Hematopathology
THIRD EDITION
A Volume in the Series: Foundations in Diagnostic Pathology
Series Editor
John R. Goldblum, MD Chairman of Pathology
Cleveland Clinic, Professor of Pathology
Cleveland Clinic Lerner College of Medicine
Cleveland, Ohio
1600 John F. Kennedy Blvd.
Ste 1800 Philadelphia, PA 19103-2899
HEMATOPATHOLOGY: A VOLUME IN THE SERIES: FOUNDATIONS IN DIAGNOSTIC PATHOLOGY, THIRD EDITION
Copyright © 2018 by Elsevier, Inc. All rights reserved.
ISBN: 978-0-323-47913-4
No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or any information storage and retrieval system, without permission in writing from the publisher. Details on how to seek permission, further information about the Publisher’s permissions policies and our arrangements with organizations such as the Copyright Clearance Center and the Copyright Licensing Agency, can be found at our website: www.elsevier.com/permissions
This book and the individual contributions contained in it are protected under copyright by the Publisher (other than as may be noted herein).
Knowledge and best practice in this field are constantly changing. As new research and experience broaden our understanding, changes in research methods, professional practices, or medical treatment may become necessary.
Practitioners and researchers must always rely on their own experience and knowledge in evaluating and using any information, methods, compounds, or experiments described herein. In using such information or methods they should be mindful of their own safety and the safety of others, including parties for whom they have a professional responsibility.
With respect to any drug or pharmaceutical products identified, readers are advised to check the most current information provided (i) on procedures featured or (ii) by the manufacturer of each product to be administered, to verify the recommended dose or formula, the method and duration of administration, and contraindications. It is the responsibility of practitioners, relying on their own experience and knowledge of their patients, to make diagnoses, to determine dosages and the best treatment for each individual patient, and to take all appropriate safety precautions.
To the fullest extent of the law, neither the Publisher nor the authors, contributors, or editors, assume any liability for any injury and/or damage to persons or property as a matter of product liability, negligence or otherwise, or from any use or operation of any methods, products, instructions, or ideas contained in the material herein.
Previous editions copyrighted 2012 and 2007.
Library of Congress Cataloging-in-Publication Data
Names: Hsi, Eric D., editor.
Title: Hematopathology / [edited by] Eric D. Hsi.
Other titles: Hematopathology (Hsi) | Foundations in diagnostic pathology.
Description: Third edition. | Philadelphia, PA : Elsevier, [2018] | Series: Foundations in diagnostic pathology | Includes bibliographical references and index.
Identifiers: LCCN 2017034574 | ISBN 9780323479134 (hardcover : alk. paper)
Subjects: | MESH: Hematologic Diseases–pathology | Hematologic Diseases–diagnosis | Lymphatic Diseases–pathology | Lymphatic Diseases–diagnosis
Classification: LCC RB145 | NLM WH 120 | DDC 616.07/561–dc23 LC record available at https://lccn.loc.gov/2017034574
Content Strategist: Michael Houston
Content Development Specialist: Meghan Andress
Publishing Services Manager: Catherine Jackson
Senior Project Manager: Rachel E. McMullen
Design Direction: Renee Duenow
To my teachers and mentors who inspired me, our trainees who constantly challenge and question, and my family who makes it all worthwhile.
John Anastasi, MD
Associate Professor Department of Pathology
University of Chicago, Pritzker School of Medicine; Director
Section of Hematopathology, Clinical Hematology Laboratory University of Chicago Medical Center Chicago, Illinois
Daniel A. Arber, MD Professor Department of Pathology University of Chicago Chicago, Illinois
Dong Chen, MD, PhD Consultant
Division of Hematopathology Mayo Clinic; Associate Professor Laboratory Medicine and Pathology College of Medicine, Mayo Clinic Rochester, Minnesota
Kudakwashe Chikwava, MB, ChB Assistant Professor Department of Pathology and Cell Biology University of Pennsylvania School of Medicine; Sydney Kimmel Medical College–Thomas Jefferson University; Pediatric Pathologist Department of Pathology
Nemours/Alfred L. duPont Hospital for Children Philadelphia, Pennsylvania
John Kim Choi, MD, PhD Director of Hematopathology Department of Pathology St. Jude Children’s Research Hospital Memphis, Tennessee
James R. Cook, MD, PhD Department of Laboratory Medicine Cleveland Clinic; Associate Professor of Pathology Cleveland Clinic Lerner College of Medicine Cleveland, Ohio
Yuri Fedoriw, MD
Associate Professor, Director of Hematopathology Department of Pathology and Laboratory Medicine University of North Carolina Chapel Hill, North Carolina
Falko Fend, MD
Professor Institute of Pathology
University Hospital Tübingen Tübingen, Germany
Judith A. Ferry, MD Professor of Pathology Department of Pathology
Harvard Medical School; Director of Hematopathology Department of Pathology Massachusetts General Hospital Boston, Massachusetts
William G. Finn, MD Medical Director
Warde Medical Laboratory Ann Arbor, Michigan
Juehua Gao, MD, PhD
Assistant Professor of Pathology Department of Pathology Northwestern University Chicago, Illinois
Tracy I. George, MD Professor
Department of Pathology
University of New Mexico School of Medicine; Vice Chair of Clinical Affairs Hematopathology Division Chief Department of Pathology University of New Mexico Health Sciences Center Albuquerque, New Mexico
Robert Paul Hasserjian, MD
Associate Professor Department of Pathology
Massachusetts General Hospital and Harvard Medical School Boston, Massachusetts
Rong He, MD Consultant, Assistant Professor Division of Hematopathology Mayo Clinic Rochester, Minnesota
James D. Hoyer, MD Consultant, Professor Division of Hematopathology Mayo Clinic Rochester, Michigan
Eric D. Hsi, MD Professor of Pathology Chair
Department of Laboratory Medicine Cleveland Clinic; Professor of Pathology
Cleveland Clinic Lerner College of Medicine Cleveland, Ohio
Rebecca L. King, MD
Assistant Professor of Laboratory Medicine and Pathology Division of Hematopathology
Mayo Clinic
Rochester, Minnesota
Kandice Kottke-Marchant, MD, PhD
Medical Director Hemostasis and Thrombosis Department of Laboratory Medicine Cleveland Clinic Foundation; Professor of Pathology Cleveland Clinic Lerner College of Medicine Cleveland, Ohio
Paul J. Kurtin, MD
Professor of Laboratory Medicine and Pathology Division of Hematopathology
Mayo Clinic Rochester, Minnesota
Steven J. Kussick, MD, PhD
Director of Hematopathology and Flow Cytometry
PhenoPath Laboratories; Clinical Assistant Professor Department of Laboratory Medicine University of Washington Seattle, Washington
Pei Lin, MD Professor
Department of Hematopathology University of Texas–MD Anderson Cancer Center Houston, Texas
William R. Macon, MD Professor of Laboratory Medicine and Pathology Division of Hematopathology Mayo Clinic Rochester, Minnesota
Sara A. Monaghan, MD
Associate Professor Department of Pathology University of Pittsburgh School of Medicine Pittsburgh, Pennsylvania
Megan O. Nakashima, MD Department of Laboratory Medicine Cleveland Clinic Cleveland, Ohio
Phuong L. Nguyen, MD
Associate Professor of Laboratory Medicine and Pathology Division of Hematopathology Mayo Clinic Rochester, Minnesota
Robert S. Ohgami, MD, PhD
Assistant Professor, Associate Program Director for Anatomic Pathology Department of Pathology Stanford University Stanford, California
Jennifer L. Oliveira, MD Consultant, Assistant Professor Division of Hematopathology
Mayo Clinic
Rochester, Minnesota
Dennis P. O’Malley, MD
Hematopathologist
Neogenomics
Aliso Viejo, California
Jennifer Lee Picarsic, MD
Assistant Professor
Department of Pathology
University of Pittsburgh School of Medicine; Attending Pathologist Division of Anatomic Pathology
Children’s Hospital of Pittsburgh of UPMC Pittsburgh, Pennsylvania
Leticia Quintanilla-Martinez, MD Professor Institute of Pathology University Hospital, Eberhard-Karls-University Tübingen Tübingen, Germany
Heesun J. Rogers, MD, PhD
Staff Hematopathologist Department of Laboratory Medicine Cleveland Clinic Foundation Cleveland, Ohio
Jonathan W. Said, MD Professor of Pathology Department of Pathology
David Geffen School of Medicine; Vice Chair Research, Chief of Hematopathology Department of Pathology
David Geffen School of Medicine and UCLA Medical Center Los Angeles, California
Graham W. Slack, MD
Pathologist
Department of Pathology and Laboratory Medicine
British Columbia Cancer Agency; Clinical Associate Professor University of British Columbia Vancouver, British Columbia, Canada
Lauren Smith, MD
Associate Professor of Pathology, Section Head, Hematopathology, Director, Hematopathology Fellowship, Director, Medical School Path of Excellence in Ethics University of Michigan Ann Arbor, Michigan
Beenu Thakral, MD
Assistant Professor
Department of Hematopathology
MD Anderson Cancer Center Houston, Texas
David
S. Viswanatha, MD Consultant, Professor
Division of Hematopathology
Mayo Clinic
Rochester, Minnesota
Sa A. Wang, MD
Associate Professor
Department of Hematopathology
MD Anderson Cancer Center
Houston, Texas
James
M. Ziai, MD Research Pathologist Genentech, Inc.
San Francisco, California
As any practicing pathologist knows, it is not possible for any single individual to master all of the skills and knowledge required to perform all areas of surgical pathology at the highest level. Providing a correct diagnosis is challenging enough, but the standard of care has now far surpassed simply providing an accurate pathologic diagnosis. We are now asked to provide an enormous amount of ancillary information, both diagnostic and prognostic, often on minute tissue fragments, a task that can be daunting even for the most skilled and experienced pathologists.
Although general surgical pathology textbooks remain useful resources, they cannot possibly cover many of the detailed aspects that pathologists need to know and include in their diagnostic reports. For this reason, Foundations in Diagnostic Pathology was initiated. This series of books is designed to cover the major areas of surgical pathology, with each volume focusing on one major topic. The goal of every book in this series, which is now in its third edition, is to provide the essential information that any pathologist—whether general or subspecialized, in training or in practice—would find useful in the evaluation of virtually any type of specimen encountered.
My colleague of many years at Cleveland Clinic, Dr. Eric Hsi, has edited this outstanding book on hematopathology that fulfills the goals and philosophy behind Foundations in Diagnostic Pathology. This third edition cuts to the essentials of what all practicing pathologists want and need to know about
this rapidly evolving group of neoplastic and non-neoplastic disorders. As always, the list of contributors is impressive and includes a who’s who of hematopathologists, all of whom were extremely generous in providing their time and expertise to this book. By design, there is uniformity in the organization of these chapters, each of which includes practical information and numerous photographs that emphasize the essential diagnostic points. Importantly, there is seamless integration of ancillary diagnostic techniques including immunohistochemistry, cytogenetics, and molecular techniques, which are essential to the modern practice of hematopathology.
The book is organized into four major sections: NonNeoplastic Disorders; Lymphomas; Leukemias, Chronic Myeloproliferative Neoplasms, and Myelodysplasia; and finally Disorders of Histiocytes, Mast Cells, Plasma Cells, Spleen, and Ancillary Techniques. This edition features 24 chapters written by leaders in the field, and each chapter cuts to the heart of what practicing pathologists need to know.
I would like to extend my deepest appreciation to Dr. Eric Hsi and all of the authors who have contributed to this outstanding third edition of the Foundations in Diagnostic Pathology series. I strongly believe this edition improves on what was already an exceptional volume in this series, and I am confident you will enjoy this edition of the Foundations in Diagnostic Pathology series.
John R. Goldblum, MD
Since the first edition of this text in 2007, classifications have evolved to incorporate much more information that has an impact on the diagnosis and therapy of hematologic diseases. I am honored to have been asked to embark on a third edition of this book. New authors have been added and chapters updated and revised. I am pleased to have incorporated a more complete treatment of coagulation that serves to differentiate this book as one that now covers the spectrum of non-neoplastic hematopathology, neoplastic hematopathology, and coagulation. I believe this text will give the reader a solid foundation for learning and practicing hematopathology.
Importantly, it incorporates information and classifications relevant to the long-awaited 2016 WHO update. As in previous editions, I am indebted to the numerous expert contributors who have given of themselves to produce excellent and concise chapters (with a minimal amount of nagging from me …). They are the true resource and talent for this book, and it has been an honor to work with this group of outstanding pathologists and educators.
Eric D. Hsi, MD
SECTION 1
Non-Neoplastic Disorders
Chapter 1
Red Blood Cell/Hemoglobin Disorders, 3
Juehua Gao, MD, PhD, and Sara A. Monaghan, MD
Chapter 2
Hemostasis and Thrombosis, 57
Heesun J. Rogers, MD, PhD, Megan O. Nakashima, MD, and Kandice Kottke-Marchant, MD, PhD
Chapter 3
Non-Neoplastic Morphologic Abnormalities of White Blood Cells and Macrophages, 106
Dennis P. O’Malley, MD, Eric D. Hsi, MD, Lauren Smith, MD, and Yuri Fedoriw, MD
Chapter 4
Reactive Lymph Nodes and Castleman Disease, 118
Judith A. Ferry, MD
Chapter 5
Bone Marrow Failure Syndromes, 167
James R. Cook, MD, PhD
Chapter 6
Benign Causes of Bone Marrow Abnormalities Including Infections, Storage Diseases, Systemic Disorders, and Stromal Changes, 184
Dennis P. O’Malley, MD, Lauren Smith, MD, and Yuri Fedoriw, MD
SECTION 2 Lymphomas
Chapter 7
Small B-Cell Lymphomas, 213
Rebecca L. King, MD, and Paul J. Kurtin, MD
Chapter 8
Diffuse Aggressive B-Cell Lymphomas, 271
Graham W. Slack, MD, and Eric D. Hsi, MD
Chapter 9
Peripheral T-Cell Lymphomas, 306
William R. Macon, MD
Chapter 10
Immunodeficiency-Related Lymphoproliferative Disorders, 340
Jonathan W. Said, MD
Chapter 11
Hodgkin Lymphoma, 363
Falko Fend, MD, and Leticia Quintanilla-Martinez, MD
SECTION 3
Leukemias, Chronic Myeloproliferative Neoplasms, and Myelodysplasia
Chapter 12
B-Cell Leukemias of Mature Lymphocytes, 397
Eric D. Hsi, MD
Chapter 13
Mature T-Cell and Natural Killer–Cell Leukemias, 414
William G. Finn, MD, and William R. Macon, MD
Chapter 14
Acute Myeloid Leukemia, 429
Daniel A. Arber, MD
Chapter 15
Precursor Lymphoid Neoplasms, 467
John Kim Choi, MD, PhD
Chapter 16
Acute Undifferentiated Leukemia and Mixed-Phenotype
Acute Leukemias, 481
John Kim Choi, MD, PhD
Chapter 17
Myeloproliferative and “Overlap” Myelodysplastic/ Myeloproliferative Neoplasms, 488
Beenu Thakral, MD, John Anastasi, MD, and Sa A. Wang, MD
Chapter 18
Myelodysplastic Syndromes, 539
Phuong L. Nguyen, MD, and Robert Paul Hasserjian, MD
Red Blood Cell/Hemoglobin Disorders*
■ Juehua Gao, MD, PhD ■ Sara A. Monaghan, MD
■ INTRODUCTION
The disorders of red blood cells can be divided into those of decreased red blood cell (RBC) mass (anemias) and those of increased RBC mass (erythrocytoses). Most of this chapter is devoted to various types of anemias, with only a short discussion of erythrocytosis.
From the pathophysiologic standpoint, anemias can be divided into three categories (Box 1.1): anemias of blood loss, anemias of decreased RBC production, and anemias caused by increased RBC destruction (hemolysis). Each of these categories can be subdivided further, as illustrated in Box 1.1 and subsequent sections.
This chapter focuses on commonly encountered conditions. Various rare causes of anemia and some disorders lacking characteristic changes in peripheral blood or bone marrow are omitted.
■ ANEMIAS OF BLOOD LOSS
Blood loss, the most common cause of anemia, can occur as obvious bleeding from trauma, occult bleeding (e.g., gastrointestinal ulcers, uterine fibroids), induced bleeding secondary to surgery, hemodialysis, or repeated diagnostic testing. Blood loss can be acute or chronic. The anemia of acute blood loss, such as that associated with major trauma, is not immediately evident based on complete blood cell count (CBC) parameters because losses consist of whole blood, and the vascular system contracts on the decreased blood volume. However, with intravascular volume repletion, the erythrocytes will be diluted and the degree of RBC and hemoglobin (Hb) loss will be manifest on the CBC. Chronic, low-level blood loss is initially compensated by increased marrow RBC production; therefore no anemia will
*We gratefully acknowledge the contribution of Steven H. Kroft, MD, who co-authored this chapter in previous editions.
be present, although reticulocytosis may be evident. With prolonged blood loss, body iron stores become depleted, the iron-deficient marrow is no longer able to compensate, and iron deficiency anemia (IDA) develops. Thus the anemia of chronic blood loss is essentially IDA (see the following section).
■ ANEMIAS OF DECREASED RED BLOOD CELL PRODUCTION
Anemias of decreased red blood cell production can be divided into those due to ineffective erythropoiesis and those due to a decrease in effective RBC production.
INEFFECTIVE ERYTHROPOIESIS
Anemias of ineffective erythropoiesis are characterized in general by a failure of the marrow to produce adequate mature erythrocytes, despite erythroid hyperplasia in the marrow, and often demonstrate morphologic abnormalities in erythroid precursors, mature RBCs, or both. The defect may be primarily one of nuclear maturation (megaloblastic anemia) or cytoplasmic maturation (iron deficiency). Myelodysplastic syndromes, although often characterized by ineffective erythropoiesis, are discussed in Chapter 18
MEGALOBLASTIC ANEMIA
The megaloblastic anemias are macrocytic anemias associated with characteristic morphologic features of hematopoietic cells. Cytopenias are due to impaired deoxyribonucleic acid (DNA) synthesis, which leads to abnormal nuclear maturation of hematopoietic precursors and ineffective hematopoiesis. The major causes of megaloblastic anemia are folate deficiency and cobalamin (Cbl; vitamin B12) deficiency. Folate in the form of tetrahydrofolate (THF) is a transporter of 1-carbon units, used for synthesis of purines, thymidine, and methionine
Box 1.1 Classification of Red Blood Cell Disorders
Anemias
Anemias of blood loss
Acute Chronic
Anemias of decreased red blood cell production
Ineffective erythropoiesis
Megaloblastic anemia
Iron deficiency
Myelodysplastic syndromes
Sideroblastic anemia
Congenital dyserythropoietic anemia
Decreased effective erythropoiesis
Stem cell defects
Marrow replacement processes
Anemia of chronic kidney disease
Anemia of chronic disease
Hemolytic anemias
Intrinsic red cell defects
Hemoglobin disorders
Structural hemoglobin disorders
Sickling diseases
Hemoglobin C disease
Thalassemias
Red cell membrane disorders
Hereditary spherocytosis
Hereditary elliptocytosis
Hereditary pyropoikilocytosis
Red cell enzyme disorders
Glucose-6-phosphate dehydrogenase deficiency
Paroxysmal nocturnal hemoglobinuria
Extrinsic red cell defects
Autoimmune hemolytic anemia
Warm
Cold
Microangiopathic hemolytic anemia
Parasitic hemolysis
Malaria
Babesia
Erythrocytosis
Relative erythrocytosis (decreased plasma volume)
Absolute erythrocytosis (polycythemia)
Primary
Polycythemia vera
Primary familial polycythemia
Secondary
Chronic hypoxia
Cardiac disorders
Pulmonary disorders
Sleep apnea
High-altitude residence
Hemoglobin abnormalities
High-oxygen-affinity hemoglobins
Increased carboxyhemoglobin (smokers)
Aberrant erythropoietin production by tumors
Other rare congenital causes of secondary polycythemia (e.g., Chuvash polycythemia)
(Fig. 1.1). Cbl is a required intermediate in an intracytoplasmic reaction that converts methyltetrahydrofolate, the main form of folate transported in plasma and taken up by cells, into THF while transferring the methyl group to homocysteine to generate methionine. Therefore either folate or Cbl deficiency impairs thymidine and methionine synthesis. Cbl also serves as a cofactor for intramitochondrial conversion of methylmalonyl-coenzyme A to succinyl-coenzyme A (not illustrated).
Because Cbl is produced in nature only by microorganisms, humans must obtain it from their diet. Food sources include red meat, seafood, and dairy products. After ingestion, Cbl is released from food in the stomach in the presence of pepsin and gastric acid and then binds to haptocorrins (R-proteins) secreted in saliva. However, in the alkaline milieu of duodenum, haptocorrins are degraded by pancreatic enzymes, permitting Cbl to bind to intrinsic factor secreted by gastric parietal cells. Intrinsic factor–Cbl binds to receptors specific for this complex on ileal enterocytes and is absorbed. A small proportion of Cbl can also passively diffuse through gastrointestinal mucosa. A fraction of Cbl is bound to the transporter protein, transcobalamin II (TC II), in the plasma and is the biologically active component available for receptormediated cellular uptake; however, a larger proportion of plasma Cbl is bound to haptocorrins. In summary, the Cbl absorption depends on adequate dietary intake, gastric acid and pepsin, pancreatic proteases, gastric-derived intrinsic factor, and ileal receptors for intrinsic factor–Cbl complex. Disruption of any factor along this pathway may lead to Cbl deficiency.
The body stores a large amount of Cbl; therefore it may take several years for deficiency to develop, even with the onset of severe dietary insufficiency or malabsorption. Cbl deficiency is most often caused by malabsorption. Autoimmune gastritis resulting in a lack of intrinsic factor (i.e., pernicious anemia) is a common cause, but malabsorption of Cbl may also be due to other gastric disease (e.g., atrophic gastritis, Helicobacter pylori infection), ileal disease (e.g., inflammatory bowel disease, tropical sprue), postsurgical malabsorption, pancreatic insufficiency, drugs (e.g., proton pump inhibitors, histamine H2–receptor agonists, biguanides), chronic alcoholism, and competition for dietary Cbl (e.g., intestinal bacterial overgrowth, fish tapeworm). Overt Cbl deficiency is rarely due to isolated dietary insufficiency, but a devastating exception to this occurs among exclusively breast-fed infants of mothers who have subclinical Cbl insufficiency that may be related to a vegan diet or occult malabsorption. These infants have increased risk for neural tube defects including spina bifida and anencephaly. In rare cases, Cbl deficiency can be caused by congenital deficiency of TC II.
In contrast to Cbl, the amount of folate body stores is sufficient for only about 4 months, and, thus folate deficiency is more often related to a poor diet. Important dietary sources for folate include leafy vegetables, fruit, cereals, and fortified flour and grains. Other causes of folate deficiency are increased demand (e.g., pregnancy, prematurity, chronic hemolytic anemia, severe exfoliative dermatitis, leukemias), loss due
One-carbon unit
Thymidylate synthase 5,10–Methylene THF
(E) (C) (B)
Dihydrofolate reductase Methyl cobalamin
Methionine synthase
5–Methyl THF (A)
Folate and cobalamin in the synthesis of DNA and methionine. Folates conjugated with multiple glutamic acid residues (folate polyglutamates) are deconjugated during absorption in the duodenum and jejunum. Folate monoglutamates then undergo conversion to N5-methyl tetrahydrofolate (N5-methyl THF), the main form of folate transported in plasma and taken up by cells (A). Demethylation of N5-methyl THF (B) yields tetrahydrofolate (THF), which undergoes polyglutamation needed for cellular retention of folate. This methyltransferase reaction requires cobalamin as an intermediate with transfer of the methyl group to homocysteine to generate methionine. THF, as an intermediate, accepts a one-carbon unit (C). The folate derivative N5,N10-methylene THF donates the one-carbon unit to the reaction catalyzed by thymidylate synthase (D). Both thymidylate (dTMP), a required component for DNA synthesis, and dihydrofolate (DHF) are produced. Dihydrofolate reductase reduces DHF back to THF (E).
to hemodialysis, drugs, and competition for dietary folates (intestinal bacterial overgrowth). Malabsorption of folate is rare unless there is extensive intestinal disease (e.g., gluteninduced enteropathy, tropical sprue).
CLINICAL FEATURES
The hematologic manifestation of Cbl and folate deficiency is megaloblastic anemia. The onset of anemia is gradual, and the severity at presentation is extremely variable. Some patients exhibit profound macrocytic anemia with Hb levels as low as 2 g/dL, whereas other patients may have a more subtle manifestation or present with symptoms other than anemia. Due to defective DNA synthesis, ineffective erythropoiesis or intramedullary hemolysis can lead to jaundice, markedly increased lactate dehydrogenase, and increased indirect bilirubin. The ineffective hematopoiesis and hemolysis may be so pronounced that haptoglobin is reduced and hemosiderinuria occurs. The mean corpuscular volume (MCV) is characteristically high, with clinically overt megaloblastic anemia, but may also become elevated before the development of detectable anemia. However, the MCV may be in the normal range if there is concurrent iron deficiency anemia or thalassemia. The reticulocyte count is low. Leukopenia and thrombocytopenia are often present because impairment of DNA synthesis affects all proliferating cells.
The effect of Cbl deficiency on epithelial cells may result in the classic “beefy red smooth tongue” (atrophic glossitis). More important, Cbl deficiency is often associated with a
peripheral neuropathy and demyelinization of the dorsal and lateral columns of the spinal cord (i.e., subacute combined degeneration), leading to paresthesias, loss of position and vibration sense, spastic ataxia, and extensor plantar reflexes. Cognitive impairment may be seen; psychosis is a rare complication. Neurologic complications of Cbl deficiency may be only partially reversible, making early recognition of this disorder essential.
PATHOLOGIC FEATURES
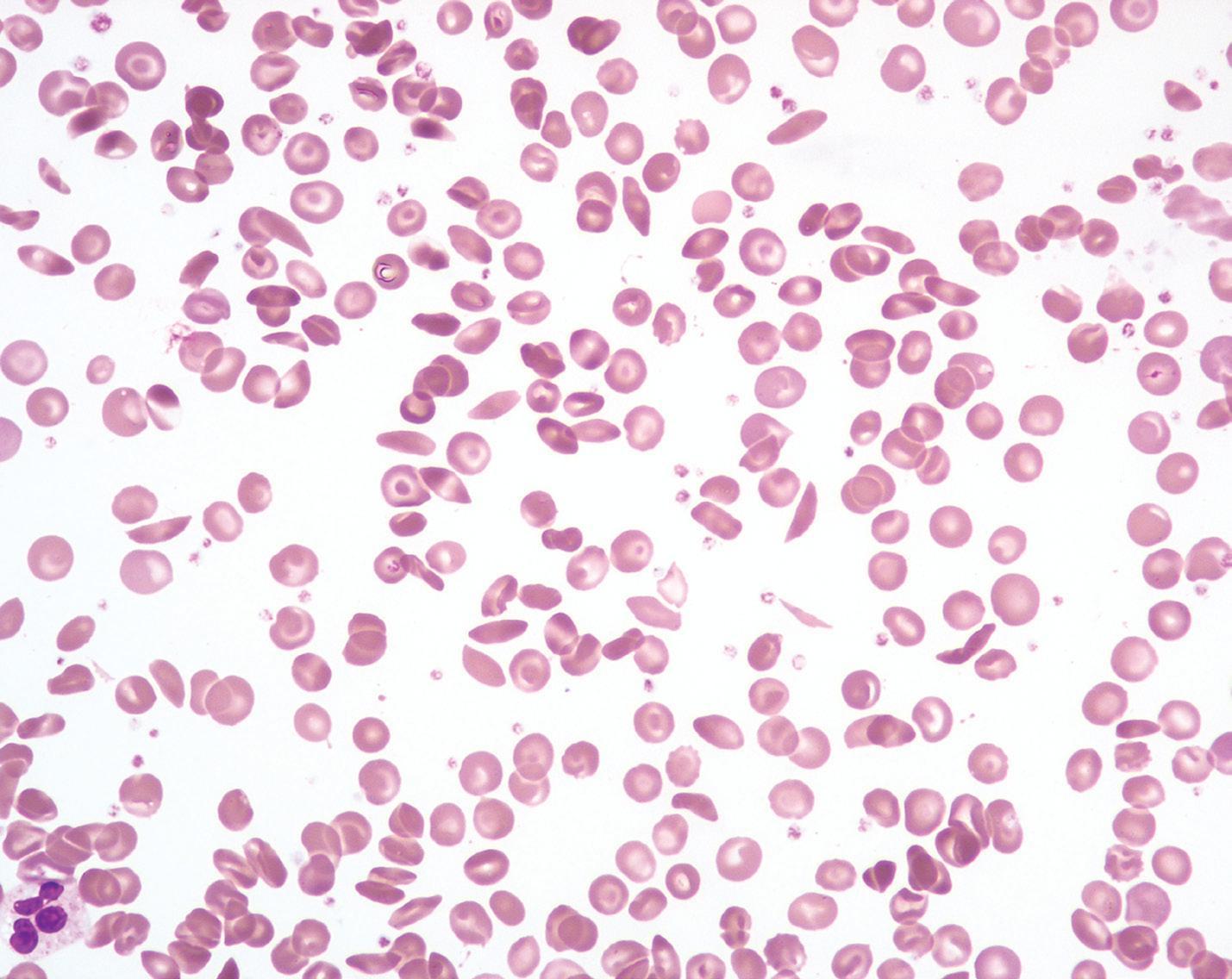
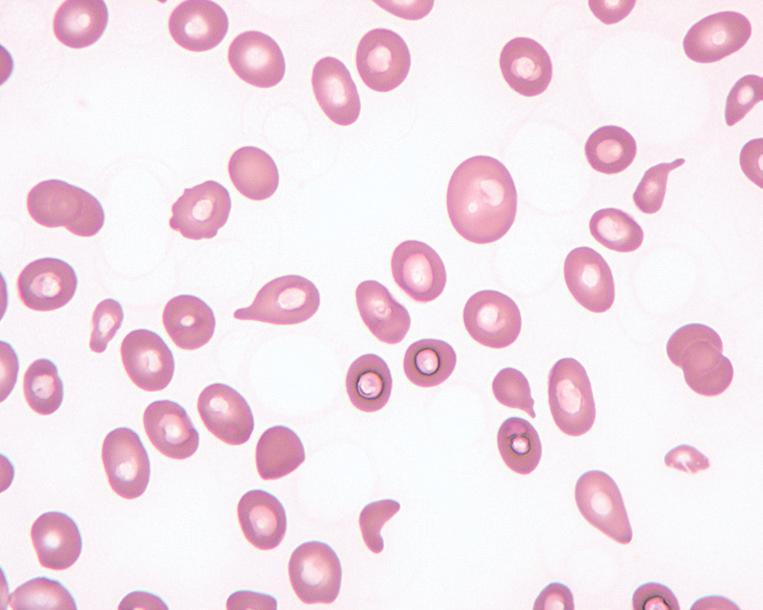
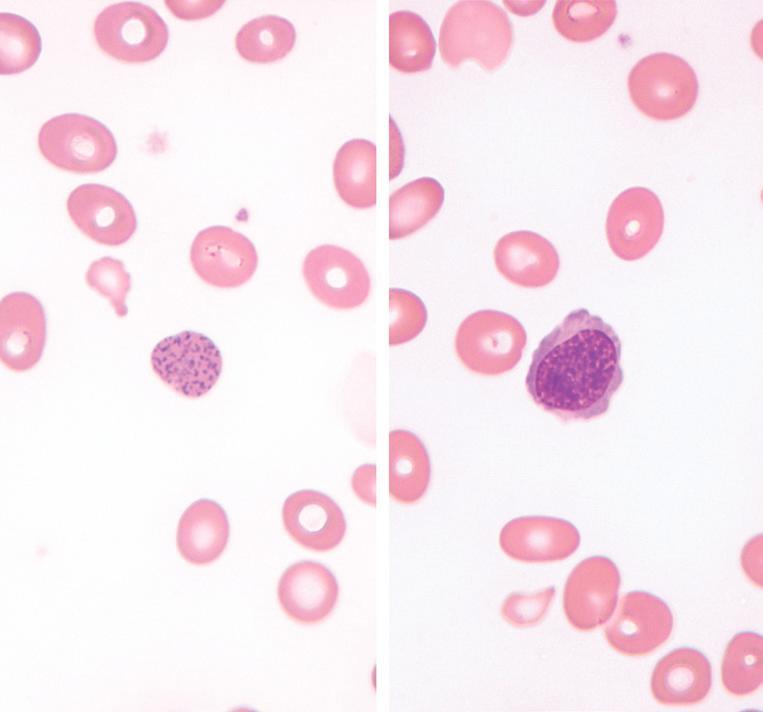
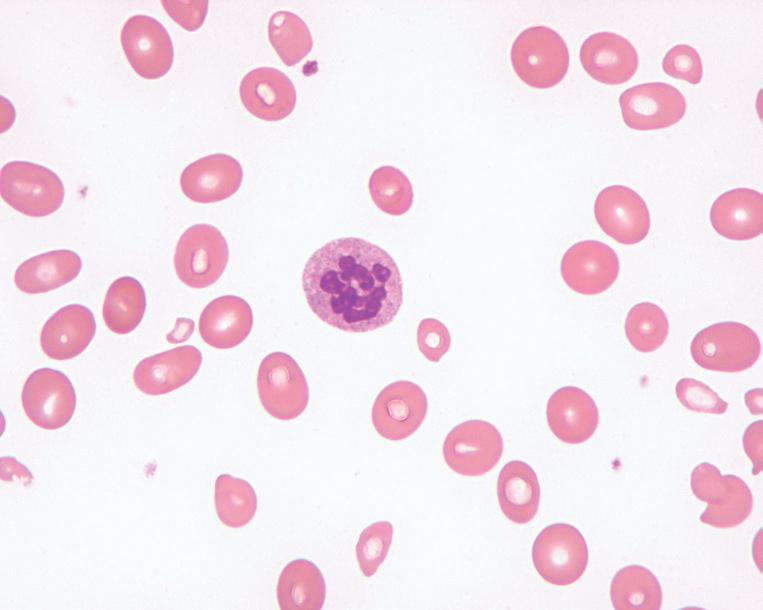
In clinically overt megaloblastic anemia, the peripheral blood film demonstrates marked variability in red cell size (anisocytosis), with large, oval red cells (macroovalocytes), teardrop cells, fragments, microcytes, and other nonspecific abnormally shaped red cells (poikilocytes; Fig. 1.2). Polychromasia is characteristically absent. With increasing severity, there is increased red cell fragmentation. Howell-Jolly bodies, nucleated RBCs with megaloblastic morphology, and basophilic stippling are often observed (Fig. 1.3). Hypersegmented neutrophils with six or more nuclear lobes, in combination with macrocytic anemia, are highly specific for megaloblastic anemia (Fig. 1.4).
Bone marrow aspirate smears typically demonstrate erythroid hyperplasia and a shift to immaturity, as well as characteristic cytologic changes in all three myeloid lineages. Maturing cells are larger than normal, with asynchronous nuclear and cytoplasmic maturation. The chromatin of the erythroid precursors appears more granular than during normal
Cobalamin
Homocysteine
Methionine
FIG. 1.1
MEGALOBLASTIC ANEMIA—FACT SHEET
Definition
■ Macrocytic anemia due to folate deficiency, cobalamin deficiency, and other causes, resulting in impaired DNA synthesis, abnormal nuclear maturation, and ineffective hematopoiesis
Incidence and Location
■ Folate deficiency is rare in countries with mandatory food fortification
■ Dietary cobalamin deficiency is rare, except among vegans and populations with low intake of animal-source foods
Morbidity and Mortality
■ Increased risk of fetal neural tube defects in folate/Cbl-deficient pregnancy
■ Peripheral neuropathy, subacute combined degeneration of the spinal cord, and cognitive abnormalities that may be only partially reversible (cobalamin deficiency)
■ Increased risk of gastric adenocarcinoma and carcinoid tumor (patients with pernicious anemia)
Prevalence
■ Prevalence estimates based on low red blood cell (<140 ng/mL) and serum (<3 ng/mL) folate levels among women of childbearing age in the United States is 4.5% and 0.3%,
respectively (2005–2006 National Health and Nutrition Examination Survey)
■ Increased incidence of cobalamin deficiency in breast-fed infants and young children among populations at risk of dietary insufficiency (strict vegans and developing countries)
■ Prevalence of cobalamin deficiency owing to malabsorption increases with age
Clinical Features
■ Gradual onset of macrocytic anemia
■ Evidence of hemolysis (jaundice, elevated lactate dehydrogenase, elevated total bilirubin, low haptoglobin)
■ Atrophic glossitis
■ Neuropsychiatric effects (cobalamin deficiency)
– Paresthesias
– Loss of position and vibration sense
– Ataxia
– Cognitive impairment (impaired memory, dementia)
– Psychosis (rare)
Prognosis and Therapy
■ Treatment of the underlying cause of the deficiency
■ Intramuscular injection (or large oral doses) of vitamin B12
■ Oral folate supplementation (parenteral administration needed in some cases)
Megaloblastic anemia, peripheral blood findings. This blood smear from a patient with severe megaloblastic anemia demonstrates prominent anisopoikilocytosis, including large oval cells (oval macrocytes), microcytes, and teardrop cells.
FIG. 1.2
FIG. 1.3
Megaloblastic anemia, peripheral blood findings. Coarse basophilic stippling (left) and circulating nucleated red cells (right) are common in megaloblastic anemia. The red cell precursor is relatively mature, judging by the pink-gray quality of the cytoplasm. However, the chromatin is quite open, with an abnormal stippled appearance, indicating megaloblastic change.
FIG. 1.4
Megaloblastic anemia, peripheral blood findings. A hypersegmented neutrophil in the peripheral blood of a patient with megaloblastic anemia. Neutrophils are generally considered hypersegmented when they contain six or more lobes.
MEGALOBLASTIC ANEMIA—PATHOLOGIC FEATURES
Peripheral Blood
■ Anemia, often pancytopenia
■ Prominent anisocytosis
■ Poikilocytosis
– Macroovalocytes
– Teardrop cells
– Fragments
■ Lack of polychromasia
■ Hypersegmented neutrophils
■ Howell-Jolly bodies, nucleated red cells, basophilic stippling
Bone Marrow
■ Hypercellular marrow with trilineage expansion
■ Left-shifted hematopoietic precursors
■ Asynchronous nuclear to cytoplasmic maturation
– Megaloblastic erythroid precursors
– Giant metamyelocytes
– Larger than normal megakaryocytes with abnormal nuclei
Differential Diagnosis
■ Other causes of megaloblastic hematopoiesis
■ Drugs
■ Some myelodysplastic syndromes
■ Nonmegaloblastic macrocytosis
– Reticulocytosis
– Liver disease
– Alcohol
– Hypothyroidism
– Drugs
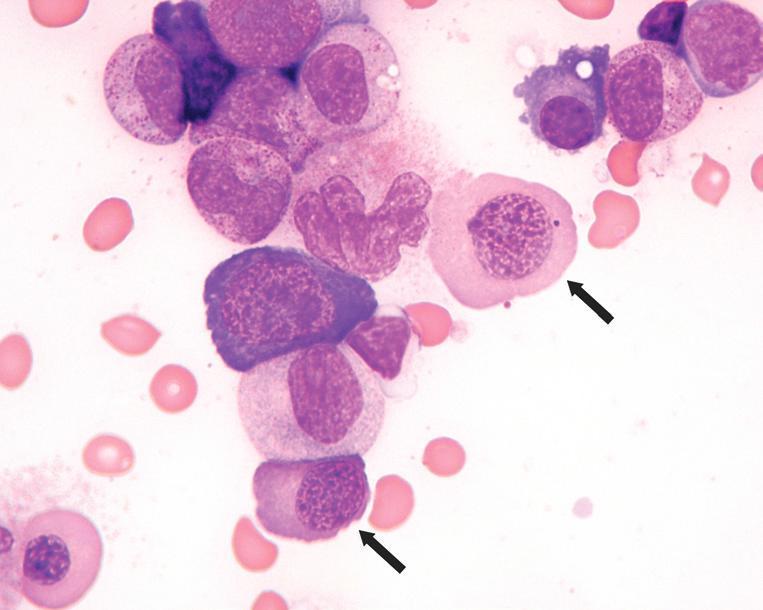
Megaloblastic anemia, bone marrow aspirate smear findings. This marrow aspirate smear image from a patient with severe megaloblastic anemia demonstrates two late-stage erythroid precursors (based on an advanced degree of cytoplasmic hemoglobinization) that are abnormally large and have dispersed, stippled megaloblastic chromatin (arrows). Also present is a giant granulocyte at approximately the metamyelocyte stage with abnormally dispersed chromatin (immediately to the left of upper arrowed megaloblast).
maturation, and the most mature erythroid precursors display a prominent fenestrated nuclear appearance that contrasts with the expected complete condensation of chromatin at this stage (Fig. 1.5). Erythroid precursors with abnormally shaped, multiple, budding, or fragmented nuclei are often seen (Fig. 1.6), resembling those seen in myelodysplastic syndromes. Giant metamyelocytes and bands with abnormally open chromatin are also characteristic (Figs. 1.5 and 1.7), whereas larger than normal megakaryocytes with unattached nuclear lobes or hyperlobated nuclei reflect similar abnormalities in megakaryocytic maturation. Because of the pronounced ineffective hematopoiesis, markedly increased iron stores are seen with a Prussian blue iron stain unless there is concurrent iron deficiency. Bone marrow core biopsy specimens demonstrate a markedly hypercellular marrow with expanded and left-shifted trilineage hematopoiesis (Fig. 1.8). In particular, islands of immature erythroid precursors with finely stippled chromatin and distinct, irregular, eosinophilic nucleoli are often prominent (Fig. 1.9).
ANCILLARY STUDIES
Serum Cbl is fairly sensitive and specific for clinically overt Cbl deficiency. Causes of falsely low serum Cbl include pregnancy, use of oral contraceptives, anticonvulsant administration, human immunodeficiency virus infection, and folate deficiency. Mixed Cbl/folate deficiency complicates the
FIG. 1.5