The esc textbook of sports cardiology 1st edition antonio pelliccia 2024 scribd download
The ESC Textbook of Sports Cardiology
1st Edition Antonio Pelliccia
Visit to download the full and correct content document: https://ebookmass.com/product/the-esc-textbook-of-sports-cardiology-1st-edition-anto nio-pelliccia/
More products digital (pdf, epub, mobi) instant download maybe you interests ...
The ESC Textbook of Cardiovascular Medicine (Third Edition)
Edited by A. John Camm, Thomas F. Lüscher, Gerald Maurer and Patrick W. Serruys
The ESC Textbook of Intensive and Acute Cardiovascular Care (Second Edition)
Edited by Marco Tubaro, Pascal Vranckx, Susanna Price, and Christiaan Vrints
The ESC Textbook of Cardiovascular Imaging (Second Edition)
Edited by Jose Luis Zamorano, Jeroen Bax, Juhani Knuuti, Udo Sechtem, Patrizio Lancellotti, and Luigi Badano
The ESC Textbook of Preventive Cardiology
Edited by Stephan Gielen, Guy De Backer, Massimo Piepoli, and David Wood
The EHRA Book of Pacemaker, ICD, and CRT Troubleshooting: Case-based learning with multiple choice questions
Edited by Haran Burri, Carsten Israel, and Jean-Claude Deharo
The EACVI Echo Handbook
Edited by Patrizio Lancellotti and Bernard Cosyns
The ESC Handbook of Preventive Cardiology: Putting prevention into practice
Edited by Catriona Jennings, Ian Graham, and Stephan Gielen
The EACVI Textbook of Echocardiography (Second Edition)
Edited by Patrizio Lancellotti, José Luis Zamorano, Gilbert Habib, and Luigi Badano
The EHRA Book of Interventional Electrophysiology: Case-based learning with multiple choice questions
Edited by Hein Heidbuchel, Mattias Duytschaever, and Haran Burri
The ESC Textbook of Vascular Biology
Edited by Robert Krams and Magnus Bäck
The ESC Textbook of Cardiovascular Development
Edited by José M. Pérez-Pomares and Robert Kelly
The EACVI Textbook of Cardiovascular Magnetic Resonance
Edited by Massimo Lombardi, Sven Plein, Steffen Petersen, Chiara BucciarelliDucci, Emanuela Valsangiacomo Buechel, Cristina Basso, and Victor Ferrari
The ESC Textbook of Sports Cardiology
Edited by Antonio Pelliccia, Hein Heidbuchel, Domenico Corrado, Mats Börjesson, and Sanjay Sharma
Forthcoming
The ESC Handbook on Cardiovascular Pharmacotherapy
Edited by Juan Carlos Kaski and Keld Kjeldsen
For a full listing of all ESC Educational Publications please visit: https://www.escardio.org/Education/Textbooks
The ESC Textbook of Sports Cardiology
Edited by Antonio Pelliccia
Hein Heidbuchel
Domenico Corrado
Mats
Börjesson
Sanjay Sharma
Great Clarendon Street, Oxford, OX2 6DP, United Kingdom
Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and in certain other countries
The moral rights of the authors have been asserted
Impression: 1
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by licence or under terms agreed with the appropriate reprographics rights organization. Enquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above
You must not circulate this work in any other form and you must impose this same condition on any acquirer
Published in the United States of America by Oxford University Press 198 Madison Avenue, New York, NY 10016, United States of America
British Library Cataloguing in Publication Data
Data available
Library of Congress Control Number: 2018951253
ISBN 978–0–19–108506–2
Printed in Great Britain by Bell & Bain Ltd., Glasgow
Oxford University Press makes no representation, express or implied, that the drug dosages in this book are correct. Readers must therefore always check the product information and clinical procedures with the most up-to-date published product information and data sheets provided by the manufacturers and the most recent codes of conduct and safety regulations. The authors and the publishers do not accept responsibility or legal liability for any errors in the text or for the misuse or misapplication of material in this work. Except where otherwise stated, drug dosages and recommendations are for the non-pregnant adult who is not breast-feeding
Links to third party websites are provided by Oxford in good faith and for information only. Oxford disclaims any responsibility for the materials contained in any third party website referenced in this work.
Contents
Abbreviations viii
Contributors xi
SECTION 1
Physiology of cardiovascular response to exercise and cardiac remodelling
1.1 Cardiovascular response induced by exercise 3
1.1.1 Physiology of exercise 3
Andrew D’Silva and Sanjay Sharma
1.2 Long-term adaptation to exercise: athlete’s heart and vascular adaptations 9
1.2.1 Structural and functional adaptations in the athlete’s heart 9
Antonio Pelliccia and Stefano Caselli
1.2.2 Impact of sporting discipline, gender, ethnicity, and genetics on the athlete’s heart 20 Nabeel Sheikh
1.2.3 The athlete’s heart in children and adolescents 32
Graham Stuart and Guido E. Pieles
1.2.4 Vascular remodelling 41
Stephan Gielen, M. Harold Laughlin, and Dirk J. Duncker
SECTION 2
Clinical evaluation of the athlete’s heart
2.1 History and physical examination 51
2.1.1 History and physical examination 51
Maurizio Schiavon, Alessandro Zorzi, and Domenico Corrado
2.2 The electrocardiogram in the athlete 57
2.2.1 The electrocardiogram in the athlete 57 Alessandro Zorzi and Domenico Corrado
2.2.2 Common ECG patterns in the athlete’s heart 68
Ricardo Stein and Victor Froelicher
2.2.3 Overlap ECG patterns in the athlete’s heart and cardiomyopathies 77
Harshil Dhutia and Michael Papadakis
SECTION 3
Additional testing in the evaluation of the athlete’s heart
3.1 Exercise testing 87
3.1.1 Protocols of exercise testing in athletes and cardiopulmonary testing: assessment of fitness 87
Marco Guazzi and Paolo Emilio Adami
3.1.2 Evaluation of ischaemia, blood pressure, QT interval, and arrhythmias 98
Frédéric Schnell and François Carré
3.2 Arrhythmia registration 107
3.2.1 Ambulatory (24-hour Holter monitoring, event recorders) and signal-averaged ECG for arrhythmia registration in the athlete’s heart 107 Mahdi Sareban and Josef Niebauer
3.2.2 Class 1 anti-arrhythmic drug provocation test 114
Matthias Antz
3.2.3 Electrophysiological study 116
Matthias Antz
3.3 Imaging the athlete’s heart: anatomical and functional 120
3.3.1 Echocardiogram: morphological and functional evaluation including new echocardiographic techniques 120
Stefano Caselli and Flavio D’Ascenzi
3.3.2 Cardiac magnetic resonance imaging 140
Guido Claessen and André La Gerche
3.3.3 Coronary computed tomography 153
Stefan Möhlenkamp
3.3.4 Nuclear imaging 159
Stefan Möhlenkamp
3.3.5 Coronary angiography 162
Stefan Möhlenkamp
3.4 Genotyping 166
3.4.1 Indications for genetic testing in athletes and its application in daily practice 166
Andrea Mazzanti, Katherine Underwood, and Silvia G. Priori
SECTION 4
Cardiac diseases of interest in sports cardiology
4.1 Myocardial and coronary diseases 179
4.1.1 Hypertrophic cardiomyopathy in athletes 179
Aneil Malhotra and Sanjay Sharma
4.1.2 Arrhythmogenic cardiomyopathy and sudden death in young athletes: causes, pathophysiology, and clinical features 184
Gaetano Thiene, Kalliopi Pilichou, Stefania Rizzo, and Cristina Basso
4.1.3 Myocarditis in athletes 201
Martin Halle
4.1.4 Differentiating athlete’s heart from left ventricular non-compaction cardiomyopathy 209
Andrew D’Silva and Sanjay Sharma
4.1.5 Congenital coronary artery anomalies 217
Cristina Basso, Carla Frescura, Stefania Rizzo, and Gaetano Thiene
4.2 Valvular and aortic disease 226
4.2.1 Mitral valve prolapse in relation to sport 226
Christian Schmied and Sanjay Sharma
4.2.2 Bicuspid aortic valve disease and competitive sports: key considerations and challenges 233
Benjamin S. Wessler and Natesa G. Pandian
4.2.3 The athlete with congenital heart disease 238
Guido E. Pieles and Graham Stuart
SECTION 5
Rhythm disorders of interest in sports cardiology
5.1 Channelopathy in athletes 253
Nicole M. Panhuyzen-Goedkoop and Arthur A.M. Wilde
5.2 Ventricular tachyarrhythmias 265
Eduard Guasch and Lluís Mont
5.3 Supraventricular tachyarrhythmias 277
Matthias Wilhelm
5.4 Pre-excitation and conduction abnormalities 288
Pietro Delise
SECTION 6
Sudden cardiac death in athletes
6.1 Incidence of sudden cardiac death in athletes 299
Jonathan A. Drezner and Kimberly G. Harmon
6.2 Cardiovascular causes of sudden death in athletes 309
Cristina Basso, Stefania Rizzo, and Gaetano Thiene
6.3 The risk, aetiology, clinical features, management, and prevention of exercise-related sudden cardiac death and acute cardiac events in adult athletes 321
Paul D. Thompson
6.4 Less frequent causes of sudden cardiac death 328
6.4.1 Less frequent causes of SCD (commotio cordis): non-cardiac causes (drug abuse, hyperpyrexia, rhabdomyolysis, sickle cell anaemia)— Part 1 328
Erik Ekker Solberg and Paolo Emilio Adami
6.4.2 Less frequent causes of SCD (aortic rupture): non-cardiac causes (asthma, extreme environmental conditions (heat, cold, altitude))—Part 2 332
Erik Ekker Solberg and Paolo Emilio Adami
6.5 Pre-participation screening of young competitive athletes 339
Domenico Corrado and Alessandro Zorzi
6.6 Cardiovascular screening of adult/senior competitive athletes 352
Luc Vanhees and Mats Börjesson
6.7 Cardiovascular screening of children and adolescent athletes (<14 years) 359
Massimo Chessa, Werner Budts, and Javier Fernandez Sarabia
SECTION 7
Sports eligibility in athletes with cardiac abnormalities
7.1 Criteria and considerations relative to safe participation in sport for athletes with cardiac abnormalities 369
Antonio Pelliccia, Hein Heidbuchel, Domenico Corrado, Sanjay Sharma, and Mats Börjesson
SECTION 8
Exercise prescription for cardiovascular health
8.1 Physical activity and leisure-time exercise prescription for sedentary/untrained individuals 381
Massimo F. Piepoli, Mikael Dellborg, and Mats Börjesson
8.2 Monitoring exercise programmes and improving cardiovascular performance 389
Stephan Mueller, Flavia Baldassarri, Julia Schoenfeld, and Martin Halle
SECTION 9
Cardiac safety at sports facilities
9.1 Resuscitation on the field: basic and advanced life support and automatic external defibrillators 403
Mark S. Link and Mark Estes III
9.2 Cardiac safety at sports events: the medical action plan 411
Luis Serratosa, Efraim Kramer, and Mats Börjesson
9.3 Cardiac safety at fitness centres 419
Erik Ekker Solberg, Jakob Johansson, and Alessandro Biffi
SECTION 10
Cardiovascular effects of substances of abuse/doping
10.1 World Anti-Doping Agency (WADA) and International Olympic Committee (IOC) list of prohibited substances and methods and their cardiovascular effects 427
Josef Niebauer and Carl Johan Sundberg
10.2 Nutrition and ergogenic aids prescription for competitive athletes 433
Ronald J. Maughan and S.M. Shirreffs
SECTION 11
Hypertension in athletes
11.1 Diagnosis and management of hypertension in athletes 447
Stefano Caselli and Josef Niebauer Index 453
Abbreviations
AAS androgenic anabolic steroids
AC arrhythmogenic cardiomyopathy
ACC American College of Cardiology
ACE angiotensin-converting enzyme
ACEi angiotensin convertase inhibitor
ACS acute coronary syndrome
ACSM American College of Sports Medicine
AECVP Association for European Cardiovascular Pathology
AED automated external defibrillator
AF atrial fibrillation
AFL atrial flutter
AHA American Heart Association
ALCAPA anomalous left coronary artery from the pulmonary artery
ALS advanced life support
AMI acute myocardial infarction
AMS acute mountain sickness
ANS autonomic nervous system
Ao aorta
AoV aortic valve
APB atrial premature beat
APMHR age-predicted maximal heart rate
ARB angiotensin receptor blocker
ARVC arrhythmogenic right ventricular cardiomyopathy
ARVD/C arrhythmogenic right ventricular dysplasia/cardiomyopathy
T-EPS trans-oesophageal electrophysiological study
TGA transposition of the great arteries
TLOC transient loss of consciousness
TOE trans-oesophageal echocardiography
TUE therapeutic use exemption
TV tricuspid valve
TWI T-wave inversion
VA ventricular arrhythmia
VCO2 carbon dioxide production
Vd ventilatory dead space
VE minute ventilation
VO2 oxygen consumption
VOC venue operations centre
VPB ventricular premature beat
VR virtual reality
VSD ventricular septal defect
Vt tidal volume
VT ventilatory threshold, ventricular tachycardia
VUS variant of uncertain significance
WADA World Anti-Doping Agency
WPW Wolff–Parkinson–White
Contributors
Paolo Emilio Adami
Sports Medicine and Science Institute of the Italian Olympic Committee, Rome, Italy
Matthias Antz
Department of Electrophysiology, Hospital Braunschweig, Braunschweig, Germany
Flavia Baldassarri
Department of Prevention, Rehabilitation and Sports Medicine, Technical University of Munich (TUM), Munich, Germany
Cristina Basso
Cardiovascular Pathology Unit, Department of Cardiac, Thoracic, and Vascular Sciences, University of Padua Medical School, Padua, Italy
Alessandro Biffi
Institute of Sport Medicine and Science, Rome, Italy
Mats Börjesson
Institute of Neuroscience and Physiology and Institute of Food and Nutrition, and Sports Science, University of Gothenburg, and Sahlgrenska University Hospital/Östra, Gothenburg, Sweden
Werner Budts
Cardiovascular Diseases, University Hospitals Leuven, Leuven, Belgium
François Carré
Department of Sports Medicine, Pontchaillou Hospital, Rennes, and Department of Physiology, Rennes 1 University, Rennes, France
Stefano Caselli
Institute of Sports Medicine and Science, Rome, Italy
Massimo Chessa
Paediatric and Adult Congenital Heart Centre, IRCCSPoliclinico San Donato University Hospital, Milan, Italy
Guido Claessen
Department of Cardiovascular Medicine, University Hospital Leuven, Leuven, Belgium
Domenico Corrado
Department of Cardiac, Thoracic, and Vascular Sciences, University of Padua Medical School, Padua, Italy
Flavio D’Ascenzi
Department of Medical Biotechnologies, Division of Cardiology, University of Siena, Siena, Italy
Pietro Delise
Department of Cardiovascular, Neurological, and Kidney Diseases, Division of Cardiology, Hospital of Conegliano, Veneto, Italy
Mikael Dellborg
Department of Medicine, Sahlgrenska Academy, University of Gothenburg, and Adult Congenital Heart Unit, Sahlgrenska University Hospital/Östra, Gothenburg, Sweden
Harshil Dhutia
Cardiovascular and Cell Sciences Research Centre, St George’s University of London, London, UK
Jonathan Drezner
Center for Sports Cardiology, Department of Family Medicine, University of Washington, Seattle, WA, USA
Andrew D’Silva
St George’s University of London and St George’s University Hospital NHS Foundation Trust, London, UK
Dirk J. Duncker
Department of Cardiology, Thoraxcenter, Erasmus
University Medical Centre, Rotterdam, The Netherlands
Mark Estes III
University of Pittsburgh Medical Center, Pittsburgh, PA, USA
Javier Fernandez Sarabia
Paediatric and Adult Congenital Heart Centre, IRCCS-Policlinico san Donato University Hospital, Milan, Italy
Carla Frescura
Cardiovascular Pathology Unit, Department of Cardiac, Thoracic, and Vascular Sciences, University of Padua Medical School, Padua, Italy
Victor Froelicher
Sports Cardiology Clinic and Center for Inherited Cardiovascular Disease, Stanford School of Medicine, Stanford, CA, USA
Stephan Gielen
Department of Cardiology, Angiology, and Intensive Care Medicine, Klinikum Lippe, Detmold, Germany, and
Martin-Luther-University Halle/Wittenberg, Medical Faculty, Halle/Saale, Germany
Eduard Guasch
Hospital Clínic de Barcelona—IDIBAPS, University of Barcelona, Barcelona, Spain
Marco Guazzi
University of Milan, Italy
Martin Halle
Department of Prevention, Rehabilitation, and Sports Medicine, Technical University of Munich (TUM), Munich, Germany; and DZHK (German Centre for Cardiovascular Research), partner site Munich Heart Alliance, Munich, Germany
Kimberly Harmon
Center for Sports Cardiology, Department of Family Medicine, University of Washington, Seattle, WA, USA
Hein Heidbuchel
Antwerp University and Department of Cardiology, Antwerp University Hospital, Antwerp, Belgium, and Hasselt University, Hasselt, Belgium
Jakob Johansson
Institute of Medicine and Sports Science, Rome, Italy
Efraim Kramer
University of the Witwatersrand, Johannesburg, South Africa
André La Gerche
Department of Cardiovascular Medicine, University Hospital Leuven, Leuven, Belgium; Department of Sports
Cardiology, Baker IDI Heart and Diabetes Institute, Melbourne, Australia, and Department of Cardiology, Alfred Hospital, Melbourne, Australia
M. Harold Laughlin
Departments of Biomedical Sciences and Medical Pharmacology and Physiology, University of Missouri, Columbia, MO, USA
Mark Link
UT Southwestern Medical Center, Dallas, TX, USA
Aneil Malhotra
St. George’s University of London, Cardiology Clinical Academic Group, London, UK
Ronald J. Maughan
School of Medicine, University of St Andrews, St Andrews, UK
Clinic of Cardiology and Intensive Care Medicine, Bethanien Hospital Moers, Moers, Germany
Lluís Mont
Hospital Clínic de Barcelona—IDIBAPS, University of Barcelona, Barcelona, Spain
Stephan Mueller
Department of Prevention, Rehabilitation, and Sports Medicine, Technical University of Munich (TUM), Munich, Germany
Josef Niebauer
Institute of Sports Medicine, Prevention and Rehabilitation and Research Institute of Molecular Sports Medicine and Rehabilitation, Paracelsus
Medical University Salzburg; Institute of Sports Medicine of the State of Salzburg; and Sports Medicine of the Olympic Centre Salzburg-Rif, Salzburg, Austria
Natesa G. Pandian
Tufts Medical Center, Tufts University School of Medicine, Boston, MA, USA, and Hoag Hospital, Newport Beach, CA, USA
Nicole M. Panhuyzen-Goedkoop
Heart Centre, Department of Clinical and Experimental Cardiology, Amsterdam University Medical Centre, Amsterdam, The Netherlands
Michael Papadakis
Cardiovascular and Cell Sciences Research Centre, St George’s University of London, London, UK
Antonio Pelliccia
Institute of Sport Medicine and Science, Rome, Italy
Guido E. Pieles
Congenital Heart Unit, Bristol Royal Hospital for Children/Bristol Heart Institute, University Hospital Bristol NHS Trust, Bristol, UK
Massimo F. Piepoli
Heart Failure Unit, Cardiology Department, G. Da Saliceto Hospital, ASL Piacenza, Italy
Kalliopi Pilichou
Department of Cardiac, Thoracic, and Vascular Sciences, Cardiovascular Pathology Unit, University of Padua Medical School, Padua, Italy
Silvia G. Priori
Molecular Cardiology, IRCCS Salvatore Maugeri Foundation, Pavia, Italy; Department of Molecular Medicine, University of Pavia, Pavia, Italy; and Fundación Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain
Stefania Rizzo
Cardiovascular Pathology, Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, Padua, Italy
Mahdi Sareban
Paracelsus Medical University, Salzburg, Austria
Maurizio Schiavon
Sports Medicine and Physical Activities Unit, National Health Service—ULSS 6 Euganea of Padua, Italy
Christian Schmied
Department of Cardiology, University Hospital Zurich, Switzerland
Frédéric Schnell
Department of Sports Medicine, Pontchaillou Hospital, Rennes, and Department of Physiology, Rennes 1 University, Rennes, France
Julia Schoenfeld
Department of Prevention, Rehabilitation, and Sports Medicine, Technical University of Munich (TUM), Munich, Germany
Luis Serratosa
Hospital Universitario Quironsalud Madrid, Spain; and Ripoll & De Prado Sport Clinic, FIFA Medical Centre of Excellence, Spain
Sanjay Sharma
St George’s University of London, Cardiology Clinical Academic Group, London, UK
Nabeel Sheikh
Department of Cardiology and Division of Cardiovascular Sciences, Guy’s and St Thomas’ Hospitals and King’s College London, UK
S.M. Shirreffs
School of Medicine, University of St Andrews, St Andrews, UK
Erik Ekker Solberg Diakonhjemmet Hospital, Oslo, Norway
Ricardo Stein Internal Medicine Division, Hospital de Clínicas de Porto Alegre - Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Graham Stuart
Congenital Heart Unit, Bristol Royal Hospital for Children/Bristol Heart Institute, University Hospital Bristol NHS Trust, Bristol, UK
Carl-Johan Sundberg
Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden
Gaetano Thiene
Cardiovascular Pathology Unit, Department of Cardiac, Thoracic, and Vascular Sciences, University of Padua Medical School, Padua, Italy
Paul D. Thompson
Hartford Healthcare Cardiovascular Institute, Hartford, CT, USA
Department of Rehabilitation Sciences, Biomedical Sciences, University of Leuven, Leuven, Belgium
Benjamin S. Wessler
Tufts Medical Center, Tufts University School of Medicine, Boston, MA, USA
Arthur A.M. Wilde
Heart Centre, Department of Clinical and Experimental Cardiology, Amsterdam University Medical Centre, Amsterdam, The Netherlands
Matthias Wilhelm
Department of Cardiology, Interdisciplinary Center for Sports & Exercise Medicine, University Hospital Bern, Bern, Switzerland
Alessandro Zorzi
Department of Cardiac, Thoracic, and Vascular Sciences, University of Padua Medical School, Padua, Italy
SECTION 1
Physiology of cardiovascular response to exercise and cardiac remodelling
1.1 Cardiovascular response induced by exercise 3
1.1.1 Physiology of exercise 3
Andrew D’Silva and Sanjay Sharma
1.2 Long-term adaptation to exercise: athlete’s heart and vascular adaptations 9
1.2.1 Structural and functional adaptations in the athlete’s heart 9
Antonio Pelliccia and Stefano Caselli
1.2.2 Impact of sporting discipline, gender, ethnicity, and genetics on the athlete’s heart 20
Nabeel Sheikh
1.2.3 The athlete’s heart in children and adolescents 32
Graham Stuart and Guido E. Pieles
1.2.4 Vascular remodelling 41
Stephan Gielen, M. Harold Laughlin, and Dirk J. Duncker
Cardiovascular response induced by exercise
Contents
1.1.1 Physiology of exercise
1.1.1 Physiology of exercise
Andrew D’Silva and Sanjay Sharma
Introduction
Exercise physiology incorporates the study of both the acute responses and chronic adaptation of the human body to physical activity. The normal resting cardiac output is approximately 5L/min. The liver, kidneys, muscle, and brain account for the largest consumption of blood volume at approximate proportions of 27%, 22%, 20%, and 14%, respectively. The situation changes dramatically during strenuous exercise when the cardiac output is capable of increasing up to 25–35L/min. Skeletal muscle demands approximately 84% of this blood volume when exerting strenuously, and this is critical to satisfy the oxygen demand for ATP generation and carbon dioxide removal necessary for acid–base homeostasis [1]. Athletes capable of sustaining the highest cardiac outputs during prolonged exercise achieve this through chronic adaptation, particularly of the heart and cardiovascular system. Through training, the heart is capable of adapting in structure and function to meet the demands of an increasing workload. Despite this, maximal cardiac output remains a major limitation on oxygen delivery and therefore limits exercise capacity. For the purposes of this chapter, the heart should be considered as both a pump for delivering oxygen to the tissues and carbon dioxide to the lungs, and a highly specialized striated muscle with substantial energy demands.
We begin by outlining the respiration requirements of every muscle cell, including cardiac and skeletal muscle and how the demands of exercise are met.
Exercise and the muscle cell
Fundamentally, exercise is an energetic process that reflects an upregulation of the essential oxidation of metabolic substrate to support physical activity. Oxygen facilitates oxidative chemical reactions to create high energy bonds in compounds, chiefly in the terminal phosphate bond of ATP, which is the cellular currency for energy transformation. Exercise demands that chemical energy is converted into kinetic energy at a high rate and mass scale. When energy is borrowed from alternative anaerobic sources it must also be repaid.
At the level of striated muscle, ATP is split to release energy to generate mechanical force through the interaction between the myosin head and actin. Occurrence of this process on a large scale, throughout the entire muscle, results in fibre shortening and muscle contraction downstream. Exercise demands high levels of ATP and therefore high oxygen consumption, which is necessary to power this contractile machinery.
Even when physical activity is initiated abruptly there is no delay in achieving sufficient ATP levels, as one would anticipate if ATP were sourced from the oxidation of metabolic substrates. From an evolutionary perspective it is prudent to be able to summon energy immediately to escape an unanticipated threat. This is achieved by having stores of high energy phosphate bonds in the form of phosphocreatine, which are rapidly hydrolysed in early exercise by creatine kinase to release inorganic phosphate and generate ATP. However, as this becomes rapidly depleted, sustained exercise depends on the aerobic oxidation of metabolic substrates, predominantly glycogen and fatty acids for the continued regeneration of ATP.
Andrew D’Silva and Sanjay Sharma 3
Human skeletal muscle contains approximately 15–18g of glucose per kilogram stored as glycogen, which can be broken down into glucose for utilization through glycogenolysis. The liver also has a fluctuating glycogen reserve that can be converted into glucose and released into the bloodstream when required. In addition, during severe and prolonged exercise the liver ensures that plasma glucose levels do not fall and maintains a constant glucose supply from the conversion of non-carbohydrate precursors through gluconeogenesis. Both glycogenolysis and gluconeogenesis are stimulated by a rise in circulating adrenaline. When glucose is metabolized as the metabolic fuel for energy, through glycolysis, the net yield is 36 molecules of ATP per molecule of glucose. Six oxygen molecules are consumed and six carbon dioxide molecules are produced for every single glucose molecule, and the result is a respiratory quotient of 1.0 for the oxidation of glucose:
Fatty acids are also catabolized as an energy substrate. Fat metabolism provides more energy and greater storage economy than carbohydrate; however, it is more expensive in terms of oxygen consumption. Therefore, during conditions of strenuous exercise where oxygen transport is a limiting factor, muscle metabolism preferentially utilizes carbohydrate as a fuel, which yields ATP at a rate four times faster than the oxidation of fatty acids [2].
When exercise is sufficiently intense and prolonged, oxygen delivery to the muscle mitochondria becomes a limiting factor and is unable to match the rate of ATP consumption. At this point, the body is able to continue to generate energy for muscle contraction in the absence of oxygen through anaerobic glycolysis, albeit at a lower yield of ATP and at greater metabolic cost due to the development of lactic acidosis. Muscle groups experiencing inadequate oxygen flow to meet demand will initiate anaerobic cellular metabolism to augment ATP production once aerobic metabolism reaches maximal capacity. The net gain of ATP is only three molecules from each molecule of glucose metabolized into two lactate molecules. This pales in comparison with the 36 ATP molecules generated from the complete oxidation of glucose to carbon dioxide and water via the aerobic pathway.
Acid–base homeostasis is of vital importance in the cell as acidosis can denature proteins, altering their molecular structure and affecting enzymatic activity. Severe acidosis reduces myocardial contractility directly and by causing resistance to beta-adrenergic stimulation. Although the effects of acidosis are predominantly negative, mild acidosis promotes aerobic cellular respiration by allowing oxygen to dissociate from haemoglobin more readily and increasing
the availability of oxygen to the muscle. Hence the body regulates pH extremely tightly through the use of multiple buffering systems, including bicarbonate ions, and by elimination of carbon dioxide from the lungs. When bicarbonate ions accept protons from acid they form carbonic acid, which is catalysed to carbon dioxide and water by the enzyme carbonic anhydrase, which is present in erythrocytes. Elimination of carbon dioxide from the lungs ensures that carbonic acid dissociation leaves water as the only byproduct. The bicarbonate buffer system is described by
In the early stages of exercise, energy is generated by the most efficient method, aerobic glycolysis, where oxygen consumption is coupled to ATP production. As exercise progresses, a point is reached where oxygen transport to muscle cells is insufficient to meet the demands required to maintain energy production and anaerobic metabolism begins to contribute. Beyond this anaerobic threshold, an oxygen debt accumulates and lactic acid is generated. In this phase, oxygen uptake continues to rise throughout exercise, persisting into the recovery phase where the oxygen debt must be repaid. The ceiling value of oxygen uptake achieved at exhaustive exercise is termed the peak oxygen consumption, or peak VO2. Peak oxygen consumption is a key determinant of exercise performance and reflects the integrative function of muscle metabolism, oxygen transport through the cardiovascular system, and pulmonary ventilation. An impediment in any of these systems will limit peak oxygen consumption, which can be expressed as the product of cardiac output (CO) and systemic arteriovenous oxygen difference (C(a-v)O2), described in the Fick equation:
The Fick equation defines the entire oxygen transport and utilization capacity of the system. The principles discussed here, at the cellular level, are fundamental to understanding and interpreting cardiopulmonary exercise tests. In a cardiopulmonary exercise test, the respiratory exchange ratio is a useful parameter for assessing whether maximal effort was achieved. At rest, the body metabolizes both carbohydrate and fat to generate ATP. As fat metabolism yields a respiratory quotient of 0.71, this means that, at rest, oxygen consumption (VO2) is slightly greater than carbon dioxide production (VCO2), resulting in a respiratory exchange ratio of 0.8–0.9. On commencing exercise, active muscle exclusively metabolizes carbohydrate for the generation of ATP. As the respiratory quotient of carbohydrate is balanced at 1.0, carbon dioxide production is equal to oxygen consumption in early exercise, when glycolysis is entirely aerobic. This
results in a rise of the respiratory exchange ratio to 1.0. With more prolonged and strenuous exercise anaerobic glycolysis of carbohydrate ensues, generating lactic acid which must be buffered to maintain cellular pH. Ultimately, bicarbonate ions accept protons to form carbonic acid, which upon dissociation results in a substantial rise in carbon dioxide relative to oxygen consumption, and this requires ventilatory elimination. In this phase of exercise, the respiratory exchange ratio rises above 1.0 and a ratio of greater than 1.15 indicates excellent effort with confidence that the anaerobic threshold was exceeded.
Understanding these principles permits calculation of the ventilatory anaerobic threshold (AT); the point during exercise where anaerobic metabolism contributes to energy production and lactic acid is produced. The AT can be determined by plotting oxygen consumption (VO2) against carbon dioxide production (VCO2) and determining the point of intersection of two lines of best fit. This is known as the V-slope method and is commonly used in cardiopulmonary exercise testing to determine the AT (% Fig. 1.1.1.1). The AT usually occurs at 50–60% of the peak oxygen consumption in normal individuals, although it can be increased to 80% of peak oxygen consumption in endurance-trained athletes.
Exercise and the cardiovascular system
Fig. 1.1.1.1 Carbon dioxide output (VCO2) and heart rate (HR) vs oxygen uptake (VO2). Carbon dioxide output increases linearly with oxygen uptake with a slope of 1 up to the anaerobic threshold. After the anaerobic threshold carbon dioxide output increases more rapidly, with steepening of the slope depending on the rate of lactic acid buffering. Heart rate increases linearly with oxygen uptake. The V-slope method of determining ventilator anaerobic threshold (AT) is demonstrated by intersecting lines. The respiratory compensation threshold is indicated by RC.
At rest, a proportion of muscle capillaries have little or no blood flow. The role of these dormant capillaries and arterioles is to vasodilate during strenuous exercise, allowing greater diffusion of oxygen and nutrients into contracting muscles. Vasodilatation is triggered by local tissue depletion of oxygen, which is rapidly consumed by active muscle. Oxygen deficiency stimulates the release of vasodilator substances including adenosine, which promotes vasodilatation in early exercise, and potassium ions, ATP, lactic acid, and carbon dioxide maintain this process throughout the rest of exercise. Local vasodilatation is accompanied by activation of the sympathetic nervous system, which results in a sustained increase in cardiac output and arterial pressure. At the onset of exercise, the brain initiates movement from the motor cortex and simultaneously recruits the vasomotor centre which activates the sympathetic nervous system. Sympathetic activity on the heart results in greater contractile force (inotropy) and an increase in heart rate (chronotropy). As cardiac output is the product of stroke volume and heart rate, adequate function of the sympathetic nervous system is essential to achieving high performance exercise. Sympathetic vasoconstrictor nerves exert their action through the release of noradrenaline at their nerve endings and innervate most arterioles of the peripheral circulation. In concert with this, the adrenal medulla secretes large amounts of noradrenaline and adrenaline into the circulation during exercise, and the consequence is widespread vasoconstriction of peripheral blood vessels except for arterioles in active muscles, which are overwhelmed by the local vasodilator effects described above. This diversion of blood is responsible for increasing muscle blood flow by approximately 2L/min. The coronary and cerebral circulations are central circulatory systems, vital for sustaining prolonged exercise, and are spared this vasoconstrictor effect as they possess little vasoconstrictor innervation. Approximately two-thirds of the total blood volume resides within the veins and their distensible capacitance vessels. This vascular bed is the seat of the most profound vasoconstrictor effects. Vasoconstriction of the muscular venous walls produces a significant rise in the systemic venous filling pressure, which increases venous return of blood to the heart that is critical for increasing cardiac output and eliminating carbon dioxide. These combined effects of the sympathetic nervous system raise the systemic arterial pressure, which drives muscle perfusion. Considering the circulation as a closed hydraulic circuit, in order to enhance oxygen transport blood flow rates to exercising muscles must be increased. A high flow rate within the arteries and arterioles supplying muscle is maintained by
an elevated systemic arterial pressure, which also stretches the vessel walls, increasing vessel cross-sectional area to further augment blood flow. Elevation of systemic blood pressure during exercise and arterial distensibility are crucial for those endurance athletes who demand up to 35L/ min of cardiac output over a sustained period to maintain physical activity at a high level.
With respect to the heart, cardiac output is increased by increasing stroke volume and raising the heart rate. Stroke volume is regulated during exercise by intrinsic forces and extrinsic factors. The intrinsic force is governed by the Frank–Starling relationship. During exercise venous return rises, resulting in an increase in ventricular volume loading and an increased diastolic filling pressure. The magnitude of myocardial fibre stretch in diastole governs the contractile force and stroke volume. Therefore intrinsic regulation of the myocardial contractile force allows automatic adjustment of stroke volume to match venous return. In addition, extrinsic factors also increase the myocardial contractile force, generating positive inotropy independent of myocardial fibre tension, which is achieved by the sympathetic nervous system. The myocardium has a high proportion of β1-adrenoreceptors, which become activated by circulating adrenaline and noradrenaline. On cardiopulmonary exercise testing, the oxygen pulse is a surrogate of stroke volume. Oxygen pulse is calculated by dividing oxygen uptake by heart rate and therefore represents the stroke volume multiplied by the arteriovenous blood oxygen content difference:
1.1.1.2 Heart rate (HR) and oxygen pulse (VO2/HR) vs time (t).
Oxygen pulse, which is a surrogate of stroke volume, rises first and reaches a plateau earlier, followed by heart rate. The vertical dashed lines represent the ventilatory anaerobic threshold (AT) and the respiratory compensation threshold (RC). The normal range of maximal heart rate is indicated (HR Max).
between successive action potentials to be reduced. The atrioventricular node is also under the influence of sympathetic innervation and will permit faster conduction when stimulated by noradrenaline.
When initial rises in stroke volume alone are insufficient to meet the demands of intense exercise, sympathetic nerves arising from the thoracic sympathetic chain stimulate an increase in heart rate. Whilst the heart rate is capable of rising substantially during exercise, efficiency at higher rates is compromised due to shortening of ventricular filling time in diastole, which results in an impairment of stroke volume. In health, heart rate rises in direct proportion to oxygen uptake to the point of exhaustion, where the physiological limit to heart rate is reached. Achievement of an estimated maximum predicted heart rate based on age can provide some supportive evidence of exhaustive effort during exercise (% Fig. 1.1.1.2).
Positive chronotropy is achieved by noradrenergic stimulation of the sinoatrial node, which increases the permeability of the pacemaker cells to sodium and calcium ions. This results in an increase in the slope of the action potential, allowing the sinoatrial node pacemaker cells to reach the activation threshold more quickly and the interval
Exercise and the respiratory system
Oxygen is the key to unlocking chemical energy in the cell and regenerating ATP. Ventilation is a crucial factor not only for providing an adequate source of oxygen but also for the excretion of carbon dioxide, which is essential for maintaining acid–base homeostasis. In health, the lungs are easily able to meet the ventilatory requirements for efficient gas exchange, even during maximal exercise when ventilation can increase from approximately 6L/min at rest to over 100L/min. An increase in the depth and rate of ventilation during exercise allows not only replenishment of oxygen but, more importantly, the elimination of carbon dioxide. The control of breathing during exercise is under the influence of many complex integrating neuroanatomical pathways and reflexes.
The accumulation of lactic acid beyond the anaerobic threshold is initially buffered by bicarbonate ions, generating an excess of carbon dioxide. This increase in carbon
Fig.
dioxide level is sensed by chemoreceptors, located in the carotid bodies, which stimulate an increase in ventilation via the medullary respiratory centre. Once the lactic acid buffering capacity of the blood is exceeded during strenuous maximal exercise, the blood pH falls and this is an additional extremely potent trigger for chemoreceptor-mediated stimulation to drive dramatic hyperventilation. The considerable rise in ventilation can be detected on the cardiopulmonary exercise test and is termed either respiratory compensation for acidosis or the ventilatory compensation threshold (% Fig. 1.1.1.3(a)).
At low intensity exercise, ventilation increases initially by an increased depth of breathing or tidal volume. After the tidal volume reaches approximately 50–60% of the vital capacity, further increases in ventilation are achieved by increases in respiratory rate. Minute ventilation (VE) is the product of tidal volume and respiratory rate, which rises continuously throughout exercise approaching the maximum breathing capacity. Gas exchange becomes even more efficient during exercise, through increased pulmonary capillary perfusion and greater recruitment of alveoli, resulting in greater uniformity in ventilation–perfusion matching. The improvement in ventilation–perfusion matching depends not only on increasing the alveolar surface area for
gas exchange but also on the adequacy of the circulation, providing sufficient venous return to the right ventricle, good right ventricular function, and responsive pulmonary vasculature.
Although oxygen is consumed at a high rate during strenuous exercise and the arteriovenous oxygen content difference becomes greater, the ability of the lungs to source sufficient oxygen and enrich the blood is not a limiting factor in healthy individuals. Increases in minute ventilation are driven primarily by carbon dioxide production, acidosis, and central command from the brain. Ventilatory efficiency is best represented by the rise in minute ventilation per litre of carbon dioxide output. This relationship is linear below the ventilatory compensation threshold and the normal VE/ VCO2 slope is approximately 25 (% Fig. 1.1.1.3(b)). Any condition that prevents air taken in during a breath from participating in gas exchange increases the physiological dead space in the airways. This may be a pulmonary condition that disturbs alveolar function or a circulatory disorder limiting alveolar perfusion and the delivery of carbon dioxide to the lungs. The result in either case is a steepening of the VE/VCO2 slope with high minute ventilation relative to the carbon dioxide output. This manifests clinically as
(a)
ventilation
and work (Power) vs time. The vertical dashed lines represent the ventilatory anaerobic threshold (AT) and the respiratory compensation threshold (RC). The horizontal blue line indicates the estimated maximal ventilatory volume (MVV). (b) Minute volume (VE) vs carbon dioxide output (VCO2). The relationship of this plot is linear until the respiratory compensation threshold is reached. In health the slope is less than 30. The horizontal blue line indicates the estimated maximal ventilatory volume (MVV). The ventilatory anaerobic threshold and respiratory compensation threshold are indicated by AT and RC, respectively. The normal range of vales is indicated (VE/VCO2).
Fig. 1.1.1.3
Minute
(VE) (t)
uncomfortable dyspnoea, which limits functional capacity in disease states.
Alveolar ventilation and pulmonary perfusion both increase during exercise, which results in greater uniformity in ventilation–perfusion matching. End-tidal oxygen levels are relatively stable during early exercise and rise at the ventilatory compensation threshold due to metabolic acidosis-induced hyperventilation. End-tidal carbon dioxide levels increase minimally in the early stages of incremental exercise, resulting from the increase in production from substrate metabolism coupled with the release from the bicarbonate buffering of lactic acid. Once the ventilatory compensation threshold is passed in heavy exercise, acidosis-driven hyperventilation dramatically augments minute ventilation, resulting in a fall in end-tidal carbon dioxide levels.
In summary, exercise reflects the ability of the human body to summon skeletal muscle activity with immediacy and for prolonged periods. From an evolutionary perspective, the ability to fight or flee effectively is a critical determinant of species survival. Physical activity is the culmination of an impeccably orchestrated arrangement incorporating the brain, nervous system, heart, circulation, lungs, adrenal glands, and muscles. Each system has a part to play in energy generation, oxygen uptake, and carbon dioxide elimination, with the ability to respond and modulate outputs akin to fine tuning. It is remarkable how cardiac output and pulmonary
ventilation are so precisely adjusted to meet the metabolic demands of exercise until fatigue. The versatility of how chemical energy can be sourced from various substrates, stored, and borrowed permits the necessary transformation from chemical to kinetic energy, harnessing locomotive power. Exercise performance is limited by the role of the cardiovascular system in oxygen transport, although athletes demonstrate considerable functional remodelling of the heart to deliver greater cardiac output and better accommodate the repeated physiological stresses of exercise.
Further reading
Jones NL, Killian KJ. Exercise limitation in health and disease. N Engl J Med 2000; 343(9): 632–41.
Laughlin MH. Cardiovascular response to exercise. Am J Physiol 1999; 277 (6 Pt 2): S244–59.
Wasserman K, Hansen J, Sue D, et al. Principles of Exercise Testing and Interpretation (4th edn) Philadelphia, PA: Lippincott Williams & Wilkins, 2005.
References
1. McArdle W, Catch F, Catch V. Functional capacity of the cardiovascular system. Exercise Physiology: Nutrition, Energy, and Human Performance (8th edn), Chapter 17. Baltimore, MD: Wolters Kluwer, 2014 , p. 347.
2. Jones NL, Killian KJ. Exercise limitation in health and disease. N Engl J Med 2000; 343(9): 632–41.
1.2 Long-term adaptation to exercise: athlete’s heart and vascular adaptations
Contents
1.2.1 Structural and functional adaptations in the athlete’s heart Antonio Pelliccia and Stefano Caselli 9
1.2.2 Impact of sporting discipline, gender, ethnicity, and genetics on the athlete’s heart Nabeel Sheikh 20
1.2.3 The athlete’s heart in children and adolescents Graham Stuart and Guido E. Pieles 32
1.2.4 Vascular remodelling Stephan Gielen, M. Harold Laughlin, and Dirk J. Duncker 41
1.2.1 Structural and functional adaptations in the athlete’s heart
Antonio Pelliccia and Stefano Caselli
Historical perspective: from chest percussion to 3D imaging
The human body has an extraordinary ability to respond to environmental changes in a way that guarantees vital function and survival under many circumstances. Sport activity is an example of this impressive adaptation capacity, with a series of integrated changes in structure and function of different organs and systems within the body which allow the accomplishment of greater exercise performance.
The cardiovascular system goes through many adaptations as a consequence of exposure to chronic exercise training, which have been described by scientists for more than a century. Indeed, the first observations regarding cardiac changes associated with high intensity exercise training date back to 1899, when Henshen, from the University of Uppsala, in his publication ‘A study in sports medicine;
skiing and competitive skiing’, stated that ‘skiing causes an enlargement of the heart, and this enlarged heart can perform more work than a normal heart. There is, therefore, a physiologic enlargement of the heart due to athletic activity, i.e. the ‘Athlete’s Heart’ [1]. This accurate definition is impressive considering that the assessment was conducted solely by physical examination consisting of carefully performed chest percussion.
Subsequently, radiological techniques to assess the heart volume in athletes were introduced by the Scandinavian scientists Rohrer and Kahlstorf [2]. Cardiac dimensions were measured using X-rays in athletes participating in the 1928 Olympic Games in Amsterdam, and it was found that cardiac size varied in athletes engaged in different sports, with athletes engaged in endurance disciplines showing the most enlarged cardiac silhouette [3,4]. Subsequently, Reindell and Hollman, using chest X-rays, reported that an increased heart volume was associated with improved physical performance, with athletes with the greatest increase in X-ray cardiac dimension also showing the highest aerobic capacity, as expressed by maximum oxygen uptake [5,6].
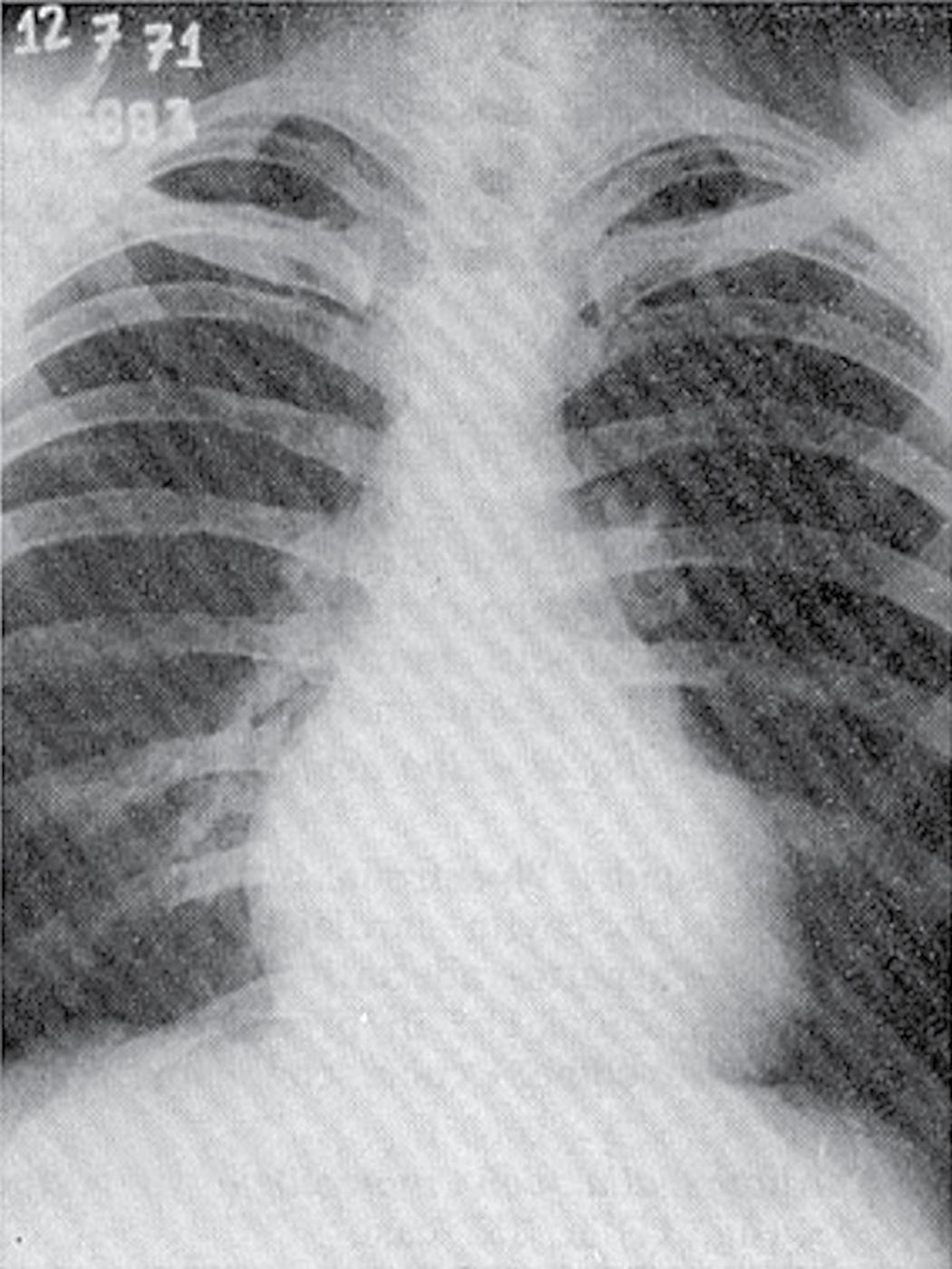
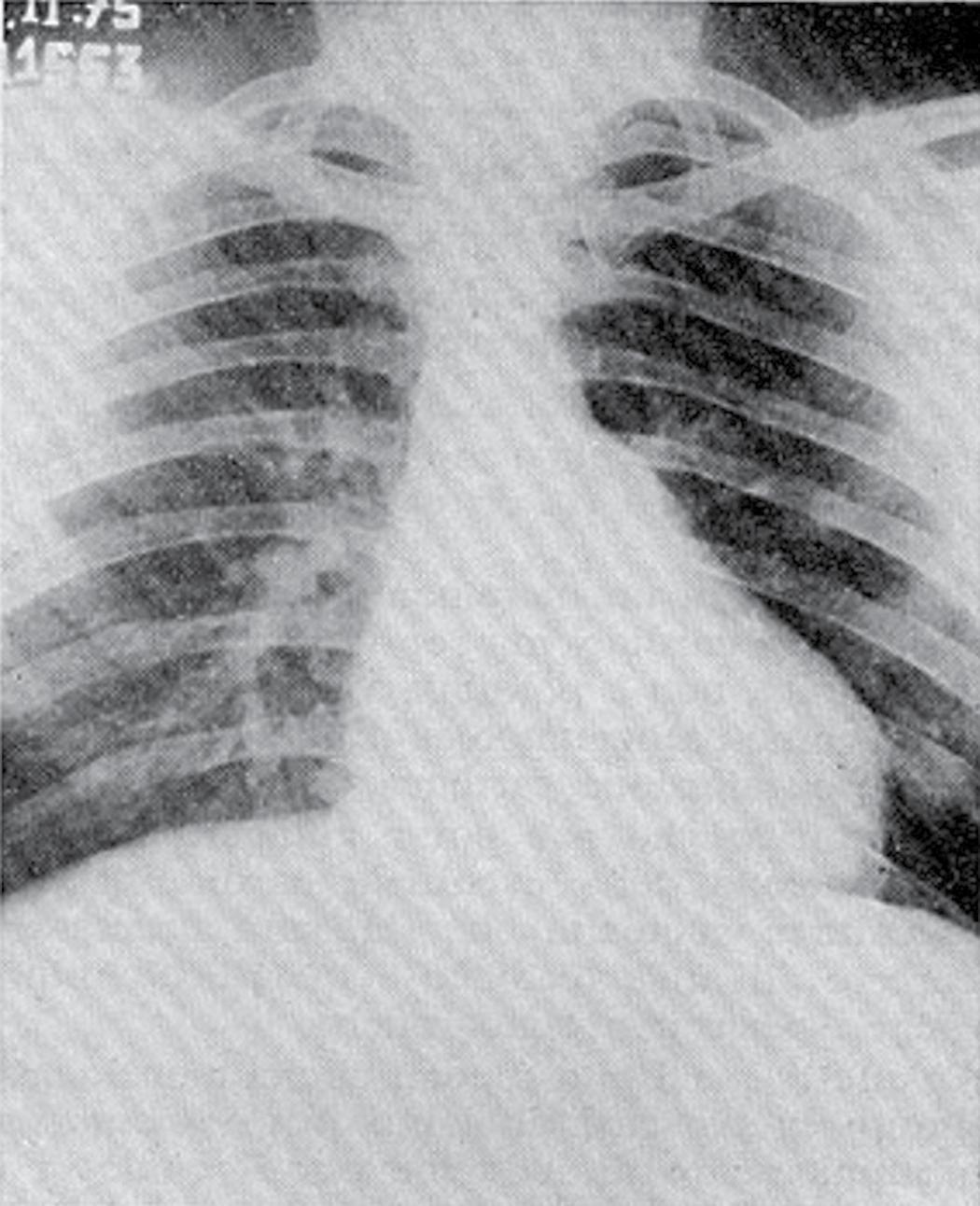
Substantial contributions to the clinical issues surrounding the ‘athlete’s heart’ originated in Italy in the 1930s, when Cassinis published Controllo Medico dello Sport (Medical Evaluation in Sport) [7], the first manual dedicated to the evaluation of athletes. Subsequently, Venerando and colleagues at the Institute of Sports Medicine in Rome performed a series of radiological and electrocardiographic studies of athletes participating in the 1960 Olympic Games [8]. They made several intriguing observations: a variety of cardiac X-ray silhouettes were described in trained athletes (% Fig. 1.2.1.1), in selected cases mimicking those found in patients with valvular or congenital heart disease which raised questions regarding
Fig. 1.2.1.1 Chest X-rays from (a) a 27-year-old male professional cyclist, showing a symmetrically enlarged cardiac silhouette, and (b) a 24-year-old male elite water polo player, showing an enlarged cardiac silhouette suggestive of distinctly enlarged LV dimensions. An increased pulmonary vascular tree is also evident in both X-ray images,.
differential diagnosis between cardiac disease and physiological heart adaptation [9].
Echocardiography, introduced in clinical cardiovascular practice at the end of the 1970s, gave a great impulse to investigation of the morphological and functional features of the ‘athlete’s heart’. In 1975 the first comparative echocardiographic study of athletes engaged in endurance-type and strength-type sports was published by Morganroth et al. in the Annals of Internal Medicine [10]. This study compared M-mode echocardiographic measurements of untrained subjects, strength-trained athletes, and endurance-trained athletes. The authors observed that athletes engaged in endurance training showed eccentric left ventricular (LV) remodelling (as a consequence of volume overload), while those in strength training showed concentric hypertrophy (as consequence of pressure overload). Therefore they hypothesized that morphological adaptations in athletes could be related to the type of haemodynamic overload associated with the specific exercise training.
This investigation pioneered a large series of studies assessing the structural changes, upper limits. and clinical correlates of cardiac remodelling in trained athletes. In this context, in 1991 Pelliccia et al. published a pivotal study in the New England Journal of Medicine describing the characteristics and upper limits of LV hypertrophy derived from a large athlete cohort from the database of Institute of Sports Medicine and Science [11]. A few years later, in 1995, the largest investigation describing the characteristics of ‘athlete’s heart’ in women appeared in the Journal of the American Medical Association [12], and in 1999 a report on the morphological features and clinical correlates of LV dilatation was published in the Annals of Internal Medicine [13]. Since then, reports of several subsequent studies have been published describing the determinants of cardiovascular adaptations to exercise training according to age, gender, ethnicity, and the sport participated in [14–17].
In 1976, two episodes of sudden cardiac death in young competitive athletes occurred within 8 weeks of each other, one due to aortic rupture caused by Marfan syndrome and
(a) (b)
one secondary to hypertrophic cardiomyopathy (HCM). These tragic events attracted the attention of the media and raised the consciousness of the medical community about the potential harm of intense exercise activity in individuals with inherent, often silent, cardiovascular disease [18,19]. These and other detrimental events in subsequent years resulted in a major drive towards the timely identification of cardiac diseases capable of causing sudden cardiac death (SCD) in competitive athletes, and the utility of implementing pre-participation screening programmes to prevent such catastrophes. As a consequence, a large literature has been produced regarding the criteria for identifying the risk of cardiac disease in athletes and the differentiation of physiological cardiac remodelling from cardiac pathology.
Of particular note, a study from the Veneto region of Italy published by Corrado et al. in 2006 [20] demonstrated that, after implementing the mandatory preparticipation screening (including 12-lead ECG), the incidence of SCD in screened athletes progressively declined, mainly due to the ability to identify athletes with underlying cardiomyopathies. Many studies have been performed subsequently in order to emphasize the importance of ECG screening in athletes as a model for identifying pathological conditions at risk of SCD.
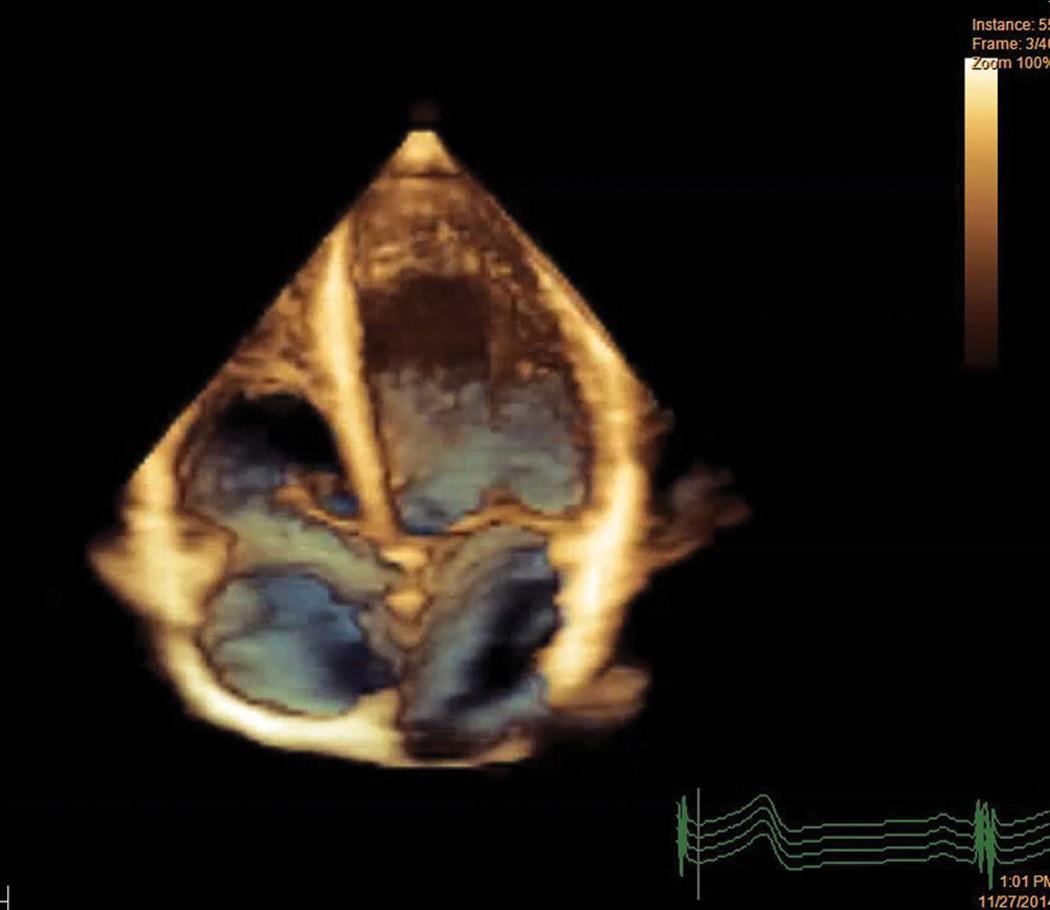
In this scenario, cardiac imaging has continuously evolved with substantial advances in cardiac magnetic resonance (CMR) and newer echocardiographic techniques (such as 3D echocardiography (% Fig. 1.2.1.2) and speckle tracking imaging). These highly sophisticated techniques have led to further
clarification of the geometrical characteristics of cardiac chamber remodelling in athletes and further insight into the physiology of myocardial contraction by making a relevant contribution to the differential diagnosis of physiological cardiac remodelling from cardiovascular disorders, and are part the complete evaluation of athletes in the modern era [21–24].
Structural and functional adaptations of cardiac chambers
The cardiovascular system undergoes several modifications as a consequence of exercise training, with acute responses, which occur within few seconds of intensive exercise, and chronic adaptations that include more profound structural remodelling and are a result of long-term conditioning [25,26]
The acute response to exercise training includes substantial increases in heart rate, stroke volume, cardiac output, systolic blood pressure, and maximum oxygen consumption. Chronic cardiovascular adaptations to exercise training determine structural remodelling of the cardiac chambers and vessels which facilitates an increased capacity to deliver oxygen to the working muscles during prolonged bouts of exercise. Expression of cardiac remodelling consists of an increase in left and right ventricular and left atrial cavity size associated with normal (or even improved) diastolic function.
LV wall stress seems to be the most important determinant of LV remodelling, and most changes in LV geometry can be explained by the Laplace law:
LVwallstress
(LVpressure radius )/(2 LV wall thickness)
= ××
This law describes the factors that determine LV wall stress, which is a major determinant of myocardial hypertrophy. LV wall stress is the force acting against the myocardial cells. This is directly proportional to the ventricular pressure and radius, and inversely proportional to the wall thickness. LV pressure increases in conditions with high afterload, such as systemic hypertension and aortic valve stenosis, while LV radius increases in states of high preload, such as aortic or mitral regurgitation. An increase in LV wall thickness is necessary to normalize wall stress in situations associated with an increase in preload and/or afterload (% Fig. 1.2.1.3) [27].
The biomechanical stress induced by the haemodynamic overload stimulates the release of angiotensin II which, in association with other humoral factors and hormones (growth factors, insulin, IGF-1) leads to ‘beneficial’ adaptive changes in the cardiac myocyte. Experimental models show that exercise training is associated with an over-expression of the isoform α of the myosin heavy chain, which is associated with enhanced cardiac contractility [28]. However, the
Fig. 1.2.1.2 Three-dimensional echocardiographic evaluation of the athlete’s heart. This new technology allows accurate morphological assessment of cardiac structures as well as evaluation of size and function of all cardiac chambers. Notice the global and symmetric enlargement of all cavities, with preserved global left and right cavity shape.
Fig. 1.2.1.3 Cardiac adaptations to athletic training can be explained by the Laplace law: the increase in LV thickness reduces myocardial wall tension that occurs with increased chamber size at a given pressure.
adult cardiomyocyte is a post-mitotic cell without regeneration capacity, and therefore myocardial hypertrophy may occur only through an increase in cellular size [29].
At the molecular level, physiological training is associated with an over-expression of the isoform α of the myosin heavy chain, which enhances cardiac contractility. The expression of mRNA for the α myosin heavy chain is an early event in the adaptation to chronic exercise and occurs before cardiac growth is evident [29,30]. In addition to the hypertrophic growth of contractile fibres, experimental studies suggest that improved LV function in trained animals can be ascribed to an alteration of the Ca2+ regulatory systems involved in the excitation–contraction coupling and relaxation processes [31–33].
Consistent with the increase in myocardial mass, adaptations in coronary circulation have been described, including increased vessel size and number and improved endothelial function which ultimately lead to increased coronary flow and myocardial oxygen supply [27,34].
Determinants of cardiac hypertrophy
Macroscopic structural remodelling of the athlete’s heart has been extensively studied using echocardiography for almost four decades and this has enabled an appreciation of the determinants and upper limits of the cardiac adaptation in highly trained athletes. It is now well recognized that age, sex, ethnicity, body size, type of sport, and amount of training (with a dose–effect relation) are the main determinants of remodelling, and therefore structural adaptation may vary considerably between one athlete and another. However, the relative contribution of demographic and environmental or genetic determinants of LV remodelling in trained athletes has long been a subject of controversy [35].
Multivariate analysis of data obtained from large athlete populations shows that 75% of variability in LV cavity size is attributable to non-genetic factors, such as body size, type of sport, gender, age, and anthropometric features [12]. The remaining 25% of variability is otherwise unexplained, and is possibly due in part to genetic factors [12,35].
The pattern and magnitude of physiologically increased LV mass varies with respect to the type and intensity of exercise training. The most extreme increases in LV cavity dimension and/or wall thickness are observed in athletes engaged in endurance sports such as rowing, cross-country skiing, cycling, and swimming (% Fig. 1.2.1.4). However, other than the sport discipline itself, cardiovascular adaptation may rely on the intrinsic characteristics of training. For instance, the
of
L.D. RUNNING
Fig.
Effects on LV wall thickness
haemodynamic load may vary even within the same discipline; the paradigm is soccer where the goalkeeper is subject to a different haemodynamic load compared with the striker or midfield players. Indeed, the individual characteristics of the athlete (age, gender, body size, and composition) significantly affect the extent of cardiac remodelling.
For didactic purposes, exercise physiology can be dichotomously divided into isotonic (dynamic) and isometric (static) components [36,37]. Chronic isotonic exercise imparts a volume overload to all cardiac chambers which induces cavity enlargement, whereas isometric exercise is associated with short bursts of a significant increase in afterload. Although the afterload may be very intense, its shorter duration has a lower impact on LV dimensions compared with isotonic exercise [38]. However, this dichotomous classification cannot be applied to the majority of sports disciplines, which may include a varying mixture of components of dynamic
and static exercise. Therefore knowledge of the specific exercise training of the athlete is mandatory to improve understanding of the characteristics of cardiac remodelling .
Left heart dimensions and function in athletes
LV wall thickness in athletes varies with regard to sex, ethnicity, and type of sport, but in the vast majority of cases remains within normal limits. The greatest degree of LV hypertrophy is usually seen in male athletes, but values >12mm are observed in less than 2% of Caucasian athletes (% Fig. 1.2.1.5). Conversely, in female athletes the degree of hypertrophy is usually milder, with values extending up to 11mm. Interestingly, while white athletes rarely demonstrate an increase in wall thickness in the range overlapping with pathological LV hypertrophy such as HCM, a larger
Fig. 1.2.1.5 (a) Distribution of LV wall thickness in a large population of Italian athletes. An increase in wall thickness >12mm (dotted line) is observed in 2% of male and 0% of female athletes. (b) Distribution of LV cavity size in a large population of Italian athletes. Of the overall population, 14% had values >60mm and up to 70mm. (c) Distribution of left atrial dimension in a group of highly trained Italian athletes. The dotted line represents the cut-off of 40mm; approximately 20% of individuals showed an increase in left atrial size, with only 2% showing marked left atrial enlargement >45mm. (d) Distribution of aortic root dimensions in a group of highly trained Italian athletes. The dotted line represents the cut-off of 40mm.
proportion of black male athletes (up to 18%) may present with LV wall thickness >12mm (% Fig. 1.2.1.6) [11,16,39,40].
An increased LV chamber dimension in trained athletes has been well documented. LV cavity dimensions vary widely with respect to type of sport and gender, and may be strikingly enlarged, with end-diastolic values ≥ 60mm in almost 15% of highly trained athletes [13] ( % Fig. 1.2.1.5). This chamber enlargement may be accompanied by a small increase in absolute LV wall thickness exceeding upper normal limits (range 13–15mm) when endurance training is associated with components of strength training [11].
In a 3D echocardiography study, LV end-diastolic volume in endurance athletes was on average 50% than in untrained subjects. An interesting and practical observation is that the increase in LV cavity size is always associated with a consistent increase in LV mass, suggesting that in the physiological adaptation to exercise training there is always a balanced and homogeneous remodelling, with consistency in terms of increased LV volume and mass [17] (% Fig. 1.2.1.7).
Because of the large LV cavity volume, the heart of an athlete is capable of a high stroke volume. The increased stroke volume, coupled with increased vagal tone, explains why trained athletes usually have low heart rates. The LV