Chapter12-NeuralnetworkpotentialsJinzheZeng &LiqunCao&TongZhu
https://ebookmass.com/product/chapter-12-neural-networkpotentials-jinzhe-zeng-liqun-cao-tong-zhu/
Instant digital products (PDF, ePub, MOBI) ready for you
Download now and discover formats that fit your needs...
Elsevier Weekblad - Week 26 - 2022 Gebruiker
https://ebookmass.com/product/elsevier-weekbladweek-26-2022-gebruiker/
ebookmass.com
Jock Seeks Geek: The Holidates Series Book #26 Jill Brashear
https://ebookmass.com/product/jock-seeks-geek-the-holidates-seriesbook-26-jill-brashear/
ebookmass.com
The New York Review of Books – N. 09, May 26 2022 Various Authors
https://ebookmass.com/product/the-new-york-review-ofbooks-n-09-may-26-2022-various-authors/
ebookmass.com
New Perspectives on HTML5, CSS3, and JavaScript 6th Edition Patrick Carey
https://ebookmass.com/product/new-perspectives-on-html5-css3-andjavascript-6th-edition-patrick-carey/
ebookmass.com
A Curse of Queens Amanda Bouchet https://ebookmass.com/product/a-curse-of-queens-amanda-bouchet-2/
ebookmass.com
Sociology (9th Edition) Anthony Giddens
https://ebookmass.com/product/sociology-9th-edition-anthony-giddens/
ebookmass.com
Bring Me Your Midnight 1st Edition Rachel Griffin
https://ebookmass.com/product/bring-me-your-midnight-1st-editionrachel-griffin/
ebookmass.com
Tamed by Air: Book 4 of the Nature Hunters Academy Series Quinn Loftis
https://ebookmass.com/product/tamed-by-air-book-4-of-the-naturehunters-academy-series-quinn-loftis/
ebookmass.com
Singing and Survival: The Music of Easter Island Dan Bendrups
https://ebookmass.com/product/singing-and-survival-the-music-ofeaster-island-dan-bendrups/
ebookmass.com
Why https://ebookmass.com/product/why-we-fly-kimberly-jones/
ebookmass.com
Neuralnetworkpotentials JinzheZenga,b,LiqunCaoa,b,andTongZhua,b,
aShanghaiEngineeringResearchCenterofMolecularTherapeutics&NewDrugDevelopment, SchoolofChemistryandMolecularEngineering,EastChinaNormalUniversity,Shanghai,China bNYU-ECNUCenterforComputationalChemistryatNYUShanghai,Shanghai,China
∗Correspondingauthor:E-mailaddress:tzhu@lps.ecnu.edu.cn
Abstract Recently,artificialneuralnetwork-basedmethodsfortheconstructionofpotentialenergysurfacesandmoleculardynamics(MD)simulationsbasedonthemhavebeenincreasinglyusedinthefieldoftheoreticalchemistry.Theneuralnetworkpotentials(NNP)strikeagoodbalancebetweenaccuracyandcomputational efficiencyrelativetoquantumchemicalcalculationsandMDsimulationsbasedonclassicalforcefields.Thus, NNPisbecomingapowerfultoolforstudyingthestructureandfunctionofmolecules.Inthischapter,we introducethebasictheoryofNNP.TheconstructionstepsandtheusageofNNParealsointroducedindetail withtheMDsimulationofmethanecombustionasanexample.Wehopethatthischaptercanhelpthose readerswhoarenewbutinterestedinenteringthisfield.
Keywords: Neuralnetwork,Potentialenergysurface,Moleculardynamicsimulation,Chemicalreaction
Introduction Moleculardynamics(MD)simulationhasbeenakeytheoreticaltoolforstudyingthestructuralanddynamicalpropertiesofawiderangeofchemicalsystems.OneoftheessentialelementsdeterminingthereliabilityofMDsimulationsistheunderlyingpotentialenergy surface(PES),whichdescribestheintra-andintermolecularinteractions.Therearetwomain waystoconstructthePESforMD.Thefirstoneistoperformquantummechanical(QM)calculationon-the-fly,whichisalsoknownasabinitioMDsimulation(AIMD) [1].Theotherone istheempiricalinteratomicpotentials(forcefields)whicharebasedonclassicalmolecular mechanics(MM) [2,3].Theforcefieldsundoubtedlyhavetheadvantageintermsofcomputationalefficiency.However,thesefunctionalformsofforcefieldscanonlyaccuratelydescribeclassicalphysicalinteractionsandusuallydonothaveagoodaccuracywhentaking
intoaccountthemotionofelectrons.Thus,theiraccuracyisoftenquestionedastheylackimportantquantumeffectssuchaspolarizationandchargetransfer.Thisisalsothereasonthat onlyafewforcefieldscandescribechemicalreactions.TheAIMDismoreaccurateinprinciple.However,itsapplicationsaresignificantlylimitedbyitscomputationalcost.Itseemsto bedifficulttoensurebothefficiencyandaccuracyofPESsatthesametime.
Inthepasttwodecades,thedevelopmentofmachinelearningpotentials(MLP)provides thepossibilitytosolvethiscontradiction.MLPemploysmachinelearningmodelsratherthan empiricalformulasorSchr € odingerequationtoestablishtherelationshipbetweenthepotentialenergyofthesystemandthestructureaswellasthechemicalinformation.Compared withempiricalformulas,MLmodelshaveabetterfitpowerandarethereforemoreaccurate. Theyhavebeenusedsincethe1990s [4–9].FollowingtheworkofBehlerandParrinello [10],a seriesofMLPconstructionapproacheshavebeenproposedforawidevarietyofchemical systems.Forexample,Csa ´ nyiandco-workers [11] proposedtheGaussianApproximationPotentials,Mulleretal.proposedtheGDML,DTNNandtheSchNetmethods [12–14],Hammer etal.proposedthekCONmodel [15],Eandco-workersdevelopedtheDeepPotential (DeePMD)model [16,17],Jiangetal.developedtheEANNmodel [18],andDralandcoworkersproposedtheKREGmethod [19,20].Thereareseveralmachinelearningmethods thatcanbeusedtobuildthePES.Amongthem,theartificialNNiswidelyusedduetoits bettercomputationalscalingforperformanceandmemoryrequirements [21,22].Recently, universalNNPshavebeenproposedformanyelements [23,24]
ComparedwithAIMD,theefficiencyoftheM LPissubstantiallyimproved(typicallyby morethan3ordersofmagnitude).Withcodeoptimizationorondedicatedhardware,MD simulationwithMLPhasbeenabletosimulatesystemscontaining100millionatoms [17]. However,thereisstillanorderofmagnitude differenceintheefficiencyofMLPcompared toclassicalforcefields.Therefore,itwasma inlyusedtostudyscientificproblemsthat traditionalforcefieldsarenotcapableoforrequirehighaccuracy.Forexample,thedesign/discoveryofnewmaterials,homogeneouscatalysis,etc [25– 39].Thelatestdevelopmentsandapplicationsinthisfieldcanbefoundinseveralrecentcomprehensivereviews [40– 51].
Recently,wehaveusedtheneuralnetwork-basedpotentials(NNP)tosimulateanumber oftypicalchemicalsystems,suchasthehydrationofzincions [52],metalloproteins [53],and combustionreactions [54,55] (Fig.1).Metals,especiallysometransitionmetals,areimportant cofactorsintheregulationofproteinstructureandfunction.However,importantquantum effectsassociatedwithmetalcoordinationareoftennotaccuratelydescribedbyclassicalforce fields.Wefoundthatbenefittingfromitspowerfulfittingability,theNNPcaneasilyachieve higheraccuracythanclassicalforcefieldsinthesesystems.Combustionisanevenmorecomplexchemicalsystemthatinvolvesthousandsofchemicalreactionsandgenerateshundreds ofmolecularspeciesduringtheprocess.Traditionally,MDsimulationsbasedonreactive forcefieldssuchasReaxFF [56] wereusedtoinvestigatethecombustionreactionprocesses. However,theaccuracyoftheReaxFFhasoftenbeencriticized [57,58].Wehavedeveloped NNPsasaccurateasDFTmethodtoachieveefficientsimulationsofmethanecombustion andlong-chainalkanepyrolysis.FromtheMDtrajectories,onecannotonlyobtaindetailed reactionnetworksbutalsodiscovernewreactions.Withthischapter,wehopetopassonour experiencetohelpmorebeginnersbetterunderstandanduseNNPandtheMDsimulations basedonit.
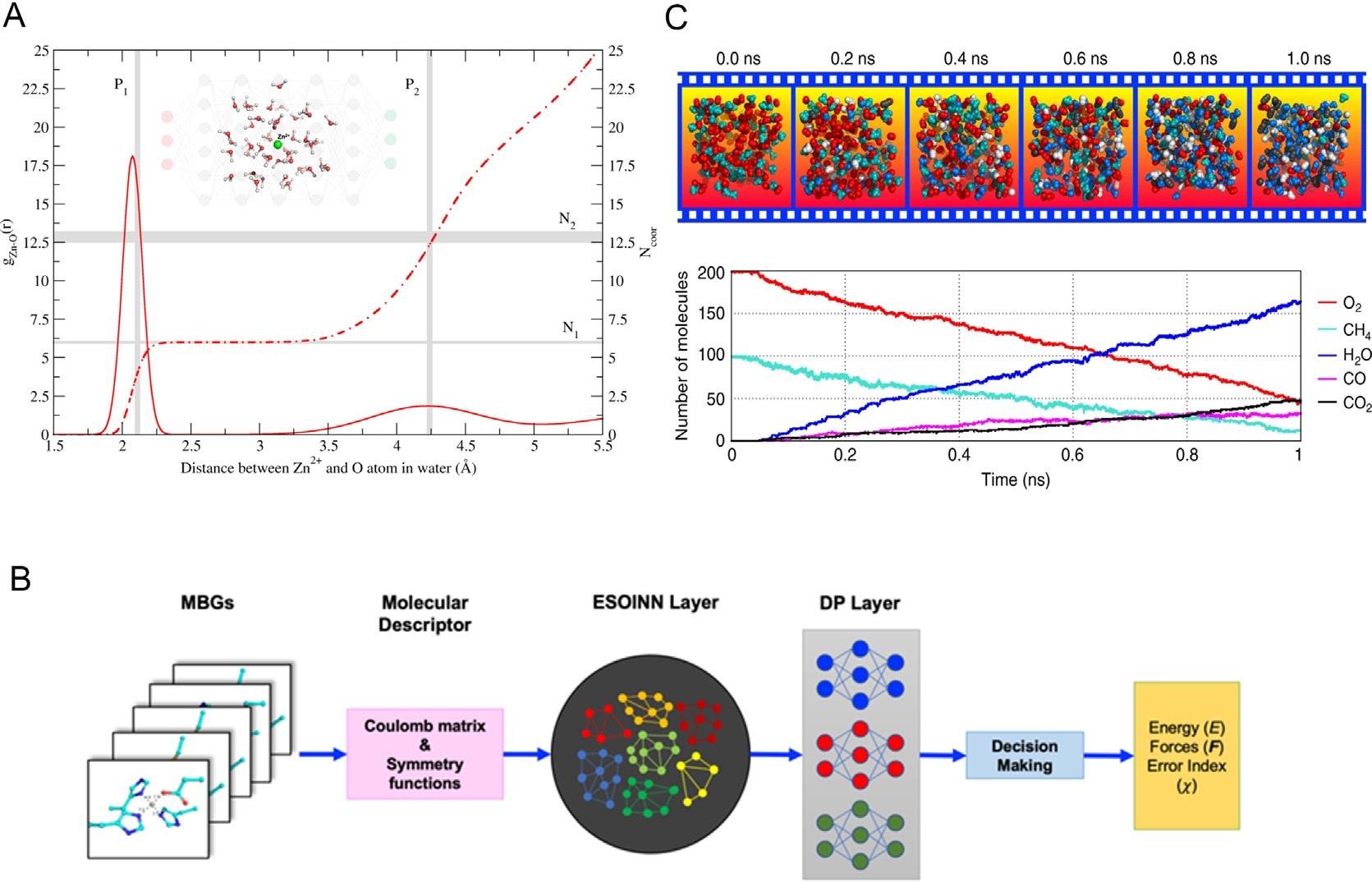
FIG.1 (A)RadialdistributionfunctionofZn Odistance(solidlines)andcorrespondingintegrationofradialdistributionfunction(RDF, dashedlines)calculatedfromthetrajectorysimulatedbyNNP.The gray areaistheexperimentalmeasurementofRDF [52].TheP1 andP2 denote,respectively,thepeakexperimentalradialdistributionof thefirstandsecondsolvationshells,whileN1 andN2 denote,respectively,theexperimentalcoordinationnumber ofthefirstandsecondsolvationshells.(B)TheworkflowtoconstructNNPESforzincproteins [53]. (C)SnapshotsofthepartialcombustionsystemextractedfromthereactiveMDsimulationofmethanecombustion [54] Panel(A)ThisfigureisreprintedwithpermissionfromM.Xu,T.Zhu,J.Z.H.Zhang,Moleculardynamicssimulationof zincioninwaterwithanabinitiobasedneuralnetworkpotential,J.Phys.Chem.A123(2019)6587–6595.Copyright2019 AmericanChemicalSociety.Panel(B)ThisfigureistakenfromM.Xu,T.Zhu,J.Z.H.Zhang,Automaticallyconstructedneural networkpotentialsformoleculardynamicssimulationofzincproteins,Ftont.Chem.9(2021)692200.Panel(C)Thisfigureis takenfromJ.Zeng,L.Cao,M.Xu,T.Zhu,J.Z.H.Zhang,Complexreactionprocessesincombustionunraveledbyneural network-basedmoleculardynamicssimulation,Nat.Commun.11(2020)5713.
Methods AgoodNNPmodelshouldmeetthefollowingrequirements:(1)Itsaccuracymustbevery closetothequantumchemistrymethodthatusedtolabelthedata.(2)Theonlyinputneeded bythemodelshouldbetheelementinformation,atomiccoordinate,chargesandspinstate. (3)IfusedforMDsimulation,themodelshouldbesmoothanddifferentiable.(4)Inmost cases,thePESmodelshouldpreservethetranslational,rotational,andpermutationalsymmetriesofthesystem,whichisessentialtoguaranteethetransferabilityofthemodel.
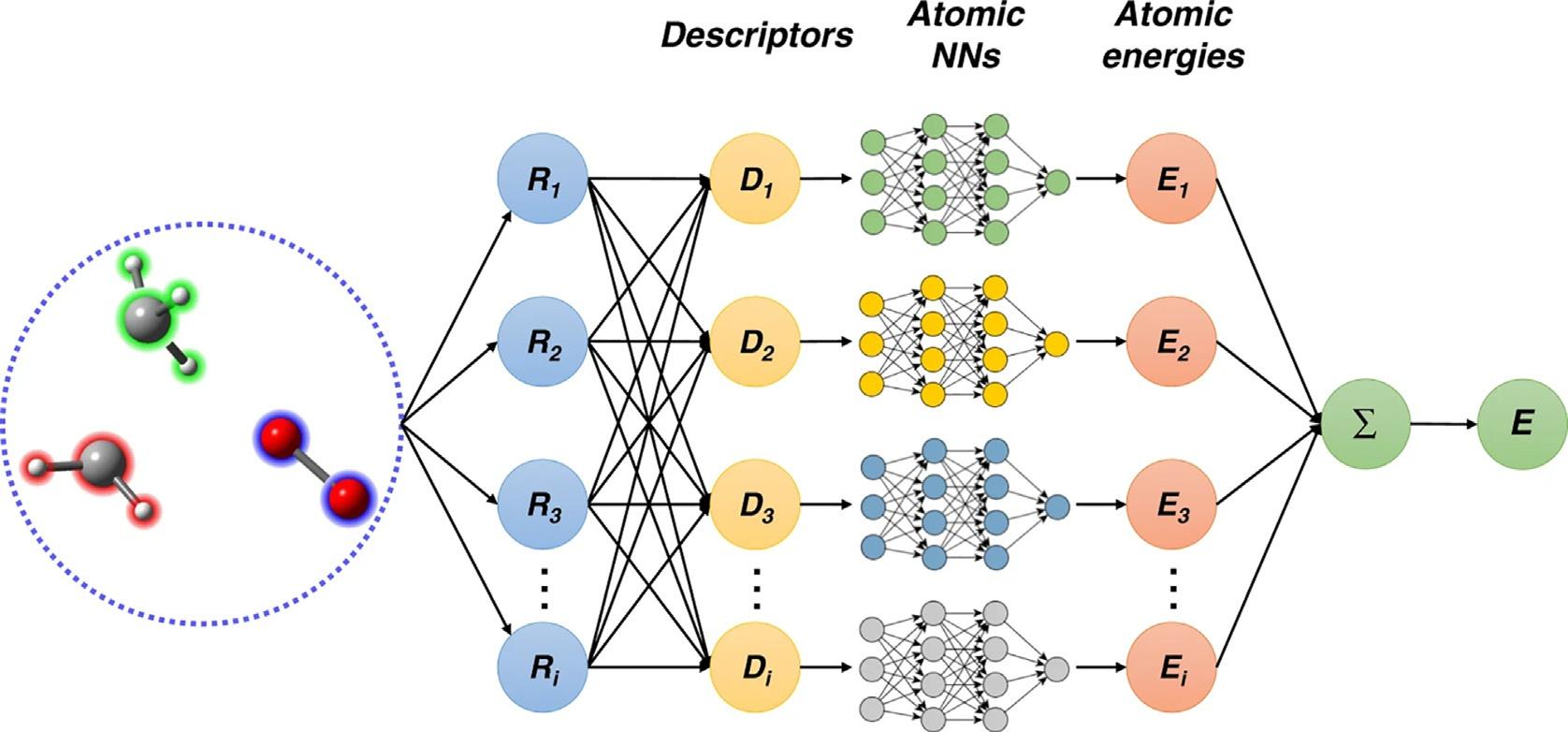
Inmost,butnotall,NNPmodels,thepotentialenergyofthesystemisexpressedasthesum ofatomiccontributionstotheenergy(Fig.2)
FIG.2 TheneuralnetworkmodelthatgeneratesthepotentialenergysurfaceforMDsimulation. Thisfigureistaken fromJ.Zeng,L.Cao,M.Xu,T.Zhu,J.Z.H.Zhang,Complexreactionprocessesincombustionunraveledbyneuralnetworkbasedmoleculardynamicssimulation,Nat.Commun.11(2020)5713.
Whiletheenergyofatom i isdeterminedbyitschemicalenvironment.Therelationship betweenthemisdescribedbyaneuralnetwork N :
Thechemicalenvironmentreferstotheelementinformationandrelativepositions e Ri ofthe atom i andallatomswithinapre-defineddistance/angularcut-offwhichcenteredatatom i. Tosatisfytherequirement4mentionedabove,weusemathematicsormachinelearning methodstotransformtheCartesiancoordinatesofatomsinthechemicalenvironment. Themathematicalrepresentationafterthetransformationiscalledamoleculardescriptor D. Adeepneuralnetworkfunction N isthecompositionofmultiplelayers Li :
Alayer L isusuallycomposedby y ¼L x; w, b ðÞ¼ ϕ xT w + b ,(4) where x istheinputvectorand y istheoutputvector. w and b areweightsandbiases,respectively,bothofwhicharetrainable. ϕ istheactivationfunction,suchasReLU,softplus,sigmoid,tanh,GeLU,andetc.
OneofthesimplestdescriptorsistheCoulombmatrix [59]:
where Z isthenuclearcharge.Sinceallelementsinthematrixaredeterminedonlybythe relativepositionsofatoms,thetranslationalandrotationalsymmetriescanbewellpreserved, whilethepermutationinvariancecanbeachievedbysortingtherowsandcolumnsbytheir respectivenorm.However,thesortedCoulombmatrixcannotensurethesmoothnessand differentiabilityofthePESmodel,anditcanbeonlyusedwhenthetotalenergyislearned directly,nodecompositionintoatomiccontributions.
Currently,therearealotofmoleculardescriptorsusedintheconstructionofNNP.Some ofthem [18] arementionedorintroducedinotherchaptersofthisbook(suchas Chapters11, 12,and 19).Herewewillintroducethreeofthem,theatom-centeredsymmetryfunctions (ACSFs) [60],theSchNetmodel [14],andtheDeepPotential-smoothedition(DeepPot-SE) [61] method.
Atomic-centeredsymmetryfunctions Theatomic-centeredsymmetryfunctions(ACSFs)wereproposedbyBehlerandParrinllo in2007 [10],whichcontainsaseriesofGaussian-typefunctions:
1 and G2 areradicalandangularterm,respectively.
where Rs, λ, η,and ζ arehyperparameters,
,andthecut-offfunction fc isdefinedby
RC istheuser-definedcut-off.Thecut-offfunctionmakesatomsoutofthecut-offradius havenocontributiontoeither G1 or G2 . Onthisbasis,Gasteggeretal.proposedtheweightedatomic-centeredsymmetryfunctions (wACSFs)descriptor [62],whichintroducedweightingfunctionsofthechemicalelementsto symmetryfunctions(Eqs. 7–8):
where g(Zj) ¼ Zj and h(Zj, Zk) ¼ ZjZk.
wACSFsovercometheundesirablescalingofACSFswithanincreasingnumberofdifferentelements.Meanwhile,toobtainacomparablespatialresolutionofthemolecularstructures,asubstantiallysmallernumberofwACSFsthanACSFsisneeded.
Intheirpreviouswork [63],Smithetal.alsooptimizedtheangulartermofACSFsandproposedtheANAKIN-ME(ANI)model.TheyaddtwonewhyperparameterstoEq.(8):
whichgivesthedescriptoranobviouslymoreexpressivepower.
SchNet ItisworthtomentionthattheACSFs(anditsvariants)arefixedbeforetraininganNNP.In fact,thereareothermoleculardescriptorssuchasSchNet [14] andDeepPot-SE [61] whichcan belearnedinthetrainingprocess.SchNetcontainsatrainableconvolutionalNN.Itsdescriptor D isaNNfunction N ofthecoordinates Rij andatomictype α ofthegivenchemical system.
TheNNfunctioncontainsboththedenselayers Ld andtheconvolutionallayers Lc :
where x istheinputtensorsthatcouldbeeither α ortheoutputfromthelastlayer, n isa hyperparametertodecidehowmanytimes I isappliedto x, ϕ isthesoftplus,andthe convolutionallayers Lc aregivenby
where W istrainable. Ld canbefoundinEq. 4.DetailsofthismodelcanbefoundinRef. [14]
DeepPot-SE InDeepPot-SE,theexpressionfortheatomiccontribution Ei isaneuralnetworkconsisting ofthreehiddenlayers.TheinputlayeristhemoleculardescriptorD Ri ,whichdetermined bythe“environmentmatrix” e Ri ,the“embedding”matrix Gi ,andareduceddimensionembeddingmatrix G< i [64].
s(Rij)isaswitchedreciprocaldistancefunctionthatcontrolstherangeofthechemicalenvironmenttobeconsidered.Ifanatomisseparatedfromatom i byadistancegreaterthan Roff, thentheatomisnotincludedinthechemicalenvironmentofatom i.Ifaneighboringatomis withinadistanceof Ron,thenitisgivenfullweightinthedescriptor.Theweightcansmoothly changefrom Ron to Roff.Theenvironmentmatrixisa Nneigh 4array,where Nneigh isthenumberofatomswithin Roff
The“embedding”matrix Gi isactuallyanotherneuralnetwork N whichiscalled “embeddingnetwork”: Gi ¼N sRij (20)
Theequationoftheneuralnetwork N isgiveninEq. 3.Usually,weput3hiddenlayersin theembeddingnetwork.Eachrowoftheembeddingmatrixcorrespondstoaneighbor.If thereare M3 nodesinthethirdlayerof Gi ,thesizeoftheembeddingmatrixis Nneigh M3. Thereduceddimensionalembeddingmatrixhasthesamevaluesas Gi ,butonlythefirst M0 columnsarestored,where M0 issmallerthan M3.Themainpurposeoftheusageof G< i istoreducethecomputationalcost.
Intheirrecentwork [22],PinheiroJr.etal.systematicallybenchmarkedaseriesofmoleculardescriptorsindifferentapplicationcontextsandgiveselectionrecommendationsbased onthetargetandthesizeofthetrainingset.Westronglyrecommendthatreadersofthisbook readthisworkbeforeconstructtheirownNNPs.
Exampleofsimulationwithneuralnetworkpotentials Afterselectingthemoleculardescriptor,oneonlyneedtofocusontwoissues,thepreparationofthereferencedataset(includingthetrainingandtestset)andthetrainingoftheneuralnetwork.Amongthem,thetrainingofneuralnetworksisrelativelystraightforward, currentlymainlybasedonopen-sourceframeworkssuchasTensorFloworPyTorch.On thecontrary,theconstructionofareferencedatasetthatcoversthetargetchemicalspace
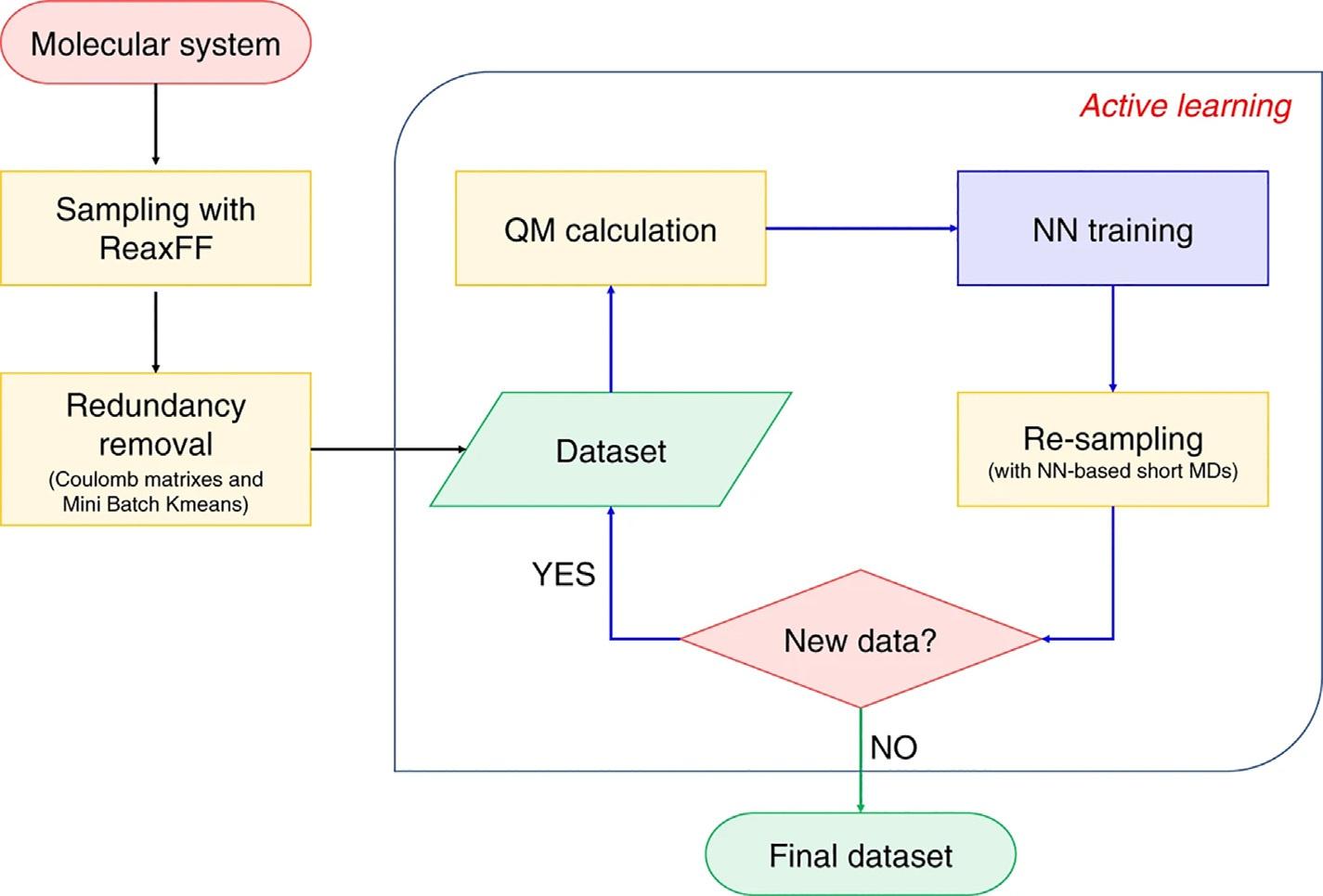
FIG.3 Theworkflowofreferencedatasetconstruction. ThisfigureistakenfromJ.Zeng,L.Cao,M.Xu,T.Zhu,J.Z.H. Zhang,Complexreactionprocessesincombustionunraveledbyneuralnetwork-basedmoleculardynamicssimulation,Nat. Commun.11(2020)5713.
whilebeingassmallaspossibleisthemostcriticalanddifficult.Whenonecannotpredictthe chemicalspacethatMDwillexplore,suchassimulatingcombustionreactions,theconstructionofreferencedatasetsbecomesevenmoredifficult.AfeasibleapproachistorunMDsimulationwhilesampling.PreviousstudiessuggestedusingmultipleNNPmodelstoidentify poorlysampledregionsoftheconfigurationspace [65,66].Inthismethod,severalNNP modelswillbetrainedbasedonthesamereferencedataset.DuringtheMDsimulation,a largenumberoftrialconfigurationsareevaluatedbyallofthesemodels.Ifagivenstructure differsobviouslyfromalltraineddata,thepredictionsofthesemodelsshouldbesignificantly different.Thenoneneedtoaddthisstructureintothedataset.Conversely,ifthetraining setalreadycontainssimilarstructuresofthegivenone,thepredictedresultsofthesemodels shouldbeconsistent.Thisalgorithmwascalled“activelearning”or“learningon-the-fly”and hasbeenusedbymanyworks [66–76],see Chapter14 Fig.3 showsan“activelearning” workflowweusedinthesimulationofmethanecombustion.
Firstly,weneedtoprepareaninitialdatasetbyusingashortMDsimulationwiththe ReaxFFforcefield.Foreachatomineachsnapshotofthetrajectory,webuiltamolecularclusterwhichcontainsthisatomandspeciesthatwithinaspecifiedcut-offcenteredonit.Thenthe MiniBatchKMeans [77] isusedtoremovetheredundancy.Startingfromtheinitialdataset, fourdifferentNNPmodelsweretrainedbasedonthedatasetfromthelaststep.Thenseveral shortMDsimulationswereperformedbasedononeoftheseNNPmodels.Duringthesimulation,theatomicforcesofthecentralatominthemolecularclusterareevaluatedbyallfour NNPmodelssimultaneously.Foraspecificatom,ifthepredictedforcesbythesefourmodels
areconsistentwitheachother,thensimilarmolecularclustersshouldalreadybeincludedin thedataset.Onthecontrary,iftheresultsofthesefourmodelsareinconsistentwitheachother andtheerrorbetweenthemisinaspecificrange,thecorrespondingmolecularclusterwillbe addedintothedataset.Theupdateofthedatasetwillbecontinueduntilthepredictionsofthe fourmodelsarealwaysconsistentorthetargetlengthoftheMDsimulationisreached.More techniquedetailscanbefoundinourpreviousstudy [54].
Casestudies Simulationoftheoxidationofmethane Inthissection,wewilltakethesimulationofmethanecombustionasanexampleandintroducetheprocedureofNNP-basedMDsimulation.Allfilesneededinthissectioncanbe downloadedfromthefollowinglink:https://github.com/tongzhugroup/Chapter13tutorial.
Attheverybeginning,oneneedstoconsiderwhichsoftwaretouse.Thereareseveral open-sourceandwidelyusedNNPbuildingsoftware,suchasTensorMol [38],MLatom [19,20],EANN [18],andDeePMD-kit [78].Forbeginners,thereislittledifferencebetween thesesoftwarepackages.TheMLatomandEANNmethodareintroducedinotherchapters ofthisbooksuchas Chapters13 and 19,respectively.HereweemploytheDeePMD-kit.The followingstepsareusedtotraintheNNPandperformMDsimulation.
Step1:Preparingthereferencedataset Inthereferencedatasetpreparationprocess,onealsohastoconsidertheexpectaccuracyof thefinalmodel,oratwhatQMleveloneshouldlabelthedata.InRef.54,theGaussian16 [79] softwarewasusedtocalculatethepotentialenergyandatomicforcesofthereferencedataat theMN15/6-31G** level.TheMN15functionalwasemployedbecauseithasgoodaccuracy forbothmulti-referenceandsingle-referencesystems [80],whichisessentialforoursystemas wehavetodealwithalotofradicalsandtheirreactions.Here,weassumethatthedatasetis preparedinadvance,whichcanbedownloadedfromtheabovelink.
Step2.TrainingtheNNPES Beforethetrainingprocess,weneedtoprepareaninputfilecalled methane_param.json whichcontainsthecontrolparameters.Thetrainingcanbedonebythefollowingcommand: $$deepmd_root/bin/dptrainmethane_param.json
Thereareseveralparametersweneedtodefineinthe methane_param.json file.The type_map referstothetypeofelementsincludedinthetraining,andtheoptionof rcut isthecut-offradiuswhichcontrolsthedescriptionoftheenvironmentaroundthecenteratom.Thetypeof descriptoris“se_a”inthisexample,whichrepresentstheDeepPot-SEmodel.Thedescriptor willdecaysmoothlyfrom rcut_smth (Ron)tothecut-offradius rcut (Roff).Here rcut_smth and rcut aresetto1.0A ˚ and6.0A ˚ ,respectively.The sel definesthemaximumpossiblenumberof neighborsforthecorrespondingelementwithinthecut-offradius.Theoptions neuron in descriptor and fitting_net isusedtodeterminetheshapeoftheembeddingneuralnetworkand thefittingnetwork,whicharesetto(25,50,100)and(240,240,240)respectively.Thevalueof
axis_neuron representsthesizeoftheembeddingmatrix,whichwassetto12.Andtheoptions start_lr, decay_rate,and decay_steps inthe learning_rate modulecontrolsthelearningratefor n th batchaccordingtothefollowingformula:
Theinitiallearningratewassetto0.001anditwilldecayevery400steps.Thelossfunction isdefinedas:
The Δε and Δ Fi inthelossfunctionrepresentthedifferencebetweenthepredictionofNNP inthelabeledenergyandforce,respectively. pε and pf areprefactorsthatdecaysexponentially duringtraining.Byassigningdifferentrandomseedsintheneuralnetworkinitializationprocess,onecantrainmultiplemodelsatthesametime.
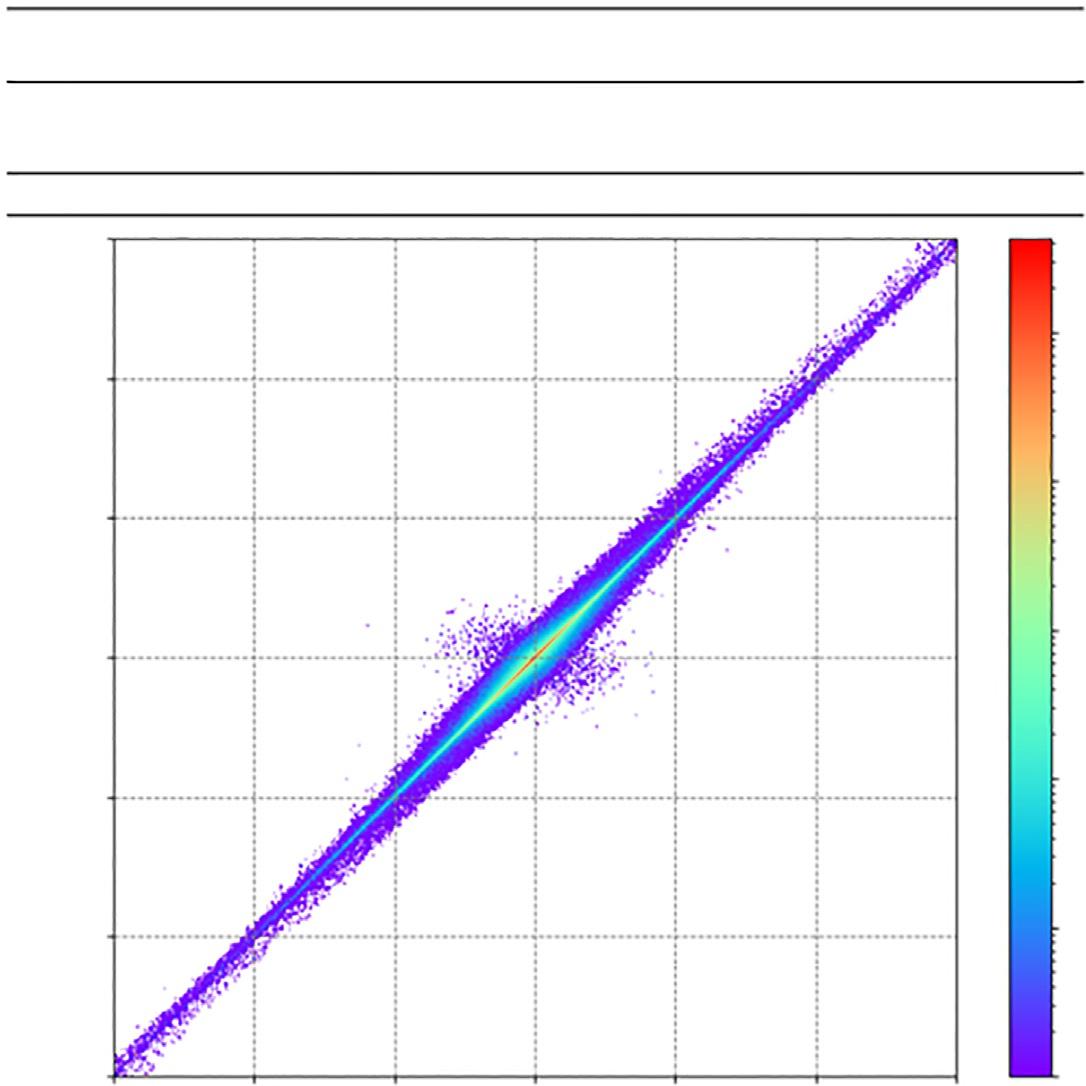
Afterthetrainingiscompleted,thepredictivepowerofthemodelmustbecheckedfirst.As showninthe Fig.4,themeanabsoluteerrors(MAE)ofpotentialenergyareonly0.04eV/atom
Training Set / Test Set
FIG.4 (A)Energypredictionerrorsforthereferenceset.Themeanabsoluteerrors(MAEs)androotmeansquared errors(RMSEs)areineV/atom.(B).ThecorrelationofatomicforcesbetweenNNpredictionsandDFTcalculations. ThisfigureistakenfromJ.Zeng,L.Cao,M.Xu,T.Zhu,J.Z.H.Zhang,Complexreactionprocessesincombustionunraveledby neuralnetwork-basedmoleculardynamicssimulation,Nat.Commun.11(2020)5713.
and0.14eV/atominthetrainingsetandthetestset,respectively.Asfortheatomicforces,the correlationcoefficientbetweenthepredictedandlabeledvaluesis0.999andtheMAEis 0.12eV/A ˚ .
Step3:Freezethemodel Thisstepistoextractthetrainedneuralnetworkmodel.Tofreezethemodel,thefollowing commandwillbeexecuted:
$$deepmd_root/bin/dpfreeze-ograph.pb
Afilecalledgraph.pbcanbefoundinthetrainingfolder.Thenthefrozenmodelcanbe compressed [81]:
$$deepmd_root/bin/dpcompress-igraph.pb-ograph_compressed.pb-tmethane_param.json
Step4:RunningMDsimulationbasedontheNNP ThefrozenmodelcanbeusedtorunreactiveMDsimulationstoexplorethedetailedreactionmechanismofmethanecombustion.TheMDengineisprovidedbytheLAMMPSsoftware [82].Hereweusethesamesystemfromourpreviouswork [54],whichcontains100 methaneand200oxygenmolecules.TheMDwillbeperformedundertheNVTensemble at3000Kfor1ns.TheLAMMPSprogramcanbeinvokedbythefollowingcommand:
$$deepmd_root/bin/lmp-iinput.lammps
The input.lammps istheinputfilethatcontrolstheMDsimulationindetail,techniquedetailscanbefoundinthemanualofLAMMPS(https://docs.lammps.org/).TousetheNNP, the pair_style optioninthisinputshouldbespecifiedasfollows: pair_styledeepmdgraph_compressed.pb pair_coeff **
Step5:Analysisofthetrajectory Afterthesimulationisdone,wecanusetheReacNetGenerator [83] softwarewhichwas developedinourpreviousstudytoextractthereactionnetworkfromthetrajectory.Allspeciesandreactionsinthetrajectorywillbeputonaninteractivewebpagewherewecananalyzethembymouseclicks.Eventuallyweshouldbeabletoobtainreactionnetworksthat consistentwith Figs.2–4 inRef.[54].
$ reacnetgenerator-imethane.lammpstrj-aCHO–dump
Themostexpensivestepsarestep2(trainingtheNNP)andstep4(RunningMDsimulation basedontheNNP).Performanceofthesetwostepsisbenchmarkedusingdifferenthardware asshownin Table1.
TABLE1 PerformanceoftrainingandMDsimulationsusingdifferenthardware(unit:ms/step)a
NVIDIAGeForceRTX3080Ti16.3910.773.1643.325
NVIDIAGeForceRTX309024.2626.786.7386.965
NVIDIATeslaV10027.0719.175.8635.558
a TrainingisconductedwiththemethanecombustiontrainingsetgivenintheGitHubrepository,usingtheDeePMD-kitsoftware.MD simulationsareconductedwithamethanecombustionsystemof900atoms,usingLAMMPSinterfacetotheDeePMD-kit.
Conclusionsandoutlook Inthischapter,weintroducethebasicconceptsandideasthatabouttheconstructionand usageofNNPfromabeginner’sperspective.Inthepastyears,machinelearning-basedmoleculardynamicssimulationshavegainedsignificantdevelopmentsandadvancesthathave changedtheresearchparadigmthroughoutthetheoreticalchemistrycommunity.Intheprocessofgrowingfrombeginnertoexpert,webelievethatthefollowingissuesareneededtobe considered [84,85].(1)Dataisattheheartofallmachinelearningmethods.Therefore,the questforbettermethodstobuildreferencedatasetswillneverstop.Somerecentusefuldiscussionscanbefoundintheliterature [86,87].(2)Theselectionofpropermoleculardescriptorswhichcanrepresentthechemicalenvironmentwell,whilewithminimalcomplexity. Althoughwell-establishedmoleculardescriptorssuchasACSFs,EANNandDeepPot-SE arealreadyavailable,onemaystillwanttodesignnewdescriptorsforspecificsystemsofinteresttofurtherenhancethetransferabilityoftheNNPmodelandimprovetheaccuracyand efficiencyofthetrainingprocessandMDsimulation.(3)Theselectionofhyper-parameters forNN.Therearemanyhyper-parameterssuchaslearningrateandnetworkstructureparameterswhichhavehugeimpactontheaccuracyandefficiencyofNNPs.Althoughsome automatedmethodshavebeenproposed [60,87,88],thechoiceoftheseparametersisstill largelyempiricallydominated.Whencomputationalresourcesarelimited,oneneedstocarefullycompareandselecttheappropriatesetofparameters.(4).Thetreatmentoflong-range interactions.Asmentionedabove,distancecut-offs(usuallyaround5A ˚ )areusedindefining thechemicalenvironment.Thepurposeofthisistoavoidhavingtoomanydegreesoffreedominthechemicalenvironment,whichwouldgreatlyincreasethedifficultyofthetraining processaswellasthesizeofthereferencedataset.Forsmallsystemswiththeperiodicboundary,thelong-rangeinteractionscanbeeffectivelyincludedintheshort-rangeinteractions. However,forlargeand/ornon-periodicsystemsincondensedphases,suchasbiomolecules likeproteins,long-rangeinteractionsmustbeexplicitlyconsidered.Onlyveryrecentlysome encouragingprogresshasbeenmade [89,90]
Acknowledgment ThisworkwassupportedbytheNationalNaturalScienceFoundationofChina(GrantsNo.22173032,21933010).J.Z. wassupportedinpartbytheNationalInstitutesofHealth(GM107485)underthedirectionofDarrinM.York.Wealso thanktheECNUMultifunctionalPlatformforInnovation(No.001)andtheExtremeScienceandEngineeringDiscoveryEnvironment(XSEDE),whichissupportedbyNationalScienceFoundationGrantACI-1548562.56(specifically,theresourcesEXPANSEatSDSCthroughallocationTG-CHE190067),forprovidingsupercomputertime.
References [1] R.Iftimie,P.Minary,M.E.Tuckerman, Abinitio moleculardynamics:concepts,recentdevelopments,andfuture trends,Proc.Natl.Acad.Sci.U.S.A.102(2005)6654–6659.
[2] J.A.Harrison,J.D.Schall,S.Maskey,P.T.Mikulski,M.T.Knippenberg,B.H.Morrow,Reviewofforcefieldsand intermolecularpotentialsusedinatomisticcomputationalmaterialsresearch,Appl.Phys.Rev.5(2018),031104.
[3] P.E.Lopes,O.Guvench,A.D.MacKerellJr.,Currentstatusofproteinforcefieldsformoleculardynamicssimulations,MethodsMol.Biol.1215(2015)47–71.
[4] T.B.Blank,S.D.Brown,A.W.Calhoun,D.J.Doren,Neuralnetworkmodelsofpotentialenergysurfaces,J.Chem. Phys.103(1995)4129–4137.
[5] G.E.Moyano,M.A.Collins,Molecularpotentialenergysurfacesbyinterpolation:strategiesforfaster convergence,J.Chem.Phys.121(2004)9769–9775.
[6] M.J.T.Jordan,K.C.Thompson,M.A.Collins,Convergenceofmolecularpotentialenergysurfacesbyinterpolation:applicationtotheOH+H2 ! H2O+Hreaction,J.Chem.Phys.102(1995)5647–5657.
[7] K.C.Thompson,M.J.T.Jordan,M.A.Collins,Polyatomicmolecularpotentialenergysurfacesbyinterpolationin localinternalcoordinates,J.Chem.Phys.108(1998)8302–8316.
[8] G.G.Maisuradze,D.L.Thompson,Interpolatingmovingleast-squaresmethodsforfittingpotentialenergysurfaces:illustrativeapproachesandapplications,J.Phys.Chem.A107(2003)7118–7124.
[9] C.Qu,Q.Yu,J.M.Bowman,Permutationallyinvariantpotentialenergysurfaces,Annu.Rev.Phys.Chem. 69(2018)151–175.
[10] J.Behler,M.Parrinello,Generalizedneural-networkrepresentationofhigh-dimensionalpotential-energysurfaces,Phys.Rev.Lett.98(2007),146401.
[11] A.P.Bartok,M.C.Payne,R.Kondor,G.Csanyi,Gaussianapproximationpotentials:theaccuracyofquantum mechanics,withouttheelectrons,Phys.Rev.Lett.104(2010),136403.
[12] S.Chmiela,A.Tkatchenko,H.E.Sauceda,I.Poltavsky,K.T.Schutt,K.R.Muller,Machinelearningofaccurate energy-conservingmolecularforcefields,Sci.Adv.3(2017),1603015.
[13] K.T.Schutt,F.Arbabzadah,S.Chmiela,K.R.Muller,A.Tkatchenko,Quantum-chemicalinsightsfromdeeptensorneuralnetworks,Nat.Commun.8(2017),13890.
[14] K.T.Schutt,H.E.Sauceda,P.J.Kindermans,A.Tkatchenko,K.R.Muller,SchNet-adeeplearningarchitecturefor moleculesandmaterials,J.Chem.Phys.148(2018),241722.
[15] X.Chen,M.S.Jorgensen,J.Li,B.Hammer,Atomicenergiesfromaconvolutionalneuralnetwork,J.Chem.TheoryComput.14(2018)3933–3942.
[16] L.F.Zhang,J.Q.Han,H.Wang,R.Car,E.Weinan,Deeppotentialmoleculardynamics:ascalablemodelwiththe accuracyofquantummechanics,Phys.Rev.Lett.120(2018),143001.
[17] D.Lu,H.Wang,M.Chen,L.Lin,R.Car,E.Weinan,W.Jia,L.Zhang,86PFLOPSdeeppotentialmoleculardynamicssimulationof100millionatomswithabinitioaccuracy,Comput.Phys.Commun.259(2021),107624.
[18] Y.Zhang,C.Hu,B.Jiang,Embeddedatomneuralnetworkpotentials:efficientandaccuratemachinelearning withaphysicallyinspiredrepresentation,J.Phys.Chem.Lett.10(2019)4962–4967.
[19] P.O.Dral,F.Ge,B.X.Xue,Y.F.Hou,M.PinheiroJr.,J.Huang,M.Barbatti,MLatom2:anintegrativeplatformfor atomisticmachinelearning,Top.Curr.Chem.379(2021)27.
[20] P.O.Dral,MLatom:aprogrampackageforquantumchemicalresearchassistedbymachinelearning,J.Comput. Chem.40(2019)2339–2347.
[21] J.Behler,Perspective:machinelearningpotentialsforatomisticsimulations,J.Chem.Phys.145(2016),170901.
[22] M.Pinheiro,F.Ge,N.Ferre,P.O.Dral,M.Barbatti,Choosingtherightmolecularmachinelearningpotential, Chem.Sci.12(2021)14396–14413.
[23] J.S.Smith,B.T.Nebgen,R.Zubatyuk,N.Lubbers,C.Devereux,K.Barros,S.Tretiak,O.Isayev,A.E.Roitberg, Approachingcoupledclusteraccuracywithageneral-purposeneuralnetworkpotentialthroughtransferlearning,Nat.Commun.10(2019)2903.
[24] C.Devereux,J.S.Smith,K.K.Davis,K.Barros,R.Zubatyuk,O.Isayev,A.E.Roitberg,Extendingtheapplicability oftheANIdeeplearningmolecularpotentialtosulfurandhalogens,J.Chem.TheoryComput.16(2020) 4192–4202.
[25] J.Behler,Firstprinciplesneuralnetworkpotentialsforreactivesimulationsoflargemolecularandcondensed systems,Angew.Chem.Int.Edit.56(2017)12828–12840.
[26] T.Morawietz,J.Behler,Afull-dimensionalneuralnetworkpotential-energysurfaceforwaterclustersuptothe hexamer,Z.Phys.Chem.227(2013)1559–1581.
[27] T.Morawietz,J.Behler,Adensity-functionaltheory-basedneuralnetworkpotentialforwaterclustersincluding vanderWaalscorrections,J.Phys.Chem.A117(2013)7356–7366.
[28] M.delCueto,X.Zhou,L.Zhou,Y.Zhang,B.Jiang,H.Guo,NewperspectivesonCO2–Pt(111)interactionwitha high-dimensionalneuralnetworkpotentialenergysurface,J.Phys.Chem.C124(2020)5174–5181.
[29] C.Hu,Y.Zhang,B.Jiang,DynamicsofH2OadsorptiononPt(110)-(1 2)basedonaneuralnetworkpotential energysurface,J.Phys.Chem.C124(2020)23190–23199.
[30] X.Lu,X.Wang,B.Fu,D.Zhang,TheoreticalinvestigationsofratecoefficientsofH+H2O2 ! OH+H2Oona full-dimensionalpotentialenergysurface,J.Phys.Chem.A123(2019)3969–3976.
[31] H.Wang,W.Yang,Towardbuildingproteinforcefieldsbyresidue-basedsystematicmolecularfragmentation andneuralnetwork,J.Chem.TheoryComput.15(2019)1409–1417.
[32] H.Wang,W.T.Yang,Forcefieldforwaterbasedonneuralnetwork,J.Phys.Chem.Lett.9(2018)3232–3240.
[33] O.T.Unke,D.Koner,S.Patra,S.K€ aser,M.Meuwly,High-dimensionalpotentialenergysurfacesformolecular simulations:fromempiricismtomachinelearning,Mach.Learn.:Sci.Technol.1(2020),013001.
[34] A.Jonayat,A.C.VanDuin,M.J.Janik,Discoveryofdescriptorsforstablemonolayeroxidecoatingsthroughmachinelearning,ACSAppl.EnergyMater.1(2018)6217–6226.
[35] A.C.Rajan,A.Mishra,S.Satsangi,R.Vaish,H.Mizuseki,K.-R.Lee,A.K.Singh,Machine-learning-assistedaccuratebandgappredictionsoffunctionalizedMXene,Chem.Mater.30(2018)4031–4038.
[36] G.Pilania,J.E.Gubernatis,T.Lookman,Multi-fidelitymachinelearningmodelsforaccuratebandgappredictionsofsolids,Comput.Mater.Sci.129(2017)156–163.
[37] R.Jinnouchi,F.Karsai,G.Kresse,On-the-flymachinelearningforcefieldgeneration:applicationtomelting points,Phys.Rev.B100(2019),014105.
[38] K.Yao,J.E.Herr,D.W.Toth,R.McKintyre,J.Parkhill,TheTensorMol-0.1modelchemistry:aneuralnetwork augmentedwithlong-rangephysics,Chem.Sci.9(2018)2261–2269.
[39] S.KondatiNatarajan,T.Morawietz,J.Behler,Representingthepotential-energysurfaceofprotonatedwater clustersbyhigh-dimensionalneuralnetworkpotentials,Phys.Chem.Chem.Phys.17(2015)8356–8371.
[40] B.Jiang,J.Li,H.Guo,High-fidelitypotentialenergysurfacesforgas-phaseandgas–surfacescatteringprocesses frommachinelearning,J.Phys.Chem.Lett.11(2020)5120–5131.
[41] S.Manzhos,T.Carrington,Neuralnetworkpotentialenergysurfacesforsmallmoleculesandreactions,Chem. Rev.121(2021)10187–10217.
[42] M.Meuwly,Machinelearningforchemicalreactions,Chem.Rev.121(2021)10218–10239.
[43] J.Behler,Fourgenerationsofhigh-dimensionalneuralnetworkpotentials,Chem.Rev.121(2021)10037–10072.
[44] M.Ceriotti,C.Clementi,O.AnatolevonLilienfeld,Introduction:machinelearningattheatomicscale,Chem. Rev.121(2021)9719–9721.
[45] V.L.Deringer,A.P.Barto ´ k,N.Bernstein,D.M.Wilkins,M.Ceriotti,G.Csa ´ nyi,Gaussianprocessregressionfor materialsandmolecules,Chem.Rev.121(2021)10073–10141.
[46] A.Glielmo,B.E.Husic,A.Rodriguez,C.Clementi,F.Noe,A.Laio,Unsupervisedlearningmethodsformolecularsimulationdata,Chem.Rev.121(2021)9722–9758.
[47] B.Huang,O.A.vonLilienfeld,Abinitiomachinelearninginchemicalcompoundspace,Chem.Rev.121(2021) 10001–10036.
[48] F.Musil,A.Grisafi,A.P.Barto ´ k,C.Ortner,G.Csa ´ nyi,M.Ceriotti,Physics-inspiredstructuralrepresentationsfor moleculesandmaterials,Chem.Rev.121(2021)9759–9815.
[49] A.Nandy,C.Duan,M.G.Taylor,F.Liu,A.H.Steeves,H.J.Kulik,Computationaldiscoveryoftransition-metal complexes:fromhigh-throughputscreeningtomachinelearning,Chem.Rev.121(2021)9927–10000.
[50] O.T.Unke,S.Chmiela,H.E.Sauceda,M.Gastegger,I.Poltavsky,K.T.Schutt,A.Tkatchenko,K.-R.Muller,Machinelearningforcefields,Chem.Rev.121(2021)10142–10186.
[51] J.Westermayr,P.Marquetand,Machinelearningforelectronicallyexcitedstatesofmolecules,Chem.Rev.121 (2021)9873–9926.
[52] M.Xu,T.Zhu,J.Z.H.Zhang,Moleculardynamicssimulationofzincioninwaterwithanabinitiobasedneural networkpotential,J.Phys.Chem.A123(2019)6587–6595.
[53] M.Xu,T.Zhu,J.Z.H.Zhang,Automaticallyconstructedneuralnetworkpotentialsformoleculardynamicssimulationofzincproteins,Front.Chem.9(2021),692200.
[54] J.Zeng,L.Cao,M.Xu,T.Zhu,J.Z.H.Zhang,Complexreactionprocessesincombustionunraveledbyneural network-basedmoleculardynamicssimulation,Nat.Commun.11(2020)5713.
[55] J.Zeng,L.Zhang,H.Wang,T.Zhu,Exploringthechemicalspaceoflinearalkanepyrolysisviadeeppotential GENerator,EnergyFuel35(2021)762–769.
[56] A.C.VanDuin,S.Dasgupta,F.Lorant,W.A.Goddard,ReaxFF:areactiveforcefieldforhydrocarbons,J.Phys. Chem.A105(2001)9396–9409.
[57] E.Wang,J.Ding,Z.Qu,K.Han,Developmentofareactiveforcefieldforhydrocarbonsandapplicationtoisooctanethermaldecomposition,EnergyFuel32(2017)901–907.
[58] T.Cheng,A.Jaramillo-Botero,W.A.Goddard,H.Sun,AdaptiveacceleratedReaxFFreactivedynamicswithvalidationfromsimulatinghydrogencombustion,J.Am.Chem.Soc.136(2014)9434–9442.
[59] M.Rupp,A.Tkatchenko,K.R.Muller,O.A.vonLilienfeld,Fastandaccuratemodelingofmolecularatomization energieswithmachinelearning,Phys.Rev.Lett.108(2012),058301.
[60] J.Behler,Atom-centeredsymmetryfunctionsforconstructinghigh-dimensionalneuralnetworkpotentials,J. Chem.Phys.134(2011),074106.
[61] L.Zhang,J.Han,H.Wang,W.A.Saidi,R.Car,E.Weinan,End-to-endsymmetrypreservinginter-atomicpotentialenergymodelforfiniteandextendedsystems,NIPS’18:Proceedingsofthe32ndInternationalConferenceon NeuralInformationProcessingSystems,CurranAssociatesInc.,RedHook,NY,USA,2018,pp.4441–4451.
[62] M.Gastegger,L.Schwiedrzik,M.Bittermann,F.Berzsenyi,P.Marquetand,wACSF-weightedatom-centered symmetryfunctionsasdescriptorsinmachinelearningpotentials,J.Chem.Phys.148(2018),241709.
[63] J.S.Smith,O.Isayev,A.E.Roitberg,ANI-1:anextensibleneuralnetworkpotentialwithDFTaccuracyatforce fieldcomputationalcost,Chem.Sci.8(2017)3192–3203.
[64] J.Zeng,T.J.Giese,S.Ekesan,D.M.York,Developmentofrange-correcteddeeplearningpotentialsforfast,accuratequantummechanical/molecularmechanicalsimulationsofchemicalreactionsinsolution,J.Chem.TheoryComput.17(2021)6993–7009.
[65] J.Behler,Neuralnetworkpotential-energysurfacesinchemistry:atoolforlarge-scalesimulations,Phys.Chem. Chem.Phys.13(2011)17930–17955.
[66] J.S.Smith,B.Nebgen,N.Lubbers,O.Isayev,A.E.Roitberg,Lessismore:samplingchemicalspacewithactive learning,J.Chem.Phys.148(2018),241733.
[67] L.F.Zhang,D.Y.Lin,H.Wang,R.Car,E.Weinan,Activelearningofuniformlyaccurateinteratomicpotentials formaterialssimulation,Phys.Rev.Mater.3(2019),023804.
[68] W.Wang,T.Yang,W.H.Harris,R.Gomez-Bombarelli,Activelearningandneuralnetworkpotentialsaccelerate molecularscreeningofether-basedsolvateionicliquids,Chem.Commun.(Camb.)56(2020)8920–8923.
[69] Q.Lin,Y.Zhang,B.Zhao,B.Jiang,Automaticallygrowingglobalreactiveneuralnetworkpotentialenergysurfaces:atrajectory-freeactivelearningstrategy,J.Chem.Phys.152(2020),154104.
[70] S.J.Ang,W.Wang,D.Schwalbe-Koda,S.Axelrod,R.Go ´ mez-Bombarelli,Activelearningacceleratesabinitio moleculardynamicsonreactiveenergysurfaces,Chem(2021)738–751.
[71] Z.Li,J.R.Kermode,A.DeVita,Moleculardynamicswithon-the-flymachinelearningofquantum-mechanical forces,Phys.Rev.Lett.114(2015),096405.
[72] B.Huang,O.A.vonLilienfeld,Quantummachinelearningusingatom-in-molecule-basedfragmentsselectedon thefly,Nat.Chem.12(2020)945–951.
[73] E.V.Podryabinkin,A.V.Shapeev,Activelearningoflinearlyparametrizedinteratomicpotentials,Comput.Mater.Sci.140(2017)171–180.
[74] N.J.Browning,R.Ramakrishnan,O.A.vonLilienfeld,U.Roethlisberger,Geneticoptimizationoftrainingsetsfor improvedmachinelearningmodelsofmolecularproperties,J.Phys.Chem.Lett.8(2017)1351–1359.
[75] Y.Zhang,H.Wang,W.Chen,J.Zeng,L.Zhang,H.Wang,E.Weinan,DP-GEN:aconcurrentlearningplatform forthegenerationofreliabledeeplearningbasedpotentialenergymodels,Comput.Phys.Commun.253(2020), 107206.
[76] Q.Lin,L.Zhang,Y.Zhang,B.Jiang,Searchingconfigurationsinuncertaintyspace:activelearningofhighdimensionalneuralnetworkreactivepotentials,J.Chem.TheoryComput.17(2021)2691–2701.
[77] D.Sculley,Web-scalek-meansclustering,in:Proceedingsofthe19thinternationalconferenceonWorldWide Web,2010,ACM,2010,pp.1177–1178.
[78] H.Wang,L.Zhang,J.Han,E.Weinan,DeePMD-kit:adeeplearningpackageformany-bodypotentialenergy representationandmoleculardynamics,Comput.Phys.Commun.228(2018)178–184.
[79] M.Frisch,G.Trucks,H.Schlegel,G.Scuseria,M.Robb,J.Cheeseman,G.Scalmani,V.Barone,G.Petersson,H. Nakatsuji,Gaussian16,RevisionA.03,GaussianInc.,WallingfordCT,2016.
[80] S.Y.Haoyu,X.He,S.L.Li,D.G.Truhlar,MN15:aKohn–Shamglobal-hybridexchange–correlationdensityfunctionalwithbroadaccuracyformulti-referenceandsingle-referencesystemsandnoncovalentinteractions, Chem.Sci.7(2016)5032–5051.
[81] D.Lu,W.Jiang,Y.Chen,L.Zhang,W.Jia,H.Wang,M.Chen,DPTrain,thenDPCompress:ModelCompression inDeepPotentialMolecularDynamics,arXivpreprint,2021.arXiv:2107.02103.
[82] H.M.Aktulga,J.C.Fogarty,S.A.Pandit,A.Y.Grama,Parallelreactivemoleculardynamics:numericalmethods andalgorithmictechniques,ParallelComput.38(2012)245–259.
[83] J.Zeng,L.Cao,C.H.Chin,H.Ren,J.Z.H.Zhang,T.Zhu,ReacNetGenerator:anautomaticreactionnetworkgeneratorforreactivemoleculardynamicssimulations,Phys.Chem.Chem.Phys.22(2020)683–691.
[84] V.Botu,R.Batra,J.Chapman,R.Ramprasad,Machinelearningforcefields:construction,validation,and outlook,J.Phys.Chem.C121(2017)511–522.
[85] J.Behler,Constructinghigh-dimensionalneuralnetworkpotentials:atutorialreview,Int.J.QuantumChem.115 (2015)1032–1050.
[86] J.F.Xia,Y.L.Zhang,B.Jiang,Efficientselectionoflinearlyindependentatomicfeaturesforaccuratemachine learningpotentials,Chin.J.Chem.Phys.34(2021)695–703.
[87] M.Xu,T.Zhu,J.Z.H.Zhang,Automatedconstructionofneuralnetworkpotentialenergysurface:theenhanced self-organizingincrementalneuralnetworkdeeppotentialmethod,J.Chem.Inf.Model.61(2021)5428–5437.
[88] G.Imbalzano,A.Anelli,D.Giofre,S.Klees,J.Behler,M.Ceriotti,Automaticselectionofatomicfingerprintsand referenceconfigurationsformachine-learningpotentials,J.Chem.Phys.148(2018),241730.
[89] T.W.Ko,J.A.Finkler,S.Goedecker,J.Behler,Afourth-generationhigh-dimensionalneuralnetworkpotential withaccurateelectrostaticsincludingnon-localchargetransfer,Nat.Commun.12(2021)398.
[90] A.Grisafi,M.Ceriotti,Incorporatinglong-rangephysicsinatomic-scalemachinelearning,J.Chem.Phys.151 (2019),204105.
Other documents randomly have different content From Plato’s thoughts their Attic dress, To charm an era rude, He tore away, and in its stead A meaner garb indued.
But unto eyes, o’er which no film By ignorance is thrown, His dreams those garments only grace In which at birth they shone.
Of bright Cadmean rune he wove A rich asbestic web; Sometimes its woof like sunset glows, Of gold and purple thread;
Sometimes with rosy spring it vies— Then flowers inwoven shine; Sometimes diaphonous as oil; Than Coan gauze more fine.
And thus each imaged thought, that sprung From his sciential brain, A fluent drapery received, To make its beauty plain.
Here pilgrims dwell, strange sights that saw On many a foreign strand— He born beneath the Doge’s rule, Beloved of Kubla Khan,
And Mandeville, who journeyed far Against the Eastern wind, The sacred Capital to see, And miracles of Ind.
None ever wore the sandal shoon More marvellous than he; For then the world had far away
Its realms of mystery The giant Roc then winnowed swift The morning-cradled breeze, And happy islands glittered o’er
The Occidental seas.
Upon Saint Michael’s happy morn How throbbed his glowing brow, When towards the ancient Orient His galley turned her prow!
Already in the wind he smells
Hyblæan odors blown From isles invisible, afar Amid the Indian foam.
The turbaned millions, dusky, wild, Already meet his eyes— The domes of Islam crescent-crowned In long perspective rise,
Mid waving palms, o’er level sands, With skyey verges low, Where from his eastern tent, the Sun Spreads wide a saffron glow.
The golden thrones of Asian kings, Their empery supreme, Their capitals Titanic, laved By many a famous stream;
The cities, desolate and lone, Where desert monsters prowl, Where spiders film the royal throne, And shrieks the nightly owl;
Enormous Caf, the mountain wall
Of ancient Colchian land—
Where dragon-drawn Medea gave The Argonaut her hand;
Nysæan Meros, mid whose rifts
The viny God was born,—
The empyreal sky its summit cleaves, In shape a golden horn;
And o’er its top reclining swim
In zones of windless air
The slumbrous deities of Ind, Removed from earthly care;
The Ammonian phalanx round its base
In festal garments ranged, Their brows with ivied chaplets bound, Their swords to thyrsi changed;
The ravenous gryphons, brooding o’er
The desert’s gleaming gold,
The auroral Chersonese, that shines With treasures manifold;
The groves of odorous scent, that line
The green Sabæan shore, Whence wrapped in cerements dipt in balm, His sire the Phœnix bore;
The Persian valley famed in song, Where gentle Hafiz strayed;
The Indian Hollow far beyond, By mountains tall embayed;
Whose virgins boast a richer bloom
Than peaches of Cabool, And nymph-like fall their marble urns With fountain-waters cool;
Whose looms produce a gorgeous web, That with the rainbow vies, So delicate its downy woof, So deep its royal dyes.
The motionless Yogee, who stands In wildernesses lone, His sleepless eye forever fixed On Brahma’s airy throne, In blue infinity to melt His troubled soul away, And of the sunny Monad form A portion and a ray.
The tales, Milesian-like, that charm The vacant ear at eve, Wherein the Orient fabulists Their marvels interweave;
Of wondrous realms beyond the reach Of mortal footstep far, Whose maidens, winged with pinions light, Outstrip the falling star;
Whose forests bear a vocal fruit, With human tongues endowed, That mid the autumn-laden boughs Are querulous and loud;
Of sparry caves in musky hills, Which sevenfold seas surround, Where ancient kings enchanted lie, In dreamless slumber bound;
Of potent gems, whose hidden might Can thwart malignant star; Of Eblis’ pavement saffron-strewn
’Neath fallen Istakhar; All these in long succession rose, Illumed by fancy’s ray, As swiftly towards the Morning lands His galley ploughed her way.
Elfin Land. ���� ��.
But far the greatest miracle Which Elfin land can show, A hostel is, like that which stood In Eastcheap long ago.
Before the entrance, in the blast There swings a tusky sign; And when at night the Elfin moon And constellations shine,
A ruddy glow illumes the panes, And looking through you see, With merry faces seated round, A famous company.
Prince Hal the royal wassailer, And that great fount of fun, Diana’s portly forester, The merry knight Sir John,
With all their losel servitors, Mirth-shaken cheek by cheek; Cambysean Pistol, Peto, Poins, And Bardolph’s fiery beak.
A grove there is in Elfin land, Where closely intertwine The Grecian myrtle’s branches light With Gothic oak sublime.
Beneath its canopy of shade,
Their temples bound with bays, Are grouped the minstrels, that adorn
The mediæval days.
The laurelled Ghibelline, who saw The Stygian abyss, The fiery mosques and walls, that gird The capital of Dis;
The realms of penance, and the rings Of constellated light, Whose luminous pavilions hold The righteous robed in white;
Uranian groves and spheral vales, Saturnian academes, Where sainted theologues abide, Discoursing mystic themes;
The Paradisal stream, that winds Through Heaven’s unfading bowers, And on its banks the beauteous maid, Who culled celestial flowers.
Him next the sweet Vauclusian swan, Love’s Laureate, appears, Who bathed his mistress’ widowed urn With Heliconian tears;
Certaldo’s storied sage,—a bard, Though round his genius rare The golden manacles of verse He did not choose to wear.
Those rosy morns, that usher in Each festal-gladdened day, His prose depicts in hues as bright
As could the poet’s lay
His ultramontane brother, born In Albion’s shady isle; Dan Chaucer, of his tameless race Apollo’s eldest child;
The Medecean banqueter, Whose Fescennines unfold The deeds of heathen Anakim Restored to Peter’s fold;
Ferrara’s Melesigenes, Who o’er a wide domain Of haunted forests, mounts, and seas, Exerts his magic reign;
A glowing Mœnad, with her locks Dishevelled in the wind, His fancy wantons far and near, From Thule unto Ind;
Now from her griffin steed alights Alcina’s palace near, Now in the Patmian prophet’s car Ascends the lunar sphere;
Or with Rinaldo wanders through The Caledonian wood, Amid whose shades and coverts green Heroic trophies glowed;
Or paints the mighty Paladin Transformed to monster gross, Whose mistress drank in Ardennes lone The lymph of Anterōs.
Next hapless Tasso, pale and wan, Released from dungeon grates; The sacred legions of the cross
His genius celebrates;
Armida’s mountain paradise
Amid the western seas; Her dragon-yoke, whose nimble hooves Could run upon the breeze.
The sombre forest, where encamped Dark Eblis’ minions lay, With shapes evoked from Orcus’ gloom To fright his foes away.
Lo, marble pontifices spring, To arch illusive streams, And swans and nightingales rehearse Their moist melodious threnes!
The centuried trees are cloven wide, And forth from every plant A maiden steps, whose tears might melt A heart of adamant.
A sudden darkness veils the sky, And fortresses of fire, With ruddy towers of pillared flame, Above the woods aspire.
Transfigured in the morning beam. On Zion’s holy height
Rinaldo puts the dusky swarms Of Erebus to flight.
Nor absent from the shining throng That dainty bard, I ween, Who hung the maiden empress throne With garlands ever green.
The Elfin Court’s Demodocus,