Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and certain other countries.
Published in the United States of America by Oxford University Press 198 Madison Avenue, New York, NY 10016, United States of America.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by license, or under terms agreed with the appropriate reproduction rights organization. Inquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above.
You must not circulate this work in any other form and you must impose this same condition on any acquirer.
CIP data is on file at the Library of Congress ISBN 978–0–19–999099–3
This material is not intended to be, and should not be considered, a substitute for medical or other professional advice. Treatment for the conditions described in this material is highly dependent on the individual circumstances. And, while this material is designed to offer accurate information with respect to the subject matter covered and to be current as of the time it was written, research and knowledge about medical and health issues is constantly evolving and dose schedules for medications are being revised continually, with new side effects recognized and accounted for regularly. Readers must therefore always check the product information and clinical procedures with the most up-to-date published product information and data sheets provided by the manufacturers and the most recent codes of conduct and safety regulation. The publisher and the authors make no representations or warranties to readers, express or implied, as to the accuracy or completeness of this material. Without limiting the foregoing, the publisher and the authors make no representations or warranties as to the accuracy or efficacy of the drug dosages mentioned in the material. The authors and the publisher do not accept, and expressly disclaim, any responsibility for any liability, loss or risk that may be claimed or incurred as a consequence of the use and/or application of any of the contents of this material.
9 8 7 6 5 4 3 2 1
Printed by Sheridan Books, Inc., United States of America
Preface vii
Acknowledgments ix
Abbreviations xi
Part I Common Issues in the Newborn
1. Hypotonia 3
2. Intrauterine Growth Restriction 11
3. Overgrowth 17
4. Twins 25
5. Non-Immune Hydrops 31
6. Teratogenic Agents 37
Part II Cardiovascular System
7. Cardiac Defects 49
8. Heterotaxy 57
Part III Craniofacial System
9. Ear Anomalies 63
10. Eye Anomalies 69
11. Cleft Lip 79
12. Cleft Palate 85
13. Craniosynostoses 91
Part IV Central Nervous System
14. Macrocephaly and Megalencephaly 103
15. Microcephaly 109
16. Cerebellar Anomalies 115
17. Holoprosencephaly 121
18. Hydrocephalus 127
19. Neural Tube Defects 133
20. Perinatal Arterial Stroke 139
Part V Gastrointestinal System
21. Diaphragmatic Hernia 147
22. Gastroschisis 153
23. Omphalocele 157
24. Anorectal Malformations 163
25. Hirschsprung Disease 167
Part VI Genitourinary System
26. Renal and Urinary Tract Anomalies 173
27. Hypospadias 183
Part VII Skeletal System
28. Arthrogryposis 191
29. Clubfoot 197
30. Upper Extremity Anomalies 203
31. Lower Extremity Anomalies 211
32. Polydactyly 217
33. Syndactyly 223
Part VIII Skeletal Dysplasias
34. Skeletal Dysplasias: Overview 231
35. Skeletal Dysplasias: Life-Limiting 235
36. Skeletal Dysplasias: Viable 241
37. Skeletal Dysplasias: Fractures in Infancy 249
Part IX Skin System
38. Skin: Ectodermal Dysplasias 257
39. Skin: Epidermolysis Bullosa 261
40. Skin: Ichthyoses 267
41. Skin: Vascular Malformations 273
42. Skin: Other Disorders 277
Appendix: Syndromes That Commonly Present in the Newborn 283
1S. Trisomy 21 285
2S. Trisomy 18 289
3S. Trisomy 13 293
4S. Turner Syndrome 297
5S. Wolf–Hirschhorn Syndrome 301
6S. Chromosome 5p Deletion Syndrome 305
7S. Chromosome 22q11.2 Deletion Syndrome 309
8S. Achondroplasia 313
9S. Beckwith–Wiedemann Syndrome 317
10S. CHARGE Syndrome 321
11S. Cornelia de Lange Syndrome 325
12S. Diabetic Embryopathy 329
13S. Fetal Alcohol Spectrum Disorder 333
14S. Incontinentia Pigmenti 337
15S. Prader–Willi Syndrome 341
16S. Noonan Syndrome and Related Disorders 345
17S. Smith–Lemli–Opitz Syndrome 349
18S. VATER/VACTERL Association 353
19S. Williams Syndrome 357 Index 361
Preface
In recent decades, stunning advances in neonatal medicine have dramatically reduced the morbidity and mortality of prematurity, sepsis, and respiratory distress. However, as the outcome for these conditions has improved, neonatal medicine has increasingly focused on addressing the needs of patients with congenital anomalies. Concurrently, astonishing progress in clinical genetics and genomics has facilitated diagnosis and delineation of both common and rare malformations and syndromes.
This book was written to assist clinicians who care for newborns with congenital abnormalities in their diagnosis, genomic testing, and management. Our goal was to make the evaluation of common neonatal anomalies and genetic syndromes accessible and understandable. In addition, we hoped that this book might serve as an initial guide for practitioners in areas where clinical genetic expertise is not readily available. As we wrote this book, the testing paradigm shifted to a genomic approach: Chromosome analysis gave way to microarrays, and single gene testing was largely replaced by gene panels and exome sequencing. This book, which was initially intended as a clinical primer, of necessity became a resource for gene-based information as well. Despite our frequent revisions, advances in genetic and genomic testing are certain to outpace the editing process.
We carefully considered how to simplify this complex subject matter to make it easier to use in a busy inpatient intensive care nursery. The 42 main chapters, written in a bulleted format, each focus on a different malformation or common clinical problem, such as hydrocephalus, hypotonia, or the growth-restricted infant, in which genetic diagnoses feature prominently in the differential diagnosis. Each chapter begins with a story—a brief case history illustrating a learning point. Malformations are defined and characterized as isolated or syndromic. Background information is followed by a differential diagnosis and a short guide to evaluation and management. Syndromic descriptions are brief, but the interested reader can obtain additional, updated information through links and references. For this purpose, we have included the unique MIM number that identifies each single gene disorder in the online database Mendelian Inheritance in Man. Most chapters contain “pearls” of practical information gleaned
from our combined experience that have rarely been reported in the medical literature. The book concludes with short summaries of 19 conditions that are commonly diagnosed in neonates, focusing on features that make these syndromes recognizable as well as key points in their management. We used photographs of affected infants, whenever possible, to illustrate the phenotypes at this young age. We are grateful to the many colleagues and patient support organizations that contributed photographs to this effort. Space constraints forced us to omit some and limit other topics. The differential diagnoses are broad but not encyclopedic. Some anomalies that might have filled a chapter of their own (e.g., duodenal atresia, tracheoesophageal fistula, vertebral anomalies and seizures) have been reviewed in an abbreviated form in a differential diagnosis or were left for possible inclusion in a future edition. Similarly, we could not include many
biochemical and metabolic disorders, which could have filled their own book.
Six years after we began, we delivered this book to our patient publisher. The effort involved in writing this book far exceeded our projections, but it is clear to both of us that this joint effort, marked by mutual dedication and hard work, is a better book than either of us could have written alone. This book is not a substitute for a comprehensive genetic consultation by an experienced clinical geneticist or dysmorphologist, but it may provide the framework for initiating a complex workup in the nursery, neonatal intensive care unit, or office setting. We hope it offers timely assistance to you, your precious patients, and their deserving families.
Cynthia J. Curry, MD, and Robin D. Clark, MD May 2018
Acknowledgments
We thank our generous colleagues who provided expert opinions, guidance, and advice during the preparation of this book: John C. Carey, Dian Donnai, William Dobyns, Donna J. Eteson, Jamie Fisher, June-Anne Gold, Karen Gripp, Judith G. Hall, Julie Hoover-Fong, Louanne Hudgins, Kathreen Johnston, Kenneth L. Jones, Marilyn Jones, Jennifer Kalish, Deborah Krakow, Subhadra Ramanathan, Bianca Russell, Eric Villain, Andrew Wilkie, Elizabeth Woods, and any others we may have inadvertently omitted. We owe more than can be acknowledged here to our ever-inspiring mentors: Michael Baraitser, Bryan Hall, Charles Epstein,* David Rimoin,* and Robin Winter.* Our colleagues and genetic support groups who kindly provided photographs are acknowledged in each chapter.
We offer our thanks to the many neonatologists whose referrals and timely photos made this book possible, including David Aguilar, Douglas Deming, Stephen Elliott, Isabel Escalante, Angela Flores, Andrew Hopper, Alok Kumar, Shahriar Mokrian, Juanito Novales, Richard Peverini, William Phaklides, Anand Rajani, Krishna Rajani, Allan Wolpe.
Special thanks goes to Francis Fung, graphic designer at UCSF/Fresno, and Emily Kishibay for scanning countless slides and photographs.
We are grateful to the staff at Oxford University Press for their patience and flexibility, with particular thanks to our editors, Chad Zimmerman and Chloe Layman, for their unfailing encouragement, ready availability, and sound advice.
This book would not exist without our newborn patients. It has been and continues to be a privilege to work with deeply committed and caring families and their inspiring children during what is often the most difficult times of their lives. Through the decades, they have been our teachers as well as our patients. Our gratitude goes to them all.
Last here but first in our hearts, we thank our families: spouses Bjorn Nilson (CJC) and Terry Long (RDC) and our children and grandchildren. May they forgive us for many late dinners, lost evenings and weekends, and missed family events. Their love and support have made this long effort possible.
Robin D. Clark and Cynthia J. Curry August 2018
*Deceased.
* Syndromes that appear with an asterisk (*) in the text are discussed more extensively in their own chapters. The authors ask that readers consult the table of contents for maximum clinical utility in these cases.
Abbreviations
4p-, 5p-, 22q- chromosome deletion syndromes for 4p, 5p, or 22q
UPD unipaternal disomy; maternal (matUPD) or paternal (patUPD)
US ultrasound
VCUG vesicourethrogram
VDRL Venereal Disease Research Laboratory (test for syphilis)
VLCAD very long-chain acyl-CoA dehydrogenase deficiency
VPA valproic acid
VSD ventricular septal defect
VZIG varicella zoster-specific immunoglobulin G
VZV varicella zoster virus
WES whole exome sequencing
WHO World Health Organization
WHS Wolf–Hirschhorn syndrome
Part I
Common Issues
in the Newborn
1
Hypotonia
Clinical Consult
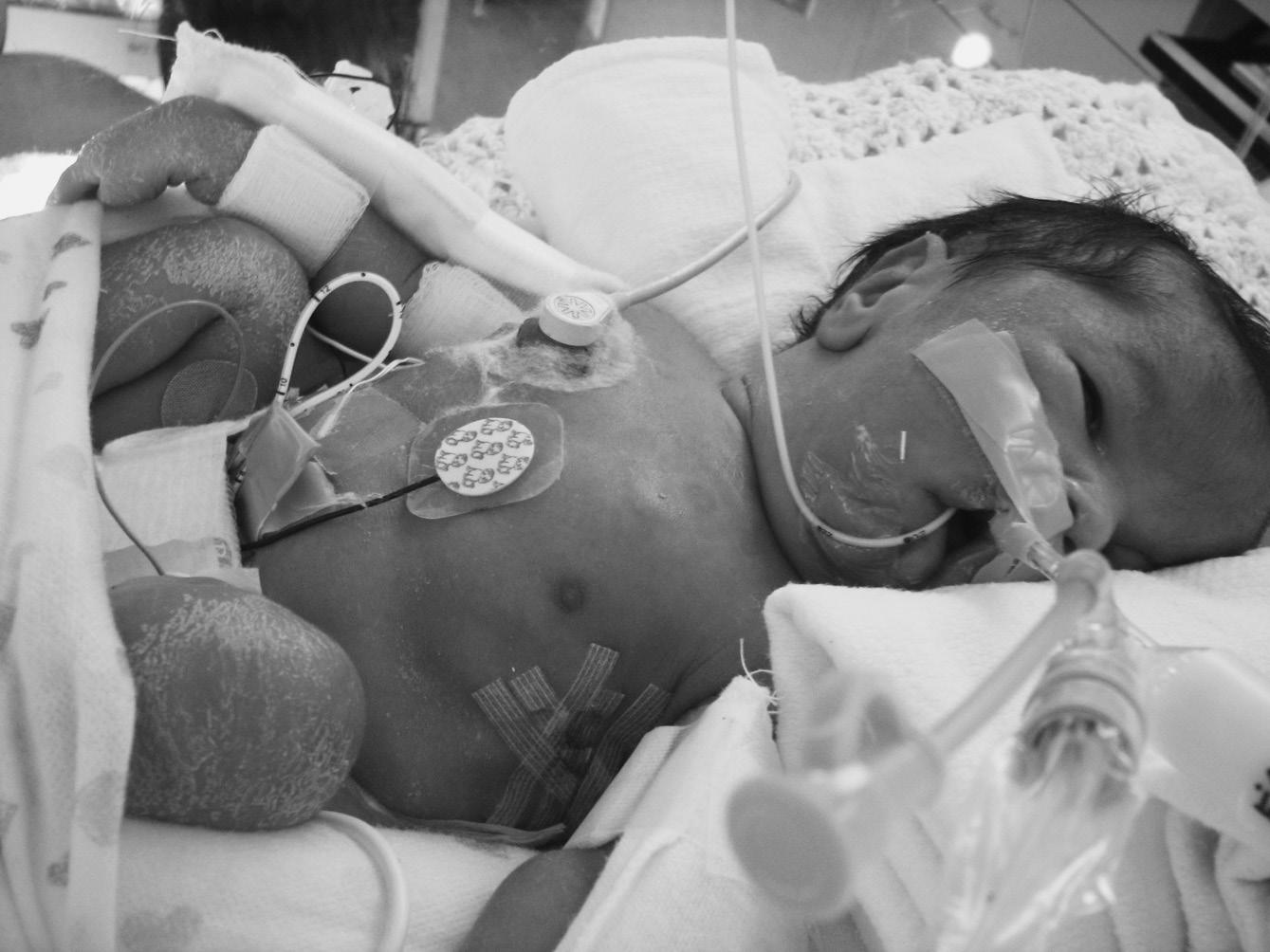
A term infant with hypotonia and bilateral metatarsus adductus presented with relative pulmonary hypoplasia requiring full ventilatory support. The mother had mild polyhydramnios and reported decreased fetal activity. The physical exam was challenging due to multiple tubes and monitoring devices. His mouth was tented (Figure 1.1). He had hypoactive reflexes and minimal spontaneous movements of arms, legs, and fingers. The initial diagnosis was a disorder with Fetal Akinesia sequence, and exome sequencing was considered.
The family history was pertinent for three healthy children. Another child died 2 years previously with presumptive hypoxic ischemic encephalopathy. An autopsy did not reveal an underlying cause. The parents had been given a low recurrence risk. The mother reported no history of weakness or difficulty releasing a grasped object. She had subtle facial weakness. No grasp myotonia was elicited on initial examinations by genetics and neurology consultants, but careful reassessment revealed weak eyelids and suggestive grasp myotonia. Molecular testing for myotonic dystrophy in the infant revealed an expansion of 1,200 CTG repeats in DMPK1, consistent with congenital myotonic dystrophy
Symptoms of myotonic dystrophy type 1 in affected mothers may be subtle and mild. Even experienced consultants can miss this diagnosis, which was the case when the first severely affected child was born to this mildly affected mother.
Definition
• Hypotonia is low muscle tone for age, often caused by weakness or abnormalities of the central nervous system (CNS).
▪ Clinical features: reflexes hypoactive, lack of antigravity movements
• Prenatal findings often associated with hypotonia
⚬ Decreased fetal movements
⚬ Polyhydramnios
⚬ Breech presentation
Differential Diagnosis
• We outline only a few of the hundreds of genetic conditions that can cause neonatal hypotonia. Many chromosomal and microarray abnormalities, single gene disorders, various metabolic diseases, and numerous complex syndromes cause congenital hypotonia. Multiple brain malformations are an important cause of neonatal hypotonia, but these are discussed in other chapters.
• The pace of new gene discoveries in infants with hypotonis is astonishing and as molecular pathways are elucidated, therapeutic targets are emerging.
• Using all tools available, a diagnosis can be achieved in ~90% of hypotonic infants. A detailed physical examination and thorough history remain essential for diagnosis, even in the genomic era.
⚬ More than 50% of patients can be diagnosed by exam and history alone.
⚬ In the remaining patients, a well-defined clinical phenotype facilitates choice of diagnostic tests and interpretation of molecular results.
• Chromosome disorders
⚬ Down syndrome* (MIM 190685)
▪ Clinical features: characteristic facial findings, small ears < 3%, theatrical grimace (when crying), short broad hands with fifth finger clinodactyly, sandal gap, congenital heart defects in almost half
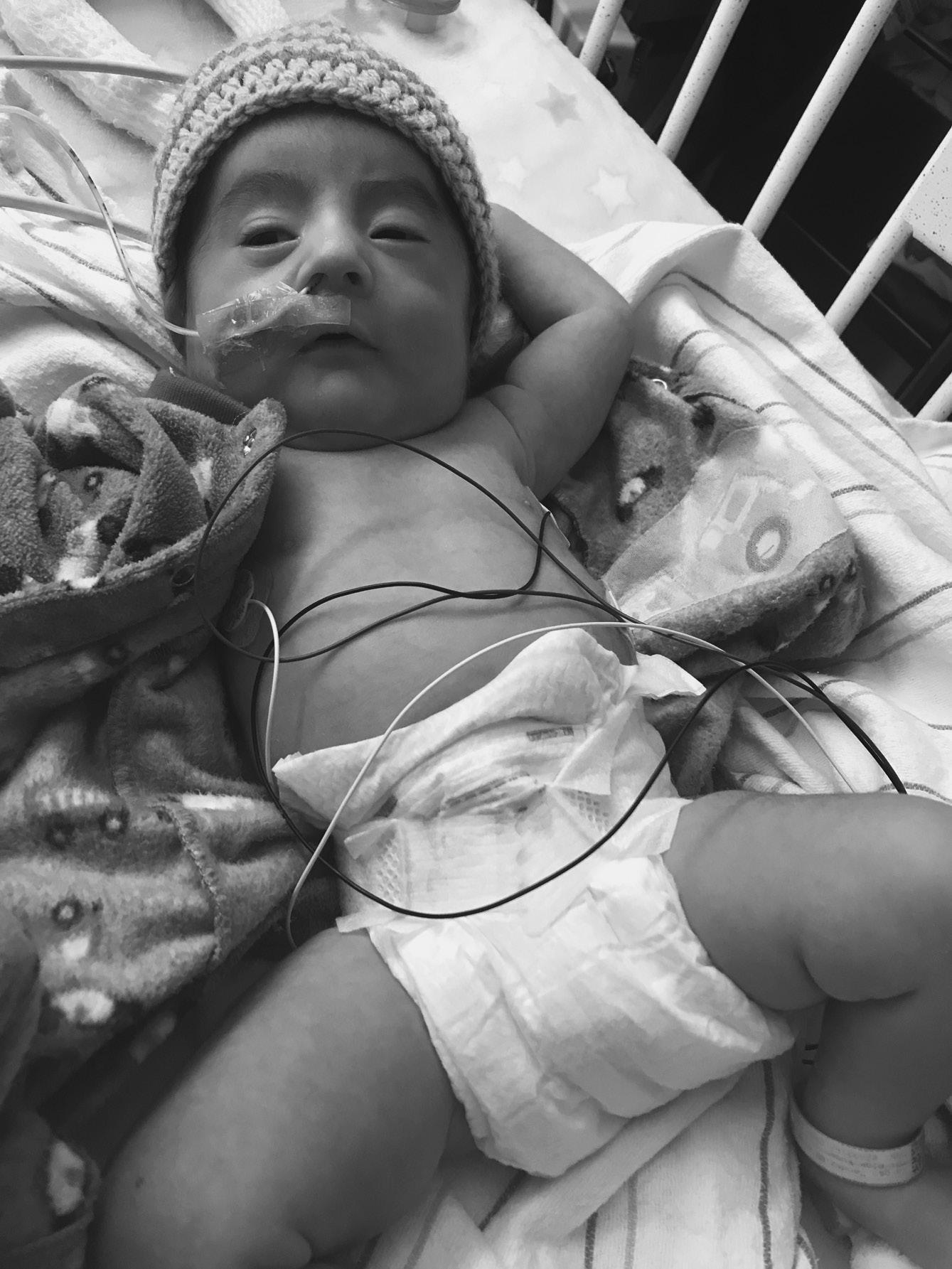
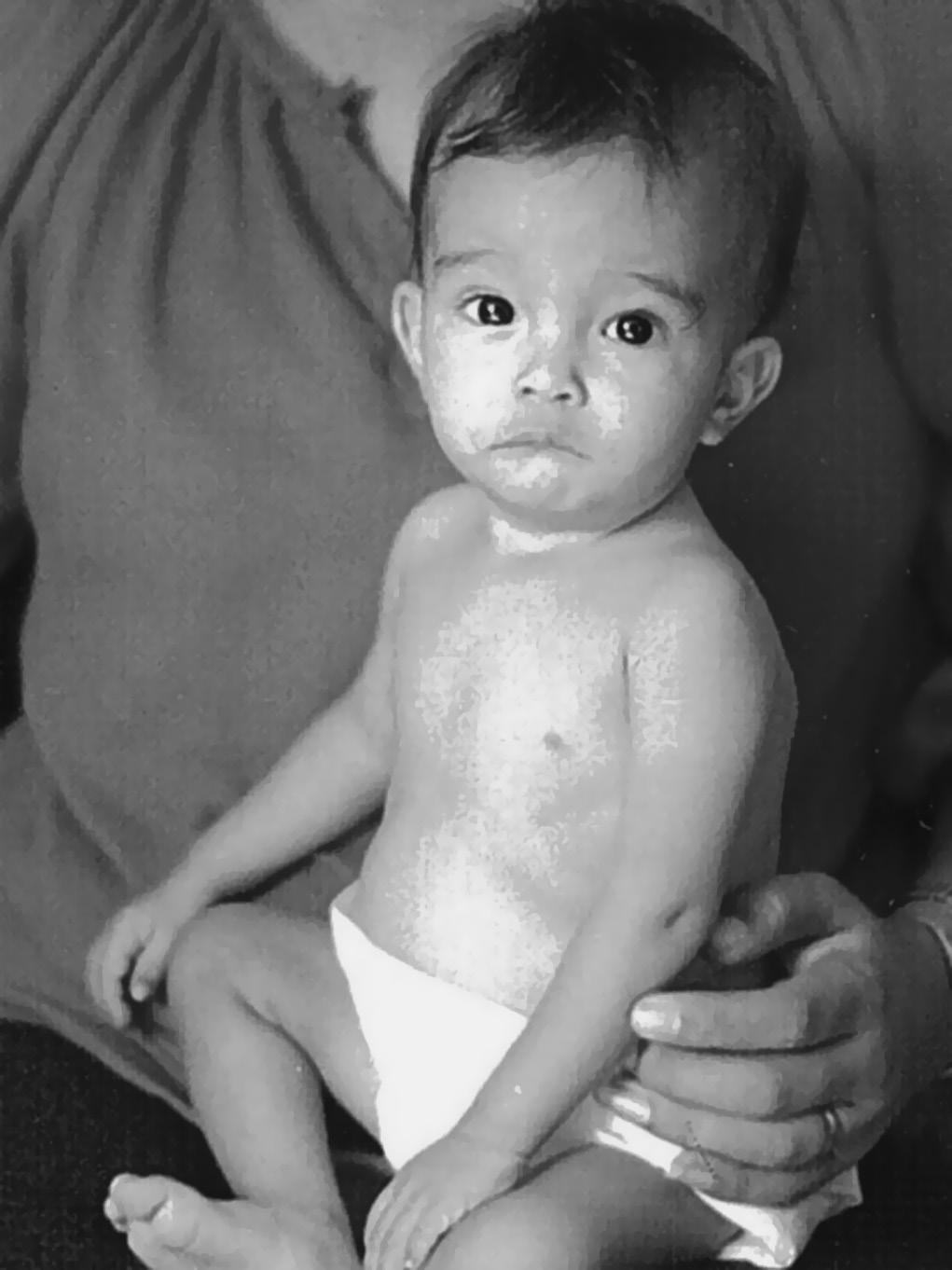
⚬ Prader–Willi syndrome* (MIM 176270)
▪ Missing or inactive paternal contribution at chr 15q11.2
▪ Clinical features: congenital generalized hypotonia a constant feature (Figure 1.2), poor feeding, absent or reduced suck, frog leg positioning, reflexes present, genital hypoplasia, clitoral hypoplasia (often overlooked in females—a helpful sign), cryptorchidism, decreased pigmentation for family background
Pearl: Unexplained poor feeding in a term infant without congenital anomalies warrants a brain MRI. If the MRI is normal, follow with DNA methylation study to rule out Prader–Willi syndrome.
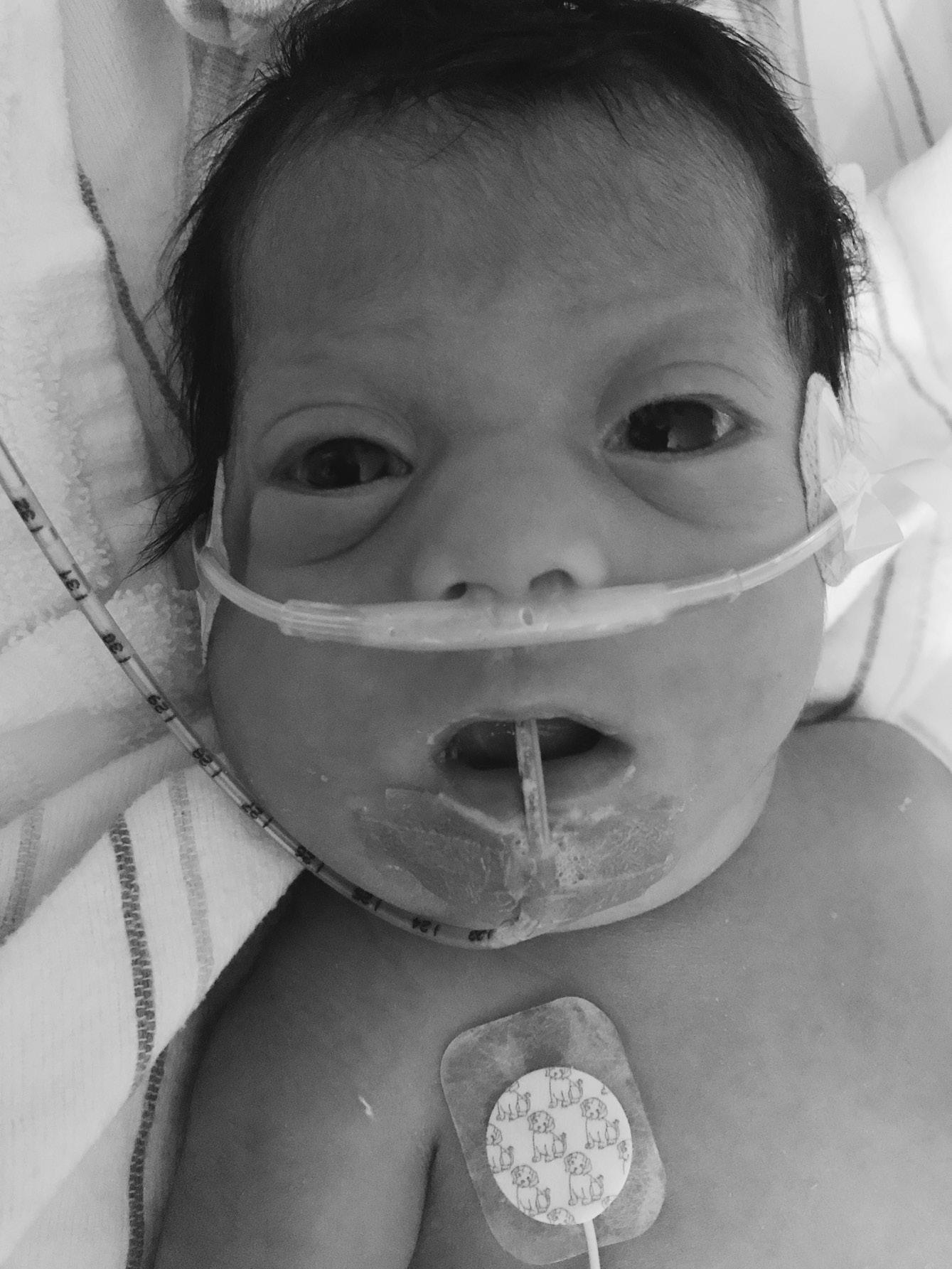
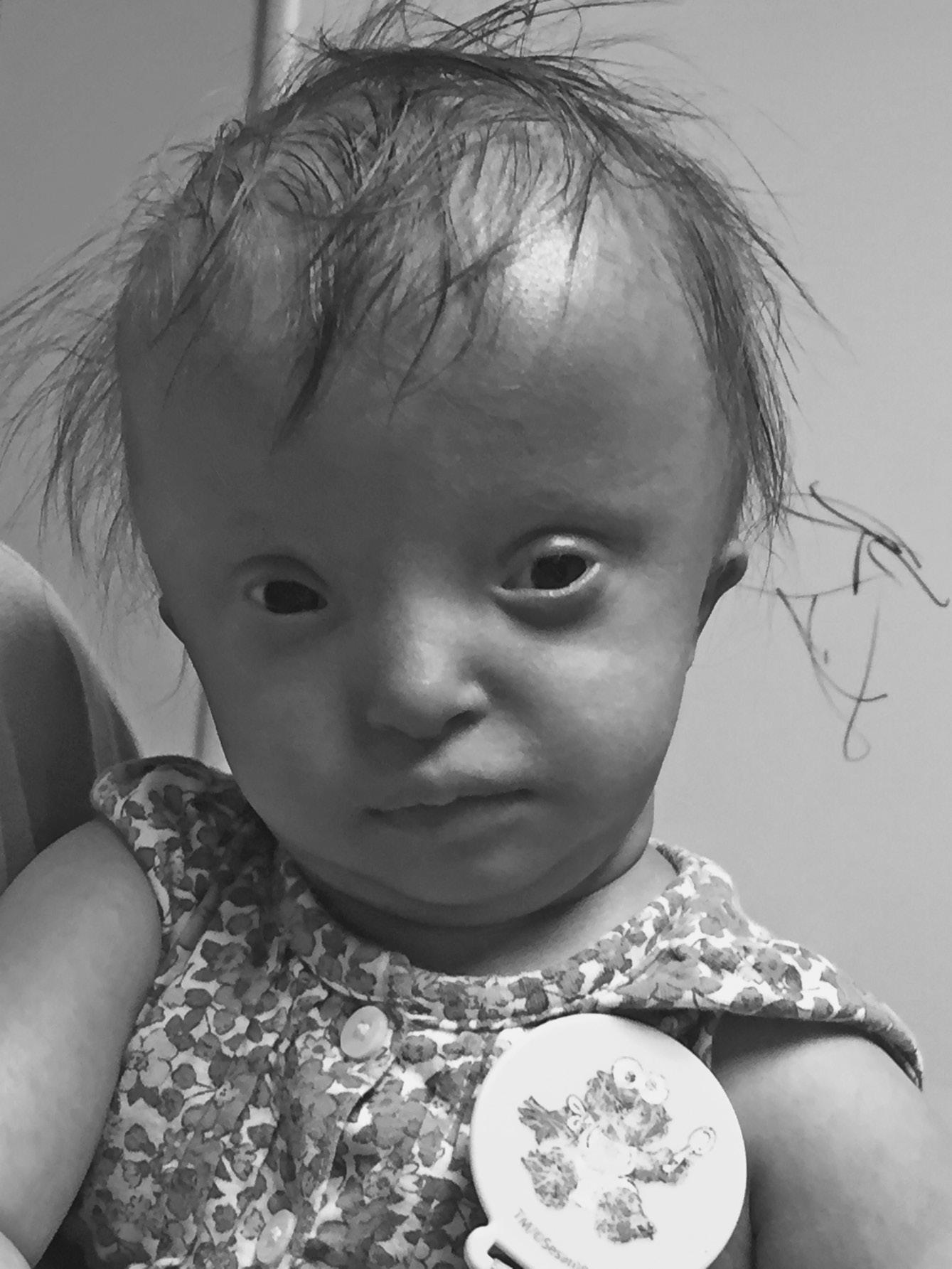
⚬ Smith–Magenis syndrome (MIM 182290)
▪ Deletion of 3.7-Mb interstitial deletion in chromosome 17p11.2
• 10% caused by mutation in the RAI1 gene at 17p11.2
FIGURE 1.1 Congenital myotonic dystrophy in a neonate with pulmonary hypoplasia, tented mouth, facial edema, and hypotonic posture due to
FIGURE 1.2 Hypotonia in baby with Prader–Willi syndrome. Note frog leg position of comfort and typical facial features.
FIGURE 1.3 Smith–Magenis syndrome in an infant with dysmorphic features including small ears, downturned mouth corners, and lower canthal folds Note copious oral secretions reflecting decreased oral–motor tone.
▪ Clinical features: small ears, brachycephaly, midface hypoplasia, prognathism, hoarse cry, cardiac and other defects, seizures, intellectual disability (ID) (Figure 1.3)
▪ Later: sleep problems and characteristic behaviors
▪ Mosaic marker chromosome consisting of two copies of short arm of chr 12; not present in all tissues
▪ Clinical features: congenital diaphragmatic hernia, small ears, bitemporal alopecia, characteristic face and upper lip (see Figure 3.2), feeding problems, seizures, severe ID
⚬ MECP2 duplication at Xq28 (MIM 300260)
▪ Variable, mostly small <1-Mb duplications on chr Xq28, diagnosed on chromosome single nucleotide polymorphism (SNP) microarray; usually diagnosed in males
▪ Clinical features: severe ID, hypotonia and spasticity, recurrent respiratory infections, neonatal renal calculi
• Dysmorphic single gene syndromes
⚬ Kabuki syndrome (MIM 147920)
▪ Autosomal dominant disorder, caused by heterozygous variants in MLL2 (KMT2D)
▪ Clinical features: cleft palate, cardiac defects, genitourinary defects, mildly myopathic face with long palpebral fissures, blue sclerae, everted lateral third of lower eyelids, prominent fingertip pads, short fifth fingers (see Figure )
⚬ Smith–Lemli–Opitz syndrome* (MIM 270400)
▪ Autosomal recessive disorder of cholesterol metabolism caused by variants in DCHR7
▪ A multiple congenital anomaly syndrome with cleft palate, polydactyly, genital ambiguity, cardiac anomalies, and intrauterine growth restriction. Characteristic face with ptosis and anteverted nares.
▪ Hypotonia a constant finding
⚬ Rett syndrome variant (MIM 613454)
▪ Autosomal dominant, caused by de novo heterozygous variant in FOXG1
▪ Autosomal dominant, caused by de novo heterozygous variants in AXL1
▪ Clinical findings: distinctive facial features, variable microcephaly, nevus flammeus, hypertrichosis, severe myopia, unusual posture of arms with flexion at elbows and wrists, hypotonia, severe feeding problems with vomiting, severe ID
▪ Increased risk for Wilms tumor; ultrasound (US) surveillance indicated
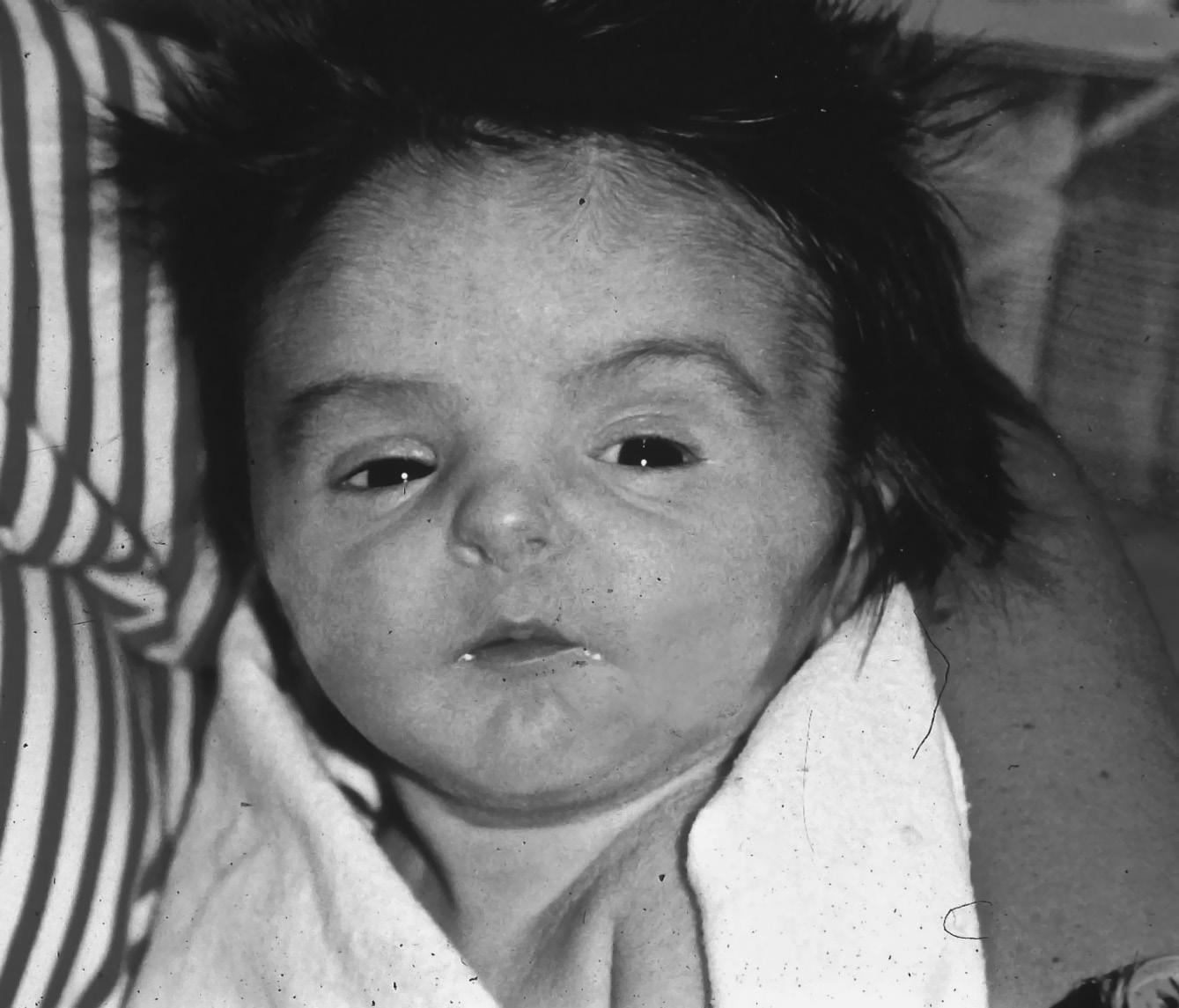
▪ Lethal autosomal recessive disorder, caused by homozygous or compound heterozygous variants in PEX1 and other peroxisome biogenesis genes
▪ Clinical features: characteristic face with high forehead, large fontanelles (Figure 1.4), severe progressive hypotonia, hepatomegaly, liver disease, poor feeding and seizures
⚬ Congenital disorders of glycosylation (CDG-1A, MIM 212605)
▪ Expanding group of autosomal recessive disorders of protein glycosylation
• CDG-1A is the most common type, caused by biallelic variants in PMM2.
▪ Two main groups based on type of biochemical error: type I CDG and type II CDG
▪ Clinical features: highly variable: liver disease, failure to thrive, microcephaly, developmental delay, dysmorphic features, abnormal fat distribution on buttocks and elsewhere
• Brain MRI: cerebellar hypoplasia and other CNS lesions
⚬ Primary coenzyme Q10 (CoQ10 deficiency type I, MIM 607426)
▪ Autosomal recessive disorder, caused by biallelic variants in nine genes involved in synthesis of CoQ
▪ Clinical features: multisystem disease that may present as fatal neonatal encephalopathy with hypotonia
• Steroid-resistant nephrotic syndrome may be initial manifestation. This occasionally may be an isolated finding.
• Hypertrophic cardiomyopathy, retinopathy, optic atrophy, hearing loss
• Later onset forms: slowly progressive multiple system disorder with parkinsonism, cerebellar ataxia, pyramidal dysfunction, dystonia, spasticity, seizures, ID
▪ Labs: biochemical demonstration on frozen muscle homogenates of reduced levels of CoQ10 (ubiquinone) in skeletal muscle or of complex I + III and II + III of the mitochondrial respiratory chain
▪ Treat with oral high-dose CoQ10
• Neonatal death is frequent. There is occasional long survival with late-onset renal disease and neurologic and autonomic symptoms.
Pearl: Serum coenzyme Q levels reflect dietary intake. CoQ10 levels in a tissue biopsy (preferably skeletal muscle) are needed to make the diagnosis of deficiency.
⚬ Other metabolic disorders
▪ Most are autosomal recessive and many are detected on newborn screening.
• Rare inborn errors of metabolism are identified with exome sequencing or targeted panels.
▪ Aminoacidurias
• Methylmalonic aciduria (MIM 251000)
• Maple syrup urine disease (MIM 248600)
• Propionic acidemia (MIM 606054)
▪ Lysosomal storage diseases
• Pompe disease (glycogen storage disease type II, MIM 232300)
• Congenital lower motor neuron diseases
⚬ Spinal muscular atrophy I (Werdnig–Hoffman disease, SMA1, MIM 255300)
▪ Autosomal recessive disorder: >95% of patients have a homozygous deletion in SMN1 on chromosome 5q.
FIGURE 1.4 Infant with Zellweger syndrome. Note tented mouth, hypertelorism, and high forehead.
• Relentless, progressive weakness due to lower motor neuron dysfunction, with eventual respiratory failure
▪ Treatment
• Multiple clinical trials are in progress to ameliorate disease process; see https://clinicaltrials.gov.
• Spinraza (Nusinersen) was approved by the U.S. Food and Drug Administration (FDA) in late 2016. Clinical trials suggest that motor milestones may be maintained when it is given to presymptomatic infants with later infancy onset forms of spinal muscular atrophy.
⚬ Spinal muscular atrophy with respiratory distress (SMARD1, MIM 604320)
▪ Autosomal recessive disorder, caused by biallelic variants in IGHMBP2
⚬ Clinically and genetically heterogeneous group of disorders, with inconsistent and complex terminology and classification systems. Pathogenic variants in the same gene can produce variable phenotypes in both the myopathy and muscular dystrophy categories.
• Autosomal recessive trait, caused by biallelic mutations in one of three collagen VI genes: COL6A1, COL6A2, COL6A3
• Clinical features: most severe end of spectrum has striking joint hypermobility of hands and feet, congenital hip dislocation, clubfeet, elbow and knee contractures, kyphoscoliosis, torticollis, progressive weakness
• Respiratory insufficiency, especially at night
• Normal IQ
▪ Bethlem myopathy (MIM 158810)
• Autosomal dominant trait, allelic to Ullrich congenital muscular dystrophy but milder, caused by heterozygous mutations in same three collagen VI genes
Pearl: A baby with both congenital hypotonia or joint laxity can present with contractures (e.g., SMA1, Marfan syndrome, Loeys–Dietz syndrome, and Ullrich muscular dystrophy).
⚬ Congenital myotonic dystrophy type 1 (MIM 160900) (see Clinical Consult)
▪ Autosomal dominant disorder, caused by expanded number of trinucleotide (CTG) repeats in DMPK on chromosome 19p
• Congenital presentation typically has >1,000 CTG repeats and is inherited from an affected mother, who may be mildly affected or asymptomatic.
▪ Prenatal findings: polyhydramnios, decreased fetal activity, hydrops of upper body
• Affected mothers often deny symptoms. Ask about grasp myotonia in several ways: “Do you ever have difficulty letting go of a hairbrush or the steering wheel?” and “Do your hands ever get ‘stuck’ after picking up a heavy object?”
Pearl: The electromyogram (EMG), diagnostic of myotonic dystrophy in older children and adults, is usually not helpful in neonates.
• Brain MRI: a range from cobblestone (type II) lissencephaly to more focal polymicrogyria; cerebellar malformations, hypoplasia of midline brain structures, ventricular dilatation, Dandy–Walker malformation, posterior occipital encephalocele
▪ Labs: strikingly elevated CK
▪ Variable course from neonatal lethal to death in the first year, to less severe presentations
▪ Autosomal recessive disorder, caused by biallelic variants in RYR1, which encodes the sarcoplasmic reticulum calcium release channel
• Recessive pathogenic variants usually result in a neonatal presentation.
▪ Clinical features: facial weakness, early onset severe progressive scoliosis
• Ophthalmoplegia in some forms of RYR1 myopathy (central core and minicore myopathy) but may be absent in the congenital presentation
▪ May require gastrostomy and nighttime ventilator support
• Nongenetic causes:
⚬ Most are not addressed here, including prematurity, congenital infection, botulism, hypothyroidism, cardiac failure, anemia, hypoxic and hemorrhagic brain lesions, hypothyroidism, and perinatal stroke. Hypoxic ischemic encephalophathy is discussed briefly.
⚬ Hypoxic ischemic encephalopathy (HIE)
▪ HIE is caused by acute or chronic in utero or perinatal asphyxia.
▪ Incidence 1–2/1,000 live births
▪ Common clinical features: cord blood pH <7.0; base excess >12; Apgar score <3 for greater than 5 minutes; early onset moderate or severe encephalopathy; seizures within 12 hours of life, unresponsive to pyridoxine; multi- organ dysfunction: liver, kidney, lung
• Magnetic resonance imaging (MRI)/computerized tomography (CT) show characteristic findings consistent with asphyxia; early brain MRI helps date injury
• Other disease processes must be excluded.
Pearl: Low Apgar scores are neither necessary nor sufficient to make a diagnosis of HIE. Many other conditions cause low Apgar scores.
Evaluation and Management
• Review pregnancy history: polyhydramnios, commonly due to reduced fetal swallowing, abnormal fetal lie, and decreased fetal activity.
⚬ Note delivery complications, Apgar scores, resuscitation status at birth, and cord gases.
• Document the family history: consanguinity, infant death, weakness.
• Examine the parents for weakness and, especially in the mother, for slow grip release and other findings of myotonic dystrophy.
• Examine for distinctive features. Careful phenotyping will increase diagnostic yield.
⚬ Dysmorphic features: e.g. cleft palate, micrognathia, large fontanelles (e.g. Zellweger syndrome)
⚬ Eye exam for retinal dystrophy: e.g. in muscular dystrophy with eye and brain anomalies (Walker–Warburg) syndrome
⚬ Genitalia: e.g. small phallus, clitoral hypoplasia (e.g. Prader–Willi syndrome*), cryptorchidism, genital ambiguity (e.g. Smith–Lemli–Opitz syndrome*)
⚬ Neurological exam: Consult pediatric neurology to refine the neurologic phenotype.
▪ Reflexes (absent in spinal muscular atrophy), extra ocular movements, seizures, head control, spontaneous movement of extremities, muscle bulk and so on.
• Imaging
⚬ Skeletal survey for epiphyseal stippling in suspected peroxisomal disorders
▪ Order lateral view of the foot for heel stippling.
⚬ Abdominal US for organomegaly
⚬ Echocardiogram for cardiomegaly (Pompe disease MIM 232300)
⚬ Neuroimaging establishes the diagnosis in ~25%.
▪ MRI gives best definition of anatomy.
▪ Brain CT scan is useful for hemorrhagic events.
• Genetic testing
⚬ Chromosome analysis (for suspected trisomy 21); SNP microarray in all others. Excessive homozygosity may direct further testing.
⚬ DNA methylation for chr 15q11.2 for suspected Prader–Willi syndrome*
⚬ Metabolic studies for hypotonia without an identified cause
Pearl: Elevated CK narrows the differential diagnosis. Include it in initial round of tests.
⚬ For suspected congenital disorders of glycosylation
▪ Serum carbohydrate-deficient transferrin analysis and plasma N-glycan profile
⚬ For suspected peroxisomal disorders, begin with biochemical testing.
▪ Plasma very long-chain fatty acids (VLCFAs)
• Elevated C26:0 and C26:1 and C24/C22 and C26/ C22 ratios
• When VLCFA is abnormal, consult with reference laboratory for further testing.
• Consider peroxisomal disorders sequencing panel to expedite diagnosis
• Patients with mild Zellweger syndrome may have (near) normal biochemical tests in plasma and urine, and further tests in fibroblasts or a gene panel may be needed.
⚬ Molecular genetic testing should be guided by clinical findings.
⚬ Single gene tests
▪ Spinal muscular atrophy: exonic deletion in SMN1
▪ Congenital myotonic dystrophy: increased CTG trinucleotide repeats in DMPK
⚬ Gene panels or exome sequencing trio testing (child and both parents if possible) for complex or unusual phenotypes
▪ Consult genetic experts to interpret results and variants of unknown significance.
Pearl: Trinucleotide repeat disorders such as congenital myotonic dystrophy cannot be diagnosed with exome sequencing.
⚬ Locate genetic laboratories at
▪ https://www.genetests.org
▪ https://www.ncbi.nlm.nih.gov/gtr
⚬ Reserve invasive testing (EMG, nerve conduction, and muscle biopsy) for infrequent select cases.
▪ Additional tests on muscle biopsy may include immunohistochemistry staining, electron
microscopy, and respiratory chain enzyme analysis of mitochondrial DNA.
▪ Include CoQ10 levels in skeletal muscle homogenates.
▪ Increasingly, molecular testing (panels, exomes) is replacing invasive testing.
Further Reading
Bushby KM, Collins J, Hicks D. (2014) Collagen type VI myopathies. Adv Exp Med Biol. 802:185–99. PMID 24443028
Falsaperla R, Praticò AD, Ruggieri M, et al. (2016) Congenital muscular dystrophy: from muscle to brain. Ital J Pediatr. 42(1):78. PMID 27576556
Gonorazky HD, Bönnemann CG, Dowling JJ. (2018) The genetics of congenital myopathies. Handb Clin Neurol.148:549–564. PMID 29478600
Groen EJN, Talbot K, Gillingwater TH. (2018) Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 14(4):214–24. PMID 29422644
Jungbluth H, Ochala J, Treves S, Gautel M. (2017) Current and future therapeutic approaches to the congenital myopathies. Semin Cell Dev Biol. 64:191–200. PMID 27515125
Salviati L, Trevisson E, Doimo M, Navas P. (2017) Primary coenzyme Q10 deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2017. PMID 28125198
Sparks SE, Krasnewich DM. (2017) Congenital disorders of N-linked glycosylation and multiple pathway overview. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2017. PMID 20301507
Tanaka AJ, Bai R, Cho MT et al. (2015) De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol Case Stud. E pub PMID 27148565
Tarailo-Graovac M, Wasserman WW, Van Karnebeek CD. (2017) Impact of next-generation sequencing on diagnosis and management of neurometabolic disorders: Current advances and future perspectives Expert Rev Mol Diagn. 17:307–9. PMID 28277145
2
Intrauterine Growth
Restriction
Clinical Consult
An 800-g, 33-week gestation infant born to first cousin Palestinian parents had been in the neonatal intensive care unit (NICU) for 6 weeks. He had a benign course, but his growth was poor at less than the third percentile. Consultation revealed an extremely small baby with unusual craniofacial features: prominent eyes, a beaked nose, and microcephaly (Figure 2.1). As the infant’s features became more noticeable and distinct over time, his mother researched her extensive family history and found a history of two other individuals with severe short stature and early death in her multiply consanguineous family. This history, consistent with autosomal recessive inheritance, plus this child’s emerging facial characteristics over time allowed the diagnosis of Majewski (microcephalic) osteodysplastic primordial dwarfism II (MOPD II) (Figure 2.2). The diagnosis was later confirmed by demonstration of a mutation in the causative gene, PCNT, encoding pericentrin. He has had a very complicated course with severe short stature, delayed development, and multiple strokes due to Moyamoya malformation, a known complication of MOPD II.
Definition
• Intrauterine growth restriction (IUGR) is the term used to describe infants who are below –2 SD of that expected for gestational age.
• May be genetic or environmental, including
⚬ Fetal/infant, placental, or maternal causes
Differential Diagnosis
placental Factors
• Common etiology for IUGR
• Placental lesions
⚬ Structural abnormalities are usually associated with mild IUGR.
▪ Single umbilical artery
Maternal Factors
• Maternal weight and nutrition
⚬ Maternal and paternal size and weight; mother’s small size more predictive of IUGR than father’s size
⚬ Maternal weight gain; low birth weight common with famine
⚬ Mother’s birth weight; mothers who were IUGR themselves have an increased risk for IUGR babies.
⚬ Confined placental mosaicism. A trisomic or abnormal cell line is confined to the placenta. Most pregnancy outcomes are normal but IUGR can result
⚬ Small deletions or duplications: 4p-*, 5p-*, 19q13.11, and many other copy number variants detected on microarray
⚬ Uniparental disomy (UPD) for chromosomes 6, 7, 11, 14, 16. Both involved chromosomes originate from one parent only.
▪ Many cases are the result of trisomy or monosomy rescue that is more common with advanced maternal age.
▪ Isodisomy, when both chromosomes are identical, can be detected on SNP microarray, but heterodisomy, when non-identical chromosomes are inherited from one parent, requires specific UPD testing.
• Genomic imprinting errors
⚬ Russell–Silver syndrome (Silver–Russell syndrome, RSS or SRS, MIM 180860)
▪ Sporadic disorder caused by various complex epigenetic mechanisms
• Hypomethylation of chromosome 11p15 imprinting center 1 (ICR1) ~60%
• Maternal uniparental disomy 7 (matUPD7) ~18%
FIGURE 2.1 Infant with Majewski osteodysplastic dysplasia II as a newborn. Note prominent nose, prominent eyes, and small jaw.
FIGURE 2.2 Same child as shown in Figure 2.1 at 8 months of age. Features are more recognizable over time.
• Chromosome 11p15 rearrangements
• CDKN1C duplications or gain-of-function variants
• Heterozygous mutations in HMGA2 and paternally inherited variants in IGF2 account for rare familial cases of RSS.
▪ Diagnosis based on compilation of features that can lead to both overdiagnosis and underdiagnosis
• Clinical features: prenatal growth restriction, height and weight <–2 SD, relative macrocephaly at birth, body asymmetry (frequent, not invariable) (Figure 2.3)
▪ Cornelia de Lange syndrome*: limb anomalies, diaphragmatic hernia
• Congenital Infection: <5% of IUGR
⚬ Toxoplasmosis, cytomegalovirus, malaria, syphilis, Zika, and herpes are most common. Bacterial infections (e.g., tuberculosis and Listeria) are rare.
• Endocrine and metabolic: rare
⚬ Laron dwarfism (MIM 262500)
▪ Autosomal recessive disorder due to pathogenic variants in growth hormone receptor gene
• Primordial dwarfism: In these syndromes, infants have low birth weights and in general remain small throughout life. Unlike the primary skeletal dysplasias, they are proportionate. Microcephaly accompanies several of these rare conditions.
⚬ Microcephalic osteodysplastic dwarfism II (MOPD, MIM 210720) (See Clinical Consult)
▪ Autosomal recessive due to biallelic PCNT variants
▪ Severe IUGR
▪ Unusual facial features, including prominent eyes, beaked nose, a high squeaky voice; small and often dysplastic or missing teeth; usually mild ID but can be severe
⚬ Seckel syndrome (MIM 2106000)
▪ Autosomal recessive disorder with significant clinical and molecular heterogeneity. Eight genes to date involving cell cycle regulation.
▪ IUGR (not as severe as MOPD II), microcephaly, distinctive facial features; ID not as severe as predicted by head size
⚬ Meier–Gorlin syndrome (ear, patella, short stature syndrome, MIM 224690)
▪ Autosomal recessive syndrome: multiple genes in the DNA pre-initiation complex including ORC1, ORC4, ORC6, and CDC45 and others
▪ Clinically variable, with prenatal IUGR, microcephaly, and classic features of microtia, short stature, and hypoplasia/ absence of patellae seen in ~80%. Intelligence can be normal or impaired (Figure 2.5)
• Lipodystrophies and neonatal progeroid syndromes
▪ Rare syndrome caused by de novo heterozygous variants in PIK3R1
▪ Clinical features: IUGR, short stature, lack of subcutaneous fat, Rieger eye anomaly in some
• Triangular face, thin alae nasi (Figure 2.6)
• Hearing loss common
Pearl: Downregulation of genes in the PIK–AKT–mTOR pathway is the basis of SHORT syndrome and related overlapping phenotypes. This syndrome is a “mirror image” of the activating mutations in this pathway that cause macrocephaly/hemimegalencephaly/overgrowth syndromes.
• Cutis Laxa IIA (CLIIA, MIM 219200)
⚬ Autosomal recessive disorder with a defect in N- and Oglycosylation due to variants in ATP6V0A2. Other types do not have demonstrable metabolic error.
⚬ Clinical features: IUGR, failure to thrive, enlarged anterior fontanel, congenital hip dislocation, inguinal hernia, high myopia
▪ Loose skin, joint hypermobility, long philtrum
FIGURE 2.5 Meier–Gorlin syndrome. Note sparse scalp hair and eyebrows, and full lips. Ears were also very small.
FIGURE 2.6 SHORT syndrome. Note thin alae nasi and small jaw and mouth.
▪ Cortical and cerebellar malformations (cobblestone lissencephaly) in most is associated with severe developmental delay, seizures, and neurologic regression.
• Cutis laxa IIIA—DeBarsy syndrome (MIM 219150)
⚬ Autosomal recessive, pathogenic variants in ALDH18A1
⚬ Clinical features: IUGR, failure to thrive, corneal clouding, cataracts
▪ Progeroid appearance, pinched nose, small mouth, loose skin, hyperextensibility
⚬ Autosomal recessive disorder; recent report of biallelic variants in POL3RA
⚬ Clinical features: pseudohydrocephalic appearance with prominent forehead vessels
▪ Late-onset IUGR in utero; failure to thrive; sparse hair, brows, and lashes; natal teeth
▪ Variable ID and early death
• Skeletal dysplasias*
⚬ Disproportionately short long bones and abnormal findings on skeletal X-rays can be subtle in newborns who present primarily with IUGR.
• SHOX deficiency (MIM 312865)
⚬ X-linked gene in the pseudoautosomal region of the short arm of the X chromosome at Xp22.3. Both males and females have functional SHOX genes so the inheritance is pseudoautosomal dominant. Variants cause non-specific short stature and IUGR. Loss of the SHOX gene is the probable cause of short stature in Turner syndrome.*
⚬ Leri–Weill dyschondrosteosis (MIM 127300) is a mild skeletal dysplasia caused by heterozygous variants or deletion (80%) in SHOX
▪ Clinical features: mesomelic short stature (short forearms), Madelung deformity of the wrist. More severe in females. Skeletal changes usually not seen in infancy
⚬ Langer mesomelic dysplasia (MIM 249700)
▪ A more severe skeletal dysplasia with extreme mesomelic shortening caused by homozygous variants in SHOX
Pearl: SHOX deficiency, which causes 2–15% of idiopathic IUGR, has a prevalence of at least 1:1000 births.
⚬ Treatment with growth hormone is effective, so early diagnosis is important.
⚬ Check parents’ stature and examine them for mesomelia and Madelung deformity, more severe in females.
• Spondyloepiphyseal dysplasia congenita (MIM 183900) and Three M syndrome (MIM 273750) are skeletal dysplasias that may present primarily with IUGR. The skeletal features may be subtle in newborns (see Skeletal Dysplasias-Viable*).
Evaluation and testing
• Prenatal and family histories
⚬ Document known risk factors, parental heights, and birth weights.
• Physical examination
⚬ Measure skeletal proportions to detect disproportionate skeletal dysplasia that may present as IUGR (span, upper/ lower segment ratio)
⚬ Examine for limb asymmetry, which is common in Russell–Silver syndrome.
⚬ Subcutaneous fat stores: Decreased fat may suggest malnutrition in utero or a lipodystrophy.
⚬ Note dysmorphic facial features and other anomalies, which may suggest chromosomal aneuploidy or syndrome.
• Genetic testing
⚬ Gross and histologic evaluation of placenta is indicated in all cases of IUGR.
⚬ Chromosomal microarray is indicated in IUGR, with or without dysmorphic features. Lack of striking dysmorphic features does not rule out a chromosomal cause (especially ring chromosomes or microdeletions).
⚬ Other tests depend on clinical findings and course.
⚬ Perform skeletal radiographs when short long bones are suspected.
⚬ Perform cranial imaging with IUGR and microcephaly (non-sedated MRI optimal).
⚬ When placenta is abnormal and thrombophilia is suspected, consider testing mother for antiphospholipid antibody syndrome, factor V Leiden, prothrombin mutation G20210A, protein C activity, and protein S activity levels (defer last test to 6 weeks postpartum because pregnancy lowers levels).
▪ In neonates: factor V Leiden and prothrombin 20210 only
⚬ For suspected IMAGe syndrome, rapid assessment and treatment for adrenal insufficiency, CDKN1C sequencing and deletion/duplication analysis to confirm diagnosis.
⚬ For suspected Russell–Silver syndrome, start with 11p15 methylation studies and microarray; more studies may be indicated, especially if a parent is also affected (HMGA2, paternally inherited IGF2).
⚬ For suspected SHORT syndrome, consider clinical sequencing of PIK3R1 or exome sequencing. Follow chronologically before testing.
⚬ For suspected SHOX deficiency, perform SNP microarray first and then, if negative, consider SHOX sequence analysis or gene panel testing.
⚬ For suspected IGF1 spectrum disorders, order growth hormone levels and other endocrine investigations: IGF1 and IGF3.
⚬ Intrauterine growth restriction sequencing panels or microcephalic primordial dwarfism gene panels may be