Buy ebook Recent advances in understanding and design of efficient hydrogen evolution electrocatalys
Recent advances in understanding and design of efficient hydrogen evolution electrocatalysts for water splitting: A comprehensive review Bashir Adegbemiga Yusuf
Recent advances in understanding and design of efficient hydrogen evolution electrocatalysts for water splitting: A comprehensive review
Bashir Adegbemiga Yusuf a, b , Waleed Yaseen a , Meng Xie a, * , Rabi Sanusi Zayyan e ,
Atika Ibrahim Muhammad c , Rosalynn Nankya d , Jimin Xie a , Yuanguo Xu a, b, *
a School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China
b School of Materials Science & Engineering, Jiangsu University, Zhenjiang 212013, China
c The University of Manchester, UK
d Rice University, Houston, Texas 77005, USA
e Department of Basic and Applied Sciences, College of Science and Technology, Hassan Usman, Katsina Polytechnic. Katsina state, Nigeria
ARTICLE INFO
Keywords:
Intrinsic effect
HER
Water-splitting reaction
Extrinsic effect
Catalyst fabrication
Electrocatalysis
ABSTRACT
An unsustainable reliance on fossil fuels is the primary cause of the vast majority of greenhouse gas emissions, which in turn lead to climate change. Green hydrogen (H2), which may be generated by electrolyzing water with renewable power sources, is a possible substitute for fossil fuels. On the other hand, the increasing intricacy of hydrogen evolution electrocatalysts that are presently being explored makes it more challenging to integrate catalytic theories, catalytic fabrication procedures, and characterization techniques. This review will initially present the thermodynamics, kinetics, and associated electrical and structural characteristics for HER electrocatalysts before highlighting design approaches for the electrocatalysts. Secondly, an in-depth discussion regarding the rational design, synthesis, mechanistic insight, and performance improvement of electrocatalysts is centered on both the intrinsic and extrinsic influences. Thirdly, the most recent technological advances in electrocatalytic water-splitting approaches are described. Finally, the difficulties and possibilities associated with generating extremely effective HER electrocatalysts for water-splitting applications are discussed.
1. Introduction
The agreement for the sustainable advancement of society is to change the energy system and improve the amount of renewable energy in the overall structure due to the deepening of the conventional energy crisis and the rising prominence of environmental issues. The generation and use of hydrogen energy from renewable sources has emerged as one of the most important future directions for the global development goal [1–4]. H2 energy is considered the energy source of the future due to its exceptional characteristics of zero pollution, great effectiveness, numerous supplies, and a vast array of applications. In particular, the generation of hydrogen through water electrolysis using renewable power is recognized as one of the most promising approaches due to its extremely low pollutant output, zero carbon dioxide emissions, and environmental friendliness [5–8].
The hydrogen evolution reaction (HER), which is one of the halfreactions in the process of electrocatalytic water splitting, has gotten a lot of attention in the past few decades, leading to the discovery and
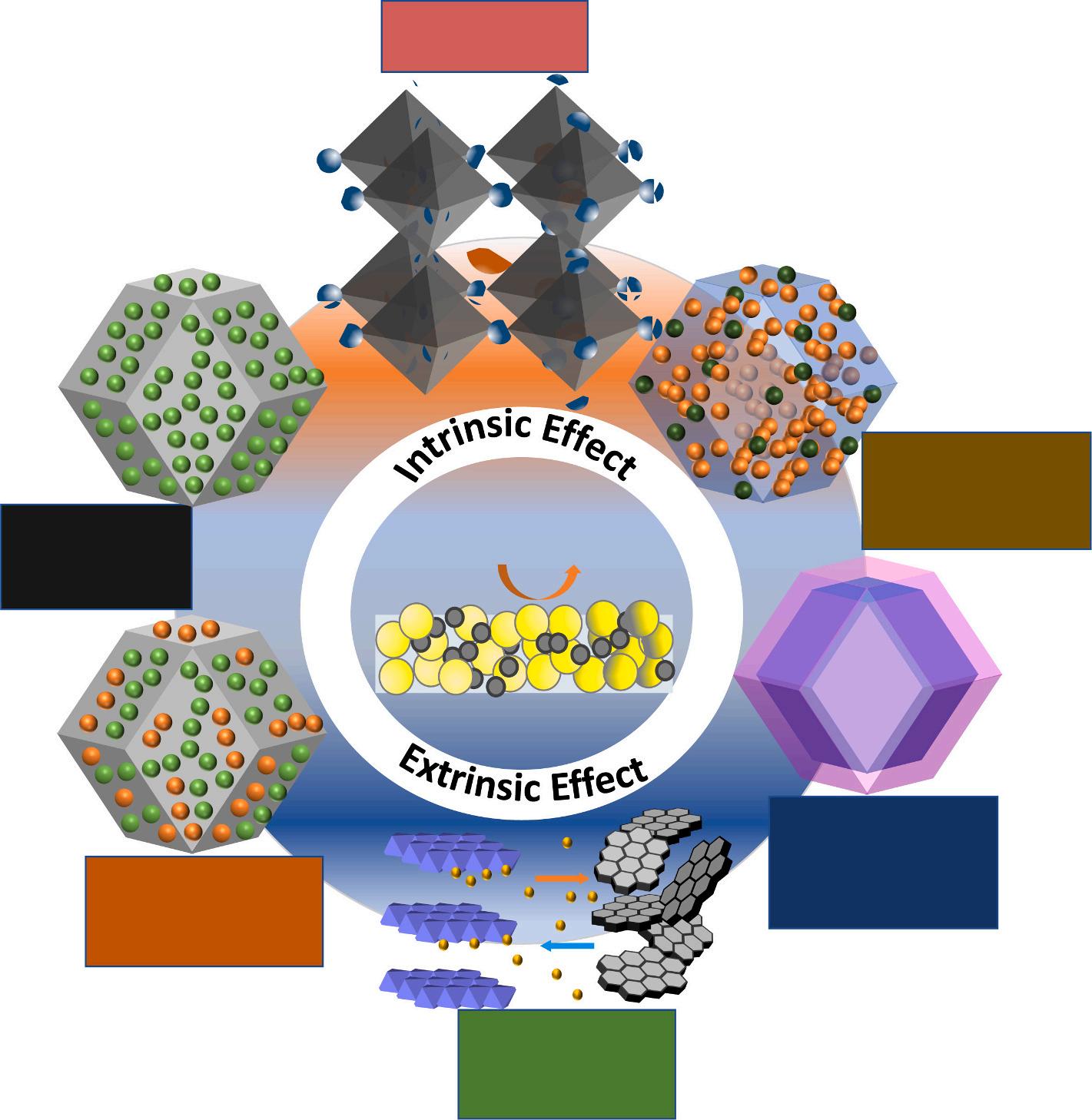
design of many active catalysts [9–19]. As a result, it is not hard to find out through detailed research that catalysts with great performance tend to have high intrinsic performance, a large surface area, and rapid electron transfer. This means that improving these characteristics should be a primary target during the design and preparation of the catalyst. Based on the intrinsic impacts and extrinsic aids of the catalysts themselves, the fabrication approaches of catalysts can be tailored to the aforementioned characteristics (Fig. 1). The implementation of the fabrication approach of intrinsic or extrinsic impacts does not often influence the structural or electrical performance of the catalytic materials individually, but rather enhances both concurrently. For instance, the nano-structuring impact and narrow size distribution have been shown to be effective for increasing both the interfacial area (due to the structural impact) and the number of catalytically active sites that are inherently exposed (electronic impact), leading to an increase in electrocatalytic effectiveness [20].
To further maximize the inherent overall catalytic efficiency, catalytic materials with all essential components cohesively bonded were
* Corresponding authors at: School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China.
developed in accordance with the concepts of catalyst configuration. These components, which optimized the electronic characteristics of the catalyst, included phase modification, surface chemistry, and heteroatomic infusion defects. Even though there are various exceptional studies pertinent to HER [21–31], there is not yet a thorough review that takes into account all of the internal and external effects, such as alloyimpact, strain-impact, and support-effect, for HER. Herein, this article provides a summary of current breakthroughs in the intrinsic impact and the extrinsic influence of enhanced electrocatalytic materials toward HER. These electrocatalytic materials include those centered on noblemetal and transition-metal nanomaterials. First, we begin with a concise discussion of the reaction mechanism of HER, followed by a discussion of both conventional and cutting-edge descriptors for hydrogen evolution reactions. The subsequent step involves an in-depth discussion of the rational design, fabrication, mechanistic insight, and continuous improvement of electrocatalytic materials. In the following section, current developments in the technological advancements of proton exchange membranes and anion exchange membrane fuel cell systems are further presented in a concise manner. Finally, the difficulties and prospects of the hydrogen evolution reaction are discussed. We anticipate that this review will provide useful guidance and new perspectives on the development and application of improved electrocatalytic materials for water-splitting approaches.
2. Fundamental mechanistic concepts of hydrogen evolution reactions
2.1. Catalytic pathways for hydrogen evolution reactions
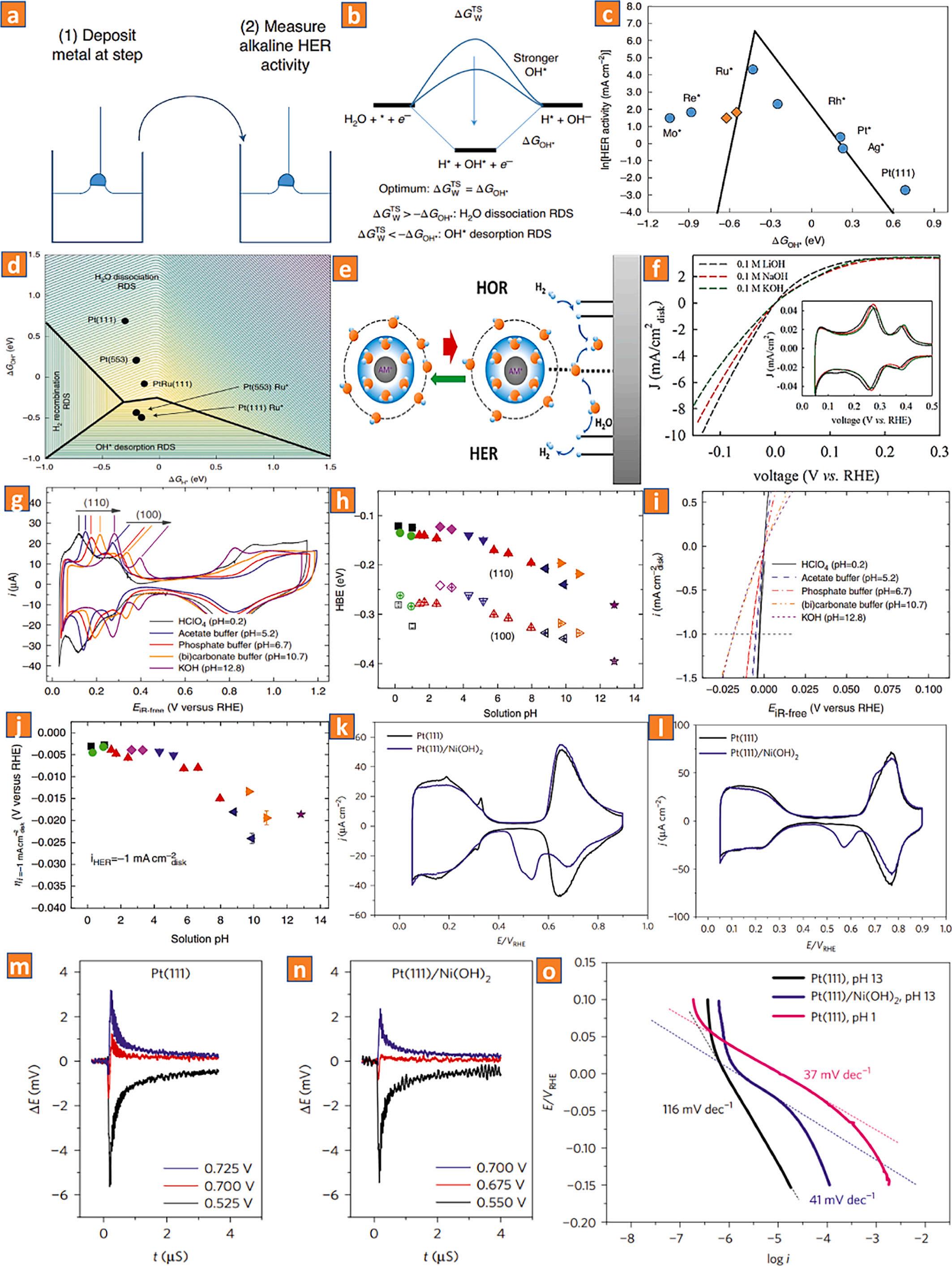
The generation of H2 through the HER process in an alkaline solution and the invention of an alternative to renewable fuels for diverse energy technologies are presently the subjects of intense research. Owing to an additional phase of electrocatalytic water dissociation, the reaction rate of this procedure is sluggish. Therefore, current catalysts operate well in an acidic condition but underperform in an alkaline solution. According to theoretical investigations, two parameters influence the effectiveness of the catalysts in an alkaline solution. These parameters are the disintegration of H2O and the binding energy of H2. As a result, any catalysts with a higher potential to split H2 molecules with a high adsorption potential and generate a H2 molecule will improve the HER catalytic reaction in a basic environment [32]. In regards to the reaction effectiveness, very few electrocatalytic materials competent with platinum in an alkaline medium have been described. Hence, it is necessary to verify the fundamentals of electrode kinetic parameters and to investigate the response process. This will lay the groundwork for future researchers to investigate novel and effective electrocatalysts [33]. Various mechanisms govern the catalytic processes, as indicated in Figs. 2 and 3. In an acidic solution, the reaction follows the Volmer mechanism, which involves the combination of an e from the electrode interface with a H+ from the electrolyte. The Tafel mechanism is another combination of the present H-atom with the nearby one. The Heyrovsky mechanism refers to the combination of another H+ from the electrolyte and an e from the interface of the electrode. In an alkaline
Fig. 1. Diverse impacts on the hydrogen evolution reaction catalysts.
Electrode deposited catalyst surface area
Electrode deposited catalyst surface area
representa on
e+eOHH2O+ + Volmer step
environment, however, H+ are missing from electrolytes. The reaction therefore begins with the breakdown of a H2O molecule, a process known as the Volmer mechanism. Subsequently, either the Tafel or Heyrovsky mechanism is utilized to produce H2 The excessive H2O dissociation phase in the basic electrolyte suggests that the same catalytic material performs less efficiently in an alkaline environment than in an acidic environment [34]. Furthermore, the approach can describe the HER process on the interface of the catalyst-deposited electrode. The HER in the alkaline solution is considered a combination of three basic steps: one chemical and two electrochemical. The first phase consists of a chemical reduction of H2O to produce an H2 molecule, which is subsequently deposited on the interface of the electrode by the Volmer mechanism. The deposited H2 then undergoes an electrochemical phase to generate hydrogen, followed by the Heyrovsky process or a chemical process, i.e., the Tafel response [35].
Additionally, the Tafel slope measurements illustrate the HER process by defining the potential difference (PD) needed to change the current density. When the Volmer or discharging response is rapid and the chemical dissociation is the rate-determining phase, the b = 29 mV/ dec is determined by the formula b = 2.3RT/2F = 0.029 V/dec @ 25 ◦ C. Therefore, if the electrochemical dissociation, or Heyrovsky process, is the rate-determining phase and the discharging response is quick, the b = 39 mV/dec and determined by b = 2.3RT/2F = 0.039 V/dec @ 25 ◦ C. Lastly, assuming the discharging response is slow, b = 116 mV/dec can be calculated as b = 2.3RT/2F = 0.116 V/dec @ 25 ◦ C.
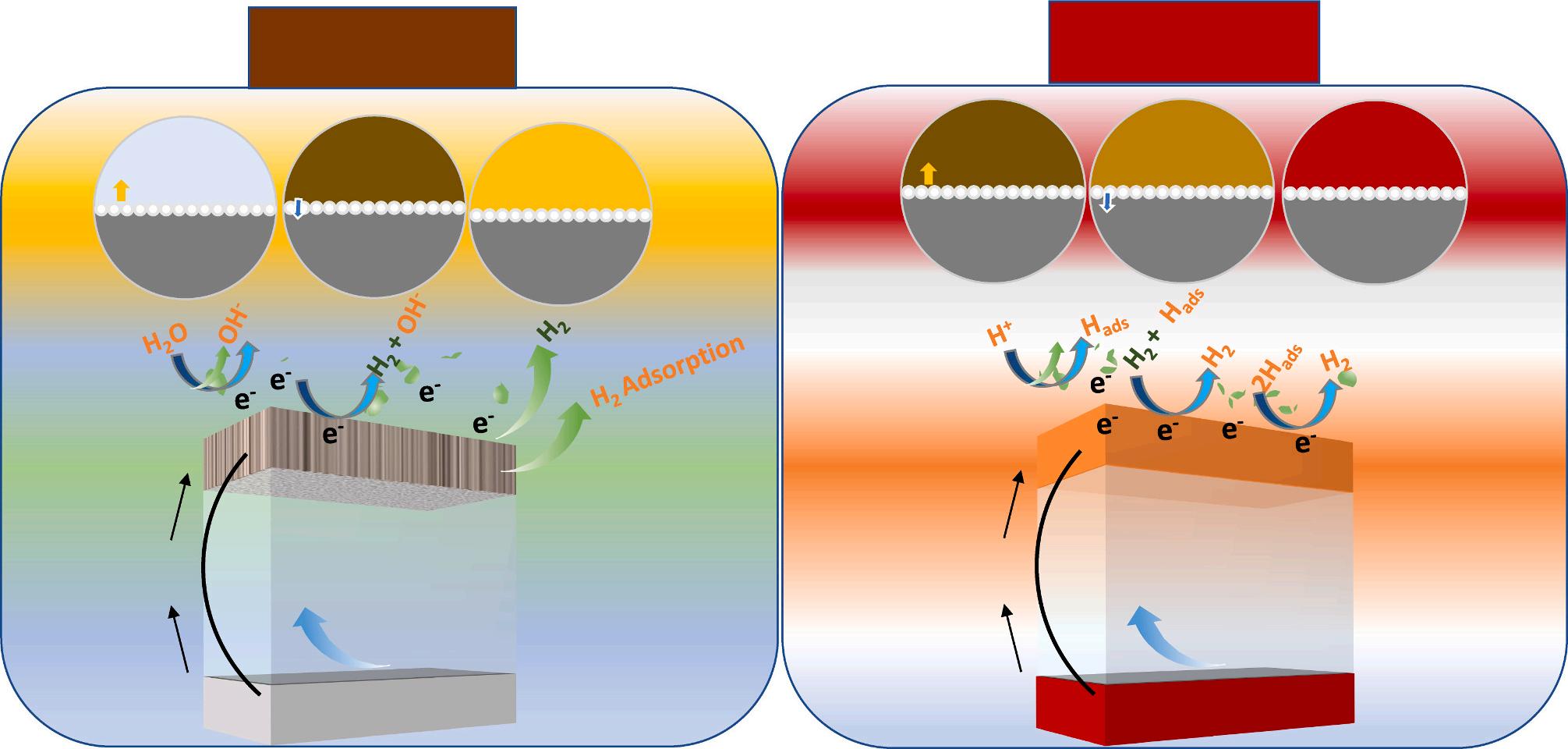
The actual reaction process is considerably more complex than these phases suggest. Current perspectives, such as the water-splitting hypothesis [36,37], the H2 binding energy theory [38,39], and the surface H2O and/or anion transport principle [40], have typically addressed the crucial themes in the system of alkaline hydrogen evolution reaction, as to whether water dissociation or H2-adsorption is the key component to accelerate the reaction mechanism of alkaline hydrogen evolution. According to water-splitting theory, the function of the hydroxide (OH) is one of the most perplexing questions in the alkaline hydrogen evolution process. Current findings have shown that OH* deposition neither contributes to nor impacts the Volmer stage of the alkaline hydrogen evolution process nor its effectiveness [41,42]. Koper and coworker described a bi-functional pathway using OHads in alkaline HER processes [43]. They demonstrate a volcano-like link between the rate of alkaline H2 development and the OH binding potential. The authors also discover that platinum with ruthenium at the phase’s border is 65-times more effective for HER than uncoated platinum. Models of electrochemical H2O dissolution demonstrate that the activation energy corresponds with the OHads strength, even when the deposited OH is not an outcome precursor, resulting in a computed volcano slope that corresponds to the experimental plot (Fig. 4 a-d). Similarly, Jia and coworkers show that OHads (water)-alkali M+ (where M+ represents metal cation) derivates perform catalytic roles in the alkaline hydrogen evolution process and the hydrogen oxidation approach reaction mechanism [44]. This is supported by the observation that increasing the abundance of OHads at the surface benefits the HER/HOR, whereas increasing the intensity of alkali M+ only improves the hydrogen evolution process, and altering the specificity of alkali M+ influences both the hydrogen evolution process and the hydrogen oxidation approach, respectively. According to the hard-soft acid-base principle, the formation of OHads (water)-alkali M+ in the double-layer region facilitates the withdrawal of OHads into the mass and the generation of OH (water)-alkali M+ , which then promotes the HER (Fig. 4e, f).
Despite current debate about the possible role of precipitated hydroxide in the hydrogen evolution process in alkaline environments, hydrogen binding energy theory has been demonstrated to be the most accurate description of hydrogen evolution reaction performance. Since the reaction pathways for HER in basic and acidic solutions are similar, some researchers think that the factors that control HER performance are the same in all pH media and mostly depend on how hydrogen and
e- Adsorp on
Volmer Reac on e- Desorp on
Heyrovsky Reac on e- Recombina on
Tafel Reac on
e- Adsorp on
Volmer Reac on e- Desorp on
Heyrovsky Reac on e- Recombina on
Tafel Reac on
Fig. 2. Schematic representations of the Volmer-Heyrovsky and Volmer-Tafel reactions taking place on the surface of a catalyst in both acidic and basic environments.
Fig. 3. Overall schematic illustrations of the Volmer-Heyrovsky and VolmerTafel reactions. B.A.
Fig. 4. (a) A schematic diagram depicting the process of measuring and decorating HER performance. (b) The reaction energy chart illustrates the reaction mechanism of the HER reaction. (c) Theoretical (black line) and experimental (blue dots) HER process kinetics on Pt (553), Pt (553 with Mo*, Re*, Ru*, Rh*, and Ag* deposited at phase, and Pt (111) vs the DFT predicted OHads energy at 0 Vs. RHE. (d) A chart illustrating a 3D model of the HER active volcano. Reproduced with permission [43]. Copyright 2017, Nature Publishing Group. (e) Schematic depiction of the catalytic functions of OHads in the reaction mechanism of alkaline HER/ HOR. (f) HER/HOR polarization curves of a polycrystalline platinum electrode in 0.1 M solutions of sodium hydroxide, potassium hydroxide, and lithium hydroxide that are saturated with H2 Reproduced with permission [44]. Copyright 2019, American Chemical Society. (g) A comparative study of the Hupd characteristic peaks of polycrystalline platinum in electrolytes of varying pH. (h) pH of the solution in relation to HBE for both Pt (110) and Pt (100). (i) HER on Pt across the entire pH spectrum of solutions. (j) The overpotential of the HER of Pt in all electrolytes with a pH buffer. Reproduced with permission. ref Copyright 2015, Nature Publishing Group. (k) Cyclic voltammetry was employed to measure the interaction of Pt(111) with a buffered solution with pH=10 adjusted by a submonolayer quantity of Ni (OH)2 (l) Measurements of cyclic voltammetry and laser-induced temperature jumps at pH=13. (m) Coulostatic potential transients created by laser for the Pt(111) electrode. (n) Laser-induced coulostatic potential transients acquired for the Ni(OH)2-coated Pt(111) electrode. (o) H2 evolution and reaction mechanism on Pt(111) in the presence of Ni(OH)2 Reproduced with permission [47]. Copyright 2017, Nature Publishing Group.
the targeted catalyst interact [45,46]. Yan and colleagues discovered a link between HOR/HER activities and empirically observed HBE for polycrystalline Pt tested in many buffer solutions over a broad pH scale of 0–13 [38]. The HOR/HER performance measured by the rotating disc electrode technique decreases with increasing pH, whereas the HBE measured by cyclic voltammograms grows linearly with increasing pH. Attributing the HOR/HER processes to the HBE yields a monotonically decreasing HOR/HER interaction with the HBE, providing excellent evidence for the theory that HBE is the only response descriptor for the HOR/HER operation (Fig. 4g-j). This indicates that the HBE dependency hypothesis is not always applicable to all situations. In the meantime, a significant amount of work has been put into the development of useful compounds (poly-crystalline and stepped single-crystal platinum). It is quite unlikely that the Hupd spectra are indirectly related to the *OH, and it is important to note that the influence of oxygenated molecules adsorbed must also be properly considered. To put it differently, the deposited OH may interact with the reactive substrate at the same interface locations and/or enhance its uptake capacity, which will ultimately have an effect on the reaction mechanisms of the HER. In addition, the electrochemical surface architecture at the atomic level must be considered (for instance, the potential of zero total charge). Koper and colleagues reported direct experimental proof of the adsorption of a modest quantity of nickel (II) hydroxide on platinum (111) [47]. They hypothesized that the influence of the potential of zero total charge on the activation threshold of Hads is the energy loss caused by the rearrangement of interfacial water in order to support electron transport via the electrocatalytic double-layer support. Specifically, HER and the Hads region occur in an alkaline environment away from the potential of zero total charge. In an alkaline condition, the interfacial H2O network at the HER and the Hads region potentials interacts significantly with the good interfacial electromagnetic field, resulting in a more complex and rigorous rearrangement during the flow of charges via the electrocatalytic double-layer support (Fig. 4k-o).
2.2. The thermodynamics and reaction kinetics of the hydrogen evolution reaction
2.2.1. The thermodynamics of the HER process
The hydrogen evolution process can proceed in two possible ways: through a Volmer-Tafel or a Volmer-Heyrovsky pathway. Hads and hydrogen synthesis are thus two sequential steps in the conversion of hydrogen ions to hydrogen molecules. The complexity of the targeted catalyst for starting the process is often assessed using two equivalent theoretical descriptors, Hads energy and the Gibbs free energy for adsorbing H-atoms (ΔGH*). The Nernst effect is an additional parameter that contributes to the HER mechanism. This potential represents the likelihood of thermodynamic equilibrium occurring throughout the electrochemical process. HER activation under equilibrium conditions occurs infrequently in real-world contexts. This indicates that the vast majority of electrochemical processes must surpass a preset activation energy barrier. The magnitude of the energy barrier that must be overcome is significantly affected by the nature of the contact at which the reaction occurs. Therefore, electrochemical reactions often demand
a larger quantity of energy than thermodynamics predicts.
2.2.2. The kinetics of the HER process
The overpotential (η): is the most important criterion for evaluating the HER performance of an electrocatalyst. A catalyst that is suitable for the hydrogen evolution process must be able to reduce the overpotential, as a minimal overpotential value is the cause of strong catalytic performance. The theoretic estimate of the cell potential for the total water splitting pathway is 1.23 V (0 V for HER). Due to impediments, the HER requires additional capacity to generate responses. Extra potential, also known as overpotential, is a critical component in estimating the catalyst’s effectiveness. Typically, the overpotential values of various electrocatalysts are correlated at a constant current density level, such as 100 mA/cm2 and/or 10 mA/cm2 The ideal HER electrocatalyst must be able to catalyze the electrochemical process at a potential of 100 mV or lower.
The Tafel slope (b) and exchange current density (j0): Re-plotting the polarisation data into Tafel slope plotlines (overpotential" versus "lg| current density|) allows the deduction of two key variables from the Tafel slope system (η = a + b lg|j|), namely the Tafel slope (b) and the exchange current density (j0). Both of these variables may be found in the Tafel calculations.
The Tafel formula depicts the direct proportionality involving overpotential and lg|j|, where g stands for overpotential, a denotes for the Tafel constant, b stands for the Tafel slope, and j stands for current density. It should be noted that a linear correlation is developed when the overpotential is greater than or equal to 59 mV. Typically, the Tafel slope is designed to evaluate catalytic performance and potential hydrogen evolution process pathways. When the gradient of the Tafel curve is less steep, optimizing the current density needs a considerable low overpotential, indicating a rapid charge transport reaction mechanism.
When g is considered to be 0, the exchange current density, denoted by j0, is calculated as the result. This result is dependent on the inherent catalytic properties of the electrode material itself when parameters are in steady state. If the exchange current density is significantly high, then the electron transport rate will be higher, and the reaction efficiency will be even better. In summary, the ideal characteristics of an effective electrocatalytic material are a small overpotential (η), a reduced Tafel slope (b), and a significant exchange current density (j0).
Electrochemically active surface area (ECSA): In order to evaluate the effectiveness of various nanosized electrocatalysts, it is important to calculate the electrochemically active surface area (ECSA). The equivalent cross-sectional region of a nanosized electrode is typically more than the geometrical region of the equivalent planar electrode. The ECSA of the hydrogen evolution reaction electrocatalytic materials can typically be evaluated utilizing two distinct methodologies: the Coulombic charge of a predominant surface Faradaic reaction including H2 underpotential accumulation, underpotential accumulation of copper, carbo monoxide stripping, and oxidation/reduction of interface metal; and the electrical double-layer capacitance. Both methodologies are described in more detail below (Cdl). Furthermore, the approach based on the evaluation of Cdl is relatively often employed to obtain the
B.A. Yusuf et al.
ECSAs of all various types of HER electrocatalytic materials because almost all electrocatalytic materials exhibit the characteristics of Cdl when subjected to cyclic voltammetry scans.
Cdl is calculated in the same manner as catalytic evaluation, by spinning the electrodes in non-Faradaic regions (i.e., at the potential areas where no charge transport response happens but adsorptiondesorption mechanisms can exist). The variation in non-Faradaic current density (j) while cycling at different sweep rates (m) should be directly proportional to the scan rate, resulting in the electrical doublelayer capacitances per the slopes (Cdl = j/m).
Faradaic efficiency (FE): is an additional criterion for assessing the performance of an electrocatalytic material. This is modified so that an assessment can be made on the number of electrons involved in the intended reaction rather than the reaction product. Faradaic efficiency is the ratio of empirically detected hydrogen to hypothetical hydrogen calculated from current density at 100 percent faradaic yield. This is the ratio between the empirically produced quantity of hydrogen and the predicted quantity of hydrogen in the HER system. The quantity of generated hydrogen can be estimated using gas chromatography (GC) or the H2O displacement approach. The conceptual modelling of hydrogen is performed under the conditions of potentiostatic or galvanostatic electrolytic set-ups, and comparing it to an experimental result allows the calculation of the faradaic efficiency. In the hydrogen evolution process, 100 percent faradaic efficiency is typically recorded, and a higher faradaic efficiency indicates increased HER mechanism sensitivity in alkaline conditions.
The turnover frequency (TOF): is a metric that describes the quantity of reactants that are transformed into the specific product at each catalytic domain in a given amount of time by the catalytic material. At the moment, the system which is mostly aimed at the HER electrocatalytic TOF values is primarily based on TOF = jA/4nF, where A denotes the region of the working electrode and n illustrates the electroactive material number of moles, which is aimed from the interface area of the electrocatalyst. However, it is not possible to find precise TOF values for the majority of the solid-state catalytic materials, particularly for the few developing complexes. The explanation for this is that not all of the atoms on the catalytic material’s interface are equally accessible or catalytically activated at the same time. In addition, bubbles are generated by the hydrogen evolution process on the interface of the electrode, which results in a higher overpotential due to the degradation of the electronic conductivity. Even if the TOF values are inaccurate, they can still be used when attempting to compare the catalytic performance of multiple catalysts, particularly in the context of identical systems [48].
Hydrogen bonding energy (HBE): The optimum electrocatalytic material for HER is one that demonstrates an HBE that is neither too strong nor underly weak. The hydrogen evolution process demonstrates that the initial protons diffuse and then are attached to the interface of the electrocatalytic material. Ultimately, the proton is converted to hydrogen. Instead, the low HBE value suggests that there is a relatively low concentration of deposited protons on the electrocatalytic material. When the HBE concentration is high, the catalysts deteriorate into a poisonous state due to the constant coverage of the catalytically active sites by deposited hydrogen ions. Since the value of the SHE is assumed to be 0, the magnitude of ΔGH* must also be assumed to be zero for an ideal HER electrocatalytic material. When attempting to demonstrate the effectiveness of HER, the current density is another highly significant factor to consider. Therefore, electrocatalytic materials that are somewhat close to the peak of the Sabatier volcano curves are the ones that demonstrate enhanced performance for the hydrogen evolution process [49].
The relationship between the electrochemical active surface area and the kinetics of the HER process: The relationship between the electrochemical active surface area and the kinetics of the HER process, namely two intermediates (*H and *OH), determines the HER catalytic performance in alkaline medium [50–52]. In contrast to the complex multi-
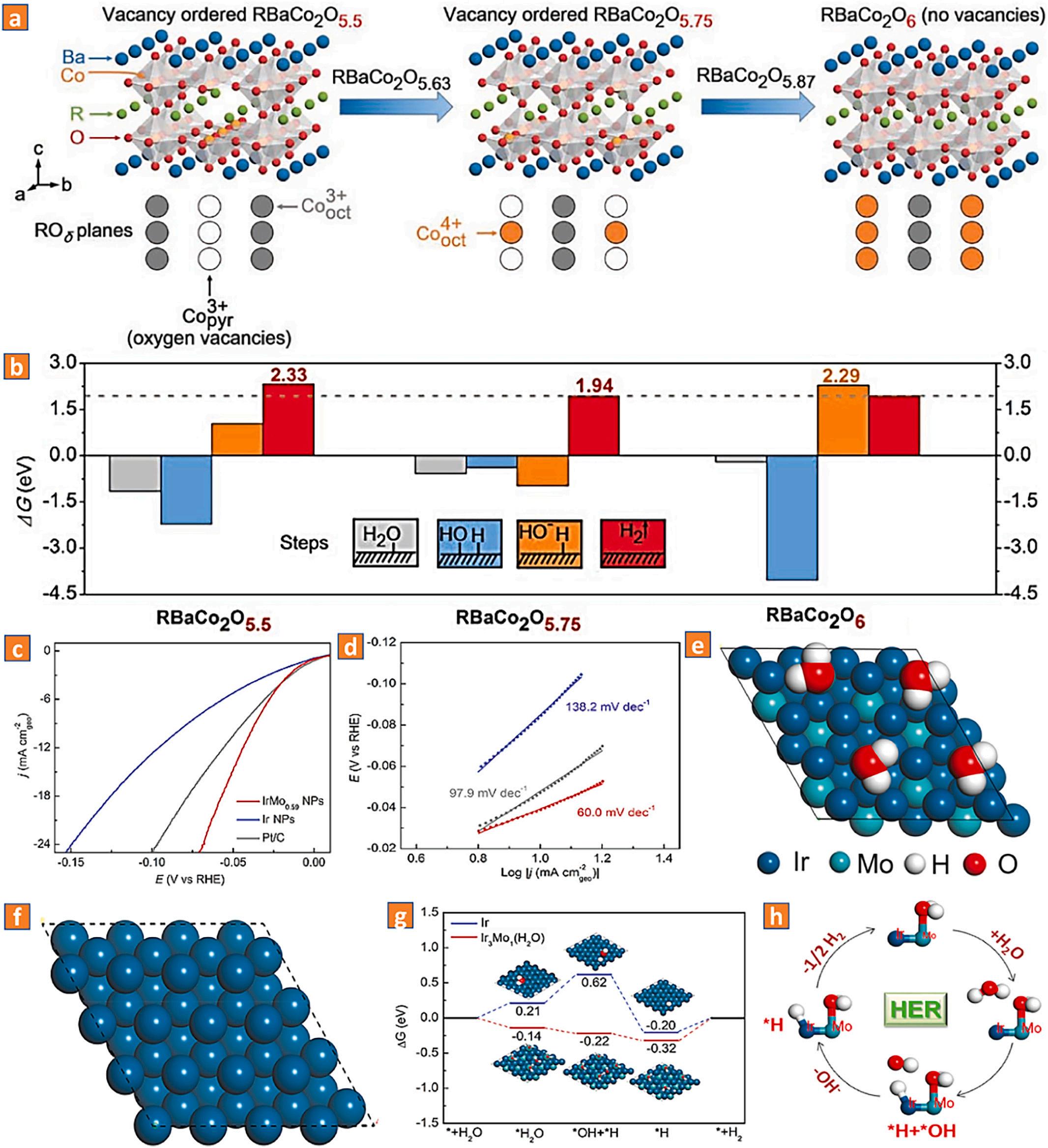
step processes of the oxygen evolution reaction, HER performance may therefore be simply modified [51,53]. In basic medium, cobaltbased oxides are excellent materials for the oxygen evolution reaction, whereas pure TM-oxides are ineffective for the HER owing to their unfavorable H2 adsorption/desorption energy values [52,54,55]. Owing to the generation of O 2p ligand vacancies from the robust bonding between the three-dimensional transition metals and O 2p states, highvalence TM-ions (such as Fe4+ , Cu3+ , Co4+, and Ni3+) exhibit significant metallic characteristics, suggesting their possible participation in hydrogen desorption [54]. Moreover, oxygen vacancies in oxides enhance the adsorption of H2O [56]. Motivated by these ideals, Shao and colleagues established two important criteria for fabricating excellent HER electrocatalytic materials based on bulk TM-oxides: (1) exceptional electrochemical active surface area with district catalytic active centers for the two-step HER process and (2) short reaction routes [54]. To demonstrate the fabrication approach, the study utilized A-siteordered perovskite oxides RBaCo2O5.5+δ (R = gadolinium, samarium, and lanthanum). By precisely manipulating the amount and dispersion of oxygen vacancies at pyramidal Co3+ sites and octahedral Co4+ sites, a near-ideal reaction route and relative-optimal synergistic interactions could be established for the HER process (Fig. 5a, b). Significantly, both catalytic active sites were incorporated concurrently into the structured perovskite oxide lattice. Thus, the optimum sample (Gd0.5La0.5) BaCo2O5.75 significantly surpassed the standard platinum/C catalyst. Chen and colleagues also investigated double perovskite oxides as HER catalytic materials in basic environments. The authors discovered that the orthorhombic double perovskite PrBaCo2O5+δ (δ ≈ 0.52) demonstrated superior HER activity compared to the cubic Pr0.5Ba0.5CoO3 and tetragonal PrBaCo2O5+δ (δ ≈ 0.76). The significant level of structural misfit, increased electrochemically active surface area, lattice-oxygen species, and reduced charge-transfer resistance all contribute to the robust performance. Additionally, the occurrence of the redox pair Co3+/Co4+ is favorable for the HER kinetic process [57].
Remarkably, 4d and 5d TMs with greater valence can also serve as HER catalytic active sites. For instance, Luo and colleagues, for instance, described the synthesis of IrMo nanomaterials as excellent bifunctional electrocatalysts for HER and hydrogen oxidation processes in alkaline conditions [58]. Notably, improved IrMo0.59 demonstrated the best catalytic performance, nearly 10 times that of conventional platinum/C and its equivalent iridium. Simulations based on DFT revealed that the initial phase of the HER reaction (H2O + e + * → OH + H* (the Volmer reaction, electrochemical adsorption, ≈120 mV dec 1)) preferred to take place at the molybdenum site of IrMo. H2O-adsorbed molybdenum may substantially improve the interface concentrations of *H2O and *OH molecules, with *OH species participating actively in the HER kinetic process. Furthermore, Gibbs free energy values were determined for the various reactive intermediates of the HER kinetic process of iridium and Ir3Mo(H2O), displaying that the H2O-adsorption/ dissociation procedures were more advantageous for IrMo(H2O) than for iridium, suggesting the synergistic impact of molybdenum(H2O) and iridium on the reported HER kinetic process (Fig. 5c-h).
2.3. The conventional hydrogen evolution reaction descriptors
2.3.1. The hydrogen adsorption Gibbs free energy (ΔGH*)
The ΔGH* is a concise expression for bonding strength that is frequently employed in the determination of the hydrogen evolution performance of the electrocatalytic reactive sites [59–62]. In accordance with the Sabatier hypothesis [63], for the reaction pathways to be able to be induced and the byproducts to be easily removed, the electrocatalytic reactive sites must maintain a bonding strength that is adequate for the essential intermediates [64]. This bond must neither be too weak nor too robust. Platinum is acknowledged for demonstrating the superior efficacy of HER and its ability to achieve higher response rates with low overpotential (η). Platinum is placed quite close to the volcano chart’s peak and exhibits an almost thermally independent
B.A.
Fig. 5. (a) Crystal structure of the oxygen vacancies and A-site ordered double perovskites. (b) Calculated free energies at the various steps of the alkaline HER process on RBaCo2O5.5+δ (δ = 0, 0.25, and 0.5). Reproduced with permission [54]. Copyright 2019, Wiley-VCH. (c) HER polarization curves. (d) The Tafel of IrMo0.59 NPs, Ir NPs, and Pt/C catalysts in Ar-saturated 0.1 M KOH with a scan rate of 10 mV s 1 at the rotating speed of 1600 rpm. (e, f) The geometric configurations of Ir3Mo1(H2O) (a) and Ir (b) (111) surface. (g) Calculated free energy diagram of the series of reaction intermediates of the HER process of Ir and Ir3Mo1(H2O). (h) Schematic diagram showing the HER mechanism of Ir3Mo1(H2O). Reproduced with permission [58]. Copyright 2020, American Chemical Society.
ΔGH* Consequently, platinum is commonly utilized as a standard. If the catalyst is positioned to the left of platinum (ΔGH* less than 0), the initial Volmer phase is straightforward, but H-atom desorption is challenging. This hampers the subsequent Heyrovsky or Tafel processes, which ultimately causes the electrocatalyst contact to be poisonous. In contrast,
Roger Parson reported the relationship that exists between the exchange current for electrochemical HER and the capability of the electrode to bind H-atoms [65]. The author observed that when the catalyst is positioned on the right side of the platinum (ΔGH* greater than 0), the hydrogen molecule binding ability is quite poor. Therefore, a greater
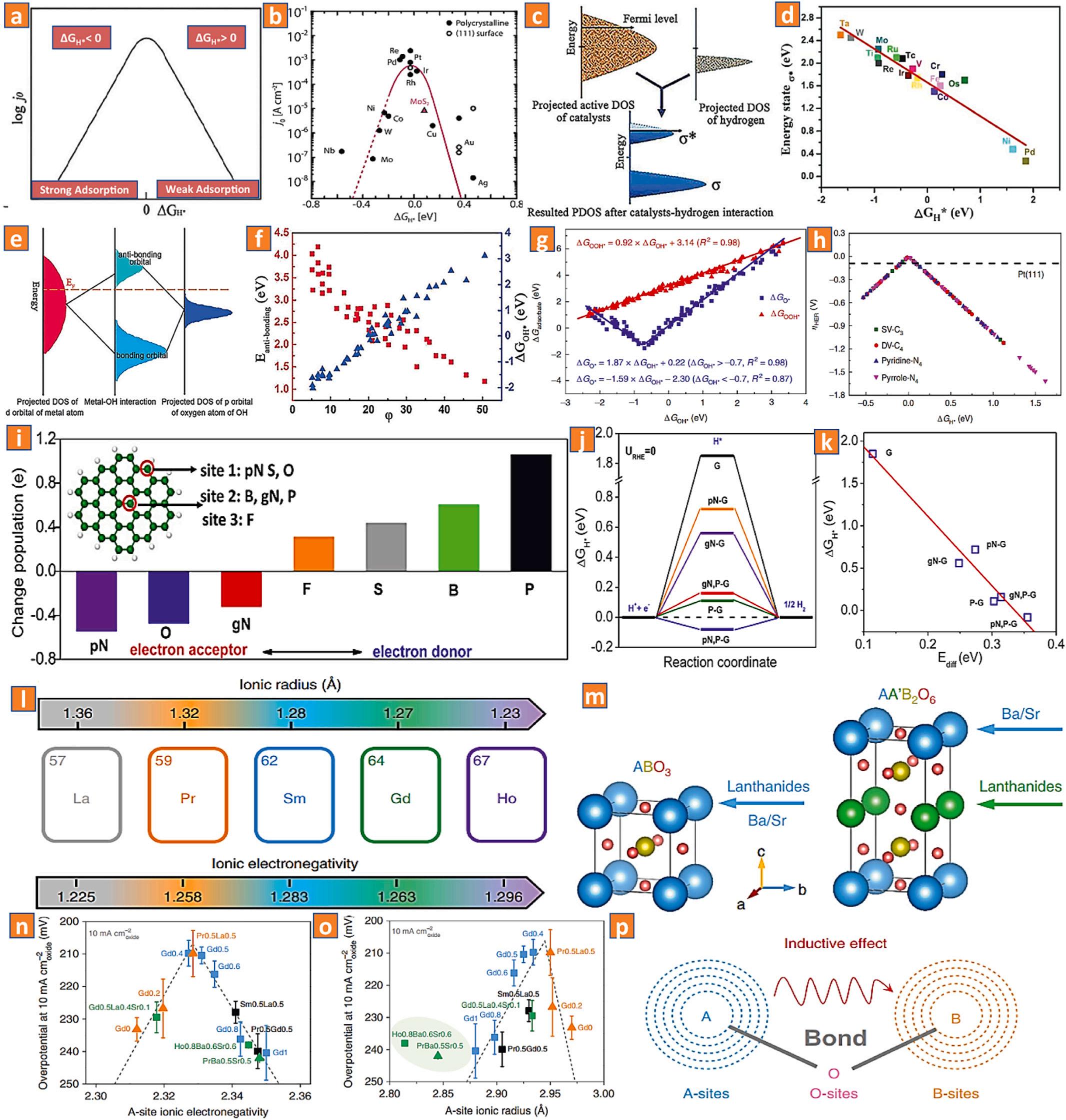
amount of energy is needed to activate the Volmer phase, which decreases the overall efficiency of the catalytic process (Fig. 6a). The volcano plot can reveal which kinds of catalytic materials are most effective for a particular category of catalysts. Nevertheless, as shown in (Fig. 6b), despite the fact that molybdenum disulfide reaches a value close to the ideal ΔGH* the exchange current density of molybdenum disulfide is quite low. This is because the volcano plot is totally dependent on a thermodynamic descriptor, whereas the precise reaction process is quantitatively dictated by other kinetic characteristics. The active volcano plot does not shift to the left-side or right-side but rather upward and downward, indicating that the descriptor can still identify the bonding characteristics of the optimal HER catalytic material despite the variation of phases [66].
2.3.2. The d-band center theory
The composition of the metal species is the primary factor that determines the inherent performance of a specific catalyst. As a result of this, metal-based descriptors have been established to evaluate the interaction between the features of the active site and the catalytic performance [67,68]. Electronic interaction between catalysts and adsorption processes is the origin of the vast majority of catalytic efficiency, such as the activation of reactants and the adsorption of intermediates. Therefore, certain hypothetical characteristics of metal catalytic materials have been provided for the scalability relationship to the adsorption intensity of chemical components and precursors on the metal interface. These hypothetical parameters include the d-band center [69], filling (eg) [70], and valence-and conduction-band [71].
The d-band theory was utilized in order to provide an explanation for the genesis of electrocatalytic performance, as can be seen in (Fig. 6c). After catalyst-H bonding, the valence orbital d2 z of electrocatalysts can cleave into bonding and anti-bonding orbitals, denoted by the symbols (σ) and (σ*), respectively. The greater the d-band position of a metal nanomaterial, the greater the anti-bonding (σ*) orbital was filled by fewer electrons. This increased H-adsorption on the catalytic active interface, resulting in a lower H-adsorption free energy (ΔG*H). As a consequence of this, the energy of the anti-bonding state, also known as (E*σ), which was shown to be in a linear proportion to ΔG*H, emerged as an effective descriptor for the HER mechanism (Fig. 6d) [72]. Based on this scalability correlation, the optimum HER effectiveness (ΔG*H = 0) on the volcano plot was matched to the ideal d-band center of the metal ion pivot. This ensured that the H-adsorption was neither excessively strong nor excessively weak.
In a similar manner, as can be seen in (Fig. 6e, f), the descriptor that originated from the d-band concept was also extended to the interaction between the M-atom (where M is a metal) catalyst and the intermediary hydroxyl species, which served as the most important precursor. Because more electrons were able to occupy the (σ*) orbital as a function of catalyst-hydroxyl coupling, the amount of hydroxyl-adsorption was lowered when the energy intensity of the d-band center moved in the direction of a lower value [68]. Consequently, specific volcano plots connected with descriptors have been constructed in order to supply useful insights for the effective computational assessment and experimentation [73,74]. In addition, Zeng and colleagues have established a unified descriptor, φ, which can allow access to the catalytic properties of the hydrogen evolution process with the basic characteristics of active centers, namely the electronegativity of M-atoms, the amount and quantity of coordinating atoms of catalytic active sites, and the number of total reactive centers. For the classification and regulation of M ion centers, it is important that the optimal electronic configuration be found in the active ion and that it be close to or higher than the maximum level of the volcano (Fig. 6g, h) [68].
2.3.3. The valence band model
The valence band model is appropriate for non-metallic carbonbased material composites if the d-band center concept is directed towards M-atoms of catalytic active sites. Qiao and coworkers studied the
sources of the diverse Hads responses observed in doped-graphene nanomaterials [75]. As demonstrated in (Fig. 6i, j, k), the ΔGH* values of all tested models exhibit a consistent trend in relation to Evar (Evar, signifies the difference between the minimum valence orbital energy of (Cs) catalytic active sites and the maximum valence orbital intensity of the as-doped graphene aggregate). In addition, the author demonstrates how the valence band (v) of the activated atom interacts with the deposited H* to form the bonding (v-σ) and anti-bonding (v-σ) * phases that constitute the basis for this interaction. Lower catalytic active site valence orbitals may promote (v-σ) state filling, hence strengthening the bond between H* and catalytic active Cs and reducing ΔGH* This equates to the smallest number of ΔGH* and, thus, the maximum predicted hydrogen evolution reaction performance.
2.3.4. Ionic electronegativity
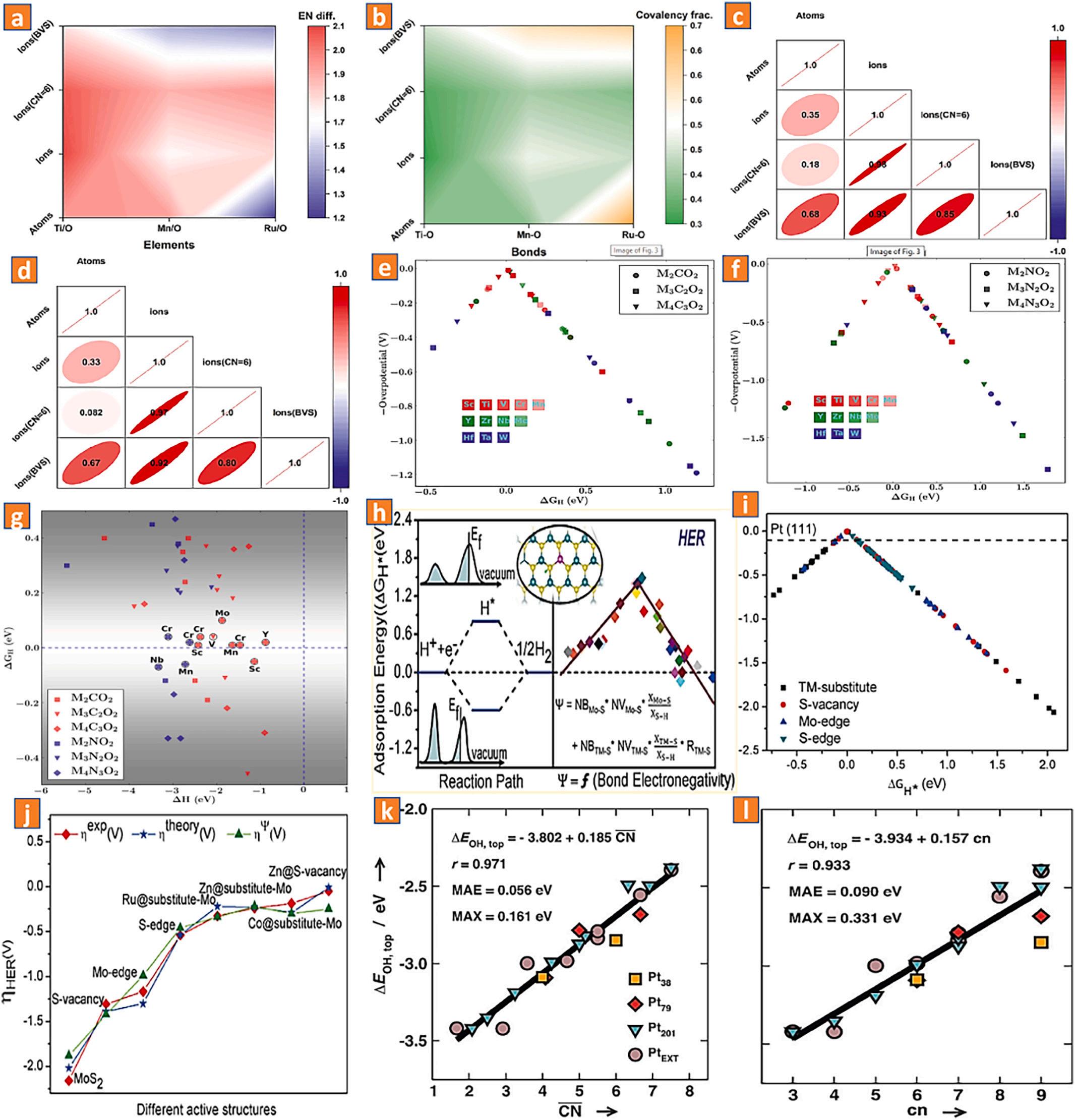
Pauling and co-workers were the first to introduce the idea of electronegativity (abbreviated as EN) [76], which is regarded as a chemical attribute that characterizes an atoms or functional chemical units’ propensity to draw electrons towards itself. The atomic number and the separation between its valence electrons and ionic centers always have an impact on the EN [77]. In order to systematically explain the bond ionic/covalent proportion and the transition metal ionic ENs with varying coordination numbers, oxidation states, and spinning states in the complex oxide-based compounds, several scalings of ionic electronegativity and bridge electronegativity have been created [78–81]. In fact, according to the bond valence sum (BVS) model, the inherent chemical electron orbitals for the majority of materials, particularly for the transition metal ions, are never unitary. Thus, it stands to reason that the electronegativity values of transition metal compounds perovskite oxide-based should be better determined by the bond valence, which has a strong correlation with the chemical reaction strengths [82–84].
In order to anticipate the hydrogen evolution reaction performances of Co-based perovskite materials, Zhou and colleagues use coordination theories to present A-site ionic electronegativity (AIE) as an effective integrative descriptor [85]. In comparison to earlier descriptors based on B-sites of perovskite materials, the authors employ adjacent combinations of active B-sites to quickly and logically screen effective perovskite materials for the hydrogen evolution process. The authors show that AIE, as a single unified interactive descriptor, has the highest prediction accuracy for distinguishing an extremely hydrogen evolutionefficient oxide-based material from more than ten distinct Co-based perovskite materials (produced under the same conditions). This is done specifically by correlating structural or thermodynamic variables (such as A-site ionic radius and A-O bond length) (Fig. 6l-p). Furthermore, Bai and colleagues reported that bond valence sums (BVS) increased ionic electronegativity (EN) scales for effectively integrating the double perovskite materials La2CuMO6-x (Metal = titanium, manganese, ruthenium) with a heterogeneous bonding propensity [86]. In order to correlate structural and physical properties, the coordinated Bsite sub-assembled deformation and the metal oxygen bonded covalency percentage are also considered. The authors hypothesized that both the d(metal)-p(oxygen) orbital hybridization and the metal-oxygen bond covalency could be regulated by adopting the metal cation with the suitable ionic electronegativity values (Fig. 7a-d). Moreover, this engineering concept is applicable to MXenes and transition M-disulfide (bond electronegativity) as well. Thygesen and coworkers discovered that the thicknesses of the covalently bonded MXene mono-shells have a significant impact and may be used to adjust the functionality of MXenes for the hydrogen evolution reaction by utilizing the free energy of Hads as a major descriptor (Fig. 7e-g) [87]. In addition, Liu and colleagues have described a generalized hydrogen evolution reaction performance design technique for transition M-disulfides, atom valence electron numbers and bond electronegativity [88]. By identifying the electron transport potential, these variables are used to predict catalytic performance. According to the findings of the authors, the charge transport efficiency of the centralized model next to adsorption sites is a factor
Fig. 6. (a) The graphical illustration showing that the deposited atoms follow a Langmuir isotherm involves exchange current at an H2 electrode and the standard free energy of H-adsorption on the electrode interface. Reproduced with permission [65]. Copyright 1958, Royal Society of Chemistry. (b) HER volcanic scheme involving metals and molybdenum disulfide. Reproduced with permission [66]. Copyright 2017, American Association for the Advancement of Science. (c) The orbital hybridization between the d-band of the M-catalyst and the s-band of the H-atom (σ = bonding, and σ* = antibonding phase orbital). (d) The linear relationship between Gibbs free energy (ΔGH*) and the condition of anti-bonding (σ*). Reproduced with permission [72]. Copyright 2019, Wiley-VCH. (e) Orbital hybridization between an M-atom and a hydroxyl adsorbate. EF represents the Fermi level. (f) The anti-bonding orbital Eanti-bonding center and the descriptor r φ Vs. ΔGOH* (g) The relative adsorption free energies of OOH and O compared to those of OH on each individual transition metal atom supported by graphene. (h) Hydrogen evolution reaction: hypothetical overpotential in comparison to adsorption free energies denoted by ΔGH* for each and every single transition metal atom supported by graphene. Reproduced with permission [68]. Copyright 2018, Nature Publishing Group. (i) Analysis of the abundance of nonmetallic heteroatoms in graphene matrix. (j) The HER at equilibrium potential models (ΔGH*) diagram. (k) The connection between (ΔGH*) and Evar for different models. Reproduced with permission [75]. Copyright 2022, American Chemical Society. (l) Graphical representation of values of ionic radius and ionic electronegativity. (m) Doping lanthanides and alkaline-earth metal cations into solitary perovskites centered on Co. (n) A-site ionic electronegativity. (o) A-site ionic radius for single and double perovskites. (p) Molecular orbital theory predicting inductive interactions and electron exchange exchanges between A-and B-sites in perovskites. Reproduced with permission [85]. Copyright 2019, Nature Publishing Group.
Fig. 7. Graphical representations of heat images of EN variations between M and oxygen elements (a) Metal-oxygen bond covalency percentages. (b) Heat images of various elemental EN levels with both 6-fold coordination and BVS-modified charge state. (c) Relationship graphs of EN variations, and bond covalency percentages. (d) for the four EN levels. Reproduced with permission [86]. Copyright 2022, Elsevier B.V. (e, f) Negative plot of carbide and nitride MXene overpotentials versus hydrogen adsorption free energy (ΔGH*). (g) The graphical representation of ΔGH* versus ΔGH plot for MXenes. The circled data points represent compounds with |Δ GH| greater than or equal to 0.1 eV and hence the potential to operate as excellent hydrogen evolution materials. Reproduced with permission [87]. Copyright 2017, American Chemical Society. (h) Graphical representation of bond electronegativity as a HER Descriptor. (i) HER hypothetical overpotential vs adsorption free energies ΔGH* for all TMs doped in molybdenum disulfide with varied defect configurations. (j) Conceptual, experimental, and estimated by descriptor overpotentials for HER relative to the descriptor Ψ Reproduced with permission [88]. Copyright 2020, American Chemical Society. (k) The generalized adsorption energies of *OH above and (l) the coordination numbers of the locations to which the adsorbate is attached. Reproduced with permission [92]. Copyright 2014, Wiley-VCH.
that defines the level of catalytic characteristics (Fig. 7h-j).
2.3.5. Generalized coordination number In chemistry, coordination numbers are employed as an estimation of
zeroth order to represent the electronic framework whereby an atom is situated. In the majority of face-centered cubic crystals, maximal coordination is achieved when each atom is surrounded by its twelve closest neighbors. Coordination numbers lower than twelve are characteristic of
interfaces and relate to atoms with a propensity to establish interactions that address the lack of interaction [89,90]. According to the bond-order preservation concept [91], there is indeed a proportional correlation between the poor coordination of interface states and their responsiveness to the generation of strong bonds. To illustrate this relationship, the E-ads of hydroxyl above were represented by the 2 kinds of coordination numbers for a variety of adsorbent centers on platinum38, platinum79, platinum201, and extending interfaces (Fig. 7k, l) [92].
Typical coordination numbers (cn) and their first-order generalization, which we shall refer to as "generalized coordination numbers" (CN), represent the patterns in (Fig. 6k, l). To determine the generalized coordination number (CN) of an atom (i) having ni (closest neighbors), the neighbors’ normal coordination numbers are recorded and weighed Equation (1):
CN (i) = ∑ni j=1 ( cn(j)nj /cnmax ) (1)
As a result, rather than accounting for each neighbor of atom (i) with a value of 1, as is customary, nj/cnmax is used instead (cnmax being equal to 12 for a face-centered cubic crystal). The generalized coordination number of the interface metal atom for the topmost location on a (111) interface is CN = (9 X 6 + 12 x 3)/12 = 7:50. (as there are 6 and 3 neighbors in the initial and secondary shell, respectively, with typical coordination numbers of 9 and 12. Furthermore, Table 1 provides a summary of several exemplary theoretical descriptors utilized for HER.
3. Enhancement of HER electrocatalysts
3.1. Intrinsic impact
3.1.1. Alloying
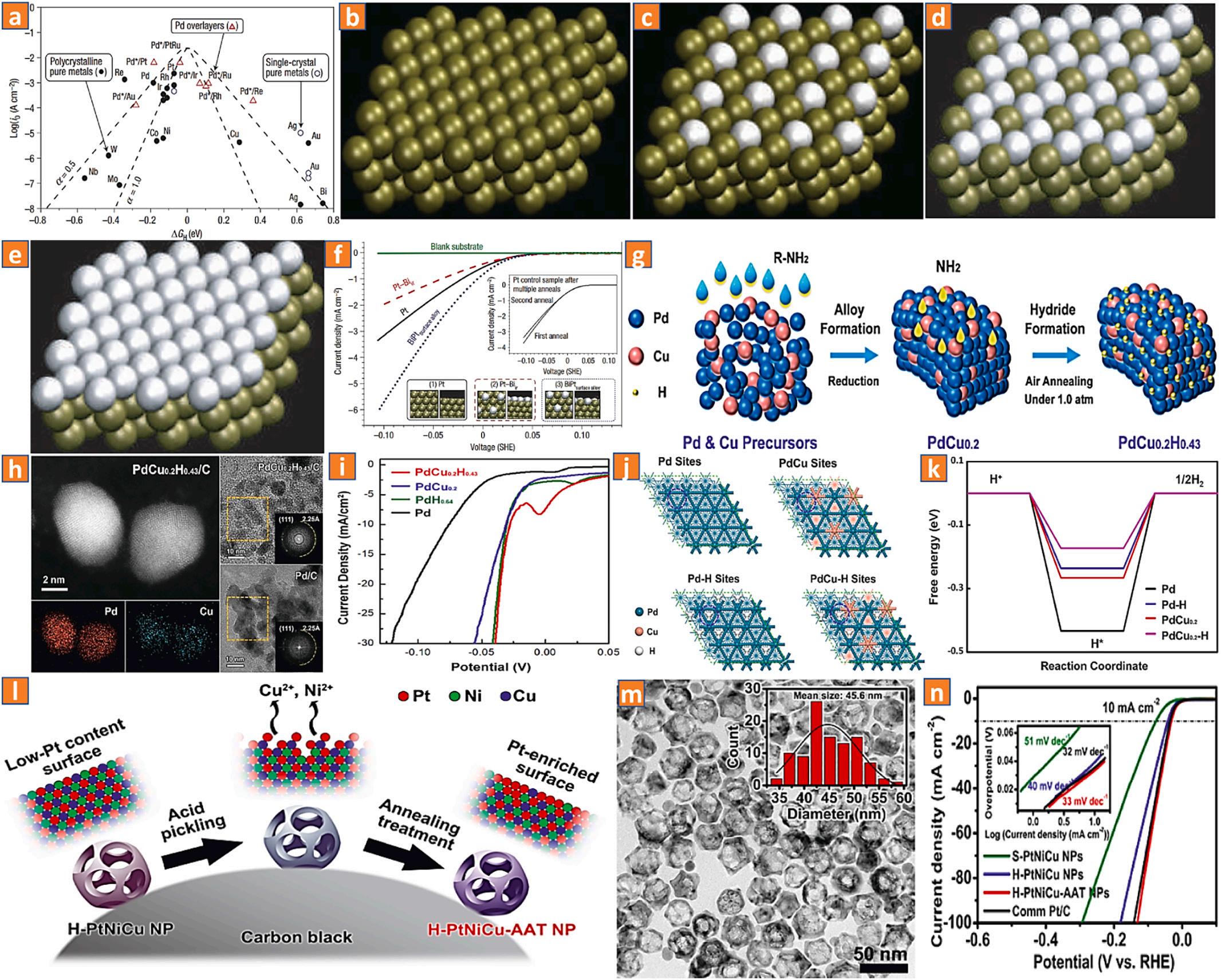
It has been demonstrated that alloying noble metals with 3d TMs increases their inherent activity while decreasing their consumption. In these instances, the chemical composition of the different metallic interfaces can be altered by modifying the maximum interface d-band energy and spacing as a result of total overall strain and binding site impacts through multiple atomic bond configurations and d-spacing modification, thereby optimizing the Hads uptake capacity to boost HER performance [93–95]. Nrskov and co-workers initially achieved a robust evaluation system density functional theory to effectively analyze the
Table 1
Some exemplary theoretical descriptors for HER.
Descriptor Category Trends of catalytic performances Applicable
ΔGH* Electronic descriptor ΔGH* < 0 a relatively strong H molecule binding ability; ΔGH* > 0 a relatively weak H molecule binding ability
d-band center theory Electronic descriptor Downward/upward shift of the d-band center. Weakening/ enhancing the metalH bond strength
Valence band theory Electronic descriptor Increase/decrease of the filling of the bonding states enhancing/ weakening the metal-H bond strength
HER operations and degradability of over 700 binary interfacial alloys, as depicted in (Fig. 8a-f) [62], and discovered that bismuth-platinum alloy could be a reasonable alternative with even superior electrocatalytic performance than conventional platinum. The quest for robust HER noble metal-based alloy catalytic materials with diverse morphologies has made significant advancements over the last decade. Using the noble metal platinum-based alloy as an illustration, we will now explore the alloying impact on the results of the hydrogen evolution process. By relocating the platinum d-band centers, alloying platinum with 3d-TMs (iron, cobalt, nickel, copper, and titanium) considerably improves the catalytic efficiency of platinum-based alloyed nanostructures. The calculated electronegativity of platinum and TMs such as copper, nickel, iron, titanium, and cobalt are 2.20, 1.90, 1.91, 1.83, 1.88, and 1.30 eV, respectively, creating an electron transport route from TMs to platinum, giving rise to the massive negative charges on platinum’s interface, which will undoubtedly enhance the H2 evolution reaction performance. Dai and coworkers devised a simple approach to intercalate intermittent H-atoms into palladium-copper alloy nanomaterials in order to improve HER activity [96]. Owing to the suitable Hads free energy and the reduced metal dispersibility, the resulting palladium-copper-hydride catalyst possesses good HER durability. The enhancement of Hads intensity on as-fabricated palladium-copper-hydride nanostructures is mostly attributable to the copper-doped precursor and interaction with an intermediate H-atom, which serve to decrease the H* coupling and increase the HER performance (Fig. 8g-k), In addition to the affordable cost, considerable enhancements in catalytic performance and morphological durability revealed the viability of incorporating an additional TM into a binary equivalent nanomaterial. For instance, platinum-silver-cobalt, platinum-rhodium-cobalt, and platinum-nickelcobalt. Cheng and coworkers fabricated nanomaterials with a reduced platinum concentration, a hollowed core, and exposed interfaces [97]. The synthesis of hollowed platinum-nickel-copper nanomaterials is a result of atom migration and a galvanic replacement interaction involving copper-nickel nanoparticles and platinum atoms that were generated in-situ. Incessant activation through etching and calcination removes unwanted copper and nickel from the interface, leading to an abundance of platinum. Owing to the advantages of alloy-based impact, hollowed and porous architecture, the resultant catalytic material exhibits exceptional HER electrochemical characteristics (Fig. 8l, m, n).
3.1.2. Chemical compounds and phase transformation
All catalyst
Metal and metal alloy/oxide
Nonmetallic carbon materials
Cobalt-based perovskites transition metal disulfide
Metals and metal alloys single-atom electrocatalysts
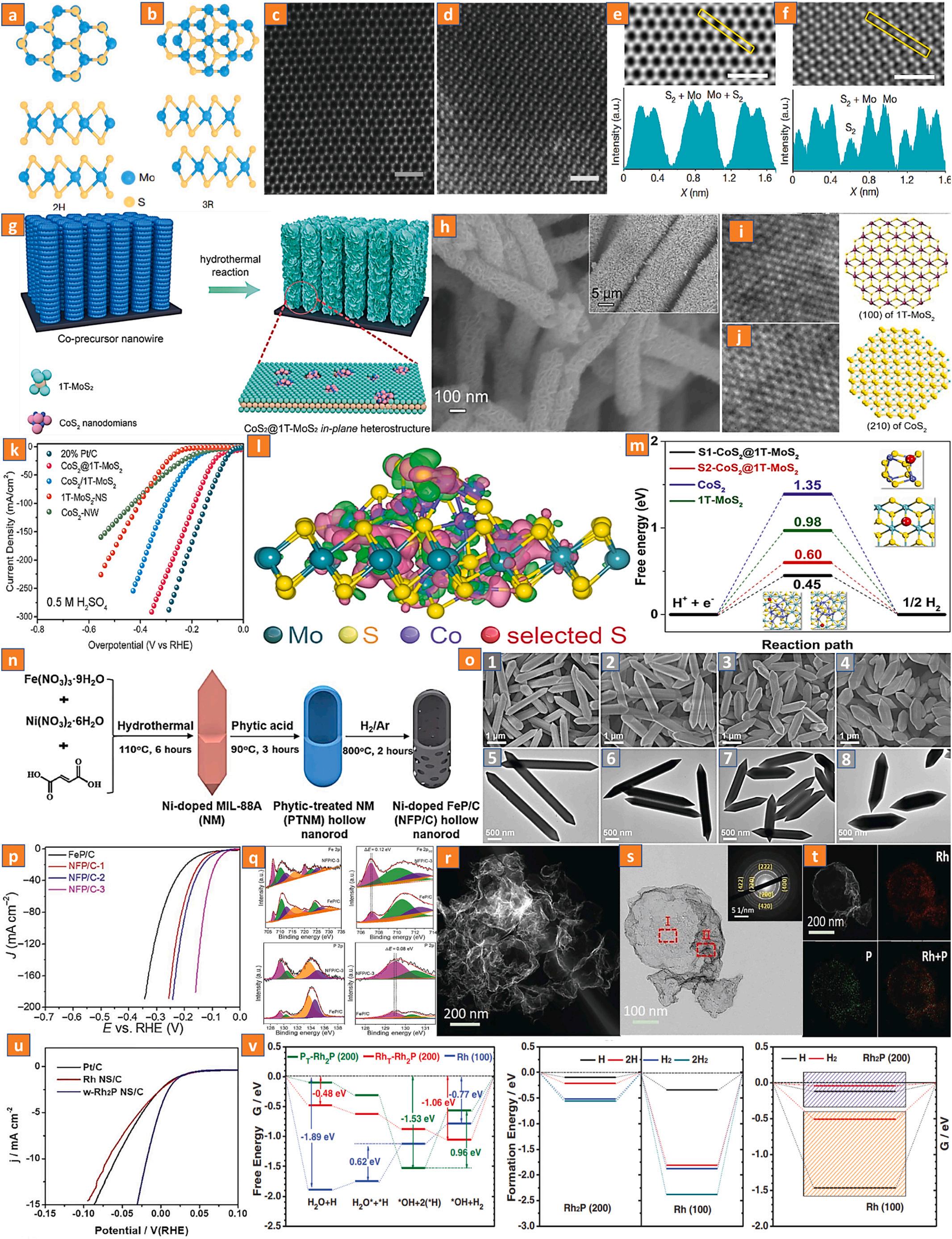
In the areas of the hydrogen evolution process, TM compounds, including S2 , C2 2 , N 3 , and P 3-based precursors, as well as their distinctive phase transitions, have garnered considerable interest. Some TM dichalcogenides (TMDs), such as molybdenum disulfide [98–101], rhenium disulfide [102], and nickel disulfide [103], as rapidly evolving 2-dimensional TMDs, have received great attention due to their exceptional physical and chemical characteristics. Experimentation and density functional theory (DFT) simulations show that the majority of extremely active catalytic sites in TM sulphides are localized at the limiting edges, whereas the bulk activation is weak. This is the case for all of the TM sulphides. Molybdenum disulfide is a good example since it has a tiny size and/or numerous defects, both of which are favorable to the active edge being exposed to its full potential. In addition to merely exposing a significant number of defect sites, the focus was also placed on designing unsaturated sulphur edges and exceptionally low interlayer conductivity [104]. In its natural state, molybdenum disulfide is capable of forming three distinct crystalline phases. These frameworks are designated as the rhombic hexahedron 3R phase, the hexagon shaped 2H phase, and the tetrahedron 1T-phase, and they differ in terms of the layer thickness and the interaction of the molybdenum atoms. In the 2H phase, the d-orbital will split into the defective phases, leaving an energy band gap of approximately 1 eV (Fig. 9a-f) [105]. In the 1T-phase, the dorbitals will disintegrate, which results in the emergence of six electrons occupying the e2g orbital [106]. Metallic characteristics are produced by only partially filling the orbitals, whereas semiconducting
Fig. 8. (a) HER volcano plot for a variety of pure metals and metal overlayers. Geometrical representations of interfacial alloys at solute coverages for which computations are performed. (b) pure metal. (c) An alloy layer with solute saturation= 1/3 ML (d) An alloy layer with solute saturation= 2/3 ML. (e) 1 ML of solute atoms that form a coating. (f) H2 evolution after every phase of BiPt interfacial alloy formation on a fluorine-doped tin-oxide layer. Reproduced with permission [62]. Copyright 2006, Nature Publishing Group. (g) Schematic representation of the PdCu0.2H0.43 nanomaterial synthesis. (h) Atomic-scale HAADF-STEM and elemental EDS images of PdCu0.2H0.43/C. (insert) HRTEM images and FFT patterns matching to PdCu0.2H0.43/C and Pd/C. (i) Linear sweep voltammetry plots. (j) DFT structures of Pd (111), Pd-H (111), PdCu0.2 (111), and PdCu0.2-H (111) interfaces. (k) A diagram illustrating the HER’s free energy on various Pd-based catalyst interfaces. Reproduced with permission [96]. Copyright 2022, American Chemical Society. (l) Graphical representation of the formation of trimetallic hollow PtNiCu nanomaterials with exposed and Pt-enriched interfaces. (m) TEM images with distinctive magnifications. (n) LSV curves and Tafel plots (inset) of S-PtNiCu, H-PtNiCuAAT, H-PtNiCu nanomaterials, and commercial Pt/C catalysts. Reproduced with permission [97]. Copyright 2020, American Chemical Society.
characteristics are produced by completely filling the orbitals (2H).
In comparison to the HER reaction that occurs on semiconducting 2H-molybdenum disulfide, the hydrogen evolution reaction that occurs on metallic 1T-molybdenum disulfide maintains exceptional kinetics due to the efficient electron transport and abundance of catalytically active sites on the surface and edges [39,104]. In contrast, the 1T-molybdenum disulfide phase exhibits thermodynamic metastability and easily converts into the 2H-molybdenum disulfide phase, especially during calcination procedures. Therefore, research into the method of stabilizing 1T-molybdenum disulfide and the development of durable nanomaterials will be advantageous to the process of boosting the catalytic performance. It is possible to incorporate the necessary dopants into the 1T-system in order to resolve this issue and achieve the required results. M-dopants and non-metal dopants are the two most frequently employed types of phase-induced doping agents. M-dopants, which often serve as electron donors, maintain high electron states in the d-
orbital of molybdenum ions through their own oxidation states of (4+ and 3+), thereby optimizing 1T-molybdenum disulfide (Fig. 9g-m) [107].
In relation to hydrogen evolution reactions, interest in TM phosphides (TMPs) has increased as a result of their low resistivity, greater mechanical properties, and exceptional thermal durability compared to other TM nanomaterials [108–114]. The effect that TMPs have on H2 evolution reactions is highly dependent on their composition. Owing to the significant contact between precipitated H2 and the transition metal interface, most metals (for example, nickel) have low H2 evolution performance. When P permeates the crystalline phase of a metal, its incorporation can disperse the M-atoms while maintaining the metal’s intrinsic electrical characteristics, hence reducing the free energy of Hads, which is favorable to Hdes [114–123]. M-phosphides, as compared to M-sulphides, are present in their bulk state and are reactive. In this research direction, certain novel approaches, like the doping of hetero
B.A.
(caption on next page)
Fig. 9. The stacking of bilayer MoS2 interlayers. (a, b) Schematic depictions of 2H (a) and 3R (b) stacking, respectively. (c, d) HAADF-STEM images of 2H and 3R bilayer MoS2 in planar view, respectively. (d, e) High-resolution HAADF-STEM images of bilayer 2H and 3R MoS2 Reproduced with permission [105]. Copyright 2022, Springer Nature Limited. (g) Schematic representation of the CoS2@1T-MoS2 fabrication. (h) SEM image, (i, j) magnification of the selected locations and associated lattice diagram. (k) LSV plot in acid (0.5 M H2SO4). (l) Heterostructure deformation charge density of CoS2@1T-MoS2 (m) Hydrogen absorption freeenergy (ΔGH) illustration. Reproduced with permission [107]. Copyright 2019, Elsevier B.V. (n) Schematic depiction of the synthesis of Ni-doped FeP/C nanomaterials. (o) FESEM and TEM images. (p) LSV curves. (q) XPS spectra of Fe 2p, Fe 2p3/2 and P 2p. Reproduced with permission [124]. Copyright 2019, American Association for the Advancement of Science. (r) HAADF-STEM image. (s) TEM image. (t) STEM-EDX elemental mapping of wrinkled Rh2P nanomaterials. (u) LSV plots and (v) Free energy pathways (ΔG) for HER. Reproduced with permission [126]. Copyright 2018, Wiley-VCH.
atoms, were implemented to expedite HER reactivity. Lou and coworkers fabricated a symmetrical hollow nanostructure of nickel-doped iron phosphide nanomaterials using a carbon-conjugated technique
[124]. X-Ray photoelectron spectroscopy surface analysis revealed that the positive shift for P 2p and the negative shift for Fe 2p spectra indicate the presence of electron transport from phosphorus atom to iron atom,
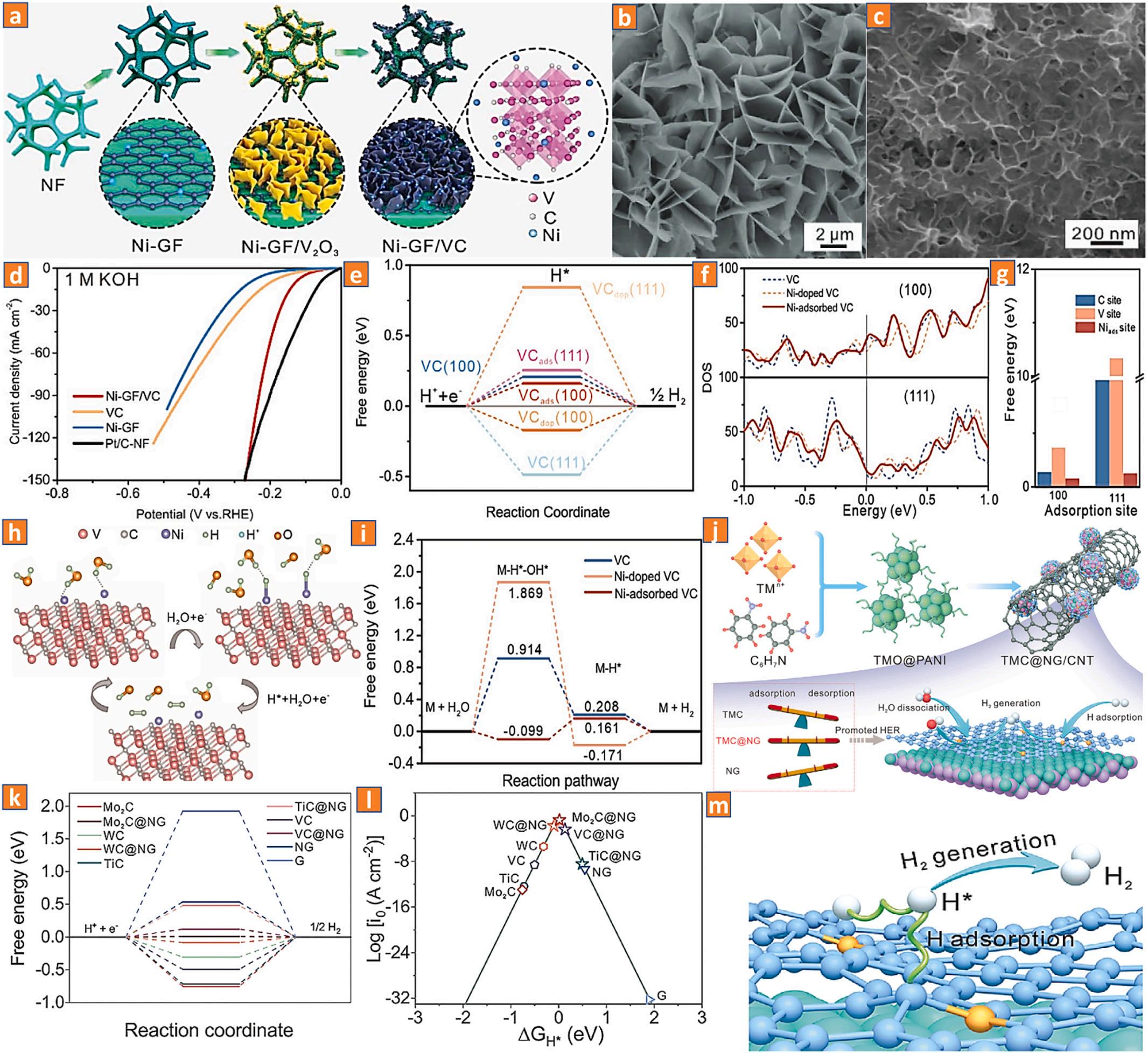
Fig. 10. (a) A schematic representation of the fabrication procedure of the Ni-GF/VC nanomaterials. (b, c) SEM images. (d) The iR-corrected LSV curves. (e) Computed ΔGH* for HER. (f) The overall DOS configurations of (100) and (111) surfaces. (g) the computed ΔGH* of the Ni-deposition VC catalyst at different Hads sites for HER. (h, i) Schematic illustration and free energy chart for the suggested HER route on the Ni-GF/VC catalyst interface in alkaline conditions. Reproduced with permission [138]. Copyright 2020, Wiley-VCH. (j) Schematic representation of the fabrication procedures and the predicted Hads and Hdes for various TMC@NG electrocatalysts. (k) Diagrams of free energy derived from DFT simulations of HER performance on several TMC@NG model electrocatalysts. (l) A volcano plot representing the HER activities on TMC@NG model electrocatalysts. (m) Schematic illustration of HER mechanism pathways in alkaline media. Reproduced with permission [143]. Copyright 2022, Wiley-VCH.
B.A.
and nickel doping iron phosphide results in the generation of higher levels for catalytically effective phosphides, which remarkably optimize the electrical properties and consequently result in outstanding HER performance (Fig. 9n-q). Numerous studies have shown that platinumgroup, rhodium-group, and ruthenium-group noble metal P-based catalysts [125–129] exhibit exceptional HER performances comparable to or even superior to conventional Pt/C. For example, Guo and coworkers established a novel category of crumpled ultrathin Rh2P nanomaterials for improving HER reactivity [126]. These nanomaterials are extremely fine and wrinkled. According to the results of theoretical calculations, the P-3p band orbital, which has a robust porous layer effect, makes Rh4d band more conducive to enhanced proton-electron charge transfer and advantageous to the HER mechanism (Fig. 9r-v).
Due to its distinctive "Pt-like functionality," transition metal carbide (TMC) has been identified as one of the most viable options to substitute platinum-based electrocatalytic materials [130–134]. The bonding between the d-orbitals of a TM and the s-orbital/p-orbital of a C-atom in the associated carbide-based materials greatly broadens the d-band of a TM within a TMC. The d-band configuration of a transition metal carbide is remarkably comparable to that of platinum metal [135]. Typically, doping with active TMs and non-metallic elements (such as iron [136], cobalt [137], nickel [138], nitrogen [139], phosphorous [140], boron [141], and sulphur [142]) has a considerable impact on the abundance of electrons at the M-sites, leading to a downshift of the dband centers and a weakening of hydrogen-binding towards enhancing TMCs. Nickel adsorption on vanadium carbide catalytic material shown increased hydrogen evolution performance and reduced overpotential in basic solution. Li and coworkers have observed nickel activation of transition metal carbides (M = vanadium) by adsorption of Ni-atoms on the nickel-transition metal carbide interface [138]. The authors found the lowest ΔGH* values for nickel atom deposition on the (100) and (111) surfaces of vanadium carbide catalytic materials. This indicates that nickel atom deposition on vanadium carbide is the most active process. Due to the fact that the electronic characteristics of the nickeldeposited vanadium carbide catalytic material around the Fermi level are much higher than those of the pure vanadium carbide and nickeldoped vanadium carbide nanomaterials, its exceptional electronic conductance can be assumed (Fig. 10a-e). Moreover, a relatively large density levels of carbon and vanadium on the adsorbent surface for (100) and (111) of the nickel deposited vanadium carbide catalytic materials compared to those of the pure vanadium carbide and nickeldoped vanadium carbide nanomaterials at the Fermi level suggests that both carbon and vanadium sites are induced in the nickel deposited vanadium nanomaterial along with a significant increase in charge density (Fig. 10f-i). Yang and coworkers presented a generalized "balancing effect" technique involving the introduction of N-doped graphene (NG) to reduce the interplay of transition metal carbides (M = vanadium) with H* [143]. The DFT binding energies for H2 indicate that the TMCs combined with N-doped graphene are thermoneutral. Due to the dissimilar activities of TMCs and N-doped graphene, incomplete electrons are transferred from the TMC interface to the N-doped graphene interface, leading to the optimization of the electronic configuration of the as-fabricated electrocatalysts. These improved electronic architectures achieve an equilibrium between Hads and Hdes, resulting in exponentially increased HER rates (Fig. 10j-m).
3.1.3.
Defects effect
The term "defect" was originally identified by Latham and coworkers in 1897 as a deficiency or omission of something fundamental to wholeness [144]. In modern scientific nomenclature, defects are deviations from the diffraction pattern in complete crystals [145]. According to their arrangement, defects in crystalline nanomaterials can be divided into bulk and interface defects. In order to change the electronic composition and surface characteristics of electrocatalytic materials, interface defects, such as point, line, and plane defects, have been intensively used. In electrocatalytic processes, point defects (including
vacancy, interstitial, anti-site, and replacement defects), which deviate from the usual crystallographic configuration in the nodes or neighboring states, are highly efficient and widely employed. Recent studies have found that defects are advantageous for generating electrocatalytic materials with distinctive electronic configurations and for altering their interface nano-environments in order to improve their effectiveness. Oxygen vacancies [146,147] in metal oxides and sulphur vacancies [148–151] in 2-dimensional transition metal dichalcogenides (TMDs) materials are two examples of the frequent point defects known as vacancy defects. By adjusting the density of vacancies, the ΔGH* might be made to function optimally, using the sulphur vacancies as an example. Additionally, the formation of sulphur vacancies causes the leftover electrons to preferentially migrate to the nearby M-atoms, resulting in an electron-rich area on the M-atoms. More specifically, the delocalized electrons surrounding M-atoms support higher H2 affinity and enhance Hads on interfaces, increasing the number of charge carriers on the inert lattice. Voiry and colleagues investigated the hydrogen evolution performance of defective multidimensional molybdenum disulfide across a broad range of interface S vacancy ratios [149]. The authors discovered that the HER process on defective molybdenum disulfide is classified into 2 phases, correlating to (i) "point" defects at small concentrations of interface sulphur-vacancies and (ii) under-organized molybdenum areas induced by S-atom extraction at increased concentrations of interface defects (Fig. 11a-f).
Linear defects (consisting of dislocations and stepping-phases) are regular structural breakdowns that form along a path close to the dislocated-atoms. For instance, Chen and co-workers used the thermal shock approach at a non-equilibrium high temperature in order to create numerous dislocations in platinum nanostructures that they referred to as Dr-Pt (Fig. 11g-j) [152]. During the crystallization process, dislocations that are generated by thermomechanical stresses crystallize interactively at a rapid processing temperature. This process takes place in a matter of milliseconds (Fig. 11k, l, m). According to the author’s findings, the efficiency of the hydrogen evolution process can be greatly improved by the addition of abundant dislocations. A crystal is frequently divided into several smaller subdomains by certain surfaces, which have stronger atomic configuration stability and extreme atom distortion adjacent to the surfaces between the subdomains. These defects include twin defects, layered defects, stacking defects and grain boundary-defects. Grain boundary-defects are typical examples of interface defects found in polycrystalline-based materials that are ultrathin or 2-dimensional. Recent research has demonstrated that the occurrence of grain boundary defects can increase the electrocatalytic performance of metallic materials [153,154]. For instance, Liu and coworkers generated wafer-sized nanostructured ultrathin films with extremely high grain boundary densities (Fig. 11n, o). Owing to the existence of grain boundaries, domains on the normally hydrogen evolution inert particle surface of the molybdenum disulfide were activated, leading to a better hydrogen evolution efficiency in acidic media in the final nano-grain layer (Fig. 11p, q) [155].
Bulk defects are crystal defects in the 3D region, such as cracks, pores, unwanted inclusions, as well as other components that are typically generated during treatment and synthesis procedures. Porous structures, the most common type of metal bulk defect, contain a huge quantity of pore structures. The pore structure is distinctly visible in high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images owing to the darker contrast of porous locations, which have fewer atoms. For instance, Yang and colleagues, for instance, demonstrated the fabrication of mesoporous copper nanoribbons using in-situ electrochemical reduction of copper-based MOFs (Fig. 12a, b, c) [156]. The HAADF-STEM image displayed a dense distribution of mesopores across the nanoribbons (Fig. 12d), as numerous dark pores were observed on the bright copper nanoribbons. The evidence could be obtained further from the HAADF-STEM image, which displayed pores distinctly. For instance, the intensity profile was derived from a HAADF-STEM image to corroborate the pores (Fig. 12e).
Another random document with no related content on Scribd:
The Project Gutenberg eBook of Memorie della
vita di Giosue Carducci (1835-1907)
This ebook is for the use of anyone anywhere in the United States and most other parts of the world at no cost and with almost no restrictions whatsoever. You may copy it, give it away or re-use it under the terms of the Project Gutenberg License included with this ebook or online at www.gutenberg.org. If you are not located in the United States, you will have to check the laws of the country where you are located before using this eBook.
Title: Memorie della vita di Giosue Carducci (1835-1907)
Author: Giuseppe Chiarini
Release date: June 23, 2024 [eBook #73895]
Language: Italian
Original publication: Firenze: Barbèra, 1907
Credits: Barbara Magni and the Online Distributed Proofreading Team at http://www.pgdp.net (This file was produced from images made available by The Internet Archive) *** START OF THE PROJECT GUTENBERG EBOOK MEMORIE DELLA VITA DI GIOSUE CARDUCCI (1835-1907) ***
VITA DI GIOSUE CARDUCCI.
MEMORIE DELLA VITA
DI GIOSUE CARDUCCI
(1835-1907)
RACCOLTE DA UN AMICO (GIUSEPPE CHIARINI).
Seconda edizione corretta e accresciuta.
FIRENZE, G. BARBÈRA, EDITORE. 1907.
FIRENZE, 706-1907. — Tipografia Barbèra
ALFANI E VENTURI proprietari.
Compiute le formalità prescritte dalla Legge, i diritti di riproduzione e traduzione sono riservati.
INDICE
ALLA MEMORIA DI ADRIANO LEMMI.
PROEMIO.
Feci la conoscenza personale del Carducci nell’estate del 1855. Lo avea veduto tre anni avanti a San Giovannino delle Scuole Pie alle lezioni di filosofia, dove andavo qualche volta benchè avessi terminati l’anno innanzi gli studi. Egli entrò ch’era già cominciata la lezione, entrò con passo ardito e franco e con la testa alta, e andò a mettersi al suo posto nei gradi più bassi dell’anfiteatro. Io aveva sentito parlare di lui con ammirazione dai suoi compagni di scuola, da alcuno dei quali ebbi copia di qualche sua poesia, che mi parve molto bella. Più tardi uno di quelli stessi compagni mi diede a leggere manoscritta la canzone su Dante che il Carducci aveva composta nel 1854 per una Accademia delle Scuole Pie, della quale dovrò parlare più avanti. La canzone avea fatto un po’ di chiasso, specie fra i giovani, e ne corsero delle copie manoscritte, una delle quali appunto fu data a me. Io ne restai vivamente ammirato, e me ne crebbe il desiderio, che già avevo, di conoscere di persona l’autore. Avevo fatta da poco la conoscenza di Enrico Nencioni, stato condiscepolo del Carducci alla scuola di retorica del Padre Barsottini, e suo amicissimo. Esposi a lui il mio desiderio, ch’egli fu lieto di sodisfare. Il Carducci faceva allora il secondo anno di studi alla Scuola Normale Superiore di Pisa; ma veniva spesso nei giorni di vacanza a Firenze, dove aveva parenti, presso alcuno dei quali andava ad alloggiare. Quando lo andai a trovare in compagnia del Nencioni, egli abitava presso una zia, in via Borgognissanti. Andammo di mattina (era di domenica) fra le nove e le dieci. Egli era prevenuto, sapeva che io era un grande ammiratore, anzi adoratore, del Leopardi, che amavo i classici, che facevo dei versi, che ammiravo grandemente i suoi. Ci venne incontro in maniche di
camicia; ci demmo subito del tu, come s’usa fra giovani, si cominciò a parlare di letteratura, si parlò del Leopardi, del Giordani; io gli chiesi qualche cosa di suo, egli mi trascrisse lì per lì sopra un grande foglio di carta gli ultimi due sonetti da lui composti, quello che comincia Poichemalquesta sonnacchiosaetadee l’altro Aisepolcri deigrandiitalianiinSantaCroce; dopo di che ci lasciammo, ed io me ne tornai lieto e contento come se portassi meco un tesoro.
Ci rivedemmo qualche volta nei giorni appresso in compagnia del Nencioni, di Ottaviano Targioni Tozzetti, di Giulio Cavaciocchi e di altri amici del Carducci e del Nencioni, che diventarono anche amici miei. Egli tornò indi a poco a Pisa, poi andò a passare le vacanze autunnali in famiglia a Pian Castagnaio, dove suo padre era medico condotto; io dovei per ragione d’impiego andare ad Arezzo, dove stetti fino ai primi del 1856. Avevamo promesso di scriverci: io fui il primo; egli mi rispose da Pian Castagnaio il 4 settembre 1855, dicendomi che là infieriva il colèra (il quale infieriva pure ad Arezzo), e che aveva dovuto lasciare gli studi per curare gli ammalati. Fino dai primi casi egli con un suo fratello ed altri due giovani senesi avean prestato volontari l’opera loro; in seguito di che il Municipio aveva composto di essi e di altri tre una commissione di assistenza gratuita, incaricando lui della direzione.[Vedi note pag. 433]
Al riaprirsi dell’anno accademico 1855-56 il Carducci tornò alla Scuola Normale; io ai primi del 1856 tornai, come ho accennato, a Firenze. D’allora in poi le mie relazioni con lui e cogli altri amici fiorentini furono continue, quasi giornaliere. Ma prima di proseguire, sarà buono dire qualche cosa della infanzia e della prima giovinezza del poeta. Il che farò, lasciando quanto più è possibile la parola a lui stesso, e giovandomi delle testimonianze altrui. A questa parte della Vita saranno dedicati i due primi capitoli: di ciò che narrerò negli altri fui testimone io stesso. E poichè l’antica amicizia ha mantenuto fra noi una corrispondenza epistolare che va senza interruzione dall’anno 1855 a questo in cui scrivo, attingerò, oltre che dalla memoria, da questa specie di domestico archivio ciò che mi parrà conferir meglio a delineare viva e vera la figura dell’uomo e dello scrittore.
CAPITOLO I. (1835-1854.)
Ricordo d’infanzia: «Via, via, brutto te.» — La famiglia
Carducci e il padre del poeta. — I primi anni e i primi studi a Bolgheri e a Castagneto. — La famiglia Carducci a Firenze e Giosue alle Scuole Pie. — Giosue a retorica dal
Padre Barsottini — Il Carducci e il Nencioni. — Passione di Giosue pei libri. — Il primo passo. — Il Carducci a Celle. —
Primi sonetti satirici. — Accademia dei Risoluti e Fecondi. — Canzone del Carducci su Dante letta all’Accademia.
«Io della mia infanzia, scrive il Carducci, non ho memorie nè belle nè buone nè curiose.
«Il mio più antico ricordo mi pone subito, ahimè, inrelazioneconun essere dell’altro sesso, come si direbbe con la lingua d’un certo uso, che, secondo i manzoniani, dovrebbe anche essere la lingua del buon gusto. Mi ritrovo in un luogo nè bello nè brutto — forse un giardinetto presso la casa ove nacqui, — a una giornata nè di primavera nè d’inverno nè d’estate nè d’autunno. Mi pare che tutto, cielo e terra, sopra, sotto, e d’intorno, fosse umido, grigio, basso, ristretto, indeterminato, penoso.
«Io con una bambina dell’età mia, della quale non so chi sia o chi sia stata, dondolavamo, tenendola per i due capi, una fune; e mi pare che così dicevamo o credevamo di fare il serpente. Quando a un tratto ci si scoperse tra i piedi una bella bodda: è il nome, nel dialetto della Versilia, d’un che di simile al rospo.
«Grandi ammirazioni ed esclamazioni di noi due creature nuove su quell’antica creatura.
«Le esclamazioni pare fossero un po’ rumorose. Perchè un grave signore, con gran barba nera e con un libro in mano, si fece in sull’uscio a sgridarci, o meglio a sgridarmi. Non era mio padre: era, seppi molto tempo dopo, un marito putativo d’una moglie altrui alloggiata per certo caso ivi presso.
«Io brandendo la fune, come fosse un flagello, me gli feci incontro gridandogli: Via, via, brutto te!
»D’allora in poi ho risposto sempre così ad ogni autorità che sia venuta ad ammonirmi, con un libro in mano e un sottinteso in corpo, a nome della morale.»[1]
Questo aneddoto mostra già nel fanciullo una delle qualità più caratteristiche dell’uomo. Perciò l’ho messo qui, affinchè sia come il battesimo della vita del nostro poeta.
Il piccolo ribelle nacque il 27 luglio dell’anno 1835 alle ore 11 di sera in Val di Castello, frazione del comune di Pietrasanta, da Michele Carducci e Ildegonda Celli. Gli furono dati all’atto del battesimo, ch’ebbe luogo due giorni dopo, i nomi di Giosue, Alessandro, Giuseppe, essendo compare un suo zio Natale Carducci.[Vedi l’atto di nascita nelle note a pag. 434]
La famiglia Carducci, stabilita da gran tempo fra Serravezza e Pietrasanta, discendeva dai Carducci di Firenze; e il nonno del poeta, Francesco Giuseppe, andava orgoglioso di tale discendenza, e si compiaceva molto, nella intimità della famiglia e degli amici, di evocarne le gloriose memorie. Ma quelle memorie, troppo lontane, non ebbero alcuna influenza sui suoi sentimenti politici: e nemmeno la familiarità sua col poeta repubblicano Giovanni Fantoni, di cui era grande ammiratore. Egli era e rimase un fedele suddito del Granduca
di Toscana; e come tale odiò le novità e le rivoluzioni. Il padre del poeta invece, il dottore Michele, avea nel sangue l’istinto della battaglia e della libertà. Fin da scolare prese parte alle cospirazioni politiche, fu carbonaro, e dei pochi Toscani che pei fatti del 1831 patirono relegazione e prigionia. Quando gli nacque il primo figliuolo Giosue, egli a Val di Castello era medico di una società francese che aveva assunto l’escavazione di certe miniere di piombo argentifero, poste tra Val di Castello e Serravezza. Ma o fosse l’indole sua irrequieta, o che non gli piacesse, o non gli convenisse, l’ufficio presso la società mineraria francese, o che, come altri dice, gli desse fastidio la sospettosa vigilanza della polizia, ben presto abbandonò la Versilia per la maremma toscana, e verso il 1838 andò medico condotto a Bolgheri, frazione di Castagneto, e feudo dei Conti della Gherardesca.
Tra Bolgheri e Castagneto la famiglia Carducci passò ben undici anni, che il poeta chiama la sua triste primavera e dei quali parla egli stesso così: «Mio padre era un manzoniano fervente.... Ridottosi a vivere in condotta in uno dei più oscuri paeselli della maremma, viveva coi contadini, e, nelle ore di riposo o di sosta, con alcuni pochi libri di storia e letteratura che, oltre i non pochi dell’arte sua, aveva raccolti ed amava. Figuravano tra questi bellissime le opere del Manzoni, con i giudizi del Goethe, le analisi critiche del Fauriel, i commenti del Tommaseo; e quei volumi, rilegati con certa pretensione di lusso, mostravano impressi nelle costole a oro certi fregi che rendean figura come di casette con due alberetti davanti. Io, ragazzo di circa dieci anni, credevo che quella fosse la canonica di Don Abbondio; e leggevo e rileggevo Ipromessisposi. Perchè fino a quattordici anni non ebbi quasi altro maestro che mio padre, il quale altro non m’insegnava che latino; ma, un po’ per l’indole sua, un po’ per i doveri di medico, mi lasciava molta libertà e molto tempo per leggere.
«E io insieme alle opere del Manzoni lessi l’Iliade, l’Eneide, la Gerusalemme, e la Storia Romana del Rollin, e la Storia della RivoluzioneFrancese del Thiers; i poemi con ineffabile rapimento, le storie con un serio oblio di tutto il resto: e, aiutato da qualche conversazione di mio padre con certi amici ed ospiti, per ragazzo ne intendevo anche troppo. Invasato così di ardore epico e di furore repubblicano e rivoluzionario, io sentivo il bisogno di traboccare il mio idealismo nell’azione; e per ciò in brigata co’ miei fratelli e con altri ragazzi del vicinato organizzavo sempre repubbliche, e repubbliche sempre nuove, ora rette ad arconti ora a consoli ora a tribuni, pur che la rivoluzione fosse la condizion normale dell’essere, e cosa di tutti i giorni l’urto tra i partiti e la guerra civile.
«La nostra repubblica consisteva di ragunanze tumultuose e di battaglie a colpi di sassi e bastoni, con le quali intendevamo riprodurre i più bei fatti de’ bei tempi di Roma e della rivoluzione francese. In coteste rappresentazioni, del resto, il rispetto alla storia non era certo spinto a quegli eccessi pedanteschi che soglion guastare o raffreddare l’effetto vivo drammatico. Che benedette sassate applicai un giorno a Cesare il quale era su ’l passare il Rubicone! Per quel giorno il tiranno dovè rifugiarsi non so dove con le sue legioni, e la repubblica fu salva. Ma il dì appresso Cesare mi colse in una macchia, affermando sè essere Opimio e quello il luco delle furie: invano io protestai contro l’anacronismo e per la mia qualità di Scipione Emiliano: egli mi fece togliere in mezzo da’ suoi cretensi come un Gracco qualunque e flagellare, mentre io chiedevo che almeno rispettasse la storia lasciandomi libero di farmi uccidere al mio schiavo. Come picchiavano e rideano quei cretensi! Me ne vendicai, per altro ed in breve, e storicamente, quando, presa d’assalto una rimessa che facea da Tuileries, stimai bene di lasciar libero il corso al furor popolare su gli svizzeri prezzolati di Luigi XVI. «Ma il rumore di questi grandi fatti giungeva qualche volta alle orecchie del mio manzoniano padre, il quale allora, nulla commosso dalle mie oneste ferite, mi condannava pur troppo a lunghe prigionie; in mezzo alle quali egli di quando a quando riappariva per rivedermi il latino, e mi lasciava tre libri su ’l tavolo, dicendomi serio
ed asciutto: — Leggete qui, e persuadetevi che il taratantaraclassico non è più per questi tempi. — I tre libri erano: la Moralecattolica di Alessandro Manzoni, i Doveri dell’uomo di Silvio Pellico, e la Vita di San Giuseppe Calasanzio scritta da certo padre Tosetti (parmi) del secolo passato.
«Che idea fosse quella del manzoniano mio padre di dare a leggere la Moralecattolicaa un ragazzo, io non so: so che d’allora in poi per un gran pezzo morale cattolica e frati, doveri dell’uomo e santini, furono per me la stessa cosa; e odiai, odiai quei libri, d’un odio catilinario. Essi mi rappresentavano la mortificazione, la solitudine, la privazione di libertà e d’aria e di combattimento, la fame delle grandi letture, un nuovo carcere tulliano. Trovavo uno sfogo ad affacciarmi alla finestra, declamando la parte di Guglielmo de’ Pazzi:
Soffrire, ognor soffrire? altro consiglio
Darmi, o padre, non sai? Ti sei tu fatto
Schiavo or così che del mediceo giogo
Non senti il peso e i gravi oltraggi e l’onte?
Dispetto! i cretensi e gli svizzeri eran sotto la finestra, e ridevano, e mi gettavano pomi.»[2]
Queste battaglie e le letture non erano i soli svaghi del selvatico fanciullo, dice il Borgognoni: «e’ si teneva in casa e allevava con grande amore una civetta, un falco e (imaginate!) anche un lupacchiotto.» Ma il padre, cui tutto ciò non andava a genio, «un bel giorno ammazzò il falco e regalò a un tale di Livorno il lupo.» Giosue ne fu così addolorato, che «scappò di casa e passava le giornate intere errando pei boschi in riva al mare e su pei colli cretacei.»[3] Aveva intorno a dieci anni; e cominciò fin d’allora a sentirsi tentare dalla smania di far versi; scrisse prima alcune ottave sulla presa di Bolgheri, poi alcune terzine sulla morte di Cesare e un sonetto per la morte della sua civetta. In questo tempo essendoglisi messa addosso una febbre maremmana, che gli durò due anni, il padre (che stava allora a Bolgheri), per vedere di guarirlo, lo mandò a Castagneto in casa d’un collega. Qui Giosue, sentendosi pienamente
libero, cominciò, dice il Borgognoni, a farla da uomo; si legò in amicizia con un tale Alessandro Scalzini repubblicano, e nella casa di lui spiegava ai molti che accorrevano a sentirle, le poesie del Giusti, che allora andavano attorno manoscritte. Fu richiamato a casa dal padre nel 1846; ma due anni dopo, nella primavera del 1848, tutta la famiglia si trasferì a Castagneto, e Giosue vi riprese la vita di prima. «Allorchè, scrive il Borgognoni, fu affisso a Castagneto il bollettino che annunziava lo statuto largito da Carlo Alberto, il Carducci ci scrisse sotto col lapis:
Esecrato Carignano
Va il tuo nome in ogni gente, con quanta approvazione dello Scalzini non è da chiedere. Seguì in quei giorni una dimostrazione nel paese. Se non che non si vedeva bene dove la dimostrazione andasse a parare. Allora il Carducci persuase lo Scalzini e i suoi a levare il primo grido: Abbassotuttiire! vivalarepubblica!Lo Scalzini gridò, tutti gridarono: la dimostrazione era riuscita.»[4]
Intanto il padre, che, uomo libero e battagliero, come s’è detto, avea preso parte fin dal principio ai moti del 1848, e tal parte che ben presto la dimora di Bolgheri non gli parve più sicura per lui, aveva trasportato le sue tende a Castagneto; ma anche qui non potè durare lungamente. In quelli anni gli umori e le voglie erano così diverse e divise, che un uomo poco prudente correva rischio d’attaccare lite ad ogni momento. Dovè dunque l’anno dipoi, al tempo del governo provvisorio del Guerrazzi, abbandonare anche Castagneto: andò come medico interinoa Laiatico, ma ci stette ben poco, costretto a fuggire dalla reazione moderata toscana, che ivi lo colse. I contadini, dice il Borgognoni, lo costrinsero a baciare un busto in gesso di Leopoldo II, e lo bastonarono anche, pare, un pochino. Egli allora riparò a Firenze, dove avea parenti da parte della
moglie, dove gli era più facile passare inosservato, e dove poteva provvedere meglio all’educazione dei figliuoli. Ben presto la famiglia andò a raggiungerlo, e si allogarono in una povera casa in fondo di via Romana, segnata del n. 1843. I figliuoli, che allora eran tre, Giosue di 14 anni, Dante di 13, Valfredo di 8, furono subito messi alle Scuole Pie.
Il corso classico secondario si compieva allora in tutte le scuole di Toscana in sei anni; tre anni di grammatica (1ª, 2ª, 3ª grammatica), un anno di umanità e due di retorica. Per quelli che volevano proseguire gli studi, c’era un corso di scienze (filosofia, matematiche, fisica) che durava un anno. Giosue fu inscritto nel maggio 1849 alla classe di umanità, i due fratelli a classi inferiori. Era maestro di umanità un Padre Michele Benetti, uomo colto e studiosissimo di Dante e dei classici antichi, dei quali aveva, con un discorso a stampa, preso le difese contro il Padre Ventura, che giudicava pericoloso lo studio di essi nelle scuole. Non pare ch’ei facesse speciale attenzione al Carducci: e si capisce; egli avea una classe di 62 scolari parecchio indisciplinati; e il Carducci era stato da lui poco più di tre mesi. Ma nei registri, che ancora si conservano, il maestro aveva notato che il giovinetto era «assai bene istruito», e perciò fra gli scolari «da passarsi», e che aveva tenuto una condotta irreprensibile.
Dei 62 scolari del Benetti soli 8 avevano in quell’anno ottenuto la nota di irreprensibile; ed i 62 erano all’esame ridotti 43, avendone il maestro dovuti espellere 8 e costringere altri a ritirarsi, per gravi ragioni d’indisciplina. Ciò che dimostra, osserva il Padre E. Pistelli, alla cui cortesia devo queste notizie,[5] che gli scolari d’oggi non sono peggiori di quelli d’allora. «Io, soggiunge il Pistelli, in 22 anni d’insegnamento ho dovuto espellere uno scolaro solo.» Gli esami furono dati dal 21 al 31 agosto; e il Carducci fu approvato con queste classificazioni: nei due latini scritti mediocre, nella spiegazione a voce di Virgilio (En., l. V) ottimo, nei precetti di letteratura bene, nella sfera armillare mediocre, nella storia toscana bene.
«Da vecchio il Benetti, dice il Pistelli, parlava sempre dei suoi scolari, e ne aveva avuti, a Firenze, a Cortona, a Siena nel Collegio Tolomei, una infinità. Ma non ho memoria d’avergli sentito ricordar mai il Carducci. Era uomo molto pio e scrupolosissimo, e forse non ricordava volentieri chi in quegli anni era chiamato il cantore di Satana.»
Mentre Giosue stava per finire la scuola di umanità, il padre ebbe per un momento l’idea di farlo entrare nel Liceo Militare e ne fece domanda; ma poi, quale ne fosse la ragione, abbandonò quell’idea; e terminate le vacanze scolastiche, il giovane riprese i suoi studi alle Scuole Pie.[Vedi le note a pag. 434]
Il corso di retorica durava ordinariamente due anni, e il Carducci li fece tutti due (1849-50, 1850-51). Era maestro il Padre Geremia Barsottini, «anima ardente e, a quei giorni, liberale», dice il Pistelli; un buono e brav’uomo davvero, dalla figura maestosa e imponente, che contrastava in modo singolare con la sua faccia sempre piena di sorriso e le sue maniere cortesi e carezzevoli. Era amato e stimato, non pure dagli scolari, ma da tutta la cittadinanza; ebbe fama di valente oratore; e di un suo volume di versi, un po’ rugiadosi e sdolcinati, che piacevano molto alle signore, furono fatte più edizioni. Pubblicò anche un’antologia omerica «Le bellezze d’Omero», e fu studioso dei classici; ma era e si mantenne sempre romantico. Perciò il Carducci, che pure gli volle bene, non lo tenne mai per un modello di insegnante; e perciò egli, per quanto sentisse l’ingegno del Carducci, gli preferì il Nencioni, romantico come lui.
Nel primo anno di retorica il Carducci ebbe tra i condiscepoli Torquato Gargani, ma non il Nencioni; il quale, per quanto appare dai registri, fu con lui soltanto il secondo anno (1850-51).
Pel passaggio dal primo al secondo anno di retorica non c’erano esami. Negli esami alla fine del secondo anno il Carducci fu
approvato con queste note: prosa italiana, bene assai; versione scritta dal latino, ottimo; versione dal classico a voce, bene; versione scritta dall’italiano in latino, ottimo; precetti, ottimo. L’esame di greco, ch’era libero, il Carducci non lo diede. Se l’esame del Carducci fu buono, quello del Nencioni fu migliore, avendo egli avuto la nota di ottimoanche nel latino orale, e di benenella prosa italiana, senza l’assai, che attenua. Del qual fatto è, secondo il Pistelli, da cercare la ragione nell’essere il Nencioni romantico come il maestro. Non bisogna credere però che il buon Barsottini non avesse nei due anni ch’ebbe sotto di sè il Carducci, misurato tutto il valore di lui. Attesta il Pistelli nelle sue note di avere una sola volta parlato del Carducci al Barsottini, quando questi era vecchio e malato; e il Barsottini gli disse: «Ha mantenuto tutto quello che prometteva, ed era tanto!»
Io poi ricordo. S’era negli anni fra il 1865 e il 1870: il Carducci, venuto da Bologna a Firenze a passare alcuni giorni, era con me in via Larga, non lungi da San Giovannino. A un tratto ci viene incontro il Padre Barsottini, con la faccia illuminata da quel suo sorriso gioviale, che pareva volere abbracciare e proteggere le persone colle quali egli parlava. Si fermò, prese per le mani il Carducci, e guardandolo affettuosamente, non senza un po’ di soggezione, gli disse: «Bravo il mio Giosue, tu sei diventato un gran professorone.»
Più che nelle parole, c’era nel tuono della voce e in tutta l’espressione del viso del buon frate la sodisfazione del maestro che diceva fra sè: Ed io l’ho avuto scolare! Non parmi che il Carducci rispondesse con molta espansione alla espansione del maestro; egli era allora la bestia nera dei moderati toscani; e ciò lo metteva subito di cattivo umore tutte le volte che s’incontrava con qualche toscano non dei pochissimi (cinque o sei) suoi intimi. A me quell’incontro fece molto piacere; e ne serbai viva memoria. Non credo che il Carducci abbia poi riveduto più il Barsottini. Per essere ammesso al corso di scienze il Carducci dovè l’anno appresso (1851-52) dare un esame di aritmetica, nel quale fu approvato; e si inscrisse alle lezioni di geometria, di fisica e di filosofia. Di geometria e di filosofia era insegnante, o, come allora dicevano, lettore, il Padre Celestino Zini, che fu più tardi direttore
delle Scuole Pie, e morì arcivescovo di Siena; di fisica, il Padre Filippo Cecchi, che avea fin d’allora buon nome fra gli scienziati e che poi s’acquistò fama per lavori importanti di meteorologia e di fisica. Nel registro del Padre Zini il Carducci è notato irreprensibile per la condotta, notabileper il profitto; ma nella colonna della frequenza è scritto: «quasi assiduo»: nell’esame fu approvato a pieni voti e pluralità di plauso, come risulta dall’attestato che conservasi nell’archivio della R. Scuola Normale Superiore di Pisa.[Vedi le note a pag. 435]
Gli anni dal 1850 a tutto il 1852 furono i tre anni che il Nencioni passò, come dice nel suo Consule Planco, «in continua compagnia, in fraterna comunanza di studj e di affetti»[6] col Carducci.
Alla quasi assidua frequenza alle lezioni di filosofia può servir di commento il fatto accennato dal Borgognoni,[7] e confermatomi dagli stessi Carducci e Nencioni, delle grandi passeggiate che i due amici andavano a fare insieme pei colli di Firenze, saltando la scuola. Anche mi raccontarono come essi, specialmente il Carducci, per fare imbroncire il buon Padre Zini, lo aspettassero talora alla porta dell’aula, quando usciva od entrava, e gli esprimessero la loro ammirazione per la filosofia del Leopardi, dicendogli all’orecchio: —
Padre Zini, evviva il Leopardi. — Naturalmente il buon frate attribuiva alla inesperienza e al bollor giovanile queste scappatelle, e pur rimproverandole le compativa.
«Quante cose, dice il Nencioni nello scritto che sopra ho citato, potrei raccontare della vita domestica e scolastica del Carducci!» Giacchè anche lui, come il Gargani, ci ha abbandonati innanzi tempo, e a me non è dato supplire che in piccola parte alla copia di notizie che loro avrebbero potuto dare sulla prima giovinezza dell’amico nostro, riferirò qui due pagine dello scritto del Nencioni, note certamente a molti, ma che tutti rileggeranno volentieri.
«Mi par di vederlo ancora, a scuola di retorica, un sabato che si doveva spiegare qualche frammento di classico latino ad libitum, escir dal suo posto, traversare impettito e fiero la scuola, e presso la cattedra del maestro levarsi di tasca con meraviglia di tutti noi un libriccino in carta pecora, un vecchio elzeviro, e cominciare a leggere.... Era un Persio senza note. Stupore nella scolaresca, e un certo imbarazzo nel nostro buono e bravo maestro, Padre Geremia Barsottini. — Lesse, costruì, tradusse, commentò, franco, preciso, sicuro, e se ne tornò al suo posto fra un silenzio d’ammirazione. — Da quel giorno fu il dittatore della scuola. Lo vedo ancora arrivare le mattine d’inverno quasi sempre in ritardo, in giacchetta di panno turchino con bottoni d’ottone, con berrettino militare, senza paletot, senza mantello, senza sciarpe, sfidando i geli, come Souvarow.
«L’adolescenza e la prima gioventù del Carducci sono state veramente spartane: quelli anni così ridenti per tutti, furon per lui anni di sacrifizj, di perseveranza, di lavoro ostinato, di dignitosi silenzi, di nobili e alteri rifiuti. E conosco una povera casa in Firenze, in fondo di via Romana, che fu testimone di giornaliere ignote lotte, — consolate solo dalle pure gioje della poetica ispirazione, da entusiasmi di ammirazioni artistiche, dalla lettura di qualche libro prestato — povera casa dove il Carducci ha scritto i primi suoi versi, le Odi oraziane, — e che a me ha insegnato più e meglio di tutti i palazzi Strozzi e Farnese, che cosa sono le realtà e le idealità della vita.
«Legato a lui fin d’allora di fraterna amicizia, gli procuravo dei libri ed ebbi così la fortuna di fargli conoscere alcuni poeti stranieri, lo Schiller, fra gli altri, — e di italiani il Leopardi; i cui Canti (vecchia edizione Piatti) da me prestati al Carducci, destarono nel futuro poeta delle Odi barbare un vero fanatismo. Ricopiò, mi rammento, più della metà del volume; e il Bruto Minore, e la Saffo, gli imparò subito a mente. — Guido Mannering e altri romanzi dello Scott, il Guglielmo Tell, alcune scene del Fausto, lo colpirono vivamente fin d’allora: di Byron, a quel tempo, ammirava più la vita che le poesie (è vero che lo leggeva tradotto in barbara prosa). Lamartine non gli andò mai giù. Gli scritti clandestini di Giuseppe Mazzini, che riceveva
da un suo stretto parente, lo facevan ruggire.... Anche l’Ortis è un libro su cui l’ho visto fremere e piangere.
«Cosa singolare! i libri di erudizione, particolarmente filologica, erano per lui letture gradite, e avidamente cercate, quasi quanto i poeti. Mi ricordo che dopo avere nitidamente trascritto, con una diligenza da benedettino, le sue imitazioni da Orazio — una quarantina di odi, di cui due solamente sono restate negli Juvenilia, — egli cedè volentieri il volumetto manoscritto, in baratto con una vecchia edizione del Malmantileannotato dal Biscioni, e tornò a casa glorioso e trionfante col grosso polveroso volume, prezioso per lui più per le note erudite che per l’arguto testo fiorentino.»
A proposito della sua passione pei libri il Nencioni mi raccontò che il giorno che egli riuscì ad avere, non so se comperate o donategli, le poesie del Foscolo, per le quali spasimava da lungo tempo, tornando a casa, salì in ginocchione la scala che dall’uscio di strada conduceva diritta al povero quartiere, e giunto nella stanza dov’era sua madre, presentatole il libro, volle s’inginocchiasse a baciarlo. La mattina di poi, quando il Gargani andò da lui, lo trovò non ancora finito di vestire, che gli veniva incontro, e lì in cima alla scala, senza lasciarlo entrare, gli lesse ad alta voce, commentando con le sue ammirazioni, una gran parte dei Sepolcri.
Nell’anno 1852, mentre studiava, o a dir meglio (sono sue parole) non studiava affatto filosofiadagli Scolopii, il Carducci fece ilprimo passo verso il numero deipiù, cioèdegli uoministampati. «Lo feci presto, dice, e da buon italiano, con un sonetto, un sonetto d’occasione, e quale occasione! per i coristi del Teatro di Borgo Ognissanti, o salvo il vero, della Piazza Vecchia.... Stavo vicino di casa in via Romana con Emilio Torelli stampatore, e già dei fedeli, dei veramente e onestamente fedeli, di F. D. Guerrazzi. Egli mi chiese il sonetto. Come dir di no a un democratico del ’48, che aveva tale una
franca impostatura tra di soldato e di ciompo (egli fu capitano dei municipali, e sua madre era piemontese), e portava sempre uno smisurato cappello o di felpa o di paglia, all’ombra delle cui grandi ale poteva riparare una cospirazione? Diedi il sonetto; e fu stampato, anonimo. Non me ne ricordo, ma ci doveva essere qualche frase di Armonide Elideo, o, meno arcadicamente, d’Angelo Mazza.
«Il vero primo passo per altro, e questo con la ferma intenzione di peccare, solamente non seguìta dall’effetto, lo avevo mosso qualche mese innanzi. In quegli anni io scrivevo sempre: ammiravo il bello da per tutto, cioè non capivo nulla. Ebbi in una giornata di luglio il coraggio di mettere assieme in tutti i metri che mi corsero per la testa (nessun barbaro: allora, al più, rifacevo alcaiche sul modello del Fantoni) una novella romantica. L’intitolai Amore e Morte. C’era dentro un po’ di tutto — un torneo in Provenza — e il rapimento della regina del torneo fatto da un cavaliere italiano vincitore — e una fuga con dialoghi al lume di luna tra gli abeti — e il fratello della vergine non più vergine che raggiungeva gli amanti in Napoli — e un duello — e la morte del vago — e la monacazione della vaga — e un successivo impazzamento — e l’annessa morte dopo la confessione in
Endecasillabi
Catullïani
Dolci per facili
Modi toscani.
(ROSSETTI, Veggenteinsolitudine.)
Ricordo.... due strofe, quando la regina del torneo posava una ghirlanda su ’l capo del vincitore, che s’era tratto l’elmo:
Qui la bella di Tolosa
Del baron gli occhi fisò, Poi tremante e vergognosa
Chinò gli occhi e sospirò.
Ma una fiamma al roseo volto, Una fiamma le salì
Quando il nero crin disciolto Fra le dita errar sentì.
«Finita che ebbi la novella verso le quattro di sera, e il caldo era grande (come scrivevano i vecchi cronisti), pensai a farla stampare. Perchè no? Leggevo stampati tutti i giorni tanti versi che mi parevano peggio dei miei. L’abate Stefano Fioretti pistoiese compilava allora certo foglio teatrale e letterario, intitolato non ricordo più se l’Arpao il Liutoo il Trovatore o il Menestrello, o quale altro de’ nomi d’oggetti di spogliatoio melodrammatico che usavano ancora su quegli sgoccioli del romanticismo. Mi manca il tempo e la serenità dell’animo a raccogliere e rendere i tratti di ciò ch’era allora l’abate toscano: non prete del tutto, ma nè men secolare: molto arcadicamente o romanticamente letterato: il cappello lungo; cravattina simulante il collare sotto al solino imbiancato co ’l turchinetto; abito moderatamente talare tenuto aperto per lasciar vedere una catenella d’argento a mezzo la sottoveste abbottonata fin molto in su; tutto in nero, s’intende; nero ed argento; in argento legate possibilmente le lenti, pomo d’argento o d’altro metallo biancheggiante nella canna d’India; infine andatura un po’ solenne, ma con passi di minuetto e naso all’aria. Il Fioretti del resto era persona piacente, e galantuomo, e buon compagno: aveva l’ufficio del giornale in un de’ vicoli che rameggiano da via Calzaioli. Salgo le scale con grande trepidazione; il direttore non c’era, c’era la governante, o la cameriera, o la nipote; non so in somma che cosa fosse precisamente. Il che mi piacque, non mica per la cameriera o governante o nipote — che era del resto un bel pezzo di ragazza, tipo fiorentino del Ghirlandaio un po’ volgarizzato ma io, figuratevi, ero troppo fresco dell’AmoreeMortee della mia creazione di Gilda. Mi piacque perchè così potei scrivere una lettera al direttore (a parlare mi sarei imbrogliato), con la quale gli lasciavo e raccomandavo la mia novella: sarei tornato il giorno dopo per la risposta. Tornai; e il piacente abate con squisita cortesia mi fece
capire che la mia novella era troppo lunga e troppo letteraria per un foglio come il suo.
«Rividi poi, circa il 59, e più volte, l’abate Fioretti; e finimmo buoni amici. Mi dava o mi mandava certe sue cantate storiche. Una mi ricordo: Gli Orti Oricellari a tempo dell’ultima cacciata dei Medici da Firenze, fu musicata dal Mabellini per i parentali a Niccolò Machiavelli celebrati in Pistoia la sera del 26 luglio 1863. E me ne ricordo un’aria a più voci tra Palla Rucellai, il Machiavelli figliolo e Zanobi Buondelmonti.
PALLA.
Ah.... del ribelle moto
Côrremo i frutti amari.
MACHIAVELLI.
Ai Medici devoto
Vedrem l’Oricellari?
PALLA.
Tutti i tiranni abomino
Detesto al par di te;
Ma nella plebe instabile
Non so ripor la fe’.
BUONDELMONTI.
Torna a regnare il popolo
Che plebe vil non è.
»Io gli lodai quella cantata. Sicuro! Gli ero debitore dell’avermi risparmiato la stampa della novella. Immaginatevi se i critici italiani avessero poi scoperto che a sedici anni feci una poesia romantica!»[8]
* * *
Finiti nel 1852 gli studi agli Scolopii, il Carducci andò colla famiglia a raggiungere il padre, che fino dall’anno innanzi era andato medico condotto a Celle nel Montamiata. Quivi passò quasi tutto il 1853, riordinando e compiendo con assoluta libertà, che lo condusse ad un classicismo assoluto, i suoi studi letterari. Non li aveva tralasciati mai neppure a Firenze nell’ultimo anno mentre studiava filosofia. Anzi specialmente in quell’anno le conversazioni col Nencioni, i libri ch’ei gli procurava da leggere, quelli che cercava da sè nelle biblioteche fiorentine, dove volle vedere anche i codici, contribuirono a svolgere, non senza un po’ di confusione, le attitudini sue svariatissime, di erudito insieme e d’artista. Dalla lettura dei lirici dei primi secoli nel Manuale del Nannucci, nella raccolta del Valeriani e nei codici della Riccardiana, passava a scrivere odi saffiche o alcaiche ad imitazione d’Orazio, sonetti burleschi e satirici, e, come s’è visto, anche poemetti romantici.
De’ sonetti satirici ne avea composti fin da retorica. I primi a provare la mordacità della sua musa giovanile furono i suoi compagni di scuola. Mi ricordo che il Nencioni, raccontandomi le loro inimicizie e guerricciole di scolari, mi recitava dei pezzi di sonetti carducciani veramente feroci. Il Carducci poi, mandandomi nel settembre del 1860 da Pistoia la intiera raccolta manoscritta di tutti i suoi sonetti burleschi e satirici, mi scriveva: «Ho voluto conservare due di quelli fatti da ragazzo per un’ambizioncella di mostrare come pensavo e sentivo ec. ec., e come presto incominciai l’arringo satirico, pel quale veramente sarei fatto più che per ogni altro.» E prometteva di mandarmi poi altri saggi di poesia satirica puerile, «sciatti saggi, diceva, ma che pur dicono qualche cosa, molto più certo delle poesie serie che facevo a quel tempo.»
Non ricordo bene; ma credo che fra quei sonetti ce ne fosse uno su Celle (ad imitazione di quello del Berni su Verona) il quale cominciava così:
Questa Celle è una terra di Toscana,
ed uno contro un suo compagno di scuola, che avea parlato male di lui.
Quei sonetti li rimandai dopo parecchi anni al Carducci, che ne fece una scelta per la edizione definitiva degli Juvenilia. * * *
Mentre il nostro era a Celle avvenne un fatto che determinò la scelta della sua carriera nella vita.
Fra gli scolari di retorica delle Scuole Pie, in quelli anni che c’era appunto il Carducci, esisteva un’Accademia, che avea nome dei «Risoluti e Fecondi», della quale era come Presidente il Padre Barsottini. Ne facevano parte i migliori alunni, e tra questi naturalmente dei primi il Carducci, il Nencioni e il Gargani. Per una delle tornate di cotesta Accademia, che fu tenuta nel 1853, il Carducci, narra il Borgognoni, mandò alcuni suoi versi sulle Crociate, «i quali, dice egli, oltre che al Padre Barsottini, ebbero la ventura di piacere non poco e a Leopoldo Cempini (stato già amico del Giusti) e al canonico Sbragia, prete di parte moderata scappato nel 1848 in Piemonte col Giorgini, i quali due si trovarono tra gli astanti. Lo Sbragia anzi disse al Barsottini persuadesse il Carducci a concorrere alla Scuola Normale, di cui egli era in allora il rettore. Il Barsottini non si fece pregare, come non si fece pregare il Carducci, il quale concorse, ottenne e andò.»[9] Così avvenne che il Carducci prese la via dell’insegnamento.
Anche entrato alla Scuola Normale, Giosue seguitò ad appartenere all’Accademia, la quale tenne una delle sue più famose sedute l’8 settembre 1854. Ne fu stampato il programma (Firenze, coi tipi Calasanziani, 1854) con questo titolo: «Il Genio cristiano | del medio Evo | in Italia | Trattenimento letterario | dato | nella Sala delle Scuole Pie fiorentine | dagli Accademici | Risolutie Fecondi| la sera