed or incurred as a consequence of the use and/or application of any of the contents of this material.
9 8 7 6 5 4 3 2 1
Printed byMarquis, Canada
This Book Is Dedicated to
Ellen, whose unwavering support makes everything possible and whose love makes every day a treasure.
AEM
To the memory of Dr. Izabella Rozenfeld, dear “Bella,” whose knowledge and unbridled passion for neurology guide me still.
TMD
To my mentors, who have given me the priceless gift of their knowledge and time; and to my patients who have trusted me to share the journey of illness with them, and who are so much more than a case in a book.
MTF
To my dad, my first and forever mentor, whom I can always count on for my “what do I do now?” questions.
IKS
24 Confusion, Muscle Cramps, and Weight Loss 127
25 The Unforgettable Encephalitis 133
26 The Peppered Brainstem 139
27 A Case of a Fall After a Trip 143
28 A Case of Relapsing Visual Loss 149
29 Checks and Balances 153
30 A Case of Intractable Vomiting 157
31 A Case of Cognitive Change in HIV 163
32 A Case of Progressive Myelopathy 167
33 A Case of Cranial Neuropathies and Leptomeningeal Enhancement 173
34 A Case of Diabetes Insipidus and Enhancing Brain Lesions 177
35 A Case of Optic Neuritis and Leg Weakness 183
36 A Man with Multiple Sclerosis and Continuing Disease Activity on Disease-Modifying Therapy 189
37 A Case of Syncope and Impaired Micturition 195
38 A Case of Longitudinally Extensive Transverse Myelitis 201
39 A Perplexing Pattern of Weakness 205
40 Progressive Numbness and Tingling 209
Index 215
Preface
In the six years since the publication of the first edition of Neuroimmunology in the What Do I Do Now? series, novel immunological disorders of the nervous system have been discovered, and understanding of this fascinating class of diseases has burgeoned. New therapies based on large clinical trials for well-known disorders such as MS have proliferated, whereas treatment for less common disorders remains anecdotal, albeit based on increasing experience of experts.
In this second expanded edition of Neuroimmunology, our goal remains to provide a useful framework for clinicians to use when approaching challenging immune-mediated diseases of both the central and peripheral nervous systems. Each chapter begins with a very brief case presentation, including just enough information to challenge your thinking. The subsequent discussions focus on localization, differential diagnosis, and treatment specific to the case and disorder itself. We have presented a range of conditions, including both those frequently encountered such as multiple sclerosis and those much more rarely seen, such as several unique autoimmune encephalopathies. In MS, the availability of new therapies has created a variety of scenarios, only a few of which could be presented in this small volume. Our recommendations for diagnostic testing and therapy rest, whenever possible, on available current evidence, as reflected in the updated key references for this edition. Key clinical points are highlighted in the conclusion of each chapter. In select chapters, tables and figures are provided to illustrate cardinal teaching points such as diagnostic criteria.
We trust this volume will serve as an accessible and practical tool for neurologists and other clinicians, both those in practice and those still in training. We hope it will be an important and useful resource for both clinicians and their patients in helping to facilitate better recognition and management of an ever-increasing number of neurological disorders.
Aaron E. Miller, MD
Tracy M. DeAngelis, MD
Michelle Fabian, MD
Ilana Katz Sand, MD
Acknowledgments
Many of the cases described in this book have involved challenging presentations of neuroimmunological disorders requiring the expertise and support of a multidisciplinary clinical care team at Mount Sinai Hospital or North Shore University Hospital. These patients have been the subject of extensive discussion among our colleagues at the Corinne Goldsmith Dickinson Center for Multiple Sclerosis at Mount Sinai or at Neurological Associates of Long Island. These consummate professionals have all shared their wisdom and experience, in addition to their passionate provision of excellent, compassionate patient care.
In addition, we acknowledge the invaluable collaboration with our colleagues in the fields of neuroradiology, neuro-ophthalmology, rheumatology, internal medicine, and neuro-critical care, among others. We would also like to recognize the residents and fellows at the front lines of managing such difficult cases for their diligence, commitment, and dedication that inspire us daily. We especially thank Drs. Bradley Delman, Amit Aggarwal, and Lara Marcuse for sharing their expertise and providing some of the images, and Drs. David Podwall, Denis Ostrovsky, Michael Han, and Vincent DeOrchis for their respective case contributions and neuromuscular knowledge.
Finally, we extend our sincerest gratitude to the team at Oxford University Press, Tiffany Lu and Craig Panner, for their help, support, and persistence in shepherding this volume to completion.
Aaron E. Miller, MD
Tracy M. DeAngelis, MD
Michelle Fabian, MD
Ilana Katz Sand, MD
Neuroimmunology
1 A Case of Monocular Blurred Vision
A 26-year-old white woman consults you one month after she was diagnosed with right optic neuritis by an ophthalmologist. She reports that initially she “could not see much out of the right eye.” She denied pain on eye movement, but she did experience supraorbital headache. She was treated with three days of intravenous methylprednisolone, followed by oral taper of prednisone over the next two weeks. Her vision markedly improved, though she does not feel it is quite back to normal. Her past medical and neurological history is negative.
The patient’s neurological examination is normal except that she notes that a bright red stimulus appears less intense with the right eye than with the left, and she has a right afferent pupillary defect (APD).
What do you do now?
MULTIPLE SCLEROSIS
This patient presents with optic neuritis, which may be a monophasic illness, but is very commonly the initial manifestation of multiple sclerosis (MS). Most cases of optic neuritis occur in the retrobulbar portion of the nerve so that, acutely, the fundus appears normal. Eventually some degree of optic atrophy may become evident as pallor of the optic disk. An APD is demonstrated by the “swinging flashlight” test. When the flashlight is initially shone in the affected right eye, both direct and consensual pupillary constriction occur.
The light is then shone in the left eye, and both pupils remain constricted. However, when the flashlight swings back to the right eye, the pupils both dilate because the intensity of the light stimulus is now perceived as less in the eye that had the optic neuritis. This sign often persists even in an eye with normal visual acuity after recovery from optic neuritis, indicating residual damage to the optic nerve.
A definite diagnosis of MS depends on the demonstration of clinical symptoms and signs that indicate a disease process that involves the central nervous system (CNS; brain, spinal cord, and optic nerves), is disseminated in space (meaning involvement of two or more non-contiguous areas of the CNS) and time (meaning two or more episodes separated by at least a month), and the absence of a better clinical explanation. It usually presents in young to middle-aged adults, but neither young children nor older adults are completely spared. This patient has had no prior clinical events, but modern diagnostic criteria allow evidence for dissemination in space or time (or both) to be obtained with magnetic resonance imaging (MRI).
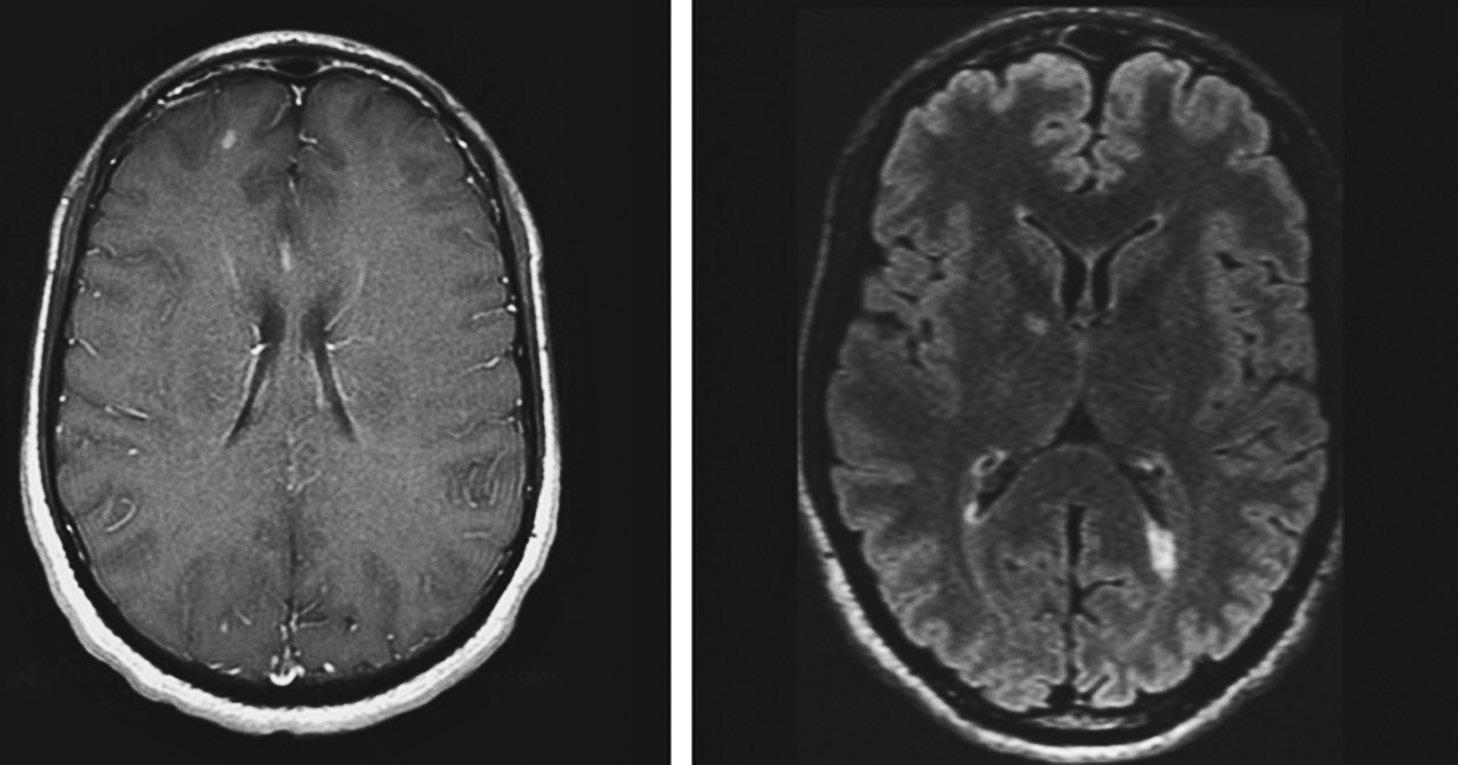
At this point, therefore, you should order a brain MRI with and without contrast administration. You should also order dedicated fat-saturated orbital imaging to evaluate the optic nerve, mainly to exclude an alternative diagnosis. In this patient, the brain MRI showed multiple T2 hyperintense lesions. In addition, she had many gadolinium-enhancing (GdE) lesions (Figure 1.1). Today, the most widely used criteria for making the diagnosis of MS are known as the “McDonald criteria,” with the most recent revision in 2017. Although these criteria allow for the traditional clinical diagnosis, their use often enables an earlier diagnosis to be made by allowing MRI findings to satisfy criteria for either dissemination in space or time, or both.
FIGURE 1.1 This patient met criteria for definite MS by the 2017 revision of the McDonald criteria because, at the time of his presentation with a clinically isolated syndrome, his MRI showed both T2 hyperintense lesions (FLAIR sequence, right) and a gadolinium-enhancing lesion in the right frontal lobe (left).
The current criteria require the presence of both symptoms suggestive of involvement of the CNS and objective signs on the neurological examination. Dissemination in space (DIS) could be satisfied by the presence on MRI of at least one lesion in at least two of the following locations:
Periventricular
Juxtacortical or cortical
Infratentorial
Spinal cord
Dissemination in time (DIT) can be satisfied by the appearance of any new lesion on a subsequent MRI performed after a previous MRI. Alternatively, if both GdE and other non-enhancing lesions appear on the initial MRI, the criteria for DIT are met.
Cerebrospinal fluid (CSF) studies are also extremely helpful in confirming the diagnosis of MS. Routine studies on the CSF are usually normal, or may show a mild mononuclear pleocytosis (under 50 cells) or a mildly elevated protein. In most cases (as many as 90% in some published series, but probably closer to 60–70% in early cases in routine clinical practice), the CSF shows evidence of abnormal immunoglobulin production, demonstrated by either the presence of oligoclonal bands or elevation of the IgG index of IgG synthesis rate. While these abnormal CSF findings (oligoclonal bands
are more specific) are not entirely pathognomonic of MS, they usually help to eliminate alternative diagnoses. In a major change with the 2017 revision of the McDonald criteria, the presence of two or more oligoclonal bands in the CSF of a patient who presents with a typical clinically isolated syndrome substitutes for DIT, even when criteria for DIT are not met on the MRI. Therefore, a patient who presents with an initial event typical of MS, meets MRI criteria for DIS, and has oligoclonal bands in the CSF may be diagnosed with definite MS. In all cases, however, it is imperative that there be no better clinical explanation for the patient’s symptoms (Table 1.1).
Optic neuritis is also a major manifestation of neuromyelitis optica spectrum disorder (NMOSD), a condition discussed in Chapter 30. The availability of serological testing for anti-aquaporin4 antibody has greatly facilitated the diagnosis of this condition and has a sensitivity of more than 70%, with high specificity. Although this patient’s brain MRI is much more typical of MS than of NMOSD, some experts now routinely order aquaporin4 antibody testing in anyone presenting with optic neuritis.
TABLE 1.1 2017 Revised McDonald Criteria for Determination of Dissemination in Space and Time
Attacks Lesions Additional Requirements
2 or more 2 or more None
2 or more 1
1 2
1 1
0 1
DIS:
❑ Further attack or ❑ MRI* (≥1 T2 lesions in ≥2 of 4 areas1)
DIT:
❑ Further attack or
❑ MR (presence of GdE and non-GdE lesions) or CSF-OCB
DIS and DIT
❑ >1 year of progression and ❑ 2 of 3 of:
◦ T2 hyperintense lesions in brain—1 of 3 areas2
◦ T2 hyperintense lesions in spinal cord—≥2 areas
◦ CSF-OCB
After the MRI has been completed, you should inform this patient that she has a diagnosis of definite relapsing-remitting MS (RRMS). MS is currently described, according to its temporal profile, as RRMS, secondary progressive (SPMS), or primary progressive (PPMS). Approximately 80–85% of patients begin with RRMS, in which they have episodes of neurological symptoms, typically lasting days to weeks, followed by recovery or improvement before the development of another attack (also called exacerbation or relapse). Conventionally, an attack is considered new when it occurs at least 30 days after the onset of symptoms in the previous exacerbation. Most of the remaining 15–20% of patients have PPMS, in which symptoms and signs—most often those of spastic paraparesis—progress gradually without any acute exacerbations. Many patients—often estimated at about 50% or even more—who begin with RRMS eventually change to a course that is gradually worsening. Such a course is then labeled SPMS. This insidiously progressive course may or may not be accompanied by superimposed exacerbations. Recently, an international committee has recommended that any type of MS be further characterized by the presence or absence of activity (by either clinical or imaging evidence), always placed within a temporal context (e.g., one year). Progressive forms of MS may plateau, so these phenotypes should be further modified by the description “with or without progression,” again within a temporal framework.
When informing the patient of the diagnosis of MS, you should do so in the most hopeful terms possible, a position currently justified by the availability of a variety of medications to treat RRMS and the promise of additional drugs in the near future. You should always inform the patient in person, rather than in a telephone conversation or message. You should tell the patient specifically that he or she has MS and avoid the use of euphemisms, such as “demyelinating disease.” The diagnosis should be accompanied by an explanation of the condition in terms that the patient can readily understand.
After giving the patient the diagnosis of MS, you should recommend initiation of treatment with a disease-modifying agent. However, generally, you should defer the discussion of the specific therapeutic options to a subsequent meeting with the patient. Receiving the diagnosis of MS, even if it was not totally unexpected, is an emotionally upsetting experience, and it
will be difficult for the patient to focus on the details of the therapy discussion at that time.
A wide variety of disease-modifying agents is currently available. The oldest medications, including several interferon beta preparations and glatiramer acetate, are administered by injection (mainly subcutaneous). These agents are modestly effective, but extremely safe. Newer medications are generally more effective but convey additional risks, including those of serious infection. Oral agents include fingolimod, teriflunomide, and dimethyl fumarate. Probably the most efficacious disease-modifying agents are monoclonal antibodies that are administered by intravenous infusion. These include natalizumab, administered every four weeks; ocrelizumab, given every six months; and alemtuzumab (which, in the United States, is recommended only for patients failing or not tolerating multiple other agents), administered for five days initially and then for three days a year later. Detailed discussion of the disease-modifying agents’ respective advantages and disadvantages is beyond the scope of this chapter, but is available in the suggested readings.
KE y POINTS TO REMEMBER
• Multiple sclerosis is an inflammatory demyelinating disease of the CNS that usually affects young to middle-aged adults and women much more often than men.
• The key to diagnosis is the demonstration of DIS and DIT, for which the MRI is extremely helpful.
• CSF analysis is very helpful in MS, with most patients showing at least two oligoclonal bands (OCB). In the 2017 McDonald criteria, the presence of OCB may substitute for DIT.
• The disease phenotype is described as RRMS, SPMS, or PPMS.
• Acute attacks are usually treated with high-dose corticosteroids.
• Many disease-modifying agents are available to treat relapsing forms of MS, differing in route of administration, efficacy, tolerability, and risks.
Further Reading
Bermel RA, Fox RJ. MRI in multiple sclerosis. Continuum: Lifelong Learn Neurol. 2010;16(5):37–57.
Cree BAC. Diagnosis and differential diagnosis of multiple sclerosis. Continuum: Lifelong Learn Neurol. 2010;16(5):19–36.
Graber JJ, McGraw CA, Kimbrough D, Dhib-Jalbut S. Overlapping and distinct mechanisms of action of multiple sclerosis therapies. Clin Neurol Neurosurg. 2010;112(7):583–591.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the “McDonald criteria.” Ann Neurol. 2011;69(2):292–302.
2 A Case of Multifocal Neurological Symptoms
A 25-year-old man presents with two days of double vision, unsteady gait, and mild confusion. He denies headache, fever, recent infection, or travel, but reports receiving a flu vaccination two weeks prior. On examination, he is oriented to self and place but does not know the date, and his mentation is slow. There is a left internuclear ophthalmoplegia and a left adduction palsy. Strength is full, but he has diffuse hyperreflexia and a right extensor plantar response. Vibration and joint position sense involving his right foot are impaired. Gait is moderately dystaxic.
MRI demonstrates multiple T2 hyperintense white matter lesions and a lesion in the left rostral midbrain. The majority demonstrate gadolinium enhancement, some with a distinct open-ring pattern. There are two enhancing spinal cord lesions. CSF analysis shows 26 WBC (lymphocytes), protein 55, negative HSV-2 PCR, Lyme serology, VDRL, and normal cytology. Oligoclonal bands are absent.
What do you do now?
ACUTE DISSEMINATED ENCEPHALOM y ELITIS
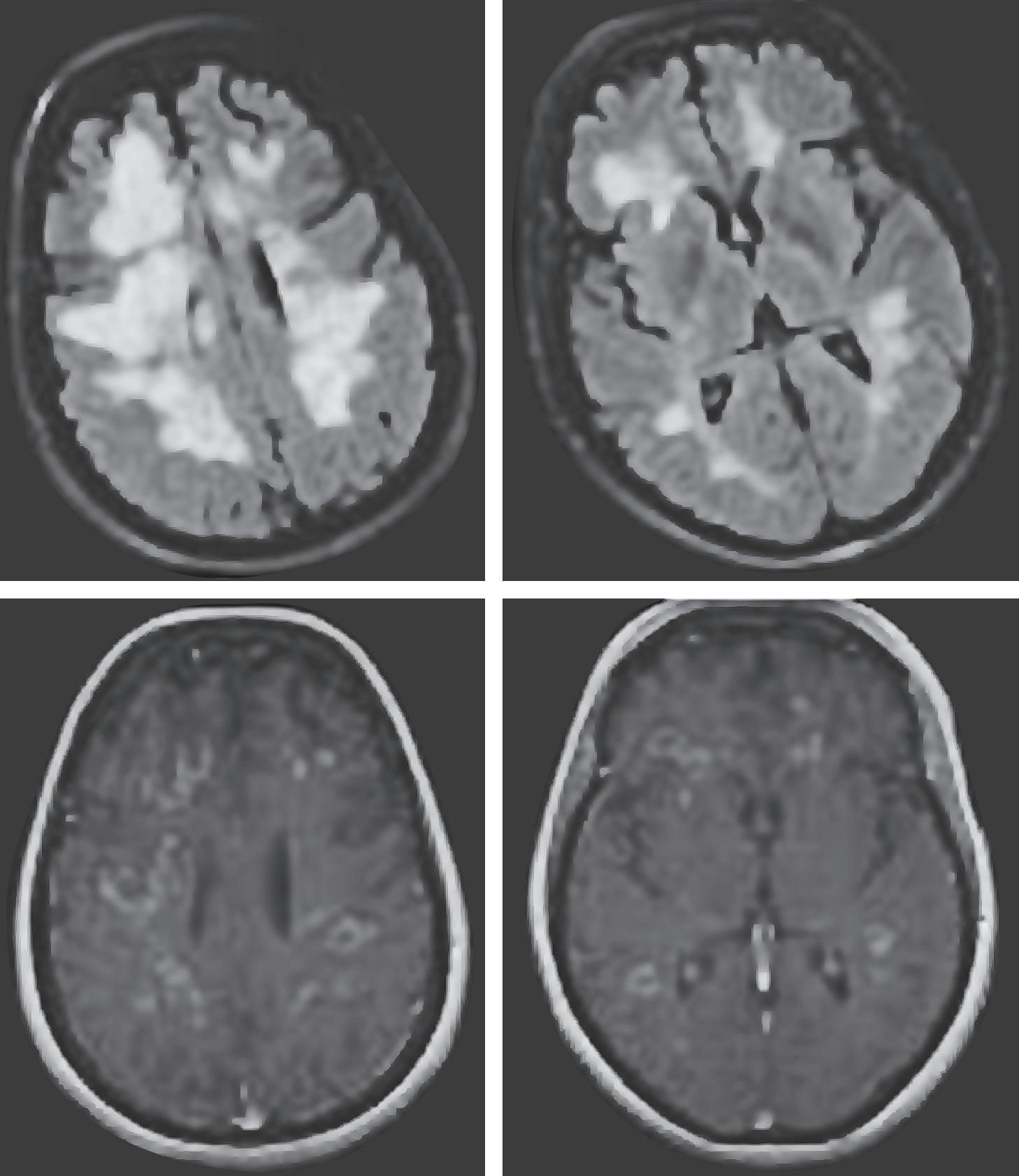
This patient presents with a post-vaccinal neurological syndrome with multifocal symptomatology and deficits with supratentorial and infratentorial involvement, as well as encephalopathy. MR imaging demonstrates multiple T2/FLAIR hyperintense white matter abnormalities involving both the brain and spinal cord, the majority of which demonstrate gadolinium enhancement, including an open-ring enhancement pattern radiographically characteristic of demyelination (Figure 2.1). This presentation is
FIGURE 2.1 MRI from a patient with acute disseminated encephalomyelitis showing contrast enhancement in nearly all lesions (lower panels). The upper panels show the lesions hyperintense on T2-weighted imaging. Reprinted with permission from Bermel RA, Fox RJ. MRI in multiple sclerosis. Continuum: Lifelong Learning in Neurology. 2010;16(5):46.
(A)
(B)
(C)
(D)
highly suspicious for acute disseminated encephalomyelitis (ADEM), an immune-mediated demyelinating disorder characterized by multifocal CNS involvement and encephalopathy. Children are preferentially affected. Various vaccines and systemic infections have been linked to ADEM, including influenza, varicella zoster, herpes simplex, measles, mumps, rubella, Borrelia burgdorfei, mycoplasma, and Epstein-Barr virus. Of note, however, ADEM, more often than not, develops in the absence of any antecedent infection. In addition to multifocal CNS involvement and encephalopathy, 2013 diagnostic criteria require MRI lesions typical for demyelination as well as the lack of new symptoms/signs more than three months after onset.
The onset of ADEM is generally rapid, and peak neurological dysfunction manifests within several days. Clinical symptoms are multifocal and varied, including encephalopathy, cerebral hemispheric deficits such as hemiparesis and hemisensory loss, brain stem dysfunction, and myelopathy, as well as seizures. The degree of encephalopathy can range from mild lethargy and confusion to frank coma. Rarely, a hyperacute, severe variant of ADEM occurs, referred to as acute hemorrhagic leukoencephalitis (AHLE), which pathologically involves petechial hemorrhage and venular necrosis. In cases of AHLE, CSF analysis generally demonstrates increased opening pressure, elevated protein, and a pleocytosis of both red and white blood cells. As such, the differential diagnosis here would include hemorrhagic, necrotizing infectious processes such as herpes simplex meningoencephalitis, brain abscesses, and neoplasms such as metastatic melanoma. Peripheral nervous system involvement has been described, in the form of acute polyradiculoneuropathies, more commonly in adult rather than pediatric presentations.
ADEM can be difficult to distinguish from a severe, fulminant first episode of multiple sclerosis (MS), but differentiating these two entities has critical long-term therapeutic implications. ADEM is typically a self-limited monophasic illness and does not require the chronic immunomodulatory or immunosuppressive therapies of MS. Factors suggested as supportive of ADEM include the presence of encephalopathy, seizures, radiographic presence of poorly marginated, large, sometimes tumefactive-sized lesions, deep gray matter and cortical lesions, and a predominance of gadoliniumenhancing lesions. Both incomplete and complete ring-shaped patterns of