CyclizedHelicalPeptides:Synthesis,Properties, andTherapeuticApplicationsZigangLi
https://ebookmass.com/product/cyclized-helical-peptidessynthesis-properties-and-therapeutic-applications-zigang-li/
Instant digital products (PDF, ePub, MOBI) ready for you
Download now and discover formats that fit your needs...
Silicon Containing Hybrid Copolymers: Synthesis, Properties, and Applications Zibiao Li
https://ebookmass.com/product/silicon-containing-hybrid-copolymerssynthesis-properties-and-applications-zibiao-li/
ebookmass.com
Renewable Polymers and Polymer-Metal Oxide Composites: Synthesis, Properties, and Applications Sajjad Haider
https://ebookmass.com/product/renewable-polymers-and-polymer-metaloxide-composites-synthesis-properties-and-applications-sajjad-haider/
ebookmass.com
Polar Organometallic Reagents - Synthesis, Structure, Properties and Applications Wheatley Andrew E.H. (Ed.)
https://ebookmass.com/product/polar-organometallic-reagents-synthesisstructure-properties-and-applications-wheatley-andrew-e-h-ed/ ebookmass.com
Rescuing Stitch (Guardian Hostage Rescue Specialists: CHARLIE Team Book 2) Ellie Masters
https://ebookmass.com/product/rescuing-stitch-guardian-hostage-rescuespecialists-charlie-team-book-2-ellie-masters/ ebookmass.com
The Overproduction of Truth: Passion, Competition, and Integrity in Modern Science Gianfranco Pacchioni
https://ebookmass.com/product/the-overproduction-of-truth-passioncompetition-and-integrity-in-modern-science-gianfranco-pacchioni/
ebookmass.com
Electrical Properties of Materials (10th Edition) L. Solymar
https://ebookmass.com/product/electrical-properties-of-materials-10thedition-l-solymar/
ebookmass.com
The Varieties of Spiritual Experience: 21st Century Research and Perspectives David B. Yaden
https://ebookmass.com/product/the-varieties-of-spiritualexperience-21st-century-research-and-perspectives-david-b-yaden/
ebookmass.com
The Girl They All Forgot Martin Edwards
https://ebookmass.com/product/the-girl-they-all-forgot-martin-edwards/
ebookmass.com
Garner's Modern English Usage: The Authority on Grammar, Usage, and Style, Fifth Edition Bryan A. Garner
https://ebookmass.com/product/garners-modern-english-usage-theauthority-on-grammar-usage-and-style-fifth-edition-bryan-a-garner-2/
ebookmass.com
Digital Transformation Success: Achieving Alignment and Delivering Results with the Process Inventory Framework
1st Edition Michael Schank
https://ebookmass.com/product/digital-transformation-successachieving-alignment-and-delivering-results-with-the-process-inventoryframework-1st-edition-michael-schank/ ebookmass.com
CyclizedHelicalPeptides
CyclizedHelicalPeptides Synthesis,Properties,andTherapeuticApplications
ZigangLi
HuiZhao
ChuanWan
Authors
Prof.ZigangLi
ShenzhenGraduateSchoolofPeking University ChemicalBiologyandBiotechnology
LishuiRoad
XiliTown 518055Shenzhen,NanshanDisctrict China
Dr.HuiZhao
ShenzhenGraduateSchoolofPeking University ChemicalBiologyandBiotechnology
LishuiRoad
XiliTown NanshanDisctrict 518055Shenzhen China
Dr.ChuanWan
ShenzhenGraduateSchoolofPeking University ChemicalBiologyandBiotechnology
LishuiRoad
XiliTown NanshanDistrict 518055Shenzhen China
CoverDesign: Wiley
CoverImage: ©Molekuul/iStockphoto; ©ivanastar/iStockphoto
Allbookspublishedby WILEY-VCH arecarefullyproduced.Nevertheless, authors,editors,andpublisherdonot warranttheinformationcontainedin thesebooks,includingthisbook,to befreeoferrors.Readersareadvised tokeepinmindthatstatements,data, illustrations,proceduraldetailsorother itemsmayinadvertentlybeinaccurate.
LibraryofCongressCardNo.: appliedfor
BritishLibraryCataloguing-in-Publication Data
Acataloguerecordforthisbookis availablefromtheBritishLibrary.
Bibliographicinformationpublishedby theDeutscheNationalbibliothek TheDeutscheNationalbibliotheklists thispublicationintheDeutsche Nationalbibliografie;detailed bibliographicdataareavailableonthe Internetat <http://dnb.d-nb.de>.
©2021WILEY-VCHGmbH,Boschstr. 12,69469Weinheim,Germany
Allrightsreserved(includingthoseof translationintootherlanguages).No partofthisbookmaybereproducedin anyform–byphotoprinting, microfilm,oranyothermeans–nor transmittedortranslatedintoa machinelanguagewithoutwritten permissionfromthepublishers. Registerednames,trademarks,etc. usedinthisbook,evenwhennot specificallymarkedassuch,arenotto beconsideredunprotectedbylaw.
PrintISBN: 978-3-527-34342-3
ePDFISBN: 978-3-527-34347-8
ePubISBN: 978-3-527-34345-4
oBookISBN: 978-3-527-34343-0
Typesetting Straive,Chennai,India
PrintingandBinding
Printedonacid-freepaper 10987654321
Contents 1Introduction 1
1.1Protein–ProteinInteractionsandTheirSmall-MoleculeModulators 1
1.1.1CharacteristicsofProtein–ProteinInteractions 1
1.1.2InterventionofProtein–ProteinInteractionsUsingSmallMolecules 3
1.1.2.1LeukocyteFunction-AssociatedAntigen-1 5
1.1.2.2InhibitorofApoptosisProteins 6
1.1.2.3Bromodomains 6
1.1.2.4HumanImmunodeficiencyVirusIntegrase 6
1.1.2.5B-CellLymphoma-2Family/B-CellLymphoma-2Homology3Proteins Interaction 7
1.1.2.6MouseDoubleMinute2–p53Interaction 7
1.2FeaturesofPeptideasMolecularTools 8
1.2.1AdvantagesofPeptidesasMolecularTools 8
1.2.2DisadvantagesofPeptidesasMolecularTools 11
1.3HelicalStructuresandTheirCharacterization 12
1.3.1DifferentTypesofHelices 12
1.3.1.1 α-Helix 12
1.3.1.2310 -Helix 12
1.3.1.3 π-Helix 13
1.3.2CharacterizationofHelicalPeptides 15
1.3.2.1CircularDichroism 15
1.3.2.2X-rayCrystallography 16
1.3.2.3NuclearMagneticResonance(NMR) 17
1.4StabilizationofPeptides 18
1.4.1PeptideStabilizationviaCyclization 18
1.4.1.1Monocyclization 18
1.4.1.2Multicyclization 19
1.4.2PeptideStabilizationviaBackboneReconstruction 21
1.4.2.1Methylation 21
1.4.2.2Foldamers 23 References 27
2ConstructionofConstrainedHelices 35
2.1Side-ChainCross-linking 35
2.1.1DisulfideBond 35
2.1.2AmideandEster 37
2.1.3All-HydrocarbonStapledPeptide 42
2.1.4Thioether 48
2.1.5Azole 56
2.2EndNucleation 57
2.2.1Macrocycle-BasedN-capTemplates 58
2.2.2HydrogenBondSurrogateApproaches 60
2.2.3N-TerminalSideChainConstrainsasHelix-Nucleating Templates 61 References 62
3PropertiesofStabilizedPeptides 69
3.1Helicity 69
3.1.1RingSize 69
3.1.2Rigidity 74
3.1.3Comparison 79
3.2BindingAffinity 80
3.2.1Helicity 80
3.2.2CyclizationPosition 81
3.2.3Substitution 84
3.3CellPermeability 84
3.3.1Amphiphilicity:HydrophobicityandIsoelectricPoint 84
3.3.2Helicity 87
3.3.3Summary 89
3.4NonspecificToxicity 89
3.5Stability 91
3.5.1ProteolyticStability 91
3.5.2PharmacokineticProperties 95
3.6AdditionalFeatures 98 References 103
4ApplicationsofConstrainedHelices 107
4.1Cancer 107
4.1.1MDM2/X 107
4.1.2B-CellLymphoma2
(MCL-1/BCL-2/BCL-XL)-BID/Noxa/BAX/BIM/PUMA 110
4.1.3NOTCH 114
4.1.4InsulinReceptorSubstrate1 118
4.1.5Ras 120
4.1.6Rab 124
4.1.7 β-CateninBCL-9/AXIN 126
4.1.8EpidermalGrowthFactorReceptor 130
4.1.9EstrogenReceptor α 135
4.1.10Hypoxia-InducibleFactor 138
4.1.11EmbryonicEctodermDevelopment – EnhancerofZeste Homolog2 140
4.2InfectiousDisease 143
4.2.1HIV 143
4.2.2RespiratorySyncytialVirusF(RSV) 148
4.3MetabolicDisease 150
4.3.1Glucokinase-Phospho-BAD 150 References 152
5StabilizedPeptideCovalentInhibitors 159
5.1MethodologiesofPeptideCovalentInhibitor 159
5.1.1CovalentWarhead 160
5.1.2StaplingMethod 162
5.2Applications 162
5.2.1BCL-2FamilyProteinsAsTarget 163
5.2.2MDM2andMDM4AsTarget 164
5.2.3SulfoniumTetheredPeptide 168
5.2.4Others 176 References 177
6StabilizedPeptidePROTAC 181
6.1Proteolysis-TargetingChimera(PROTAC) 181
6.2DesignofPeptidePROTAC 181
6.2.1ExploitationofE3UbiquitinLigase-RecruitingLigand 182
6.2.2DesignofStabilizedPeptideLigand 183
6.2.3ImpactofLinker 184
6.3TherapeuticApplicationsofStabilizedPeptidePROTAC 184
6.3.1TargetingERα 185
6.3.2Targeting β-Catenin 188 References 188
7StabilizedPeptideforDrugDelivery 191
7.1Cell-PenetrayingPeptides(CPPs) 191
7.1.1ClassificationofCPPs 192
7.1.2MechanismofCellPenetrationofCPPs 193
7.1.3ApplicationsofCPPs 194
7.2Cell-PermeableCyclicPeptides(CyclicCPPs) 196
7.2.1CyclicCPPs-MediatedDrugDelivery 198
7.2.2Cyclo-RGD 202
7.3Co-assemblyNanocarrierSystem 204
7.4ExamplesofStabilizedPeptideDrugs 206 References 208
viii Contents
8Outlook 213
8.1TheDevelopmentofPeptide-StabilizingMethods 213
8.2ApplicationsofStabilizedHelicalPeptides 218 References 220
Index 223
1.1Protein–ProteinInteractionsandTheir Small-MoleculeModulators
1.1.1CharacteristicsofProtein–ProteinInteractions
Proteinsthatworkanddegradeinhighlycongestedandcomplexenvironmentsmust befoundbytheirpartnersinalargenumberofnon-partners.Itisestimatedthat humanbeingshave650000differentpairsofinteractions,whichareresponsible foranumberofkeybiomolecularprocesses[1].Thesurfaceofsolubleproteinsis coveredbyhydrophobicandhydrophilicresidues,aswellasbyhydrophilicbackbone.Thehighlyspecificphysicalcontactbetweentwoormoreproteinmolecules ismainlyrelatedtohydrophobicinteractions,salt,andhydrogenbonds.
Protein–proteininteractionshavedifferentaffinityandlongevity.Somecomplexes areweaklyandinstantaneouslyclustered;somemaycontinuetoformpartofalarger proteincomplex,stabilizedthroughmultipleinteractions;somereversiblesignal complexeshavehighpairingaffinity,butonlylimitedtime;somecomplexesarestable,buthavebuilt-intimers;thepresenceofantibodiesandantigensandprotease andinhibitorcomplexescantakeuptoaday,someofwhichmaybecategorizedas irreversible[2].
Inaddition,protein–proteininteractionscanbecategorizedaccordingtothe structuralcharacteristics(Figure1.1)[3]:theinteractionbetweenglobularprotein pairs,theinteractionsbetweenglobularproteinsandindividualpeptidechains withcontinuousordiscontinuoustableposition,andtheinteractionbetweentwo segmentsofpeptidechains.Correspondingly,thepolypeptidethatparticipatesin protein–proteininteractionsmayadoptacombinationofstructures:theextension structureinthegroove, β-sheet, α-helix,andeventhepoly-prolinehelix. Thereiscertainregularityinthepresenceofaminoacidresiduesinproteins[3a]. Inthegeneralinterface,leucineisthemostcommonresidue,followedbyarginine. Furthermore,chargedresiduesaremorecommonthanpolarresidues,andboth, exceptforarginineandhistidine,aregenerallyabundantonthesurface.Aromatic aminoacids,exceptfortryptophan,haveaverylowabundanceonthesurfacebut haveahighabundanceattheinterface.Asismentionedabove,thefrequencyof CyclizedHelicalPeptides:Synthesis,Properties,andTherapeuticApplications, FirstEdition. ZigangLi,HuiZhao,andChuanWan. ©2021WILEY-VCHGmbH.Published2021byWILEY-VCHGmbH.
1Introduction PPI class
Globular protein-helical peptide, discontinuous epitope
Globular protein-peptide, continuous epitope
Globular protein-peptide, continuous epitope
Globular protein-peptide, anchor residue
Globular protein-globular protein, discontinuous epitope
DescriptionSimplified illustrationExamples (targetdisplaced)
Helix with a discontinuous epitope binding into a groove
Continuous epitope on β-sheet or β-strand and loops binding into surface with pockets
Binding into pocket in a β-propeller
Peptide with an anchor residue owing to post-translational modification binding into a pocket
Two proteins both presenting discontinuous epitopes
Examples structure
MDM2-p53
BCL-XL–BAK*
BCL-XL–BAD and ZipA–FtsZ S100B–p53
β–Catenin–TCF3–TCF4
Protein Data Bank (PDB) ID: 2xa0
MCL1–BH3 SUR2–ESX
XIAP–SMAC* HIV integrase–LEDGF Integrins RAD51–BRCA2 PDZ domains
NRP1–VEGFA Menin–MLL
KEAP1–NRF2* WDRS–MLL
Bromodomains* PDEδ–KRAS SH2 domains PLK1 PBD–peptide VHL–HIF1a
IL-2-IL-2R* TNF–TNF E2–E1
PDBID: 1g73
Peptide-peptide
A pair of helices with an elongated binding interaction
MYC–MAX* NEMO–IKK Annexin II–P11 (also known as S100A10)
PDBID: 2dyh
PDBID: 3uvvv
PDBID: 1z92
PDBID: 1nkp
BAD, BCL-2-associated agonist of cell death: BAK, BCL-2 bomologousantagonist/killer: BCL B cell lymphoma: BH3, BCL-2 bolology domain 3: BRCAZ, breast Cancer type 2 susceptibility protein: HIF1a, hypoxia-inducible factor 1α: IL-Z, interleukin-2: IL-2R, interleukin-2 receptor: IKK, inhibitor of nuclear factor x8 kinase: KEAP1, kelch-like ECH- associated protein 1: LEDGF, lens epitheliurn-derived growth factor: MAX MYC-associated factor X: MCL1, myeloid cell leukaemia 1: MLL, kinase 1 polo box domain: S100A10, S100 calcium-binding protein A10: SH2, SRC homology 2: SMAC, second mitochondria- derived activator caspase:SUR2, mediator of RNA polymerase II transcription subunit 23: TCF3, HMG box trnscription factor 3: TNF, tumor necrosis factor: VEGFA vascular endothelial growth factor: VHL, Von Hippel-Lindau disease tumor suppressor: WDRS, WD repeat-containing protein 5: XIAP, X-linked inhibitor of apoptosis protein *Example structure illustrated in the column to the right.
mixed-lineage leukaemia: NEMO, nuclear factor κB essential modulator: NRF2, nuclear factor erythroid 2-related factor 2: NRP1, neuropilin 1: PLK1 PBD, polo-like
Figure1.1 Classificationofprotein–proteininteractionsandexamples[3b].Source:Scott etal.[3b].©2016,SpringerNature.
occurrenceofhydrophobicresiduesisgenerallyhighattheinterfaceandislowon thesurface.Cysteineisparticularlyrarebothonthesurfaceandattheinterface. Inaddition,basedontheresultsofalaninescanningmutagenesis,theresidual basethathasagreatinfluenceonthebindingaffinityiscalled“hotspot”[4].Hot spotsarealmostalwaysburiedinthecenterofthecore,notincontactwithsolvents. Thehotspotprocessesthehighestsequenceconservation[5].Tryptophan,arginine, andtyrosinearethemostcommon,accountingformorethanhalfofthetotal,ashot spots.Thesethreeversatileresidueswereabletoformhydrophobic,aromatic,and polarinteractions,allofwhichcanbewrappedincomplementarysurfacestomeet unpairedhydrogen-bondeddonorsandreceptors.Inaddition,thepolar“π-cation” bondbetweenarginineandtryptophanortyrosinewasfoundinmorethan50%hot spots[6].Apartfroma“π-cation”bondwitharginine,thetraditionalsidechaininteractionismorecommonfortyrosine.Bycontrast,themostcommonresidualatthe interface,leucine,israrelyfoundinhotspots,whileisoleucineisrich. Inthecomplexesintheproteindatabase,62%hasahelixontheinterface[7]. However,thepresenceofahelixattheinterfacedoesnotmeanthatthehelixplaysa
1.1Protein–ProteinInteractionsandTheirSmall-MoleculeModulators 3 keyrole.Analysisshowsthatinabout60%oftheinterface,thehotresidueislocated ononesideofthehelix,one-thirdofthecomplexeswiththehotspotsontwofaces ofthehelix,andabout10%ofthecomplexwithallthreefacesparticipatinginthe interactionwiththetargetprotein.Intheproteindatabase,thefirstfourmajortypes offunctionofprotein–proteininteractions,wherehelicesareinvolved,aregeneregulation,enzymefunction,cellcycle,andsignaltransduction.
Analysisofthecontributionofeachhelixresiduetotheinteractionshowsthat leucineappearsmostintheinterfacearea.Thisisnotsurprising,becauseingeneral,leucineisalsothemostcommonresidueinproteins.Afterthenormalization ofnaturalabundance,aromaticaminoacids,arginine,andleucineareofthehighest frequenciesatthehelixinterfaceascomparedwithpolarresidues[4,8].Inaddition, polarandchargedresiduesarealsoimportantcontributorstotheinterface.
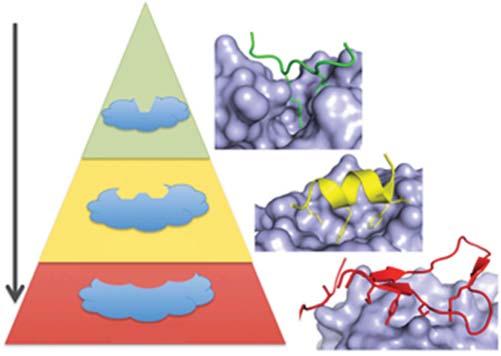
1.1.2InterventionofProtein–ProteinInteractionsUsingSmall Molecules Abnormalprotein–proteininteractionsarethebasisofmultiplediseases,andan increasingnumberofresearchersarecommittedtodevelopingmoleculestomodulateproteininteractionsfortherapeuticpurposes.Smallmoleculeisaclassofentity withpotentiallyidealtherapeuticpotentials.However,thecontactsurfaceofsomeof theproteininteractionsislargeandshallow(about1000–6000Å2 ),especiallythose featuredbyalinearpeptideepitope1–4aminoacidslong,comparedtothetraditionallysmallanddeepsmall-moleculebindingpockets[9](Figure1.2).Therefore,theinterfacebetweenproteinsissometimesregardedasatargetof“undruggable.”Inestablishingguidelinesforthediscoveryofprotein-proteininteraction (PPI)inhibitors,clinicalsuccesscasesshouldbeconsideredinthecontextofthe typeofinterface.
Workinrecentyearshasbeguntoshowthatsomeprotein–proteininteractions areabletobesuppressedbysmallmolecules.Mostofthedevelopedinhibitors targetPPIs,wherehotspotresiduesarerestrictedtosmallbindingpockets (250–900Å2 )[11].Somesmall-moleculeinhibitorsdisrupttheinteractionbetween aglobularproteinandasinglepeptidechainwithasecondaryortertiarystructure, throughbindingtothepocketontheglobularprotein.Itisnoteworthythatthe secondarystructuralfeaturesprocessedbythepeptidechain,suchas α-helices and β-strands,haveimportantimplicationsforthedesignofinhibitorsthatmimic andreplacethesepeptides.Withabetterunderstandingofthestructuralbiology oftheprotein–proteininteractions,itseemsmorepromisingandreasonableto discoverdrugstargetingprotein–proteininteractionswithdefinedstructures.In addition,thehotspotsoftheinteractioninterfacecanbetargetedbyinhibitors ofprotein–proteininteractions.Theinteractionoftherigidglobularproteinwith apolypeptidemaybemoresuitableforsmall-moleculeinterruptionbecausethe polypeptidecancontributemoretothebindingenergyandbereplacedbythesmall moleculewithgooddesign.Atpresent,therearemanystrategiestodiscoverhitsor leadsthatinterferewithprotein–proteininteractions,themostnotableofwhichis high-throughputscreening,fragmentscreening,andoptimization.
Figure1.2 ThecomplexityofthePPIinterfaceaffectsdruggabilityPPIscanbeclassified bywhetheronesideoftheinterfaceconsistsofaprimary(linear)proteinsequence(green), asingleregionofsecondarystructure(suchasan α-helix,yellow),ormultiplesequences requiringtertiarystructure(red).Therearefewerexamplesofsmall-moleculeinhibitorsof PPIsastheinterfacebecomesmorecomplex(fromprimarytosecondarytotertiary epitopes).StructuresshownareBRDt/histone(green;ProteinDataBank[PDB]:2WP1), MDM2/p53(yellow;PDB:1YCR),andIL-2/IL-2Ra(red;PDB:1Z92)[10].Source:Arkinetal. [10].©2014,Elsevier.
High-throughputscreeningisaneffectivewaytofindahitinatraditionaldrug target.Mostofthehigh-throughputscreeningstrategiesrelyonassayssuchasfluorescenceresonanceenergytransfer,amplifiedluminescentproximityhomogeneous assayscreen,surfaceplasmonresonance,orfluorescencepolarizationbecausethey arehighlyefficient,sensitive,andreagent-available[12].However,thesemethods canusuallydisruptenzymeactivityandleadtomorefalse-positivesignals.Another methodisbasedonthelabel-freestrategy,includingtherefractiveindexpropertiesandmassspectrometry[12b].Theirapplicationsmaybemoreextensive,more quicklydeveloped,androbustbecausetheyeliminatethestepsassociatedwithintroducingandobservingtags.Despitetheseestablishedmethods,itisstilldifficultto effectivelygenerateprotein–proteininteractioninhibitorsthroughhigh-throughput screeningsincethecompoundsusedforscreeningaremainlytargetingtraditional drugtargets.Traditionalhigh-throughputscreeningfacessomechallengesindealingwithprotein–proteininteractions–lowhitratio,lowactivity,andhardtoeliminatefalsepositives[12b].However,high-throughputscreeninghasbeensuccessfully appliedinthediscoveryoftheanalogofdiscontinuousepitopeonan α-helix.
Thefragment-baseddrugdiscoveryisastrategytodiscovermoleculesfrom smallerfragmentofdrugsorfunctionalgroupswithlowaffinity,whichcaneffectivelyexplorethechemicalspace[13].Thesefragmentscansimplifythecalculation andanalysisofligandbindingtoimproveaffinity.Thediscoveryofdrugfragments inthepasthasbecomeaneffectivewaytotargetprotein–proteininteractions.Many proteininterfaceshaveanchoredresiduestooccupythepocketsofproteins,such astyrosine,phenylalanine,tryptophan,orleucine[14].Thepocketsoftheshort
1.1Protein–ProteinInteractionsandTheirSmall-MoleculeModulators 5 peptidewithwell-definedstructurecaneffectivelybecomethetargetofthedrug discoverybasedonfragments[15].Fragmentdrugdiscoveryscreeningusually consistsoftwosteps.Thefirststepinvolvesusingasurfaceplasmonresonance ordifferentialscanningfluorescenceforapreliminaryrapidscreening[16].The secondstepincludesmoretargetedvalidationofthehitmolecule,theuseofX-ray crystallographyorprotein-basednuclearmagneticresonance(NMR)todefine thespatialaspectsofthebindingsite,thethermodynamicparametersdefined byisothermalcalorimetry,andthesurfaceplasmonresonancetodefinekinetics [15].Fragmentdiscoverymethodscombinefragmentspacewiththeenhanced hitratioforlowercomplexitymolecule,makingitselfapowerfulleadgeneration tool.Comparedwithhigh-throughputscreening,fragment-baseddrugdiscovery cancapturemorechemicalstructureswithdifferenthits,providingmorehitsfora largernumberofproteintargets,higherrecognitionratesandfewerfalsepositives, andsimplerandmorereliabledetectionmethods[17].However,theneedfora largenumberofproteinsisalsoaproblemthatfragment-baseddrugdiscovery needstoaddress.Inaddition,fragmentscombinecomputationalanalysisaspects, requiringnewhardwaredesignornewconceptsandgreatprogress.
Virtualscreeningbasedonstructureisanimportanttooltohelpthediscoveryand optimizationofpotentialleadinafastandcost-effectivewaybasedonstructural drugdiscovery.Thevirtualscreeningbasedonstructureisusedtoselectthe large-classdrugcompoundlibrary.Then,thescreenedoutpromisingcompounds wereselectedforexperimentaltesting.Inthemethodofdenovodesign,the three-dimensional(3D)structureofreceptorsisusedtodesignnovelmolecules thathaveneverbeensynthesizedbeforeusingligandgrowingprogramsandthe intuitionofmedicinalchemists[18].Comparedwithhigh-throughputscreening, thediscoveryofcomputer-assisteddrugshastheadvantageofpredictingnew bioactivecompoundsandtheirreceptor-bindingstructures,andinsomecases havingagreaterhitrate.
Sofar,usingtheabovemethods,anumberofsmall-moleculecompoundstargeting protein–proteininteractionshaveenteredclinicaltrials.Herearesomesuccessful examplesofsmall-moleculeinhibitorsthatinterferewithprotein–proteininteractions.
1.1.2.1LeukocyteFunction-AssociatedAntigen-1 Leukocytefunction-associatedantigen-1isa β2integrinthatparticipatesinthe activationandadhesionofTcellsandisatargetintheweakeningofinflammatory immuneresponse[19].Lymphocytefunction-associatedantigen1,theheterodimer consistingofan α-chainanda β-chain,bindstoitsligandintercellularadhesion molecule-1,whichisimportantforTcell–Tcellinteractions.Theanti-lymphocyte function-associatedantigen1antibodyefalizumab,animmunosuppressantthat inhibitslymphocyteactivationandcellmigrationbybindingtotheCD11asubunit oflymphocytefunction-associatedantigen1,hadbeenapprovedforpsoriasis andthenwithdrawnforimmunosuppression-inducedfatalviralinfections[20]. Anotherleukocytefunction-associatedantigen-1antagonistlifitegrast,whichwas discoveredbySunesis,andthendevelopedclinicallybySARcode/Shire,hasbeen
approvedastheonlydrugforthetreatmentofdryeyedisease[21].Themechanism ofactionofthemoleculeisunderdebate,whichistheinhibitionofleukocyte function-associatedantigen-1frombindingtointercellularadhesionmolecule1, associatedwitheitherintercellularadhesionmoleculesiteontheIdomainorthe relatedsiteontheI-likedomain[22]
1.1.2.2InhibitorofApoptosisProteins
Apoptosisisaprogrammedcelldeathmediatedbycaspasesactivation.Inhibitor ofapoptosisproteinshasbeenexpressedintumorcellsbyinhibitingtheactivity ofapoptosis-inducingprotease,whichregulatesthefateofcells,includingdeath andimmunityofapoptoticcells.X-linkedinhibitorofapoptosisproteinsisthe mosteffectiveinhibitorofapoptosisproteins,whichinteractwiththeinitiator caspase-9throughtheBaculoviralIAPrepeat3structuredomainandcaspase-3/7 throughtheBaculoviralIAPrepeat1/2domain[23].Discoveringnewcompounds thatinhibittheinteractionbetweenX-linkedinhibitorofapoptosisproteins andenzymesisthoughttobeapromisingstrategyforcancertreatment.Smac isanaturalproteininhibitorofX-linkedinhibitorofapoptosisproteins,which competeswithcaspasebindingtoBaculoviralIAPrepeatdomainthroughthe alanine–valine–proline–isoleucinetetra-peptideatthenitrogenend[24].Smac proteinshaveattractedtheattentionofacademicsandpharmaceuticalcompanies tothedesignofsmall-moleculesmacsimulators[25].Therearecurrentlytwo typesofinhibitors,includingunivalentandbivalentinhibitors.Someofthemhave enteredphaseIIclinicalstage,suchasLCL-161byNovartisandDebio-1143by Debiopharm[12a].
1.1.2.3Bromodomains Theacetylationoflysineresiduesorthemethylationoflysineandarginineresidues canbe“read,”whichundertakescentralrolesinepigeneticregulation.Bromodomainsandhistoneinteractionsareimportantincontrollinggeneexpressionand DNArepairandinregulatinginflammationandcancer.Thebromodomainssharea conservativestructureconsistingoffour α-helicalbundles,whichareconnectedby differentcyclicregionsofvariablechargeandlength.Ahydrophobicpocketincludes aconservativeasparticamideandfivewatermoleculesthatcanidentifyacetylation oflysine[26].Aquinazolinecompound,apabetalonebyResverlogix,whichisable toincreasetranscriptionoftheApoA-Igenebyinhibitingbromodomainandextra terminaldomainproteins,especiallybromodomain-containingprotein4,isinphase IIIclinicaltrials,forthepotentialtreatmentofdiabetesmellitus,renalimpairment, andcardiovasculardiseases[12a].
1.1.2.4HumanImmunodeficiencyVirusIntegrase
Thehomotetramericproteinhumanimmunodeficiencyvirusintegraseintegrates theviralgenomeintohumanDNA,whichisvitalforhumanimmunodeficiencyvirusreplication.Theprotein–proteininteractionsbetweenthehuman immunodeficiencyvirus1integraseandthegrowthfactor/p75ofthehostprotein lensepitheliumarekeytothisprocess,whichmakesintegraseatargetforhuman immunodeficiencyvirus.Thestructureofhumanimmunodeficiencyvirusintegrase
1.1Protein–ProteinInteractionsandTheirSmall-MoleculeModulators 7 consistsofthreedomains,nitrogen-terminalDNAbindingdomains,catalyticcore domains,andcarbon-terminalDNAbindingdomains.Thecatalyticcoredomain hasseveralpocketsselectedassmallmoleculartargetforinhibitionofenzymeactivity[27].Nowseveralsmallmoleculestargetingtheenzymehavebeenapprovedfor thetreatmentofhumanimmunodeficiencyvirusinfections,suchasraltegravirby Merck,dolutegravirbyGSK,andelvitegravirbyTobacco,whichinhibittheenzyme activitybybindingtotheactivesite.Furthermore,aseriesof2-(quinolin-3-yl)acetic acidderivatives,includingclinicalcompoundBI-224436byGilead,havebeendevelopedtoblocktheintegrationstep,byinhibitinglensepithelium-derivedgrowth factor/p75-integraseinteraction,whichdisplaysadifferentresistanceprofile[28].
1.1.2.5B-CellLymphoma-2Family/B-CellLymphoma-2Homology3Proteins Interaction
B-celllymphomafamilyproteins,includingmembersofthefamilypromoting apoptosisandresistancetofamilymembers,arethecentraleffectorsofcell apoptosis.B-celllymphoma-2homology3-containingpromotesapoptoticproteins, suchasB-celllymphoma-2homologousantagonistkiller,throughasinglehelix bindingtohydrophobicpocketsthatinhibittheapoptosisofB-celllymphoma-2 proteins.Theuseofsmall-moleculecompoundstosimulatetheB-celllymphoma-2 homology3domainhasshownsignificanttherapeuticpotential[29].Several B-celllymphoma-2homology3simulationsweredeterminedbytheselectionof NMR-basedfragmentsandtheoptimizationofstructures.Forexample,ABT-737 hasanaffinityforB-celllymphoma-2andwithnanometermolerange[30].This compoundoccupiesthesamehydrophobicbagasaB-celllymphoma-2homologousantagonistkiller-derivedpeptide,whichhasthesamebindingpositionas B-celllymphoma-2homologousantagonistkiller’sLeu78andIle85tobindtothe keyresidues.Anothersmall-moleculetherapeuticVenetoclax,whichwasgranted BreakthroughTherapyDesignationbyUSFDA,hasbeenapprovedforthetreatment ofchroniclymphocyticleukemia.
1.1.2.6MouseDoubleMinute2–p53Interaction Theinteractionbetweenp53anditsnegativeregulatoryproteinmousedouble minute2(MDM2)orMDMXisthetargetofanticancertreatment,anditisalsoa commonmodelsystemtoevaluatethenewmethodofprotein–proteininteraction inhibition.Thisinteractionismediatedbytheshort α-helixpeptidesequenceofp53, whichbindstotheglobulardomainofMDM2orMDMX.Structurally,N-terminal domainofMDM2bindstoashort15-residual α-helixpeptideofp53,wherethree hydrophobicresiduesofp53occupyawell-definedhydrophobicpocketonMDM2 [31].ThesestructuralcharacteristicsmakethestrategyoftargetingMDM2–p53 protein–proteininteractionfeasible.Variousmethodshadbeenusedtodetermine theinhibitorsofmdm2–p53interactions,andaseriesof cis-imidazolineanalogs, nutlins,weredeterminedbyscreeningthecomplexlibrary[32].Thesemoleculesare abletoinhibittheinteractionbetweenp53andmdm2andadoptthesamebinding modeasthekeyresiduesofp53.Amongtheseanalogs,idasanutlinbyRochewasin phaseIIIclinicaltrialforthepotentialtreatmentofacutemyelogenousleukemia.
1.2FeaturesofPeptideasMolecularTools 1.2.1AdvantagesofPeptidesasMolecularTools
Naturalorartificialpeptidesorproteinsplayacentralroleinmolecularprocesses, thankstotheirstrongmolecularrecognitioncapabilities.Thestrongrecognition abilityofpeptidescanbeexplainedbyalargenumberofdifferenttypesoffunctionalgroups,whichareeasytoconstruct.Aminoacidsarerichinphysicochemical properties.Bypolarity,theycanbeclassifiedasbasic,acidic,nonpolar,orpolar aminoacids,whichprovidehydrogen-bondeddonors,receptors,orhydrophobic cores.Accordingtorigidity,someaminoacidsareflexible,suchasglycine,whileothersprocessfixedanglessuchasproline.Thisrichbuildingblock,combinedwithan easycombinationofamidekeys,makespolypeptidesdiverseandcomplexenoughto formmacromoleculesofaparticularnatureandmediateimportantmolecularprocessesaccordingly.Inaddition,alargenumberofposttranslationalmodifications andunnaturalaminoacidsgreatlyenhancetheirpotentialfunction.
Inaddition,peptide-mediatedidentificationprocessesareubiquitousinnature, includingthosebetweenproteins/peptidesandproteins/peptides,betweenproteins/peptidesandnucleicacids,andbetweenproteinsandlipids,allofwhich involveallprocessesofbiologicalsystems.Thesestructuralinformationare knownorreadilyavailableandcanbedesignedaccordingtocomplexstructures ofpolypeptidemodulatorsorfurtherexplorethedevelopmentofsmall-molecule inhibitors.
Further,polypeptidesandproteinsareeasytoscreenandevolve.Becausethe amidebondiseasytoconstruct,thepeptidecombinatoriallibrarycanbeeasilyused inthescreeningofactivesequences.Protein/polypeptideislocatedattheendofthe centralcode,sotheirmolecularevolutioncaneasilybeachievedthroughtheappropriatesizeofDNAlibraries,biologicalsystems,andDarwinianchoices.Incontrast, thedirectevolutionofsmallmoleculesisdifficult.AnoverviewofthemostcommontechnologiesispresentedinTable1.1.Alltechniquesaresomewhatrelated andsharecommonsteps.Thecommontechnicalstrategiesforpeptidescreening aredescribedbelow.
Multi-peptidearraysweresynthesizedbyspeckletechnique.Asahigh-throughput researchtool,peptidearraysareanewtypeofbiochipthatusesautomatedinstrumentation,insitusynthesis,todesignhundredsoforeventhousandsofpolypeptides inveryhighdensity.Thispeptidechipcanbeincubateddirectlywithavarietyofdifferentbiologicalsamples.Afterseveralwashingsteps,asecondaryantibody,whichis typicallylabeledbyafluorescentlabelandcanbedetectedbyafluorescentscanner, isapplied[44].
Protein/peptideevolutiontechniquesarefurthermodifiedtomeetmoreapplicationneeds.TheuseofpowerfultechniquestogenerateandscreenDNA-encoded proteinlibrarieshelpspromoteproteindevelopmentasadrugligand.However,their useasdrugligandsislimitedbytheirintrinsiccharacteristics.Twointrinsiclimitationsincluderotationalflexibilityofthepolypeptidebackboneandalimitednumber
Table1.1 Summaryofkeyfeaturesofscreeningtechnologiescommonlyusedforcyclic peptidediscovery.
Librarysizeand itsrestriction Screening hostCyclic
One-bead-onepeptide[33]
106,library construction
InvitroVarious chemistries
SICLOPPS[34]109, Transformation efficiency In cellulo
Peptideon plasmid[35]
Prokaryotic [36]
Head-to-tail cyclizationvia split-intein chemistry
109,transformation efficiency
109,transformation efficiency
Nonnaturalamino acids/PTMs/backbone modification
Yes–allresin compatible chemistries
Possiblewith ambercodon suppression
BacteriaPossible
Eukaryotic[37]107,transformation efficiency Yeast
BacteriaYes–commonly bypost translational cysteine alkylation
Phage[38]109,transformation efficiency Phage
CIS[39]1014,translation scale
Ribosome[39] mRNA[40]
RaPID [41]/TRAP[42]
Source:Obexeretal.[43].©2017,Elsevier.
InvitroYes–commonlyYes–possible
Yes–commonly N -acetyl chloride chemistry
Extensive reprogramming usingtheFIT system
(20)ofnaturalaminoacids.However,theserestrictionscanbeovercomebyusing chemicalmodifications.
Intheone-bead-one-peptide(1B1P)method,pioneeredbyLamandSalmonin 1991[45],splitandpoolsynthesistechniquesweredevelopedtogeneratediverse librariesofbeads(upto107compoundscurrently),eachcoatedwithmultiple copiesofauniquepeptide.Resin-compatiblechemistrieshadbeenexploitedto makediversebackbones,peptoids, D-aminoacids,andpeptidecyclizationsaccessible.PeiLabpresentedamethodforsynthesizingandscreeningacomplex1B1C Libraryofcyclicpeptidesforbiologicaltargets,suchasproteins.IntheTentagel micro-beads,upto10milliondifferentcyclicpeptidesweresynthesizedrapidly bysplit-poolsynthesis,andfollowedbymultistagescreeningscheme,including fluorescentactivatedcellsorting,magneticselection,theenzyme-linkedreaction onbeads,andtheanalysisofcyclicpeptidesinsolutionbyfluorescenceanisotropy. Finally,themostactivehitsaredeterminedbythepartialEdmandegradation-mass spectrometry[33].
Split-inteincircularligationofpeptidesandproteins(SICLOPPS),aninvivo methodfordiscoveringhead-to-tailcyclicpeptides,isfreefromgeneticcode reprogramming.Themethodappliessplit-inteinchemistrytocyclizerandomized peptidesequences.Thecyclicpeptidelibrarycanpotentiallybeofanysize,andthe peptideitselfmaycontainunlimitedrandomresidues,includingunnaturalamino acids[46].Plasmidpropagationwithincellsbridgegenotypesandphenotypes. Accordingly,transformationefficiencylimitstheachievablelibrarysizeto1079. Apartfrombeingimplementedin Escherichiacoli,SICLOPPShassincebeen extendedtoeukaryoticcells[47].
PhagedisplaywasforthefirsttimereportedbyGeorgeP.Smithin1985[38a]. Phagedisplaytechnologyisconsideredasafastandeffectivemethodforscreeningsmallpeptides.Inthistechnique,ageneencodesaninterestproteinintothe phageshellproteingene,andthephagedisplaystheproteinoutsideofitforbinding forcescreening.Phagedisplayisausefultoolfordrugdiscovery,buttherearesome deficiencies.First,thelibrary’scapacitycanonlyreach109,whichislimitedbytransfectionefficiency.Second,weneedtosolvethediversityproblemofthepolypeptide library.Third,asmallamountofpeptideduetoitshydrophobicityorbecauseofthe foldingoftheoutermembraneproteincannotbedisplayedonthephagesurface. Duringphagedisplay,chemicalepoxidationcanbeincorporatedtodirectlyevolve thecyclicpeptide,includingthedirectevolutionofpolycyclicpolypeptideandhelicalpeptides.Forexample,insitucyclizationiseasilyrealizedbydisulfidebridging oralkylationviacysteineorenzyme-mediatedmodifications[48].
Ribosomedisplaytechniquesareusedtoperformproteinevolutioninvitro, producingproteinsthatcanbindtoanidealligand[40].Thistechniqueoptimizes theinteractionoffunctionalproteinsthroughPlückthunlaboratories.TheribosomeshowsthebeginningofthepolypeptideencodedfromtheDNAsequence’s originallibrary.Eachsequenceistranscribedandthentranslatedintoadultforeign peptides.TheDNAlibraryisfusedtoalackofastopcodonintervalsequence.The absenceofastopcodonpreventsthereleasefactorfrombinding,triggeringthe dispersalofthetranscriptioncomplex.Therefore,thisintervalsequenceremains connectedtothepeptidetRNA,occupyingtheribosometunnel,whichmakes theproteinofinterestprotrudingfromtheribosomeandfolds.Theresulting mRNA,ribosomes,andproteincomplexescanbescreened.ThefilteredmRNA wasthentranscribedtothecDNAandamplifiedbypolymerasechainreaction (PCR).Sinceitiscarriedoutcompletelyinvitro,therearetwomainadvantages overotheralternativetechniques.First,thediversityofthelibraryisnotlimited bythetransfectionefficiencyofbacterialcellbutisonlyaffectedbythenumber ofribosomesanddifferentmRNAmoleculesinthetesttube.Second,random mutationscanbeeasilyintroducedaftereverychoice,makingproteinsevolveover severalgenerations.
Traditionally,reprogrammingwasachievedviastopcodonsuppressionor removalofcanonicalaminoacids.Moreextensivegeneticcodereprogrammingis particularlyfacilewhencarryingoutinvitrodisplaytechniques,whereenhanced librarydiversitycanbeachievedthroughreconstitutedtranslationsystemsgivingcompositionalfreedom[49].Theflexibleinvitrotranslation(FIT)method
1.2FeaturesofPeptideasMolecularTools 11 remainsthemostversatileandleastlabor-intensiveprocedureforgeneticcode reprogramminginvitro.Furthermore,integratingtheFITsystemwithmRNA displaygaverisetotherandomnonstandardpeptideintegrateddiscovery(RaPID) system[50],theversatilityofwhichisreflectedinitsbroaduseinthepharmaceuticalindustry.Bashiruddinetal.createdtricycles,bycombiningbicycle bridgingmoietywiththeclassic N -acetylchloridecyclisationoftheRaPID system[51].
Theligandisanoligonucleotideorpeptidemoleculethatbindstoaspecifictarget molecule.Theaptamerisusuallycreatedbyselectingthemfromalargerandom sequencepool,butthenaturalligandsalsoexistinriboswitches.Polypeptide ligandsareartificialproteinchoicesoraredesignedtobindtospecifictarget molecules[52].Theseproteinsarerepresentedbyoneormorepolypeptideloops byavariablesequenceofproteinscaffolds.Theyareusuallyseparatedfromthe combinatoriallibraryandareoftenmodifiedbydirectionalmutationorbymutation andselectionofthevariableregion.Invivo,peptideligandscanbindtocellprotein targetsandexertbiologicaleffects,includinginterferingwiththenormalprotein interactionsbetweentheirtargetmoleculesandotherproteins.Thelibraryof polypeptideligandsisusedasa“mutation.”In2013,ShekhtmanLaboratories developedamethodtobuildacombinatoriallibraryofimprovedpeptideadaptation (clips)ofhighcomplexity,containingmorethan3 × 1010 independentclonesas moleculartoolsforthestudyofbiologicalpathways[53].Theproteinskeletonwas modifiedtoimproveitssolubility,andtheaggregationofpeptidewaseliminated. Clipsisusedinyeasttwo-hybridscreeningtodeterminethepeptideadaptation tothelateglycationendproductofreceptorsindifferentdomains.Cellfunction detectionshowedthat,inadditiontodirectinterferencewiththeknownbinding sites,thecombinationofpolypeptideanddistalandligandsitesinhibitedthesignal transductionofrageligand-inducedsignaling.Thefindingshighlightthepotential ofusingfragmentstoselectbiologicaltargetinginhibitors.
1.2.2DisadvantagesofPeptidesasMolecularTools Comparedwiththesmallmolecule,thepolypeptidehassomedisadvantagesin thepropertiesofproprietarymedicines,whichlimiteditsbioavailability.First, amidebondsarefragileinvivo,makingstabilityafatalweaknessofnaturallinear peptides.Therearemanywaystoimproveitsstability,suchastheinsertionofa looporunnaturalmodule.InChapter2,thestabilitymethodofhelixpeptideis summarizedanddiscussed.Furthermore,thepenetrationofthepeptidesislimited. Thecompartmentsintheorganismaremainlycomposedoflipophilicsubstances, whichprovidethebasisforthetimeandspacecontrolofbiologicalprocesses, whilepeptidesaregenerallyhydrophilicinnature,whichmakesitdifficulttocross compartments.Therefore,thepenetrationofpeptidesaswellasabsorptionisoften problematic.Regulatingthephysicochemicalpropertiesofpeptides,suchassubstitution,modification,toincreasetheirinteractionwithbiofilms,canimprovetheir penetrability.InChapter3,thefactorsaffectingthepermeabilityofpolypeptides arediscussed.
1.3HelicalStructuresandTheirCharacterization 1.3.1DifferentTypesofHelices
1.3.1.1 ��-Helix
Atypical α-helixturniscomposedofanaverageof3.6aminoacidresidueswith dihedralangles(��, �� )inbackbonescloseto 55∘ and 45∘ ,respectively.Asariseof 1.5Å/residueor5.4Å/turn,only i + 4and i + 7positionscanmakethesidechains ofagivenresidueatthe i positionandtheotherresidueatthe i + n positionareon thesameface. α-Helicesarestabilizedbyintramolecular i → i + 4hydrogenbonds betweenacarbonylgroupoftheresidueatposition i andanamideprotonatposition i + 4inthemainchain,withabout2.72Åinthedistanceofnitrogen–oxygenandthe sidechainspointingawayfromthehelixaxis[54].
Protein–proteininteractionsareinvolvedinlotsofbiologicalprocessessuchas transcription,signaltransduction,exocytosis,andsoon[55]. α-Helixisthemost abundantsecondarystructuremotifinproteins,accountingforover30%innature. Meanwhile, α-helixisinvolvedatinterfacesofdiverseprotein–proteininteractions, whichwasknownfor α-helix-mediatedprotein–proteininteractions.Foritssignificantproportionfoundinproteins’structures,itisnotsurprisingtotellthat α-helix isthemostfundamentalrecognitionmotifsindiverseprotein–proteininteractions. Accordingtothestudyofhelicalinterfacesinprotein–proteininteractionsbasedon theProteinDataBank(PDB),about13%multi-proteinsystemscontainedthehelix interface,rangingfromenzymaticactivitiestoproteinassociationsbyclassification oftheirfunctions,suchasenergymetabolism,proteinsynthesis,transcription,DNA binding,signaling,transport,immunesystem,andsoon[56].
Thestructuralcharacteristicof α-helixforcesresiduesespeciallytheirsidechains toextendouttothesurroundingenvironmentforselectiveandspecificrecognition,makingittobeatemplatefordesigningsmall-moleculeinhibitorsoractivatorstowardprotein–proteininteractions.Thesimplestsystemfor α-helix-mediated protein–proteininteractionsbetweentwoproteinsisthatonepartnerbindstoits partnerproteinbyformingashorthelicalmotif.
1.3.1.2310 -Helix Besidesclassical α-helixand β-sheetconformations,the310 -helixisanother importantsecondarystructuralmotifoccurringinnaturalproteins,whichalso playssignificantrolesinstabilizingproteins’conformationsandmaintainingtheir biologicalfunctions.Taylorfirstproposedthe310 -helixstructurein1941.Sincethen thisstructuregainedmuchattentionandwasstudiedfully[57].Theshortname of310 -heliximpliesthatthenumberofresiduesperturnis3andthenumberof atomscontainedineachintramolecularhydrogenbondis10,whichindicatesthat 310 -helixismoretightlypackedthan α-helix(alsocalled3.613 -helix).Thebackbone torsionangles(��, �� )in310 -helicesareapproximately 60∘ and 30∘ ,respectively, whichareveryclosetothatin α-helices(�� =−55∘ , �� =−45∘ ).However,310 -helices displaysignificantlydistincthydrogen-bondingpatternof i → i + 3,while α-helices arestabilizedby i → i + 4intramolecularhydrogenbonds[58].The310 -helixis
1.3HelicalStructuresandTheirCharacterization 13 lessstablethanthe α-helixbecauseofitslessfavorablevanderWaalsenergyand nonoptimalhydrogenbondgeometry[59].However,onaccountofthehighstructuralsimilaritybetweenthe α-helixand310 -helix,itisproposedthatthe α-helixcan beturnedintothe310 -helixwhensidechaininteractionshappen.Indeed,310 -helices arenotrareandcouldbefoundinglobularproteinslikeaconitase,dienelactone hydrolase,andphageT4 lysozyme.BarlowandThorntonanalyzedglobularprotein crystalstructuresinthedatabaseandsuggestedthatatleast3.4%oftheresidues areinvolvedin310 -helices.Theyalsofoundthatthelocationof310 -helicesisoften closetotheN-orC-terminalofan α-helix[60].Marshalletal.provedthatAib (α-aminoisobutyricacidorCα,α -dimethyl-glycine)canpromotetheformationof 310 -helicesbycalculationsin1971.Sincethen,manyX-raydiffractionstructures ofpeptidesinvolvingrichAibindicatetheirstructurepreferenceof310 -helices [61].Itisworthnotingthatan α-helicalpeptiderequestsatleastsevenaminoacid residues,whiletheformationof310 -helicalpeptideshasnodependenceonmain chainlength[62].
1.3.1.3 ��-Helix
Sofaronlythreehelixtypes α-,310 -,and π-helixwerefoundinproteinstructures. Comparedwith α-helix(30%)and310 -helix(4%)innature, π-helixseemsparticularlyrare,whichcouldbeattributedtotheinstabilityofcorrespondingstructures. Tobespecific,valuesofdihedralanglesin π-helixwereveryclosetotheallowed minimumenergyrequirementsindicatedbyRamachandranplotandproventobe unfavorable[63].Meanwhile,itwassuggestedthattherequiredenergycostforstabilizingtheintramolecular i → i + 5hydrogenbondtoformahelixwashuge[64]. Therefore,manypeoplebelievethat π-helicesareunstableinnature.However,as researcheson π-helixaremovingforward,traditionalconceptsabout π-helixare broke.Researchersfoundtheformationof π-helicesinmoleculardynamicssimulationsofpeptides[65].Moreimportantly, π-heliceswereobservedinmanyprotein structures.Mostofnaturallyoccurring π-helicescontainatleastsevenaminoacid residuesandminimumtwo i → i + 5hydrogenbondsandmaximumsevenH-bonds. Alongwith α-helixand310 -helix, π-helixcanstablyexistandmayplayimportant rolesinmaintaininglotsofbiologicalfunctions.
π-Helicesarealsocalled4.416 -helixwhere4.4isthenumberofresiduesineach turnand16isthenumberofatomsinvolvedinahydrogenbond[66]. π-Helicesare stabilizedbyintramolecular i → i + 5hydrogenbondsbetweenacarbonylgroupof theresidueatposition i andanamideprotonatposition i + 4inthemainchain. α-Helicesand310 -helicesarestabilizedbyrepeating i → i + 4and i → i + 3hydrogen bonds,respectively.Therefore,minimalnumberofresiduesinasingle π-helixisone morethanthatinan α-helixandtwomorethanthatina310 -helix.Accordingtothe structuralanalysisof π-helicesinproteinsinPDB,themeanvaluesofdihedralangles (��, �� )observedin π-helicescouldbearound 76∘ and 41∘ ,respectively.However, itcouldhaveslightdistinctionsaccordingtodifferentmodelsonstructuredefinition of π-helices.Besides,valuesof1.2Åinanaverageunitrise,4.4residuesineachturn, and83∘ inanaverageunittwistwereobservedinthehelicalgeometryof π-helices. Like α-helices, π-heliceshaveitsfeaturedaminoacidpreferenceinsequences.The
distributionsofaminoacidresiduesfor π-helicesshowedthataromaticresidueslike Tyr,Trp,Phe,andHis,aswellasbulkyaliphaticresidueslikeIleandLeuhavehigher propensities,whilesmallaminoacidslikeAla,Gly,andProarelesspreferential. Also,thereareaminoacidresiduepreferencesintheirpositionsinsequences.For example,bulkyresiduessuchasPhe,Tyr,Trp,Ile,andLeuaremorelikelytobe locatedatthebeginningandattheendof π-helices.Besideshydrogenbondinteractions,otherfactorsfacilitatedthestabilizationof π-helices.Comparedwiththe α-helix,the π-helixhadalowerunitrise(1.2Å),whosesidechainswouldbecloser toeachotherinspace.Therefore,otherinteractionsbetweensidechainssuchasthe vanderWaals,aromaticringstacking,andelectrostaticinteractionsbecameimportantcontributorsforthestabilizationof π-helices.Thisiswhyaromaticandlarge aliphaticaminoacidshavehigherpropensitiesin π-helices[67].
Itisworthnotingthatsomeresearchesrevealedthattheremaybeanevolutionary relationshipbetween α-helicesand π-helices.Basedonthefamiliesofstructurally similarproteins(FSSP)surveyonallknown π-helix-containingproteinstructures indatabases[68],in106proteinswith π-helices,88werefoundtoexhibitthe α-helix withonelessaminoacidresidue,accountingforover80%,whichsuggestedthat nature π-helicesmayoriginatefromtheinsertionofoneresidueintothecorresponding α-helicesduringevolution.Meanwhile,atleastthreeresiduescouldbefoundin designated α-helicesinover95%oftheanalyzed π-helicesbyFSSPmethod,which alsosuggestedthattherewasastrongassociationbetween α-helicesand π-helices [69].Thehypothesisontheoriginalityof π-heliceswasfurtherconfirmedbythephenomenonof α-helix-to-π-helixconversioninsomeproteinfamilies.Forexample,in mercuricionreductases,an α-helix-to-π-helixconversion,whichwasattributedto theinsertionofasingleresiduecomparedtoitsancientreductasemember,occurred andputacatalyticTyrresidueintothebindingsiteandtriggeredtheHg2+ detoxificationbymercuricreductase[70].
Functionof ��-Helix Cellularfunctiondependsonhighlyspecificinteractions betweenbiomolecules(proteins,RNA,DNA,andcarbohydrates).Abasiclimitation ofdrugdevelopmentistheinabilityoftraditional“small-molecule”pharmaceuticalstospecificallytargetlargeproteininterfaces,manyofwhicharedesirabledrug targets. α-Helices,ubiquitouselementsofproteinstructures,playfundamental rolesinmanyprotein–proteininteractions.Stablemimicsof α-helicesthatcan predictablydisrupttheseinteractionswouldbeinvaluableastoolsinchemical biologyandasleadsindrugdiscovery.Therehasbeenexcitingprogressinthe moleculardesignoftheseproteindomainmimeticsandtheirremarkablepotential toinhibitchallenginginteractionsinthepastdecade.Keychallengesinthefield includingidentificationofsuitabletargetsandbioavailabilityofmedium-sized moleculesdonotconformtoempiricalrulesfollowedintraditionaldrugdesign. Stabilized α-helicesavoidsomeofthestrictlimitationsthathavebeenplacedon drugdiscovery.Whendesigningpotentialdrugcandidates,medicinalchemists oftenadheretotheLipinskirules,whichstipulatethatthemolecularmassofadrug shouldnotexceed500Da.Recentfindingssuggestthatlargesynthetic α-helices cantrafficintothecellandefficientlycompetewithcellularprotein–protein
1.3HelicalStructuresandTheirCharacterization 15 interactions,contrarytopredictionsbasedontheLipinskirules.Althoughthese moleculeshaveundoubtedlyproventheirvalueasprobesfordecodingbiological complexity,thenextbigquestioniswhetherthesemoleculescanbecometherapeutics.Thischapterdiscussestheapplicationsofconstrainedhelices,basedon propertiesofprotein–proteininteractions.
Although π-heliceshadlowerfrequencyobservedinproteinstructurecompared withotherhelixtypes,moreandmoreresultsrevealedthat π-helicescouldexhibit distinctfunctionalimportance,especiallyinthechelatasefamilyofproteins.For example,ferrochelatasesfrom Bacillussubtilis,whichcatalyzestheinsertionofFe2+ intoprotoporphyrintogenerateheme,containeda10aminoacidresidue-length π-helix.
This π-helixwaslocatedneartheactivesiteoftheenzymeatthedistanceof10Å, whoseresidueswereproventohaveinteractionswithahydratedmagnesiumcomplexinitsboundstate[71].Similarasferrochelatasesfrom B.subtilis chelatase,the cobaltchelatase,whichwasresponsiblefortheinsertionofCo2+ intoprecorin-2to generatecobalt-precorin,alsopossesseda π-helixofsevenaminoacidresiduelength [72].Dioxygenaseisanotherexample,whosethreehistidineresiduesH494,H499, andH504onthesurfaceofthe π-helixwereinvolvedintheformationofametal bindingsite,whichsuggestedthat π-helicesaretheuniquestructuralelementfor metalbindingenzymes.
Besidesthechelatasefamily, π-helicescontributetodiverseproteinfunctions. Forexample,an π-helixconformationwasfoundinthehydroxylasecomponentof methanemonooxygenasehydroxylase(MMOH),whichparticipatesinthebinding ofaproductanalog6-bromohexan-1-olattheactivesite.Thelong π-helixcontains two π-helicesnamedpiBandpiD,respectively.Intheprocessofligandbinding, aperistalticshiftofpiDinanesophagealperistalsis-likemannerwasperformed intheC-terminaldirection[73].Similarphenomenonof π-helicalshiftcouldbe observedintherelatedtoluene-4-mono-oxygenase(ToMO)hydroxylasestructure duringitsbindingoftheregulatorycomponent(ToMOD)[74],whichmayprovide anewinsightintomechanismsfortheactivationofbacterialmulticomponent monooxygenases(BMMs)bytheirregulatorysubunits[69].
1.3.2CharacterizationofHelicalPeptides Thestudyofthemolecularstructurecangivefinedetailsabouttheinterfacethat enablestheinteractionbetweenproteins.Themolecularstructuresofmanyprotein complexeshavebeenunlockedbythetechniqueofX-raycrystallography[75].Later, NMRalsostartedtobeappliedwiththeaimofunravelingthemolecularstructure ofproteincomplexesanditisadvantageousforcharacterizingweakprotein–protein interactions[76].
1.3.2.1CircularDichroism Circulardichroism(CD)isasimpleandconvenientmethodtoanalyzesecondary structuresofproteinsandpeptides.Onlychiralmoleculescanexhibitfeatured absorptionbandsintheirCDspectrawhilerandomlyorientedsystemshaveno CDintensity.Thisiswhyproteinsandpeptides,whicharecomposedofchiral
aminoacidresidues,showcharacteristicCDabsorptionaccordingtotheirdifferent secondarystructures.BecauseofthesensitivityandconvenienceofCDmeasurement,CDhasbecomearoutinetoolinmanyapplications,especiallyinthestudyof determiningthehelicalcontentofproteinsandpeptides[77].Generallyspeaking, proteinsandpeptidesinrandomordenaturedstructuresusuallyhaveveryweak signalsintheirCDspectra,whileproteinsandpeptideswithwell-definedandstable conformationssuchas α-helixcangivesignificantCDintensitiesandcharacteristics inCDspectra.Thefeaturedstructuralcharacteristicsin α-helix,including i/i + 4 hydrogen-bondingpattern,3.6aminoacidresiduesperturn,100∘ turnand1.5Å translationbetweentwopeptideunits,make α-helixbedistinguishedfromother structuralmotifsinproteinsandpeptides,whichexhibitsdistinctbandabsorptions inCDspectra.Absorptionsatdifferentwavelengthsindicatedifferentkindsof electrontransitionsuchasn → π * , π → π * ,n → σ * occurringat220,207–190,and 175nminCDsignals,respectively.The α-helicesdisplaytwoseparatenegative maximumsignalsatboth222nm(then → π * transition)and208nm(partofthe π → π * transition)aswellasanotherpositivesignalat195nm(partofthe π → π * transition).Comparedwith α-helices’featuredabsorptions,theothercommon structuralunit β-sheetsshowanegativebandatabout220nmwithanotherpositive bandatabout200nminthecorrespondingCDspectra.Itisworthnotingthatthe predictionof α-helicesinproteinsandpeptidesbyaCDtoolisverysensitiveand hasveryhighaccuracy.
1.3.2.2X-rayCrystallography
X-raycrystallographyisatechniqueusedfordeterminingtheatomicandmolecularstructureofacrystal,inwhichthecrystallineatomscauseabeamofincident X-raystodiffractintomanyspecificdirections.Themethodhasrevealedthestructureandfunctionofmanybiologicalmolecules,includingvitamins,drugs,proteins, andnucleicacids.Thecrystalstructuresofproteinswerefirstreportedinthelate 1950s,beginningwiththestructureofspermwhalemyoglobin.X-raycrystallographyisnowusedroutinelybyscientiststodeterminehowapharmaceuticaldrug interactswithitsproteintargetandtounveilthedetailedinterfaceofprotein–protein interactions[78].NavitoclaxisaninhibitorofbothBCL-2andBCL-XL ,buttheconcomitanton-targetthrombocytopeniacausedbyBCL-XL inhibitionlimitstheefficacyachievablewiththisagent.Basedonthecrystalstructureofthiscompoundin complexwithBCL-2familyproteins,AndrewJ.Souersetal.re-engineeredandoptimizednavitoclaxtocreateahighlypotent,orallybioavailableandBCL-2-selective inhibitorABT-199,whichshowedpotentinhibitionactivityonBCL-2-dependent tumorsinvivoandspareshumanplatelets[79].
Thetechniqueofsingle-crystalX-raycrystallographyhasthreebasicsteps.The firststepistoobtainanadequatecrystalofthemoleculeunderstudy.Thecrystal shouldbelargeenough(typicallylargerthan0.1mminalldimensions),regularin structure,andpureincomposition,withnosignificantinternalimperfectionssuch ascracksortwinning,andthisisoftenthemostdifficultstep.Inthesecondstep, thecrystalisilluminatedwithafinelyfocusedmonochromaticbeamofX-rays,producingadiffractionpatternofregularlyspacedspotsknownasreflections.Asthe
1.3HelicalStructuresandTheirCharacterization 17 crystalisgraduallyrotated,previousreflectionsdisappear,andnewonesappear,the intensityofeveryspotisrecordedateveryorientationofthecrystal.Inthethirdstep, the2Dimagestakenatdifferentorientationsareconvertedintoa3Dmodelofthe densityofelectronswithinthecrystalusingthemathematicalmethodofFourier transforms;thesedataarecombinedcomputationallywithcomplementarychemicalinformationtoproduceandrefineamodelofthearrangementofatomswithin thecrystal.Thecrystalstructurescouldgiveessentialdetailsaboutthestructuresof amoleculeormacromolecularcomplex.
1.3.2.3NuclearMagneticResonance(NMR) NMRisanotherfastandpowerfulmethodtodetermineconformationsofproteins andpeptidesinsolution.In1Dand2DNMRspectroscopies,nuclearoverhauser effects(NOEs),temperaturedependenceoftheNMRspectrum,andcouplingconstantsarethreeimportantindicatorsforthedeterminationofpeptides’secondary structures[80].Besides,measurementsofthechemicalshiftindexandtherateof exchangewithdeuteronsarealsotworegularmethodsfortheconformationanalysis ofpeptides.
ThereisaveryclosecorrelationbetweenNOEsandthedistancebetweenprotons. Generallyspeaking,NOEsignalsbetweentwoprotonscanarisewhentheirdistance isunder5Åinspace,indicatingstrongevidencesofsecondarystructures[81]. Thecross-peaksina2Dscalar-correlated(COSY)spectrumindicatethecoupling patternsfortheresonancesassignedtospecificprotonsofaminoacidresidues. Specifically,sequentialconnectivitiesandNH–NHaswellasNH–CHcross-peaks in2D-ROESYspectraindicatekeymedium-andlong-rangeROEs,whichstrongly supportstheidentificationofsecondarystructures.Somerepresentativenonsequentialmediumcross-peakssuchas dαN (i, i + 4), dαN (i, i + 3),and dαβ (i, i + 3)in ROESYspectrasupporttheadoptionofhelicalconformations[82].
Thetemperaturedependenceofamideprotonresonancesisutilizedfordeterminingwhetherthebackboneaminoacidresiduesareinvolvedintheintramolecular hydrogen-bondingformation.Generallyspeaking,amideprotonresonancesexhibit highertemperaturecoefficientswithincreasingtemperature,whichsuggestsno involvementofhydrogenbondsandthecompleteexpositiontosolvents.Avalueof temperaturecoefficient(< 4.0ppb/K)indicatesthespecificamideproton’sparticipationinthehydrogen-bondinginH2 O,whichissocriticalforthestabilizationof an α-helix.Valuesoftemperaturecoefficientforindicatingthehydrogen-bonding aredifferentinvarioushydrogen-bondingsolvents.ComparedwithH2 O,temperaturegradientsforthechemicalshiftoftheNH-signalisconsideredtobeunder 3ppb/Kindimethylsulfoxide(DMSO).
Vicinalcouplingconstant 3 J hasacloserelationshipwithdihedralangleoriginatedfromtheKarplusequationsandisveryimportanttoanalyzeconformations ofproteinsandpeptides.Amongthesedifferenttypesofcoupling,thehomonuclear 3 J H NCαH iseasytobemeasuredandthusparticularlyimportant.However,according totheKarpluscurve,thereisnoone-to-onecorrespondencebetweenmeasuredcouplingconstantsandbondanglesinspiteofthedependenceof 3 J H NCαH ontheangle ��.Indeed,inmostcases,onecouplingconstantcanrespondtofourdifferentbond
angles ��.Therefore,itisambiguousandhardtoachievecompellingconclusions from 3 J H NCαH measurements,thusviewedasacomplementapproachforanalyzing conformationsofpeptidesbasedonthebondangle �� [83].Itisgenerallyacknowledgedthatthecouplingconstant 3 J H NCαH under6Hzforaminoacidresiduesindicatestheadoptionofhelicalconformations.
Besides,thechemicalshiftisanequallyimportantindexforthedetermination ofsecondarystructuresofpeptidesandproteins.Itisfoundthat 1 HNMRchemical shiftshaveaverycloseconnectionwiththecharacterofsecondarystructures.Comparedwiththerandomcoilvalue,the 1 HNMRchemicalshiftofthe α-CHproton ofallnatural20aminoacidsshowsanupfieldshiftwhentheyareinvolvedinthe helicalconformation.Conversely,adownfieldshiftofthecorresponding 1 HNMR chemicalshiftisobservedwhentheyadoptthe β-strandconformations.Therefore, theseobservationsof 1 HNMRchemicalshiftcharactersareveryhelpfultodeterminethelocation,aswellascomparecontentsofdifferentsecondarystructural motifsinproteinsandpeptides.ComparedwithtraditionalNOE-basedmethodsfor secondarystructuredetermination,thismethodsimplyrequeststhemeasurement of α-CH 1 Hresonanceassignmentsandwasproventobeaccurateandusefulbya lotofexamples[84].
Finally,therateofexchangewithdeuteronslikeaddedD2 Oisaconvenient methodformeasuringmoleculardynamicsofpeptides.Specifically,theexchange ofamideprotonswiththedeuteronsolventcantellwhethertheseprotonsare involvedinformingintramolecularhydrogenbondsbymeasuringthecorrespondingexchangerate.Generallyspeaking,theamideprotonparticipatingin intramolecularhydrogenbondsisexpectedtoexhibitlowerexchangeratein deuteronsolventswhilethoseprotonsabsentfromhydrogenbondsareexchanged morerapidly[85].
1.4StabilizationofPeptides Thissectiondescribesthemethodsofstabilizingpeptides,includingthecyclicand mainalteration.
1.4.1PeptideStabilizationviaCyclization
1.4.1.1Monocyclization
Macrocyclizationusesadditionalcovalentbondstolimitthedistancebetweentwo points.Thecyclicincreaseofpeptideresistancetoproteasereducestheexposureof hydrogenbonddonorsorreceptorstoregulatethephysicalandchemicalproperties ofpeptides.
Disulfidebondssensitivetoredoxreactionsarecommoninnature.Thespecific patternandlengthoftheartificialdisulfidebondcanbeusedtostabilizethehelix, hairpin,orloop.Theirreversiblepropertiescanbeusedtostudythefoldingofpeptidesortheuptakeofcells[86].Atthesametime,thebiologicalsystemcandirectly evolvedisulfidebonds.Similarly,thehydrophilicamideoresterbondiswidelyused
1.4StabilizationofPeptides 19 intheconstructionofpeptideanalogasabridgeconnectingthenaturalclock.Inparticular,thelysine-asparticacidstablehelixshowsthehighesthelicityinthewater.
Asakindofhighstabilitybridgeconnectingmode,thethioletherbondisoften usedasthesubstitutionofdisulfidebondstorestrainpeptideconformation.The thioletherbondcanbeconstructedbyusinganucleophilicsubstitutionreaction ofhalidesorbyusingafreeradicalreactionofunsaturatedhydrocarbons.Inaddition,comparedwithacomponentbindingstrategy,thetwo-componentstabilization strategiescangivethepeptidewithspecialcharacteristics,suchasreversibilityand modifyingpotentials[87].Likedisulfide,thetwo-ingredientstrategybasedoncysteinecanbeusedinmicro-proteinsorcombinedwithascreeningsystem.
Asanonpolarconnection,theall-hydrocarbonstrategyincreasesthehydrophobicityofpeptides.Thestapledpolypeptide,theall-hydrocarbonsidechainstabilized helix,isconstructedbyring-closingmetathesis.Varioustypesofstaplingstrategies havebeendevelopedandwidelyused.Inaddition,allhydrocarbonchainscanbe usedinreplaceofhydrogenbondasanN-terminaltemplate.Inaddition,azoles,constructedusingbio-orthogonalreactions,canbeusedtostabilizethepeptide,includingthehelixandhairpinstructure[88].Inone-componentway,theringaddition canderiveafluorescentbridge.Whilefortwo-componentstrategy,thefunctional stapledpolypeptidecanbeestablishedtofurtherlabel[89].
1.4.1.2Multicyclization Increasingthenumberofpolypeptiderings,suchasconstructionofbicyclicoreven polycyclicpeptides,canfurtherimprovethetherapeuticperformanceofpeptides andevenshowtheoraldosingpotentials.Therearemanypolycyclicpeptidesin nature,andintheprocessofstudyingthesepolycyclicpeptides,manyvaluable techniquesandmoleculeshavebeenobtained.Herewewillfocusonthepolycyclic polypeptideinnature.Therearemanybioactivepolycyclicpeptidesinnature, includingbacterialsecretions,suchaslantibiotics,toxicpeptides,suchasamatoxin andphallotoxin,andpeptidetoxinsofanimalsecretions,suchas α-toxins.
Lantibioticsisanimportantpolypeptideantibioticthatisfoundandproducedbya largenumberofGram-positivebacteria(including Streptococcus and Streptomyces) andattacksotherGram-positivebacteria.Thepeptidecontainsthecharacteristics ofmulticycliclanthionineormethyllanthionineandsomeunsaturatedaminoacid dehydroalanineand2-aminoisobutyricacidasthebasicbuildingblocks.Lanthionine,abbreviationforlanthionine-containingpeptideantibiotics,isoneoftheimportantbuildingblocksoflantibiotics,consistingoftwoalanineresidues,whichare connectedbysulfurtotheir β-carbonatoms[90].Mostofthelantibioticsareconsideredtobeaclassofbacteriocinssynthesizedbyribosomebiosynthesis,withapilot polypeptidesequencethatisremovedwhenthemoleculeistransportedoutofthe syntheticcell.Therearefourenzymesinvolvedintheconstructionoflanthionine rings.Thisprocessdiffersfrommostoftheothernaturalantibioticswehavepreviouslyknown[91].LipidII,aglycoproteinofthisgram-positivebacterium,isthe targetforalargenumberoflantibiotics.Somelantibioticscausesdestructivepores orinhibitsthebiosynthesisofpeptides.Nisinisalantibioticswidelyusedintoday’s commercialpreservatives.