Chemoinformaticsand Bioinformaticsinthe Pharmaceutical Sciences
Editedby NavneetSharma
HimanshuOjha
PawanKumarRaghav
RameshK.Goyal
AcademicPressisanimprintofElsevier 125LondonWall,LondonEC2Y5AS,UnitedKingdom 525BStreet,Suite1650,SanDiego,CA92101,UnitedStates 50HampshireStreet,5thFloor,Cambridge,MA02139,UnitedStates TheBoulevard,LangfordLane,Kidlington,OxfordOX51GB,UnitedKingdom
Copyright © 2021ElsevierInc.Allrightsreserved.
Nopartofthispublicationmaybereproducedortransmittedinanyformorbyany means,electronicormechanical,includingphotocopying,recording,oranyinformation storageandretrievalsystem,withoutpermissioninwritingfromthepublisher.Detailson howtoseekpermission,furtherinformationaboutthePublisher’spermissionspolicies andourarrangementswithorganizationssuchastheCopyrightClearanceCenterandthe CopyrightLicensingAgency,canbefoundatourwebsite: www.elsevier.com/permissions .
Thisbookandtheindividualcontributionscontainedinitareprotectedundercopyright bythePublisher(otherthanasmaybenotedherein).
Notices
Knowledgeandbestpracticeinthis fieldareconstantlychanging.Asnewresearchand experiencebroadenourunderstanding,changesinresearchmethods,professional practices,ormedicaltreatmentmaybecomenecessary.
Practitionersandresearchersmustalwaysrelyontheirownexperienceandknowledgein evaluatingandusinganyinformation,methods,compounds,orexperimentsdescribed herein.Inusingsuchinformationormethodstheyshouldbemindfuloftheirownsafety andthesafetyofothers,includingpartiesforwhomtheyhaveaprofessional responsibility.
Tothefullestextentofthelaw,neitherthePublishernortheauthors,contributors,or editors,assumeanyliabilityforanyinjuryand/ordamagetopersonsorpropertyasa matterofproductsliability,negligenceorotherwise,orfromanyuseoroperationofany methods,products,instructions,orideascontainedinthematerialherein.
LibraryofCongressCataloging-in-PublicationData
AcatalogrecordforthisbookisavailablefromtheLibraryofCongress
BritishLibraryCataloguing-in-PublicationData
AcataloguerecordforthisbookisavailablefromtheBritishLibrary
ISBN:978-0-12-821748-1
ForinformationonallAcademicPresspublicationsvisitour websiteat https://www.elsevier.com/books-and-journals
Publisher: AndreWolff
AcquisitionsEditor: ErinHill-Parks
EditorialProjectManager: BillieJeanFernandez
ProductionProjectManager: MariaBernadetteVidhya
CoverDesigner: MarkRogers
Contributors
TanmayArora
SchoolofChemicalandLifeSciences(SCLS),JamiaHamdard,NewDelhi,Delhi, India;DivisionofCBRNDefence,InstituteofNuclearMedicine&AlliedSciences, DRDO,NewDelhi,Delhi,India
ShereenBajaj
DivisionofCBRNDefence,InstituteofNuclearMedicine&AlliedSciences, DRDO,NewDelhi,Delhi,India
PrernaBansal
DepartmentofChemistry,RajdhaniCollege,UniversityofDelhi,NewDelhi,Delhi, India
AmanChandraKaushik
WuxiSchoolofMedicine,JiangnanUniversity,Wuxi,Jiangsu,China
RamanChawla
DivisionofCBRNDefence,InstituteofNuclearMedicine&AlliedSciences, DRDO,NewDelhi,Delhi,India
GuruduttaGangenahalli
StemCellandGeneTherapyResearchGroup,InstituteofNuclearMedicine& AlliedSciences(INMAS),DefenceResearchandDevelopmentOrganisation (DRDO),NewDelhi,Delhi,India
SrishtyGulati
NucleicAcidResearchLab,DepartmentofChemistry,UniversityofDelhi,North Campus,NewDelhi,Delhi,India
MonikaGulia
SchoolofMedicalandAlliedSciences,GDGoenkaUniversity,Gurugram, Haryana,India
VikasJhawat
SchoolofMedicalandAlliedSciences,GDGoenkaUniversity,Gurugram, Haryana,India
DivyaJhinjharia
SchoolofBiotechnology,GautamBuddhaUniversity,GreaterNoida,India
JayadevJoshi
GenomicMedicine,LernerResearchInstitute,ClevelandClinicFoundation, Cleveland,OH,UnitedStates
RitaKakkar
ComputationalChemistryLaboratory,DepartmentofChemistry,Universityof Delhi,NewDelhi,Delhi,India
AmanChandraKaushik
WuxiSchoolofMedicine,JiangnanUniversity,Wuxi,Jiangsu,China
ShrikantKukreti
NucleicAcidResearchLab,DepartmentofChemistry,UniversityofDelhi,North Campus,NewDelhi,Delhi,India
ShwetaKulshrestha
DivisionofCBRNDefence,InstituteofNuclearMedicine&AlliedSciences, DRDO,NewDelhi,Delhi,India
RajeshKumar
DepartmentofComputationalBiology,IndraprasthaInstituteofInformation Technology,NewDelhi,Delhi,India;BioinformaticsCentre,CSIR-Instituteof MicrobialTechnology,Chandigarh,India
SubodhKumar
StemCellandGeneTherapyResearchGroup,InstituteofNuclearMedicine& AlliedSciences(INMAS),DefenceResearchandDevelopmentOrganisation (DRDO),NewDelhi,Delhi,India
HirdeshKumar
LaboratoryofMalariaImmunologyandVaccinology,NationalInstituteofAllergy andInfectiousDiseases,NationalInstitutesofHealth,Bethesda,MD,United States
VinodKumar
DepartmentofComputationalBiology,IndraprasthaInstituteofInformation Technology,NewDelhi,Delhi,India;BioinformaticsCentre,CSIR-Instituteof MicrobialTechnology,Chandigarh,India
AnjaliLathwal
DepartmentofComputationalBiology,IndraprasthaInstituteofInformation Technology,NewDelhi,Delhi,India
AsrarA.Malik
SchoolofChemicalandLifeSciences(SCLS),JamiaHamdard,NewDelhi,Delhi, India
GandharvaNagpal
DepartmentofBioTechnology,GovernmentofIndia,NewDelhi,Delhi,India
HimanshuOjha
CBRNProtectionandDecontaminationResearchGroup,DivisionofCBRN Defence,InstituteofNuclearMedicineandAlliedSciences,NewDelhi,Delhi, India
MallikaPathak
DepartmentofChemistry,MirandaHouse,UniversityofDelhi,NewDelhi,Delhi, India
PawanKumarRaghav
DepartmentofComputationalBiology,IndraprasthaInstituteofInformation Technology,NewDelhi,Delhi,India
ShaktiSahi
SchoolofBiotechnology,GautamBuddhaUniversity,GreaterNoida,Uttar Pradesh,India
ManishaSaini
CBRNProtectionandDecontaminationResearchGroup,DivisionofCBRN Defence,InstituteofNuclearMedicineandAlliedSciences,NewDelhi,Delhi, India
ManishaSengar
DepartmentofZoology,DeshbandhuCollege,UniversityofDelhi,NewDelhi, Delhi,India
MamtaSethi
DepartmentofChemistry,MirandaHouse,UniversityofDelhi,NewDelhi,Delhi, India
V.G.ShanmugaPriya
DepartmentofBiotechnology,KLEDr.M.S.SheshgiriCollegeofEngineeringand Technology,Belagavi,Karnataka,India
VidushiSharma
DelhiInstituteofPharmaceuticalEducationandResearch,NewDelhi,Delhi, India
AnilKumarSharma
SchoolofMedicalandAlliedSciences,GDGoenkaUniversity,Gurugram, Haryana,India
MaltiSharma
DepartmentofChemistry,MirandaHouse,UniversityofDelhi,NewDelhi,Delhi, India
NavneetSharma
DepartmentofTextileandFiberEngineering,IndianInstituteofTechnology,New Delhi,Delhi,India
MdShoaib
NucleicAcidResearchLab,DepartmentofChemistry,UniversityofDelhi,North Campus,NewDelhi,Delhi,India
AnjuSingh
NucleicAcidResearchLab,DepartmentofChemistry,UniversityofDelhi,North Campus,NewDelhi,Delhi,India;DepartmentofChemistry,RamjasCollege, UniversityofDelhi,NewDelhi,Delhi,India
JyotiSingh
DepartmentofChemistry,HansrajCollege,UniversityofDelhi,NewDelhi,Delhi, India
KailasD.Sonawane
StructuralBioinformaticsUnit,DepartmentofBiochemistry,ShivajiUniversity, Kolhapur,Maharashtra,India;DepartmentofMicrobiology,ShivajiUniversity, Kolhapur,Maharashtra,India
RakhiThareja
DepartmentofChemistry,St.Stephen’sCollege,UniversityofDelhi,NewDelhi, Delhi,India
NishantTyagi
StemCellandGeneTherapyResearchGroup,InstituteofNuclearMedicine& AlliedSciences(INMAS),DefenceResearchandDevelopmentOrganisation (DRDO),NewDelhi,Delhi,India
YogeshKumarVerma
StemCellandGeneTherapyResearchGroup,InstituteofNuclearMedicine& AlliedSciences(INMAS),DefenceResearchandDevelopmentOrganisation (DRDO),NewDelhi,Delhi,India
SharadWakode
DelhiInstituteofPharmaceuticalEducationandResearch,NewDelhi,Delhi, India
Impactof chemoinformatics approachesandtoolson currentchemicalresearch
RajeshKumar1, 3, a,AnjaliLathwal1, a,GandharvaNagpal2,VinodKumar1, 3 , PawanKumarRaghav1, a
1DepartmentofComputationalBiology,IndraprasthaInstituteofInformationTechnology,New Delhi,Delhi,India; 2DepartmentofBioTechnology,GovernmentofIndia,NewDelhi,Delhi,India;
3BioinformaticsCentre,CSIR-InstituteofMicrobialTechnology,Chandigarh,India
1.1 Background
Biologicalresearchremainsatthecoreoffundamentalanalysisinthequestto understandthemolecularmechanismoflivingthings.Biologicalresearchersproduceenormousamountsofdatathatcriticallyneedtobeanalyzed.Bioinformatics isanintegrativesciencethatarisesfrommathematics,chemistry,physics,statistics, andinformatics,whichprovidesacomputationalmeanstoexploreamassive amountofbiologicaldata.Also,bioinformaticsisamultidisciplinarysciencethat includestoolsandsoftwaretoanalyzebiologicaldatasuchasgenes,proteins, molecularmodelingofbiologicalsystems,molecularmodeling,etc.ItwasPauline Hogeweg,aDutchsystembiologist,whocoinedthetermbioinformatics.Afterthe adventofuser-friendlySwissportmodels,theuseofbioinformaticsinbiological researchhasgainedmomentumatunparalleledspeed.Currently,bioinformatics hasbecomeanintegralpartofalllifescienceresearchthatassistsclinicalscientists andresearchersinidentifyingandprioritizingcandidatesfortargetedtherapies basedonpeptides,chemicalmolecules,etc.
Chemoinformaticsisaspecializedbranchofbioinformaticsthatdealswiththe applicationofdevelopedcomputationaltoolsforeasydataretrievalrelatedtochemicalcompounds,identificationofpotentialdrugtargets,andperformanceofsimulationstudies.Theseapproachesareusedtounderstandthephysical,chemical,and biologicalpropertiesofchemicalcompoundsandtheirinteractionswiththebiological systemthatcanhavethepotentialtoserveasaleadmoleculefortargetedtherapies. Althoughthesensitivityofthecomputationalmethodsisnotasreliableasexperimentalstudies,thesetoolsprovideanalternativemeansinthediscoveryprocess becauseexperimentaltechniquesaretimeconsumingandexpensive.Theprimary
ChemoinformaticsandBioinformaticsinthePharmaceuticalSciences. https://doi.org/10.1016/B978-0-12-821748-1.00001-4 Copyright © 2021ElsevierInc.Allrightsreserved.
applicationofadvancedchemoinformaticsmethodsandtoolsisthattheycanassist biologicalresearcherstoarriveatinformeddecisionswithinashortertimeframe. Amoleculewithdrug-likenesspropertiesh astopassphysicochemicalproperties suchastheLipinskiruleoffiveandabsorption,distribution,metabolism,excretion,andtoxicity(ADMET)propertiesbef oresubmittingitforclinicaltrials.If anycompoundfailstopossessreliableADMETproperties,itislikelytobe rejected.So,intheprocessofacceleratingthedrugdiscoveryprocess,researchers canusedifferentinsilicochemoinformaticscomputationalmethodsforscreening alargenumberofcompoundsfromchemicallibrariestoidentifythemostdruggablemoleculebeforelaunchingintoc linicaltrials.Asimilarapproachcanbe employedfordesigningsubunitvaccinecandidatesfromalargenumberofprotein sequencesofpathogenicbacteria.
Intheliterature,severalotherreviewarticlesfocusonspecializedpartsofbioinformatics,butthereisnosucharticledescribingtheuseofbioinformaticstoolsfor nonspecialistreaders.Thischapterdescribestheuseofdifferentbiologicalchemoinformaticstoolsanddatabasesthatcouldbeusedforidentifyingandprioritizing drugmolecules.Thekeyareasincludedinthischapteraresmallmoleculedatabases, proteinandliganddatabases,pharmacophoremodelingtechniques,andquantitative structure activityrelationship(QSAR)studies.Organizationofthetextineach sectionstartsfromasimplisticoverviewfollowedbycriticalreportsfromtheliteratureandatabulatedsummaryofrelatedtools.
1.2 Ligandandtargetresourcesinchemoinformatics
Currently,therehasbeenanenormousincreaseindatarelatedtochemicalsand medicinaldrugs.Theavailableexperimentallyvalidateddatacanbeutilizedin computer-aideddrugdesignanddiscoveryofsomenovelcompounds.However,most oftheresourceshavingsuchdatabelongstoprivatedomainsandlargepharmaceutical industries.Theseresourcesmainlyhousedatainformofchemicaldescriptorsthatmay beusedtobuilddifferentpredictivemodels.Acompleteoverviewofthechemical descriptors/featuresanddatabasescanbefoundin Tables1.1and1.2.Abriefdescriptionofeachtypeofdatabasecanbefoundinthesubsequentsubsectionsofthischapter.
1.2.1 Smallmoleculecompounddatabases
Smallmoleculecompounddatabasesholdinformationonactiveorganicandinorganicsubstances,whichcanshowsomebiologicaleffect.Thelargestrepository ofactivesmallmoleculecompoundsistheAvailableChemicalDirectory(ACD), whichstoresalmost300,000activesubstances.TheACD/Labsdatabaseprovides informationonthephysicochemicalpropertiessuchaslogP,logS,andpKavalues ofactivecompounds.AnothersuchdatabaseistheSPRESIwebdatabasecontaining morethan4.5millioncompoundsand3.5millionreactions.Anotherdatabase, CrossFireBeilstein,hasmorethan8millionorganiccompoundsand9millionactive biochemicalreactionsalongwithavarietyofproperties,includingvariousphysical properties,pharmacodynamics,andenvironmentaltoxicity.
Table1.1 Tablerepresentingstandardfeaturesandtheirtypeutilizedin quantitativestructure activityrelationship.
Descriptor typeBasisExample
Theoreticaldescriptors
0DStructuralcountMolecularweight,numberofbonds,numberof hydrogenbonds,aromaticandaliphaticbonds
1DChemicalgraph theory
2DTopological properties
3DGeometrical structural properties
Numbersoffunctionalgroups,fragmentcounts, disulfidebonds,ammoniumbond
Randicindex,wienerindex,molecularwalkcount, kappashapeindex
Autocorrelation,3D-Morse,fingerprints
4DConformationalGRID,raptor,sampleconformation
Experimentaldescriptors
ElectronicElectrostatic properties
Dissociationconstant,hammettconstant
StericStericpropertiesChartonconstant
HydrophobicHydrophobic properties logP,hydrophobicconstant
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/ scoringfunctions/descriptionavailabilitywithuniformresourcelocator(URL) andsupportedplatforms.
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
Commonlyuseddatabasesinchemoinformatics
Available Chemical Directory(ACD)
CrossFire Beilstein
Accesstometiculously examinedexperimental NuclearMagneticResonance (NMR)data,completewith assignedstructuresand referencesofmillionsof chemicalcompounds
https://www. acdlabs.com/ products/dbs/nmr_ db/index.php
Dataonmorethan320million scientificallymeasured propertiesofchemical compounds.Thelargest databaseinorganicchemistry. www. crossfirebeilstein. com
SpresiWebDataregardingmillionsof chemicalmoleculesand reactionsextractedfrom researcharticles
https://www. spresi.com/ indexunten.htm
32681440
11604014
24160861 Continued
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
ChEMBLApproximately2.1million chemicalcompoundsfrom nearly1.4millionassays
PubChemContains9.2million compoundswithactivity information
CARLSBADContainsactivityinformationof 0.43millionactivecompounds
DrugcentralProvidesinformationon4,444 pharmaceuticalingredients with1,605humanprotein targets
repoDBAstandarddatabasefordrug repurposing
PharmGKBAdatabaseforexploringthe effectofgeneticvariationon drugtargets
ZINCAcommerciallyavailable databaseforvirtualscreening
https://www.ebi. ac.uk/chembl/ 21948594
https://pubchem. ncbi.nlm.nih.gov
26400175
http://carlsbad. health.unm.edu/ carlsbad/ 23794735
http://drugcentral. org 27789690
http://apps. chiragjpgroup.org/ repoDB/ 28291243
https://www. pharmgkb.org 23824865
http://zinc.docking. org 15667143
Databasesforexploringprotein ligandinteraction
ProteinData Bank(PDB)
Cambridge Structural Database(CSD)
ProteinLigand Interaction Database(PLID)
ProteinLigand Interaction Clusters(PLIC)
Providesinformationon 166,301crystallographic identifiedstructuresof macromolecules
Providesinformationonnearly 0.8millioncompounds
Aresourceforexploringthe protein ligandinteraction fromPDB
Arepositoryforexploring nearly84,846protein ligand interactionsderivedfromPDB
CREDOAresourceprovidingprotein ligandinteractioninformation fordrugdiscovery
https://www.rcsb. org/ 10592235
https://www.ccdc. cam.ac.uk/ solutions/csdsystem/ components/csd/ 27048719
http://203.199. 182.73/gnsmmg/ databases/plid/ 18514578
http://proline. biochem.iisc.ernet. in/PLIC 24763918
http://www-cryst. bioc.cam.ac.uk/ credo 19207418
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
PDBbindAresourceforthebinding affinityofnearly5,897 protein ligandcomplexes
http://www. pdbbind.org/ 15943484
Databaseforexploringmacromolecularinteractions
DOMININOAdatabaseforexploringthe interactionbetweenprotein domainsandinterdomains
PIMAdbAresourceforexploring interchaininteractionamong proteinassemblies
PDB-eKBAcommunity-driven knowledgebaseforfunctional annotationandpredictionof PDBdata
CATHAdatabaseforclassificationof proteindomains
LIGANDAcompositedatabaseof chemicalcompounds, reactions,andenzymatic information
TheMolecular Interaction Database(MINT)
Databaseof Interacting Proteins(DIP)
The Biomolecular Interaction Network Database(BIND)
Providesinformationon experimentallyverifiedprotein proteininteractions
Adatabaseforexploring, prediction,andevolutionof protein proteininteraction, andidentificationofanetwork ofinteractions
http://dommino. org 22135305
http://caps.ncbs. res.in/pimadb 27478368
https://www.ebi. ac.uk/pdbe/pdbekb 31584092
http://www. cathdb.info 20368142
http://www. genome.ad.jp/ ligand/ 11752349
https://mint.bio. uniroma2.it/mint/ 17135203
http://dip.doe-mbi. ucla.edu 10592249
Adatabaseforexploring biomolecularinteractions www.bind.ca 2519993
Softwareusedforpharmacophoremodeling
PharmerAcomputationaltoolfor pharmacophoresearching usingbloomfingerprint
PharmaGistAserverforligand-based pharmacophoresearchingby utilizingtheMtreealgorithm
smoothdock.ccbb. pitt.edu/pharmer/ 21604800
http://bioinfo3d.cs. tau.ac.il/ PharmaGist/ 18424800
Continued
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
LiSiCAAsoftwareforligand-based virtualscreening
ZINCPharmerAtoolforpharmacophore searchingfromtheZINC database
LigandScoutAtoolforgeneratinga3D pharmacophoremodelusing sixtypesofchemicalfeatures
SchrodingerThephasefunctionof schrodingercanbeutilizedfor ligandandstructure-based pharmacophoremodeling
VirtualToxLabAllowsrationalizingprediction atthemolecularlevelby analyzingthebindingmodeof thetestedcompoundfor targetproteinsinreal-time3D/ 4D
ToolsusedinQSARmodeldevelopment
http://insilab.org/ lisica/ 26158767
http://zincpharmer. csb.pitt.edu/ 22553363
http://www. inteligand.com/ download/ InteLigand_ LigandScout_4.3_ Update.pdf 15667141
https://www. schrodinger.com/ phase 32860362
http://www. biograf.ch/index. php?id¼projects& subid¼virtualtoxlab 32244747
DPubChemSoftwareforautomated generationofaQSARmodel www.cbrc.kaust. edu.sa/dpubchem 29904147
QSAR-CoOpen-sourcesoftwarefor QSAR-basedclassification modeldevelopment https://sites. google.com/view/ qsar-co 31083984
DTClabAsuiteofsoftwareforcurating andgeneratingaQSARmodel forvirtualscreening https://dtclab. webs.com/ software-tools 31525295
EzqsarAstandaloneprogramsuitefor QSARmodeldevelopment https://github.com/ shamsaraj/ezqsar 29387275
DataWarriorAnintegratedcomputertool forgenerationandvirtual screeningofaQSARmodel http://www. openmolecules. org/datawarrior/ 30806519 6 CHAPTER1
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
FeatureselectionalgorithmusedinbuildingaQSARmodel
Waikato Environmentfor Knowledge Analysis(WEKA)
Ageneral-purpose environmentforautomatic classification,regression, clustering,andfeature selectionofcommondata miningproblemsin bioinformaticsresearch
http://www.cs. waikato.ac.nz/ml/ weka 15073010
DWFSAweb-basedtoolforfeature selection https://www.cbrc. kaust.edu.sa/dwfs/ 25719748
SciKitAPython-basedframeworkfor featureselectionandmodel optimization https://scikit-learn. org/stable/ modules/feature_ selection.html 32834983
Dockingsoftwarecommonlyusedinchemoinformatics
Autodock4GA;LGA;SA/empiricalfree energyforcefield http://autodock. scripps.edu 19399780
AutodockVinaGA,PSO,SA,Q-NM/X-Score http://vina.scripps. edu 19499576
BDTAutoGridandAutoDock http://www. quimica.urv.cat/ wpujadas/BDT/ index.html 16720587
BetaDockGA http://voronoi. hanyang.ac.kr/ software.htm 21696235
CDockerSA http://accelrys. com/services/ training/lifescience/ StructureBased Design Description.html 11922947
DARWINGA http://darwin.cirad. fr/product.php 10966571
DOCKIC/ChemScore,SAsolvation scoring,DockScore http://dock. compbio.ucsf.edu 19369428
DockoMaticAutoDock https:// sourceforge.net/ projects/ dockomatic/ 21059259
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
DockVisionMC,GA http://dockvision. sness.net/ overview/overview. html 1603810
eHiTSRBDoffragmentsfollowedby reconstruction/eHiTS www.simbiosys.cs/ ehits/index.html 16860582
FINDSITECOMB SP-score http://cssb.biology. gatech.edu/ findsitelhm 19503616
FITTEDGA/RankScore http://www.fitted. ca 17305329
FleksyFlexibleapproachtoIFD http://www.cmbi. ru.nl/software/ fleksy/ 18031000
FlexXIC/FlexXScore,PLP,Screen Score,DrugScore https://www. biosolveit.de 10584068
FlipDockGA http://flipdock. scripps.edu 17523154
FREDRBD/ScreenScore,PLP, Gaussianshapescore, ChemScore,ScreenScore, Chemgauss4scoringfunction https://docs. eyesopen.com/ oedocking/fred. html 21323318
GalaxyDockGalaxyDockBP2Score http://galaxy. seoklab.org/ softwares/ galaxydock.html
23198780, 24108416
GEMDOCKEA/empiricalscoringfunction http://gemdock. life.nctu.edu.tw/ dock/ 15048822
GlamDockMC/SA http://www.chil2. de/Glamdock.html 17585857
GlideHierarchicalfiltersandMC/ GlideScore,glidecomp https://www. schrodinger.com/ glide/ 15027865
GOLDGA/GoldScore,chemScore https://www.ccdc. cam.ac.uk/ solutions/csddiscovery/ components/gold/ 12910460
GriDockAutoDock4.0 http://159.149.85. 2/cms/index.php? Software_projects: GriDock
20623318
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools Algorithm/scoring functions/descriptionWebsiteURLPMIDs
HADDOCKSA/HADDOCKScore
HYBRIDCGO/Ligand-basedscoring function
iGEMDOCKGA/Simpleempiricalscoring functionanda pharmacophore-based scoringfunction
LeadFinderGA
LigandFitMonteCarlosampling/Lig Score,PLP,PMF, hammerhead
Mconf-DOCKDOCK5
http://haddock. science.uu.nl/ services/ HADDOCK2.2/ 12580598
https://docs. eyesopen.com/ oedocking/hybrid. html 17591764
http://gemdock. life.nctu.edu.tw/ dock/igemdock. php 15048822
http://moltech.ru 19007114
https://www. phenix-online.org/ documentation/ reference/ligandfit. html 12479928
http://www.mti. univ-paris-diderot. fr/recherche/ plateformes/ logiciels 18402678
MOEGaussianfunction http://www. chemcomp.com/ MOE-Molecular_ Operating_ Environment.htm 19075767
MolegroVirtual Docker Evolutionaryalgorithm http://www. scientificsoftwaresolutions.com/ product.php? productid¼17625 16722650
POSITSHAPEFIT https://docs. eyesopen.com/ oedocking/posit_ usage.html 21323318
RosettaLigandRosettascript https://www. rosettacommons. org/software 22183535
Continued
Table1.2 Commonlyusedtoolsandsoftwarecategorizedonalgorithms/scoring functions/descriptionavailabilitywithuniformresourcelocator(URL)andsupported platforms. cont’d
Databases/ tools
Algorithm/scoring functions/descriptionWebsiteURLPMIDs
Surflex-DockIC/Hammerhead https://omictools. com/surflex-docktool 22569590
VLifeDockGA/PLPscore,XCscore,and Steric þ Electrostaticscore http://www. vlifesciences.com/ products/ VLifeMDS/ VLifeDock.php 30124114
CommonlyusedMDsimulationstools
AbaloneSuitableforlongsimulations http://www. biomolecularmodeling.com/ Abalone/index.html 26751047
ACEMDFastestMDengine https://www. acellera.com/ products/ moleculardynamicssoftware-GPUacemd/ 26616618
AMBERUsedforsimulations http://ambermd. org/ 16200636
CHARMMAllowsmacromolecular simulations http://yuri.harvard. edu/ 31329318
DESMONDPerformshigh-performance MDsimulations https://www. deshawresearch. com/resources_ desmond.html 16222654
GROMACSWidelyusedwithexcellent performance http://www. gromacs.org/ 21866316
LAMMPSAcoarse-graintool, specificallydesignedfor materialMDsimulations http://lammps. sandia.gov/ 31749360
MOILAcompletesuiteforMD simulationsandmodeling http://clsbweb. oden.utexas.edu/ moil.html 32375019
NAMDProvidesauser-friendly interfaceandpluginsto performlargesimulations http://www.ks. uiuc.edu/ Research/namd/ 29482074
TINKERPerformsbiomoleculeand biopolymerMDsimulations http://dasher. wustl.edu/tinker/ 30176213
1.2.2 Proteinandligandinformationdatabases
3Dinformationofaligandanditsbindingresidueswithinthepocketofitstarget proteinisanessentialrequirementwhiledeveloping3D-QSAR-basedmodels. Thus,thedatabasesholdinginformationaboutmacromoleculestructuresareofgreat importanceforpharmaceuticalindustriesandresearchers.TheProteinDataBank (PDB)(Roseetal.,2017)isonesuchopen-sourcelargerepositorycontainingstructuralinformationidentifiedviacrystallographicandNuclearMagneticResonance (NMR)experimentaltechniques.ThecurrentversionofPDBholdsstructuralinformationon166,301abundantmacromolecularcompounds.ThePDBisupdated weeklywitharateofalmost100structures.Anothersuchextensivedatabaseis theCambridgeStructuralDatabase(Groometal.,2016),whichprovidesstructural informationonlargemacromoleculessuchasproteins.
1.2.3 Databasesrelatedtomacromolecularinteractions
Oftenthebiologicalactivityofaproteincanbemodulatedbybindingaligandmoleculewithinitsactivesite.Thus,identificationofmolecularinteractionsamong ligand proteinandprotein proteinisofutmostimportance.Moreover,thebiologicalpathwaysandchemicalreactionsoccurringattheprotein ligandinterfaceare alsoessentialinunderstandingdiseasepathology.LIGANDisadatabasethatprovidesinformationonenzymaticreactionsoccurringatthemacromolecularlevel (Gotoetal.,2000).Severalotherdatabases,suchastheDatabaseofInteractingProteins,BiomolecularInteractionNetworkDatabase,andMolecularInteraction Network,arealsopresentintheliterature,whichincludesinformationonprotein proteininteractions.
1.3 Pharmacophoremodeling
Theprocessofdrugdesigningdatesbackto1950(NewmanandCragg,2007).Historically,theprocessofdrugdesigningfollowsahit-and-missapproach.Ithasbeen observedthatonlyoneortwotestedcompoundsoutof40,000reachclinicalsettings,suggestingalowsuccessrate.Oftenthedevelopedleadmoleculelacks potencyandspecificity.Thetraditionaldrugdesignprocessmaytakeupto7 12 years,andapproximately$1 2billioninlaunchingasuitabledrugintothemarket. Allthissuggeststhatfindingadrugmoleculeistimeconsuming,expensive,and needstobeoptimizedinadifferentwaytoidentifythecorrectleadmolecule.These limitationsalsosignifythatthereshouldbesomenovelalternativewaystoidentify hitsthatmayleadtodrugmolecules.Soonafterdiscoveringcomputationalmethods todesignandscreenlargechemicaldatabases,theprocessofdrugdiscoveryhas primarilyshiftedfromnaturaltosynthetic(Lourencoetal.,2012).Therationalstrategiesforcreatingactivepharmaceuticalcompoundshavebecomeanexcitingarea ofresearch.Industriesandresearchinstitutionsarecontinuouslydevelopingnew
toolsthatcanaccelerateandspeedupthedrugdiscoveryprocess.Themethodology involvesidentifyingactivemoleculesvialigandoptimizationknownaspharmacophoremodelingorthestructure activityrelationshipapproach.Thissectionofthe chapterdescribesligand-basedpharmacophoremodelingindetailtofindtheactive compoundwithdesiredbiologicaleffects.
Apharmacophoreissimplyarepresentationoftheligandmolecules’structural andchemicalfeaturesthatarenecessaryforitsbiologicalactivity.Accordingtothe InternationalUnionofPureandAppliedChemistry,apharmacophoreisan ensembleofstericandelectrostaticfeaturesrequiredtoensureoptimalinteractions withspecificbiologicaltargetstoblockitsresponse.Thepharmacophoreisnota realleadmolecule,butanensembleofcommonmoleculardescriptorssharedby activeligandsofdiverseorigins.Thisway,pharmacophoremodelingcanhelpidentifytheactivefunctionalgroupswithinligandbindingsitesoftargetproteinsand providecluesonnoncovalentinteractions.Theactivepharmacophorefeatureincludeshydrogenbonddonor,acceptor,cationic,aromatic,andhydrophobiccomponentsofaligandmolecule,etc.Thecharacteristicfeaturesofactiveligandsareoften describedin3Dspacebytorsionalangle,locationdistance,andotherfeatures. Severalsoftwaretoolsareavailabletodesignthepharmacophoremodel,suchas thecatalyst,MOE,LigandScout,Phases,etc.
1.3.1 Typesofpharmacophoremodeling
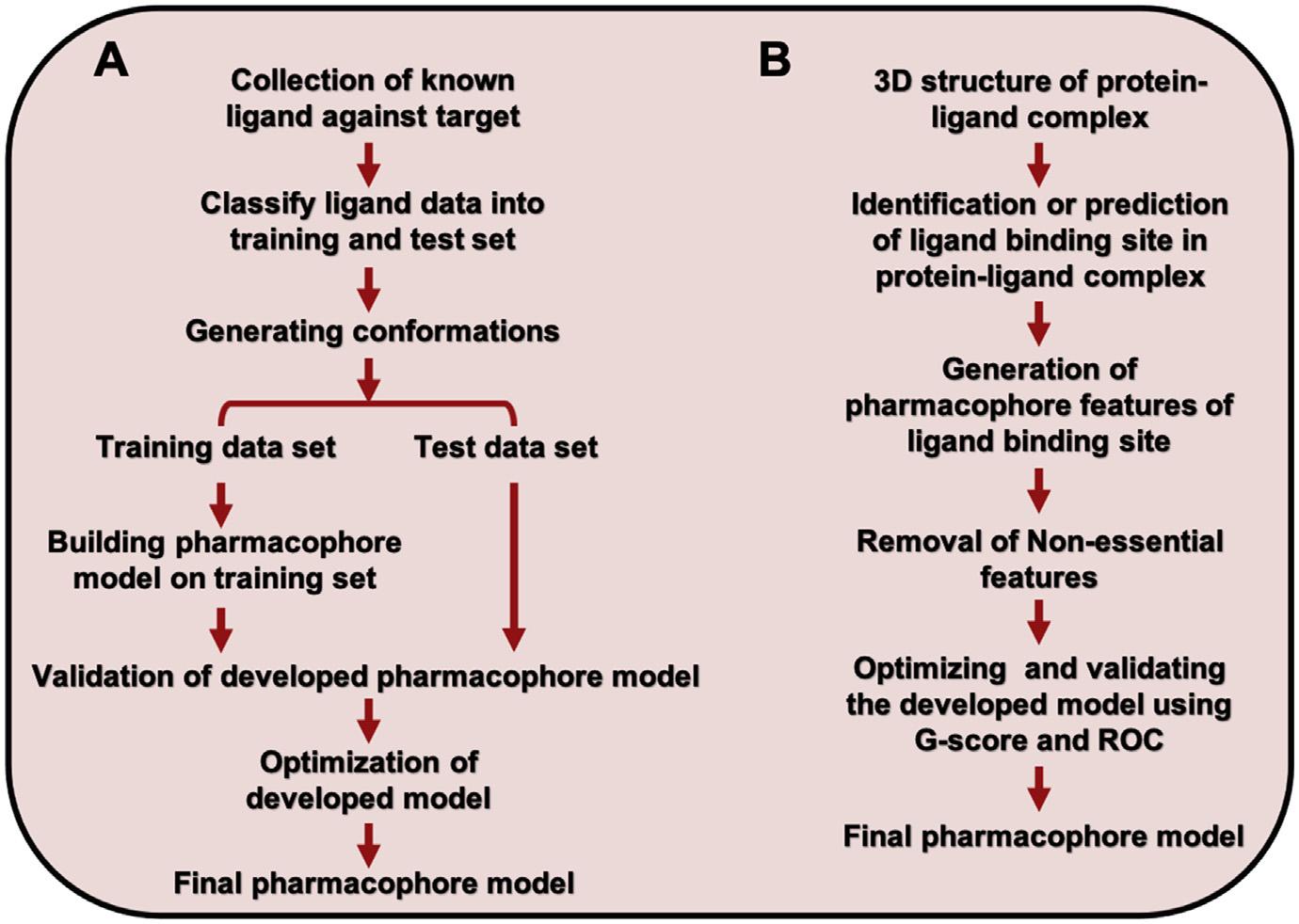
Pharmacophoremodelingisbroadlyclassifiedintotwocategories:ligand-basedand structure-basedpharmacophoremodeling.Abriefaboutthemethodologyadopted byeachtypeofmodelingisshownin Fig.1.1.However,structure-basedpharmacophoremodelingexclusivelydependsonthegenerationofpharmacophoremodels basedonthereceptor-bindingsite.Still,forligand-basedpharmacophoremodeling, thebioactiveconformationoftheligandisusedtoderivethepharmacophoremodel. Thebestapproachistoconsiderthereceptor ligandcomplexandgeneratethepharmacophoremodelsfromthere.Thisprovidesexclusionvolumesthatrestrictthe ligandduringvirtualscreeningtothetargetsiteandthusisquitesuccessfulinvirtual screeningoflargechemicaldatabaselibraries.
1.3.2 Scoringschemeandstatisticalapproachesusedin pharmacophoremodeling
Severalparametersassessthequalityofdevelopedpharmacophoremodels,suchas predictivepower,identifyingnovelcompounds,costfunction,testsetprediction, receiveroperatingcharacteristic(ROC)analysis,andgoodnessoffitscore.Generally,atestsetapproachisusedtoestimatethepredictivepowerofadevelopedpharmacophoremodel.Atestsetisagroupoftheexternaldatasetofstructurallydiverse compounds.Itcheckswhetherthedevelopedmodelcanpredicttheunknown instance.Ageneralobservationisthatifadevelopedmodelshowsacorrelation coefficientgreaterthan0.70onbothtrainingandtestset,itisofgoodquality.
FIGURE1.1
Overallworkflowofthemethodologyusedindevelopingthepharmacophoremodel.(A) Ligand-basedpharmacophoremodel.(B)Structure-basedpharmacophoremodel. ROC,Receiveroperatingcharacteristic.
Thecommonlyusedstatisticalparameter,cost functionanalysis,isintegratedinto theHypoGenprogramtovalidatethepredictivepowerofthedevelopedmodel.The optimalqualitypharmacophoremodelgenerallyhasacostdifferencebetween40 and60bits.Thecostvaluesignifiesthepercentageofprobabilityofcorrelating thedatapoints.Thevaluebetween40and60bitsmeansthatthedevelopedpharmacophoremodelshowsa75% 90%probabilityofcorrelatingthedatapoints.The ROCplotgivesvisualaswellasnumericalrepresentationofthedevelopedpharmacophoremodel.Itisaquantitativemeasuretoassessthepredictivepowerofadevelopedpharmacophoremodel.TheROCcurvedependsonthetruepositive,true negative,falsepositive,andfalsenegativepredictedbythedevelopedmodel.The ROCplotcanbeplottedusing1-specificity(falsepositiverate)ontheX-axisand sensitivity(truepositiverate)ontheY-axisofthecurve.
Thedevelopedpharmacophoremodelhashugetherapeuticadvantagesinthe screeningoflargechemicaldatabases.Theidentifiedpharmacophoreutilizedby themethodologyjustmentionedandstatisticalapproachesmayservethebasisof designingactivecompoundsagainstseveraldisorders.Successfulexamplesinclude novelCXCR2agonistsagainstcancer(Cheetal.,2018),acortisolsynthesisinhibitordesignedagainstCushingsyndrome(Akrametal.,2017),designingofACE2
inhibitors(Rellaetal.,2006),andchymaseinhibitors(Aroojetal.,2013).Various softwaretoolsthatareavailablefordesigningthecorrectpharmacophoreareshown in Table1.2.Overall,wecansaythatmedicinalchemistsandresearcherscanuse pharmacophoreapproachesascomplementarytoolsfortheidentificationand optimizationofleadmoleculesforacceleratingthedrugdesigningprocess.
AQSARmodelcanbedevelopedusingessentialstatisticssuchasregression coefficientsofQSARmodelswithsignificanceatthe95%confidencelevel,the squaredcorrelationcoefficient(r2),thecross-validatedsquaredcorrelationcoefficient(Q2),thestandarddeviation(SD),theFisher’sF-value(F),andtheroot meansquarederror.Theseparameterssuggestbetterrobustnessofthepredicted QSARmodelbasedondifferentalgorithmslikesimulatedannealingandartificial neuralnetwork(ANN).Thealgorithm-basedacceptableQSARmodelisrequired tohavestatisticalparametersofhighervalueforthesquareofcorrelationcoefficient (r 2 nearto1),andFisher’sF-value(F ¼ max),whilethevalueislowerforstandard deviation(SD ¼ low).TheintercorrelationoftheseindependentparametersgeneratedfordescriptorsisrequiredtodeveloptheQSARmodel.
1.4 QSARmodels
Itisofutmostimportancetoidentifythedrug-likenessofthecompoundsobtained afterpharmacophoremodelingandvirtualscreeningofthechemicalcompound databases.QSAR-basedmachinelearningmodelsarecontinuouslybeingusedby thepharmaceuticalindustriestounderstandthestructuralfeaturesofachemical thatcaninfluencebiologicalactivity(KausarandFalcao,2018).TheQSARbasedmodelsolelydependsonthedescriptorsofthechemicalcompound.Descriptorsarethenumericalfeaturesextractedfromthestructureofacompound.The QSARmodelattemptstocorrelatebetweenthedescriptorsofthecompounds withitsbiologicalactivity.AbriefoverviewoftheQSARmethodologyusedin pharmaceuticalindustriesandresearchlaboratoriesfollows.
1.4.1 MethodologiesusedtobuildQSARmodels
TheprimarygoalofallQSARmodelsistoanalyzeanddetectthemoleculardescriptorsthatbestdescribethebiologicalactivity.Thedescriptorsofchemicalcompoundsaremainlyclassifiedintotwocategories:theoreticaldescriptorsand experimentaldescriptors(Loetal.,2018).
Thetheoreticaldescriptorsareclassifiedinto0D,1D,2D,3D,and4Dtypes, whereastheexperimentaldescriptorsareofthehydrophobic,electronic,andsteric parametertypes.Abriefdescriptionofdescriptortypesisshownin Table1.1.
Thedescriptorsusedasinputforthedevelopmentofmachinelearning-based modelspredictthepropertyofthechemicalcompound.QSARmethodsarenamed afterthetypeofdescriptorsusedasinput,suchas2D-QSAR,3D-QSAR,and4DQSARmethods.AbriefdescriptionofeachQSARmethodfollows.
1.4.2 Fragment-based2D-QSAR
Inrecentyears,theuseof2D-QSARmodelstoscreenandpredictbioactivemoleculesfromlargedatabaseshasgainedmomentuminpharmaceuticalindustriesdue totheirsimple,easy-to-use,androbustnature.ItallowsthebuildingofQSAR modelsevenwhenthe3Dstructureofthetargetismainlyunknown.AhologrambasedQSARmodelwasthefirst2D-QSARmethoddevelopedbyresearchersthat didnotdependonthealignmentbetweenthecalculateddescriptorsofacompound. First,theinputcompoundissplitintoallpossiblefragmentsfedtotheCRCalgorithm,whichthenhashesthefragmentsintobins.Thesecondstepinvolvesthe correlationanalysisofgeneratedfragmentbinswiththebiologicalactivity.The basisofthefinalmodelispartialleastregressionthatidentifiesthecorrelationof fragmentbinswithbiologicalactivity(IC50, Vmax).
1.4.3 3D-QSARmodel
3D-QSARmodelsarecomputationallyintensive,bulky,andimplementcomplexalgorithms.Theyareoftwotypes:alignmentdependentandalignmentindependent, andbothtypesrequire3Dconformationoftheligandtobuildthefinalmodel. Comparativemolecularfieldanalysis(CoMFA)andcomparativemolecularsimilarityindicesanalysis(CoMSIA)arethepopularlyused3D-QSARmethodsutilizedby pharmaceuticalindustriesformodelbuilding.TheCoMFAmethodconsidersthe electrostaticandstericfieldsinthegenerationandvalidationofa3Dmodel,while theCoMSIAutilizeshydrogenbonddonor acceptorinteractions.Then,stericand electrostaticinteractionsaremeasuredateachgridpoint.Subsequently,partialleast squaresregressionanalysiscorrelatesthemoleculardescriptorsoftheligandwith thebiologicalactivitiestomakeafinalQSARmodel.
1.4.4 Multidimensionalor4D-QSARmodels
Totacklethelimitationsof3D-QSARmethods,multidimensionalQSARmodelsare heavilyusedinthepharmaceuticalindustries.Theessentialrequirementforthe developmentof4D-QSARmethodsisthe3Dgeometryofthereceptorsandligand. Onesuch4D-QSARmethodisHopfinger’s,whichisdependentontheXMAPalgorithm.ThecommonlyusedsoftwaretoolsfordevelopingmultidimensionalQSAR modelsareQuasarandVirtualToxLabsoftware.
Beforeapplyingmachinelearning-basedQSARmodeling,thefeatureselection processfordimensionalityreductionmustensurethatonlyrelevantandbestfeatures shouldbeusedasinputinthemachinelearningprocess.Otherwise,thedeveloped QSARmodelonallrelevantandirrelevantfeatureswilldecreasethemodel’s performance.Themostwidelyusedopen-sourcefeatureselectiontoolsare WEKA,scikitinPython,DWS,FEASTinMatlab,etc.Acompletelistoffeature extractionalgorithmscommonlyusedinpharmaceuticalindustriesisshownin Table1.2.Theselectedfeaturesoftheactiveandinactivecompoundswere usedasinputfeaturesfordevelopingtheQSAR-basedmachinelearningmodel.
Machinelearning-basedstrategiestrytolearnfromtheinputstructuralfeaturesand predictthecompounds’biologicalproperties.ThefinaldevelopedQSARmodelcan beappliedtothelargechemicalcompoundlibrariestoscreenthecompoundsand predicttheirbiologicalproperties.Allthefeatureselectionprogramsutilizeone orotheralgorithms,namelystepwiseregression,simulatedannealing,geneticalgorithm,neuralnetworkpruning,etc.
1.4.5 StatisticalmethodsforgenerationofQSARmodels
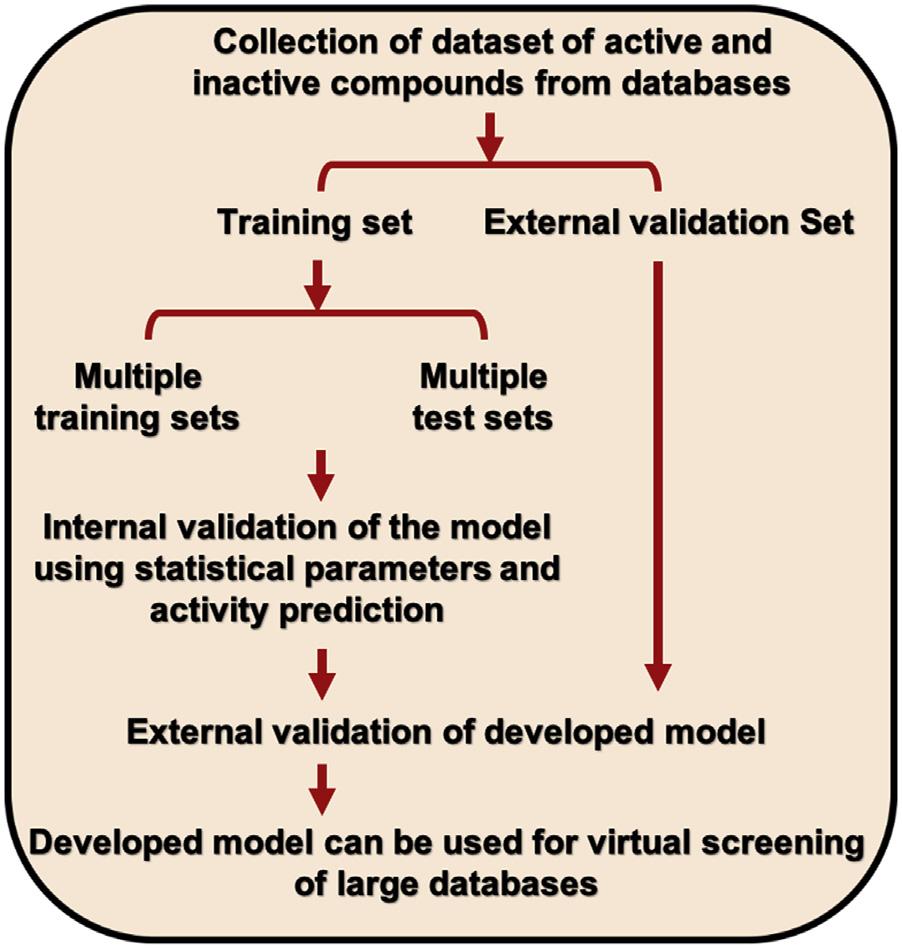
Themachinelearning-basedQSARmodelingapproachhastwosubcategories.The firstoneincludesregression-basedmodeldevelopment,andthesecondoneprovides classificationtechniquesbasedonthepropertiesofthedata.Theregression-based statisticalmethodsimplementalgorithms,suchasmultivariatelinearregression (MLR),principalcomponentanalysis,partialleastsquare,etc.Atthesametime, classificationtechniquesincludelineardiscriminantanalysis, k-nearestneighboralgorithm,ANN,andclusteranalysisthatlinkqualitativeinformationtoarriveat property structurerelationshipsforbiologicalactivity.Eachalgorithmhasits uniquefunctionandscoringschemeforbuildingthepredictiveQSARmodel (Haoetal.,2010).ThegeneralworkflowandstatisticaldetailsofMLRareshown in Fig.1.2.
FIGURE1.2
Overallworkflowofthepredictivequantitativestructure activityrelationshipmodel development.
1.4.6 Multivariatelinearregressionanalysis
TheregressionanalysismoduleoftheMLRalgorithmestimatesthecorrelation betweenthebiologicalactivitiesofligands/compoundswiththeirmolecularchemicaldescriptors.Theessentialandfirststepincludesthefindingofdatapointsfrom descriptorsthatbestsuittheperformanceoftheQSARmodel.Next,aseriesofstepwisefiltersisapplied,whichreducesthedimensionalityofdescriptorstoarriveat minimumdescriptorsthatbestfitthemodel.Thiswillincreasethepredictivepower ofthealgorithmaswellasmakeitlesscomputationallyexhaustive.Cross-validation estimatesthepredictivepowerofthedevelopedmodel.Themathematicaldetailsof theprocedure,asalreadymentioned,aredescribedasfollows.Let X bethedata matrixofdescriptors(independentvariable),and Y bethedatavectorsofbiological activity(dependentvariable).Then,regressioncoefficient b canbecalculatedas:
¼ðX0 XÞ 1X0 Y
Thestatisticalparametertotalsumofsquaresisawayofrepresentingtheresult obtainedfromMLRanalysis.Anexamplesethereshowsallthemathematicalequations.Forexample,thedevelopmentofaQSARmodelforpredictingtheantiinflammatoryeffectsoftheCOX2compoundisdonewiththehelpoftheScigressExplore method.Thecorrelationbetweentheactualinhibitoryvalue(r 2 ¼ 0.857)andpredictedinhibitoryvalues(r 2CV ¼ 0.767)isgoodenough,provingthatthepredicted modelisofgoodquality.Thefeaturesusedindevelopingthepredictivemodelsare asexplainedinthefollowingequation:
Predictedantiinflammatoryactivitylog(LD50) ¼þ0.167357 Dipole vector (Debye) þ 0.00695659 Stericenergy(kcal/mol) 0.00249368 Heat offormation(kcal/mol) þ 0.852125 Sizeofsmallestring 1.1211 Group count(carboxyl) 1.24227
Here, r 2 definestheregressioncoefficient.ForbetterQSARmodeldevelopment, themeandifferencebetweenactualandpredictedvaluesshouldbeminimum.Ifthe valueof r 2 variesalot,thenthemodelisoverfitted.AbriefofthegeneralmethodologyusedinbuildingtheQSARmodelisillustratedin Fig.1.2.
TraditionalQSAR-basedmodelingonlypredictsthebiologicalnatureofthe compoundandiscapableofscreeningthenewmoleculebasedonthelearning. However,thisapproachhasseverallimitations;allthepredictedcompoundsdo notfitintothecriteriaoftheLipinskiruleoffiveandthusmayhavecytotoxicproperties,etc.ModernQSAR-basedstrategiesshouldemployvariousotherfiltration processessuchastheincorporationofempiricalrules,pharmacokineticandpharmatoxicologicalprofiles,andchemicalsimilaritycutoffcriteriatohandletheaforementionedissues(Cherkasovetal.,2014).Thisway,aligandwithpotentialdruggability andADMETpropertiescanbemadeinatime-efficientmanner.Severalsoftware toolslikeclick2drug,SWISS-ADME,andADMET-SARcansolvetheuser’s problemsinpredictingthedesiredADMETpropertiesofacompound.
1.5 Dockingmethods
Dockingisanessentialtoolindrugdiscoverythatpredictsreceptor ligandinteractionsbyestimatingitsbindingaffinity(Mengetal.,2012),duetoitslowcostand timesavingthatworkswellonapersonalcomputercomparedtoexperimental assays.Thesignificantchallengesindockingarearepresentationofreceptor,ligand, structuralwaters,side-chainprotonation,flexibility(fromside-chainrotationsto domainmovement),stereoisomerism,inputconformation,solvation,andentropy ofbinding(Torresetal.,2019).However,recentadvancesinthefieldofdrug designinghavebeenreportedaftertheadventofdockingandvirtualscreening (Lounnasetal.,2013).Receptor ligandcomplexstructuregenerationusing insilicodockingapproachesinvolvestwomaincomponents:posingandscoring. Dockingisachievedthroughligandorientationalandconformationalsamplingin thereceptor-activesite,whereinscoringpredictsthebestnativeposeamongthe rankligands(ChaputandMouawad,2017).Dockinginvolvesthestructureofligandsforposeidentificationandligandbindingtendencytopredictaffinity(Clark etal.,2016).Thisimpliesthatsearchmethodsofligandflexibilityarecategorized intosystematicstrategiesbasedonincrementalconstruction(Rareyetal.,1996), conformationalsearch,anddatabases(DOCKandFlexX).Thestochasticorrandom approachesusegenetic,MonteCarlo,andtabusearchalgorithmsimplementedin GOLD,AutoDock,andPRO_LEADS,respectively.Atthesametime,simulation methodsareassociatedwithmoleculardynamics(MD)simulationsandglobal energyminimization(DOCK)(Yurievetal.,2011).
Thereceptorisrepresentedasa3DstructureindockingobtainedfromNMR, X-raycrystallography,threading,homologymodeling,anddenovomethods.Nevertheless,ligandbindingisadynamiceventinsteadofastaticprocess,whereinboth ligandandproteinexhibitconformationalchanges.
Severaldockingsoftwareandvirtualscreeningtools(Table1.2)areavailableand widelyused.Nonetheless,onesuchsoftwarethatexplicitlyaddressesreceptorflexibilityisRosettaLigand,whichusesthestochasticMonteCarloapproach,whereina simulatedannealingprocedureoptimizesthebindingsiteside-chainrotamers(Davis etal.,2009).Anothersoftware,Autodock4,completelymodelstheflexibilityofthe selectedproteinportioninwhichselectedsidechainsoftheproteincanbeseparated andexplicitlytreatedduringsimulationsthatenablerotationthroughoutthe torsionaldegreeoffreedom(Biancoetal.,2016).Alternatively,theproteincanbe madeflexiblebytheInsightIIside-chainrotamerlibraries(Wangetal.,2005).Besides,theInducedFitDocking(IFD)workflowofSchrodingersoftwarerelieson rigiddockingusingtheGlidemodulecombinedwiththeminimizationofcomplexes andhomologymodeling.IFDhasbeenusedforkinases(Zhongetal.,2009),HIV-1 integrase(Barrecaetal.,2009),heatshockprotein90(Lauriaetal.,2009),and monoacylglycerollipase(Kingetal.,2009)studies.Furthermore,atomreceptor flexibilityintodockingwasintroducedusingMDsimulations,whichmeasuredits effectontheaccuracyofthistoolbycross-docking(Armenetal.,2009).The bestcomplexmodelsareobtainedbasedonflexiblesidechainsandmultipleflexible backbonesegments.
Incontrast,thebindingofdockedcomplexescontainingflexibleloopsand entirelyflexibletargetswasfoundlessaccuratebecauseofincreasednoisethat affectsitsscoringfunction.InternalCoordinateMechanics(ICM),a4D-docking protocol,wasreportedwherethefourthdimensionrepresentsreceptorconformation (AbagyanandTotrov,1994).ICMaccuracywasfoundtobeincreasedusingmultiplegridsthatdescribedmultiplereceptorconformationscomparedtosinglegrid methods.Agradient-basedoptimizationalgorithmwasimplementedinalocalminimizationtoolusedtocalculatetheorientationalgradientbyadjustingparameters withoutalteringmolecularorientation(Fuhrmannetal.,2009).Thedocking approachesarecomputationallycostlyforcreatingdockerligandlibraries,receptor ensembles,anddevelopingindividualligandsagainstlargerensembles(Huangand Zou,2006).Normalmodeanalysisusedtogeneratereceptorensemblesisoneofthe bestalternativestoMDsimulations(Moroyetal.,2015).Theelasticnetworkmodel (ENM)methodinduceslocalconformationalchangesinthesidechainsandprotein backbone,whichsignifiesitsimportancemoreefficientlythanMDsimulations.
Asmallchangeintheligandconformationcausessignificantvariationsinthe scoresofdockedposesandgeometries.Thissuggeststhatnomethodorligandgeometryproducesthemostprecisedockingpose(Mengetal.,2012).Ligandconformationaltreatmenthasbeenprecomputedthroughseveralavailablemethodslikethe generationofligandconformations(TrixXConformerGenerator)(Grieweletal., 2009),systematicsampling(MOLSDOCKandAutoDock4)(Vijietal.,2012), incrementalconstruction(DOCK6),geneticalgorithms(Jonesetal.,1997), Lamarckiangeneticalgorithm(FITTEDandAutoDock),andMonteCarlo(RosettaLigandandAutoDock-Vina).
1.5.1 Scoringfunctions
Dockingsoftwareandwebserversarevalidatedbyproducing“correct”binding modesbasedontheranking,whichidentifiesactiveandinactivecompoundsstillunderstudy.Thus,severalattemptshavebeenmadetoimprovescoringfunctionslike entropy(Lietal.,2010),desolvationeffects(Fongetal.,2009),andtargetspecificity.Mainly,fourtypesofscoringfunctionshavebeencategorizedandimplementedinforcefields:classical(D-Score,G-Score,GOLD,AutoDock,and DOCK)(Heveneretal.,2009);empirical(PLANTSCHEMPLP,PLANTSPLP) (Korbetal.,2009),RankScore2.0,3.0,and4.0(EnglebienneandMoitessier, 2009),Nscore(TarasovandTovbin,2009),LUDI,F-Score,ChemScore,and X-SCORE(Chengetal.,2009);knowledge(ITScore/SE)(HuangandZou,2010), PoseScore,DrugScore(Lietal.,2010),andMotifScorebased;andmachinelearning (RF-Score,NNScore)(DurrantandMcCammon,2010).
DockingcalculationsofentropiesareincludedwithintheMolecularMechanics/ Poisson-BoltzmannSurfaceArea(MM/PBSA),whereinitisamodifiedformof framework,andtheentropylossiscalculated.Thisiscorrespondinglyassessedafter ligand receptorbindingbasedonthelossofrotational,torsional,translational, vibrational,andfreeenergies.Themodificationincludesthefreeenergychange